self-consistent gw calculations for single-molecule transport: bridging the theory-experiment gap in...
TRANSCRIPT

Self-consistent GW calculations for single-molecule transport:
Bridging the theory-experiment gap in single-molecule transport
Kristian S. Thygesen
Center for Atomic-scale Materials Design (CAMD)
Department of Physics
Technical University of Denmark

A single molecule
HOMO
LUMO
En
erg
y
)1()( 00 NENE
)1()( 10 NENE
)()1( 00 NENE
)1()( 20 NENE
)()1( 01 NENE

EF EF
HOMO
LUMO
En
erg
y
Closed shell
Weak coupling to electrodes

Chemical bond with electrodesE
nerg
y
EFEF

Finite bias voltageE
nerg
y
EF
EF

En
erg
y
EF
EF
How does electron-electron interactions and the nonequilibrium
conditions affect the electronic structure and transport properties of
molecular junctions?
Finite bias voltage

Outline
DFT Conductance of BDT/BDA junctions
GW-transport scheme
Free molecules
Molecule-solid interface
Finite bias
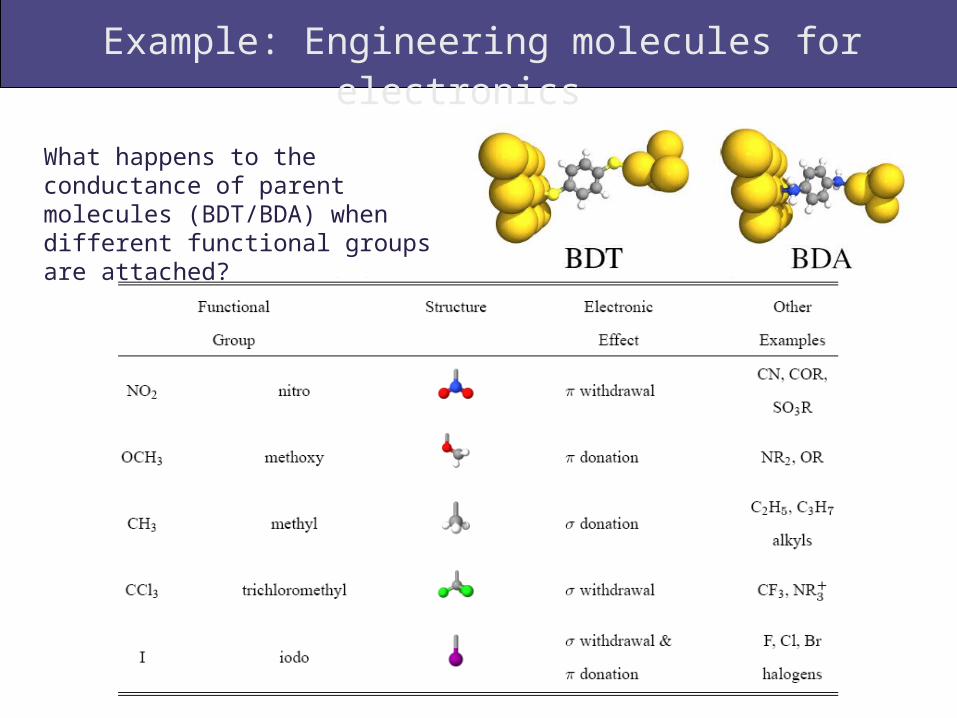
Example: Engineering molecules for electronics
What happens to the conductance of parent molecules (BDT/BDA) when different functional groups are attached?

DFT-based conductance calculations
Qualitative effect as expected, but…
Effect of side groups very weak, and …
Calculated conductance 10-100 times larger than experimental values!
D. J. Mowbray, G. Jones, and K. S. Thygesen, JCP 128, 111103 (2008)

Effect of side group on HOMO levels
Correction for SI errors and image charge interactions.
Image charge effect partially saves the day for DFT.
D. J. Mowbray, G. Jones, and K. S. Thygesen, JCP 128, 111103 (2008)
Correction for SI errors

Comparison to experiment (BDA@Au)
D. J. Mowbray, G. Jones, and K. S. Thygesen, JCP 128, 111103 (2008)

M. Strange et al., PRL 101, 096804 (2008)
W. H. Thijssen et al. PRL 96, 026806 (2006)
M. Strange, et al. JCP 128, 114714 (2008)
Some success stories
Why does DFT work well in some cases while it fails in other cases?

Beyond the single-particle approximation
Time-dependent DFT Stefanucci and Almbladh, Euro. Phys. Lett. 67, 14 (2004)
Di Ventra and Todorov, J. Phys.:Cond.Mat. 16, 8025 (2004)
Linear response Kubo formulaBokes, Jung and Godby, Phys. Rev. B 76, 125433 (2007)
Rate equations + exact diagonalizationHettler, Wenzel, Wegewijs, Schoeller, Phys. Rev. Lett. 90, 076805 (2003)
Many-body perturbation theoryDarancet, Ferretti, Mayou and Olevano Phys. Rev. B 75, 075102 (2007)
Thygesen and Rubio, J. Chem. Phys. 126, 091101 (2007)
Ferretti, Calzolari, Di Felice, and Manghi, Phys. Rev. B 72, 125114 (2005)

The band gap problem of DFT
DFT + local xc-functionals underestimate
HOMO-LUMO gaps
Hartree-Fock is good for small molecules
(SI-free), but overestimates the gap for
extended systems
GW includes screening in the exchange
and this solves the gap problem.
Hartree-Fock exchange Screening correction
Schilfgaarde, Kotani, and Faleev, PRL 96, 226402 (2006)

Many-body approach to quantum transport: GW in the central region
Thygesen and Rubio, J. Chem. Phys 126, 091101 (2007) ; Phys. Rev. B 77, 115333 (2008)
vxc
GW
Two problems:
Conventional GW gives quasi-particle excitations of the groundstate, but transport is a nonequilibrium phenomenon
How to deal with interactions in infinite, non-periodic systems?
Two (possible) solutions:
Formulate GW on the Keldysh contour
Assume that leads can be described at the mean-field (DFT) level, and include correlations only in a central region

Hamiltonian
Non-interacting part:
Kohn-Sham Hamiltonian:
Interactions:MB Hamiltonian:

Non-equilibrium GFs and current
The retarded GF of the central region:
Embedding self-energies
Interaction self-energyCorrelation functions:
Current from lead α into central region:
Symmetrized current:
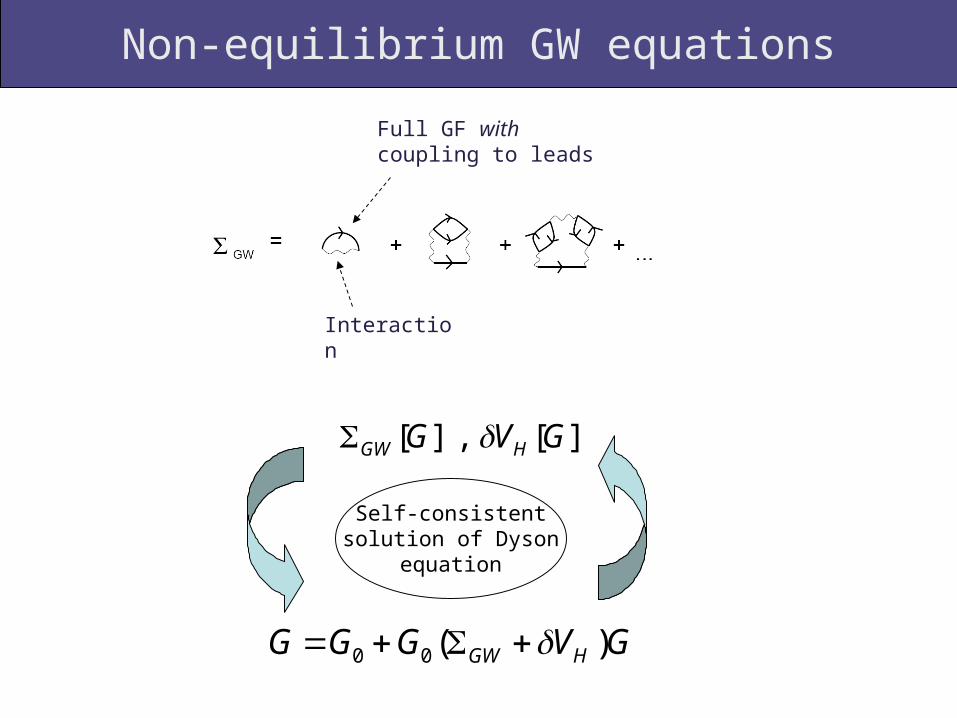
Non-equilibrium GW equations
Interaction
Full GF with coupling to leads
][,][ GVG HGW
GVGGG HGW )(00
Self-consistent solution of Dyson
equation

Program- flow

Keldysh nonequilibrium formalism
Dyson equation solved fully self-consistent with GW self-energy (charge conservation, no G0 dependence)
Full dynamical dependence of all quantities (no plasmon-pole approximation, no linearized quasi-particle equation)
All quantities calculated in real time/frequency (no analytic continuation)
Valence-core exchange included (known in PAW)
Non-conventional features:
Overview of GW-transport method
Localized atomic orbital basis.
Dynamical dependence sampled uniformly in real frequency/time.
Ions described by PAW + frozen core approximation
Product basis technique to reduce size of four-index quantities (W and P).
Parallelization over orbitals and time/frequency grid.

PBE underestimates position of occupied states due to self-interaction errors.
PBE0 improves PBE slightly.
HF yields too deep-lying levels because of neglect of orbital relaxations
Self-consistent GW corrects the HF energies by including dynamical screening (orbital relaxation).
Small molecules (2-8 atoms) containing :
H, C, N, Cl, O, F, S, P, Na, Li, Si
Calculated HOMO level for 35 gas molecules
C. Rostgaard, K. W. Jacobsen, and K. S. Thygesen, submitted

Calculated HOMO level for 35 gas molecules
GW shifts occupied levels up in energy compared to HF
GW systematically improves HF energies

Limitations: Strong vs. weak correlations
Spectral functions:
Local interactions (Hubbard) -> strong correlations
Long-range interactions (1/R) -> weak correlations
Entropy of reduced density matrix:
Degree of correlation:
Θ = S/Smax , 0< Θ <1
Θ=0.52
Θ=0.11

Molecule@surface: Dynamic polarization effects
S. Kubatkin et al. Nature, 425, 698 (2003)
Experiments on OPV5 molecule transistors.
J. Repp et al. PRL, 94, 026803 (2005)
STS of pentacene adsorbed at NaCl/Cu thin films.

Energy cost of adding an electron to the LUMO is given by spectral function:
Molecule@surface: Dynamic polarization effects

Microscopic model of metal-molecule interface
VHHH molmetˆˆˆˆ

Dependence on metal-molecule interaction
Gap reduction due to screening in metal (image charge formation).
Open squares: Exact difference in total groundstate energy with an extra electron (hole) on the molecule.
All many-body eigenstates are single Slater determinants: weakly correlated system
Vanishing thyb (weak physisorption)
Thygesen and Rubio, Phys. Rev. Lett. 102, 046802 (2009)

Dependence on metal band width
Vanishing thyb (weak physisorption)
Small t Large metal DOS at EF Large density response Efficient screening
GW quasiparticle is not just total energy difference, i.e. the QP has overlap with excited N+1 particle states of the metal.

Dependence on metal-molecule hopping
The density response of the molecule increase with the coupling.
Intra-molecular screening occurs via charge-transfer to the metal.
Suggests a direct correlation between chemisorption bond strength and HOMO-LUMO gap reduction.
Thygesen and Rubio, Phys. Rev. Lett. 102, 046802 (2009)

First-principles GW calculations: Physisorbed benzene
z=4.5 Å G0W0 calculations performed with the Yambo code(*).
Yambo:
G0W0 LDA, Plane wave basis, norm-conserving pseusopotentials, plasmon pole approximation.
(*) A. Marini, C. Hogan, M. Grüning, D. Varsano, arXiv:0810.3118 (2009)
See also: J. B. Neaton et al. Phys. Rev. Lett. 97, 216405 (2006)

GW and LDA benzene HOMO-LUMO gaps on different surfaces
LDA gaps are independent of substrate
GW gaps show large variation across different surfaces
GW gap sensitive to atomistic details, e.g. surface plane (BaO)
J.M.Garcia, A. Rubio and KST, submitted
4.5 Å

Classical image charge model
)1(
)1(
)(4)(
0
2
r
rimg zz
qzV
Best-fit values for and z0:
Electrostatic energy of point charge above a polarizable medium:
Classical model describes the physics of the gap reduction qualitatively.
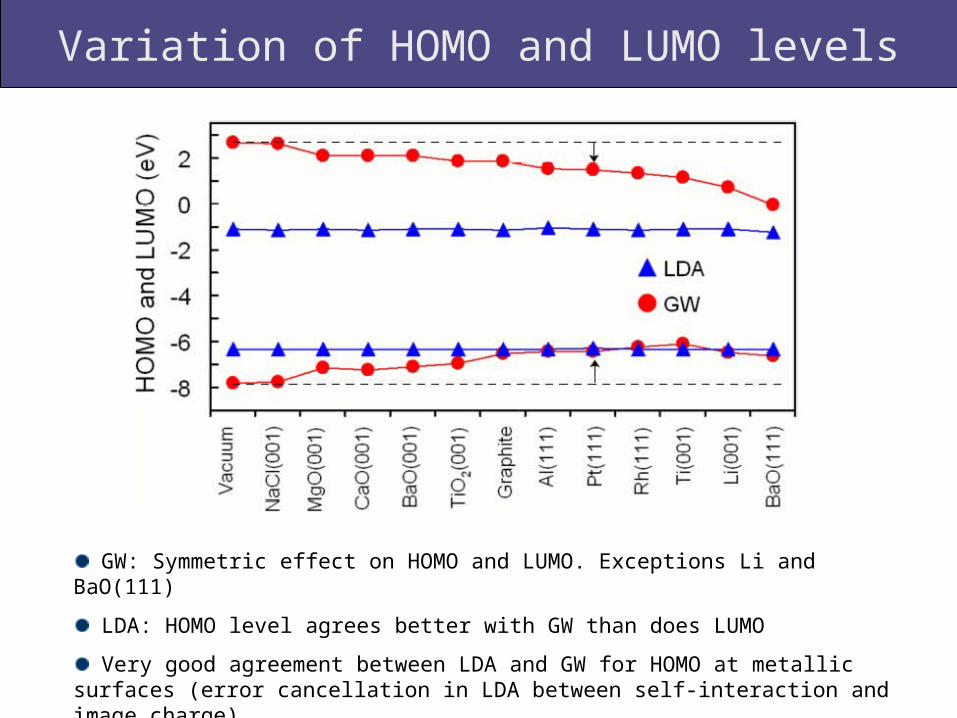
Variation of HOMO and LUMO levels
GW: Symmetric effect on HOMO and LUMO. Exceptions Li and BaO(111)
LDA: HOMO level agrees better with GW than does LUMO
Very good agreement between LDA and GW for HOMO at metallic surfaces (error cancellation in LDA between self-interaction and image charge)
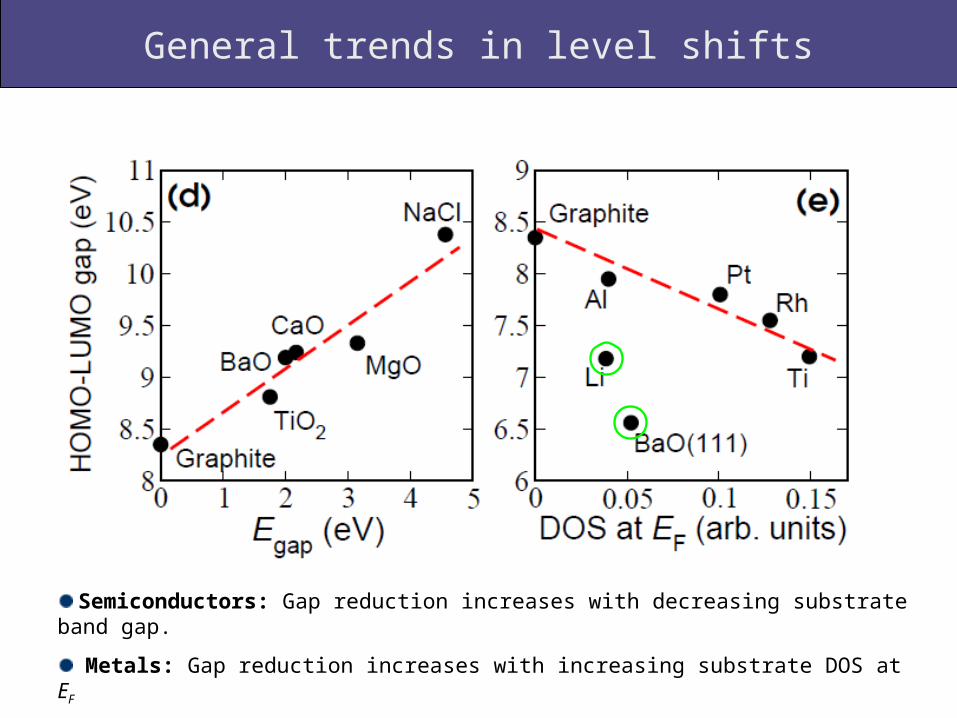
General trends in level shifts
Semiconductors: Gap reduction increases with decreasing substrate band gap.
Metals: Gap reduction increases with increasing substrate DOS at EF
Li and BaO(111) deviate from general trend!

GW self-energy to second order in V
Renormalization of single electronic level, , by non-local
interactions with substrate electrons (time-ordered quantities):
Substrate joint density of states weighted by particle-hole transitions

Quasiparticle self-consistent equation:
Graphical solution to QP equation (“generic” ∆ corresponding to constant Vkk’):
Retarded self-energy:
Effective interaction strength:
Microscopic origin of general trends
Substrate joint density of states weighted by particle-hole transitions
Trends for both metals and semiconductors can be explained by assuming constant and system independent Vkk’

Pt-H2-Pt: Density of states
GW/HF
SZ/DZP basis set.
Image charge effect

Pt-H2-Pt: Transmission
More Pt atoms in GW region
Basis set
General trend: GDFT > GGW > GHF
Dynamical screening (image charge effects)

Applying a bias voltage
The bias can affect the HOMO-LUMO gap
The bias can affect the resonance width
These effects must be taken into account to describe the IV
Molecular DOS
Current:

Impact of exchange-correlation on IV
Peaks in dI/dV:
Position and width influenced by the bias!

Evolution of HOMO/LUMO levels in Hartree (crosses), HF (triangles), and GW (circles).
HF and GW agree at low bias
Quasi-particle scattering reduces electronic lifetimes at finite bias
Enhanced dynamic screening reduces GW gap
Impact of exchange-correlation on IV
Thygesen, Phys. Rev. Lett. 100, 166804 (2008)

Acknowledgements
Carsten Rostgaard
Karsten W. Jacobsen
Juan Maria Garcia Lastra
Angel Rubio
Collaborators:
CAMD, Technical University of Denmark
University of the Basque Country, Spain
Funding:The Lundbeck Foundation
Danish Center for Scientific Computing (DCSC)

Conclusions
Bridging the ”experiment – theory gap” in single-molecule transport
rely on proper incorporation of correlation effects beyond the mean-
field (DFT) approximation.
Band gap problem of DFT: Why does DFT-transport work for some
systems?
HF works well for isolated molecules
Dynamical screening important at molecule-surface interfaces ->
DFT levels may not be that wrong (error cancellations)
Importance of correlation effects increase out of equilibrium

Anderson model