transcriptional targeting of lentiviral vectors by long terminal repeat enhancer replacement
TRANSCRIPT

JOURNAL OF VIROLOGY, Apr. 2002, p. 3996–4007 Vol. 76, No. 80022-538X/02/$04.00�0 DOI: 10.1128/JVI.76.8.3996–4007.2002Copyright © 2002, American Society for Microbiology. All Rights Reserved.
Transcriptional Targeting of Lentiviral Vectors by Long TerminalRepeat Enhancer Replacement
Francesco Lotti,1 Emilio Menguzzato,1 Claudia Rossi,1 Luigi Naldini,2 Laurie Ailles,2†Fulvio Mavilio,3,4 and Giuliana Ferrari1*
TIGET, Istituto Scientifico H. San Raffaele,1 and Genera S.p.A.,4 20132 Milan, Institute for Cancer Research and Treatment,University of Torino Medical School, 10060 Candiolo (Turin),2 and Department of Biomedical Sciences, University of Modena
School of Medicine, 41100 Modena,3 Italy
Received 9 August 2001/Accepted 21 January 2002
Gene therapy of many genetic diseases requires permanent gene transfer into self-renewing stem cells andrestriction of transgene expression to specific progenies. Human immunodeficiency virus (HIV)-derived len-tiviral vectors are very effective in transducing rare, nondividing stem cell populations (e.g., hematopoietic stemcells) without altering their long-term repopulation and differentiation capacities. We developed a strategy fortranscriptional targeting of lentiviral vectors based on replacing the viral long terminal repeat (LTR) enhancerwith cell lineage-specific, genomic control elements. An upstream enhancer (HS2) of the erythroid-specificGATA-1 gene was used to replace most of the U3 region of the LTR, immediately upstream of the HIV type 1(HIV-1) promoter. The modified LTR was used to drive the expression of a reporter gene (the green fluorescentprotein [GFP] gene), while a second gene (a truncated form of the p75 nerve growth factor receptor[�LNGFR]) was placed under the control of an internal constitutive promoter to monitor cell transduction,or to immunoselect transduced cells, independently from the expression of the targeted promoter. Thetranscriptionally targeted vectors were used to transduce cell lines, human CD34� hematopoietic stem-progenitor cells, and murine bone marrow (BM)-repopulating stem cells. Gene expression was analyzed in thestem cell progeny in vitro and in vivo after xenotransplantation into nonobese diabetic-SCID mice or BMtransplantation in coisogenic mice. The modified LTR directed high levels of transgene expression specificallyin mature erythroblasts, in a TAT-independent fashion and with no alteration in titer, infectivity, and genomicstability of the lentiviral vector. Expression from the modified LTR was higher, better restricted, and showedless position-effect variegation than that obtained by the same combination of enhancer-promoter elementsplaced in a conventional, internal position. Cloning of the woodchuck hepatitis virus posttranscriptionalregulatory element at a defined position in the targeted vector allowed selective accumulation of the genomictranscripts with respect to the internal RNA transcript, with no loss of cell-type restriction. A critical advantageof this targeting strategy is the use of a spliced, major viral transcript to express a therapeutic gene and thatof an internal, independently regulated promoter to express an additional gene for either cell marking or invivo selection purposes.
Retroviral vectors provide a safe and relatively efficient genetransfer system for many gene therapy applications (reviewedin references 5, 17, and 27). These vectors are still the onlyavailable tool to achieve stable genomic integration of a ther-apeutic gene, and they are therefore largely used in clinicaltrials for gene therapy of genetic diseases. A significant limi-tation of retroviral vectors derived from the Moloney murineleukemia virus (Mo-MLV) backbone is their inability to inte-grate into the genome of nondividing cells, which essentiallylimits their use to ex vivo applications and to cells which can beinduced to replicate in culture without losing a specific, ther-apeutically relevant phenotype. As an example, hematopoieticstem cells (HSCs) are very difficult to maintain in culture in anactive mitotic state without compromising their self-renewingand bone marrow (BM)-repopulating capacity and are there-fore difficult to transduce with retroviral vectors at an efficiencycompatible with clinical applications. A number of groups have
recently reported that human immunodeficiency virus type 1(HIV-1)-derived lentiviral vectors can integrate into the ge-nome of nondividing cells in vivo and ex vivo (reviewed inreferences 24 and 28) and are much more efficient than Mo-MLV-derived vectors in transducing HSCs without compro-mising their repopulation capacity upon transplantation (10,15, 29). Several years of research have drastically improvedboth design and packaging of lentiviral vectors and have vir-tually abolished the safety concerns originally raised by theidea of transducing human cells with a vector derived fromHIV (6, 31, 32).
Correction of genetic disorders affecting a specific progenyof HSCs (e.g., hemoglobinopathies or thalassemias) requiresrestricting expression of the therapeutic gene in a cell lineage-specific fashion. In these cases, transcriptional targeting of thetransferred gene is mandatory. Appropriate transgene regula-tion in the framework of a retroviral vector is a difficult task,due to a number of technical factors such as limited vectorcapacity, transcriptional interference between the viral longterminal repeat (LTR) and internal enhancers-promoters, andgenetic instability of complex regulatory sequences (e.g., locuscontrol regions) in this context (4). We had previously pro-
* Corresponding author. Mailing address: TIGET-H. San Raffaele,Via Olgettina 58, 20132 Milan, Italy. Phone: 39-02-26434705. Fax:39-02-26434668. E-mail: [email protected].
† Present address: Department of Pathology and DevelopmentalBiology, Stanford University School of Medicine, Stanford, CA 94305.
3996
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 11
Feb
ruar
y 20
22 b
y 18
8.92
.209
.147
.

posed a transcriptional targeting strategy for Mo-MLV-derivedretroviral vectors, consisting of the addition of tissue- or lin-eage-specific regulatory elements into the LTR, thereby over-coming some of the problems associated with the use of inter-nal transcriptional units (9, 11). Transcriptional targeting oflentiviral vectors has been attempted by placing the gene ofinterest under the control of restricted internal promoters (16)in the framework of self-inactivating (SIN) vectors, in whichthe LTR is disabled by deletion of the U3 region (enhancerand promoter) for safety reasons (31). Modification of the HIVLTR could theoretically be accomplished by the same strategyused for the Mo-MLV LTR, although the presence of a TAT-dependent promoter and a TAT-responsive element (TAR) inthe primary transcript might add complexity to the vector de-sign.
We attempted the development of transcriptionally targetedlentiviral vectors by replacing the viral LTR U3 enhancer ele-ments with a DNase hypersensitive region (HS2) upstream ofthe erythroid-specific, GATA-1 zinc finger transcription factorgene (8, 25). This element contains an autoregulatory en-hancer able to restrict transcription of a heterologous pro-moter to human or murine erythroblastic cell lines (18, 26) andto confer erythroid lineage specificity to a Mo-MLV-derivedvector upon transduction of human and murine HSCs (11).The LTR modification was introduced in lentiviral vectorsencoding the green fluorescence protein (GFP) as reportergene within the major genomic transcript. A second gene, thecDNA for a truncated form of the p75 nerve growth factorreceptor (�LNGFR) (21), was placed under the control of aninternal, constitutive phosphoglycerokinase (PGK) promoterto monitor cell transduction and immunoselect transducedcells, independently from the expression of the targeted pro-moter. The vectors contained a central polypurine tract(cPPT), to enhance transduction of nondividing cells (10), andthe woodchuck hepatitis virus posttranscriptional regulatoryelement (WPRE) (30) to increase the expression of the tar-geted transgene. The vectors were used to transduce hemato-poietic cell lines and human and murine HSCs, and transgeneexpression was analyzed in the differentiated progenies in vitroand in vivo. The transcriptionally targeted HIV LTR directedhigh levels of transgene expression specifically in mature eryth-roblasts, in a TAT-independent fashion and with no alterationin titer, infectivity, and genomic stability of the lentiviral vec-tor. Expression from the targeted LTR was higher, better re-stricted, and showed less position-effect variegation than thatobtained with the same combination of enhancer-promoterelements placed in the conventional, internal position. Inser-tion of the WPRE upstream of the internal promoter allowedselective accumulation of the genomic transcripts with respectto the internal RNA transcript, with no loss of cell-type restric-tion. These data indicate that enhancer replacement is aneffective strategy to achieve efficient and targeted expression ofa transgene carried by a lentiviral vector while maintaining anindependently regulated, second transcriptional unit to expressadditional gene functions.
MATERIALS AND METHODS
Plasmids. An EcoRI/SacI fragment containing part of the 3� LTR of theHIV-1-derived lentiviral vector pHR2 (6) was subcloned into the PvuII site of thepUC-19 plasmid. The plasmid was digested with EcoRV and BanII, self-ligated
to generate a �418 to �40 deletion (where �1 is the transcription start site) inthe U3 region of the LTR, or ligated with a 200-bp BamHI fragment containingthe GATA-1 HS2 human genomic fragment (�856 to �655) (18, 26) to obtaina chimerical LTR. The two LTRs were then introduced in the pRRL.SIN vectorbackbone (31) to obtain the pRRL.sin-40.GFP (�40) and the pRRL.GATA.GFP (G) vectors, respectively. A second, independent transcriptional cassette(PGK.�LNGFR) was introduced into the HindII/SacII sites to generate thevectors pRRL.sin-40.GFP.PGK.�N (�40/P) and pRRL.GATA.GFP.PGK.�N(G/P). The WPRE (30) was cloned in the filled EcoRI or HindII sites of the G/Pvector to generate the pRRL.GATA.GFP.W.PGK.�N. (G.W/P) or pRRL.GATA.GFP.PGK. �N.W (G/P.W) vectors, respectively. To generate the pRRL.sin-18.GATA.GFP (�18/G) vector, a fragment of 370 bp containing the GATA-1HS2 and a portion of the HIV-1 LTR (�40 to �75) was inserted in the filledBamHI site of the pRRL.GFP.sin-18 vector. For some experiments, the G.W/Pvector was further modified by the insertion of a 118-bp HpaII/ClaI fragmentcontaining the HIV-1 cPPT, as previously described (10).
Vector production. Viral stocks pseudotyped with the vesicular stomatitis Gprotein (VSV-G) were prepared by transient cotransfection of 293T cells usinga three-plasmid system (the transfer vector, the pCMV�R8.74 encoding Gag,Pol, Tat, and Rev, and the pMD.G plasmid encoding VSV-G), as previouslydescribed (6). In some cases, viral preparations were concentrated by ultracen-trifugation to increase titer. The viral p24 antigen concentration was determinedby using an HIV-1 p24 Core Profile enzyme-linked immunosorbent assay (NENLife Science Products). Viral titers (infectious particles) were determined bytransduction of HeLa or HEL cells with serial dilution of the vector stocks.Transduction efficiency was evaluated by scoring GFP and/or �LNGFR trans-gene expression by flow cytometry (FACScan; Becton Dickinson, MountainView, Calif.).
Transduction of stable cell lines. HeLa cells were maintained in Dulbecco’smodified Eagle’s medium (EuroClone), supplemented with 10% fetal bovineserum (FBS; HyClone), and transduced in six-well plates as described previously(6). Human erythroleukemic (HEL), myeloblastic (K562), and monocytic (U937)cell lines (all from American Type Culture Collection [ATCC], Rockville, Md.)were grown in RPMI 1640 (EuroClone) supplemented with 10% FBS and trans-duced with viral supernatant at different multiplicities of infection (MOIs) in thepresence of 4 �g of Polybrene/ml. On day 4 after transduction, cells wereharvested and analyzed by flow cytometry for transgene expression.
Transduction and maintenance of human HSCs. Human CD34� stem-pro-genitor cells were purified from the Ficoll mononuclear cell fraction of umbilicalcord blood by positive selection using the CD34 magnetic cell isolation kit(MiniMACS; Milthenyi, Auburn, Calif.). CD34� cells were infected with viralstocks at an MOI of 50 to 200 in serum-free Iscove’s modified Dulbecco’smedium (IMDM; BioWhittaker, Verviers, Belgium) containing BIT serum sub-stitute (Stem Cell Technologies, Vancouver, British Columbia, Canada), 20 ng ofrecombinant human interleukin-6 (rhIL-6) per ml (R&D Systems), 20 ng ofthrombopoietin/ml (PeproTech, Rocky Hill, N.Y.), 100 ng of stem cell factor(rhSCF) per ml (R&D Systems), and 100 ng of Flt3-L/ml (PeproTech) for 24 h.Transduced CD34� cells were washed in IMDM with 10% FBS (EuroClone),plated at a density of 1,000 cells/ml in methylcellulose medium (1% methylcel-lulose [Stem Cell Technologies], 30% FBS, 2 mM L-glutamine, 10�4 M �-mer-captoethanol, 1% bovine serum albumin in IMDM) containing 4 U of erythro-poietin per ml (EPREX), 10 ng of granulocyte-macrophage colony-stimulatingfactor per ml, 10 ng of rhIL-3 (R&D Systems) per ml, and 50 ng of rhSCF/ml,and scored by light and fluorescence microscopy 10 to 14 days after plating.Alternatively, transduced CD34� cells were grown in liquid culture under threedifferent conditions: (i) in the same medium used for transduction to maintain anundifferentiated phenotype; (ii) in X-VIVO-10 (BioWhittaker) serum-free me-dium supplemented with 20 ng of rhSCF per ml and 4 U of recombinant humanerythropoietin per ml to induce erythroid differentiation; and (iii) in IMDMsupplemented with 20% FBS, 20 ng of both rhIL-3 and rhIL-6 per ml to inducemyeloid differentiation. Cell phenotypes and transduction levels were deter-mined by flow cytometry.
Transplantation in NOD-SCID mice. A breeding colony of nonobese diabetic(NOD)-SCID mice (Jackson Laboratory, Bar Harbor, Maine) was housed in apositive-airflow ventilated rack and bred and maintained in microisolators underspecific-pathogen-free conditions. Mice to be transplanted were sublethally ir-radiated at 6 to 8 weeks of age with 350 cGy of total body irradiation from acesium source. Transduced human CD34� cells (1.5 � 105 to 3 � 105) wereadministered within 24 h of irradiation in a single intravenous (tail vein) injec-tion. Mouse BM was analyzed by flow cytometry 5 to 11 weeks posttransplant.BM cells were plated at a density of 0.5 � 105 to 2 � 105 cells/ml in methylcel-lulose preparation preferentially supporting the growth of human colonies (1).
VOL. 76, 2002 TRANSCRIPTIONAL TARGETING OF LENTIVIRAL VECTORS 3997
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 11
Feb
ruar
y 20
22 b
y 18
8.92
.209
.147
.

Transduction of murine BM cells and BM transplantation. Murine BM cellswere harvested from C57BL/6 mice (Charles River) by flushing femurs and tibiaeand infected for 24 h with viral stocks at an MOI of 200 in serum-free IMDMcontaining BIT serum substitute and 50 ng of mouse SCF (muSCF)/ml, 10 ng ofmuIL-3/ml, 10 ng of human Flt3-L (huFlt3-L)/ml, and 20 ng of huIL-6/ml.Transduced cells were injected (5 � 106 cells/mouse) into the tail vein of recip-ient 8- to 12-week-old male C57Bl/6-Ly-5.1 mice (B/6.SJL-CD45a-Pep3b; JacksonLaboratories) and irradiated with two split doses of 400 cGy 4 h apart. Nine to11 weeks after BM transplantation, animals were sacrificed and hematopoietictissues were collected for fluorescence-activated cell sorter (FACS) analysis andclonogenic assay.
Flow cytometry analysis. �LNGFR expression in transduced cells was moni-tored by flow cytometry or epifluorescence microscopy using an indirect stainingwith the anti-human p75-NGFR monoclonal antibody (MAb) 20.4 (ATCC) andR-phycoerythrin (RPE)-conjugated goat anti-mouse serum (Southern Biotech-nology Associates, Birmingham, Ala.). Human cell surface phenotype and hu-man cell engraftment in transplanted NOD-SCID mice were determined by flowcytometry using RPE-conjugated anti-human CD45 (DAKO, Glostrup, Den-mark) to detect total human leukocytes, CD34 (Becton Dickinson, San Jose,Calif.) for stem-progenitor cells, CD19 (DAKO) for B lymphocytes, CD14(DAKO) for monocytes, CD13 (Caltag Laboratories, Burlingame, Calif.) formyeloid cells, and GpA (DAKO) for erythrocytes. Indirect staining with biotin-ylated anti-p75-NGFR MAb and Tricolor-conjugated streptavidin (Caltag Lab-oratories) was used to monitor transduction efficiency in each BM cell subpopu-lation. Mouse cell phenotyping was carried out using PE-conjugated anti-mouseTER-119 and Gr-1 (PharMingen) antibodies. The fluorescein isothiocyanate(FITC)-conjugated anti-mouse CD45.2 MAb (PharMingen) was used to evaluatedonor-host chimerism in BM-transplanted mice. Isotype-matched nonspecificantibodies were used as controls (Becton Dickinson). For cytofluorometry anal-ysis of nonerythroblastic cell populations, BM cells were lysed with 8.3% ammo-nium chloride to remove erythroid cells and washed with phosphate-bufferedsaline containing 0.1% bovine serum albumin and 0.01% sodium azide. Cellswere then resuspended at 1 � 106 to 2 � 106 cells/ml and incubated with mouseimmunoglobulin G (IgG; Sigma, St. Louis, Mo.) and 5% human serum to blocknonspecific binding to the Fc receptor.
Southern and Northern blot analyses. Genomic DNA was isolated by lysiswith TNE (10 mM Tris [pH 7.5], 100 mM NaCl, 1% sodium dodecyl sulfate),proteinase K treatment, phenol-chloroform extraction, and ethanol precipita-tion. Ten micrograms of DNA was digested overnight with AflII and EcoRI, runon a 0.8% agarose gel, transferred to a nylon membrane (Hybond-N; Amer-sham) by Southern capillary transfer, probed with 2 � 107 dpm of a 32P-labeledGFP probe, and exposed to X-ray film. Total cellular RNA was extracted byguanidine-isothiocyanate, poly(A)�-selected by oligo(dT)-cellulose chromatog-raphy, size-fractionated on 1% agarose-formaldehyde gel, blotted onto nylonmembranes, hybridized to 107 dpm of 32P-labeled NGFR, GFP, and glyceralde-hyde-3-phosphate dehydrogenase (GAPDH) probes, and exposed to X-ray film(22).
RESULTS
Replacement of the U3 enhancer of the HIV LTR with theGATA-1 HS2 element restricts lentiviral gene expression toerythroblastic cell lines. To restrict lentiviral transgene expres-sion to a specific cell context, we developed a transcriptionaltargeting strategy based on replacing the U3 enhancer of theHIV LTR with cell-specific, genomic transcriptional controlelements. The U3 region of the 3� LTR of the pHR2 lentiviralvector (6), from position �418 to �40 with respect to thetranscription start site, was removed and replaced by a 200-bphuman genomic fragment containing an autoregulatory en-hancer of the GATA-1 erythroid-specific transcription factorgene (GATA-1 HS2) (18, 26). The �40-deleted LTR and theGATA-1/HIV-1 chimerical LTR (GATA-LTR) were used toreplace the 3� LTR of the pRRL.SIN vector carrying GFP as areporter gene (31). The resulting vectors, pRRL.sin-40.GFP(�40) and pRRL.GATA.GFP (G), were pseudotyped withVSV-G by a second-generation lentiviral packaging system (6)and preliminarily tested for their transcriptional properties by
transducing bulk cultures of human and murine stable cell lines(MOI, 10). FACS analysis of GFP expression in transducedhuman erythroblastic (K562 and HEL) and nonhematopoietic(HeLa) cell lines and murine fibroblastic (NIH 3T3) cellsshowed no expression from the �40 vector, and expression wasrestricted to the HEL and K562 cells from the targeted Gvector (Fig. 1). These data showed that the enhancerlessHIV-1 LTR is virtually inactive in the absence of TAT, aspreviously reported for two similar U3 deletions (�45 and�36) in the context of lentiviral SIN vectors (31). Transcrip-tional activity is restored by the insertion of the GATA-1 HS2enhancer, but it is restricted to the GATA-1-expressing K562and HEL cells.
A modified LTR is more efficient than an internal promoterto drive transgene expression in the context of a lentiviralvector. Retargeting the specificity of the LTR promoter al-lowed us to exploit the efficiency of the viral machinery indirecting gene expression from the spliced, major genomictranscript rather than from an intronless, internal transcrip-tional unit. In order to test the advantage of this design, wecompared the activity of the G vector with that of an RRL.sin-18.GATA.GFP (�18/G) vector containing the same combina-tion of enhancer-promoter in the conventional internal posi-tion in the context of a SIN vector (Fig. 2A, left panel). HELcells were transduced with the G and �18/G vectors at thesame MOI (10), maintained in culture as bulk populations, andmonitored for GFP expression by FACS analysis every 2 weeksfor 2 months. Transduction efficiency was very similar for bothvectors (virtually 100%), but GFP expression was fourfoldhigher in cells transduced with the G vector than in those
FIG. 1. Analysis of GFP expression in cell lines transduced with theG lentiviral vector, carrying a GFP gene under the control of theGATA-1 HS2-modified LTR (see Fig. 2A), and with a control �40vector, carrying a GFP gene under the control of an enhancerless LTR.Human erythroblastic HEL and K562 cells, nonhematopoietic HeLacells, and murine fibroblastic NIH 3T3 cells were infected at an MOIof 10, grown as bulk cultures, and analyzed for GFP expression bysingle-channel cytofluorimetry (x axis). Expression profiles are indi-cated by a thin (�40 vector) or a bold (G vector) line, respectively.
3998 LOTTI ET AL. J. VIROL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 11
Feb
ruar
y 20
22 b
y 18
8.92
.209
.147
.

transduced with the �18/G vector (mean fluorescence inten-sity [MFI], 2,300 versus 560; Fig. 2A, right panel). Southernblot analysis of DNA restricted with AflII showed that trans-duced cells harbored stably integrated vectors with intact re-combinant LTRs at a comparable vector copy number (Fig.2B). Restriction with EcoRI indicated essentially random in-tegration in both cell populations, although a distinct band ofunknown origin appeared over the hybridization smear in both
cases (Fig. 2B). This does not reflect oligoclonality of the bulkpopulations, as demonstrated by subsequent analysis of indi-vidual clones (see below). Northern blot analysis showed thatgenomic transcripts from the G vector were accumulated inHEL cells at a level fivefold higher than that of the subgenomictranscript from the �18/G vector, as quantitated by phospho-rimaging after normalization for GAPDH mRNA content(Fig. 2C). GFP expression, number of vector copies, and level
FIG. 2. (A, Left) Schematic maps of the RRL.GATA.GFP (G) and the RRL.sin-18.GATA.GFP (�18/G) vectors in their proviral forms. HS2indicates the GATA-1 HS2 autoregulatory enhancer replacing the HIV LTR U3 region in the G vector. An arrow indicates the transcription startsite of the chimerical LTR promoter. A crossed arrow indicates the disabled LTR promoter in the �18/G vector. SD, splice donor; SA, spliceacceptor; RRE, REV responsive element. Restriction sites used for the Southern analysis shown in panel B are indicated. (Right) Single-channelflow cytometry of GFP expression in HEL cells transduced with the G (upper panel) or �18/G (lower panel) vectors at an MOI of 10, as analyzed4 weeks after transduction. Distributions of transduced and control, untransduced cells are indicated by white and black peaks, respectively. MFIof GFP expression is in arbitrary units. (B) Southern blot analysis of genomic DNA extracted from HEL cell lines transduced with the G or �18/Gvector, digested with AflII or EcoRI, and hybridized to a GFP probe. Molecular size markers are reported in kb. (C) Northern blot analysis ofpoly(A)� RNA extracted from HEL cells transduced with the G or �18/G vector and hybridized with GFP (upper panel) and GAPDH (lowerpanel) probes.
VOL. 76, 2002 TRANSCRIPTIONAL TARGETING OF LENTIVIRAL VECTORS 3999
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 11
Feb
ruar
y 20
22 b
y 18
8.92
.209
.147
.

of vector-derived transcripts were also analyzed in single cellclones derived from the transduced bulk cultures. Cells werecloned at 0.2 to 0.3 cells/well, and single clones were isolatedafter 2 weeks of culture and grown up to 2 months for furtheranalysis. Overall, GFP expression levels were consistentlyhigher in clones transduced with the G vector than in thosetransduced with the �18/G vector, as analyzed by Southernblotting of each individual clone and normalization for thevector copy number (data not shown). In both groups ofclones, Southern blot analysis showed no evidence of commonor preferential sites of proviral integration. Interestingly, GFPexpression levels varied considerably among clones transducedwith the �18/G vector (MFI, 63 to 407) and correlated with thenumber of integrated proviruses (2 to 9), while in clones trans-duced with the G vector GFP levels were uniformly higher(MFI, 852 to 1,275) and essentially independent from the vec-tor copy number (4 to 18). Furthermore, 2 out of 18 HELclones transduced with the �18/G vector lost transgene expres-sion during prolonged culture, while all 16 clones transducedwith the G vector showed sustained transgene expressionthroughout the 2 months. These results indicate that a lentivi-ral vector carrying a modified LTR directs higher levels oftargeted gene expression at low vector copy number than avector containing the same enhancer-promoter combination inan internal position, and that its expression could be less sub-jected to position-effect variegation in the target cell genome.
LTR modification allows expression of two independentlyregulated transcriptional units. In order to monitor cell trans-duction and immunoselect transduced cells independentlyfrom the transcriptional activity of the targeted LTR, the�LNGFR cDNA was cloned downstream from the GFP genein both vectors as a second reporter gene, under the control ofa constitutive PGK promoter. The resulting vectors, RRL.sin-40.GFP.PGK.�N (�40/P) and RRL.GATA.GFP.PGK.�N(G/P) (Fig. 3A), were packaged at titers ranging from 5 � 106
to 5 � 107 transducing units/ml, as determined by infection ofHeLa cells with serial dilutions of viral supernatants and FACSanalysis of �LNGFR expression. To study the expression char-acteristics of the two transcriptional units, human myeloblastic(Kasumi-1), myelomonocytic (U937), erythroblastic (K562 andHEL), and nonhematopoietic (HeLa) cell lines and murineerythroblastic (MEL) and fibroblastic (NIH 3T3) cell lineswere infected by �40/P and G/P vectors (MOI, 5 to 10) andgrown as bulk cultures. Transduction efficiency ranged be-tween 67 and 96%, depending on the cell line, as analyzed byFACS analysis of LNGFR expression. Southern blot analysisof genomic DNA showed stable integration of intact provirusesand the polyclonal nature of all tested cell populations (datanot shown). Expression of vector-derived transcripts was ana-lyzed by Northern blotting of poly(A)� RNA and hybridizationto a �LNGFR-specific probe. As expected, accumulation ofLTR-driven transcripts was never detected in cells transducedwith the �40/P vector (Fig. 3B). In cells transduced with theG/P vector, transcripts derived from the GATA-LTR weredetected in K562, HEL (Fig. 3B), and MEL (data not shown)cells, but not in U937, HeLa (Fig. 3B), Kasumi-1, and NIH 3T3(data not shown) cells. Subgenomic transcripts derived fromthe internal PGK promoter were present in all cell lines trans-duced with both vectors (Fig. 3B and data not shown). At theprotein level, expression of both GFP and �LNGFR was quan-
titatively analyzed by double-fluorescence flow cytometry afterstaining with a PE-conjugated anti-LNGFR antibody (Fig. 4).Expression of �LNGFR from the internal PGK promoter wasobserved in all transduced cell lines, while expression of GFPfrom the GATA-LTR was detected only in erythroblastic cells(HEL and K562; Fig. 4). The two reporter genes were coex-pressed in �90% of transduced erythroblastic cells. These dataindicate that the use of a modified LTR and an internal pro-moter allows the expression of two independently regulatedgenes in the context of a lentiviral vector.
Targeted gene expression is selectively enhanced by specificpositioning of a WPRE. Northern blot analysis showed thattranscripts from the modified LTR, although restricted, wereaccumulated at a low level in K562 and HEL cells transduced
FIG. 3. (A) Schematic map of the RRL.sin-40.GFP.PGK.�N(�40/P) and the RRL.GATA.GFP.PGK.�N (G/P) lentiviral vectors.In the �40/P vector, the LTR carries a �418 to �40 deletion removingthe U3 enhancer, while in the G/P vector the same region is replacedby the GATA-1 HS2 autoregulatory enhancer. The arrows indicate thetranscription start sites of the LTR or the internal PGK promoter. SD,splice donor; SA, splice acceptor; RRE, REV responsive element;(A)n, polyadenylation site. (B) Northern blot analysis of poly(A)�
RNA extracted from K562, HEL (erythroblastic), U937 (myelomono-cytic), and HeLa (nonhematopoietic) cell lines transduced with the�40/P or the G/P vector and hybridized with GFP (upper panel) andGAPDH (lower panel) probes. Transcripts originating from the viralLTR or the internal PGK promoter are indicated on the left.
4000 LOTTI ET AL. J. VIROL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 11
Feb
ruar
y 20
22 b
y 18
8.92
.209
.147
.

with the double-expression cassette G/P vector compared tocells transduced with the single-cassette vector G (compareFig. 3 and 2C), suggesting a negative interference of the inter-nal promoter upon transcription of the GATA-LTR. In orderto improve the level of LTR-driven transcripts in a selectivefashion, we cloned a 500-bp WPRE at alternative positions inthe G/P vector, downstream of the GFP gene (Fig. 5A, G.W/Pvector) or the �LNGFR gene (Fig. 5A, G/P.W vector). As aresult, the WPRE was contained exclusively in the LTR-driven,viral genomic transcript in the G.W/P vector or in both thegenomic and the subgenomic PGK-driven transcripts in theG/P.W vector. Viral stocks derived from these vectors wereused to transduce HEL, K562, and U937 cell lines (MOI, 10).In K562 and HEL cells transduced with the G/P.W vector, thelevel of both the LTR- and the PGK-driven transcripts wasapproximately twofold higher than that in cells transduced withthe G/P vector, as quantitated by phosphorimaging of North-ern blots of poly(A)� RNA extracted from bulk cultures (Fig.5B, lane G/P.W versus G/P). Conversely, in cells transducedwith the G.W/P vector the level of the LTR-driven genomictranscripts was selectively increased (about fourfold) with re-spect to cells transduced with the G/P vector, while the level ofthe PGK-driven, subgenomic transcripts remained essentiallyunchanged (Fig. 5B, lane G.W/P versus G/P). Interestingly,addition of the WPRE in either configuration did not rescueaccumulation of genomic transcripts in nonerythroid cells (Fig.5B, U937) and had therefore no effect on the transcriptionaltargeting of the GATA-LTR. Protein expression data paral-
leled the results obtained at the RNA accumulation level.PGK-driven, �LNGFR expression increased slightly in bothHEL and K562 cells transduced with the G/P.W vector com-pared to cells transduced with the G/P and the G.W/P vectors(MFI in HEL cells, 1,336 versus 759 and 1,051, respectively;MFI in K562 cells, 1,345 versus 1,010 and 1,116, respectively, ina representative experiment). Conversely, GFP expression in-creased three- to fivefold in HEL and K562 cells transducedwith the G/P.W vector compared to cells transduced with G/Pand G.W/P (MFI in HEL cells, 345 versus 78 and 72, respec-tively; MFI in K562 cells, 260 versus 68 and 107, respectively).Transduced U937 cells never showed expression of GFP, while�LNGFR levels increased slightly in cells transduced withG/P.W (MFI, 422) with respect to cells transduced with G/P orG.W/P (MFI, 318 and 303, respectively). These results dem-onstrate that the presence of WPRE within a transcript selec-tively enhances its accumulation in transduced cells, thus al-lowing us to differentially modulate the expression levels of twodifferent genes carried by the same lentiviral vector. TheG.W/P vector was therefore chosen to evaluate transcriptionaltargeting of the GATA-LTR in human primary HSCs
Expression of the GATA-LTR is restricted to the erythro-
FIG. 4. Double-immunofluorescence FACS analysis of GFP (xaxis) and �LNGFR expression (y axis) in K562, HEL, U937, and HeLacells transduced with the G/P vector (Fig. 3) at an MOI of 10. Cellswere grown as bulk culture and analyzed 3 days after infection afterstaining with a PE-conjugated anti-LNGFR antibody.
FIG. 5. (A) Schematic maps of the RRL.GATA.GFP.PGK.�N.W(G/P.W) and RRL.GATA.GFP.W.PGK.�N (G.W/P) vectors in theirproviral forms. See Fig. 3 for abbreviations. The WPRE (W) is insertedat alternative positions in the vectors, both derived from the G/P vectordescribed in the legened to Fig. 3. (B) Northern blot analysis ofpoly(A)� RNA extracted from K562, HEL, and U937 cells transducedwith the G/P, G/P.W, or G.W/P vectors and hybridized to LNGFR(upper panels) or GAPDH (lower panels) probes. Transcripts origi-nating from the viral LTR or the internal PGK promoter are indicatedon the left.
VOL. 76, 2002 TRANSCRIPTIONAL TARGETING OF LENTIVIRAL VECTORS 4001
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 11
Feb
ruar
y 20
22 b
y 18
8.92
.209
.147
.

blastic progeny of human HSCs. Human cord blood-derivedCD34� stem-progenitor cells were cultured for 24 h in serum-free medium supplemented with cytokines (SCF, Flt-3L, IL-6,IL-3) and transduced with the G.W/P vector at an MOI of 50to 100. Three days after infection, cells were analyzed by flowcytometry for CD34, LNGFR, and GFP expression. More than60% of CD34� cells were transduced, as measured by theexpression of LNGFR driven by the internal PGK promoter,while only 23% of them expressed GFP from the GATA-LTR(Fig. 6, left panels). To test the activity of the GATA-LTR indifferentiated cells, transduced CD34� progenitors were in-duced to differentiate in liquid culture into either myeloid orerythroid lineages. Ten days after induction, expression ofGFP and LNGFR was evaluated by triple-immunofluores-cence FACS analysis of cells stained with antibodies againsteither erythroid (GpA) or myeloid (CD13) lineage-specificsurface markers. The proportion of GpA� cells expressingLNGFR and GFP was comparable (69 and 73%, respectively),indicating that the GATA-LTR was active in virtually all trans-duced erythroid cells (Fig. 6, right panels). Conversely, GFPexpression was barely detectable in a small fraction (9%) of theCD13� myeloid cells transduced at a level of 74% (Fig. 6,middle panels). Under both differentiation conditions, a frac-tion of GFP� cells stained negative for both GpA and CD13,indicating the presence of a residual, immature subpopulationderived from CD34� cells and maintaining GATA-LTR activ-ity. At a quantitative level, LNGFR expression was higher inundifferentiated (CD34�) progenitors than in differentiated(GpA� and CD13�) progeny (MFI, 174, 58, and 68, respec-tively), whereas GFP expression increased fivefold in theGpA� progeny of CD34� cells (MFI, 52 to 255), showing thatexpression from the GATA-LTR increases with erythroid cell
differentiation and suggesting that in an appropriate cell con-text the activity of a targeted LTR is higher than that of aconstitutive internal promoter.
To analyze transgene expression at the level of singlecolonies, CD34� cells were transduced with the G.W/P vec-tor, immunoselected for �LNGFR expression by immuno-magnetic cell sorting, and directly scored for GFP expres-sion by fluorescence microscopy in methylcellulose clonalcultures. The introduction of the immunoselection step al-lowed us to analyze GATA-LTR-driven gene expressiononly in cells differentiating from transduced progenitors.Colonies were morphologically scored as erythroid-burstforming units (BFU-E), granulocyte-macrophage-colonyforming units (CFU-GM), or granulocyte-erythroid-macro-phage-megakaryocyte-colony forming units (CFU-GEMM)between 10 and 14 days from plating, and no difference incolony-forming efficiency was found in control, mock-trans-duced cells versus G.W/P-transduced and immunoselectedcells. In the representative experiment shown in Fig. 7, mostBFU-E colonies (54 out of 57) scored strongly positive for GFPby fluorescence microscopy, while only rare CFU-GM colonies(2 out of 49) appeared weakly positive by the same assay.
Overall, these results show that transcription from theGATA-LTR is active at low levels in a fraction of the CD34�
hematopoietic progenitors, strongly down-regulated duringmyelomonocytic differentiation, and activated and maintainedat high levels in cells undergoing erythroid differentiation.
Expression of the GATA-LTR is restricted to the erythroidprogeny of human SRCs. The lineage-specific expression of theGATA-LTR was tested in human hematopoietic stem-progen-itor cells in vivo in the NOD-SCID mouse model. Cord bloodCD34� cells were transduced with the G.W/P vector, further
FIG. 6. Expression of GFP (upper panels) and �LNGFR (lower panels) in liquid culture of human CD34� hematopoietic stem-progenitor cellstransduced with the G.W/P vector (Fig. 5) and analyzed 3 days after infection (left panels) or 10 days after induction of myelomonocytic orerythroblastic differentiation (center and right panels). Cells were analyzed by triple-immunofluorescence flow cytometry after staining with aTC-conjugated anti-LNGFR antibody and PE-conjugated antibodies against specific surface markers for undifferentiated progenitors (CD34, rightpanels) or myeloid (CD13, center panels) and erythroid (GpA, right panels) progeny. Values in the gated areas indicate the percentage of cellsdouble positive for each surface marker and either GFP (upper panels) or �LNGFR (lower panels) expression.
4002 LOTTI ET AL. J. VIROL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 11
Feb
ruar
y 20
22 b
y 18
8.92
.209
.147
.

modified by the addition of the HIV-1 cPPT sequence, whichwas recently reported as essential for high efficiency of genetransfer into SCID-repopulating cells (SRCs) (10). A total of10 mice in three different experiments were sublethally irradi-ated, inoculated with 1.5 � 105 to 3 � 105 CD34� cells trans-duced at different MOIs (100 or 200), and sacrificed 5 to 11weeks after transplantation. Transduction efficiency of CD34�
cells ranged from 49 to 76%, as assayed by �LNGFR expres-sion 4 days after infection (Table 1). Engraftment of humancells ranged from 1.5 to 58%, as indicated by the proportion ofhuman CD45� cells in the mouse BM. Gene transfer efficiencyin engrafted human cells ranged from 35 to 72%, as assayed by�LNGFR expression (Table 1). A representative analysis ofthe BM of a NOD-SCID mouse that received a transplantationdose of 3 � 105 CD34� cells transduced at a proportion of76% is shown in Fig. 8. Here, 58% of the BM cells were of
human origin (CD45�) and 72% of human CD45� cells ex-pressed �LNGFR. Further staining with antibodies against thelineage markers CD19, CD13, and CD34 showed the presenceof human lymphoid, myeloid, and undifferentiated progenitorcells. LNGFR� cells were found in similar percentages in alltested lineages (Fig. 8). GFP expression was detectable in1% of human cells recovered from BM of all but one of the10 analyzed mice. In a single mouse (mouse number 1.3 inTable 1), GFP expression was detected in 33, 40, and 36% oftransduced CD45�, CD34�, and CD19� cells, respectively(data not shown), suggesting the presence in this animal of ahigh proportion of immature GATA-1-expressing multilineageand lymphoid progenitors. Since human erythroid differentia-tion occurs at a very low level in vivo in the NOD-SCID model(data not shown), expression of the GATA-LTR in the eryth-roblastic progeny of transduced SRCs was analyzed by plating
FIG. 7. Erythroid-specific expression of GFP in methylcellulose colonies derived from CD34� human hematopoietic progenitors transducedwith the G.W/P lentiviral vector (Fig. 5) and immunoselected for �LNGFR expression before plating. (A and C) Bright-field view of fully matureBFU-E and CFU-GM colonies 14 days after plating. (B and D) Green fluorescence view of the same fields (GFP-specific filters). Magnification,�10.
TABLE 1. Transduction of SRCs with the G.W/P vectora
Group Mouse Wk aftertransplantation MOI TE FACS
(%)Cell dose
(105)
Human cellengraftment(% CD45�)
LNGFR�
human cells(%)
GFP� BFU-E colonies
(%)b
GFP� CFU-GM colonies
(%)b
I 1.1 5 200 76 3 2.5 62 ND ND1.2 5 200 76 3 3 63 61 (8/13) 0 (0/21)1.3 9 200 76 3 58 72 73 (29/40) 0 (0/91)
II 2.1 9 200 68 2 50.5 44 52 (16/31) 0 (0/63)2.2 9 200 68 2 6.5 64 ND ND2.3 9 200 68 2 9.4 54 ND ND2.4 9 200 68 2 23.7 43 46 (11/24) 0 (0/43)
III 3.1 11 100 49 1.5 5.7 47 54 (6/11) 0 (0/24)3.2 11 100 49 1.5 4 35 ND ND3.3 11 100 49 1.5 1.5 45 ND ND
a G.W/P lentiviral vector confers erythroid-specific expression in BFU-E colonies differentiating from transduced SRCs. The results from three different experimentsare shown. MOI indicates MOI for CD34� cells. TE indicates transduction efficiency assessed by FACS 4 days after transduction. ND, not done.
b Ratios in parentheses indicate GFP� colonies/total counted colonies.
VOL. 76, 2002 TRANSCRIPTIONAL TARGETING OF LENTIVIRAL VECTORS 4003
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 11
Feb
ruar
y 20
22 b
y 18
8.92
.209
.147
.

BM cells from transplanted and control NOD-SCID mice inclonal cultures under conditions that preferentially supportoutgrowth of human progenitors (1). At 11 to 14 days afterplating, single colonies were counted and GFP� colonies werescored under an inverted fluorescence microscope. Overall, 70out of 119 BFU-E colonies were GFP� (average, 59%), while0 out of a total of 232 CFU-GM colonies showed detectableGFP expression (Table 1). Figure 9A to D shows GFP expres-sion in fully differentiated human erythroid colonies grownfrom the BM of a transplanted NOD-SCID mouse. Murinecolonies derived from control, mock-transplanted NOD-SCIDmice were few, GFP�, and morphologically distinguishablefrom human colonies obtained from transplanted animals.
Expression of the GATA-LTR is restricted to the erythroid
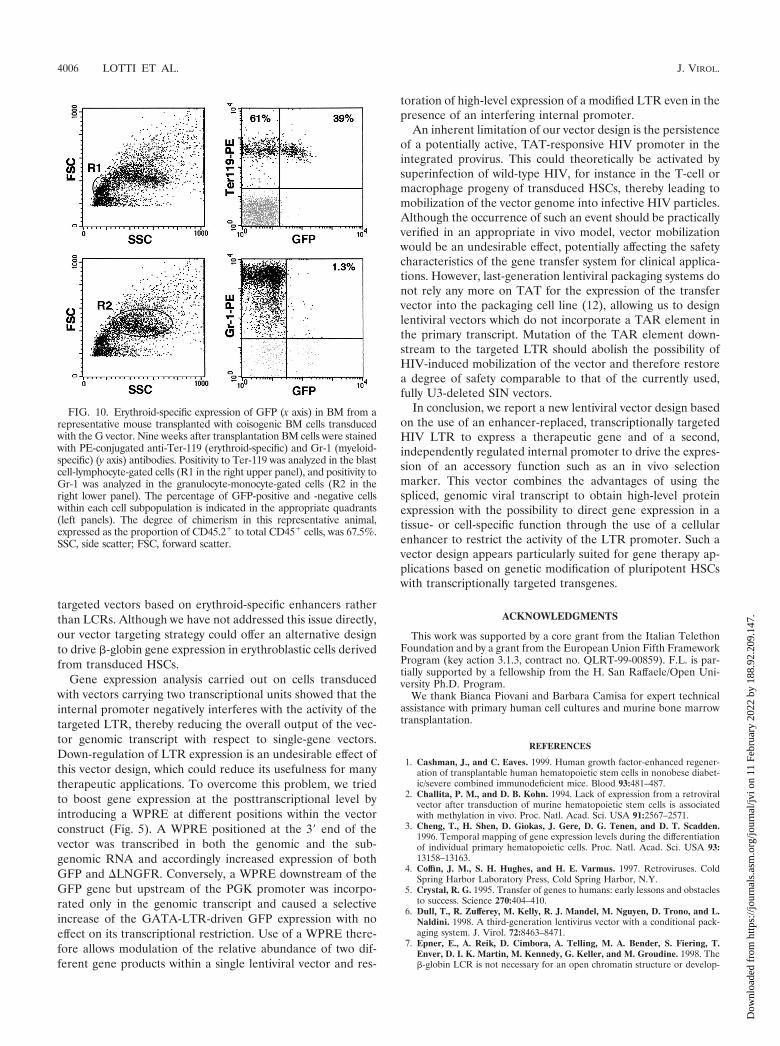
progeny of murine repopulating HSCs. To test the lineage-specific expression of the GATA-LTR in a transplantationmodel supporting the terminal differentiation of all progeniesderived from transduced HSCs, 5 � 106 BM cells fromC57BL/6 (CD45.2�) donor mice were transduced with the Gvector, further modified by the addition of the HIV-1 cPPTand WPRE sequences (MOI, 200), and transplanted into le-thally irradiated C57BL/6-Ly-5.1 (CD45.1�) recipient mice.Transduction efficiency of murine progenitors, expressed asthe percentage of GFP� BFU-E colonies in a methylcelluloseassay, averaged 75%. Nine to 11 weeks after transplantation,BM cells from recipient animals were analyzed for expressionof GFP and erythroid (TER-119) and myeloid (Gr-1) lineage-specific markers. Engraftment of donor cells in the BM, indi-cated by the proportion of CD45.2� to total CD45� cells,ranged from 26 to 94% (data not shown). GFP� cells weredetected at a significant level (3.0 to 39%) only in the eryth-roblastic, TER119� subpopulation of BM cells (Fig. 10, upperright panel). Conversely, in myeloblastic Gr-1� cells GFP ex-pression was virtually undetectable (0.6 to 1.3%) (Fig. 10,lower right panel). After normalization for the proportion ofengrafted donor cells in BM, TER119�/GFP� cells rangedfrom 3.2 to 96%. These results indicate that expression of theGATA-LTR is restricted to the fully differentiated erythroblas-tic progeny of transduced mouse repopulating stem cells invivo.
DISCUSSION
Gene therapy of most blood genetic disorders (e.g., SCID,chronic granulomatous disease, thalassemia) requires ex vivogene transfer into transplantable, self-renewing HSCs and reg-ulation of transgene expression in one or more differentiatedcell lineages. Lentiviral vectors are emerging as the vectors ofchoice for transducing HSCs without compromising their re-population capacity after BM transplantation (10, 15, 29).However, most preclinical studies carried out so far have reliedon the use of internal, constitutive promoters to drive trans-gene expression, while no systematic attempt has been under-taken to regulate transcription in a tissue- or cell-specific fash-ion (24, 28). One way to target transgene transcription is todrive an internal expression cassette with specific enhancer-promoter combinations (16). As an alternative strategy, wehave attempted to redirect the activity of the HIV-1 LTRpromoter by replacing most of the U3 region (from position�418 to �40 from the transcription start site) with a 200-bpautoregulatory enhancer (HS2) of the erythroid-specificGATA-1 transcription factor. In this design, the modified LTRcontrols the expression of the gene of interest (GFP in thisstudy), while an internal PGK promoter drives the expressionof a constitutive, independently regulated reporter gene(�LNGFR). Human cord blood-derived CD34� stem-progen-itor cells and murine repopulating HSCs were transduced athigh efficiency (up to 70%) with this type of vector, as shown bythe expression of the constitutively regulated reporter gene inundifferentiated progenitors as well as in the differentiatedprogenies (myeloid, lymphoid, and erythroid) obtained in vitroand in vivo upon transplantation into NOD-SCID (humancells) or coisogenic (murine cells) mice. Expression of thetransgene driven by the modified LTR was successfully re-
FIG. 8. Multilineage reconstitution of the BM of a NOD/SCIDmouse transplanted with cord blood-derived human CD34� hemato-poietic stem/progenitor cells transduced with the G.W/P lentiviral vec-tor (Fig. 5) at an MOI of 200. Transduction efficiency in the trans-planted cells was measured by FACS analysis of �LNGFR expression4 days after infection (upper left panel). Cells were transplanted in 10different NOD/SCID mice (see Table 1 for complete data). Engraft-ment was analyzed by FACS analysis of the human-specific CD45marker in bone marrow cells 9 weeks after transplantation (lower leftpanel). Transduction efficiency in the total engrafted cell population(CD45�, lower left panel) and in the progenitor (CD34�), B lymphoid(CD19�), and myeloid (CD13�) subpopulations (right panels) wereanalyzed by �LNGFR expression. The percentage of LNGFR-positiveand -negative cells within each cell subpopulation is indicated in theappropriate quadrants.
4004 LOTTI ET AL. J. VIROL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 11
Feb
ruar
y 20
22 b
y 18
8.92
.209
.147
.

stricted to the erythroblastic progeny of CD34� progenitors invitro and of human SCID repopulating cells and murine HSCsin vivo. Transcription from the chimerical LTR was active at alow level in a subpopulation of transduced CD34� cells (prob-ably cycling progenitors), down-regulated in myelomonocyticcells, and up-regulated and maintained at sustained levels inerythroblastic cells, strictly paralleling the expression patternof the GATA-1 gene during differentiation of human primaryhematopoietic cells (3, 23). Modification of the LTR had noeffects on titer, infectivity, and genomic stability of the vector.Interestingly, expression from the hybrid LTR does not requirecoexpression of TAT and appears therefore to be relativelyinsensitive to the presence of a TAR secondary structure in thegenomic transcript produced by the integrated provirus.
As a general strategy, modification of the viral LTR offers anumber of potential advantages. First, it provides a more effi-cient vector design, which allows the use of the major viraltranscriptional unit to express a gene of interest under theform of a genomic, spliceable RNA. In this study, we havedirectly compared the expression of a GFP gene driven by themodified LTR with that obtained by the same enhancer-pro-moter combination placed in a conventional, internal position,and we showed that expression from the genomic transcript isindeed higher and better restricted than that obtained from anunspliced transcript driven by an internal promoter. Second, itallows the use of a second, independently regulated internalpromoter to control additional vector functions. For most ge-netic diseases that could be cured by transplantation of genet-ically modified HSCs, providing an in vivo selection function totransduced cells (e.g., a drug resistance or a modified growthfactor receptor gene) is mandatory. In our vectors, the internalpromoter is transcribed at approximately constant levels inundifferentiated HSCs (CD34� progenitors or SRCs) and alltheir differentiated progenies in vitro and in vivo, while tran-
scription from the modified LTR is effectively restricted to theerythroblastic lineage. This design is therefore suitable forexpressing a constitutive in vivo selection marker and a tran-scriptionally targeted therapeutic gene in a single vector.Third, the presence of two copies of a genomic enhancer flank-ing the integrated provirus could reduce the chances of chro-matin-mediated inactivation of transcription, which is knownto affect the long-term maintenance of retroviral transgeneexpression in vivo, particularly in stem cells (2). Indeed, weshow that the expression driven by the GATA-LTR is persis-tent at uniformly high levels and independently from the num-ber of integrated proviruses in individual clones of transducedHEL cells, showing remarkably less position-effect variegationthan a vector based on the same enhancer-promoter combina-tion placed in an internal position. This vector design couldtherefore be indicated in all cases in which long-term persis-tence of gene expression, tight gene regulation, and high-levelprotein expression are required in a specific progeny of trans-duced HSCs, such as in gene therapy of thalassemia. A majorbreakthrough in this field has been obtained by the use of a full�-globin gene cloned in opposite transcriptional orientation ina lentiviral vector containing a reduced version of the �-globinlocus control region (LCR), which allowed the synthesis ofpotentially therapeutic levels of adult hemoglobin in a murinemodel of thalassemia (14). However, LCR sequences do notappear to confer position-independent expression to the viral-encoded transgene, even in this configuration (14). Indeed, therole of the LCR in maintaining an open chromatin conforma-tion to the �-globin locus has been challenged by several au-thors (7, 20). Binding of erythroid-specific transcription fac-tors, such as GATA-1, to enhancers of erythroid-specific genesearly in development or differentiation could instead be a keyfactor in initiation and maintenance of active chromatin struc-tures (13, 19), providing a rationale for using transcriptionally
FIG. 9. Erythroid-specific expression of GFP in methylcellulose colonies derived from human NOD-SCID mouse repopulating cells (SRCs)transduced with the G.W/P lentiviral vector (Fig. 5) and analyzed ex vivo 9 weeks after transplantation. (A and C) Bright-field view of fully matureBFU-E and CFU-GM colonies 14 days after plating. (B and D) Green fluorescence view of the same fields (GFP-specific filters). Magnification:�4 in panels A and B; �10 in panels C and D.
VOL. 76, 2002 TRANSCRIPTIONAL TARGETING OF LENTIVIRAL VECTORS 4005
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 11
Feb
ruar
y 20
22 b
y 18
8.92
.209
.147
.
targeted vectors based on erythroid-specific enhancers ratherthan LCRs. Although we have not addressed this issue directly,our vector targeting strategy could offer an alternative designto drive �-globin gene expression in erythroblastic cells derivedfrom transduced HSCs.
Gene expression analysis carried out on cells transducedwith vectors carrying two transcriptional units showed that theinternal promoter negatively interferes with the activity of thetargeted LTR, thereby reducing the overall output of the vec-tor genomic transcript with respect to single-gene vectors.Down-regulation of LTR expression is an undesirable effect ofthis vector design, which could reduce its usefulness for manytherapeutic applications. To overcome this problem, we triedto boost gene expression at the posttranscriptional level byintroducing a WPRE at different positions within the vectorconstruct (Fig. 5). A WPRE positioned at the 3� end of thevector was transcribed in both the genomic and the sub-genomic RNA and accordingly increased expression of bothGFP and �LNGFR. Conversely, a WPRE downstream of theGFP gene but upstream of the PGK promoter was incorpo-rated only in the genomic transcript and caused a selectiveincrease of the GATA-LTR-driven GFP expression with noeffect on its transcriptional restriction. Use of a WPRE there-fore allows modulation of the relative abundance of two dif-ferent gene products within a single lentiviral vector and res-
toration of high-level expression of a modified LTR even in thepresence of an interfering internal promoter.
An inherent limitation of our vector design is the persistenceof a potentially active, TAT-responsive HIV promoter in theintegrated provirus. This could theoretically be activated bysuperinfection of wild-type HIV, for instance in the T-cell ormacrophage progeny of transduced HSCs, thereby leading tomobilization of the vector genome into infective HIV particles.Although the occurrence of such an event should be practicallyverified in an appropriate in vivo model, vector mobilizationwould be an undesirable effect, potentially affecting the safetycharacteristics of the gene transfer system for clinical applica-tions. However, last-generation lentiviral packaging systems donot rely any more on TAT for the expression of the transfervector into the packaging cell line (12), allowing us to designlentiviral vectors which do not incorporate a TAR element inthe primary transcript. Mutation of the TAR element down-stream to the targeted LTR should abolish the possibility ofHIV-induced mobilization of the vector and therefore restorea degree of safety comparable to that of the currently used,fully U3-deleted SIN vectors.
In conclusion, we report a new lentiviral vector design basedon the use of an enhancer-replaced, transcriptionally targetedHIV LTR to express a therapeutic gene and of a second,independently regulated internal promoter to drive the expres-sion of an accessory function such as an in vivo selectionmarker. This vector combines the advantages of using thespliced, genomic viral transcript to obtain high-level proteinexpression with the possibility to direct gene expression in atissue- or cell-specific function through the use of a cellularenhancer to restrict the activity of the LTR promoter. Such avector design appears particularly suited for gene therapy ap-plications based on genetic modification of pluripotent HSCswith transcriptionally targeted transgenes.
ACKNOWLEDGMENTS
This work was supported by a core grant from the Italian TelethonFoundation and by a grant from the European Union Fifth FrameworkProgram (key action 3.1.3, contract no. QLRT-99-00859). F.L. is par-tially supported by a fellowship from the H. San Raffaele/Open Uni-versity Ph.D. Program.
We thank Bianca Piovani and Barbara Camisa for expert technicalassistance with primary human cell cultures and murine bone marrowtransplantation.
REFERENCES
1. Cashman, J., and C. Eaves. 1999. Human growth factor-enhanced regener-ation of transplantable human hematopoietic stem cells in nonobese diabet-ic/severe combined immunodeficient mice. Blood 93:481–487.
2. Challita, P. M., and D. B. Kohn. 1994. Lack of expression from a retroviralvector after transduction of murine hematopoietic stem cells is associatedwith methylation in vivo. Proc. Natl. Acad. Sci. USA 91:2567–2571.
3. Cheng, T., H. Shen, D. Giokas, J. Gere, D. G. Tenen, and D. T. Scadden.1996. Temporal mapping of gene expression levels during the differentiationof individual primary hematopoietic cells. Proc. Natl. Acad. Sci. USA 93:13158–13163.
4. Coffin, J. M., S. H. Hughes, and H. E. Varmus. 1997. Retroviruses. ColdSpring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
5. Crystal, R. G. 1995. Transfer of genes to humans: early lessons and obstaclesto success. Science 270:404–410.
6. Dull, T., R. Zufferey, M. Kelly, R. J. Mandel, M. Nguyen, D. Trono, and L.Naldini. 1998. A third-generation lentivirus vector with a conditional pack-aging system. J. Virol. 72:8463–8471.
7. Epner, E., A. Reik, D. Cimbora, A. Telling, M. A. Bender, S. Fiering, T.Enver, D. I. K. Martin, M. Kennedy, G. Keller, and M. Groudine. 1998. The�-globin LCR is not necessary for an open chromatin structure or develop-
FIG. 10. Erythroid-specific expression of GFP (x axis) in BM from arepresentative mouse transplanted with coisogenic BM cells transducedwith the G vector. Nine weeks after transplantation BM cells were stainedwith PE-conjugated anti-Ter-119 (erythroid-specific) and Gr-1 (myeloid-specific) (y axis) antibodies. Positivity to Ter-119 was analyzed in the blastcell-lymphocyte-gated cells (R1 in the right upper panel), and positivity toGr-1 was analyzed in the granulocyte-monocyte-gated cells (R2 in theright lower panel). The percentage of GFP-positive and -negative cellswithin each cell subpopulation is indicated in the appropriate quadrants(left panels). The degree of chimerism in this representative animal,expressed as the proportion of CD45.2� to total CD45� cells, was 67.5%.SSC, side scatter; FSC, forward scatter.
4006 LOTTI ET AL. J. VIROL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 11
Feb
ruar
y 20
22 b
y 18
8.92
.209
.147
.

mentally regulated transcription of the native mouse �-globin locus. Mol.Cell 2:447–455.
8. Evans, T., and G. Felsenfeld. 1989. The erythroid-specific transcription fac-tor Eryf1: a new finger protein. Cell 58:877–885.
9. Ferrari, G., G. Salvatori, C. Rossi, G. Cossu, and F. Mavilio. 1995. Aretroviral vector containing a muscle-specific enhancer drives gene expres-sion only in differentiated muscle fibers. Hum. Gene Ther. 6:733–742.
10. Follenzi, A., L. E. Ailles, S. Bakovic, M. Geuna, and L. Naldini. 2000. Genetransfer by lentiviral vectors is limited by nuclear translocation and rescuedby HIV-1 pol sequences. Nat. Genet. 25:217–222.
11. Grande, A., B. Piovani, A. Aiuti, S. Ottolenghi, F. Mavilio, and G. Ferrari.1999. Transcriptional targeting of retroviral vectors to the erythroblasticprogeny of transduced hematopoietic stem cells. Blood 93:3276–3285.
12. Klages, N., R. Zufferey, and D. Trono. 2000. A stable system for the high-titerproduction of multiply attenuated lentiviral vectors. Mol. Ther. 2:170–176.
13. Martin, D. I., S. Fiering, and M. Groudine. 1996. Regulation of beta-globingene expression: straightening out the locus. Curr. Opin. Genet. Dev. 6:488–495.
14. May, C., S. Rivella, J. Callegari, G. Heller, K. M. Gaensler, L. Luzzatto, andM. Sadelain. 2000. Therapeutic haemoglobin synthesis in beta-thalassaemicmice expressing lentivirus-encoded human beta-globin. Nature 406:82–86.
15. Miyoshi, H., K. A. Smith, D. E. Mosier, I. M. Verma, and B. E. Torbett. 1999.Transduction of human CD34� cells that mediate long-term engraftment ofNOD/SCID mice by HIV vectors. Science 283:682–686.
16. Moreau-Gaudry, F., P. Xia, G. Jiang, N. P. Perelman, G. Bauer, J. Ellis,K. H. Surinya, F. Mavilio, C. K. Shen, and P. Malik. 2001. High-levelerythroid-specific gene expression in primary human and murine hemato-poietic cells with self-inactivating lentiviral vectors. Blood 98:2664–2672.
17. Mulligan, R. C. 1993. The basic science of gene therapy. Science 260:926–932.
18. Nicolis, S., C. Bertini, A. Ronchi, S. Crotta, L. Lanfranco, E. Moroni, B.Giglioni, and S. Ottolenghi. 1991. An erythroid specific enhancer upstreamto the gene encoding the cell-type specific transcription factor GATA-1.Nucleic Acids Res. 19:5285–5291.
19. Orkin, S. H. 1995. Regulation of globin gene expression in erythroid cells.Eur. J. Biochem. 231:271–281.
20. Reik, A., A. Telling, G. Zitnik, D. Cimbora, E. Epner, and M. Groudine.1998. The locus control region is necessary for gene expression in the human�-globin locus but not the maintenance of an open chromatin structure inerythroid cells. Mol. Cell. Biol. 18:5992–6000.
21. Ruggieri, L., A. Aiuti, M. Salomoni, E. Zappone, G. Ferrari, and C. Bor-dignon. 1997. Cell-surface marking of CD(34�)-restricted phenotypes ofhuman hematopoietic progenitor cells by retrovirus-mediated gene transfer.Hum. Gene Ther. 8:1611–1623.
22. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: alaboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, ColdSpring Harbor, N.Y.
23. Sposi, N. M., L. I. Zon, A. Care, M. Valtieri, U. Testa, M. Gabbianelli, G.Mariani, L. Bottero, C. Mather, S. H. Orkin, et al. 1992. Cell cycle-depen-dent initiation and lineage-dependent abrogation of GATA-1 expression inpure differentiating hematopoietic progenitors. Proc. Natl. Acad. Sci. USA89:6353–6357.
24. Trono, D. 2000. Lentiviral vectors: turning a deadly foe into a therapeuticagent. Gene Ther. 7:20–23.
25. Tsai, S. F., D. I. Martin, L. I. Zon, A. D. D’Andrea, G. G. Wong, and S. H.Orkin. 1989. Cloning of cDNA for the major DNA-binding protein of theerythroid lineage through expression in mammalian cells. Nature 339:446–451.
26. Tsai, S. F., E. Strauss, and S. H. Orkin. 1991. Functional analysis and in vivofootprinting implicate the erythroid transcription factor GATA-1 as a posi-tive regulator of its own promoter. Genes Dev. 5:919–931.
27. Verma, I. M., and N. Somia. 1997. Gene therapy–-promises, problems andprospects. Nature 389:239–242.
28. Vigna, E., and L. Naldini. 2000. Lentiviral vectors: excellent tools for exper-imental gene transfer and promising candidates for gene therapy. J. GeneMed. 2:308–316.
29. Woods, N. B., C. Fahlman, H. Mikkola, I. Hamaguchi, K. Olsson, R.Zufferey, S. E. Jacobsen, D. Trono, and S. Karlsson. 2000. Lentiviral genetransfer into primary and secondary NOD/SCID repopulating cells. Blood96:3725–3733.
30. Zufferey, R., J. E. Donello, D. Trono, and T. J. Hope. 1999. Woodchuckhepatitis virus posttranscriptional regulatory element enhances expression oftransgenes delivered by retroviral vectors. J. Virol. 73:2886–2892.
31. Zufferey, R., T. Dull, R. J. Mandel, A. Bukovsky, D. Quiroz, L. Naldini, andD. Trono. 1998. Self-inactivating lentivirus vector for safe and efficient in vivogene delivery. J. Virol. 72:9873–9880.
32. Zufferey, R., D. Nagy, R. J. Mandel, L. Naldini, and D. Trono. 1997. Multiplyattenuated lentiviral vector achieves efficient gene delivery in vivo. Nat.Biotechnol. 15:871–875.
VOL. 76, 2002 TRANSCRIPTIONAL TARGETING OF LENTIVIRAL VECTORS 4007
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 11
Feb
ruar
y 20
22 b
y 18
8.92
.209
.147
.