languedoc - roussillon - montpellier · languedoc - roussillon - montpellier clinical research...
TRANSCRIPT

Discipline: Radiation Oncology Dr David AZRIA
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
C O N F I D E N T I A L
1
CLINICAL RESEARCH REGIONAL DEPARTMENT
Languedoc - Roussillon - Montpellier
Clinical Research Protocol
PHRC 2005
- National Grant -
Dr David AZRIA
Discipline
Radiation Oncology
Project Title: Multicenter prospective evaluation of a test to predict late toxicities after
radiotherapy by measuring the radiation-induced CD8 lymphocyte apoptosis rate.
Application to breast and prostate cancer.
Discipline: Radiation Oncology Dr David AZRIA
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
C O N F I D E N T I A L
2
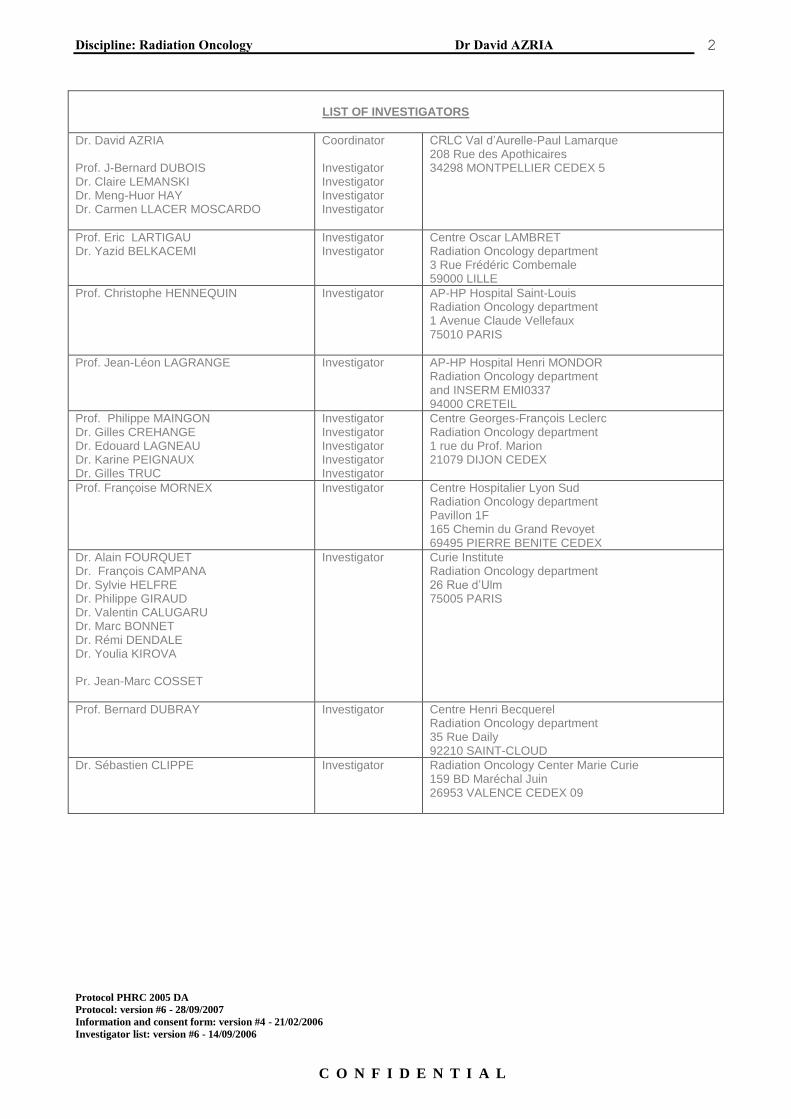
LIST OF INVESTIGATORS
Dr. David AZRIA Prof. J-Bernard DUBOIS Dr. Claire LEMANSKI Dr. Meng-Huor HAY Dr. Carmen LLACER MOSCARDO
Coordinator Investigator Investigator Investigator Investigator
CRLC Val d’Aurelle-Paul Lamarque 208 Rue des Apothicaires 34298 MONTPELLIER CEDEX 5
Prof. Eric LARTIGAU Dr. Yazid BELKACEMI
Investigator Investigator
Centre Oscar LAMBRET Radiation Oncology department 3 Rue Frédéric Combemale 59000 LILLE
Prof. Christophe HENNEQUIN Investigator AP-HP Hospital Saint-Louis Radiation Oncology department 1 Avenue Claude Vellefaux 75010 PARIS
Prof. Jean-Léon LAGRANGE Investigator AP-HP Hospital Henri MONDOR Radiation Oncology department and INSERM EMI0337 94000 CRETEIL
Prof. Philippe MAINGON Dr. Gilles CREHANGE Dr. Edouard LAGNEAU Dr. Karine PEIGNAUX Dr. Gilles TRUC
Investigator Investigator Investigator Investigator Investigator
Centre Georges-François Leclerc Radiation Oncology department 1 rue du Prof. Marion 21079 DIJON CEDEX
Prof. Françoise MORNEX Investigator Centre Hospitalier Lyon Sud Radiation Oncology department Pavillon 1F 165 Chemin du Grand Revoyet 69495 PIERRE BENITE CEDEX
Dr. Alain FOURQUET Dr. François CAMPANA Dr. Sylvie HELFRE Dr. Philippe GIRAUD Dr. Valentin CALUGARU Dr. Marc BONNET Dr. Rémi DENDALE Dr. Youlia KIROVA Pr. Jean-Marc COSSET
Investigator
Curie Institute Radiation Oncology department 26 Rue d’Ulm 75005 PARIS
Prof. Bernard DUBRAY Investigator Centre Henri Becquerel Radiation Oncology department 35 Rue Daily 92210 SAINT-CLOUD
Dr. Sébastien CLIPPE Investigator Radiation Oncology Center Marie Curie 159 BD Maréchal Juin 26953 VALENCE CEDEX 09

Discipline: Radiation Oncology Dr David AZRIA
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
C O N F I D E N T I A L
3
Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
4
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
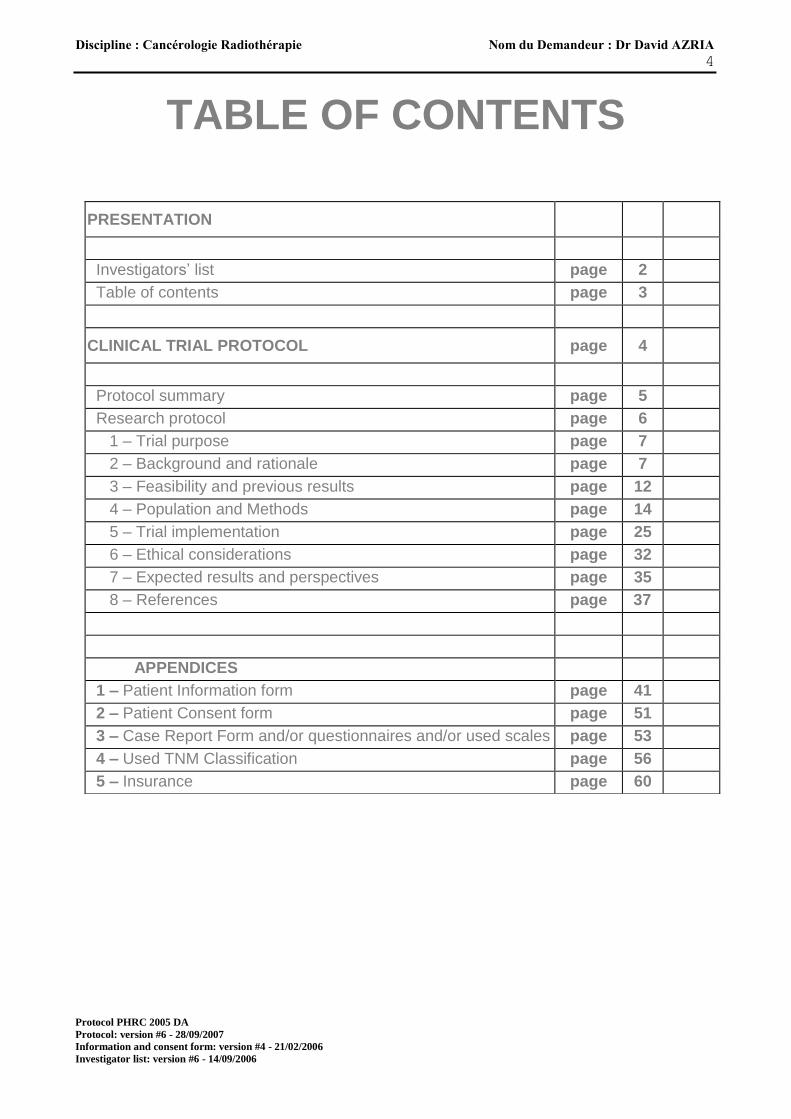
TABLE OF CONTENTS
PRESENTATION
Investigators’ list page 2
Table of contents page 3
CLINICAL TRIAL PROTOCOL page 4
Protocol summary page 5
Research protocol page 6
1 – Trial purpose page 7
2 – Background and rationale page 7
3 – Feasibility and previous results page 12
4 – Population and Methods page 14
5 – Trial implementation page 25
6 – Ethical considerations page 32
7 – Expected results and perspectives page 35
8 – References page 37
APPENDICES
1 – Patient Information form page 41
2 – Patient Consent form page 51
3 – Case Report Form and/or questionnaires and/or used scales page 53
4 – Used TNM Classification page 56
5 – Insurance page 60

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
5
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
PROTOCOL

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
6
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
Protocol summary
Project Rationale: The direct correlation between total dose and oncologic benefit is well known in external radiotherapy, especially in breast and prostate cancer. However, dose escalation leads to late toxicities that are often irreversible. The rate of radiation-induced CD8 lymphocyte apoptosis seems to predict the patients at risk of severe late toxicities in our preliminary studies. Objectives: The primary objective is to prospectively evaluate the prediction of radiotherapy late toxicities by measuring the rate of radiation-induced CD8 lymphocyte apoptosis nation-wide. The concerned pathologies are i) intermediate-risk prostate adenocarcinoma treated with conformational radiotherapy with or without intensity modulation, and ii) breast adenocarcinoma treated with postoperative radiotherapy after breast-conserving surgery in patients < 60 year/old. The identification of the patients at risk of severe late toxicities (about 5% of all patients) would allow the administration of high dose radiotherapy to the other patients (about 95%) at lower risk of such toxicities. Trial design: prospective multicenter trial to assess a predictive test of late toxicities Methodology: 1/ Population: Prostate adenocarcinoma of intermediate risk and breast adenocarcinoma (breast-conserving surgery and age < 60 years). Inclusion Criteria: Intermediate-risk prostate cancer, T ≥ T1c-T2a and < T3b; T1b or c with PSA ≤ 10 ng/ml; T1b or c with Gleason ≥ 6; N0; strictly < grade 2 initial state (CTC v3.0). Breast cancer (conservative surgery and aged < 60 years or > 60 years with boost indication), breast-conserving surgery; primary tumor T1, T2; sentinel node negative, N0, N1 or N2; M0; strictly < grade 2 clinical signs (CTC v3.0). Primary endpoint: Late complication for prostate cancer is defined as the occurrence, at least three months after the beginning of treatment, of a grade ≥ 2 toxicity (CTC v3.0) among the following events: digestive (diarrhea and rectitis), urinary (cystitis, incontinence, stenosis, urinary frequency) and sexual (erectile dysfunction). Late complication for breast cancer is defined as the occurrence, at least three months after the beginning of treatment, of a grade ≥ 2 toxicity (CTC v3.0) among the following events: atrophy, pigmentation, fibrosis and telangiectasia. 2/ Sample size: 862 patients (breast cancer: 494 patients, prostate cancer: 368 patients). 3/ Treatment: measurement of the radiation-induced CD8 lymphocyte apoptosis rate before radiotherapy (RT). Toxicities will be monitored and graded according to the NCI-CTC v3.0 classification on a weekly basis during RT, at one, three and six months after RT end (M1, M3, M6), and every six months up to three years (M12, M18, M24, M30, M36). 4) Statistical analyses: The cumulative incidence of complications according to the prognostic variables will be estimated using a non-parametric method. The primary statistical analysis includes a multivariate analysis using a competitive risks model. Perspectives: This study will allow identifying the subset of patients at high risk of developing severe late toxicities and thus will permit to adapt their treatment. Curative high dose radiotherapy may be delivered to the majority of patients with a lower risk and without compromising their therapeutic index.
Key words
« Radiotherapy » « late side-effects » « predictive assay »
Disciplines Radiation oncology Radiobiology
Concerned pathologies Breast cancer Prostate cancer
Project nature Therapeutic trial Translational research predictive test

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
7
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
RESEARCH PROTOCOL

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
8
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
1) TRIAL PURPOSE The primary purpose of this trial is to prospectively evaluate the prediction of radiotherapy
late toxicities by measuring the rate of radiation-induced CD8 lymphocyte apoptosis.
This multicenter trial concerns patients with prostate cancer requiring dose escalation and
patients with breast cancer needing a local boost.
This study will allow identifying the subset of patients at high risk of developing severe late
toxicities and thus will permit to adapt their treatment. Curative high dose radiotherapy
may be delivered to the majority of patients with a lower risk and without compromising
their therapeutic index.
2) BACKGROUND
Radiation therapy success depends primarily on the total dose homogeneously delivered
to the tumor. However, the total radiation dose is limited by the healthy tissue tolerance in
the irradiated area (Emami et al., 1991).
Radiotherapy induces multiple side effects (Withers et al., 1988), without the possibility to
individually identify the toxicity risk. Indeed, radiotherapy regimens are prescribed
according to general guidelines without considering individual phenotypes or genotypes
(Buchholz, 1999).
Two types of radio-induced side effects (acute and late) are usually monitored in
radiotherapy settings; however, in this project, only the late side effects will be considered
because they are irreversible and strongly affect the patients’ quality of life (Lartigau et al.,
1997). Indeed, acute side effects occur during or shortly after radiotherapy and usually
disappear without sequelae within the first few months following the treatment (Lartigau et
al., 1997).
2.1) Radiation-induced side effects:
Healthy tissues are generally affected by doses lower than those required to sterilize the
tumor. Following radiotherapy, late side effects usually appear within three months;
however, the length of this interval is still debated (some authors reported a 6-month
delay).
Symptoms may be moderate or severe and may worsen over time.

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
9
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
Late side effects usually occur in tissues with slow cell renewal, such as subcutaneous
tissue, adipose tissue, muscles, or in tissues with quick cell renewal, such as the digestive
tract wall (Stone et al., 2003).
Late radiation-induced histological lesions are numerous and include fibrosis, necrosis,
atrophy and vascular damage (Stone et al., 2003).
The mechanisms of appearance of late radiation damage are still poorly understood, but
several lines of research support the hypothesis of a continuous local production of
cytokines and growth factors. Moreover, the physiopathology of radiation-induced late
lesions in normal tissues is characterized by the direct destruction of stem cells, vascular
damage and progressive development of interstitial fibrosis (Lartigau et al., 1997). Such
radiation-induced fibrosis, almost constant in the irradiated area, corresponds to an
abnormal accumulation of extracellular matrix. Matrix remodeling is continuous and
progressively worsens spontaneously due to the destruction of the local homeostatic
balance. Its clinical translation is extremely variable over time (from the inflammatory
phase to « late » fibrosis) and in intensity (from simple induration to invalidating retractile
sclerosis) (Delanian et al., 1999; Delanian et al., 2003; Dubray et al., 1997a).
2.2) Factors of normal tissue radiosensitivity
Radiosensitivity factors can be subdivided in two groups that remain closely linked during
a radiotherapy treatment: i) factors related to the treatment and ii) factors related to the
patient.
2.2.1) Treatment-related factors:
The risk, severity and nature of late reactions depend intimately on the irradiation
modalities. Indeed, four determinants influence these reactions:
- The total dose: Late secondary effects are mostly determinist and appear above a
threshold of irradiation. Moreover, the slope of the dose-effect curve is steeper for
late responding than for early responding normal tissues (Thames et al., 1989).
Thus, a moderated reduction of the dose delivered to healthy tissues can have a
significant impact on the incidence and severity of late lesions.

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
10
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
- Fractioning and interval between fractions: The dose per fraction is usually of
1.8 - 2 Gy. Late responding normal tissues are particularly sensitive to modifications
of the dose per fraction. Indeed, the use of a dose per fraction lower than 1.8 Gy
(hyperfractionation) allows reducing the incidence and severity of radiation-induced
sequelae, while maintaining the same therapeutic total dose. Moreover, the use of
hyperfractionation allows delivering a higher total dose to the tumor without
increasing the late toxicity.
The optimal interval between fractions has not been exactly determined, but many
experimental and clinical data confirm that an interval of at least six hours between
fractions allows the repair of at least 50% of radiation-induced lesions. The length of
this interval is a major factor of late toxicity due to the correlation between the
important capacity to repair radiation-induced DNA lesions and the sensitivity of
healthy tissues to late responses (Dubray, 1995).
- Distribution: or overall treatment time; it has very little influence on the risk of late
damage to normal tissues. Conversely, accelerating a treatment can cause very
important acute toxicity events and, thus, late reactions as a consequence of this
hyper-reaction (« consequential late effect ») (Stone et al., 2003).
- Irradiated volume: The volume of healthy tissue that has been irradiated can
influence the risk of complications in radiotherapy. Many mathematical models have
been employed to assess the dose–volume–effect relationship in order to predict
the risk of complications of a given irradiation regimen. The most developed models
concern the late effects of radiotherapy. Some complications depend directly on the
irradiated volume: it is the case for post-radiation hepatitis the severity of which
depends on the irradiated liver volume. For lung, it is possible to predict the risk of
post-radiation clinical pneumonitis of grade 2 (according to the classification of the
Radiation Therapy Oncology Group, RTOG) in function of the dose–volume
histograms. Besides the effects on the differentiated cell compartment, the
irradiated volume can also affect the functional reserve of an organ. For instance,
for hematopoietic bone marrow and salivary glands, it is the volume of normal
tissue spared from radiation exposure that will allow avoiding the complication. This
type of volume–effect direct relationship concerns parallel organs. Its role is
considered to be minor, even non-existent, for serial organs (Huchet et al., 2003).
2.2.2) Patient-related factors:

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
11
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
Two major contexts can be envisaged in this paragraph:
- Genetic context: some genetic diseases are characterized by defects in the cell repair
mechanisms of radiation-induced molecular lesions and present hypersensitivity of normal
tissues. For instance, patients harboring a mutation of the ATM (Ataxia- Telangiectasia)
gene have an increased risk of late radiation-induced toxicity (Cesaretti et al., 2005;
Iannuzzi et al., 2002).
- Risk factors: the risk of complications or of sequelae after radiation therapy seems to be
increased by a number of factors, such as advanced age, association with chemotherapy
(Lartigau et al., 1997) or hormone therapy (Azria et al., 2004), smoking, microvascular
problems (diabetes, arterial hypertension) and generalized scleroderma (Dubray et al.,
1997a).
2.3) Predictive texts of toxicity
Anti-cancer treatments, including radiation therapy, are essentially limited by the tolerance
dose of normal tissues, particularly late responding tissues. Moreover, these doses are
badly known and are based on rather old estimates (Emami et al., 1991). It is clear that,
within a population treated homogeneously from a technical point of view and assessed by
the same team, the radiosensitivity of normal tissues can be intrinsically different. The
hypothesis of a gene deficit or modification is emerging and could allow the identification of
a small part of the population at risk.
Technically, the study of the radiobiological features of normal tissues is, in principle, less
problematic than for tumors, mainly due to the tissue accessibility, the quantity of available
material and the cell population homogeneity. Moreover, in the case of the hypothesis of a
genetic abnormality at the origin of the individual hypersensitivity, the diagnosis could be
done using circulating lymphocytes or fibroblasts from a skin biopsy (Dubray et al., 1997b).
Lymphocytes seem to be the tissue of choice due to the easy of sampling and the
available quantity. Radiation-induced lymphocyte apoptosis has been evaluated already
for a few years and it is a type of cell death that is sensitive and reproducible for our study
(Radford & Murphy, 1994; Trowell, 1952). Moreover, CD4 and CD8 lymphocytes can be
perfectly isolated from the other lymphocyte sub-types by using a flow cytometry-based
technique (Crompton & Ozsahin, 1997; Ozsahin et al., 1997). Other cell types (mainly
fibroblasts) and several assessment methods (micronuclei, lethal chromosomal

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
12
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
aberrations) have been considered, but the results in terms of prediction of late secondary
effects remain contradictory (Dewey et al., 1995; Floyd & Cassoni, 1994; Hoeller et al.,
2003; Johansen et al., 1996; Kiltie et al., 1999). Moreover, the first works on the
lymphocyte apoptosis assay have shown that this test is fast and reproducible, differently
from the other previously mentioned methods (Crompton et al., 2001; Ozsahin et al., 1997;
Slonina & Gasinska, 1997).
2.4) Assessment of late toxicities
The detection and assessment of radiation-induced late adverse effects is a key objective
of many on-going studies (Trotti et al., 2003). The «National Cancer Institute (NCI)
Common Toxicity Criteria system (CTC v1.0)» was put in place for the first time in 1983 to
help assessing and grading chemotherapy adverse effects. It was updated and improved
in 1998 (version 2: CTC v2.0) by focusing always on the acute adverse effects (Trotti et
al., 2000). Recently and with the aim of setting up a single classification that includes also
late toxicities, the NCI has developed a third version of the CTC: the CTCAE v3.0 (i.e.,
Common Terminology Criteria for Adverse Events version 3.0).
Table 1 (below) summarizes the different classifications used by the different
chemotherapy or radiotherapy protocols since 1979:
Classification Criteria Organs Modalities Evaluation
WHO (1979) 28 9 Chemo-induced Acute
CTC (1983) 18 13 Chemo-induced Acute
RTOG/EORTC-Acute (1984) 14 13 Radio-induced Acute
RTOG/EORTC-Late (1984) 16 13 Radio-induced Late
Lent (1995) 152 22 Radio-induced Late
CTC v2.0 (1998) 260 22 Radio and chemo-induced Acute
CTC v3.0 (2002) 370 All Radio and chemo-
induced
Acute and Late
The CTC v3.0 (Trotti et al., 2003) has several advantages compared to the previous
scales for toxicity assessment:
- The RTOG/EORTC, LENT-SOMA and CTC v2.0 scales have shown assessment
discrepancies, justifying the interest of a common classification (Denis et al., 2003).

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
13
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
- Acute and late adverse effects are listed in a single classification without taking into
account the delay of appearance. Each investigator can thus describe and grade
the observed adverse effect at each visit without worrying about the « time »
parameter.
- This new classification represents a large international consensus panel of
oncologists and has been validated by all clinical research groups. The participants
to its elaboration have been numerous and of many different origins, including the
Radiation Therapy Oncology Group (RTOG), European Organization for Research
and Treatment of Cancer (EORTC), American College of Surgeons Oncology
Group (ACOSOG), European Society for Therapeutic Radiology and Oncology
(ESTRO) and American Society for Therapeutic Radiology and Oncology (ASTRO).
- It is easy and fast to use in the daily clinical practice. Other evaluation techniques of
late toxicities exist, notably in mammary pathology, but they are difficult to use on a
larger scale and do not necessarily reflect our possibilities in the clinic (Dubray et
al., 1997a).
Altogether, these data endorse the CTC v3.0 as the classification of choice for our study.
In order to avoid as far as possible the subjectivity of each investigator, we ask an
independent assessment of the toxicities by two physicians at each visit. The
statistical analysis will take into account the highest score for each assessment (see,
Statistical analyses chapter).
3) FEASIBILITY AND PREVIOUS RESULTS
- Based on the pre-clinical and clinical (retrospective) data described in chapter 2.3, we
have demonstrated in a prospective study the predictive value of the assay to measure the
radiation-induced apoptosis rate of CD8 lymphocytes. In this prospective study, we
assessed the prediction of the late adverse effects after radiotherapy based on the in vitro
rate of radiation-induced apoptosis of CD8 lymphocytes. Briefly, a blood sample was
collected in a 5ml heparin tube from each patient (n=399) before external beam
radiotherapy (RT) as curative treatment. Each blood sample was prepared and then
irradiated (8 Gy). Forty-eight hours after irradiation, lymphocytes were isolated and their
apoptosis rate measured by flow cytometry. All patients were followed regularly and late
toxicities were recorded and graded according to the RTOG/EORTC classification. Six

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
14
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
patients refused RT after blood sampling and were excluded from the analyses. The area
under the curve (AUC) of the ROC analyses was used to calculate the late toxicity
prediction based on the lymphocyte apoptosis rate. The competitive risk analysis allowed
estimating the cumulative incidence of late adverse effects in function of the radiation-
induced apoptosis.
Patients had mainly breast cancer (n=149, 147 women and 2 men), ORL cancer (n=75) or
prostate cancer (n=36). The proportion of late toxicities of grade 2 and 3 was respectively
31% (121/393) and 7% (28/393). A low lymphocyte apoptosis rate was significantly
correlated with the percentage of late toxicities of grade ≥ 2 (p<0.0001). A lymphocyte
apoptosis rate higher than 24 was found in all patients who did not have toxicity of grade 3
(p<0.0001). The AUC was 0.827 for the patients with late toxicity of grade ≥ 2. The positive
predictive value was 83% for a lymphocyte apoptosis rate ≤ 16% and the negative
predictive value was 86% for a lymphocyte apoptosis rate > 24%. The cumulative
incidence rate of late toxicities of grade ≥ 2 at two years was 70, 32 and 12%, respectively,
for lymphocyte apoptosis rates ≤ 16, 16-24 and > 24%.
This study was accepted for publication in Clinical Cancer Research:
M Ozsahin, NEA Crompton, S Gourgou, A Kramar, L Li, YQ Shi, W Jeanneret-Sozzi, A
Zouhair, RO Mirimanoff, D Azria. CD4 and CD8 T-Lymphocyte Apoptosis Can Predict
Radiation-Induced Late Toxicity: A Prospective Study in 399 Patients. Clin Cancer Res,
2005; 11:7426-33.
- Among the 399 patients of the study described above, the 147 patients with breast
cancer were treated by conservative surgery and adjuvant radiotherapy and 90
started tamoxifen (20 mg/day) before the beginning of radiotherapy. The rate of
survival without fibrosis at two years was 51% in the radiotherapy + tamoxifen group
versus 80% in the group without tamoxifen (p = 0.029). Moreover, this difference
was very clear in patients at risk of developing late adverse effects. Indeed, in the
sub-group of 147 patients with breast cancer, the rate of survival without fibrosis
was significantly lower in patients with a low radiation-induced lymphocyte
apoptosis rate. Analysis of survival without fibrosis in function of the treatment with
or without tamoxifen and of the radiation-induced lymphocyte apoptosis rate
showed that the rate of survival without fibrosis was only 20% in patients with a
radiation-induced lymphocyte apoptosis rate < 16% and who received

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
15
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
concomitantly tamoxifen + radiotherapy. Conversely, patients with an elevated
radiation-induced lymphocyte apoptosis rate were not very sensitive to the
concomitant prescription of tamoxifen and radiotherapy.
This article was published in the British Journal of Cancer in 2004.
D Azria, S Gourgou, WJ Sozzi, A Zouhair, RO Mirimanoff, A Kramar, C Lemanski, JB
Dubois, G Romieu, A Pèlegrin, M Ozsahin. Concomitant use of Tamoxifen with
Radiotherapy enhances subcutaneous breast fibrosis in hypersensitive patients. Br J
Cancer, 2004; 91:1251-1260.
- Currently, a randomized phase II study is assessing the risk of late skin toxicities
following the concurrent or sequential association of radiotherapy and an anti-aromatase
(Letrozole) in 150 patients treated by conservative surgery for localized breast cancer. In
this study there are several stratification factors, including the rate of radiation-induced
apoptosis of CD8 lymphocytes. Patients’ inclusion started at the beginning of January
2005 with a provisional duration of 15 months.
These first results strongly encouraged us to develop this assay in clinical practice.
However, no large-scale multicenter study is available at the moment. The study proposed
for this PHRC (Hospital Program of Clinical Research) will allow answering to several
questions:
- The pertinence of this test in two pathologies that require dose escalation (breast
and prostate cancer, see chapter 4).
- The use at different sites of the same toxicity classification (CTC v3.0) with an
independent evaluation by two physicians at each visit.
- The feasibility of a simple biological assay (centralized in Montpellier during the first
stage of assay development) and the involvement of French oncologists in this type
of study.
4) POPULATION AND METHODS
The objective of this study is to identify by using a biological assay (the rate of radiation-
induced apoptosis of CD8 lymphocytes) the patients at risk of late toxicity. The two

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
16
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
concerned pathologies are intermediate-risk prostate adenocarcinoma treated by
conformational radiation therapy with or without intensity modulation and breast
adenocarcinoma treated by adjuvant radiotherapy after breast-conserving surgery in
patients younger than 60. These two populations treated by external beam radiotherapy
show a rate of local control that depends directly on the total dose and are thus more at
risk of developing late toxicity. Moreover, as the long-term remission rate is very elevated
in these two pathologies, it is justified to develop research protocols aiming at decreasing
the occurrence of late toxicities.
4.1) The chosen population:
4.1.1) Patients with intermediate-risk prostate cancer
They are defined by:
A clinical stage: T ≥ T2a and < T3b (Annex 3)
Or T1b or c with PSA ≥ 10 ng/ml (normal value=4 ng/ml)
Or T1b or c with Gleason ≥ 7.
4.1.2) Patients with breast cancer (conservative surgery and age < 60 years)
They are defined by:
Breast-conserving surgery, extension assessment: negative, healthy resection borders
and T1, T2 tumor; negative sentinel lymph nodes, N0, N1 or N2; M0 (TNM 2002, Annex 3).
4.2) Recruitment modality, place and feasibility:
Patients’ recruitment will be carried out at each initial consultation by the different
investigators. During this visit, patients will receive oral information and a written letter
describing in detail this study. The informed consent will be collected the day of blood
sampling, preferentially a Monday morning.
The recruitment place corresponds to each declared investigation center: «Detailed
Composition of the Groups».
The recruitment feasibility has been already evaluated with all the teams involved in this
study. The inclusion of 862 patients should be achieved within the time imposed by the
study given the important number of investigator centers and the very high frequency of
the two selected cancers (prostate and breast cancer). Moreover, the fact that this study

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
17
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
does not modify the daily clinical practices of treatment should facilitate the inclusion flow.
Finally, the follow-up will be carried out thanks to the PHRC support after the end of the
radiotherapy for all included patients. The PHRC maximum duration of three years is thus
theoretically justified. Supplementary funding may be required at the end of these three
years to continue the follow-up and this outside this PHRC.
4.3) Inclusion/exclusion criteria:
4.3.1) Patients with intermediate-risk prostate cancer
Neoadjuvant or concurrent treatment by hormone therapy is permitted but must be mentioned in the case report form (CRF). INCLUSION CRITERIA:
- Cancer localized to the prostate, histologically proven
- Absence of metastases (M0): normal bone scintigraphy.
- Absence of radiological lymph node invasion (N0). Bilateral lymph node dissection
(curettage) is left to the investigator’s choice in function of the risk of lymph node
invasion higher than 10% according to the Partin table.
- Clinical stage: T ≥ T1c-T2a and < T3b
Or T1b or c with PSA ≤ 10 ng/ml
Or T1b or c with Gleason ≥ 6
- PSA < 30 ng/ml.
- Toxicity signs and symptoms according to the CTC v3.0 classification < grade 2
- ECOG performance index ≤ 1
- No hip prosthetic implant
- No prostatic stent
- Patient older than 18 and younger than 80 years
- Patient affiliated to a social security organism
- Dated and signed written informed consent.
EXCLUSION CRITERIA
- History of invasive cancer (except if treated more than five years ago and without
progression) with the exception of a basal cell carcinoma
- Positive seminal vesicle biopsy

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
18
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
- PSA ≥ 30 ng/ml at two successive measurements
- History of pelvic radiotherapy
- History of radical anterior prostatectomy for cancer
- Patients with another non-stabilized systemic disease (cardiovascular, kidney, liver,
pulmonary embolism, etc.) or generalized scleroderma
- Patients known as HIV seropositive (no specific test required to define eligibility)
- Known homozygous ATM (Ataxia telangiectasia) gene mutation
- Impossibility of correct follow-up of the patients (for social, family, geographical
reasons………)
- Patient cannot give his/her consent, protected person of full age, vulnerable person
- Patient enrolled in another clinical study.
4.3.2) Patients with breast cancer (conservative surgery and age > or < 60 years) Adjuvant treatment by hormone therapy and/or chemotherapy is permitted but must be mentioned in the CRF. INCLUSION CRITERIA
- Breast-conserving surgery
- Extension assessment: negative
- Healthy resection margins
- T1, T2 tumor; Negative sentinel lymph nodes, N0, N1 or N2; M0.
- Hormone receptors: irrelevant
- Toxicity signs and symptoms according to the CTC v3.0 classification < grade 2
- Patient older than 18 and younger than 60 or older than 60 years and with
indication for radiotherapy boost.
- Patient affiliated to a social security organism
- Dated and signed written informed consent.
EXCLUSION CRITERIA
- Patients with metastases
- Bilateral breast cancer (concomitant or previous) except in situ
- T4 or N3 tumor or treated by mastectomy
- Patient with neoadjuvant chemotherapy or hormone therapy

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
19
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
- Patients who already had another cancer (other than a breast tumor) before or
concomitantly in the last five years BUT skin basal cell carcinoma or neck
carcinoma in situ. Patients who previously had another cancer must be without
relapse for at least five years
- Patients with another non-stabilized systemic disease (cardiovascular, kidney, liver,
pulmonary embolism, etc.) or generalized scleroderma
- Pregnant or breastfeeding women
- Known homozygous ATM (Ataxia telangiectasia) gene mutation
- Patients known to be HIV seropositive (no specific test required to define eligibility)
- Patient cannot give his/her consent, protected person of full age, vulnerable person
(art. L1121-6, L. 1121-7, L .1211-8, L.1211-9)
- Patient enrolled in another clinical study.
4.4) Technical features of the radiation therapy:
4.4.1) Intermediate-risk prostate cancer
Treatments will be carried out by using high energy (greater or equal to 10 MV) x-ray
beams. All beams are used at each session.
The practical implementation of the radiotherapy is in accordance with the GETUG
14/0207 protocol.
- Target volumes, beam limits and doses:
The contours of the target volumes and normal organs at risk of complications are
identified on the CT sections obtained in the treatment position before radiation therapy.
The seminal vesicles are included in the initial clinical target volume (CTV), but they
receive only 46 Gy.
The first CTV (CTV1) includes prostate and seminal vesicles.
The second CTV (CTV2) is limited to prostate and excludes the seminal vesicles.
The planning target volumes « PTV » 1 and 2 are defined by adding a margin of 1cm
around the corresponding CTV; this margin is reduced at the back to 0.5 cm to protect the
rectum. This margin takes into account the uncertainties of patient’s positioning under the
treatment machine and the internal motion of the target volumes.

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
20
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
The beam limits (primary and secondary beam collimation) are defined by adding an
additional margin around the PTVs. The dimensions of this margin are in function of the
beam penumbra.
The dose will be targeted to the center or to the central part of the PTV2 (ICRU reference
point) and delivered by daily fractions of 2 Gy, five fractions per week for a total of 8 weeks
of radiation therapy. The total doses will be: 46 Gy to the PTV1 and 34 Gy to the PTV2. In
order to obtain a homogeneous irradiation, the doses received by each point of the PTVs
must be higher than 95% and lower than 107% of the dose received by the ICRU point.
These constraints can be applied with the intensity modulation. To respect the constraints
concerning the doses received by healthy tissues in classical conformational radiotherapy,
it is admitted that at least 85% of the PTV2 must receive at least 95% of the prescribed
dose; the minimum dose received by the PTV2 must be at least 90% of the prescribed
dose.
The maximum volume of rectum receiving a dose higher or equal to 72 Gy must remain
smaller than 25% of the total volume. An occasional maximum dose of 76 Gy to the rectal
wall must not be exceeded. The maximum volume of bladder receiving a dose higher or
equal to 70 Gy must be smaller than 50% of the total volume. The dose of 80 Gy must not
be exceeded in any point of the bladder volume. The volume of each femur head (femur
neck included) receiving a dose higher or equal to 55 Gy must remain smaller than 5% of
the total volume.
The minimum dose to the PTV2 should be ≥ 72 Gy and at least 85% of the volume should
receive a dose ≥ 76 Gy.
The occasional maximum dose to the rectum must be ≤ 76 Gy and not more than 25% of
the rectum volume must receive a dose ≥ 72 Gy.
The occasional maximum dose to the bladder must be ≤ 80 Gy and not more than 50% of
the bladder volume must receive a dose ≥ 70 Gy.
Not more than 5% of the volume of the femur heads must receive a dose ≥ 55 Gy.
Taking into account the uncertainties allows a protocol tolerance of 2% concerning the
volumes and doses defined above.
- Treatment preparation, dosimetric planning and quality control:
Patient’s positioning is free, but its reproducibility with a tolerance of 5 mm with or without
positioning/immobilization devices must be verified by each team. Bladder and rectum

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
21
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
should be emptied about one hour before the planning CT scan and before each
radiotherapy session.
CT image acquisition is done in the treatment position, with the positioning/immobilization
devices. An intravenous injection of contrast agent should be carried out in the absence of
contraindications. The injection of 10 cc of air through an intrarectal urinary catheter allows
the good visualization of the rectal walls and the anal canal. Contiguous CT slices of 5 mm
in thickness at most will be acquired between the base of the sacroiliac joints and the
lesser trochanters.
The CTVs are outlined by the radiotherapist on all CT slices. The contour does not include
the periprostatic areas unless there is a clinical or paraclinical indication that suggests a
zone of invasion. Each investigator can choose the method to designate the apex. The
volume of the seminal vesicles stops at the last slice where it is visible.
The organs at risk are also delineated on the CT slices. The investigator must take into
account the external contour of the rectum and of the anal canal up to 2 cm above and
below the CTV1 as well as the external contour of the bladder on all the slices where it is
visible. The rectal volume is the volume included between the external contour and
an internal contour of 5 mm. This volume is defined by a negative expansion in 2D. The
bladder volume is the volume included between the external contour and an internal
contour at 7 mm from the former. This volume is defined by a negative expansion in 2D.
The penile bulb is outlined on the slices where it is visible.
The beams are defined and positioned by virtual simulation with automatic delimitation of
the areas to be protected by the leaves of the multi-leaf collimator. A digital image of each
simulated field can be obtained.
The dose distribution is calculated in the three dimensions (3D) and is represented as
dose-volume histograms for the organs at risk and the target volumes. The dose is
calculated by taking into account the density heterogeneities after suppression of the
bladder opacification effect on density measurement.
Each participant site must ensure the quality of the treatments by means of periodic quality
controls concerning all the machines and software systems used for the treatment
procedure.
4.4.2) Breast cancer (conservative surgery and age < 60 years)

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
22
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
The treatment will be carried out by using linear particle accelerators with energies lower
or equal to 6 Mev.
- Target volumes, beam limits and doses:
Data are systematically acquired by pre-treatment CT imaging and will serve as the basis
for the provisional dosimetry study. The slice thickness and spacing should allow a good
image reconstruction. The acquisition area goes from the upper limit of the clavicle to 4
cm below the lower limit of the breast by taking care of covering the entire lung. The
mammary gland, localized if possible by external marks during the CT image acquisition, is
delineated on all the slices where it is visible. The internal mammary chain and the apical
lymph nodes are highlighted if their position can be determined by CT imaging.
The lung is outlined in its totality as well as the heart in the case of the left breast.
The tumorectomy area, localized preferentially by a clip positioned during surgery, is
demarcated.
The dose is delivered by fractions of 2 Gy per day according to the ICRU 62
recommendations. The prescription is of 50 Gy on the breast in total. The addition of a
boost of 10-16 Gy to the surgical area is decided by the investigator, but it is
recommended for patients younger than 60 years (Bartelink et al., 2001). The sternal and
supraclavicular fields are treated according to the modality of each participant center and
these data will be recorded in the CRF.
Dose prescription will be based on an isodose line chosen by the radiotherapist for the
breast and at the level of the circled lymph nodes for the other beams. If the lymph nodes
cannot be detected by CT imaging, dose prescription will be done at a depth of 3 cm.
- Treatment preparation, dosimetric planning and quality control:
A system to guarantee the patient’s positioning reproducibility must be used during the
preparation and treatment phases.
The investigator will be able to choose the irradiation technique, but this must be put in
place by virtual simulation starting from the 3D volume reconstructed by integrating the
target volumes and the organs at risk. The regions of sub-dosage (cold triangle) should be
reduced as much as possible.

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
23
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
The classical treatment ballistics includes tangential fields for the treatment of the
mammary gland and direct beams for the lymph node areas. The treatment with inclusion
of the internal mammary chain in the tangential fields is authorized.
Beam modification devices will be put in place to protect the organs at risk, matching
adjacent fields or to homogenize the dose in the target volume.
The boost (10-16 Gy) is carried out using an electron beam for a dose complementary to
the one received in the tumorectomy area by the tangential fields up to a total dose of 60-
66 Gy. The provisional dosimetry study will be done based on the CT images and the dose
calculation will be carried out in 3D with incorporation or not of the heterogeneities.
The quality control of the patient’s positioning will be done by portal imaging during the first
three days of treatment for all the fields and at each beam modification. An image of the
tangential fields will then be acquired once per week. These images will be compared with
the reference DRR images derived from the dosimetry.
4.4.3) Justification of the choice of irradiation dose
The choice of irradiation dose is based on the type of treated population.
Concerning intermediate-risk prostate cancer, a more elevated total dose has been
correlated with a better survival without biochemical failure in two randomized trials. The
randomized study of the MD Anderson in Houston demonstrated that a dose increase from
70 to 78 Gy results in a significant improvement of survival without biochemical failure in
intermediate and high risk patients (Pollack et al., 2000b). Very recently, at the ASTRO
2004 meeting, Zietman et al. reported a biological failure rate at 5 years of 19% and 37%
for irradiation doses of 70.2 Gy and 79.2 Gy, respectively, without hormone therapy
(Zietman et al., 2004). Other non-randomized studies confirmed the interest of dose
escalation in terms of survival without biochemical failure for patients with intermediate-risk
prostate cancer (Jacob et al., 2005; Pollack et al., 2000a).
Concerning breast irradiation after conservative surgery, it is established that the reference
treatment must include the irradiation of the mammary gland with 50 Gy in 25 fractions
(Arriagada et al., 1985). A boost is discussed in function of some prognostic factors, such
as the patient’s age, tumor size and tumor palpability (Maingon et al., 2004). Two
randomized trials evaluated the interest of a boost after irradiation of 50 Gy on the whole
mammary gland. The trial 22881 of the European Organization for Research and
Treatment of Cancer (EORTC) (Bartelink et al., 2001) reported rates of local recurrence of

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
24
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
4.3% and 7% in the 50 Gy + 16 Gy boost arm and in the 50 Gy without boost arm,
respectively (p<0.001). The multifactorial analysis found three variables that were
significantly linked to the occurrence of a local recurrence: the presence of a palpable
tumor, the absence of progesterone receptors and the patient’s age (patients older than 60
years benefit less of this boost). The trial in Lyon (Romestaing et al., 1997) showed a
reduction of the estimated rate of local recurrence in the 50 Gy + 10 Gy boost arm
compared to the 50 Gy without boost arm (3.6% versus 4.5%, respectively; p=0.044).
Contrary to the EORTC 22881 trial, no multifactorial analysis was carried out.
4.5) Primary end-point:
The aim of this study is to identify by using a biological assay (rate of radiation-induced
apoptosis of CD8 lymphocytes) the patients at risk of late toxicity.
Late toxicity for breast cancer is defined as the appearance at least three months after the
beginning of the treatment of a toxicity of grade ≥ 2 among the following events: atrophy,
pigmentation, fibrosis and telangiectasia. Dermatitis, as defined in the CTC v3.0
classification and described in Annex 5, must be considered as an acute toxicity event.
Late toxicity for prostate cancer is defined as the appearance at least three months after
the beginning of the treatment of a toxicity of grade ≥ 2 among the following events:
digestive (diarrhea and rectitis), urinary (cystitis, incontinence, stenosis, urinary frequency)
and sexual (erectile dysfunction) in the CTC v3.0 classification (Annex 3).
This interval of three months allows the clear separation between late complications and
acute toxicities, notably the late toxicities of the « consequential late effect » type that
correspond to late reactions resulting from very important acute secondary effects.
Other [secondary] end-points will be taken into account, such as local recurrences, distant
metastases and overall survival. These criteria must be evaluated because they determine
the attainment of the primary end-point (see Number of patients required, paragraph 4.6,
and Statistical Analysis, paragraph 5.5).
4.6) Number of patients required:
The number of individuals required for this study will be determined based on a
generalization of the usual formulas for the study of a continuous prognostic variable

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
25
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
(Hsieh & Lavori, 2000), and for an end-point that is a competing risk (Latouche et al.,
2004). In a previous study (Azria et al., 2004), the patients’ CD8 lymphocyte apoptosis
rates were categorized in three classes according to the 33% quantiles. As this distribution
may not be suitable for a larger population, this variable will be treated as a quantitative
variable. Moreover, we want to evaluate the relationship between CD8 lymphocyte
apoptosis rates and cumulative incidence of complications, rather than the hazard rate of
complications. For this reason, we will use a competing risk model rather than a specific
risk model.
Competing risk and quantitative variable
The appearance of a late complication can be considered as a competing risk in the
presence of other events (such as recurrence or death) that can occur before the
complication. The formulas routinely used to calculate the number of individuals in the
context of this type of events are based on a sub-distribution hazards model and not on
modeling the cause-specific hazard (Latouche et al., 2004). The following formula is used
to calculate the total number of events E required:
E = (Z1- /2+Z1- )²[ ²(log )²]-1
where ² corresponds to the variance of the variable X; corresponds to the ratio of the
logarithms of the complement of the cumulative incidence functions associated with one
unit change in the value of the continuous variable X (Hsieh & Lavori, 2000). The total
number of individuals is obtained by using the formula N=E/ , where corresponds to the
proportion of expected events. These formulas can be applied in the presence of other
competing events, such as recurrence or death.
In the multivariate case, the variable X is adjusted relative to other prognostic variables,
leading to an increase in the number of individuals required by a variance inflation factor fv
=1/(1-R²). The total number of individuals is thus obtained by N*fv (Hsieh & Lavori, 2000).
Rate of radiation-induced CD8 lymphocyte apoptosis and late complications
- Breast Cancer
To test the prognostic value of the radiation-induced CD8 lymphocyte apoptosis rate on
the occurrence of complications in breast cancer, we start from the results of a preliminary
study (Azria et al., 2004). We consider the variable X=log(CD8) that has shown an effect
on the cumulative incidence of complications with an estimated relative risk of 5.5, which
corresponds to the risk of complications associated with one unit change of X. For a

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
26
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
relative risk estimated by the competing risk model with =log(5.5)=1.7, we estimate that a
relative risk equal to exp( )=exp(0.9)=2.4 should be detected. In this study, a regression
analysis of the variable X with potential prognostic factors (age, type of surgery, tumor
volume, adjuvant hormone therapy and adjuvant chemotherapy) gives a value of R²=0.13.
Based on ²=0.54, an estimated complication rate of =15%, a two-tailed error =0.05
and a error =0.05 (power=0.95), 430 patients must be included. As these estimates have
been obtained on a population who received a boost, while the current study plans to
include only 85% of patients with boost, the number of patients is increased by 15% for a
total number of 494 patients.
- Prostate Cancer
To test the prognostic value of the radiation-induced CD8 lymphocyte apoptosis rate on
the occurrence of genitourinary and gastrointestinal complications in prostate cancer, we
use as a base the results of the study by Zelefsky et al. (Zelefsky et al., 2002). By
considering the same parameters as in breast cancer, with the exception of a relative risk
for the competing risk model estimated at =1.7 and an estimated rate of gastrointestinal
complications of =5% and of genitourinary complications of 10%, a two-tailed error
=0.05 and a error =0.05 (power=0.95), we must include 368 patients.
With a total of 862 patients in the two populations, we will have enough statistical power to
determine the value of the radiation-induced CD8 lymphocyte apoptosis rate for the
prediction of the occurrence of the late complications under study.
4.7) Study exit criteria:
A patient could be withdrawn from the study for the following reasons:
- Tumor progression
- Consent withdrawal
- Unavailability of the basal value of radiation-induced apoptosis in CD8 lymphocytes
(technical, transport problem…)
5) TRIAL IMPLEMENTATION

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
27
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
5.1) Biological assay before the beginning of radiotherapy:
Each biological assay will be carried out before the beginning of radiotherapy.
At the moment of blood sampling, patients may have already started the hormone
therapy (population: breast and prostate) or have had an adjuvant chemotherapy
(population: breast).
Blood sampling will be carried out on a Monday morning and sent by express mail
(dedicated DHL account number) on the same day to the main investigation center at the
following address:
Mickaël Coelho, Laboratoire d’Immunociblage des Tumeurs et Ingénierie des
Anticorps, Bâtiment de Recherche, 1er étage (André Pèlegrin), CRLC Val d’Aurelle,
Rue croix verte, 34298 Montpellier cedex 05.
A single blood sample of 5 ml in a heparin tube is required. Samples will be handled and
prepared for the assay at the Laboratoire d’Immunociblage des Tumeurs et Ingénierie des
Anticorps (Tumor Immunotargeting and Antibody Engineering Laboratory), Montpellier.
This procedure will start the same day upon reception of the tube in the laboratory. The
other participant centers do not need to do any preparation before sending the tube
to Montpellier.
An anonymization procedure will be implemented: A self-adhesive label (provided by
the coordination center) must be stuck on each tube. The name of the participant center,
the sampling date and the first three letters of the patient’s surname and name will be
mentioned on this label. Each participant center will have an inclusion file that will allow
matching each sample to the clinical file.
If the sample cannot be used for technical problems (transport, storage problem …), the
local investigator will propose to the patient to give another blood sample. If the patient
refuses, he/she will be considered as withdrawn from the study and will receive the initially
planned treatment without any modification.
Sample handling for the apoptosis assay will be carried out only in Montpellier upon
reception of the tubes as follows:
- 200 µl of blood are distributed in small flasks or 6-well Petri dishes (3 cm in
diameter).

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
28
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
- 2 ml of RPMI1640/20% FCS are added to each flask to dilute the blood (1:10
dilution). Each assay is carried out in TRIPLICATE (for each patient, irradiation with
0 Gy and 4 Gy).
- After 24h, flasks are irradiated (4 Gy) or not (0 Gy, control). Flasks must be
removed from the irradiation room during the irradiation of other flasks.
- Immediately afterwards, the irradiated (or not) flasks are incubated at 37°C, 5%
CO2 for 48 h.
- Then the following protocol is carried out. A sheet of blue paper and a bleach-
resistant liquid waste container are placed in the radioactivity-dedicated hood. The
entire content of each flask is transferred to a FACS tube (5 ml) and centrifuged at
1450 rpm for 5 min. Some bleach is added to the (non-radioactive) liquid waste
container. The supernatant of the tubes is aspirated and pellets are resuspended in
200 µl of 1X PBS. Then, 10 µl of anti-human CD4-FITC antibody or 10 µl of anti-
human CD8-FITC antibody are added and after mixing, tubes are incubated in the
dark for 20 min. Four ml of Lysis Buffer diluted 1:10 with water are added to each
tube. After mixing, tubes are incubated in the dark for 20 min and then centrifuged
at 1450 rpm for 5 min. The supernatant is then removed. If blood is still present in
the pellet (red pellet instead of pink or white), pellets are resuspended in lysis buffer
(1 to 4 ml per tube for 5 to 20 min) and centrifuged at 1450 rpm for 5 min. Pellets
are then resuspended in 4 ml PBS, mixed and centrifuged again at 1450 rpm for 5
min. If the pellet is now white, it is resuspended in 200 µl PBS and 5 µl of Propidium
Iodide (PI) and 5 µl of RNase are added. Tubes are then stored at 4°C if the FACS
analysis with a FACSCAN is not carried out immediately.
5.2) Radiotherapy:
Radiotherapy will be carried out according to the modalities described in chapter 4.4.
In any case, the 5 ml blood samples will be collected before the beginning of radiotherapy
and sent to the main investigation center (see modalities in chapter 5.1).
No change in the routine practice is asked to the participant centers.
Toxicity monitoring during and after radiotherapy is described in chapter 5.4.
5.3) Inclusion work-up and cancer follow-up

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
29
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
Neither inclusion work-up nor specific cancer follow-up is required for this study. The
routine cancer management of each participant center is accepted.
5.4) Toxicity monitoring protocol:
A specific difficulty concerning the study of late irradiation toxicity events is their
appearance delay and progressive worsening over time (Dubray et al., 1997a). The
appearance delay of the clinical manifestations requires a prolonged follow-up of all
patients and the use of appropriate statistical methods. The interval between irradiation
and observation of late side effects can be of several years, but overall the literature data
indicate that after three years the majority of late secondary effects can be evaluated. For
instance, in a Danish study, 90% of sub-cutaneous fibroses were recorded at 3.2 years
(confidence interval at 95%: 2.3-3.9 years) after irradiation (Dubray et al., 1997a).
All late side effects will be evaluated independently by two physicians who have
been trained in their classification in order to minimize the assessment subjectivity. In
case of disagreement, the highest score will be retained. In all cases, both evaluators
will not be aware of the radiation-induced CD8 lymphocyte apoptosis rates.
It is planned that all patients will be monitored regularly according to the guidelines
described below: toxicity assessment before the first radiotherapy session (RT beginning),
once per week during the entire radiotherapy treatment, a month after the end of
radiotherapy (M1), three months after the end of radiotherapy (M3), six months after the
end of radiotherapy (M6) and then every six months up to three years after the end of
radiotherapy (i.e., M12, M18, M24, M30 and M36).
Data will be recorded in the CRF by the person designed by the investigator of each
center.
This follow-up schema is for guidance and a modification is authorized within the 15 days
following each initially planned date. If one visit of the provisional calendar could not be
carried out, the follow-up will continue according to the date of the next visit. If two
consecutive visits are missed, the participant center must inform the coordination center in
M36 M30 M24 M18 M12 M6 M3 M1 RT end RT beginning

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
30
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
order to confirm the inclusion of the patient in the study. The investigator must then call the
patient for a follow-up visit in the month following this decision and then continue according
to the initially fixed calendar.
5.5) Statistical analysis:
For the evaluation of late effects, all time intervals will be calculated from the beginning of
the treatment. The cumulative incidences of complications in function of the prognostic
variables will be calculated using a non-parametric model (Pepe & Mori, 1993). The main
statistical analysis will include a multivariate analysis using the Fine et al model of
competing risks (Fine, 2001) for the assessment of the impact of the radiation-induced
CD8 lymphocyte apoptosis rate on the occurrence of late complications in the presence of
other events (such as relapse or death) that are considered as competing risks in each of
the two pathologies. For breast cancer, the factors to take into account in the analysis are
the following: adjuvant hormone therapy, adjuvant chemotherapy and the different
radiotherapy parameters (irradiation volume, boost, dose at the breast surface, dose
delivered to the breast with or without lymph nodes, dose per fraction). We estimate that
60% of patients will receive adjuvant hormone therapy (RH+), 70% of RH+ patients will
receive adjuvant chemotherapy, 85% of RH patients will receive adjuvant chemotherapy
and 85% of patients will receive a boost (the remaining 15% are patients aged between 50
and 60 years who will not receive any boost in some participant centers in function of the
local guidelines).
For prostate cancer, the factors to be taken into account in the analysis are the following:
hormone therapy, diabetes, irradiated rectal volume and maximum dose delivered to the
rectum.
A multivariate ROC curve analysis will be carried out to assess the sensitivity and
specificity of the radiation-induced CD8 lymphocyte apoptosis rate by selecting patients
who have at least two years of follow-up in order to well differentiate the populations with
or without a late complication.

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
31
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
The data from the different participating centers will be sent to the Biostatistic Department
of Montpellier: Dr Andrew KRAMAR, Unités de Biostatistique, CRLC Val d’Aurelle, 34298
Montpellier cedex 05.
5.6) Data collection and processing
Data will be collected in a case report form (CRF) specific for this study. Each investigator
is responsible for all the information recorded in the CRF. The CRFs will be sent in
batches according to the status of each patient: at the end of the treatment, at the first and
third month of follow-up, and then each six months during three years.
The computer database will be updated with the CRF data using input fields specific for
the type of uploaded variable. Before uploading, a manual control of the files to be entered
is carried out to record the reception of the CRF sheets and to identify major incongruities.
Data uploading is carried out gradually during the study progression at the data
management center by a single operator for each project. The objective is to obtain a
computer file that is the exact copy of the documents to be entered (CRF). Each detected
problem is commented by the operator and recorded in a data entry form.
After each batch of data uploading, data are validated with control programs that are
defined beforehand, according to the protocol and the CRF, and that detect missing data,
consistency anomalies and accuracy anomalies.
If an error is detected, a liaison form between the management unit and the
investigator/ARC (clinical research associate) (DRF=Discrepancy report form) is prepared
to collect the correction. The correction is entered straight after the signed and dated DRF
has been sent back. This process is repeated up to when no error is left.
Problematic data are recorded in a glossary with the associated solutions and the retained
rules.
Each patient’s data are listed as data listings (lists by chapter) and « patient’s profile »
(summary of the main data for each patient).
The database is locked for the validated data. Data locking is done by dated copy of the
database and then by selection of the observations matching the criterion. This process is
repeated until all the validated data are recovered.
The Capture System software will be used for data management and the Stata software
for the statistical analysis.

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
32
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
5.7) Study termination/suspension:
The study may be stopped at the promoter’s request.
5.8) Adverse events:
An adverse event during a clinical study is the occurrence of any non-desired medical
effect in an individual who received a treatment, independently from a causality relation.
Cases of pregnancy must be reported for further studies.
Adverse events are recorded after the patient has been enrolled. If a patient shows an
adverse event after having signed the informed consent (study entry), but before the
attribution of any treatment (enrollment), the event will not be recorded, unless the
investigator thinks that such event might be due to a protocol procedure.
Before enrollment, the personnel of the site will record the medical condition (appearance
and nature) of each individual.
During the study, the personnel will record also any modification as well as the
appearance and nature of any adverse event.
Any clinical or biological, intercurrent event must be reported to the promoter and recorded
in the CRF in the pages provided for this.
Definition of serious adverse effects:
Are considered as serious adverse effects:
- events resulting in death,
- events requiring hospitalization or prolonging an existing hospitalization,
- life-threatening events,
- events resulting in disability or severe or permanent incapacity,
- a congenital anomaly.
A clinical outcome (for instance death) is not considered to be a serious event, unless the
investigator suspects a causality link with the treatment administered during the study.
Serious events occurring after study exit will not be recorded, unless the investigator thinks
that such event is linked to the used treatment or to a procedure carried out during the
protocol.
What to do in case of serious event:

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
33
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
In the case of a serious intercurrent event, the investigator must inform the promoter by
telephone or fax within the 24 hours following the observation. The investigator must fill in
the page in the CRF provided for intercurrent events and the "report form of serious event
occurring during clinical trials" joined to the CRF. The first two pages of this form must be
sent back to the trial promoter within five days after the initial telephone call and the last
page is kept in the CRF.
Concomitantly with the form, the investigator will prepare, if required, a written report on
this serious event and on its consequences.
Report of serious adverse events:
In accordance with the article L.1123-8 of the French law n. 88-1138 of the 20 December
1988 and its subsequent texts and the recommendations concerning the reporting of
serious events likely to be due to biomedical research carried out on a drug or related
product, the promoter will declare to the Agence du Médicament (French Health Products
Safety Agency) the deaths and the life-threatening events at latest within seven calendar
days following the date of the initial information, and for all the other cases at latest within
fifteen calendar days.
If applicable, also the trial coordinator, the different investigators, the Trial Supervisory
Board and the CCPPRB (Advisory Committee for People Protection in Medical Research)
should be informed.
If the treatment had to be stopped or the dose reduced in a patient, because of an adverse
effect, the circumstances and the data that led to this decision must be clearly stated in the
CRF.
If in some cases, the investigator reports an unexpected benefit for the patient, the
personnel of the site must mention «unexpected benefit» with the description of the
relevant event.
6) ETHICAL CONSIDERATIONS
6.1) Data ownership
Any information resulting from this trial is considered as confidential, at least until
completion of the appropriated analyses and controls by the promoter and the coordinating

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
34
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
investigator. The results can be published or presented only after the promoter’s
authorization.
The order of appearance of the principal investigators and/or the investigators in
communications will be in function of the number of patients who could be evaluated in the
program and the role played during the study implementation. For each center, one or
more investigators may be listed in the communications in function of the number of
inclusions and of the involvement in the study. Centers that included fewer than five
patients will be excluded from the publications.
6.2) Ethical and regulatory aspects
The clinical trial must be carried out in accordance with the ethical principles of the
Helsinki declaration of 1964 revised in Edinburgh in 2000, with the Good Clinical Practice
of the International Conference on Harmonization (ICH–E6, 17/07/96), with the European
Directive (2001/20/CE) on the management of clinical trials, with the amended Huriet’s law
(20/12/98) concerning the Protection of People volunteering for Biomedical Research as
well as with the rules laid down by the Commission Nationale Informatique et Libertés
(French National Commission for IT and Civil Liberties) (law n. 94-548 of the 1/07/94
completing the law n. 78-17 of the 6/01/78).
6.3) Comité consultatif pour la Protection des Personnes dans la Recherche Biomédicale
(CCPPRB; Advisory Committee for People Protection in Medical Research)
The clinical trial protocol as well as the different amendments or any information or
document considered necessary by the promoter will be submitted by the trial coordinating
investigator to a CCPPRB for advisory opinion (art. L. 1223-6). This duly constituted and
independent committee must authorize the trial implementation. Neither investigators, nor
persons who are not independent from this assay can take part in this decision. The
different original documents of the CCPPRB written answers must be sent by the
coordinator to the promoter.
A copy of the CCPPRB approval letter is sent to all participant centers.
The CCPPRB is informed about any new significant event concerning the product under
trial. The investigator is responsible for the transmission to the CCPPRB of any new
significant event concerning the product under trial.

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
35
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
6.4) Patients’ information and consent
Before carrying out biomedical research on a person, his/her free, informed and explicit
consent must be collected after this person has been informed by the investigator or the
investigator’s representative about the research objective(s), the implementation and
duration of the study, the trial benefits, potential risks and constraints as well as the nature
of the product under study and the CCPPRB opinion (art. L. 1122-1).
The consent form will be dated and signed personally by the patient and by the
investigator or physician who represents the investigator. Two originals will be signed; one
will be given to the patient or his/her legal representative and the other will be archived by
the investigator.
The information and informed consent forms for the patient must be associated in the
same document in order to avoid any risk of dispute about the content of the given
information.
6.5) Investigators’ responsibilities
The principal investigator pledges to carry out the clinical trial according to the protocol
that has been approved by the CCPPRB. The investigator must not introduce any
modification in the protocol without the promoter’s authorization and without the CCPPRB
favorable opinion on the proposed modifications.
The principal investigator is responsible for:
- giving to the promoter his/her curriculum vitae as well as those of the co-investigators,
- identifying the members of his/her group who will take part in the trial and defining their
responsibilities,
- starting the patients’ recruitment after the promoter’s authorization,
- ensuring the required number of patients within the limits of the planned recruitment
period.
Each investigator is responsible for:
- obtaining the informed consent dated and signed personally by the patient before any
specific selection procedure,

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
36
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
- filling in regularly the CRF for each patient included in the trial and giving direct access
to the source documents to the Clinical Research Associate (Assistant de Recherche
Clinique; ARC) for CRF data validation,
- correcting and signing the corrections in the CRF for each patient included in the study,
- accepting the ARC’s regular visits and, if applicable, visits from auditors authorized by
the promoter or from inspectors of the supervising organisms.
Any document concerning the study (protocol, consent forms, CRFs, investigator’s file,
etc…), and the original documents (laboratory results, radiology results, consultation
reports, reports of the clinical exams carried out, etc.) must be stored in a safe place and
considered as confidential material. Data storage will be under the investigator’s
responsibility according to the current legislation.
The investigator must store the data as well as the patients’ identification list for at least 30
years after the study end.
6.6) Promoter’s responsibilities
According to the Huriet’s law, the promoter is responsible for:
- taking out an insurance that guarantees the promoter’s civil liability in case of harmful
consequences of the research for the person who takes part in this research (art.
L.1121-7),
- paying to the competent Direction Régionale des Affaires Sanitaires et Sociales
(DRASS; Regional Office for Health and Social Affairs) the fixed duty for consulting a
CCPPRB (decree of the 7/5/91 and of 13/12/01),
- sending to the Agence Française de Sécurité Sanitaire des Produits de Santé
(AFSSAPS; French Health Products Safety Agency) the initial letter of intent that
describes the essential research data together with the CCPPRB opinion (art.L.1123-8),
- informing the Directors (art. L.1123-10, R.5124) and Pharmacists (art. L.5125-19,
R.5124-1) of the health care facilities,
- communicating to the investigators all the information required to carry out the research
(art. R.5122),
- informing, as soon as aware of it, the AFSSAPS about any serious adverse reaction
likely to be caused by the research as well as any premature study
termination/suspension (art. L.1123-8).

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
37
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
The promoter must also make a statement concerning the personal and electronic medical
data to the Comité Consultatif et de la Commission Nationale Informatique et Liberté
(CNIL), according to the terms and conditions of the simplified procedure (law n. 94-548 of
the 1/07/94 completing the law n. 78-17 of the 6/01/78).
The promoter must guarantee the storage of the essential documents concerning the
study implementation in conditions that ensure their safety for the minimum period
specified by the good clinical practice (i.e., 15 years after the research end).
7) EXPECTED RESULTS AND PERSPECTIVES
The pre-treatment identification of the radiobiological features of healthy tissues might
ultimately allow envisaging a tailored radiotherapy approach to optimize the therapeutic
index.
Indeed, it is tempting to think that most patients might be «under-treated» due to concerns
about complications that occur only among the most sensitive among them. This study
might allow the identification of a big portion of the population who is not at risk of late
toxicities and thus to propose to them dose-intensified radiotherapy. Moreover, the
identification of the “sensitive” patients will thus allow increasing the irradiation dose for
some organs at risk in the patients considered as «resistant».
In summary, patients treated by curative radiotherapy receive standard doses (i.e., the
same dose for all). An assay that allows the radiotherapist to determine the radiosensitivity
of the patient’s normal tissues by measuring the circulating lymphocyte apoptosis rate
could help tailoring the dose of radiotherapy in hypersensitive patients and increasing the
total dose in the other patients. Theoretically, the local control can be improved by
increasing moderately the total dose of radiotherapy. It is thought that 5 to 10% of patients
treated by curative radiotherapy develop severe complications. These patients are thus
considered as hypersensitive.
Moreover, as we already showed for hormone therapy (Azria et al., 2004), the use of such
assay might allow associating some drugs with radiotherapy in an optimized manner
relative to the risk of late toxicity.

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
38
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
8) REFERENCES
Arriagada, R., Mouriesse, H., Sarrazin, D., Clark, R.M. & Deboer, G. (1985). Radiotherapy
alone in breast cancer. I. Analysis of tumor parameters, tumor dose and local control:
the experience of the Gustave-Roussy Institute and the Princess Margaret Hospital. Int
J Radiat Oncol Biol Phys, 11, 1751-7.
Azria, D., Gourgou, S., Sozzi, W.J., Zouhair, A., Mirimanoff, R.O., Kramar, A., Lemanski,
C., Dubois, J.B., Romieu, G., Pelegrin, A. & Ozsahin, M. (2004). Concomitant use of
tamoxifen with radiotherapy enhances subcutaneous breast fibrosis in hypersensitive
patients. Br J Cancer, 91, 1251-60.
Bartelink, H., Horiot, J.C., Poortmans, P., Struikmans, H., Van den Bogaert, W., Barillot, I.,
Fourquet, A., Borger, J., Jager, J., Hoogenraad, W., Collette, L. & Pierart, M. (2001).
Recurrence rates after treatment of breast cancer with standard radiotherapy with or
without additional radiation. N Engl J Med, 345, 1378-87.
Buchholz, T.A. (1999). Finding our sensitive patients. Int J Radiat Oncol Biol Phys, 45,
547-8.
Cesaretti, J.A., Stock, R.G., Lehrer, S., Atencio, D.A., Bernstein, J.L., Stone, N.N.,
Wallenstein, S., Green, S., Loeb, K., Kollmeier, M., Smith, M. & Rosenstein, B.S.
(2005). ATM sequence variants are predictive of adverse radiotherapy response among
patients treated for prostate cancer. Int J Radiat Oncol Biol Phys, 61, 196-202.
Crompton, N.E. & Ozsahin, M. (1997). A versatile and rapid assay of radiosensitivity of
peripheral blood leukocytes based on DNA and surface-marker assessment of
cytotoxicity. Radiat Res, 147, 55-60.
Crompton, N.E., Shi, Y.Q., Emery, G.C., Wisser, L., Blattmann, H., Maier, A., Li, L.,
Schindler, D., Ozsahin, H. & Ozsahin, M. (2001). Sources of variation in patient
response to radiation treatment. Int J Radiat Oncol Biol Phys, 49, 547-54.
Delanian, S., Balla-Mekias, S. & Lefaix, J.L. (1999). Striking regression of chronic
radiotherapy damage in a clinical trial of combined pentoxifylline and tocopherol. J Clin
Oncol, 17, 3283-90.
Delanian, S., Porcher, R., Balla-Mekias, S. & Lefaix, J.L. (2003). Randomized, placebo-
controlled trial of combined pentoxifylline and tocopherol for regression of superficial
radiation-induced fibrosis. J Clin Oncol, 21, 2545-50.
Denis, F., Garaud, P., Bardet, E., Alfonsi, M., Sire, C., Germain, T., Bergerot, P., Rhein,
B., Tortochaux, J., Oudinot, P. & Calais, G. (2003). Late toxicity results of the GORTEC

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
39
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
94-01 randomized trial comparing radiotherapy with concomitant radiochemotherapy for
advanced-stage oropharynx carcinoma: comparison of LENT/SOMA, RTOG/EORTC,
and NCI-CTC scoring systems. Int J Radiat Oncol Biol Phys, 55, 93-8.
Dewey, W.C., Ling, C.C. & Meyn, R.E. (1995). Radiation-induced apoptosis: relevance to
radiotherapy. Int J Radiat Oncol Biol Phys, 33, 781-96.
Dubray, B. (1995). [Late complications of radiotherapy. Role of the time factor]. Bull
Cancer Radiother, 82, 98-100.
Dubray, B., Delanian, S. & Lefaix, J.L. (1997a). [Late effects of mammary radiotherapy on
skin and subcutaneous tissues]. Cancer Radiother, 1, 744-52.
Dubray, B., Pavy, J.J., Giraud, P., Danhier, S. & Cosset, J.M. (1997b). [Predictive tests of
response to radiotherapy. Assessment and perspectives in 1997]. Cancer Radiother, 1,
473-83.
Emami, B., Lyman, J., Brown, A., Coia, L., Goitein, M., Munzenrider, J.E., Shank, B., Solin,
L.J. & Wesson, M. (1991). Tolerance of normal tissue to therapeutic irradiation. Int J
Radiat Oncol Biol Phys, 21, 109-22.
Fine, J.P. (2001). Regression modeling of competing crude failure probabilities.
Biostatistics, 2, 85-97.
Floyd, D.N. & Cassoni, A.M. (1994). Intrinsic radiosensitivity of adult and cord blood
lymphocytes as determined by the micronucleus assay. Eur J Cancer, 30A, 615-20.
Hoeller, U., Borgmann, K., Bonacker, M., Kuhlmey, A., Bajrovic, A., Jung, H., Alberti, W. &
Dikomey, E. (2003). Individual radiosensitivity measured with lymphocytes may be used
to predict the risk of fibrosis after radiotherapy for breast cancer. Radiother Oncol, 69,
137-44.
Hsieh, F.Y. & Lavori, P.W. (2000). Sample-size calculations for the Cox proportional
hazards regression model with nonbinary covariates. Control Clin Trials, 21, 552-60.
Huchet, A., Caudry, M., Belkacemi, Y., Trouette, R., Vendrely, V., Causse, N., Recaldini,
L., Atlan, D. & Maire, J.P. (2003). [Volume-effect and radiotherapy [II]. Part II: volume-
effect and normal tissue]. Cancer Radiother, 7, 353-62.
Iannuzzi, C.M., Atencio, D.P., Green, S., Stock, R.G. & Rosenstein, B.S. (2002). ATM
mutations in female breast cancer patients predict for an increase in radiation-induced
late effects. Int J Radiat Oncol Biol Phys, 52, 606-13.
Jacob, R., Hanlon, A.L., Horwitz, E.M., Movsas, B., Uzzo, R.G. & Pollack, A. (2005). Role
of prostate dose escalation in patients with greater than 15% risk of pelvic lymph node
involvement. Int J Radiat Oncol Biol Phys, 61, 695-701.

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
40
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
Johansen, J., Bentzen, S.M., Overgaard, J. & Overgaard, M. (1996). Relationship between
the in vitro radiosensitivity of skin fibroblasts and the expression of subcutaneous
fibrosis, telangiectasia, and skin erythema after radiotherapy. Radiother Oncol, 40, 101-
9.
Kiltie, A.E., Ryan, A.J., Swindell, R., Barber, J.B., West, C.M., Magee, B. & Hendry, J.H.
(1999). A correlation between residual radiation-induced DNA double-strand breaks in
cultured fibroblasts and late radiotherapy reactions in breast cancer patients. Radiother
Oncol, 51, 55-65.
Lartigau, E., Dubray, B. & Mornex, F. (1997). [Biological mechanisms of late effects of
ionizing radiations]. Cancer Radiother, 1, 669-76.
Latouche, A., Porcher, R. & Chevret, S. (2004). Sample size formula for proportional
hazards modelling of competing risks. Stat Med, 23, 3263-74.
Maingon, P., Chapet, O., Barillot, I. & Romestaing, P. (2004). [To boost the tumor bed: the
age of reason]. Cancer Radiother, 8, 33-8.
Ozsahin, M., Ozsahin, H., Shi, Y., Larsson, B., Wurgler, F.E. & Crompton, N.E. (1997).
Rapid assay of intrinsic radiosensitivity based on apoptosis in human CD4 and CD8 T-
lymphocytes. Int J Radiat Oncol Biol Phys, 38, 429-40.
Pepe, M.S. & Mori, M. (1993). Kaplan-Meier, marginal or conditional probability curves in
summarizing competing risks failure time data? Stat Med, 12, 737-51.
Pollack, A., Smith, L.G. & von Eschenbach, A.C. (2000a). External beam radiotherapy
dose response characteristics of 1127 men with prostate cancer treated in the PSA era.
Int J Radiat Oncol Biol Phys, 48, 507-12.
Pollack, A., Zagars, G.K., Smith, L.G., Lee, J.J., von Eschenbach, A.C., Antolak, J.A.,
Starkschall, G. & Rosen, I. (2000b). Preliminary results of a randomized radiotherapy
dose-escalation study comparing 70 Gy with 78 Gy for prostate cancer. J Clin Oncol,
18, 3904-11.
Radford, I.R. & Murphy, T.K. (1994). Radiation response of mouse lymphoid and myeloid
cell lines. Part III. Different signals can lead to apoptosis and may influence sensitivity to
killing by DNA double-strand breakage. Int J Radiat Biol, 65, 229-39.
Romestaing, P., Lehingue, Y., Carrie, C., Coquard, R., Montbarbon, X., Ardiet, J.M.,
Mamelle, N. & Gerard, J.P. (1997). Role of a 10-Gy boost in the conservative treatment
of early breast cancer: results of a randomized clinical trial in Lyon, France. J Clin
Oncol, 15, 963-8.

Discipline : Cancérologie Radiothérapie Nom du Demandeur : Dr David AZRIA
41
Protocol PHRC 2005 DA
Protocol: version #6 - 28/09/2007
Information and consent form: version #4 - 21/02/2006
Investigator list: version #6 - 14/09/2006
Slonina, D. & Gasinska, A. (1997). Intrinsic radiosensitivity of healthy donors and cancer
patients as determined by the lymphocyte micronucleus assay. Int J Radiat Biol, 72,
693-701.
Stone, H.B., Coleman, C.N., Anscher, M.S. & McBride, W.H. (2003). Effects of radiation on
normal tissue: consequences and mechanisms. Lancet Oncol, 4, 529-36.
Thames, H.D., Hendry, J.H., Moore, J.V., Ang, K.K. & Travis, E.L. (1989). The high
steepness of dose-response curves for late-responding normal tissues. Radiother
Oncol, 15, 49-53.
Trotti, A., Byhardt, R., Stetz, J., Gwede, C., Corn, B., Fu, K., Gunderson, L., McCormick,
B., Morrisintegral, M., Rich, T., Shipley, W. & Curran, W. (2000). Common toxicity
criteria: version 2.0. an improved reference for grading the acute effects of cancer
treatment: impact on radiotherapy. Int J Radiat Oncol Biol Phys, 47, 13-47.
Trotti, A., Colevas, A.D., Setser, A., Rusch, V., Jaques, D., Budach, V., Langer, C.,
Murphy, B., Cumberlin, R., Coleman, C.N. & Rubin, P. (2003). CTCAE v3.0:
development of a comprehensive grading system for the adverse effects of cancer
treatment. Semin Radiat Oncol, 13, 176-81.
Trowell, O.A. (1952). The sensitivity of lymphocytes to ionising radiation. J Pathol
Bacteriol, 64, 687-704.
Withers, H.R., Taylor, J.M. & Maciejewski, B. (1988). Treatment volume and tissue
tolerance. Int J Radiat Oncol Biol Phys, 14, 751-9.
Zelefsky, M.J., Fuks, Z., Hunt, M., Yamada, Y., Marion, C., Ling, C.C., Amols, H.,
Venkatraman, E.S. & Leibel, S.A. (2002). High-dose intensity modulated radiation
therapy for prostate cancer: early toxicity and biochemical outcome in 772 patients. Int J
Radiat Oncol Biol Phys, 53, 1111-6.
Zietman, A.L., Desilvio, M., Slater, J.D., Rossi, C.J., Yonemoto, L.T., Slater, J.M., Berkey,
B., Adams, J.A. & Shipley, W.U. (2004). A Randomized trial comparing conventional
dose (70.2 Gy) and High-dose (79.2 Gy) conformal radiation in early stage
adenocarcinoma of the prostate: results of an interim analysis of PROG 95-09. Proc
ASTRO, 60, 131 (Abst 4).