phosphorylation by the c-abl protein tyrosine kinase inhibits … · phosphorylation by the c-abl...
TRANSCRIPT

Phosphorylation by the c-Abl protein tyrosinekinase inhibits parkin’s ubiquitination andprotective functionHan Seok Koa,b, Yunjong Leea,c, Joo-Ho Shina,b, Senthilkumar S. Karuppagoundera,b, Bharathi Shrikanth Gadada,b,Anthony J. Kolesked, Olga Pletnikovae, Juan C. Troncosob,e, Valina L. Dawsona,b,c,f, and Ted M. Dawsona,b,f,1
aNeuroregeneration and Stem Cell Programs, Institute for Cell Engineering, Departments of bNeurology, cPhysiology, and ePathology, Division ofNeuropathology, and fSolomon H. Snyder Department of Neuroscience, The Johns Hopkins University School of Medicine, Baltimore, MD 21205; anddDepartment of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06520
Edited* by Ann M. Graybiel, Massachusetts Institute of Technology, Cambridge, MA, and approved August 11, 2010 (received for review May 7, 2010)
Mutations in PARK2/Parkin, which encodes a ubiquitin E3 ligase,causeautosomal recessiveParkinsondisease (PD).Herewe showthatthenonreceptor tyrosine kinase c-Abl phosphorylates tyrosine 143 ofparkin, inhibiting parkin’s ubiquitin E3 ligase activity and protectivefunction. c-Abl is activated by dopaminergic stress and by dopami-nergic neurotoxins, 1-methyl-4-phenylpyridinium (MPP+) in vitroand in vivo by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP),leading to parkin inactivation, accumulation of the parkin substratesaminoacyl-tRNA synthetase-interacting multifunctional protein type2 (AIMP2) (p38/JTV-1) and fuse-binding protein 1 (FBP1), and celldeath. STI-571, a c-Abl-family kinase inhibitor, prevents the phos-phorylation of parkin, maintaining parkin in a catalytically activeand protective state. STI-571’s protective effects require parkin, asshRNAknockdownofparkinpreventsSTI-571protection.Conditionalknockout of c-Abl in the nervous systemalso prevents the phosphor-ylation of parkin, the accumulation of its substrates, and subsequentneurotoxicity in response to MPTP intoxication. In human postmor-tem PD brain, c-Abl is active, parkin is tyrosine-phosphorylated, andAIMP2 and FBP1 accumulate in the substantia nigra and striatum.Thus, tyrosine phosphorylation of parkin by c-Abl is a major post-translational modification that inhibits parkin function, possibly con-tributing to pathogenesis of sporadic PD. Moreover, inhibition ofc-Abl may be a neuroprotective approach in the treatment of PD.
AIMP2 | Parkinson disease | STI-571 | ubiquitin
Parkinson disease (PD) is a common neurodegenerative dis-order characterized by the loss of dopamine (DA) neurons and
protein accumulation in intracellular inclusions designated asLewy bodies and Lewy neurites (1). Although the majority ofPD is sporadic in nature, rare familial mutations are providinginsight into this chronic, progressive neurodegenerative disease.Mutations in α-synuclein and LRRK2 cause autosomal-dominantPD, whereas mutations in DJ-1, PINK1, and parkin result inautosomal-recessive PD (2). Parkin mutations are the mostcommon cause of autosomal-recessive PD and, for the most part,PD due to parkin mutations is indistinguishable from sporadic PD(3). Parkin is a ubiquitin E3 ligase, and familial mutations arethought to impair the E3 ligase activity of parkin (4, 5).Parkin ubiquitinates proteins via monoubiquitination or poly-
ubiquitination using either lysine 48 (K48) or lysine 63 (K63).Monoubiquitination by parkin is thought to regulate receptor traf-ficking (6). Polyubiquitination by parkin via K48 is thought to me-diate proteasomaldegradation (7, 8),whereas polyubiquitination byK63may be involved in inclusion formation (9). Parkin’s differentialubiquitination properties are likely to be regulated by differentubiquitin-conjugating E2s and other associated proteins or regula-toryprocesses (3).Anumberofputativeparkin substrateshavebeenidentified (for a review, see ref. 3). Aminoacyl-tRNA synthetase-interacting multifunctional protein type 2 (AIMP2) (p38/JTV-1)and fuse-binding protein 1 (FBP1) are parkin substrates that appearto be regulated by K48 ubiquitination and proteasomal degradationbecause they accumulate in parkin exon 7 knockout (KO)mice andin autosomal-recessive PD brains due to parkin mutations (8).AIMP2 may play a pathogenic role, as it is selectively toxic to DA
neurons (8). Parkinmay also regulate the biologic function of othersubstrates (3).The nonreceptor tyrosine kinase c-Abl localizes to the nucleus
and cytoplasm and is activated by cellular stress (10). c-Abl playsa prominent role in tumorigenesis, as c-Abl is a homolog of thetransforming element of the Abelson murine leukemia virus (11).In brain, c-Abl is involved in neuronal plasticity, neurite outgrowth,and neurogenesis (12). Activation of c-Abl may also play a rolein neurologic disorders such as Alzheimer’s disease (13) andNiemann–Pick type-2 disease (14) through aberrant activation.Parkin’s E3 ligase activity and protective function are regulated
by posttranslational modifications including S-nitrosylation (15),phosphorylation (16, 17), and dopamine conjugation (18). Herewe report functional regulation of parkin activity by phosphory-lation of tyrosine 143 by c-Abl. Phosphorylation of parkin by c-Ablinhibits parkin’s E3 ligase activity and protective function in bothin vitro and in vivomodels of PD. In human postmortem PD brain,c-Abl is active, parkin is tyrosine-phosphorylated, and AIMP2 andFBP1 accumulate, suggesting a pathophysiologic regulation ofparkin by c-Abl in sporadic PD.
ResultsParkin Interacts with c-Abl. To determine whether parkin directlyinteracts with c-Abl, a glutathione S-transferase (GST) pull-downassay was performed with recombinant GST-parkin and recom-binant c-Abl (Fig. 1A). c-Abl interacts with GST-parkin as de-termined by immunoblot with an anti-c-Abl antibody, whereasc-Abl does not interact with GST alone (Fig. 1A). Immunopre-cipitation of parkin from mouse brain coimmunoprecipitatesc-Abl, indicating that c-Abl and parkin interact in vivo. c-Abl failsto coimmunoprecipitate with mouse IgG, indicating that the in-teraction between parkin and c-Abl is specific (Fig. 1B). To furtherdetermine the specificity of the interaction, immunoprecipitiationwas compared between wild-type (WT) and parkin KO mice.Antibodies to parkin coimmunoprecipitate c-Abl from WT brainbut not parkin KO brain (Fig. S1A). Cotransfection experimentsin SH-SY5Y cells indicate that V5-tagged parkin (V5-parkin)interacts with GFP-tagged c-Abl (GFP-c-Abl) via coimmunopre-cipitation of either V5 or GFP (Fig. 1 C and D). Interestingly,a kinase-dead (KD) (lysine 290 arginine) version of c-Abl (19) failsto interact with parkin (Fig. 1 C and D). To determine the domainof c-Abl that interacts with parkin, different domains of c-Abl werecotransfected with V5-parkin. Constructs of c-Abl that contain theSH3 domain interact with parkin, whereas c-Abl constructs lackingthe SH3 domain fail to interact with parkin (Fig. 1E and Fig. S1B).Thus, the c-Abl SH3 domain is required for the interaction with
Author contributions: H.S.K., V.L.D., and T.M.D. designed research; H.S.K., Y.L., J.-H.S., S.S.K.,and B.S.G. performed research; A.J.K., O.P., and J.C.T. contributed new reagents/analytictools; H.S.K., Y.L., J.-H.S., S.S.K., V.L.D., and T.M.D. analyzed data; and H.S.K., V.L.D., andT.M.D. wrote the paper.
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.1To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1006083107/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1006083107 PNAS | September 21, 2010 | vol. 107 | no. 38 | 16691–16696
NEU
ROSC
IENCE
Dow
nloa
ded
by g
uest
on
June
1, 2
020

parkin. To ascertain the domain of parkin that interacts with c-Abl,different myc-tagged domains of parkin were cotransfected withGFP-c-Abl. c-Abl interacts with the RING finger and in-betweenRING finger domains of parkin (Fig. 1F).
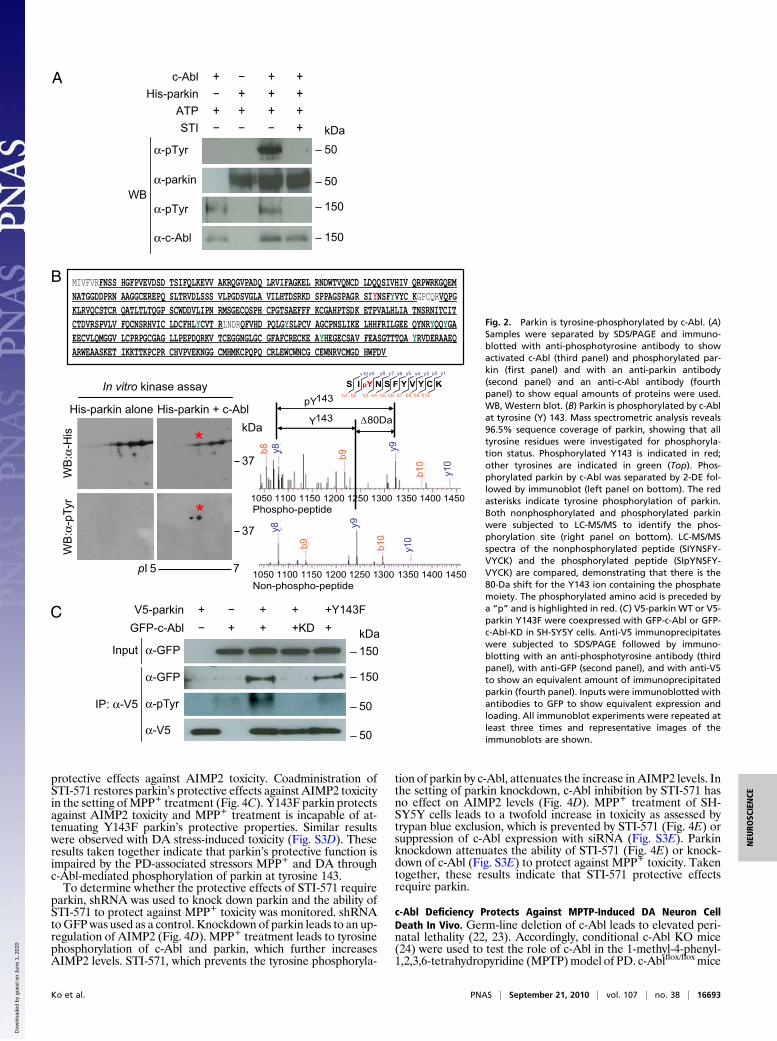
c-Abl Tyrosine Phosphorylates Parkin. An in vitro kinase assay wasperformed using recombinant c-Abl and recombinant His6-taggedparkin to determine whether c-Abl is able to phosphorylate parkin.Phosphorylation was monitored with an anti-phosphotyrosine (anti-p-tyrosine) antibody. c-Abl tyrosine-phosphorylates parkin, and thec-Abl-family kinase inhibitor STI-571 prevents the tyrosine phos-phorylation of parkin by c-Abl (Fig. 2A). To determine the site ofphosphorylation of parkin by c-Abl, two-dimensional (2-DE) gelelectrophoresis was used to separate phosphorylated from non-phosphorylated parkin (Fig. 2B). c-Abl-phosphorylated parkin wassubmitted for mass spectrometry for determination of the phos-phorylation site. Mass spectrometric analysis provided 96.5% se-quence coverageof parkin, andall tyrosine residueswere investigatedfor phosphorylation status. Only tyrosine 143 is phosphorylated,whereas none of the other tyrosines are phosphorylated (Fig. 2B). Toconfirm that tyrosine 143 was the sole site of phosphorylation byc-Abl, tyrosine 143 in parkin was mutated to phenylalanine to createa Y143F parkin mutant. Cotransfection experiments in SH-SY5Ycells indicate that WT parkin is phosphorylated by c-Abl but notc-Abl-KD. c-Abl is not able to tyrosine-phosphorylate parkin Y143F(Fig. 2C). These results taken together indicate that c-Abl phos-phorylates parkin on tyrosine 143.
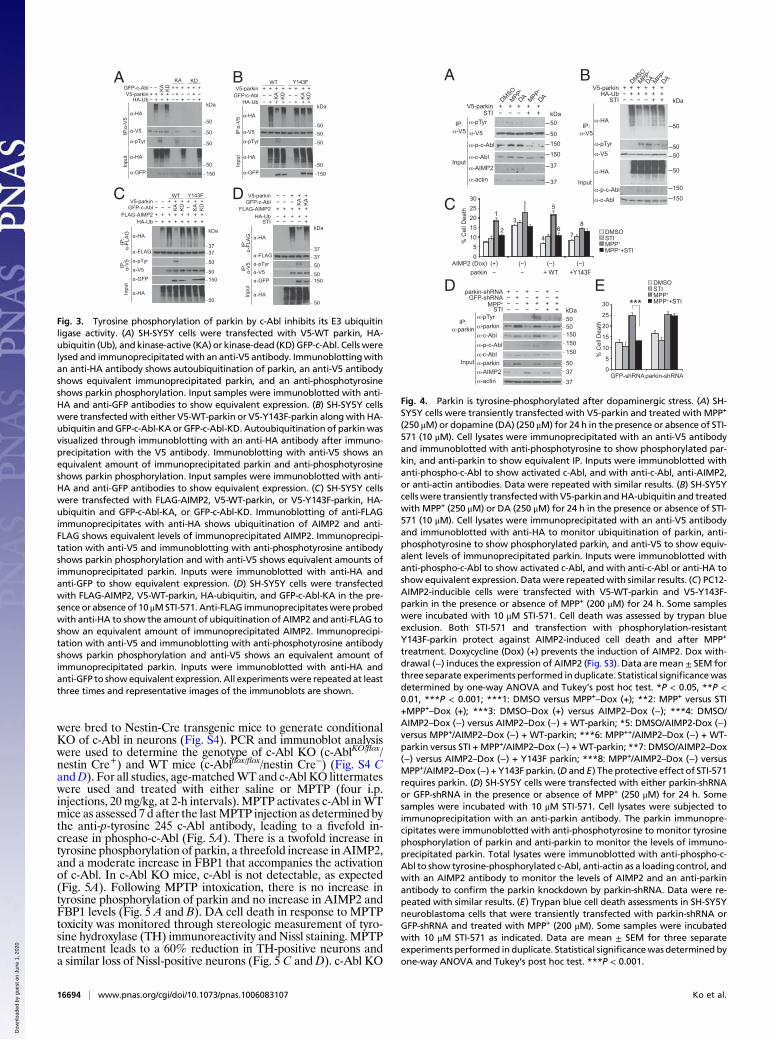
Tyrosine Phosphorylation of Parkin Inhibits Its Ubiquitin E3 LigaseActivity. To determine whether c-Abl phosphorylation of parkinmodulates parkin’s ubiquitination activity, autoubiquitination ofparkin was monitored in the presence of kinase-active c-Abl(c-Abl-KA) or c-Abl-KD. SH-SY5Y cells were cotransfected withV5-tagged parkin and HA-tagged ubiquitin followed by immu-noprecipitation of parkin. Parkin is autoubiquitinated, as detectedby anti-HA immunoreactivity as previously reported (Fig. 3A)(4, 5). Kinase-active c-Abl, which tyrosine-phosphorylates parkin,almost completely inhibits the autoubiquitination of parkin,whereas c-Abl-KD has no effect (Fig. 3A). To ascertain whethertyrosine 143 is required for the c-Abl-mediated inhibition ofparkin autoubiquitination, WT parkin and Y143F parkin autou-biquitination were monitored in the presence of c-Abl-KA orc-Abl-KD. WT parkin ubiquitination is inhibited by c-Abl-KA,whereas it has no effect on parkin Y143F ubiquitination (Fig. 3B).c-Abl-KD has no effect on WT parkin or Y143F parkin autou-biquitination (Fig. 3B). Next, the effects of c-Abl on parkinubiquitination of AIMP2 were monitored. Parkin ubiquitinatesAIMP2 as previously described (8, 20) and Y143F parkin alsoubiquitinates AIMP2 (Fig. 3C). c-Abl-KA impairs WT parkinubiquitination of AIMP2, but c-Abl-KD has no effect (Fig. 3C).Y143F parkin ubiquitination of AIMP2 is not affected by eitherc-Abl-KA or c-Abl-KD (Fig. 3C). To pharmacologically confirmthe role of c-Abl in tyrosine-phosphorylating parkin and inhibitingits ubiquitination function, AIMP2 ubiquitination by parkin wasevaluated in the presence and absence of STI-571, a c-Abl-familykinase inhibitor. The c-Abl-mediated tyrosine phosphorylation ofparkin and the inhibition of parkin’s ubiquitination of AIMP2 areprevented by STI-571 (Fig. 3D). To determine whether c-Ablaffects parkin’s ubiquitination of other substrates, the ubiquiti-nation of FBP1 was monitored. FBP1 ubiquitination by parkin isinhibited by c-Abl-KA, and STI-571 inhibits the c-Abl-mediatedtyrosine phosphorylation of parkin and restores the ubiquitinationof FBP1 (Fig. S2).
Dopaminergic Stress Activates c-Abl Inhibiting Parkin’s E3 LigaseActivity and Protective Function. Different types of genotoxicstress are known to activate c-Abl (21). To determine whetherPD-associated stressors activate c-Abl, we monitored c-Abl acti-vation with an anti-p-tyrosine antibody to tyrosine 245 of c-Abl inSH-SY5Y cells transfected with V5-parkin. TheDA neurotoxin 1-methyl-4-phenylpyridinium (MPP+) (250 μM) or DA (250 μM)activates c-Abl, as determined by the anti-p-tyrosine 245 c-Ablantibody (Fig. 4A). c-Abl activation byMPP+ or DA is attenuatedby the c-Abl-family kinase inhibitor STI-571 (Fig. 4A). Accom-panying the activation of c-Abl by MPP+ or DA is tyrosinephosphorylation of parkin, as detected with an anti-p-tyrosineantibody against immunoprecipitated parkin. The tyrosine phos-phorylation of parkin is completely attenuated by STI-571 (Fig.4A) or suppression of c-Abl expression with siRNA (Fig. S3A).MPP+ or DA also increases the level of the parkin substrateAIMP2, and this increase is blocked by STI-571 (Fig. 4A) orsuppression of c-Abl expression with siRNA (Fig. S3A). To de-termine whether activation of c-Abl by MPP+ or DA impairsparkin’s ubiquitination activity, parkin autoubiquitination wasmonitored in SH-SY5Y cells transfected with V5-parkin. BothMPP+ and DA reduce parkin autoubiquitination, which is at-tenuated by STI-571 (Fig. 4B) or knockdown of c-Abl expressionwith siRNA (Fig. S3B).The susceptibility of PC-12 cells to AIMP2-induced cell death
was used to determine whether tyrosine phosphorylation of par-kin by c-Abl regulates parkin’s protective function. A Tet-off–inducible PC-12 cell line that overexpresses AIMP2 in the absenceof doxycycline was developed to monitor AIMP2 toxicity (Fig.S3C). In the absence of doxycycline, AIMP2 is substantially in-duced, leading to a greater than twofold increase in cell death asmonitored by trypan blue exclusion (Fig. 4C). In noninduced cells,MPP+ treatment leads to a greater than threefold increase in celldeath that is significantly inhibited by STI-571 (Fig. 4C). AIMP2toxicity is unaffected by STI-571, and STI-571 treatment alone hasno substantial effect on cell viability in noninduced cells (Fig. 4C).MPP+ treatment enhances AIMP2 toxicity, and this enhancementis inhibited by STI-571 (Fig. 4C). Parkin overexpression attenu-ates AIMP2 toxicity, but MPP+ treatment prevents parkin’s
A
++ +KDGFP-c-Abl
Input
++
++
++KD
V5-parkinGFP-c-Abl
Input -GFP
B
DC
+ +++V5-parkinE
SH
3S
H2
DN
A-B
D
F1F2F3
lbA-cTW
c-AblWT
+ ++
Input
kDa
WT
Pull-down
IgGH
150
50
kDa kDa
kDa
PonceauS
++V5-parkin
IP
IgG
150Input
WB
+++ +GFP-c-Abl kDaInput
+
++
myc-parkinF
150kDa
+
R2
GST
GST-parkin
-c-Abl
WB: -c-Abl
-c-Abl
-parkin
-parki
n
IP:-GFP
-V5
-V5
-GFP
50
50
150
IP:-V5 -V5
-GFP
50
150
150
IP:-V5
-c-Abl
-c-Abl
-V5
TK Dom
ain
Act
inbi
ndin
g
50150
150
50
F1 F2 F3
465395R2
UBL R1 IBR
77UBL 465R1-IBR 403220
564TW 1
-GFP
-myc
-GFP
IP:-myc
50
150
150
WT UBLR1-I
BR
R2
GSTGST-pa
rkin
Fig. 1. Parkin interacts with c-Abl. (A) GST-parkin immobilized on gluta-thione-Sepharose beads pulls down recombinant c-Abl. Pull downs wereimmunoblotted with anti-c-Abl. (B) Mouse brain lysates were immunopre-cipitated with anti-parkin and anti-IgG, followed by immunoblot with anti-parkin or anti-c-Abl antibodies. (C) Lysates from SH-SY5Y cells transfectedwith GFP-c-Abl or a kinase-dead (KD) (lysine 290 arginine) version of c-Abl(GFP-c-Abl-KD) and V5-parkin were subjected to immunoprecipitation (IP)with anti-GFP, followed by anti-V5 immunoblotting (Middle) or with anti-GFP antibody (Bottom) to show an equivalent amount of immunoprecipi-tated c-Abl. (D) Lysates from SH-SY5Y cells transfected with V5-parkin andGFP-c-Abl and GFP-c-Abl-KD subjected to IP with anti-V5, followed by anti-GFP (Middle) or anti-V5 (Bottom) immunoblotting to show an equivalentamount of immunoprecipitated parkin. (E) Lysates from SH-SY5Y cellstransfected with V5-parkin and c-Abl domain constructs were subjected to IPwith anti-V5, followed by anti-c-Abl (Middle) or anti-V5 (Bottom) immuno-blotting. The arrow highlights the F3 domain of c-Abl. The deletion domainsof c-Abl used are shown at the bottom of the panel. (F) Lysates from SH-SY5Y cells transfected with GFP-c-Abl and myc-tagged fragments of parkinsubjected to IP with anti-myc antibodies, followed by anti-GFP (Middle) oranti-myc (Bottom) immunoblotting. A schematic representation of the dif-ferent parkin fragments used is shown. All experiments were repeated atleast three times and representative images of the immunoblots are shown.
16692 | www.pnas.org/cgi/doi/10.1073/pnas.1006083107 Ko et al.
Dow
nloa
ded
by g
uest
on
June
1, 2
020
protective effects against AIMP2 toxicity. Coadministration ofSTI-571 restores parkin’s protective effects against AIMP2 toxicityin the setting ofMPP+ treatment (Fig. 4C). Y143F parkin protectsagainst AIMP2 toxicity and MPP+ treatment is incapable of at-tenuating Y143F parkin’s protective properties. Similar resultswere observed with DA stress-induced toxicity (Fig. S3D). Theseresults taken together indicate that parkin’s protective function isimpaired by the PD-associated stressors MPP+ and DA throughc-Abl-mediated phosphorylation of parkin at tyrosine 143.To determine whether the protective effects of STI-571 require
parkin, shRNA was used to knock down parkin and the ability ofSTI-571 to protect against MPP+ toxicity was monitored. shRNAtoGFPwas used as a control. Knockdown of parkin leads to an up-regulation of AIMP2 (Fig. 4D). MPP+ treatment leads to tyrosinephosphorylation of c-Abl and parkin, which further increasesAIMP2 levels. STI-571, which prevents the tyrosine phosphoryla-
tion of parkin by c-Abl, attenuates the increase in AIMP2 levels. Inthe setting of parkin knockdown, c-Abl inhibition by STI-571 hasno effect on AIMP2 levels (Fig. 4D). MPP+ treatment of SH-SY5Y cells leads to a twofold increase in toxicity as assessed bytrypan blue exclusion, which is prevented by STI-571 (Fig. 4E) orsuppression of c-Abl expression with siRNA (Fig. S3E). Parkinknockdown attenuates the ability of STI-571 (Fig. 4E) or knock-down of c-Abl (Fig. S3E) to protect against MPP+ toxicity. Takentogether, these results indicate that STI-571 protective effectsrequire parkin.
c-Abl Deficiency Protects Against MPTP-Induced DA Neuron CellDeath In Vivo. Germ-line deletion of c-Abl leads to elevated peri-natal lethality (22, 23). Accordingly, conditional c-Abl KO mice(24) were used to test the role of c-Abl in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)model of PD. c-Ablflox/flox mice
++
++
++KD
+Y143F+
α-pTyr
WB
kDa
V5-parkinGFP-c-Abl
Input
*
*
In vitro kinase assay
His-parkin alone His-parkin + c-Abl
pI 5 7
37
37
kDa
+ +++ATP
α-parkin
α-c-Abl
α-pTyr
50
50
150
150
WB
:α-p
Tyr
WB
:α-H
is
α-GFP
α-GFP
α-pTyr
α-V5
kDa
50
50
150
150
IP: α-V5
c-Abl +++ −+++His-parkin −
+STI − − −
−−
MIVFVRFNSS HGFPVEVDSD TSIFQLKEVV AKRQGVPADQ LRVIFAGKEL RNDWTVQNCD LDQQSIVHIV QRPWRKGQEMNATGGDDPRN AAGGCEREPQ SLTRVDLSSS VLPGDSVGLA VILHTDSRKD SPPAGSPAGR SIYNSFYVYC KGPCQRVQPGKLRVQCSTCR QATLTLTQGP SCWDDVLIPN RMSGECQSPH CPGTSAEFFF KCGAHPTSDK ETPVALHLIA TNSRNITCITCTDVRSPVLV FQCNSRHVIC LDCFHLYCVT RLNDRQFVHD PQLGYSLPCV AGCPNSLIKE LHHFRILGEE QYNRYQQYGAEECVLQMGGV LCPRPGCGAG LLPEPDQRKV TCEGGNGLGC GFAFCRECKE AYHEGECSAV FEASGTTTQA YRVDERAAEQARWEAASKET IKKTTKPCPR CHVPVEKNGG CMHMKCPQPQ CRLEWCWNCG CEWNRVCMGD HWFDV
∆80Da
pY143
Y143
1050 1100 1150 1200 1300 1350 1400 1450
1050 1100 1150 1200 1250 1300 1350 1400 1450Non-phospho-peptide
Phospho-peptide
S I pY N S F Y V Y C Ky1y2y3y4y5y6y7y8y9y10
b10b9b8b7b6b5b4b3b2b1
b8 y8 b9
y9
b10
y10
y8
b9
y9
b10
y10
1250
A
B
C
Fig. 2. Parkin is tyrosine-phosphorylated by c-Abl. (A)Samples were separated by SDS/PAGE and immuno-blotted with anti-phosphotyrosine antibody to showactivated c-Abl (third panel) and phosphorylated par-kin (first panel) and with an anti-parkin antibody(second panel) and an anti-c-Abl antibody (fourthpanel) to show equal amounts of proteins were used.WB, Western blot. (B) Parkin is phosphorylated by c-Ablat tyrosine (Y) 143. Mass spectrometric analysis reveals96.5% sequence coverage of parkin, showing that alltyrosine residues were investigated for phosphoryla-tion status. Phosphorylated Y143 is indicated in red;other tyrosines are indicated in green (Top). Phos-phorylated parkin by c-Abl was separated by 2-DE fol-lowed by immunoblot (left panel on bottom). The redasterisks indicate tyrosine phosphorylation of parkin.Both nonphosphorylated and phosphorylated parkinwere subjected to LC-MS/MS to identify the phos-phorylation site (right panel on bottom). LC-MS/MSspectra of the nonphosphorylated peptide (SIYNSFY-VYCK) and the phosphorylated peptide (SIpYNSFY-VYCK) are compared, demonstrating that there is the80-Da shift for the Y143 ion containing the phosphatemoiety. The phosphorylated amino acid is preceded bya “p” and is highlighted in red. (C) V5-parkin WT or V5-parkin Y143F were coexpressed with GFP-c-Abl or GFP-c-Abl-KD in SH-SY5Y cells. Anti-V5 immunoprecipitateswere subjected to SDS/PAGE followed by immuno-blotting with an anti-phosphotyrosine antibody (thirdpanel), with anti-GFP (second panel), and with anti-V5to show an equivalent amount of immunoprecipitatedparkin (fourth panel). Inputs were immunoblotted withantibodies to GFP to show equivalent expression andloading. All immunoblot experiments were repeated atleast three times and representative images of theimmunoblots are shown.
Ko et al. PNAS | September 21, 2010 | vol. 107 | no. 38 | 16693
NEU
ROSC
IENCE
Dow
nloa
ded
by g
uest
on
June
1, 2
020
were bred to Nestin-Cre transgenic mice to generate conditionalKO of c-Abl in neurons (Fig. S4). PCR and immunoblot analysiswere used to determine the genotype of c-Abl KO (c-AblKO/flox/nestin Cre+) and WT mice (c-Ablflox/flox/nestin Cre−) (Fig. S4 CandD). For all studies, age-matchedWT and c-Abl KO littermateswere used and treated with either saline or MPTP (four i.p.injections, 20mg/kg, at 2-h intervals).MPTP activates c-Abl inWTmice as assessed 7 d after the lastMPTP injection as determined bythe anti-p-tyrosine 245 c-Abl antibody, leading to a fivefold in-crease in phospho-c-Abl (Fig. 5A). There is a twofold increase intyrosine phosphorylation of parkin, a threefold increase inAIMP2,and a moderate increase in FBP1 that accompanies the activationof c-Abl. In c-Abl KO mice, c-Abl is not detectable, as expected(Fig. 5A). Following MPTP intoxication, there is no increase intyrosine phosphorylation of parkin and no increase in AIMP2 andFBP1 levels (Fig. 5 A and B). DA cell death in response to MPTPtoxicity was monitored through stereologic measurement of tyro-sine hydroxylase (TH) immunoreactivity and Nissl staining. MPTPtreatment leads to a 60% reduction in TH-positive neurons anda similar loss of Nissl-positive neurons (Fig. 5 C and D). c-Abl KO
DC
BAV5-parkin
GFP-c-Abl
HA-UbkDa
α-HA
α-V5
α-pTyr
α-HA
α-GFP
50
50
150
50
50
+ + + + + +
+ + ++ + +
+ +WT Y143F
KA
KD− − KA
KD−
− −
−
FLAG-AIMP2
−
HA-Ub
V5-parkin
Input
kDaα-HA
α-V5
α-pTyr
α-HA
α-GFP
IP:
α -V
5
α-FLAG
IP:
α -F
LA
G
37
37
150
50
50
50
GFP-c-Abl+ + +
+
+
+ + ++ + +
+ +
+ + + +
+
+ + +
WT Y143F
KA
KD− − KA
KD−
−
−−
STIkDa
α-HA
α-V5
α-pTyr
α-HA
α-GFP
α-FLAG37
37
50
50
150
50
Input
IP:
α -V
5IP
:α -
FLA
G
FLAG-AIMP2
−
HA-Ub
V5-parkinGFP-c-Abl
+
+ + ++
+
+ +
+ + + + +
KA−
−
− KA−
−−−−
−
Input
IP:α
-V5
Input
+ ++V5-parkin++ +−
+
−−
− −
+
+
+−
+
+−
+
−−
+
+−
+
+−
HA-Ub
KA
KA
KD
KD
kDa
GFP-c-Abl
α-HA
α-V5
α-pTyr
α-HA
α-GFP
IP:α
-V5
50
50
150
50
50
Fig. 3. Tyrosine phosphorylation of parkin by c-Abl inhibits its E3 ubiquitinligase activity. (A) SH-SY5Y cells were transfected with V5-WT parkin, HA-ubiquitin (Ub), and kinase-active (KA) or kinase-dead (KD) GFP-c-Abl. Cells werelysed and immunoprecipitatedwith an anti-V5 antibody. Immunoblottingwithan anti-HA antibody shows autoubiquitination of parkin, an anti-V5 antibodyshows equivalent immunoprecipitated parkin, and an anti-phosphotyrosineshows parkin phosphorylation. Input samples were immunoblotted with anti-HA and anti-GFP antibodies to show equivalent expression. (B) SH-SY5Y cellswere transfectedwith either V5-WT-parkin or V5-Y143F-parkin alongwith HA-ubiquitin andGFP-c-Abl-KA orGFP-c-Abl-KD. Autoubiquitination of parkinwasvisualized through immunoblotting with an anti-HA antibody after immuno-precipitation with the V5 antibody. Immunoblotting with anti-V5 shows anequivalent amount of immunoprecipitated parkin and anti-phosphotyrosineshows parkin phosphorylation. Input samples were immunoblotted with anti-HA and anti-GFP antibodies to show equivalent expression. (C) SH-SY5Y cellswere transfected with FLAG-AIMP2, V5-WT-parkin, or V5-Y143F-parkin, HA-ubiquitin and GFP-c-Abl-KA, or GFP-c-Abl-KD. Immunoblotting of anti-FLAGimmunoprecipitates with anti-HA shows ubiquitination of AIMP2 and anti-FLAG shows equivalent levels of immunoprecipitated AIMP2. Immunoprecipi-tation with anti-V5 and immunoblotting with anti-phosphotyrosine antibodyshows parkin phosphorylation and with anti-V5 shows equivalent amounts ofimmunoprecipitated parkin. Inputs were immunoblotted with anti-HA andanti-GFP to show equivalent expression. (D) SH-SY5Y cells were transfectedwith FLAG-AIMP2, V5-WT-parkin, HA-ubiquitin, and GFP-c-Abl-KA in the pre-sence or absence of 10 μMSTI-571.Anti-FLAG immunoprecipitateswereprobedwith anti-HA to show the amount of ubiquitination of AIMP2 and anti-FLAG toshow an equivalent amount of immunoprecipitated AIMP2. Immunoprecipi-tation with anti-V5 and immunoblotting with anti-phosphotyrosine antibodyshows parkin phosphorylation and anti-V5 shows an equivalent amount ofimmunoprecipitated parkin. Inputs were immunoblotted with anti-HA andanti-GFP to showequivalent expression. All experiments were repeated at leastthree times and representative images of the immunoblots are shown.
V5-parkin ++
A
STI +++++
Input
DkDa
kDa
STIMPP+
DMSO
+Y143Fparkin
1
2
3
4
5
67
8
C
AIMP2 (Dox)
+ WT
-pTyr
-V5
-p-c-Abl
-c-Abl
-AIMP2
-actin
IP:
-V5
Input
50
50
150
150
37
37
% C
ell
De
ath
(+) ( ) ( ) ( )
MPP++STI
parkin-shRNA ++ +
+ ++ +MPP+
+ +STI
++ GFP-shRNA +
50
50
150
150
37
150
50
37
-pTyr
-p-c-Abl
-c-Abl
-AIMP2
-actin
IP:
-parkin-parkin
-c-Abl
-parkin
DM
SO
MPP
+
DA
MPP
+
DA
Input
B
E***
parkin-shRNA
% C
ell
De
ath
0
5
10
20
25
30
15
kDa
GFP-shRNA
IP:
-V5
-HA
-pTyr
-V5
-HA
-p-c-Abl
-c-Abl
50
50
50
50
150
150
STIMPP+
DMSO
MPP++STI
++++++V5-parkinHA-Ub +++++
+STI +
DM
SO
MPP
+
DA
MPP
+
DA
30
25
20
15
10
5
0
Fig. 4. Parkin is tyrosine-phosphorylated after dopaminergic stress. (A) SH-SY5Y cells were transiently transfected with V5-parkin and treated with MPP+
(250 μM) or dopamine (DA) (250 μM) for 24 h in the presence or absence of STI-571 (10 μM). Cell lysates were immunoprecipitated with an anti-V5 antibodyand immunoblotted with anti-phosphotyrosine to show phosphorylated par-kin, and anti-parkin to show equivalent IP. Inputs were immunoblotted withanti-phospho-c-Abl to show activated c-Abl, and with anti-c-Abl, anti-AIMP2,or anti-actin antibodies. Data were repeated with similar results. (B) SH-SY5Ycells were transiently transfectedwith V5-parkin andHA-ubiquitin and treatedwith MPP+ (250 μM) or DA (250 μM) for 24 h in the presence or absence of STI-571 (10 μM). Cell lysates were immunoprecipitated with an anti-V5 antibodyand immunoblotted with anti-HA to monitor ubiquitination of parkin, anti-phosphotyrosine to show phosphorylated parkin, and anti-V5 to show equiv-alent levels of immunoprecipitated parkin. Inputs were immunoblotted withanti-phospho-c-Abl to show activated c-Abl, and with anti-c-Abl or anti-HA toshow equivalent expression. Data were repeated with similar results. (C) PC12-AIMP2-inducible cells were transfected with V5-WT-parkin and V5-Y143F-parkin in the presence or absence of MPP+ (200 μM) for 24 h. Some sampleswere incubated with 10 μM STI-571. Cell death was assessed by trypan blueexclusion. Both STI-571 and transfection with phosphorylation-resistantY143F-parkin protect against AIMP2-induced cell death and after MPP+
treatment. Doxycycline (Dox) (+) prevents the induction of AIMP2. Dox with-drawal (−) induces the expression of AIMP2 (Fig. S3). Data are mean ± SEM forthree separate experiments performed in duplicate. Statistical significancewasdetermined by one-way ANOVA and Tukey’s post hoc test. *P < 0.05, **P <0.01, ***P < 0.001; ***1: DMSO versus MPP+–Dox (+); **2: MPP+ versus STI+MPP+–Dox (+); ***3: DMSO–Dox (+) versus AIMP2–Dox (−); ***4: DMSO/AIMP2–Dox (−) versus AIMP2–Dox (−) + WT-parkin; *5: DMSO/AIMP2-Dox (−)versus MPP+/AIMP2–Dox (−) + WT-parkin; ***6: MPP++/AIMP2–Dox (−) + WT-parkin versus STI + MPP+/AIMP2–Dox (−) + WT-parkin; **7: DMSO/AIMP2–Dox(−) versus AIMP2–Dox (−) + Y143F parkin; ***8: MPP+/AIMP2–Dox (−) versusMPP+/AIMP2–Dox (−) + Y143F parkin. (D and E) The protective effect of STI-571requires parkin. (D) SH-SY5Y cells were transfected with either parkin-shRNAor GFP-shRNA in the presence or absence of MPP+ (250 μM) for 24 h. Somesamples were incubated with 10 μM STI-571. Cell lysates were subjected toimmunoprecipitation with an anti-parkin antibody. The parkin immunopre-cipitates were immunoblotted with anti-phosphotyrosine to monitor tyrosinephosphorylation of parkin and anti-parkin to monitor the levels of immuno-precipitated parkin. Total lysates were immunoblotted with anti-phospho-c-Abl to show tyrosine-phosphorylated c-Abl, anti-actin as a loading control, andwith an AIMP2 antibody to monitor the levels of AIMP2 and an anti-parkinantibody to confirm the parkin knockdown by parkin-shRNA. Data were re-peated with similar results. (E) Trypan blue cell death assessments in SH-SY5Yneuroblastoma cells that were transiently transfected with parkin-shRNA orGFP-shRNA and treated with MPP+ (200 μM). Some samples were incubatedwith 10 μM STI-571 as indicated. Data are mean ± SEM for three separateexperiments performed in duplicate. Statistical significancewas determined byone-way ANOVA and Tukey’s post hoc test. ***P < 0.001.
16694 | www.pnas.org/cgi/doi/10.1073/pnas.1006083107 Ko et al.
Dow
nloa
ded
by g
uest
on
June
1, 2
020
significantly prevents the loss of TH-positive and Nissl-positiveneurons followingMPTP intoxication (Fig. 5C andD). Equivalentlevels of MPP+, the active metabolite of MPTP, are observed inWT and c-Abl KO animals, indicating that the protective effectobserved in c-Abl KO mice is not due to altered bioavailability ofMPP+ (Fig. 5E). Striatal TH-positive fiber density was assessed inWT and c-Abl KO mice after MPTP administration. MPTP leadsto a 50% reduction in TH-positive fiber density in the striatum ofWT mice. KO of c-Abl significantly protects against the loss ofstriatal TH-positive fibers (Fig. S5A andB). Taken together, thesedata indicate that c-Abl activation plays a role in DA cell lossfollowing MPTP intoxication.
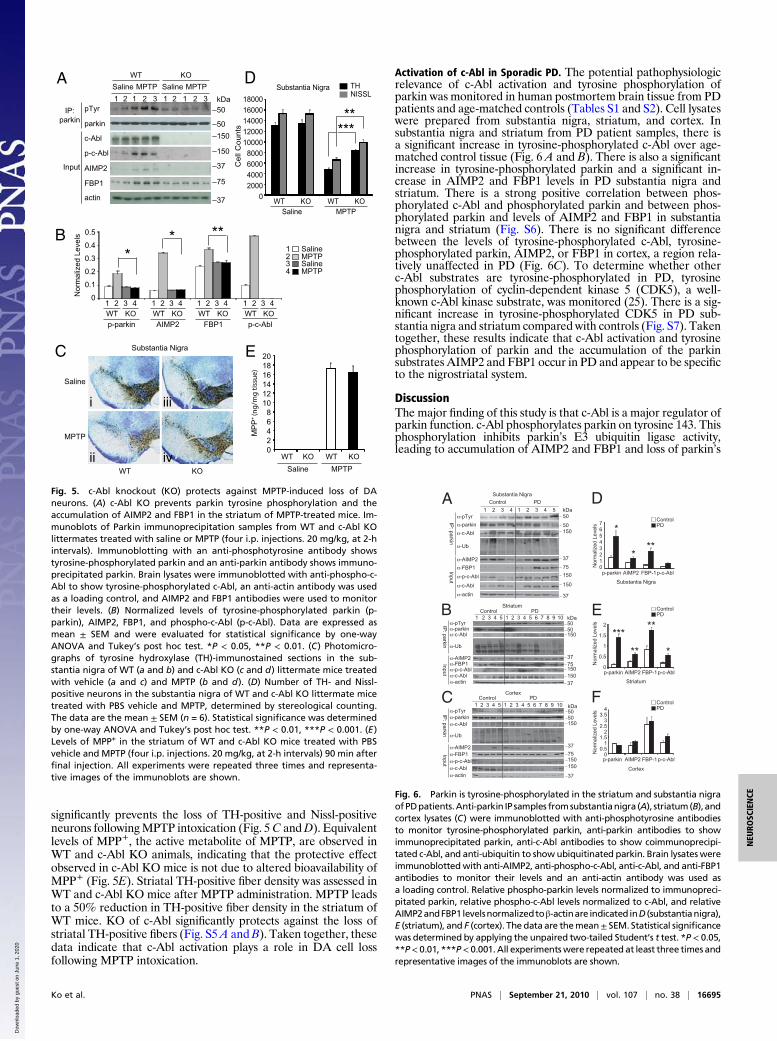
Activation of c-Abl in Sporadic PD. The potential pathophysiologicrelevance of c-Abl activation and tyrosine phosphorylation ofparkin was monitored in human postmortem brain tissue from PDpatients and age-matched controls (Tables S1 and S2). Cell lysateswere prepared from substantia nigra, striatum, and cortex. Insubstantia nigra and striatum from PD patient samples, there isa significant increase in tyrosine-phosphorylated c-Abl over age-matched control tissue (Fig. 6 A and B). There is also a significantincrease in tyrosine-phosphorylated parkin and a significant in-crease in AIMP2 and FBP1 levels in PD substantia nigra andstriatum. There is a strong positive correlation between phos-phorylated c-Abl and phosphorylated parkin and between phos-phorylated parkin and levels of AIMP2 and FBP1 in substantianigra and striatum (Fig. S6). There is no significant differencebetween the levels of tyrosine-phosphorylated c-Abl, tyrosine-phosphorylated parkin, AIMP2, or FBP1 in cortex, a region rela-tively unaffected in PD (Fig. 6C). To determine whether otherc-Abl substrates are tyrosine-phosphorylated in PD, tyrosinephosphorylation of cyclin-dependent kinase 5 (CDK5), a well-known c-Abl kinase substrate, was monitored (25). There is a sig-nificant increase in tyrosine-phosphorylated CDK5 in PD sub-stantia nigra and striatum compared with controls (Fig. S7). Takentogether, these results indicate that c-Abl activation and tyrosinephosphorylation of parkin and the accumulation of the parkinsubstrates AIMP2 and FBP1 occur in PD and appear to be specificto the nigrostriatal system.
DiscussionThe major finding of this study is that c-Abl is a major regulator ofparkin function. c-Abl phosphorylates parkin on tyrosine 143. Thisphosphorylation inhibits parkin’s E3 ubiquitin ligase activity,leading to accumulation of AIMP2 and FBP1 and loss of parkin’s
Substantia Nigra
Saline
MPTP
C
WT
Saline MPTP
pTyr
parkin
c-Abl
p-c-Abl
AIMP2
FBP1
actin
IP:
parkin
Input
1 2 1 2
B
3
KO
1 2 1 2 3 kDa
50
75
37
50
A
i
ii
iii
iv
* MPTPSalineMPTP
Saline1234
0
0.1
0.2
0.3
0.4
0.5sle
ve
Ld
ezil
amr
oN
* **
p-parki n AIMP 2 FBP 1 p-c-Abl
3 4 3 4
W T K O W T K O W T K O WT
E
0
2
8
14
20
4
6
10
12
16
18
PP
M+
)eussitg
m /gn(
WT KO WT KO
Saline MPTP
THNISSL
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
**
Substantia NigraD
stn
uo
Cll
eC
***
W T K O W T KO
Salin e MPTP
WT KO
150
150
37
1 2 1 2 3 41 2 3 41 2
KO
Saline MPTP
Fig. 5. c-Abl knockout (KO) protects against MPTP-induced loss of DAneurons. (A) c-Abl KO prevents parkin tyrosine phosphorylation and theaccumulation of AIMP2 and FBP1 in the striatum of MPTP-treated mice. Im-munoblots of Parkin immunoprecipitation samples from WT and c-Abl KOlittermates treated with saline or MPTP (four i.p. injections. 20 mg/kg, at 2-hintervals). Immunoblotting with an anti-phosphotyrosine antibody showstyrosine-phosphorylated parkin and an anti-parkin antibody shows immuno-precipitated parkin. Brain lysates were immunoblotted with anti-phospho-c-Abl to show tyrosine-phosphorylated c-Abl, an anti-actin antibody was usedas a loading control, and AIMP2 and FBP1 antibodies were used to monitortheir levels. (B) Normalized levels of tyrosine-phosphorylated parkin (p-parkin), AIMP2, FBP1, and phospho-c-Abl (p-c-Abl). Data are expressed asmean ± SEM and were evaluated for statistical significance by one-wayANOVA and Tukey’s post hoc test. *P < 0.05, **P < 0.01. (C) Photomicro-graphs of tyrosine hydroxylase (TH)-immunostained sections in the sub-stantia nigra of WT (a and b) and c-Abl KO (c and d) littermate mice treatedwith vehicle (a and c) and MPTP (b and d). (D) Number of TH- and Nissl-positive neurons in the substantia nigra of WT and c-Abl KO littermate micetreated with PBS vehicle and MPTP, determined by stereological counting.The data are the mean ± SEM (n = 6). Statistical significance was determinedby one-way ANOVA and Tukey’s post hoc test. **P < 0.01, ***P < 0.001. (E)Levels of MPP+ in the striatum of WT and c-Abl KO mice treated with PBSvehicle and MPTP (four i.p. injections. 20 mg/kg, at 2-h intervals) 90 min afterfinal injection. All experiments were repeated three times and representa-tive images of the immunoblots are shown.
Control
Control
Control
PD
PD
Striatum
5050150
37
75150
150
37
kDa
0.5
1
1.5
2
***
**
**
*
Striatum
No
rma
lize
d L
eve
ls-pTyr-parkin
-AIMP2
-p-c-Abl
-c-Abl
-FBP1
-actin-c-Abl
Inp
ut
IP: p
ark
in
DA
E
F
-Ub
-pTyr
-parkin
-Ub
-AIMP2
-p-c-Abl
-c-Abl
-FBP1
-actin
-c-Abl
Inp
ut
IP: p
ark
in
C5050150
37
75150
150
37
kDa
00.5
11.5
22.5
33.5
4
No
rma
lize
d L
eve
ls
Cortex
Cortex
-pTyr
-parkin
-Ub
-AIMP2
-p-c-Abl
-c-Abl
-FBP1
-actin
-c-Abl
Inp
ut
IP: p
ark
in
B
50
50150
37
75
150
150
37
kDa
p-parkin01234567
No
rma
lize
d L
eve
ls
Substantia Nigra
Substantia Nigra
PDControl
PDControl
PDControl
*
***
p-c-AblFBP-1AIMP2
p-parkin p-c-AblFBP-1AIMP2
p-parkin p-c-AblFBP-1AIMP2
PD
1 2 3 4 1 2 3 4 5
1 2 3 4 1 2 3 45 5 6 7 8 9 10
1 2 3 4 1 2 3 45 5 6 7 8 9 10
0
Fig. 6. Parkin is tyrosine-phosphorylated in the striatum and substantia nigraofPDpatients.Anti-parkin IP samples fromsubstantianigra (A), striatum(B), andcortex lysates (C) were immunoblotted with anti-phosphotyrosine antibodiesto monitor tyrosine-phosphorylated parkin, anti-parkin antibodies to showimmunoprecipitated parkin, anti-c-Abl antibodies to show coimmunoprecipi-tated c-Abl, and anti-ubiquitin to showubiquitinated parkin. Brain lysateswereimmunoblottedwith anti-AIMP2, anti-phospho-c-Abl, anti-c-Abl, and anti-FBP1antibodies to monitor their levels and an anti-actin antibody was used asa loading control. Relative phospho-parkin levels normalized to immunopreci-pitated parkin, relative phospho-c-Abl levels normalized to c-Abl, and relativeAIMP2andFBP1levelsnormalizedtoβ-actinare indicated inD (substantianigra),E (striatum), and F (cortex). The data are themean± SEM. Statistical significancewas determined by applying the unpaired two-tailed Student’s t test. *P < 0.05,**P< 0.01, ***P< 0.001.All experimentswere repeated at least three times andrepresentative images of the immunoblots are shown.
Ko et al. PNAS | September 21, 2010 | vol. 107 | no. 38 | 16695
NEU
ROSC
IENCE
Dow
nloa
ded
by g
uest
on
June
1, 2
020

cytoprotective function and cell death. The c-Abl-family kinaseinhibitor STI-571 maintains parkin in a catalytically active andneuroprotective state by preventing the tyrosine phosphorylationof parkin. shRNA knockdown of parkin attenuates STI-571 pro-tection, indicating that the protective effect of STI-571 is parkin-dependent. c-Abl is active in sporadic PD substantia nigra andstriatum, correlating with the increase in phosphorylated parkinand accumulation of AIMP2 and FBP1. These data implicatec-Abl as an important regulator of parkin’s E3 ligase activity andcytoprotective function and identify c-Abl as a potentially impor-tant therapeutic target for the treatment of PD.Our findings provide further support to the idea that parkin is
inactivated in sporadic PD. Previous studies suggest that parkin isinactivated in sporadic PD through S-nitrosylation (15, 26), oxi-dative stress (27), and dopamine conjugation (18). Here we de-scribe another posttranslational modification of parkin that ispresent in sporadic PD. Thus, parkin inactivation is emerging asa common event in sporadic PD. Maintaining parkin functionthrough inhibition of S-nitrosylation, dopamine conjugation, orphosphorylation by c-Abl is an attractive therapeutic target.Parkin inactivation by c-Abl may be a dominant pathway, becauseinhibition of c-Abl with STI-571 or knockdown and knockout ofc-Abl prevents the inactivation of parkin by MPTP and DA.The mechanisms underlying c-Abl activation in sporadic PD
await further characterization. One of the possible mechanisms,however, can be partially explained by oxidative stress, which isprevalent in sporadic PD brain, because various oxidative stres-sors activate c-Abl (28). In addition, as shown here, dopami-nergic stress also increases c-Abl activity. In sporadic PD, c-Ablactivation and parkin inactivation and accumulation of parkin
substrates appear to be limited to the striatum and substantianigra, regions of the brain that are primarily affected in PD, asthese changes are not observed in the cerebral cortex of PDpatients. Thus, activation of c-Abl, tyrosine phosphorylation ofparkin, and accumulation of parkin substrates may contribute tothe pathogenesis of sporadic PD.c-Abl inhibitors are widely used in the treatment of chronic
myeloid leukemia. Because the c-Abl inhibitor maintains parkin ina catalytically active neuroprotective state and KO of c-Abl pro-tects against dopaminergic cell loss following MPTP intoxication,the testing and use of brain-permeable c-Abl inhibitors as a neu-roprotective treatment for PD are reasonable and potentiallyexciting objectives.
Materials and MethodsAll methods used in this article are routinely used in our laboratories and arethus referenced (7, 8, 24, 29) and are described in SI Materials and Methods.For protein interactions, standard coimmunoprecipitation experiments andubiquitination assays were used as previously described (7, 8). For LC-MS/MS,excised 2-DE spots were subjected to a modified in-gel trypsin digestionprocedure. Protein identity was determined by the software programSequest (Thermo Finnigan). Conditional c-Abl knockouts were used, andMPTP intoxication studies and assessments in mice were performed as pre-viously described (24, 29).
ACKNOWLEDGMENTS. This work was supported by NIH/NINDS GrantsNS38377, NS048206, NS051764, and NS39475, and the Bachmann StraussDystonia and Parkinson’s Disease Foundation. Y.L. is supported by the Sam-sung Scholarship Foundation. T.M.D. is the Leonard and Madlyn AbramsonProfessor in Neurodegenerative Diseases.
1. Savitt JM, Dawson VL, Dawson TM (2006) Diagnosis and treatment of Parkinsondisease: Molecules to medicine. J Clin Invest 116:1744–1754.
2. Gasser T (2009) Molecular pathogenesis of Parkinson disease: Insights from geneticstudies. Expert Rev Mol Med 11:e22.
3. Dawson TM, Dawson VL (2010) The role of parkin in familial and sporadic Parkinson’sdisease. Mov Disord 25 (Suppl 1):S32–S39.
4. Shimura H, et al. (2000) Familial Parkinson disease gene product, parkin, isa ubiquitin-protein ligase. Nat Genet 25:302–305.
5. Zhang Y, et al. (2000) Parkin functions as an E2-dependent ubiquitin-protein ligaseand promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1.Proc Natl Acad Sci USA 97:13354–13359.
6. Fallon L, et al. (2006) A regulated interaction with the UIM protein Eps15 implicatesparkin in EGF receptor trafficking and PI(3)K-Akt signalling. Nat Cell Biol 8:834–842.
7. Ko HS, Kim SW, Sriram SR, Dawson VL, Dawson TM (2006) Identification of farupstream element-binding protein-1 as an authentic Parkin substrate. J Biol Chem281:16193–16196.
8. Ko HS, et al. (2005) Accumulation of the authentic parkin substrate aminoacyl-tRNAsynthetase cofactor, p38/JTV-1, leads to catecholaminergic cell death. J Neurosci 25:7968–7978.
9. Lim KL, et al. (2005) Parkin mediates nonclassical, proteasomal-independentubiquitination of synphilin-1: Implications for Lewy body formation. J Neurosci 25:2002–2009.
10. Hantschel O, Superti-Furga G (2004) Regulation of the c-Abl and Bcr-Abl tyrosinekinases. Nat Rev Mol Cell Biol 5:33–44.
11. Reddy EP, Smith MJ, Srinivasan A (1983) Nucleotide sequence of Abelson murineleukemia virus genome: Structural similarity of its transforming gene product toother onc gene products with tyrosine-specific kinase activity. Proc Natl Acad Sci USA80:3623–3627.
12. Moresco EM, Koleske AJ (2003) Regulation of neuronal morphogenesis and synapticfunction by Abl family kinases. Curr Opin Neurobiol 13:535–544.
13. Alvarez AR, Sandoval PC, Leal NR, Castro PU, Kosik KS (2004) Activation of theneuronal c-Abl tyrosine kinase by amyloid-β-peptide and reactive oxygen species.Neurobiol Dis 17:326–336.
14. Alvarez AR, et al. (2008) Imatinib therapy blocks cerebellar apoptosis and improvesneurological symptoms in a mouse model of Niemann-Pick type C disease. FASEB J 22:3617–3627.
15. Chung KK, et al. (2004) S-nitrosylation of parkin regulates ubiquitination andcompromises parkin’s protective function. Science 304:1328–1331.
16. Avraham E, Rott R, Liani E, Szargel R, Engelender S (2007) Phosphorylation of Parkinby the cyclin-dependent kinase 5 at the linker region modulates its ubiquitin-ligaseactivity and aggregation. J Biol Chem 282:12842–12850.
17. Rubio de la Torre E, et al. (2009) Combined kinase inhibition modulates parkininactivation. Hum Mol Genet 18:809–823.
18. LaVoie MJ, Ostaszewski BL, Weihofen A, Schlossmacher MG, Selkoe DJ (2005)Dopamine covalently modifies and functionally inactivates parkin. Nat Med 11:1214–1221.
19. Kawai H, Nie L, Yuan ZM (2002) Inactivation of NF-κB-dependent cell survival, a novelmechanism for the proapoptotic function of c-Abl. Mol Cell Biol 22:6079–6088.
20. Corti O, et al. (2003) The p38 subunit of the aminoacyl-tRNA synthetase complex isa Parkin substrate: Linking protein biosynthesis and neurodegeneration. Hum MolGenet 12:1427–1437.
21. Kharbanda S, et al. (1995) Activation of the c-Abl tyrosine kinase in the stress responseto DNA-damaging agents. Nature 376:785–788.
22. Schwartzberg PL, et al. (1991) Mice homozygous for the ablm1 mutation show poorviability and depletion of selected B and T cell populations. Cell 65:1165–1175.
23. Tybulewicz VL, Crawford CE, Jackson PK, Bronson RT, Mulligan RC (1991) Neonatallethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell 65:1153–1163.
24. Moresco EM, Donaldson S, Williamson A, Koleske AJ (2005) Integrin-mediateddendrite branch maintenance requires Abelson (Abl) family kinases. J Neurosci 25:6105–6118.
25. Zukerberg LR, et al. (2000) Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosinephosphorylation, kinase upregulation, and neurite outgrowth. Neuron 26:633–646.
26. Yao D, et al. (2004) Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc Natl Acad Sci USA101:10810–10814.
27. Wang C, et al. (2005) Stress-induced alterations in parkin solubility promote parkinaggregation and compromise parkin’s protective function. Hum Mol Genet 14:3885–3897.
28. Sun X, et al. (2000) Activation of the cytoplasmic c-Abl tyrosine kinase by reactiveoxygen species. J Biol Chem 275:17237–17240.
29. Andres-Mateos E, et al. (2007) DJ-1 gene deletion reveals that DJ-1 is an atypicalperoxiredoxin-like peroxidase. Proc Natl Acad Sci USA 104:14807–14812.
16696 | www.pnas.org/cgi/doi/10.1073/pnas.1006083107 Ko et al.
Dow
nloa
ded
by g
uest
on
June
1, 2
020