ncx3 regulates mitochondrial ca handling through the...
TRANSCRIPT

Journ
alof
Cell
Scie
nce
NCX3 regulates mitochondrial Ca2+ handling throughthe AKAP121-anchored signaling complex andprevents hypoxia-induced neuronal death
Antonella Scorziello1,*, Claudia Savoia1,*, Maria Jose Sisalli1,*, Annagrazia Adornetto1, Agnese Secondo1,Francesca Boscia1, Alba Esposito1, Elena V. Polishchuk2, Roman S. Polishchuk2, Pasquale Molinaro1,Annalisa Carlucci3, Luca Lignitto3, Gianfranco Di Renzo1, Antonio Feliciello3 and Lucio Annunziato1,4,`
1Division of Pharmacology, Department of Neuroscience, Reproductive and Dentistry Sciences, School of Medicine, Federico II University of Naples-National Institute of Neuroscience, Italy2Telethon Institute of Genetics and Medicine, Institute of Protein Biochemistry, Naples, Italy3Department of Molecular Medicine and Medical Biotechnology, Federico II University of Naples, Italy4Fondazione IRCCS SDN, Naples, Italy
*These authors contributed equally to this work`Author for correspondence ([email protected])
Accepted 9 September 2013Journal of Cell Science 126, 5566–5577� 2013. Published by The Company of Biologists Ltddoi: 10.1242/jcs.129668
SummaryThe mitochondrial influx and efflux of Ca2+ play a relevant role in cytosolic and mitochondrial Ca2+ homeostasis, and contribute to theregulation of mitochondrial functions in neurons. The mitochondrial Na+/Ca2+ exchanger, which was first postulated in 1974, has beenprimarily investigated only from a functional point of view, and its identity and localization in the mitochondria have been a matter ofdebate over the past three decades. Recently, a Li+-dependent Na+/Ca2+ exchanger extruding Ca2+ from the matrix has been found in the
inner mitochondrial membrane of neuronal cells. However, evidence has been provided that the outer membrane is impermeable to Ca2+
efflux into the cytoplasm. In this study, we demonstrate for the first time that the nuclear-encoded NCX3 isoform (1) is located on the outermitochondrial membrane (OMM) of neurons; (2) colocalizes and immunoprecipitates with AKAP121 (also known as AKAP1), a member
of the protein kinase A anchoring proteins (AKAPs) present on the outer membrane; (3) extrudes Ca2+ from mitochondria throughAKAP121 interaction in a PKA-mediated manner, both under normoxia and hypoxia; and (4) improves cell survival when it works in theCa2+ efflux mode at the level of the OMM. Collectively, these results suggest that, in neurons, NCX3 regulates mitochondrial Ca2+ handling
from the OMM through an AKAP121-anchored signaling complex, thus promoting cell survival during hypoxia.
Key words: Mitochondria, Ca2+ flux, NCX3, AKAP121, AKAP1
IntroductionMitochondria play a pivotal role in neuronal Ca2+ signaling by
sensing and shaping cytosolic Ca2+ transients. In turn, intra-
mitochondrial Ca2+ also has a central role in neuronal
metabolism, regulating the activity of matrix dehydrogenases
involved in ATP production (Denton, 2009). By contrast,
mitochondrial Ca2+ (Ca2+m) overload causes the opening of
the permeability transition pore (PTP), thereby influencing
mitochondrial control of programmed neuronal death (Denton
and McCormack, 1980; Gunter et al., 2000; Kroemer, 1997).
Moreover, Ca2+m overload, occurring during ischemia, leads to
mitochondrial dysfunction and ultimately neuronal death
(Starkov et al., 2004). Thus, the mitochondrial Ca2+ influx and
efflux pathways might have a relevant role in maintaining
cytosolic and mitochondrial Ca2+ homeostasis. The existence of a
mitochondrial Na+/Ca2+ exchanger, first postulated in 1974
(Carafoli et al., 1974), has been primarily investigated from a
functional point of view. Its identity and localization have been a
matter of debate for the past three decades. Recently, among the
Ca2+-efflux mechanisms, NCLX, a member of the CCX
superfamily, has been found on the cristae of the inner
mitochondrial membrane (IMM), where it mediates Na+- and
Li+-dependent Ca2+ efflux from the mitochondrial matrix (Palty
et al., 2010). By contrast, mounting evidence has suggested that
the outer mitochondrial membrane (OMM) is not passively
permeable to Ca2+ fluxes into the cytoplasm, but rather plays a
key role in controlling mitochondrial function and Ca2+ cycling
(Bathori et al., 2006; Jonas et al., 1999; Szabadkai and Duchen,
2008). More specifically, the OMM serves as a permeability
barrier not only to Ca2+ influx but also to Ca2+ efflux (Baines
et al., 2007; Crompton et al., 2002; Moran et al., 1992). This
evidence suggests that an additional Na+-dependent Ca2+
extruding mechanism operates between the intermembrane
space and the cytoplasmic compartment.
In the present study, we explored the possibility that a member
of the NCX family, coded by nuclear DNA, as is the case for the
majority of mitochondrial proteins, might be present on
mitochondria (Stojanovski et al., 2003). Moreover, we also
investigated whether an interaction between NCX and other
OMM proteins could constitute a possible mechanism that is
involved in the regulation of mitochondrial Ca2+ efflux and
neuronal survival. We focused particularly on AKAP121 (also
5566 Research Article

Journ
alof
Cell
Scie
nce
known as AKAP1), a member of the protein kinase A (PKA)
anchoring protein (AKAP) family that controls mitochondrial
metabolism and neuronal survival (Carlucci et al., 2008b) by
interacting with several mitochondrial proteins and biochemical
signaling pathways. Briefly, AKAP121, as well as its human
homolog AKAP149, besides having an RNA-binding motif
(KH domain) that binds several mRNAs of nuclear-encoded
mitochondrial proteins, also assembles an mRNA and PKA
complex on mitochondria. This multicomponent system, which
represents an important signal crossroad regulating translation
and mitochondrial protein import, also encompasses cAMP
phosphodiesterase (PDE4A), Ser/Thr phosphatase (PP1), and an
Src-associated tyrosine phosphatase (PTPD1).
Here, we demonstrate for the first time that the nuclear
encoded NCX3 isoform (1) is located on the outer mitochondrial
membrane of neurons; (2) colocalizes and immunoprecipitates
with AKAP121; (3) extrudes Ca2+ from mitochondria through
AKAP121 interaction in a PKA-mediated manner, both under
normoxia and hypoxia; and (4) improves cell survival when it
works in the Ca2+-efflux mode at the level of the outer
mitochondrial membrane.
ResultsBiochemical identification and localization of NCX3
isoform on the OMM in cortical neurons and BHK cells
We found that the NCX3 protein, apart from being present at the
plasma membrane (PM), as previously described (Secondo et al.,
2007), was also localized on the mitochondrial membranes. By
contrast, NCX1 and NCX2 isoforms were not detected (Fig. 1A).
Experiments performed with whole mitochondria or with
mitochondrial fractions of baby hamster kidney (BHK) NCX3-
transfected cells revealed that NCX3 was located on the OMM
Fig. 1. Biochemical identification and
localization of NCX3 isoform on the
OMM in BHK transfected cells.
(A) Expression of NCX3, NCX1 and NCX2
within membrane (Membr), cytosolic (Cyt)
and mitochondrial (Mito) fractions obtained
from BHK-Wt and BHK cells stably
transfected with NCX1, NCX2 or NCX3
cells. Membr. refers to the precipitate
obtained after the first centrifugation
performed on BHK cell lysates in a buffer
containing mannitol, EDTA and Hepes.
This fraction contains membranes but does
not contain the intracellular organelles
including mitochondria. (B) Localization of
NCX3 isoform on the OMM. Whole
mitochondria of BHK cells stably
transfected with NCX3 were digested with
trypsin (20 mg/ml) or Triton X-100 (0,2%)
or treated with digitonin (500 mM, 10 or
20 minutes). (C) Distribution of NCX3 in
mitochondria (arrows), PM and ER and
quantification of NCX3 immunogold
density in BHK-Wt and BHK cells stably
transfected with NCX3 along the
mitochondria. Scale bars: 200 nm.
(D) Absence of ER, Golgi, lysosomes and
MAM contamination in the mitochondrial
fraction in BHK cells stably transfected
with NCX3.
Mitochondrial Ca2+ and cell death 5567

Journ
alof
Cell
Scie
nce
(Fig. 1B,C). Indeed, when the OMM was removed with trypsin
(20 mg/ml) (Sardanelli et al., 2006), NCX3 immunoreactivity
disappeared. Importantly, immunoreactivity for voltage-
dependent anion channel protein 1 (VDAC) and AKAP121,
two proteins specific to the outer mitochondrial membrane, were
undetectable after treatment of fractionated mitochondria with
trypsin. Conversely, Mn-SOD immunoreactivity, a marker of the
mitochondrial matrix (MM), and COX-IV, a marker of the IMM,
remained unaffected (Fig. 1B). Similar results were obtained
by treating mitochondria with digitonin (500 mM) (Fig. 1B).
Interestingly, confocal microscopy showed that some NCX3
immunoreactivity in the cell overlapped with Mito-Tracker- or
Mito-RFP-stained mitochondria, along neurites of cortical
neurons (Fig. 2A, panels e, f and g). Furthermore, double
immunofluorescence experiments with NCX3 and Mito-RFP
indicated that, whereas Mito-RFP stained the entire
mitochondria, the NCX3 signal localized in punctuate spots,
which mostly aligned along mitochondria edges (arrows, Fig. 2A,
panel f). This punctuate distribution pattern, which we observed
with both the anti-NCX3 and the anti-FLAG antibody in cells
transfected with FLAG-tagged NCX3 (NCX3F), is similar to that
described for other proteins located on the mitochondria
(Margineantu et al., 2002). The calculated percentage of NCX3
immunoreactivity in mitochondria corresponded to 25% of
the total NCX3 signal (Fig. 2A, panel h). Immunoelectron
microscopy, performed both in NCX3-transfected BHK cells
(Fig. 1C) and in neurons (Fig. 2B) demonstrated that NCX3
labeling was detected on the PM, endoplasmic reticulum (ER)
and mitochondria (Fig. 1C), whereas it was undetectable in other
compartments such as Golgi stacks and endosomes (Table 1).
Fig. 2. Mitochondrial distribution of
NCX3 in cortical neurons.
(A) Distribution and quantification of the
NCX3 immunosignal. A representative
cortical neuron double-labeled with NCX3
(green) and MitoTracker (a–e) or Mito-
RFP (f) (red). (e) Colocalization of the
NCX3 punctate staining (green) with
mitochondria (red) along a single neurite.
(f) Higher magnification image depicting
the punctate NCX3 distribution on
mitochondria (arrows). (g) Spatial profile
plot of pixel intensity of NCX3 (green) and
MitoTracker (red) along the line indicated
in panel e, demonstrating the spatial
overlap of individual puncta positive for
NCX3 and MitoTracker. (h) Quantification
of the amount of colocalization of NCX3
immunosignal at the mitochondrial level
measured as the percentage of the total
NCX3 immunosignal. Scale bars: 20 mm
(a–d), 5 mm (e), 2.5 mm (f).
(B) Distribution of NCX3 in mitochondria
(arrows), PM, and ER, and quantification
of the endogenous NCX3 immunogold
density in Ncx3+/+ and Ncx32/2 neurons
along the mitochondria. Arrows in insets
show NCX3 mainly on OMM. Scale bar:
200 nm. (C) Localization of NCX3 within
membrane (Membr), cytosolic (Cyto) and
mitochondrial (Mito) fractions obtained
from total mouse brain of Ncx3+/+ and
Ncx32/2 mice. Membr. refers to the
precipitate obtained after the first
centrifugation, performed on tissue lysates
in a buffer containing mannitol, EDTA and
Hepes. This fraction contains membranes
but does not contain the intracellular
organelles including mitochondria.
Table 1. Quantification of the density of gold particles (in arbitrary units) associated with NCX3 in different
subcellular compartments
Cells Mitochondria Endoplasmic reticulum Plasma membrane Golgi complex Endosome-lysosome
Neurons 0.1960.02 0.2460.06 0.2360.08 0.0560.03 0.0460.02NCX3F-transfected
BHK cells0.2560.03 0.3260.04 0.9860.04 0.0660.03 0.0560.04
Journal of Cell Science 126 (24)5568
Journ
alof
Cell
Scie
nce
These findings were supported by immunocytochemistry
experiments performed in NCX3F-transfected BHK cells
(supplementary material Fig. S1). In particular, NCX3
labeling on mitochondria was mainly on the OMM (77.6%
in BHK cells and 90.7% in neurons, Fig. 1C). Fig. 1D shows
that the mitochondrial fraction was not contaminated by other
membranes, as immunoblotting detected the absence of
calnexin, LAMP1 and GM130, markers of the ER,
lysosomes, and Golgi compartments, respectively, as well as
IP3R3, a specific marker of the mitochondrial-associated
membrane (MAM) (Hood et al., 2003; Liu et al., 2012;
Mendes et al., 2005; Szabadkai et al., 2006). Consistently,
mitochondria obtained from whole brain extracts of
Ncx3+/+ (Slc8a3+/+) mice displayed an obvious NCX3
immunoreactivity (Fig. 2C). Conversely, mitochondria
obtained from Ncx32/2 (Slc8a3–/–) mice failed to reveal any
NCX3 immunoreactivity (Fig. 2B).
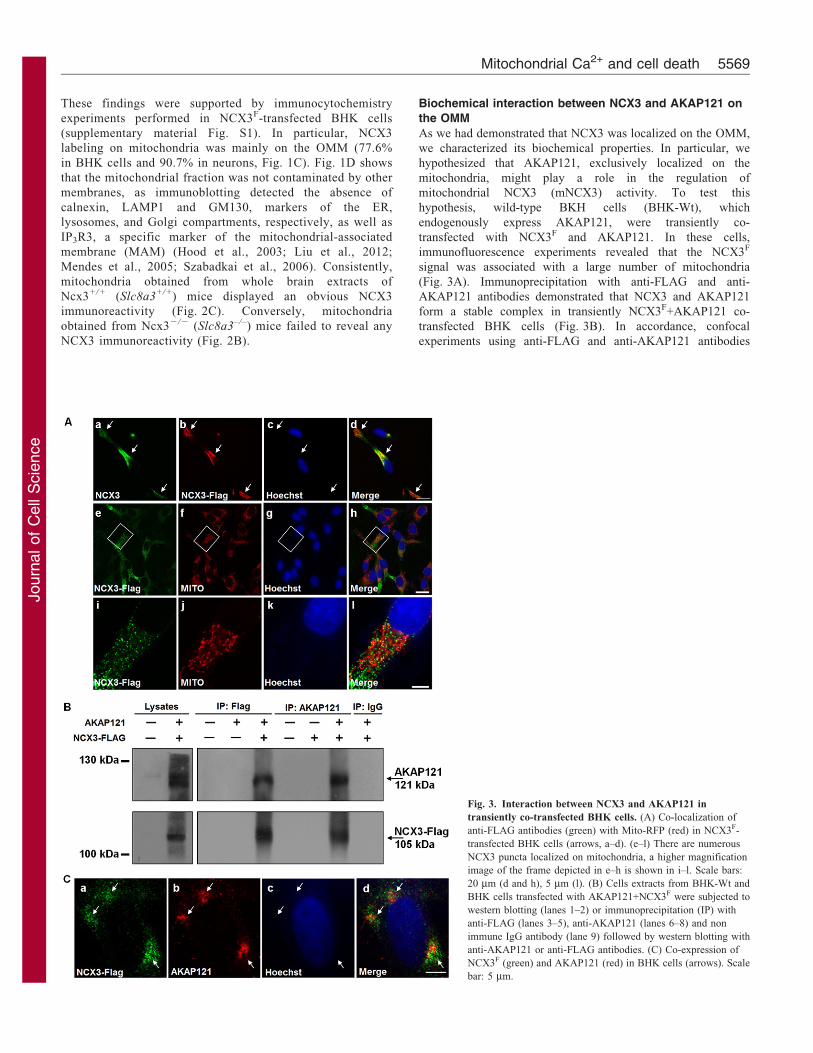
Biochemical interaction between NCX3 and AKAP121 on
the OMM
As we had demonstrated that NCX3 was localized on the OMM,
we characterized its biochemical properties. In particular, we
hypothesized that AKAP121, exclusively localized on the
mitochondria, might play a role in the regulation of
mitochondrial NCX3 (mNCX3) activity. To test this
hypothesis, wild-type BKH cells (BHK-Wt), which
endogenously express AKAP121, were transiently co-
transfected with NCX3F and AKAP121. In these cells,
immunofluorescence experiments revealed that the NCX3F
signal was associated with a large number of mitochondria
(Fig. 3A). Immunoprecipitation with anti-FLAG and anti-
AKAP121 antibodies demonstrated that NCX3 and AKAP121
form a stable complex in transiently NCX3F+AKAP121 co-
transfected BHK cells (Fig. 3B). In accordance, confocal
experiments using anti-FLAG and anti-AKAP121 antibodies
Fig. 3. Interaction between NCX3 and AKAP121 in
transiently co-transfected BHK cells. (A) Co-localization of
anti-FLAG antibodies (green) with Mito-RFP (red) in NCX3F-
transfected BHK cells (arrows, a–d). (e–l) There are numerous
NCX3 puncta localized on mitochondria, a higher magnification
image of the frame depicted in e–h is shown in i–l. Scale bars:
20 mm (d and h), 5 mm (l). (B) Cells extracts from BHK-Wt and
BHK cells transfected with AKAP121+NCX3F were subjected to
western blotting (lanes 1–2) or immunoprecipitation (IP) with
anti-FLAG (lanes 3–5), anti-AKAP121 (lanes 6–8) and non
immune IgG antibody (lane 9) followed by western blotting with
anti-AKAP121 or anti-FLAG antibodies. (C) Co-expression of
NCX3F (green) and AKAP121 (red) in BHK cells (arrows). Scale
bar: 5 mm.
Mitochondrial Ca2+ and cell death 5569

Journ
alof
Cell
Scie
nce
revealed that NCX3F immunosignal was colocalized withAKAP121 (Fig. 3C, arrowheads).
Functional interaction between mNCX3 and AKAP121 incortical neurons and in BHK cells
To demonstrate that the molecular interaction between NCX3 and
AKAP121 might be responsible for Ca2+m handling, we
coexpressed NCX3F and cytosolic (cytoAEQ) or mitochondria-targeted (mtAEQ) aequorin-based Ca2+ probes in BHK cells,
endogenously expressing AKAP121, and evaluated global andorganellar Ca2+ responses to agonist stimulation. Afterreconstitution with aequorin cofactor coelenterazine, cells were
challenged with histamine (100 mM), and luminescence wasmeasured and converted into a value for [Ca2+]. As reported inFig. 4A, the mitochondrial Ca2+ concentration ([Ca2+]m) waslower in cells transfected with NCX3F than that measured in
BHK-Wt cells. Interestingly, when the cells were transientlyco-transfected with NCX3F and siRNA against AKAP121(siAKAP121), [Ca2+]m was higher compared to cells transiently
co-transfected with NCX3F (Fig. 4A,B). To exclude the possibilitythat AKAP121 might affect Ca2+
m through its ability to modifymitochondrial membrane potential and not NCX3 activity, further
experiments measuring mitochondrial membrane potential wereperformed in BHK-Wt, and in NCX3F, siAKAP121, andNCX3F+siAKAP121 transiently transfected cells. The resultsobtained demonstrated that in BHK cells transfected with
siAKAP121, mitochondrial membrane potential was lowercompared to BHK cells transfected with NCX3F orNCX3F+siAKAP121 (Fig. 4A, inset), thus suggesting that the
effect of AKAP121 on [Ca2+]m is independent of changes inmitochondrial membrane potential but depends on its ability tomodulate NCX3 activity. Indeed, when NCX3F and siAKAP121
are co-transfected, the mitochondrial membrane is hyperpolarizedand [Ca2+]m is higher than when NCX3F is transfected alone(Fig. 4A and inset). Similar results were obtained when the
catalytic subunit of PKA was inhibited by the PKI construct, whichcontains a PKA pseudophosphorylation site (data not shown). Thespecificity of siRNA for AKAP121 is reported in Fig. 4C. Todemonstrate that [Ca2+]m is not indirectly affected by the changes
in cytosolic Ca2+ level due to variation in PM NCX3 expression,we measured cytosolic Ca2+ concentrations in each of the above-mentioned experimental conditions and we did not detect any
differences (Fig. 4D). The capacity of NCX3 to enhancemitochondrial Ca2+ efflux independently of PM NCX3 was alsoconfirmed in digitonin-permeabilized BHK cells transiently
transfected with NCX3F and mtAEQ (Fig. 4F). Finally, toexclude any possible effect of NCX3 expression in the ER, thesame experiments were performed in the presence of thapsigargin.
These experiments demonstrated that in cells pretreated withthapsigargin for 15 minutes, the effect of histamine on [Ca2+]m
was the same as that observed in the absence of thapsigargin, thussuggesting that the inhibition of SERCA does not affect [Ca2+]m
(Fig. 4G). This evidence suggests that the activation of non-selective plasma membrane cationic channels elicited by histamineincreases the intracellular Ca2+ concentration ([Ca2+]i) in these
cells, as previously reported (Zanner et al., 2002).
To demonstrate the role of endogenous NCX3 in themodulation of Ca2+
m extrusion, cortical neurons treated with
siRNA against NCX3 (siNCX3) and siAKAP121 were exposedto thapsigargin (Tg, 1 mM, 100 seconds) to allow ER Ca2+
depletion (Secondo et al., 2000) and consequently [Ca2+]m
loading (Baumgartner et al., 2009). Cells were then treated withthe mitochondrial uncoupler FCCP (300 nM for 1 minute) to
induce mitochondrial depolarization and Ca2+ extrusion (Medlerand Gleason, 2002). After thapsigargin treatment, the Ca2+
release into cytoplasm upon FCCP exposure was measured as
[Ca2+]i increase. In particular, when FCCP-induced Ca2+ releaseinto the cytoplasm is low, a higher activity of the Ca2+
m effluxpathway is occurring and vice versa. As reported in Fig. 5A–C,siNCX3 or siAKAP121 induced an enhancement of [Ca2+]m
compared to untreated neurons. Interestingly, [Ca2+]i levels weresignificantly affected only by siNCX3, thus demonstrating thespecificity of siAKAP121 in the modulation of mNCX3 activity
(Fig. 5D).
The functional role of endogenous mNCX3 was supported byquantitative colocalization analysis of NCX3 with MitoTracker
and Mito-RFP in cortical neurons exposed to oxygen and glucosedeprivation (OGD, 3 hours) followed by reoxygenation (Rx,24 hours). As shown in Fig. 6A,B, double-labeling experiments
with anti-NCX3 antibody in MitoTracker-stained and in Mito-RFP-infected neurons revealed that NCX3 mitochondrialimmunosignal decreased during OGD and returned to the basal
level after Rx. Western blotting on isolated mitochondriasupported this finding (Fig. 6C). In accordance with theseresults, [Ca2+]m significantly increased when neurons wereexposed to OGD, whereas it decreased following OGD-Rx
(Fig. 6D). Interestingly, when NCX3 was knocked down withsiRNA, an impairment in Ca2+
m extrusion was recorded underboth basal and OGD-Rx conditions, whereas no alteration
occurred in Ca2+m extrusion during OGD, a condition in which
the NCX3 mitochondrial immunosignal decreased.
Effect of chemical hypoxia on [Ca2+]m and cell survival inBHK cells co-transfected with NCX3F and AKAP121
The [Ca2+]m increase elicited by chemical hypoxia in BHK-Wtcells was reduced when these cells were transfected with NCX3F,thus showing that mNCX3 works as a Ca2+ efflux pathway(Fig. 6E). Moreover, the silencing of endogenous AKAP121 in
NCX3F-transfected BHK cells reduced the Ca2+ efflux activity ofthe mNCX, as demonstrated by the [Ca2+]m increase caused bychemical hypoxia (Fig. 6E). In agreement with these results, cell
survival following chemical hypoxia was higher in BHK cellstransfected with NCX3F and NCX3F plus AKAP121 (Fig. 6F).Moreover, the silencing of constitutively expressed AKAP121
completely reverted the prosurvival effect exerted by NCX3F
(Fig. 6F). By contrast, the silencing of endogenous AKAP121 inwild-type BHK cells not expressing NCX3 (third column of
Fig. 6F) had no protective action.
DiscussionThis study provides evidence that the nuclear-encoded NCX3 isthe only Na+/Ca2+ exchanger isoform localized within the OMMof neurons, where it plays a relevant role in controlling Ca2+
m
homeostasis under both basal and hypoxic conditions. Furtherresults also revealed that mNCX3 colocalizes with AKAP121 – amember of PKA anchoring protein expressed on the OMM – and
that this interaction modulates mNCX3 activity. This finding ledto the hypothesis that the interplay between mNCX3 andAKAP121 regulates [Ca2+]m and contributes to cell survival.
Indeed, when the constitutively expressed AKAP121 wassilenced, [Ca2+]m increases and the prosurvival effect exertedby these two proteins was reverted.
Journal of Cell Science 126 (24)5570

Journ
alof
Cell
Scie
nce
Fig. 4. Functional interaction between mitochondrial NCX3 and AKAP121 in BHK cells. (A) Ca2+ measurement with recombinant mtAEQ ([Ca2+]mt) in
BHK-Wt cells and BHK cells transiently transfected with NCX3F, and NCX3F+siAKAP121 and stimulated with histamine at the indicated point. *P,0.05 versus
BHK-Wt cells and NCX3F+siAKAP. The inset shows the DYm in BHK-Wt cells and BHK cells transiently transfected with NCX3F, siAKAP and
NCX3F+siAKAP121. *P,0.05 versus BHK-Wt; **P,0.05 versus BHK-Wt+siAKAP. (B) Quantification of [Ca2+]mt in BHK-Wt cells and BHK cells transiently
transfected with NCX3F, and NCX3F+siAKAP121. *P,0.05 versus BHK-Wt cells. The AUC of [Ca2+]m signal induced by histamine in BHK-Wt cells transiently
transfected with NCX3F and NCX3F+siAKAP121 was calculated with the trapezioidal rule and is expressed as mM/second. *P,0.05 versus BHK-Wt cells;
**P,0.05 versus NCX3F. (C) Effect of siAKAP on AKAP121 protein expression in BHK-Wt and NCX3F-transfected BHK cells. *P,0.05 versus BHK-Wt.
(D) Ca2+ measurement with recombinant cytoAEQ ([Ca2+]cyto) in BHK-Wt cells and BHK cells transiently transfected with NCX3F, and NCX3F+siAKAP121. All
the cells transfected with mtAEQ or cytoAEQ were reconstituted by incubating for 2 hours with 5 mM of the wild-type aequorin prosthetic group coelenterazine
(Molecular Probes) in KRB as reported in the Materials and Methods. Cells were stimulated with 100 mM histamine in the same KRB solution. Each bar
represents the mean6s.e.m. of different experimental values studied in three independent experimental sessions. (E) AUC of the [Ca2+]cyto signal induced by
histamine in BHK-Wt cells transiently transfected with NCX3F and NCX3F+siAKAP121 calculated with the trapezioidal rule and expressed as mM/second.
(F) [Ca2+]mt in digitonin-permeabilized BHK-Wt and NCX3F-transfected BHK cells. *P,0.05 versus BHK-Wt cells. (G) Quantification of Ca2+ measurement
with recombinant mtAEQ after stimulation with histamine (100 mM) in the presence of thapsigargin (Tg, 1 mM) expressed as percentage of the BHK-Wt value in
BHK cells transiently transfected with NCX3F, and NCX3F+siAKAP121. The rate of mitochondrial Ca2+ release is expressed as the ratio between the time of
recovery from the histamine-induced Ca2+m peak to the basal level in each group and the time of recovery in BHK-Wt cells. *P ,0.05 versus BHK-Wt,
**P,0.05 versus NCX3F.
Mitochondrial Ca2+ and cell death 5571

Journ
alof
Cell
Scie
nce
An interesting finding emerging from our studies is that, among
the three nuclear-encoded Na+/Ca2+ exchanger isoforms, only
NCX3 was localized on mitochondria. In particular, mNCX3 was
specifically localized on the OMM, as its lysis completely
eliminated mNCX3 immunoreactivity. In this regard, an even
more compelling result is that ablation of the gene encoding NCX3
(Slc8a3) induced not only the disappearance of the protein from
the OMM but also the accumulation of Ca2+m in cortical neurons.
Immunocytochemistry and electron microscopy experiments
performed in both neurons and NCX3F-transfected BHK cells
further support the hypothesis that, among the subcellular
compartments, NCX3 is localized mainly to mitochondrial.
Moreover, in neurons, the NCX3 mitochondrial localization was
also particularly evident along the neurites and in the neuropils
close to the PM, where ATP is necessary to drive the activity of
those proteins involved in the regulation of ionic homeostasis
(Blaustein et al., 2002; Guerini et al., 2005; Lytton, 2007).
Although a recent paper reports that all three NCX isoforms,
NCX1, NCX2 and NCX3, can be detected on mitochondria in
both neurons and glial cells (Gobbi et al., 2007), in this study,
NCX2 expression was detected by an anti-NCX2 antibody,
which, in fact, cross-reacts strongly and not specifically with an
unidentified glial protein different from NCX2 (Thurneysen et al.,
2002). Additionally, the molecular mass of the NCX2 protein
detected with this anti-NCX2 antibody differs from those
reported in the literature (Boscia et al., 2009; Secondo et al.,
2007; Sirabella et al., 2009). Regarding the presence of NCX1
protein on mitochondria reported by Gobbi et al. (Gobbi et al.,
2007), it should be underlined that in their study the exchanger
isoform was detectable at very low levels, most likely because
detection occurred only after immunoprecipitation. More
convincingly, our data demonstrated that even when NCX1 and
NCX2 were overexpressed in BHK cells, no immunoreactivity
was detected for either one of these proteins in the whole
mitochondrial fraction. In a very recent study, another component
of the NCX family, NCLX, has been identified in neurons at the
level of the IMM, particularly within the cristae (Palty et al.,
2010). Moreover, this study revealed that this Li+-sensitive
protein, which is both phylogenetically and functionally distinct
from NCX and NCKX family members also participates in the
mitochondrial Na+/Ca2+ exchange activity in neuronal cells. This
novel finding fully agrees with our own results, in that we
reasoned that the Na+/Ca2+ exchange in mitochondria requires
two consecutive steps. The first, operated by the Na+-sensitive
NCLX, mediates Ca2+ transport from the matrix to the
intermembrane space, and the second, operated by mNCX3,
promotes Ca2+ efflux from the intermembrane space to the
cytosol. This interpretation is in line with the recent physiological
role attributed to the OMM in the control of Ca2+m cycling.
Indeed, the outer surface of the membrane is not a passive
permeable membrane as previously considered (Babcock et al.,
1997; Gunter and Pfeiffer, 1990), but it does constitute a
permeability barrier not only to Ca2+ influx but also to Ca2+
efflux (Baines et al., 2007; Jonas et al., 1999; Moran et al., 1992;
Szabadkai and Duchen, 2008). In addition, it has been recently
demonstrated that VDAC, localized on OMM and whose opening
depends on OMM potential and cytosolic Ca2+ levels, promotes
Ca2+ influx from the cytosol to the intermembrane space (Bathori
et al., 2006). In the light of these findings, it is conceivable that
VDAC, together with NCLX, could generate a driving force for
NCX3 to extrude Ca2+ from the intermembrane space to the
cytosol by increasing [Ca2+] in the interspace compartment.
Another novel finding emerging in this paper is the physical,
biochemical and functional interaction between mNCX3 and the
PKA anchoring protein AKAP121 in cortical neurons. We
previously demonstrated that AKAP121 regulates the activity of
the components belonging to the mitochondrial respiratory chain,
thus promoting DYm hyperpolarization and improving the
Fig. 5. Fura-2AM-detected [Ca2+]cyto in cortical
neurons transfected with siNCX3 and
siAKAP121. (A) Superimposed single-cell traces
representative of the effect of Tg+FCCP (1 mM;
300 nM) on [Ca2+]i in cortical neurons in control
conditions (CTL) or individually treated with
siNCX3 or siAKAP. (B) Quantification of the
effect of the above mentioned conditions on [Ca2+]i
expressed as percentage of control. Each bar
represents the mean6s.e.m. of almost 20–30
neurons recorded in three independent experimental
sessions. *P,0.05 versus cortical neurons alone.
(C) Rate of [Ca2+]i increase induced by Tg+FCCP.
*P,0.05 versus cortical neurons alone. (D) Effect
of siAKAP and siNCX3 on basal [Ca2+]i recorded
in the experiments described. *P,0.05 versus
cortical neurons alone.
Journal of Cell Science 126 (24)5572

Journ
alof
Cell
Scie
nce
oxidative synthesis of ATP in a PKA-dependent manner (Livigni
et al., 2006). Similarly, in the present study, immunoprecipitation
assays and confocal microscopy experiments revealed that
mNCX3 interacts with AKAP121, thus suggesting the
involvement of the anchoring protein in promoting mNCX3
Ca2+ extrusion activity under basal and hypoxic conditions. This
finding suggests that the endogenous AKAP121 plays a role in
the regulation of mNCX3 efflux activity, most likely mediated by
its ability to anchor PKA on mitochondria, given that the
silencing of AKAP121 or the inhibition of the kinase by the co-transfection of a specific PKI construct, which contains a PKApseudophosphorylation site, abolished the effects of AKAP121
on [Ca2+]m.
The physiological importance of this interaction is even more
evident given its role in preventing hypoxic neuronal death.Indeed, the silencing of AKAP121 significantly reduced mNCX3
Fig. 6. Effect of hypoxia on mNCX3 activity in cortical neurons and in NCX3- and AKAP121-transfected BHK cells. (A) Confocal double
immunofluorescence images displaying both NCX3 (green) and Mito (red) immunosignals in cortical neurons under control (CTL) conditions (a–c, j) and
following OGD (d–f, k) or OGD-Rx (g–I, l). Superimposed images displaying colocalizing pixels (white) in panels c, f, i, j, k and l. Scale bars: 20 mm (i),
20 mm (l). (B) Quantification of the number of NCX3 and Mito colocalizing points (white pixels) in each condition as a percentage of control. Each bar represents
the mean6s.e.m. of data obtained from ten microscope fields per group in three independent experimental sessions. *P,0.05 versus CTL; **P,0.05 vs OGD.
(C) NCX3 protein expression in membrane, cytosolic, and mitochondrial fractions obtained from cortical neurons after OGD and OGD-Rx. These fractions
were prepared by differential centrifugation as reported in the Materials and Methods. (D) [Ca2+]i measured by Fura-2-AM in cortical neurons transiently
transfected with siNCX3 and exposed to OGD and OGD-Rx. *P,0.05 versus CTL; **P,0.05 versus OGD; ***P,0.05 versus OGD-Rx. (E) [Ca2+]i in single
BHK-Wt, and in BHK cells transfected with NCX3F and NCX3F+siAKAP121 cells. *P,0.05 versus control BHK-Wt cells; **P,0.05 versus BHK-Wt
exposed to chemical hypoxia; ‘P,0.05 versus NCX3F-transfected BHK cells exposed to chemical hypoxia. (F) Effect of chemical hypoxia plus Rx on cell
viability in BHK-Wt and BHK cells transfected with siAKAP121, NCX3F, and NCX3F+siAKAP121. *P,0.05 versus control BHK-Wt cells; **P,0.05 versus
BHK-Wt exposed to hypoxia plus Rx; ‘P,0.05 versus BHK cells transfected with NCX3F exposed to hypoxia plus Rx. Each bar represents the
mean6s.e.m. of different experimental values studied in three independent experimental sessions.
Mitochondrial Ca2+ and cell death 5573

Journ
alof
Cell
Scie
nce
Ca2+-efflux activity and, consequently, increased cell death during
hypoxia. Accordingly, we reasoned that the activation of mNCX3
by AKAP121-anchored PKA on the OMM regulates Ca2+m
handling, thus boosting mitochondrial metabolism and, in
the process, cellular survival. These results reveal a close link
with our previous data demonstrating that, in BHK cells, NCX3
significantly contributes to the maintenance of [Ca2+]i homeostasis
during experimental conditions mimicking ischemia, thereby
preventing DYm collapse and cell death (Secondo et al., 2007).
Moreover, the results obtained in neurons exposed to OGD and
OGD-Rx demonstrated that the endogenous mNCX3 also plays a
relevant role in the regulation of Ca2+m extrusion. Indeed, when
neurons were exposed to OGD, [Ca2+]m significantly increased
while NCX3 expression was reduced. By contrast, when neurons
were exposed to OGD-Rx, [Ca2+]m decreased, whereas NCX3
expression returned to the basal values. Interestingly, when NCX3
was knocked down, an impairment in Ca2+m extrusion was
recorded under both basal and OGD-Rx conditions, whereas no
alteration in Ca2+m extrusion occurred during OGD. These results
might be related to changes in the expression of endogenous NCX3
and AKAP121 during OGD and OGD-Rx (Carlucci et al., 2008a;
Sirabella et al., 2009). Indeed, we have already shown in a previous
study that during hypoxia NCX3 and AKAP121 undergo a rapid
degradation through the ubiquitin-proteosome pathway in neuronal
culture. In particular, for AKAP121, the responsible ubiquitin
ligase is SIAH2, a hypoxia-induced protein. The resultant decrease
in AKAP121 significantly reduces mitochondrial activity
(Carlucci et al., 2008a). These experiments strongly suggest that
the mitochondrial-localized fraction of NCX3 is responsible for
the cellular events observed. However, it should be underlined that
NCX3 expression in mitochondria of neurons exposed to OGD-Rx
is higher compared to that observed in neurons in basal conditions.
Therefore, it is possible to speculate that NCX3 might translocate
to mitochondria as needed, thus contributing to improved
mitochondrial function during OGD-Rx.
The identification of NCX3 isoform as a molecular target of PKA
on the OMM of neurons represents the first evidence for a functional
relationship between AKAP121 and those mitochondrial proteins
able to regulate Ca2+m efflux. This finding might have important
physiological and pathophysiological implications. Specifically,
a considerable crosstalk between the bioenergetic function and
Ca2+ homeostasis occurs within the mitochondria, for Ca2+ is
necessary to activate mitochondrial oxidative metabolism and to
promote mitochondrial respiration (Denton, 2009; Denton and
McCormack, 1980; McCormack et al., 1990). However, if [Ca2+]m
increases over its buffering capacity, ATP production will decrease,
causing the mitochondrial PTP to open, and, eventually, cells to die
by apoptosis (Jeong and Seol, 2008; Kim et al., 2003; Krieger and
Duchen, 2002). Recently, Ghandi et al. have demonstrated that
PINK1, a mitochondrial kinase whose gene mutation is involved in
Parkinson’s disease, participates in the regulation of an unidentified
mitochondrial Na+/Ca2+ exchanger. This hypothesis, based on the
observation that mitochondria from neurons with PINK1 knocked-
out display Ca2+ accumulation due to the impairment of Ca2+ efflux
mechanisms, suggests that the accumulation of Ca2+m is indeed
responsible for increased neuronal vulnerability (Gandhi et al.,
2009).
In summary, we have identified NCX3 as the putative mNCX
isoform and demonstrated that mNCX3, by interacting with
AKAP121 on the outer mitochondrial membrane of neurons,
might participate in the control of Ca2+m efflux and cell survival
in a PKA-sensitive manner.
Materials and MethodsCell cultureWild-type and BHK cells stably transfected with canine cardiac NCX1, or rat brainNCX2 or NCX3 (Linck et al., 1998) were grown on plastic dishes in a mix ofDMEM and Ham’s F12 media (1:1) (Invitrogen) supplemented with 5% FBS,100 U/ml penicillin and 100 mg/ml streptomycin (Sigma). For confocal and Ca2+
imaging experiments, cells were plated on glass coverslips (Fisher) coated withpoly-D-lysine (100 mg/ml) (Sigma), and used at least 24 hours after seeding.
Mixed cultures of cortical neurons from 2–4-day-old Wistar rat pups (CharlesRiver), were prepared as previously described (Abramov et al., 2007). Primarycultures from Ncx32/2 (Slc8a3–/–) mice were obtained as previously reported(Molinaro et al., 2008; Sokolow et al., 2004). The experiments on primary corticalneurons were performed according to the procedures described in experimentalprotocols approved by Ethical Committee of the ‘Federico II’ University ofNaples.
For immunocytochemistry, confocal and Ca2+ imaging experiments, cells wereplated on glass coverslips coated with poly-D-lysine (100 mg/ml).
Plasmids and transfectionMouse pCEP4-AKAP121 cDNA was a gift from Dr C. Rubin (Albert EinsteinCollege of Medicine, NY). Vectors encoding the CMV promoter and AKAP121protein are as previously described (Affaitati et al., 2003). OmicLink ExpressionM14 vector expressing NCX3F was engineered by ligating an oligonucleotideencoding a 3XFLAG epitope to the C-terminus of mouse NCX3 (Slc8a3) cDNA(GeneCopeia). No deletion mutants of NCX3 were used. To knock-down NCX3, the18 nucleotides sequence spanning from +124 to +142, referring to transcriptionalstart site (+1), of rat NCX3 cDNA (GenBank accession no. U53420) was inserted inthe mammalian expression vector pSUPER.retro.puro (OligoEngine). All theseconstructs were transiently transfected using Lipofectamine 2000 (Invitrogen). ThesiRNAs were transiently transfected using Lipofectamine 2000 (Invitrogen) at a finalconcentration of 250 pmol/ml of culture medium.
Mitochondrial extractsExtraction with differential centrifugationBHK-Wt and NCX3-transfected cells were washed in phosphate-buffered salinemedium, and trypsinized with 1 ml of trypsin solution per 10-cm plate. Thetrypsinization was stopped by adding 6 ml of growth medium. As reported insupplementary material Fig. S2A, the cell suspension was collected in a 15-mlfalcon tube and pelleted by centrifugation at 112 g for 3 minutes. The pellet wasresuspended in 300 ml of homogenization buffer solution (Buffer A) containing thefollowing (mM): 250 mannitol, 0.5 EGTA, 5 HEPES (pH 7.4), 1.5 MgCl2, 0.1%aprotinin, 0.7 mg/ml pepstatin, and 1 mg/ml leupeptin, and gently disrupted bypassing ten times through a 26-gauge needle. The suspension was centrifuged at500 g for 5 minutes at 4 C. After the first centrifugation, pellets, corresponding tothe fraction containing membranes but not intracellular organelles includingmitochondria, were separated from supernatants and measured for proteinconcentrations. The supernatant, was centrifuged at 500 g for 5 minutes at 4 C.After this centrifugation the pellet was discarded, and the supernatantcorresponding to the cytosolic fraction containing the organelles, was furthercentrifuged at 19,000 g for 10 minutes at 4 C in order to separate themitochondrial from the cytosolic fraction. Supernatants (Cytosol) were thenremoved and assessed for protein content. Next, the pellets containingmitochondria were lyzed in 50 ml of lysis buffer containing (mM): 20 Tris-HCl(pH 7.5), 10 NaF, 150 NaCl, 1 PMSF, 1% Nonidet P-40, 1 Na3VO4, 0.1%aprotinin, 0.7 mg/ml pepstatin, and 1 ml/mg leupeptin, and kept on ice for15 minutes. Finally, samples were purified again by centrifugation (18,000 g,10 minutes) and supernatants (Mitochondria) were assessed for protein content byBradford’s assay (Bradford, 1976). The three fractions obtained were used forwestern blotting (WB). All the procedure was carried out at 4 C to minimize theaction of proteases and phospholipases.
The purity of the mitochondrial preparation was assessed by evaluating theexpression of the proteins GM131, a Golgi marker, calnexin, an ER marker,LAMP1, a lysosome marker, (Hood et al., 2003; Liu et al., 2012; Szabadkai et al.,2006), and the IP3R3 receptor, a MAM marker (Mendes et al., 2005) on thefractions relative to membranes, cytosol and mitochondria.
Extraction of brain mitochondria by the Percoll gradient methodThe Percoll gradient method was used to purify mitochondria from tissues (Hoviuset al., 1990). Ncx3+/+ and Ncx32/2 mice were killed by decapitation according tothe experimental procedures approved by the Ethical Committee of the ‘FedericoII’ University of Naples and the brains were rapidly removed. Each brain waswashed twice in cold PBS in order to remove the blood and was cut into smallpieces using scissors. The brain pieces were transferred into a 50-ml glass/teflonPotter Elvehjem homogenizer and the homogenization buffer solution (Buffer A)
Journal of Cell Science 126 (24)5574

Journ
alof
Cell
Scie
nce
was added in the ratio of 5 ml of buffer per gram of tissue. The homogenizationwas performed by using a teflon pestle with a slow movement to preserve theintegrity of mitochondria. The homogenate was passed through a 22-gauge needleonce and then through a 26-gauge needle five times to accomplish thehomogenization of the brain. Subsequently, it was centrifuged four times at 500g for 5 minutes and once again at 18,000 g for 10 min. The supernatant wasremoved and assayed for protein content. As depicted in supplementary materialFig. S2B, the pellet containing crude mitochondria was suspended in 1 ml buffer Aand then stratified in 3 ml of Percoll medium containing the following (mM): 30%(v/v) Percoll, 250 mannitol, 0.5 EGTA, 5 HEPES (pH 7.4), and centrifuged at95,000 g for 30 min at 4 C in a Beckman 60Ti rotor. The fraction containing puremitochondria, lying under the Percoll band (supplementary material Fig. 2B, darkgrey), was removed, suspended in buffer A, and then centrifuged twice (7000 g for10 minutes). The pellet obtained was re-suspended in a 50 ml lysis buffer, kept onice for 10 minutes, and centrifuged (18,000 g for 10 minutes). The supernatant wasfinally assessed for protein content and used for western blot analysis. Allprocedures were carried out at 4 C to minimize the action of proteases andphospholipases.
Western blotting
50 mg of protein samples were analyzed on 8% SDS-PAGE and electrotransferredto Hybond ECL nitrocellulose paper (Amersham). Membranes were first blockedwith 5% non-fat dry milk in 0.1% Tween-20 (TBS-T; 2 mM Tris–HCl, 50 mMNaCl, pH 7.5) for 2 hours at room temperature. They were then incubatedovernight at 4 C in the blocked buffer with the 1:1000 anti-NCX1 (rabbitpolyclonal antibody, Swant), 1:1000 anti-NCX2 (rabbit polyclonal antibody,Alpha Diagnostic), and 1:5000 anti-NCX3 rabbit polyclonal antibody (kindlyprovided by Kenneth D. Philipson, UCLA, Los Angeles, California, USA), andsubsequently incubated with the secondary antibodies for 1 hour (1:5000;Amersham). Immunoreactive bands were detected with the ECL kit(Amersham). Discrimination among the distinct types of extracts was ensuredby running parallel western blots with the endogenous protein tubulin (localized tothe PM), Mn-SOD (localized to the MM), VDAC and AKAP121 (localized to theOMM) (Cardone et al., 2004; Ginsberg et al., 2003; Liu et al., 2012), and COX-IV(localized to the IMM) (Mattei et al., 2011).
Immunocytochemistry in cortical neurons and BHK cells
BHK wild-type cells, BHK cells stably transfected with the NCX3 isoform andcortical neurons were cultured on glass coverslips [BHK cells for 48 hours andcortical neurons for 11 days in vitro (DIV)]. The cells were rinsed twice in cold0.01M PBS at pH 7.4 and fixed at room temperature in 4% (w/v)paraformaldehyde for 20 minutes. Following three washes in PBS, cells wereblocked in PBS containing 10% FBS and the following antibodies: anti-NCX3(rabbit, kindly supplied by Kenneth D., Philipson, UCLA, Los Angeles, California,USA, dilution 1:4000), anti-FLAG (mouse, Sigma, dilution 1:500), and anti-AKAP121 (rabbit, Carlucci et al., 2008a), anti-LAMP1 (kindly supplied byPolishchuk EV, dilution 1:200), anti-VAP-A (kindly supplied by Maria AntoniettaDe Matteis, Telethon Institute of Genetics and Medicine, Naples, Italy, dilution1:300). They were then incubated overnight at 4 C. Next, slides were washed inPBS, incubated with anti-rabbit Cy2-conjugated antibody (Jackson, dilution 1:200)and anti-mouse Cy3-conjugated antibody (Jackson, dilution 1:200) for 1 hour atroom temperature under dark conditions, and washed again with PBS. Finally, theywere mounted with a SlowFadeTM Antifade Kit (Molecular Probes-Invitrogen) andanalyzed by confocal microscopy (Boscia et al., 2012). The cells were infectedwith CellLight Reagents BacMam 2.0 Mito-RFP (Molecular probes) according tothe manufacturer’s protocol to stain mitochondria.
Cells were analyzed for colocalization between Mito (red) and NCX3 (green) byusing the ‘co-localization highlighter’ plug-in for ImageJ Software (NIH,Bethesda, MA, USA). Before colocalization analysis, threshold settings for eachimage were determined, and quantification was achieved by counting the numberof NCX3 and Mito colocalized points (white) per microscope field. Results wereexpressed as a percentage of colocalization (Molinaro et al., 2011).
Immunoelectron microscopy analysis in cortical neurons and BHK cells
Ncx3+/+, Ncx32/2 cortical neurons, and wild-type and transfected BHK cells werefirst fixed with a mixture of 4% paraformaldehyde and 0.05% glutaraldehyde andthen labeled with a polyclonal antibody against NCX3 using the Nanogoldprotocol. Next, they were embedded in Epon-812 and finally cut in thin sections(Polishchuk et al., 2003). Electron microscopy images were acquired from the thinsections using a JEOL JEM-1011 electron microscope (Tokyo, Japan) equippedwith a Morada CCD digital camera.
Surface gold density (arbitrary units, AU) was estimated according to themethod of Griffiths and Hoppeler (Griffiths and Hoppeler, 1986). Following thismethod, a morphometric grid with definitive size (100 nm) was applied to allimages acquired and then the number of gold particles present on the membranesof interest (mitochondria, ER, PM, Golgi stacks and endosomes) was counted.Next, the number of intersections between the organelle membranes and the gridwas calculated. In the case of mitochondria, only the outer membrane was counted.
Finally, the number of gold particles was divided by the number of intersections togive the gold density value. At total of 30 structures were analyzed for eachcompartment of interest. Surface gold density for NCX3 in mitochondriacorresponded to 0.19 AU in neurons and to 0.21 AU in BHK NCX3-transfectedcells; surface gold density for NCX3 in ER corresponded to 0.24 AU in neuronsand to 0.32 AU in BHK NCX3-transfected cells; the plasma membrane surfacegold density for NCX3 corresponded to 0.23 AU in neurons and to 0.98 AU inBHK NCX3-transfected cells (Fig. 1C; Fig. 2B; Table 1).
Ca2+ measurement with recombinant aequorin
Transfected cytoAEQ and mtAEQ were reconstituted by incubating BHK cells for2 hours with a 5 mM concentration of the wild-type aequorin prosthetic groupcoelenterazine (Molecular Probes) in KRB (Krebs–Ringer modified buffer:125 mM NaCl, 5 mM KCl, 1 mM Na3PO4, 1 mM MgSO4, 5.5 mM glucose and20 mM Hepes, pH 7.4) supplemented with 1 mM CaCl2 at 37 C. During theexperiment, the cells were continuously perfused with KRB medium. Agonist wasadded to the same medium. In our experiments, we induced a rise in [Ca2+] bystimulating the cells with histamine 100 mM. All aequorin measurements wereterminated by lyzing the cells with 100 mM digitonin in a hypotonic Ca2+-richsolution (10 mM CaCl2 in H2O) thus discharging the remaining AEQ pool. Theaequorin luminescence data were calibrated off-line into [Ca2+] values using acomputer algorithm based on the Ca2+ response curve of wild-type aequorin aspreviously reported (Brini et al., 1995; Rizzuto et al., 1995). In the experimentswith permeabilized cells, a buffer mimicking the cytosolic ionic composition[intracellular buffer (IB)] was employed: 140 mM KCl, 10 mM NaCl, 1 mMK3PO4, 5.5 mM glucose, 2 mM MgSO4, 1 mM ATP, 2 mM Na+ succinate, and20 mM Hepes (pH 7.5 at 37 C) supplemented with EGTA 1 mM. BHK cells werepermeabilized by a 5-minute incubation with 20 mM digitonin (added to IB withEGTA) before luminescence measurements (Rapizzi et al., 2002). Changes in[Ca2+]m and [Ca2+]cyto were followed as previously reported (Staiano et al., 2009)and the area under the curve (AUC) was calculated using the trapezoidal rule(Bailer and Piegorsch, 1990). The rate of mitochondrial Ca2+ release was measuredas the time of recovery from the histamine-induced Ca2+
m peak to the basal level.These data were reported as the ratio between the time of recovery in each group(NCX3F-transfected BHK cells and BHK cells transfected with NCX3F+siAKAP)and that measured in BHK-Wt cells.
Imaging of mitochondrial membrane potential
Mitochondrial membrane potential was assessed using the fluorescent dye tetramethylrhodamine ethyl ester (TMRE) in the ‘redistribution mode’. Cells were loaded withTMRE (20 nM) for 30 minutes in a medium containing 156 mM NaCl, 3 mM KCl,2 mM MgSO4, 1.25 mM KH2PO4, 2 mM CaCl2, 10 mM glucose and 10 mM Hepes.The pH value was adjusted to 7.35 with NaOH. At the end of the incubation period,cells were washed in the same medium containing TMRE (20 nM) and allowed toequilibrate. A decline in the mitochondrial fluorescence intensity was indicative ofmitochondrial membrane depolarization (Livigni et al., 2006).
Confocal images were obtained using a Zeiss inverted 510 confocal laserscanning microscopy and a 636 oil immersion objective. The illuminationintensity of the 543 Xenon laser, used to excite TMRE fluorescence, was kept to aminimum of 0.5% of laser output to avoid phototoxicity.
Imaging of cytosolic Ca2+ in cortical neurons and BHK cells
[Ca2+]i was measured by single-cell computer-assisted videoimaging as previouslydescribed (Secondo et al., 2007). Briefly, cells, grown on glass coverslips, wereloaded with 10 mM Fura-2 acetoxymethyl ester (AM) (Fura-2AM) for 30 minutesat room temperature in normal Krebs solution containing (in mM) 5.5 KCl, 160NaCl, 1.2 MgCl2, 1.5 CaCl2, 10 glucose and 10 Hepes–NaOH, pH 7.4. At the endof the Fura-2-AM loading period, the coverslips were placed into a perfusionchamber (Medical System, Co. Greenvale, NY, USA) mounted onto a ZeissAxiovert 200 microscope (Carl Zeiss, Germany). The experiments were carried outwith a digital imaging system composed of a MicroMax 512BFT cooled CCDcamera (Princeton Instruments, Trenton, NJ, USA), LAMBDA 10-2 filter wheeler(Sutter Instruments, Novato, CA, USA), and Meta-Morph/MetaFluor ImagingSystem software (Universal Imaging,West Chester, PA, USA). All the results arepresented as cytosolic Ca2+ concentration, assuming that the Kd for FURA-2 was224 nM. The amount of Ca2+ extruded in the cytoplasm upon FCCP exposure wasmeasured as [Ca2+]i elevations. This method is widely used to measure [Ca2+]m
content (Medler and Gleason, 2002). The rate of [Ca2+]i increase after Tg+FCCPexposure was calculated as the peak of [Ca2+]i against time, averaged over threemeasurements, and expressed as mean6s.e.m.
Immunoprecipitation and immunoblot analyses
Cells were homogenized in lysis buffer containing (mM) the following: 50 Tris-HCl pH 7.4, 150 NaCl, 1 EDTA, 1% Triton X-100, 100 NaF, 100 Na3VO4, 5 mg/ml aprotinin, 10 mg/ml leupeptin and 2 mg/ml pepstatin. The lysates were clearedby centrifugation (15,000 g, 15 min). Two milligrams of cell lysate wasimmunoprecipitated with either anti-FLAG mouse antibody (1:100), anti
Mitochondrial Ca2+ and cell death 5575

Journ
alof
Cell
Scie
nce
AKAP121 (1:200) or non immune IgG antibody. One hundred micrograms eitherof total cell lysate or of immunoprecipitates was resolved by SDS-PAGE gel andtransferred to nitrocellulose membrane. Immunoblot analysis was performed usinganti AKAP121 or anti FLAG (Cardone et al., 2004; Carlucci et al., 2008a).
Chemical hypoxia and oxygen and glucose deprivation and reoxygenation
Chemical hypoxia was reproduced by adding 5 mg/ml oligomycin plus 2 mM 2-deoxyglucose to cells in glucose-free medium for 45 minutes. The medium wascomposed of (in mM): 145 NaCl, 5.5 KCl, 1.2 MgCl2, 1.5 CaCl2, and 10 Hepes,pH 7.4, as already described (Secondo et al., 2007). Control cells were exposed forthe same amount of time to normal Krebs medium composed of (in mM): 145NaCl, 5.5 KCl, 1.2 MgCl2, 1.5 CaCl2, 10 glucose and 10 Hepes, pH 7.4.Reoxygenation was performed in 16 MEM plus 25 mM NaHCO3 and 22 mMdextrose at 37 C.
In cortical neurons, OGD insult was reproduced by exposing cells for 3 hours toa medium containing (in mM): 116 NaCl, 5.4 KCl, 0.8 MgSO4, 26.2 NaHCO3, 1NaH2PO4, 1.8 CaCl2, 0.01 glycine and 0.001 w/v Phenol Red (Carlucci et al.,2008a; Sirabella et al., 2009). Hypoxic conditions were maintained using a hypoxiachamber (Billups Rothemberg Inc. Del Mar.) (temperature 37 C, under anatmosphere of 5% CO2 and 95% N2). Reoxygenation was obtained by incubatingthe cells for 24 hours in the presence of normal levels of glucose and oxygen.
Determination of cell death
To quantify cell death after the experimental procedures, cells were washed withnormal Krebs and stained with 36 mM fluorescein diacetate (FDA) and 7 mMpropidium iodide (PI) for 5 minutes at 37 C in PBS (Secondo et al., 2007). Celldeath was determined by the ratio of the number of PI-positive cells to PI+FDA-positive cells (Wei et al., 2000).
Statistical analysis
Data were generated from three independent experiments. Ca2+ measurementswere performed at least in 20 cells for each of the three independent experimentalsessions. Data are expressed as mean6s.e.m. Statistical analysis was performedwith ANOVA followed by Newman-Keul’s test. Statistical significance wasaccepted at the 95% confidence level (P#0.05).
AcknowledgementsWe thank Paola Merolla for the editorial revision, Vincenzo Grillofor technical support, and Telethon Electron Microscopy CoreFacility and Integrated Microscopy Facility for electron microscopyassistance. We also thank Maria Antonietta De Matteis (TelethonInstitute of Genetics and Medicine, Naples) for the anti-VAP-Aantibody. We are particularly grateful to Michael Duchen andGyorgy Szabadkai of University College London for the AEQconstructs.
Author contributionsA. Scorziello, L.A., G.D.R. and A.F. conceived of the experiments;A. Scorziello, L.A. and G.D.R. wrote the manuscript; C.S., M.J.S.,A.G., A. Secondo, F.B., A.E., P.M., A.C., L.L., E.V.P and R.S.P.performed experiments; A. Scorziello, C.S., M.J.S., A.G., A.Secondo, F.B., A.E. and P.M. analyzed data; E.V.P and R.S.P.provided VAP-A and LAMP-1 antibodies for immunocytochemstry.
FundingThis work was supported by the following grants: COFIN2008 [grantnumber 20089BARSR_002]; Ricerca Sanitaria [grant number RF-FSL352059]; Ricerca Oncologica 2006 [grant number RF-IDI-2006-367185]; Progetto Strategico 2007 [grant number RF-06711];Progetto Ordinario 2007 [grant number RF-SDN-2007-666932];Programma Operativo Nazionale [grant number PON_01602] all toL.A.; and the Associazione Italiana Ricerca sul Cancro [grantnumber IG 11788] to A.F.
Supplementary material available online at
http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.129668/-/DC1
ReferencesAbramov, A. Y., Scorziello, A. and Duchen, M. R. (2007). Three distinct mechanisms
generate oxygen free radicals in neurons and contribute to cell death during anoxiaand reoxygenation. J. Neurosci. 27, 1129-1138.
Affaitati, A., Cardone, L., de Cristofaro, T., Carlucci, A., Ginsberg, M. D., Varrone,
S., Gottesman, M. E., Avvedimento, E. V. and Feliciello, A. (2003). Essential roleof A-kinase anchor protein 121 for cAMP signaling to mitochondria. J. Biol. Chem.
278, 4286-4294.
Babcock, D. F., Herrington, J., Goodwin, P. C., Park, Y. B. and Hille, B. (1997).Mitochondrial participation in the intracellular Ca2+ network. J. Cell Biol. 136, 833-844.
Bailer, A. J. and Piegorsch, W. W. (1990). Estimating integrals using quadraturemethods with an application in pharmacokinetics. Biometrics 46, 1201-1211.
Baines, C. P., Kaiser, R. A., Sheiko, T., Craigen, W. J. and Molkentin, J. D. (2007).Voltage-dependent anion channels are dispensable for mitochondrial-dependent celldeath. Nat. Cell Biol. 9, 550-555.
Bathori, G., Csordas, G., Garcia-Perez, C., Davies, E. and Hajnoczky, G. (2006).Ca2+-dependent control of the permeability properties of the mitochondrial outermembrane and voltage-dependent anion-selective channel (VDAC). J. Biol. Chem.
281, 17347-17358.
Baumgartner, H. K., Gerasimenko, J. V., Thorne, C., Ferdek, P., Pozzan, T.,
Tepikin, A. V., Petersen, O. H., Sutton, R., Watson, A. J. and Gerasimenko, O. V.(2009). Calcium elevation in mitochondria is the main Ca2+ requirement formitochondrial permeability transition pore (mPTP) opening. J. Biol. Chem. 284,20796-20803.
Blaustein, M. P., Juhaszova, M., Golovina, V. A., Church, P. J. and Stanley, E. F.
(2002). Na/Ca exchanger and PMCA localization in neurons and astrocytes:functional implications. Ann. N. Y. Acad. Sci. 976, 356-366.
Boscia, F., Gala, R., Pannaccione, A., Secondo, A., Scorziello, A., Di Renzo, G. and
Annunziato, L. (2009). NCX1 expression and functional activity increase inmicroglia invading the infarct core. Stroke 40, 3608-3617.
Boscia, F., D’Avanzo, C., Pannaccione, A., Secondo, A., Casamassa, A., Formisano, L.,Guida, N., Sokolow, S., Herchuelz, A. and Annunziato, L. (2012). Silencing orknocking out the Na+/Ca2+ exchanger-3 (NCX3) impairs oligodendrocyte differentiation.Cell Death Differ. 19, 562-572.
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation ofmicrogram quantities of protein utilizing the principle of protein-dye binding. Anal.
Biochem. 72, 248-254.
Brini, M., Marsault, R., Bastianutto, C., Alvarez, J., Pozzan, T. and Rizzuto,
R. (1995). Transfected aequorin in the measurement of cytosolic Ca2+ concentration([Ca2+]c). A critical evaluation. J. Biol. Chem. 270, 9896-9903.
Carafoli, E., Tiozzo, R., Lugli, G., Crovetti, F. and Kratzing, C. (1974). The releaseof calcium from heart mitochondria by sodium. J. Mol. Cell. Cardiol. 6, 361-371.
Cardone, L., Carlucci, A., Affaitati, A., Livigni, A., DeCristofaro, T., Garbi, C.,Varrone, S., Ullrich, A., Gottesman, M. E., Avvedimento, E. V. et al. (2004).Mitochondrial AKAP121 binds and targets protein tyrosine phosphatase D1, a novelpositive regulator of src signaling. Mol. Cell. Biol. 24, 4613-4626.
Carlucci, A., Adornetto, A., Scorziello, A., Viggiano, D., Foca, M., Cuomo, O.,
Annunziato, L., Gottesman, M. and Feliciello, A. (2008a). Proteolysis of AKAP121regulates mitochondrial activity during cellular hypoxia and brain ischaemia. EMBO
J. 27, 1073-1084.
Carlucci, A., Lignitto, L. and Feliciello, A. (2008b). Control of mitochondria dynamicsand oxidative metabolism by cAMP, AKAPs and the proteasome. Trends Cell Biol.
18, 604-613.
Crompton, M., Barksby, E., Johnson, N. and Capano, M. (2002). Mitochondrialintermembrane junctional complexes and their involvement in cell death. Biochimie
84, 143-152.
Denton, R. M. (2009). Regulation of mitochondrial dehydrogenases by calcium ions.Biochim. Biophys. Acta 1787, 1309-1316.
Denton, R. M. and McCormack, J. G. (1980). On the role of the calcium transportcycle in heart and other mammalian mitochondria. FEBS Lett. 119, 1-8.
Gandhi, S., Wood-Kaczmar, A., Yao, Z., Plun-Favreau, H., Deas, E., Klupsch,
K., Downward, J., Latchman, D. S., Tabrizi, S. J., Wood, N. W. et al. (2009).PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell 33, 627-638.
Ginsberg, M. D., Feliciello, A., Jones, J. K., Avvedimento, E. V. and Gottesman,M. E. (2003). PKA-dependent binding of mRNA to the mitochondrial AKAP121protein. J. Mol. Biol. 327, 885-897.
Gobbi, P., Castaldo, P., Minelli, A., Salucci, S., Magi, S., Corcione, E. and Amoroso,S. (2007). Mitochondrial localization of Na+/Ca2+ exchangers NCX1-3 in neurons andastrocytes of adult rat brain in situ. Pharmacol. Res. 56, 556-565.
Griffiths, G. and Hoppeler, H. (1986). Quantitation in immunocytochemistry:correlation of immunogold labeling to absolute number of membrane antigens.J. Histochem. Cytochem. 34, 1389-1398.
Guerini, D., Coletto, L. and Carafoli, E. (2005). Exporting calcium from cells. Cell
Calcium 38, 281-289.
Gunter, T. E. and Pfeiffer, D. R. (1990). Mechanisms by which mitochondria transportcalcium. Am. J. Physiol. 258, C755-C786.
Gunter, T. E., Buntinas, L., Sparagna, G., Eliseev, R. and Gunter, K. (2000).Mitochondrial calcium transport: mechanisms and functions. Cell Calcium 28, 285-296.
Hood, J. L., Logan, B. B., Sinai, A. P., Brooks, W. H. and Roszman, T. L. (2003).Association of the calpain/calpastatin network with subcellular organelles. Biochem.
Biophys. Res. Commun. 310, 1200-1212.
Hovius, R., Lambrechts, H., Nicolay, K. and de Kruijff, B. (1990). Improved methodsto isolate and subfractionate rat liver mitochondria. Lipid composition of the innerand outer membrane. Biochim. Biophys. Acta 1021, 217-226.
Journal of Cell Science 126 (24)5576

Journ
alof
Cell
Scie
nce
Jeong, S. Y. and Seol, D. W. (2008). The role of mitochondria in apoptosis. BMB Rep.
41, 11-22.
Jonas, E. A., Buchanan, J. and Kaczmarek, L. K. (1999). Prolonged activation ofmitochondrial conductances during synaptic transmission. Science 286, 1347-1350.
Kim, J. S., He, L. and Lemasters, J. J. (2003). Mitochondrial permeability transition: acommon pathway to necrosis and apoptosis. Biochem. Biophys. Res. Commun. 304,463-470.
Krieger, C. and Duchen, M. R. (2002). Mitochondria, Ca2+ and neurodegenerativedisease. Eur. J. Pharmacol. 447, 177-188.
Kroemer, G. (1997). Mitochondrial implication in apoptosis. Towards an endosymbionthypothesis of apoptosis evolution. Cell Death Differ. 4, 443-456.
Linck, B., Qiu, Z., He, Z., Tong, Q., Hilgemann, D. W. and Philipson, K. D. (1998).Functional comparison of the three isoforms of the Na+/Ca2+ exchanger (NCX1,NCX2, NCX3). Am. J. Physiol. 274, C415-C423.
Liu, L., Feng, D., Chen, G., Chen, M., Zheng, Q., Song, P., Ma, Q., Zhu, C., Wang,
R., Qi, W. et al. (2012). Mitochondrial outer-membrane protein FUNDC1 mediateshypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 14, 177-185.
Livigni, A., Scorziello, A., Agnese, S., Adornetto, A., Carlucci, A., Garbi, C.,
Castaldo, I., Annunziato, L., Avvedimento, E. V. and Feliciello, A. (2006).Mitochondrial AKAP121 links cAMP and src signaling to oxidative metabolism. Mol.
Biol. Cell 17, 263-271.
Lytton, J. (2007). Na+/Ca2+ exchangers: three mammalian gene families control Ca2+
transport. Biochem. J. 406, 365-382.
Margineantu, D. H., Brown, R. M., Brown, G. K., Marcus, A. H. and Capaldi, R. A.
(2002). Heterogeneous distribution of pyruvate dehydrogenase in the matrix ofmitochondria. Mitochondrion 1, 327-338.
Mattei, V., Matarrese, P., Garofalo, T., Tinari, A., Gambardella, L., Ciarlo, L.,
Manganelli, V., Tasciotti, V., Misasi, R., Malorni, W. et al. (2011). Recruitment ofcellular prion protein to mitochondrial raft-like microdomains contributes to apoptosisexecution. Mol. Biol. Cell 22, 4842-4853.
McCormack, J. G., Halestrap, A. P. and Denton, R. M. (1990). Role of calcium ionsin regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 70, 391-425.
Medler, K. and Gleason, E. L. (2002). Mitochondrial Ca2+ buffering regulates synaptictransmission between retinal amacrine cells. J. Neurophysiol. 87, 1426-1439.
Mendes, C. C., Gomes, D. A., Thompson, M., Souto, N. C., Goes, T. S., Goes, A. M.,
Rodrigues, M. A., Gomez, M. V., Nathanson, M. H. and Leite, M. F. (2005). Thetype III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+
signals into mitochondria. J. Biol. Chem. 280, 40892-40900.
Molinaro, P., Cuomo, O., Pignataro, G., Boscia, F., Sirabella, R., Pannaccione, A.,
Secondo, A., Scorziello, A., Adornetto, A., Gala, R. et al. (2008). Targeteddisruption of Na+/Ca2+ exchanger 3 (NCX3) gene leads to a worsening of ischemicbrain damage. J. Neurosci. 28, 1179-1184.
Molinaro, P., Viggiano, D., Nistico, R., Sirabella, R., Secondo, A., Boscia, F.,
Pannaccione, A., Scorziello, A., Mehdawy, B., Sokolow, S. et al. (2011). Na+-Ca2+
exchanger (NCX3) knock-out mice display an impairment in hippocampal long-termpotentiation and spatial learning and memory. J. Neurosci. 31, 7312-7321.
Moran, O., Sciancalepore, M., Sandri, G., Panfili, E., Bassi, R., Ballarin, C. and
Sorgato, M. C. (1992). Ionic permeability of the mitochondrial outer membrane. Eur.
Biophys. J. 20, 311-319.
Palty, R., Silverman, W. F., Hershfinkel, M., Caporale, T., Sensi, S. L., Parnis, J.,
Nolte, C., Fishman, D., Shoshan-Barmatz, V., Herrmann, S. et al. (2010). NCLXis an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci.
USA 107, 436-441.
Polishchuk, E. V., Di Pentima, A., Luini, A. and Polishchuk, R. S. (2003). Mechanismof constitutive export from the golgi: bulk flow via the formation, protrusion, and enbloc cleavage of large trans-golgi network tubular domains. Mol. Biol. Cell 14, 4470-4485.
Rapizzi, E., Pinton, P., Szabadkai, G., Wieckowski, M. R., Vandecasteele, G., Baird,
G., Tuft, R. A., Fogarty, K. E. and Rizzuto, R. (2002). Recombinant expression ofthe voltage-dependent anion channel enhances the transfer of Ca2+ microdomains tomitochondria. J. Cell Biol. 159, 613-624.
Rizzuto, R., Brini, M., Bastianutto, C., Marsault, R. and Pozzan, T. (1995).Photoprotein-mediated measurement of calcium ion concentration in mitochondria ofliving cells. Methods Enzymol. 260, 417-428.
Sardanelli, A. M., Signorile, A., Nuzzi, R., Rasmo, D. D., Technikova-Dobrova, Z.,
Drahota, Z., Occhiello, A., Pica, A. and Papa, S. (2006). Occurrence of A-kinaseanchor protein and associated cAMP-dependent protein kinase in the innercompartment of mammalian mitochondria. FEBS Lett. 580, 5690-5696.
Secondo, A., Taglialatela, M., Cataldi, M., Giorgio, G., Valore, M., Di Renzo, G. and
Annunziato, L. (2000). Pharmacological blockade of ERG K+ channels and Ca2+
influx through store-operated channels exerts opposite effects on intracellular Ca2+
oscillations in pituitary GH(3) cells. Mol. Pharmacol. 58, 1115-1128.Secondo, A., Staiano, R. I., Scorziello, A., Sirabella, R., Boscia, F., Adornetto, A.,
Valsecchi, V., Molinaro, P., Canzoniero, L. M., Di Renzo, G. et al. (2007). BHKcells transfected with NCX3 are more resistant to hypoxia followed by reoxygenationthan those transfected with NCX1 and NCX2: Possible relationship withmitochondrial membrane potential. Cell Calcium 42, 521-535.
Sirabella, R., Secondo, A., Pannaccione, A., Scorziello, A., Valsecchi, V., Adornetto,A., Bilo, L., Di Renzo, G. and Annunziato, L. (2009). Anoxia-induced NF-kappaB-dependent upregulation of NCX1 contributes to Ca2+ refilling into endoplasmicreticulum in cortical neurons. Stroke 40, 922-929.
Sokolow, S., Manto, M., Gailly, P., Molgo, J., Vandebrouck, C., Vanderwinden,
J. M., Herchuelz, A. and Schurmans, S. (2004). Impaired neuromusculartransmission and skeletal muscle fiber necrosis in mice lacking Na/Ca exchanger 3.J. Clin. Invest. 113, 265-273.
Staiano, R. I., Granata, F., Secondo, A., Petraroli, A., Loffredo, S., Frattini, A.,
Annunziato, L., Marone, G. and Triggiani, M. (2009). Expression and function ofNa+/Ca2+ exchangers 1 and 3 in human macrophages and monocytes. Eur. J.
Immunol. 39, 1405-1418.Starkov, A. A., Chinopoulos, C. and Fiskum, G. (2004). Mitochondrial calcium and
oxidative stress as mediators of ischemic brain injury. Cell Calcium 36, 257-264.Stojanovski, D., Johnston, A. J., Streimann, I., Hoogenraad, N. J. and Ryan, M. T.
(2003). Import of nuclear-encoded proteins into mitochondria. Exp. Physiol. 88, 57-64.
Szabadkai, G. and Duchen, M. R. (2008). Mitochondria: the hub of cellular Ca2+
signaling. Physiology (Bethesda) 23, 84-94.Szabadkai, G., Bianchi, K., Varnai, P., De Stefani, D., Wieckowski, M. R., Cavagna,
D., Nagy, A. I., Balla, T. and Rizzuto, R. (2006). Chaperone-mediated coupling ofendoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 175, 901-911.
Thurneysen, T., Nicoll, D. A., Philipson, K. D. and Porzig, H. (2002). Sodium/calcium exchanger subtypes NCX1, NCX2 and NCX3 show cell-specific expressionin rat hippocampus cultures. Brain Res. Mol. Brain Res. 107, 145-156.
Wei, H., Leeds, P. R., Qian, Y., Wei, W., Chen, R. and Chuang, D. (2000). beta-amyloid peptide-induced death of PC 12 cells and cerebellar granule cell neurons isinhibited by long-term lithium treatment. Eur. J. Pharmacol. 392, 117-123.
Zanner, R., Hapfelmeier, G., Gratzl, M. and Prinz, C. (2002). Intracellular signaltransduction during gastrin-induced histamine secretion in rat gastric ECL cells. Am.
J. Physiol. 282, C374-C382.
Mitochondrial Ca2+ and cell death 5577