tau protein and tau aggregation · pdf filetau protein and tau aggregation inhibitors bruno...
TRANSCRIPT

lable at ScienceDirect
Neuropharmacology 59 (2010) 276e289
Contents lists avai
Neuropharmacology
journal homepage: www.elsevier .com/locate/neuropharm
Review
Tau protein and tau aggregation inhibitors
Bruno Bulic a,1, Marcus Pickhardt b, Eva-Maria Mandelkowb, Eckhard Mandelkowb,*
aCenter for Advanced European Studies and Research, Ludwig-Erhard-Allee 2, 53175 Bonn, GermanybMax-Planck-Unit for Structural Molecular Biology, 22607 Hamburg, Germany
a r t i c l e i n f o
Article history:Received 30 October 2009Received in revised form20 January 2010Accepted 26 January 2010
Keywords:Aggregation inhibitorsAlzheimer's diseaseAmyloidsNeurodegenerationTau protein
* Corresponding author. c/o DESY, Notkestrasse 85Fax: þ49 (0) 40 89716810.
E-mail addresses: [email protected] (B. Bu(E. Mandelkow).
1 Fax: þ49 22896569401.
0028-3908/$ e see front matter � 2010 Elsevier Ltd.doi:10.1016/j.neuropharm.2010.01.016
a b s t r a c t
Alzheimer disease is characterized by pathological aggregation of two proteins, tau and Ab-amyloid, bothof which are considered to be toxic to neurons. In this review we summarize recent advances on smallmolecule inhibitors of protein aggregation with emphasis on tau, with activities mediated by the directinterference of self-assembly. The inhibitors can be clustered in several compound classes according totheir chemical structure, with subsequent description of the structureeactivity relationships, showingthat hydrophobic interactions are prevailing. The description is extended to the pharmacological profileof the compounds in order to evaluate their drug-likeness, with special attention to toxicity andbioavailability. The collected data indicate that following the improvements of the in vitro inhibitorypotencies, the consideration of the in vivo pharmacokinetics is an absolute prerequisite for the devel-opment of compounds suitable for a transfer from bench to bedside.
� 2010 Elsevier Ltd. All rights reserved.
1. Introduction
In protein aggregation diseases such as Alzheimer, Parkinson,Huntington and others, it is known that aggregation inhibitors ofa specific amyloidogenic peptide are also potential aggregationinhibitors of a wide variety of other amyloidogenic peptides (Heiseret al., 2002; Jin et al., 2003; Porat et al., 2006; Taniguchi et al., 2005).Histological dyes such as Thioflavin S or Congo red bind to aggre-gates in at least fifteen genetically unrelated disorders (Kelly, 1996).Indeed, amyloid fibrils, irrespective of their amino acid sequence,share a common X-ray diffraction pattern showing a characteristic4.6e4.8 Å meridional reflection (Fig. 1). It corresponds to thespacing between the peptide chains forming cross-b structure, suchthat the b-strands of the proteins are arranged perpendicular to thefibril axis (Fig. 1b). AFM microscopy of Ab42 aggregates in recon-stituted membranes revealed a remarkable supramolecular ion-channel like structure, (Lashuel and Lansbury, 2006; Quist et al.,2005), a striking feature shared by most amyloids such as a-synu-clein, Ab42, IAPP and others. Irrespective of their amino acidsequence, some of the soluble oligomeric amyloids also havesimilar structures, which are recognised by oligomer-specificantibodies independently of the amino acid sequence (Glabe, 2004;
, 22607 Hamburg, Germany.
lic), [email protected]
All rights reserved.
Kayed et al., 2007, 2003). It is noteworthy that even whena compound class is reported to be inhibitor of a specific amyloidtype, its inhibitory potency on other amyloid types can hardly bepredicted, due to variations in the interactions with the amino acidresidues forming the peptide backbone.
Neurodegenerative diseases associated with tau proteins whichself-assemble into abnormal fibers (“paired helical filaments” orPHFs) are known as “tauopathies” (Goedert et al., 1998). Thesefilaments can form higher order aggregates (“neurofibrillarytangles”, “neuropil threads”) in neurons or other cell types of thebrain (Ballatore et al., 2007; Kidd, 1963; Leroy et al., 2007; Sawayaet al., 2007; Vieira et al., 2007). PHFs have the appearance of twofilaments twisted around one other with a cross-over repeat of80 nm and an apparent width varying between w10 and w22 nm.The most common tauopathy is Alzheimer's disease, but taudeposits also occur in frontotemporal dementias (FTDP-17), Pick'sdisease, Parkinson's disease, progressive nuclear palsy and otherconditions (Arima, 2006; Guerrero et al., 2008).
The neurodegenerative processes involved in AD are still poorlyunderstood, however, protein aggregates and oligomers are rec-ognised as major elements of cellular toxicity. Therefore, smallmolecule inhibitors of protein aggregation are anticipated drugcandidates, and preliminary data on a phase II clinical trial with thedrug candidate methylene blue chloride (MTC) has been claimed tosupport the concept of tau aggregation inhibition as a means toaddress Alzheimer's disease (Wischik et al., 1996, 2007).
A significant advance in understanding tau's behaviour camewhen it was recognised that the protein contains isolated short
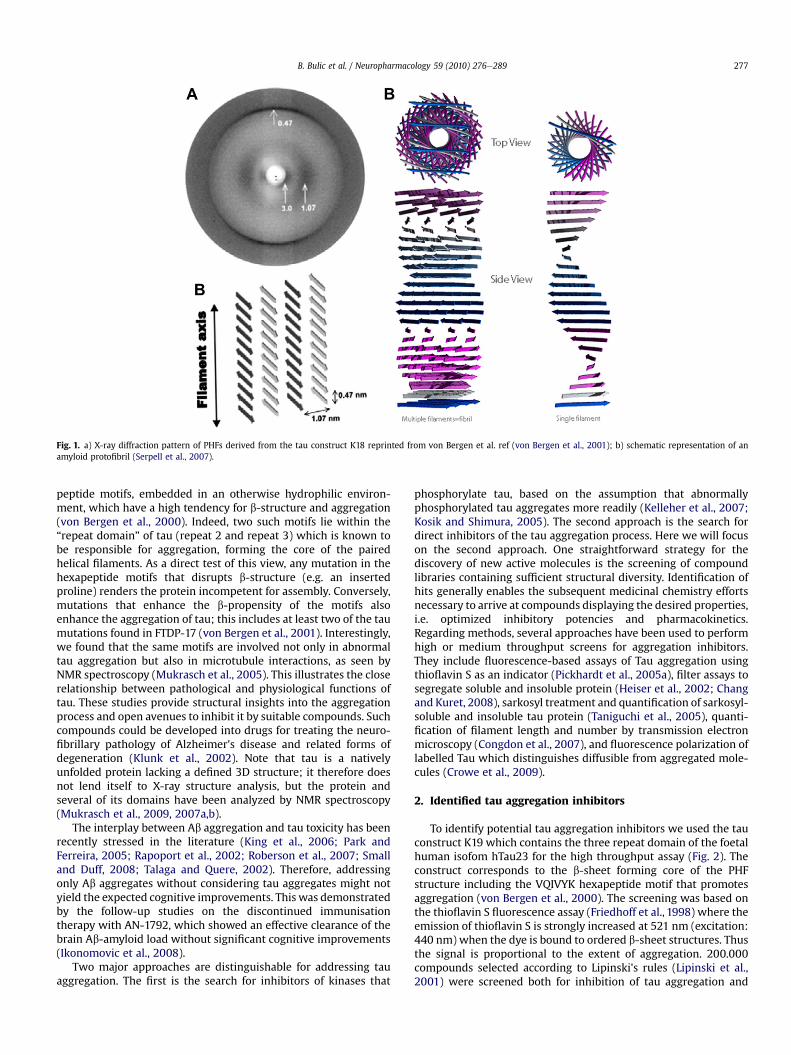
Fig. 1. a) X-ray diffraction pattern of PHFs derived from the tau construct K18 reprinted from von Bergen et al. ref (von Bergen et al., 2001); b) schematic representation of anamyloid protofibril (Serpell et al., 2007).
B. Bulic et al. / Neuropharmacology 59 (2010) 276e289 277
peptide motifs, embedded in an otherwise hydrophilic environ-ment, which have a high tendency for b-structure and aggregation(von Bergen et al., 2000). Indeed, two such motifs lie within the“repeat domain” of tau (repeat 2 and repeat 3) which is known tobe responsible for aggregation, forming the core of the pairedhelical filaments. As a direct test of this view, any mutation in thehexapeptide motifs that disrupts b-structure (e.g. an insertedproline) renders the protein incompetent for assembly. Conversely,mutations that enhance the b-propensity of the motifs alsoenhance the aggregation of tau; this includes at least two of the taumutations found in FTDP-17 (von Bergen et al., 2001). Interestingly,we found that the same motifs are involved not only in abnormaltau aggregation but also in microtubule interactions, as seen byNMR spectroscopy (Mukrasch et al., 2005). This illustrates the closerelationship between pathological and physiological functions oftau. These studies provide structural insights into the aggregationprocess and open avenues to inhibit it by suitable compounds. Suchcompounds could be developed into drugs for treating the neuro-fibrillary pathology of Alzheimer's disease and related forms ofdegeneration (Klunk et al., 2002). Note that tau is a nativelyunfolded protein lacking a defined 3D structure; it therefore doesnot lend itself to X-ray structure analysis, but the protein andseveral of its domains have been analyzed by NMR spectroscopy(Mukrasch et al., 2009, 2007a,b).
The interplay between Ab aggregation and tau toxicity has beenrecently stressed in the literature (King et al., 2006; Park andFerreira, 2005; Rapoport et al., 2002; Roberson et al., 2007; Smalland Duff, 2008; Talaga and Quere, 2002). Therefore, addressingonly Ab aggregates without considering tau aggregates might notyield the expected cognitive improvements. This was demonstratedby the follow-up studies on the discontinued immunisationtherapy with AN-1792, which showed an effective clearance of thebrain Ab-amyloid load without significant cognitive improvements(Ikonomovic et al., 2008).
Two major approaches are distinguishable for addressing tauaggregation. The first is the search for inhibitors of kinases that
phosphorylate tau, based on the assumption that abnormallyphosphorylated tau aggregates more readily (Kelleher et al., 2007;Kosik and Shimura, 2005). The second approach is the search fordirect inhibitors of the tau aggregation process. Here we will focuson the second approach. One straightforward strategy for thediscovery of new active molecules is the screening of compoundlibraries containing sufficient structural diversity. Identification ofhits generally enables the subsequent medicinal chemistry effortsnecessary to arrive at compounds displaying the desired properties,i.e. optimized inhibitory potencies and pharmacokinetics.Regarding methods, several approaches have been used to performhigh or medium throughput screens for aggregation inhibitors.They include fluorescence-based assays of Tau aggregation usingthioflavin S as an indicator (Pickhardt et al., 2005a), filter assays tosegregate soluble and insoluble protein (Heiser et al., 2002; Changand Kuret, 2008), sarkosyl treatment and quantification of sarkosyl-soluble and insoluble tau protein (Taniguchi et al., 2005), quanti-fication of filament length and number by transmission electronmicroscopy (Congdon et al., 2007), and fluorescence polarization oflabelled Tau which distinguishes diffusible from aggregated mole-cules (Crowe et al., 2009).
2. Identified tau aggregation inhibitors
To identify potential tau aggregation inhibitors we used the tauconstruct K19 which contains the three repeat domain of the foetalhuman isofom hTau23 for the high throughput assay (Fig. 2). Theconstruct corresponds to the b-sheet forming core of the PHFstructure including the VQIVYK hexapeptide motif that promotesaggregation (von Bergen et al., 2000). The screening was based onthe thioflavin S fluorescence assay (Friedhoff et al., 1998) where theemission of thioflavin S is strongly increased at 521 nm (excitation:440 nm) when the dye is bound to ordered b-sheet structures. Thusthe signal is proportional to the extent of aggregation. 200.000compounds selected according to Lipinski's rules (Lipinski et al.,2001) were screened both for inhibition of tau aggregation and

Fig. 2. Diagram of the full-length tau isoform htau40 and the repeat domain constructused in the PHF inhibition assay (construct K19, repeat domain with 3 repeats, R2absent). PHF6 is the hexapeptide motif that nucleates the aggregation of construct K19.
B. Bulic et al. / Neuropharmacology 59 (2010) 276e289278
for induced disassemblyof tau aggregates (Pickhardt et al., 2005a,b).Tau protein was incubated overnight under conditions favoringaggregation in the presence of the screened compound (Fig. 3, upperpart). The amount of aggregated tau protein was then quantifiedby fluorescence after addition of thioflavine S. In a similar approachthe disassembly of tau fibrils was performed by incubation ofpreformed PHFs in the presence or absence of compounds overnightat 37 �C and then measuring the ThS-fluorescence of the remainingfibrils (Fig. 3, lower part). The obtained Z-factor value of 0.81confirmed the reliability of the assay (Zhang et al., 1999). The resultswere verified with other assays (electron microscopy, filter/pellet-ing-assay) as well as with other tau constructs and isoforms.
The property of phosphorylated tau to detach from microtu-bules and to aggregate into fibrils depends on a complexphosphorylation pattern. In fact, phosphorylation could eithercontribute or hinder the aggregation, depending on its extent andlocalisation. Therefore the screening was performed with unphos-phorylated recombinant tau which provided fibers highly similar tothose purified from Alzheimer brain tissues and whose structurehas been previously assessed by electron microscopy and
Fig. 3. Thioflavin S (ThS) fluorescence assay for compounds that inhibit aggregation oftau into PHFs (upper part), and compounds that disaggregate preformed PHFs (lowerpart) (Bulic et al., 2007).
spectroscopic methods, indicating that the in vitro aggregation isa reliable PHF model (Barghorn et al., 2004; Schneider et al., 1999).
To avoid false-positive ThS-signals (and therefore false inhibi-tory effects) the compounds were initially screened for theirpotential self-fluorescence at the given excitation- and emissionwavelengths. All compounds which showed a higher fluorescencesignal in absence of protein than in the presence were excludedfrom further testing. To exclude possible quenching or ThS-displacement effects the substances were also tested in 'dye-free'assays such as pelleting and filter assays, intrinsic tryptophanfluorescence or electron microscopy.
The ability not only to inhibit tau aggregation but also to disas-semble preformed aggregates was included in the selectionparameters for the compound screen, leading to the elimination of99.96% of the library compounds. The identified hits were able toinduce the disassembly of PHFs with up to 80% efficiency at 60 mMcompound concentration. These hits were classified in clustersaccording to their chemical structures. Details on the inhibitoryactivities obtained from the N-phenylamines, anthraquinones,phenylthiazolyl-hydrazides (PTHs) and thioxothiazolidinones (rho-danines) were described previously (Bulic et al., 2007; Khlistunovaet al., 2007; Larbig et al., 2007; Pickhardt et al., 2007a, 2005b).
A chemistry primarily aimed at elucidating the requirements forimproved inhibitory potencies allowed the deduction of thestructureeactivity relationship (SAR) of the two last mentionedcompound clusters (PTHs and rhodanines). Selected compoundswere subsequently tested on a neuronal cell model of tau aggre-gation (Fig. 4).
2.1. Rhodanine-based inhibitors
The rhodanine based compounds (Fig. 5) are members of anappealing hit class. The scaffold is found in various bioactivecompounds that were reported as antimalarial, antituberculous,antibiotic, anticancer, antitoxin and hypoglycaemic by targetingrespectively the Enoyl-acyl Carrier Protein Reductase, peptidedeformylase (PDF), RNA-polymerase, JNK-stimulating phosphatase1 (JSP-1), bacterial proteases, PPAR-receptor and GSK-3 (Ahn et al.,2006; Cutshall et al., 2005; Gualtieri et al., 2006; Hu et al., 2008;Irvine et al., 2008; Johnson et al., 2008; Kumar et al., 2007;Martinez et al., 2005; Russell et al., 2009; Villain-Guillot et al.,2007; Zervosen et al., 2004). When focusing on Alzheimer'sdisease, the pleiotropic effect of rhodanines might be of interestsince both PPARg and GSK-3 are potential targets in AD.
Although derivatives are suspected to undergo conjugate addi-tion in vivo that might reduce their plasma half-life, (Carlson et al.,2006) they are frequently employed in medicinal chemistry withno apparent side effects. Their bioavailability and tolerability arereported in a long-term clinical study with epalrestat (3 years,150 mg/day oral administration), an aldose reductase inhibitorindicated for diabetic neuropathy (Fig. 6) (Hotta et al., 2006). The invitro permeability (rat jujenum) was observed at Papp ¼ 4.34 �1.78 � 10e6 cm/s. The extracted data indicate a good permeabilitywith an excellent bioavailability (100 � 22% bioavailability, peakplasma concentration of 4.0 � 0.9 mg/ml at 1.7 h after administra-tion of a 50 mg oral dose to adults) and a good plasma stability(half-life t1/2: 102.5 min) (Sturm et al., 2006). Epalrestat was foundto be highly protein bound in plasma with a 90% binding rate.Derivatives based on the rhodanine scaffold were submitted to invitro pharmacodynamic analyses, showing their adequate perme-ability, non-toxicity on HepG2 cells, moderate effect on cytochromeP450s and hERG, but variable stability with liver microsomes,depending on the rhodanine substituents (Johnson et al., 2008). Onthe other side, the parent thiazolidinedione heterocycle present inPPAR agonists such has troglitazone is associated with high

Fig. 4. Tau expression, aggregation and inhibition in a cellular model affected by compounds (Larbig et al., 2007; Pickhardt et al., 2007b; Bulic et al., 2007).
B. Bulic et al. / Neuropharmacology 59 (2010) 276e289 279
hepatotoxicity that might be however related to the substitutentssince pioglitazone and rosiglitazone are considered safe (Fig. 6)(Park et al., 2005; Tolman and Chandramouli, 2003).
The rhodanine core for tau aggregation inhibition has beeninvestigated via the synthesis of a focused library. In these exper-iments, rhodanines (R1 ¼ S and R2 ¼ S, Fig. 6b), thiohydantoin(R1 ¼ S and R2 ¼ N), thioxooxazolidine (R1 ¼ S and R2 ¼ O), oxa-zolidinedione (R1 ¼ O and R2 ¼ O), and hydantoin (R1 ¼ O andR2 ¼ N) were synthesised and screened for activity on tau aggre-gation inhibition and disaggregation of preformed tau aggregates.The following trend in the depolymerisation of tau aggregates wasobserved: rhodanine (IC50/DC50 (mM); 0.8/0.1) > thiohydantoin(6.1/0.4)>> oxazolidinedione (3.5/2.2)¼ thioxooxazolidinone (3.1/2.4) >> hydantoin (22.6/54.3). The IC50 and DC50 values representrespectively the assembly-inhibiting and disassembly-inducinghalf-maximal concentrations measured in vitro. The rhodanineheterocycle appeared to be the most potent, underlining theimportance of the thioxo group in rhodanines. The rhodanine corehas been reported as a carboxylic acid bioisoster by size, lowelectronegativity, and ability to engage in hydrogen bonds (Pataniand LaVoie, 1996).
Fig. 5. Variation of rhodanine inhibitor structure. Illustration of the variations on thecore (R1 and R2) and on the flanking substituents (R3 and R4) (Bulic et al., 2007).
Besides the central rhodanine core, the substitution patterns onR1 and R3 (Fig. 5) showed that hydrogen bond acceptors in the formof a nitro group, carboxylic acids, phenols, sulfonates/sulfonamidesare required, in line with observations from other known amyloidaggregation inhibitors (Bulic et al., 2007). Noteworthy, the totallength of the molecule proved to be of importance, as also reportedbelow (par. 2.3) for curcumin. Variations of the length of the linkerbetween the carboxylic acid and the rhodanine core (red part,Fig. 5) revealed that increasing the distance up to two carbon bondsresulted in an appreciable increase in the compound's inhibitorypotency indicating an optimal positioning of the inhibitor towardits binding site. The heteroaromatic side chain (R3, Fig. 5) toleratedvariations, but modifications on the furan heterocycle showed thatthe electron density distribution on the molecule is important, asreplacement of the furan ring in 16 for thiophene in 22 (Fig. 7)lowered the potency, as well as the pyridine in 27 compared to thefuran in 19.
Moreover, the presence of an aromatic side chainwas important,supporting an hydrophobic interaction and/or p-stacking on thisfragment (Waters, 2002). After optimization, an interesting 170 nMIC50 (compound 19) could be obtained.
The compounds were further tested on neuronal cell models oftau aggregation, with activities depicted in Fig. 12. This also illus-trated the slight discrepancy between potencies observed in vitroand in the cell-based assay, reflecting the need for further optimi-zation of this compound class with respect to ADME parameters(absorption/distribution/metabolism/excretion) relevant for the invivo activity, and in the first place their membrane permeability forcell models. Whereas the toxicity was restricted to a safe range(2e8%, LDH assay in N2A cells, incubation 24 h with 10 mMcompound, Fig. 12), the negatively charged carboxylate present onmost compounds may limit the membrane permeability. The betterefficiency on cells of charge-neutral benzimidazole-containing

Fig. 6. Rhodanine inhibitor of tau aggregation (01) and parent drug compounds.
B. Bulic et al. / Neuropharmacology 59 (2010) 276e289280
compound 14 (Fig. 7) might therefore be correlated with its betterpermeability, a feature that would allow optimization of the otherinhibitors for in vivo potency by modification of the carboxylatewith charge-neutral bioisosters.
Because a discussion about toxicity of small oligomers hasemerged there may be concerns about the toxicity of fragmentsgenerated by the disassembly of fibrils (Lashuel and Grillo-Bosch,2005; Necula et al., 2007). Nevertheless, studies using compound14 allowed observing the effect of tau filament disassembly oncellular viability. A clear reversal of the toxicity caused by tauaggregation in the cytosol was seen (Khlistunova et al., 2007). Also,recent reports on compounds binding to Ab fibrils indicated thatthey might also bind to oligomers and further support neuro-protective effects (Hong et al., 2007; Maezawa et al., 2008).
2.2. Phenylthiazolyl-hydrazide inhibitors
In parallel to the rhodanine compounds, a hit from HTS-screenwas selected for lead optimization and SAR investigation (Larbiget al., 2007). The application of in silico scaffold hopping providedthe phenylthiazolyl-hydrazide (PTH) scaffold that was furtherdeveloped through synthetic derivatisation of R1eR3 (Fig. 8) inorder to improve its aggregation inhibition potency. This is inagreement with the pharmacophore model obtained by scaffoldhopping which stipulated two aromatic rings at R1 and R3,a hydrophobic region on the thiazole ring and a hydrogen bondacceptor on the carboxyl amide (Fig. 8).
Through the implementation of a hydrogen binding substituenton R3 a further improvement of the inhibitory potency wereobserved, e.g. in BSc3094 (Fig. 9). In parallel, the hydrophobic or p-stacking interactions on R1 appeared to contribute to targetbinding, as confirmed by STD-NMR experiments revealing a stronginteraction of the tau construct K18 with the R1 aromatic ring(Fig. 9). Moreover, the STD-NMR experiments showed that thebinding is specific with 62 mM dissociation constant (Pickhardtet al., 2007b).
The comparison of in vitro activities of compounds mustconsider possible differences in cell permeabilities which canbalance their efficiency in cells, e.g. the PTHs compared to theabove-mentioned rhodanines. Their activity in cells appeared to bein the same range than rhodanines as depicted in Fig. 12. Never-theless, these phenylthiazolyl-hydrazides also had a somewhathigher cytotoxicity (Fig. 12), in good agreement with hydrazide-containing substances from the literature. Indeed, numeroussubstances presenting the thiazolyl-hydrazides scaffold are repor-ted with analgesic, antibacterial, antifungal and antiproliferativeactivities (Hafez and El-Gazzar, 2008; Sonar and Crooks, 2009;Vijaya Raj et al., 2007). The mode of actions and targets are notentirely identified, although their antimalarial activity is thought tobe due to the inhibition of the cysteine protease cruzain (Leite et al.,2006). Focusing on the thiazole moiety (in blue in Fig. 10),a Monoamine Oxidase (MAO) irreversible inhibition has beenreported (Raciti et al., 1995). Thiazoles are widespread heterocycles,
commonly observed in medicinal chemistry and naturalcompounds. The most prominent examples are thiopeptide anti-biotics such as thiostrepton (Jin, 2009; Lentzen et al., 2003;Nicolaou et al., 2009) (Fig. 10) or metal-chelating siderophores(Roy et al., 1999). Their metabolic stability is high, but theirsubstitution pattern might have a stabilising/destabilising effecttoward their oxidative ring scission catalysed by the cyp P450, asobserved with sudoxicam (Fig. 10) (Obach et al., 2008).
On the other hand, the hydrazine/hydrazides moiety (in red,Fig. 10) display a more ambiguous in vivo behaviour, most probablyrelated to their metabolites and the potential production of radicaloxydative species (ROS) and strongly reducing agents resultingfrom the oxydative cleavage of the hydrazine NeN bond. Theformation of reactive acyl radicals has been reported for the anti-tuberculosis isoniazid (Fig. 10), accounting also in part for thenumerous side effects (Preziosi, 2007; Timmins and Deretic, 2006).The hepatotoxicity is unfortunately often observed with antide-pressant hydrazides/hydrazines targeting the monoamine oxidase(MAO). Iproclozide (Fig. 10) was withdrawn from the market afterreported cases of fulminant hepatitis, (Pessayre et al., 1978) andiproniazid after pronounced hepatotoxicity (Fagervall and Ross,1986; Park et al., 2005). The least hepatotoxic isocarboxazid(Fig. 10) is readily absorbed and reaches its peak plasma concen-tration at 4 � 1 h (Koechlin et al., 1962). In addition, the carcino-genic activity of hydrazines must be closely monitored (Kean et al.,2006; Toth, 1980, 2000). Noteworthy, the above-mentioned MAOinhibition might be a valuable pleiotropic effect with these tauaggregation inhibitors.
2.3. N-Phenylamines, phenothiazines and benzothiazoles
Further tau aggregation inhibitors were reported for the classesof N-phenylamines, phenothiazines and benzothiazoles (Neculaet al., 2005; Pickhardt et al., 2007a, 2005a). The SAR on the N-phenylamines is close to the one described above for rhodaninesand PTHs, with hydrogen bonding substituents such as nitro/carboxylic acids on the N-phenylamines (Fig. 11) and hydrophobicbinding domains provided by the aromatic rings.
In contrast to the two compound classes discussed above (rho-danines and phenylthiazolyl-hydrazides), the N-phenylaminesdisplayed far lower potencies in vitro and in cells, probably due tothe non-planar conformation of the molecule. The aromatic ringsconnected to the nitrogen atom have indeed a complete freedom ofrotation around the carbonenitrogen axis. The N-phenlyaminestructure overlaps strongly with anthranilic acids used as non-steroidal anti-inflammatory drugs (meclofenamic acid, niflumicacid). The higher cytotoxicity observed in vitro (Fig. 12) and thesignificant hepatotoxicity that has been reported for anthranilicacid derivatives would not recommend the use of this compoundclass in vivo.
The phenothiazines, e.g. thionin (Fig. 11) are tricyclic structuresthat incorporate a sulphur and nitrogen on the central heterocycle,and are close to the above-mentioned N-phenylamine inhibitors,
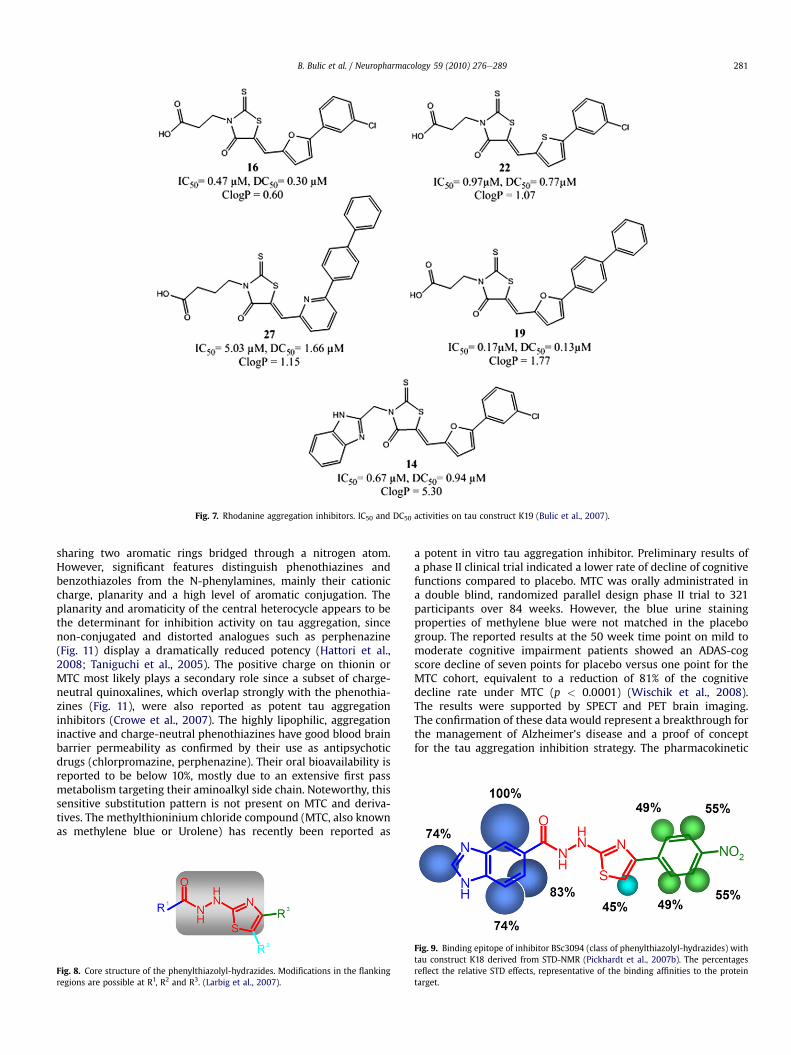
Fig. 7. Rhodanine aggregation inhibitors. IC50 and DC50 activities on tau construct K19 (Bulic et al., 2007).
B. Bulic et al. / Neuropharmacology 59 (2010) 276e289 281
sharing two aromatic rings bridged through a nitrogen atom.However, significant features distinguish phenothiazines andbenzothiazoles from the N-phenylamines, mainly their cationiccharge, planarity and a high level of aromatic conjugation. Theplanarity and aromaticity of the central heterocycle appears to bethe determinant for inhibition activity on tau aggregation, sincenon-conjugated and distorted analogues such as perphenazine(Fig. 11) display a dramatically reduced potency (Hattori et al.,2008; Taniguchi et al., 2005). The positive charge on thionin orMTC most likely plays a secondary role since a subset of charge-neutral quinoxalines, which overlap strongly with the phenothia-zines (Fig. 11), were also reported as potent tau aggregationinhibitors (Crowe et al., 2007). The highly lipophilic, aggregationinactive and charge-neutral phenothiazines have good blood brainbarrier permeability as confirmed by their use as antipsychoticdrugs (chlorpromazine, perphenazine). Their oral bioavailability isreported to be below 10%, mostly due to an extensive first passmetabolism targeting their aminoalkyl side chain. Noteworthy, thissensitive substitution pattern is not present on MTC and deriva-tives. The methylthioninium chloride compound (MTC, also knownas methylene blue or Urolene) has recently been reported as
Fig. 8. Core structure of the phenylthiazolyl-hydrazides. Modifications in the flankingregions are possible at R1, R2 and R3. (Larbig et al., 2007).
a potent in vitro tau aggregation inhibitor. Preliminary results ofa phase II clinical trial indicated a lower rate of decline of cognitivefunctions compared to placebo. MTC was orally administrated ina double blind, randomized parallel design phase II trial to 321participants over 84 weeks. However, the blue urine stainingproperties of methylene blue were not matched in the placebogroup. The reported results at the 50 week time point on mild tomoderate cognitive impairment patients showed an ADAS-cogscore decline of seven points for placebo versus one point for theMTC cohort, equivalent to a reduction of 81% of the cognitivedecline rate under MTC (p < 0.0001) (Wischik et al., 2008).The results were supported by SPECT and PET brain imaging.The confirmation of these data would represent a breakthrough forthe management of Alzheimer's disease and a proof of conceptfor the tau aggregation inhibition strategy. The pharmacokinetic
Fig. 9. Binding epitope of inhibitor BSc3094 (class of phenylthiazolyl-hydrazides) withtau construct K18 derived from STD-NMR (Pickhardt et al., 2007b). The percentagesreflect the relative STD effects, representative of the binding affinities to the proteintarget.

Fig. 10. Thiazole (blue) and hydrazine (red) containing structures. IC50 and DC50 for BSc3094 are for the tau construct K19 (Pickhardt et al., 2007b). (For interpretation of thereferences to colour in this figure legend, the reader is referred to the web version of this article.)
B. Bulic et al. / Neuropharmacology 59 (2010) 276e289282
profile of methylene blue and parent phenothiazine derivatives isabundantly documented because of their long-standing use, mostprominently as antimalarial agents (Wainwright and Amaral,2005). The ADME data analysis suggests a fairly high oralbioavailability at 72% despite its hydrophilic character (DiSanto and
Fig. 11. Structures of N-phenylamine-derived compounds, benzothiazoles and phenothiazinet al., 2007; Necula et al., 2005; Pickhardt et al., 2007a; Taniguchi et al., 2005).
Wagner, 1972a,b,c; Walter-Sack et al., 2009) due in part to thedispersion of the positive charge on the molecule by resonance.Investigations on the pharmacokinetics of methylene blue,administrated as prophylaxis of ifosfamide-associated encepha-lopathy indicate an half-life of 5 h, an high volume distribution and
es. IC50s are for aggregation inhibition of tau isoforms (Crowe et al., 2007; Khlistunova

Fig. 12. Summary of cytotoxicity (B, black bars) and aggregation-inhibitory activity in cells (B, red bars), half-maximal concentration value for assembly-inhibition (IC50; A, greenbars) and disassembly-induction (DC50; A, brown bars). Cytotoxicity assay (LDH) was performed over 24 h in the presence of 10 mM compound. For testing the inhibitory compound-activity in cells the induced cells were treated with 15 mM compound over 5 days. The remaining aggregate-positive cells were quantified by ThS-staining. (Bulic et al., 2007;Khlistunova et al., 2007; Pickhardt et al., 2005a; Pickhardt et al., 2007b). (For interpretation of the references to colour in this figure legend, the reader is referred to the webversion of this article.)
Fig. 13. Structures of benzothiazole aggregation inhibitors and binders.
B. Bulic et al. / Neuropharmacology 59 (2010) 276e289 283
significant brain penetration (Peter et al., 2000). Nevertheless thebioavailability after oral administration was found to be stronglyreduced because of extensive liver accumulation, as also observedfor cyanine dyes described below (Meijer et al., 1988). The longterm administration of methylene blue as antimalarial agent didnot induce severe side effects, as indicator of its safety (Wainwrightand Amaral, 2005). Noteworthy, the pleiotropic action of methyleneblue might play an important role in the observed clinical effects inAD patients, since it has been reported to also behave as an MAOinhibitor, NO-synthase inhibitor and its redox potential mightrescue mitochondrial dysfunction (Atamna et al., 2008; Brucheyand Gonzalez-Lima, 2008). This last antioxidative property ofmethylene blue implies the presence of two chemical entities (theoxidized cationic form and the reduced neutral form also calledleuco form) (Bruchey and Gonzalez-Lima, 2008) depending on theredox potential of the cellular environment (the leuco form beingbetter compatiblewith thehighmembranepermeability observed invivo). Despite apparent favourable pharmacokinetics, it would be ofprime importance toestimate theratiosof each forminvivo, since thereduced leuco formmost probably does not display anti-aggregationabilities on tau because of its obvious structural similarity withaggregation inactive phenothiazines such as perphenazine, chlor-promazine (Hattori et al., 2008; Taniguchi et al., 2005).
Last but not least, benzothiazoles (Fig. 11) share with pheno-thiazines the characteristic positive charge and extensivearomatic conjugation. Representative example of benzothiazole-based inhibitors with excellent inhibitory potencies have beenreported (N744 IC50 ¼ 300 nM, Fig. 13) (Congdon et al., 2009;Necula et al., 2005). The required moieties supporting thehydrophobic interactions commonly observed on aggregationinhibitors are provided by the benzothiazole heterocycles. Thisfeature probably accounts for the pronounced inhibitory activityin comparison with thioflavins like ThS or ThT which are in use asreporters of the b-structure in the aggregated state, withoutaffecting assembly as such (Necula et al., 2005). The thio-carbocyanines present a characteristic cationic charge that mayinteract with the target by chargeecharge interactions, but mostprobably only contribute to the planarity of the structure, asdescribed above also for phenothiazines. Indeed, the planar butcharge-neutral ThT analogue, Pittsburgh compound B (PiB,Fig. 13), was found to have an 45 fold increased affinity toward Ab(1e40) compared to ThT (Ki(PiB) ¼ 20 nM vs. Ki(ThT) ¼ 890 nM)
(Ikonomovic et al., 2008; Klunk et al., 2005, 2001; Mathis et al.,2002; Mathis et al., 2007).
A loss of inhibitory activity with N744 has been reported at highconcentration, caused by the aggregation of the compound leadingto H-aggregates which consist of face-to-face columnar stacks ofmolecules. By contrast, the N744 inhibitor has a monomeric ordimeric structure at low concentration which is considered to bethe active forms of the inhibitor (Congdon et al., 2007). A modifi-cation of the N744 compound to provide a structure that couldreadily form a dimer through a closed clamshell structure(compound 2, Fig. 13) ruled out the dimeric conformation as theactive form of the inhibitor (Honson et al., 2007).
The pharmacology of benzothiazoles is relatively well docu-mented, owing to the use of PiB (Fig. 13) for imaging of in vivoamyloid deposition (Klunk et al., 2005; Mathis et al., 2002, 2007)and the prescription of the benzothiazole drug Riluzole (Fig. 14) foramyotrophic lateral sclerosis (NMDA receptor antagonist).
Riluzole presents a satisfactory 60% bioavailabilty with a 2 hpeak plasma concentration. The half-life at 12 h gives evidence ofits good stability (van Kan et al., 2005). The BBB permeability hasbeen confirmed by HPLC analysis on brain extracts in mice,however riluzole was found to be a substrate for PgP efflux trans-porter (Milane et al., 2007). Positively charged analogue furtherconjugated to heteroaromatics, also known as cyanine dyes (N744,

Fig. 14. Benzothiazole-containing drug (riluzole), tau aggregation inhibitor (C11) andparent cyanine dye (Cy3).
B. Bulic et al. / Neuropharmacology 59 (2010) 276e289284
Fig. 13), might be prone to complexation with DNA and thereforemutagenic (Clifton and Leikin, 2003; Hilal and Taylor, 2008).Numerous cyanine dyes from this compound class family, such asCy3 and Cy5 (Fig. 14), are extensively used in laboratories asmolecular probes for nucleic acid staining. The delocalisation of thepositive charge through the molecule improves their lipophilicityand thus their membrane permeability. Cyanine dyes as well asclosely related positively charged redox active substances such asphentothiazines are also known as mitochondrion selective stains(Hassan and Fridovich, 1979; Johnson, 2001). Therefore, specialattention must be paid during the design of this promising class ofcompounds in order to avoid potential toxic side effects. Promisingresults have been achieved with the cyanine analogue C11 (Fig. 14)on organotypic slice culture model, strongly supporting theprevious in vitro observation (Congdon et al., 2009; Chang et al.,2009) A pharmacophore mapping of the anthracyclines as Abaggregation inhibitors yielded a three point model with thearomatic rings serving as hydrophobic regions and the sugarmoiety as hydrogen bond donor and acceptor (Fig. 15).
Another compound class, the polyphenols, has been presentedabove as tau aggregation inhibitors (myrcetin, Fig. 14). Thesecompounds show inhibitory activity on a variety of amyloids suchas a-synuclein, IAPP, Ab40, PrPsc or tau (Klunk et al., 2001, 2002;Mathis et al., 2002, 2007).
2.4. Polyphenols and anthraquinones
The chemical class of polyphenole is characterized by thepresence of several phenols functionalities i.e. one or morehydroxyl-OH bound to aromatic rings (Fig. 15). These substancesare often observed in higher plants, such as ginkgo biloba, tea bush,grape or turmeric. Polyphenols are often perceived as naturallyoccurring curative substances, in line with numerous claims ontheir therapeutical benefits linked to antibiotic, antiviral, anti-cancer, antidiabetic and neuroprotective activities (Han et al.,2007). However, conclusions on their in vivo effects with longterm administration are risky, for the reason that pharmacological
Fig. 15. Classification
investigations defining their bioavailability and organ distributionare often lacking. Polyphenols can be categorized according to theirstructure in flavonoids, phenolic acids and stilbenes (Fig. 15). Mostof naturally occurring dietary polyphenols are found as glycosy-lated forms which are highly hydrophilic and thus poorly absorbedfrom the intestine. Only aglycones resulting from the hydrolysis bythe intestinal flora are detected in the plasma, though their residualpolarity and hydrophilicity due to the heavy decoration of themolecule with polar hydroxyls impedes their throughout passagefrom the intestine to the blood compartments. Their bioavailabilityis therefore classified as low (5e20% range) (Hu, 2007). The poly-phenol fraction present in the plasma is further extensivelymetabolised through methylation, sulfation and glucuronidation,reducing further their bioavailability (Manach et al., 2004). Thecorrelation with their neuroprotective effects observed in patientssuffering from neurodegeneration requires therefore furtherinvestigation to clarify their bloodebrain barrier permeability andbrain concentration (Ono et al., 2003; Singh et al., 2008). Furtherinvestigations should determine if the required polyphenolsconcentration in brain is compatible with patient safety concerns.There is indeed increasing evidence pointing at acute polyphenoltoxicity at high doses, as reported for the green tea extract Poly-phenon� (Kapetanovic et al., 2009; Mennen et al., 2005). A prom-inent example of the ambiguous properties of polyphenols isdocumented for phytoestogen isoflavones whose similarity withoestrogens might confer a carcinogenic oestrogenic activity (Allredet al., 2004; Anupongsanugool et al., 2005; Dixon, 2004).
Numerous polyphenols show inhibitory activity on a variety ofamyloids such as a-synuclein, IAPP, Ab40, PrPsc. Myrcetin has beenreported as tau aggregation inhibitors with a 1.2 mM IC50 and the invitro data indicate that they interfere with the elongation phase offibril assembly (Howlett et al., 1999; Porat et al., 2006; Taniguchiet al., 2005; Yang et al., 2005).
The structure activity-relationship for polyphenolic aggregationinhibitors on tau and Ab points toward an essential role of thehydroxyls on the phenolic moiety, (Ono et al., 2003) since etheranalogues displayed reduced activities. Phenolic hydroxyls areknown for their greater acidity than aliphatic hydroxyls, and fortheir ability to form hydrogen bonds. The requirement for hydrogenbonding was also observed for scyllo-inositol inhibitors (McLaurinet al., 2000). Symmetrical structures such those of rosmarinic acidand curcumin and an optimal molecular length of 16e19 Å isreported to be most favourable on Ab40 aggregation inhibition(Reinke and Gestwicki, 2007). Notewothy, the planarity of thestructure appears to be of high importance, as also described abovefor other aggregation inhibitors, deduced from the comparativeaggregation-inhibitory potencies between the planar cyanidin andthe non-planar and inactive catechin (Fig. 16) (Hattori et al., 2008;Taniguchi et al., 2005).
Anthracyclines are not classified as polyphenols, yet are closeanalogues which also display aggregation inhibition abilities on tauand Ab40 (Howlett et al., 1999; Taniguchi et al., 2005). The
of polyphenols.

B. Bulic et al. / Neuropharmacology 59 (2010) 276e289 285
anthraquinones all share a tricyclic structure with one or morephenolic moieties (Fig. 17) also observed in polyphenols asdescribed above.
The anthracyclines are known as intercalating cytostatics thatare successfully employed in anticancer therapies, but for the samereason they present a hazardous toxicological profile for long termadministration in AD. The side effects with adriamycin (Fig. 17) arewell-known, including alopecia and congestive heart failure. Theformation of reactive oxygen species (ROS) is well documented,resulting from adriamycin iron chelation (Xu et al., 2005). Our LDHcytotoxicity data (Fig. 12) confirm substantial toxicity in vitro. Theglycones anthracyclines are generally administrated intravenously,because of their low bioavailability due to the high hydrophylicityof the daunosamine sugar (blue on ring D, Fig. 11). The agylcones,such as emodin (Fig. 17), have a better but insufficient bioavail-ability (22.5%) due to the high residual hydrophilicity (Teng et al.,2007). The ingestion of anthraquinones is one of the majorcurrent self-medications, due to the laxative properties of thecompounds (Mueller et al., 1999; van Gorkom et al., 1999). Theover-the-counter herbal extracts contain principally emodins(Fig. 17) and sennosides which exert their laxative effects bydamaging colonic epithelial cells. Their use has been correlatedwith a higher incidence of colon cancer and others (Mueller et al.,1998; van Gorkom et al., 1999).
The five compounds depicted on Fig. 17 were able to inhibit theaggregation of the K19 tau construct and also induced the disag-gregation of preformed aggregates, with IC50 values between1.1 mM and 2.4 mM and DC50 values between 2.2 mM and 3.8 mM(Pickhardt et al., 2005a). One exception is compound PHF005which showed a markedly lower activity, probably due to theflexibility of the structure compared to closed tricyclic structures(rotation axis depicted by the arrow in Fig. 17). Moreover, thesubstitutions on the ring A (daunorubicin, Fig. 17) did not appearto play a crucial role, since different patterns yielded similarinhibitory potencies. Noteworthy, the ring D in daunorubicin andadriamycin bearing the sugar does not give a competitive advan-tage compared to PHF016, indicating that the compounds are onlymoderately sensitive to the substitutions on that ring andaccommodate even bulky substitutions such as sugars. A phar-macophore mapping of the anthracyclines as Ab aggregationinhibitors yielded a three point model with the aromatic ringsserving as hydrophobic regions and the sugar moiety as hydrogenbond donor and acceptor (Howlett et al., 1999).
Fig. 16. Planar (cyanidin) and non-planar (catechin) polyphenols. IC50s for
3. Binding mode
Folding pathways are complex and started from non-toxicnative monomeric peptides to fibrils implying the presence ofvarious potential targets for aggregation inhibitors and dis-aggregators (Howlett, 2001; LeVine, 2002). The numerous ques-tions about the mechanisms and targets are still awaiting answers.As reported for Ab, it probably involves the trapping of the mono-mer or an assembly intermediate involved in the dynamic equi-librium between fibril and monomer (Harper and Lansbury, 1997;Matsuoka et al., 2003; von Bergen et al., 2005). The compoundsmay also be able to interact directly with the aggregated structureand disturb the proteineprotein p-stacking arrangement of thefibrils. A different binding mode and different targets for inhibitionand disaggregation on the complex pathway of aggregation fromthe native non-toxic peptide tofibrils could therefore rationalize thedifferences in potencies between observed IC50s and DC50s. Thebinding epitopes obtained by STD-NMR between a PTH inhibitorand a monomeric tau peptide depicted in Fig. 9 illustrate that thedirect interaction of small molecules with the monomeric peptidemight be a common feature among aggregation inhibitors anddisaggregation promoters. However, binders from the thioflavinfamily such as ThT have been reported to only bind to pre-aggre-gates and aggregate forms, showing that multiple binding modesare likely (LeVine, 1993).
Noteworthy, several compounds listed above as aggregationinhibitors have been observed to form also unordered micellarstructures inherent to their hydrophobic nature, and are thereforesuspected to be unspecific through colloidal inhibition (Feng et al.,2008).
4. Summary and outlook
A close interplay between Ab-amyloids and tau aggregation isemerging, long after the histopathological observations on post-mortem brains pointing at an intimate correlation between theload of neurofibrillary tangles within neurons and cellular degen-eration (King et al., 2006; Park and Ferreira, 2005; Rapoport et al.,2002; Roberson et al., 2007; Small and Duff, 2008; Talaga andQuere, 2002). Even though the elucidation of the cascade ofevents leading to tau aggregation is not yet achieved, several linesof evidence suggest that Ab oligomers or aggregates, commonlyconsidered as the major culprits of AD, might also exert their toxic
tau aggregation inhibition (Hattori et al., 2008; Taniguchi et al., 2005).

Fig. 17. Structures of anthraquinone-derived compounds. The glycoside parts aredepicted in blue. (For interpretation of the references to colour in this figure legend,the reader is referred to the web version of this article.)
B. Bulic et al. / Neuropharmacology 59 (2010) 276e289286
function via tau as a constitutive intracellular component (Kinget al., 2006; Park and Ferreira, 2005; Rapoport et al., 2002;Roberson et al., 2007; Talaga and Quere, 2002). Oligomeric taucan also play a role in the pathogenic mechanism of AD, possiblyeven via extracellular pathways (Frost et al., 2009; Clavaguera et al.,2009), and putative inhibitors must be testet with regard of theirability to inhibit possible oligomer formation. Our own observa-tions using a charge-neutral compound of moderate in vitroactivity such as 14 (Fig. 7) allowed the study of the effects of taufilament disassembly on cellular viability, which resulted in theprevention or reversal of the toxicity caused by tau aggregation inthe cytosol (Khlistunova et al., 2007; Meyer-Luehmann et al., 2008;Winklhofer et al., 2008). Therefore, apart from providing a reliablebiomarker for Alzheimer's disease diagnostic, tau aggregationinhibitors and disaggregators might complement the currenttherapeutic approaches, as suggested by the recent clinical trialswith methylene blue and the mitigated results obtained after Ab-immunisation alone (Holmes et al., 2008; Wischik et al., 1996,2007).
In view of the neuronal toxicity mediated by small oligomers(Lashuel and Grillo-Bosch, 2005; Necula et al., 2007), it has beenrecently suggested that interventions that reduce amyloid load butincrease small oligomers could be harmful (Cheng et al., 2007). Inprinciple, it is therefore necessary to determine the binding prop-erties of the amyloid inhibitor compounds to the small oligomericamyloid precursors. Noteworthy, recent findings indicate thatseveral Ab-amyloid binders and inhibitors also bind to Ab oligo-mers and have neuroprotective effects (Hong et al., 2007; Maezawaet al., 2008).
Shared structural features between the compound classes startto emerge, with the presence of aromatic/hydrophobic patches andhydrogen bonding elements along flat extended structures. Thedevelopment of improved aggregation inhibitors will be linked tothe understanding of their binding mode at the molecular level,and the ability to integrate crucial elements of pharmacology at anearly stage of the compounds development to address the in vivotoxicity, brain permeability and plasma stability. Especially thenegative or positive charges shared by most aggregation inhibitorsimpede membrane permeability and therefore their use in vivo.The successful compound optimization toward charge-neutral cellpermeable structures is illustrated by the design of imaging agentsfor diagnosis such as Pittsburgh compound B (PIB) or methoxy-X04,inspired by the poorly permeable thioflavins or Congo redanalogues (Cai et al., 2007; Kinosian et al., 2000; Klunk et al., 2001,2005, 2002, 1989; Mathis et al., 2007).
The ability to form H- or J-aggregates by many of the aggrega-tion inhibitors might indicate a specific binding mode involving theself-assembly of the compounds to form a supramolecular struc-ture (Piekarska et al., 1996; Skowronek et al., 2000; Spolnik et al.,2007; Von Berlepsch et al., 2000) distinct from the unorderedmicelles typically observed with promiscuous inhibitors (Fenget al., 2008; McGovern et al., 2003). The determination of theactive form of the inhibitors and their rational design will presentan exciting challenge to reach potential drug candidates.
Acknowledgements
We are grateful to Prof. Herbert Waldmann (Max-Planck-Insti-tute Dortmund), and Prof. Boris Schmidt (Technical UniversityDarmstadt) for continuous discussions and encouragementthroughout this project. This work was supported by MPG, DFG,VW Foundation, EU-FP7/Memosad.
References
Ahn, J.H., Kim, S.J., Park, W.S., Cho, S.Y., Ha, J.D., Kim, S.S., Kang, S.K., Jeong, D.G.,Jung, S.K., Lee, S.H., Kim, H.M., Park, S.K., Lee, K.H., Lee, C.W., Ryu, S.E., Choi, J.K.,2006. Synthesis and biological evaluation of rhodanine derivatives as PRL-3inhibitors. Bioorg. Med. Chem. Lett. 16, 2996e2999.
Allred, C.D., Allred, K.F., Ju, Y.H., Goeppinger, T.S., Doerge, D.R., Helferich, W.G., 2004.Soy processing influences growth of estrogen-dependent breast cancer tumors.Carcinogenesis 25, 1649e1657.
Anupongsanugool, E., Teekachunhatean, S., Rojanasthien, N., Pongsatha, S.,Sangdee, C., 2005. Pharmacokinetics of isoflavones, daidzein and genistein,after ingestion of soy beverage compared with soy extract capsules in post-menopausal Thai women. BMC Clin. Pharmacol. 5, 2.
Arima, K., 2006. Ultrastructural characteristics of tau filaments in tauopathies:immuno-electron microscopic demonstration of tau filaments in tauopathies.Neuropathology 26, 475e483.
Atamna, H., Nguyen, A., Schultz, C., Boyle, K., Newberry, J., Kato, H., Ames, B.N., 2008.Methylene blue delays cellular senescence and enhances key mitochondrialbiochemical pathways. FASEB J. 22, 703e712.
Ballatore, C., Lee, V.M., Trojanowski, J.Q., 2007. Tau-mediated neurodegeneration inAlzheimer's disease and related disorders. Nat. Rev. Neurosci. 8, 663e672.
Barghorn, S., Davies, P., Mandelkow, E., 2004. Tau paired helical filaments fromAlzheimer's disease brain and assembled in vitro are based on beta-structure inthe core domain. Biochemistry 43, 1694e1703.
von Bergen, M., Friedhoff, P., Biernat, J., Heberle, J., Mandelkow, E.M., Mandelkow, E.,2000. Assembly of tau protein into Alzheimer paired helical filaments dependson a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc.Natl. Acad. Sci. U S A 97, 5129e5134.
von Bergen, M., Barghorn, S., Li, L., Marx, A., Biernat, J., Mandelkow, E.M.,Mandelkow, E., 2001. Mutations of tau protein in frontotemporal dementiapromote aggregation of paired helical filaments by enhancing local beta-structure. J. Biol. Chem. 276, 48165e48174.
von Bergen, M., Barghorn, S., Biernat, J., Mandelkow, E.M., Mandelkow, E., 2005. Tauaggregation is driven by a transition from random coil to beta sheet structure.Biochim. Biophys. Acta 1739, 158e166.
Bruchey, A.K., Gonzalez-Lima, F., 2008. Behavioral, physiological and biochemicalhormetic responses to the autoxidizable dye methylene blue. Am. J. Pharmacol.Toxicol. 3, 72e79.
Bulic, B., Pickhardt, M., Khlistunova, I., Biernat, J., Mandelkow, E.M., Mandelkow, E.,Waldmann, H., 2007. Rhodanine-based tau aggregation inhibitors in cell modelsof tauopathy. Angew. Chem. Int. Ed. Engl. 46, 9215e9219.
Cai, L., Innis, R.B., Pike, V.W., 2007. Radioligand development for PET imaging ofbeta-amyloid (Abeta) e current status. Curr. Med. Chem. 14, 19e52.
Carlson, E.E., May, J.F., Kiessling, L.L., 2006. Chemical probes of UDP-galactopyranosemutase. Chem. Biol. 13, 825e837.
Chang, E., Congdon, E.E., Honson, N.S., Duff, K.E., Kuret, J., 2009. Structure-activityrelationship of cyanine tau aggregation inhibitors. J. Med. Chem. 52,3539e3547.
Chang, E., Kuret, J., 2008. Detection and quantification of tau aggregation usinga membrane filter assay. Anal. Biochem. 372 (2), 330e336.
Cheng, I.H., Scearce-Levie, K., Legleiter, J., Palop, J.J., Gerstein, H., Bien-Ly, N.,Puolivali, J., Lesne, S., Ashe, K.H., Muchowski, P.J., Mucke, L., 2007. Acceleratingamyloid-beta fibrillization reduces oligomer levels and functional deficits inAlzheimer disease mouse models. J. Biol. Chem. 282, 23818e23828.
Clavaguera, F., Bolmont, T., Crowther, R.A., Abramowski, D., Frank, S., Probst, A.,Fraser, G., Stalder, A.K., Beibel, M., Staufenbiel, M., Jucker, M., Goedert, M.,Tolnay, M., 2009. Nat. Cell Biol. 11 (7), 909e913.
Clifton 2nd, J., Leikin, J.B., 2003. Methylene blue. Am. J. Ther. 10, 289e291.Congdon, E.E., Necula, M., Blackstone, R.D., Kuret, J., 2007. Potency of a tau fibrilli-
zation inhibitor is influenced by its aggregation state. Arch. Biochem. Biophys.465, 127e135.

B. Bulic et al. / Neuropharmacology 59 (2010) 276e289 287
Congdon, E.E., Figueroa, Y.H., Wang, L., Toneva, G., Chang, E., Kuret, J., Conrad, C.,Duff, K.E., 2009. Inhibition of tau polymerization with a cyanine dye in twodistinct model systems. J. Biol. Chem. 284, 20830e20839.
Crowe, A., Ballatore, C., Hyde, E., Trojanowski, J.Q., Lee, V.M., 2007. High throughputscreening for small molecule inhibitors of heparin-induced tau fibril formation.Biochem. Biophys. Res. Commun. 358, 1e6.
Crowe, A., Huang, W., Ballatore, C., Johnson, R.L., Hogan, A.-M.L., Huang, R.,Wichterman, J., McCoy, J., Huryn, D., Auld, D.S., Smith III, A.B., Inglese, J.,Trojanowski, J.Q., Austin, C.P., Brunden, K.R., Lee-, V.M.-Y., 2009. Identification ofaminothienopyridazine inhibitors of tau assembly by quantitative high-throughput screening. Biochemistry 48, 7732e7745.
Cutshall, N.S., O'Day, C., Prezhdo, M., 2005. Rhodanine derivatives as inhibitors ofJSP-1. Bioorg. Med. Chem. Lett. 15, 3374e3379.
DiSanto, A.R., Wagner, J.G., 1972a. Pharmacokinetics of highly ionized drugs. 3.Methylene blue e blood levels in the dog and tissue levels in the rat followingintravenous administration. J. Pharm. Sci. 61, 1090e1094.
DiSanto, A.R., Wagner, J.G., 1972b. Pharmacokinetics of highly ionized drugs. I.Methylene blue e whole blood, urine, and tissue assays. J. Pharm. Sci. 61,598e602.
DiSanto, A.R., Wagner, J.G., 1972c. Pharmacokinetics of highly ionized drugs. II.Methylene blue e absorption, metabolism, and excretion in man and dog afteroral administration. J. Pharm. Sci. 61, 1086e1090.
Dixon, R.A., 2004. Phytoestrogens. Annu. Rev. Plant Biol. 55, 225e261.Fagervall, I., Ross, S.B., 1986. Inhibition of monoamine oxidase in monoaminergic
neurones in the rat brain by irreversible inhibitors. Biochem. Pharmacol. 35,1381e1387.
Feng, B.Y., Toyama, B.H., Wille, H., Colby, D.W., Collins, S.R., May, B.C., Prusiner, S.B.,Weissman, J., Shoichet, B.K., 2008. Small-molecule aggregates inhibit amyloidpolymerization. Nat. Chem. Biol. 4, 197e199.
Friedhoff, P., Schneider, A., Mandelkow, E.M., Mandelkow, E., 1998. Rapid assemblyof Alzheimer-like paired helical filaments from microtubule-associated proteintau monitored by fluorescence in solution. Biochemistry 37, 10223e10230.
Frost, B., Jacks, R.L., Diamond, M.I., 2009. Propagation of tau misfolding from theoutside to the inside of the cell. J. Biol. Chem. 284 (19), 12845e12852.
Glabe, C.G., 2004. Conformation-dependent antibodies target diseases of proteinmisfolding. Trends Biochem. Sci. 29, 542e547.
Goedert, M., Crowther, R.A., Spillantini, M.G., 1998. Tau mutations cause fronto-temporal dementias. Neuron. 21, 955e958.
van Gorkom, B.A., de Vries, E.G., Karrenbeld, A., Kleibeuker, J.H., 1999. Reviewarticle: anthranoid laxatives and their potential carcinogenic effects. Aliment.Pharmacol. Ther. 13, 443e452.
Gualtieri, M., Bastide, L., Villain-Guillot, P., Michaux-Charachon, S., Latouche, J.,Leonetti, J.P., 2006. In vitro activity of a new antibacterial rhodanine derivativeagainst Staphylococcus epidermidis biofilms. J. Antimicrob. Chemother. 58,778e783.
Guerrero, R., Navarro, P., Gallego, E., Avila, J., de Yebenes, J.G., Sanchez, M.P., 2008.Park2-Null/Tau transgenic mice reveal a functional relationship between parkinand tau. J. Alzheimers Dis. 13, 161e172.
Hafez, H.N., El-Gazzar, A.B., 2008. Design and synthesis of 3-pyrazolyl-thiophene,thieno[2,3-d]pyrimidines as new bioactive and pharmacological activities.Bioorg. Med. Chem. Lett. 18, 5222e5227.
Han, X., Shen, T., Lou, H., 2007. Dietary polyphenols and their biological significance.Int. J. Mol. Sci. 8, 950e998.
Harper, J.D., Lansbury Jr., P.T., 1997. Models of amyloid seeding in Alzheimer's diseaseand scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 66, 385e407.
Hassan, H.M., Fridovich, I., 1979. Intracellular production of superoxide radical andof hydrogen peroxide by redox active compounds. Arch. Biochem. Biophys. 196,385e395.
Hattori, M., Sugino, E., Minoura, K., In, Y., Sumida, M., Taniguchi, T., Tomoo, K.,Ishida, T., 2008. Different inhibitory response of cyanidin and methylene bluefor filament formation of tau microtubule-binding domain. Biochem. Biophys.Res. Commun. 374, 158e163.
Heiser, V., Engemann, S., Brocker, W., Dunkel, I., Boeddrich, A., Waelter, S.,Nordhoff, E., Lurz, R., Schugardt, N., Rautenberg, S., Herhaus, C., Barnickel, G.,Bottcher, H., Lehrach, H., Wanker, E.E., 2002. Identification of benzothiazoles aspotential polyglutamine aggregation inhibitors of Huntington's disease by usingan automated filter retardation assay. Proc. Natl. Acad. Sci. U. S. A. 99 (Suppl. 4),16400e16406.
Hilal, H., Taylor, J.A., 2008. Cyanine dyes for the detection of double stranded DNA. J.Biochem. Biophys. Methods 70, 1104e1108.
Holmes, C., Boche, D., Wilkinson, D., Yadegarfar, G., Hopkins, V., Bayer, A., Jones, R.W.,Bullock, R., Love, S., Neal, J.W., Zotova, E., Nicoll, J.A., 2008. Long-term effects ofAbeta42 immunisation in Alzheimer's disease: follow-up of a randomised,placebo-controlled phase I trial. Lancet 372, 216e223.
Hong, H.S., Maezawa, I., Yao, N., Xu, B., Diaz-Avalos, R., Rana, S., Hua, D.H., Cheng, R.H.,Lam, K.S., Jin, L.W., 2007. Combining the rapid MTT formazan exocytosis assayand the MC65 protection assay led to the discovery of carbazole analogs as smallmolecule inhibitors of Abeta oligomer-induced cytotoxicity. Brain Res. 1130,223e234.
Honson, N.S., Jensen, J.R., Darby, M.V., Kuret, J., 2007. Potent inhibition of taufibrillization with a multivalent ligand. Biochem. Biophys. Res. Commun. 363,229e234.
Hotta, N., Akanuma, Y., Kawamori, R., Matsuoka, K., Oka, Y., Shichiri, M., Toyota, T.,Nakashima, M., Yoshimura, I., Sakamoto, N., Shigeta, Y., 2006. Long-term clinical
effects of epalrestat, an aldose reductase inhibitor, on diabetic peripheralneuropathy: the 3-year, multicenter, comparative aldose reductase inhibitor-diabetes complications trial. Diabetes Care 29, 1538e1544.
Howlett, D.R., 2001. Ab oligomerisation: a therapeutic traget for Alzheimer'sdisease. Curr. Med. Chem. Immunol, Endocr. Metab. Agents 1, 25e38.
Howlett, D.R., George, A.R., Owen, D.E., Ward, R.V., Markwell, R.E., 1999. Commonstructural features determine the effectiveness of carvedilol, daunomycin androlitetracycline as inhibitors of Alzheimer beta-amyloid fibril formation. Bio-chem. J. 343 (Pt 2), 419e423.
Hu, M., 2007. Commentary: bioavailability of flavonoids and polyphenols: call toarms. Mol. Pharm. 4, 803e806.
Hu, M., Li, J., Yao, S.Q., 2008. In situ “click” assembly of small molecule matrixmetalloprotease inhibitors containing zinc-chelating groups. Org. Lett. 10,5529e5531.
Ikonomovic, M.D., Klunk, W.E., Abrahamson, E.E., Mathis, C.A., Price, J.C.,Tsopelas, N.D., Lopresti, B.J., Ziolko, S., Bi, W., Paljug, W.R., Debnath, M.L.,Hope, C.E., Isanski, B.A., Hamilton, R.L., DeKosky, S.T., 2008. Post-mortemcorrelates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer'sdisease. Brain 131, 1630e1645.
Irvine, M.W., Patrick, G.L., Kewney, J., Hastings, S.F., MacKenzie, S.J., 2008.Rhodanine derivatives as novel inhibitors of PDE4. Bioorg. Med. Chem. Lett.18, 2032e2037.
Jin, Z., 2009. Muscarine, imidazole, oxazole and thiazole alkaloids. Nat. Prod. Rep.26, 382e445.
Jin, L.W., Claborn, K.A., Kurimoto, M., Geday, M.A., Maezawa, I., Sohraby, F.,Estrada, M., Kaminksy, W., Kahr, B., 2003. Imaging linear birefringence anddichroism in cerebral amyloid pathologies. Proc. Natl. Acad. Sci. U. S. A. 100,15294e15298.
Johnson, I.D., 2001. Cellular functionprobes. Curr. Protoc. Cytom. (Chapter 4), Unit 4.4.Johnson, S.L., Chen, L.H., Harbach, R., Sabet, M., Savinov, A., Cotton, N.J., Strongin, A.,
Guiney, D., Pellecchia, M., 2008. Rhodanine derivatives as selective proteaseinhibitors against bacterial toxins. Chem. Biol. Drug Des. 71, 131e139.
van Kan, H.J., Groeneveld, G.J., Kalmijn, S., Spieksma, M., van den Berg, L.H.,Guchelaar, H.J., 2005. Association between CYP1A2 activity and riluzole clear-ance in patients with amyotrophic lateral sclerosis. Br. J. Clin. Pharmacol. 59,310e313.
Kapetanovic, I.M., Crowell, J.A., Krishnaraj, R., Zakharov, A., Lindeblad, M.,Lyubimov, A., 2009. Exposure and toxicity of green tea polyphenols in fastedand non-fasted dogs. Toxicology 260, 28e36.
Kayed, R., Head, E., Thompson, J.L., McIntire, T.M., Milton, S.C., Cotman, C.W.,Glabe, C.G., 2003. Common structure of soluble amyloid oligomers impliescommon mechanism of pathogenesis. Science 300, 486e489.
Kayed, R., Head, E., Sarsoza, F., Saing, T., Cotman, C.W., Necula, M., Margol, L., Wu, J.,Breydo, L., Thompson, J.L., Rasool, S., Gurlo, T., Butler, P., Glabe, C.G., 2007. Fibrilspecific, conformation dependent antibodies recognize a generic epitopecommon to amyloid fibrils and fibrillar oligomers that is absent in prefibrillaroligomers. Mol. Neurodegener. 2, 18.
Kean, T., Miller, J.H., Skellern, G.G., Snodin, D., 2006. Acceptance criteria for levels ofhydrazine in substances for pharmaceutical use and analytical methods for itsdetermination. Pharmeur. Sci. Notes, 23e33.
Kelleher, I., Garwood, C., Hanger, D.P., Anderton, B.H., Noble, W., 2007. Kinaseactivities increase during the development of tauopathy in htau mice. J. Neu-rochem. 103, 2256e2267.
Kelly, J.W., 1996. Alternative conformations of amyloidogenic proteins govern theirbehavior. Curr. Opin. Struct. Biol. 6, 11e17.
Khlistunova, I., Pickhardt, M., Biernat, J., Wang, Y., Mandelkow, E.M., Mandelkow, E.,2007. Inhibition of tau aggregation in cell models of tauopathy. Curr. AlzheimerRes. 4, 544e546.
Kidd, M., 1963. Paired helical filaments in electron microscopy of Alzheimer'sdisease. Nature 197, 192e193.
King, M.E., Kan, H.M., Baas, P.W., Erisir, A., Glabe, C.G., Bloom, G.S., 2006. Tau-dependent microtubule disassembly initiated by prefibrillar beta-amyloid.J. Cell Biol. 175, 541e546.
Kinosian, B.P., Stallard, E., Lee, J.H., Woodbury, M.A., Zbrozek, A.S., Glick, H.A., 2000.Predicting 10-year care requirements for older people with suspected Alz-heimer's disease. J. Am. Geriatr. Soc. 48, 631e638.
Klunk, W.E., Pettegrew, J.W., Abraham, D.J., 1989. Quantitative evaluation of congored binding to amyloid-like proteins with a beta-pleated sheet conformation.J. Histochem. Cytochem. 37, 1273e1281.
Klunk, W.E., Wang, Y., Huang, G.F., Debnath, M.L., Holt, D.P., Mathis, C.A., 2001.Uncharged thioflavin-T derivatives bind to amyloid-beta protein with highaffinity and readily enter the brain. Life Sci. 69, 1471e1484.
Klunk, W.E., Bacskai, B.J., Mathis, C.A., Kajdasz, S.T., McLellan, M.E., Frosch, M.P.,Debnath, M.L., Holt, D.P., Wang, Y., Hyman, B.T., 2002. Imaging Abeta plaques inliving transgenic mice with multiphoton microscopy and methoxy-X04,a systemically administered Congo red derivative. J. Neuropathol. Exp. Neurol.61 (9), 797e805.
Klunk, W.E., Lopresti, B.J., Ikonomovic, M.D., Lefterov, I.M., Koldamova, R.P.,Abrahamson, E.E., Debnath, M.L., Holt, D.P., Huang, G.F., Shao, L., DeKosky, S.T.,Price, J.C., Mathis, C.A., 2005. Binding of the positron emission tomographytracer Pittsburgh compound-B reflects the amount of amyloid-beta in Alz-heimer's disease brain but not in transgenic mouse brain. J. Neurosci. 25,10598e10606.
Koechlin, B.A., Schwartz, M.A., Oberhaensli, W.E., 1962. Metabolism of C-14-ipro-niazid and C-14-isocarboxazid in man. J. Pharmacol. Exp. Ther. 138, 11e20.

B. Bulic et al. / Neuropharmacology 59 (2010) 276e289288
Kosik, K.S., Shimura, H., 2005. Phosphorylated tau and the neurodegenerative fol-dopathies. Biochim. Biophys. Acta 1739, 298e310.
Kumar, G., Parasuraman, P., Sharma, S.K., Banerjee, T., Karmodiya, K., Surolia, N.,Surolia, A., 2007. Discovery of a rhodanine class of compounds as inhibitors ofPlasmodium falciparum enoyl-acyl carrier protein reductase. J. Med. Chem. 50,2665e2675.
Larbig, G., Pickhardt, M., Lloyd, D.G., Schmidt, B., Mandelkow, E., 2007. Screening forinhibitors of tau protein aggregation into Alzheimer paired helical filaments:a ligand based approach results in successful scaffold hopping. Curr. AlzheimerRes. 4, 315e323.
Lashuel, H.A., Grillo-Bosch, D., 2005. In vitro preparation of prefibrillar intermedi-ates of amyloid-beta and alpha-synuclein. Methods Mol. Biol. 299, 19e33.
Lashuel, H.A., Lansbury Jr., P.T., 2006. Are amyloid diseases caused by protein aggre-gates that mimic bacterial pore-forming toxins? Q. Rev. Biophys. 39, 167e201.
Leite, A.C., de Lima, R.S., Moreira, D.R., Cardoso, M.V., Gouveia de Brito, A.C., Farias DosSantos, L.M., Hernandes, M.Z., Kiperstok, A.C., Soares, M.B., 2006. Synthesis, dock-ing, and invitroactivityof thiosemicarbazones, aminoacyl-thiosemicarbazidesandacyl-thiazolidones against Trypanosoma cruzi. Bioorg. Med. Chem. 14, 3749e3757.
Lentzen, G., Klinck, R., Matassova, N., Aboul-ela, F., Murchie, A.I., 2003. Structuralbasis for contrasting activities of ribosome binding thiazole antibiotics. Chem.Biol. 10, 769e778.
Leroy, K., Bretteville, A., Schindowski, K., Gilissen, E., Authelet, M., De Decker, R.,Yilmaz, Z., Buee, L., Brion, J.P., 2007. Early axonopathy preceding neurofibrillarytangles in mutant tau transgenic mice. Am. J. Pathol. 171, 976e992.
LeVine 3rd, H., 1993. Thioflavine T interaction with synthetic Alzheimer's diseasebeta-amyloid peptides: detection of amyloid aggregation in solution. ProteinSci. 2, 404e410.
LeVine, H., 2002. The challenge of inhibiting Abeta polymerization. Curr. Med.Chem. 9, 1121e1133.
Lipinski, C.A., Lombardo, F., Dominy, B.W., Feeney, P.J., 2001. Experimental andcomputational approaches to estimate solubility and permeability in drugdiscovery and development settings. Adv. Drug Deliv. Rev. 46, 3e26.
Maezawa, I., Hong, H.S., Liu, R., Wu, C.Y., Cheng, R.H., Kung, M.P., Kung, H.F., Lam, K.S.,Oddo, S., Laferla, F.M., Jin, L.W., 2008. Congo red and thioflavin-T analogs detectAbeta oligomers. J. Neurochem. 104, 457e468.
Manach, C., Scalbert, A., Morand, C., Remesy, C., Jimenez, L., 2004. Polyphenols: foodsources and bioavailability. Am. J. Clin. Nutr. 79, 727e747.
Martinez, A., Alonso, M., Castro, A., Dorronsoro, I., Gelpi, J.L., Luque, F.J., Perez, C.,Moreno, F.J., 2005. SAR and 3D-QSAR studies on thiadiazolidinone derivatives:exploration of structural requirements for glycogen synthase kinase 3 inhibi-tors. J. Med. Chem. 48, 7103e7112.
Mathis, C.A., Bacskai, B.J., Kajdasz, S.T., McLellan, M.E., Frosch, M.P., Hyman, B.T.,Holt, D.P., Wang, Y., Huang, G.F., Debnath, M.L., Klunk, W.E., 2002. A lipophilicthioflavin-T derivative for positron emission tomography (PET) imaging ofamyloid in brain. Bioorg. Med. Chem. Lett. 12, 295e298.
Mathis, C.A., Lopresti, B.J., Klunk, W.E., 2007. Impact of amyloid imaging on drugdevelopment in Alzheimer's disease. Nucl. Med. Biol. 34, 809e822.
Matsuoka, Y., Saito, M., LaFrancois, J., Gaynor, K., Olm, V., Wang, L., Casey, E., Lu, Y.,Shiratori, C., Lemere, C., Duff, K., 2003. Novel therapeutic approach for thetreatment of Alzheimer's disease by peripheral administration of agents withan affinity to beta-amyloid. J. Neurosci. 23, 29e33.
McGovern, S.L., Helfand, B.T., Feng, B., Shoichet, B.K., 2003. A specific mechanism ofnonspecific inhibition. J. Med. Chem. 46, 4265e4272.
McLaurin, J., Golomb, R., Jurewicz, A., Antel, J.P., Fraser, P.E., 2000. Inositol stereo-isomers stabilize an oligomeric aggregate of Alzheimer amyloid beta peptideand inhibit abeta-induced toxicity. J. Biol. Chem. 275, 18495e18502.
Meijer, D.K., Weert, B., Vermeer, G.A., 1988. Pharmacokinetics of biliary excretion inman. VI. Indocyanine green. Eur. J. Clin. Pharmacol. 35, 295e303.
Mennen, L.I., Walker, R., Bennetau-Pelissero, C., Scalbert, A., 2005. Risks and safetyof polyphenol consumption. Am. J. Clin. Nutr. 81, 326Se329S.
Meyer-Luehmann, M., Spires-Jones, T.L., Prada, C., Garcia-Alloza, M., de Calignon, A.,Rozkalne, A., Koenigsknecht-Talboo, J., Holtzman, D.M., Bacskai, B.J., Hyman, B.T.,2008. Rapid appearance and local toxicity of amyloid-beta plaques in a mousemodel of Alzheimer's disease. Nature 451, 720e724.
Milane, A., Fernandez, C., Vautier, S., Bensimon, G., Meininger, V., Farinotti, R., 2007.Minocycline and riluzole brain disposition: interactions with p-glycoprotein atthe bloodebrain barrier. J. Neurochem. 103, 164e173.
Mueller, S.O., Stopper, H., Dekant, W., 1998. Biotransformation of the anthraqui-nones emodin and chrysophanol by cytochrome P450 enzymes. Bioactivation togenotoxic metabolites. Drug Metab. Dispos. 26, 540e546.
Mueller, S.O., Schmitt, M., Dekant, W., Stopper, H., Schlatter, J., Schreier, P., Lutz, W.K.,1999. Occurrence of emodin, chrysophanol and physcion invegetables, herbs andliquors. Genotoxicity and anti-genotoxicity of the anthraquinones and of thewhole plants. Food Chem. Toxicol. 37, 481e491.
Mukrasch, M.D., Biernat, J., von Bergen, M., Griesinger, C., Mandelkow, E.,Zweckstetter, M., 2005. Sites of tau important for aggregation populate {beta}-structure and bind to microtubules and polyanions. 2005 Jul 1. J. Biol. Chem.280 (26), 24978e24986.
Mukrasch, M.D., Markwick, P., Biernat, J., Bergen, M., Bernado, P., Griesinger, C.,Mandelkow, E., Zweckstetter, M., Blackledge, M., 2007a. Highly populated turnconformations in natively unfolded tau protein identified from residual dipolarcouplings and molecular simulation. J. Am. Chem. Soc. 129, 5235e5243.
Mukrasch, M.D., von Bergen, M., Biernat, J., Fischer, D., Griesinger, C., Mandelkow, E.,Zweckstetter, M., 2007b. The “jaws” of the tau-microtubule interaction. J. Biol.Chem. 282, 12230e12239.
Mukrasch, M.D., Bibow, S., Korukottu, J., Jeganathan, S., Biernat, J., Griesinger, C.,Mandelkow, E., Zweckstetter, M., 2009. Structural polymorphism of 441-residuetau at single residue resolution. PLoS Biol. 7, e34.
Necula, M., Chirita, C.N., Kuret, J., 2005. Cyanine dye N744 inhibits tau fibrillizationby blocking filament extension: implications for the treatment of tauopathicneurodegenerative diseases. Biochemistry 44, 10227e10237.
Necula, M., Kayed, R., Milton, S., Glabe, C.G., 2007. Small molecule inhibitors ofaggregation indicate that amyloid beta oligomerization and fibrillizationpathways are independent and distinct. J. Biol. Chem. 282, 10311e10324.
Nicolaou, K.C., Chen, J.S., Edmonds, D.J., Estrada, A.A., 2009. Recent advances in thechemistry and biology of naturally occurring antibiotics. Angew. Chem. Int. Ed.Engl. 48, 660e719.
Obach, R.S., Kalgutkar, A.S., Ryder, T.F., Walker, G.S., 2008. In vitro metabolism andcovalent binding of enol-carboxamide derivatives and anti-inflammatoryagents sudoxicam and meloxicam: insights into the hepatotoxicity of sudox-icam. Chem. Res. Toxicol. 21, 1890e1899.
Ono, K., Yoshiike, Y., Takashima, A., Hasegawa, K., Naiki, H., Yamada, M., 2003.Potent anti-amyloidogenic and fibril-destabilizing effects of polyphenols invitro: implications for the prevention and therapeutics of Alzheimer's disease. J.Neurochem. 87, 172e181.
Park, B.K., Kitteringham, N.R., Maggs, J.L., Pirmohamed, M., Williams, D.P., 2005. Therole of metabolic activation in drug-induced hepatotoxicity. Annu. Rev. Phar-macol. Toxicol. 45, 177e202.
Park, S.Y., Ferreira, A., 2005. The generation of a 17 kDa neurotoxic fragment: analternative mechanism by which tau mediates beta-amyloid-induced neuro-degeneration. J. Neurosci. 25, 5365e5375.
Patani, G.A., LaVoie, E.J., 1996. Bioisosterism: a rational approach in drug design.Chem. Rev. 96, 3147e3176.
Pessayre, D., de Saint-Louvent, P., Degott, C., Bernuau, J., Rueff, B., Benhamou, J.P.,1978. Iproclozide fulminant hepatitis. Possible role of enzyme induction.Gastroenterology 75, 492e496.
Peter, C., Hongwan, D., Kupfer, A., Lauterburg, B.H., 2000. Pharmacokinetics andorgan distribution of intravenous and oral methylene blue. Eur. J. Clin. Phar-macol. 56, 247e250.
Pickhardt, M., Gazova, Z., von Bergen, M., Khlistunova, I., Wang, Y., Hascher, A.,Mandelkow, E.M., Biernat, J., Mandelkow, E., 2005a. Anthraquinones inhibit tauaggregation and dissolve Alzheimer's paired helical filaments in vitro and incells. J. Biol. Chem. 280, 3628e3635.
Pickhardt, M., von Bergen, M., Gazova, Z., Hascher, A., Biernat, J., Mandelkow, E.M.,Mandelkow, E., 2005b. Screening for inhibitors of tau polymerization. Curr.Alzheimer Res. 2, 219e226.
Pickhardt, M., Biernat, J., Khlistunova, I., Wang, Y.P., Gazova, Z., Mandelkow, E.M.,Mandelkow, E., 2007a. N-phenylamine derivatives as aggregation inhibitors incell models of tauopathy. Curr. Alzheimer Res. 4, 397e402.
Pickhardt, M., Larbig, G., Khlistunova, I., Coksezen, A., Meyer, B., Mandelkow, E.M.,Schmidt, B., Mandelkow, E., 2007b. Phenylthiazolyl-hydrazide and its deriva-tives are potent inhibitors of tau aggregation and toxicity in vitro and in cells.Biochemistry 46, 10016e10023.
Piekarska, B., Skowronek, M., Rybarska, J., Stopa, B., Roterman, I., Konieczny, L., 1996.Congo red-stabilized intermediates in the lambda light chain transition fromnative to molten state. Biochimie 78, 183e189.
Porat, Y., Abramowitz, A., Gazit, E., 2006. Inhibition of amyloid fibril formation bypolyphenols: structural similarity and aromatic interactions as a commoninhibition mechanism. Chem. Biol. Drug Des. 67, 27e37.
Preziosi, P., 2007. Isoniazid: metabolic aspects and toxicological correlates. Curr.Drug Metab. 8, 839e851.
Quist, A., Doudevski, I., Lin, H., Azimova, R., Ng, D., Frangione, B., Kagan, B., Ghiso, J.,Lal, R., 2005. Amyloid ion channels: a common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. U S A 102, 10427e10432.
Raciti, G., Mazzone, P., Raudino, A., Mazzone, G., Cambria, A., 1995. Inhibition of ratliver mitochondrial monoamine oxidase by hydrazine-thiazole derivatives:structureeactivity relationships. Bioorg. Med. Chem. 3, 1485e1491.
Rapoport, M., Dawson, H.N., Binder, L.I., Vitek, M.P., Ferreira, A., 2002. Tau isessential to beta -amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. U S A 99,6364e6369.
Reinke, A.A., Gestwicki, J.E., 2007. Structureeactivity relationships of amyloid beta-aggregation inhibitors based on curcumin: influence of linker length andflexibility. Chem. Biol. Drug Des. 70, 206e215.
Roberson, E.D., Scearce-Levie, K., Palop, J.J., Yan, F., Cheng, I.H., Wu, T., Gerstein, H.,Yu, G.Q., Mucke, L., 2007. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science 316, 750e754.
Roy, R.S., Gehring, A.M., Milne, J.C., Belshaw, P.J., Walsh, C.T., 1999. Thiazole andoxazole peptides: biosynthesis and molecular machinery. Nat. Prod. Rep. 16,249e263.
Russell, A.J., Westwood, I.M., Crawford, M.H., Robinson, J., Kawamura, A.,Redfield, C., Laurieri, N., Lowe, E.D., Davies, S.G., Sim, E., 2009. Selective smallmolecule inhibitors of the potential breast cancer marker, human arylamine N-acetyltransferase 1, and its murine homologue, mouse arylamine N-acetyl-transferase 2. Bioorg. Med. Chem. 17, 905e918.
Sawaya, M.R., Sambashivan, S., Nelson, R., Ivanova, M.I., Sievers, S.A., Apostol, M.I.,Thompson, M.J., Balbirnie, M., Wiltzius, J.J., McFarlane, H.T., Madsen, A.O.,Riekel, C., Eisenberg, D., 2007. Atomic structures of amyloid cross-beta spinesreveal varied steric zippers. Nature 447, 453e457.
Schneider, A., Biernat, J., von Bergen, M., Mandelkow, E., Mandelkow, E.M., 1999.Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214)

B. Bulic et al. / Neuropharmacology 59 (2010) 276e289 289
also protects it against aggregation into Alzheimer paired helical filaments.Biochemistry 38, 3549e3558.
Serpell, L.C., Benson, M., Liepnieks, J.J., Fraser, P.E., 2007. Structural analyses offibrinogen amyloid fibrils. Amyloid 14 (3), 199e203.
Singh, M., Arseneault, M., Sanderson, T., Murthy, V., Ramassamy, C., 2008. Chal-lenges for research on polyphenols from foods in Alzheimer's disease:bioavailability, metabolism, and cellular and molecular mechanisms. J. Agric.Food Chem. 56, 4855e4873.
Skowronek, M., RotermanKonieczny, L., Stopa, B., Rybarska, J., Piekarska, B.,Gorecki, A., Krol, M., 2000. The conformational characteristics of Congo red,Evans blue and Trypan blue. Comput. Chem. 24, 429e450.
Small, S.A., Duff, K., 2008. Linking Abeta and tau in late-onset Alzheimer's disease:a dual pathway hypothesis. Neuron 60, 534e542.
Sonar, V.N., Crooks, P.A., 2009. Synthesis and antitubercular activity of a series ofhydrazone and nitrovinyl analogs derived from heterocyclic aldehydes. J.Enzym. Inhib. Med. Chem. 24, 117e124.
Spolnik, P., Stopa, B., Piekarska, B., Jagusiak, A., Konieczny, L., Rybarska, J., Krol, M.,Roterman, I., Urbanowicz, B., Zieba-Palus, J., 2007. The use of rigid, fibrillarCongo red nanostructures for scaffolding protein assemblies and inducing theformation of amyloid-like arrangement of molecules. Chem. Biol. Drug Des. 70,491e501.
Sturm, K., Levstik, L., Demopoulos, V.J., Kristl, A., 2006. Permeability characteristicsof novel aldose reductase inhibitors using rat jejunum in vitro. Eur. J. Pharm. Sci.28, 128e133.
Talaga, P., Quere, L., 2002. The plasma membrane: a target and hurdle for thedevelopment of anti-abeta drugs? Curr. Drug Targets CNS Neurol. Disord. 1,567e574.
Taniguchi, S., Suzuki, N., Masuda, M., Hisanaga, S., Iwatsubo, T., Goedert, M.,Hasegawa, M., 2005. Inhibition of heparin-induced tau filament formationby phenothiazines, polyphenols, and porphyrins. J. Biol. Chem. 280, 7614e7623.
Teng, Z.H., Zhou, S.Y., Yang, R.T., Liu, X.Y., Liu, R.W., Yang, X., Zhang, B.L., Yang, J.Y.,Cao, D.Y., Mei, Q.B., 2007. Quantitation assay for absorption and first-passmetabolism of emodin in isolated rat small intestine using liquid chromatog-raphy-tandem mass spectrometry. Biol. Pharm. Bull. 30, 1628e1633.
Timmins, G.S., Deretic, V., 2006. Mechanisms of action of isoniazid. Mol. Microbiol.62, 1220e1227.
Tolman, K.G., Chandramouli, J., 2003. Hepatotoxicity of the thiazolidinediones. Clin.Liver Dis. 7, 369e379 (vi).
Toth, B., 1980. Actual new cancer-causing hydrazines, hydrazides, and hydrazones. J.Cancer Res. Clin. Oncol. 97, 97e108.
Toth, B., 2000. A review of the natural occurrence, synthetic production and use ofcarcinogenic hydrazines and related chemicals. In Vivo 14, 299e319.
Vieira, M.N., Forny-Germano, L., Saraiva, L.M., Sebollela, A., Martinez, A.M.,Houzel, J.C., De Felice, F.G., Ferreira, S.T., 2007. Soluble oligomers from a non-
disease related protein mimic Abeta-induced tau hyperphosphorylation andneurodegeneration. J. Neurochem. 103, 736e748.
Vijaya Raj, K.K., Narayana, B., Ashalatha, B.V., Suchetha Kumari, N., Sarojini, B.K.,2007. Synthesis of some bioactive 2-bromo-5-methoxy-N'-[4-(aryl)-1,3-thiazol-2-yl]benzohydrazide derivatives. Eur. J. Med. Chem. 42, 425e429.
Villain-Guillot, P., Gualtieri, M., Bastide, L., Roquet, F., Martinez, J., Amblard, M.,Pugniere, M., Leonetti, J.P., 2007. Structureeactivity relationships of phenyl-furanyl-rhodanines as inhibitors of RNA polymerase with antibacterial activityon biofilms. J. Med. Chem. 50, 4195e4204.
Von Berlepsch, H., Böttcher, C., Ouart, A., Burger, C., Dähne, S., Kirstein, S., 2000.Supramolecular structures of J-aggregates of carbocyanine dyes in solution. J.Phys. Chem. B 104, 5255e5262.
Wainwright, M., Amaral, L., 2005. The phenothiazinium chromophore and theevolution of antimalarial drugs. Trop. Med. Int. Health 10, 501e511.
Walter-Sack, I., Rengelshausen, J., Oberwittler, H., Burhenne, J., Mueller, O.,Meissner, P., Mikus, G., 2009. High absolute bioavailability of methylene bluegiven as an aqueous oral formulation. Eur. J. Clin. Pharmacol. 65, 179e189.
Waters, M.L., 2002. Aromatic interactions in model systems. Curr. Opin. Chem. Biol.6, 736e741.
Winklhofer, K.F., Tatzelt, J., Haass, C., 2008. The two faces of protein misfolding:gain- and loss-of-function in neurodegenerative diseases. EMBO J. 27, 336e349.
Wischik, C.M., Edwards, P.C., Lai, R.Y., Roth, M., Harrington, C.R., 1996. Selectiveinhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc.Natl. Acad. Sci. U S A 93, 11213e11218.
Wischik, C.M., Rickard, J.E., Harrington, C.R., Horsley, D., Storey, J.M.D., Marshall, C.,Sinclair, J.P., 2007. Preparation of phenothiazine derivatives for treatment oftauopathy. PCT Int. Appl. WO 2007110630.
Wischik, C.M., Bentham, P., Wischik, D.J., Seng, K.M. Tau Aggregation Inhibitors (TAI)Therapy with Remembertm Arrests Disease Progression in Mild and ModerateAlzheimer's Disease over 50 Weeks, Abstract 03-04-07, International Confer-ence on Alzheimer's Disease U.S.A., Chicago, 2008.
Xu, X., Persson, H.L., Richardson, D.R., 2005. Molecular pharmacology of the inter-action of anthracyclines with iron. Mol. Pharmacol. 68, 261e271.
Yang, F., Lim, G.P., Begum, A.N., Ubeda, O.J., Simmons, M.R., Ambegaokar, S.S.,Chen, P.P., Kayed, R., Glabe, C.G., Frautschy, S.A., Cole, G.M., 2005. Curcumininhibits formation of amyloid beta oligomers and fibrils, binds plaques, andreduces amyloid in vivo. J. Biol. Chem. 280, 5892e5901.
Zervosen, A., Lu, W.P., Chen, Z., White, R.E., Demuth Jr., T.P., Frere, J.M., 2004.Interactions between penicillin-binding proteins (PBPs) and two novel classesof PBP inhibitors, arylalkylidene rhodanines and arylalkylidene iminothiazoli-din-4-ones. Antimicrob. Agents Chemother. 48, 961e969.
Zhang, J.H., Chung, T.D., Oldenburg, K.R., 1999. A simple statistical parameter for usein evaluation and validation of high throughput screening assays. J. Biomol.Screen. 4, 67e73.