molecular biology-2016 1 - university of...
TRANSCRIPT

Molecular Biology-2016 1
http://mysite.science.uottawa.ca/jbasso/molecular/home.htm

Molecular Biology-2016 2
GENERAL DIRECTIVES
1. Attendance is mandatory. Please be on time.
2. Shoes and appropriate dress must be worn at all times.
3. Leave outerwear, backpacks, and any other extraneous materials in the lockers outside of the
lab. It is strongly recommended that you have a lock. We are not responsible for lost or stolen
items.
4. Wear a lab coat and gloves at all times while working in the lab.
5. Remove your gloves anytime you walk out of the lab.
6. Remove your gloves when using either our or your own computers.
7. Always dispose of used pipettes, tips, microcentrifuge tubes, and other materials in the
biohazard bags provided so that they can be disposed of properly. Do NOT throw trash in the
autoclave bag.
8. Never lick your fingers, or put your fingers in your mouth.
9. No eating or drinking in the lab.
10. No radios, MP3 players, or CD players in the lab.
11. No use of cell phones or texting in the lab.
12. Notify the T.A. or instructor of any accident, no matter how minor.
13. Notify the T.A. or instructor of any breakage or malfunction of the equipment supplied.
Material you MUST have to work in the molecular biology lab:
A lab coat
A thin tipped permanent, preferably black, marker for labelling.
A note book to record your results. Any type is acceptable. Do not waste your money.
A USB key to save your pictures
A calculator. The use of cell phone calculators is not allowed
Optional but strongly recommended:
Notify the instructor of any safety or medical concerns so that appropriate accommodations can be
taken. For example, allergies, diabetes, hypoglycemia, epilepsy, exposed wounds, color blindness,
etc..
Notify the instructor of any special needs you may require so that appropriate accommodations can
be taken. For example, if you write your exams with SASS.

Molecular Biology-2016 3
Schedule
Exercise 1 Jan. 12-15 Concentrations and Dilutions
Restriction digests & agarose gel electrophoresis
Exercise 2 Jan. 19-22 Project I: Verifying the restriction map of a DNA insert
Project II: Site directed mutagenesis of GFP
Plasmid DNA isolation
PCR
Exercise 3 Jan. 26-29 Project I: Verifying the restriction map of a DNA insert
Project II: Site directed mutagenesis of GFP
Electrophoresis of GFP PCR amplicons
Digestion of GFP PCR amplicons and pGFPuv
Ligation of GFP amplicons
Exercise 4 Feb. 2-5 Project I: Verifying the restriction map of a DNA insert (XhoI)
Project II: Site directed mutagenesis of GFP
Transformation of ligation mixtures
Project III: Genomic fingerprinting
Isolation of human genomic DNA from cheek cells
PCR amplification of ApoC2 VNTR
PCR amplification of ApoB RFLP
Exercise 5 Feb. 9-12 Project II: Site directed mutagenesis of GFP
PCR screening of transformants
Analysis of colony PCR products
Patching
Project III: Genomic fingerprints
Project IV: Transcriptional control of GFP by the LacZ promoter
E.coli RNA isolation
STUDY BREAK Feb. 14-20
MIDTERM Feb. 23-26

Molecular Biology-2016 4
Exercise 6 Mar. 1-4 Project IV: Transcriptional control of GFP by the LacZ promoter
RNA gel electrophoresis
Northern transfer
Exercise 7 Mar. 8-11 Project IV: Transcriptional control of GFP by the LacZ promoter
Northern hybridizations
Project V: RT-PCR of the yeast ribosomal protein URP1
Yeast RNA isolation
RT-PCR
Exercise 8 Mar. 15-18 Project II: Site directed mutagenesis of GFP
Evaluation of GFP activity
Project IV: Transcriptional control of GFP by the LacZ promoter
Immunodetection of northern blots
Project V: RT-PCR of the yeast ribosomal protein URP1
Analysis of RT-PCR reactions
PRACTICAL FINAL EXAM Mar. 29-Apr. 1
THEORETICAL FINAL EXAM
Molecular Biology-2016 5
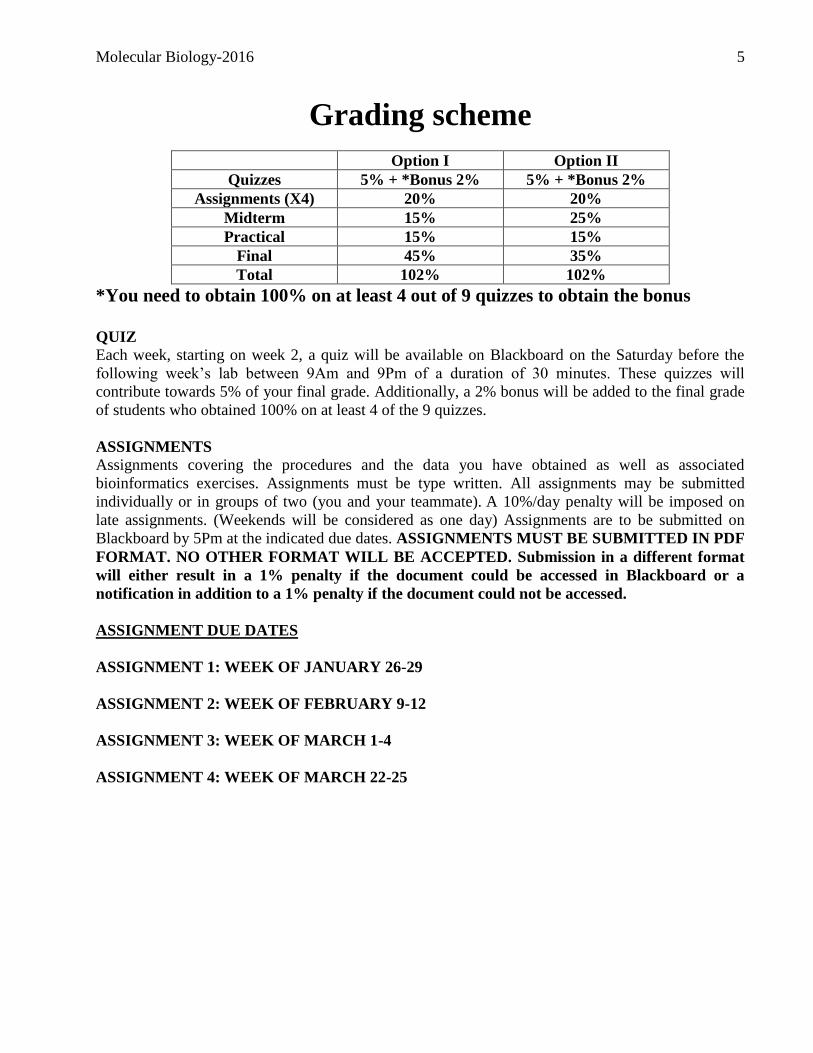
Grading scheme
Option I Option II
Quizzes 5% + *Bonus 2% 5% + *Bonus 2%
Assignments (X4) 20% 20%
Midterm 15% 25%
Practical 15% 15%
Final 45% 35%
Total 102% 102%
*You need to obtain 100% on at least 4 out of 9 quizzes to obtain the bonus
QUIZ
Each week, starting on week 2, a quiz will be available on Blackboard on the Saturday before the
following week’s lab between 9Am and 9Pm of a duration of 30 minutes. These quizzes will
contribute towards 5% of your final grade. Additionally, a 2% bonus will be added to the final grade
of students who obtained 100% on at least 4 of the 9 quizzes.
ASSIGNMENTS
Assignments covering the procedures and the data you have obtained as well as associated
bioinformatics exercises. Assignments must be type written. All assignments may be submitted
individually or in groups of two (you and your teammate). A 10%/day penalty will be imposed on
late assignments. (Weekends will be considered as one day) Assignments are to be submitted on
Blackboard by 5Pm at the indicated due dates. ASSIGNMENTS MUST BE SUBMITTED IN PDF
FORMAT. NO OTHER FORMAT WILL BE ACCEPTED. Submission in a different format
will either result in a 1% penalty if the document could be accessed in Blackboard or a
notification in addition to a 1% penalty if the document could not be accessed.
ASSIGNMENT DUE DATES
ASSIGNMENT 1: WEEK OF JANUARY 26-29
ASSIGNMENT 2: WEEK OF FEBRUARY 9-12
ASSIGNMENT 3: WEEK OF MARCH 1-4
ASSIGNMENT 4: WEEK OF MARCH 22-25

Molecular Biology-2016 6
Written Exams
All exams are open book. Access to the internet is allowed during both the final and midterm
examination.
The breakdown of the midterm exam (2.5 hours) will be as follows:
8 calculation problems (16 points)
1 calculation problem including a practical component. (4 points)
5 bioinfo exercises (5 points)
5 theoretical questions on bioinfo and molecular procedures (5 points)
2 out of 3 problems with an emphasis on data analysis and experimental design (10 points)
The final exam is cumulative and the breakdown (3 hours) will be as follows:
5 calculation problems (10 points)
10 bioinfo exercises (10 points)
5 theoretical questions on bioinfo and molecular procedures (5 points)
3 out of 4 problems with an emphasis on data analysis and experimental design (15 points)
Practical Exam
As with all exams, this exam is open book and will be given over a 2 hour period. Students will be
required to come to the lab, ON AN INDIVIDUAL BASIS, to carry out tasks which were routinely
performed during the semester. These may include, the preparation of PCR reactions, the preparation
of solutions, performing restriction digests, DNA isolations, migration of agarose gels, etc.. Grading
will be based on the results obtained and not on the approach used or their performance during the
course of the exam period.

Molecular Biology-2016 7
Exercise 1
What we are doing today!
Concentrations and dilutions
Restriction digests & agarose gel electrophoresis

Molecular Biology-2016 8
Introduction to concentrations One very important property of solutions that must be addressed is concentration. Concentration
generally refers to the amount of solute contained in a certain amount of solution. To deal with
concentration you must keep in mind the distinctions between solute, solvent and solution. Because
varying amounts of solute can be dissolved in a solution, concentration is a variable property and we
often need to have a numerical way of indicating how concentrated a solution happens to be. Over
the years a variety of ways have been developed for calculating and expressing the concentration of
solutions.
That can be done with percentages using measurements of weight (mass) or volume or both. It can
also be done using measurements that more closely relate to ways that chemicals react with one
another (moles).
In the pages that follow, several concentration types will be presented. They include volume
percent, weight percent, weight/volume percent, molarity (the workhorse of chemical
concentrations), and weight/volume.
You will get experience with more than one way of establishing the concentration of solutions. You
can prepare a solution from scratch and measure each of the components that go into the solution.
You can prepare a solution by diluting an existing solution. If an existing solution is colored, you can
determine its concentration by measuring the color intensity using colorimetry.
PERCENTAGE
The use of percentages is a common way of expressing the concentration of a solution. It is a
straightforward approach that refers to the amount of a component per 100. Percentages can be
calculated using volumes as well as weights, or even both together. One way of expressing
concentrations, with which you might be familiar, is by volume percent. Another is by weight
percent. Still another is a hybrid called weight/volume percent.
Volume percent is usually used when the solution is made by mixing two liquids.
For example, rubbing alcohol is generally 70% by
volume isopropyl alcohol. That means that 100
mL of solution contains 70 mL of isopropyl
alcohol. That also means that a litre (or 1000 mL)
of this solution has 700 mL of isopropyl alcohol
plus enough water to bring it up a total volume of
1 litre, or 1000 mL.
Volume percent = volume of solute
volume of solution x 100

Molecular Biology-2016 9
Mass percent is a way of expressing the concentration of a solution as the mass of solute/mass of
solution.
To calculate the mass percent of a solution, you must divide the mass of the solute by the mass of the
solution (both the solute and the solvent together) and then multiply by 100 to change it into percent.
Mass/volume percent
Another variation on percentage concentration is mass/volume percent. This variation measures the
amount of solute in grams but measures the amount of solution in millilitres. An example would be a
5% (m/v) NaCl solution. It contains 5 g of NaCl for every 100 mL of solution.
Volume percent = mass of solute (in g)
volume of solution (in mL) x 100
This is the most common way that percentage solutions are expressed in this lab course.
Mass percent = mass of solute
mass of solution x 100
As an example, let's consider a 12% by
weight sodium chloride solution. Such a
solution would have 12 grams of sodium
chloride for every 100 grams of solution. To
make such a solution, you could weigh out 12
grams of sodium chloride, and then add 88
grams of water, so that the total mass for the
solution is 100 grams. Since mass is
conserved, the masses of the components of
the solution, the solute and the solvent, will
add up to the total mass of the solution.
12 % NaCl solution = 12 g NaCl
100 g solution
12 g NaCl
(12 g NaCl + 88 g water) = 12% NaCl solution

Molecular Biology-2016 10
MOLARITY
Another way of expressing a concentration is
called molarity. Molarity is the number of moles
of solute dissolved in one litre of solution. The
units, therefore are moles per litre, specifically
it's moles of solute per litre of solution.
Molarity = moles of solute
litre of solution
Rather than writing out moles per litre, these units are abbreviated as M. So when you see M it
stands for molarity, and it means moles per litre (not just moles). You must be very careful to
distinguish between moles and molarity. "Moles" measures the amount or quantity of material you
have; "molarity" measures the concentration of that material. So when you're given a problem or
some information that says the concentration of the solution is 0.1 M that means that it has 0.1 moles
for every litre of solution; it does not mean that it is 0.1 moles.
WEIGHT/VOLUME
This means of expressing concentrations is very similar to that of percentages and is one of the most
popular ways used by molecular biologists. In contrast to percent, the concentration is expressed as a
mass per any volume the user wishes to use. Most commonly, these concentrations are expressed per
one measuring unit. For example, per 1 mL, 1 µL or 1L, etc. Essentially these expressions represent
the mass of solute present in a given amount of solution. For example a solution at a concentration of
1mg/mL contains 1 mg of solute in 1 mL of solution.
RATIOS
All the ways described above to express concentrations are done as a function of the total volume of
the solution which is the volume of the solvent and that of the solute. A common method used by
many molecular biologists and chemists is to express concentrations as ratios. In this case, the
relationship between the solvent and the solute is expressed independently of one another. For
example, we could say that the ratio between a solute and its solvent is 2:1. This indicates that for
two parts of the solute there is one part of solvent. Thus three parts total of solution.

Molecular Biology-2016 11
Dilutions and the use of micropipettors
Dilutions: Being able to prepare dilutions is essential for the preparation of several reagents, reaction mixes,
and solutions used in a molecular biology lab. Basically, dilutions serve to reduce the initial
concentration of a compound in order to reach a new desired concentration. Solutions prepared by
the formulation of dilutions may be composed of one or more ingredients. The number of ingredients
within the solution does not in any way influence the formulation of dilutions. A brief overview of
the formulation of dilutions is presented below. Make sure that you fully understand their
preparation, since you will be called upon to prepare them throughout the semester as well as on the
final exam.
To comprehend how dilutions are prepared, you must grasp the following three concepts:
Concentration, dilution factor, and the dilution.
A concentration is defined as the quantity of a given element for a total volume of solution. Since a
quantity may be expressed in several different ways, concentrations are expressed as the unit of
measure of the quantity of the element/total final volume.
Ex. Grams/Litre
Molecules/Litre
Moles/Litre
Etc.
The dilution factor represents the multiple by which an initial concentration must be divided by in
order to obtain the desired final concentration. For example, if a solution contains 30g of caffeine per
litre of solution and you wish to reduce the caffeine concentration to 0.3 g/L, then you will have to
divide the initial concentration by 100, which represents the dilution factor. You can use the
following formula in order to determine a dilution factor.
Dilution Factor = Initial Concentration or What I have
Final Concentration What I want
The dilution represents the fraction of the component being investigated. For example, in the
previous problem a dilution of 1/100 was prepared. The dilution is expressed as a fraction of 1 over
the dilution factor. That is to say that the initial solution is represented as a fraction of the original
over the total. For example if you determined that a 1/100 dilution has to be prepared, this means that
one hundredth of the new solution must be represented by the original solution. Therefore for a total
volume of let’s say 2mL, 0.02mL must be represented by the original solution.

Molecular Biology-2016 12
Preparing a solution that requires the dilution of more than one ingredient:
The basis for the formulation of a solution that requires the dilution of more than one ingredient is
the same as that of a solution with only one ingredient. The only difference is the volume of solvent
that must be added.
Let’s take as an example that we wish to prepare 10mL of a 0.1M solution from a stock solution of
2M.
To calculate the dilution factor: What I have = 2M = 20
What I want 0.1M
Thus the required dilution is 1/20, which means that one twentieth of the total must be represented by
the original solution.
Therefore to prepare 10mL we must add 0.5mL of the stock solution and make up the volume with
9.5mL of solvent.
If the solution to be prepared includes a second ingredient; let’s say that the final concentration of
this one must be 0.5M and that the stock solution is 3M.
Once again to calculate the dilution factor: What I have = 3M = 6
What I want 0.5M
Thus the required dilution is 1/6, which means that one sixth of the total volume must be represented
by the original solution.
Therefore, to prepare 10mL one must add 0.5mL of the first solution, 1.7mL of the second solution
and complete to the total volume with 7.8mL of solvent.
If the above explanations are not sufficient you can always consult the following web site:
http://www.wellesley.edu/Biology/Concepts/Html/dilutions.html

Molecular Biology-2016 13
Use of micropipettors
Micropipettes are indispensable tools in modern biology laboratories. How accurate are yours? How
precise are yours? What is the difference between accuracy and precision?
Accuracy refers to performance with respect to a standard value. Precision refers to the reliability or
repeatability of performance and doesn’t necessarily depend on a standard at all. These two features
are independent of each other. It is possible to have an instrument that is precisely inaccurate (or
accurately imprecise)!
Target analogy to illustrate accuracy and precision
Here, the bull’s-eye represents the "standard" against which accuracy is judged. To simultaneously
evaluate the accuracy and precision of your pipettes you must conduct multiple measurements and
compute the % Error of the Mean and Standard Deviation of the measurements for each pipette.
Directions for use of Gilson micropipettors
You have 3 sizes of micropipette. The P-1000, the P-200 and the P-20. The model numbers refer to
the maximum volume in microliters. Initially you will need to determine which pipette to use in a
given circumstance. For example, if you need to transfer 0.18 ml you would probably need to use the
P-200 because 0.18 ml = 180 µL and because the P-200 will measure this volume with greater
accuracy and precision than the P-1000 will.
Rules:
Always use a disposable tip. The P-20 and P-200 use the smaller tips and the P-1000 uses the
bigger ones.
Never draw any fluid into the white barrel of the pipette itself.
Never lay a pipette down while there is fluid in the tip. The fluid may accidentally find its
way into the barrel.
Never turn the adjustment scale below or above the full range settings.
To maximize precision, Always use the smallest volume pipette for a given total volume.
1. Set the desired volume:
Turn the volume up just a bit past the desired setting, then back down.
2. Attach a tip:
Press it on firmly, with a slight twisting motion. The tip must make an air-tight seal
with the pipette barrel.
Accurate & Precise Precise & Inaccurate Imprecise & Inaccurate

Molecular Biology-2016 14
3. Depress the plunger to first stop.
4. Insert the tip in the liquid you want to transfer. Not far, just a bit below the surface.
5. Slowly release plunger.
6. As you withdraw the tip, touch it to the side wall of the tube to remove excess fluid from the
exterior.
7. To dispense, depress plunger slowly to the first stop; then depress all the way.
Never dispense a small volume into thin air. Always dispense into a liquid or onto the
wall of a tube so that adhesion will draw the expelled liquid off the tip.
8. With the plunger still fully depressed, remove tip from the liquid.
REMEMBER THAT YOUR MICROPIPETTORS ARE EXPENSIVE PIECES OF
EQUIPMENT!
Dilutions exercise with micropipettors (TO BE PERFORMED BY EACH STUDENT)
These exercises are included so that each individual will be familiar with the basics of solution
preparation, micropipetting, and the use of tips. (ALWAYS LABEL YOUR TUBES!! ).
Materials: Solution I (1% Compound “A” (m/v); M.W. 800g/mole)
Solution II (1.2M Compound “B” M.W. 60g/mole; Density of compound “B”: 1.6g/mL)
Method:
1. Prepare 1mL of each of the following solutions from the above stock solutions.
a. A 1.5mM solution of compound “A”.
b. A 0.36% (m/v) solution of compound “B”.
c. A 6% (v/v) solution of solution I.
d. A solution containing 0.5mg of compound “A” and 0.1% (v/v) of compound “B”.
e. A solution representing the following ratio: solution I: solution II : water : 2:1:2
2. Transfer 100 µL from each of the solutions to the appropriate wells of a 96 well plate as shown
below:
96 well microtiter plate layout (One plate/table)
Soln. a Soln. b Soln. c Soln. d Soln. e Person 1 Group 1
Soln. a Soln. b Soln. c Soln. d Soln. e Person 2
Soln. a Soln. b Soln. c Soln. d Soln. e Person 1 Group 2
Soln. a Soln. b Soln. c Soln. d Soln. e Person 2
Soln. a Soln. b Soln. c Soln. d Soln. e Person 1 Group 3
Soln. a Soln. b Soln. c Soln. d Soln. e Person 2
Soln. a Soln. b Soln. c Soln. d Soln. e Person 1 Group 4
Soln. a Soln. b Soln. c Soln. d Soln. e Person 2
Molecular Biology-2016 15
More dilutions: Determining the concentration of DNA (Groups of 2) As with most compounds, the concentration of nucleic acids can be determined
spectrophotometrically. To do so, the absorbance of the compound is determined at the wavelength
for maximal absorption. In the case of nucleic acids, this is in the UV range at a wavelength of
260nm. In the following exercise you will prepare samples at different known concentrations of
DNA and then use the absorbance values obtained to generate a standard curve representing
absorbance Vs DNA concentration (NOT AMOUNT). The standard curve generated will then allow
you to determine the concentration of an unknown DNA sample as well as determining the
relationship between DNA concentration and absorbance at a wavelength of 260nm. Specifically you
want to determine what concentration of DNA (in µg/mL or ng/µL) is equal to an absorbance of 1.0.
Materials
Salmon sperm DNA (200µL at 0.5mg/mL)
Salmon sperm DNA (200µL of unknown concentration)
Method:
1. Prepare 500µL in water of each of the following DNA standard solutions from the DNA sample
of known concentration: 0.0, 0.05, 0.025, 0.01, 0.005 and 0.0025mg/mL.
2. Prepare 500µL samples in water representing 1/4 and 1/10 dilutions of the unknown DNA
sample.
3. Transfer 200µL of each of the standard DNA solutions to a microtiter plate as indicated in the
plan below.
4. Transfer 200µL of each of the unknown DNA solutions to a microtiter plate as indicated in the
plan below.
5. Measure absorbance at 260nm.
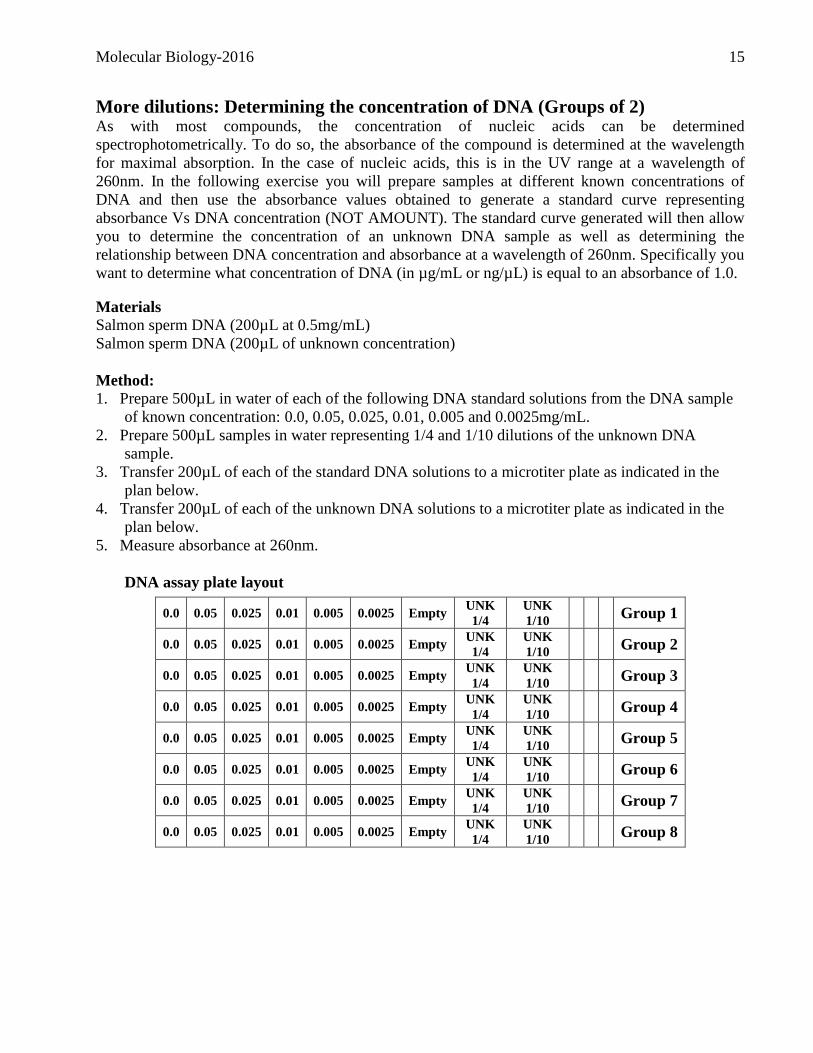
DNA assay plate layout
0.0 0.05 0.025 0.01 0.005 0.0025 Empty UNK
1/4
UNK
1/10 Group 1
0.0 0.05 0.025 0.01 0.005 0.0025 Empty UNK
1/4
UNK
1/10 Group 2
0.0 0.05 0.025 0.01 0.005 0.0025 Empty UNK
1/4
UNK
1/10 Group 3
0.0 0.05 0.025 0.01 0.005 0.0025 Empty UNK
1/4
UNK
1/10 Group 4
0.0 0.05 0.025 0.01 0.005 0.0025 Empty UNK
1/4
UNK
1/10 Group 5
0.0 0.05 0.025 0.01 0.005 0.0025 Empty UNK
1/4
UNK
1/10 Group 6
0.0 0.05 0.025 0.01 0.005 0.0025 Empty UNK
1/4
UNK
1/10 Group 7
0.0 0.05 0.025 0.01 0.005 0.0025 Empty UNK
1/4
UNK
1/10 Group 8

Molecular Biology-2016 16
Restriction digests & agarose gel electrophoresis (Groups of 2) The use of restriction endonucleases to cleave DNA at specific sites was a key breakthrough in
opening up the field of molecular biology in the mid-1970's. There are now several hundred different
restriction enzymes that are commercially available, and you can find detailed information about
some (including the ones that you will be using in this course) in catalogues from commercial
suppliers. Their specificity makes them very useful for several tasks including but not limited to the
mapping of DNA, cloning and subcloning of DNA, etc. The goal of this exercise is to introduce
you to restriction enzyme mapping and analysis. At the end of this exercise you should be able
to answer the following questions:
Which enzymes cut within the plasmid insert?
Which enzymes do not cut within the plasmid insert?
What is the size of the plasmid insert?
What are the possible positions of the different restriction sites?
The DNA fragment was inserted in which restriction site within the MCS?
For more information on restriction enzymes and their use consult the following web site.
http://askabiologist.asu.edu/expstuff/mamajis/restriction/restriction.html
Agarose gel electrophoresis
Agarose gel electrophoresis is the most commonly used technique to answer these questions. This
technique involves the separation of DNA molecules based on their size and conformation through
an agarose gel using an electric current. The electrophoretic migration of a DNA fragment through
agarose is inversely proportional to the logarithm of its molecular weight (for certain size classes
under defined conditions). [Tip! When you are estimating the sizes of restriction fragments, be sure
to keep in mind the accuracy that is warranted - i.e. the number of significant digits.] The
concentration of agarose used depends on the sizes of the DNAs being studied; for separating linear
DNAs in the 0.5 kb to 10 kb range, a 1% agarose gel is commonly used. After electrophoresis the
gels will be viewed under ultraviolet light and a digital picture will be taken. The intensity of
fluorescence is proportional to the amount (and length) of linear DNA and this method can be used
for a rough estimate of the quantity of DNA in the samples.
Agarose gel electrophoresis is performed at voltage levels that are potentially hazardous. The gel
boxes have a safety interlock feature and the leads must be removed before opening the lid.
ALWAYS TURN THE POWER SUPPLY OFF BEFORE DISCONNECTING THE LEADS.
Ethidium bromide, which is used in staining agarose gels to visualize DNA under ultraviolet light, is
a potential carcinogen; so always wear gloves when handling anything containing it. The UV light
source is also extremely hazardous to skin and particularly your eyes, so be sure to use proper
protection (gloves, lab coat, face mask) when viewing your agarose gels.

Molecular Biology-2016 17
Each gel apparatus has two gel casting plates and three combs (8-wells, 10 wells, and 14-wells
holding about 14L, 12L, and 6L respectively). Remember that the 8 and 10-wells gels require
about twice as much DNA as the 14-wells one because of the difference in well size. Also if you
wish to visualize fairly small fragments (<500bp) which are less intensely stained you may consider
using more DNA and perhaps a higher percentage of agarose (e.g. 1.5% instead of the more standard
1%) for a better resolution.
Method:
A. Preparation of an agarose gel (8 well comb)
Materials:
Agarose
10X TBE
1. Prepare 200mL TBE buffer at a final concentration of 1X in your graduated cylinder. Mix well.
2. Mix the appropriate amount of agarose to obtain a final concentration of 1.0% m/v) in 25 mL of
1X TBE.
3. To dissolve the agarose, microwave (25-45 sec.). Loosely cover the mouth of the flask with
plastic to prevent evaporation. Once the agarose is totally dissolved, allow the flask to cool to 50-
60 oC (5 minutes or so).
4. Add 50 L of a stock solution of ethidium bromide at 1mg/mL (CAUTION! CARCINOGEN!)
and mix well.
5. Pour into the gel tray. After pouring the gel, place the 8 well comb (Well capacity approx. 14 L)
and remove any air bubbles (e.g. with a small tip). Allow to solidify at least 15-20 minutes.
6. Once the gel has solidified, remove the dams. Pour a sufficient amount of 1X TBE to cover the
gel by approx. 0.5cm.
7. Carefully remove the comb.

Molecular Biology-2016 18
B. Checking the good operation of your gel
8. Attach electrodes so that the cathode (negative electrode) is at the "origin" end of the gel and the
samples will run through the gel towards the anode (positive electrode). (CAUTION! HIGH
VOLTAGE!)
9. Set the power supply at 100V. Turn on the power supply. Verify that the current is in the 40-55
mA range. If it is not there is a problem.
Possible problems:
The buffer is at the wrong concentration
The buffer in the gel is at the wrong concentration
You forgot to put buffer in the gel
C. Analysis of restriction digests
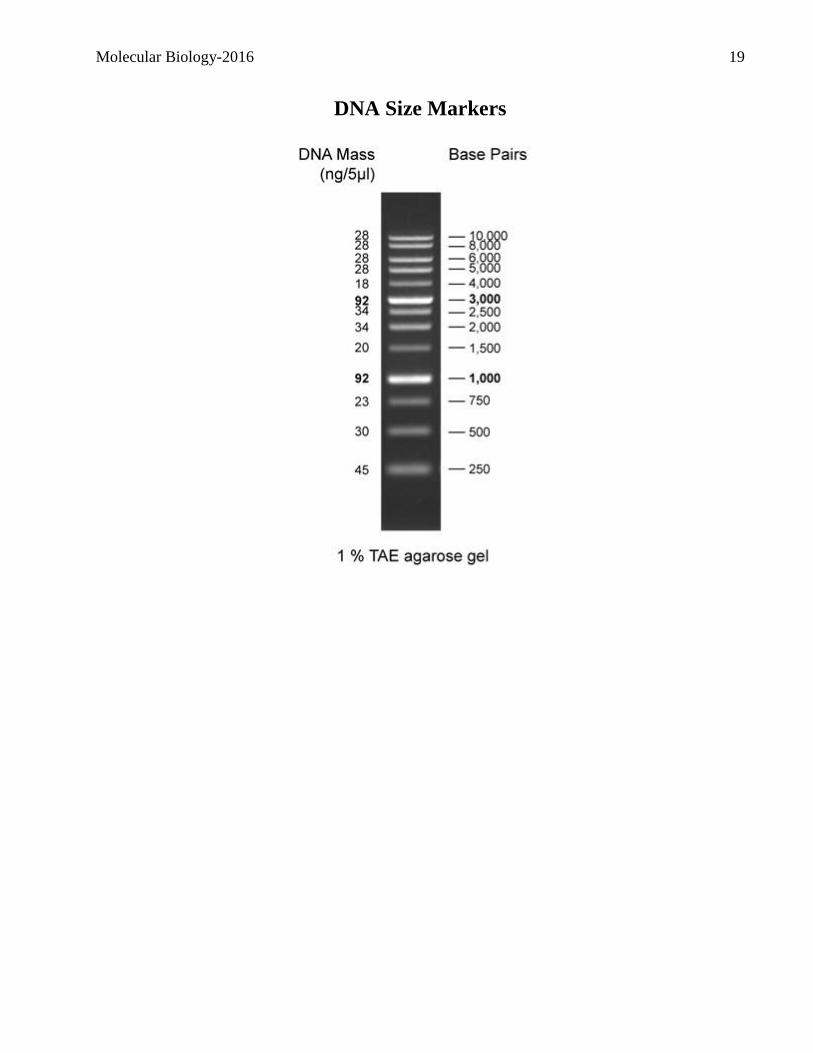
10. Load the following DNA samples. The unknown is a recombinant with pUC9 as the vector:
a. 1Kbp molecular weight markers (5 µL)
b. Recombinant pUC9 plasmid DNA, 5 µL
c. Recombinant pUC9 plasmid cut with BamHI, 5 µL
d. Recombinant pUC9 plasmid cut with EcoRI, 5 µL
e. Recombinant pUC9 plasmid cut with HindIII, 5 µL
f. Recombinant pUC9 plasmid cut with EcoRI + HindIII, 5 µL
g. Recombinant pUC9 plasmid cut with PstI, 5 µL
h. Plasmid pUC9 cut with BamHI, 5 µL
11. Carry out the electrophoresis at 100V for approx. 45 minutes and ask a teaching assistant to take
a picture.
Molecular Biology-2016 19
DNA Size Markers
Molecular Biology-2016 20

Molecular Biology-2016 21
Exercise 2
What we are doing today!
Project I: Verifying the restriction map of a DNA insert
Project II: Site directed mutagenesis of GFP
Plasmid DNA isolation
PCR

Molecular Biology-2016 22
Restriction enzymes and agarose gel electrophoresis Following most restriction digest reactions it is necessary to answer the following questions:
Was the DNA digested?
Was the digest complete or partial?
Is there partially digested DNA?
Is there partially undigested DNA?
Have the DNA samples completely digested? If there is only partial cleavage of the DNA (for
example - low enzyme activity due to inappropriate reaction conditions or impurities in the DNA that
inhibit the enzyme), the slower migrating species (representing uncleaved fragments containing a site
for the enzyme used) are usually present in sub-stoichiometric amounts. In other words, because the
UV fluorescence intensity is proportional to the amount of ethidium bromide bound (and therefore
the amount/length of DNA), these species are not as intense as they would be if present in equimolar
amounts to the completely digested fragments.
Are the calculated sizes of restriction fragments internally consistent? If you are running different
digests of cloned DNA, it is important to check that the calculated sizes of the fragments add up to
approximately the same value in each of the different lanes (that is, the size of the intact DNA
molecule). Because there are inaccuracies in estimating sizes from the standard markers (and those in
the non-linear part of the curve have greater error), there usually is only approximate agreement
(perhaps 200-300 bp differences for a 5 Kbp recombinant plasmid). If there are greater discrepancies,
consider the following: Might there be co-migrating species? (Clues from relative fluorescence
intensity) Are partial digestion products being included in the calculation? Might there be multiple
low MW fragments (of correspondingly lower intensity that you cannot easily see)? Could you be
using the size of fragments which are the result of a partial digest in your calculation? Are linear size
markers being used to size linear (as opposed to non-linear) DNA molecules? Remember that linear,
relaxed circular and supercoiled double-stranded DNA molecules have different migration properties
under our conditions of agarose gel electrophoresis. Because the size markers that we are using are
linear ones, can they be used to estimate the size of an uncut recombinant plasmid?

Molecular Biology-2016 23
Project I: Verifying the restriction map of a DNA insert (Groups of 2) The goal of this project is to give you an understanding of the technique of restriction mapping. In
the next few weeks you will be asked to use both experimental and bioinformatics approaches to
characterize a cloned DNA fragment. Each group of two will be working with a plasmid that
contains an insert representing one of the genes listed under the heading "sequences" > Unknown
"genes" on this course's web site. Amongst a group of four (per end of table) each group of two will
have the same unknown but in different orientations. These inserts were all obtained from a genomic
library created in the cloning vector pUC19. Briefly, genomic DNA was isolated from an organism,
digested, and the resulting fragments were then ligated into the vector pUC19 which was linearized
with an appropriate restriction enzyme whose site is within the multiple cloning site. (See figure on
next page).
The goals of this project are:
Using an experimental approach to: o Determine the insertion site
o Verify the size of the insert
o Verify the orientation of the insert
o Determine the restriction map
Tips for working with restriction enzymes
Always keep the enzyme stocks on ice when they're out of the -20oC freezer, and plan your setup for
that time to be as short as possible.
The enzyme is always the last component added to a reaction mixture, and it is added directly into
the solution in the bottom of the tube rather than as a drop on the side of the tube.
Make sure the solution is well mixed (e.g. "flicking" the tube with your finger) and if any drops are
along the side of the tube, spin them down using the microcentrifuge. Alternatively, the sample can
be mixed by vortexing (unless you are using high molecular weight genomic DNA in which case
there is the danger of shearing it).
Never touch the end of the plastic pipette tips with your fingers or anything except the solution that
you are transferring.
Always use a clean tip for each operation and dispose of the used tips at once.
In compliance with the BIOHAZARD GUIDELINES, all disposable items (micropipette tips,
microcentrifuge tubes, etc.) used when working with recombinant DNAs and bacterial host cells
must be placed in special waste containers (that is, the disposal boxes at your work stations which
you will then transfer to orange bags) to be autoclaved before disposal.

Molecular Biology-2016 24
The following enzymes do not cut pUC19 : AdeI, AloI, ApaI, AscI, BaeI, BbvCI, BclI, BcuI, BglII, BoxI, BpiI, BplI, Bpu10I,
Bpu1102I, BsaAI, BsaBI, BseRI, BsgI, BshTI, BsmFI, Bsp68I, Bsp119I, Bsp120I,
Bsp1407I, BspTI, Bst1107I, BstXI, Bsu15I, BtrI, Cfr42I, CpoI, DsaI, Eco32I,
Eco47III, Eco52I, Eco72I, Eco81I, Eco91I, Eco105I, Eco130I, Eco147I, FseI, Kpn2I,
KspAI, MlsI, MluI, Mph1103I, MssI, MunI, Mva1269I, NcoI, NheI, NotI, PacI, PauI,
PdiI, Pfl23II, PsiI, Psp5II, PsyI, SacII, SanDI, SexAI, SfiI, SgfI, SgrAI, SmiI,
SrfI, SstII, Van91I, XagI, XcmI, XhoI, XmaJI.
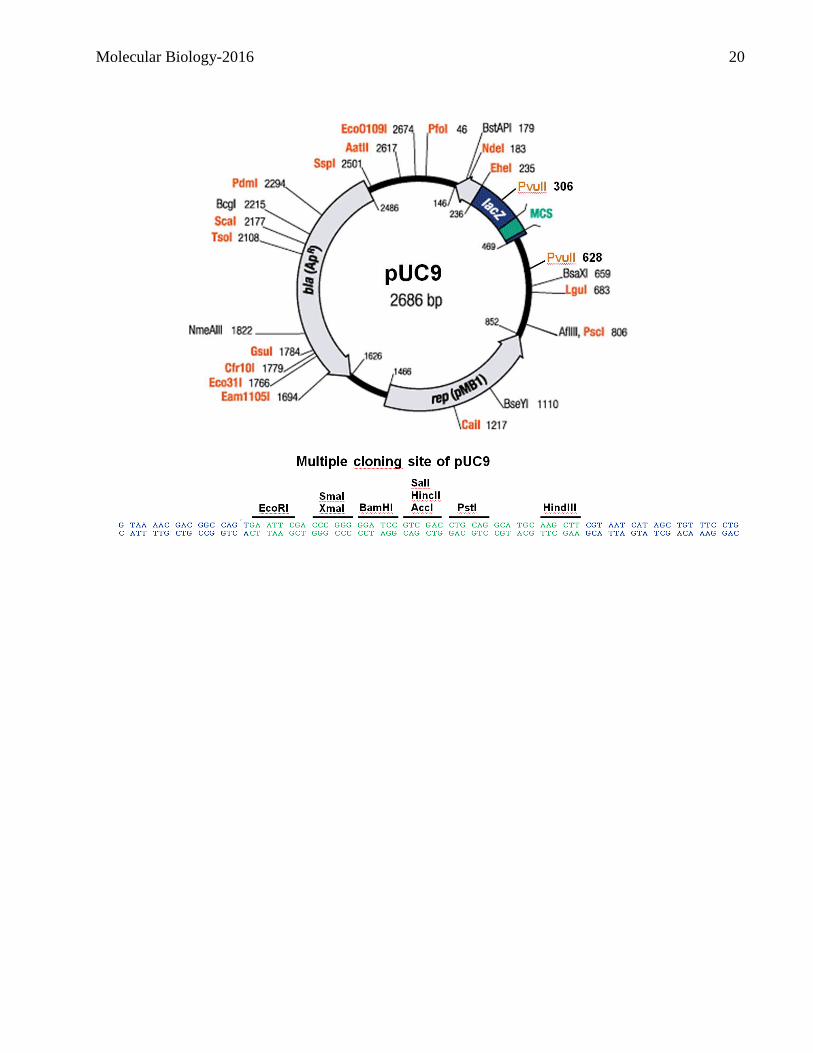
The pUC vectors are small, high copy number, plasmids that have a multiple cloning site (MCS), the
pMB1 origin of replication responsible for the replication of the plasmid (source – plasmid pBR322),
and the bla gene, coding for beta-lactamase, which confers resistance to ampicillin (source – plasmid
pBR322). Note that all the restriction sites within the multiple cloning site occur only once.
Amorce univ. reverse
PvuII
PvuII
Univ. forward primer
Univ. reverse primer

Molecular Biology-2016 25
Tips for restriction enzyme cleavage of plasmid DNA
Amount of DNA to use:
The ease of detection of your restriction fragments (by UV fluorescence after ethidium bromide
staining where fluorescence intensity is proportional to the mass of DNA in a fragment) will depend
on the amount of DNA used (and also parameters such as gel well width, sharpness of bands, etc).
Typically, 200-600ng plasmid DNA/lane is used for digests with a single enzyme. For digests that
result in many fragments you may need to restrict 400-1,000ng so that the smallest fragment can be
visualized after staining (WHY??)
After taking DNA samples from the freezer, be sure to completely thaw them before pipetting (to
obtain the correct amount of DNA in a given volume).
Your digestions will be carried out in the presence of a commercial buffer from Fermentas.
Restriction enzymes vary in their preferred salt concentration and this can be achieved by using
buffers with different salt concentrations.
A few enzymes require an incubation temperature other than 37oC. Also note that restriction
enzymes having the same recognition sequence are called isoschizomers (e.g. SacII and SstII) and
that some enzymes with very similar names (EcoRI, EcoRV) have distinctively different recognition
sequences (so be sure to read the labels on the enzyme tubes carefully).
In principle, 1 unit of restriction enzyme digests 1 g of purified DNA to completion in 1 hr.
However, because we are often using crude DNA preparations, we routinely increase the amount of
enzyme used and for high complexity genomic DNAs, incubation times are usually extended as well.
Typically restriction enzymes are supplied in concentrations of 5-10 units/L.
The final volume of enzyme should not exceed 1/10 the total reaction volume, because the
glycerol in enzyme stocks, added to prevent freezing upon storage in the freezer, may inhibit the
reaction. Also note that solutions containing glycerol are more difficult to pipette accurately than
aqueous solutions, so carefully monitor the volumes in the pipette tips.
For an overview on restriction mapping visit the following web sites:
http://faculty.plattsburgh.edu/donald.slish/RestMap/RestMapTutorial.html
http://www.vivo.colostate.edu/hbooks/genetics/biotech/enzymes/maps.html
http://wps.prenhall.com/esm_klug_essentials_5/17/4576/1171606.cw/content/index.html

Molecular Biology-2016 26
List of restriction enzymes available
ENZYME SITE
BamHI G▼GATCC
HindIII A▼AGCTT
PstI CTGCA▼G
PvuII CAG▼CTG
ScaI AGT▼ACT
XhoI C▼TCGAG
▼- indicates cleaved phosphodiester linkage

Molecular Biology-2016 27
Method & Materials:
Some of the restriction enzymes you will be using can be found in your freezer box. If they
are not in your freezer box they are in the freezer box of the group facing you on the same
end of bench as you.
Your unknown recombinant plasmid, at a concentration of 100g/mL is in your freezer box.
10X Restriction enzyme buffers are in your freezer box.
Setting up your restriction digests: Since different enzymes require different buffers, you will need to be flexible in making a restriction
digest. Prepare a chart listing all the components that you intend to add and their volumes before you
begin. As you add a component to your labelled tube, cross out the entry on your list. Choose the
restriction buffer which gives a 100% activity with the chosen enzyme. (Consult the table on the next
page)
Reaction mix for a typical single digest in a 30µL volume:
Ingredients Final Concentration/µL
DNA (100ng/µL) 250ng total
10X restriction buffer 1X
Enzyme (approx. 10 units/µL) 10 units
Water Complete to 30µL
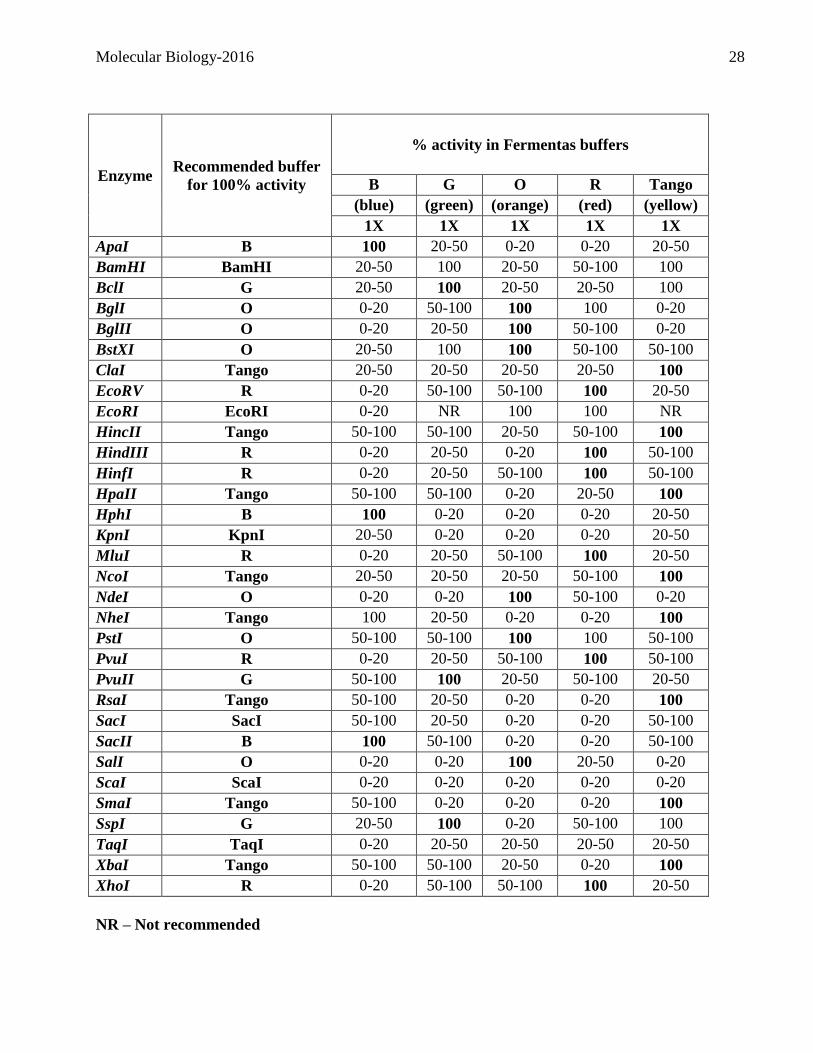
Molecular Biology-2016 28
Enzyme Recommended buffer
for 100% activity
% activity in Fermentas buffers
B G O R Tango
(blue) (green) (orange) (red) (yellow)
1X 1X 1X 1X 1X
ApaI B 100 20-50 0-20 0-20 20-50
BamHI BamHI 20-50 100 20-50 50-100 100
BclI G 20-50 100 20-50 20-50 100
BglI O 0-20 50-100 100 100 0-20
BglII O 0-20 20-50 100 50-100 0-20
BstXI O 20-50 100 100 50-100 50-100
ClaI Tango 20-50 20-50 20-50 20-50 100
EcoRV R 0-20 50-100 50-100 100 20-50
EcoRI EcoRI 0-20 NR 100 100 NR
HincII Tango 50-100 50-100 20-50 50-100 100
HindIII R 0-20 20-50 0-20 100 50-100
HinfI R 0-20 20-50 50-100 100 50-100
HpaII Tango 50-100 50-100 0-20 20-50 100
HphI B 100 0-20 0-20 0-20 20-50
KpnI KpnI 20-50 0-20 0-20 0-20 20-50
MluI R 0-20 20-50 50-100 100 20-50
NcoI Tango 20-50 20-50 20-50 50-100 100
NdeI O 0-20 0-20 100 50-100 0-20
NheI Tango 100 20-50 0-20 0-20 100
PstI O 50-100 50-100 100 100 50-100
PvuI R 0-20 20-50 50-100 100 50-100
PvuII G 50-100 100 20-50 50-100 20-50
RsaI Tango 50-100 20-50 0-20 0-20 100
SacI SacI 50-100 20-50 0-20 0-20 50-100
SacII B 100 50-100 0-20 0-20 50-100
SalI O 0-20 0-20 100 20-50 0-20
ScaI ScaI 0-20 0-20 0-20 0-20 0-20
SmaI Tango 50-100 0-20 0-20 0-20 100
SspI G 20-50 100 0-20 50-100 100
TaqI TaqI 0-20 20-50 20-50 20-50 20-50
XbaI Tango 50-100 50-100 20-50 0-20 100
XhoI R 0-20 50-100 50-100 100 20-50
NR – Not recommended

Molecular Biology-2016 29
Your digests:
1. Setup the following restriction digests of 0.25g of DNA.
BamHI
HindIII
PstI
PvuII
ScaI
2. Prepare a digestion control, which contains all the components except for enzyme.
3. Incubate at 37oC for 60 minutes.
4. While your digestions are incubating, prepare a 1.0% agarose gel containing ethidium bromide.
5. Following the incubation period, transfer 5µL of your digestions to new appropriately labelled
tubes and then add 5X DNA loading buffer to each of them in order to obtain a final
concentration of 1X or more.
6. Store the remainder of each of your digests (properly labelled) at –20oC.
7. Load the samples containing the loading buffer in the gel.
8. Load 5μL of the digested pUC9 vector which has been prepared for you.
9. Load 5µL of the molecular weight ladder.
10. Also load on this gel 5μL, not including the loading buffer, of the plasmid pGFPuv isolated by
the alkaline lysis and the Qiagen methods (performed later)
11. Carry out the electrophoresis at 100V.
12. Following the electrophoresis, examine your gel under the UV light and take a picture for
analysis.
Restriction analysis with double digests Following the analysis of your digests, you should have a list of restriction enzymes that cut within
the cloned fragment of your plasmid and the sizes of the fragments generated by these digests. Based
upon this information you will generate a preliminary restriction map (or maps if there was no unique
unambiguous map).
Next week you are to build upon that data and test the validity of the map that you proposed. Your
goal is to use double digests to confirm as precisely as possible the position of the restriction sites
within the insert and to resolve any ambiguity.
A simple example will illustrate this approach. Suppose after the first round of digestions with single
enzymes you found a single restriction site in the insert for the enzyme X and no restriction sites for
EcoRI. Knowing that EcoRI can be found in the polylinker, then a double digest with EcoRI + X will
give the distance from X to the known EcoRI site.
For next week's lab you must determine which double digests will allow you to most precisely map
all restriction sites located within your insert. Note that you may use a combination of any of the
restriction enzymes used in this week's lab.

Molecular Biology-2016 30
Project II: Site directed mutagenesis of GFP
Plasmid DNA isolation (Groups of 2)
Most methods used to isolate DNA rely on the disruption of cells in the presence of strong
denaturants. Disruption may be by freezing and fracturing cells by grinding or blending or by
chemical lysis with strong alkali. The denaturants are essential to inactivate exogenous and
endogenous nucleases, which would otherwise degrade the DNA. Examine the components of the
DNA extraction buffers and determine the purpose of each chemical.
Plasmids are non-obligate, circular, extrachromosomal bacterial replicons. Plasmid DNA isolation
requires separation of this DNA from the chromosomal DNA in the bacterial cell as well as from the
polysaccharides, lipids and proteins that constitute the cell. Subsequent manipulation, especially
enzymatic modification, of the plasmid DNA requires that it be free of these impurities.
Purifying plasmid DNA by alkaline lysis
In this protocol, cells are lysed by strong alkali (NaOH) and the proteins are denatured by a strong
alkali and a strong detergent (SDS). The detergent complexes are then precipitated with a
neutralizing salt (KOAC). The plasmid is separated from the bacterial DNA by virtue of the
plasmid's relative stability in alkali. Leaving the plasmid preparation in alkali for too long will
destroy the plasmid DNA as well. The chromosome is also attached to the membranes and will be
precipitated by the salt and detergent. It is therefore important not to mix the solution too vigorously
and release the chromosomal DNA from it trap. The plasmid is smaller and will remain free in
solution. The plasmid solution is then separated from the cellular debris by centrifugation and further
concentrated by an alcohol precipitation.

Molecular Biology-2016 31
A. Preparing your solutions:
Prepare 1mL of solutions I & II as well as TE from the following stock solutions:
10M NaOH
10% (m/v) SDS
0.5M glucose
1M Tris-Cl (pH 8.0)
0.5M EDTA
3M KOAc pH 5
Isopropanol
RNase 10mg/mL
Solution I:
50mM Glucose (buffer)
25mM Tris-Cl (pH 8.0) (buffer)
10mM EDTA (pH 8.0) (Chelator)
Solution II:
0.2M NaOH (from 10M stock) (Alkali)
1% (m/v) SDS (Detergent)
T.E.:
10mM Tris-Cl pH 8.0
1mM EDTA pH 8.0
B. Protocol for isolation of pGFPuv plasmid:
1. Spin at maximal speed 1.5mL of the plasmid containing E.coli suspension given to you in the
microcentrifuge for 1 minute.
2. Carefully pour off supernatant without disturbing the cell pellet. Use a micropipettor to remove
any remaining supernatant.
3. Add 200L of Solution I to the pellet and suspend the pellet by vortexing.
4. Add 400L of solution II. Cap the tube and mix by inversion.
5. Add 300L of ice cold KOAc. Mix well and keep on ice for 5 minutes.
6. Spin tube in the microcentrifuge at maximum speed for 5-10 minutes. This pellets the proteins
and chromosomal DNA along the side of the tube.
7. Transfer 700µL of the supernatant to a new microcentrifuge tube. Add an equal volume of
isopropanol. Close cap and mix well by rapidly inverting the tubes.
8. Spin tube at maximum speed for 5 minutes. Pour off supernatant carefully.
9. The white pellet at the bottom of the tube contains plasmid DNA and RNA.
10. Suspend the pellet in 50L TE pH8.0.
11. Add 1L of an RNAse solution at 10mg/mL and incubate at 37oC 5-10min.
12. You will need a sample of this preparation in another section of this lab exercise.

Molecular Biology-2016 32
Plasmid DNA isolation with the QIAGEN kit Another method for the isolation of plasmid DNA makes use of commercial kits such as the one
offered by Qiagen. Fundamentally, this technique is based on the alkaline lysis procedure that you
previously performed. In order to compare the two, you will repeat the DNA purification from the
same bacterial culture with the Qiagen kit. At the end of this procedure you are expected to be able to
compare these two methods with respect to their similarities and differences.
Step 1
Steps 2-5
Steps 6-7
Steps 8-9
Step 10
Bacterial pellet
Wash
Elute
Bind
Suspend
Lyse
Neutralize
Pure plasmid DNA

Molecular Biology-2016 33
Protocol for isolation of the pGFPuv plasmid: 1. Obtain the 1.5mL plasmid containing E.coli culture that was prepared for you and harvest the
cells by centrifugation at maximum speed for 1 minute. Decant supernatant.
2. Suspend the cell pellet in 250L of Buffer P1 (+ RNAse) and transfer to a microcentrifuge tube.
No cell clumps should be visible after suspension of the pellet.
3. Add 250L of Buffer P2 (containing NaOH/SDS) and gently invert the tube 4-6 times to mix. Do
not vortex, as this will result in shearing of genomic DNA. If necessary, continue inverting the
tube until the solution becomes viscous and slightly clear, but do not extend the time longer than
5 min because the plasmid DNA may become irreversibly denatured.
4. Add 350L of Buffer N3 (neutralization, high salt buffer) and immediately invert the tube gently
4-6 times. The solution should become cloudy.
5. Centrifuge at maximum speed for 10 min. A compact white pellet will form with the “cleared
lysate” above it.
6. Using a pipette, transfer the supernatant from step 5 to the QIAprep column placed in a 2mL
collection tube.
7. Centrifuge 60 sec at maximum speed. Discard the flow-through volume in an organic waste
container.
8. Wash the QIAprep spin column by adding 0.375mL of Buffer PE (containing ethanol) and
centrifuge 60 sec. at maximum speed. Discard the flow-through (organic waste). Repeat a second
time.
9. Discard the flow-through (organic waste), transfer to a centrifuge tube with the cap cut off and
centrifuge for an additional 1 min to remove residual wash buffer.
IMPORTANT: Residual ethanol from Buffer PE may inhibit subsequent enzymatic reactions so
steps 8 and 9 are therefore very important.
10. Place the QIAprep column in a clean 1.5mL microcentrifuge tube with the cap cut off. To elute
DNA, add 50L of Buffer EB (elution buffer = 10mM Tris-HCl, pH 8.5) and centrifuge for 1
min.
11. Transfer to a new microcentrifuge tube. Make sure to appropriately label and store this
preparation since you will need it for subsequent experiments and the final practical exam! Store in your freezer box at -20oC
Molecular Biology-2016 34
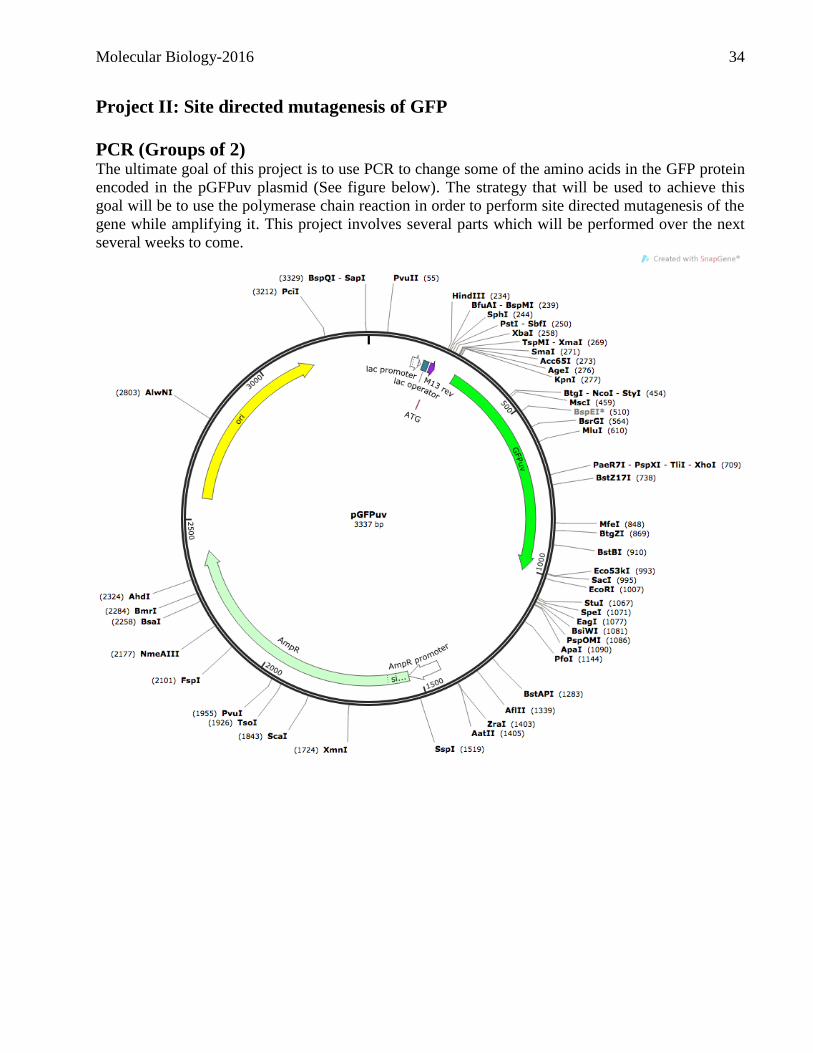
Project II: Site directed mutagenesis of GFP
PCR (Groups of 2) The ultimate goal of this project is to use PCR to change some of the amino acids in the GFP protein
encoded in the pGFPuv plasmid (See figure below). The strategy that will be used to achieve this
goal will be to use the polymerase chain reaction in order to perform site directed mutagenesis of the
gene while amplifying it. This project involves several parts which will be performed over the next
several weeks to come.

Molecular Biology-2016 35
Primers have been designed to mutagenize and amplify part of the coding sequence of the GFP gene
located in the plasmid pGFPuv. The primers you will use are indicated below. Note that several
characteristics have been included to facilitate the cloning and the subsequent screening. Consult the
following web pages to refresh your knowledge on PCR:
http://www.maxanim.com/genetics/PCR/PCR.htm
http://www.dnalc.org/resources/animations/pcr.html
“Forward” primer: GFPfor
GCCAAGCTTGCATGCCTGCAGATCGAC
Characteristics:
A HindIII site is indicated in italics and underlined and will be used for cloning.
A one base change, indicated in red, underlined and in bold, represents a silent mutation which
abolishes a SalI restriction site.
“Reverse” primers:
GFPrev-1
TGTACTCGAGTTTGTGTCGGAGAATGTTTCCATC
GFPrev-2
TGTACTCGAGTTTGTGTCAGAGAATGTTTCCATC
GFPrev-3
TGTACTCGAGTTTGTGTGCGAGAATGTTTCCATC
GFPrev-4
TGTACTCGAGTTTGTG*CCGAGAATGTTTCCATC Characteristics:
An XhoI site is indicated in italics and underlined and will be used for cloning.
GFPrev-1 contains a one base change, indicated in red, underlined and in bold, which changes a
glycine codon to arginine.
GFPrev-2 contains a one base change, indicated in red, underlined and in bold, which creates a stop
codon.
GFPrev-3 contains a one base change, indicated in red, underlined and in bold, which changes a
glycine codon to alanine.
GFPrev-4 contains a one base deletion, indicated in red, underlined and in bold, which creates a -1
frameshift.
Molecular Biology-2016 36
Method:
Preparation of the pGFPuv DNA template: 1. Obtain the pGFPuv preparation you isolated by the Qiagen method.
2. Dilute a sample of your preparation by a factor of 100.
3. Use 5 µL of the diluted template for your PCR reaction.
PCR reaction setup:
Prepare your PCR reactions in a total reaction volume of 50L. In labelled PCR tubes (use 200L
thin walled PCR tubes) add the following ingredients in the order listed in the table below. For each
component use a new autoclaved tip. Note: The Taq polymerase will be added by the T.A.
Ingredients Stock Conc. Final Conc. Volume
Water Complete to 50µL
Taq PCR buffer 10X 1X
GFPfor primer 2µM 0.2µM
*Assigned GFPrev primer 2µM 0.2µM
MgCl2 50mM 1.5mM
dNTP 2mM 200µM
pGFPuv - 5 µL
Taq polymerase 5 units/µL 0.05 units/µL
* Make sure that you record which GFP reverse primer you were assigned!
Once your reaction setup is completed give them to your teaching assistant. Your samples will be
returned to you next week.
PCR amplification conditions:
1. 1 cycle of 5min, 94oC to denature;
2. 30 cycles of 30sec 94oC to denature, 1 min at 68oC to anneal and extend.
3. 1 cycle of 5min, 72oC.
4. Cool to 4oC indefinitely.

Molecular Biology-2016 37
Exercise 3
What we are doing today!
Project I: Verifying the restriction map of a DNA insert
Project II: Site directed mutagenesis of GFP
Electrophoresis of GFP PCR amplicons
Digestion of GFP PCR amplicons and pGFPuv
Ligation of GFP amplicons

Molecular Biology-2016 38
Project I: Verifying the restriction map of a DNA insert You should already have determined which double digests you wish to perform for this week's
experiment. Note that for each double digest, you should also run the corresponding single digests on
your gel. For example if you wish to do a BamHI-EcoRI double digest, your gel should also include
BamHI and EcoRI single digests to facilitate the analysis.
Setting up your restriction digests: 1. Setup your restriction digests as previously. Note that since different enzymes require different
buffers you will need to be flexible in making your restriction digests. If both enzymes cut within
the same buffer, simply add 1.0L of each enzyme. However, if the two enzymes cut in different
buffers consult the compatibilities table in order to determine the best buffer for maximal activity
of the two enzymes.
2. Incubate your digests for 60 minutes at 37oC.
3. Fractionate your digests as previously on a 1% agarose gel. Make sure to use the appropriate
comb; that is the one that will provide the minimum number of required wells.

Molecular Biology-2016 39
Project II: Site directed mutagenesis of GFP (Groups of 2)
Electrophoresis of GFP PCR amplicons Last week you performed a PCR amplification to amplify and mutagenize the GFP gene. This week
you will use agarose gel electrophoresis to determine whether your amplification was successful
before initiating the cloning.
1. Obtain your PCR reactions.
2. Transfer 8 L of your PCR products into new tubes and add 2.5L of 5X loading buffer. Return
the remainder on ice for subsequent experiments.
3. Load your samples on a 1% agarose gel, that has been prepared for you, containing ethidium
bromide and on which appropriate DNA size markers have been loaded.
4. Examine under UV light and take a picture for analysis.
Digestion of GFP PCR amplicons and pGFPuv The PCR products you amplified and analyzed by gel electrophoresis can now be cloned. Recall that
during the PCR reaction, restriction sites included to allow the directional cloning of the PCR
product. We will now have to digest both the pGFPuv plasmid and the PCR products in order to
generate compatible ends for the subsequent ligation reaction. Since the digestions and ligation
reactions are performed in different buffers, we will need to purify the DNA between these steps in
order to optimize the next reaction. The order of the steps is:
PCR reactions (stored in the freezer)
Digestion of amplicons and of pGFPuv
Purification of the PCR products digested with HindIII and XhoI
Ligation of amplicons in pGFPuv for subsequent transformation

Molecular Biology-2016 40
Purification with the QIAQUICK purification kit of digested amplicons
Method: (Assigned groups of 2)
1. Add 5 volumes of Buffer PB to 1 volume of the PCR reaction and mix by inversion.
2. Place a QIAQUICK spin column in a 2mL collection tube.
3. To bind DNA, apply the sample to the QIAQUICK spin column and centrifuge for 1 minute.
4. Discard the flow-through to ORGANIC WASTE and place the QIAQUICK spin column back
into the same tube.
5. To wash, add 0.375mL of Buffer PE to the column and centrifuge for 1 minute.
6. Discard the flow-through to ORGANIC WASTE.
7. Wash again by adding 0.375mL of Buffer PE to the column and centrifuge for 1 minute.
8. Discard the flow-through to ORGANIC WASTE. Place the QIAQUICK spin column back into
the same tube. Centrifuge for 1 minute. This second centrifugation removes any residual ethanol
from Buffer PE that may elute with the DNA and interfere with subsequent steps.
9. Place the QIAQUICK spin column into a new labelled 1.5mL microcentrifuge tube.
10. To elute DNA add 30L of Buffer EB (10mM Tris-HCl, pH 8.5) to the centre of the QIAQUICK
spin column, wait 1 minute and then centrifuge for 1 minute.
11. Using a new pipette tip, transfer the recovered liquid BACK onto the center of the QIAQUICK
spin column and centrifuge for 1 minute.
12. Transfer the recovered liquid into a labelled 1.5mL microcentrifuge tube and store until needed.
(If you were unable to recover 30.0L, add elution buffer to bring the volume up to 30.0L).
Digestion of purified GFP amplicons or of pGFPuv vector
Method: (Assigned groups of 2)
One of the groups of two will perform the digestion of the GFP amplicon whereas the other
group of two will perform the digestion of the pGFPuv plasmid.
1. Setup as follows a reaction mixture of 50µL to perform HindIII-XhoI double digests of both the
GFP PCR product and the plasmid pGFPuv that you purified by the Qiagen method in week 2.
Volume PCR Volume Vector
GFP PCR product 10µL ---------
OR pGFPuv plasmid --------- 5µL
Red 10X Restriction buffer 1X 1X
HindIII 1µL 1µL
XhoI 1µL 1µL
Water Complete to 50µL Complete to 50µL
2. Perform the digestion at 37oC for 1 hour.

Molecular Biology-2016 41
Purification with QIAQUICK purification kit of digested GFP amplicons or pGFPuv vector
Method: (Assigned groups of 2)
1. Add 5 volumes of Buffer PB to 1 volume of the restriction mixture and mix by inversion.
2. Place a QIAQUICK spin column in a 2mL collection tube.
3. To bind DNA, apply the sample to the QIAQUICK spin column and centrifuge for 1 minute.
4. Discard the flow-through to ORGANIC WASTE and place the QIAQUICK spin column back
into the same tube.
5. To wash, add 0.375mL of Buffer PE to the column and centrifuge for 1 minute.
6. Discard the flow-through to ORGANIC WASTE.
7. Wash again by adding 0.375mL of Buffer PE to the column and centrifuge for 1 minute.
8. Discard the flow-through to ORGANIC WASTE. Place the QIAQUICK spin column back into
the same tube. Centrifuge for 1 minute. This second centrifugation removes any residual ethanol
from Buffer PE that may elute with the DNA and interfere with subsequent steps.
9. Place the QIAQUICK spin column into a new labelled 1.5mL microcentrifuge tube.
10. To elute DNA add 30L of Buffer EB (10mM Tris-HCl, pH 8.5) to the centre of the QIAQUICK
spin column, wait 1 minute and then centrifuge for 1 minute.
11. Using a new pipette tip, transfer the recovered liquid BACK onto the center of the QIAQUICK
spin column and centrifuge for 1 minute.
12. Transfer the recovered liquid into a labelled 1.5mL microcentrifuge tube and store until needed.
(If you were unable to recover 30.0L, add elution buffer to bring the volume up to 30.0L).
Molecular Biology-2016 42
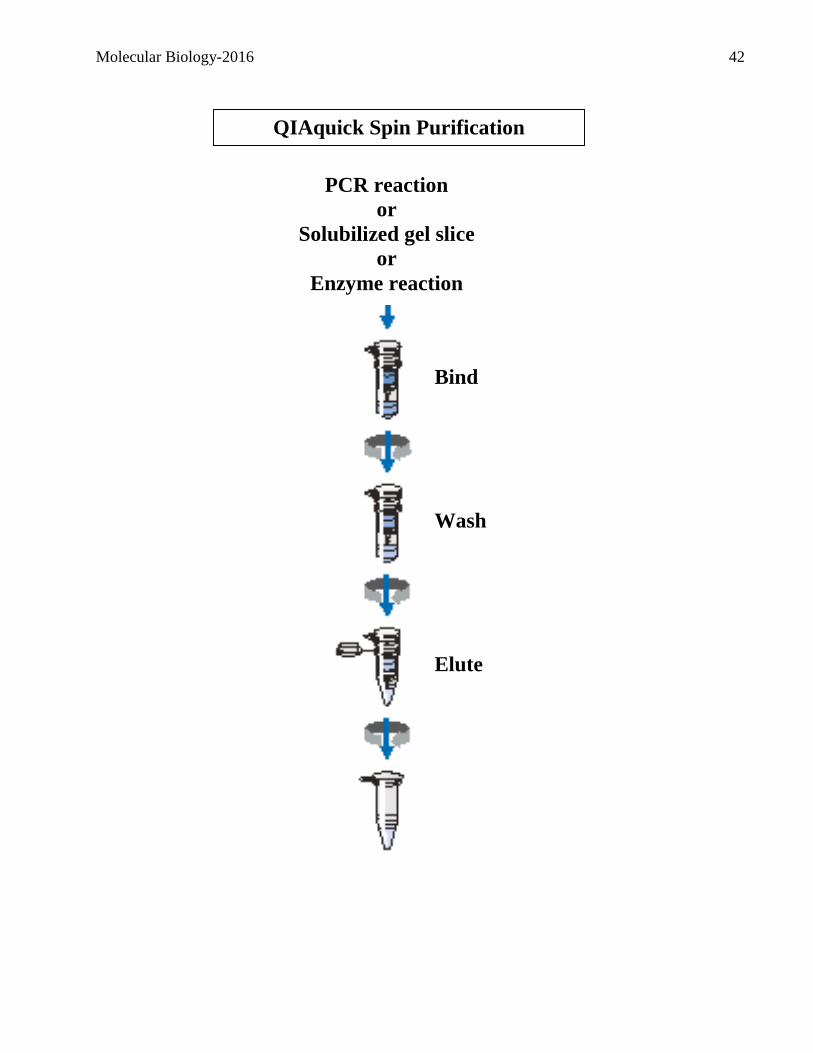
QIAquick Spin Purification
Procedure
PCR reaction
or
Solubilized gel slice
or
Enzyme reaction
Bind
Wash
Elute

Molecular Biology-2016 43
Project II: Site directed mutagenesis of GFP (Groups of 4)
Ligation of GFP amplicons
Having digested the GFP PCR amplicon and the pGFPuv vector you will now proceed with the
ligation. Your ultimate goal is to substitute the wild type GFP sequence in the plasmid with the one
you mutagenized by PCR.
Ingredient Tube 1 Tube 2
Digested pGFPuv plasmid 5.0L 5.0L
10 X ligase buffer 2.0L 2.0L
GFP PCR amplicon 5.0L 0.0L
Water add water to complete the volume to 19.0L
Ligase 1.0L 1.0L
GIVE YOUR LABELED SAMPLES TO THE DEMONSTRATOR
The ligations will be incubated at room temperature until tomorrow and then stored at –20oC

Molecular Biology-2016 44
Exercise 4
What we are doing today!
Project I: Verifying the restriction map of a DNA insert (XhoI)
Project II: Site directed mutagenesis of GFP
Transformation of ligation mixtures
Project III: Genomic fingerprinting
Isolation of human genomic DNA from cheek cells
PCR amplification of ApoC2 VNTR
PCR amplification of ApoB RFLP

Molecular Biology-2016 45
Project I: Verifying the restriction map of a DNA insert (XhoI) Following the analysis of your results you were able to precisely map all restriction sites within your
insert. You will now use this information to map a novel restriction site; XhoI. It was found that this
enzyme cuts once within your insert. Design a proper and appropriate experiment to determine the
precise position of the XhoI site.
Project II: Site directed mutagenesis of GFP
Transformation of ligation mixtures (Groups of 4)
Last week you did ligations to substitute the wild type GFP sequence in the plasmid with the one you
mutagenized by PCR. To isolate, amplify, and maintain the desired recombinants you will now
introduce these plasmid recombinants into E.coli. You will transform Escherichia coli XL-1 cells
with your ligation mixtures from last week. Normally, E. coli does not readily take up DNA but these
have been treated with CaCl2 to alter the bacterial cell wall so that the cells you receive are now
COMPETENT for DNA uptake. However, even with this treatment only a small percentage of cells
uptake DNA. In order to identify these, a selectable marker, ampicillin resistance, is included in the
vector. Consequently, only cells which uptake a circular plasmid that can be replicated and
maintained will express this resistance and therefore grow on plates containing ampicillin. All cells
that do not uptake DNA or that uptake linear DNA, which cannot be replicated or maintained, will
not express this resistance and therefore will die on selective plates.
Materials:
Competent E.coli XL-1 Cells
YT-AMP plates (Supplemented with 50 g/mL ampicillin for selection).
Method:
1. Appropriately label 2 disposable tubes (15-mL polypropylene snap-cap tubes) and store on ice.
2. Dispense 90L of cells into each of the tubes, being careful not to contaminate the sides of the
tube with the micropipettor.
3. Add 5L of the correct ligation mixture to the labelled tube. Mix by swirling gently, and keep on
ice for 30 minutes.
4. Incubate for exactly 45 seconds in a 42oC water bath. This heat shock step is necessary to get
good transformation efficiency but if you hold the cells at 42oC too long you will kill the cells.
Then immediately transfer on ice.
5. Add 0.9 mL of pre-warmed (37oC) SOC broth. Incubate at 37oC for 1 hour with shaking to allow
the cells to start expressing the antibiotic resistance gene.
6. Spread 100μL & 200L of each transformation mixture onto YT-AMP plates and incubate
overnight at 37oC. Make sure your plates are clearly labelled and placed upside down in the 37oC
incubator overnight.
The following transformation controls will be performed by your demonstrators:
Cells treated with plasmid DNA (~5 pg of circular pUC)
Always use biohazard waste containers to dispose of microbial or recombinant DNA waste.

Molecular Biology-2016 46
Project III: Genomic fingerprinting (Groups of 2) The analysis of Polymorphisms (SNP) has become a current trend to identify disease and non-disease
genotypes.
VNTR
The evolutionary principle of variation within a population is a cornerstone in biology. This variation
results from subtle differences in the DNA sequence in individuals of a given species. Variation
commonly originates by the mistaken duplication of a small sequence of nucleotides when only one
copy was present before replication. This results in a tandem repeat of the original sequence. If this
mistake occurs again in another round of replication, then three copies of a sequence will be in
tandem (figure). These tandem repeats are part of our chromosomes and as such, they will be
inherited according to Mendelian genetics. Over the centuries, the number of tandem repeat units has
increased, therefore each of us has inherited a variable number of tandem repeats (VNTRs) at many
loci scattered throughout our genomes. A VNTR can be thought of as a locus with each particular
number of repeated units being analogous to different alleles. Therefore, each human (except for
identical twins) carries a unique combination of VNTRs and these alleles can be used in population
studies or to identify a particular individual.
Figure. Illustration of variable number of tandem repeats (VNTRs). Single strands of DNA from the
same locus from three different individuals are shown. Within this region, the trinucleotide repeat
CAT is present once, twice, or three times which results in alleles of three different lengths.
RFLP
Another type of polymorphism which occurs is called restriction length Polymorphisms (RFLP).
These polymorphisms occur when a single nucleotide change occurs in a restriction site, thus
abolishing or changing it. Consequently, a given region of the genome may possess a restriction site
that is absent from the same region in another copy of the genome or in the genome of a different
individual.
In this project, you will determine your genotype of two polymorphism, a VNTR and an RFLP,
associated with the genes ApoC2 and ApoB respectively.

Molecular Biology-2016 47
Isolation of human genomic DNA from cheek cells:
1. One person from each group of two should use a sterile cotton swab to gently scrape the inside of
one cheek six times. Without rotating the swab, move the swab directly over to the inside of the
other cheek and gently scrape six times.
2. Insert the cotton portion of the swab into a 1.5mL microcentrifuge tube. Then, break off the stick
just above the cotton so that the cotton part falls into the tube.
3. Add 400µL of 1X PBS pH 7.4 to your sample tube.
4. Add 20µL of proteinase K followed by 400µL buffer AL. Mix immediately by vortexing for 15
sec. Do not add proteinase K directly to buffer AL.
5. Incubate at 56oC for 10 min. Spin briefly to remove drops from tube walls and lid.
6. Add 400µL 99% ethanol. Vortex 8 to 10 sec to mix. Spin briefly to remove drops from tube walls
and lid.
7. Apply 600µL of your mixture to the column. Cap to avoid aerosols and centrifuge at 8k rpm for
1min. Change collection tube.
8. Apply remaining mixture to the column. Cap to avoid aerosols and centrifuge at 8k rpm for 1min.
Change collection tube.
9. Add 500µL AW1 buffer. Cap and centrifuge at 8k rpm for 1min. Change collection tube.
10. Add 500µL AW2 buffer. Cap and centrifuge at maximum rpm for 3min. Change collection tube.
11. Centrifuge at maximum rpm for 1 min to remove residual buffer and reduce carryover.
12. Place the QIAprep column in a clean 1.5-mL microcentrifuge tube with the cap cut off. Add
150µL AE buffer to the column. Wait 1 min and then centrifuge at 8k rpm for 1 min to elute.
13. Transfer eluted DNA to a new properly labelled tube and store for future use.
Molecular Biology-2016 48
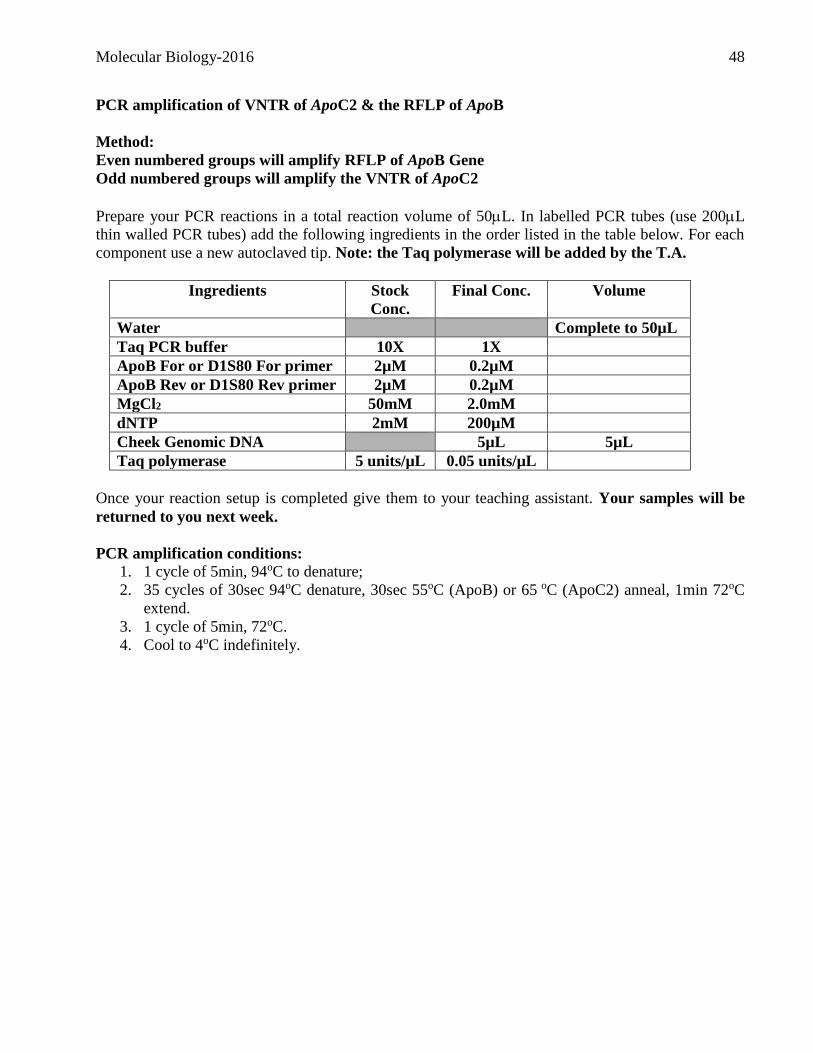
PCR amplification of VNTR of ApoC2 & the RFLP of ApoB
Method:
Even numbered groups will amplify RFLP of ApoB Gene
Odd numbered groups will amplify the VNTR of ApoC2
Prepare your PCR reactions in a total reaction volume of 50L. In labelled PCR tubes (use 200L
thin walled PCR tubes) add the following ingredients in the order listed in the table below. For each
component use a new autoclaved tip. Note: the Taq polymerase will be added by the T.A.
Ingredients Stock
Conc.
Final Conc. Volume
Water Complete to 50µL
Taq PCR buffer 10X 1X
ApoB For or D1S80 For primer 2µM 0.2µM
ApoB Rev or D1S80 Rev primer 2µM 0.2µM
MgCl2 50mM 2.0mM
dNTP 2mM 200µM
Cheek Genomic DNA 5µL 5µL
Taq polymerase 5 units/µL 0.05 units/µL
Once your reaction setup is completed give them to your teaching assistant. Your samples will be
returned to you next week.
PCR amplification conditions:
1. 1 cycle of 5min, 94oC to denature;
2. 35 cycles of 30sec 94oC denature, 30sec 55oC (ApoB) or 65 oC (ApoC2) anneal, 1min 72oC
extend.
3. 1 cycle of 5min, 72oC.
4. Cool to 4oC indefinitely.

Molecular Biology-2016 49
Exercise 5
What we are doing today!
Project II: Site directed mutagenesis of GFP
PCR screening of transformants
Analysis of colony PCR products
Patching
Project III: Genomic fingerprints
Project IV: Transcriptional control of GFP by the LacZ promoter
E.coli RNA isolation

Molecular Biology-2016 50
Project II: Site directed mutagenesis of GFP
PCR screening of transformants (Groups of 2)
If your transformations were successful, you will observe many colonies on your plates representing
independent E.coli clones that harbour plasmid. Your first task will be to enumerate these colonies
on all the transformation plates, including the ligation controls, as well as the transformation
controls. Record the number of colonies on each plate.
MAKE SURE TO TAKE AN ADEQUATE RECORD OF THE RESULTS; THESE WILL
NOT BE GIVEN OUT TO YOU AFTERWARDS!!
Once you have recorded your counts you will use PCR and restriction digestion to screen the
recombinants for inserts (and view PCR products after electrophoresis).
Method:
1. Label 8 microcentrifuge tubes from 1-8 and draw a grid on a YT+ amp plate labelled 1-8.
2. Add 50L of water to each of the tubes. Using a sterile pipette tip, pick one of the colonies on
your plate representing the transformation of plasmid + insert. Gently streak it on the quadrant of
the plate labelled 1 and then place the tip in the corresponding tube labelled 1.
3. Repeat the above procedure for 7 more colonies. Place your agar plates at the designated area so
that they may be incubated.
4. Briefly vortex each of the 8 tubes containing the pipette tip, to suspend the colony (Hold the tip
while vortexing).
5. Remove the tip from each tube, close the tube, and boil for 5 minutes. Place on ice for 1 minute.
6. Centrifuge at maximum speed for 5 minutes. You will be using the supernatant for the PCR
analysis.
7. Prepare a PCR cocktail for 9 reactions of 20L. KEEP ON ICE. Below is the recipe for a single
20L reaction.
PCR Reaction
Solution Stock conc. Final conc.
Water L to a final volume of 20 L
Taq PCR buffer 10 X 1 X
MutScreenFor primer 2 M 0.2 M
MutScreenRev primer 2 M 0.2 M
MgC12 50 mM 2.5 mM
dNTPs 2 mM 200 M
Taq DNA Pol. 5 U/L 2.5 U
8. Distribute 19L of cocktail to each of 8 appropriately labelled PCR tubes (ON ICE)
9. Add 1L of each of the colony supernatants you obtained in step 6 to each of the appropriate
PCR tubes.
10. Prepare a 1.25% agarose gel containing ethidium bromide.

Molecular Biology-2016 51
PCR amplification conditions:
i. 1 cycle of 5min, 94oC to denature;
ii. 30 cycles of 30sec 94oC to denature, 1 min at 68oC to anneal and extend.
iii. 1 cycle of 5min, 72oC.
iv. Cool to 4oC indefinitely.
Restriction digestion of colony PCR products:
1. Obtain your PCR reactions.
2. Prepare 8 SalI digestions, one for each of your PCR reactions, in a final volume of 20µL
containing 5µL of each of your PCR reactions.
3. Prepare an undigested control, using any of the 8 PCR reactions.
4. Digest for one hour.
4. Following the digestion, add 5µL loading buffer to each sample.
5. Load 10L of each mixture, as well as the molecular weight ladder, on your 1.25% agarose.
Migrate for 1 to 1.5 hours at 100 V.
6. Take a picture of your gel.
Project III: Genomic fingerprints (Groups of 2)
ApoB gene:
1. Obtain your PCR reactions for the ApoB gene.
2. Use 10µL of the PCR reaction to set up a restriction digest with the enzyme EcoRI in a final
volume of 20µL.
3. One group in the class will prepare an undigested control as well.
4. After having performed the digest for one hour, add loading dye and load on the pre-poured gel.
5. Once everyone has loaded the gels, these will be migrated and a picture taken for your analysis.
ApoC2 gene 1. Obtain your PCR reactions for the ApoC2 gene.
2. Add loading dye to a 10µL sample of the PCR reaction.
3. Load on the pre-poured gel.
4. Once everyone has loaded the gels, these will be migrated and a picture taken for your analysis.

Molecular Biology-2016 52
Project IV: Transcriptional control of GFP by the LacZ promoter
E.coli RNA isolation This week you will initiate experiments which will allow you to examine the transcriptional
regulation of the GFP gene which is under the control of the LacZ promoter in pGFPuv. You will
examine the relative abundance of the GFP transcript in E.coli cells grown under different
conditions. The method which will be used is referred to as a northern analysis. This technique
involves the isolation, fractionation and transfer of RNA.
As with all experiments, the isolation of the starting material in a pure form is crucial. You will be
isolating RNA from E.coli cells and then use spectroscopy to estimate the RNA concentration and
purity.
Working with RNA is more demanding than working with DNA as RNA is more susceptible to
degradation. Thus you must be very careful not to contaminate any solution.
Always wear gloves!! Method:
Each group of 2 will be assigned one of the following growth conditions from which you will isolate
RNA:
Culture grown in LB.
Culture grown in LB + glucose.
Culture grown in LB + lactose.
Culture grown in LB + IPTG.
1. Centrifuge at maximum speed for 1 minute 1.5mL of the assigned bacterial culture.
2. Remove supernatant and then add 0.5mL RNAzol reagent. Resuspend pellet by vortexing.
3. Incubate for 5 min at 65oC.
4. Add 0.2 mL chloroform, vortex vigorously for 15 sec and keep at R.T. for 2-3 min.
5. Centrifuge at maximum speed for 5 min.
6. Transfer approximately 75% of the upper aqueous phase (without disturbing the phases) to a new
microcentrifuge tube and add 1 volume of isopropanol.
7. Store at room temperature for 5 minutes.
8. Centrifuge at maximum speed for 5 minutes. Remove supernatant.
9. Briefly air-dry the pellet.
10. Resuspend the pellet in formamide.
RNAzol:
1.86 M guanidine isothiocyanate
12 mM sodium citrate
87 mM sodium acetate pH 4.0
0.37% sarcosyl
42 mM 2-mercaptoethanol

Molecular Biology-2016 53
Determining the RNA concentration and your recovery by spectroscopy
1. Prepare a 1/20 dilution in water in a final volume of 100l of an aliquot of your RNA and
measure the absorbance at both 260nm and 280nm against a blank. Sore the remainder of your
sample in an appropriately labelled tube in your freezer box until next week.
2. You can estimate the RNA concentration using the formula (for single-stranded RNA) that an
absorbance of 1.0 at 260nm is equivalent to an RNA concentration of 40g/mL. If the ratio of
A260/A280 is >1.90 the RNA is relatively pure and free from proteins.
3. Calculate the amount of RNA you have recovered.

Molecular Biology-2016 54
Exercise 6
What we are doing today!
Project IV: Transcriptional control of GFP by the LacZ promoter
RNA gel electrophoresis
Northern transfer

Molecular Biology-2016 55
Project IV: Transcriptional control of GFP by the LacZ promoter
RNA gel electrophoresis Northern blot analysis can be used to detect an individual RNA species within a population of total
RNA from an organism. Before a northern blot can be performed the RNA molecules are resolved as
a function of their size on a denaturing agarose gel. The most common type of RNA gel is
formaldehyde. This type of gel provides denaturing conditions that prevent base-pairing and thus
reduce RNA secondary structure. This is important because unlike double-stranded DNA fragments,
which have the same conformation regardless of length, single-stranded RNAs can adopt different
conformations due to intra-strand base-pairing. This would affect electrophoretic migration and make
size estimation inaccurate.
Materials:
Ribonucleases are everywhere! To minimize the danger of RNA degradation, wear gloves and keep
samples on ice unless otherwise specified.
Formaldehyde and formamide are toxic chemicals. Wear gloves when handling these solutions and
work in the fume hood whenever possible.
Methods:
A. Preparation of an agarose-formaldehyde gel (One gel per groups of 4) 1. To prepare a 1.5% agarose-formaldehyde gel (25mL) (using the 8 well comb), combine the
following:
5X MOPS buffer 5 mL
H20 15.5 mL
Agarose 0.375 g
2. Dissolve agarose in microwave oven. Cool slightly; then add 4.5mL of formaldehyde (37%
solution) (WORK IN THE FUMEHOOD. CAUTION! VOLATILE FUMES). Swirl to mix
and cast the gel immediately.
3. Close the cover and let the gel set for at least 30 minutes.

Molecular Biology-2016 56
B. Preparation of RNA samples (per groups of 4)
You will prepare four different RNA samples. It is important that equal amounts of RNA be
loaded in each lane.
You will need the following RNA samples:
Total RNA sample from cultures grown in LB.
Total RNA sample from cultures grown in LB + glucose.
Total RNA sample from cultures grown in LB + lactose.
Total RNA sample from cultures grown in LB + IPTG.
1. To prepare the RNA samples prior to loading on the gel, mix the following components using
labelled tubes KEPT ON ICE:
Final volume = 30 L
RNA sample: L (This volume of sample should contain 5g in a maximum volume of 9
L)
Loading buffer: 21L (RNA loading buffer not the DNA loading buffer)
Water to 30 L: L
2. Incubate your samples at 700C for 10 minutes and then place the samples on ice. If there is liquid
on the side of the tube, spin in the microcentrifuge for a few seconds.
3. Each group of 4 should load a gel as follows: Load 14L aliquots of each of the RNA samples
from the different growth conditions on one gel as follows:
LANE SAMPLE 1 Empty
2 Empty
3 RNA from cultures grown in LB.
4 RNA from cultures grown in LB + glucose.
5 RNA from cultures grown in LB + lactose.
6 RNA from cultures grown in LB + IPTG.
7 Empty
8 Empty
This will result in one gel containing samples from two groups of 4 and duplicate gels per table.

Molecular Biology-2016 57
C. Gel Electrophoresis and Northern Transfer 1. The gel running buffer will be 1X MOPS buffer. Run the gel at 80 V (current ~80 mA) until the
bromophenol blue dye is further than half way down the gel.
2. Remove the gel from the electrophoresis chamber, observe under U.V. and take a picture.
NB: Formaldehyde gels are MORE FRAGILE than non-denaturing gels so be extra careful!
3. Wash the gel 2 times with 5 volumes of water, then 1 time with 2 volumes of 10 X SSC for about
5 min each time. This is necessary to wash away the formaldehyde which otherwise would
prevent hybridization.
D. Capillary transfer (per groups of 4)
You will be provided with a nylon membrane as well as filter paper that were cut to the same size as
the gel. Avoid as much as possible handling the membrane with your fingers.
1. Label the membrane on one end with your lab day, group number, and “RNA”.
2. Wet membrane in a tray of distilled water, and then in 2X SSC.
3. Set up your northern transfer as shown on the next page. Make sure to surround the periphery of
your gel with Saran wrap so as to avoid a short circuit.
4. Following the transfer, the membrane is dried, labelled, UV cross-linked and stored in a plastic
bag.
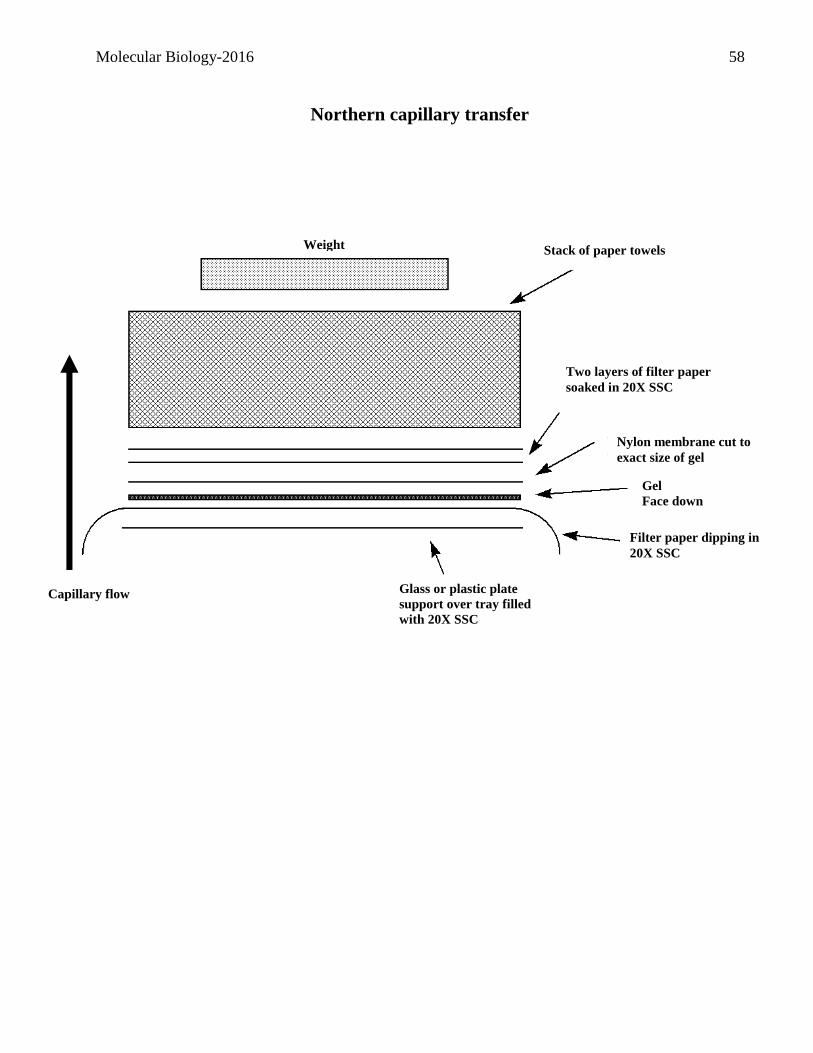
Molecular Biology-2016 58
4.
5.
6.
7.
8.
9.
10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. 24. 25. 26. 27. 28. 29. 30.
SOUTHERN BLOT SETUP
Nylon membrane cut to
exact size of gel
Gel
Face down
Two layers of filter paper
soaked in 20X SSC
Filter paper dipping in
20X SSC
Glass or plastic plate
support over tray filled
with 20X SSC
Stack of paper towels Weight
Capillary flow
Northern capillary transfer

Molecular Biology-2016 59
Exercise 7
What we are doing today!
Project IV: Transcriptional control of GFP by the LacZ promoter
Northern hybridizations
Project V: RT-PCR of the yeast ribosomal protein URP1
Yeast RNA isolation
RT-PCR

Molecular Biology-2016 60
Project IV: Transcriptional control of GFP by the LacZ promoter
Northern hybridizations (Groups of 4) In past weeks you have generated a Northern. This week you will prepare these membranes for
hybridizations with a Dig-labelled probe.
General information about hybridization of nucleic acids:
Before you hybridize your probe to a membrane, a prehybridization step is performed. This step is
necessary to reduce both background and non-specific hybridization. Background is defined as
non-specific binding of your probe to the membrane and does not involve hybridization. Non-
specific hybridization represents hybridization of you probe to sequences that possess a low degree
of identity. In order to reduce both of these events the membranes are initially exposed to a blocking
reagent that usually includes a high concentration of non-specific DNA, such as salmon sperm DNA.
The purpose of this reagent is to saturate any binding sites on the membrane as well as non-specific
hybridization sites on the nucleic acids.
Following the prehybridization step, the membranes are exposed to the labelled probe, once again in
the presence of the blocking reagent. Depending on the stringency conditions, the probe will easily
out-compete non-specific DNA binding and thus hybridize specifically to complementary sequences.
Excess probe, which remained unhybridized, or that is weakly hybridized to poorly complementary
sequences is then removed by a series of washes of varying stringency. Note that stringency is
determined by both temperature and salt concentration. Both of these parameters determine the level
of homology required to allow hybridization. The lower the stringency the more permissive the
hybridization conditions and thus the lower the degree of complementarity required to allow the
formation of stable hybrids. Inversely, the higher the stringency the less permissive are the
hybridization conditions and thus the higher the degree of complementarity that is required to allow
the formation of stable hybrids.

Molecular Biology-2016 61
Prehybridization
You should have duplicate northern membranes on each bench.
Prehybridization/Hybridization Solution:
5X SSC (20X SSC: 3M NaCl et 0.3M NaCitrate)
0.02% sodium dodecyl sulfate (SDS)
20 mM sodium maleate, pH 7.5
4M Urea
100μg/mL Salmon sperm DNA
1. Place your northern blot in a 50mL Falcon tube that has been clearly labelled with your group’s
name. Add 10mL of prehybridization mix to the tube and close tightly.
2. Place your tube in the hybridization oven glass tube. Be careful not to scratch the inside of the
glass tube.
3. Clamp the glass tube into the rotator wheel in the hybridization oven. Ensure that the tubes are
balanced by placing a second glass tube on the opposite side of the rotator.
4. Rotate gently (setting #3) at 42oC for a minimum of 1 hour.
Hybridizations:
You are now ready to initiate the hybridization with the either a probe against the GFP gene
(experimental) or the bla gene (internal control). But before you can do the hybridization, the
labelled DNA must be denatured. Make sure to record which probe you were assigned!
1. Boil the probe you were assigned for 3 minutes and then quickly chill by placing the tube in an
ice bath. Spin in a microcentrifuge to bring any liquid to the bottom.
Setting up you hybridizations
2. Decant the prehybridization solution from the tube containing your northern membrane.
3. Immediately add 10 mL fresh hybridization mix to each tube.
4. Then, carefully add 50L of either the denatured GFP or bla probe (as assigned) directly into the
hybridization mix of the tube with the northern membrane. Avoid direct contact of the
concentrated probe with the membrane. Cap the tubes tightly and place them in the hyb oven as
previously.
5. Carry out the hybridization of your northern membranes at 42oC overnight.
FOLLOWING THE HYBRIDIZATION, THE MEMBRANES WILL BE WASHED BY THE
T.A.S AND THEN STORED FOR LATER USE

Molecular Biology-2016 62
Project V: RT-PCR of the yeast ribosomal protein URP1 (Groups of 2)
Yeast RNA isolation (Steps 1-3 have been done for you)
1. Obtain 10mL of a yeast culture.
2. Centrifuge for 10 minutes at 7 000 rpm.
3. Discard supernatant.
4. Suspend pellet in 200μL lysis buffer.
5. Transfer to a screw cap tube containing glass beads and 200μL phenol:chloroform:isoamyl
alcohol (25:24:1).
6. Vortex 2 minutes and then place on ice two minutes.
7. Repeat step 6 two more times.
8. Centrifuge at maximum speed for 5 minutes. Transfer the upper aqueous phase to a new tube.
9. Add 0.1 volumes of 3M NaOAc and 1mL 99% ETOH. Mix by inversion and centrifuge at
maximum speed for 2 minutes.
10. Discard supernatant and suspend pellet in 100μL DEPC treated water.
11. Determine yield and purity.
RT-PCR Another method that can be used to compare the relative abundance of a transcript under different
conditions is RT-PCR. In contrast to northern analysis, this method is more sensitive and ideal for
weakly expressed transcripts.
In contrast to standard PCR reactions, such as the one you performed from genomic DNA, RT-PCR
is an amplification that uses RNA rather than DNA as template. However, since the thermostable
DNA polymerase that is used in the PCR reaction can only use DNA as a template, the RNA of
interest must first be transcribed into DNA. This can be accomplished by the RNA-dependent DNA
polymerase reverse transcriptase (RT). Initially, reverse transcriptase is used to synthesize a single-
stranded complementary DNA (cDNA) strand of the RNA of interest. An oligo dT primer will be
used for this reaction. Once the cDNA has been generated, a standard PCR reaction can be used to
amplify the DNA sequence complementary to the mRNA.
Consult the following web site for an overview of RT-PCR:
http://www.bio.davidson.edu/courses/Immunology/Flash/RT_PCR.html

Molecular Biology-2016 63
Method:
Each group will perform the following 5 reactions:
PCR reaction of DNAse treated RNA not treated with RT
PCR reaction of RT reaction on DNAse treated RNA
PCR reaction of RT reaction on RNA NOT treated with DNAse
PCR reaction of RNA NOT treated with either DNAse or RT
PCR reaction without RNA (No template)
Preparation of RNA samples for RT reactions:
Prepare a dilution mixture of your RNA as follows. This dilution will then be used as your source of
RNA for all the required reactions described in this section.
4.0g of your RNA sample
5.0L of 10X DNAse I Buffer
Water to a final volume of 48L
DNAse treatment of total RNA:
1. Before you initiate the RT reaction, you want to ensure that you eliminate any contaminating
DNA from your RNA. In a final volume of 25L add the following:
24L of your diluted RNA sample
1L DNAse I
2. Incubate at room temperature for 15 minutes. Following the DNAse I treatment, inactivate the
enzyme by adding 2.5L of 25mM EDTA and heating at 65oC for 10 minutes. Chill on ice.
Reverse transcriptase reactions:
3. Prepare your RT reactions in PCR tubes as indicated below. Use a new autoclaved tip for each
component added.
4. Add 19L of DEPC treated water to each of two labelled PCR tubes labelled as follows:
+DNAse and -DNAse
5. Add 4.0L of the oligo-dT primer to each tube.
6. Add 4.0L of a 2mM dNTP mixture to each tube.
7. Add 2L of your DNAase treated RNA to the tube labelled + DNAse.
8. Add 2L of your untreated RNA dilution to the tube labelled -DNAse.
9. Allow annealing of the oligo-dT primer by incubating your RT pre-reaction mixture at 70oC for 5
minutes.
10. Let your tubes slowly cool down on the bench for 5 minutes.
11. Add 8.0L of 5X RT buffer to each tube.
12. Add 2.0L of 100mM DTT for a final concentration of 5mM to each tube.
13. Add 1.0 L MMLV reverse transcriptase at 200U/L for a final concentration of 5U/L to each
tube.
14. Incubate at 42oC for 60 minutes.
15. Inactivate the RT enzyme by incubating the reaction mixture at 70oC for 15 minutes.

Molecular Biology-2016 64
PCR reactions: 1. Following the RT reactions, prepare the following PCR reactions in 6 appropriately labelled
PCR tubes:
PCR reaction of yeast genomic DNA (Will be provided)
PCR reaction of DNAse treated RNA not treated with RT
PCR reaction of RT reaction on DNAse treated RNA
PCR reaction of RT reaction on RNA NOT treated with DNAse
PCR reaction of RNA NOT treated with either DNAse or RT
PCR reaction without RNA (No template)
Primers:
URP (FOR) GTGCAATTTCAGGCATTACCG
URP (REV) CTGGGGCCAAAGTTTGAGGAAC
Sample Stock conc. Final conc.
Water L to a final volume of 49L
Taq PCR buffer 10 X 1X
URP (For) primer 2M 0.2M
URP (Rev) primer 2M 0.2M
MgCl2 50 mM 2.0mM
dNTP 2mM 200M
Taq Polymerase 5units/µL 2.5units/reaction
2. Add 1L of each of the appropriate templates or water to each of the appropriate PCR mixes.
Once your PCR reactions have been prepared, place them in the ice bucket at the front for the
PCR to be performed.
3. Your PCR reactions will be returned to you next week.
Your PCR reactions will be performed under the following conditions:
i. 1 cycle: 5 min, 94oC to denature;
ii. 30 cycles: 30sec@94oC to denature, 30sec@ 58oC to anneal, 1.0 min 72oC to extend.
iii. 1 cycle: 5 min, 72oC.
iv. Cool to 4oC.

Molecular Biology-2016 65
Exercise 8
What we are doing today!
Project II: Site directed mutagenesis of GFP
Evaluation of GFP activity
Project IV: Transcriptional control of GFP by the LacZ promoter
Immunodetection of northerns
Project V: RT-PCR of the yeast ribosomal protein URP1
Analysis of RT-PCR reactions

Molecular Biology-2016 66
Project II: Site directed mutagenesis of GFP
Evaluation of GFP activity
Project IV: Transcriptional control of GFP by the LacZ promoter
Immunodetection of northern blots Having performed the hybridizations of the northern membranes you will now carry out a procedure
to detect the hybrids. Note that the membranes have been washed for you to remove free as well as
weakly hybridized probe.
All treatments are carried out at room temperature
1. Place the membrane in a small plastic bag.
2. Add 15 mL of maleic acid buffer 1X. Incubate with shaking for 15 min. Discard.
3. Add 20 mL of blocking solution. Incubate with shaking for 30 min. Discard.
4. Add 10 mL of antibody solution. Incubate with shaking for 30 min. Discard.
5. Transfer membranes to Tupperware containers. DO NOT LET MEMBRANES DRY! Add 50
mL of washing buffer and incubate with shaking for 15 min. Discard and repeat step 5 once
more.
6. Add 50 mL of maleic acid buffer 1X and incubate with shaking for 15 min. Discard.
7. Add 10 mL of detection buffer and incubate with shaking for 5 min. Discard.
8. The next steps must be completed quickly and in a coordinated fashion. Inform your teaching
assistant or the lab technician before proceeding. Put 2.5mL of the CDP-Star solution in a corner
of the container and allow it to flow over the membrane until it is covered. DO NOT PUT the
solution directly on the membrane. Incubate for 5 minutes in the dark in your drawer
9. Place your membrane, RNA side up, on a Whatman filter paper.
10. Place 2 membranes, RNA side up, in the photography apparatus. Have a picture taken of the
chemiluminescence reaction (exposure 2 to 10 minutes).

Molecular Biology-2016 67
Project V: RT-PCR of the yeast ribosomal protein URP1
Analysis of RT-PCR reactions 1. Pour a 1.5% agarose gel.
2. Obtain your RT-PCR reactions from the freezer.
3. Add the appropriate amount of DNA loading buffer to 10µL samples of each of your reactions.
4. Load and fractionate at 80V.
5. Following the migration, visualize under UV and take a picture.
Molecular Biology-2016 68
Appendices
Molecular Biology-2016 69
Amino acid 3 letter code One letter code
alanine ala A
arginine arg R
asparagine asn N
aspartic acid asp D
asparagine or aspartic acid asx B
cysteine cys C
glutamic acid glu E
glutamine gln Q
glutamine or glutamic acid glx Z
glycine gly G
histidine his H
isoleucine ile I
leucine leu L
lysine lys K
methionine met M
phenylalanine phe F
proline pro P
serine ser S
threonine thr T
tryptophan try W
tyrosine tyr Y
valine val V
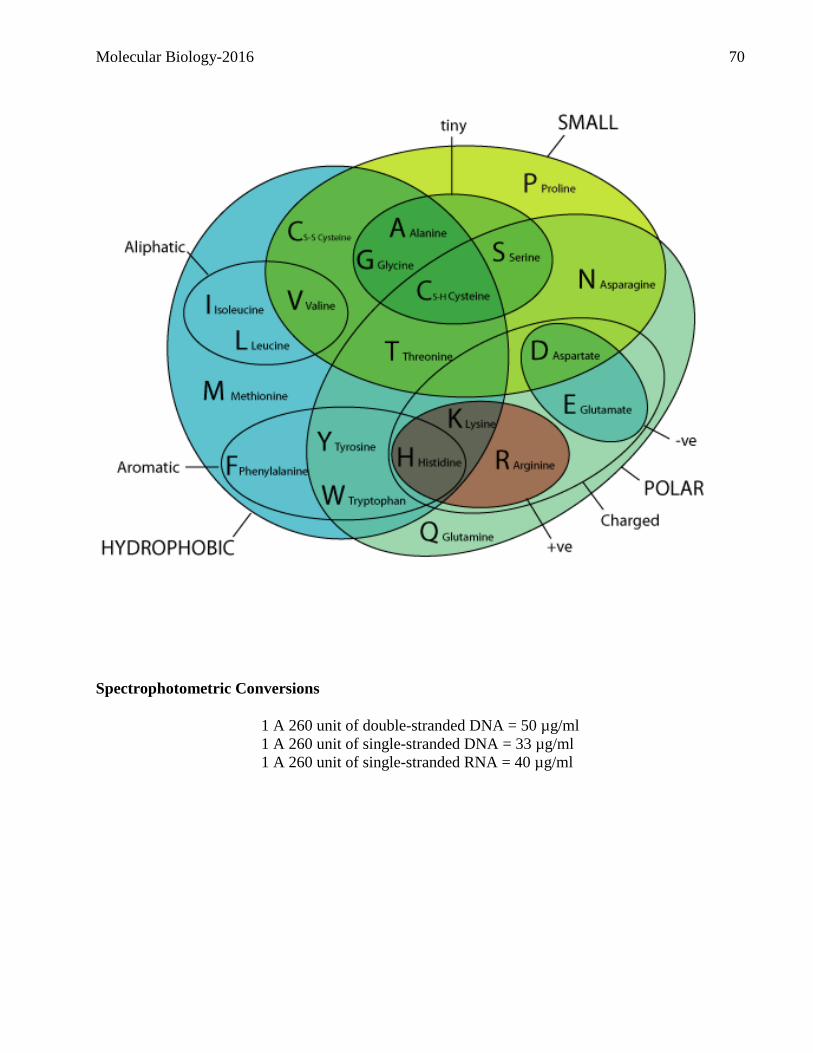
Molecular Biology-2016 70
Spectrophotometric Conversions
1 A 260 unit of double-stranded DNA = 50 µg/ml
1 A 260 unit of single-stranded DNA = 33 µg/ml
1 A 260 unit of single-stranded RNA = 40 µg/ml