oulu 2019 d 1528 university of oulu p.o. box 8000 fi-90014
TRANSCRIPT

UNIVERSITY OF OULU P .O. Box 8000 F I -90014 UNIVERSITY OF OULU FINLAND
A C T A U N I V E R S I T A T I S O U L U E N S I S
University Lecturer Tuomo Glumoff
University Lecturer Santeri Palviainen
Senior research fellow Jari Juuti
Professor Olli Vuolteenaho
University Lecturer Veli-Matti Ulvinen
Planning Director Pertti Tikkanen
Professor Jari Juga
University Lecturer Anu Soikkeli
Professor Olli Vuolteenaho
Publications Editor Kirsti Nurkkala
ISBN 978-952-42-2343-8 (Paperback)ISBN 978-952-42-2344-5 (PDF)ISSN 0355-3221 (Print)ISSN 1796-2234 (Online)
U N I V E R S I TAT I S O U L U E N S I S
MEDICA
ACTAD
D 1528
AC
TAT
iia Honkanen
OULU 2019
D 1528
Tiia Honkanen
MORE EFFICIENT USE OFHER TARGETING AGENTSIN CANCER THERAPY
UNIVERSITY OF OULU GRADUATE SCHOOL;UNIVERSITY OF OULU,FACULTY OF MEDICINE;MEDICAL RESEARCH CENTER OULU;OULU UNIVERSITY HOSPITAL


ACTA UNIVERS ITAT I S OULUENS I SD M e d i c a 1 5 2 8
TIIA HONKANEN
MORE EFFICIENT USE OF HER TARGETING AGENTS IN CANCER THERAPY
Academic dissertation to be presented with the assent ofthe Doctoral Training Committee of Health andBiosciences of the University of Oulu for public defencein Auditorium 7 of Oulu University Hospital, on 18October 2019, at 12 noon
UNIVERSITY OF OULU, OULU 2019

Copyright © 2019Acta Univ. Oul. D 1528, 2019
Supervised byDocent Jussi Koivunen
Reviewed byDocent Minna TannerDocent Maria Sundvall
ISBN 978-952-62-2343-8 (Paperback) ISBN 978-952-62-2344-5 (PDF)
ISSN 0355-3221 (Printed)ISSN 1796-2234 (Online)
Cover DesignRaimo Ahonen
JUVENES PRINTTAMPERE 2019
OpponentProfessor Jorma Isola

Honkanen, Tiia, More efficient use of HER targeting agents in cancer therapy. University of Oulu Graduate School; University of Oulu, Faculty of Medicine; MedicalResearch Center Oulu; Oulu University HospitalActa Univ. Oul. D 1528, 2019University of Oulu, P.O. Box 8000, FI-90014 University of Oulu, Finland
Abstract
Cancer treatments have remarkably improved over the past years since targeted therapies andimmunotherapy have been introduced to the field of oncology. The benefit of these new therapiesis often limited, however, by de novo or acquired therapy resistances, which should be noticedwhen making clinical decisions.
In this current work, we studied the prognostic and predictive values of several immunologicalmarkers in metastatic HER2-positive breast cancer treated with trastuzumab, because trastuzumabis still given to patients according to the HER2 status only, without certainty of tumor response.We also determined the role of HER2 and HER3 for cancer stem cells (CSC) in ALK translocatednon-small cell lung cancer (NSCLC) cell lines since the CSCs are causing therapy resistance andcancer recurrence.
The results demonstrated that a high number of cytotoxic T cells, together with a high numberof M1-like macrophages in the center of the tumor (CT), are promising and independentprognostic factors in HER2-positive breast cancer. These markers together also can predict theprogression of the disease and the length of trastuzumab discontinuation in tumor response.Expression of HER2 and HER3 increased the stem-like properties of ALK translocated NSCLCcells, which were decreased when the expressions were downregulated. HER2-HER3-dependentCSCs also mediated the ALK therapy resistance.
In conclusion, this study suggests that patients with a favorable immunological tumor profile(high number of cytotoxic T cells and M1-like macrophages in the CT) could be treated in a less-intensive manner, that trastuzumab discontinuation could be feasible for these patients, and thattargeting of HER2 and HER3 receptors can lead to more effective killing of cancer stem-like cellsand should be further studied.
Keywords: ALK, breast cancer, cancer stem cell, ERBB, HER2, HER3, NSCLC,trastuzumab, tumor-infiltrating lymphocytes


Honkanen, Tiia, HER-proteiineja kohdentavien lääkkeiden tehokkaampi käyttösyöpähoidoissa. Oulun yliopiston tutkijakoulu; Oulun yliopisto, Lääketieteellinen tiedekunta; Medical ResearchCenter Oulu; Oulun yliopistollinen sairaalaActa Univ. Oul. D 1528, 2019Oulun yliopisto, PL 8000, 90014 Oulun yliopisto
Tiivistelmä
Syöpähoidot ovat kehittyneet huomattavasti, kun kohdennetut hoidot ja immunologiset hoidotovat tulleet perinteisten hoitojen rinnalle. Usein näiden hoitojen hyötyä kuitenkin rajoittaa joolemassa oleva lääkeresistenssi tai sen kehittyminen, mikä tulisi ottaa huomioon hoitoja suunni-teltaessa.
Tässä työssä tutkittiin immunologisia merkkiaineita, joilla voitaisiin ennustaa trastutsumabi-hoidon vastetta sekä potilaiden ennustetta levinneessä HER2-positiivisessa rintasyövässä. Tällähetkellä trastutsumabi-hoitopäätös tehdään pelkän HER2-geenimonistuman mukaan ilman var-muutta siitä, hyötyykö potilas oikeasti hoidosta. Lisäksi tutkimme HER2- ja HER3-reseptorienmerkitystä syövän kantasoluille ALK-translokoituneessa ei-pienisoluisessa keuhkosyövässä(NSCLC), sillä syövän kantasolut ovat yksi merkittävimmistä tekijöistä lääkeresistenssin kehit-tymisessä ja syövän uusiutumisessa.
Työssä havaittiin, että kasvaimen keskellä oleva suuri määrä sytotoksisia T-soluja sekä M1-tyypin makrofageja on yhteydessä potilaiden parempaan ennusteeseen ja että kyseiset merkkiai-neet ovat toisistaan riippumattomia. Merkkiaineet pystyivät ennustamaan myös taudin etenemis-tä sekä trastutsumabi-hoitokeskeytyksen pituutta. HER2- ja HER3-proteiinien tuotto lisäsi ALK-translokoituneiden NSCLC-solujen kantasolumaisia ominaisuuksia, jotka puolestaan vähenivät,kun proteiinien tuotto estettiin. Lisäksi HER2-HER3 -riippuvaiset syövän kantasolut säätelivätlääkeresistenssiä kyseisessä taudissa.
Työn tulokset viittaavat siihen, että potilaita, joilla on suotuisa kasvaimen immunoprofiili(suuri määrä sytotoksisia T-soluja ja M1-tyypin makrofageja kasvaimen keskellä) pystyttäisiinhoitamaan keveimmillä hoidoilla ja HER2-hoitokeskeytys voisi olla mahdollinen näillä potilail-la. Lisäksi työ korostaa HER2- ja HER3-reseptorien kohdentamista syövän kantasolumaistensolujen tehokkaamman tuhoamisen saavuttamiseksi.
Asiasanat: ALK, ERBB, HER2, HER3, kasvaimeen infiltroituneet lymfosyytit,NSCLC, rintasyöpä, syövän kantasolut, trastutsumabi


“It’s when you cry just a little bit, but you laugh in the middle that you’ve made it – And don’t it feel alright –
And don’t it feel so nice – Lovely” Jason Mraz

8

9
Acknowledgements
This study was carried out at the Department of Oncology and Radiotherapy, Oulu
University Hospital during the years 2015–2019.
I would like to express my deepest gratitude to my supervisor Docent Jussi
Koivunen, MD, PhD, for his great expertise and guidance throughout these past
years. Thank you for introducing me to this great field of science, it has been a
pleasure to work with you and learn from you. I truly admire the enthusiasm you
have for research and work. Thank you for all your support and encouragement. I
was extremely lucky to have you as my supervisor.
I would like to thank the members of my follow-up group, Professor Outi
Kuittinen, MD, PhD, Minna Jääskeläinen, MD, PhD and Nina Kokkonen, PhD for
their expertise and great comments. I appreciate and thank the pre-examiners of
this thesis Docent Minna Tanner, MD, PhD and Docent Maria Sundvall, MD, PhD
for taking the time to examine my thesis and for your great comments, which
improved my work.
I wish to thank all the co-authors and collaborators for their contributions to
my projects: Docent Peeter Karihtala, MD, PhD, Juha Väyrynen, MD, PhD,
Professor Markus Mäkinen, MD, PhD, Professor Peppi Karppinen, MD, PhD,
Docent Päivi Auvinen, MD, PhD, Satu Tiainen, MD, Emmi Wilenius, MSc and
Antti Tikkanen, MB. I also thank Virpi Glumoff, PhD, Elitsa Dimova, PhD, Hanna-
Riikka Teppo, MD, PhD and Ms. Riitta Vuento for their help and technical
assistance in my projects. Special thanks to Peeter and Juha for their statistical and
pathological guidance.
I would like to express my warmest thanks to our research community for
sharing this time, all the struggles and successes with me: Professor Taina
Turpeenniemi-Hujanen, MD, PhD, Professor Outi Kuittinen, MD, PhD and all the
past and current fellow researchers. Special thanks to my dear co-worker Ms. Anne
Bisi. Thank you for everything; for the technical assistance and all the support. You
made the working days so much better.
I want to express my gratitude to my dear friends Meira, Krista, Emmi, Heikki,
Kati, Mikko, Anna and Fawzi, who are also my colleagues. Thank you for
everything; all the time we have spent, all the laughs and adventures, every advice
you have given me, and all the sympathy and support during these years. I would
also like to thank my non-academic friends, especially Suvi and Janne, who have
made it easier to relax and escape the pressure of the scientific world. Suvi, my

10
dear friend, thank you for sticking by me all this time, ever since the childhood.
I’m lucky to have a friend like you.
Special thanks to my family, my brothers Juha and Tomi, and my parents Tuovi
and Pentti. I appreciate the warm relationship I have with both of my brothers. We
support each other no matter what, I love you both. I also want to thank Päivi, it
has been a real pleasure getting to know you. My deepest gratitude goes to my
parents. Without you I would not be where I am now. Thank you for showing me
how to love and truly care about someone. Thank you for always believing in me
and supporting me, I love you. Finally, I want to thank my beloved partner, Toni.
Thank you for all the support and love you have given me during these years. You
mean the world to me. I would also like to thank the whole Valtanen-family for
their care and support.
I acknowledge the financial support of this thesis provided by the Cancer
Foundation Finland, Sigrid Juselius Foundation, Ida Montin foundation, the
University of Oulu Graduate School and the University of Oulu Scholarship
Foundation.
Oulu, August 2019 Tiia Honkanen

11
Abbreviations
4E-BP1 eukaryotic translation initiation factor 4E-binding protein 1
ADCC antibody-dependent cellular cytotoxicity
ADCP antibody-dependent cellular phagocytosis
ALDH1 aldehyde dehydrogenase 1
ALK anaplastic lymphoma kinase
APC antigen presenting cell
ATP adenosine triphosphate
AUC area under the curve
BRCA breast cancer susceptibility gene
BSA bovine serum albumin
CCL2 chemokine ligand 2
CD cluster of differentiation
ChT chemotherapy
CRISPR clustered regularly interspaced short palindromic repeats
CSC cancer stem cell
CSF cerebrospinal fluid
CSLC cancer stem-like cell
CT center of the tumor
DC dendritic cell
DNA deoxyribonucleic acid
EDTA ethylenediaminetetraacetic acid
EGF epidermal growth factor
EGFR epidermal growth factor receptor
EMA European medicines agency
EML4 echinoderm microtubule-associated protein-like 4
EMT epithelial to mesenchymal transition
ER estrogen receptor
ERBB avian erythroblastosis oncogene B
ERK extracellular signal-regulated kinase
ET endocrine therapy
FcγR fragment C gamma receptor
FDA food and drug administration
FoxP3 forkhead box P3
Grb2 growth factor receptor-bound protein 2
GDP guanosine diphosphate

12
GFP green fluorescent protein
GTP guanosine triphosphate
HB-EGF heparin binding EGF-like growth factor
H&E hematoxylin and eosin
HER human epidermal growth factor receptor
HRP horseradish peroxidase
IDO1 indoleamine 2,3-dioxygenase 1
IFN-γ interferon-gamma
IL-10 interleukin-10
IM invasive margin
iNOS inducible nitric oxide synthase
M1 M1-like macrophages
M2 M2-like macrophages
mAb monoclonal antibody
MAPK mitogen-activated protein kinase
MDSC myeloid-derived suppressor cell
MEK mitogen-activated protein kinase kinase
MHC major histocompatibility complex
mRNA messenger RNA
miRNA microRNA
mTOR mammalian target of rapamycin
NK natural killer
NKT natural killer T cell
NRG neuregulin
NSCLC non-small cell lung cancer
OS overall survival
PCR polymerase chain reaction
PD-1 programmed cell death 1
PD-L1 programmed cell death ligand 1
PI3K phosphoinositide 3-kinase
PIP2 phosphatidylinositol 4,5-bisphosphate
PIP3 phosphatidylinositol (3,4,5)-trisphosphate
PTB phosphotyrosine-binding
PTEN phosphatase and tensin homolog
PVDF polyvinylidene difluoride
RB1 retinoblastoma 1
RNA ribonucleic acid

13
ROC receiver operating characteristic
RT room temperature / radiotherapy
RTK receptor tyrosine kinase
S6K1 ribosomal protein S6 kinase 1
SDS-PAGE sodium dodecyl sulfate-polyacrylamide gel electrophoresis
SH2 Src homology-2
Shc Src homology-2 domain containing
shRNA short hairpin RNA
sgRNA single guide RNA
SOS son of sevenless
SOX2 sex determining region Y-box 2
TAM tumor-associated macrophage
T-DM1 trastuzumab emtansine
Th T helper
TGF-β transforming growth factor beta
TIL tumor infiltrating lymphocytes
TKI tyrosine kinase inhibitor
TLS tertiary lymphoid structure
TNF-α tumor necrosis factor alpha
TP53 tumor protein 53
TRAIL TNF-related apoptosis-inducing ligand
Treg regulatory T cell

14

15
Original publications
This thesis is based on the following publications, which are referred throughout
the text by their Roman numerals:
I Honkanen, T.J.*, Moilanen, T.*, Karihtala, P., Tiainen, S., Auvinen, P., Väyrynen, J.P., Mäkinen, M. & Koivunen, J.P. (2017). Prognostic and predictive role of spatially positioned tumour infiltrating lymphocytes in metastatic HER2 positive breast cancer treated with trastuzumab. Sci Rep, 7(1), 18027.
II Honkanen, T.J., Tikkanen, A., Karihtala, P., Mäkinen, M., Väyrynen, J.P. & Koivunen, JP. (2019). Prognostic and predictive role of tumour-associated macrophages in HER2 positive breast cancer. Sci Rep, 9(1), 10961.
III Honkanen, T., Wilenius, E., Koivunen, P., & Koivunen, J.P. (2017). HER2 regulates cancer stem-like cell phenotype in ALK translocated NSCLC. Int J Oncol, 51(2), 599-606.
IV Honkanen T.J. & Koivunen J.P. (2019). HER3 regulates cancer stem-like cell properties in ALK translocated NSCLC. Manuscript.
*Equal contribution

16

17
Table of contents
Abstract
Tiivistelmä
Acknowledgements 9
Abbreviations 11
Original publications 15
Table of contents 17
1 Introduction 19
2 Review of the literature 21
2.1 Cancer ..................................................................................................... 21
2.1.1 Cancer genetics ............................................................................. 21
2.1.2 Cancer epigenetics ........................................................................ 23
2.1.3 Cancer biology ............................................................................. 23
2.1.4 Main signaling pathways altered in cancer ................................... 27
2.2 Tumor immunology ................................................................................ 29
2.2.1 Innate immune system and cancer ................................................ 31
2.2.2 Adaptive immune system and cancer ........................................... 33
2.2.3 Tumor immune profile and Immunoscore .................................... 34
2.3 Cancer stem cells .................................................................................... 35
2.3.1 Hierarchical and stochastic cancer stem cell models .................... 36
2.3.2 Signaling pathways related to cancer stem cell phenotype........... 38
2.3.3 Markers used to identify cancer stem cells ................................... 38
2.4 HER family ............................................................................................. 39
2.4.1 Structure and function .................................................................. 39
2.4.2 HERs and cancer .......................................................................... 41
2.4.3 HER targeted therapies ................................................................. 43
2.5 HER2+ breast cancer .............................................................................. 46
2.5.1 Treatment of HER2+ breast cancer .............................................. 47
2.5.2 Tumor-infiltrating lymphocytes in HER2+ breast cancer ............ 49
2.6 ALK in cancer ......................................................................................... 50
2.6.1 ALK translocated NSCLC ............................................................. 50
2.6.2 ALK targeting and resistance mechanisms in cancer ................... 51
3 Aims of the study 53
4 Materials and methods 55
4.1 Publications I and II ................................................................................ 55
4.1.1 Patient material ............................................................................. 55

18
4.1.2 Immunohistochemistry and immune cell counting ....................... 55
4.1.3 Statistics ........................................................................................ 57
4.1.4 Ethics ............................................................................................ 57
4.2 Publications III and IV ............................................................................ 58
4.2.1 Cell lines, inhibitors and growth factors ....................................... 58
4.2.2 Lentiviral knockdown and retroviral overexpression ................... 59
4.2.3 CRISPR-Cas9 knockdown ........................................................... 59
4.2.4 DNA extraction, PCR and sequencing .......................................... 60
4.2.5 Western blot .................................................................................. 61
4.2.6 Colony formation assay ................................................................ 61
4.2.7 Tumor sphere formation assay ...................................................... 62
5 Results 65
5.1 Immunological markers in metastatic HER2+ breast cancer .................. 65
5.1.1 Patient characteristics ................................................................... 65
5.1.2 Staining and image analysis of the studied markers ..................... 66
5.1.3 Association of T cell subsets with clinical data ............................ 68
5.1.4 Association of macrophage subsets with clinical data .................. 70
5.1.5 Association of other markers with clinical data ............................ 70
5.1.6 Combined analysis of CD8 and M1.............................................. 71
5.1.7 CD8 and M1 infiltration and trastuzumab benefit ........................ 71
5.2 The role of HER2 and HER3 for cancer stem-like cell phenotype ......... 73
5.2.1 Overexpression and knockdown of HER2 and HER3 .................. 73
5.2.2 The effects of genetic alterations to cancer stem-like cell
marker expression ......................................................................... 73
5.2.3 The effects of the genetic alterations to cytotoxic response
to ALK inhibition ......................................................................... 74
5.2.4 Alterations in the Akt and ERK1/2 downstream signaling ........... 75
5.2.5 Compensatory expression of other HER family members ........... 75
6 Discussion 77
6.1 Immunological markers in HER2+ breast cancer ................................... 77
6.2 The role of HER2 and HER3 for cancer stem-like cells in ALK
translocated NSCLC ................................................................................ 81
6.3 Implications of the results and future perspectives ................................. 83
6.4 Limitations of the study .......................................................................... 84
7 Conclusions 87
References 89
Original publications 117

19
1 Introduction
Cancer has a major impact on society across the world and is one of the leading
causes of death. Cancer incidence has increased in recent years. An estimated 14.1
million new cancer cases worldwide were seen in 2012, whereas the estimation was
increased to 18.1 million cases in 2018 (Bray et al., 2018; Torre et al., 2015).
Fortunately, cancer therapies have been developed over the years with targeted
therapies and immunotherapy taking their place alongside surgery, chemotherapy
and radiation therapy. The new therapies are expensive, however, and have
increased the economic impact of cancer care (Tangka et al., 2010; Torkki et al.,
2018).
In the era of personalized medicine, the cancer therapies are selected for
patients according to their tumor’s genetic status. For example, ALK-positive lung
cancers containing oncogenic alterations of ALK are treated with ALK inhibitors,
because the cancer cells depend on ALK signaling and are thus extremely sensitive
to the receptor’s inhibition (Peters et al., 2017; Shaw et al., 2013; Soda et al., 2007).
However, patient selection still needs improvements, since some patients selected
for a specific treatment do not benefit or respond to the therapy. The
unresponsiveness can be caused by an existing resistance to the therapy that blocks
the therapeutic effects of the treatment. Patients selected for monoclonal antibody
(mAb) therapies, such as trastuzumab, are a great example. Even if the patient and
the tumor seem suitable for a specific mAb treatment, like the presence of HER2
amplification for trastuzumab, the efficacy of the therapy requires a functional
immune system. If the effector immune cells essential for the therapeutic effect of
the treatment are suppressed, the patients will likely not respond. Some of the
HER2-positive breast cancer patients have extreme responses to trastuzumab (over
10 years), but still a major part of these patients are either primary refractory for
the treatment or will develop a resistance against it (Cantini et al., 2018). Selecting
patients more efficiently can increase the tumor responses, and better treatment
options can be designed for the nonresponding patients.
Even if the selected patients respond to the targeted therapy or immunotherapy,
a major limiting factor of these therapies is the emergence of acquired resistance.
Resistance mechanisms for different cancer therapies have been discovered, one of
which is the presence of cancer stem cells (Doebele et al., 2012; Sasaki et al., 2011;
Wang, Qu, & Wang, 2017). Cancer stem cells are able to initiate and sustain the
growth of a tumor and are highly tumorigenic. These cells have been shown to
cause therapy resistance for various cancer treatments (Creighton et al., 2009;

20
McLendon et al., 2006; Phillips, McBride, & Pajonk, 2006; Wang et al., 2017).
Several cancer stem cell targeting agents have been developed and are in clinical
trials (Li, Atkinson, & Zhang, 2017), but the clinical utility of these agents is still
quite unknown. Protein members of the human epidermal growth factor receptor
family have been shown to be tumorigenic and are often linked to cancer stem cells
(Ithimakin et al., 2013; Korkaya, Paulson, Iovino, & Wicha, 2008; Lee et al., 2014;
Shi et al., 2018). Their targeting in the context of cancer stem cell targeting is still
in its infancy, and more studies are required. Since many HER targeting agents are
already in clinical use and are constantly being developed, HER dependency of
cancer stem cells could lead to a rapid clinical testing of the agents in the context
of cancer stem-like cell targeting. The utilization of already existing medical drugs
is not a new idea, since metformin, a mainstay therapy for diabetes mellitus, is now
being tested in clinical trials for cancer stem cell targeting (Buckanovich et al., 2017;
Pernicova & Korbonits, 2014).

21
2 Review of the literature
2.1 Cancer
Cancer is one of the leading causes of death worldwide. 2018 saw an estimated
18.1 million new cancer cases and 9.6 million cancer-related deaths. The numbers
are probably going to increase due to population growth, longer life expectancies
and poor lifestyle behaviors, such as smoking, bad diet and physical inactivity. Over
100 different cancer types exist, but lung and breast cancer are the most common
cancers diagnosed and causing cancer-related deaths in men and women,
respectively (Bray et al., 2018).
Cancer is a genetic disease, meaning that cancer is caused by changes in our
genes, our DNA. These changes occur and accumulate over one’s lifetime; some
can be even inherited from our parents. However, our cells’ behavior is extremely
tightly controlled, and multiple steps are required for cells to become cancerous
and tumors to be developed. This chapter briefly describes these changes.
2.1.1 Cancer genetics
The DNA of both normal and neoplastic cells is continuously altered either by
misincorporation of nucleotides during the DNA replication or by exposure to
exogenous or endogenous mutagens. Most of these changes are repaired by the
cell’s own repair mechanisms; however, some of the changes escape from the repair
machinery, leading to the development of mutations (Stratton, Campbell, & Futreal,
2009). Most mutations required for normal, healthy cells to become cancerous cells
are somatic, nongermline mutations that accumulate over the lifetime of a cancer
patient before the tumor is formed. Some germline mutations and genes, however,
are known to cause heritable cancer, for example, BRCA1 and BRCA2 genes (Hall
et al., 1990; Wooster et al., 1994).
Multiple types of somatic mutations are seen in the cancer genome, but the
most common mutations are single-base substitutions, which means that one
nucleotide is replaced by another one (Vogelstein et al., 2013). The cancer genome
might carry following somatic mutations, in addition to the single-base
substitutions: deletions or insertions of a small or larger DNA segments,
rearrangement of DNA sequence, and gene copy number increases or depletions
(Stratton et al., 2009; Vogelstein et al., 2013).

22
Some of the mutations found in the cancer genome are essential for tumor
progression and maintenance and are thus called the “driver” mutations. Other
mutations seen in the cancer genome are called the “passenger” mutations, which
have neutral effects and do not provide any growth advantages to the cancer cells.
Since the driver mutations give growth advantages to the cancer cells, these
mutations occur in proto-oncogenes and/or tumor suppressor genes that are
essential for cell proliferation and cell survival. Proto-oncogenes are present in
normal cells; they encode for proteins that positively control the cell proliferation
and survival. These genes activate the cell cycle and protect the cells from apoptosis.
Proto-oncogenes become oncogenes after being activated by a mutation or
translocation. Oncogenes can be classified into six groups based on their biological
functions: growth factors, growth factor receptors, signal transducers, chromatin
remodelers, transcription factors and apoptosis regulators (Croce, 2008).
Tumor suppressor genes, conversely, have a negative impact on cell
proliferation. The products of these genes limit cell proliferation and protect the
cells from damages that could lead to uncontrolled cell proliferation; in other words,
they prevent the development of cancer and are, thus, commonly dysregulated in
tumors. Tumor suppressor genes encode various proteins, such as receptors or
signal transducers that inhibit the proliferation (e.g. TGF-β); proteins that control
the cell cycle (e.g. retinoblastoma); cell cycle checkpoint proteins that trigger cell
cycle arrest if the DNA is damaged or there are defects in the chromosomes (e.g.
BRCA1); proteins involved in repairing mistakes in the DNA (e.g. p53 and DNA
mismatch repair protein 2); and apoptosis inducing proteins (e.g. p53) (Hanahan &
Weinberg, 2011; Wang, Li-Hui, Wu, Rajasekaran, & Shin, 2018). However, it
should be noticed that the division of cancer-driving genes into oncogenes and
tumor suppressor genes is not always black and white, since tumor suppressor
genes, such as p53, can also act as oncogenes (Soussi & Wiman, 2015).
In addition to the genetic alteration of protein-encoding oncogenes and tumor
suppressor genes, small non-coding RNAs, microRNAs (miRNA), have been
shown to be involved in cancer development. These small miRNAs play an
important role in cell proliferation, differentiation and survival by binding
complimentary to their target messenger RNAs (mRNAs) and causing mRNA
degradation or inhibition of their translation. miRNAs can be dysregulated in
tumors in a way that they cannot inhibit the translation of proteins, providing
growth advantages to the tumor, or the expression of miRNA can be increased to
inhibit the translation of growth-suppressing proteins (Rupaimoole & Slack, 2017).

23
2.1.2 Cancer epigenetics
Tumors consist of a heterogenous cell population, meaning that one tumor can
contain cancer cells that carry different genetic mutations. The genetic changes
alone, however, cannot explain all the diversity seen within a cancer cell population.
Epigenetic changes can control the activity of genes without altering the genetic
code and can explain why two genetically identical cells can behave differently.
Epigenetic modifications regulate all DNA-based processes such as DNA repair,
replication and transcription of the genes. The most well-known epigenetic
modification is DNA methylation, which means addition of methyl groups into
cytosines preceding guanines, thus preventing gene transcription (Esteller, 2008).
DNA hypomethylation, the loss of DNA methylation, was the first identified
epigenetic modification seen in tumors (Feinberg & Vogelstein, 1983). Several
years after the discovery of hypomethylation in tumors, the tumor suppressor genes
were found to be hypermethylated, causing inactivation of the genes (Greger,
Passarge, Höpping, Messmer, & Horsthemke, 1989).
2.1.3 Cancer biology
Hanahan & Weinberg have described the six hallmarks of cancer that give the
functional abilities for cancer cells to survive, proliferate and disseminate:
sustaining proliferative signaling, evading growth suppressors, resisting cell death,
enabling replicative immortality, inducing angiogenesis and activating invasion
and metastasis. Two hallmarks are emerging, which are not novel findings but are
added to the original six hallmarks: reprogramming of energy metabolism and
evading immune destruction. Genome instability and tumor-promoting
inflammation are two characteristics that also enable cancer cells to gain all the
above-mentioned hallmarks (Hanahan & Weinberg, 2011). This chapter further
examines these cancer characteristics (Fig. 1).
The six hallmarks of cancer
One of the most fundamental characteristics of cancer cells is their ability to sustain
proliferative signaling. The proliferation is usually initiated by growth factors that
bind to their receptors found on the cell surface after which the signals are
transmitted inside the cell, leading to cell proliferation. These proliferative signals
are extremely strictly controlled in normal cells, for example, by limiting the

24
availability of growth factors or the number of growth factor receptors. Cancer cells
can bypass these growth-limiting signals by, for example, producing the growth
factors themselves or stimulating the neighboring cells, even normal cells, to
secrete the growth factors for them (Bhowmick, Neilson, & Moses, 2004; Hanahan
& Weinberg, 2011). Cancer cells may also increase the number of growth factor
receptors, or the receptors can be functionally altered by activating mutations to
reach ligand-independent signaling. Furthermore, the signaling components acting
downstream of the receptors may also be functionally altered, allowing receptor-
independent signaling. Cancer cells might also deregulate negative-feedback
signals, which limit the signaling transmissions, keeping them transient, in normal
conditions (Hanahan & Weinberg, 2011).
Cell proliferation is controlled not only by limiting the growth stimulating
signals but also by activating programs that negatively regulate cell proliferation.
The main negative regulating programs are dependent on the tumor suppressor
genes, of which RB1 and TP53 are the two most well-known. The protein products
of these genes decide whether the cell enters the cell cycle or whether senescence
or apoptosis programs are activated. These negative regulators are commonly
suppressed in tumors to enable uncontrolled proliferation (Hanahan & Weinberg,
2011; Sherr & McCormick, 2002).
Cells with uncontrolled proliferation will be eliminated in normal conditions
by triggering programmed cell death, apoptosis. Cells have two apoptotic programs,
extrinsic and intrinsic; both will activate a complex proteolytic signaling cascade
by regulating the balance of anti- and pro-apoptotic proteins. Cancer cells have
multiple ways to overcome apoptosis. They can damper the damage-detecting
sensors, increase the expression of anti-apoptotic factors or survival signals,
downregulate pro-apoptotic regulators or impair the receptors used to deliver stress
signals outside of the cell (Fernald & Kurokawa, 2013; Hanahan & Weinberg,
2011).
Uncontrolled and unlimited proliferation in normal cells is also restricted by
the length of telomeric DNA, which allows the cell to pass only a certain number
of cell divisions before entering cell senescence or cell crisis that lead to cell death.
Cancer cells have increased the expression of telomerase enzymes, which adds
telomere repeat segments to the ends of telomeric DNA to overcome this issue
(Hanahan & Weinberg, 2011).
Even if cancer cells have bypassed all above-mentioned restrictions and can
proliferate limitlessly, they need to sustain the distribution of oxygen and nutrients
and the removal of metabolic waste and carbon dioxide by angiogenesis.

25
Angiogenesis occurs in normal cells only transiently, for example, during the
embryogenesis or wound healing, but the angiogenic switch is continuously on
during tumor progression (Bergers & Benjamin, 2003; Hanahan & Weinberg, 2011).
A key feature of tumor progression is dissemination of tumor cells by invasion
or metastasis. It is implicated that the invasion and metastasis of carcinomas is
regulated by the epithelial-mesenchymal transition (EMT) program that is
orchestrated by various transcriptional factors, such as Snail, Slug, Twist and
Zeb1/2. These transcriptional factors associate with the loss of adherens junctions,
morphological changes from epithelial to fibroblastic morphology, increased
mobility, expression of matrix-degrading enzymes and increased resistance to
apoptosis (Hanahan & Weinberg, 2011). Indeed, it has been shown that some highly
aggressive carcinomas have downregulated the expression of cell-cell and cell-
extracellular matrix adhesion molecules, such as E-cadherin, and upregulated
adhesion molecules like N-cadherin that are normally seen during embryogenic cell
migrations (Cavallaro & Christofori, 2004). However, it should be noted that the
stromal cells, in other words, the non-malignant cells around the tumor, can also
affect the dissemination of tumor cells, for example, by stimulating invasive
behavior in tumor cells or by supplying matrix-degrading enzymes to facilitate the
invasion (Hanahan & Weinberg, 2011).
The two emerging hallmarks
Cancer cells use glycolysis over oxidative phosphorylation to produce ATP even in
the presence of oxygen. It has been implicated that tumor cells prefer glycolysis,
because it allows the utilization of glycolytic intermediates to generate the
nucleosides and amino acids required for assembling new cells. To compensate the
lower efficiency of ATP production, tumor cells have been shown to upregulate
glucose transporters, which increases the glucose import into the cell (DeBerardinis,
Lum, Hatzivassiliou, & Thompson, 2008; Hanahan & Weinberg, 2011).
Cancer cells must also avoid the immunological destruction barrier to survive.
Tumors can either alter their recognition by immune cells, or they can impair the
immune effector cells. The tumors can change their antigen presenting behavior to
evade the detection by immune cells, for example, by losing the molecules required
for immune cells to detect them, such as major histocompatibility complex I (MHC
I). Cancer cells can impair the immune cells by expressing immune cell inhibitory
molecules on their cell surface, such as programmed cell death ligand-1 (PD-L1),
or by secreting immunosuppressive cytokines or enzymes, which affect the

26
functions of both innate and adaptive immune cells. (Hanahan & Weinberg, 2011;
Vesely, Kershaw, Schreiber, & Smyth, 2011).
The two enabling characteristics
Cancer cells require various mutations, types of which are mentioned in chapter
2.1.1., to orchestrate tumorigenesis and to gain the above-mentioned hallmarks of
cancer. However, the rate of spontaneous mutations is extremely low due to a cell’s
own genome maintenance system, which recognizes and solves defects of the DNA.
Genome instability will provide a higher rate of mutations; cancer cells damage
one or several components of the genome maintenance system to achieve that. The
favorable targets in the maintenance system are genes called the genome’s
“caretakers”. The products of these genes are the ones that detect the DNA damage,
activate the repair machinery or repair the DNA directly. Caretaker gene products
can also inactivate the mutagens even before the DNA is damaged (Hanahan &
Weinberg, 2011).
Inflammation is another enabling characteristic that has been accepted to be
involved in tumorigenesis. The inflammatory conditions can exist before the tumor
has formed, but the cancer cells themselves can also create an inflammatory
microenvironment. The tumor microenvironment contains both innate and adaptive
immune cells as a result of inflammation. Although immune cells destroy tumor
cells, they also provide tumor-promoting factors such as growth factors, survival
factors, proangiogenic factors and matrix-modifying enzymes and, thus, enable the
acquisition of the hallmarks of cancer (Aggarwal, Vijayalekshmi, & Sung, 2009;
Grivennikov, Greten, & Karin, 2010; Hanahan & Weinberg, 2011; Mantovani,
Allavena, Sica, & Balkwill, 2008).

27
Fig. 1. The hallmarks of cancer. Modified from Hanahan & Weinberg 2011.
2.1.4 Main signaling pathways altered in cancer
Intercellular communications are essential for cells functions and survival. Signal
transduction is initiated largely by growth factors binding to their receptors, which
leads to activation of intracellular signaling cascades. Several classes of receptors
are found on the cell surface, such as receptor tyrosine kinases (RTKs), cytokine
receptors and G-protein -coupled receptors (Mendelsohn, Gray, Howley, Israel, &
Thompson, 2014).
RTKs are key regulators of essential cell processes such as proliferation,
differentiation, migration and survival, and their activating oncogenic alterations
are frequently seen in tumors. Human RTKs consist of 20 different subfamilies,
including epidermal growth factor receptors, insulin receptors and vascular
endothelial growth factor receptors. All RTKs share a similar structure: an
extracellular region, a transmembrane helix, and a cytoplasmic region. Binding of
a specific ligand to the extracellular region of its receptor initiates a signaling
cascade by creating receptor dimerization or oligomerization, which in turn leads
to activation of the tyrosine kinase domain in the cytoplasmic region, followed by
autophosphorylation of tyrosine residues in the carboxy terminal tail. These
phosphotyrosines function as recruitment sites for downstream signaling molecules
Sustainingproliferativesignaling Evading
growthsuppressors
Inducingangiogenesis
Activatinginvasion and metastasis
Evadingimmunedestruction
Reprogrammingenergy metabolism
Enablingreplicativeimmortality
Resisting cell death Genomic instabilityTumor-promoting inflammation
Enabling characteristics

28
containing either Src homology-2 (SH2) or phosphotyrosine-binding (PTB)
domains. Downstream signaling molecules can be also recruited to the receptor via
docking proteins that are phosphorylated by the receptor. The downstream
signaling proteins eventually become activated, and the signaling cascade
continues (Lemmon & Schlessinger, 2010; Mendelsohn et al., 2014).
Two well-known RTK signaling pathways are the Ras/Raf/MEK/ERK and
PI3K/Akt/mTOR pathways, which are commonly altered in tumors. Figure 2
presents a simplified scheme of these two pathways. In the Ras/Raf/MEK/ERK
pathway, the phosphotyrosines of an activated RTK recruit the Src homology-2
domain containing (Shc) adaptor protein to the receptor, which instead recruits
growth factor receptor-bound protein 2 (Grb2) and son of sevenless (SOS) protein.
SOS is a guanine nucleotide exchange factor, which activates Ras, a membrane-
bound G protein, by catalyzing the exchange of GDP to GTP. Activated Ras recruits
Raf to the membrane where it will be activated. Raf phosphorylates and activates
mitogen-activated protein kinase kinases 1 and 2 (MEK1/2), which in turn
phosphorylate and activate extracellular signal-regulated kinases 1 and 2 (ERK1/2),
which have multiple targets, both in the cytosol and in the nucleus (McCubrey et
al., 2011; Mendelsohn et al., 2014). Ras and Raf proteins are frequently mutated in
human cancers (Bos, 1989; Davies et al., 2002; Karnoub & Weinberg, 2008),
whereas MEK and ERK mutations have been reported in cancers but are less
common (Nikolaev et al., 2012; Ojesina et al., 2014).
PI3K/Akt/mTOR signaling pathway is initiated when phosphatidylinositol-3-
kinase (PI3K) associates with phosphorylated tyrosine residues on an activated
receptor and becomes activated. PI3K phosphorylates phosphatidylinositol 4,5-
bisphosphate (PIP2), generating phosphatidylinositol 3,4,5-trisphosphate (PIP3),
which functions as a second messenger and is converted back to PIP2 by tumor
suppressor PTEN. PIP3 then attracts Akt proteins to the membrane, where they are
activated via phosphorylation, after which Akt proteins phosphorylate a series of
substrates in the cytosol and in the nucleus. One well-known target of Akt is
mammalian target of rapamycin complex 1 (mTORC1), targets of which include
protein synthesis regulators ribosomal protein S6 kinase 1 (S6K1) and eukaryotic
translation initiation factor 4E-binding protein 1 (4E-BP1). Many components of
the PI3K/Akt/mTOR pathway are mutated or amplified in tumors, most commonly
PI3K and PTEN, but alterations of Akt have also been reported (Liu, Cheng,
Roberts, & Zhao, 2009; McCubrey et al., 2011; Mendelsohn et al., 2014).

29
Crosstalk between signaling pathways
Different signaling pathways interact with each other to create a complex and
dynamic signaling network. The signaling molecules of one pathway can, for
example, activate molecules of another pathway, as figure 2 shows, with GTP-
bound Ras, which can bind and allosterically activate PI3K (Suire, Hawkins, &
Stephens, 2002). The crosstalk between the signaling pathways is highlighted when
components of a specific pathway are blocked. It has been shown that inhibition of
PI3K/Akt/mTOR-pathway leads to activation of Ras/Raf/MEK/ERK-pathway and
vice versa (Carracedo et al., 2008; Won et al., 2012).
Fig. 2. RTK signaling pathways PI3K/Akt/mTOR and Ras/Raf/MEK/ERK.
2.2 Tumor immunology
The most important function of the immune system is to prevent or eradicate
infections. The first idea that the immune system can control cancer arose in the
early 1900s but the whole concept of immunosurveillance was under a constant
debate for almost a century until mouse models were improved enough, and it was
shown that mice lacking adaptive immune cells and IFNγ responsiveness were
more susceptible to cancer development (Schreiber, Old, & Smyth, 2011;
Shankaran et al., 2001). However, the roles of the immune system in tumorigenesis
and in the tumor microenvironment are paradoxical. Even though immune cells
destroy cancer cells, this eradication leads to a selection of a poorly immunogenic
population of cancer cells that are more capable of surviving in an
Grb2 SOS
RasGDP
PPP
P
P
Raf
MEK
ERK
PI3K
PIP2PIP3PIP3
mTORC1
S6K1 4E-BP1
P PP P PP P P
AktP
P P
Nucleus
Shc
Ras
Ras
GTP
GTP

30
immunocompetent host (De Visser, Eichten, & Coussens, 2006; Shankaran et al.,
2001).
Cells of the immune system are guarding throughout the human body and are
also found within the malignant tissue. The cell types of tumor-infiltrated immune
cells vary between different diseases and from patient to patient, and they are
usually located in the center of the tumor, in the stroma or in the tertiary lymphoid
structures (TLS) found near the tumors (Fridman, Pagès, Saut`s-Fridman, & Galon,
2012). The human immune system can be classically divided into the innate and
the adaptive immune system. This chapter will describe the dual roles of both innate
and adaptive immune cells in tumors (Fig. 3).
Fig. 3. Different subtypes of immune cells and their predominant contributions to tumor
progression or tumor suppression. DC=dendritic cells, M=macrophages,
MDSC=myeloid-derived suppressor cells, NK=natural killer cells, NKT=natural killer T
cells, Th=T helper cells, Treg=regulatory T cells.
Tumor suppression Tumor progression
CD8+ T
Th1DC1
Neutrophils
NKM1
NKT
B cells
Treg
DC2
M2
MDSC
Th2
Neutrophils
Mast cells Mast cells
Treg
Th2 Th17Th17

31
2.2.1 Innate immune system and cancer
The first line of defense against foreign pathogens or an injury is the innate immune
system, which comprises of different types of immune cells. The functions of the
innate immune cells are commonly altered in cancer.
Neutrophils, which reflect the state of inflammation, can be involved in tumor
initiation, proliferation and metastasis (Ocana, Nieto-Jiménez, Pandiella, &
Templeton, 2017; Szczerba et al., 2019), and indeed, high levels of neutrophils,
especially in the peripheral blood but also in the tumor, have been associated with
worse patient outcomes in many solid cancers (Jensen et al., 2009; Leibowitz-Amit
et al., 2014; Templeton et al., 2014; Wang, J. et al., 2014). However, the role of
neutrophils in cancer is complex, probably due to a wide scale of different
neutrophil populations (Coffelt, Wellenstein, & De Visser, 2016). Some studies
have shown that neutrophils might have antagonizing effects on metastasis and are
associated with better survival in some cancers (Galdiero et al., 2016; Granot et al.,
2011).
The presence of mast cells, which are both positive and negative regulators of
the immunity (Galli, Grimbaldeston, & Tsai, 2008), has been linked to tumor
development in many studies; their infiltration into tumors has been associated with
poor patient outcomes (Johansson et al., 2010; Nonomura et al., 2007; Ribatti et al.,
2003; Tth, Tth-Jakatics, Jimi, Takebayashi, & Kawamoto, 2000). However,
evidence also exists about a protective role of mast cells in human cancers
(Fleischmann et al., 2009; Rajput et al., 2008).
Dendritic cells, important antigen-presenting cells, are largely defective in
tumors; their differentiation, activation and immune-response stimulation are often
impaired. Indeed, decreased density of tumor-infiltrating dendritic cells is linked to
poor prognosis (Inoshima et al., 2002; Seigo Kashimura et al., 2012; Veglia &
Gabrilovich, 2017). However, evidence also exists that tumor cells can drive the
tumor-associated dendritic cells towards an immunosuppressive phenotype, and
some types of the tumor-infiltrating dendritic cells have been associated with poor
prognosis (Lombardi, Khaiboullina, & Rizvanov, 2015; Saadeh, Kurban, & Abbas,
2016; Tesone et al., 2016).
Natural killer (NK) cells, which have attributes of both innate and adaptive
immunity, recognize malignant cells and kill them by secreting the toxic content of
their cytosolic granules. NK cells also produce cytokines, such as IFNγ and TNF-
α, which will instead, for example, activate adaptive immune responses. Tumor-
infiltrating NK cells have been linked to better prognosis in a variety of cancers;

32
however, the NK cells are commonly suppressed in tumors by the tumor cells
themselves or by immunosuppressive macrophages, dendritic cells or T cells. The
maturation, proliferation and function of NK cells can be directly suppressed via
different immunosuppressive cytokines and other molecules, such as transforming
growth factor beta (TGF-β) and indoleamine 2,3-dioxygenase 1 (IDO1) (Della
Chiesa et al., 2006; Ghiringhelli et al., 2005; Lee, Kang, & Cho, 2017). Still, NK
cells are one of the key players of antibody-dependent cellular cytotoxicity (ADCC),
which is suggested to be the main mechanism of action of the therapeutic
monoclonal antibodies (Arnould et al., 2006; Clynes, Towers, Presta, & Ravetch,
2000; Tian et al., 2017). It was thought for a long time that NK cells were solely
antitumorigenic, but it has become increasingly clear that the NK cells can also
possess tumor promoting effects (Bruno, Ferlazzo, Albini, & Noonan, 2014).
Macrophages are known for their immunomodulatory effects. Macrophages
display variable phenotypes, which form a continuum from M1-like state to M2-
like state. Classically activated M1-like macrophages are pro-inflammatory and
antitumoral, while alternatively activated M2-like macrophages possess anti-
inflammatory and immunosuppressive effects and are, thus, protumoral (Biswas &
Mantovani, 2010; Brown, Recht, & Strober, 2017). Tumor-associated macrophages
(TAMs) have been associated with prognosis and therapy response in many solid
tumors. M1-like TAMs are linked to a better disease course and, conversely, M2-
like TAMs are associated with more adverse outcomes (Biswas, Allavena, &
Mantovani, 2013; Fridman, Zitvogel, Sautès-Fridman, & Kroemer, 2017). M2-like
TAMs are known to suppress innate and adaptive immune responses by promoting
the activity of immunosuppressive T cells, by secreting inhibitory cytokines such
as TGF-β and IL-10 and by expressing/producing inhibitory molecules like PD-L1
and IDO1. M2-like macrophages also promote angiogenesis and facilitate tumor
invasion (Biswas & Mantovani, 2010; Biswas et al., 2013; Mantovani, Marchesi,
Malesci, Laghi, & Allavena, 2017). TAMs are also known to contribute to
antibody-dependent cellular phagocytosis (ADCP), which is induced in TAMs by
therapeutic monoclonal antibodies and leads to phagocytosis of antibody-coated
tumor cells by TAMs (Kurdi et al., 2018; Shi et al., 2015; Vermi et al., 2018).
Yet another cell type of the innate immunity found in tumors is myeloid-
derived suppressor cells (MDSC), which comprise of myeloid cells, immature
dendritic cell, immature granulocytes or immature macrophages. MDSCs suppress
various T cell functions and modulate the cytokine production of macrophages by
converting them into M2-like immunosuppressive state (Gabrilovich & Nagaraj,

33
2009). MDSCs have been shown to be associated with a poor prognosis in many
solid cancers (Zhang et al., 2016).
2.2.2 Adaptive immune system and cancer
The next line defenses against foreign pathogens or an injury are the adaptive
immune responses, which are activated after antigen recognition. The adaptive
immune system comprises B and T lymphocytes. B lymphocytes mediate a
humoral immunity and can recognize various types of molecules -- both soluble
and cell surface-bound forms -- whereas T lymphocytes mediate cell-mediated
immunity and recognize only peptide fragments of proteins introduced to them via
specific receptors. Lymphocytes have an essential role in tumor eradication, and
they are also commonly altered in cancer for immune evasion (Nelson, 2010;
Sharma & Allison, 2015).
B lymphocytes are typically located in conventional lymphoid tissues such as
spleen, lymph node or blood, but they can also be found in other nonlymphoid
tissues, usually in tertiary lymphoid structures and in aggregates with other immune
cells (Drayton, Liao, Mounzer, & Ruddle, 2006). Increasing evidence has shown
that many tumors are infiltrated with B lymphocytes, but the exact effects of B cells
in tumors is unclear. Many B cell subtypes have diverse functions that complicate
the effects of B cells in tumorigenesis and in tumors overall. B cells act directly
through tumor-reactive antibodies or cytotoxic pathways, or they can influence T
cell responses through secretion of chemokines and cytokines, or by facilitating the
formation of TLS, or they can act as antigen-presenting cells (Flynn,
Somasundaram, Arnold, & Sims-Mourtada, 2017; Nelson, 2010). The complex
functions of B cells are also reflected in their clinical significance, since tumor-
infiltrating B cells have been shown to associate with both poor and good prognoses
in many solid cancers (Flynn et al., 2017).
T cells are one of the most abundant immune cells found in the tumors. T
lymphocytes can be divided according to their effector functions into cytotoxic T
cells (CD8+) and helper T cells (CD4+), which comprise Th1, Th2, Th17, regulatory
T (Treg) cells, and natural killer T cells (Grivennikov et al., 2010). The activation
of CD8+ or CD4+ naïve T cells requires not only the recognition of tumor antigens
by T cell receptors but also costimulatory signals via engagement of B7 molecules
(CD80, CD86) to the CD28 receptor found on T cells. The tumor antigens can be
presented to T cells through MHC I or II receptors, which are expressed by many
cell types, but the costimulatory B7 molecules are found mainly on antigen-

34
presenting cells (APCs). The T cells can proliferate, differentiate further and traffic
to the tumor site after activation (Greenwald, Freeman, & Sharpe, 2005; Sharma &
Allison, 2015; Townsend & Allison, 1993).
The antitumoral effects of T cells are mediated through cytokines, chemokines
and direct cytotoxicity, but these T cell actions are often disrupted in cancer by
multiple mechanisms (Lin, Karin, Pearce, & Kleyman, 2007; Swann & Smyth,
2007). First, it is critical that the T cells in the tumor microenvironment are in close
contact with the tumor cells, but their infiltration is often blocked. Second, even if
T cells can infiltrate into tumors, they must overcome additional barriers. The
antitumoral actions of T cells can be suppressed by the tumor cells themselves, by
MDSCs, by M2-like macrophages and by other immunosuppressive cells, even
regulatory T cells. The immunosuppressive effects can be transmitted through
inhibitory cytokines, such as TNFα or TRAIL, inhibitory enzymes like IDO1 or
other inhibitory molecules (Joyce & Fearon, 2015; Sharma & Allison, 2015). One
extremely well-known, inhibitory molecule expressed by various of cell types,
including cancer cells and macrophages, is PD-L1, which binds to PD-1 receptor
on T cells, leading to inhibition of their cytotoxic activities and induction of
apoptosis (Freeman et al., 2000).
T cells have been shown to possess both antitumoral and tumor-promoting
effects. CD8+ cytotoxic T cells and Th1 helper T cells have been shown to correlate
with better survival in many cancer types, whereas Tregs, Th2 and Th17 cells have
contradictory roles (Fridman et al., 2012; Fridman et al., 2017). Tregs are
extensively studied immunosuppressive cells that are essential for developing and
maintaining self-tolerance but are often recruited to the tumor site. They have
multiple ways to inhibit the functions of cytotoxic T cells, such as producing
inhibitory cytokines like IL-10 or expressing inhibitory molecules like PD-L1
(Terabe & Berzofsky, 2004). High densities of Tregs are associated with poor
prognosis in numerous cancer types, but in colorectal and gastric cancer, Tregs have
been linked to favorable prognosis. Th2 and Th17 cells have been similarly
associated with both good and poor survival (Fridman et al., 2012; Fridman et al.,
2017).
2.2.3 Tumor immune profile and Immunoscore
Three different immune profiles of the tumor can be distinguished according to
their inflammatory state. The first profile is the immune-inflamed phenotype,
which is characterized by high infiltration of immune cells, especially CD4+ and

35
CD8+ T cells and high PD-L1 expression. The second profile is the immune-
excluded phenotype, which is characterized by an abundance of immune cells
which, however, are retained in the stroma surrounding the tumor and are unable
to infiltrate into the tumor. The third profile is called the immune-desert phenotype,
which is characterized by a lack of T cells and other immune cells in the stroma
and in the tumor. These tumor-immune profiles correlate with a patient’s response
to anti-PD-1/PD-L1 therapy (Chen & Mellman, 2017).
The significance of the tumor-infiltrating immune cells’ location has been
shown in many studies with different cancer types, demonstrating that the
prognostic value of the tumor-infiltrating immune cells depends on both the
location and the type of the immune cells (Galon et al., 2006; Hermans et al., 2014;
Zhou et al., 2018). Combinatory analysis of CD3+ and CD8+ T cells in the center
of the tumor (CT) and in the invasive margin (IM) has provided a clinically useful
prognostic marker for colorectal cancer. This analysis is described as the
Immunoscore, which categorizes tumors according to their CD3+ and CD8+
immune-cell densities in both CT and IM. Tumors are scored from Immunoscore
0, where the density of both cell types in both locations is low, to Immunoscore 4,
where the density of both cell types in both locations is high. Immunoscore has
been suggested to be included in the classification of colorectal cancer (Galon et
al., 2014; Galon et al., 2006). A recent study with over 2,500 colorectal cancer
patients from 14 different cancer centers and 13 different countries showed that the
Immunoscore (3-tiered: low, mediate, high Immunoscore) provided an even better
estimate of risk for all clinical parameters than the TNM classification (Pagès et al.,
2018). Whether the use of the Immunoscore could be expanded into other cancer
types remains to be seen.
2.3 Cancer stem cells
Cancer cells within one tumor can exist in distinct phenotypic states, thus creating
tumor heterogeneity. Some of the tumor cells have features similar to normal stem
cells and are, therefore, called cancer stem cells (CSCs). These cells can self-renew
and generate various types of non-CSC progenies, which form the rest of the tumor
(Eun, Ham, & Kim, 2017). The first CSCs were confirmed in acute myeloid
leukemia in the 1990s (Bonnet & Dick, 1997), whereas the first CSCs in solid
cancers were found in 2003 in breast cancer (Al-Hajj, Wicha, Benito-Hernandez,
Morrison, & Clarke, 2003). To date multiple cancer types, such as colon, ovarian
and lung cancer, has been shown to contain CSC subpopulations (Medema, 2013).

36
CSCs can evade cell death and metastasize, and their presence in tumors has been
linked to an aggressive disease course and poor survival (Beier et al., 2008;
Charafe-Jauffret et al., 2010; Chiou et al., 2008; Ginestier et al., 2007; Liu et al.,
2007; Pallini et al., 2008). Furthermore, CSCs have been shown to be resistant to
the conventional cancer therapies, targeted therapies and immunotherapies
(Creighton et al., 2009; Maccalli, Parmiani, & Ferrone, 2017; McLendon et al.,
2006; Phillips et al., 2006; Reim et al., 2009; Wang et al., 2017). Thus, these cells
are extremely important also from the clinical viewpoint.
2.3.1 Hierarchical and stochastic cancer stem cell models
Two models -- hierarchical and stochastic -- have been proposed to describe the
tumor progression and heterogeneity driven by the CSCs (Fig. 4). The hierarchical
model states that carcinogenesis occurs when a normal stem cell evades regulation
and becomes a tumorigenic stem cell, thus CSC, and can initiate tumor growth. The
CSCs are defined as a totally distinct population in this model and are the only cells
that can give rise to differentiated cell populations with limited proliferative
capacity. However, this model rules out the interchange between stem-like and
differentiated states within one cell (Melzer, von der Ohe, Lehnert, Ungefroren, &
Hass, 2017; Plaks, Kong, & Werb, 2015; Takebe, Harris, Warren, & Ivy, 2011).
Conversely, the stochastic model suggests that carcinogenesis can be initiated
by any normal cell that has randomly (i.e. stochastically) acquired oncogenic
mutations, epigenetic changes, etc. This model states that every cancer cell within
one tumor is equally capable of maintaining and promoting the tumor growth. The
tumor heterogeneity in this model is mediated through genetic changes or by
extrinsic and intrinsic factors such as tumor microenvironment and different
signaling pathways (Melzer et al., 2017; Plaks et al., 2015; Takebe et al., 2011).
These two models are not mutually exclusive, because the phenotypical states
of the cells in a hierarchical organization are more transitory than earlier believed,
and the stochastic events are also able to generate hierarchical cell populations
(Plaks et al., 2015). To date it is known that a differentiated cell, either a cancer cell
or a normal cell, can dedifferentiate back to the stem-like state (Chaffer et al., 2011;
Gupta et al., 2011; Schwitalla et al., 2013). This dedifferentiation of the cell state
can be either inherited (hierarchical model) or caused by mutations that lead to the
acquisition of stem-like cell properties (stochastic model) (Plaks et al., 2015). This
cancer plasticity is extremely valid to be considered in the clinical targeting of
cancer stem cells.
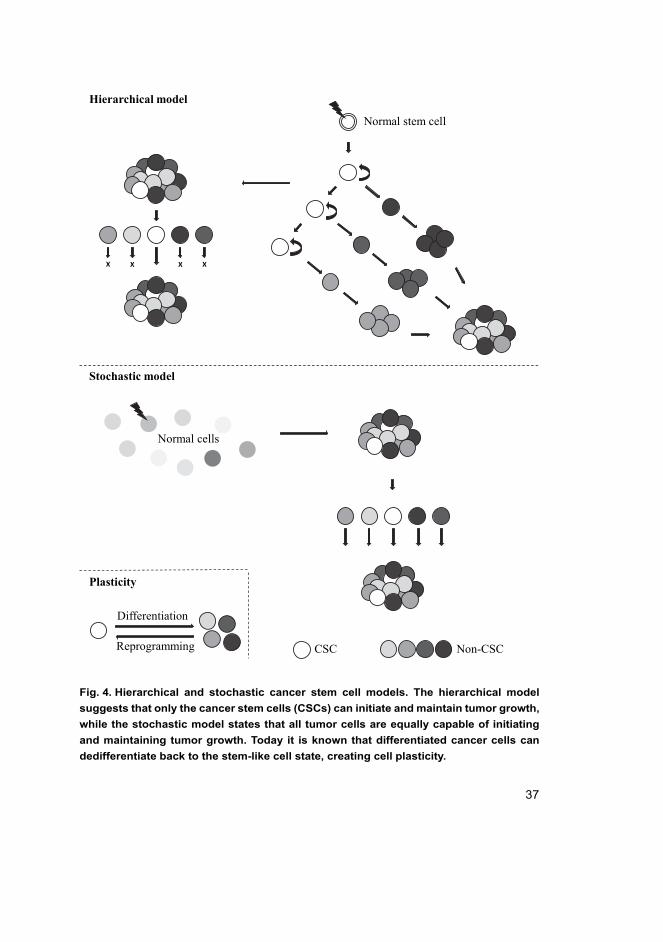
37
Fig. 4. Hierarchical and stochastic cancer stem cell models. The hierarchical model
suggests that only the cancer stem cells (CSCs) can initiate and maintain tumor growth,
while the stochastic model states that all tumor cells are equally capable of initiating
and maintaining tumor growth. Today it is known that differentiated cancer cells can
dedifferentiate back to the stem-like cell state, creating cell plasticity.
X XX X
CSC Non-CSC
Hierarchical model
Stochastic model
Normal cells
Plasticity
Differentiation
Reprogramming
Normal stem cell

38
2.3.2 Signaling pathways related to cancer stem cell phenotype
CSCs are regulated by specialized microenvironments called CSC niches; several
studies have suggested that CSCs require these niches to maintain their stem cell
properties. The cells and molecules of the CSC niche affect the signaling of CSCs
(Plaks et al., 2015). Normal stem cells and CSCs share similar signaling pathways,
of which the most fundamental ones for the maintenance of the stemness are Notch,
Hedgehog and Wnt/β-catenin pathways (Takebe et al., 2011).
Notch pathway has a critical role in controlling the cell-to-cell interactions in
embryogenesis, cell proliferation and differentiation, and apoptosis (Artavanis-
Tsakonas, Rand, & Lake, 1999). Notch can also affect the self-renewal of stem cells
(Dontu et al., 2004; McKay et al., 2006). Hedgehog pathway acts in the embryonic
development by controlling the tissue polarity and by participating in the
maintenance of the stem cells and is, therefore, often hyperactivated in tumors
(Ingham & McMahon, 2001; Takebe et al., 2011). Wnt signaling has also a vital
role in embryogenesis, as it regulates the development of a variety of organ systems.
In adults wnt regulates tissue self-renewal, for example, in the hair follicles
(Clevers, 2006; Grigoryan, Wend, Klaus, & Birchmeier, 2008). It has been shown
that CSCs require β-catenin to maintain their tumorigenic phenotype (Kim et al.,
2018; Malanchi et al., 2008; Zimmerli et al., 2018).
All the aforementioned pathways have vital roles in the embryogenesis, and
indeed, EMT, an essential process for the embryonic development, occurs also
during tumorigenesis and allows CSCs to become metastatic (Shibue & Weinberg,
2017; Shook & Keller, 2003).
2.3.3 Markers used to identify cancer stem cells
Cancer stem cell markers are commonly proteins that can identify cell populations
enriched within a tumor and that fulfill the functional definition of cancer stem-like
cells. Two key characteristics are used to define CSCs: their enhanced
tumorigenicity and their capacity to self-renew or differentiate. Numerous CSC
markers have been identified, of which the most broadly used are high CD133
expression, high CD44 expression, and high aldehyde dehydrogenase 1 (ALDH1)
expression or activity (Medema, 2013).
CD133, also called prominin-1, is a transmembrane glycoprotein that is used
to identify CSCs in many solid cancers, including breast, lung, brain and colorectal
cancers (Bidlingmaier, Zhu, & Liu, 2008; Cioffi et al., 2015; Liu et al., 2013;

39
Wright et al., 2008). CD44 is also a transmembrane glycoprotein that has been
shown to be potential CSC marker in various cancer types, such as breast, colorectal,
lung and gastric cancers (Morath, Hartmann, & Orian-Rousseau, 2016). ALDH1,
however, is a cytosolic enzyme that is widely used to isolate CSCs in many cancers
(Carpentino et al., 2009; Ginestier et al., 2007; Jiang et al., 2009; van den Hoogen
et al., 2010). In addition to these markers, transcription factors governing
pluripotency of embryonic stem cells, such as SOX2, have been linked to CSCs
and tumor initiation and are potential CSC markers (Leis et al., 2012; Masui et al.,
2007; Zhu et al., 2017).
Universal CSC markers do not exist, because CSCs are an extremely
heterogeneous cell population. A marker identified in one tumor may not be able to
distinguish CSCs in another tumor, even in a tumor of the same origin (Plaks et al.,
2015; Vlashi & Pajonk, 2014). Hence, more than one CSC marker is commonly
used, for example, in breast cancer CD44high/CD24low marker (Wright et al., 2008).
2.4 HER family
Human epidermal growth factor receptor (HER) proteins are essential during
development and in normal adult physiology, and they are expressed in various
neuronal, epithelial and mesenchymal tissues (Hynes & Lane, 2005). HERs belong
to the RTK protein family. As mentioned earlier in section 2.1.3., the RTKs are
crucial to numerous cell functions such as proliferation, differentiation and survival.
The HER family is among the most studied cell-signaling families in biology.
2.4.1 Structure and function
The HER family comprises four members: EGFR/HER1/ERBB1, HER2/ERBB2,
HER3/ERBB3 and HER4/ERBB4. All HER proteins share a nearly similar
structure. They have an extracellular domain that contains four distinct parts
(subdomains I-IV), a single transmembrane segment, an intracellular segment
containing juxtamembrane, a protein kinase domain and a carboxyterminal (C-
terminal) tail (Roskoski, 2014; Ullrich et al., 1984).
The signaling through HER proteins is initiated by the binding of a specific
ligand. Both EGFR and HER4 have seven ligands, whereas HER3 has two ligands,
and HER2 has none (Table 1) (Hynes & MacDonald, 2009). The subdomains I and
III of the receptors participate in the ligand binding. Bivalent ligand, for example,
epidermal growth factor (EGF), binds to the subdomains I and III on a single

40
receptor. The ligand binding initiates major conformational changes in the
extracellular part of the receptor, revealing a dimerization arm in the subdomains
II and IV (Ogiso et al., 2002). The HER2 receptor, however, already exists in this
open conformation where the dimerization arm is not buried (Cho et al., 2003),
which could explain its lack of ligands. After the dimerization arm is revealed, the
HER proteins can form either hetero- or homodimers (or even oligomers), which
will lead to an allosteric activation of the tyrosine kinase domains in the
intracellular parts of the receptors (Hubbard & Miller, 2007; Roskoski, 2014;
Zhang, Gureasko, Shen, Cole, & Kuriyan, 2006). Active kinases then
phosphorylate tyrosine residues on the C-terminal tails of the receptors, which
creates docking sites for the downstream signaling molecules (Roskoski, 2014).
The kinase domain of the HER3 protein is catalytically impaired, but it can still act
as an efficient phosphotyrosine scaffold, and it can activate its dimeric partners via
allosteric activation mechanism (Shi, Telesco, Liu, Radhakrishnan, & Lemmon,
2010).
As mentioned earlier in section 2.1.3., the main downstream signaling
pathways of RTKs are the PI3K/Akt/mTOR and Ras/Raf/MEK/ERK pathways, of
which the former has an important role for cell survival and the latter participates
in cell proliferation (Yarden & Pines, 2012). The HER3 receptor is a major player
in cell survival, since it has several docking sites for the p85 subunit of PI3K. Other
HER proteins are also able to activate the PI3K pathway; HER4 has one binding
site for PI3K, but EGFR and HER2 cannot directly bind the enzyme (Kainulainen
et al., 2000; Roskoski, 2014; Schulze, Deng, & Mann, 2005).
The signaling through HER and other RTK proteins is downregulated by taking
the receptor-ligand complex inside the cell by endocytosis. The receptor-ligand
complex will be transported via endosomes to lysosomes for degradation. Before
the endocytosis, the receptor is ubiquitinated, which is essential for the trafficking
of the endosome to lysosomes. This downregulation is crucial for terminating the
proliferative signals produced by the activated receptors (Katzmann, Odorizzi, &
Emr, 2002; Mizuno et al., 2005).

41
Table 1. Human epidermal growth factor receptor ligands. Modified from Hynes &
MacDonald 2009.
EGFR HER2 HER3 HER4
EGF - NRG1 Betacellulin
TGF-α NRG2 HB-EGF
Amphiregulin Epiregulin
Epigen NRG1
Betacellulin NRG2
HB-EGF NRG3
Epiregulin NRG4
HB-EGF=heparin binding EGF-like growth factor, NRG=neuregulin
2.4.2 HERs and cancer
The HER proteins and their functions are often altered in cancer. The HER genes
can be amplified or mutated, the ligands may be overproduced by the tumor cells
or the surrounding stromal cells, or the downregulation of the receptor may be
impaired. All these alterations can lead to constitutively activated receptors and
prolonged signaling through them, causing tumor progression (Arteaga &
Engelman, 2014; Hu et al., 2015). Activating oncogenic alterations of EGFR or
HER2 are commonly seen in several cancers, while activating alterations of HER3
and HER4 are less frequent, but their altered expressions have been reported in
cancer (Mishra, Hanker, & Garrett, 2017).
In the 1980s Kawamoto et al. showed by blocking EGFR in A431 cells that the
receptor was able to drive the cancer cell growth (Kawamoto et al., 1983). Since
then EGFR amplification, overexpression and activating mutations have been
discovered in several carcinomas (Birkman et al., 2016; Shan et al., 2015). Mutated
EGFR, which lacks exons 2–7 in the extracellular domain and is called EGFRvIII,
is found mainly in gliomas but also in a fraction of lung, breast and ovarian cancers
and leads to enhanced tumorigenicity (Moscatello et al., 1995; Nishikawa et al.,
1994; Sugawa, Ekstrand, James, & Collins, 1990). EGFR mutations were
discovered in the early 2000s in non-small cell lung cancer (NSCLC), further
highlighting the role of the receptor in tumorigenesis (Lynch et al., 2004; Paez et
al., 2004). The two most common EGFR mutations seen in NSCLC are deletion of
exon 19 and leucine-to-arginine substitution at amino acid position 858 (L858R),
which are both located in the kinase domain of the receptor (Pao & Chmielecki,
2010).

42
The most common oncogenic alteration of HER2 is its amplification. In 1987
it was discovered that around 20% of breast cancers have HER2 amplification
which is associated with poor patient outcomes (Slamon et al., 1987). It has been
seen that metastatic mammary tumors are generated when wild-type HER2 is
controlled by a mammary-specific promoter in transgenic mice (Andrechek et al.,
2000; Finkle et al., 2004). HER2 amplification has also been discovered in other
cancers, such as gastric, esophageal and lung cancers (Bang et al., 2010; Cappuzzo
et al., 2005). Activating mutations of HER2 are less common and are usually found
in cancers without HER2 amplification. HER2 mutations have been reported in
several cancers, such as lung adenocarcinomas, breast and colorectal cancer
(Arteaga & Engelman, 2014; Bose et al., 2013; Hanker et al., 2017; Kavuri et al.,
2015; Suzuki et al., 2015).
HER3 has also been linked to several cancers, mainly through its role in
promoting signaling from oncogenic HER2 or EGFR by activating PI3K (Mishra,
Patel, Alanazi, Yuan, & Garrett, 2018). Blocking HER3 has been shown to inhibit
the growth of HER2-dependent tumors in xenograft models, which highlights the
essential role of HER3 for HER2-driven tumorigenesis (Garrett et al., 2013; Lee-
Hoeflich et al., 2008). HER3 alterations have also been discovered, but the
oncogenic activity of HER3 may still be dependent on HER2 or other HER proteins
(Jaiswal et al., 2013; Mishra et al., 2017).
HER4 is much less extensively studied than the other members of the HER
family, and its activating mutations are less frequent in tumors. However, HER4
alterations have been observed in different types of cancer, mostly in melanoma,
and HER4 signaling has been shown to play an important role for cancer recurrence
in lung cancer (Hegde et al., 2013; Kurppa, Denessiouk, Johnson, & Elenius, 2016;
Mishra et al., 2017).
HERs and CSCs
The expression of EGFR, HER2 and HER3 has been linked to cancer stem-like cell
(CSLC) properties in several types of cancer. EGFR expression can identify a
CSLC population, induce CSLC properties, and promote their survival (Mazzoleni
et al., 2010; Shi et al., 2018). HER2 expression is able to drive tumorigenesis and
can maintain and regulate a CSLC population (Ithimakin et al., 2013; Jiang et al.,
2012; Korkaya et al., 2008; Nakanishi et al., 2010; Rainusso et al., 2012).
Neuregulin-1 (NRG1), a HER3 and HER4 ligand, has been shown to induce CSLC
characteristics in breast cancer by triggering HER3, since the NRG1-induced

43
CSLC characteristics are blocked when HER3 is inhibited (Jeong et al., 2014; Lee
et al., 2014). The expression of EGFR, HER2 and HER3 has also been linked to
poor clinical outcomes (Brabender et al., 2001; Cao, Chen, Xiong, & Chen, 2016;
Carey et al., 2007; Cheang et al., 2009; Day et al., 2017; Slamon et al., 1987; Yan
et al., 2018). A connection between HER4 expression and CSLC properties,
however, is quite unexplored. Some studies have shown that HER4 can promote
CSLC tumor progression, although one study showed that HER4 expression alone
was unable to induce tumorigenesis in vivo but was still able to enhance the survival
and growth of Ras- and/or wnt-driven tumors (Jardé et al., 2016; Williams et al.,
2015). HER4 expression is often linked to better prognosis, which supports the idea
that HER4 alone is unable to induce tumorigenesis. However, some studies have
linked the HER4 expression to poor survival (Munk et al., 2013; Wang et al., 2016;
Zhao et al., 2014).
2.4.3 HER targeted therapies
When tumors are addicted to a specific oncogene, they can be easily targeted by
inhibiting the oncogenic product. Multiple EGFR and HER2 targeting therapies
have been developed, some of which are already in clinical use. HER3 targeting
therapies are being developed, whereas HER4 is quite unexplored in the field of
therapeutic targeting. HER targeting agents are commonly either monoclonal
antibodies (mAbs) or tyrosine kinase inhibitors (TKIs). Monoclonal antibodies are
highly specific for their target and are always administered intravenously, whereas
TKIs are usually given orally and are not as specific as the mAbs. TKIs bind the
kinase domain either reversibly or irreversibly, and they can be ATP competitors
binding the ATP pocket directly or they can bind the kinase domain allosterically,
leading to modifications of the ATP pocket. However, it is important to notice that
even though oncogene addicted tumors are easily targeted, the tumors are clever
and will usually develop resistance to the given treatment if it does not already exist
(Arteaga et al., 2011). This chapter briefly describes the HER targeted therapies
and the most common resistance mechanisms.
EGFR
Three generations of small molecule EGFR inhibitors, TKIs, have been developed.
First-generation inhibitors, erlotinib and gefitinib which are reversible and
competitive for ATP binding, have shown drastic clinical responses in EGFR-

44
mutated cancer patients (Lynch et al., 2004; Mok et al., 2009; Rosell et al., 2012).
The mutated EGFRs have a higher affinity for erlotinib and gefitinib than the wild-
type EGFR (Carey et al., 2006; Lynch et al., 2004; Yun et al., 2007). These
inhibitors are in clinical use, but tumors almost inevitably develop acquired
resistance to these first-generation inhibitors. The most common resistance
mechanism is a gatekeeper mutation T790M in the EGFR receptor (Pao et al., 2005).
The second-generation EGFR TKIs, such as ATP-competitive afatinib, bind
irreversibly to the receptor and were developed to overcome acquired gefitinib and
erlotinib resistance. Even though these second-generation inhibitors are shown to
target the EGFRT790M, they also target the wild-type EGFR. Afatinib also binds
HER2 and HER4 proteins. In clinical trials, however, afatinib was unable to
overcome the EGFR TKI resistances and is currently used mainly as an alternative
agent for 1st generation inhibitors in metastatic, EGFR mutant NSCLC (Arteaga &
Engelman, 2014; Li et al., 2008; Miller et al., 2012).
Third-generation inhibitors are much more potent towards the mutated EGFR,
including T790M, than towards the wild-type. AZD9291, also called osimertinib,
has been shown to be well tolerated and has a high rate of clinical responses (Cross
et al., 2014; Jänne et al., 2015; Mok et al., 2017; Soria et al., 2018). Osimertinib is
an irreversible, ATP-competitive inhibitor and has received EMA and FDA
approval, but resistance towards osimertinib has been discovered (Cross et al., 2014;
Thress et al., 2015).
EGFR-neutralizing antibodies have been developed in addition to TKIs, of
which cetuximab and panitumumab are in clinical use, mainly to treat colorectal
cancer and head and neck cancer (Arteaga et al., 2011). Cetuximab in combination
with afatinib has been studied to treat NSCLC, because the combination was shown
to target the EGFRT790M in mice, and the combination has shown some clinical
activity, but the studies are still ongoing (Horn et al., 2017; Janjigian et al., 2014;
Regales et al., 2009).
HER2
Both TKIs and antibodies have been developed to target the HER2 protein.
Trastuzumab (Herceptin) is a monoclonal antibody that binds HER2 on the C-
terminal portion of subdomain IV and is used in the clinic for treatment of HER2-
positive cancers (Cho et al., 2003). Trastuzumab inhibits a cleavage of HER2
ectodomain, uncouples HER2 homodimers leading to a partial inhibition of
downstream signaling, but its main mechanism of action is to induce antibody-

45
dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular
phagocytosis (ADCP) (Clynes et al., 2000; Ghosh et al., 2011; Molina et al., 2001;
Shi et al., 2015). ADCC and ADCP are regulated via fragment c gamma receptors
(FcγR) on immune cells, and the effects of trastuzumab are remarkably diminished
when FcγR are absent (Clynes et al., 2000). These actions of trastuzumab are
mainly mediated by NK cells and macrophages, but it has been shown that T cells
are necessary for its full activation (Arnould et al., 2006; Gennari et al., 2004; Park
et al., 2010). Tumors can develop resistance to trastuzumab via multiple
mechanisms. The binding of trastuzumab can be impeded by expression of
truncated HER2, which lacks the receptor’s extracellular domains. The tumors can
overcome the inhibition of the HER2 signaling by increasing the expression of
EGFR, HER3 or other RTKs, or by altering the downstream signaling molecules.
The PI3K/Akt/mTOR pathway has often been linked to trastuzumab resistance. The
immunomodulatory functions of trastuzumab can be affected by FcγR
polymorphisms and by immunosuppressive cells, which inhibit the function of NK
cells, macrophages and T cells (Arteaga et al., 2011; Elster et al., 2015; Musolino
et al., 2008). However, a recent study has shown that the ADCP in macrophages,
induced by trastuzumab itself, can increase the expression of PD-L1 and IDO1,
leading to immunosuppression (Su et al., 2018).
Pertuzumab is a monoclonal antibody that binds to subdomain II of HER2
protein blocking heterodimerization of the receptor and, thus, leads to partial
inhibition of the receptor signaling and also to activation of the ADCC and ADCP
(Agus et al., 2002). Pertuzumab is given together with trastuzumab, because they
target the different domains and inhibit together all HER2-containing dimers. This
combinatory treatment has shown synergy in preclinical studies and in clinical trials
and is now used as a treatment for HER2-positive breast cancer (Baselga et al.,
2012; Gianni et al., 2012; Scheuer et al., 2009). The resistance mechanisms for
pertuzumab treatment are much less known than for trastuzumab.
Trastuzumab emtansine (T-DM1) is yet another HER2 targeting treatment
containing trastuzumab antibody, which is covalently linked to three to four non-
cleavable maytansinoid molecules that inhibit microtubule polymerization. When
T-DM1 binds to HER2 and the receptor is internalized, the T-DM1 comes along
and will be released after lysosomatic degradation, leading to cell lysis (Lewis
Phillips et al., 2008). The functions of T-DM1 are, otherwise, quite similar to
trastuzumab (Junttila, Li, Parsons, Phillips, & Sliwkowski, 2011). T-DM1 is in
clinical use for HER2-positive breast cancer, but resistance is eventually developed
(Barok, Joensuu, & Isola, 2014; Verma et al., 2012). The suggested resistance

46
mechanisms include impaired lysosomal activity, defects in the system inducing
mitosis and differential endocytosis (Ríos-Luci et al., 2017; Sabbaghi et al., 2017;
Sung et al., 2018).
TKI used for HER2-positive cancers (mainly breast cancer) is lapatinib, which
blocks the HER2 signaling by binding reversibly to the ATP pocket. Lapatinib is
used in clinical practice and has shown activity in HER2-positive breast cancers
that have progressed after trastuzumab. Lapatinib binds also the inactive formation
of EGFR but has not shown activity against EGFR-mutated cancers (Arteaga &
Engelman, 2014; Geyer et al., 2006; Konecny et al., 2006; Wood et al., 2004).
Resistance to lapatinib is proposed to occur via activation of compensatory survival
pathways, for example, increasing signaling trough estrogen receptors and
activating mutations of PI3K (Garrett & Arteaga, 2011).
Neratinib, an irreversible, ATP-competitive TKI that targets EGFR, HER2 and
HER4, has recently been approved for the treatment of early stage HER2-positive
breast cancer patients to follow trastuzumab-based adjuvant therapy (Feldinger &
Kong, 2015; Martin et al., 2017).
HER3 and HER4
Targeting agents for HER3 and HER4 are less studied than for EGFR and HER2.
The signaling through HER3 has been shown to be involved in the resistance to
EGFR and HER2 targeting therapeutic agents, and HER3 targeting inhibitors are
being developed. Since the tyrosine kinase of HER3 is impaired, TKI targeting of
HER3 is unlikely to be effective. The therapeutic approaches for HER3 targeting
focus on inhibiting the ligand binding and dimerization of the receptor, including
the internalization of HER3, and on trapping the receptor in its inactive
conformation (Karachaliou, Lazzari, Verlicchi, Sosa, & Rosell, 2017). Targeting
agents for HER4 are quite unexplored. Few monoclonal antibodies that target
HER4 in vitro and in vivo have been developed (Hollmén, Määttä, Bald,
Sliwkowski, & Elenius, 2009; Okazaki et al., 2016).
2.5 HER2+ breast cancer
HER2 gene is amplified or overexpressed in ~10–20% of all breast cancers (Cortet
et al., 2018; Kohler et al., 2015; Köninki, Tanner, Auvinen, & Isola, 2009). HER2
positivity is associated with large tumor size, lymph-node involvement, high-grade
tumors, high rates of proliferation, higher recurrence rates and poor survival. Anti-

47
HER2 therapies have remarkably improved the patient outcomes of HER2-positive
breast cancer (Andrulis et al., 1998; Baselga et al., 2012; Gonzalez-Angulo et al.,
2009; Marty et al., 2005; Ménard et al., 2002; Payandeh, Shahriari-Ahmadi,
Sadeghi, & Sadeghi, 2016; Slamon et al., 1987; Slamon et al., 1989). The addition
of trastuzumab alone in the treatment practice has made a great difference, but when
HER2-positive metastatic breast cancer patients received pertuzumab in addition
to trastuzumab and docetaxel (a chemotherapeutic agent), an improvement of 15.7
months in the overall survival (OS) was seen reaching a median OS of 56.5 months
(Swain et al., 2015).
2.5.1 Treatment of HER2+ breast cancer
Local disease
Figure 5 shows a scheme of the basic treatment options for local HER2+ breast
cancer. Surgery is the primary treatment for local disease. Mastectomy is a standard
option if the breast conserving surgery is not feasible and the patient has no wish
to conserve the breast. However, if the patient wishes to conserve the breast,
neoadjuvant therapy can be given to downsize the tumor. Neoadjuvant therapy is
also given for patients with inoperable T4 (T, tumor volume) and locally advanced
diseases. Regimens of the neoadjuvant therapies are planned individually for each
patient, but trastuzumab is usually given to all HER2+ breast cancers.
Chemotherapy can be given as neoadjuvant therapy, and endocrine therapy is given
according to the estrogen receptor status of the tumor (Finnish Breast Cancer Group,
2019; Senkus et al., 2015).
Two to six weeks after surgery, adjuvant therapies are planned for each patient
according to the risk of relapse, the predicted sensitivity to the treatment and the
benefit from them. Postoperative radiation therapy is given after breast-conserving
surgery, for patients with locally advanced disease and after mastectomy for T3–
T4 non-advanced tumors. HER2+ breast cancer patients will commonly receive
chemotherapy and trastuzumab therapy and possible endocrine therapy as an
adjuvant therapy. Guidelines exist about which treatments can be given
concomitantly and which cannot, for example, trastuzumab should not be given
together with anthracyclines due to its cardiotoxicity (Finnish Breast Cancer Group,
2019; Senkus et al., 2015). However, much uncertainty exists about the duration of
trastuzumab treatment. Some studies have shown that longer trastuzumab

48
treatments (2 years) do not give additional benefits, but other studies have been
unable to demonstrate that (Goldhirsch et al., 2013; Joensuu et al., 2009; Pivot et
al., 2013). The standard duration of trastuzumab treatment is currently one year. It
should be noted that these therapies are just examples, and every therapy is planned
separately for each patient. The things that must be considered when making the
final decision about the therapies are the predicted late effects of the treatment, the
general health status of the patient, the patient’s biological age and the patient’s
preferences (Finnish Breast Cancer Group, 2019; Senkus et al., 2015).
Fig. 5. Treatment options for early HER2+ breast cancer. Neoadjuvant therapy can be
given before surgery, surgery will be either breast conserving or mastectomy, and
adjuvant therapy will be given after the surgery. ChT=chemotherapy, ET=endocrine
therapy, RT=radiation therapy, T=trastuzumab, P=pertuzumab. Figure modified from
Senkus et al., 2015.
Early HER2+ breast cancer
Small tumorand/or surgery
feasible
Inoperable T4 orlocally advanced tumor
or patient wishes for breast conservation,
but tumor is too large
No wish for breastconservation, or it is
not possible
Neoadjuvant therapy(ChT + (ET) + T + P)
Unsatisfactoryresponse
Sat
isfa
ctor
yre
spon
se
Breast conservingsurgery
Mastectomy
Adjuvant therapy(ChT + (ET) + T)
+ postoperative RT

49
Metastatic disease
The treatment of metastatic disease depends on the tumor/metastasis status. If the
amplification/overexpression of HER2 gene is present, the standard first-line
therapy in the metastatic setting is the combination of chemotherapy, trastuzumab
and pertuzumab. Common chemotherapeutic agents used with the trastuzumab +
pertuzumab combination are docetaxel or paclitaxel. If the first-line therapy
provides benefit, anti-HER2 and ET therapies can be given as maintenance therapy.
According to the guidelines, the maintenance therapy should be used until
progression. However, expert opinion has stated that optimal duration of HER2
therapy is unknown for patients with complete response, and stopping may be
considered if a retreatment option is available. If the disease progresses after
trastuzumab-based therapy, the second-line therapy is T-DM1. Trastuzumab +
lapatinib can be a reasonable treatment option for some patients (Cardoso et al.,
2018).
2.5.2 Tumor-infiltrating lymphocytes in HER2+ breast cancer
Lymphocyte infiltration into HER2-positive breast cancer was first observed in
1990. The significance of tumor-infiltrating lymphocytes (TILs) in HER2+ breast
cancer has been widely studied, mainly by evaluating the abundance of total TILs
present in the tumor or in the stroma, and the high infiltration is associated with
remarkably improved outcomes and increased trastuzumab benefit (Denkert et al.,
2018; Lee et al., 2015; Loi et al., 2014; Luen et al., 2017; Salgado et al., 2015).
TILs may also predict the efficacy of chemotherapy in HER2+ breast cancer
(Denkert et al., 2015; Ingold Heppner et al., 2016).
TILs in HER2+ breast cancer are commonly studied by evaluating the
morphology of the immune cells from hematoxylin and eosin-stained tumor tissues,
even though distinct immune cells may have different impacts on the tumor
progression as section 2.2. described. Some of the most recent studies on HER2+
breast cancer have evaluated the impact of the immune cells to patient outcomes
and treatment responses by analyzing cytotoxic T cells (CD8), regulatory T cells
(FoxP3) and M2 macrophages (CD163 + CD68) separately. High stromal and
intratumoral infiltration of CD8+ cells and a high ratio of CD8/FoxP3 have been
linked to improved progression-free survival, overall survival and objective
response rate of trastuzumab-pertuzumab-docetaxel -treatment (Hou, Nitta, Wei,

50
Banks, Lustberg et al., 2018; Liu et al., 2017; Takada et al., 2018; van Rooijen et
al., 2018).
2.6 ALK in cancer
Anaplastic lymphoma kinase (ALK) is an RTK and a member of the insulin
receptor superfamily. ALK is involved in a normal development and function of the
nervous system, which has been shown to be mediated via MAPK signaling
pathway (Iwahara et al., 1997; Motegi, Fujimoto, Kotani, Sakuraba, & Yamamoto,
2004; Souttou, Carvalho, Raulais, & Vigny, 2001). ALK has been found to be
altered in many solid and hematological tumors and was first identified in
anaplastic large cell lymphoma, after which the ALK is named. The best-
characterized alterations of ALK are the translocations of the gene, but ALK can
also be mutated or amplified (Grande, Bolós, & Arriola, 2011).
2.6.1 ALK translocated NSCLC
Lung cancer is the most common type of cancer and has a high mortality. Lung
cancers are divided into two major classes: non-small cell lung cancer (NSCLC),
which accounts for over 80% of all lung cancers, and small cell lung cancer.
NSCLC is further divided into two categories, squamous cell carcinoma and
nonsquamous cell carcinoma, which include adenocarcinoma, large cell carcinoma
and other cell types. Around 60% of all NSCLC are adenocarcinomas (Cao et al.,
2019; Ettinger et al., 2017).
The most widely recognized translocation of ALK, the echinoderm
microtubule-associated protein-like 4 (EML4)-ALK fusion, was discovered in
NSCLC in 2007 (Grande et al., 2011; Lin, Riely, & Shaw, 2017; Soda et al., 2007).
EML4-ALK translocations are rare in NSCLC, only 3 to 7% of NSCLCs harbor the
fusion (Shaw et al., 2009), but the total amount of these cancers is still rather high
considering the high incidence of lung cancer worldwide. At least 15 different
variants of EML4-ALK translocation have been identified, but the variants 1, 2 and
3a/b are the most common ones. All EML4-ALK translocations contain the kinase
domain of the ALK protein, while the EML4 part differs between the variants.
These translocations lead to a constitutively and ligand-independently activated
ALK kinase, which signals through Ras/Raf/MEK/ERK, MAPK and JAK/STAT-
pathways. The EML4-fusion also changes the location of the ALK kinase, since it
is no longer located at the cell membrane but in the cytosol (Sabir, Yeoh, Jackson,

51
& Bayliss, 2017). ALK rearrangements are a classical example of an oncogene
addiction in cancers, and they can be easily targeted.
2.6.2 ALK targeting and resistance mechanisms in cancer
The first-developed ALK targeting inhibitor was crizotinib, a reversible ATP-
competitive TKI that has gained FDA and EMA approvals for ALK-rearranged
NSCLC and has been shown to be superior to the first- and second-line
chemotherapy in advanced ALK-rearranged NSCLC (Kwak et al., 2010; Shaw et
al., 2013; Solomon et al., 2014; Zou et al., 2007). Like with other TKIs, ALK TKI
treated patients will inevitably progress due to acquired resistance. Second-
generation ALK inhibitors, ceritinib, alectinib and brigatinib, are reversible ATP-
competitive inhibitors and have been developed to overcome crizotinib resistance.
They are approved for the treatment of ALK-rearranged NSCLCs previously treated
with crizotinib (Lin et al., 2017).
The two major classes of ALK TKI resistance mechanisms are ALK-dependent
mechanisms, including ALK secondary mutations or ALK amplification, and ALK-
independent mechanisms, including activation of bypass signaling pathways. ALK
secondary mutations causes around 20–30% of crizotinib resistances and around
56% of the second-generation ALK TKI resistances (Lin et al., 2017). The spectrum
of these ALK secondary mutations is rather broad, but the three most well-known
mutations are L1196M, G1269A and G1202R, which hinder the binding of the TKI
to ALK (Lin & Shaw, 2016). The G1202R mutated ALK has been shown to be
highly resistant to the first- and second-generation ALK TKIs, but the resistance
can be overcome with third-generation ALK TKI lorlatinib, which gained FDA
approval in 2018 (Gainor et al., 2016; Zou et al., 2015). Around half of the ALK
TKI resistances are caused independently of ALK. Numerous bypass signaling
activations have been shown to cause ALK TKI resistance, of which the first-
identified bypass mechanism was the activation of EGFR. The activation of other
HER proteins has also been shown to drive the ALK TKI resistance (Lin et al.,
2017; Wilson et al., 2015). Furthermore, crizotinib resistance can be driven by
pharmacokinetic resistance due to a low penetration of crizotinib through
cerebrospinal fluid (CSF), reducing the effect of crizotinib in the central nervous
system (Costa et al., 2011; Metro et al., 2015).

52

53
3 Aims of the study
This current work focused on more efficient use of HER targeting agents in cancer
therapy. Our aim was to find prognostic and predictive markers for metastatic
HER2-positive breast cancer patients treated with trastuzumab and determine the
role of HER2 and HER3 for cancer stem-like cells in ALK translocated non-small
cell lung cancer. More specifically the main aims of the study were:
1. To study the impact of immunosuppressive molecules and several distinct
immune cell subtypes in different tumor locations for the prognosis of
metastatic HER2-positive breast cancer patients and whether these
immunological markers could predict the benefit from trastuzumab.
2. To investigate the role of HER2 and HER3 receptors for cancer stem-like cells
in ALK translocated NSCLC by altering the gene expression of these proteins
and evaluate the effects of the genetic alteration to the cells.

54

55
4 Materials and methods
4.1 Publications I and II
The following materials and methods were used in publications I and II.
4.1.1 Patient material
All patients who had received at least one dose of intravenous trastuzumab for the
treatment of metastatic HER2-positive breast cancer in Oulu University Hospital
in 2009–2014 (n=54) were retrospectively identified from the pharmacy records
and were included in publications I and II. The studies were limited to patients who
had prechemotherapy samples available (n=48). In publication II the number of
adequate tumor samples was decreased to 40 samples, and the analysis of IDO1 in
publication II was limited to 25 samples due to a limited tumor sample availability
at the time of analysis.
The following patient data were previously (Moilanen, Mustanoja, Karihtala,
& Koivunen, 2017) collected from the electronic patient records: Patients’ age, date
of diagnosis, date of metastatic disease, histological subtype, TNM staging, tumor
grade, Ki-67 staining, estrogen- and progesterone receptor status,
adjuvant/metastatic treatment regimens, treatment durations and therapy responses.
The patients’ HER2 positivity was characterized by the presence of HER2
amplification in chromogenic in situ hybridization. Survival in metastatic disease
was defined as the time from the identification of metastatic disease to death or end
of follow-up. HER2 therapy was interrupted with patients whose HER2 therapy
response lasted >12 months or who had a therapy response but severe, suspected
HER2 therapy-related adverse effects. The length of HER2 therapy discontinuation
was determined from the date of the last administration of trastuzumab (in the
longest therapy interruption period) to the date of trastuzumab re-initiation, death,
or end of follow-up.
4.1.2 Immunohistochemistry and immune cell counting
Immunohistochemistry was conducted on 3.5 μm sections cut from paraffin-
embedded specimens. The sections were deparaffinized in xylene or in HistoClear
(IDO1) and rehydrated through graded alcohols. Antigen retrieval was conducted
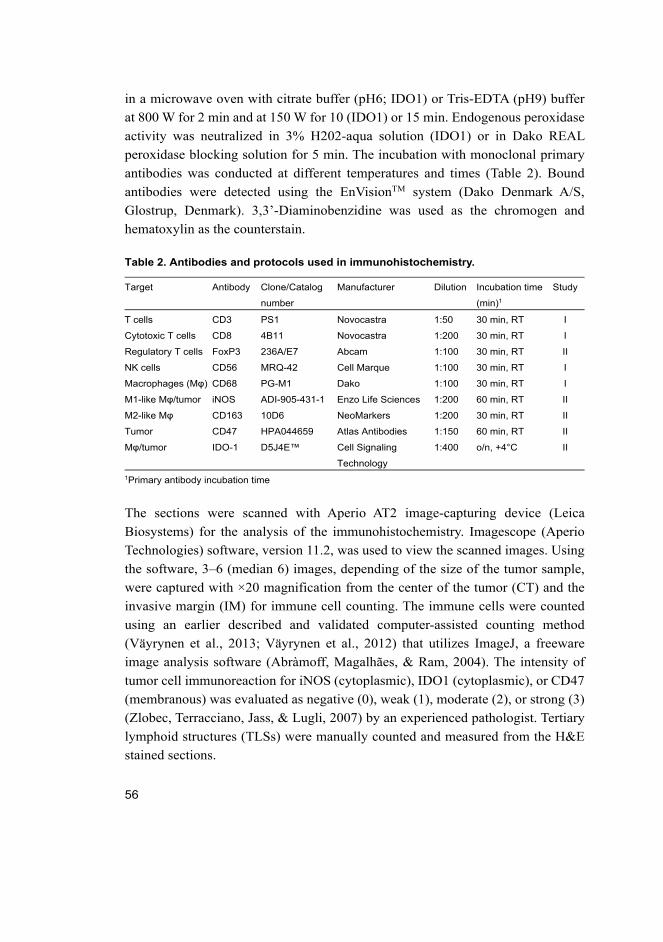
56
in a microwave oven with citrate buffer (pH6; IDO1) or Tris-EDTA (pH9) buffer
at 800 W for 2 min and at 150 W for 10 (IDO1) or 15 min. Endogenous peroxidase
activity was neutralized in 3% H202-aqua solution (IDO1) or in Dako REAL
peroxidase blocking solution for 5 min. The incubation with monoclonal primary
antibodies was conducted at different temperatures and times (Table 2). Bound
antibodies were detected using the EnVisionTM system (Dako Denmark A/S,
Glostrup, Denmark). 3,3’-Diaminobenzidine was used as the chromogen and
hematoxylin as the counterstain.
Table 2. Antibodies and protocols used in immunohistochemistry.
Target Antibody Clone/Catalog
number
Manufacturer Dilution Incubation time
(min)1
Study
T cells CD3 PS1 Novocastra 1:50 30 min, RT I
Cytotoxic T cells CD8 4B11 Novocastra 1:200 30 min, RT I
Regulatory T cells FoxP3 236A/E7 Abcam 1:100 30 min, RT II
NK cells CD56 MRQ-42 Cell Marque 1:100 30 min, RT I
Macrophages (Mφ) CD68 PG-M1 Dako 1:100 30 min, RT I
M1-like Mφ/tumor iNOS ADI-905-431-1 Enzo Life Sciences 1:200 60 min, RT II
M2-like Mφ CD163 10D6 NeoMarkers 1:200 30 min, RT II
Tumor CD47 HPA044659 Atlas Antibodies 1:150 60 min, RT II
Mφ/tumor IDO-1 D5J4E™ Cell Signaling
Technology
1:400 o/n, +4°C II
1Primary antibody incubation time
The sections were scanned with Aperio AT2 image-capturing device (Leica
Biosystems) for the analysis of the immunohistochemistry. Imagescope (Aperio
Technologies) software, version 11.2, was used to view the scanned images. Using
the software, 3–6 (median 6) images, depending of the size of the tumor sample,
were captured with ×20 magnification from the center of the tumor (CT) and the
invasive margin (IM) for immune cell counting. The immune cells were counted
using an earlier described and validated computer-assisted counting method
(Väyrynen et al., 2013; Väyrynen et al., 2012) that utilizes ImageJ, a freeware
image analysis software (Abràmoff, Magalhães, & Ram, 2004). The intensity of
tumor cell immunoreaction for iNOS (cytoplasmic), IDO1 (cytoplasmic), or CD47
(membranous) was evaluated as negative (0), weak (1), moderate (2), or strong (3)
(Zlobec, Terracciano, Jass, & Lugli, 2007) by an experienced pathologist. Tertiary
lymphoid structures (TLSs) were manually counted and measured from the H&E
stained sections.

57
4.1.3 Statistics
Statistical analyses were performed with IBM SPSS Statistics 24.0 for Windows
(IBM Corporation, Armonk, NY, USA). Receiver operating characteristics (ROC)
analysis for the whole patient material was used to determine optimal cut-off scores
of CD3, CD8, FoxP3, CD56, CD68, iNOS, CD163, IDO1 and TLS density for
discriminating the survivors from the non-survivors (Table 3). Area under the curve
(AUC) describes the accuracy of a test and also how well the test can separate the
survivors from the non-survivors. AUC values range from 0.5 to 1, and the closer
the value is to 1, the more discriminatory power the test has. The AUC values in
our studies were rather low because of the small number of patients in the studies
(Table 3). The associations between the studied markers and the survival of the
patients were analyzed with Kaplan-Meier method using the log-rank test to
compare the survival in different groups. Multivariate analyses were performed
with Cox regression analysis to test whether the studied markers were independent
prognostic markers. Correlations between different variables were performed using
Spearman’s correlation (2-tailed) and scatter plot analysis. Probability values below
0.05 were considered significant.
4.1.4 Ethics
The patient data collection and tumor analyses were carried out under permissions
from the Northern Ostrobothnia Hospital District ethical committee (114/2011,
amendment 23.02.2015), National Supervisory Authority for Welfare and Health
(9850/05.01.00.06/2010), and the medical director of Oulu University Hospital
(study no. 60/2015).

58
Table 3. ROC analysis AUC and cut-off values of the studied immune cell markers.
Marker Location AUC (CI) Cut-off (cells/mm2)1 Publication
CD3 IM 0.620 (0.409–0.831) 700 I
CT 0.672 (0.457–0.887) 300 I
CD8 IM 0.644 (0.441–0.846) 300 I
CT 0.695 (0.488–0.903) 120 I
CT 0.627 (0.401–0.853) 120 II
FoxP3 IM 0.556 (0.298–0.813) 134 II
CT 0.626 (0.384–0.868) 82 II
CD56 IM 0.683 (0.461–0.905) 17 I
CT 0.722 (0.505–0.938) 12 I
CD68 IM 0.563 (0.347–0.779) 646 I
CT 0.717 (0.544–0.889) 647 I
iNOS IM 0.594 (0.371–0.818) 58 II
CT 0.702 (0.498–0.906) 37 II
CD163 IM 0.504 (0.263–0.744) 307 II
CT 0.459 (0.216–0.703) 241 II
IDO IM 0.632 (0.333–0.930) 62 II
CT 0.706 (0.473–0.939) 19 II
TLS - 0.519 (0.277–0.762) 6.5 I
1Cut-off value for TLS is lymphoid follicles/2x field
4.2 Publications III and IV
The following materials and methods were used in publications III and IV.
4.2.1 Cell lines, inhibitors and growth factors
The cell lines used in the studies included ALK translocated non-small cell lung
cancer (NSCLC) cell lines H3122 and H2228. Both cell lines were further modified
to overexpress HER2 or knockdown HER2 or HER3. The original cell lines were
kind gifts from Dr. Pasi Jänne (Dana-Farber Cancer Institute, Boston, MA, USA).
The cell lines were cultured in RPMI-1640 medium supplemented with 10% fetal
bovine serum and 100 IU/ml penicillin and streptomycin, which were all purchased
from HyClone (Logan, UT, USA). The cells were incubated at 37˚C with 5% CO2
in the atmosphere. Table 4 shows all inhibitors and growth factors used to treat the
cells in studies III and IV. Inhibitors were diluted in dimethyl sulfoxide (DMSO)
and stored at -20˚C, while growth factors were diluted in sterile, distilled water and
stored at -80˚C.

59
Table 4. Used inhibitors and growth factors.
Molecule Target Used concentration Manufacturer
Inhibitor
TAE684 ALK 0.1 M Axon Medchem,
Groningen, Netherlands
Crizotinib ALK 1 M LC Laboratories,
Woburn, MA, USA
Lapatinib HER2, EGFR 1 M Alexis Biochemicals,
Lausen, Switzerland
Afatinib EGFR, HER2, HER4 1 M LC Laboratories,
Woburn, MA, USA
Growth factor
NRG1 HER3, HER4 50 ng/ml Sino Biological Inc.,
Beijing, China
EGF EGFR 50 ng/ml Sino Biological Inc.,
Beijing, China
4.2.2 Lentiviral knockdown and retroviral overexpression
Lentiviral and retroviral vectors were used to achieve knockdown and
overexpression of GFP and HER2 in H3122 and H2228 cell lines in publication III.
HER2 shRNA vector was purchased from Sigma-Aldrich (St. Louis, MO, USA),
while GFP shRNA vector and both retroviral vectors were kind gifts from Dr. Pasi
Jänne (Dana-Farber Cancer Institute). GFP was used as a control for the
overexpression and knockdown. 293T cells were transfected with lenti-/retroviral
expression vectors and packaging plasmids using FuGENE 6 reagent (Promega,
Madison, WI, USA). The packaging plasmids used were Gag&Pol, Rev and VSV-
G with lentiviral vectors and pCL-Eco with retroviral vectors. All packaging
plasmids were gifts from Dr. Pasi Jänne. Lentiviral supernatants were collected 24h
and retroviral supernatants 48h after transfection. Both supernatants were filtered
through 0.45 μm filter and applied to the target cells in the presence of polybrene
(Sigma-Aldrich). After 48h of infections, the target cells were selected with
puromycin (Sigma-Aldrich) for 72–96h.
4.2.3 CRISPR-Cas9 knockdown
HER3 knockdown in publication IV was performed using the CRISPR-Cas9
genome editing system. Single guide RNA (sgRNA) design and CRISPR-Cas9

60
transfection plasmid were purchased from Sigma-Aldrich. A plasmid with non-
targeting sgRNA was a kind gift from Dr. Peppi Karppinen, and the plasmid was
used to create a control knockdown cell line, referred to now on as NEG. Table 5
shows information about the transfection plasmids; vectors, sgRNA sequences and
targeted exons. HER3 has two known transcript variants (National Center for
Biotechnology Information) both of which were targeted with the same plasmid.
Table 5. CRISPR-Cas9 transfection plasmids.
Gene Vector Target site Target exon
HER3 U6gRNA-Cas9-2A-GFP ACTGTACAAGCTCTACGAGAGG 2
NEG pSpCas9(BB)-2A-GFP GCACTACCAGAGCTAACTCA -
H3122 cells were plated on 24-well plates, 25,000 cells/well, allowed to attach for
one day, after which they were transfected with CRISPR-Cas9 plasmids using
Fugene HD (Promega) transfection reagent. Transfection was performed according
to the manufacturer’s instructions using 1 µg of DNA per well. The transfected,
GFP-positive cells were single cell sorted 44–48 hours after transfection into 96-
well plates from where single cell colonies were further grown and analyzed. The
knockdown was verified by sequencing the genomic DNA and analyzing the
changes in protein expression with western blot.
4.2.4 DNA extraction, PCR and sequencing
Genomic DNA was isolated from cultured cells using the NucleoSpin® Tissue Kit
(Macherey-Nagel, Düren, Germany). Target sequences were amplified with
polymerase chain reaction (PCR) using MyTaqTM HS Red Mix (Bioline, London,
UK) with specific primers designed to cover the CRISPR-Cas9 sgRNA sequence
in HER3 (Table 6). The primers were purchased from Sigma-Aldrich. Sizes of the
PCR products were checked on 2% agarose gel, and the products were extracted
from the gel using the GeneJET Gel Extraction Kit (Thermo Fisher Scientific
Baltics UAB, Vilnus, Lithuania) with an extra washing step and pre-warmed elution
buffer. The PCR products were then sequenced with ABI3500xL Genetic Analyzer
by Biocenter Oulu Sequencing Center, and the sequences were analyzed with
CodonCode Aligner (CodonCode Corporation, Centerville, MA, USA).

61
Table 6. PCR primers and their sequences.
Primer Sequence Amplicon length (bp)
HER3 forward 5’-AAA GCT CTC AGG CCA CTA CA-3’ 519
HER3 reverse 5’-GCT ACA ACA GTG AGA CCA TAG G-3’
4.2.5 Western blot
Cultured cells were plated on 6-well plates, allowed to attach for 1–2 days, and
treated with the desired drugs for 5h or 5 days, after which the cells were lysed with
NP-40 lysis buffer (20 mM Tris-HCl pH 8.0, 137 mM NaCl, 10% glycerol, 2 mM
EDTA, 1 mM sodium orthovanadate, 1% igepal CA-630, 10 μg/ml aprotinin and
10 μg/ml leupeptin). Protein concentrations were measured with Bio-Rad Protein
assay (Bio-Rad Laboratories, Hercules, CA, USA). Concentrations between
samples were equalized, after which Laemmli buffer was added, and samples were
boiled and stored at -80˚C.
Equal amounts of proteins were separated on sodium dodecyl sulfate
polyacrylamide gel electrophoresis (SDS-PAGE), proteins were transferred to
polyvinylidene difluoride (PVDF) membrane, blocked with 5% BSA (1x PBS, 0.1%
Tween-20, 0.005% sodium azide) and incubated with primary antibodies overnight
at 4˚C. The membranes were incubated with horseradish peroxidase (HRP)-linked
secondary antibodies for 1h at room temperature, after which the membranes were
developed using chemiluminescence and exposed to radiographic films. The
quantification of western blot images was prepared using ImageJ software version
1.4.3.67, measuring the intensities (pixel percentages) for each sample in the same
membrane with an equal, manually selected area.
Antibodies (Table 7) were diluted in 5% BSA and used at 1:1,000, 1:20,000 (-
actin) or 1:3,000 (secondary antibodies) dilutions. Antibodies were purchased from
Cell Signaling Technology (Danvers, MA, USA), except -actin (Novus
Biologicals, Abingdon, UK) and ALDH1 (BD Transduction Laboratories, Franklin
Lakes, NJ, USA).
4.2.6 Colony formation assay
Cells were plated on 24-well plates, 600–1,000 cells/well, allowed to attach for 1–
2 days, after which they were treated with the desired drugs for 7 days. Table 4
displays the used inhibitors, growth factors and final concentrations. The cells were
then allowed to recover and form several colonies before they were fixed with ice-
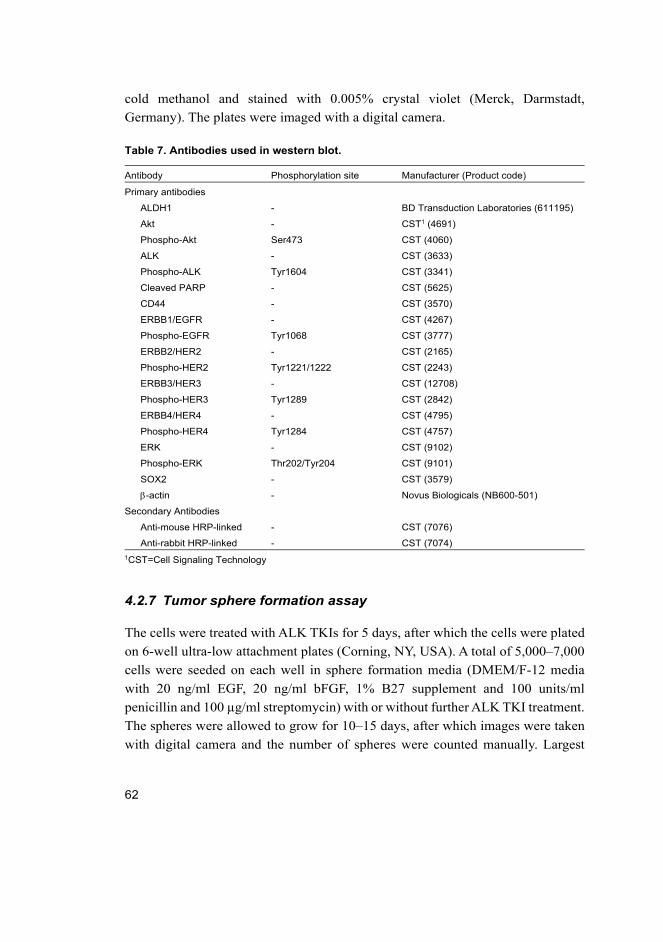
62
cold methanol and stained with 0.005% crystal violet (Merck, Darmstadt,
Germany). The plates were imaged with a digital camera.
Table 7. Antibodies used in western blot.
Antibody Phosphorylation site Manufacturer (Product code)
Primary antibodies
ALDH1 - BD Transduction Laboratories (611195)
Akt - CST1 (4691)
Phospho-Akt Ser473 CST (4060)
ALK - CST (3633)
Phospho-ALK Tyr1604 CST (3341)
Cleaved PARP - CST (5625)
CD44 - CST (3570)
ERBB1/EGFR - CST (4267)
Phospho-EGFR Tyr1068 CST (3777)
ERBB2/HER2 - CST (2165)
Phospho-HER2 Tyr1221/1222 CST (2243)
ERBB3/HER3 - CST (12708)
Phospho-HER3 Tyr1289 CST (2842)
ERBB4/HER4 - CST (4795)
Phospho-HER4 Tyr1284 CST (4757)
ERK - CST (9102)
Phospho-ERK Thr202/Tyr204 CST (9101)
SOX2 - CST (3579)
-actin - Novus Biologicals (NB600-501)
Secondary Antibodies
Anti-mouse HRP-linked - CST (7076)
Anti-rabbit HRP-linked - CST (7074)
1CST=Cell Signaling Technology
4.2.7 Tumor sphere formation assay
The cells were treated with ALK TKIs for 5 days, after which the cells were plated
on 6-well ultra-low attachment plates (Corning, NY, USA). A total of 5,000–7,000
cells were seeded on each well in sphere formation media (DMEM/F-12 media
with 20 ng/ml EGF, 20 ng/ml bFGF, 1% B27 supplement and 100 units/ml
penicillin and 100 µg/ml streptomycin) with or without further ALK TKI treatment.
The spheres were allowed to grow for 10–15 days, after which images were taken
with digital camera and the number of spheres were counted manually. Largest

63
spheres from each well were imaged with phage contrast microscope using 10x
magnification in publication III.

64
65
5 Results
5.1 Immunological markers in metastatic HER2+ breast cancer
Immunological markers were studied in publications I and II.
5.1.1 Patient characteristics
As a result of a systematic search, we identified 54 patients who had received
intravenous trastuzumab in 2009–2014 for the treatment of breast cancer and had
metastatic disease at the time of diagnosis or relapsed disease unsuitable for
curative treatment. Of these patients, prechemotherapy samples were available for
48 patients, and further analyses were limited to this group. However, the amount
of adequate tumor samples was decreased gradually, first to 40 samples and finally
to 25 samples. Table 8 demonstrates the types of samples available that were used
in the analyses.
Table 8. Types of tumor specimens.
Total n (publication) Surgical, n (%) Biopsy, n (%)
48 (I) 31 (64.6) 17 (35.4)
40 (I, II)1 25 (62.5) 15 (37.5)
25 (II)2 21 (84.0) 4 (16.0)
1CD56 and CD68 staining in publication I and all in publication II, 2IDO1 staining
Table 9 illustrates the patient demographics relevant to the studies. The patients had
generally an aggressive disease, since most of them had a high-grade tumor and
high Ki-67 value, which describes the proliferation rate of the tumor. Over one-
third of the patients had primary metastatic disease, and 30–40% were identified to
have undergone trastuzumab treatment interruption in a prolonged tumor response.
66
Table 9. Patient demographics.
Factor n (%)1 n (%)2 n (%)3
Histology
Ductal 41 (85.4) 33 (82.5) 20 (80)
Lobular 5 (10.4) 5 (12.5) 5 (20)
Unknown 2 (4.2) 2 (5.0) 0 (0)
Estrogen receptor status
Positive 33 (68.8) 27 (67.5) 16 (64)
Negative 15 (31.3) 13 (32.5) 9 (36)
Progesterone receptor status
Positive 19 (39.6) 18 (45.0) 13 (52)
Negative 29 (60.4) 22 (55.0) 12 (48)
Bone metastasis only
Yes 16 (33.3) 14 (35.0) 12 (48)
No 32 (66.7) 26 (65.0) 13 (52)
Ki-67
Low (5–15%) 5 (10.4) 4 (10.0) 3 (12)
Intermediate (16–30%) 11 (22.9) 10 (25.0) 6 (24)
High (>30%) 30 (62.5) 24 (60.0) 16 (64)
Unknown 2 (4.2) 2 (5.0) 0 (0)
Grade
II 16 (33.3) 10 (25.0) 6 (24)
III 30 (62.5) 28 (70.0) 19 (76)
Unknown 2 (4.2) 2 (5.0) 0 (0)
Adjuvant trastuzumab
No 9 (18.8) 8 (20.0) 6 (24.0)
Yes 20 (41.7) 17 (42.5) 10 (40.0)
Primary metastatic disease 19 (39.6) 15 (37.5) 9 (36.0)
HER2 therapy discontinuation
Yes 14 (29.2) 12 (30.0) 10 (40)
No 34 (70.8) 28 (70.0) 15 (60)
1total n=48, 2total n=40, 3total n=25
5.1.2 Staining and image analysis of the studied markers
The tumors were stained with multiple markers, illustrated in tables 10 and 11.
Most of the staining images can be seen in the original publications (I and II). The
quantitative analysis of the markers was successful, and the stained immune cells
were analyzed separately from the invasive margin (IM) and center of the tumor
(CT). The density of tertiary lymphoid structures (TLSs) could not be analyzed
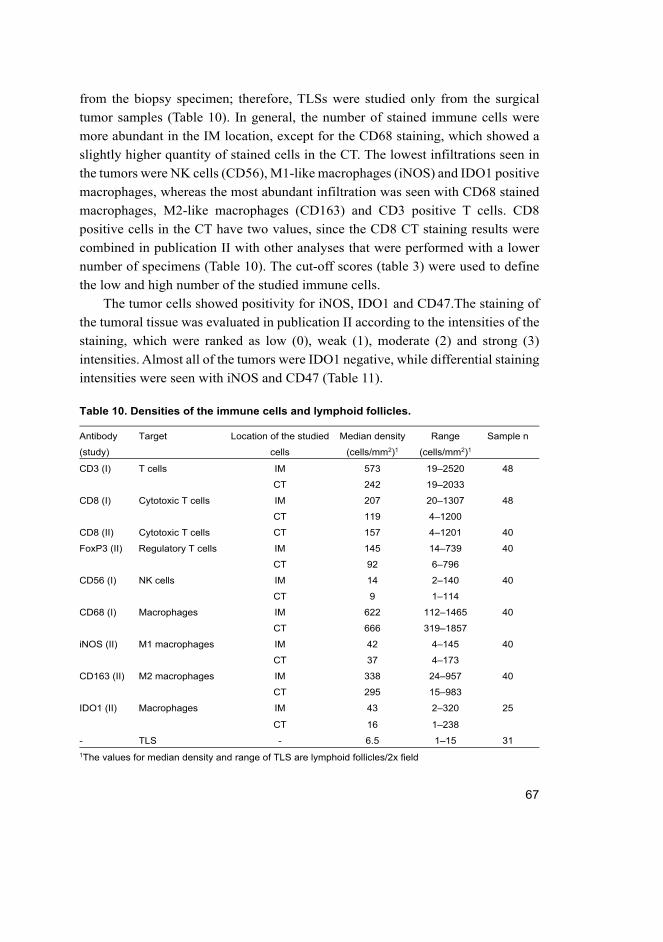
67
from the biopsy specimen; therefore, TLSs were studied only from the surgical
tumor samples (Table 10). In general, the number of stained immune cells were
more abundant in the IM location, except for the CD68 staining, which showed a
slightly higher quantity of stained cells in the CT. The lowest infiltrations seen in
the tumors were NK cells (CD56), M1-like macrophages (iNOS) and IDO1 positive
macrophages, whereas the most abundant infiltration was seen with CD68 stained
macrophages, M2-like macrophages (CD163) and CD3 positive T cells. CD8
positive cells in the CT have two values, since the CD8 CT staining results were
combined in publication II with other analyses that were performed with a lower
number of specimens (Table 10). The cut-off scores (table 3) were used to define
the low and high number of the studied immune cells.
The tumor cells showed positivity for iNOS, IDO1 and CD47.The staining of
the tumoral tissue was evaluated in publication II according to the intensities of the
staining, which were ranked as low (0), weak (1), moderate (2) and strong (3)
intensities. Almost all of the tumors were IDO1 negative, while differential staining
intensities were seen with iNOS and CD47 (Table 11).
Table 10. Densities of the immune cells and lymphoid follicles.
Antibody
(study)
Target Location of the studied
cells
Median density
(cells/mm2)1
Range
(cells/mm2)1
Sample n
CD3 (I) T cells IM 573 19–2520 48
CT 242 19–2033
CD8 (I) Cytotoxic T cells IM 207 20–1307 48
CT 119 4–1200
CD8 (II) Cytotoxic T cells CT 157 4–1201 40
FoxP3 (II) Regulatory T cells IM 145 14–739 40
CT 92 6–796
CD56 (I) NK cells IM 14 2–140 40
CT 9 1–114
CD68 (I) Macrophages IM 622 112–1465 40
CT 666 319–1857
iNOS (II) M1 macrophages IM 42 4–145 40
CT 37 4–173
CD163 (II) M2 macrophages IM 338 24–957 40
CT 295 15–983
IDO1 (II) Macrophages IM 43 2–320 25
CT 16 1–238
- TLS - 6.5 1–15 31
1The values for median density and range of TLS are lymphoid follicles/2x field

68
Table 11. Staining intensities of the tumor tissues.
Antibody (study) Location Negative (0)
n (%)
Weak (1)
n (%)
Moderate (2)
n (%)
Strong (3)
n (%)
Sample n
iNOS (II) cytoplasm 7 (17.5) 14 (35.0) 10 (25.0) 9 (22.5) 40
CD47 (II) membrane 9 (22.5) 7 (17.5) 12 (30.0) 12 (30.0) 40
IDO1 (II) cytoplasm 20 (80.0) 0 (0.0) 3 (12.0) 2 (8.0) 25
5.1.3 Association of T cell subsets with clinical data
We studied whether the different T cell subsets present in the tumor before any
treatment had effects on the patient outcomes using the Kaplan-Meier survival
analysis. As assumed, a high number of regulatory T cells (Tregs) identified with
FoxP3 nuclear positivity showed a tendency towards worsened survival in both IM
and CT but without reaching statistical significance, however. CD3, a protein part
of the T cell receptor complex, recognizes all sets of T cells, and a high number of
CD3 positive cells was associated with improved survival in the CT (p = 0.007) but
not in the IM. Similarly, a high number of CD8 positive cells recognizing cytotoxic
T cells were associated with improved survival in the CT (p = 0.001) but not in the
IM.
A combined analysis of both CD3 and CD8 positive cells in the IM and in the
CT is a promising prognostic parameter in colorectal cancer, which has been
reported to predict improved survival better than either of the markers alone (Galon
et al., 2014; Galon et al., 2006; Pagès et al., 2018). This parameter is called the
Immunoscore, which categorizes the densities of each of these cell types in both
IM and CT as low (0 points) or high (1 point), yielding a five-tiered classification
from Immunoscore 0 to Immunoscore 4. The significance of the Immunoscore in
breast cancer is quite unknown, and we decided to carry out this combined analysis
of CD3 and CD8 in IM and CT in our material. However, due to a rather small
number of patients, we categorized the Immunoscore values into three groups
instead of five; 0 (n=13), 1–2 (n=24), 3–4 (n=11). A high Immunoscore value
showed a tendency towards better survival but did not reach statistical difference.
We also conducted a combined analysis of CD3 and CD8 only in the CT by using
a two-class grouping: 0–1 low (n=32), 2 high (n=16). A significant association of
CT Immunoscore with survival was seen (p = 0.001); however, it was not higher
than the significance seen with CD8 positivity in the CT alone.
The abundance of CD8 positive cells in the CT was the most prominent
indicator of improved survival, and it was proven to remain as an independent
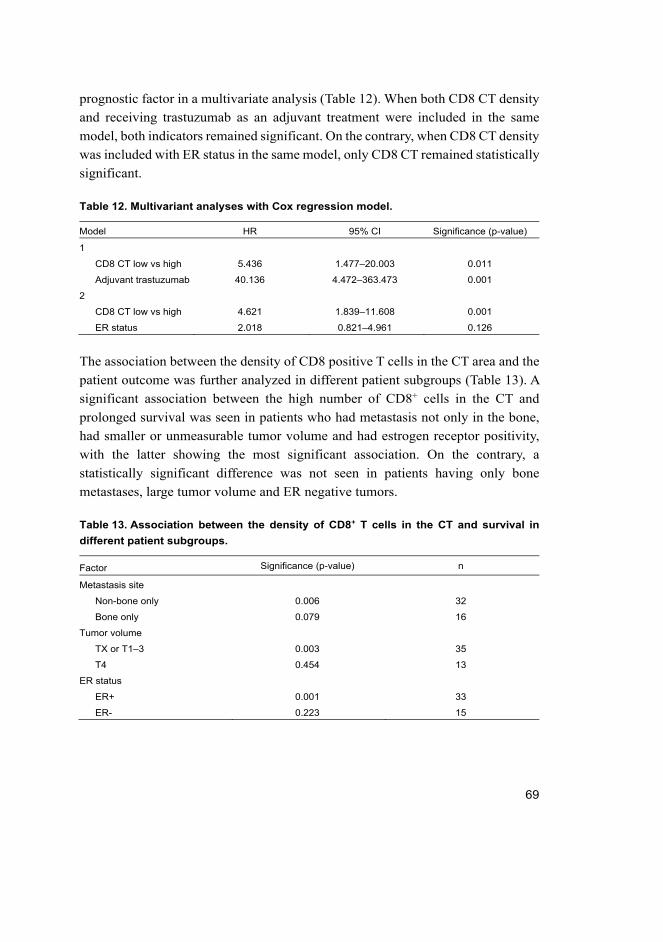
69
prognostic factor in a multivariate analysis (Table 12). When both CD8 CT density
and receiving trastuzumab as an adjuvant treatment were included in the same
model, both indicators remained significant. On the contrary, when CD8 CT density
was included with ER status in the same model, only CD8 CT remained statistically
significant.
Table 12. Multivariant analyses with Cox regression model.
Model HR 95% CI Significance (p-value)
1
CD8 CT low vs high 5.436 1.477–20.003 0.011
Adjuvant trastuzumab 40.136 4.472–363.473 0.001
2
CD8 CT low vs high 4.621 1.839–11.608 0.001
ER status 2.018 0.821–4.961 0.126
The association between the density of CD8 positive T cells in the CT area and the
patient outcome was further analyzed in different patient subgroups (Table 13). A
significant association between the high number of CD8+ cells in the CT and
prolonged survival was seen in patients who had metastasis not only in the bone,
had smaller or unmeasurable tumor volume and had estrogen receptor positivity,
with the latter showing the most significant association. On the contrary, a
statistically significant difference was not seen in patients having only bone
metastases, large tumor volume and ER negative tumors.
Table 13. Association between the density of CD8+ T cells in the CT and survival in
different patient subgroups.
Factor Significance (p-value) n
Metastasis site
Non-bone only 0.006 32
Bone only 0.079 16
Tumor volume
TX or T1–3 0.003 35
T4 0.454 13
ER status
ER+ 0.001 33
ER- 0.223 15

70
5.1.4 Association of macrophage subsets with clinical data
When macrophages were studied with general macrophage marker CD68, only a
tendency towards improved survival was seen with a high density of CD68 positive
cells both in the IM and CT. However, this marker recognizes both M1- and M2-
like macrophages and also other types of immune cells, such as myeloid cells and
dendritic cells. Thus, we decided to study the M1- and M2-like phenotypes of the
macrophages separately using iNOS and CD163 markers. A high number of M1-
like macrophages in the IM (p = 0.009) and CT (p = 0.009) was associated with
improved survival, whereas a high number of M2-like macrophages in the CT but
not in the IM displayed a tendency towards poor survival, not reaching statistical
significance. Some tumor cells showed positivity for iNOS expression; however,
this did not provide any prognostic value.
5.1.5 Association of other markers with clinical data
We investigated also other immunological markers besides T cells and
macrophages. The number of NK cells, identified by CD56 positivity, was low in
the tumors, which was reflected in the survival studies. There was only a tendency
towards a better survival seen in patients with a high number of NK cells in the CT,
but it was not statistically significant. We also analyzed the density of lymphoid
follicles in the adjacent area of the tumors, called tertiary lymphoid structures
(TLSs), from H&E stained sections, which was shown to be associated with
improved survival (p = 0.021).
Furthermore, we wanted to study a few immunosuppressive molecules
expressed by the tumor cells to reveal if the immune cells were directly suppressed
by the tumors. CD47 is a molecule on the surface of tumor cells that binds to
phagocytosing cells to inhibit their phagocytic effects. CD47 intensities were
divided into two groups, low (int 0 and 1) and high (int 2 and 3) intensities, where
high CD47 intensity predicted worsened survival (p = 0.046). The intensities of
IDO1, an enzyme catalyzing the degradation of tryptophan, were also divided into
two groups, similar to CD47. A high intensity of IDO1 was associated with poor
survival (p = 0.018). In addition to the tumoral expression, IDO1 was observed also
in macrophages in the IM and CT; however, IDO1 expression in macrophages did
not provide any prognostic value.
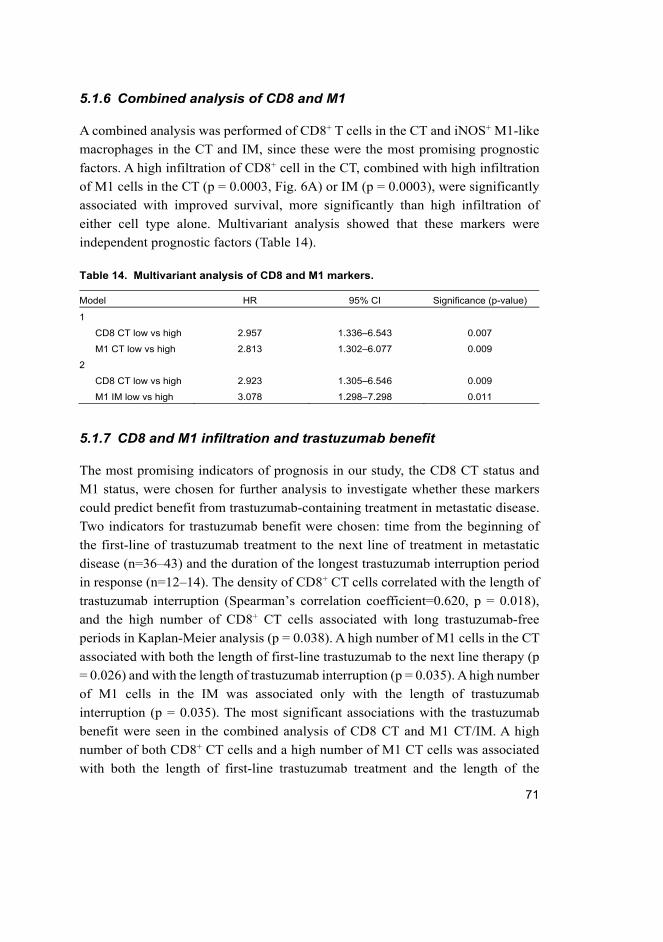
71
5.1.6 Combined analysis of CD8 and M1
A combined analysis was performed of CD8+ T cells in the CT and iNOS+ M1-like
macrophages in the CT and IM, since these were the most promising prognostic
factors. A high infiltration of CD8+ cell in the CT, combined with high infiltration
of M1 cells in the CT (p = 0.0003, Fig. 6A) or IM (p = 0.0003), were significantly
associated with improved survival, more significantly than high infiltration of
either cell type alone. Multivariant analysis showed that these markers were
independent prognostic factors (Table 14).
Table 14. Multivariant analysis of CD8 and M1 markers.
Model HR 95% CI Significance (p-value)
1
CD8 CT low vs high 2.957 1.336–6.543 0.007
M1 CT low vs high 2.813 1.302–6.077 0.009
2
CD8 CT low vs high 2.923 1.305–6.546 0.009
M1 IM low vs high 3.078 1.298–7.298 0.011
5.1.7 CD8 and M1 infiltration and trastuzumab benefit
The most promising indicators of prognosis in our study, the CD8 CT status and
M1 status, were chosen for further analysis to investigate whether these markers
could predict benefit from trastuzumab-containing treatment in metastatic disease.
Two indicators for trastuzumab benefit were chosen: time from the beginning of
the first-line of trastuzumab treatment to the next line of treatment in metastatic
disease (n=36–43) and the duration of the longest trastuzumab interruption period
in response (n=12–14). The density of CD8+ CT cells correlated with the length of
trastuzumab interruption (Spearman’s correlation coefficient=0.620, p = 0.018),
and the high number of CD8+ CT cells associated with long trastuzumab-free
periods in Kaplan-Meier analysis (p = 0.038). A high number of M1 cells in the CT
associated with both the length of first-line trastuzumab to the next line therapy (p
= 0.026) and with the length of trastuzumab interruption (p = 0.035). A high number
of M1 cells in the IM was associated only with the length of trastuzumab
interruption (p = 0.035). The most significant associations with the trastuzumab
benefit were seen in the combined analysis of CD8 CT and M1 CT/IM. A high
number of both CD8+ CT cells and a high number of M1 CT cells was associated
with both the length of first-line trastuzumab treatment and the length of the
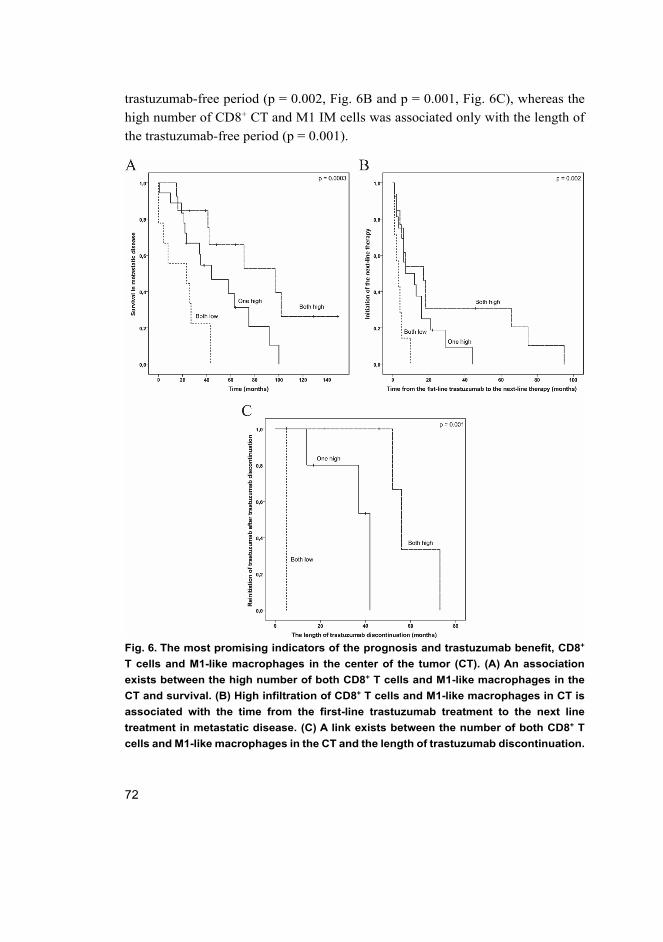
72
trastuzumab-free period (p = 0.002, Fig. 6B and p = 0.001, Fig. 6C), whereas the
high number of CD8+ CT and M1 IM cells was associated only with the length of
the trastuzumab-free period (p = 0.001).
Fig. 6. The most promising indicators of the prognosis and trastuzumab benefit, CD8+
T cells and M1-like macrophages in the center of the tumor (CT). (A) An association
exists between the high number of both CD8+ T cells and M1-like macrophages in the
CT and survival. (B) High infiltration of CD8+ T cells and M1-like macrophages in CT is
associated with the time from the first-line trastuzumab treatment to the next line
treatment in metastatic disease. (C) A link exists between the number of both CD8+ T
cells and M1-like macrophages in the CT and the length of trastuzumab discontinuation.

73
5.2 The role of HER2 and HER3 for cancer stem-like cell phenotype
The role of HER2 and HER3 for cancer stem-like cell phenotype in publications
III and IV was studied using ALK translocated NSCLC cell lines H2228 and H3122.
5.2.1 Overexpression and knockdown of HER2 and HER3
H3122, a cell line that is extremely sensitive to ALK inhibition, was altered to
overexpress HER2 with retroviral expression vector. H2228 cell line, which is only
modestly sensitive to ALK inhibition and expresses HER2 basally, was altered by
knocking down HER2 with shRNA vector. HER3 was knocked down only in the
H3122 cell line, because the HER3 protein expression levels of H2228 control cells
were extremely low, and we were unable to detect it with western blot assay. The
knockdown of HER3 in H3122 line was performed with more advanced gene
editing system called CRISPR-Cas9, and the knockdown cell line was derived from
a single cell. All the genetic alterations were validated and were successful. GFP-
altered cell lines were used as control lines for the overexpression and knockdown
of HER2, whereas a control CRISPR-Cas9 transfection plasmid containing non-
targeting sgRNA was used for the HER3 knockdown.
5.2.2 The effects of genetic alterations to cancer stem-like cell
marker expression
We have previously shown that cancer stem-like cells (CSLCs), identified with
specific markers ALDH1 (H3122) and CD44/ALDH1 (H2228), can mediate ALK
tyrosine kinase inhibitor (TKI) resistance (Jokinen, Laurila, Koivunen, & Koivunen,
2014). Thus, we assessed whether the genetic alterations of HER2 and HER3 had
an effect on the CSLCs by examining the expression of CSLC markers using
western blot analysis. As seen previously, ALK inhibition increased the expression
of CSLC markers, suggesting CSLC-mediated resistance. In the H3122 line, the
overexpression of HER2 did not change the basal ALDH1 expression but led to a
more pronounced increase in the expression in response to ALK inhibition. Basal
expression of CD44 was unaltered when HER2 was knocked down in H2228,
whereas the basal expression of ALDH1 was markedly downregulated. The
expression of CD44, however, was downregulated when ALK was inhibited. HER3
knockdown in H3122 did not change the basal expression of ALDH1 but
downregulated the basal expression of SOX2. The induction of ALDH1 expression

74
in response to ALK inhibition was blocked when HER3 was knocked down, and
the induction of SOX2 was slightly downregulated by the knockdown.
The expression of the CSLC markers was even further increased in the control
cells and HER2 overexpressing cells when NRG1, a HER3 and HER4 ligand,
linked to tumor initiating cells, was introduced simultaneously with ALK inhibition.
A slight recovery of the CSLC marker downregulation in the HER2 knockdown
line was seen by the addition of NRG1 to ALK inhibition, but no changes were
seen in the HER3 knockdown cells, as expected.
5.2.3 The effects of the genetic alterations to cytotoxic response to
ALK inhibition
We investigated whether HER2 and HER3 alterations affected the cytotoxic
response of the cells to ALK inhibition by exploring their colony formation ability.
The main cytotoxic responses to ALK TKI remained unaffected by the alterations;
however, the overexpression of HER2 resulted in a slight increase in the number of
surviving colonies, and knockdown of HER2 or HER3 resulted in a slightly
decreased number of surviving colonies compared to the controls. The increased
survival in HER2 overexpressing cells was blocked by the addition of lapatinib
(targets EGFR and HER2) or afatinib (targets EGFR, HER2 and HER4) together
with the ALK TKI. No differences in the surviving colonies were seen between the
HER3 knockdown and control cells when afatinib or lapatinib was combined with
ALK TKI; however, the number of surviving colonies in HER2 knockdown cells
was decreased compared to the control cells.
More pronounced differences in the cytotoxic responses were seen when HER3
(and HER4) was activated with NRG1 treatment. NRG1 by itself did not alter the
colony formation abilities of the studied cells, however, when it was combined with
ALK TKI differences were seen between the HER2/HER3 altered cells and the
control cells. The addition of NRG1 to ALK TKI treatment increased markedly the
number of surviving colonies in the HER2 overexpressing cells, whereas only a
minor increase was seen in the control cells. No differences were seen between the
HER2 knockdown cells and the control cells when NRG1 was combined with ALK
TKI. The increased colony formation ability seen in the control cells in response to
NRG1 and ALK TKI was blocked by the knockdown of HER3.
The changes in the cytotoxic response to ALK TKI after HER2 alterations were
studied also with tumor sphere formation assay, which is a commonly used assay
to identify CSLCs in vitro. However, the assay was more like a colony formation

75
assay with a CSLC-inducing environment for the H3122 cells, because the formed
spheres were cell rafts instead of spheres due to the cells’ ability to stick together
and difficulty in separating them properly. Despite that, the results from the assay
were similar to colony formation assay and CSLC protein expression patterns,
showing that HER2 knockdown diminished the survival of CSLCs if ALK was
inhibited, and the overexpression of HER2 increased the survival of CSLCs even
when ALK was continuously inhibited for 15 days.
5.2.4 Alterations in the Akt and ERK1/2 downstream signaling
The changes of HER2/HER3 alterations to the most common downstream signaling
pathways of RTKs, PI3K and MAPK were examined by studying the
phosphorylation of Akt and ERK1/2 proteins at 5h or 5 days after the initiation of
ALK inhibition. The alteration did not affect the initial (5h) response to ALK TKI;
instead, the differences were seen in the long-term (5 days) responses. HER2
overexpression in H3122 resulted in reformed downstream RTK signaling with
major upregulation of both phosphorylated Akt and ERK1/2, which was not seen
in the control cells. H2228 control cells were able to recover the downstream
signaling pathways after 5-day inhibition of ALK, which was blocked by the
knockdown of HER2. HER3 knockdown, however, hampered the Akt signaling
recovery, whereas ERK1/2 was able to recover similar to the control cells.
We examined the impact of NRG1 also on the downstream signaling pathway
recoveries. The addition of NRG1 with ALK TKI recovered the downstream
signaling of Akt and ERK1/2 in H3122 control cells, and the expression of
phosphorylated Akt was further increased in the HER2 overexpression cell line.
These signaling recoveries were blocked by the knockdown of HER3.
5.2.5 Compensatory expression of other HER family members
We examined whether the alterations of HER2 and HER3 affected the expression
and activation of the other HER family members. HER2 overexpression in H3122
cells led to an increased activation of EGFR, HER3 and HER4 proteins, of which
the activation of HER3 mimicked the expression patterns of the ALDH1, since the
most prominent activation of HER3 was seen when ALK was inhibited and NRG1
was provided, which also showed the most prominent expression of ALDH1. We
were unable to see activation of the HER proteins in the H2228 cell lines, but the
knockdown of HER2 downregulated the expression of HER3. In H3122 the HER3

76
knockdown increased the phosphorylation of EGFR, but we were unable to see
activations of the other HER proteins in the control and in the HER3 knockdown
cells. The activation of EGFR was unable to recover the Akt signaling in the HER3
knockdown cells.
The addition of NRG1 was able to induce the activation of HER3 in HER2
overexpressed cells even when ALK was not inhibited, but more pronounced
activation was seen with a combinatory treatment of ALK TKI and NRG1, which
was also seen in H3122 control cells. Furthermore, ALK TKI together with NRG1
induced the strongest expression of CSLC markers.

77
6 Discussion
There are many types of cancer treatments these days, such as surgery,
chemotherapy, radiation therapy, targeted therapy, and the latest newcomer,
immunotherapy. It is now well understood that one type of therapy is not suitable
for every patient, and precision medicine has taken its place in the field of oncology.
Targeted therapies, including small-molecule inhibitors and monoclonal antibodies,
and immunotherapy can be given to patients according to their tumor’s genetic
status. However, even though the genetics of the tumors appear to be suitable for
some specific therapy, not every patient will respond to the therapy, or the disease
will progress soon after the response due to development of an acquired resistance.
Many resistance mechanisms for different therapies have been discovered, for
example, the involvement of cancer stem-like cells that are resistant to different
therapies and are extremely tumorigenic. Unfortunately, cancer stem-like cells are
difficult to target, and there are no targeting agents available in the clinic. Therefore,
further research is needed to improve precision medicine in order to enhance patient
selection and develop new targeting agents or utilize the ones that are already
available.
6.1 Immunological markers in HER2+ breast cancer
The disease outcomes of HER2-positive breast cancers have dramatically improved
after HER2 targeting agents, like trastuzumab, became available (Marty et al., 2005;
Swain et al., 2015; Von Minckwitz et al., 2009). However, since its approval,
trastuzumab has been given according to the tumoral HER2 status only, and other
predictive factors for trastuzumab sensitivity or resistance are currently quite
uncharacterized. The mechanisms of action of trastuzumab are based on its ability
to bind the extracellular domain of HER2 and partially inhibit the signaling through
it, but the main effects of trastuzumab have been shown to be the activation of the
immune system through ADCC and ADCP (Clynes et al., 2000; Nami, Maadi, &
Wang, 2018; Petricevic et al., 2013). Some trastuzumab-resistance mechanisms
have been suggested, of course, such as alterations that abolish the signal
transduction inhibition of trastuzumab, like p95 variant of HER2, or alterations of
PTEN and PI3KCA and also compromised host immunoactivation, which prevents
the main effects of the antibody (Gagliato, Jardim, Marchesi, & Hortobagyi, 2016).
Among the metastatic HER2-positive breast cancers, great variation is seen in
treatment outcomes, with some patients surviving even more than 10 years with the

78
disease (Moilanen et al., 2017). This is similar to what is seen with new cancer
immunotherapies; therefore, we speculated that the immunological status of the
tumor would play an important role in patient outcomes.
Our studies assessed whether the tumor-infiltrating immune cells evaluated by
using a quantitative computer-assisted counting method could be used as
prognostic and predictive markers in metastatic HER2-positive breast cancer
treated with trastuzumab. A larger number of patients could have been included in
the study if all the HER2-positive breast cancer patients were included. We selected
to study metastatic disease only, however, since this provides a more homogenous
population and better possibilities to study treatment effects. Furthermore, our
institute has pioneered in trastuzumab discontinuation in responding patients,
therefore providing a unique patient cohort to study factors associated with long
trastuzumab-free periods.
ADCC is mediated mainly through NK and T cells (Clynes et al., 2000; Park
et al., 2010), which were both studied in our retrospective patient material. Previous
studies have shown NK cell infiltration and activation in tumors after the
administration of trastuzumab to be associated with good trastuzumab responses
(Arnould et al., 2006; Tian et al., 2017). We studied the role of NK cells in
pretreatment samples and saw only a minor infiltration of the cells in our tumor
samples. A higher number of NK cells showed a tendency towards a better
prognosis, but the NK cell infiltration may not be optimal for predicting the clinical
outcomes in this subset of patients. It would have been interesting to see whether
the NK cells were able to penetrate the tumor tissue after administration of
trastuzumab, but unfortunately only a few patients had post-treatment samples
available, and the analysis was not performed. Furthermore, treatment decisions are
made based on the available patient and tumor characteristics, and post-treatment
markers provide less information for the clinical decision making.
T cell infiltration has been studied in many cancer types, and in colorectal
cancer the Immunoscore characterizing the abundance and location of CD3 and
CD8 positive T cells has been shown to be a powerful prognostic parameter (Galon
et al., 2014; Galon et al., 2006; Pagès et al., 2018). At the time of our studies, tumor-
infiltrating lymphocytes in HER2-positive breast cancers were studied mainly
using a morphological evaluation of the immune cells or measuring the immune
cell infiltrations in the whole tumor area. We studied the quantity of CD3 and CD8
positive T cells in the center of the tumors (CT) and in the invasive margin (IM)
separately and saw that a high number of the center tumoral CD3 or CD8 positive
T cells were significantly associated with improved outcomes, with the latter being

79
a more powerful prognostic factor. These results highlight the important role of
cytotoxic T cells in cancer eradication and that T cells in the CT and IM might
reflect different types of host immunoreaction to the tumor, which requires further
investigation. During the past few years, more studies on HER2-positive breast
cancer have been examining specific immune cell subtypes in different tumor
locations (in the center of the tumor or in the stroma) in patients treated with
trastuzumab (Hou, Nitta, Wei, Banks, Parwani et al., 2018; Liu et al., 2017; Takada
et al., 2018; van Rooijen et al., 2018). These studies have also confirmed the
importance of cytotoxic T cells in the disease.
Tumor associated macrophages (TAMs) are known to have clinical impact in
the tumor microenvironment, and the ADCP induced by trastuzumab is mainly
mediated by TAMs (Shi et al., 2015). However, little is known about the importance
and prognostic role of macrophages in HER2-positive breast cancer. We first
studied the TAMs using the CD68 marker, which did not give any significant
association with survival, although a high number of CD68 in the CT showed a
tendency towards improved prognosis. The CD68 marker also recognizes other
types of immune cells, not just macrophages, and because it is now well known that
there is a continuum of different macrophage phenotypes shifting from antitumoral
M1-like to pro-tumoral M2-like phenotype (Biswas & Mantovani, 2010; Brown et
al., 2017), we continued our studies using two distinct markers. Our studies
revealed that a high number of M1 polarized macrophages in both IM and CT show
a strong association with better outcomes and are consistent with previous works
studying breast, colorectal, ovarian and gastric cancers (Biswas et al., 2013;
Fridman et al., 2017), whereas the high number of M2 polarized macrophages
showed a tendency towards a poorer prognosis. Thus, our study supports the idea
of targeting the macrophage polarization in HER2-positive breast cancer, but
further investigation is required.
The tumor cells themselves can suppress the functions of the immune cells
required for the proper function of trastuzumab and other monoclonal antibodies,
and our results showed that the low tumoral expression of CD47 and IDO1 are
associated with improved patient outcomes, which were inconsistent with other
reports. CD47 is a commonly known inhibitor of phagocytosis, whereas IDO1 is
associated with an increased M2/M1 macrophage ratio, both of which lead to
immunosuppression and further highlight the importance macrophage-mediated
phagocytosis in tumor eradication (Jaiswal et al., 2009; Wang, X et al., 2014). Anti-
CD47 antibodies are already in clinical trials and have shown promising effects on

80
lymphomas with a favorable safety profile (Advani et al., 2018), while the clinical
trials are still ongoing in solid cancer.
One study (Loi et al., 2014) has shown that adjuvant trastuzumab efficacy is
limited to patients with high TILs. We therefore studied whether our most
promising prognostic markers, high number of CD8+ T cells in the CT and high
number of M1-like macrophages in the IM and CT, could predict the benefit from
trastuzumab in a metastatic setting. This analysis was rather difficult, since the
patients of our retrospective material were treated with trastuzumab at varying
points of their disease. Nevertheless, we selected the treatment length of the 1st
trastuzumab-containing regimen in a metastatic setting (surrogate of progression-
free survival) and the length of trastuzumab interruption in response as indicators
for the treatment benefit, based on the evidence from randomized trials and our
oncologists’ personal experience. In contrast to the recommended clinical protocols,
our institute has aimed to discontinue trastuzumab treatment if a metastatic HER2+
breast cancer patient has achieved a prolonged radiological response, and we have
seen that these patients can experience long trastuzumab-free periods (median of
51 months) without progression (Moilanen et al., 2017). We were unable to see a
connection between the length of the 1st trastuzumab treatment and the number of
CD8 positive CT cells alone, whereas M1 macrophages in the CT showed a
significant association. However, we saw an even more remarkable association
when we combined these markers. When we studied the patients whose
trastuzumab treatment was interrupted, a high number of all the three markers (CD8
CT, M1 CT, M1 IM) were associated with the length of trastuzumab discontinuation,
and the strongest association was seen with a combined analysis of CD8 CT and
M1 CT or IM status. Our results suggest that trastuzumab interruption could be
feasible for patients with a high CD8 CT and M1 status and that CD8 CT combined
with M1 CT should be further studied in the context of trastuzumab efficacy.
Our results support the idea of using distinct immune cell types in specific
tumor locations as prognostic and predictive markers for HER2-positive breast
cancer. However, it should be noted that different cancer therapeutics can alter the
immune cell composition of the tumors, for example, in a favorable way by
increasing the influx of cytotoxic cells but also in an unfavorable way by increasing
the influx of immunosuppressive cells into the tumor (Arnould et al., 2006;
DeNardo et al., 2011). A recent study additionally revealed that ADCP, induced in
TAMs by trastuzumab treatment, increases the expression of PD-L1 and IDO1 in
TAMs leading to immunosuppression (Su et al., 2018). This is quite
counterintuitive when considering the high impact and well-established benefit of

81
monoclonal antibodies in cancer therapies and should definitely be further studied.
Studying the immune cell infiltrations in trastuzumab-treated patients can be
challenging, because trastuzumab is received in combination with multiple
chemotherapeutic agents, which in turn can induce immunogenic cell death and
modulate the composition of immune cell prolife (Casares et al., 2005; García-
Martínez et al., 2014; Obeid et al., 2007; Parikh et al., 2014; Takahashi, Sakakura,
Mito, Ida, & Chikamatsu, 2016). Nevertheless, in the era of personalized medicine,
we need to be able to identify the patients who will or will not benefit from a
specific treatment. Trastuzumab is given to every breast cancer patient with HER2
amplification, even though it cannot be guaranteed that they will really benefit from
the therapy. Since adjuvant trastuzumab received in localized disease is a predictor
of poor prognosis in a metastatic setting (Moilanen et al., 2017; Murthy et al., 2012;
Rier et al., 2017), suggesting that acquired resistance to trastuzumab has been
developed, additional predictive markers for the patient selection are required.
6.2 The role of HER2 and HER3 for cancer stem-like cells in ALK
translocated NSCLC
HER proteins have been linked to cancer stem-like cells (CSLCs) in many cancer
types. NRG1, a HER3/HER4 ligand, has also been shown to induce CSLC features
in breast cancer (Ithimakin et al., 2013; Jeong et al., 2014; Korkaya et al., 2008;
Lee et al., 2014). CSLCs, in turn, have been linked to radiation therapy,
chemotherapy, targeted therapy and immunotherapy resistances, causing cancer
recurrence. CSLCs can prevent the therapeutic effects of monoclonal antibodies,
such as trastuzumab, by secreting immunosuppressive cytokines and modifying the
immune responses (Creighton et al., 2009; Maccalli et al., 2017; McLendon et al.,
2006; Phillips et al., 2006; Reim et al., 2009; Wang et al., 2017). The molecular
mechanisms behind the CSLC acquire are quite unknown, even though some
signaling pathways, like wnt/β-catenin, TGF-β and the HER2, have been linked to
it (Anido et al., 2010; Korkaya et al., 2009; Takebe et al., 2011). Several preclinical
studies and clinical trials are studying potential cancer stem-like cell targeting
agents, but none have been approved for cancer therapy (Li et al., 2017). A Phase
II study with ovarian cancer patients treated with chemotherapy and metformin, a
suggested CSLC targeting agent, showed a decreased number of ALDH+ CSLCs in
the tumors and increased sensitivity of the tumors to cisplatin in vitro (Buckanovich
et al., 2017). However, more data on the clinical utility of CSLC targeting agents
are urgently needed for the development of more efficient cancer treatments.

82
We assessed the significance of HER2 and HER3 for CSLCs in ALK
translocated NSCLC in studies III and IV. We chose to study ALK translocated
NSCLC because this disease represents a subgroup of patients who are extremely
sensitive to ALK inhibition but will inevitably develop a resistance to the treatment
within 1–2 years, which is common for targeted therapies (Peters et al., 2017;
Solomon et al., 2014). In addition, we have previously shown that CSLCs can
mediate the ALK tyrosine kinase inhibitor (TKI) resistance in the disease, and
several reports have linked the activation of bypass signaling via HER receptors to
the ALK TKI resistance (Doebele et al., 2012; Jokinen et al., 2014; Katayama et al.,
2012; Sasaki et al., 2011). Our results highlighted the importance of HER2 and
HER3 for CSLC-mediated ALK TKI resistance. Overexpression of HER2 resulted
in more pronounced expression of CSLC markers in response to ALK TKI, whereas
the knockdown of HER2 or HER3 inhibited this TKI-induced expression of CSLC
markers. The strongest expression of CSLC markers was seen when HER3 was
activated by NRG1, suggesting that CSLC-driven ALK TKI resistance could be
mediated via HER3 signaling. Even though NRG1 also activates HER4, it wasn’t
enough to restore the CSLC marker expression when HER3 was knocked down.
Moreover, our results pointed towards functionality of HER2 and HER3 to cancer
stem-like properties assessed by colony and sphere formation. HER2 and HER3
activation led to increased colony and sphere formation abilities, whereas the
knockdowns downregulated these capabilities. Our results support the idea of
blocking HER2-HER3 signaling to target CSLCs in order to inhibit the tumor
progression after cancer treatments.
HER2-HER3 is an important dimer signaling through PI3K and MAPK
pathways to sustain survival and proliferation of the cells. The HER3 part of the
dimer is essential for the PI3K pathway, since it can directly bind PI3K with its
several phospho-tyrosine residues, and HER2-amplified cancers have been shown
to depend on the HER3 signaling (Garrett et al., 2013; Lee-Hoeflich et al., 2008;
Schulze et al., 2005). HER2 and HER3 alteration of our studies did affect the
signaling through PI3K and MAPK pathways, with a more sustainable impact on
the former one. The overexpression of HER2 was able to reactivate the Akt and
ERK1/2 signaling after long-term exposure to ALK inhibition. This reactivation of
the signaling pathways, especially the Akt signaling, was blocked by the
knockdown of HER2 or HER3. In ALK translocated NSCLC, the Akt and ERK1/2
signaling are mainly driven by the ALK, and the recovery of the signaling pathways
after long exposure to ALK TKI is not entirely known. Our results demonstrate the
importance of HER2 and HER3 in this signaling recovery. Additionally, the

83
signaling recovery, after a long exposure to ALK inhibitor, is likely to reflect a
CSLC acquire in comparison to acute signaling changes that mirror the cytotoxic
responses, further highlighting the role of HER2 and HER3 for the CSLC
phenotype. The exact mechanisms of how HER2 and HER3 are activated after ALK
inhibition are unknown. It might be that the suppression of HER2 and HER3 by
negative feedback pathways is simultaneously inhibited when the downstream
signaling of ALK is blocked. Akt, for example, has been shown to mediate
suppression of HER3, and inhibition of Akt leads to increased expression and
phosphorylation of HER3 (Chandarlapaty et al., 2011). Moreover, the HER2 and
HER3 might not be the only RTKs that are upregulated when ALK is inhibited, as
seen in previous studies (Doebele et al., 2012; Katayama et al., 2012), and this
could be further studied.
Studies by others have shown that inhibition of one HER family member leads
to compensatory activation of other HER family members (Engelman et al., 2007;
Ritter et al., 2007; Yonesaka et al., 2011), which could be a major problem when
targeting these receptors therapeutically. Our results showed that HER2
overexpression can orchestrate the other members of the protein family.
Knockdown of HER3 increased the activity of EGFR; however, it was not enough
to reactivate the Akt signaling blocked by the knockdown. Our studies suggest that
targeting HER2 together with HER3 could be feasible for more efficient killing of
the cancer stem cells.
6.3 Implications of the results and future perspectives
The results of these studies provide suggestions for more efficient use of anti-HER
targeting agents. Our results with metastatic HER2-positive breast cancer patients
suggest that patients with a favorable tumor immune profile (low tumor expression
of CD47 and IDO1, high number of tumor-infiltrating CD8+ T cells and M1 TAMs
in the CT) might be treated in a less intensive manner and remain progression-free
during trastuzumab interruption, while patients with non-favorable immune profile
could be candidates for experimental immunotherapeutic approaches and more
effective cancer therapies should be developed for them (Fig. 7). These hypotheses,
however, must be confirmed in larger prospective studies but, if applicable, this
improved patient selection could lead to increased response rates and economical
savings. Our future studies will investigate further the impact of cytotoxic T cells
and M1-like macrophages utilizing in vitro ADCC and ADCP model. Because the
main mechanisms of action of trastuzumab depends on the activation of the

84
immune system, evaluation of chemokine profile in the tumor microenvironment
and the peripheral blood could be an interesting future study on the subject. A recent
work showed that HER2 signaling can recruit tumor-infiltrating immune cells by
inducing CCL2, which was important for the function of trastuzumab (Triulzi et al.,
2019). Another interesting perspective could be to study the gut microbiota, which
is extremely important for the proper function of the immune system and has been
shown to affect the immunotherapy responses (Routy et al., 2018; Vétizou et al.,
2015).
Since HER2 targeting agents have already been developed and are currently in
clinical use, and HER3 targeting agents are constantly being developed, the HER2-
HER3 dependency of CSLC-driven resistance revealed in our studies supports the
idea of investigating anti-HER agents as stem-like cell targeting agents in different
cancer types. This is not the first time HER proteins have been linked to CSLCs,
but our studies further support their targeting in combination with other treatments
to improve patient outcomes (Fig. 8). Studies combining HER3 targeting or NRG1
inhibitory agents with HER2 targeting agents should be studied in vitro and in vivo
in the future, and the significance of HER2-HER3 dimer could also be studied in
other diseases as well.
6.4 Limitations of the study
Our studies had some limitations. The number of tumor samples was low in
studies I and II, and patients were retrospectively collected from a single academic
cancer center. Patients included in studies I and II were all treated with trastuzumab,
which made the evaluation of the predictive values more challenging. Additionally,
most of the immune cell infiltrations were studied using only a single marker;
however, well characterized and widely used markers were selected for the studies.
Studies III and IV were performed only for two different cell lines, and only in vitro
models were used due to a restricted availability of cell line models of ALK
translocated NSCLC. The results of these studies should be confirmed with in vivo
models, even though it will be challenging.
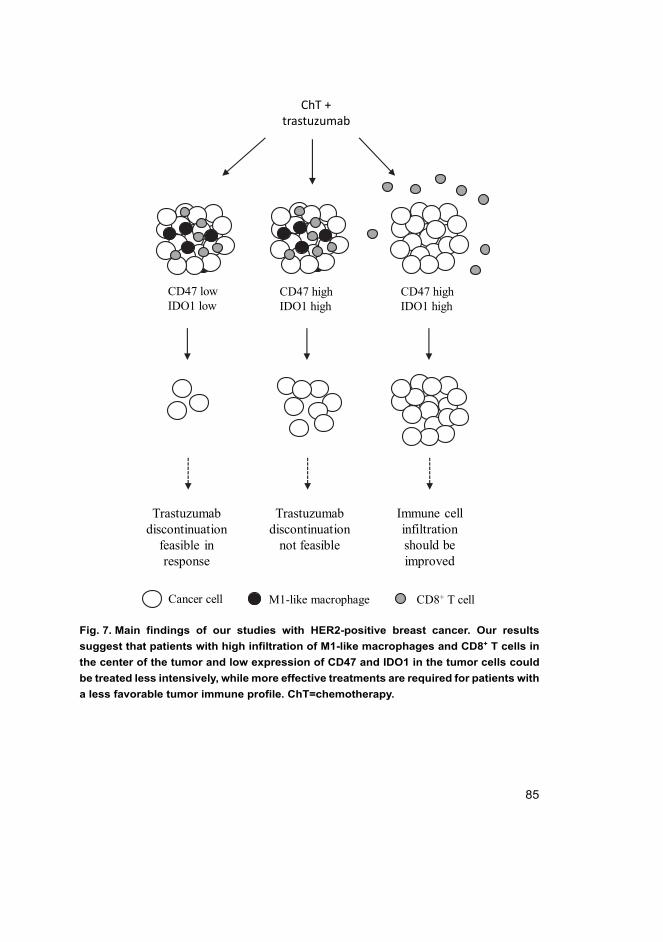
85
Fig. 7. Main findings of our studies with HER2-positive breast cancer. Our results
suggest that patients with high infiltration of M1-like macrophages and CD8+ T cells in
the center of the tumor and low expression of CD47 and IDO1 in the tumor cells could
be treated less intensively, while more effective treatments are required for patients with
a less favorable tumor immune profile. ChT=chemotherapy.
CD47 highIDO1 high
CD47 lowIDO1 low
CD47 highIDO1 high
ChT +trastuzumab
Trastuzumabdiscontinuation
feasible in response
Trastuzumabdiscontinuation
not feasible
Immune cellinfiltrationshould beimproved
Cancer cell M1-like macrophage CD8+ T cell

86
Fig. 8. Schematic representation of dual targeting of ALK and HER2-HER3 dimer. Our
results suggest that expression of HER2 and HER3 is linked to cancer stem-like cells,
which are causing resistance to ALK TKI therapy and cancer recurrence. Dual inhibition
of ALK and HER2-HER3 dimer could improve cancer cell killing.
ALK TKI
HER2
HER2HER3
HER3
ALK TKI+ anti-HER2/HER3
HER2
HER2
Stem-like cell
Cancer cell

87
7 Conclusions
The current study investigated the possibilities of using HER targeting agents more
efficiently by selecting the HER2-positive breast cancer patients more accurately
for the trastuzumab treatment and by utilizing already available targeting agents to
target cancer stem-like cells. The following conclusions were made based on the
results:
1. A high density of cytotoxic T cells and/or M1-like macrophages in the center
of the tumor (CT) in the pretreatment tumor samples predicts a more favorable
outcome in metastatic HER2-positive breast cancer. The most significant
prognostic value is seen with a combinatory analysis of cytotoxic T cells and
M1-like macrophages.
2. A high density of cytotoxic T cells and M1-like macrophages in the CT can
predict the disease progression and the possibility of interrupting trastuzumab
treatment in metastatic HER2-positive breast cancer. This suggests that
patients with a favorable immune profile could be treated less intensively and
that more efficient treatments should be developed for patients with a low
density of cytotoxic T cells and M1-like macrophages in the CT.
3. An intense expression of CD47 and IDO1 in the tumor cells of HER2-positive
breast cancer is associated with more adverse outcomes, whereas a low
expression predicts more favorable outcomes.
4. HER2 and HER3 have significant roles for the cancer stem-like cell phenotype
in ALK translocated non-small cell lung cancer.
5. ALK tyrosine kinase inhibitor resistance can be mediated by HER2-HER3-
dependent cancer stem-like cells in ALK translocated NSCLC suggesting that
targeting the dimer together with the ALK inhibitor could be beneficial for
overcoming ALK TKI resistances and improving cancer treatments.

88

89
References
Abràmoff, M. D., Magalhães, P. J., & Ram, S. J. (2004). Image processing with imageJ. Biophotonics International, 11(7), 36-41.
Advani, R., Flinn, I., Popplewell, L., Forero, A., Bartlett, N. L., Ghosh, N., . . . Smith, S. M. (2018). CD47 blockade by Hu5F9-G4 and rituximab in non-hodgkin's lymphoma. The New England Journal of Medicine, 379(18), 1711-1721. doi:10.1056/NEJMoa1807315
Aggarwal, B. B., Vijayalekshmi, R. V., & Sung, B. (2009). Targeting inflammatory pathways for prevention and therapy of cancer: Short-term friend, long-term foe. Clinical Cancer Research, 15(2), 425-430. doi:10.1158/1078-0432.CCR-08-0149
Agus, D. B., Akita, R. W., Fox, W. D., Lewis, G. D., Higgins, B., Pisacane, P. I., . . . Sliwkowski, M. X. (2002). Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell, 2(2), 127-137.
Al-Hajj, M., Wicha, M. S., Benito-Hernandez, A., Morrison, S. J., & Clarke, M. F. (2003). Prospective identification of tumorigenic breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America, 100(7), 3983-8. doi:10.1073/pnas.0530291100
Andrechek, E. R., Hardy, W. R., Siegel, P. M., Rudnicki, M. A., Cardiff, R. D., & Muller, W. J. (2000). Amplification of the neu/erbB-2 oncogene in a mouse model of mammary tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America, 97(7), 3444-3449. doi:10.1073/pnas.050408497
Andrulis, I. L., Bull, S. B., Blackstein, M. E., Sutherland, D., Mak, C., Sidlofsky, S., . . . McCready, D. (1998). Neu/erbB-2 amplification identifies a poor-prognosis group of women with node-negative breast cancer. toronto breast cancer study group. Journal of Clinical Oncology, 16(4), 1340-1349. doi:10.1200/JCO.1998.16.4.1340
Anido, J., Sáez-Borderías, A., Gonzàlez-Juncà, A., Rodón, L., Folch, G., Carmona, M. A., . . . Seoane, J. (2010). TGF-β receptor inhibitors target the CD44(high)/Id1(high) glioma-initiating cell population in human glioblastoma. Cancer Cell, 18(6), 655-668. doi:10. 1016/j.ccr.2010.10.023
Arnould, L., Gelly, M., Penault-Llorca, F., Benoit, L., Bonnetain, F., Migeon, C., . . . Coudert, B. (2006). Trastuzumab-based treatment of HER2-positive breast cancer: An antibody-dependent cellular cytotoxicity mechanism? British Journal of Cancer, 94(2), 259-267. doi:10.1038/sj.bjc.6602930
Artavanis-Tsakonas, S., Rand, M. D., & Lake, R. J. (1999). Notch signaling: Cell fate control and signal integration in development. Science, 284(5415), 770-776. doi:10.1126/ science.284.5415.770
Arteaga, C. L., & Engelman, J. A. (2014). ERBB receptors: From oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell, 25(3), 282-303. doi:10.1016/j.ccr.2014.02.025
Arteaga, C. L., Sliwkowski, M. X., Osborne, C. K., Perez, E. A., Puglisi, F., & Gianni, L. (2011). Treatment of HER2-positive breast cancer: Current status and future perspectives. Nature Reviews. Clinical Oncology, 9(1), 16-32. doi:10.1038/nrclinonc. 2011.177

90
Bang, Y., Van Cutsem, E., Feyereislova, A., Chung, H. C., Shen, L., Sawaki, A., . . . Kang, Y. (2010). Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet, 376(9742), 687-697. doi:10.1016/S0140-6736(10)61121-X
Barok, M., Joensuu, H., & Isola, J. (2014). Trastuzumab emtansine: Mechanisms of action and drug resistance. Breast Cancer Research, 16(2), 209. doi:10.1186/bcr3621
Baselga, J., Cortés, J., Kim, S., Im, S., Hegg, R., Im, Y., . . . Swain, S. M. (2012). Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. The New England Journal of Medicine, 366(2), 109-119. doi:10.1056/NEJMoa1113216
Beier, D., Wischhusen, J., Dietmaier, W., Hau, P., Proescholdt, M., Brawanski, A., . . . Beier, C. P. (2008). CD133 expression and cancer stem cells predict prognosis in high-grade oligodendroglial tumors. Brain Pathology, 18(3), 370-377. doi:10.1111/j.1750-3639. 2008.00130.x
Bergers, G., & Benjamin, L. E. (2003). Tumorigenesis and the angiogenic switch. Nature Reviews Cancer, 3(6), 401-410. doi:10.1038/nrc1093
Bhowmick, N. A., Neilson, E. G., & Moses, H. L. (2004). Stromal fibroblasts in cancer initiation and progression. Nature, 432(7015), 332-337. doi:10.1038/nature03096
Bidlingmaier, S., Zhu, X., & Liu, B. (2008). The utility and limitations of glycosylated human CD133 epitopes in defining cancer stem cells. Journal of Molecular Medicine, 86(9), 1025-1032. doi:10.1007/s00109-008-0357-8
Birkman, E., Ålgars, A., Lintunen, M., Ristamäki, R., Sundström, J., & Carpén, O. (2016). EGFR gene amplification is relatively common and associates with outcome in intestinal adenocarcinoma of the stomach, gastro-oesophageal junction and distal oesophagus. BMC Cancer, 16, 406. doi:10.1186/s12885-016-2456-1
Biswas, S. K., Allavena, P., & Mantovani, A. (2013). Tumor-associated macrophages: Functional diversity, clinical significance, and open questions. Seminars in Immunopathology, 35(5), 585-600. doi:10.1007/s00281-013-0367-7
Biswas, S. K., & Mantovani, A. (2010). Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nature Immunology, 11(10), 889-896. doi:10.1038/ni.1937
Bonnet, D., & Dick, J. E. (1997). Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nature Medicine, 3(7), 730-737. doi:10.1038/nm0797-730
Bos, J. L. (1989). Ras oncogenes in human cancer: A review. Cancer Research, 49(17), 4682-4689.
Bose, R., Kavuri, S. M., Searleman, A. C., Shen, W., Shen, D., Koboldt, D. C., . . . Ellis, M. J. (2013). Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discovery, 3(2), 224-237. doi:10.1158/2159-8290.CD-12-0349
Brabender, J., Danenberg, K. D., Metzger, R., Schneider, P. M., Park, J., Salonga, D., . . . Danenberg, P. V. (2001). Epidermal growth factor receptor and HER2-neu mRNA expression in non-small cell lung cancer is correlated with survival. Clinical Cancer Research, 7(7), 1850-1855.

91
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., & Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer Journal for Clinicians, 68(6), 394-424. doi:10.3322/caac.21492
Brown, J. M., Recht, L., & Strober, S. (2017). The promise of targeting macrophages in cancer therapy. Clinical Cancer Research, 23(13), 3241-3250. doi:10.1158/1078-0432.CCR-16-3122
Bruno, A., Ferlazzo, G., Albini, A., & Noonan, D. M. (2014). A think tank of TINK/TANKs: Tumor-infiltrating/tumor-associated natural killer cells in tumor progression and angiogenesis. Journal of the National Cancer Institute, 106(8) doi:10.1093/jnci/dju200
Buckanovich, R. J., Brown, J., Shank, J., Griffith, K. A., Reynolds, R. K., Johnston, C., . . . Mehta, G. (2017). A phase II clinical trial of metformin as a cancer stem cell targeting agent in stage IIc/III/IV ovarian, fallopian tube, and primary peritoneal cancer. Journal of Clinical Oncology, 35(15), 5556. doi:10.1200/JCO.2017.35.15_suppl.5556
Cantini, L., Pistelli, M., Savini, A., Bastianelli, L., Della Mora, A., Merloni, F., . . . Berardi, R. (2018). Long-responders to anti-HER2 therapies: A case report and review of the literature. Molecular and Clinical Oncology, 8(1), 147-152. doi:10.3892/mco.2017. 1495
Cao, G., Chen, K., Xiong, M., & Chen, B. (2016). HER3, but not HER4, plays an essential role in the clinicopathology and prognosis of gastric cancer: A meta-analysis. PloS One, 11(8), e0161219. doi:10.1371/journal.pone.0161219
Cao, Z., Gao, Q., Fu, M., Ni, N., Pei, Y., & Ou, W. B. (2019). Anaplastic lymphoma kinase fusions: Roles in cancer and therapeutic perspectives. Oncology Letters, 17(2), 2020-2030.
Cappuzzo, F., Varella-Garcia, M., Shigematsu, H., Domenichini, I., Bartolini, S., Ceresoli, G. L., . . . Hirsch, F. R. (2005). Increased HER2 gene copy number is associated with response to gefitinib therapy in epidermal growth factor receptor-positive non-small-cell lung cancer patients. Journal of Clinical Oncology, 23(22), 5007-5018. doi:10.1200 /JCO.2005.09.111
Cardoso, F., Senkus, E., Costa, A., Papadopoulos, E., Aapro, M., André, F., . . . Winer, E. P. (2018). 4th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 4)†. Annals of Oncology, 29(8), 1634-1657. doi:10.1093/annonc/mdy192
Carey, K. D., Garton, A. J., Romero, M. S., Kahler, J., Thomson, S., Ross, S., . . . Sliwkowski, M. X. (2006). Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Research, 66(16), 8163-8171. doi:10.1158/0008-5472.CAN-06-0453
Carey, L. A., Dees, E. C., Sawyer, L., Gatti, L., Moore, D. T., Collichio, F., . . . Perou, C. M. (2007). The triple negative paradox: Primary tumor chemosensitivity of breast cancer subtypes. Clinical Cancer Research, 13(8), 2329-2334. doi:10.1158/1078-0432.CCR-06-1109

92
Carpentino, J. E., Hynes, M. J., Appelman, H. D., Zheng, T., Steindler, D. A., Scott, E. W., & Huang, E. H. (2009). Aldehyde dehydrogenase-expressing colon stem cells contribute to tumorigenesis in the transition from colitis to cancer. Cancer Research, 69(20), 8208-8215. doi:10.1158/0008-5472.CAN-09-1132
Carracedo, A., Ma, L., Teruya-Feldstein, J., Rojo, F., Salmena, L., Alimonti, A., . . . Pandolfi, P. P. (2008). Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. The Journal of Clinical Investigation, 118(9), 3065-3074. doi:10.1172/JCI34739
Casares, N., Pequignot, M. O., Tesniere, A., Ghiringhelli, F., Roux, S., Chaput, N., . . . Kroemer, G. (2005). Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. The Journal of Experimental Medicine, 202(12), 1691-1701. doi:10.1084/jem.20050915
Cavallaro, U., & Christofori, G. (2004). Cell adhesion and signalling by cadherins and ig-CAMs in cancer. Nature Reviews Cancer, 4(2), 118-132. doi:10.1038/nrc1276
Chaffer, C. L., Brueckmann, I., Scheel, C., Kaestli, A. J., Wiggins, P. A., Rodrigues, L. O., . . . Weinberg, R. A. (2011). Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proceedings of the National Academy of Sciences of the United States of America, 108(19), 7950-7955. doi:10.1073/pnas.1102454108
Chandarlapaty, S., Sawai, A., Scaltriti, M., Rodrik-Outmezguine, V., Grbovic-Huezo, O., Serra, V., . . . Rosen, N. (2011). AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell, 19(1), 58-71. doi:10.1016 /j.ccr.2010.10.031
Charafe-Jauffret, E., Ginestier, C., Iovino, F., Tarpin, C., Diebel, M., Esterni, B., . . . Wicha, M. S. (2010). Aldehyde dehydrogenase 1–Positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clinical Cancer Research, 16(1), 45-55. doi:10.1158/1078-0432.CCR-09-1630
Cheang, M. C. U., Chia, S. K., Voduc, D., Gao, D., Leung, S., Snider, J., . . . Nielsen, T. O. (2009). Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. Journal of the National Cancer Institute, 101(10), 736-750. doi:10.1093/jnci/djp082
Chen, D. S., & Mellman, I. (2017). Elements of cancer immunity and the cancer-immune set point. Nature, 541(7637), 321-330. doi:10.1038/nature21349
Chiou, S., Yu, C., Huang, C., Lin, S., Liu, C., Tsai, T., . . . Lo, J. (2008). Positive correlations of oct-4 and nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clinical Cancer Research, 14(13), 4085-4095. doi:10.1158/1078-0432. CCR-07-4404
Cho, H. S., Mason, K., Ramyar, K. X., Stanley, A. M., Gabelli, S. B., Denney, D. J., & Leahy, D. (2003). Structure of the extracellular region of HER2 alone and in complex with the herceptin fab. Nature, 421(6924), 756-760. doi:10.1038/nature01392
Cioffi, M., D'Alterio, C., Camerlingo, R., Tirino, V., Consales, C., Riccio, A., . . . Scala, S. (2015). Identification of a distinct population of CD133 + CXCR4 + cancer stem cells in ovarian cancer. Scientific Reports, 5(1), 10357. doi:10.1038/srep10357
Clevers, H. (2006). Wnt/beta-catenin signaling in development and disease. Cell, 127(3), 469-80. doi:10.1016/j.cell.2006.10.018

93
Clynes, R. A., Towers, T. L., Presta, L. G., & Ravetch, J. V. (2000). Inhibitory fc receptors modulate in vivo cytotoxicity against tumor targets. Nature Medicine, 6(4), 443-446. doi:10.1038/74704
Coffelt, S. B., Wellenstein, M. D., & De Visser, K. E. (2016). Neutrophils in cancer: Neutral no more. Nature Reviews Cancer, 16(7), 431-446. doi:10.1038/nrc.2016.52
Cortet, M., Bertaut, A., Molinié, F., Bara, S., Beltjens, F., Coutant, C., & Arveux, P. (2018). Trends in molecular subtypes of breast cancer: Description of incidence rates between 2007 and 2012 from three french registries. BMC Cancer, 18(1), 161. doi:10.1186/ s12885-018-4080-8
Costa, D. B., Kobayashi, S., Pandya, S. S., Yeo, W., Shen, Z., Tan, W., & Wilner, K. D. (2011). CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. Journal of Clinical Oncology, 29(15), 443. doi:10.1200/JCO.2010.34.1313
Creighton, C. J., Li, X., Landis, M., Dixon, J. M., Neumeister, V. M., Sjolund, A., . . . Chang, J. C. (2009). Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proceedings of the National Academy of Sciences of the United States of America, 106(33), 13820-13825. doi:10.1073/pnas.0905718106
Croce, C. M. (2008). Oncogenes and cancer. New England Journal of Medicine, 358(5), 502-511. doi:10.1056/NEJMra072367
Cross, D. A. E., Ashton, S. E., Ghiorghiu, S., Eberlein, C., Nebhan, C. A., Spitzler, P. J., . . . Pao, W. (2014). AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discovery, 4(9), 1046-1061. doi:10.1158/2159-8290.CD-14-0337
Davies, H., Bignell, G. R., Cox, C., Stephens, P., Edkins, S., Clegg, S., . . . Futreal, P. A. (2002). Mutations of the BRAF gene in human cancer. Nature, 417(6892), 949-954. doi:10.1038/nature00766
Day, K. C., Hiles, G. L., Kozminsky, M., Dawsey, S. J., Paul, A., Broses, L. J., . . . Day, M. L. (2017). HER2 and EGFR overexpression support metastatic progression of prostate cancer to bone. Cancer Research, 77(1), 74-85. doi:10.1158/0008-5472.CAN-16-1656
De Visser, K. E., Eichten, A., & Coussens, L. M. (2006). Paradoxical roles of the immune system during cancer development. Nature Reviews Cancer, 6(1), 24-37. doi:10.1038/ nrc1782
DeBerardinis, R. J., Lum, J. J., Hatzivassiliou, G., & Thompson, C. B. (2008). The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metabolism, 7(1), 11-20. doi:10.1016/j.cmet.2007.10.002
Della Chiesa, M., Carlomagno, S., Frumento, G., Balsamo, M., Cantoni, C., Conte, R., . . . Vitale, M. (2006). The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood, 108(13), 4118-4125. doi:10.1182/blood-2006-03-006700
DeNardo, D. G., Brennan, D. J., Rexhepaj, E., Ruffell, B., Shiao, S. L., Madden, S. F., . . . Coussens, L. M. (2011). Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discovery, 1(1), 54-67. doi:10.1158/2159-8274.CD-10-0028

94
Denkert, C., von Minckwitz, G., Brase, J. C., Sinn, B. V., Gade, S., Kronenwett, R., . . . Loibl, S. (2015). Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. Journal of Clinical Oncology, 33(9), 983-991. doi:10.1200/JCO.2014.58.1967
Denkert, C., von Minckwitz, G., Darb-Esfahani, S., Lederer, B., Heppner, B. I., Weber, K. E., . . . Loibl, S. (2018). Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: A pooled analysis of 3771 patients treated with neoadjuvant therapy. The Lancet. Oncology, 19(1), 40-50. doi:10.1016/S1470-2045(17)30904-X
Doebele, R. C., Pilling, A. B., Aisner, D. L., Kutateladze, T. G., Le, A. T., Weickhardt, A. J., . . . Camidge, D. R. (2012). Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clinical Cancer Research, 18(5), 1472-1482. doi:10.1158/1078-0432.CCR-11-2906
Dontu, G., Jackson, K. W., McNicholas, E., Kawamura, M. J., Abdallah, W. M., & Wicha, M. S. (2004). Role of notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Research, 6(6), R605–R615. doi:10.1186/bcr920
Drayton, D. L., Liao, S., Mounzer, R. H., & Ruddle, N. H. (2006). Lymphoid organ development: From ontogeny to neogenesis. Nature Immunology, 7(4), 344-353. doi:10.1038/ni1330
Elster, N., Collins, D. M., Toomey, S., Crown, J., Eustace, A. J., & Hennessy, B. T. (2015). HER2-family signalling mechanisms, clinical implications and targeting in breast cancer. Breast Cancer Research and Treatment, 149(1), 5-15. doi:10.1007/s10549-014-3250-x
Engelman, J. A., Zejnullahu, K., Mitsudomi, T., Song, Y., Hyland, C., Park, J. O., . . . Jänne, P. A. (2007). MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science, 316(5827), 1039-1043. doi:10.1126/science. 1141478
Esteller, M. (2008). Epigenetics in cancer. New England Journal of Medicine, 358(11), 1148-1159. doi:10.1056/NEJMra072067
Ettinger, D. S., Wood, D. E., Aisner, D. L., Akerley, W., Bauman, J., Chirieac, L. R., . . . Hughes, M. (2017). Non-small cell lung cancer, version 5.2017, NCCN clinical practice guidelines in oncology. Journal of the National Comprehensive Cancer Network, 15(4), 504-535.
Eun, K., Ham, S. W., & Kim, H. (2017). Cancer stem cell heterogeneity: Origin and new perspectives on CSC targeting. BMB Reports, 50(3), 117-125. doi:10.5483/BMBRep. 2017.50.3.222
Feinberg, A. P., & Vogelstein, B. (1983). Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature, 301(5895), 89-92. doi:10.1038/ 301089a0
Feldinger, K., & Kong, A. (2015). Profile of neratinib and its potential in the treatment of breast cancer. Breast Cancer, 7, 147-162. doi:10.2147/BCTT.S54414
Fernald, K., & Kurokawa, M. (2013). Evading apoptosis in cancer. Trends in Cell Biology, 23(12), 620-633. doi:10.1016/j.tcb.2013.07.006

95
Finkle, D., Quan, Z. R., Asghari, V., Kloss, J., Ghaboosi, N., Mai, E., . . . Erickson, S. (2004). HER2-targeted therapy reduces incidence and progression of midlife mammary tumors in female murine mammary tumor virus huHER2-transgenic mice. Clinical Cancer Research, 10(7), 2499-2511.
Finnish Breast Cancer Group. (2019). Rintasyövän valtakunnallinen diagnostiikka- ja hoitosuositus. Retrieved from https://rintasyoparyhma.yhdistysavain.fi/hoitosuositus/
Fleischmann, A., Schlomm, T., Köllermann, J., Sekulic, N., Huland, H., Mirlacher, M., . . . Erbersdobler, A. (2009). Immunological microenvironment in prostate cancer: High mast cell densities are associated with favorable tumor characteristics and good prognosis. Prostate, 69(9), 976-981. doi:10.1002/pros.20948
Flynn, N. J., Somasundaram, R., Arnold, K. M., & Sims-Mourtada, J. (2017). The multifaceted roles of B cells in solid tumors: Emerging treatment opportunities. Targeted Oncology, 12(2), 139-152. doi:10.1007/s11523-017-0481-x
Freeman, G. J., Long, A. J., Iwai, Y., Bourque, K., Chernova, T., Nishimura, H., . . . Honjo, T. (2000). Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. Journal of Experimental Medicine, 192(7), 1027-1034. doi:10.1084/jem.192.7.1027
Fridman, W. H., Pagès, F., Saut`s-Fridman, C., & Galon, J. (2012). The immune contexture in human tumours: Impact on clinical outcome. Nature Reviews Cancer, 12(4), 298-306. doi:10.1038/nrc3245
Fridman, W. H., Zitvogel, L., Sautès-Fridman, C., & Kroemer, G. (2017). The immune contexture in cancer prognosis and treatment. Nature Reviews Clinical Oncology, 14(12), 717-734. doi:10.1038/nrclinonc.2017.101
Gabrilovich, D. I., & Nagaraj, S. (2009). Myeloid-derived suppressor cells as regulators of the immune system. Nature Reviews Immunology, 9(3), 162-174. doi:10.1038/nri2506
Gagliato, D. M., Jardim, D. L. F., Marchesi, M. S. P., & Hortobagyi, G. N. (2016). Mechanisms of resistance and sensitivity to anti-HER2 therapies in HER2+ breast cancer. Oncotarget, 7(39), 64431-64446. doi:10.18632/oncotarget.7043
Gainor, J. F., Dardaei, L., Yoda, S., Friboulet, L., Leshchiner, I., Katayama, R., . . . Shaw, A. T. (2016). Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discovery, 6(10), 1118-1133. doi:10.1158/2159-8290.CD-16-0596
Galdiero, M. R., Bianchi, P., Grizzi, F., Di Caro, G., Basso, G., Ponzetta, A., . . . Jaillon, S. (2016). Occurrence and significance of tumor-associated neutrophils in patients with colorectal cancer. International Journal of Cancer, 139(2), 446-456. doi:10.1002/ijc. 30076
Galli, S. J., Grimbaldeston, M., & Tsai, M. (2008). Immunomodulatory mast cells: Negative, as well as positive, regulators of immunity. Nature Reviews Immunology, 8(6), 478-486. doi:10.1038/nri2327
Galon, J., Mlecnik, B., Bindea, G., Angell, H. K., Berger, A., Lagorce, C., . . . Pages, F. (2014). Towards the introduction of the 'immunoscore' in the classification of malignant tumours. Journal of Pathology, 232(2), 199-209. doi:10.1002/path.4287

96
Galon, J., Costes, A., Sanchez-Cabo, F., Kirilovsky, A., Mlecnik, B., Lagorce-Pages, C., . . . Pages, F. (2006). Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science, 313(5795), 1960-1964. doi:10.1126/science. 1129139
García-Martínez, E., Gil, G. L., Benito, A. C., González-Billalabeitia, E., Conesa, M. A. V., García García, T., . . . Ayala de la Peña, Francisco. (2014). Tumor-infiltrating immune cell profiles and their change after neoadjuvant chemotherapy predict response and prognosis of breast cancer. Breast Cancer Research, 16(6), 488. doi:10.1186/s13058-014-0488-5
Garrett, J. T., & Arteaga, C. L. (2011). Resistance to HER2-directed antibodies and tyrosine kinase inhibitors: Mechanisms and clinical implications. Cancer Biology & Therapy, 11(9), 793-800.
Garrett, J. T., Sutton, C. R., Kurupi, R., Bialucha, C. U., Ettenberg, S. A., Collins, S. D., . . . Arteaga, C. L. (2013). Combination of antibody that inhibits ligand-independent HER3 dimerization and a p110α inhibitor potently blocks PI3K signaling and growth of HER2+ breast cancers. Cancer Research, 73(19), 6013-6023. doi:10.1158/0008-5472. CAN-13-1191
Gennari, R., Menard, S., Fagnoni, F., Ponchio, L., Scelsi, M., Tagliabue, E., . . . Costa, A. (2004). Pilot study of the mechanism of action of preoperative trastuzumab in patients with primary operable breast tumors overexpressing HER2. Clinical Cancer Research, 10(17), 5650-5655. doi:10.1158/1078-0432.CCR-04-0225
Geyer, C. E., Forster, J., Lindquist, D., Chan, S., Romieu, C. G., Pienkowski, T., . . . Cameron, D. (2006). Lapatinib plus capecitabine for HER2-positive advanced breast cancer. The New England Journal of Medicine, 355(26), 2733-2743. doi:10.1056/ NEJMoa064320
Ghiringhelli, F., Ménard, C., Terme, M., Flament, C., Taieb, J., Chaput, N., . . . Zitvogel, L. (2005). CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-ß-dependent manner. Journal of Experimental Medicine, 202(8), 1075-1085. doi:10.1084/jem.20051511
Ghosh, R., Narasanna, A., Wang, S. E., Liu, S., Chakrabarty, A., Balko, J. M., . . . Arteaga, C. L. (2011). Trastuzumab has preferential activity against breast cancers driven by HER2 homodimers. Cancer Research, 71(5), 1871-1882. doi:10.1158/0008-5472. CAN-10-1872
Gianni, L., Pienkowski, T., Im, Y., Roman, L., Tseng, L., Liu, M., . . . Valagussa, P. (2012). Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): A randomised multicentre, open-label, phase 2 trial. The Lancet. Oncology, 13(1), 25-32. doi:10.1016/S1470-2045(11)70336-9
Ginestier, C., Hur, M. H., Charafe-Jauffret, E., Monville, F., Dutcher, J., Brown, M., . . . Dontu, G. (2007). ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell, 1(5), 555-567. doi:10.1016/j.stem.2007.08.014

97
Goldhirsch, A., Gelber, R. D., Piccart-Gebhart, M. J., de Azambuja, E., Procter, M., Suter, T. M., . . . Baselga, J. (2013). 2 years versus 1 year of adjuvant trastuzumab for HER2-positive breast cancer (HERA): An open-label, randomised controlled trial. Lancet, 382(9897), 1021-1028. doi:10.1016/S0140-6736(13)61094-6
Gonzalez-Angulo, A. M., Litton, J. K., Broglio, K. R., Meric-Bernstam, F., Rakkhit, R., Cardoso, F., . . . Hortobagyi, G. N. (2009). High risk of recurrence for patients with breast cancer who have human epidermal growth factor receptor 2-positive, node-negative tumors 1 cm or smaller. Journal of Clinical Oncology, 27(34), 5700-5706. doi:10.1200/JCO.2009.23.2025
Grande, E., Bolós, M., & Arriola, E. (2011). Targeting oncogenic ALK: A promising strategy for cancer treatment. Molecular Cancer Therapeutics, 10(4), 569-579. doi:10.1158/1535-7163.MCT-10-0615
Granot, Z., Henke, E., Comen, E., King, T., Norton, L., & Benezra, R. (2011). Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell, 20(3), 300-314. doi:10.1016/j.ccr.2011.08.012
Greenwald, R. J., Freeman, G. J., & Sharpe, A. H. (2005). The B7 family revisited. Annual Review of Immunology, 23, 515-48. doi:10.1146/annurev.immunol.23.021704.115611
Greger, V., Passarge, E., Höpping, W., Messmer, E., & Horsthemke, B. (1989). Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Human Genetics, 83(2), 155-158. doi:10.1007/BF00286709
Grigoryan, T., Wend, P., Klaus, A., & Birchmeier, W. (2008). Deciphering the function of canonical wnt signals in development and disease: Conditional loss- and gain-of-function mutations of beta-catenin in mice. Genes & Development, 22(17), 2308. doi:10.1101/gad.1686208
Grivennikov, S. I., Greten, F. R., & Karin, M. (2010). Immunity, inflammation, and cancer. Cell, 140(6), 883-899. doi:10.1016/j.cell.2010.01.025
Gupta, P., Fillmore, C., Jiang, G., Shapira, S., Tao, K., Kuperwasser, C., & Lander, E. (2011). Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell, 146(4), 633-644. doi:10.1016/j.cell.2011.07.026
Hall, J. M., Lee, M. K., Newman, B., Morrow, J. E., Anderson, L. A., Huey, B., & King, M. -. (1990). Linkage of early-onset familial breast cancer to chromosome 17q21. Science, 250(4988), 1684-1689. doi:10.1126/science.2270482
Hanahan, D., & Weinberg, R. A. (2011). Hallmarks of cancer: The next generation. Cell, 144(5), 646-674. doi:10.1016/j.cell.2011.02.013
Hanker, A. B., Brewer, M. R., Sheehan, J. H., Koch, J. P., Sliwoski, G. R., Nagy, R., . . . Arteaga, C. L. (2017). An acquired HER2T798I gatekeeper mutation induces resistance to neratinib in a patient with HER2 mutant-driven breast cancer. Cancer Discovery, 7(6), 575-585. doi:10.1158/2159-8290.CD-16-1431
Hegde, G. V., de la Cruz, Cecile C., Chiu, C., Alag, N., Schaefer, G., Crocker, L., . . . Jackson, E. L. (2013). Blocking NRG1 and other ligand-mediated Her4 signaling enhances the magnitude and duration of the chemotherapeutic response of non-small cell lung cancer. Science Translational Medicine, 5(171), 171ra18. doi:10.1126/scitranslmed.3004438

98
Hermans, C., Anz, D., Engel, J., Kirchner, T., Endres, S., & Mayr, D. (2014). Analysis of FoxP3+ T-regulatory cells and CD8+T-cells in ovarian carcinoma: Location and tumor infiltration patterns are key prognostic markers: e111757. PLoS ONE, 9(11) doi:10. 1371/journal.pone.0111757
Hollmén, M., Määttä, J. A., Bald, L., Sliwkowski, M. X., & Elenius, K. (2009). Suppression of breast cancer cell growth by a monoclonal antibody targeting cleavable ErbB4 isoforms. Oncogene, 28(10), 1309-1319. doi:10.1038/onc.2008.481
Horn, L., Gettinger, S., Camidge, D. R., Smit, E. F., Janjigian, Y. Y., Miller, V. A., . . . Groen, H. J. M. (2017). Continued use of afatinib with the addition of cetuximab after progression on afatinib in patients with EGFR mutation-positive non-small-cell lung cancer and acquired resistance to gefitinib or erlotinib. Lung Cancer, 113, 51-58. doi:10.1016/j.lungcan.2017.08.014
Hou, Y., Nitta, H., Wei, L., Banks, P. M., Lustberg, M., Wesolowski, R., . . . Li, Z. (2018). PD-L1 expression and CD8-positive T cells are associated with favorable survival in HER2-positive invasive breast cancer. The Breast Journal, 24(6), 911-919. doi:10. 1111/tbj.13112
Hou, Y., Nitta, H., Wei, L., Banks, P. M., Parwani, A. V., & Li, Z. (2018). Evaluation of immune reaction and PD-L1 expression using multiplex immunohistochemistry in HER2-positive breast cancer: The association with response to anti-HER2 neoadjuvant therapy. Clinical Breast Cancer, 18(2), e23-e244. doi:10.1016/j.clbc.2017.11.001
Hu, J., Yang, D., Zhang, H., Liu, W., Zhao, Y., Lu, H., . . . Cai, L. (2015). USP22 promotes tumor progression and induces epithelial-mesenchymal transition in lung adenocarcinoma. Lung Cancer, 88(3), 239-245. doi:10.1016/j.lungcan.2015.02.019
Hubbard, S. R., & Miller, W. T. (2007). Receptor tyrosine kinases: Mechanisms of activation and signaling. Current Opinion in Cell Biology, 19(2), 117-23. doi:10.1016/j.ceb. 2007.02.010
Hynes, N. E., & Lane, H. A. (2005). ERBB receptors and cancer: The complexity of targeted inhibitors. Nature Reviews Cancer, 5(5), 341. doi:10.1038/nrc1609
Hynes, N. E., & MacDonald, G. (2009). ErbB receptors and signaling pathways in cancer. Current Opinion in Cell Biology, 21(2), 177-184. doi:10.1016/j.ceb.2008.12.010
Ingham, P. W., & McMahon, A. P. (2001). Hedgehog signaling in animal development: Paradigms and principles. Genes & Development, 15(23), 3059-3087. doi:10.1101/ gad.938601
Ingold Heppner, B., Untch, M., Denkert, C., Pfitzner, B. M., Lederer, B., Schmitt, W., . . . Loibl, S. (2016). Tumor-infiltrating lymphocytes: A predictive and prognostic biomarker in neoadjuvant-treated HER2-positive breast cancer. Clinical Cancer Research, 22(23), 5747-5754. doi:10.1158/1078-0432.CCR-15-2338
Inoshima, N., Nakanishi, Y., Minami, T., Izumi, M., Takayama, K., Yoshino, I., & Hara, N. (2002). The influence of dendritic cell infiltration and vascular endothelial growth factor expression on the prognosis of non-small cell lung cancer. Clinical Cancer Research, 8(11), 3480-3486.

99
Ithimakin, S., Day, K. C., Malik, F., Zen, Q., Dawsey, S. J., Bersano-Begey, T. F., . . . Wicha, M. S. (2013). HER2 drives luminal breast cancer stem cells in the absence of HER2 amplification: Implications for efficacy of adjuvant trastuzumab. Cancer Research, 73(5), 1635-1646. doi:10.1158/0008-5472.CAN-12-3349
Iwahara, T., Fujimoto, J., Wen, D., Cupples, R., Bucay, N., Arakawa, T., . . . Yamamoto, T. (1997). Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene, 14(4), 439-49.
Jaiswal, B. S., Kljavin, N. M., Stawiski, E. W., Chan, E., Parikh, C., Durinck, S., . . . Seshagiri, S. (2013). Oncogenic ERBB3 mutations in human cancers. Cancer Cell, 23(5), 603-617. doi:10.1016/j.ccr.2013.04.012
Jaiswal, S., Jamieson, C. H. M., Pang, W. W., Park, C. Y., Chao, M. P., Majeti, R., . . . Weissman, I. L. (2009). CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell, 138(2), 271-285. doi:10.1016/j.cell. 2009.05.046
Janjigian, Y. Y., Smit, E. F., Groen, H. J. M., Horn, L., Gettinger, S., Camidge, D. R., . . . Pao, W. (2014). Dual inhibition of EGFR with afatinib and cetuximab in kinase inhibitor-resistant EGFR-mutant lung cancer with and without T790M mutations. Cancer Discovery, 4(9), 1036-1045. doi:10.1158/2159-8290.CD-14-0326
Jänne, P. A., Yang, J. C., Kim, D., Planchard, D., Ohe, Y., Ramalingam, S. S., . . . Ranson, M. (2015). AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. The New England Journal of Medicine, 372(18), 1689-1699. doi:10.1056/NEJMoa1411817
Jardé, T., Lloyd-Lewis, B., Thomas, M., Kendrick, H., Melchor, L., Bougaret, L., . . . Dale, T. C. (2016). Wnt and Neuregulin1/ErbB signalling extends 3D culture of hormone responsive mammary organoids. Nature Communications, 7, 13207. doi:10.1038/ ncomms13207
Jensen, H. K., Donskov, F., Marcussen, N., Nordsmark, M., Lundbeck, F., & Von Der Maase, H. (2009). Presence of intratumoral neutrophils is an independent prognostic factor in localized renal cell carcinoma. Journal of Clinical Oncology, 27(28), 4709-4717. doi:10.1200/JCO.2008.18.9498
Jeong, H., Kim, J., Lee, Y., Seo, J. H., Hong, S. R., & Kim, A. (2014). Neuregulin-1 induces cancer stem cell characteristics in breast cancer cell lines. Oncology Reports, 32(3), 1218-1224. doi:10.3892/or.2014.3330
Jiang, F., Qiu, Q., Khanna, A., Todd, N. W., Deepak, J., Xing, L., . . . Katz, R. L. (2009). Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Molecular Cancer Research, 7(3), 330-338. doi:10.1158/1541-7786.MCR-08-0393
Jiang, J., Zhang, Y., Chuai, S., Wang, Z., Zheng, D., Xu, F., . . . Chen, Z. (2012). Trastuzumab (herceptin) targets gastric cancer stem cells characterized by CD90 phenotype. Oncogene, 31(6), 671-682. doi:10.1038/onc.2011.282
Joensuu, H., Bono, P., Kataja, V., Alanko, T., Kokko, R., Asola, R., . . . Kellokumpu-Lehtinen, P. (2009). Fluorouracil, epirubicin, and cyclophosphamide with either docetaxel or vinorelbine, with or without trastuzumab, as adjuvant treatments of breast cancer: Final results of the FinHer trial. Journal of Clinical Oncology, 27(34), 5685-5692. doi:10.1200/JCO.2008.21.4577

100
Johansson, A., Rudolfsson, S., Hammarsten, P., Halin, S., Pietras, K., Jones, J., . . . Bergh, A. (2010). Mast cells are novel independent prognostic markers in prostate cancer and represent a target for therapy. American Journal of Pathology, 177(2), 1031-1041. doi:10.2353/ajpath.2010.100070
Jokinen, E., Laurila, N., Koivunen, P., & Koivunen, J. P. (2014). Combining targeted drugs to overcome and prevent resistance of solid cancers with some stem-like cell features. Oncotarget, 5(19), 9295-9307. doi:10.18632/oncotarget.2424
Joyce, J. A., & Fearon, D. T. (2015). T cell exclusion, immune privilege, and the tumor microenvironment. Science, 348(6230), 74-80. doi:10.1126/science.aaa6204
Junttila, T. T., Li, G., Parsons, K., Phillips, G. L., & Sliwkowski, M. X. (2011). Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Research and Treatment, 128(2), 347-356. doi:10.1007/s10549-010-1090-x
Kainulainen, V., Sundvall, M., Määttä, J. A., Santiestevan, E., Klagsbrun, M., & Elenius, K. (2000). A natural ErbB4 isoform that does not activate phosphoinositide 3-kinase mediates proliferation but not survival or chemotaxis. The Journal of Biological Chemistry, 275(12), 8641-8649. doi:10.1074/jbc.275.12.8641
Karachaliou, N., Lazzari, C., Verlicchi, A., Sosa, A. E., & Rosell, R. (2017). HER3 as a therapeutic target in cancer. BioDrugs, 31(1), 63-73. doi:10.1007/s40259-016-0205-2
Karnoub, A. E., & Weinberg, R. A. (2008). Ras oncogenes: Split personalities. Nature Reviews Molecular Cell Biology, 9(7), 517-531. doi:10.1038/nrm2438
Katayama, R., Shaw, A. T., Khan, T. M., Mino-Kenudson, M., Solomon, B. J., Halmos, B., . . . Engelman, J. A. (2012). Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Science Translational Medicine, 4(120), 120ra17. doi:10.1126/scitranslmed.3003316
Katzmann, D. J., Odorizzi, G., & Emr, S. D. (2002). Receptor downregulation and multivesicular-body sorting. Nature Reviews. Molecular Cell Biology, 3(12), 893-905. doi:10.1038/nrm973
Kavuri, S. M., Jain, N., Galimi, F., Cottino, F., Leto, S. M., Migliardi, G., . . . Bose, R. (2015). HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discovery, 5(8), 832-841. doi:10.1158/2159-8290.CD-14-1211
Kawamoto, T., Sato, J. D., Le, A., Polikoff, J., Sato, G. H., & Mendelsohn, J. (1983). Growth stimulation of A431 cells by epidermal growth factor: Identification of high-affinity receptors for epidermal growth factor by an anti-receptor monoclonal antibody. Proceedings of the National Academy of Sciences of the United States of America, 80(5), 1337-1341.
Kim, M. S., Cho, H. I., Yoon, H. J., Ahn, Y., Park, E. J., Jin, Y. H., & Jang, Y. K. (2018). JIB-04, A small molecule histone demethylase inhibitor, selectively targets colorectal cancer stem cells by inhibiting the wnt/β-catenin signaling pathway. Scientific Reports, 8(1), 6611.

101
Kohler, B. A., Sherman, R. L., Howlader, N., Jemal, A., Ryerson, A. B., Henry, K. A., . . . Penberthy, L. (2015). Annual report to the nation on the status of cancer, 1975-2011, featuring incidence of breast cancer subtypes by race/ethnicity, poverty, and state. Journal of the National Cancer Institute, 107(6), djv048. doi:10.1093/jnci/djv048
Konecny, G. E., Pegram, M. D., Venkatesan, N., Finn, R., Yang, G., Rahmeh, M., . . . Slamon, D. J. (2006). Activity of the dual kinase inhibitor lapatinib (GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Research, 66(3), 1630-1639. doi:10.1158/0008-5472.CAN-05-1182
Köninki, K., Tanner, M., Auvinen, A., & Isola, J. (2009). HER-2 positive breast cancer: Decreasing proportion but stable incidence in finnish population from 1982 to 2005. Breast Cancer Research, 11(3), R37. doi:10.1186/bcr2322
Korkaya, H., Paulson, A., Iovino, F., & Wicha, M. S. (2008). HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene, 27(47), 6120-6130. doi:10.1038/onc.2008.207
Korkaya, H., Paulson, A., Charafe-Jauffret, E., Ginestier, C., Brown, M., Dutcher, J., . . . Wicha, M. S. (2009). Regulation of mammary stem/progenitor cells by PTEN/akt/beta-catenin signaling. PLoS Biology, 7(6), e1000121. doi:10.1371/journal.pbio.1000121
Kurdi, A. T., Glavey, S. V., Bezman, N. A., Jhatakia, A., Guerriero, J. L., Manier, S., . . . Ghobrial, I. M. (2018). Antibody-dependent cellular phagocytosis by macrophages is a novel mechanism of action of elotuzumab. Molecular Cancer Therapeutics, 17(7), 1454-1463. doi:10.1158/1535-7163.MCT-17-0998
Kurppa, K. J., Denessiouk, K., Johnson, M. S., & Elenius, K. (2016). Activating ERBB4 mutations in non-small cell lung cancer. Oncogene, 35(10), 1283-1291. doi:10.1038/ onc.2015.185
Kwak, E. L., Bang, Y., Camidge, D. R., Shaw, A. T., Solomon, B., Maki, R. G., . . . Iafrate, A. J. (2010). Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. The New England Journal of Medicine, 363(18), 1693-1703. doi:10.1056/NEJMoa1006448
Lee, C. Y. -., Lin, Y., Bratman, S. V., Feng, W., Kuo, A. H., Scheeren, F. A., . . . Diehn, M. (2014). Neuregulin autocrine signaling promotes self-renewal of breast tumor-initiating cells by triggering HER2/HER3 activation. Cancer Research, 74(1), 341-352. doi:10. 1158/0008-5472.CAN-13-1055
Lee, H. H., Kang, H., & Cho, H. (2017). Natural killer cells and tumor metastasis. Archives of Pharmacal Research, 40(9), 1037-1049. doi:10.1007/s12272-017-0951-9
Lee, H. J., Kim, J. Y., Park, I. A., Song, I. H., Yu, J. H., Ahn, J., & Gong, G. (2015). Prognostic significance of tumor-infiltrating lymphocytes and the tertiary lymphoid structures in HER2-positive breast cancer treated with adjuvant trastuzumab. American Journal of Clinical Pathology, 144(2), 278-288. doi:10.1309/AJCPIXUYDVZ0RZ3G
Lee-Hoeflich, S. T., Crocker, L., Yao, E., Pham, T., Munroe, X., Hoeflich, K. P., . . . Stern, H. M. (2008). A central role for HER3 in HER2-amplified breast cancer: Implications for targeted therapy. Cancer Research, 68(14), 5878-5887. doi:10.1158/0008-5472. CAN-08-0380

102
Leibowitz-Amit, R., Templeton, A. J., Omlin, A., Pezaro, C., Atenafu, E. G., Keizman, D., . . . Joshua, A. M. (2014). Clinical variables associated with PSA response to abiraterone acetate in patients with metastatic castration-resistant prostate cancer. Annals of Oncology, 25(3), 657-662. doi:10.1093/annonc/mdt581
Leis, O., Eguiara, A., Lopez-Arribillaga, E., Alberdi, M. J., Hernandez-Garcia, S., Elorriaga, K., . . . Martin, A. G. (2012). Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene, 31(11), 1354-1365. doi:10.1038/onc.2011.338
Lemmon, M. A., & Schlessinger, J. (2010). Cell signaling by receptor tyrosine kinases. Cell, 141(7), 1117-1134. doi:10.1016/j.cell.2010.06.011
Lewis Phillips, G. D., Li, G., Dugger, D. L., Crocker, L. M., Parsons, K. L., Mai, E., . . . Sliwkowski, M. X. (2008). Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Research, 68(22), 9280-9290. doi:10.1158/0008-5472.CAN-08-1776
Li, D., Ambrogio, L., Shimamura, T., Kubo, S., Takahashi, M., Chirieac, L. R., . . . Wong, K. -. (2008). BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene, 27(34), 4702-4711. doi:10.1038/onc. 2008.109
Li, Y., Atkinson, K., & Zhang, T. (2017). Combination of chemotherapy and cancer stem cell targeting agents: Preclinical and clinical studies. Cancer Letters, 396, 103-109. doi:10.1016/j.canlet.2017.03.008
Lin, J. J., Riely, G. J., & Shaw, A. T. (2017). Targeting ALK: Precision medicine takes on drug resistance. Cancer Discovery, 7(2), 137-155. doi:10.1158/2159-8290.CD-16-1123
Lin, J. J., & Shaw, A. T. (2016). Resisting resistance: Targeted therapies in lung cancer. Trends in Cancer, 2(7), 350-364. doi:10.1016/j.trecan.2016.05.010
Lin, W., Karin, M., Pearce, D., & Kleyman, T. R. (2007). A cytokine-mediated link between innate immunity, inflammation, and cancer. Journal of Clinical Investigation, 117(5), 1175-1183. doi:10.1172/JCI31537
Liu, P., Cheng, H., Roberts, T. M., & Zhao, J. J. (2009). Targeting the phosphoinositide 3-kinase pathway in cancer. Nature Reviews Drug Discovery, 8(8), 627-644. doi:10.1038/ nrd2926
Liu, R., Wang, X., Chen, G. Y., Dalerba, P., Gurney, A., Hoey, T., . . . Clarke, M. F. (2007). The prognostic role of a gene signature from tumorigenic breast-cancer cells. The New England Journal of Medicine, 356(3), 217-226. doi:10.1056/NEJMoa063994
Liu, S., Chen, B., Burugu, S., Leung, S., Gao, D., Virk, S., . . . Nielsen, T. O. (2017). Role of cytotoxic tumor-infiltrating lymphocytes in predicting outcomes in metastatic HER2-positive breast cancer: A secondary analysis of a randomized clinical trial. JAMA Oncology, 3(11), e172085. doi:10.1001/jamaoncol.2017.2085
Liu, T. J., Sun, B. C., Zhao, X. L., Zhao, X. M., Sun, T., Gu, Q., . . . Liu, N. (2013). CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene, 32(5), 544-553. doi:10.1038/onc.2012.85

103
Loi, S., Michiels, S., Salgado, R., Sirtaine, N., Jose, V., Fumagalli, D., . . . Sotiriou, C. (2014). Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: Results from the FinHER trial. Annals of Oncology, 25(8), 1544-1550. doi:10.1093/annonc/mdu112
Lombardi, V. C., Khaiboullina, S. F., & Rizvanov, A. A. (2015). Plasmacytoid dendritic cells, a role in neoplastic prevention and progression. European Journal of Clinical Investigation, 45(s1), 1-8. doi:10.1111/eci.12363
Luen, S. J., Salgado, R., Fox, S., Savas, P., Eng-Wong, J., Clark, E., . . . Loi, S. (2017). Tumour-infiltrating lymphocytes in advanced HER2-positive breast cancer treated with pertuzumab or placebo in addition to trastuzumab and docetaxel: A retrospective analysis of the CLEOPATRA study. The Lancet. Oncology, 18(1), 52-62. doi:10.1016/ S1470-2045(16)30631-3
Lynch, T. J., Bell, D. W., Sordella, R., Gurubhagavatula, S., Okimoto, R. A., Brannigan, B. W., . . . Haber, D. A. (2004). Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. The New England Journal of Medicine, 350(21), 2129-2139. doi:10.1056/NEJMoa040938
Maccalli, C., Parmiani, G., & Ferrone, S. (2017). Immunomodulating and immunoresistance properties of cancer-initiating cells: Implications for the clinical success of immunotherapy. Immunological Investigations, 46(3), 221-238. doi:10.1080/08820139. 2017.1280051
Malanchi, I., Peinado, H., Kassen, D., Hussenet, T., Metzger, D., Chambon, P., . . . Huelsken, J. (2008). Cutaneous cancer stem cell maintenance is dependent on beta-catenin signalling. Nature, 452(7187), 650-653. doi:10.1038/nature06835
Mantovani, A., Allavena, P., Sica, A., & Balkwill, F. (2008). Cancer-related inflammation. Nature, 454(7203), 436-444. doi:10.1038/nature07205
Mantovani, A., Marchesi, F., Malesci, A., Laghi, L., & Allavena, P. (2017). Tumour-associated macrophages as treatment targets in oncology. Nature Reviews Clinical Oncology, 14(7), 399-416. doi:10.1038/nrclinonc.2016.217
Martin, M., Holmes, F. A., Ejlertsen, B., Delaloge, S., Moy, B., Iwata, H., . . . Chan, A. (2017). Neratinib after trastuzumab-based adjuvant therapy in HER2-positive breast cancer (ExteNET): 5-year analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet. Oncology, 18(12), 1688-1700. doi:10.1016/S1470-2045(17)30717-9
Marty, M., Cognetti, F., Maraninchi, D., Snyder, R., Mauriac, L., Tubiana-Hulin, M., . . . Extra, J. (2005). Randomized phase II trial of the efficacy and safety of trastuzumab combined with docetaxel in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer administered as first-line treatment: The M77001 study group. Journal of Clinical Oncology, 23(19), 4265-4274. doi:10.1200/JCO.2005.04. 173
Masui, S., Nakatake, Y., Toyooka, Y., Shimosato, D., Yagi, R., Takahashi, K., . . . Niwa, H. (2007). Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nature Cell Biology, 9(6), 625-635. doi:10.1038/ncb1589

104
Mazzoleni, S., Politi, L. S., Pala, M., Cominelli, M., Franzin, A., Sergi, L. S., . . . Galli, R. (2010). Epidermal growth factor receptor expression identifies functionally and molecularly distinct tumor-initiating cells in human glioblastoma multiforme and is required for gliomagenesis. Cancer Research, 70(19), 7500-7513. doi:10.1158/0008-5472.CAN-10-2353
McCubrey, J. A., Steelman, L. S., Kempf, C. R., Chappell, W. H., Abrams, S. L., Stivala, F., . . . Martelli, A. M. (2011). Therapeutic resistance resulting from mutations in raf/MEK/ERK and PI3K/PTEN/akt/mTOR signaling pathways. Journal of Cellular Physiology, 226(11), 2762-2781. doi:10.1002/jcp.22647
McKay, R. D. G., Kittappa, R., Leker, R. R., Soldner, F., Poser, S. W., Rueger, M. A., . . . Androutsellis-Theotokis, A. (2006). Notch signalling regulates stem cell numbers in vitro and in vivo. Nature, 442(7104), 823-826. doi:10.1038/nature04940
McLendon, R. E., Hjelmeland, A. B., Dewhirst, M. W., Shi, Q., Rich, J. N., Wu, Q., . . . Hao, Y. (2006). Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature, 444(7120), 756-760. doi:10.1038/nature05236
Medema, J. P. (2013). Cancer stem cells: The challenges ahead. Nature Cell Biology, 15(4), 338-344. doi:10.1038/ncb2717
Melzer, C., von der Ohe, J., Lehnert, H., Ungefroren, H., & Hass, R. (2017). Cancer stem cell niche models and contribution by mesenchymal stroma/stem cells. Molecular Cancer, 16(1), 28. doi:10.1186/s12943-017-0595-x
Ménard, S., Balsari, A., Casalini, P., Tagliabue, E., Campiglio, M., Bufalino, R., & Cascinelli, N. (2002). HER-2-positive breast carcinomas as a particular subset with peculiar clinical behaviors. Clinical Cancer Research, 8(2), 520-525.
Mendelsohn, J., Gray, J. W., Howley, P. M., Israel, M. A., & Thompson, C. B. (2014). The molecular basis of cancer: Fourth edition. The molecular basis of cancer: Fourth edition (pp. 1-863)
Metro, G., Lunardi, G., Floridi, P., Pascali, J. P., Marcomigni, L., Chiari, R., . . . Gori, S. (2015). CSF concentration of crizotinib in two ALK-positive non-small-cell lung cancer patients with CNS metastases deriving clinical benefit from treatment. Journal of Thoracic Oncology, 10(5), 26. doi:10.1097/JTO.0000000000000468
Miller, V. A., Hirsh, V., Cadranel, J., Chen, Y., Park, K., Kim, S., . . . Yang, J. C. (2012). Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-lung 1): A phase 2b/3 randomised trial. The Lancet. Oncology, 13(5), 528-538. doi:10.1016/S1470-2045(12)70087-6
Mishra, R., Hanker, A. B., & Garrett, J. T. (2017). Genomic alterations of ERBB receptors in cancer: Clinical implications. Oncotarget, 8(69), 114371-114392. doi:10.18632/ oncotarget.22825
Mishra, R., Patel, H., Alanazi, S., Yuan, L., & Garrett, J. T. (2018). HER3 signaling and targeted therapy in cancer. Oncology Reviews, 12(1), 355. doi:10.4081/oncol.2018.355

105
Mizuno, E., Iura, T., Mukai, A., Yoshimori, T., Kitamura, N., & Komada, M. (2005). Regulation of epidermal growth factor receptor down-regulation by UBPY-mediated deubiquitination at endosomes. Molecular Biology of the Cell, 16(11), 5163-5174. doi:10.1091/mbc.E05-06-0560
Moilanen, T., Mustanoja, S., Karihtala, P., & Koivunen, J. P. (2017). Retrospective analysis of HER2 therapy interruption in patients responding to the treatment in metastatic HER2+ breast cancer. ESMO Open, 2(3) doi:10.1136/esmoopen-2017-000202
Mok, T. S., Wu, Y., Ahn, M., Garassino, M. C., Kim, H. R., Ramalingam, S. S., . . . Papadimitrakopoulou, V. A. (2017). Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. The New England Journal of Medicine, 376(7), 629-640. doi:10.1056/NEJMoa1612674
Mok, T. S., Wu, Y., Thongprasert, S., Yang, C., Chu, D., Saijo, N., . . . Fukuoka, M. (2009). Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. The New England Journal of Medicine, 361(10), 947-957. doi:10.1056/NEJMoa0810699
Molina, M. A., Codony-Servat, J., Albanell, J., Rojo, F., Arribas, J., & Baselga, J. (2001). Trastuzumab (herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells. Cancer Research, 61(12), 4744-4749.
Morath, I., Hartmann, T. N., & Orian-Rousseau, V. (2016). CD44: More than a mere stem cell marker. International Journal of Biochemistry and Cell Biology, 81(Pt A), 166-173. doi:10.1016/j.biocel.2016.09.009
Moscatello, D. K., Holgado-Madruga, M., Godwin, A. K., Ramirez, G., Gunn, G., Zoltick, P. W., . . . Wong, A. J. (1995). Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Research, 55(23), 5536-5539.
Motegi, A., Fujimoto, J., Kotani, M., Sakuraba, H., & Yamamoto, T. (2004). ALK receptor tyrosine kinase promotes cell growth and neurite outgrowth. - PubMed - NCBI. Journal of Cell Science, 117(15), 3319-29.
Munk, M., Memon, A., Poulsen, S. S., Borre, M., Nexo, E., & Sorensen, B. S. (2013). The HER4 isoform JM-a/CYT2 relates to improved survival in bladder cancer patients but only if the estrogen receptor α is not expressed. Scandinavian Journal of Clinical and Laboratory Investigation, 73(6), 503-513. doi:10.3109/00365513.2013.818706
Murthy, R. K., Varma, A., Mishra, P., Hess, K. R., Young, E. J., Murray, J. L., . . . Esteva, F. J. (2012). Impact of adjuvant trastuzumab on outcomes of HER2-positive breast cancer patients treated with HER2-targeted therapy in the metastatic setting. Journal of Clinical Oncology, 30(15), 527. doi:10.1200/jco.2012.30.15_suppl.527
Musolino, A., Naldi, N., Bortesi, B., Pezzuolo, D., Capelletti, M., Missale, G., . . . Ardizzoni, A. (2008). Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. Journal of Clinical Oncology, 26(11), 1789-1796. doi:10.1200/JCO.2007. 14.8957

106
Nakanishi, T., Chumsri, S., Khakpour, N., Brodie, A. H., Leyland-Jones, B., Hamburger, A. W., . . . Burger, A. M. (2010). Side-population cells in luminal-type breast cancer have tumour-initiating cell properties, and are regulated by HER2 expression and signalling. British Journal of Cancer, 102(5), 815-826. doi:10.1038/sj.bjc.6605553
Nami, B., Maadi, H., & Wang, Z. (2018). Mechanisms underlying the action and synergism of trastuzumab and pertuzumab in targeting HER2-positive breast cancer. Cancers, 10(10) doi:10.3390/cancers10100342
Nelson, B. H. (2010). CD20+ B cells: The other tumor-infiltrating lymphocytes. Journal of Immunology, 185(9), 4977-4982. doi:10.4049/jimmunol.1001323
Nikolaev, S. I., Rimoldi, D., Iseli, C., Valsesia, A., Robyr, D., Gehrig, C., . . . Antonarakis, S. E. (2012). Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nature Genetics, 44(2), 133-139. doi:10.1038/ng.1026
Nishikawa, R., Ji, X. D., Harmon, R. C., Lazar, C. S., Gill, G. N., Cavenee, W. K., & Huang, H. J. (1994). A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proceedings of the National Academy of Sciences of the United States of America, 91(16), 7727-7731.
Nonomura, N., Takayama, H., Nishimura, K., Oka, D., Nakai, Y., Shiba, M., . . . Okuyama, A. (2007). Decreased number of mast cells infiltrating into needle biopsy specimens leads to a better prognosis of prostate cancer. British Journal of Cancer, 97(7), 952-956. doi:10.1038/sj.bjc.6603962
Obeid, M., Tesniere, A., Ghiringhelli, F., Fimia, G. M., Apetoh, L., Perfettini, J., . . . Kroemer, G. (2007). Calreticulin exposure dictates the immunogenicity of cancer cell death. Nature Medicine, 13(1), 54-61. doi:10.1038/nm1523
Ocana, A., Nieto-Jiménez, C., Pandiella, A., & Templeton, A. J. (2017). Neutrophils in cancer: Prognostic role and therapeutic strategies. Molecular Cancer, 16(1) doi:10. 1186/s12943-017-0707-7
Ogiso, H., Ishitani, R., Nureki, O., Fukai, S., Yamanaka, M., Kim, J., . . . Yokoyama, S. (2002). Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell, 110(6), 775-787. doi:10.1016/S0092-8674(02)00963-7
Ojesina, A. I., Lichtenstein, L., Freeman, S. S., Pedamallu, C. S., Imaz-Rosshandler, I., Pugh, T. J., . . . Meyerson, M. (2014). Landscape of genomic alterations in cervical carcinomas. Nature, 506(7488), 371-375. doi:10.1038/nature12881
Okazaki, S., Nakatani, F., Masuko, K., Tsuchihashi, K., Ueda, S., Masuko, T., . . . Nagano, O. (2016). Development of an ErbB4 monoclonal antibody that blocks neuregulin-1-induced ErbB4 activation in cancer cells. Biochemical and Biophysical Research Communications, 470(1), 239-244. doi:10.1016/j.bbrc.2016.01.045
Paez, J. G., Jänne, P. A., Lee, J. C., Tracy, S., Greulich, H., Gabriel, S., . . . Meyerson, M. (2004). EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science, 304(5676), 1497-1500. doi:10.1126/science.1099314
Pagès, F., Mlecnik, B., Marliot, F., Bindea, G., Ou, F., Bifulco, C., . . . Galon, J. (2018). International validation of the consensus immunoscore for the classification of colon cancer: A prognostic and accuracy study. The Lancet, 391(10135), 2128-2139. doi:10.1016/S0140-6736(18)30789-X

107
Pallini, R., Ricci-Vitiani, L., Banna, G. L., Signore, M., Lombardi, D., Todaro, M., . . . De Maria, R. (2008). Cancer stem cell analysis and clinical outcome in patients with glioblastoma multiforme. Clinical Cancer Research, 14(24), 8205-8212. doi:10.1158 /1078-0432.CCR-08-0644
Pao, W., & Chmielecki, J. (2010). Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nature Reviews. Cancer, 10(11), 760. doi:10.1038/nrc2947
Pao, W., Miller, V. A., Politi, K. A., Riely, G. J., Somwar, R., Zakowski, M. F., . . . Varmus, H. (2005). Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Medicine, 2(3), e73. doi:10.1371/journal.pmed.0020073
Parikh, F., Duluc, D., Imai, N., Clark, A., Misiukiewicz, K., Bonomi, M., . . . Sikora, A. G. (2014). Chemoradiotherapy-induced upregulation of PD-1 antagonizes immunity to HPV-related oropharyngeal cancer. Cancer Research, 74(24), 7205-7216. doi:10.1158/ 0008-5472.CAN-14-1913
Park, S., Jiang, Z., Mortenson, E. D., Deng, L., Radkevich-Brown, O., Yang, X., . . . Fu, Y. (2010). The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer Cell, 18(2), 160-170. doi:10.1016/j.ccr.2010.06.014
Payandeh, M., Shahriari-Ahmadi, A., Sadeghi, M., & Sadeghi, E. (2016). Correlations between HER2 expression and other prognostic factors in breast cancer: Inverse relations with the ki-67 index and P53 status. Asian Pacific Journal of Cancer Prevention, 17(3), 1015-1018.
Pernicova, I., & Korbonits, M. (2014). Metformin--mode of action and clinical implications for diabetes and cancer. Nature Reviews. Endocrinology, 10(3), 143-156. doi:10.1038/ nrendo.2013.256
Peters, S., Camidge, D. R., Shaw, A. T., Gadgeel, S., Ahn, J. S., Kim, D., . . . Mok, T. (2017). Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. The New England Journal of Medicine, 377(9), 829-838. doi:10.1056/NEJMoa1704795
Petricevic, B., Laengle, J., Singer, J., Sachet, M., Fazekas, J., Steger, G., . . . Bergmann, M. (2013). Trastuzumab mediates antibody-dependent cell-mediated cytotoxicity and phagocytosis to the same extent in both adjuvant and metastatic HER2/neu breast cancer patients. Journal of Translational Medicine, 11(1) doi:10.1186/1479-5876-11-307
Phillips, T. M., McBride, W. H., & Pajonk, F. (2006). The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. Journal of the National Cancer Institute, 98(24), 1777. doi:10.1093/jnci/djj495
Pivot, X., Romieu, G., Debled, M., Pierga, J., Kerbrat, P., Bachelot, T., . . . Kramar, A. (2013). 6 months versus 12 months of adjuvant trastuzumab for patients with HER2-positive early breast cancer (PHARE): A randomised phase 3 trial. The Lancet. Oncology, 14(8), 741-748. doi:10.1016/S1470-2045(13)70225-0
Plaks, V., Kong, N., & Werb, Z. (2015). The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell, 16(3), 225-238. doi:10.1016/j.stem.2015.02.015

108
Rainusso, N., Brawley, V. S., Ghazi, A., Hicks, M. J., Gottschalk, S., Rosen, J. M., & Ahmed, N. (2012). Immunotherapy targeting HER2 with genetically modified T cells eliminates tumor-initiating cells in osteosarcoma. Cancer Gene Therapy, 19(3), 212-217. doi:10.1038/cgt.2011.83
Rajput, A. B., Turbin, D. A., Cheang, M. C., Voduc, D. K., Leung, S., Gelmon, K. A., . . . Huntsman, D. G. (2008). Stromal mast cells in invasive breast cancer are a marker of favourable prognosis: A study of 4,444 cases. Breast Cancer Research and Treatment, 107(2), 249-257. doi:10.1007/s10549-007-9546-3
Regales, L., Gong, Y., Shen, R., de Stanchina, E., Vivanco, I., Goel, A., . . . Pao, W. (2009). Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. The Journal of Clinical Investigation, 119(10), 3000-3010. doi:10.1172/JCI38746
Reim, F., Dombrowski, Y., Ritter, C., Buttmann, M., Häusler, S., Ossadnik, M., . . . Wischhusen, J. (2009). Immunoselection of breast and ovarian cancer cells with trastuzumab and natural killer cells: Selective escape of CD44high/CD24low/HER2low breast cancer stem cells. Cancer Research, 69(20), 8058-8066. doi:10.1158/0008-5472.CAN-09-0834
Ribatti, D., Vacca, A., Ria, R., Marzullo, A., Nico, B., Filotico, R., . . . Dammacco, F. (2003). Neovascularisation, expression of fibroblast growth factor-2, and mast cells with tryptase activity increase simultaneously with pathological progression in human malignant melanoma. European Journal of Cancer, 39(5), 666-674. doi:10.1016/ S0959-8049(02)00150-8
Rier, H. N., Levin, M. D., van Rosmalen, J., Bos, M., Drooger, J. C., de Jong, P., . . . Jager, A. (2017). First-line palliative HER2-targeted therapy in HER2-positive metastatic breast cancer is less effective after previous adjuvant trastuzumab-based therapy. Oncologist, 22(8), 901-909.
Ríos-Luci, C., García-Alonso, S., Díaz-Rodríguez, E., Nadal-Serrano, M., Arribas, J., Ocaña, A., & Pandiella, A. (2017). Resistance to the antibody-drug conjugate T-DM1 is based in a reduction in lysosomal proteolytic activity. Cancer Research, 77(17), 4639-4651. doi:10.1158/0008-5472.CAN-16-3127
Ritter, C. A., Perez-Torres, M., Rinehart, C., Guix, M., Dugger, T., Engelman, J. A., & Arteaga, C. L. (2007). Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clinical Cancer Research, 13(16), 4909-4919. doi:10.1158/1078-0432.CCR-07-0701
Rosell, R., Carcereny, E., Gervais, R., Vergnenegre, A., Massuti, B., Felip, E., . . . Paz-Ares, L. (2012). Erlotinib versus standard chemotherapy as first-line treatment for european patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. The Lancet. Oncology, 13(3), 239-246. doi:10.1016/S1470-2045(11)70393-X
Roskoski, R. (2014). The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacological Research, 79, 34-74. doi:10.1016/j.phrs.2013.11.002

109
Routy, B., Le Chatelier, E., Derosa, L., Duong, C. P. M., Alou, M. T., Daillère, R., . . . Zitvogel, L. (2018). Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science, 359(6371), 91-97. doi:10.1126/science.aan3706
Rupaimoole, R., & Slack, F. J. (2017). MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nature Reviews Drug Discovery, 16(3), 203-221. doi:10.1038/nrd.2016.246
Saadeh, D., Kurban, M., & Abbas, O. (2016). Plasmacytoid dendritic cell role in cutaneous malignancies. Journal of Dermatological Science, 83(1), 3-9. doi:10.1016/j.jdermsci. 2016.05.008
Sabbaghi, M., Gil-Gómez, G., Guardia, C., Servitja, S., Arpí, O., García-Alonso, S., . . . Albanell, J. (2017). Defective cyclin B1 induction in trastuzumab-emtansine (T-DM1) acquired resistance in HER2-positive breast cancer. Clinical Cancer Research, 23(22), 7006-7019. doi:10.1158/1078-0432.CCR-17-0696
Sabir, S. R., Yeoh, S., Jackson, G., & Bayliss, R. (2017). EML4-ALK variants: Biological and molecular properties, and the implications for patients. Cancers, 9(9) doi:10.3390/ cancers9090118
Salgado, R., Denkert, C., Campbell, C., Savas, P., Nuciforo, P., Nucifero, P., . . . Loi, S. (2015). Tumor-infiltrating lymphocytes and associations with pathological complete response and event-free survival in HER2-positive early-stage breast cancer treated with lapatinib and trastuzumab: A secondary analysis of the NeoALTTO trial. JAMA Oncology, 1(4), 448-454. doi:10.1001/jamaoncol.2015.0830
Sasaki, T., Koivunen, J., Ogino, A., Yanagita, M., Nikiforow, S., Zheng, W., . . . Jänne, P. A. (2011). A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Research, 71(18), 6051-6060. doi:10.1158/0008-5472.CAN-11-1340
Scheuer, W., Friess, T., Burtscher, H., Bossenmaier, B., Endl, J., & Hasmann, M. (2009). Strongly enhanced antitumor activity of trastuzumab and pertuzumab combination treatment on HER2-positive human xenograft tumor models. Cancer Research, 69(24), 9330-9336. doi:10.1158/0008-5472.CAN-08-4597
Schreiber, R. D., Old, L. J., & Smyth, M. J. (2011). Cancer immunoediting: Integrating immunity's roles in cancer suppression and promotion. Science, 331(6024), 1565-1570. doi:10.1126/science.1203486
Schulze, W. X., Deng, L., & Mann, M. (2005). Phosphotyrosine interactome of the ErbB-receptor kinase family. Molecular Systems Biology, 1, 2005.0008. doi:10.1038/ msb4100012
Schwitalla, S., Fingerle, A., Cammareri, P., Nebelsiek, T., Göktuna, S., Ziegler, P., . . . Greten, F. (2013). Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell, 152(1-2), 25-38. doi:10.1016/j.cell.2012.12.012
Seigo Kashimura, Saze, Z., Terashima, M., Soeta, N., Ohtani, S., Osuka, F., . . . Gotoh, M. (2012). CD83+ dendritic cells and Foxp3+ regulatory T cells in primary lesions and regional lymph nodes are inversely correlated with prognosis of gastric cancer. Gastric Cancer, 15(2), 144-153. doi:10.1007/s10120-011-0090-9

110
Senkus, E., Kyriakides, S., Ohno, S., Penault-Llorca, F., Poortmans, P., Rutgers, E., . . . Cardoso, F. (2015). Primary breast cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Annals of Oncology, 26 Suppl 5, 8. doi:10.1093/ annonc/mdv298
Shan, L., Wang, Z., Guo, L., Sun, H., Qiu, T., Ling, Y., . . . Ying, J. (2015). Concurrence of EGFR amplification and sensitizing mutations indicate a better survival benefit from EGFR-TKI therapy in lung adenocarcinoma patients. Lung Cancer, 89(3), 337-342. doi:10.1016/j.lungcan.2015.06.008
Shankaran, V., Ikeda, H., Bruce, A. T., White, J. M., Swanson, P. E., Old, L. J., & Schreiber, R. D. (2001). IFNγ, and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature, 410(6832), 1107-1111. doi:10.1038/35074122
Sharma, P., & Allison, J. P. (2015). The future of immune checkpoint therapy. Science, 348(6230), 56-61. doi:10.1126/science.aaa8172
Shaw, A. T., Kim, D., Nakagawa, K., Seto, T., Crinó, L., Ahn, M., . . . Jänne, P. A. (2013). Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. The New England Journal of Medicine, 368(25), 2385-2394. doi:10.1056/NEJMoa1214886
Shaw, A. T., Yeap, B. Y., Mino-Kenudson, M., Digumarthy, S. R., Costa, D. B., Heist, R. S., . . . Iafrate, A. J. (2009). Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. Journal of Clinical Oncology, 27(26), 4247-4253. doi:10.1200/JCO.2009.22.6993
Sherr, C. J., & McCormick, F. (2002). The RB and p53 pathways in cancer. Cancer Cell, 2(2), 103-112. doi:10.1016/S1535-6108(02)00102-2
Shi, F., Telesco, S. E., Liu, Y., Radhakrishnan, R., & Lemmon, M. A. (2010). ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proceedings of the National Academy of Sciences of the United States of America, 107(17), 7692-7697. doi:10.1073/pnas.1002753107
Shi, Y., Fan, X., Deng, H., Brezski, R. J., Rycyzyn, M., Jordan, R. E., . . . An, Z. (2015). Trastuzumab triggers phagocytic killing of high HER2 cancer cells in vitro and in vivo by interaction with fcγ receptors on macrophages. Journal of Immunology, 194(9), 4379-4386. doi:10.4049/jimmunol.1402891
Shi, Y., Liu, N., Lai, W., Yan, B., Chen, L., Liu, S., . . . Tao, Y. (2018). Nuclear EGFR-PKM2 axis induces cancer stem cell-like characteristics in irradiation-resistant cells. Cancer Letters, 422, 81-93. doi:10.1016/j.canlet.2018.02.028
Shibue, T., & Weinberg, R. A. (2017). EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nature Reviews. Clinical Oncology, 14(10), 611-629. doi:10.1038/nrclinonc.2017.44
Shook, D., & Keller, R. (2003). Mechanisms, mechanics and function of epithelial–mesenchymal transitions in early development. Mechanisms of Development, 120(11), 1351-1383. doi:10.1016/j.mod.2003.06.005
Slamon, D. J., Clark, G. M., Wong, S. G., Levin, W. J., Ullrich, A., & McGuire, W. L. (1987). Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science, 235(4785), 177-182. doi:10.1126/science.3798106

111
Slamon, D. J., Godolphin, W., Jones, L. A., Holt, J. A., Wong, S. G., Keith, D. E., . . . Ullrich, A. (1989). Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science, 244(4905), 707-712.
Soda, M., Choi, Y. L., Enomoto, M., Takada, S., Yamashita, Y., Ishikawa, S., . . . Mano, H. (2007). Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature, 448(7153), 561-566. doi:10.1038/nature05945
Solomon, B. J., Mok, T., Kim, D., Wu, Y., Nakagawa, K., Mekhail, T., . . . Blackhall, F. (2014). First-line crizotinib versus chemotherapy in ALK-positive lung cancer. The New England Journal of Medicine, 371(23), 2167-2177. doi:10.1056/NEJMoa1408440
Soria, J., Ohe, Y., Vansteenkiste, J., Reungwetwattana, T., Chewaskulyong, B., Lee, K. H., . . . Ramalingam, S. S. (2018). Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. The New England Journal of Medicine, 378(2), 113-125. doi:10.1056/NEJMoa1713137
Soussi, T., & Wiman, K. G. (2015). TP53: An oncogene in disguise. Cell Death and Differentiation, 22(8), 1239-1249. doi:10.1038/cdd.2015.53
Souttou, B., Carvalho, N. B., Raulais, D., & Vigny, M. (2001). Activation of anaplastic lymphoma kinase receptor tyrosine kinase induces neuronal differentiation through the mitogen-activated protein kinase pathway. Journal of Biological Chemistry, 276(12), 9526-31.
Stratton, M. R., Campbell, P. J., & Futreal, P. A. (2009). The cancer genome. Nature, 458(7239), 719-724. doi:10.1038/nature07943
Su, S., Zhao, J., Xing, Y., Zhang, X., Liu, J., Ouyang, Q., . . . Song, E. (2018). Immune checkpoint inhibition overcomes ADCP-induced immunosuppression by macrophages. Cell, 175(2), 44-457.e23. doi:10.1016/j.cell.2018.09.007
Sugawa, N., Ekstrand, A. J., James, C. D., & Collins, V. P. (1990). Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proceedings of the National Academy of Sciences of the United States of America, 87(21), 8602.
Suire, S., Hawkins, P., & Stephens, L. (2002). Activation of phosphoinositide 3-kinase gamma by ras. Current Biology, 12(13), 1068-1075.
Sung, M., Tan, X., Lu, B., Golas, J., Hosselet, C., Wang, F., . . . Loganzo, F. (2018). Caveolae-mediated endocytosis as a novel mechanism of resistance to trastuzumab emtansine (T-DM1). Molecular Cancer Therapeutics, 17(1), 243-253. doi:10.1158/ 1535-7163.MCT-17-0403
Suzuki, M., Shiraishi, K., Yoshida, A., Shimada, Y., Suzuki, K., Asamura, H., . . . Tsuta, K. (2015). HER2 gene mutations in non-small cell lung carcinomas: Concurrence with Her2 gene amplification and Her2 protein expression and phosphorylation. Lung Cancer, 87(1), 14-22. doi:10.1016/j.lungcan.2014.10.014
Swain, S. M., Baselga, J., Kim, S., Ro, J., Semiglazov, V., Campone, M., . . . Cortés, J. (2015). Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. The New England Journal of Medicine, 372(8), 724-734. doi:10.1056/ NEJMoa1413513

112
Swann, J. B., & Smyth, M. J. (2007). Immune surveillance of tumors. The Journal of Clinical Investigation, 117(5), 1137-1146. doi:10.1172/JCI31405
Szczerba, B. M., Castro-Giner, F., Vetter, M., Krol, I., Gkountela, S., Landin, J., . . . Aceto, N. (2019). Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature, doi:10.1038/s41586-019-0915-y
Takada, K., Kashiwagi, S., Goto, W., Asano, Y., Takahashi, K., Takashima, T., . . . Ohira, M. (2018). Use of the tumor-infiltrating CD8 to FOXP3 lymphocyte ratio in predicting treatment responses to combination therapy with pertuzumab, trastuzumab, and docetaxel for advanced HER2-positive breast cancer. Journal of Translational Medicine, 16(1), 86. doi:10.1186/s12967-018-1460-4
Takahashi, H., Sakakura, K., Mito, I., Ida, S., & Chikamatsu, K. (2016). Dynamic changes in immune cell profile in head and neck squamous cell carcinoma: Immunomodulatory effects of chemotherapy. Cancer Science, 107(8), 1065-1071. doi:10.1111/cas.12976
Takebe, N., Harris, P. J., Warren, R. Q., & Ivy, S. P. (2011). Targeting cancer stem cells by inhibiting wnt, notch, and hedgehog pathways. Nature Reviews Clinical Oncology, 8(2), 97-106. doi:10.1038/nrclinonc.2010.196
Tangka, F. K., Trogdon, J. G., Richardson, L. C., Howard, D., Sabatino, S. A., & Finkelstein, E. A. (2010). Cancer treatment cost in the united states: Has the burden shifted over time? Cancer, 116(14), 3477-3484. doi:10.1002/cncr.25150
Templeton, A. J., McNamara, M. G., Šeruga, B., Vera-Badillo, F. E., Aneja, P., Ocaña, A., . . . Amir, E. (2014). Prognostic role of neutrophil-to-lymphocyte ratio in solid tumors: A systematic review and meta-analysis. Journal of the National Cancer Institute, 106(6) doi:10.1093/jnci/dju124
Terabe, M., & Berzofsky, J. A. (2004). Immunoregulatory T cells in tumor immunity. Current Opinion in Immunology, 16(2), 157-62. doi:10.1016/j.coi.2004.01.010
Tesone, A. J., Rutkowski, M. R., Brencicova, E., Svoronos, N., Perales-Puchalt, A., Stephen, T. L., . . . Conejo-Garcia, J. R. (2016). Satb1 overexpression drives tumor-promoting activities in cancer-associated dendritic cells. Cell Reports, 14(7), 1774-1786. doi:10.1016/j.celrep.2016.01.056
Thress, K. S., Paweletz, C. P., Felip, E., Cho, B. C., Stetson, D., Dougherty, B., . . . Oxnard, G. R. (2015). Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nature Medicine, 21(6), 560-562. doi:10.1038/nm.3854
Tian, X., Wei, F., Wang, L., Yu, W., Zhang, N., Zhang, X., . . . Ren, X. (2017). Herceptin enhances the antitumor effect of natural killer cells on breast cancer cells expressing human epidermal growth factor receptor-2. Frontiers in Immunology, 8(OCT) doi:10.3389/fimmu.2017.01426
Torkki, P., Leskelä, R., Linna, M., Mäklin, S., Mecklin, J., Bono, P., . . . Karjalainen, S. (2018). Cancer costs and outcomes in the finnish population 2004-2014. Acta Oncologica, 57(2), 297-303. doi:10.1080/0284186X.2017.1343495
Torre, L. A., Bray, F., Siegel, R. L., Ferlay, J., Lortet-Tieulent, J., & Jemal, A. (2015). Global cancer statistics, 2012. CA Cancer Journal for Clinicians, 65(2), 87-108. doi:10.3322/caac.21262

113
Townsend, S. E., & Allison, J. P. (1993). Tumor rejection after direct costimulation of CD8+ T cells by B7-transfected melanoma cells. Science, 259(5093), 368-370. doi:10.1126/science.7678351
Triulzi, T., Forte, L., Regondi, V., Di Modica, M., Ghirelli, C., Carcangiu, M. L., . . . Tagliabue, E. (2019). HER2 signaling regulates the tumor immune microenvironment and trastuzumab efficacy. Oncoimmunology, 8(1), e1512942. doi:10.1080/2162402X. 2018.1512942
Tth, T., Tth-Jakatics, R., Jimi, S., Takebayashi, S., & Kawamoto, N. (2000). Cutaneous malignant melanoma: Correlation between neovascularization and peritumor accumulation of mast cells overexpressing vascular endothelial growth factor. Human Pathology, 31(8), 955-960. doi:10.1053/hupa.2000.16658
Ullrich, A., Coussens, L., Hayflick, J. S., Dull, T. J., Gray, A., Tam, A. W., . . . Seeburg, P. H. (1984). Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature, 309(5967), 418-425. doi:10.1038/309418a0
van den Hoogen, C., van der Horst, G., Cheung, H., Buijs, J. T., Lippitt, J. M., Guzmán-Ramírez, N., . . . van der Pluijm, G. (2010). High aldehyde dehydrogenase activity identifies tumor-initiating and metastasis-initiating cells in human prostate cancer. Cancer Research, 70(12), 5163-5173. doi:10.1158/0008-5472.CAN-09-3806
van Rooijen, J. M., Qiu, S., Timmer-Bosscha, H., van der Vegt, B., Boers, J. E., Schröder, C. P., & de Vries, Elisabeth G. E. (2018). Androgen receptor expression inversely correlates with immune cell infiltration in human epidermal growth factor receptor 2-positive breast cancer. European Journal of Cancer, 103, 52-60. doi:10.1016/ j.ejca.2018.08.001
Väyrynen, J. P., Vornanen, J. O., Sajanti, S., Böhm, J. P., Tuomisto, A., & Mäkinen, M. J. (2012). An improved image analysis method for cell counting lends credibility to the prognostic significance of T cells in colorectal cancer. Virchows Archiv, 460(5), 455-465. doi:10.1007/s00428-012-1232-0
Väyrynen, J. P., Tuomisto, A., Klintrup, K., Mäkelä, J., Karttunen, T. J., & Mäkinen, M. J. (2013). Detailed analysis of inflammatory cell infiltration in colorectal cancer. The British Journal of Cancer, 109(7), 1839-1847. doi:10.1038/bjc.2013.508
Veglia, F., & Gabrilovich, D. I. (2017). Dendritic cells in cancer: The role revisited. Current Opinion in Immunology, 45, 43-51. doi:10.1016/j.coi.2017.01.002
Verma, S., Miles, D., Gianni, L., Krop, I. E., Welslau, M., Baselga, J., . . . Blackwell, K. (2012). Trastuzumab emtansine for HER2-positive advanced breast cancer. The New England Journal of Medicine, 367(19), 1783-1791. doi:10.1056/NEJMoa1209124
Vermi, W., Micheletti, A., Finotti, G., Tecchio, C., Calzetti, F., Costa, S., . . . Cassatella, M. A. (2018). Slan+ monocytes and macrophages mediate CD20-dependent b-cell lymphoma elimination via ADCC and ADCP. Cancer Research, 78(13), 3544-3559. doi:10.1158/0008-5472.CAN-17-2344
Vesely, M. D., Kershaw, M. H., Schreiber, R. D., & Smyth, M. J. (2011). Natural innate and adaptive immunity to cancer. Annual Review of Immunology, 29(1), 235-271. doi:10.1146/annurev-immunol-031210-101324

114
Vétizou, M., Pitt, J. M., Daillère, R., Lepage, P., Waldschmitt, N., Flament, C., . . . Zitvogel, L. (2015). Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science, 350(6264), 1079-1084. doi:10.1126/science.aad1329
Vlashi, E., & Pajonk, F. (2014). Cancer stem cells, cancer cell plasticity and radiation therapy. Seminars in Cancer Biology, 31, 28-35. doi:10.1016/j.semcancer.2014.07.001
Vogelstein, B., Papadopoulos, N., Velculescu, V. E., Zhou, S., Diaz Jr., L. A., & Kinzler, K. W. (2013). Cancer genome landscapes. Science, 340(6127), 1546-1558. doi:10.1126/ science.1235122
Von Minckwitz, G., Du Bois, A., Schmidt, M., Maass, N., Cufer, T., De Jongh, F. E., . . . Loibl, S. (2009). Trastuzumab beyond progression in human epidermal growth factor receptor 2-positive advanced breast cancer: A german breast group 26/breast international group 03-05 study. Journal of Clinical Oncology, 27(12), 1999-2006. doi:10.1200/JCO.2008.19.6618
Wang, A., Qu, L., & Wang, L. (2017). At the crossroads of cancer stem cells and targeted therapy resistance. Cancer Letters, 385, 87-96. doi:10.1016/j.canlet.2016.10.039
Wang, J., Jia, Y., Wang, N., Zhang, X., Tan, B., Zhang, G., & Cheng, Y. (2014). The clinical significance of tumor-infiltrating neutrophils and neutrophil-to-CD8+ lymphocyte ratio in patients with resectable esophageal squamous cell carcinoma. Journal of Translational Medicine, 12(1) doi:10.1186/1479-5876-12-7
Wang, J., Yin, J., Yang, Q., Ding, F., Chen, X., Li, B., & Tian, X. (2016). Human epidermal growth factor receptor 4 (HER4) is a favorable prognostic marker of breast cancer: A systematic review and meta-analysis. Oncotarget, 7(47), 76693-76703. doi:10.18632/ oncotarget.12485
Wang, L., Wu, C., Rajasekaran, N., & Shin, Y. K. (2018). Loss of tumor suppressor gene function in human cancer: An overview. Cellular Physiology and Biochemistry, 51(6), 2647-2693. doi:10.1159/000495956
Wang, X. -., Wang, H. -., Wang, H., Zhang, F., Wang, K. -., Guo, Q., . . . Du, J. (2014). The role of indoleamine 2,3-dioxygenase (IDO) in immune tolerance: Focus on macrophage polarization of THP-1 cells. Cellular Immunology, 289(1-2), 42-48. doi:10.1016/ j.cellimm.2014.02.005
Williams, C. S., Bernard, J. K., Demory Beckler, M., Almohazey, D., Washington, M. K., Smith, J. J., & Frey, M. R. (2015). ERBB4 is over-expressed in human colon cancer and enhances cellular transformation. Carcinogenesis, 36(7), 710-718. doi:10.1093/ carcin/bgv049
Wilson, F. H., Johannessen, C. M., Piccioni, F., Tamayo, P., Kim, J. W., Van Allen, E. M., . . . Garraway, L. A. (2015). A functional landscape of resistance to ALK inhibition in lung cancer. Cancer Cell, 27(3), 397-408. doi:10.1016/j.ccell.2015.02.005
Won, J., Yang, H. W., Shin, S., Lee, J. H., Heo, W. D., & Cho, K. (2012). The crossregulation between ERK and PI3K signaling pathways determines the tumoricidal efficacy of MEK inhibitor. Journal of Molecular Cell Biology, 4(3), 153-163. doi:10.1093/jmcb/mjs021

115
Wood, E. R., Truesdale, A. T., McDonald, O. B., Yuan, D., Hassell, A., Dickerson, S. H., . . . Shewchuk, L. (2004). A unique structure for epidermal growth factor receptor bound to GW572016 (lapatinib): Relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Research, 64(18), 6652-6659. doi:10.1158/0008-5472.CAN-04-1168
Wooster, R., Neuhausen, S. L., Mangion, J., Quirk, Y., Ford, D., Collins, N., . . . Stratton, M. R. (1994). Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science, 265(5181), 2088-2090. doi:10.1126/science.8091231
Wright, M. H., Calcagno, A. M., Salcido, C. D., Carlson, M. D., Ambudkar, S. V., & Varticovski, L. (2008). Brca1 breast tumors contain distinct CD44+/CD24 and CD133+ cells with cancer stem cell characteristics. Breast Cancer Research, 10(1), R10. doi:10.1186/bcr1855
Yan, Q., Guo, K., Feng, G., Shan, F., Sun, L., Zhang, K., . . . Ruan, S. (2018). Association between the overexpression of Her3 and clinical pathology and prognosis of colorectal cancer: A meta-analysis. Medicine, 97(37), e12317. doi:10.1097/MD.00000000000 12317
Yarden, Y., & Pines, G. (2012). The ERBB network: At last, cancer therapy meets systems biology. Nature Reviews. Cancer, 12(8), 553-563. doi:10.1038/nrc3309
Yonesaka, K., Zejnullahu, K., Okamoto, I., Satoh, T., Cappuzzo, F., Souglakos, J., . . . Jänne, P. A. (2011). Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Science Translational Medicine, 3(99), 99ra86. doi:10.1126/scitranslmed.3002442
Yun, C., Boggon, T. J., Li, Y., Woo, M. S., Greulich, H., Meyerson, M., & Eck, M. J. (2007). Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell, 11(3), 217-227. doi:10.1016/j.ccr.2006.12.017
Zhang, S., Ma, X., Zhu, C., Liu, L., Wang, G., & Yuan, X. (2016). The role of myeloid-derived suppressor cells in patients with solid tumors: A meta-analysis. PLoS ONE, 11(10) doi:10.1371/journal.pone.0164514
Zhang, X., Gureasko, J., Shen, K., Cole, P. A., & Kuriyan, J. (2006). An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell, 125(6), 1137-1149. doi:10.1016/j.cell.2006.05.013
Zhao, W., Zhuang, C., Xu, J., Wang, M., Zhang, Z., Tu, L., . . . Zhang, Z. (2014). HER4 is a novel prognostic biomarker in gastrointestinal stromal tumor specifically originated from stomach. American Journal of Cancer Research, 4(6), 838-849.
Zhou, C., Wu, Y., Jiang, L., Li, Z., Diao, P., Wang, D., . . . Yang, J. (2018). Density and location of CD3+ and CD8+ tumor‐infiltrating lymphocytes correlate with prognosis of oral squamous cell carcinoma. Journal of Oral Pathology & Medicine, 47(4), 359-367. doi:10.1111/jop.12698
Zhu, F., Qian, W., Zhang, H., Liang, Y., Wu, M., Zhang, Y., . . . Li, Y. (2017). SOX2 is a marker for stem-like tumor cells in bladder cancer. Stem Cell Reports, 9(2), 429-437. doi:10.1016/j.stemcr.2017.07.004

116
Zimmerli, D., Cecconi, V., Valenta, T., Hausmann, G., Cantù, C., Restivo, G., . . . van den Broek, M. (2018). WNT ligands control initiation and progression of human papillomavirus-driven squamous cell carcinoma. Oncogene, 37(27), 3753-3762. doi:10.5167/uzh-151334
Zlobec, I., Terracciano, L., Jass, J. R., & Lugli, A. (2007). Value of staining intensity in the interpretation of immunohistochemistry for tumor markers in colorectal cancer. Virchows Archiv, 451(4), 763-769. doi:10.1007/s00428-007-0466-8
Zou, H. Y., Friboulet, L., Kodack, D. P., Engstrom, L. D., Li, Q., West, M., . . . Smeal, T. (2015). PF-06463922, an ALK/ROS1 inhibitor, overcomes resistance to first and second generation ALK inhibitors in preclinical models. Cancer Cell, 28(1), 70-81. doi:10.1016/j.ccell.2015.05.010
Zou, H. Y., Li, Q., Lee, J. H., Arango, M. E., McDonnell, S. R., Yamazaki, S., . . . Christensen, J. G. (2007). An orally available small-molecule inhibitor of c-met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Research, 67(9), 4408-4417. doi:10.1158/0008-5472.CAN-06-4443

117
Original publications
I Honkanen, T.J.*, Moilanen, T.*, Karihtala, P., Tiainen, S., Auvinen, P., Väyrynen, J.P., Mäkinen, M. & Koivunen, J.P. (2017). Prognostic and predictive role of spatially positioned tumour infiltrating lymphocytes in metastatic HER2 positive breast cancer treated with trastuzumab. Scientific Reports, 7(1), 18027.
II Honkanen, T.J., Tikkanen, A., Karihtala, P., Mäkinen, M., Väyrynen, J.P. & Koivunen, JP. (2019). Prognostic and predictive role of tumour-associated macrophages in HER2 positive breast cancer. Scientific Reports, 9(1), 10961.
III Honkanen, T., Wilenius, E., Koivunen, P., & Koivunen, J.P. (2017). HER2 regulates cancer stem-like cell phenotype in ALK translocated NSCLC. International Journal of Oncology, 51(2), 599-606.
IV Honkanen T.J. & Koivunen J.P. (2019). HER3 regulates cancer stem-like cell properties in ALK translocated NSCLC. Manuscript.
Reprinted with permission from Springer Nature (I, II) and Spandidos Publications
(III).
Original publications are not included in the electronic version of the dissertation.

118

A C T A U N I V E R S I T A T I S O U L U E N S I S
Book orders:Granum: Virtual book storehttp://granum.uta.fi/granum/
S E R I E S D M E D I C A
1513. Ollila, Meri-Maija (2019) The role of polycystic ovary syndrome (PCOS) andoverweight/obesity in women’s metabolic and cardiovascular risk factors andrelated morbidities
1514. Kotiaho, Antti (2019) Radiation dose determination using MOSFET and RPLdosimeters in x-ray imaging
1515. Hänninen, Sandra Lynn (2019) Transcriptional control of muscle cell excitation-contraction coupling : the role of activity and mitochondrial function
1516. Väyrynen, Otto (2019) Factors affecting aggressive oral tongue cancer invasionand development of in vitro models for chemoradiotherapy assay
1517. Euro, Ulla (2019) Risk factors for sciatica
1518. Lotvonen, Sinikka (2019) Palvelutaloon muuttaneiden ikääntyneiden fyysinentoimintakyky, sen muutos ja toimintakykyyn yhteydessä olevat tekijät ensim-mäisen asumisvuoden aikana
1519. Tuomikoski, Anna-Maria (2019) Sairaanhoitajien opiskelijaohjausosaaminen jaohjaajakoulutuksen vaikutus osaamiseen
1520. Raza, Ghulam Shere (2019) The role of dietary fibers in metabolic diseases
1521. Tiinanen, Suvi (2019) Methods for assessment of autonomic nervous systemactivity from cardiorespiratory signals
1522. Skarp, Sini (2019) Whole exome sequencing in identifying genetic factors inmusculoskeletal diseases
1523. Mällinen, Jari (2019) Studies on acute appendicitis with a special reference toappendicoliths and periappendicular abscesses
1524. Paavola, Timo (2019) Associations of low HDL-cholesterol level and prematurecoronary heart disease with functionality and phospholipid composition of HDLand with plasma oxLDL antibody levels
1525. Karki, Saujanya (2019) Oral health status, oral health-related quality of life andassociated factors among Nepalese schoolchildren
1526. Szabo, Zoltan (2019) Modulation of connective tissue growth factor and activinreceptor 2b function in cardiac hypertrophy and fibrosis
1527. Karttunen, Markus (2019) Lääkehoidon turvallinen toteuttaminen ikääntyneidenpitkäaikaishoidossa hoitohenkilöstön arvioimana

UNIVERSITY OF OULU P .O. Box 8000 F I -90014 UNIVERSITY OF OULU FINLAND
A C T A U N I V E R S I T A T I S O U L U E N S I S
University Lecturer Tuomo Glumoff
University Lecturer Santeri Palviainen
Senior research fellow Jari Juuti
Professor Olli Vuolteenaho
University Lecturer Veli-Matti Ulvinen
Planning Director Pertti Tikkanen
Professor Jari Juga
University Lecturer Anu Soikkeli
Professor Olli Vuolteenaho
Publications Editor Kirsti Nurkkala
ISBN 978-952-62-2343-8 (Paperback) ISBN 978-952-62-2344-5 (PDF) ISSN 0355-3221 (Print)ISSN 1796-2234 (Online)
U N I V E R S I TAT I S O U L U E N S I S
MEDICA
ACTAD
D 1528
AC
TAT
iia Honkanen
OULU 2019
D 1528
Tiia Honkanen
MORE EFFICIENT USE OFHER TARGETING AGENTSIN CANCER THERAPY
UNIVERSITY OF OULU GRADUATE SCHOOL;UNIVERSITY OF OULU,FACULTY OF MEDICINE;MEDICAL RESEARCH CENTER OULU;OULU UNIVERSITY HOSPITAL