investigation of the involvement of covalent binding in ... · in nevirapine-induced hepatic and...
TRANSCRIPT

Investigation of the Involvement of Covalent Binding in Nevirapine-Induced Hepatic and Cutaneous
Idiosyncratic Adverse Drug Reactions
by
Amy M. Sharma A thesis submitted in conformity with the requirements
for the degree of Doctor of Philosophy
Graduate Department of Pharmaceutical Sciences Faculty of Pharmacy
University of Toronto
© Copyright by Amy M. Sharma, 2013

ii
Investigation of the Involvement of Covalent Binding in Nevirapine-
Induced Hepatic and Cutaneous Idiosyncratic Adverse Drug
Reactions
Amy M. Sharma
Doctor of Philosophy
Graduate Department of Pharmaceutical Sciences
Faculty of Pharmacy
University of Toronto
2013
Abstract Nevirapine (NVP) can cause serious idiosyncratic drug reactions (IDRs); specifically, skin
rash and hepatotoxicity. Treatment of rats or mice with NVP led to covalent binding to hepatic
proteins. Studies of this covalent binding including the use of a deuterated analog of NVP leading
to a decrease in oxidation of the methyl group indicated that the metabolite responsible for
covalent binding in the liver is a quinone methide.
Covalent binding in NVP-treated rats was also observed in the epidermis but by a different
pathway. Incubation of 12-OH-NVP sulfate with homogenized human and rat skin led to
extensive covalent binding. Inhibition of sulfation in the liver significantly decreased 12-OH-NVP
sulfate in the blood, but it did not prevent covalent binding in the skin or the rash. In contrast,
topical application of a sulfotransferase inhibitor prevented covalent binding in the skin as well as
the rash, but only where it was applied. In contrast to rats, treatment of mice with NVP did not
result in covalent binding in the skin or skin rash. These findings provide compelling evidence
that 12-OH-NVP sulfate formed in the skin is responsible for the skin rash.
IL-1β and IL-18 production in the skin of rats treated with NVP were increased. An anti-
IL-1ß antibody significantly decreased rash severity. These cytokines were also produced by
incubation of human keratinocytes with 12-OH-NVP sulfate. These data indicate that 12-OH-
NVP sulfate activates the NLRP3 inflammasome, a pathway known to be responsible for contact
hypersensitivity rashes.

iii
In summary, NVP was found to produce two different reactive metabolites, a quinone
methide species in the liver, and a benzylic sulfate in the skin. Significant liver injury did not
occur, presumably due to immune tolerance. Although it is usually assumed that reactive
metabolites are responsible for most IDRs, this is the first example to actually demonstrate that a
specific reactive metabolite is responsible for an IDR. This is also the first study to show that
sulfotransferase in the skin is responsible for bioactivation of a drug leading to a skin rash. It is
likely that there are other drugs that cause skin rashes by a similar mechanism.

iv
Acknowledgments “I can't go back to yesterday because I was a different person then.”
- Lewis Carroll, Alice in Wonderland
This thesis is truly the culmination of years of work on the nevirapine project, which I
have had the privilege to study. If I am to be completely honest, it also represents my blood, sweat
and tears. Jack, I want you to know that I have the deepest respect for you, and I am eternally
grateful to you for the work you have allowed me to do in your lab. I enjoyed every moment I
spent working on my project, and without your support, this work would not be. I was just so
happy to do the science. Each day, I would embark on my 1.5 hour commute and think how lucky
I was to have such an opportunity, such a beautiful lab to work in, under such renowned
supervision. I never once took this journey for granted and each day we became smarter, wiser,
better. It will be hard to move on and work in another lab after having you Jack, an amazing
person and scientist, as my mentor. The science will of course always be with me but it is the life
lessons I will always hold near.
Thank you for never wavering in the project and remaining true to the science – in the end,
we did succeed, but it was not without much struggle. I am evermore thankful for the experience
at having failed, failed, and failed again, only to have the hard work and discipline prevail. In my
wildest dreams I never thought the project would be this interesting and so rewarding - I guess
that’s what happens when you work with pure abandon from the gut of your soul. I also want to
thank you for the mental attitude I developed through the tough times. They say your attitude
dictates 99% of your outcome in life and the resilience, problem solving and patience I developed
working for you, I am grateful for. Most of all Jack, thank you for trusting me with this work, and
giving me the freedom to pursue science to its fullest. I hope we can always collaborate in the
future and share science, advice, and laughter.
I would like to thank my committee members, Dr. Peter Wells, Dr. Mario Ostrowski, Dr.
Peter Pennefather and Dr. Neil Shear for their valuable guidance over the years. Thanks also to
Dr. Tony Hayes, as you first introduced me to the field of IDRs with that assignment on
troglitazone that changed my life – thank you for your support. Thanks also to Dr. Peter O’Brien,
Dr. Jeff Henderson, Dr. David Dubins, and Dr. Ray Reilly, who let me teach and lecture for them,
through which I gained valuable professional experience.

v
To everyone else who played a part in this story – to lab members, especially Ms. Maria
Novalen, Dr. Yan Li, and Ms. Sandrine Fischer, for their foundational work on the project. The
many animals that were sacrificed for this work, your lives were not in vain. To my good and dear
friends, Stephanie MacAllister, Lutfiya Miller, Maya Latif, and Raza Mirza, who became my
support system – thank you deeply. To Dr. Henderson, for taking me in as an honorary member of
his lab, where I would go to hide out and just think. Thank you, Jeff.
To Dr. Dana Philpott, for her invaluable input on the NLRP3 studies and becoming one of
my role models in the field of immunology. Dana you are now also my trusted colleague and
friend and I look up to you so much, such a strong female scientist who makes it all so look so
easy (Dana, you’re a superstar!). When I ‘grow up,’ I hope I am the kind of mentor and
inspiration you have been to me.
To Dr. Lance R. Pohl, I have the deepest regard for your scientific style and your work,
and I want to thank you for introducing me to the origins of our field. I hope we can collaborate in
the future and one day determine the root causes of DILI.
To my new mentor and great scientist, a person I respect and admire very much, Dr.
Ruslan M. Medzhitov - I have never met such a larger-than-life scientist so full of generosity and
humble spirit. You have welcomed me so kindly into your entire world at Yale and I am
incredibly excited to begin my next chapter in your group. I hold you in such high esteem; you are
a scientist so ahead of his time, recognizing problems in nature before others even begin to realize
they exist. I hope that we will accomplish much and I want to thank you for this golden, once-in-
a-lifetime opportunity!
To KD, thank you for your love and support in the toughest of times. You are my best
friend and I could not have done this without your encouragement.
Finally, to Nika and my parents, for knowing that I could do it, even when I questioned if I
could. Mom and Dad you both live for your children, and in turn, our success is your most revered
reward. Mom, you are my biggest fan and I am grateful for your care – your unconditional
acceptance for your children means the world to me. Dad, I know you came here with eight bucks
in your pocket but I hope you know it was worth it – thank you for the life you have given us. I
dedicate this thesis to you both. You are my inspiration for all things good and true in this world,
and I love and admire you both more than words could ever capture.

vi
Table of Contents Abstract ........................................................................................................................................... ii
Acknowledgments .......................................................................................................................... iv
List of Figures ................................................................................................................................ xi
List of Tables ................................................................................................................................ xxi
List of Schemes ........................................................................................................................... xxii
List of Abbreviations .................................................................................................................. xxiii
List of Thesis Publications ......................................................................................................... xxiv
CHAPTER 1:
INTRODUCTION ........................................................................................................................... 1
1.1 Adverse Drug Reactions – An Overview ......................................................................... 2
1.1.1 Types of Adverse Drug Reactions ................................................................................. 2
1.2 Idiosyncratic Drug Reactions (IDRs) .................................................................................... 3
1.3 Proposed Mechanisms of Idiosyncratic Drug Reactions ....................................................... 5
1.3.1 Hapten Hypothesis ......................................................................................................... 5
1.3.2 Danger Hypothesis ......................................................................................................... 8
1.3.3 Pharmacological Interaction Hypothesis ...................................................................... 10
1.4 Idiosyncratic Hepatotoxicity ............................................................................................... 12
1.4.1 Hepatic Function and Morphology .............................................................................. 12
1.4.2 Biotransformation in the Liver ..................................................................................... 12
1.4.3 Liver Immune System in Relation to Hepatotoxicity ................................................... 14
1.5 Idiosyncratic Cutaneous Toxicity ....................................................................................... 16
1.5.1 Skin Structure and Function ......................................................................................... 16
1.5.2 Metabolic Enzymes in Skin .......................................................................................... 18
1.5.3 Immune Function of the Skin ....................................................................................... 19
1.5.4 Implications of Cutaneous Biotransformation on Immune-Mediated Skin Rashes ..... 21
1.6 Role of Drug Metabolism, Reactive Metabolites, and Covalent Binding in IDRs ............ 23
1.6.1 Sulfotransferase Enzymes in Drug Metabolism and Toxicity ..................................... 23
1.7 Nevirapine Toxicity ............................................................................................................. 25
1.7.1 Animal Model of Nevirapine-Induced Skin Rash ........................................................ 26
1.7.2 Nevirapine Metabolism leading to Skin Rash .............................................................. 27
1.7.3 Role of Sulfation in Nevirapine Skin Rash .................................................................. 28
1.7.4 Nevirapine Metabolism in the Liver and Lack of Hepatotoxicity in Rats ................... 30
1.8 Research Hypotheses .......................................................................................................... 30

vii
CHAPTER 2:
Bioactivation of Nevirapine to a Reactive Quinone Methide: Implications for Liver Injury ....... 32
2.1 Abstract ............................................................................................................................... 33
2.2 Introduction ......................................................................................................................... 33
2.3 Materials and Methods ........................................................................................................ 36
2.3.1 Chemical Materials. ..................................................................................................... 36
2.3.2 Instruments and Software. ............................................................................................ 36
2.3.3 Synthesis of 12-trideutero-NVP (DNVP). ................................................................... 36
2.3.4 Production of Anti-NVP Anti-Serum in Male White New Zealand Rabbits. .............. 37
2.3.5 Animal Care. ................................................................................................................ 40
2.3.6 Treatment of Animals with NVP, 12-OH-NVP, DNVP, or ABT. ............................... 40
2.3.7 Incubations with Microsomes or Supersomes. ............................................................. 41
2.3.8 Quantification of NVP and its Metabolites from Microsomal Incubations. ................ 41
2.3.9 Mass Spectrometry Analysis. ....................................................................................... 42
2.3.10 Analysis of Covalent Binding Using SDS-PAGE and Immunoblotting. ................... 42
2.3.11 Analysis of in Vivo Covalent Binding using Immunohistochemistry. ....................... 43
2.3.12 Plasma Alanine Transaminase and Cytokine Analysis. ............................................. 43
2.4 Results ................................................................................................................................. 44
2.4.1 Characterization of the Anti-NVP-NAC-KLH Antiserum. .......................................... 44
2.4.2 Covalent Binding of NVP, DNVP, or 12-OH-NVP to Hepatic Microsomes in Vitro and
Comparison to in Vivo Hepatic Covalent Binding. ............................................................... 45
2.4.3 Covalent Binding of NVP to Expressed Rat CYP2C11 or CYP3A1 Supersomes, or of
NVP, DNVP, or 12-OH-NVP to Human Hepatic Expressed CYP3A4 Supersomes. .......... 47
2.4.4 Covalent binding of NVP or 12-OH-NVP to Hepatic Proteins from Female BN Rats
Treated with NVP or 12-OH-NVP. ....................................................................................... 49
2.4.5 Immunohistochemistry of Liver from NVP- or DNVP-Treated or NVP + ABT Co-
treated Female BN Rats. ........................................................................................................ 51
2.4.6 Oxidation of NVP or 12-OH-NVP by Rat Liver Microsomes. .................................... 52
2.4.7 Covalent Binding, Serum ALT levels, INF-γ, and IL-6 Levels in Mice...................... 52
2.4.8 Liver Histology and ALT in Male Cbl-b-/-
or C57BL/6 Mice Treated with NVP. ...... 58
2.4.9 Comparison of Hepatic Covalent Binding of NVP between Mice and Female BN Rats.
............................................................................................................................................... 60
2.5 Discussion ........................................................................................................................... 62

viii
CHAPTER 3:
Nevirapine Bioactivation and Covalent Binding in the Skin ........................................................ 66
3.1 Abstract .............................................................................................................................. 67
3.2 Introduction ........................................................................................................................ 68
3.3 Materials and methods ....................................................................................................... 71
3.3.1 Chemicals. .................................................................................................................... 71
3.3.2 Animal Care. ................................................................................................................ 71
3.3.3 Primary and Secondary Treatment of Animals with NVP or 12-OH-NVP. ................ 72
3.3.4 Separation of Dermis and Epidermis and Preparation of Homogenates of Skin Fractions
or of Whole Rat Skin. ............................................................................................................ 72
3.3.5 Preparation of Cytosol, S9, or Microsomes from NVP-treated Rat Epidermal or Dermal
Fractions. ............................................................................................................................... 73
3.3.6 Preparation of Human Skin Dermatome. ..................................................................... 73
3.3.7 Cytosolic or S9 Fractions from Rat or Human Skin or Liver. ..................................... 73
3.3.8 Incubation of Human or Rat Skin or Fractionated Skin with NVP, 12-OH-NVP, or 12-
OH-NVP Sulfate. .................................................................................................................. 74
3.3.9 In Vitro Metabolism of 12-OH-NVP and NVP. .......................................................... 74
3.3.10 Covalent Binding Using SDS PAGE and Immunoblotting. ...................................... 75
3.3.11 Preparation of BN Rat Skin for Histology. ................................................................ 76
3.4 Results ................................................................................................................................ 76
3.4.1 Attempts to Detect In Vivo Covalent Binding in Whole Skin. .................................... 76
3.4.2 Covalent Binding of NVP, 12-OH-NVP, or 12-OH-NVP Sulfate to Human or Rat Skin
in Vitro. ................................................................................................................................. 78
3.4.3 Covalent Binding of NVP to Rat Skin in Vivo. ........................................................... 81
3.4.4 Early Histological Changes in the Skin in Response to NVP Treatment. .................... 84
3.4.5 Covalent Binding of NVP to Mouse Skin in Vivo. ...................................................... 86
3.4.6 Covalent Binding of NVP to Subcellular Rat Skin Fractions in Vivo. ........................ 87
3.4.7 Covalent Binding of 12-OH-NVP to Human or Rat Liver or Skin Proteins in the
Presence or Absence of PAPS. .............................................................................................. 88
3.4.8 Covalent Binding of 12-OH-NVP or NVP to Human or Rat Liver or Skin Subcellular
Fractions in the Presence or Absence of PAPS or NADPH. ................................................. 90
3.4.9 Sulfation and Oxidation of NVP and 12-OH-NVP in Mouse and Rat Skin. ............... 94
3.4.10 Anti-NVP and Autoantibodies in NVP-Treated Rats. ............................................... 98
3.5 Discussion ........................................................................................................................ 101
3.6 Supplemental Material ...................................................................................................... 105
3.6.1 Separation of Dermis and Epidermis of the Ear and Preparation of Homogenates. .. 105

ix
3.6.2 Grading of Skin Rash. ................................................................................................ 105
3.6.3 Covalent Binding and Histology in the Ears from NVP- or 12-OH-NVP-Treated Rats.
............................................................................................................................................. 109
CHAPTER 4:
12-OH-Nevirapine Sulfate, Formed in the Skin, is Responsible for Nevirapine-Induced Skin Rash
..................................................................................................................................................... 112
4.1 Abstract ............................................................................................................................ 113
4.2 Introduction ...................................................................................................................... 114
4.3 Materials and methods ..................................................................................................... 116
4.3.1 Chemical Materials and Reagents. ............................................................................. 116
4.3.2 Animal Care. .............................................................................................................. 116
4.3.3 Quantification of NVP, 12-OH-NVP, 12-OH-NVP Sulfate, and 4-COOH-NVP in
Plasma. ................................................................................................................................ 117
4.3.4 Sulfation Inhibition Studies. ....................................................................................... 118
4.3.5 Separation of Skin Dermis and Epidermis and Preparation of Homogenates. ........... 119
4.3.6 Preparation of Human Skin Dermatome. ................................................................... 119
4.3.7 Incubation of Human Skin or Expressed Human SULT 1A1*1 with 12-OH-NVP or 12-
OH-NVP Sulfate, With or Without PAPS. ......................................................................... 120
4.3.8 Incubation of Rat Liver or Skin Cytosol or Human Liver Cytosol with 12-OH-NVP and
1-Phenyl-1-hexanol in the Presence and Absence of PAPS. .............................................. 120
4.3.9 Covalent Binding Using SDS-PAGE and Immunoblotting. ...................................... 120
4.3.10 Preparation of BN Rat Skin for Histology. .............................................................. 121
4.4 Results .............................................................................................................................. 122
4.4.1 General Scheme to Study the Role of 12-OH-NVP Sulfation on the Skin Rash. ...... 122
4.4.2 Effect of Salicylamide on 12-OH-NVP Sulfate Levels and Rash. ............................. 122
4.4.3 Effects of DHEA on NVP Metabolism and Skin Rash. ............................................. 125
4.4.4 Effects of 1-Phenyl-1-Hexanol on Covalent Binding and Rash. ............................... 126
4.4.5 In Vitro Inhibition of Covalent Binding by 1-Phenyl-1-Hexanol. ............................. 136
4.5 Discussion ........................................................................................................................ 140
4.6 Supplemental Material ...................................................................................................... 143
4.6.1 Quantification of NVP, 12-OH-NVP, 12-OH-NVP Sulfate, and 4-COOH-NVP in
Urine. ................................................................................................................................... 143
4.6.2 Grading of Skin Rash. ................................................................................................ 143

x
CHAPTER 5:
Discussion, Conclusions and Summary ...................................................................................... 148
5.1 Hypotheses Revisited ........................................................................................................ 149
5.2 Discussion and Limitations ............................................................................................... 150
5.3 Future Directions ............................................................................................................... 153
Bibliography ................................................................................................................................ 156

xi
List of Figures
CHAPTER 1
Figure 1-1. Reaction of penicillin with protein nucleophiles via spontaneous ring opening in a
hapten type mechanism.
Figure 1-2. The Hapten Hypothesis: a reactive, unmetabolized parent drug, or reactive
metabolite, conjugates to protein in a covalent manner. Upregulation of appropriate costimulatory
molecules allows for the elicitation of an immune response. Adapted from Uetrecht, 2007.
Figure 1-3. The Danger Hypothesis: stressed or damaged cells release endogenous ‘danger
signals’ which activate APCs, leading to upregulation of costimulatory molecules (B7 on APCs)
which interact with CD28 on T cells leading to an immune response. Adapted from Uetrecht,
2007.
Figure 1-4. The P-I Hypothesis: A reactive parent drug binds directly to the MHC-TCR complex
in a reversible manner (signal 1), leading to an immune response. Adapted from Uetrecht, 2007
Figure 1-5. Proposed pathway of drug bioactivation, leading to haptenation and cellular
mechanisms of hepatocyte death. Adapted from Kaplowitz, 2004.
Figure 1-6. Proposed mechanisms of DILI, including drug bioactivation leading to reactive
intermediate which may cause hepatocyte damage invoking immune mediated responses. A
balance of hepatoprotective and inflammatory factors dictate the potential for toxicity. Adapted
from Holt and Ju, 2005.
Figure 1-7. Cellular composition of the skin. Taken from Feldmeyer et. al, 2010.

xii
CHAPTER 2
Figure 2-1. Bioactivation pathway of NVP leading to liver injury.
Figure 2-2. ELISA analysis showing (A) binding of the anti-NVP-NAC-KLH antiserum to the
NVP-NAC-BSA conjugate, KLH, or BSA and (B) the effect of preincubation of the antiserum
with NVP or its metabolites on the binding of the antisera to the NVP-NAC-BSA conjugate. Data
represent the mean ± s.d. from 3 incubations.
Figure 2-3. (A) Comparison of covalent binding of 12-OH-NVP (lanes 2, 5) and DNVP (lane 3,
6) with that of NVP (lane 4, 7) after a 30 or 60 min incubation with male BN rat microsomes (1
mg/mL protein) at a drug concentration of 1 mM. For comparison, covalent binding to hepatic
proteins is shown after 8 days of treatment of female rats with 12-OH-NVP (159 mg/kg/day, lane
9) or NVP (150 mg/kg/day, lane 10). Protein loading was 15 µg for lanes 1-7 and 20 µg for lanes
8-10. (B) Comparison of covalent binding of 12-OH-NVP (lanes 2, 5) and DNVP (lanes 3, 6) with
that of NVP (lanes 4, 7) at a concentration of 1 mM after a 30 or 60 min incubation with
microsomes (1 mg/mL protein) from male C57BL/6 mice. For comparison, covalent binding to
hepatic proteins is shown after 6 weeks of treatment of C57BL/6 mice with NVP at a dose of 950
mg/kg/day in food. Protein loading was 13 µg for lanes 1-7 and 20 µg for lanes 8-9. (C)
Comparison of covalent binding of NVP to hepatic microsomes from male C57BL/6 mice (lanes
2-4) or male BN rats (lanes 6-8) after a 15, 30, or 60 min incubation at a drug concentration of 1
mM and microsome concentration of 1 mg/mL protein. Protein loading was 20 µg per lane. The
primary antiserum dilution was 1:500 and that of the secondary antisera was 1:5000.
Figure 2-4. Covalent binding of NVP to expressed male rat CYP2C11 (A) or CYP3A1 (B) in
vitro. Protein concentration for each incubation was 0.8 mg/mL with 0.5 mM of drug. For
immunoblots, protein loading was 9 µg and 7.5 µg per lane for A and B, respectively. (+)
indicates incubations containing NVP while (–) indicates incubations lacking NVP. Proteins were
resolved on 12% gels with 1:100 dilution of primary anti-serum followed by 1:2000 dilution of
secondary antisera. Comparison of covalent binding of 12-OH-NVP (lanes 2, 5), or DNVP (lanes
3, 6) with that of NVP (lanes 4, 7) to human CYP3A4 with a drug concentration of 1 mM and
protein concentration in each incubation of 1 mg/mL (C). Proteins (10 µg/lane) were resolved on
an 8% gel. Dilutions of antisera were 1:500 for the primary anti-serum and 1:5000 for the
secondary antisera.

xiii
Figure 2-5. (A) Covalent binding to hepatic proteins from female BN rats fed NVP (150 mg/kg)
or 12-OH-NVP (159 mg/kg) for 8 days. Protein loading was 12 µg per lane. Samples were
resolved on an 8% gel. A 1:500 dilution of primary anti-serum was followed by 1:5000 dilution of
secondary antisera. (B) Incubation of the anti-NVP serum with 2 mM NVP for 2 h at 37 °C
blocked most of the binding (left side of panel) to samples from livers of 12-OH or NVP treated
rats. Samples for both panels A and B were prepared, run, blocked, incubated with secondary
antibody, and imaged at the same time and protein loading was 10 µg/well of protein per lane.
Figure 2-6. Immunohistochemistry of liver sections from female BN rats; blank control, NVP
treatment (150 mg/kg/day x 7 days in food), DNVP treatment (150 mg/kg/day x 7 days in food),
ABT treatment (50 mg/kg/day x 28 days by gavage), or NVP (150 mg/kg/day) + ABT (50
mg/kg/day) x 28 days by gavage. Slides were incubated with 1:100 dilution of primary antisera
and 1:2000 dilution of the secondary antisera. The slides were counterstained with Mayer’s
hematoxylin, magnification 20x.
Figure 2-7. 4-COOH-NVP concentrations from incubations of 12-OH-NVP with microsomes
from male (n = 3) and female (n = 1) BN rats.
Figure 2-8. (A) Changes in ALT in male C57BL/6 mice treated with NVP (950 mg/kg/day) for 4
weeks. Values are based on the mean of triplicate readings per time point per animal ± S.D, n=5
treated mice or n = 4 control mice. Unpaired t-test, 7 d.f., p<0.05. (B) Corresponding covalent
binding of NVP at the same dose in male BALB/c (n=2) or C57BL/6 (n=3) mouse livers after 6
weeks of treatment. Protein loading was 20 µg per lane. Samples were resolved on an 8% gel.
Figure 2-9. (A) Plasma ALT levels in male Cbl-b-/-
mice fed NVP orally for 14 days (950
mg/kg/day). Values are based on the mean of triplicate readings per time point per animal ± S.D,
n=5 treated mice or n = 4 control mice. Unpaired t-test, 7 d.f., p<0.05. (B) Covalent binding of
NVP in the livers of the same Cbl-b-/-
mice. (C) Plasma ALT levels in NVP-treated (950
mg/kg/day) female Cbl-b-/-
mice, n=4 treated or n=4 control mice. Values are based on the mean
of triplicate readings per time point per animal ± S.D, n=5 treated mice or n = 4 control mice.
Unpaired t-test, 6 d.f., p<0.05. (D) Covalent binding of NVP in the livers of the same mice.
Protein loading was 25 µg per lane. Samples were resolved on 10-20% gradient gels. A 1:500
dilution of primary antisera followed by 1:5000 dilution of secondary antisera was used.

xiv
Figure 2-10. Serum IL-6 (A) or IFN- (B) from control and NVP-treated Cbl-b-/-
mice at day 7 of
NVP treatment. Animals showing gross necrosis are displayed separately.
Figure 2-11. H&E staining of livers from Cbl-b-/-
mice treated with NVP for 2 weeks. (A)
Untreated control liver with normal ALT; (B) liver from a NVP-treated mouse with gross necrosis
and an ALT of 271 U/L, and (C) liver from another NVP-treated mouse with gross necrosis and
ALT of 313 U/L. Areas of massive hepatocyte necrosis surrounded by viable hepatocytes are
shown in (B) and (C).
Figure 2-12. H&E staining of livers from male C57BL/6 mice treated with NVP for 3 weeks. (A)
Untreated control liver with a normal ALT; (B) liver from a NVP-treated mouse with very mild
necrosis (appearing as the thin band around the capsule) and ALT of 94 U/L, and (C) liver from
another NVP-treated mouse with an ALT of 75 U/L. Changes to the liver parenchyma due to
enlargement of hepatocytes in the periacinar regions and extensive expansion of the endoplasmic
reticulum are also present in both (B) and (C).
Figure 2-13. Comparison of covalent binding of NVP to hepatic proteins in mice and rats. NVP
was fed to rats in a time course manner from 1 to 8 days at 150 mg/kg orally in food. Mice were
given 950 mg/kg/day for 2 weeks or 10 weeks. C57BL/6 males given NVP for 2 weeks are
represented by C57.1 and C57.2. Each lane was loaded with 20 µg of protein. Samples were
resolved on a 4-20% gradient gel. A 1:500 dilution of primary antisera followed by 1:5000
dilution of secondary was used.

xv
CHAPTER 3
Figure 3-1. (A) Immunoblot showing covalent binding of NVP (150 mg/kg) to whole rat skin in
vivo with a major artifact band in each lane indicated by the arrows. From left to right: primary
treatment days 22, 24, 25, rechallenge (RCH, 7 days), or untreated control. Each lane was loaded
with 25 µg of protein. Exposure duration in the imager was 3 min. (B) Epidermis floating above
dermis from trypsin-separated skin from a rat treated for 21 days with NVP (left panel); isolated
epidermal layer (right panel).
Figure 3-2. (A) Immunoblot showing in vitro covalent binding of 1 mM each NVP, 12-OH-NVP,
or 12-OH-NVP sulfate to rat whole skin homogenate containing both dermis and epidermis after
incubation for 30 or 60 min. (B) Covalent binding of 1 mM NVP, 12-OH-NVP, or 12-OH-NVP
sulfate to isolated epidermal or dermal homogenates prepared from a control rat. (C) Covalent
binding of 1 mM NVP, 12-OH-NVP, or 12-OH-NVP sulfate to isolated epidermal or dermal
homogenate prepared from a control rat showing that preincubation of the primary antisera with 1
mM NVP for 2 h at 37 °C blocked binding of the antibody. Proteins (7.5 µg/well) were loaded in
immunoblots A-C. (D) Human dermatome skin incubated with 1 mM each of 12-OH-NVP, NVP,
or 12-OH-NVP sulfate compared to 1 mM 12-OH-NVP +/- 0.3 mM PAPS (1 mg/mL protein). (E)
Covalent binding of 1 mM NVP, 12-OH-NVP, or 12-OH-NVP sulfate to isolated human
dermatome homogenate showing that preincubation of the primary antiserum with 1 mM NVP for
2 h at 37 °C blocked binding of the antibody. Protein (12 µg/well) was loaded for blots D-E.
Figure 3-3. Immunoblots showing epidermal covalent binding in vivo after treatment with NVP
or 12-OH-NVP for either (A) 7 days or (B) 21 days. (C) Immunoblots showing that preincubation
of the primary antiserum with 1.5 mM NVP for 2 h at 37˚C blocked binding of the antibody (right
panel) to the drug-modified proteins after treatment with NVP for 21 days or after rechallenge
with NVP (RCH), left panel. Epidermal protein loading was 15 µg/well.
Figure 3-4. (A) Immunoblot showing covalent binding in the epidermis of NVP-treated female
BN rats on days 10, 15, or 21 (n = 2 animals per time point) of NVP treatment. Each lane
represents an individual animal with 15 µg/well of protein loaded in each well. (B) Representative
H&E stained rat skin sections comparing the early infiltration of immune cells into the dermis or

xvi
epidermis of NVP- or 12-OH-NVP-treated rats. Marked acanthosis (thickening of the epidermis)
combined with early lymphocyte infiltrate at the dermal-epidermal junction can be observed by
day 7 of 12-OH-NVP-treated animals. By day 21 there is an increase in the cellular infiltrate with
areas of detachment of the epidermis. Magnification 20x.
Figure 3-5. Skin histology of PD-1-/-
knockout mice. No immune infiltrate or acanthosis was
observed as was seen with NVP-treated rats.
Figure 3-6. (A) Comparison of covalent binding to S9 with that to the cytosolic fractions from the
skin of NVP-treated rats, isolated from either the dermis or epidermis. (B) Comparison of
covalent binding to S9 with that to microsomal fractions from the skin of NVP-treated rats,
isolated from either the dermis or epidermis. All animals were treated for 21 days. Protein loading
was 10 µg/well.
Figure 3-7. (A) Immunoblot of rat skin S9 or human female (second right lane) or human male
(most right lane) liver cytosol after incubation with 12-OH-NVP in the presence and absence of
PAPS. (B) Immunoblot of rat skin S9 or cytosol, or female human or rat liver cytosol after
incubation with 12-OH NVP in the presence of absence of PAPS. (C) Covalent binding of 12-
OH-NVP to human liver S9, human dermatome skin, or rat skin S9 in the presence and absence of
PAPS. Protein loading was 12 µg/well.
Figure 3-8. (A) Immunoblot comparing covalent binding of 12-OH-NVP to rat liver S9 versus rat
skin S9 in the presence or absence of either PAPS or a NADPH-regenerating system (NRS). (B)
Comparison of covalent binding of NVP to rat liver S9 versus rat skin S9 incubated in the
presence of absence of either PAPS or a NADPH-regenerating system (NRS). (C) Comparison of
covalent binding of 12-OH-NVP to rat liver S9 versus human liver S9 incubated in the presence
or absence of either PAPS or NRS. (D) Comparison of covalent binding of NVP to human liver
S9 versus rat liver S9 incubated with or without either PAPS or NRS. (E) Covalent binding of
NVP or 12-OH-NVP to human skin in the presence or absence of PAPS or NRS. Protein loading
was 12 µg/well. 1 mM of 12-OH-NVP or NVP was used for each incubation.

xvii
Figure 3-9. (A) Immunoblot comparing the covalent binding of NVP to mouse vs. rat liver S9 in
the presence of absence of either PAPS or an NADPH-generating system (NRS). (B) Comparison
of the covalent binding of 12-OH-NVP to either mouse or rat liver S9 in the presence or absence
of either PAPS or NRS. (C) Comparison of covalent binding of NVP to mouse vs rat skin S9
either in the presence or absence of PAPS or NRS. (D) Comparison of covalent binding of 12-
OH-NVP to mouse vs. rat skin S9 either in the presence or absence of PAPS or NRS. Protein
loading was 12 µg/well. 1 mM of 12-OH-NVP or NVP was present in each incubation.
Figure 3-10. Detection of anti-NVP and autoantibodies in the serum of a rat after rechallenge
with NVP (A) Liver homogenate (10 µg/lane) from an untreated (control) rat, NVP-treated rats, or
12-OH NVP-treated rats run on SDS PAGE and stained with serum (diluted 1:500) from a rat that
had been rechallenged with NVP after earlier development of a NVP-induced rash (left panel) or
with serum from an untreated control rat (right panel). A 1:4000 dilution of goat anti-rat HRP
linked antibody was used as the secondary antibody to visualize the binding. Blots were imaged
for 3 minutes on medium exposure. (B) Using serum from the same rechallenged rat, an
analogous experiment was performed using fractionated skin protein (20 µg/lane) for the
epidermis, designated ‘E’, or dermis, marked ‘D’) from untreated (control) or NVP-treated rats.
Blots were run, blocked, incubated with secondary, and imaged together.
Supplemental Figure 3S-1. (A) Method to fractionate ear using dorsal-ventral axis separation is
shown. Ear pieces were floated on 0.625% trypsin overnight at 4 °C to ensure complete
epidermal-dermal separation. (B) Immunoblot experiments comparing the epidermis from the
neck or ear from NVP- or 12-OH-NVP-treated female BN rats; 12 µg protein/well. Lane
designations are as follows: 1 & 2 = epidermis from the neck of control rats; 3 = epidermis from
the neck of a 12-OH-NVP-treated rat; 4 = epidermis from the neck of a NVP-treated animal; 5 =
epidermis from the ear of a 12-OH-NVP-treated rat; 6 = epidermis from the ear of a NVP-treated
rat. (C) H&E images of ear sections taken from each treatment group (representative slide from 1
of 4 rats per group shown). Magnification 20x.

xviii
CHAPTER 4
Figure 4-1. (A) Incidence of skin rash, (B) plasma concentrations of NVP, (C) 12-OH-NVP, and
(D) 12-OH-NVP sulfate in female Brown Norway rats treated with NVP only (100 mg/kg/day, n
= 4), in combination with oral DHEA (50 and 100 mg/kg/day) or in combination with oral
salicylamide (274 mg/kg/day).
Figure 4-2. (A) Immunoblot of the epidermis comparing individual 12-OH-NVP-treated rats to
NVP + oral salicylamide cotreated rats (N+Sal) or NVP only-treated rats, against 0.5% methyl
cellulose gavaged controls. Protein loading was 15 µg/lane. (B) Skin histology of NVP + oral
salicylamide cotreated rats, n = 4. (C) Skin histology compared between various treatment groups:
normal and gavaged controls are normal without a cellular infiltrate in the dermis, while NVP, 12-
OH-NVP and NVP + oral salicylamide treated rats display keratinocyte necrosis within the
epidermis, with marked inflammatory infiltrate at the dermal-epidermal junction. A representative
photo from one of four animals per group is shown. All rats represented in this figure were treated
for 21 days. Magnification was 20x for all slides in this figure.
Figure 4-3. (A) Diagram of the preliminary sites for administration of topical DHEA or topical 1-
phenyl-1-hexanol to determine their effect on the NVP-induced skin rash. In 2/2 animals tested,
the rash was slightly milder with DHEA, but it was completely prevented in 1-phenyl-1-hexanol-
treated areas only (photos not shown). (B) Diagram of sites employed in 2 independent trials to
test the effect of topical 1-phenyl-1-hexanol on the NVP-induced skin rash. Five animals in total
were treated with NVP (150 mg/kg/day) in food and 1-phenyl-1-hexanol (20 mg/kg/day) on the
skin. In 100% of the animals, the rash was prevented by topical 1-phenyl-1-hexanol. One
representative rat from each study is shown above. Photos showing (C) skin from the back of a
control rat, (D) skin from the back of the NVP only-treated rat, (E) vehicle versus 1-phenyl-1-
hexanol-treated areas from an inhibitor-treated rat (topical treatment).
Figure 4-4. Using skin isolated from rats cotreated with NVP and topical 1-phenyl-1-hexanol
using the schematic shown in Figure 3B, epidermal immunoblot analysis was performed. (A)
Immunoblot of epidermis from rash areas versus vehicle areas from the epidermis of inhibitor-
treated rats cotreated with NVP. (B) Immunoblot of epidermis from topical 1-phenyl-1-hexanol
areas versus vehicle areas from epidermis of inhibitor-treated rats compared with that of an

xix
untreated control and a NVP-treated control. 15 µg of protein per lane was loaded for each of A
and B.
Figure 4-5. Representative histology of rat skin isolated from rats cotreated with NVP and topical
1-phenyl-1-hexanol using the schematic shown in Figure 3B. (A) H&E stained sections from
upper neck/rash area from control (Ct), nevirapine-treated (NVP), or 1-phenyl-1-hexanol (Ph) rat
number 1 shown in 3C-E; (B) H&E stained sections from left shoulder/vehicle area from control
(Ct), nevirapine-treated (NVP), or 1-phenyl-1-hexanol (Ph) rat number 1 shown in 3C-E; (C)
H&E stained sections from right shoulder/1-phenyl-1-hexanol-treated area from control (Ct),
nevirapine-treated (NVP), or 1-phenyl-1-hexanol (Ph) rat number 1 shown in 3C-E.
Magnification 20x for all slides in this panel.
Figure 4-6. (A) Second topical schematic used to test the inhibitor 1-phenyl-1-hexanol. (B)
Immunoblot of epidermis from areas of vehicle or inhibitor treated areas using the second
schematic shown in 6A. The control is epidermis from the untreated control rat; Ph1 or Ph2 are
topical inhibitor-treated epidermal areas from rat # 1 or 2, respectively; Vh1 or Vh2 are vehicle
treated epidermal areas for each rat, and RA1 or RA2 are from rash areas with no topical
treatment. NVP is from the epidermis of the back of the neck for the NVP-treated positive control
rat. Protein loading was 15 µg/lane.
Figure 4-7. Histology with H&E staining of skin isolated from rats cotreated with NVP and
topical 1-phenyl-1-hexanol using the schematic shown in Figure 6A. The upper left slide of each
panel is from a control animal without NVP treatment, the upper right slide is from a NVP-treated
animal with no topical treatment, and the lower two slides are from animals with NVP + topical
treatment. (A) skin from upper back with no topical treatment representing the typical rash; (B)
midback where the vehicle was applied in inhibitor-treated animals only (lower two slides); (C)
the lower back were 1-phenyl-1-hexanol was applied in inhibitor-treated animals only (lower 2
slides). Magnification 20x. (D) Preincubation of the primary anti-NVP serum with 1.5 mM NVP
dissolved in DMSO for 2 h at 37 °C prevented covalent binding of the anti-serum to epidermal
samples from the samples shown in Figure 3C-E, except for one artifact band. The DMSO control
(right most lane) where the primary anti-serum was incubated with DMSO alone. Protein loading
was 15 µg/lane.

xx
Figure 4-8. (A) Immunoblot of isolated rat liver cytosol or rat skin cytosol incubated with 12-
OH-NVP or a combination of 12-OH-NVP (12-OH) and 1-phenyl-1-hexanol in vitro, in the
presence and absence of PAPS. (B) Immunoblot of rat skin cytosol versus female human liver
cytosol incubated with or without PAPS and 12-OH-NVP or 12-OH-NVP and 1-phenyl-1-
hexanol. (C) Human skin ‘dermatome’ homogenized and incubated with 12-OH-NVP or 12-OH-
NVP + 1-phenyl-1-hexanol to show the same phenomenon exists in human skin. (D) Human
SULT 1A1*1 incubated with 1 mM 12-OH-NVP (12-OH) or 12-OH-NVP and 1-phenyl-1-
hexanol +/- 0.3 mM PAPS, or 12-OH-NVP sulfate (12-Sulfate). 12 µg/well protein was loaded for
each blot.
Supplemental Figure 4S-1. Urinary excretion of (A) 12-OH-NVP, (B) 4-COOH-NVP, (C) 12-
OH-NVP sulfate, (D) 2-OH-NVP, and (E) 3-OH-NVP from rats treated with NVP (100
mg/kg/day), NVP + DHEA (100 mg/kg/day) each or NVP + salicylamide (274 mg/kg/day; n = 4
in each group). Data depicts the mean ± SD.
Supplemental Figure 4S-2. H&E stained sections comparing the histology of rat skin in response
to (A) NVP treatment, (B) NVP + topical DHEA cotreatment, or (C) control rats. Magnification =
20x.

xxi
List of Tables
CHAPTER 1
Table 1-1. Human Sulfotransferase Isoforms and Expression.
CHAPTER 3
Table 3S-1. Day 7 Skin Rash Grading.
Table 3S-2. Day 10 and 15 Skin Rash Grading.
Table 3S-3. Day 21 Skin Rash Grading.
CHAPTER 4
Table 4-1. Inhibitors of sulfation and dosing method.
Table 4-2. Comparison of results obtained from 12-OH-NVP sulfate inhibitor studies.
Table 4S-1. Day 21 Skin Rash Grading.

xxii
List of Schemes
CHAPTER 2
Scheme 2-1. Synthetic pathway of the immunogen used for induction of anti-NVP antiserum.
CHAPTER 3
Scheme 3-1. Proposed chemical mechanism of NVP-induced skin rash resulting from covalent
binding of 12-OH-NVP sulfate in the skin.
Scheme 3-2. Proposed bioactivation pathway of NVP leading to immune-mediated skin rash.
CHAPTER 4
Scheme 4-1. Depiction of schematic used to prevent rash in this study.

xxiii
List of Abbreviations
12-OH-NVP 12-hydroxynevirapine
12-OH-NVP sulfate 12-sulfatenevirapine
2-OH-NVP 2-hydroxynevirapine
3-OH-NVP 3-hydroxynevirapine
4-COOH-NVP 4-carboxynevirapine
ADR adverse drug reaction
ALT alanine aminotransferase
APC antigen presenting cell
BN rat Brown Norway rat
CTL cytotoxic T-lymphocyte
DHEA dehydroepiandosterone
DILI drug-induced liver injury
DNA deoxyribonucleic acid
FMO flavin monoxygenase
HIV human immunodeficiency virus
HLA human leukocyte antigen
HPLC high performance liquid chromatography
IDILI idiosyncratic drug-induced liver injury
IDR idiosyncratic drug reaction
IL interleukin
KC keratinocyte
LC Langerhans cell
LC/MS liquid chromatography/mass spectrometry
LTT lymphocyte transformation test
MHC major histocompatibility complex
NADPH nicotinamide adenine dinucleotide phosphate
(reduced form)
NLR nod-like receptor
NVP nevirapine
pAPC professional antigen presenting cell
P450 cytochrome P450
PAPS 3’-phosphoadenosine-5’-phosphosulfate
P-I hypothesis pharmacological interaction hypothesis
PRR pattern recognition receptor
SA salicylamide
SJS Steven’s-Johnson syndrome
SLE systemic lupus erythematous
SN2 substitution nucleophilic 2
SULT sulfotransferase
TEN toxic epidermal necrolysis
Th1 t-helper cell 1
TLR toll-like receptor
UV ultraviolet

xxiv
List of Thesis Publications
First Author:
Bioactivation of Nevirapine to a Reactive Quinone Methide: Implications for Liver Injury.
Amy M. Sharma, Yan Li, Maria Novalen, M. Anthony Hayes, and Jack Uetrecht.
Chemical Research in Toxicology, 2012. 25, 1708-1719.
Demonstration of Nevirapine Bioactivation and Covalent Binding in Skin.
Amy M. Sharma, Klaus Klarskov, Jack Uetrecht.
Chemical Research in Toxicology, 2013. 26, 410-421.
12-OH-Nevirapine Sulfate, Formed in the Skin, is Responsible for Nevirapine-Induced Skin Rash.
Amy M. Sharma, Maria Novalen, Tadatoshi Tanino, Jack P. Uetrecht.
Chemical Research in Toxicology, 2013. 26, 817-827.
Bioactivation of Drugs in the Skin and its Relationship to Skin Rashes.
Amy M. Sharma and Jack Uetrecht.
Drug Metabolism Reviews, 2013 (in submission).
A Mechanism for Cutaneous Drug-Induced Hypersensitivity: Role of the NLRP3 Inflammasome in
Nevirapine-Induced Skin Rash.
Amy M. Sharma, Dana J. Philpott, Jack Uetrecht. Journal of Investigative Dermatology, 2013 (in
preparation).
Co-author:
Animal Models of Idiosyncratic Drug Reactions.
Amy M. Sharma (co-author), Winnie Ng (first author), Alexandra R. M. Lobach, Xu Zhu, Xin Chen,
Feng Liu, et al. Advances in Pharmacology, 2012. 63, 81-135.
Identification of ‘Danger’ Signals in Nevirapine Induced Skin Rash.
Amy M. Sharma (second author), Xiaochu Zhang (first author), Jack Uetrecht.
Chemical Research in Toxicology, 2013 (in submission).
Methanol Embryopathies and Protein Oxidation in Mouse Embryo Culture Following Pre-treatment
with a Free Radical Spin Trapping Agent and Inhibitors of Prostaglandin H Synthase and NADPH
Oxidases: A Role for NADPH Oxidase-Derived ROS.
Amy M. Sharma (second author), Lutfiya Miller (first author), Peter G. Wells. 2012 (in submission).
Direct Activation of Antigen Presenting Cells by Nevirapine and its Metabolites.
Amy M. Sharma (second author), Xin Chen (first author), Jack Uetrecht. 2013 (in preparation).

1
CHAPTER 1
INTRODUCTION
‘No amount of experimentation can ever prove me right; a single experiment can prove me
wrong.’
- Albert Einstein

2
1.1 Adverse Drug Reactions – An Overview
Much progress has been made in the past 50 years in the treatment of disease and the
control of ailments and afflictions through the use of numerous drugs – but intimately tied to this
medical success remains the dark side: adverse effects to xenobiotica. Adverse drug reactions
(ADRs) are, according to the World Health Organization, “any noxious, unintended, and
undesired effect of a drug, which occur at doses used in humans for prophylaxis, diagnosis, or
therapy.” Out of all hospitalized patients each year, approximately 2,216,000 experienced a
serious ADR and approximately 106,000 per year died from an ADR.1 Fatal ADRs rank 4
th to 6
th
among the leading causes of death in the United States.1 Clearly, ADRs represent a huge burden
on the health care system in North America and elsewhere. The occurrence of ADRs may result in
a black box warning label for the offending drug or cause the drug to be removed entirely from
the market.2 This has many implications, not only on the drug companies who spend upwards of
10 years and millions of dollars to see a drug through to medical practice, but also to the majority
of patients who do not suffer from the ADR(s) and who may then be limited from using a
potentially beneficial drug. It is therefore pertinent to understand and develop ways to test or
screen for the occurrence of ADRs, both in preclinical and clinical settings.
1.1.1 Types of Adverse Drug Reactions
ADRs may be classed into seven distinct types, each based on the mode or mechanism of
occurrence. The classification system used to describe specific ADRs is as follows:3
Type A: Augmented response to a drug
Type B: Bizarre or idiosyncratic effect
Type C: Chemical effect
Type D: Delayed effect
Type E: End of treatment effect
Type F: Failure of therapy
Type G: Genetic basis effect

3
The original classification system containing simply Type A and Type B ADRs proved
insufficient to encompass a variety of side effects and has therefore evolved over time. Type A
and Type B were first introduced in 1977 by Rawlins and Thompson.4 Type A adverse reactions
represent an augmented or exaggerated response to a drug, and their occurrence is predictable
based on the known pharmacology of the drug. Typically, a clear dose-response relationship is
associated with Type A ADRs, and discontinuation of the drug is often able to halt the adverse
effect. In contrast, Type B ADRs do not display a simple dose-dependency relationship, and these
cannot be predicted based upon the known pharmacology of the drug. The Type B ‘bizarre’
reactions are also known as idiosyncratic drug reactions (IDRs). Type A effects represent ~ 80%
of all ADRs while Type B represents ~ 20% of all ADRs; however, the Type B effects are often
very severe and can be fatal. This, along with their unpredictable nature make them the most
problematic form of ADRs.5 IDRs are the major focus of this thesis and will be discussed
extensively in the following sections.
1.2 Idiosyncratic Drug Reactions (IDRs)
Idiosyncratic drug reactions are rare and unpredictable side effects of drugs, for which an
exact definition is subject to debate. In the context of this work, the term will be used to describe
any reaction which does not usually occur in humans within clinically used dosages, and does not
involve the known pharmacologic properties of the drug.6 Due to their rare and unpredictable
nature, IDRs are usually not identified during clinical trials because the sample size is insufficient
to allow for their detection. Approximately 10% of drugs released onto the American market
between the years 1975 – 2000 were withdrawn or received a black box warning label due to
adverse side effects, which did not appear during their respective clinical trials.2 The significant
financial and patient-care burden caused by IDRs has led to the need for biomarkers or identifiers
for the occurrence of IDRs in preclinical and clinical settings. Unfortunately, reproduction or
modeling of an IDR in experimental animals is difficult because the reactions occur just as rarely
in animals as they do in humans. Therefore, understanding IDRs is challenging, and to date few
animal models exist that successfully capture clinical features of IDRs as they occur in people.
IDRs can affect almost any organ system; however, the liver and skin are common
targets.7 IDRs can also often affect the bone marrow or blood cells, which possess enzymes such

4
as myeloperoxidase that, although never “designed” to metabolize drugs, are capable of doing so.
Below is a short description of IDRs as they affect each major target organ type.
The liver represents one of the most common organs afflicted by IDRs, and it is also a
primary reason for both preclinical drug candidate failure and drug withdrawal from the market.
Toxic drug effects on the liver may manifest as asymptomatic mild increases in alanine
transaminase (ALT) or other hepatic enzymes, which typically resolve over time despite
continued treatment (termed ‘adaptation’ or ‘tolerance’); however, in some cases, continuation of
the drug may lead to fulminant liver failure and ultimately death. Halothane-induced hepatitis is
one of the best characterized examples of drug-induced idiosyncratic hepatotoxicity.
The skin is another commonly afflicted organ, with drug-induced skin toxicity
representing ~3% of hospitalized patients.8 In the case of cutaneous IDRs, they may manifest as a
mild rash, with minor irritation, few lesions and scaling, or progress to anywhere from Steven’s-
Johnson syndrome (SJS) to toxic epidermal necrolysis (TEN), with complete destruction of
cutaneous integrity. SJS and TEN are associated with high mortality: 5% for SJS, ~ 30% for
TEN).9 Most adverse skin toxicities include perivascular lymphocytic infiltration into upper
dermal layers with the presence of activated macrophages and epidermal alterations, which
supports the hypothesis that drug-induced rashes are immune mediated.10
Bloor disorders or dyscrasias including aplastic anemia, thrombocytopenia, and more
commonly, agranulocytosis, are examples of drug-induced IDRs affecting the blood and bone
marrow.8 Numerous unrelated drugs can induce toxic effects on blood. Myeloperoxidase enzymes
present in granulocytes such as neutrophils are capable of metabolizing drugs. Agranulocytosis is
a major IDR where granulocytes, mostly neutrophils, are diminished to < 500 cells/µL, where the
normal neutrophil levels are 5,000 – 10,000 cells/µL in the blood.11
In patients treated with
aminopyrine who develop agranulocytosis, anti-neutrophil antibodies have been detected.12
Clozapine is the most commonly prescribed drug that is associated with a high incidence of
idiosyncratic agranulocytosis. Myeloperoxidase present in neutrophils metabolize clozapine to a
reactive nitrenium ion capable of binding to neutrophils.13
Rechallenge in patients who previously
experienced clozapine-induced agranulocytosis usually leads to a recurrence of agranulocytosis;
however, the recurrence is delayed and does not occur any faster than on first exposure.14
This
suggests a non-immune mechanism. It may also be consistent with an autoimmune mechanism,
but this has yet to be proven.11

5
Multi-organ effects, although less common, can occur. Clinically, there is a wide range of
severity in which IDRs may present themselves. Specific factors governing the severity of IDRs,
or involvement of a single organ versus multi-organs, are not known to date. There exist a few
clinical characteristics that can be used to describe a typical IDR. Delayed onset of the adverse
effect is a common characteristic of IDRs, with the adverse effect appearing one week or more
after starting the drug.15
This is presumably because these reactions are immune mediated,16
and it
takes time to activate the few T cells that have the appropriate specificity and have them
proliferate to sufficient numbers for the immune response to become clinically evident. On
rechallenge, the reaction usually occurs more rapidly because of “memory” T cells. It can present
with a more severe toxicity than on first exposure, and it can also lead to more systemic effects or
effects on other organs as well.6 Certain IDRs also involve the production of autoantibodies, such
as in the case of hydralazine- or isoniazid-induced systemic lupus erythematosus (SLE).
Autoantibodies to specific proteins have also been linked to drug hepatotoxicity; examples of this
include halothane-induced hepatitis17
or dihydralazine-induced hepatitis.8
The fundamental mechanisms of IDRs are to date unknown. Circumstantial evidence
suggests that most IDRs involve the production of reactive metabolites generated by drug
metabolism,18
and the major hypothesis is that IDRs are immune mediated.16
Host-specific factors
(gender, age, weight, concomitant disease, etc.) and genetics (HLA gene associations)19-21
appear
to play a role in dictating the occurrence of IDRs; however, much work is still required to confirm
their specific contributions, and their associations may not exist for all drugs causing IDRs.
1.3 Proposed Mechanisms of Idiosyncratic Drug Reactions
Although the exact mechanism(s) for IDRs are unknown, three major working hypotheses
attempt to explain how IDRs may occur.6 These include the Hapten Hypothesis, the Danger
Hypothesis, and the Pharmacological Interaction (P-I) Hypothesis. Each is described in detail
below.
1.3.1 Hapten Hypothesis
In 1936, Landsteiner and Jacobs coined the term ‘hapten’ in reference to low molecular
weight (< 1000 Da) chemical allergens that are by themselves non-immunogenic unless they are
bound to larger carrier macromolecules such as proteins.22,23
Pro-haptens, in a similar manner, are
chemical entities that must be metabolized to compounds capable of irreversibly binding to such

6
macromolecules.23
The hapten hypothesis is built on the classical self-nonself immunological
framework, and can be applied to drugs, most of which are less than 500 Da. A reactive,
unmetabolized drug, or more often as circumstantial evidence suggests in the case of IDRs, a
reactive metabolite of a drug, may become antigenic by binding to endogenous proteins or other
macromolecules. Following this drug-protein conjugation step is the uptake of the modified
protein(s) via professional antigen presenting cells (pAPCs), leading to antigen processing into
peptide fragments, which are then presented in the groove of the major histocompatibility
complex (MHC) on antigen-presenting cells (APCs) to T cells. If the T cell receptor of this cell
matches the peptide, it generates a signal, which is referred to as Signal 1. In the presence of other
costimulatory molecules, which are referred to as Signal 2, this can lead to activation of the T cell.
Signal 1 in the absence of Signal 2 results in immune tolerance.
Penicillin allergy is an example of a reactive parent drug, where in the absence of
metabolism, protein haptenation occurs, sometimes leading to an IgE-mediated IDR. The sp2
hybridized carbonyl group within the β–lactam ring is forced from its ideal 120º to be 90°. This
inherent ring strain of the β–lactam ring makes it susceptible to nucleophilic attack by nitrogen
and sulfur nucleophiles, thus opening the ring and relieving the strain. Antibodies have been
detected against the penicillin-modified proteins that are associated with penicillin allergy that
occurs in a small percentage of patients. These antibodies can go on to cause mast cell
degranulation, leading to the release of inflammatory mediators such as histamine and
leukotrienes. Anaphylactic shock can result and re-exposure can be life-threatening.
β-lactam ring
Benzylpenicillin
Figure 1-1. Reaction of penicillin with protein nucleophiles via spontaneous ring opening in a
hapten-type mechanism.

7
There are numerous examples of drugs known to cause hapten formation through reactive
metabolites, and this is believed to lead to immune-mediated toxicity; some of these include
aminopyrine-induced agranulocytosis,12
halothane-induced hepatotoxicity,24
and tienelic acid-
induced hepatotoxicity.25
Although numerous drugs causing IDRs form reactive metabolites
capable of haptenating proteins, the degree of protein haptenation and risk of IDRs is not well
defined or understood, although several groups have tried to correlate this relationship.
Presumably, organ-specific responses, host-immune and enzymatic factors, and basic principles
underlying the organisms’ response to noxious stimuli produced by the degree of binding all play
a role. Numerous proteins are also typically adducted by a single drug, yet no concrete association
has been established between the type of protein adducted and IDR risk. Therefore, all of these
factors mentioned must be pieced together to understand the role of haptenation in the occurrence
of IDRs.
Figure 1-2. The Hapten Hypothesis: a reactive parent drug or the reactive metabolite of a drug
covalently binds to protein. This modified protein can elicit an immune response; however, it is

8
now known that additional factors are required as discussed below. Adapted from Uetrecht,
2007.6
1.3.2 Danger Hypothesis
In 1994, Polly Matzinger challenged the field of immunology with her radical ‘Danger’
theory,26
which opposed the classical self-nonself immunological framework. She proposed that
foreign proteins do not typically elicit an immune response unless cell stress or cellular
perturbations lead to the release of ‘danger’ or stress signals from damaged or dying cells. These
signals, as the theory posits, cause upregulation of appropriate co-stimulatory molecules, such as
B7 on APCs, which interact with T cells (i.e. through CD28), stimulating the T cells. This allows
APCs to produce Signal 2, which is necessary for an immune response. Without Signal 2, the
result is immune tolerance. Signal 1 in this context is recognition of a peptide by a T cell when the
peptide has been presented in the grove of MHC II on an APC. The Danger theory also implies
that the type or nature of an immune response is dictated by the affected tissue, and in this way an
unnecessary systemic immune response is avoided. The origins of Signal 1 are not addressed by
the Danger Hypothesis, and therefore it does not exclude an immune response against a drug
itself, drug-protein conjugate, or autoantigen.
The Danger theory is attractive in the case of IDRs because reactive metabolites have the
potential to cause cellular damage and cause the release of stress signals.27
This could explain
organ or tissue-specific effects where covalent binding is present in different tissues yet an
adverse response develops in only one. Identification of danger or stress signals is still in its initial
stages, but these molecules should be endogenous compounds such as S100, HMGB1, or heat
shock proteins28
released from damaged cells. There are numerous other intracellular proteins that
may also act as danger signals, and in the case of IDRs their release may serve as a biomarker for
drug toxicity; more work is needed to confirm which molecules or patterns of response can
translate cell stress into an immune-mediated IDR.

9
Figure 1-3. The Danger Hypothesis: stressed or damaged cells release endogenous ‘danger
signals’ that activate APCs, leading to upregulation of costimulatory molecules (B7 on APCs)
which interact with CD28 on T cells leading to an immune response. Adapted from Uetrecht,
2007.6

10
1.3.3 Pharmacological Interaction Hypothesis
In 1998 Werner Pichler found that isolated T cells from patients who had developed an
IDR proliferated in the absence of drug metabolism when incubated with the drug that had
induced the IDR.29
The Pharmacological Interaction (P-I) hypothesis posits that certain drugs can
reversibly bind to the MHC-T cell receptor complex leading to an immune response, which in
some cases may lead to an IDR. Metals such as nickel and beryllium are examples where
reversible, although very tight binding to MHC is able to induce an allergic reaction. Specific
drugs such as sulfamethoxazole, carbamazepine, lamotrigine, lidocaine, etc.,30
represent a weaker
class of reversible binding to MHC than metals; these drugs can stimulate T cells in their
unmetabolized form.30
Sulfamethoxazole is the best characterized of these drugs. It becomes
bioactivated to form a hydroxylamine metabolite, which is further oxidized to a reactive nitroso
compound. T cells isolated from patients who developed a sulfamethoxazole-induced IDR
responded to the parent drug instead of the reactive metabolite,31
supporting the P-I hypothesis.
However, a more recent study found that many more lymphocytes from patients who developed
an sulfamethoxazole-induced IDR responded to the reactive nitroso metabolite than the parent
compound, which supports the role of the reactive metabolite in sulfamethoxazole-induced
toxicity.32
This highlights the mechanistic complexity of studying IDRs, and the inability of a
single hypothesis to capture such intricacy. In addition, it must be noted that the P-I hypothesis
assumes that what T cells respond to in vitro is also what induced the immune response. We have
found in the nevirapine model that this is not the case – what T cells recognize in the lymphocyte
transformation test (LTT) in vitro is not what induced the response in vivo. Thus the basis for the
P-I hypothesis is false. Nevertheless, the P-I hypothesis may be useful for small peptide-like
compounds or compounds that do not involve reactive metabolite formation, such as
ximelegatran.33

11
Figure 1-4. The P-I Hypothesis: A parent drug binds reversibly to the MHC-TCR complex
(Signal 1) leading to an immune response. Adapted from Uetrecht, 2007.6

12
1.4 Idiosyncratic Hepatotoxicity
In order to understand the role that reactive metabolites may play in causing idiosyncratic
toxicity and how the aforementioned mechanistic hypothesis may apply to this thesis, a brief
review of the enzymatic processes leading to the production of reactive metabolites is appropriate.
Both the liver and skin will be discussed in detail. The hepatic morphology and immunological
nature of the liver is also discussed in order to understand the unique distribution of hepatic
enzymes as well as mechanisms by which toxicants interact with the liver’s own ‘immune
system.’
1.4.1 Hepatic Function and Morphology
The liver is the primary site of xenobiotic metabolism and biotransformation in the body,
and the cells of the liver are exposed to significant amounts of various chemicals. Located down
stream to the intestinal tract, the liver is the first to receive nutrients, drugs, vitamins, etc., from
the portal blood.34
Metabolic and nutrient homeostasis, synthesis of clotting factors and proteins,
lipid metabolism, and production of bile and biliary secretion are only a few of the major
functions of the liver. Toxic insults have the ability to damage the functions of the liver through
acute or chronic exposure.
The structural organization in the liver is designed to facilitate the many important
functions it must perform. The liver consists of hepatic lobules, which are further divided into the
centrilobular, midzonal, and periportal regions. Hepatic zonation is important when considering
toxicological effects on the liver. For example, the oxygen saturation of hepatocytes in zone 3,
which are closest to the central vein, is only 4-5% compared to those in zone 1, which is 9-13%.34
Drug metabolizing enzymes exhibit a similar phenomenon, with Phase I enzymes situated
predominantly in zone 3, and Phase II enzymes closer to Zone I. These enzymes are described in
the following section.
1.4.2 Biotransformation in the Liver
The primary purpose of xenobiotic metabolism is to terminate biological activity and
produce more water soluble compounds through the introduction of a polar group. This occurs
primarily by oxidation or conjugation reactions, allowing for easier excretion from the body.
However, this biotransformation can sometimes lead to the formation of a chemically reactive

13
metabolite of a drug. Therefore a basic understanding of drug metabolism is necessary to
understand how this may occur.
Drug metabolism occurs through enzymatic biotransformation primarily in the liver via
enzymes that are classified as either Phase I or Phase II reactions. Phase I functionalization occurs
mostly through a superfamily of heme-containing enzymes called cytochromes P450 (abbreviated
P450 or CYP), which are the most important and common drug metabolizing enzymes. P450s are
present to the greatest degree in the centrilobular region of the liver and membrane bound in the
endoplasmic reticulum of hepatocytes. The P450 family consists of many subfamilies and
isoforms, each with preferential substrate activity. Fifty seven human P450 enzymes have been
identified to date; however, this does not mean each human expresses all isoforms.35,36
P450 enzymes directly catalyze oxidation reactions. Specifically, they catalyze four main
types of oxidation: hydroxylation, epoxidation, dehydrogenation, and heteroatom oxidation.37
The
general formula can be summarized as follows, where R = substrate and RO = product:
NADPH+ + H
+ + R + O2 NADP
+ + H20 + RO
Although there exist many types of P450 enzymes, the 3A4 family is the most significant from a
dug metabolism perspective. CYP3A4 is the most highly expressed P450 enzyme in human liver
– it is also in the human gut38
- and exhibits a great range of substrate specificity because the
active site can accommodate very large substrates (>1000 g/mol).37
Approximately half of all
marketed drugs are metabolized by hepatic CYP3A4 enzymes.37,38
Next in metabolic significance
are CYP2C9, 2D6, and 2C19, followed by 2E1, 1A2, and 2B6, all of which have fewer substrates
than CYP3A4.39
Other enzymes can also oxidize drugs including peroxidases (myeloperoxidases,
prostaglandin H synthase, horseradish peroxidase), flavin monoxygenases (FMOs), aldehyde
dehydrogenases, xanthine oxidases, aldehyde oxidase, and monoamine oxidases.
Following traditional Phase I oxidation by P450 enzymes, predominantly in the liver,
further metabolism known as Phase II may occur. Phase II metabolism involves conjugating
enzymes such as sulfotransferases and glucuronosyl transferase. The focus will be on the sulfating
enzymes which are described in detail in section 1.6.1.
All of the enzymes described in this section function to facilitate excretion of xenobiotics
and drugs from the body. However, it is an imperfect process and chemically reactive compounds
can be produced. Reactive metabolites are electrophilic in nature (electron seeking) and react with
nucleophilic groups on proteins (typically a lone pair of electrons or negative charge) or even

14
DNA, leading to the formation of neoantigens. Some common reactive species include Michael
acceptors, epoxides/arene oxides, and nitroso amines.38
This process can occur in any organ, and
numerous cell types possess metabolizing enzymes capable of drug bioactivation. As the hapten
hypothesis stipulates, the resulting covalent binding is irreversible and creates more
immunostimulatory compounds, which in turn, activate the immune system. However, the liver is
a unique organ in that it is exposed to numerous xenobiotics and forms many neoantigens on a
daily basis; how then has it found ways to regulate its immune response? This is explored in the
following section, and later in Section 1.5, contrasted to what occurs in skin following the same
covalent binding.
Figure 1-5. Proposed pathway of drug bioactivation leading to haptenation and cellular
mechanisms of hepatocyte death. Adapted from Kaplowitz, 2004.40
1.4.3 Liver Immune System in Relation to Hepatotoxicity
The dominant immune response in the liver is immune tolerance; this is due to a unique
hepatic microenvironment and ultrastructure. The tolerogenic response is thought to be a key
reason why covalent binding in the liver does not usually lead to hepatotoxic responses to drugs.
For example, architecture in the liver allows for T cell interaction with resident APCs, which
facilitates APCs, and especially dendritic cells (DCs), which are the major professional antigen
presenting cell, to become capable of inducing a immunogenic or tolerogenic response. Cytokines

15
(and other molecules) produced by hepatic APCs (DC’s, Kupffer cells, liver sinusoidal
endothelial cells, hepatic stellate cells, etc) such as interleukin-10 (IL-10), transforming growth
factor (TGF-β), prostaglandin (PG)E2, and granulocyte macrophage-colony stimulating factor
(GMCSF) influence tolerogenic outcomes in the liver.41
In addition, DCs and other hepatic APCs
express death ligands,41
which may contribute to apoptosis of activated T cells within the liver;
this has been suggested as a reason for the ease of hepatic transplantations as compared to other
organs such as skin. In addition, hepatic T cells undergoing apoptosis have been shown to release
TGF-β and IL-10, which further promotes a tolerogenic microenvironment in the liver.41
Furthermore, activation of regulatory T cells is believed to down-regulate MHC-II and
costimulatory molecules on DCs, which maintains them in an immature phenotypic state.42
Given that the liver has developed mechanisms to down regulate inflammatory immune
mediated responses, it has been very difficult to develop animal models of idiosyncratic drug-
induced liver injury. In many cases the drug is metabolized by P450 to reactive electrophiles
which, if very electrophilic, may bind directly to the enzyme from which it was formed, acting as
a suicide inhibitor for that enzyme (i.e. P450). This P450 adduct or other hepatic adducts, in
theory, could act as a hapten, inducing an immune response, and certainly in many cases where
drug-P450 haptenation occurs, a transient increase in ALT is seen in animal models. However, the
transient injury is cleared, and animals, as well as the majority of humans, adapt or tolerize to the
insult. Further, not all drugs that modify hepatic proteins induce an immune response. No clear or
convincing mechanisms specific to idiosyncratic hepatotoxicity exist; there does appear to be a
clear link to host-dependent factors which are poorly characterized.34
Thus, a good understanding
of drug-induced immune-mediated liver injury does not exist.

16
Figure 1-6. Proposed mechanisms of DILI, including drug bioactivation leading to reactive
intermediates, which may cause hepatocyte damage and invoke immune-mediated responses. A
balance of hepatoprotective and inflammatory factors dictate the potential for toxicity. Adapted
from Holt and Ju, 2005.43
1.5 Idiosyncratic Cutaneous Toxicity
The skin is a dynamic organ and truly an environment unto its own. In order to understand
how cutaneous IDRs may arise, a discussion of the integument is necessary. Little is known
regarding the relationship between the skin structure, enzymatic processes, and immunological
activity in the skin as applied to idiosyncratic cutaneous toxicity. The purpose of this mini review
is to integrate current and emerging findings into proposed mechanisms of drug bioactivation in
the skin leading to rashes.
1.5.1 Skin Structure and Function
The skin provides the body with a structural barrier to the outside world, and it is the first
line of defense against any external stimuli. A highly complex tissue, the structure of skin can

17
easily be understood through its primary protective role. Composed of three main layers: the
epidermis, dermis, and hypodermis or subcutis, the skin maintains internal homeostasis and
participates in a myriad of important physiological and cellular functions. These include, but are
not limited to, thermal regulation, electrolyte and hormonal balance, metabolic and immune
regulation, as well as defense against invading pathogens, micro-organisms, chemicals, ultraviolet
radiation, and physical-mechanical insults.34
The skin possesses a variety of sophisticated
mechanisms to perform its numerous functions. Although the structure of skin is similar in all
areas of the body, the thickness varies depending on the specific organ and location, reflecting the
requirement for increased barrier functions in certain anatomical sites (i.e. the soles of the feet).
The hypodermis is the lowest layer of the skin, therefore in closest contact to internal
organs. This portion of skin provides cushioning and insulation because it is composed primarily
of adipocytes arranged in a lobular fashion used for fat storage. Other cell types found in the
hypodermis include fibroblasts and macrophages. Blood and lymphatic vessels, nerves, and
fibrous tissues connecting the skin to the deep fasica are also components of the hypodermis.
Above the hypodermis lies the dermis, which is structurally the largest and thickest
portion of skin, comprising 90% of the entire integument.34
The dermis provides structural
integrity as well as mechanical and tensile strength due to the dense collagen and elastin
connective tissue networks produced by resident dermal fibroblasts. The collagen alone accounts
for approximately 75% of the skin’s dry weight.44
Although fibroblasts are the major cell type
found in the dermis, other cell types such as mast cells, macrophages, and T cells, are also found
here. These cells reside mostly in the surrounding vasculature and within the papillary dermis
(upper dermis).44
The reticular dermis comprises the mid to lower dermal layers, and it is much
thicker. Other key properties of the dermis include the presence of nerves and receptors, hair
follicles, sebaceous glands, eccrine sweat glands, apocrine glands, lymphatic vessels, and all-
important blood vessels.44
The epidermis is situated above the dermis and is structurally a very different tissue type
from the rest of the skin. While the dermis serves a primarily supportive function and is
structurally very sound, the epidermis is almost completely cellular and very metabolically and
immunologically active. This is due to the cell types that form the epidermis, namely
keratinocytes (KCs), Langerhans cells (LCs), and melanocytes.

18
Keratinocytes comprise approximately 95% of the epidermis and are considered the most
important in maintaining structural epidermal integrity. Keratinocytes originate as stem cells in
the basal epidermal layers (stratum basale), and as the cells mature, they differentiate and migrate
to form the upper epidermal layers. Thus the cells divide from the stratum basale to the stratum
spinosum, granulosum, lucidum, and corneum, with the stratum corneum being the outermost
layer of skin (composed of simple keratinized cells). Desmosomes are the chief epidermal
intracellular adhesion molecule, and they provide the keratinocytes with a structural framework.44
Keratinocytes possess numerous metabolic and immunologic capabilities that will be reviewed in
the following sections, but these cells also produce keratins, which provide mechanical and
cellular strength. It must be noted that mucous membrane epithelia lack the stratum corneum
(outer keratinized layer) in order to provide a lubricating lining.
Langerhans cells and melanocytes are both bone marrow-derived cells, which migrate into
the epidermis during embryogenesis. Langerhans cells are specialized cells that account for only ~
5% of epidermal cells, and are considered professional antigen presenting cells. Their role is
considered in detail in 1.5.3. Melanocytes synthesize pigment via melanosomes and are also
found in hair follicle bulbs.
1.5.2 Metabolic Enzymes in Skin
Biotransformation in the skin occurs primarily in epidermal cells such as keratinocytes and
Langerhans cells, which are metabolically more active than dermal or hypodermal cells.
Nonetheless the skin does possess both Phase I and Phase II enzymes, and their presence is
described here.
Phase I. Classical Phase I metabolism accounts for only ~ 2% that of the liver (on a per-
body weight basis).34
CYP1A has been identified in both rodent and human skin; however, the
actual protein has only been identified in keratinocytes.45
Data on the identification CYP2A6 has
been mixed, with mRNA initially undetectable in KCs, but identified in fibroblasts and
melanocytes.45
However recent data has identified CYP2A6 mRNA and CYP2B6 protein in
KCs.45
Results for CYP2C enzymes have also yielded mixed results, but CYP3A5 has been
identified in all skin samples and biopsies examined to date.45
Mixed results are thought to be due
to changes in cells with culture conditions i.e. loss of gene expression, and differences in
protocols or methods for identification of enzymes.45
Other enzymes capable of oxidation in the

19
skin that have been confirmed in biopsy samples include flavin monooxygenases, types 1 and 3,
with the mRNA detected for the former and both the mRNA and protein for the latter.45
Phase II. Various Phase II enzymes have been identified in rodent and human skin. These
include isoforms of epoxide hydrolase, UDP-glucuronosyl transferase, quinone reductase,
β-glucuronidases, N-acetyl transferases, esterases, reductases, and sulfotransferases.34
Although
these enzymes have been identified, amounts are significantly much less than expression in the
liver, and human skin has far lower expression of all Phase I and Phase II enzymes than rodent
skin overall.34
1.5.3 Immune Function of the Skin
The skin was previously thought to be an innocent bystander in immune-mediated
hypersensitivity responses. This view has since changed, and it has become increasingly clear that
the skin is immunologically active and able to respond to various challenges. Keratinocytes, for
example, although non-professional immune cells, have been termed the ‘adjuvant’ of the skin
due to their unique ability to shape innate and adaptive cutaneous immune responses.46
This is
made possible in part by keratinocyte expression of numerous functional immune molecules such
as cytokines, chemokines, MHC-II molecules, and co-stimulatory molecules.44
In addition, KCs
are known to constitutively express specific cytokines and may produce others when activated.
These include interleukins 1α/1β, 3, 6, 7, 8, 10, 12, 15, 18, and 20.44
Other factors also produced
by KCs include IL-1ra (receptor antagonist), TNF-α, IFN-α and IFN-β, TGF-α, TGF-β,
chemokine receptor 3 (CCR3), eotaxin, RANTES, macrophage-colony stimulating factor (M-
CSF), granulocyte macrophage colony stimulating factor (GM-CSF), and CD14, CD40, and toll-
like receptors 1 to 6, and 10.44
Along with TLRs, another pattern recognition receptor found on keratinocytes is the
nucleotide-binding oligomerization domain receptor (NOD-like receptors or NLRs).47
NLRs
represent a platform for innate immune sensing, and keratinocytes have been shown to express a
large multimeric form of NLRs termed ‘inflammasomes’. The cytosolic NLRP3 inflammasome
activates caspase-1, which in turn causes cleavage of pro-IL-1β and pro-IL-18 into their mature
states.48
Both cytokines are critical for the instruction of T cell responses, providing a link of
innate instruction of adaptive immunity.48
NLRP3 responds to a variety of cell stress stimuli and

20
has been shown to be involved in contact hypersensitivity conditions,49
where covalent binding of
small chemical molecules can activate the inflammasome. It is probable that drugs can also act in
this way i.e. undergo metabolic biotransformation in the skin where they may form adducts, and
the resulting cell perturbations may activate the inflammasome.
Keratinocytes do not normally express co-stimulatory molecules such as CD80/86, and
therefore, they are unlikely to be able to constitutively prime naïve T cells.50
They are also unable
to classically process and present antigen, and they cannot prime new T cell responses. However,
non-classical presentation by keratinocytes has been suggested such as presentation of glycolipids
by CD1 molecules.50
CD1d can also be induced by poison ivy, and expression of CD1d on
cultured KCs can activate natural killer T cells.50
This suggests lipid-derived ligand presentation
by KCs may occur. Keratinocytes can also induce Th1-type responses via release of IFN-γ from T
cells, monocytes, and macrophages. Similarly, IL-18 can also be up-regulated by keratinocytes,
and this is an important cytokine for Langerhans cell migration in mouse models of contact
hypersensitivity.50
IL-18, IL-1β, IL-6, and IL-12 appear to play key roles in initiation and
development of immune responses in the skin.51
Langerhans cells are localized primarily in the lower epidermal layers and function as
pAPCs, capable of epidermal antigen processing. Following the internalization of antigen, LCs
migrate to the regional lymph nodes with the help of integrin molecules to present processed
antigen through their surface MHC-II and co-stimulatory molecules.44
Recent studies have tried to
determine the mechanisms by which LC migration occurs, and it appears that proinflammatory
cytokines stimulate LC migration; specifically, allergens that cause contact hypersensitivity
induce proinflammatory cytokines such as IL-1α, IL-1β, and TNF-α, which have been associated
with hapten-induced LC migration.51
It has been proposed that these cytokines diminish E-
cadherin-mediated contacts between KCs and LCs.51
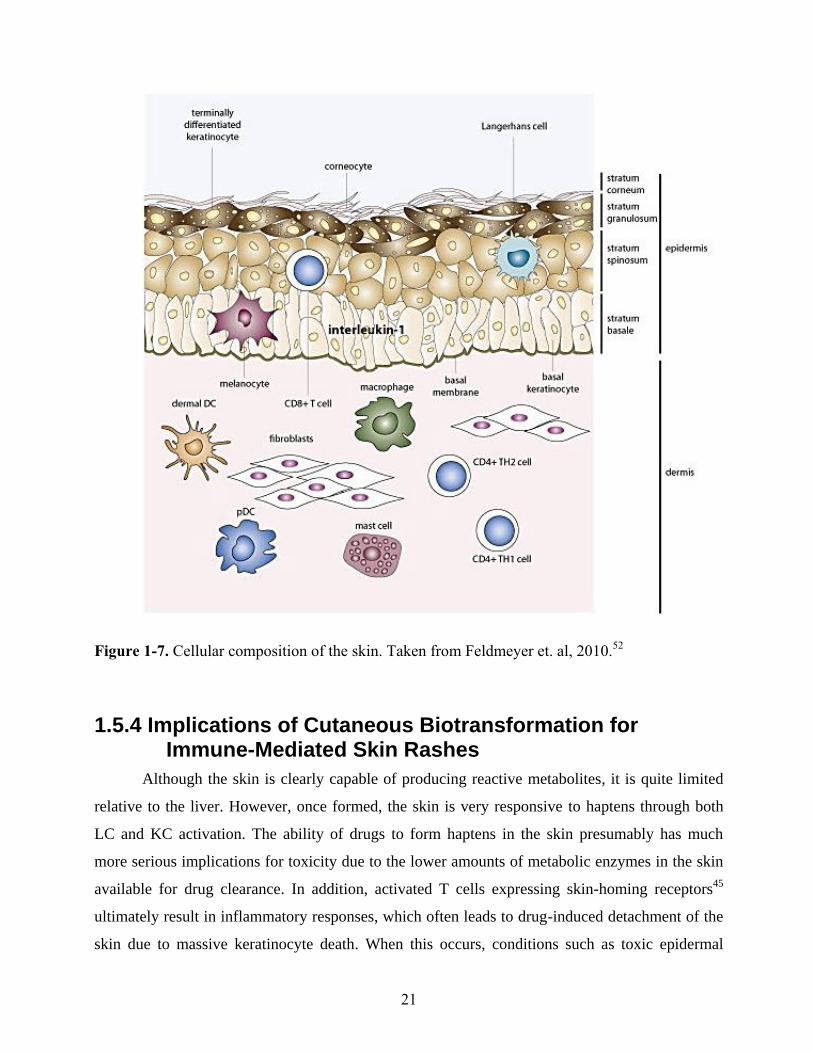
21
Figure 1-7. Cellular composition of the skin. Taken from Feldmeyer et. al, 2010.52
1.5.4 Implications of Cutaneous Biotransformation for Immune-Mediated Skin Rashes
Although the skin is clearly capable of producing reactive metabolites, it is quite limited
relative to the liver. However, once formed, the skin is very responsive to haptens through both
LC and KC activation. The ability of drugs to form haptens in the skin presumably has much
more serious implications for toxicity due to the lower amounts of metabolic enzymes in the skin
available for drug clearance. In addition, activated T cells expressing skin-homing receptors45
ultimately result in inflammatory responses, which often leads to drug-induced detachment of the
skin due to massive keratinocyte death. When this occurs, conditions such as toxic epidermal

22
necrolysis or Stevens Johnson syndrome may result.53
However, the mechanistic details of these
rashes remain to be determined.
Upregulation of Fas ligand (FasL) on keratinocytes has been observed in patients who
develop drug-induced skin rashes, and the histological portrait in these patients is one of
widespread keratinocyte apoptosis, sub-epidermal blistering, and lymphocytic/mononuclear cell
infiltrate.53
Natural killer T cells, also present during episodes of TEN, have been implicated,
along with CD8+ cytotoxic T cells (CTLs) and natural killer (NK) cells.53
Immunophenotypes of
cells present in blister fluids from five patients who developed SJS-TEN induced by
carbamazepine, phenytoin, and amoxicillin were analyzed in one study, and regardless of the
offending drug, the majority of the cells in the blister fluids of these patients were CD3+ T cells
(predominantly CD8+ CTL subset ; 33–70%), CD56
+ NK cells (48–100%), and CD8
+CD56
+ NKT
cells.54
Evidence for the involvement of CTLs in drug-induced ‘skin killing’ include the
observation that injection of granulysin directly into the skin of C3H mice induced significant
dermal and epidermal necrosis and inflammatory infiltrates in the skin that was not seen when a
similar experiment was performed with granzyme b and lysozyme.53
Granulysin is produced by
CTLs, which have been implicated in drug-induced detachment of epidermal-dermal layers.53
Despite the myriad of clues regarding the activation of drugs in the skin leading to cutaneous
immune-mediated reactions, much remains to be determined: what is the basis for specific HLA
gene associations; how does drug activation lead to granulysin secretion; how does a rash
progress from moderate severity to SJS-TEN; what is the difference between local versus
systemic drug activation in inducing skin rash, and which is more important; can granulysin be
used as a biomarker to screen for potentially bioactive drugs; and fundamentally, how do
keratinocytes sense danger?53

23
1.6 Role of Drug Metabolism, Reactive Metabolites, and Covalent Binding in IDRs
As described above, both the liver and skin are capable of the biotransformation of drugs
into reactive metabolites. The ability of reactive metabolites to conjugate to self-proteins forming
novel antigenic compounds has been explored in drug toxicity; however, the relationship between
covalent adducts and toxicity is, to date, not well understood. Whereas drugs such as halothane
covalently bind to hepatic proteins, and resulting anti-drug antibodies have been detected in
patients, acetaminophen also covalently binds in the liver and yet it does not induce an immune-
mediated hepatotoxicity.34
The skin may be different in that there is a great degree of
immunological activity, and the dominant response is not tolerance.
Covalent binding of a drug may be necessary but not sufficient to induce and sustain an
immune response. Correlation between the amount of binding and resulting toxicity has been
attempted; one study, correcting for the total daily dose of the drug to reflect hepatic exposure,
found a rough correlation in the amount of binding and risk that a drug would cause drug-induced
liver injury.55
The same study, when results were kept unadjusted, found a large overlap in DILI
and non-DILI inducing drugs regarding the amount of binding. 55
Clearly, other factors exist to
influence immune mediated toxicity.
One strategy that has been employed for screening drug candidates is the use of
radiolabeled drug analogs to test for their ability to covalently bind to proteins.38
Although
relatively simple to perform in vitro (in vivo is difficult due to the large amount of radiolabeled
drug required), false negatives may result if an enzyme capable of metabolizing the compound is
absent from the system. Another problem with screening drug candidates for potential toxicity is
the lack of metabolic similarity between rodents and humans (rodents metabolize and clear
drugs/xenobiotics much faster than humans), thus interpretation based on these studies could lead
to underestimation of risk.38
1.6.1 Sulfotransferase Enzymes in Drug Metabolism and
Toxicity
One often overlooked pathway in the mechanism of drug toxicity and covalent binding is
that of the Phase II reaction, sulfation. Sulfation occurs where sulfotransferase enzymes (SULTs)
catalyze the transfer of SO3- to appropriate substrates.
37 The sulfate donor is PAPS (3’-

24
phosphoadenosine-5’-phosphosulfate), produced from APS kinase and ATP sulfurylase, which
exists as a single bi-functional enzyme in mammals.37
Serum sulfate levels in humans are limited
to approximately 0.3 mM,37
and sulfation is considered to be a high affinity, low capacity
metabolic pathway. SULT enzymes exist in plants and all vertebrate classes examined (fish, birds,
amphibians), but limited reports of sulfation in insects exist.37
Sulfotransferases are classed into
two groups, the membrane-bound Golgi complex SULTs, which act on endogenous steroid
hormones, neurotransmitters, heparins, glucosaminoglycans, etc., and the cytosolic xenobiotic
metabolizing SULTs of ~ 300 amino acid residues, which will be discussed here.37
Table 1-1. Human Sulfotransferase Isoforms and Expression.
Isozyme Typical Substrates Expression
SULT 1A1 4-nitrophenol Liver, skin*, GI tract
SULT 1A2 4-nitrophenol
2-napthol
SULT 1A3 Dopamine Platelets, GI tract
SULT 1B1 3,3’,5’-Triiodothyronine Liver, GI tract
SULT 1C2 4-nitrophenol Fetal lung, kidney
SULT 1C4 4-nitrophenol
nonylphenol
SULT 1E1 17-β-estradiol Liver, lung, kidney
SULT 2A1 dehydroepiandosterone Liver, adrenals, skin*
SULT 2B1 dehydroepiandosterone Skin, prostate
SULT 4A1 unknown brain
Modified from P. Josephy37
and F. Oesch.56
The * indicates isoforms also found in rats.
Sulfotransferases typically sulfate alcohol or phenolic groups; the nitrogen of N-
substituted aryl and alicyclic chemicals; or pyridine N-oxides.38
Cytosolic SULT enzymes are
quite promiscuous, and there is a large overlap in substrates metabolized by various isoforms
within subfamilies.57
Although sulfation is generally regarded as a detoxification pathway, it can
lead to bioactivation to reactive intermediates, which may or may not be harmful. For example in
the case of the pyrimidine N-oxide drug minoxidil, sulfation must occur to form the N, O-sulfate
ester in order to produce the desired effects on alopecia. In other cases, sulfation is implicated in
activation of several classes of toxicants, such is seen with aryl hydroxylamines and hydroxamic
acids, which are sulfated to metabolites involved in aromatic amine carcinogenesis.37
The PPARγ agonist type-II anti-diabetic drug, troglitazone, was first released onto the
market in 1997. It was withdrawn only three years later due to idiosyncratic hepatotoxicity, which

25
appears to be multifaceted in nature. Although troglitazone liver injury is primarily and more
seriously hepatocellular, cholestatic injury also occurs. Male rats were observed to sulfate the
drug to a much greater degree than females – the male rats were also observed to be more
sensitive to troglitazone toxicity.37
The sulfate metabolite is now recognized as one toxic
metabolite of troglitazone, and is a potent inhibitor of the bile salt export pump in hepatocytes.37,58
Benzylic and allylic alcohols can also undergo sulfation leading to the production of toxic
metabolites. In this scenario, the sulfate can be cleaved heterolytically producing resonance-
stabilized electrophilic carbocation or nitrenium ions. Safrole is a good example where loss of
SO42-
leads to a resonance-stabilized allylic cation, capable of binding to DNA in rat hepatocytes.
This resonance stabilization of the remaining cation following loss of the sulfate from benzylic
and allylic alcohols produces a reactive electrophilic intermediate.
1.7 Nevirapine Toxicity
Nevirapine (NVP; ViramuneTM
)
is a non-nucleoside reverse transcriptase inhibitor
indicated for the treatment of active HIV-1 infections.59
NVP was released onto the market in
June 1996, and prescribed first in its class as part of the highly-active anti-retroviral therapy
regimen (HAART), given along with a protease inhibitor and a nucleoside reverse transcriptase
inhibitor. Nevirapine is a highly effective drug, and a single dose can limit the vertical
transmission of the virus from the mother to the fetus. Although efficacious at controlling HIV
infections, NVP is associated with two adverse toxicities: hepatic damage and rash.59
In 2000, the FDA placed a black box warning label on NVP due to hepatotoxicity, which
occurs in 6% of patients and can be life threatening.59
Liver injury normally resolves when the
drug is stopped, but it can lead to fulminant liver failure and death. In 8 - 18% of patients, NVP
hepatotoxicity can manifest as asymptomatic elevated serum alanine transaminase (ALT) levels,
which is the first indication of liver injury and typically occurs within the first six weeks of
treatment.60
There also exists evidence for increased risk of liver injury in non-HIV patients,
which may be due to higher CD4+ T cell counts.61
The initial therapeutic dose of NVP was 400 mg/kg/day; at that dose it induced skin
rashes, most of which were mild to moderate in nature, in 32- 48% of patients. In contrast, when a
lead in dose of 200 mg/kg/day for the first two weeks of treatment was introduced, the incidence

26
of skin rash was decreased to 17%. Currently the incidence of NVP skin rash is 9%; however,
16% of skin rash patients develop very severe rash in the form of SJS or TEN.59
Numerous risk factors have been reported for the development of the NVP-induced
toxicities. For example, the HLA-DRB*01 allele has been associated with the development of
hepatotoxicity in NVP patients.62,63
Risk factors associated with development of the skin rash
have been better characterized, and they include, but are not limited to, ethnicity (Chinese
populations are more sensitive), female gender, co-therapy with antihistamines and
corticosteroids, and a higher pre-therapy CD4+ T cell count.64
The first 6 weeks of NVP treatment
is the riskiest time for patients on NVP to develop liver injury or rash.59
Our laboratory has developed and well-characterized an animal model of the NVP-induced
skin rash in the female Brown Norway (BN) rat.65
This model represents the proportion of the
patients who only develop a skin rash, and it is to date one of only two excellent models of an
idiosyncratic reaction which occurs in humans (the other is D-penicillamine-induced
autoimmunity). Although patient samples would be ideal when attempting to study IDRs, for
ethical and other reasons, collection of these sample types is not always feasible. It is also not
feasible to methodically control specific variables to test hypotheses in humans. Therefore, animal
models such as this represent the next best approach in trying to understand the mechanisms of
IDRs. Using animal models as screening tools may also become viable in the future if they can be
developed. The NVP model is reviewed in the following section.
1.7.1 Animal Model of Nevirapine-Induced Skin Rash
Shenton et al. from our group first characterized the BN rat model of NVP skin rash in
2003.65
The skin rash in rats occurs over a period of about 3 weeks, with the ears turning red on
day 7, and the development of lesions and sloughing of skin between days 19-22 of treatment.65
The incidence of rash in female BN rats is 100% whereas only 20% of female Sprague-Dawley
rats develop a rash at 3 weeks or later.65
The rash in rats ranges from mild to moderate severity,
and as in humans, there appears to be quite an individual response of the rats to NVP. When rats
are removed from NVP and re-challenged, the rash occurs must faster (within 7 days) and the
animals develop a systemic sickness (lethargic, uninterested in surroundings, etc). Partial
depletion of CD4+ T cells was protective in rats, and depletion of CD8+ T cells, if anything,
appeared to make the rash worse.66
In addition, splenocytes harvested from re-challenged rats

27
were able to transfer susceptibility to NVP in naïve recipients through i.v. injection (adoptive
transfer studies).66
Pre-treatment with the immunosuppressants, tacrolimus and cyclosporine, was
able to prevent and even resolve the rash during treatment.66
Other characteristics include
increased incidence in female BN rats, increased incidence of rash with increased dose, and
tolerance induction through low dose pre-treatment.59
These characteristics are very similar to the
occurrence of rash in humans and strongly support the role of the immune system in induction of
the rash.
Morphological features of the rash are similar in humans and rats; for example, both
develop maculopapular raised lesions and general redness of the skin; some animals develop
sloughing of the skin. It is a generalized rash and can even affect mucous membranes in the
animals as well as humans. In rats, crusting of eyes and scabbing around the footpad as well as
nose have been observed. Histologically, the dermal infiltrate is also similar in humans and rats,
with a mild perivascular lymphocytic infiltrate which progresses to a more severe infiltrate later
on. A mononuclear infiltrate has been observed in both the dermis and epidermis of patients who
develop SJS and TEN;67
this infiltrate has also been seen predominantly in the dermis of rats.65
Given the numerous similarities between the animal model and humans, this model has
provided us with the unique opportunity to study the mechanism of at least one IDR in detail. It is
true that the mechanisms of IDRs for different drugs are likely very different; therefore, this is but
one piece in a large puzzle.
1.7.2 Nevirapine Metabolism leading to Skin Rash
A fundamental question in the understanding of IDRs is whether it is the parent drug or a
reactive metabolite that is responsible for causing toxicity. The NVP model has allowed us to
determine which metabolic biotransformation pathway is responsible for the rash. In 2008, Jie
Chen from our group found that the 12-hydroxylation pathway is responsible for causing the skin
rash.68
This pathway is a major route of NVP metabolism in both humans and rats.69
NVP undergoes Phase I oxidation by P450 enzymes in the liver in both humans and rats,
and it is further metabolized to glucuronide conjugates, which are the major metabolites in urine.
It can also be further oxidized to a carboxylic acid metabolite.69
CYP3A4 and CYP3A5 are,
respectively, the major and minor P450 isoforms responsible for converting NVP to the 12-
hydroxynevirapine (12-OH-NVP) metabolite in the liver of humans.70

28
Although there are many potential reactive metabolites of NVP,68
the 12-hydroxy pathway
was shown to be responsible through two key lines of evidence. The first is that feeding 12-OH-
NVP at half the standard dose of NVP required for 100% incidence (75 mg/kg vs 150 mg/kg) was
able to induce the same degree of rash in female BN rats.68
The second was using the deuterium
isotope effect. If the rate limiting step in forming the reactive metabolite is the oxidation of the
methyl group to form 12-OH-NVP, then replacement of the methyl hydrogens with deuterium
should decrease the rate of oxidation and decrease the production of the reactive metabolite, thus
decreasing the incidence of rash. The deuterated analog did cause a lower incidence of rash;
however, given at the same dose, the blood level of the deuterated analog was much lower than
that of nevirapine.68
This was unexpected, and it is proposed that the reason for this is that the
carbon radical intermediate produced from P450 oxidation can partition between oxygen rebound
to form 12-OH-NVP, or undergo hydrogen atom loss to form a reactive quinone methide
species.68
The quinone methide is a strong electrophile and inactivates P450 enzymes. Because
deuteration decreases the rate of methyl oxidation, less reactive metabolite is formed and there is
less P450 inactivation and more metabolism of the deuterated analog through other oxidative
pathways.
Despite determining that formation of 12-OH-NVP was required to produce the rash, it
was not known which chemical species ultimately induces the rash. 12-OH-NVP is the same
oxidation state as the quinone methide and cannot spontaneously rearrange into the reactive
quinone methide species. This thesis proposes the 12-OH-NVP metabolite produced from hepatic
metabolism reaches the skin through the general circulation where it is sulfated by skin-resident
sulfotransferase (SULT) enzymes, such as those present in keratinocytes (KCs). Sulfate is a good
leaving group, and attack by nucleophilic groups on cutaneous proteins could lead to cutaneous
adducts and initiate an immune response.
1.7.3 Role of Sulfation in Nevirapine-Induced Skin Rash
Both human and rat liver and skin tissue contain sulfotransferase enzymes71
capable of
sulfating the 12-OH-NVP metabolite. Production of the sulfate metabolite was confirmed by
various studies performed previously by the Uetrecht group, where the circulating plasma sulfate
levels were found to be between 1-8 µg/mL in NVP-treated rats. Previous projects had tried to
determine mechanistically if the sulfate metabolite of NVP was responsible for causing skin rash

29
through metabolic manipulation studies. Unfortunately, at the time these studies were performed,
methods to detect covalent adducts in the skin had not been successful. An overview of these
previous experiments is presented here and they were the starting point for the current work.
In previous studies, salicylamide (SA) and molybdenum were utilized to interfere with
production of 12-OH-NVP sulfate. Salicylamide is a chemical that undergoes sulfation and
depletes PAPS,72
while molybdenum interferes with the synthesis of PAPS.73
Salicylamide and
molybdenum were shown to deplete circulating 12-OH-NVP sulfate levels to below the limit of
detection for the mass spectrometer; however, rats still developed typical rash and in the case of
molybdenum, became very sick and died. It was not known at that time if binding in skin had
been prevented. Follow-up studies utilized dehydroepiandosterone (DHEA), a competitive
inhibitor of SULT 2A1, an isoform found in rat skin for which female rats exhibit a 10-fold
greater expression than males. DHEA was partially successful in preventing the skin rash;
however, it interfered with metabolism of NVP and 12-OH-NVP, making it very difficult to
interpret the results. Again, at this time it was not known what was occurring in the skin.39
All
three of these compounds were administered via gavage and presumably, at least in the case of
salicylamide and molybdenum, inhibited the production of hepatic 12-OH-NVP sulfate leading to
decreased systemic concentrations. Previous studies also indicated that the 12-OH-NVP sulfate
metabolite exhibited very low reactivity to nucleophiles in vitro, even in the addition of base;11
all
of these results taken together suggested the sulfate metabolite was not involved. However,
nucleophilic groups on cutaneous proteins may behave quite differently, and this was also
examined in this work.
A simple explanation for the discrepancies in these earlier studies may be that what is
important is the formation of the sulfate in the skin. If the turnover of salicylamide in the skin is
slow it may not deplete PAPS in the skin. The skin possesses SULT enzymes and the 12-OH-
NVP can reach the skin through the systemic circulation. Therefore, the focus of the current work
was to determine if the 12-OH-NVP sulfate formed in the skin was responsible for the rash.
Repetition of some of these previous experiments with the addition of examination of events
occurring in the skin were performed, as were direct testing of sulfate reactivity with cutaneous
proteins. Methods were also developed for isolating skin fractions. It was also determined if the
events occurring in skin correlated to the events occurring systemically in rats.

30
1.7.4 Nevirapine Metabolism in the Liver and Lack of Hepatotoxicity in Rats
The standard dose of NVP required to induce a 100% incidence of rash in female BN rats
is 150 mg/kg/day, and this dose produces a trough plasma level of 40 µg/mL.68
However, at this
dose or lower, there is no occurrence of asymptomatic ALT increases or hepatotoxicity in rats
even though metabolism to the quinone methide occurs, and binding to expressed P450 was
previously identified by Dr. Yan Li from the Uetrecht group.74
As mentioned previously, the
dominant response in the liver is immune tolerance, and this could be the key reason for lack of
hepatotoxicity. Another reason may the issue of cell stress; revisiting the Danger Hypothesis, it
could be that binding in the liver does not induce danger, while in the skin, it does.
Because of previous failed attempts at producing liver injury in rats, another focus of this
thesis was to develop a mouse model of NVP-induced liver injury. There are numerous strains of
knock-out mice available and many more reagents, stains, and procedures for use with mice. Two
knock-out strains of mice tested for increased liver toxicity in this work were the casitas-b-
lineage-lymphoma-b (Cbl-b) and programmed-cell death 1 (PD-1) knock-outs.
The Cbl-b is null for the E3 ubiquitin ligase and exhibits impaired immune tolerance
through interference with T cell receptor (TCR) and transforming growth factor-beta (TGF-β)
signaling.75
They also exhibit impaired T cell anergy and development of peripheral Foxp3+
regulatory T cells (Treg).75
Additionally, if covalent adducts are involved with causing liver
injury, than impaired protein degradation should increase the level of adducts; in this way the Cbl-
b mice may also be more sensitive. The PD-1 knock-outs lack the ability to control and maintain
peripheral T cell tolerance;76
therefore, tolerance might be overcome to initiate an immune
response. Both strains, as well as numerous wild type strains, were tested in this work to
determine the involvement of hepatic covalent adducts in NVP-induced liver damage.
1.8 Research Hypotheses
The overall hypothesis is that NVP-induced toxicity is mediated by the formation of a
reactive metabolite of NVP; in the case of the liver, a quinone methide species and in the case of
skin, the sulfate metabolite; which each go on to covalently modify self-proteins leading to an
immune response. There are five overall objectives from this hypothesis:

31
1. To detect hepatic covalent adducts in rats and mice as well as human hepatic microsomes and
examine their involvement in hepatotoxicity.
2. To develop a mouse model of NVP-induced hepatotoxicity.
3. To determine if the 12-OH-NVP sulfate metabolite can covalently bind to cutaneous proteins.
4. To determine if covalent binding occurs in the skin of animals that develop a rash.
5. To determine if inhibiting sulfation prevents covalent binding in the skin and the skin rash.

32
CHAPTER 2
Bioactivation of Nevirapine to a Reactive Quinone Methide: Implications for Liver Injury
‘Science does not know its debt to imagination.’
- Ralph Waldo Emerson
This work has been published in the following journal and is reproduced with permission:
Amy M. Sharma, Yan Li, Maria Novalen, M. Anthony Hayes, and Jack Uetrecht. Bioactivation of
Nevirapine to a Reactive Quinone Methide: Implications for Liver Injury. Chemical Research in
Toxicology, 2012. 25, 1708-1719. Epub 2012 July 13.
Reprinted with permission. Copyright 2012 American Chemical Society. All rights reserved.
In this chapter, all experiments were performed by Amy M. Sharma except Figures 2-2, 2-4 A and
B, 2-6 and 2-7.

33
2.1 Abstract
Nevirapine (NVP) treatment is associated with a significant incidence of liver injury. We
developed an anti-NVP antiserum to determine the presence and pattern of covalent binding of
NVP to mouse, rat, and human hepatic tissue. Covalent binding to hepatic microsomes from male
C57BL/6 mice and male Brown Norway rats was detected on western blots; the major protein had
a mass of ~55 kDa. Incubation of NVP with rat CYP3A1 and 2C11 or human CYP3A4 also led to
covalent binding. Treatment of female Brown Norway rats or C57BL/6 mice with NVP led to
extensive covalent binding to a wide range of proteins. Co-treatment with 1-aminobenzotriazole
dramatically changed the pattern of binding. The covalent binding of 12-hydroxy-NVP, the
pathway that leads to a skin rash, was much less than that of NVP, both in vitro and in vivo. An
analog of NVP in which the methyl hydrogens were replaced by deuterium also produced less
covalent binding than NVP. These data provide strong evidence that covalent binding of NVP in
the liver is due to a quinone methide formed by oxidation of the methyl group. Attempts were
made to develop an animal model of NVP-induced liver injury in mice. There was a small
increase in ALT in some NVP-treated male C57BL/6 mice at 3 weeks that resolved despite
continued treatment. Male Cbl-b-/-
mice dosed with NVP had an increase in ALT of >200 U/L,
which also resolved despite continued treatment. Liver histology in these animals showed focal
areas of complete necrosis while most of the liver appeared normal. This is a different pattern than
the histology of NVP-induced liver injury in humans. This is the first study to report hepatic
covalent binding of NVP and also liver injury in mice. It is likely that the quinone methide
metabolite is responsible for NVP-induced liver injury.
2.2 Introduction
Nevirapine (NVP, ViramuneTM
, Figure 2-1) is a non-nucleoside reverse transcriptase
inhibitor used for the treatment of HIV-1 infections. Treatment with NVP is associated with a
significant incidence of idiosyncratic skin rashes and/or liver toxicity.66
The incidence of skin
rashes is approximately 9%. They are usually mild to moderate in nature; however, 16% of NVP-
induced rashes are very severe, including Stevens-Johnson syndrome and toxic epidermal
necrolysis.59
In 2000, the FDA placed a black box warning on NVP due to hepatotoxicity, which
occurs in 6% of patients and can be life threatening.59
The incidence of elevated serum alanine
transaminase (ALT) in NVP-treated patients, which is the first indication of liver injury, is

34
between 8-18% and typically occurs within the first six weeks of treatment.77
Liver injury
normally resolves when the drug is stopped, but it can lead to fulminant liver failure and death.
There also exists evidence for increased risk of liver injury in non-HIV patients, which may be
due to higher CD4 cell counts.61
The mechanisms of idiosyncratic liver injury and skin rashes are currently unknown, but
most idiosyncratic drug reactions appear to be mediated by reactive metabolites. We developed an
animal model of NVP-induced skin rash in Brown Norway (BN) rats that is clearly immune-
mediated and has characteristics very similar to the rash in humans; however, the rats did not
develop liver toxicity.65,66
We postulated that the 12-hydroxylation pathway was involved in the
induction of the skin rash; therefore we replaced the hydrogens on the methyl group with
deuterium to slow down the rate of 12-hydroxylation (Figure 2-1). We found that this analog
(DNVP) did not cause a skin rash as predicted, but instead of higher blood levels because one of
the major metabolic pathways was inhibited, we found that the blood levels of DNVP were
actually much lower than those of NVP at the same dose.68
Although the reason for this was not
immediately obvious, we ultimately concluded that, in addition to oxygen rebound to form 12-
OH-NVP, the intermediate free radical in the P450-mediated oxidation could also lose a hydrogen
atom to form a reactive quinone methide (Figure 2-1). A glutathione conjugate consistent with the
quinone methide intermediate has been reported78,79
; however, it could also come from a sulfate
conjugate of the 12-OH-NVP.
In this study we used an antiserum against NVP to study the covalent binding of NVP,
DNVP, and 12-OH-NVP to hepatic proteins in mice, rats, and humans. We also studied the effects
of chronic administration of NVP to various strains of mice to determine if it causes liver injury.
In addition to C57BL/6 and BALB/c we included the Casitas B-lineage lymphoma-B (Cbl-b)
knockout mouse (Cbl-b-/-
), which is bred on a C57BL/6 background. The Cbl gene is a
mammalian gene that encodes a variety of proteins, specifically those involved in cell signaling
and protein ubiquitination. Lack of ubiquitination of NVP protein adducts could lead to more
persistent covalent binding and possibly toxicity. This also impairs immune tolerance; therefore,
if the liver injury is immune-mediated these animals should be at increased risk. These animals
also express a mouse isoform of CYP3A4 (CYP3A11); therefore, oxidative metabolism of NVP
should occur80
, and this has the potential to lead to liver injury.

35
Figure 2-1. Bioactivation pathway of NVP leading to liver injury.

36
2.3 Materials and Methods
2.3.1 Chemical Materials.
NVP was kindly supplied by Boehringer-Ingelheim Pharmaceuticals Inc. (Ridgefield, CT).
The majority of chemical reagents (1-aminobenzotriazole (ABT),
tris(hydroxymethyl)aminomethane base, methanol, DMSO, phosphate-buffered saline (PBS, pH
7.4), glycerol, silica gel, etc) were obtained from Sigma-Aldrich (Oakville, ON) unless otherwise
noted in the methods. Ammonium persulfate was obtained from Fisher Scientific (Fair Lawn, NJ).
Sodium dodecyl sulphate and Tween-20 were obtained from BioShop (Burlington, ON). Stock
acrylamide/bis solution (29:1, 3.3% C), non-fat blotting grade milk powder, and nitrocellulose
membrane (0.2 µM) were purchased from Bio-Rad (Hercules, CA). Ultra pure
tetramethylethylenediamine was purchased from Invitrogen (Carlsbad, CA). Amersham ECL Plus
Western Blotting Detection System was obtained from GE Healthcare (Oakville, ON).
Horseradish peroxidise-conjugated goat anti-rabbit IgG (H + L chains) and monoclonal GAPDH
were purchased from Sigma-Aldrich (St. Louis, Mo). Normal goat serum was obtained from
Invitrogen (Grand Island, NY). Expressed human CYP3A4, rat CYP3A1, and rat CYP2C11 (each
with P450 reductase and cytochome b5), 0.5 M potassium phosphate pH 7.4, and NADPH
regenerating system solutions A and B were purchased from BD Biosciences (Woburn, MA).
2.3.2 Instruments and Software.
AlphaEaseFC (FluorChem™ 8800) manufactured by Alpha Innotech, now Cell
Biosciences Santa Clara, California, USA was used to image blots. Integrated density values were
obtained using the SPOT DENSO function on the FluorChem™ 8800 Imager.
2.3.3 Synthesis of 12-trideutero-NVP (DNVP).
Synthesis of DNVP was carried out using the method described by Chen et al., 2008 68
. 1H
NMR (CDCl3): δ 0.31-0.41 (m, 2H), 0.83-0.90 (m, 2H), 3.60-3.64 (m, 1H), 7.06 (d, J = 4.8 Hz,
1H), 7.19 (dd, J = 4.8, 7.5 Hz, 1H), 8.01 (dd, J = 2.1, 6.6 Hz, 1H), 8.08 (d, J = 4.8 Hz, 1H), 8.50
(dd, J = 1.8, 4.8 Hz, 1H), 9.90 (bs, 1H). ESI-MS; m/z (%) 270 (MH+, 100%). The ratio of the

37
peaks at m/z 267:268:269:270 as determined by mass spectrometry was 0:0.007:0.124:0.869,
indicating only trace amounts of NVP.
2.3.4 Production of Anti-NVP Anti-Serum in Male White New Zealand Rabbits.
Scheme 2-1. Synthetic pathway of the immunogen used for induction of anti-NVP antiserum.
Synthesis of NVP-NAC Conjugate. The synthesis of the immunogen is outlined in Scheme 1. The
first step in producing the anti-NVP antiserum was to synthesize 12-OH-NVP (2) and convert this
to the benzylic chloride (12-Cl-NVP, 3). The method to produce 12-OH-NVP followed the
protocol described previously81
with minor modifications. ESI-MS; m/z (%) 283 (MH+, 100%).

38
To convert 12-OH-NVP to 12-Cl-NVP, the method of Kelly et al.82
was followed. To 12-OH-
NVP (200 mg) in dry dichloromethane (10 mL) at 0 ºC was added N,N-diisopropylethylamine
(0.14 mL) followed by thionyl chloride (3 mL) and stirred under argon at room temperature for 3
h after which the thionyl chloride was evaporated by rotary evaporation. The reaction mixture was
then extracted with ethyl acetate (3 × 10 mL). The ethyl acetate layer was washed with water (10
mL), dried over anhydrous sodium sulfate, and concentrated to yield crude product, which was
purified with open column chromatography (silica gel, pore size 60 Å, 70 - 230 mesh, column
dimensions 30 × 200 mm) eluted with 50% ethyl acetate/hexanes to yield 0.386 g of yellow solid.
ESI-MS; m/z (%) 301 (MH+, 100%).
The 12-Cl-NVP (1.78 g, 3.55 mmol) was dissolved in 18 mL of tetrahydrofuran and
reacted with N-acetylcysteine (NAC, 2.31 g, 14.18 mmol) in 5 mL of triethylamine under argon
reflux for 2 h. The crude mixture was cooled to room temperature, acidified to pH 3-4 by 1N HCl
and extracted with CHCl3. The organic layer was dried over anhydrous sodium sulfate.
Chloroform was removed under reduced pressure. Nevirapine-NAC conjugate was obtained as a
pale yellow solid (4). Formation of the nevirapine-NAC conjugate was confirmed by mass
spectrometry ESI-MS; m/z (%) 428 (MH+, 100%).
Preparation of NVP-KLH Conjugate. All reagents and glassware were dried in a vacuum
at 50 ºC. Activation of the carboxy groups on NAC of the synthesized 12-NAC-NVP occurred as
follows: to 61.4 mg 12-NAC-NVP was added 108.5 mg of N-hydroxysuccinimide and 103.9 mg
of 1-ethyl-3-(3-dimethyllaminopropyl) carbodiimide hydrochloride. Anhydrous DMF (4 mL) was
introduced via syringe at 0 ºC. The entire mixture was sealed with a rubber stopper and stirred at 0
ºC for 2 h under N2. Methylene chloride (8 mL) was added, followed by washing with water (3 ×
8 mL) and then the organic layer was partially evaporated in vacuo to yield a pale yellow solution
(0.5 mL, 5). DMF (4 mL) was added followed by Keyhole limpet hemocyanin (KLH, 8 mg) and
the mixture was stirred for 1 h at 4 ºC. The reaction mixture was then concentrated under a N2
stream and 1 mL water was added. Centrifugal filtration was performed to collect the protein
solution, which was then lyophilized. A final white powder (10.4 mg) was obtained (6) and stored
at -20 ºC. The same method was used to prepare a conjugate with bovine serum albumin (BSA)
MALDI MS; m/z 67,139 - 68,569. The hapten density of the BSA conjugate was approximately
4.5 molecules of NVP-NAC/BSA as determined by the increase in mass on mass spectrometry.

39
Production of Anti-NVP-NAC-KLH-Antiserum. Polyclonal anti-NVP-NAC-KLH
antibodies were raised in two individual 2 kg, male, pathogen-free New Zealand White rabbits
(Charles River, Quebec) housed in the animal care facility at The Division of Comparative
Medicine, University of Toronto. Each animal was immunized with the NVP-NAC-KLH
conjugate (1 mg antigen + 100 µL glycerol in 1.8 mL of phosphate buffered saline emulsified
with an equal volume of Freund’s complete adjuvant) subcutaneously at multiple sites. Injections
with 500 µg of NVP-NAC-KLH in Freund’s incomplete adjuvant divided into six to eight
subcutaneous sites were repeated 4, 6, 8, and 12 weeks after the initial immunization. The animals
were exsanguinated while under pentobarbital anesthesia 10 days after the final immunization.
The serum was heat-inactivated at 56 °C for 30 min before being stored at -80 °C.
ELISA. NVP-NAC-BSA, BSA, or KLH (100 µL, 10 µg/mL in carbonate-bicarbonate
coating buffer) were coated into the wells of a flat-bottom 96-well plate (Costar, Cambridge, MA)
and the plate was incubated overnight at 4 °C. The plates were washed with ELISA wash buffer
(50 mM tris(hydroxymethyl)aminomethane-buffered saline, pH 8.0, 0.05% Tween-20) three times
and blocked by the addition of 100 µL of post-coat solution (50 mM Tris-buffered saline, pH 8.0,
1% BSA) for 30 min at room temperature. Following the blocking step, the wells were washed
three times and various dilutions of the anti-NVP-NAC-KLH antiserum or pre-immune serum
were added to the plates, which were then incubated at room temperature for 2.5 h. The plates
were subsequently washed three times with ELISA wash buffer and horseradish peroxidase-
conjugated goat anti-rabbit IgG (diluted 1:5000 in post-coat solution; 100 µL) was added to each
well. The ELISA plates were incubated at room temperature for 2 h. Plates were then washed
three times with ELISA wash buffer. Enzyme substrate (3,3’,5,5’-tetramethylbenzidine
peroxidase substrate and peroxidase solution B, Kirkegaard & Perry Laboratories) was mixed in
equal volumes and 100 µL of the enzyme substrate was added to each well. The plate was
incubated in the dark at room temperature for 10 min. Sulfuric acid (2M, 100 µL) was added to
each well to quench the reaction. Absorbance was measured with the Basic Endpoint Option of
SoftMax® Pro 5 Software, using the SPECTRA maxPLUS384 plate reader (Molecular Devices
Technologies) set at 450 nm.

40
2.3.5 Animal Care.
Male (200 – 250 g) or female BN rats (150 – 175 g) were obtained from Charles River
(Montreal, Quebec). Rats were housed in pairs in standard cages in a 12:12 h light/dark cycle with
access to water and Agribrands powdered lab chow diet (Leis Pet Distribution, Inc. Wellesley,
Ontario) ad libidum. Following a one week acclimatization period, rats were either maintained on
control chow or started on drug containing diet (treatment groups). Drug was mixed thoroughly
with powdered lab chow if it was to be administered orally. The amount of drug administered to
animals was calculated based on body weight of the rats and their daily food intake. Rats were
sacrificed via CO2 asphyxiation.
Male Balb/c or C57BL/6 mice (6-8 weeks age) were obtained from Charles River
(Montreal, Quebec). Cbl-b-/-
knockout mice were bred in house from animals first developed by
Dr. J. Penninger at the Institute of Molecular Biotechnology of the Austrian Academy of Science,
Vienna, with his kind permission. Mice were kept 4 per cage. The average weight gain was
approximately 0.75 g per week (data not shown). NVP was administrated in lab chow following a
one week acclimatization period. Animal experiments were approved by the University of
Toronto animal care committee in accordance with guidelines of the Canadian Council on Animal
Care.
2.3.6 Treatment of Animals with NVP, 12-OH-NVP, DNVP, or ABT.
Female BN rats were treated with NVP or DNVP at 150 mg/kg/day, or 12-OH-NVP at
159 mg/kg/day (equimolar dose) orally in standard rat chow for either 8, 10, or 21 days. Dosages
were based on previous work showing induction of rash at these levels.65
Treatment of NVP or
DNVP by s.c. injection lasted 21 days with a dose of 75 mg/kg/day of either compound. ABT was
dissolved in water (20 mg/mL) and administered via gavage at a dose of 50 mg/kg/day. If ABT
was to be given to animals, the dose of NVP was 50 mg/kg/day via gavage. Methylcellulose
(0.5%) was used to suspend NVP or metabolites given to rats by gavage or s.c. injection. All mice
were started on NVP at 950 mg/kg/day in standard chow after preliminary studies showing no
apparent toxicity or mortality of mice at either 550 or 950 mg/kg/day.

41
2.3.7 Incubations with Microsomes or Supersomes.
Livers were homogenized in ice-cold 1.15% KCl using a Polytron 2100 homogenizer and
centrifuged at 26,400 × g for 10 min at 4 ˚C. The supernatant was then centrifuged at 100,000 × g
for 50 min at 4 ˚C. The pellet was homogenized in 4 volumes of glycerol-phosphate-KCl buffer
and aliquots were stored at -80 °C. The protein concentration of the prepared microsomes was
quantified using a BCA protein assay kit (Novagen, EMD Biosciences Inc.). All incubations were
performed at 37 ºC. NVP, 12-OH-NVP or DNVP stock solutions were prepared in methanol and
the final methanol concentration in the reactions did not exceed 1% for any incubation.83
The
microsomal incubations consisted of 100 mM potassium phosphate buffer (pH 7.4), a NADPH-
regenerating system (Solution A final concentrations: 1.3 mM NADP, 3.3 mM glucose-6-
phosphate, 3.3 mM MgCl2; Solution B final concentration: 0.4 Units/mL glucose-6-phosphate-
dehydrogenase), and microsomal homogenate (final protein concentration varying from 0.3
mg/mL to 15 mg/mL). EDTA·2Na (0.4 mM) was added to rat CYP3A1 and 2C11 incubations and
water was added to each incubation to reach a final volume of 400 µL for rat and mouse or 200
µL for human 3A4 incubations.84
Incubations consisting of all reaction components except the
NADPH regenerating system or drug were preincubated for 5 min. The NADPH-regenerating
system or drug was added to each of the test and control tubes after the 5 min preincubation.
Reactions were stopped by placing the sample vials on dry ice and stored at -80 °C.84
If
microsomal incubations were to be analyzed via LC/MS, 250 µL ice cold acetonitrile was used to
quench the reaction and internal standard (ethyl-NVP – a NVP derivative in which the
cyclopropyl group has been replaced with an ethyl group, 5.4 µg/mL, 50 µL) was added to each
tube, contents were centrifuged, separated by solid phase extraction (Strata® solid phase
extraction column C18-E, 100 mg, by Phenomenex), evaporated in vacuo at 50 ºC, and re-
constituted to 50 µL prior to analysis.
2.3.8 Quantification of NVP and its Metabolites from Microsomal Incubations.
Samples were re-constituted to 50 µL with mobile phase (16% acetonitrile and 84% water
with 2 mM ammonium acetate and 1% acetic acid). The samples were separated by HPLC and
analyzed by mass spectrometry. The separation was performed on an Ultracarb C18 30 X 2.0 mm,
5 µm column (Phenomenex) under isocratic conditions with a mobile phase consisting of 16%

42
acetonitrile and 84% water with 2 mM ammonium acetate and 1% acetic acid. The flow rate was
0.2 mL/min.
2.3.9 Mass Spectrometry Analysis.
Mass spectrometry was carried out using a PE Sciex API 3000 quadrupole system with an
electrospray ionizing source. The ion pairs used for this analysis were: 267.0/226.1 for NVP,
283.1/223.1 for 12-OH-NVP, 297.1/210.1 for 4-COOH-NVP, 283.1/161.0 for 2-OH-NVP,
283.1/214.0 for 3-OH-NVP, 255.1/227.2 for ethyl-NVP (positive ionization mode). Standard
curves prepared for 2-OH-NVP (0.43 – 102.9 µg/mL), 3-OH-NVP (0.36 – 86.8 µg/mL), 12-OH-
NVP (0.38 – 91.0 µg/mL), 4-COOH-NVP (0.26 – 61.8 µg/mL), and NVP (0.74 – 176.9 µg/mL)
had R2 values of > 0.99.
2.3.10 Analysis of Covalent Binding Using SDS-PAGE and Immunoblotting.
Livers homogenized in working cell lysis buffer (Cell Signaling Technologies, Pickering,
ON) containing 1X HALT Protease Inhibitor Cocktail (Pierce, Rockford, IL) with a Polytron
2100 homogenizer and centrifuged at 1000 × g for 15 min and supernatant was collected and
again centrifuged at 10 000 × g for 30 min. The supernatant was mixed with Pierce reducing
sample loading buffer in a 4:1 protein to buffer ratio and boiled for 5 min. Sodium dodecyl
sulfate polyacrylamide gel electrophoresis was performed using the Protean-3 minigel system
(BioRad, Mississauga, ON). Gels were hand-cast (8%) or bought from Bio-Rad Canada (12%),
and were run at 130 V. Electrophoresis running buffer (Bio-Rad) consisted of 25 mM Tris base,
192 mM glycine, and 0.1% sodium dodecyl sulfate, pH 8.3. Transfer to nitrocellulose membrane
(0.2 µM, BioRad) occurred at 0.13 mA for 90 min at 4 ºC using the same Protean-3 minigel
system (BioRad, Mississauga, ON). Tris-glycine transfer buffer (Bio-Rad) consisted of 25 mM
Tris, 192 mM glycine, and 20% methanol at pH 8.5. Membranes were washed twice in tris-
buffered saline tween-20 (TBST) wash solution for 5 min. Membranes were then blocked in 5%
non-fat milk blocking solution in TBST. Blocking was done for 90 min at room temperature.
Membranes were then rinsed with three changes of TBST for 5 min each and incubated with a
1:100 or 1:500 dilution of primary anti-NVP antiserum and 10% normal goat serum in TBST
overnight at 4 ºC. A 20 min wash (three changes) in TBST after overnight blocking was followed
by a 90 min incubation in secondary antisera (1:2000 or 1:5000 dilution) in TBST containing 10%
goat serum. The secondary antisera was goat anti-rabbit horseradish peroxidase antisera.

43
Membranes were washed 3 times for 20 min with TBST. All blots were incubated with enhanced
chemiluminescence stain for 5 min and analyzed with a FluorChem8800 imager. To probe for the
glyceraldehyde 3-phosphate dehydrogenase (GAPDH) loading control, membranes were stripped
of primary anti-NVP anti-serum using Pierce Restore Plus buffer (Pierce, Rockford, IL) for 15 to
20 min at room temperature followed by a one h blocking step. Membranes were then incubated
in mouse monoclonal anti-GAPDH antisera (1:40,000) and processed as above except the
secondary antisera was goat anti-mouse horseradish peroxidase antisera diluted 1:10,000 (Jackson
ImmunoResearch, Baltimore Pike, West Grove, PA.).
2.3.11 Analysis of in Vivo Covalent Binding using Immunohistochemistry.
The liver samples were fixed in 10% formalin and the paraffin block, hematoxylin/eosin
slides, or unstained sections were prepared at the Toronto Hospital for Sick Children. For
immunohistochemical staining, non-specific sites were blocked with 10% goat serum for 1 h. At
this point the anti-NVP antiserum was diluted to 1:100 in 10% goat serum and applied to each
section overnight. Following a washing step, slides were submerged in 0.3% hydrogen peroxide
in methanol for 10 min to block endogenous peroxidases followed by a washing step. Secondary
antiserum (goat anti-rabbit IgG-HRP conjugated antisera) was applied to the sections at a dilution
of 1:3000 in 10% goat serum. Sections were incubated with secondary antiserum for 2 h. After a
final washing step, Vector NovaRED stain was added as a substrate for the peroxidases following
package protocol. Sections were then counterstained with Mayer’s hematoxylin (Sigma),
dehydrated by sequential immersion in increasing concentrations of ethanol, cleared in xylenes,
and mounted using Permount mounting medium (Fisher, Markham, ON).
2.3.12 Plasma Alanine Transaminase and Cytokine Analysis.
Alanine transaminase (ALT) was assayed using the InfinityTM
ALT (glutamic pyruvate
transaminase) Liquid Stable Reagent kit by Thermo Scientific. Screening for cytokines was
performed using a Luminex immunoassay mouse cytokine/chemokine kit from Millipore
Corporation (Milliplex™ Map Kit). Homogenized liver tissue or serum samples prepared to the
kit specifications were plated and analyzed following the manufacturer’s instructions.

44
2.4 Results
2.4.1 Characterization of the Anti-NVP-NAC-KLH Antiserum.
ELISA analysis showed the anti-NVP-NAC-KLH antiserum recognized the NVP-NAC-
BSA conjugate or KLH, but not BSA alone (Figure 2-2A). The binding of the antisera to the
NVP-NAC-BSA conjugate was inhibited by preincubating the anti-NVP-NAC-KLH antiserum
with NVP or its metabolites (Figure 2-2B). Inhibition was much less with 2-OH-NVP, 3-OH-
NVP, and 4-COOH-NVP (the metabolite in which the methyl group has been oxidized to a
carboxylic acid). Binding could still be detected at an anti-serum dilution of 1/1,000,000.

45
Figure 2-2. ELISA analysis showing (A) binding of the anti-NVP-NAC-KLH antiserum to the
NVP-NAC-BSA conjugate, KLH, or BSA and (B) the effect of preincubation of the antiserum
with NVP or its metabolites on the binding of the antisera to the NVP-NAC-BSA conjugate. Data
represent the mean ± s.d. from 3 incubations.
2.4.2 Covalent Binding of NVP, DNVP, or 12-OH-NVP to Hepatic Microsomes in Vitro and Comparison to in Vivo Hepatic Covalent Binding.
When microsomes produced from male BN rats were incubated with NVP, 12-OH-NVP,
or DNVP, the greatest covalent binding observed was with NVP, and the stongest band was at
~55 kDa (Figure 2-3A), which corresponds to the mass of the male dominant P450 2C11/3A1
isoforms.85
Incubation of mouse liver microsomes with NVP produced a band of slightly higher
mass, ~57 kDa (Figure 2-3B), corresponding to the mass of the dominant murine P450 3A11.80
Significant covalent binding of 12-OH-NVP was not observed with rat microsomes, and DNVP
produced a much fainter band at 55 kDa than NVP in both rodent species tested. In vivo
experiments with either species displayed a wide range of covalently-modified bands that were
much more intense than from in vitro experiments. The covalent binding of DNVP to both rat and
mouse hepatic microsomes was also much less than that of NVP by almost 5 fold as determined
by densitometry (data not shown). The amount of binding did not increase significantly beyond 15
min (Figure 2-3C).

46

47
Figure 2-3. (A) Comparison of covalent binding of 12-OH-NVP (lanes 2, 5) and DNVP (lane 3,
6) with that of NVP (lane 4, 7) after a 30 or 60 min incubation with male BN rat microsomes (1
mg/mL protein) at a drug concentration of 1 mM. For comparison, covalent binding to hepatic
proteins is shown after 8 days of treatment of female rats with 12-OH-NVP (159 mg/kg/day, lane
9) or NVP (150 mg/kg/day, lane 10). Protein loading was 15 µg for lanes 1-7 and 20 µg for lanes
8-10. (B) Comparison of covalent binding of 12-OH-NVP (lanes 2, 5) and DNVP (lanes 3, 6) with
that of NVP (lanes 4, 7) at a concentration of 1 mM after a 30 or 60 min incubation with
microsomes (1 mg/mL protein) from male C57BL/6 mice. For comparison, covalent binding to
hepatic proteins is shown after 6 weeks of treatment of C57BL/6 mice with NVP at a dose of 950
mg/kg/day in food. Protein loading was 13 µg for lanes 1-7 and 20 µg for lanes 8-9. (C)
Comparison of covalent binding of NVP to hepatic microsomes from male C57BL/6 mice (lanes
2-4) or male BN rats (lanes 6-8) after a 15, 30, or 60 min incubation at a drug concentration of 1
mM and microsome concentration of 1 mg/mL protein. Protein loading was 20 µg per lane. The
primary antiserum dilution was 1:500 and that of the secondary antisera was 1:5000.
2.4.3 Covalent Binding of NVP to Expressed Rat CYP2C11 or CYP3A1 Supersomes, or of NVP, DNVP, or 12-OH-NVP to Human Hepatic Expressed CYP3A4 Supersomes.
Incubation of NVP with expressed rat CYP2C11 (Figure 2-4A) or CYP3A1 Supersomes
(Figure 2-4B), the dominant forms of P450 in male rats85-87
led to covalent binding with major
bands produced at ~50 kDa and ~52 kDa, respectively. In the absence of NVP as indicated in the
figures, there is a small artifact band. Binding to 2C11 and 3A4 was strongest at 30 min; a
decrease in the intensity of the P450 band was observed from 30 to 120 min.
The incubation of expressed human 3A4 displayed the greatest binding with NVP (Figure
2-4C) versus 12-OH-NVP or DNVP. However, 12-OH-NVP did bind to human CYP3A4 more
than expected, although less than NVP and there was much less binding of DNVP. The NVP-
modified band had a mass of ~57 kDa, which is the mass of CYP3A4.88

48
Figure 2-4. Covalent binding of NVP to expressed male rat CYP2C11 (A) or CYP3A1 (B) in
vitro. Protein concentration for each incubation was 0.8 mg/mL with 0.5 mM of drug. For
immunoblots, protein loading was 9 µg and 7.5 µg per lane for A and B, respectively. (+)
indicates incubations containing NVP while (–) indicates incubations lacking NVP. Proteins were
resolved on 12% gels with 1:100 dilution of primary anti-serum followed by 1:2000 dilution of
secondary antisera. Comparison of covalent binding of 12-OH-NVP (lanes 2, 5), or DNVP (lanes
3, 6) with that of NVP (lanes 4, 7) to human CYP3A4 with a drug concentration of 1 mM and
protein concentration in each incubation of 1 mg/mL (C). Proteins (10 µg/lane) were resolved on
an 8% gel. Dilutions of antisera were 1:500 for the primary anti-serum and 1:5000 for the
secondary antiserum.

49
2.4.4 Covalent binding of NVP or 12-OH-NVP to Hepatic Proteins from Female BN Rats Treated with NVP or 12-OH-NVP.
Female BN rats were treated with NVP or 12-OH-NVP for a period of 8 days at doses of
150 mg/kg/day or 159 mg/kg/day, respectively (Figure 2-5A). The pattern of covalent binding
was different for NVP and 12-OH-NVP; this difference was most prominent for the lower
molecular mass proteins (30 – 60 kDa). NVP-treated female BN rats exhibited greater covalent
binding than 12-OH-NVP-treated rats at an equimolar dose, but there was a prominent artifact
band in the 12-OH-NVP blot at about 60 kDa. Preincubation of the anti-NVP serum with NVP
blocked almost all of the binding (Figure 2-5B).

50
Figure 2-5. (A) Covalent binding to hepatic proteins from female BN rats fed NVP (150 mg/kg)
or 12-OH-NVP (159 mg/kg) for 8 days. Protein loading was 12 µg per lane. Samples were
resolved on an 8% gel. A 1:500 dilution of primary anti-serum was followed by 1:5000 dilution of
secondary antisera. (B) Incubation of the anti-NVP serum with 2 mM NVP for 2 h at 37 °C
blocked most of the binding (left side of panel) to samples from livers of 12-OH or NVP treated
rats. Samples for both panels A and B were prepared, run, blocked, incubated with secondary
antibody, and imaged at the same time and protein loading was 10 µg/well of protein per lane.

51
2.4.5 Immunohistochemistry of Liver from NVP- or DNVP-Treated or NVP + ABT Co-treated Female BN Rats.
Hepatic covalent binding of NVP and DNVP was greatest in the centrilobular area (Figure
2-6). The pattern of binding was dramatically different in rats treated with a combination of NVP
and the P450 inhibitor aminobenzotriazole; specifically, treatment with ABT blocked binding in
the centrilobular area and shifted it to the periportal area. Co-treatment with ABT also changed
the pattern of binding by western blot although there was still significant binding (data not
shown). Clearance of NVP depends on oxidative metabolism and so even if P450 is inhibited, it
causes an increase in blood levels, but ultimately NVP is oxidized.
Figure 2-6. Immunohistochemistry of liver sections from female BN rats; blank control, NVP
treatment (150 mg/kg/day x 7 days in food), DNVP treatment (150 mg/kg/day x 7 days in food),
ABT treatment (50 mg/kg/day x 28 days by gavage), or NVP (150 mg/kg/day) + ABT (50
mg/kg/day) x 28 days by gavage. Slides were incubated with 1:100 dilution of primary antisera
and 1:2000 dilution of the secondary antisera. The slides were counterstained with Mayer’s
hematoxylin, magnification 20x.

52
2.4.6 Oxidation of NVP or 12-OH-NVP by Rat Liver Microsomes.
The carboxylic acid (4-COOH-NVP) of NVP was detected in the incubation of 12-OH-
NVP with NADPH and hepatic microsomes from both male and female BN rats (Figure 2-7). No
aldehyde intermediate was detected in these reactions.
Figure 2-7. 4-COOH-NVP concentrations from incubations of 12-OH-NVP with microsomes
from male (n = 3) and female (n = 1) BN rats.
2.4.7 Covalent Binding, Serum ALT levels, INF-γ, and IL-6 Levels in Mice.
There was no change in plasma ALT in BN rats treated with NVP (data not shown).
Various strains of mice were treated with NVP to determine if it causes liver damage, covalent
binding, and/or histological changes. Male BALB/c mice treated with NVP had no increase in
ALT (data not shown) while there was an increase in ALT in male C57BL/6 mice at 3 weeks
followed by normalization of ALT levels (Figure 2-8A). Immunoblots revealed no significant
differences between the pattern and degree of binding in these two strains (Figure 2-8B). ALT
levels in both male and female Cbl-b-/-
mice increased at week 2, with a somewhat greater
increase in male mice (Figure 2-9A) than female mice (Figure 2-9C). The animals with the largest
ALT increase displayed areas of gross hepatic necrosis evident as areas of white on the surface of
the liver upon sacrifice at 2 weeks. Immunoblot analysis showed the presence of a wide range of

53
modified hepatic protein in both male (Figure 2-9B) and female (Figure 2-9D) mice. Animals
with gross necrosis appeared to have slightly more binding of NVP to lower molecular mass
proteins (Figure 2-9B, D).
Luminex analysis for a broad range of cytokines performed on serum of mice from the 2
week study on days 1, 7, and 14 of NVP treatment revealed an increase in interferon-gamma
(IFN-γ) in plasma samples of male mice on day 7 (Figure 2-10B), both in animals that developed
significant necrosis and those that did not, but the level was highest in an animal that did develop
necrosis. IL-6 was also increased at day 7 versus day 14 of NVP treatment in plasma of male mice
(Figure 2-10A). By day 14 of NVP treatment, the cytokine levels had decreased to or close to
baseline (data not shown) for the majority of animals.
Changes in cytokines were less clear for serum samples from female Cbl-b -/- mice and no
inferences could be made (data not shown). No significant changes in cytokines were observed for
GM-CSF, IL-10, 1L-12(p70), IL-13, IL-17, 1L-1β, IL-2, IL-4, IL-5, IL-7, IL-9, MCP-1, or TNF-
α.

54
Figure 2-8. (A) Changes in ALT in male C57BL/6 mice treated with NVP (950 mg/kg/day) for 4
weeks. Values are based on the mean of triplicate readings per time point per animal ± S.D, n=5
treated mice or n = 4 control mice. Unpaired t-test, 7 d.f., p<0.05. (B) Corresponding covalent
binding of NVP at the same dose in male BALB/c (n=2) or C57BL/6 (n=3) mouse livers after 6
weeks of treatment. Protein loading was 20 µg per lane. Samples were resolved on an 8% gel.

55

56
Figure 2-9. (A) Plasma ALT levels in male Cbl-b-/-
mice fed NVP orally for 14 days (950
mg/kg/day). Values are based on the mean of triplicate readings per time point per animal ± S.D,
n=5 treated mice or n = 4 control mice. Unpaired t-test, 7 d.f., p<0.05. (B) Covalent binding of
NVP in the livers of the same Cbl-b-/-
mice. (C) Plasma ALT levels in NVP-treated (950
mg/kg/day) female Cbl-b-/-
mice, n=4 treated or n=4 control mice. Values are based on the mean
of triplicate readings per time point per animal ± S.D, n=5 treated mice or n = 4 control mice.
Unpaired t-test, 6 d.f., p<0.05. (D) Covalent binding of NVP in the livers of the same mice.
Protein loading was 25 µg per lane. Samples were resolved on 10-20% gradient gels. A 1:500
dilution of primary antisera followed by 1:5000 dilution of secondary antisera was used.

57
Figure 2-10. Serum IL-6 (A) or IFN- (B) from control and NVP-treated Cbl-b-/-
mice at day 7 of
NVP treatment. Animals showing gross necrosis are displayed separately.

58
2.4.8 Liver Histology and ALT in Male Cbl-b-/- or C57BL/6 Mice Treated with NVP.
Liver histology of Cbl-b-/-
mice sacrificed after 2 weeks of NVP treatment at the time of
maximal ALT elevation is shown in Figure 2-11. The presence of gross necrosis, which was
visible on the surface of the liver as white areas was observed in 4 of 7 treated animals. Three
NVP-treated males with gross liver necrosis had ALT values ≥ 200 U/L. Two of 8 NVP-treated
female mice with minor liver necrosis had ALT values of 286 and 80 U/L. Histology in female
mice did not demonstrate as much injury as in males (data not shown). Histology of the livers of
affected males showed the presence of focal subcapsular areas of massive liver necrosis (Figure 2-
11B, 2-11C) sharply demarcated from the adjacent viable liver. Necrotic areas were surrounded
by and infiltrated by mononuclear cells, macrophages, and neutrophils. This pattern of liver
necrosis suggests an ischemic injury, but no evidence of thrombi or vasculitis was observed.
Multifocal necro-inflammatory hepatitis with neutrophil-rich inflammatory response was
observed in the absence of gross necrotic lesions in male Cbl-b-/-
mouse livers. Lower doses of
NVP were also tested with Cbl-b-/-
mice, but no injury was seen (data not shown). In contrast,
hepatic histology of C57BL/6 mice treated with NVP and sacrificed at 4 weeks displayed
hepatocyte death on the edge of the lobe in one animal, as well as small focal areas of necrosis (2-
12B). Induction of smooth endoplasmic reticulum (Figure 2-12C), presumably including P450
induction, was present in the histology of all mice strains tested, but was most prominent for Cbl-
b-/-
male mice. This marked induction may have led to greater reactive metabolite formation
contributing to the greater toxicity in this strain, and this appeared to be the case although the
difference is subtle (Figure 2-13).

59
Figure 2-11. H&E staining of livers from Cbl-b-/-
mice treated with NVP for 2 weeks. (A)
Untreated control liver with normal ALT; (B) liver from a NVP-treated mouse with gross necrosis
and an ALT of 271 U/L, and (C) liver from another NVP-treated mouse with gross necrosis and
ALT of 313 U/L. Areas of massive hepatocyte necrosis surrounded by viable hepatocytes are
shown in (B) and (C).

60
Figure 2-12. H&E staining of livers from male C57BL/6 mice treated with NVP for 3 weeks. (A)
Untreated control liver with a normal ALT; (B) liver from a NVP-treated mouse with very mild
necrosis (appearing as the thin band around the capsule) and ALT of 94 U/L, and (C) liver from
another NVP-treated mouse with an ALT of 75 U/L. Changes to the liver parenchyma due to
enlargement of hepatocytes in the periacinar regions and extensive expansion of the endoplasmic
reticulum are also present in both (B) and (C).
2.4.9 Comparison of Hepatic Covalent Binding of NVP between Mice and Female BN Rats.
Female BN rats treated with NVP for 1, 2, 4, or 8 days were sacrificed and covalent
binding was determined (Figure 2-13). In comparison with Cbl-b-/-
knockout mice at 2 or 10
weeks of treatment, or male C57BL/6 mice at 2 weeks of treatment, rats had significantly greater
binding from day 4 onwards. In all animals the presence of a modified P450 band at ~55 kDa was
prominent and represents the largest modified band in each lane. While modified proteins in rats
range from 20 to 100 kDa, it appeared that lower molecular weight proteins were modified in

61
mice (up to 70 kDa). Treatment of Cbl-b-/-
mice with NVP for 2 weeks led to greater binding than
at 10 weeks, and C57BL/6 mice displayed the least binding of the species tested.
Figure 2-13. Comparison of covalent binding of NVP to hepatic proteins in mice and rats. NVP
was fed to rats in a time course manner from 1 to 8 days at 150 mg/kg orally in food. Mice were
given 950 mg/kg/day for 2 weeks or 10 weeks. C57BL/6 males given NVP for 2 weeks are
represented by C57.1 and C57.2. Each lane was loaded with 20 µg of protein. Samples were
resolved on a 4-20% gradient gel. A 1:500 dilution of primary antisera followed by 1:5000
dilution of secondary was used.

62
2.5 Discussion
An anti-NVP antiserum was produced and used to demonstrate that NVP covalently binds
to hepatic proteins, both in vitro and in vivo. Binding occurred directly to P450 as demonstrated
by covalent binding to expressed P450s, both rat and human. We have shown that the skin rash
requires oxidation of NVP to 12-OH-NVP,68
and most recently we have shown that covalent
binding of the benzylic sulfate of this metabolite formed by sulfotransferase in the skin that is
responsible for the rash.89
In contrast, the majority of binding in the liver must involve direct
oxidation by P450 as evidenced by the marked shift in the pattern of binding from the
centrilobular region to the portal region caused by the P450 inhibitor ABT as shown in Figure 2-6.
There is also less covalent binding of 12-OH-NVP than NVP in the liver. Furthermore,
substitution of the methyl hydrogens with deuterium (DNVP) led to a marked decrease in
covalent binding. Given that oxidation of the methyl group is involved in the covalent binding,
but it does not involve 12-OH-NVP, these data provide strong evidence that the chemical species
responsible for the covalent binding in the liver is a quinone methide formed by the loss of a
hydrogen atom from the P450-generated free radical (Figure 2-1). Others have found evidence for
an epoxide reactive metabolite,79
but these data suggest that it is less important with respect to
covalent binding than the quinone methide.
Some covalent binding of 12-OH-NVP was detected in the in vitro experiments where
phase II pathways such as sulfation would not occur, and the pattern of binding was somewhat
different than that of NVP. This suggests that oxidation of 12-OH-NVP can lead to a reactive
metabolite, although the binding is less than for NVP. This could be due to oxidation of the
benzylic alcohol to an aldehyde or oxidation of some other part of the molecule. Oxidation of 12-
OH-NVP by rat hepatic microsomes led to the carboxylic acid (Figure 2-1), but the intermediate
aldehyde was not observed (Figure 2-7). This suggests that 12-OH-NVP is oxidized all the way to
the carboxylic acid by P450 without release of the intermediate aldehyde; there is precedent for
this.68
It is conceivable that some of the aldehyde could become covalently bound to P450 and be
responsible for the observed covalent binding; however, the pattern of binding was broader than
that of NVP; specifically, most of the binding was to proteins with masses different from P450.
Therefore, the aldehyde seems unlikely to be responsible for a significant amount of the covalent
binding of 12-OH-NVP. To re-emphasize, the data strongly implicate the quinone methide as
being the major species responsible for covalent binding of NVP in the liver.

63
Although we had previously observed some strange inclusion bodies in the livers of rats
treated with NVP, we did not observe an increase in ALT even though there was a significant
degree of covalent binding. This suggests that covalent binding may be necessary but not
sufficient to produce liver injury; this is consistent with an immune mechanism. We attempted to
develop an animal model of NVP-induced liver toxicity in mice. Mice metabolize NVP much
faster than rats, and even higher doses did not produce easily detectable blood levels of NVP or
outward signs of toxicity. However, even with higher doses and more rapid metabolism, the
amount of covalent binding in mice was less than in BN rats. Treatment of C57BL/6 mice with
NVP led to a small increase in ALT in some animals that resolved despite continued treatment.
This is the pattern of “adaptation” frequently observed in humans treated with a drug that can
cause more severe idiosyncratic liver injury. Liver histology in these mice revealed moderate
inflammatory nodules and areas of mild focal necrosis (Figure 2-12B, C). Although the covalent
binding in BALB/c mice was similar to that in C57BL/6 mice, no increase in ALT was observed
in BALB/c mice. We then treated Cbl-b knockout mice with NVP. Cbl-b-/-
mice lack E3 ubiquitin
ligase, which leads to impaired immune tolerance; however, the animals are phenotypically
normal. This deficiency could also lead to increased covalent binding if ubiquitin ligase is
required for clearance of modified proteins and this appeared to be the case (Figure 2-13). We
found that there was a much greater increase in ALT in some of the Cbl-b-/-
mice than in the
C57BL/6 mice, but the ALT also returned to normal despite continued treatment with the drug.
Histology performed at the time of peak ALT (14 days) showed areas of complete necrosis with a
local inflammatory response. These appeared to represent ischemic lesions because cells close to
the liver capsule were spared presumably because they could benefit from diffusion through the
liver capsule. However, no vascular lesions were evident histologically.
Luminex analysis of cytokines performed on serum samples from Cbl-b-/-
mice sacrificed
at the time of ALT peak displayed a significant increase in serum IFN-γ and IL-6 in some of the
animals (Figure 2-10A, B). This increase was most prominent on day 7 rather than day 1 or 14 in
the majority of mice, and it occurred before the ALT increase at day 14. An elevation in cytokines
or immune factors that occurs earlier than increases in other toxicity markers (i.e. ALT) is
consistent with an immune response. At the study end point of 14 days, IFN-γ in liver samples of
male Cbl-b-/-
mice was also elevated to ~100 pg/mL for two mice (data not shown) with gross
necrosis compared with 39 pg/mL for control mice. One mouse with elevated IFN-γ in the liver

64
(130 pg/mL) on day 14 also had markedly elevated plasma IFN-γ (866 pg/mL) on day 7 of
treatment. This cytokine is considered a pro-hepatotoxic mediator leading to inflammation and
tissue injury through activation of macrophages and natural killer cells.43
This is consistent with a
clinical study performed by Keane et al. that found that incubation of NVP with T cells from a
patient with NVP-induced skin rash led to the production of IFN-γ by T cells.90
Reviews regarding the difficulties with production of animal models of idiosyncratic drug
reactions are available elsewhere, but the major obstacle appears to be the development of
immune tolerance.91
This is consistent with the delayed onset of liver injury and resolution despite
continued treatment observed in these mice. We suspect that the liver injury in humans is
immune-mediated and that the reason that most humans and rats do not develop liver injury is that
the dominant response is immune tolerance. It is known that the dominant immune response in
the liver is tolerance,43
and that it is presumably why liver transplantations are relatively easy
compared to transplantation of, for example, skin. Co-treatment of Cbl-b-/-
mice with
polyinosinic:polycytidylic acid, imiquimod, and even γ-irradiation to deplete circulating
regulatory T cells was used in an attempt to break the immune tolerance and induce sustained
liver damage. All of these attempts were unsuccessful in both male and female mice (data not
shown).
A clear picture regarding the specific types of proteins covalently modified by hepatotoxic
drugs and the outcome of liver injury does not exist. Therefore, even though mice and rats display
a relatively similar pattern of NVP-induced covalent binding, other individual or species-specific
factors must play a role in the development of liver injury. In support of this, a recent clinical
study demonstrated that patients who carried the HLA-DRB*01 allele were at increased risk of
developing NVP-induced liver toxicity (the alleles associated with the risk of skin rash were
different), but there was no association with the CYP2B6 genotype, which is polymorphic and
one of the P450s involved in the metabolism of NVP.19,62
In conclusion, we have clearly demonstrated that NVP covalently binds to hepatic proteins
in mice, rats, and humans. The major chemical species responsible for this covalent binding is a
quinone methide metabolite. We have shown a mild delayed-onset liver injury in C57BL/6 mice
that may be the basis for an animal model if a method can be found to increase the liver injury.
More significant injury was observed in Cbl-b-/-
mice, but the histology suggests that the
mechanism may be different.

65
FUNDING SUPPORT. This work was supported by grants received from the Canadian Institutes
of Health Research, grant numbers
ACKNOWLEDGMENTS. The authors thank Boehringer-Ingelheim for supplying nevirapine.
A.S. is the recipient of a University of Toronto Pharmaceutical Sciences Fellowship. J.U. is the
recipient of the Canada Research Chair in Adverse Drug Reactions. The work was supported by
grants from the Canadian Institutes of Health Research. Portions of this work were parts of
presentations given by A.M. Sharma and J.P.Uetrecht at the Society of Toxicology International
Meetings in Salt Lake City, U.T., U.S.A, 2010, and Washington, D.C., USA, 2011.
ABBREVIATIONS: 1-aminobenzotriazole, ABT; Brown Norway, BN; bovine serum albumin,
BSA; 12-hydroxynevirapine, 12-OH-NVP; 12-trideutero-nevirapine, DNVP; cytochrome P450,
P450; drug-induced liver injury, DILI; glutathione, GSH; glyceraldehyde 3-phosphate
dehydrogenase, GAPDH; human immunodeficiency virus, HIV; idiosyncratic drug reaction, IDR;
interferon-gamma, IFN-γ; liquid chromatography/mass spectrometry, LC/MS; nevirapine, NVP;
tris-buffered saline tween-20, TBST.

66
CHAPTER 3
Nevirapine Bioactivation and Covalent Binding in the Skin
‘Creativity takes courage.’
- Henri Matisse
This work has been published in the following journal and is reproduced with permission:
Amy M. Sharma, Klaus Klarskov, and Jack Uetrecht. Nevirapine Bioactivation and Covalent
Binding in the Skin. Chemical Research in Toxicology, 2013. 26 (3), 410-421. Epub 2013 February 13.
Reprinted with permission. Copyright 2013 American Chemical Society. All rights reserved.
In this chapter, all experiments were performed by Amy M. Sharma.

67
3.1 Abstract
Nevirapine (NVP) treatment is associated with serious skin rashes that appear to be immune-
mediated. We previously developed a rat model of this skin rash that is immune-mediated and
very similar to the rash in humans. Treatment of rats with the major NVP metabolite, 12-OH-
NVP, also caused the rash. Most idiosyncratic drug reactions are caused by reactive metabolites;
12-OH-NVP forms a benzylic sulfate, which was detected in the blood of animals treated with
NVP or 12-OH-NVP. This sulfate is presumably formed in the liver; however, the skin also has
significant sulfotransferase activity. In this study, we used a serum against NVP to detect
covalent binding in the skin of rats. There was a large artifact band in immunoblots of whole skin
homogenates that interfered with detection of covalent binding; however, when skin was
separated into dermal and epidermal fractions, covalent binding was clearly present in the
epidermis, which is also the location of sulfotransferases. In contrast to rats, treatment of mice
with NVP did not result in covalent binding in the skin or skin rash. Although the reaction of 12-
OH-NVP sulfate with nucleophiles such as glutathione is slow, incubation of this sulfate with
homogenized human and rat skin led to extensive covalent binding. Incubations of 12-OH-NVP
with the soluble fraction from a 9,000xg centrifugation (S9) of rat or human skin homogenate in
the presence of 3’-phosphoadenosine-5’-phosphosulfate (PAPS) produced extensive covalent
binding, but no covalent binding was detected with mouse skin S9, which suggests that the reason
mice do not develop a rash is that they lack the required sulfotransferase. This is the first study to
report covalent binding of NVP to rat and human skin. These data provide strong evidence that
covalent binding of NVP in the skin is due to 12-OH-NVP sulfate, which is likely responsible for
NVP-induced skin rash. Sulfation may represent a bioactivation pathway for other drugs that
cause a skin rash.

68
3.2 Introduction
The basic mechanisms of idiosyncratic drug reactions (IDRs) are currently not well
understood. Circumstantial evidence suggests that most IDRs are caused by the formation of
reactive metabolites rather than the parent drug; however, without a valid animal model, this is
difficult to rigorously test. One such model that has allowed us to study in the mechanism of an
idiosyncratic toxicity in detail is the rat model of nevirapine (NVP)-induced skin rash.65
NVP (ViramuneTM
, Scheme 3-1) is a nonnucleoside reverse transcriptase inhibitor
indicated for the combination treatment of HIV-1 infections. Although effective, NVP was found
to induce a high incidence of skin rash or liver toxicity, and sometimes both occur in the same
patient. The incidence of skin rash is approximately 9%, most of which are mild to moderate
maculopapular rashes.59
However, 16% of NVP-induced rashes are very severe and life-
threatening, including Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN).59
Certain risk factors for development of rash have been identified, such as female gender and
higher pretreatment CD4+ T cell counts.
92
Our group has developed and characterized a novel animal model of NVP-induced skin
rash in female Brown Norway (BN) rats. This model shares many characteristics of the rash that
occurs in humans. For example, in both humans and the rat model, evidence suggests CD4+ T
cells mediate the rash.65
Additionally, there is a delay in onset of the rash upon primary NVP
treatment, but a rapid onset with secondary rechallenge in both humans and rats.65
Higher
incidence in females, increased incidence of rash with increased dose, and range in severity of
rash are all features shared by both the animal model and humans. Furthermore, lymphocytes
taken from both patients and animals after NVP-induced skin rash produce interferon-. 90,93
Using the BN rat model, we were able to show that the 12-hydroxylation pathway is
involved in the induction of the skin rash. This is based on the observation that substitution of the
NVP methyl hydrogens with deuterium markedly decreased formation of 12-OH-NVP as well as
the incidence and severity of the rash.68
Additionally, treatment with a lower dose of 12-OH-NVP
induced the same degree of skin rash as treatment with NVP itself.68
Although we know that
oxidation of NVP to 12-OH-NVP is required to induce the rash, it is not clear how it does so. 12-
OH-NVP is not chemically reactive; therefore, if the rash is caused by a reactive metabolite, 12-
OH-NVP would require further bioactivation. The oxidation state of 12-OH-NVP and the quinone
methide, which is the major species involved in covalent binding in the liver, is the same;

69
therefore, the quinone methide cannot be formed by oxidation of 12-OH-NVP. 12-OH-NVP is
oxidized to the corresponding carboxylic acid, which forms a glucuronide, but inhibition of this
oxidation does not decrease the incidence of the rash.68
The most likely candidate is the benzylic
sulfate conjugate. Sulfate is a good leaving group and numerous sulfate metabolites are known to
be reactive metabolites.94
We detected the 12-OH-NVP sulfate in the blood of BN rats treated
with NVP, which was presumably formed in the liver, and there are also sulfotransferases in the
skin.95
In the case of the 12-OH-NVP sulfate, not only is the sulfate on a benzylic position, there
is also an adjacent amide hydrogen that could be lost to form the same quinone methide as formed
by direct oxidation of the methyl group without the formation of a carbocation intermediate.
However, the 12-OH-NVP sulfate was synthesized and found to be less reactive than expected;
specifically, it reacted only very slowly with glutathione (the reaction occurred over a period of
days; unpublished results). In addition, initial attempts to detect covalent binding of NVP in the
skin of treated animals were unsuccessful. The present study was an extension of the previous
studies to test the hypothesis that 12-OH-NVP sulfate is a plausible candidate for causing NVP-
induced skin rashes.

70
Scheme 3-1. Proposed chemical mechanism of NVP-induced skin rash resulting from covalent
binding of 12-OH-NVP sulfate in the skin.

71
3.3 Materials and methods
3.3.1 Chemicals.
NVP was kindly supplied by Boehringer-Ingelheim Pharmaceuticals Inc. (Ridgefield, CT).
The majority of chemical reagents: 3’-phosphoadenosine 5’-phosphosulfate (PAPS), Tris,
methanol, DMSO, PBS (pH 7.4), glycerol, silica gel, amido black stain, were obtained from
Sigma-Aldrich (Oakville, ON) unless otherwise noted in the Methods. Ammonium persulfate was
obtained from Fisher Scientific (Fair Lawn, NJ). SDS and Tween-20 were obtained from BioShop
(Burlington, ON). Stock acrylamide/bis solution (29:1), non-fat blotting grade milk powder, and
nitrocellulose membranes (pore size 0.2 µM) were purchased from Bio-Rad (Hercules, CA).
Ultra-pure tetramethylethylenediamine and 2.5% trypsin were purchased from Invitrogen
(Carlsbad, CA). Amersham ECL Plus Western Blotting Detection System was obtained from GE
Healthcare (Oakville, ON). Horseradish peroxidase-conjugated goat anti-rabbit IgG (H + L
chains) and monoclonal GAPDH were purchased from Sigma-Aldrich (St. Louis, Mo). Normal
goat serum was obtained from Invitrogen (Grand Island, NY). The synthesis of 12-OH-NVP, 12-
OH-NVP sulfate, and preparation of NVP antiserum were described previously.68,74
Protein
concentrations were determined using a BCA protein assay kit (Novagen, EMD Biosciences Inc.).
3.3.2 Animal Care.
Female BN rats (150 – 175 g; between 8-10 weeks of age) were obtained from Charles
River (Montreal, QC). Rats were housed in pairs in standard cages in a 12:12 h light/dark cycle
with access to water and Agribrands powdered lab chow diet (Leis Pet Distribution, Inc.
Wellesley, ON) ad libitum. Following a 1 week acclimatization period, rats were either
maintained on control chow or started on drug-containing diet (treatment groups). Drug was
mixed thoroughly with powdered lab chow if it was to be administered orally. The amount of drug
administered to animals was calculated based on body weight of the rats and their daily food
intake. Rats were sacrificed via CO2 asphyxiation.
Balb/c or C57BL/6 mice (6-8 weeks age) were obtained from Charles River (Montreal,
Quebec). E3 ubiquitin ligase casitas-b-lineage-lymphoma (Cbl-b-/-
) knockout mice were bred in
house from animals first developed by Dr. J. Penninger at the Institute of Molecular
Biotechnology of the Austrian Academy of Science, Vienna, with his kind permission.
Programmed cell death-1 (PD-1-/-
) knockout mice were bred in house from animals first

72
developed by Dr. Tasuku Honjo at the Department of Immunology and Genomic Medicine,
Kyoto, Japan, with his kind permission. Mice were kept 4 per cage. NVP was administrated in lab
chow following a 1 week acclimatization period. All animal experiments were approved by the
University of Toronto Animal Care Committee in accordance with guidelines of the Canadian
Council on Animal Care.
3.3.3 Primary and Secondary Treatment of Animals with NVP or 12-OH-NVP.
Female BN rats were treated orally with NVP (150 mg/kg/day), or an equimolar dose of
12-OH-NVP (159 mg/kg/day) mixed thoroughly in rat chow for up to 21 days. For rechallenge
(secondary exposure), rats treated with NVP having a moderate to severe skin rash were removed
from drug, fed a diet of rat chow for 4 weeks so that the rash resolved, and then NVP was
resumed at the same dose until the animals developed a rash and systemic effects such as weight
loss, which usually occurred after 7-10 days.65
3.3.4 Separation of Dermis and Epidermis and Preparation of Homogenates of Skin Fractions or of Whole Rat Skin.
At sacrifice, hair was removed from the rats using an electric shaver, and the skin was
cleaned of remaining hair using PBS (1x phosphate buffered saline, pH 7.4) and Kimwipes. Skin
from the back was then excised and placed on dry ice over aluminum foil. BN rats develop
deeply pigmented skin when their fur is going through the anagen phase of the hair growth cell
cycle; the epidermis and dermis from this skin was found not to separate. Only pink (telogen
phase) skin was used to obtain proper separation of epidermis from dermis. Care was taken to
remove all hypodermis and connective tissue using blunt-tip forceps. Sections (~200 mg) of
whole skin were stretched using sharp-tipped forceps on clean petri dishes for a maximum of 2 h
at 4 °C. Once stretched, skins were floated individually overnight at 4 °C in a solution of 0.25%
trypsin.96
On day 2 the epidermis was lifted off of the dermis, the dermis was wiped clean of any
residual epidermis using Kimwipes, washed with double distilled water, and each fraction was
homogenized individually in cell lysis buffer (Cell Signaling Technologies, Pickering, ON) with
protease inhibitor (1X HALT Protease Inhibitor Cocktail, Pierce, Rockford, IL) in a 10:1 ratio
using a Polytron 2100 homogenizer (~1.5 mL working cell lysis buffer per fraction). In order to
clarify samples, dermal and epidermal fractions were centrifuged at 13,000 rpm for 2 min. The

73
supernatant was separated from insoluble debris and stored at -80 °C. Whole rat skin tissue was
prepared using the same method except dermal and epidermal layers were not separated and skin
was not floated overnight in trypsin.
3.3.5 Preparation of Cytosol, S9, or Microsomes from NVP-treated Rat Epidermal or Dermal Fractions.
Skin from 4 rats that had been treated with NVP for 21 days was excised from the back of
the neck and separated after overnight treatment with trypsin as described above. To examine
covalent binding in each subcellular fraction, the separated epidermis and dermis were used to
make either cytosolic, 9,000xg supernatant (S9), or microsomal fractions. Homogenates of
epidermis and dermis were produced as described above. The homogenates were then centrifuged
at 9,000xg for 20 min at 4 °C to obtain the S9 fraction, which was set aside for 2 of 4 rats in each
group. The S9 fractions obtained from the remaining two rats per group were further centrifuged
at 105,000xg for 1 h at 4 °C. The supernatant from these fractions contained the cytosol while the
pellet, which was homogenized in 4 volumes of glycerol-phosphate-KCl buffer (20% glycerol,
0.4% KCl, 50 nM KH2PO4 pH 7.4), contained the microsomes. All fractions were aliquoted and
stored at -80 °C until use for immunoblots.
3.3.6 Preparation of Human Skin Dermatome.
Human dermatomed skin was obtained from XenoTech LLC (Lenexa, KS, USA) 9.1 h
postmortem. The donor was a 56 year old Caucasian male who died from heart disease without
any skin diseases or infectious diseases. Skin (10X10 cm) was excised from the abdomen and
snap frozen. Skin was prepared to contain only the epidermal layers (‘dermatomed’ skin) by
XenoTech LLC. Dermatomed skin was then chopped into fine pieces, immersing in cell lysis
buffer with protease inhibitor for 15 min on ice, homogenized, and centrifuged as described
above. The supernatant was separated and stored at -80 °C.
3.3.7 Cytosolic or S9 Fractions from Rat or Human Skin or Liver.
Human liver cytosol (pool of 10, mixed gender) or S9 (pool of 50, mixed gender); IGS
Sprague-Dawley rat liver cytosol (pool of 115); skin cytosol (pool of 50); skin S9 (pool of 50); or
liver S9 (pool of 100); B6C3F1 mouse liver S9 (pool of 400); or CD1 mouse skin S9 (pool of
100) were all purchased from XenoTech LLC (Lenexa, KS, USA).

74
3.3.8 Incubation of Human or Rat Skin or Fractionated Skin with NVP, 12-OH-NVP, or 12-OH-NVP Sulfate.
Stock solutions of each compound were prepared in methanol fresh at the time of use.
Before incubation with skin, methanol was partially removed by nitrogen evaporation in order to
limit the amount of solvent in the incubation to <0.05%.83
Following this, an equal volume of
naïve human or rat whole skin, rat dermal, or rat epidermal homogenate was added to a final
concentration of either 0.8 or 1 mg/mL (see results for specific details) in each tube and vortexed.
The final concentration of all compounds tested was 1 mM. Samples were incubated at 37 °C in a
water bath and aliquots taken at time 0 (before the start of the incubation), 30, and 60 min, and
reactions were terminated by placing tubes on dry ice. Negative controls were the skin fractions
incubated at 37 °C without addition of any drug. All samples were stored at -80 °C until use for
immunoblotting experiments.
3.3.9 In Vitro Metabolism of 12-OH-NVP and NVP.
For testing sulfation, NVP or 12-OH-NVP (50 mM stock solution in methanol) was added
to Dulbecco’s PBS with MgCl2 and CaCl2 (Invitrogen, Carlsbad, CA) to a final concentration of 1
mM. Total incubation volume was 400 µL. The methanol was partially removed by nitrogen
evaporation in order to limit the amount in the incubation to <0.05%83
. Following a 5 min
preincubation with between 0.5 - 1 mg/mL of protein from skin, liver S9 or cytosol (human, rat,
or mouse; XenoTech LLC, Lenexa, KS) at 37 °C in a water bath, PAPS was added to a final
concentration of 0.3 mM. Tubes were vortexed thoroughly and incubated for 1 h. Control samples
contained all components except PAPS. Samples were frozen on dry ice to halt the reaction and
stored at -80° C until used for immunoblotting experiments.
For incubations examining P450-mediated bioactivation the procedure was the same
except the media was 100 mM potassium phosphate buffer (pH 7.4) and a NADPH-regenerating
system (Solution A final concentrations: 1.3 mM NADP, 3.3 mM glucose-6-phosphate, 3.3 mM
MgCl2; Solution B final concentration: 0.4 Units/mL of glucose-6-phosphate dehydrogenase)
replaced PAPS.

75
3.3.10 Covalent Binding Using SDS PAGE and Immunoblotting.
The supernatant from homogenized whole skin or the dermal or epidermal fractions was
mixed with Pierce 5x stock reducing sample loading buffer in a 4:1 protein to buffer ratio and
boiled for 5 min. SDS PAGE was performed using a Protean-3 minigel system (BioRad,
Mississauga, ON). Gels were hand-cast (stacking gel, 5% bisacrylamide; resolving gel, 8%
bisacrylamide) and were run at ~ 110 V. Electrophoresis running buffer (Bio-Rad) consisted of 25
mM Tris, 192 mM glycine, and 0.1% SDS, pH 8.3. Transfer to nitrocellulose membrane (pore
size 0.2 µM, BioRad) was performed at 0.13 mA for 90 min at 4 ºC using the same Protean-3
minigel system (BioRad, Mississauga, ON). Tris-glycine transfer buffer (Bio-Rad) consisted of 25
mM Tris, 192 mM glycine, and 20% methanol at pH 8.5. Membranes were washed twice in Tris-
buffered saline tween-20 (TBST) wash solution for 5 min. Membranes were then blocked in 5%
nonfat milk blocking solution in TBST for 90 min at room temperature. Membranes were then
rinsed with three changes of TBST for 5 min each and incubated with a 1:100 or 1:500 dilution of
primary NVP antiserum and 10% normal goat serum in TBST overnight at 4 ºC. A 20 min wash
(three changes) in TBST after overnight blocking was followed by a 90 min incubation in
secondary antiserum (1:2000 or 1:5000 dilution) in TBST containing 10% goat serum. In some
experiments, the primary serum was blocked by preincubation with 1 mM NVP (dissolved in
DMSO) for 2 h at 37 °C in a water bath as described previously.74
The secondary antiserum was
goat anti-rabbit horseradish peroxidase antisera. Membranes were washed 3 times for 20 min with
TBST. All blots were incubated with enhanced chemiluminescence stain for 5 min and analyzed
with a FluorChem8800 imager. To probe for the glyceraldehyde 3-phosphate dehydrogenase
(GAPDH) loading control, membranes were stripped of primary NVP antiserum using Pierce
Restore Plus buffer (Pierce, Rockford, IL) for 15 to 20 min at room temperature followed by a 1 h
blocking step. Membranes were then incubated in mouse monoclonal anti-GAPDH antiserum
(1:40 000) and processed as above except that the secondary antiserum was goat anti-mouse
horseradish peroxidase conjugated (Jackson ImmunoResearch, Baltimore Pike, West Grove, PA.)
diluted 10 000x in TBST. Amido black (Sigma) staining and de-staining was performed at room
temperature following the prescribed protocol and membranes were air-dried and visualized
immediately after.

76
3.3.11 Preparation of BN Rat Skin for Histology.
Ear or skin samples were fixed in 10% formalin. The paraffin block, hematoxylin/eosin
slides, or unstained sections were prepared at the Hospital for Sick Children in Toronto, ON
Canada.
3.4 Results
3.4.1 Attempts to Detect In Vivo Covalent Binding in Whole Skin.
Detection of covalent binding in the skin was a challenge because when whole skin was
analyzed by immunoblotting, there was a large artifact band. With much effort we were able to
produce convincing evidence of binding, but we were unable to eliminate the large artifact band
(Figure 3-1A). A number of methods to reduce background and remove the artifact band were
tried. Preincubation of the primary antiserum with skin homogenate or strips of the artifact band
obtained from a large western blot of skin from control rats failed to prevent nonspecific binding
even after a 24 h incubation. We were finally able to eliminate the artifact band by separation of
the epidermis from the dermis; binding is in the epidermis, and the artifact comes from the
dermis. A variety of methods were used to separate the skin layers, but treatment with trypsin was
found to yield the most consistent results (optimization data not shown). Therefore, when the
epidermis was separated from the dermis by overnight treatment with 0.25% trypsin to yield a
clean separation (Figure 3-1B), there was clear binding in the epidermis of NVP or 12-OH-NVP-
treated rats. However, due to the trypsin-separation method, all loading controls (GAPDH, actin,
laminin, etc) did not work, despite the fact that they work with whole skin (Figure 3-1A). Amido
black staining was therefore used to show even protein loading in each lane (data not shown).

77
Figure 3-1. (A) Immunoblot showing covalent binding of NVP (150 mg/kg) to whole rat skin in
vivo with a major artifact band in each lane indicated by the grey arrows. From left to right:
primary treatment days 22, 24, 25, rechallenge (RCH, 7 days), or untreated control. Each lane was
loaded with 25 µg of protein. Exposure duration in the imager was 3 min. (B) Epidermis floating
above dermis from trypsin-separated skin from a rat treated for 21 days with NVP (left panel);
isolated epidermal layer (right panel).

78
3.4.2 Covalent Binding of NVP, 12-OH-NVP, or 12-OH-NVP Sulfate to Human or Rat Skin in Vitro.
In order to determine if NVP, 12-OH-NVP, or 12-OH-NVP sulfate reacted with skin
proteins in vitro, each compound was incubated with skin homogenates from female BN rats or
humans. Figure 3-2 demonstrates clear covalent binding to both whole skin and isolated
epidermal homogenate prepared from a control rat after incubation with 12-OH-NVP sulfate.
Most of the proteins that were modified in the whole rat skin homogenate had masses between 40
to 70 kDa (Figure 3-2A), while modified protein from isolated epidermal homogenate varied from
20 to ~ 60 kDa (Figure 3-2B). Preincubation of the NVP antiserum with 1 mM NVP for 2 h at 37
°C blocked almost all of the covalent binding to the epidermis, showing that the antisera is
specific for proteins that have been covalently-modified by NVP metabolites (Figure 3-2C).
Incubation with 12-OH-NVP sulfate also caused covalent modification of human skin dermatome
proteins (Figure 3-2D) with modified proteins ranging from 25 – 60 kDa. When compared to 12-
OH-NVP in the presence of PAPS on the same blot, abundant binding was observed to proteins
ranging from 30 – 150 kDa. As before, when the primary antiserum was preincubated with NVP,
covalent binding of 12-OH-NVP sulfate was blocked save for the nonspecific band (Figure 3-2E),
which indicates that the binding to human dermatome proteins is specific for NVP.

79

80
Figure 3-2. (A) Immunoblot showing in vitro covalent binding of 1 mM each NVP, 12-OH-NVP,
or 12-OH-NVP sulfate to rat whole skin homogenate containing both dermis and epidermis after
incubation for 30 or 60 min. (B) Covalent binding of 1 mM NVP, 12-OH-NVP, or 12-OH-NVP
sulfate to isolated epidermal or dermal homogenates prepared from a control rat. (C) Covalent
binding of 1 mM NVP, 12-OH-NVP, or 12-OH-NVP sulfate to isolated epidermal or dermal
homogenate prepared from a control rat showing that preincubation of the primary antisera with 1
mM NVP for 2 h at 37 °C blocked binding of the antibody. Proteins (7.5 µg/well) were loaded in
immunoblots A-C. (D) Human dermatome skin incubated with 1 mM each of 12-OH-NVP, NVP,
or 12-OH-NVP sulfate compared to 1 mM 12-OH-NVP +/- 0.3 mM PAPS (1 mg/mL protein). (E)
Covalent binding of 1 mM NVP, 12-OH-NVP, or 12-OH-NVP sulfate to isolated human

81
dermatome homogenate showing that preincubation of the primary antiserum with 1 mM NVP for
2 h at 37 °C blocked binding of the antibody. Protein (12 µg/well) was loaded for blots D-E.
3.4.3 Covalent Binding of NVP to Rat Skin in Vivo.
Once covalent binding of 12-OH-NVP sulfate to skin and isolated epidermal fractions was
established in vitro, treatment of female BN rats was undertaken in order to study covalent
binding to rat skin in vivo. Treatment of female BN rats with NVP or 12-OH-NVP for 7 days led
to an average grade of skin rash of 0 in controls, 0.75 for NVP-treated rats, and 2 for 12-OH-
NVP-treated rats (see Supplemental Data for the criteria used to grade the rash). Covalent binding
in the epidermis of these animals on day 7 was minimal (Figure 3-3A), as was binding in the
dermis (data not shown). On day 21 of NVP or 12-OH-NVP treatment, the average grade of rash
was 0 for controls, 3.25 for NVP-treated rats, and 3.5 for 12-OH-NVP-treated rats. Covalent
binding for each of the NVP and 12-OH-NVP groups was marked in the epidermis on day 21
(Figure 3-3B), with modified proteins spanning from 30-60 kDa. Binding in the dermis was less
apparent as only a single prominent band was present at ~55 kDa (data not shown). Covalent
binding was also present in the epidermis of rechallenged rats, and preincubation of the antiserum
with NVP again blocked specific binding (Figure 3-3C).

82

83
Figure 3-3. Immunoblots showing epidermal covalent binding in vivo after treatment with NVP
or 12-OH-NVP for either (A) 7 days or (B) 21 days. (C) Immunoblots showing that preincubation
of the primary antiserum with 1.5 mM NVP for 2 h at 37˚C blocked binding of the antibody (right
panel) to the drug-modified proteins after treatment with NVP for 21 days or after rechallenge
with NVP (RCH), left panel. Epidermal protein loading was 15 µg/well.

84
3.4.4 Early Histological Changes in the Skin in Response to NVP Treatment.
Based on early time point experiments, it was determined that submaximal NVP covalent
binding was detectable by day 10 (Figure 3-4A). We sought to determine if the immune response
had a similar time course and this is shown in Figure 3-4B. 12-OH-NVP-treated rats appeared to
have a more advanced stage of skin degeneration at day 7 than did NVP-treated rats; it was not
until day 15 that NVP-treated rat skin displayed the same acanthosis, edema, and early
lymphocyte infiltration along the dermal-epidermal junction and papillary dermis as the 12-OH-
NVP-treated animals did on day 7.

85
Figure 3-4. (A) Immunoblot showing covalent binding in the epidermis of NVP-treated female
BN rats on days 10, 15, or 21 (n = 2 animals per time point) of NVP treatment. Each lane
represents an individual animal with 15 µg/well of protein loaded in each well. (B) Representative
H&E stained rat skin sections comparing the early infiltration of immune cells into the dermis or
epidermis of NVP- or 12-OH-NVP-treated rats. Marked acanthosis (thickening of the epidermis)
combined with early lymphocyte infiltrate at the dermal-epidermal junction can be observed by
day 7 of 12-OH-NVP-treated animals. By day 21 there is an increase in the cellular infiltrate with
areas of detachment of the epidermis. Magnification 20x.

86
3.4.5 Covalent Binding of NVP to Mouse Skin in Vivo.
NVP was administered to 4 different strains of mice (C57BL/6, BALB/c, programmed cell
death-1 knockouts, or casitas-b-lineage-lymphoma-b knockouts) at a high dose of 950 mg/kg day
in food, but all failed to develop a skin rash. The latter 2 strains were used because they have
impaired immune tolerance, and it was thought they would be more likely to develop an immune
response. Both genders of each strain were tested. In order to examine whether the lack of skin
rash was due to lack of covalent binding in skin, the anti-NVP serum was blotted against
epidermal and dermal proteins of programmed cell death-1 (PD-1-/-
) knockouts bred on a C57
background. Despite the use of a higher dose than in rats, no binding was observed in either
fraction when compared to rat epidermis (data not shown). Histological analysis in these mice
displayed no obvious alterations in the skin upon NVP treatment (Figure 3-5).
Figure 3-5. Skin histology of PD-1-/-
knockout mice. No immune infiltrate or acanthosis was
observed as was seen with NVP-treated rats.

87
3.4.6 Covalent Binding of NVP to Subcellular Rat Skin Fractions in Vivo.
BN rat skin cytosolic and S9 fractions displayed marked binding in the fractions isolated
from the epidermis (Figure 3-6A). When rat skin S9 was compared to the skin microsomal
fraction, covalent binding was observed only in the skin S9 fraction from the epidermis (Figure 3-
6B). Therefore, stronger binding was present only in the fractions containing sulfotransferases
(SULTs), i.e. S9 and cytosol from the epidermis.

88
Figure 3-6. (A) Comparison of covalent binding to S9 with that to the cytosolic fractions from the
skin of NVP-treated rats, isolated from either the dermis or epidermis. (B) Comparison of
covalent binding to S9 with that to microsomal fractions from the skin of NVP-treated rats,
isolated from either the dermis or epidermis. All animals were treated for 21 days. Protein loading
was 10 µg/well.
3.4.7 Covalent Binding of 12-OH-NVP to Human or Rat Liver or Skin Proteins in the Presence or Absence of PAPS.
The covalent binding of 12-OH-NVP to rat skin S9 was compared with that to human liver
cytosol (from a female and a male) in the presence and absence of PAPS (Figure 3-7A). Binding
of 12-OH-NVP was only observed in the presence of PAPS, which indicates that it is the sulfate
that is responsible for the binding in both. The immunoblot of rat skin S9 or cytosol compared to
the immunoblot of human and rat liver cytosol gave the same result (Figure 3-7B). Binding of 12-
OH-NVP in the presence of PAPS was also observed to proteins from incubations with human
liver S9, human dermatome skin homogenates, and rat skin S9 (Figure 3-7C).

89

90
Figure 3-7. (A) Immunoblot of rat skin S9 or human female (second right lane) or human male
(most right lane) liver cytosol after incubation with 12-OH-NVP in the presence and absence of
PAPS. (B) Immunoblot of rat skin S9 or cytosol, or female human or rat liver cytosol after
incubation with 12-OH NVP in the presence of absence of PAPS. (C) Covalent binding of 12-
OH-NVP to human liver S9, human dermatome skin, or rat skin S9 in the presence and absence of
PAPS. Protein loading was 12 µg/well.
3.4.8 Covalent Binding of 12-OH-NVP or NVP to Human or Rat Liver or Skin Subcellular Fractions in the Presence or Absence of PAPS or NADPH.
In order to examine the ability of skin to perform oxidation or sulfation of 12-OH-NVP or
NVP, various subcellular fractions of rat or human skin or liver were tested in the presence or
absence of PAPS or an NADPH-regenerating system (NRS). Covalent binding of 12-OH-NVP
was observed when incubated with rat liver or skin S9 in the presence of PAPS, but not in the
presence of NRS, as expected (Figure 3-8A). Conversely, only hepatic incubations of NVP with
NRS produced covalent binding (Figure 3-8B). The lack of covalent binding in skin subfractions
after incubation with NVP suggested that there is minimal P450 oxidase activity because
oxidation and covalent binding of NVP in the liver is extensive.74
Figure 3-8C and 3-8D produced

91
a similar pattern when comparing human liver S9 to rat liver S9. Covalent binding of NVP or 12-
OH-NVP to human skin in the presence or absence of PAPS or NRS displayed covalent binding
only with 12-OH-NVP in the presence of PAPS (Figure 3-8E), again consistent with 12-OH-NVP
sulfate being the major metabolite responsible for covalent binding in the skin.

92

93

94
Figure 3-8. (A) Immunoblot comparing covalent binding of 12-OH-NVP to rat liver S9 versus rat
skin S9 in the presence or absence of either PAPS or a NADPH-regenerating system (NRS). (B)
Comparison of covalent binding of NVP to rat liver S9 versus rat skin S9 incubated in the
presence of absence of either PAPS or a NADPH-regenerating system (NRS). (C) Comparison of
covalent binding of 12-OH-NVP to rat liver S9 versus human liver S9 incubated in the presence
or absence of either PAPS or NRS. (D) Comparison of covalent binding of NVP to human liver
S9 versus rat liver S9 incubated with or without either PAPS or NRS. (E) Covalent binding of
NVP or 12-OH-NVP to human skin in the presence or absence of PAPS or NRS. Protein loading
was 12 µg/well. 1 mM of 12-OH-NVP or NVP was used for each incubation.
3.4.9 Sulfation and Oxidation of NVP and 12-OH-NVP in Mouse and Rat Skin.
Mice do not develop a skin rash in response to NVP treatment and also have no observable
levels of covalent binding in skin. We sought to determine if this was due to differences in the
ability of mouse skin to form 12-OH-NVP sulfate or some other difference in metabolism or
distribution of 12-OH-NVP. As a positive control, rat liver S9 and mouse liver S9 incubated with
NVP in the presence of NRS displayed covalent binding, indicating formation of the quinone
methide species as we had previously shown (Figure 3-9A).74
No binding of NVP was observed
to liver S9 in the presence of PAPS, or of 12-OH-NVP to liver S9 in the presence of NRS (Figure
3-9A, B). NVP incubated with skin S9 in the presence or absence of PAPS or NRS produced no
detectable binding in rat or mouse samples, indicating a minimal role for oxidation by P450 in the

95
skin (Figure 3-9C). Incubation of 12-OH-NVP and PAPS with rat skin S9 resulted in significant
covalent binding; in contrast, although there is a very small band in the immunoblot of mouse
skin, minimal binding to mouse skin S9 was observed (Figure 3-9D). A pool of skins from 100
CD1 mice were used for Figures 3-9C & 3-9D because CD1 mice are outbred and more
representative of the general population of all mice strains, preferable in toxicology studies.

96

97
Figure 3-9. (A) Immunoblot comparing the covalent binding of NVP to mouse vs. rat liver S9 in
the presence of absence of either PAPS or an NADPH-generating system (NRS). (B) Comparison
of the covalent binding of 12-OH-NVP to either mouse or rat liver S9 in the presence or absence
of either PAPS or NRS. (C) Comparison of covalent binding of NVP to mouse vs rat skin S9
either in the presence or absence of PAPS or NRS. (D) Comparison of covalent binding of 12-
OH-NVP to mouse vs. rat skin S9 either in the presence or absence of PAPS or NRS. Protein
loading was 12 µg/well. 1 mM of 12-OH-NVP or NVP was present in each incubation.

98
3.4.10 Anti-NVP and Autoantibodies in NVP-Treated Rats.
Previously our group had found that topical administration of NVP to NVP-sensitized
animals led to the development of a generalized rash as opposed to rash only where NVP was
applied. Absorption through the skin leading to significant circulating concentrations of NVP may
have occurred. However, if the amount of NVP applied to the skin was decreased, eventually no
rash was induced, but there was no dose at which only a local response was found. This suggests
that there exists an autoimmune component to the rash so that activation of cells leads to a
systemic reaction, not just an antidrug reaction confined to drug-exposed cells. The sera from rats
rechallenged with NVP were found to contain autoantibodies in addition to anti-drug antibodies
(Figure 3-10). Comparison of binding of sera from NVP rechallenged animals and naïve animals
to hepatic proteins from naïve and NVP-treated animals demonstrates an artifact band at about 51
kDa, an antidrug antibody binding to a protein at about 35 kDa and probably 90 kDa, and an
autoantibody binding to a protein at about 42 kDa (Figure 3-10A). An analogous experiment
using epidermal proteins showed an artifact band at about 45 kDa and antidrug antibodies that
bind to proteins at about 40 kDa and 52 kDa but no antibodies to dermal proteins (Figure 3-10B).

99
Figure 3-10. Detection of anti-NVP and autoantibodies in the serum of a rat after rechallenge
with NVP (A) Liver homogenate (10 µg/lane) from an untreated (control) rat, NVP-treated rats, or
12-OH NVP-treated rats run on SDS PAGE and stained with serum (diluted 1:500) from a rat that
had been rechallenged with NVP after earlier development of a NVP-induced rash (left panel) or
with serum from an untreated control rat (right panel). A 1:4000 dilution of goat anti-rat HRP
linked antibody was used as the secondary antibody to visualize the binding. Blots were imaged
for 3 minutes on medium exposure. (B) Using serum from the same rechallenged rat, an
analogous experiment was performed using fractionated skin protein (20 µg/lane) for the
epidermis, designated ‘E’, or dermis, marked ‘D’) from untreated (control) or NVP-treated rats.
Blots were run, blocked, incubated with secondary, and imaged together.

100
Scheme 3-2. Proposed bioactivation pathway of NVP leading to immune-mediated skin rash.

101
3.5 Discussion
Most idiosyncratic drug reactions appear to involve reactive metabolites that covalently
bind to proteins.16
Consistent with previous findings, treatment of rats with 12-OH-NVP was
found to induce earlier covalent binding and histological changes, as well as a more intense rash,
than NVP itself. This again indicates that the 12-hydroxylation pathway is involved in the
induction of the rash. The most likely candidate for a reactive metabolite of 12-OH-NVP is the
benzylic sulfate. However, its chemical reactivity was less than we expected; specifically, its
reaction with glutathione was extremely slow, even under alkaline conditions. It is also less polar
than would be expected for a sulfate, and under some conditions its retention time on a reverse
phase HPLC column is actually longer than that of NVP (data not shown). This may be because
of internal hydrogen bonding of the sulfate to the adjacent amide hydrogen.
In the present study, we investigated the ability of NVP and its metabolites to covalently
bind to proteins in the skin. In contrast to its reactivity with glutathione, incubation of 12-OH-
NVP sulfate with skin homogenates led to extensive covalent binding. This presumably involves
some neighboring group effect of the proteins involved in the binding such as removal of the
adjacent amide hydrogen or neutralization of the negative charge to facilitate attack of an anionic
nucleophile. Incubation of 12-OH-NVP with the skin homogenates from rats or humans also led
to covalent binding, but only in the presence of PAPS, which indicates that it is the sulfate that is
responsible for the binding. Covalent binding of the 12-OH-NVP sulfate involved a wider range
of epidermal proteins in vitro than was observed in vivo (Figure 3-2A-E). This suggests that 12-
OH-NVP sulfate covalently binds to proteins close to where it is formed even though 12-OH-
NVP sulfate is present in significant concentrations in the blood of rats treated with NVP. In
contrast to rat and human skin homogenates, there was minimal binding of 12-OH-NVP in the
presence of PAPS to skin homogenates from mice, and this is presumably why mice do not
develop a rash when treated with NVP. Covalent binding was also observed in the skin of rats
treated with NVP, but only in the epidermal layer, which is the location of sulfotransferases. All
of these data are consistent with 12-OH-NVP sulfate being responsible for the skin rash
associated with NVP treatment.
Based on covalent binding results, the epidermis appears to be the key component of skin
affected by NVP treatment. The epidermis is primarily composed of keratinocytes (~95%), key in
the production of local immune responses. This is not surprising given that, despite their limited

102
metabolic capacity, keratinocytes express a range of enzymes such as SULTs and numerous
transporters, allowing for the uptake of drugs.70,95
In addition, keratinocytes are known to be able
to act as non-professional antigen presenting cells.50
Given that the covalent binding is in the epidermis, it is somewhat surprising that the
cellular infiltrate is in the dermis. However, the epidermis in rodents is only about 2-3 cells thick,
and the majority of the infiltrate in rats is found at the dermal-epidermal junction (Figure 3-4B).
The histology of the NVP-induced rash in humans does affect the epidermis, which is much
thicker than the rat epidermis. Havlir et al.67
examined skin biopsies of three patients who
developed rash following NVP treatment and found a perivascular lymphocytic infiltrate in the
papillary dermis in two patients, and a milder, nonspecific infiltrate in the third. This may simply
be because it is difficult for leukocytes to migrate very far from the vasculature, and the rodent
epidermis does not include blood vessels. One of the patients who developed a perivascular
infiltrate also displayed endothelial swelling, which was also observed on both the back of the
neck and the ears of NVP-treated rats (Figure 3-4B; 3S-1C).
In the studies performed to establish the progression of covalent binding and induction of
the immune response, the epidermis again appeared to play a key role. Covalent binding was not
readily detected at early time points, was relatively low at 10 days, but it was clearly present after
15 days of treatment (Figure 3-4A). This presumably reflects the half-life of the modified
proteins. The delay in the onset of an idiosyncratic drug reaction is usually explained on the basis
of the time required to develop an adaptive immune response, but in the case of NVP-induced
skin rash, the slow accumulation of modified proteins in the skin may also play a role. On day 7
of the 12-OH-NVP- and day 15 of NVP-treated groups, the observed pathology involved
acanthosis (thickening of stratum corneum, which is a nonspecific result of epidermal
inflammation/irritation or injury), dermal-epidermal edema, and early presence of lymphocytes
along the dermal-epidermal interface (Figure 3-4B). The clear spaces observed between epidermal
cells in the basal layer is termed “intercellular edema” or “spongiosis,” which is also nonspecific
but indicates that fluid from dermal edema has leaked into the epidermis. This is seen in many
forms of dermatitis, but especially if the skin is actively inflammed (e.g. bacterial dermatitis,
severe acute allergies, etc). Therefore, the early changes in skin appear to be due to inflammation,
primarily affecting the basal epidermis.

103
Based on current evidence, a proposed scheme for NVP bioactivation leading to skin rash
is presented in (Scheme 2). Oxidation of NVP in the liver leads to 12-OH-NVP, which is carried
to the skin in the general circulation. Upon arrival in the skin, 12-OH-NVP metabolism by
sulfotransferase enzymes leads to production of 12-OH-NVP sulfate in the epidermis, and
subsequent covalent binding leads to initiation of an active immune response. Acanthosis,
vacuolization, and edema are ultimately followed by a full-blown immune response. Anti-NVP
antibodies, detected in the sera of rechallenged rats (Figure 3-10) may also play a role in the
pathogenesis. The presence of antibodies against hepatic proteins may also be an indication of a
more general immune response. This is consistent with the observation that upon rechallenge, rats
present with a more systemic sickness and lethargy, which is also observed in other more
generalized immune responses such as drug-induced autoimmunity.97
The female BN rat model is a unique tool for in depth studies of one IDR. In this study we
demonstrated that the skin is capable of the bioactivation of a drug that causes a high incidence of
skin rash. Studies are currently underway to test which sulfotransferase isoforms are involved in
forming 12-OH-NVP sulfate and the specific role the sulfate conjugate has in the induction of a
skin rash.

104
FUNDING SUPPORT. This work was supported by grants received from the Canadian Institutes
of Health Research (MPO84520).
ACKNOWLEDGMENTS. The authors thank Boehringer-Ingelheim for supplying nevirapine.
The authors also thank Dr. Jeff Caswell, D.V.M., D.V.Sc., Ph.D., from the University of Guelph,
for his review of the skin pathology. A.S. is the recipient of a University of Toronto
Pharmaceutical Sciences Fellowship. J.U. is the recipient of the Canada Research Chair in
Adverse Drug Reactions. The work was supported by grants from the Canadian Institutes of
Health Research. Portions of this work were parts of presentations given by A.M. Sharma and
J.P.Uetrecht at the ISSX meeting in Japan, 2011, and the Society of Toxicology International
Meetings in Washington, D.C., USA, 2011, and San Francisco, CA., USA, 2012.
ABBREVIATIONS: Brown Norway, BN; 12-hydroxynevirapine, 12-OH-NVP; glyceraldehyde 3-
phosphate dehydrogenase, GAPDH; human immunodeficiency virus, HIV; idiosyncratic drug
reaction, IDR; NADPH-regenerating system, NRS. nevirapine, NVP; tris-buffered saline tween-
20, TBST; sulfotransferase, SULT; 3’-phosphoadenosine 5’-phosphosulfate, PAPS; keratinocyte,
KC; soluble fraction from a 9,000xg centrifugation, S9; Steven’s-Johnson syndrome, SJS; toxic
epidermal necrolysis, TEN.

105
3.6 Supplemental Material
3.6.1 Separation of Dermis and Epidermis of the Ear and Preparation of Homogenates.
At sacrifice, the entire ear was excised from the base of the skull and placed in PBS. Each
ear was sectioned into 3 parts (base of the ear, right quadrant, and left quadrant; Figure 5A) in
order to obtain full separation of dermis from epidermis. Pieces from each ear were floated
overnight at 4 °C in a solution of 40 mL of 0.625% trypsin (0.25% was found not to work well).
On day 2, the epidermis was peeled and scraped off of the dermis. The dermis was washed and
wiped clean of any residual epidermis, and each fraction was homogenized and centrifuged as
described above. The supernatant was separated and stored at -80 °C.
3.6.2 Grading of Skin Rash.
The grading scheme according to the AIDS Clinical Trial Group Protocol Management
Handbook Table for Grading Severity of Cutaneous Eruptions was used (where applicable to
rats). Grading was performed at the time of sacrifice using an area of shaved 1 x 1 inch skin on
the upper neck/back area of each rat. 92,98
(0) grade 0, normal integrity of skin is maintained
(1) grade 1, erythema with or without pruritus;
(2) grade 2, a diffuse erythematous macular or maculopapular cutaneous eruption or dry desquamation with
or without pruritus or typical target lesions without blistering, vesicles, or ulcerations in the lesions;
(3) grade 3, 1 of the following clinical presentations: urticaria; diffuse erythematous macular or
maculopapular cutaneous eruption or moist desquamation with or without pruritus together with any of the 4
constitutional findings possibly related to the drug (i.e., blistering, vesiculation, or both of cutaneous
eruptions; or any site of mucosal lesions considered related to study drug without other etiology, such as
herpes simplex or aphthous ulcer); angioedema; exfoliative dermatitis (defined as severe widespread
erythema and dry scaling of the skin and generalized superficial lymphadenopathy, with other constitutional
findings possibly related to study drug such as fever or weight loss); or diffuse rash and serum sickness-like
reactions defined as clinical symptom complex manifested as fever, lymphadenopathy, edema myalgia,
arthralgia, or a combination; and
(4) grade 4, diffuse cutaneous eruptions usually starting on the face, trunk, or back, often with prodromal
symptoms plus one of the following: cutaneous bullae, sometimes confluent with widespread sheet like
detachment of skin (Nikolsky’s sign), Stevens-Johnson syndrome, erythema multiforme major, or toxic
epidermal necrolysis, or 2 or more anatomically distinct sites of mucosal erosion or ulceration not due to
another cause. Severe rash was defined as grade 3 and grade 4 cutaneous eruptions when we used this
grading scheme. Skin biopsies were not required for categorization of rash

106
Grading Tables
Table 3S-1. Day 7 Skin Rash Grading
Key: UB = upper back; MB = mid-back; LS = left upper shoulder.
Rat Treatment Area of Skin
1x1 Inch
Description Rash Grade
Day 7 Control Rat 1 UB No visible skin
abnormality
0
Day 7 Control Rat 2 UB No visible skin
abnormality
0
Day 7 NVP Rat 1 UB 1 1
Day 7 NVP Rat 2 UB 1 1
Day 7 NVP Rat 3 UB No visible skin
abnormality
0
Day 7 NVP Rat 4 UB 1 1
Day 7 12-OH Rat 1 UB 3 SMALL 2
Day 7 12-OH Rat 2 UB 1 LARGE/DEEP 2

107
Table 3S-2. Day 10 and 15 Skin Rash Grading
Rat ID Area of Skin
1x1 Inch
Description Rash Grade
Day 10 Control UB No visible skin
abnormality
0
Day 10 NVP Rat 1 UB SLIGHT SCALING 1
Day 10 NVP Rat 2 UB SLIGHT RED 1
Day 15 Control UB CLEAN 0
Day 15 NVP Rat 1 UB 1 SMALL 2
Day 15 NVP Rat 2 UB 2 SMALL, SLIGHT
REDNESS
2

108
Table 3S-3. Day 21 Skin Rash Grading
Rat ID Area of Skin
1x1 Inch
Description Rash Grade
Day 21 Control Rat 1 UB No visible skin
abnormality
0
Day 21 Control Rat 2 UB No visible skin
abnormality
0
Day 21 Control Rat 3 UB No visible skin
abnormality
0
Day 21 Control Rat 4
UB No visible skin
abnormality
0
Day 21 12-OH-NVP
Rat 1
LS/UB DEEP LESIONS 3
Day 21 12-OH-NVP
Rat 2
UB VERY BAD; PELT
LIKE; DEEP
4
Day 21 NVP Rat 1 UB/MB VERY RED, VERY
DEEP
3
Day 21 NVP Rat 2 UB LESS LESIONS, VERY
RED
3
Day 21 NVP Rat 3 UB/MB BLOODY/SLOUGHING 4
Day 21 NVP Rat 4 UB PEELING/LESS RED 3

109
3.6.3 Covalent Binding and Histology in the Ears from NVP- or 12-OH-NVP-Treated Rats.
The ear is the first organ to turn red in response to NVP treatment in female BN rats (day
7).65
In severe cases, the rash may develop on the ear. Dorsal-ventral axis separation (Figure S-
1A) of ear epidermis and dermis from NVP- or 12-OH-NVP-treated rats (n = 4 per group)
displayed covalent binding (Figure S-1B) and a marked cellular infiltrate in the ear (Figure S-
1C).
H

110
Supplemental Figure 3S-1. (A) Method to fractionate ear using dorsal-ventral axis separation is
shown. Ear pieces were floated on 0.625% trypsin overnight at 4 °C to ensure complete
epidermal-dermal separation. (B) Immunoblot experiments comparing the epidermis from the
neck or ear from NVP- or 12-OH-NVP-treated female BN rats; 12 µg protein/well. Lane
designations are as follows: 1 & 2 = epidermis from the neck of control rats; 3 = epidermis from

111
the neck of a 12-OH-NVP-treated rat; 4 = epidermis from the neck of a NVP-treated animal; 5 =
epidermis from the ear of a 12-OH-NVP-treated rat; 6 = epidermis from the ear of a NVP-treated
rat. (C) H&E images of ear sections taken from each treatment group (representative slide from 1
of 4 rats per group shown). Magnification 20x.

112
CHAPTER 4
12-OH-Nevirapine Sulfate, Formed in the Skin, is Responsible for Nevirapine-
Induced Skin Rash
“We're all mad here.”
- Lewis Carroll, Alice in Wonderland
This work has been published in the following journal and is reproduced with permission:
Amy M. Sharma, Maria Novalen, Tadatoshi Tanino, Jack P. Uetrecht. 12-OH-Nevirapine Sulfate, Formed
in the Skin, is Responsible for Nevirapine-Induced Skin Rash. Chemical Research in Toxicology, 2013.
26, 817-827. Epub 2013 April 16.
Reprinted with permission. Copyright 2013 American Chemical Society. All rights reserved.
In this chapter, all experiments were performed by Amy M. Sharma, except Figures 4-2, 4S-1, 4S-
2.

113
4.1 Abstract
Nevirapine (NVP) treatment is associated with a significant incidence of skin rash in humans, and
it also causes a similar immune-mediated skin rash in Brown Norway (BN) rats. We have shown
that the sulfate of a major oxidative metabolite, 12-OH-NVP, covalently binds in the skin. The
fact that the sulfate metabolite is responsible for covalent binding in the skin does not prove that it
is responsible for the rash. We used various inhibitors of sulfation to test whether this reactive
sulfate is responsible for the skin rash. Salicylamide (SA), which depletes 3’-phosphoadenosine-
5’-phosphosulfate (PAPS) in the liver, significantly decreased 12-OH-NVP sulfate in the blood,
but it did not prevent covalent binding in the skin or the rash. Topical application of 1-phenyl-1-
hexanol, a sulfotransferase inhibitor, prevented covalent binding in the skin as well as the rash,
but only where it was applied. In vitro incubations of 12-OH-NVP with PAPS and cytosolic
fractions from the skin of rats or from human skin also led to covalent binding that was inhibited
by 1-phenyl-1-hexanol. Incubation of 12-OH-NVP with PAPS and sulfotransferase 1A1*1, a
human isoform that is present in the skin, also led to covalent binding, and this binding was also
inhibited by 1-phenyl-1-hexanol. We conclude that salicylamide did not deplete PAPS in the skin
and was unable to prevent covalent binding or the rash, while topical 1-phenyl-1-hexanol
inhibited sulfation of 12-OH-NVP in the skin and did prevent covalent binding and the rash.
These results provide definitive evidence that 12-OH-NVP sulfate formed in skin is responsible
for NVP-induced skin rashes. Sulfotransferase is one of the few metabolic enzymes with
significant activity in the skin, and it may be responsible for bioactivation of other drugs that
cause skin rashes.

114
4.2 Introduction
Idiosyncratic drug reactions (IDRs) are unpredictable adverse events that significantly
impact drug development and use. Many drugs that cause IDRs form reactive metabolites, and it
is usually assumed that these reactive metabolites are responsible for the IDR associated with the
drug involved.99,100
However, IDRs are difficult to study and it has not been possible to
definitively demonstrate that reactive metabolites are causal. In addition, many drugs form several
reactive metabolites and it is very difficult to test which, if any, is responsible for a specific IDR.
Furthermore, Pichler has proposed the p-i hypothesis in which a reversible interaction between the
T cell receptor – major histocompatibility complex is sufficient to trigger an IDR without the
formation of reactive metabolite.101
This is especially attractive for skin rashes because the skin
has very limited drug metabolism capacity; specifically, the levels of cytochromes P450 are very
low.102
At one point early in the NVP studies we thought that the p-i hypothesis might be relevant
for NVP-induced skin rash because we had eliminated several possible reactive metabolites;
however, we used the NVP animal model to show that the basis for the p-i hypothesis is false, and
certainly it is not the mechanism of NVP-induced skin rash.93
That does not mean that the
hypothesis itself is false; the p-i hypothesis probably is relevant for some compounds, especially
small peptidomimetic drugs such as ximelagatran.33
We have developed an animal model of
nevirapine (NVP, ViramuneTM
, TOC graphic)-induced skin rash, which is clearly immune-
mediated and has characteristics very similar to the rash that occurs in humans.65
These
characteristics include a similar time to onset, higher incidence in females, and the observation
that a low CD4+ T cell count decreases rash incidence. We used this model to test the involvement
of a reactive metabolite in the mechanism of NVP-induced skin rash.
NVP, a non-nucleoside reverse transcriptase inhibitor indicated for the treatment of HIV-1
infections, causes idiosyncratic hepatotoxicity and mild-to-severe skin rashes.61
We have
demonstrated that NVP is oxidized to a reactive quinone methide, which covalently binds in the
liver.74
However, we have not been able to demonstrate that this reactive metabolite is responsible
for the idiosyncratic liver injury caused by NVP because we were not able to produce liver injury
in animals with characteristics similar to the liver injury that occurs in humans. In contrast, we
identified the 12-hydroxylation pathway to form 12-OH-NVP is responsible for induction of the
skin rash because substitution of the methyl hydrogens of NVP with deuterium significantly
decreased the incidence and severity of the rash and treatment with 12-OH-NVP also caused a

115
skin rash.68
Because 12-OH-NVP is the same oxidation state as the quinone methide species
responsible for covalent binding in the liver, a quinone methide species could not be produced by
oxidation of 12-OH-NVP. In a recent paper we demonstrated that covalent binding of NVP in the
skin is mediated by a benzylic sulfate formed by first oxidation of NVP in the liver to 12-OH-
NVP followed by the formation of a benzylic sulfate (12-OH-NVP sulfate), which has sufficient
chemical reactivity to covalently bind to proteins.89
Both the liver and skin contain
sulfotransferases, and although chemically reactive, we were able to detect 12-OH-NVP sulfate in
the blood of rats treated with NVP.
It remained to be determined if 12-OH-NVP sulfate is responsible for NVP-induced skin
rash, and if so, whether it is sulfation in the liver or skin that is most important. We used our
animal model and various inhibitors of sulfation to answer this question.
Scheme 4-1: Depiction of schematic used to prevent rash in this study.

116
4.3 Materials and methods
4.3.1 Chemical Materials and Reagents.
NVP and ethyl-NVP (a NVP analogue where the cyclopropyl group has been replaced by
an ethyl group) were kindly supplied by Boehringer-Ingelheim Pharmaceuticals Inc. (Ridgefield,
CT). Common chemical reagents (3’-phosphoadenosine 5’-phosphosulfate (PAPS), β-
glucuronidase type IX-A, Tris, methanol, DMSO, PBS (phosphate buffered saline, 1.47 mM
KH2PO4 and 8.06 mM Na2HPO4-7H2O, pH 7.4), glycerol, silica gel, etc) were obtained from
Sigma-Aldrich (Oakville, ON) unless otherwise noted in the Methods. Ammonium persulfate was
obtained from Fisher Scientific (Fair Lawn, NJ). SDS and Tween-20 were obtained from BioShop
(Burlington, ON). Stock 30% acrylamide/bisacrylamide solution (29:1), nonfat blotting grade
milk powder, and nitrocellulose membrane (0.2 µM) were purchased from Bio-Rad (Hercules,
CA). Ultra pure tetramethylethylenediamine, frozen 2.5% trypsin, and normal goat serum were
purchased from Invitrogen (Carlsbad, CA). Amersham ECL Plus Western Blotting Detection
System was obtained from GE Healthcare (Oakville, ON). Horseradish peroxidase-conjugated
goat anti-rabbit IgG (H + L chains) was purchased from Sigma-Aldrich (St. Louis, Mo). 1-Phenyl-
1-hexanol was obtained from Tokyo Chemical Industry (Toshima, Japan) and micronized
dehydroepiandrosterone (DHEA) was obtained from PCCA (London, ON). The synthesis of 12-
OH-NVP, 12-OH-NVP sulfate, 4-carboxy-NVP (4-COOH-NVP), and the NVP anti-serum were
described previously.68,74
Human liver cytosol (pool of 10, mixed gender) or a 9,000 X
supernatant (S9) fraction containing cytosol and microsomes (pool of 50; mixed gender); rat liver
cytosol (pool of 115; female Sprague Dawley rats) or rat skin cytosol (pool of 50; female); and
recombinant human sulfotransferase (SULT) 1A1*1 expressed in Escherichia coli (E. coli) were
purchased from XenoTech LLC (Lexena, KS).
4.3.2 Animal Care.
Female BN rats (150 – 175 g; 8 to 10 weeks of age) were age-matched and obtained from
Charles River (Montreal, QC). Rats were housed in pairs in standard cages in a 12:12 h light/dark
cycle with access to water and Agribrands powdered lab chow diet (Leis Pet Distribution, Inc.
Wellesley, ON) ad libidum. Following a 1 week acclimatization period, rats were either
maintained on control chow or started on a drug-containing diet (treatment groups). For chronic
experiments, drug was mixed thoroughly with powdered lab chow to produce a NVP dose of 150

117
mg/kg/day, or an equimolar dose of 12-OH-NVP (159 mg/kg/day) for a maximum of 21 days.
The amount of drug administered to rats was calculated based on their body weight and daily food
intake. For experiments examining blood metabolite levels, drugs were ground to obtain fine
particles and NVP was gavaged at a dose of 100 mg/kg/day in 0.5% methyl cellulose. The dose
was scaled up from 50 mg/kg/day over a period of 3-5 days to avoid central nervous system
toxicity associated with high peak plasma levels of NVP. Rats were sacrificed via CO2
asphyxiation. Animal experiments were approved by the University of Toronto animal care
committee in accordance with guidelines of the Canadian Council on Animal Care.
4.3.3 Quantification of NVP, 12-OH-NVP, 12-OH-NVP Sulfate, and 4-COOH-NVP in Plasma.
Plasma (50 µL) was mixed with internal standard (ethyl-NVP, 5.4 µg/mL, 50 µL) and
concentrated with a Strata® solid phase extraction column (C18-E, 100 mg, Phenomenex,
Torrance, CA). The column was washed with 1 mL of water and the metabolites eluted with 1 mL
of methanol. The methanol was collected, dried, and reconstituted with 50 µL of the HPLC
mobile phase. The samples were separated on HPLC and analyzed by mass spectrometry. The
separation was carried out on an Ultracarb C18 30 X 2.0 mm, 5 µm column (Phenomenex) under
isocratic conditions with a mobile phase consisting of 16% acetonitrile and 84% water with 2 mM
ammonium acetate and 1% acetic acid and the flow rate of 0.2 mL/min. Mass spectrometry was
carried out using a PE Sciex API 3000 quadrupole system with an electrospray ionizing source.
The ion pairs used for quantitation in the multiple reaction monitoring/positive ion mode were:
267.0/226.1 for NVP, 283.1/223.1 for 12-OH-NVP, 297.1/210.1 for 4-COOH-NVP, 283.1/161.0
for 2-OH-NVP, 283.1/214.0 for 3-OH-NVP, 255.1/227.2 for ethyl-NVP. Standard curves
prepared for 2-OH-NVP (0.43 – 102.9 µg/mL), 3-OH-NVP (0.36 – 86.8 µg/mL), 12-OH-NVP
(0.38 – 91.0 µg/mL), 4-COOH-NVP (0.26 – 61.8 µg/mL), and NVP (0.74 – 176.9 µg/mL) had R2
values of > 0.99.
Quantification of 12-OH-NVP sulfate was done in a similar manner, the major difference
being that it was performed in the negative ion mode. Because of this the internal standard was
changed to naproxen (2.5 µg/mL, 50 µL added to the plasma). The ion pairs used for the analysis
were 361.0/96.0 for 12-OH-NVP sulfate and 229.0/169.8 for naproxen. The HPLC column used
for the separation was an Ultracarb C18 column (100 x 2 mm, 5 µm, Phenomenex) with a

118
gradient elution of 20 80% acetonitrile over a period of 10 min. The second solvent was water
with 2 mM ammonium acetate and 1% acetic acid. The flow rate was 0.2 mL/min. Standard
curves prepared for 12-OH-NVP sulfate (0.28 – 14.0 µg/mL) had R2 values of > 0.99.
4.3.4 Sulfation Inhibition Studies.
The effects of 3 sulfation inhibitors on covalent binding and skin rash were studied; see
Results for further information on specific experiments.
Table 4-1. Inhibitors of sulfation and dosing method.
Inhibitor Name Dose Vehicle Application Method
Salicylamide 274 mg/kg/day 0.5% methyl cellulose oral; 1x/ day after 5 PM
DHEA 20 mg/kg/day
topical;
50 or 100
mg/kg/day oral
50:50 oil:acetone
(topical);
0.5% methyl cellulose
(oral)
topical or oral gavage;
1x/day
1-phenyl-1-hexanol 20 mg/kg/day
topical, or 20
mg/kg/day oral
50:50 oil:acetone
(topical);
0.5% methyl cellulose
(oral)
topical or oral gavage;
1x/day
For topical inhibition studies the desired area of skin was shaved using an electric shaver,
and administration of the inhibitors was started 3 days after shaving to allow the skin barrier to
heal from any nicks that might have occurred. The skin was painted with either DHEA or 1-
phenyl-1-hexanol using a 200 µL pipette. For oral administration inhibitors were given as a co-
treatment with NVP. Controls were either fed standard lab chow or administered vehicle by
gavage if they were controls for salicylamide cotreatment groups.
For systemic inhibition studies, female BN rats were treated with NVP together with
salicylamide, DHEA, or 1-phenyl-1-hexanol. All drugs were ground, suspended in 0.5%
methylcellulose, and administered by gavage with minimal time between NVP and the inhibitor.
If NVP was to be given by gavage, the dose was escalated during the first 4 to 5 days of the study
from 50 mg/kg/day to the full dose of 100 mg/kg/day (see results for specific details). The lower
dose was given in the beginning of the study to avoid central nervous system toxicity associated
with high peak plasma levels of NVP. 12-OH-NVP was coadministered at a dose of 100
mg/kg/day by oral gavage with DHEA at doses of 25, 50, or 100 mg/kg/day by oral gavage. 1-

119
phenyl-1-hexanol was administered orally by gavage while NVP was fed in food at 150
mg/kg/day. All inhibition studies were carried out for a maximum of 28 days.
4.3.5 Separation of Skin Dermis and Epidermis and Preparation of Homogenates.
At sacrifice, hair was removed from the rats using an electric shaver, and the skin was
cleaned of remaining hair using PBS and Kimwipes. Skin from the back was then excised and
placed on dry ice over aluminum foil. Care was taken to remove all hypodermis and connective
tissue using blunt-tip forceps. Sections (~200 mg) of whole skin were stretched using sharp-tipped
forceps on clean petri dishes for a maximum of 2 h at 4 °C. Once stretched, skins were floated
individually overnight at 4 °C in a solution of 0.25% trypsin (approx. 50 mL per skin section). On
day 2, the epidermis was lifted off of the dermis, the dermis was wiped clean of any residual
epidermis using Kimwipes, and each fraction was homogenized separately in cell lysis buffer
(Cell Signaling Technologies, Pickering, ON) with protease inhibitor (HALT Protease Inhibitor
Cocktail, Pierce, Rockford, IL) in a 10:1 ratio using a Polytron 2100 homogenizer (~1.5 mL
working cell lysis buffer per fraction). In order to clarify samples, dermal and epidermal fractions
were centrifuged at 13,000 rpm each for 2 min. The supernatant was separated and stored at -80
°C. The protein concentration of the prepared homogenates was quantified using a BCA protein
assay kit (Novagen, EMD Biosciences Inc., Mississauga, ON); bovine serum albumin was used as
the standard. Whole rat skin tissue was prepared using the same method except dermal and
epidermal layers were not separated and skin was not floated overnight in trypsin.
4.3.6 Preparation of Human Skin Dermatome.
Human skin (9 g) was obtained from XenoTech LLC (Lenexa, KS, USA) from the
abdomen of a 56 year old Caucasian male 9.1 h after death due to heart disease without any skin
or infectious diseases. Skin was prepared to contain the epidermal layers only. Dermatomed skin
was then prepared by chopping into fine pieces, immersion in cell lysis buffer with protease
inhibitor for ~15 min on ice followed by homogenization via a Polytron 2100 homogenizer. Skin
was clarified of debris via centrifugation as described above. The supernatant was separated and
stored at -80 °C.

120
4.3.7 Incubation of Human Skin or Expressed Human SULT 1A1*1 with 12-OH-NVP or 12-OH-NVP Sulfate, With or Without PAPS.
Stock solutions of each compound were prepared in methanol fresh on the day of use.
Before incubation with skin or isolated SULT 1A1*1, methanol was partially removed by
nitrogen evaporation in order to limit the amount of solvent in the incubation to <0.05%.83
For
human dermatomed skin, a 5 min preincubation consisting of each chemical and Dulbecco’s PBS
with MgCl2 and CaCl2 at 37 °C in a water bath was followed by addition of dermatomed skin
(final concentration 1 mg/mL). SULT 1A1*1 pre-incubates consisted of the SULT 1A1*1 protein,
skin (final concentration 1 mg/mL), Dulbecco’s PBS with MgCl2 and CaCl2, and drug. Following
the 5 min preincubation, PAPS was added to a final concentration of 0.3 mM and the samples
were vortexed. The final concentration of all drugs tested was 1 mM. Samples were taken at time
0 (before the start of the incubation), 30, and 60 min, and reactions were terminated by placing the
samples on dry ice. Negative controls were the skin fractions incubated at 37 °C without addition
of any drug or PAPS (see results for specific details). All samples were stored at -80 °C until use
for immunoblotting experiments.
4.3.8 Incubation of Rat Liver or Skin Cytosol or Human Liver Cytosol with 12-OH-NVP and 1-Phenyl-1-hexanol in the Presence and Absence of PAPS.
To an Eppendorf tube containing Dulbecco’s PBS with MgCl2 and CaCl2 was added 12-
OH-NVP dissolved in methanol (stock 50 mM) and 1-phenyl-1-hexanol to a final concentration of
1 mM each. Methanol was partially removed by nitrogen evaporation in order to limit the amount
of solvent in the incubation to <0.05%.83
Following a 5 min preincubation with between 0.5 - 1
mg/mL each of rat skin S9 or male or female human liver cytosol (XenoTech LLC, Lenexa, KS)
at 37 °C in a water bath, PAPS was added to a final concentration of 0.3 mM. Tubes were
vortexed and incubated for 1 h. Negative controls contained all components except PAPS while
positive controls did not contain 1-phenyl-1-hexanol. All samples were frozen on dry ice to halt
the reaction and stored at -80° C until used for immunoblotting experiments.
4.3.9 Covalent Binding Using SDS-PAGE and Immunoblotting.
Whole skin, dermal, or epidermal homogenate samples, or in vitro incubates, were mixed with
Pierce reducing sample loading buffer in a 4:1 protein to buffer ratio and boiled for 5 min. SDS

121
PAGE was performed using the Protean-3 minigel system (BioRad, Mississauga, ON). Gels were
hand-cast (8% bisacrylamide) or bought from Bio-Rad Canada (12% bisacrylamide), and were
run at 130 V. Electrophoresis running buffer (Bio-Rad) consisted of 25 mM Tris, 192 mM
glycine, and 0.1% SDS, pH 8.3. Transfer to nitrocellulose membrane (0.2 µM, BioRad) occurred
at 0.13 mA for 90 min at 4 ºC using the same Protean-3 minigel system (BioRad, Mississauga,
ON). Tris-glycine transfer buffer (Bio-Rad) consisted of 25 mM Tris, 192 mM glycine, and 20%
methanol at pH 8.5. Membranes were washed twice in Tris-buffered saline tween-20 (TBST)
wash solution for 5 min. Membranes were then blocked in 5% non-fat milk blocking solution in
TBST for 90 min at room temperature. Membranes were then rinsed with 3 changes of TBST for
5 min and incubated with a 1:100 or 1:500 dilution of primary anti-NVP antiserum and 10%
normal goat serum in TBST overnight at 4 ºC. A 20 min wash (3 changes) in TBST after
overnight blocking was followed by a 90 min incubation in secondary antisera (1:2000 or 1:5000
dilution) in TBST containing 10% goat serum. The secondary anti-serum was goat anti-rabbit
horseradish peroxidase antisera. Membranes were washed 3 times for 20 min with TBST. All
blots were incubated with enhanced chemiluminescence stain for 5 min and analyzed with a
FluorChem8800 imager. To probe for the glyceraldehyde 3-phosphate dehydrogenase (GAPDH)
loading control, membranes were stripped of primary NVP antiserum using Pierce Restore Plus
buffer (Pierce, Rockford, IL) for 15 to 20 min at room temperature followed by a 1 h blocking
step. Membranes were then incubated in mouse monoclonal anti-GAPDH antisera (1:40,000) and
processed as above except the secondary antisera was goat anti-mouse horseradish peroxidase
antisera diluted 1:10,000 (Jackson ImmunoResearch, Baltimore Pike, West Grove, PA.).
4.3.10 Preparation of BN Rat Skin for Histology.
Skin samples were flattened on paper towels to prevent curling of the skin and fixed in
10% formalin. The paraffin block, hematoxylin/eosin slides, or unstained sections were prepared
at the Hospital for Sick Children in Toronto, ON Canada.

122
4.4 Results
4.4.1 General Scheme to Study the Role of 12-OH-NVP Sulfation on the Skin Rash.
The sulfation of 12-OH-NVP can occur in both the liver and skin, and it is important to
know which, if either, is involved in the NVP-induced skin rash. Salicylamide depletes PAPS and
was administered by oral gavage; it is likely to only decrease sulfation in the liver. DHEA and 1-
phenyl-1-hexanol are sulfotransferase inhibitors and were administered both orally and topically.
Previous work with 1-phenyl-1hexanol has been limited to in vitro studies, and it is likely that it is
cleared too rapidly to significantly decrease hepatic sulfation.
4.4.2 Effect of Salicylamide on 12-OH-NVP Sulfate Levels and Rash.
Cotreatment with oral salicylamide markedly decreased circulating 12-OH-NVP sulfate
levels (Figure 4-1D). It also decreased the amount of 12-OH-NVP sulfate in a 24 hour urine
sample (Figure 4S1-C, Supporting Information). However, it did not prevent covalent binding in
the skin or the skin rash (Figure 4-2). The average grade of rash for the salicylamide cotreated
group was 3.75 versus 3.25 for NVP alone (Table 4S-1, see Supporting Information for criteria
used to grade the rash and grading results). Covalent binding of NVP in the epidermis (Figure 4-
2A) and the cellular infiltrate in the skin (Figure 4-2B) were also not decreased by cotreatment
with oral salicylamide. The NVP and NVP + salicylamide cotreatment groups displayed similar
histological alterations in the skin, signifying no effect of salicylamide (Figure 4-2C). It appeared
that animals that presented with the most severe grade of skin rash (3 or 4) displayed the most
intense covalent binding from all groups tested (12-OH-NVP, NVP, or NVP + salicylamide). As
reported previously,89
essentially all of the covalent binding was in the epidermis with virtually
none in the dermis (data not shown).

123
Figure 4-1. (A) Incidence of skin rash, (B) plasma concentrations of NVP, (C) 12-OH-NVP, and
(D) 12-OH-NVP sulfate in female Brown Norway rats treated with NVP only (100 mg/kg/day, n
= 4), in combination with oral DHEA (50 and 100 mg/kg/day) or in combination with oral
salicylamide (274 mg/kg/day).
0 25 50 75 100
NVP
NVP + DHEA 50
NVP + DHEA 100
NVP + salicylamide
Incidence skin rash
0 10 20 300
20
40
60
80
NVP
NVP + DHEA 50
NVP + DHEA 100
NVP + salicylamide
Treatment day
NV
P (
g/m
l)
0 10 20 300
10
20
30
NVP
NVP + DHEA 50
NVP + DHEA 100
NVP + salicylamide
Treatment day
12-O
H-N
VP
(
g/m
l)
0 10 20 300
2
4
6
8
NVP
NVP + DHEA 50
NVP + DHEA 100
NVP + salicylamide
Treatment day
12-O
H-N
VP
su
lfate
(
g/m
l)
A
B
C
D

124

125
Figure 4-2. (A) Immunoblot of the epidermis comparing individual 12-OH-NVP-treated rats to
NVP + oral salicylamide cotreated rats (N+Sal) or NVP only-treated rats, against 0.5% methyl
cellulose gavaged controls. Protein loading was 15 µg/lane. (B) Skin histology of NVP + oral
salicylamide cotreated rats, n = 4. (C) Skin histology compared between various treatment groups:
normal and gavaged controls are normal without a cellular infiltrate in the dermis, while NVP, 12-
OH-NVP and NVP + oral salicylamide treated rats display keratinocyte necrosis within the
epidermis, with marked inflammatory infiltrate at the dermal-epidermal junction. A representative
photo from one of four animals per group is shown. All rats represented in this figure were treated
for 21 days. Magnification was 20x for all slides in this figure.
4.4.3 Effects of DHEA on NVP Metabolism and Skin Rash.
Coadministration of oral DHEA decreased plasma concentrations of NVP, 12-OH-NVP,
and 12-OH-NVP sulfate, although it did not appear to decrease the blood levels of 12-OH-NVP
sulfate as much salicylamide (Figure 4-1). It also decreased the urinary excretion of 12-OH-NVP
sulfate and other metabolites (Figure 4S-1); however, oral DHEA did prevent the skin rash.
Because of the complex effects of oral DHEA on NVP metabolism we tried topical
administration of DHEA to inhibit sulfation in the skin. It decreased the severity of the rash, but it
did not completely prevent it. This was reflected in the histology, where the dermal infiltrate and
epidermal changes resembled those of the NVP only group (Figure 4S-2A-C). Even topical
DHEA led to decreased plasma 12-OH-NVP sulfate levels near the end of treatment (day 21)

126
when compared to NVP only treated animals (data not shown). The results using oral and topical
DHEA to prevent sulfation were complicated by other effects on NVP metabolism and did not
provide a definitive answer; therefore, we abandoned its use.
4.4.4 Effects of 1-Phenyl-1-Hexanol on Covalent Binding and Rash.
In contrast to DHEA, topical 1-phenyl-1-hexanol did not significantly affect blood levels
of NVP or its metabolites (data not shown), but it completely prevented the rash where it was
applied. Specifically, topical administration of 1-phenyl-1-hexanol was performed as outlined in
Figure 3B using the left and right shoulders of the animals in order to maintain symmetry. Both
the skin rash (Figure 4-3, C-E) and covalent binding (Figure 4-4, A-B) were prevented in the 1-
phenyl-1-hexanol-treated areas in the epidermis (although there is an artifact band in the control),
and these areas had much less cellular infiltrate than vehicle-treated areas (Figure 4-5, A-C).
In order to obtain more skin, the upper and lower backs of the animals were employed in
another scheme (Figure 4-6A) to test inhibition of skin rash and covalent binding. This schematic
for application was found to be better in that rats had a hard time accessing these areas to scratch
or lick. Both skin rash and covalent binding (Figure 4-6B; photos not shown) were prevented in
the topical 1-phenyl-1-hexanol-treated areas, and these areas had much less cellular infiltrate in
the dermis than vehicle-treated areas (Figure 4-7, A-C). Oral administration of 1-phenyl-1-
hexanol did not prevent covalent binding or skin rash (data not shown). It is likely that this lack of
effect is the result of rapid clearance of 1-phenyl-1-hexanol, but we did not develop an analytical
method so that this hypothesis could be tested.
In order to show specificity of the anti-serum for NVP-modified proteins, it was
preincubated with NVP (Figure 4-7D). The preincubated anti-serum was then used as the primary
antibody in an immunoblot using the same epidermal samples shown in Figure 4-3, C-E. Covalent
binding to each of the epidermal skin fractions taken from vehicle, topical 1-phenyl-1-hexanol, or
rash areas was prevented except for the artifact band.

127

128
Figure 4-3. (A) Diagram of the preliminary sites for administration of topical DHEA or topical 1-
phenyl-1-hexanol to determine their effect on the NVP-induced skin rash. In 2/2 animals tested,
the rash was slightly milder with DHEA, but it was completely prevented in 1-phenyl-1-hexanol-
treated areas only (photos not shown). (B) Diagram of sites employed in 2 independent trials to
test the effect of topical 1-phenyl-1-hexanol on the NVP-induced skin rash. Five animals in total
were treated with NVP (150 mg/kg/day) in food and 1-phenyl-1-hexanol (20 mg/kg/day) on the
skin. In 100% of the animals, the rash was prevented by topical 1-phenyl-1-hexanol. One
representative rat from each study is shown above. Photos showing (C) skin from the back of a
control rat, (D) skin from the back of the NVP only-treated rat, (E) vehicle versus 1-phenyl-1-
hexanol-treated areas from an inhibitor-treated rat (topical treatment).

129
Figure 4-4. Using skin isolated from rats cotreated with NVP and topical 1-phenyl-1-hexanol
using the schematic shown in Figure 3B, epidermal immunoblot analysis was performed. (A)
Immunoblot of epidermis from rash areas versus vehicle areas from the epidermis of inhibitor-
treated rats cotreated with NVP. (B) Immunoblot of epidermis from topical 1-phenyl-1-hexanol
areas versus vehicle areas from epidermis of inhibitor-treated rats compared with that of an

130
untreated control and a NVP-treated control. 15 µg of protein per lane was loaded for each of A
and B.

131
Figure 4-5. Representative histology of rat skin isolated from rats cotreated with NVP and topical
1-phenyl-1-hexanol using the schematic shown in Figure 3B. (A) H&E stained sections from
upper neck/rash area from control (Ct), nevirapine-treated (NVP), or 1-phenyl-1-hexanol (Ph) rat
number 1 shown in 3C-E; (B) H&E stained sections from left shoulder/vehicle area from control
(Ct), nevirapine-treated (NVP), or 1-phenyl-1-hexanol (Ph) rat number 1 shown in 3C-E; (C)
H&E stained sections from right shoulder/1-phenyl-1-hexanol-treated area from control (Ct),
nevirapine-treated (NVP), or 1-phenyl-1-hexanol (Ph) rat number 1 shown in 3C-E.
Magnification 20x for all slides in this panel.

132
Figure 4-6. (A) Second topical schematic used to test the inhibitor 1-phenyl-1-hexanol. (B)
Immunoblot of epidermis from areas of vehicle or inhibitor treated areas using the second
schematic shown in 6A. The control is epidermis from the untreated control rat; Ph1 or Ph2 are
topical inhibitor-treated epidermal areas from rat # 1 or 2, respectively; Vh1 or Vh2 are vehicle
treated epidermal areas for each rat, and RA1 or RA2 are from rash areas with no topical
treatment. NVP is from the epidermis of the back of the neck for the NVP-treated positive control
rat. Protein loading was 15 µg/lane.

133

134
Figure 4-7. Histology with H&E staining of skin isolated from rats cotreated with NVP and
topical 1-phenyl-1-hexanol using the schematic shown in Figure 6A. The upper left slide of each

135
panel is from a control animal without NVP treatment, the upper right slide is from a NVP-treated
animal with no topical treatment, and the lower two slides are from animals with NVP + topical
treatment. (A) skin from upper back with no topical treatment representing the typical rash; (B)
midback where the vehicle was applied in inhibitor-treated animals only (lower two slides); (C)
the lower back were 1-phenyl-1-hexanol was applied in inhibitor-treated animals only (lower 2
slides). Magnification 20x. (D) Preincubation of the primary anti-NVP serum with 1.5 mM NVP
dissolved in DMSO for 2 h at 37 °C prevented covalent binding of the anti-serum to epidermal
samples from the samples shown in Figure 3C-E, except for one artifact band. The DMSO control
(right most lane) where the primary anti-serum was incubated with DMSO alone. Protein loading
was 15 µg/lane.
Table 4-2. Comparison of results obtained from 12-OH-NVP sulfate inhibitor studies.
Schematic:
Treatment group: NVP in food NVP in food + salicylamide
via gavage
NVP in food + topical 1-
phenyl-1-hexanol treatment
Effect of treatment on
blood 12-OH-NVP
sulfate levels
1-8 µg/mL Below limit of quantification 1-8 µg/mL
Is covalent binding
present with this
treatment?
Yes; epidermis Yes; epidermis Markedly decreased in
inhibitor-treated areas
Is rash present with this
treatment?
Yes Yes (same as with NVP only) None in inhibitor-treated
areas

136
4.4.5 In Vitro Inhibition of Covalent Binding by 1-Phenyl-1-Hexanol.
In order to examine the ability of 1-phenyl-1-hexanol to prevent covalent binding in vitro,
a series of studies comparing the covalent binding of 12-OH-NVP +/- PAPS alone with that of 12-
OH-NVP in combination with 1-phenyl-1-hexanol +/- PAPS were performed. As shown in Figure
4-8A, 1-phenyl-1-hexanol significantly inhibited covalent binding that occurs in the presence of
PAPS and 12-OH-NVP in vitro. This was true in cytosolic fractions of rat skin and liver. A
similar pattern was observed when rat skin cytosol was compared with human liver cytosol
(Figure 4-8B), and human skin S9 compared to human liver S9 (Figure 4-8C). Human skin
incubates were then compared to human E.coli expressed SULT 1A1*1 in order to test if SULT
1A1*1, which is found in human skin, can metabolize 12-OH-NVP. Figure 4-8D displays marked
covalent binding of 12-OH-NVP to SULT 1A1*1 in the presence of PAPS, indicating that
metabolism to the sulfate had occurred. In the absence of PAPS, no binding was observed. This
binding was also prevented by 1-phenyl-1-hexanol. SULT 1A1*1 has a mass of ~ 35 kDa, and the
multiple bands observed on the immunoblot are due to the presence of E. coli cytosol, which
contains other proteins. When binding to SULT 1A1*1 was compared to human skin on the same
immunoblot, it was observed that the same 35 kDa band was modified in both samples in the
presence of PAPS, while only background binding remained in the control lanes.

137

138

139
Figure 4-8. Immunoblot of isolated rat liver cytosol or rat skin cytosol incubated with 12-OH-
NVP or a combination of 12-OH-NVP (12-OH) and 1-phenyl-1-hexanol in vitro, in the presence
and absence of PAPS. (B) Immunoblot of rat skin cytosol versus female human liver cytosol
incubated with or without PAPS and 12-OH-NVP or 12-OH-NVP and 1-phenyl-1-hexanol. (C)
Human skin ‘dermatome’ homogenized and incubated with 12-OH-NVP or 12-OH-NVP + 1-
phenyl-1-hexanol to show the same phenomenon exists in human skin. (D) Human SULT 1A1*1
incubated with 1 mM 12-OH-NVP (12-OH) or 12-OH-NVP and 1-phenyl-1-hexanol +/- 0.3 mM
PAPS, or 12-OH-NVP sulfate (12-Sulfate). 12 µg/well protein was loaded for each blot.

140
4.5 Discussion
We previously demonstrated that 12-hydroxylation of NVP was required for the induction
of a skin rash in BN rats.68
We proposed that this was due to the formation of 12-OH-NVP
sulfate, which was expected to be chemically reactive. However, two observations argued against
this hypothesis: the chemical reactivity of the sulfate was very low, and in the first experiments to
test the involvement of the sulfate, we found that inhibition of sulfation by depletion of PAPS
with salicylamide decreased the blood levels of the circulating sulfate, but it did not prevent the
skin rash. However, in a recent paper,89
we demonstrated that 12-OH-NVP sulfate readily binds to
proteins in the epidermis, both in vitro and in vivo in rats and also in human skin homogenates.
With this ability to determine covalent binding of 12-OH-NVP sulfate in the skin, we found that
although salicylamide was able to markedly decrease blood levels of 12-OH-NVP sulfate, it did
not affect covalent binding in the skin. This is presumably because the mechanism by which
salicylamide inhibits sulfation involves depletion of PAPS, and although this works to decrease
sulfation in the liver, the turnover of salicylamide in the skin is likely to be too low to deplete
PAPS in the skin and prevent covalent binding there. Although this could be considered a
negative study, this result is important because it indicates that 12-OH-NVP sulfate formed in the
liver is not responsible for covalent binding in the skin. We tried the sulfotransferase inhibitor,
DHEA, which did prevent the skin rash, but it also affected blood levels of NVP and 12-OH-
NVP; therefore, it was impossible to determine the mechanism by which DHEA prevented the
rash. For this reason, the studies with DHEA were not pursued further.
We had previously shown that binding of 12-OH-NVP to a skin homogenate requires
PAPS but not NADPH.11
There was no binding of NVP with or without a NADPH-generating
system. Both 1-phenyl-1-hexanol, a known sulfotransferase inhibitor, and 12-OH-NVP are
benzylic alcohols and may be substrates for hydroxysteroid SULTs, which are major SULTs in
female rat skin.95
We also found that 1-phenyl-1-hexanol inhibited covalent binding of 12-OH-
NVP mediated by SULT 1A1*1, which is a polymorphic sulfotransferase found in human
skin.71,103
Treatment of rats with oral 1-phenyl-1-hexanol did not prevent covalent binding or the
rash. Although we did not develop an assay to study the metabolism of 1-phenyl-1-hexanol, it is
likely that it was cleared rapidly and did not reach inhibitory concentrations in the skin. In
contrast, topical administration of 1-phenyl-1-hexanol did prevent covalent binding of NVP in the

141
skin as well as the rash and histological changes, but only where it was applied. This, along with
the previous study showing that it is 12-OH-NVP sulfate that is responsible for NVP covalent
binding in the skin89
provides conclusive evidence that this is the species responsible for the rash.
The finding that binding also occurs to the human SULT 1A1 provides a strong link to the NVP-
induced skin rash in humans.
It is also important to note that as opposed to the liver, covalent binding in the skin may
have a direct effect on organ-specific immune responses; i.e., the skin is not a major site of
xenobiotic biotransformation and does not have the same tolerogenic mechanisms in place as the
liver.104
Rather, the epidermis is immunologically active and has been termed an ‘adjuvant,’ with
keratinocytes able to modulate numerous immune activities.50
Additionally, if covalent binding in
the skin were involved as we hypothesize, the resulting immune response and rash would take
time to develop, which is true for NVP-induced skin rash.65
This is the first study to use a valid animal model to demonstrate that a reactive metabolite
is responsible for an idiosyncratic drug reaction, in this case a reactive metabolite formed in the
skin. This mechanism is an alternative to the p-i hypothesis for the mechanism of an idiosyncratic
drug reaction in the skin. Sulfotransferase is one of the few metabolic enzymes with significant
activity in the skin, and it may be responsible for bioactivation of other drugs that cause skin
rashes.

142
FUNDING SUPPORT. This work was supported by a grant from the Canadian Institutes of
Health Research (MPO84520).
ACKNOWLEDGMENTS. The authors thank Boehringer-Ingelheim for kindly supplying
nevirapine. A.M.S. is the recipient of a University of Toronto Pharmaceutical Sciences Doctoral
Fellowship. J.U. is the recipient of the Canada Research Chair in Adverse Drug Reactions.
Portions of this work were parts of presentations given by A.M. Sharma and J.P.Uetrecht at the
ISSX meeting in Japan, 2011, and the Society of Toxicology International Meetings in San
Francisco, CA., USA, 2012 and in San Antonio, TX., USA, 2013.
ABBREVIATIONS: Brown Norway, BN; 12-hydroxynevirapine, 12-OH-NVP; 12-OH-NVP
sulfate, 12-OH-NVP Sulfate; cytochrome P450, P450; human immunodeficiency virus, HIV;
idiosyncratic drug reaction, IDR; liquid chromatography/mass spectrometry, LC/MS; nevirapine,
NVP; tris-buffered saline tween-20, TBST; dehydroepiandosterone, DHEA; 9,000 X g
supernatant, S9; salicylamide, SA; sulfotransferase, SULT; 3’-phosphoadenosine-5’-
phosphosulfate, PAPS; keratinocyte, KC; Steven’s-Johnson syndrome, SJS; Toxic epidermal
necrolysis, TEN.

143
4.6 Supplemental Material
4.6.1 Quantification of NVP, 12-OH-NVP, 12-OH-NVP Sulfate, and 4-COOH-NVP in Urine.
Urine, 50 µL from a 24-h sample was mixed with 100 µL internal standard (ethyl-NVP, 27
µg/mL in the mobile phase) and 10 µL of β-glucuronidase (approximately 10,000 U/mL in 100
mM KH2PO4 buffer, pH 7.4) and incubated overnight at 37 °C prior to concentrating on an
Strata® solid phase extraction column as per above. The samples were separated using the same
HPLC conditions as described for plasma samples. As with plasma samples, for quantification of
12-OH-NVP sulfate, the internal standard was naproxen and no preincubation with β-
glucuronidase was performed.
4.6.2 Grading of Skin Rash.
The grading scheme according to the AIDS Clinical Trial Group Protocol Management
Handbook Table for Grading Severity of Cutaneous Eruptions was used (where applicable to
rats).98
Grading was performed at the time of sacrifice using an area of shaved 1 x 1 inch skin on
the upper neck/back area of each rat.
(0) grade 0, normal integrity of skin is maintained
(1) grade 1, erythema with or without pruritus;
(2) grade 2, a diffuse erythematous macular or maculopapular cutaneous eruption or dry desquamation with
or without pruritus or typical target lesions without blistering, vesicles, or ulcerations in the lesions;
(3) grade 3, 1 of the following clinical presentations: urticaria; diffuse erythematous macular or
maculopapular cutaneous eruption or moist desquamation with or without pruritus together with any of the 4
constitutional findings possibly related to the drug (i.e., blistering, vesiculation, or both of cutaneous
eruptions; or any site of mucosal lesions considered related to study drug without other etiology, such as
herpes simplex or aphthous ulcer); angioedema; exfoliative dermatitis (defined as severe widespread
erythema and dry scaling of the skin and generalized superficial lymphadenopathy, with other constitutional
findings possibly related to study drug such as fever or weight loss); or diffuse rash and serum sickness-like
reactions defined as clinical symptom complex manifested as fever, lymphadenopathy, edema myalgia,
arthralgia, or a combination; and
(4) grade 4, diffuse cutaneous eruptions usually starting on the face, trunk, or back, often with prodromal
symptoms plus one of the following: cutaneous bullae, sometimes confluent with widespread sheet like
detachment of skin (Nikolsky’s sign), Stevens-Johnson syndrome, erythema multiforme major, or toxic
epidermal necrolysis, or 2 or more anatomically distinct sites of mucosal erosion or ulceration not due to
another cause. Severe rash was defined as grade 3 and grade 4 cutaneous eruptions when we used this
grading scheme. Skin biopsies were not required for categorization of rash

144
Grading Table
Table 4S-1: Day 21 Skin Rash Grading
Key: UB = upper back; MB = mid-back; LS = left upper shoulder.
Rat Treatment Area of Skin
1x1 Inch
Description Rash Grade
Day 21 Control Rat 1 UB No visible skin
abnormality
0
Day 21 Control Rat 2 UB No visible skin
abnormality
0
Day 21 Gavaged
Control Rat 1
UB No visible skin
abnormality
0
Day 21 Gavaged
Control Rat 2
UB No visible skin
abnormality
0
Day 21 12-OH Rat 1 LS/UB DEEP LESIONS 3
Day 21 12-OH Rat 2 UB VERY BAD; PELT
LIKE; DEEP LESIONS
4
Day 21 NVP +
Salicylamide Rat 1
UB/MB MANY LESIONS;
PEELING OF
EPIDERMIS
4
Day 21 NVP +
Salicylamide Rat 2
UB VERY RED, MANY
LESIONS
4
Day 21 NVP +
Salicylamide Rat 3
UB LARGER/MANY
LESIONS
4

145
Day 21 NVP +
Salicylamide Rat 4
UB/MB REDNESS; LESIONS 3
Day 21 NVP Rat 1 UB/MB VERY RED, VERY
DEEP
3
Day 21 NVP Rat 2 UB LESS LESIONS, VERY
RED
3
Day 21 NVP Rat 3 UB/MB BLOODY/SLOUGHING 4
Day 21 NVP Rat 4 UB PEELING/LESS RED 3

146
0 10 20 300
500
1000
1500
2000
2500NVP
NVP + salicylamide
NVP + DHEA
Treatment day
12-O
H-N
VP
(
g/2
4h
)
0 10 20 300
1000
2000
3000
4000
5000 NVP
NVP + salicylamide
NVP + DHEA
Treatment day
4-C
OO
H-N
VP
(
g/2
4h
)
0 10 20 300
500
1000
1500 NVP
NVP + salicylamide
NVP + DHEA
Treatment day
12-O
H-N
VP
su
lfate
(
g/2
4h
)
0 10 20 300
1000
2000
3000
4000
5000
NVP
NVP + salicylamide
NVP + DHEA
Treatment day
2-O
H-N
VP
(
g/2
4h
)
0 10 20 300
500
1000
1500
2000
2500NVP
NVP + salicylamide
NVP + DHEA
Treatment day
3-O
H-N
VP
(
g/2
4h
)
A
B
C
D
E
Figure 4S-1. Urinary excretion of (A) 12-OH-NVP, (B) 4-COOH-NVP, (C) 12-OH-NVP sulfate,
(D) 2-OH-NVP, and (E) 3-OH-NVP from rats treated with NVP (100 mg/kg/day), NVP + DHEA
(100 mg/kg/day) each or NVP + salicylamide (274 mg/kg/day; n = 4 in each group). Data depicts
the mean ± SD.

147
Figure 4S-2. H&E stained sections comparing the histology of rat skin in response to (A) NVP
treatment, (B) NVP + topical DHEA cotreatment, or (C) control rats. Magnification = 20x.

148
CHAPTER 5
Discussion, Conclusions, and Summary
‘I know that I am intelligent, because I know that I know nothing.’
- Socrates

149
5.1 Hypotheses Revisited
The central goal of this thesis was to study the relationship between the bioactivation and
covalent binding of NVP and the immune response to metabolites of NVP in both NVP-induced
skin rash and hepatotoxicity. The overall hypothesis, restated, is that the NVP-induced toxicity is
mediated by the formation of a reactive metabolite of NVP; in the case of the liver, a quinone
methide species, and in the case of skin, the benzylic sulfate metabolite, which each go on to
covalently modify self-proteins leading to an immune response. In the case of the skin rash, the
findings were much clearer in that the reactive benzylic sulfate was demonstrated to be
responsible for causing the rash. However, the liver proved much more difficult to study because
attempts to develop a suitable animal model failed.
The major objectives of this work focused on delineating the role of covalent adducts in
both NVP-induced liver and skin injury. Concerning the liver, we first wanted to determine if
NVP covalently binds to hepatic proteins from humans, rats, and mice, and further, to determine if
these covalent adducts are involved in hepatotoxicity. To accomplish the latter, it would require
an animal model of NVP-induced liver injury, which previous studies suggested would be
difficult to develop in rats (no liver injury was observed in rats treated with NVP). Therefore,
mice were chosen as a model species, and efforts were made to develop the model in numerous
wild type strains as well as two different knockout strains.
The immediate aim concerning the study of NVP-induced rash was to determine if the 12-
OH-NVP sulfate metabolite covalently binds to cutaneous proteins. This was a huge challenge
because of a large artifact band in the western blot, and students before me were unsuccessful.
Indeed, I spent almost 2 years attempting to accomplish this goal, and it was not until I separated
the epidermis from the dermis that I was successful. Once it was determined that 12-OH-NVP
sulfate was responsible for the covalent binding in the skin, we sought to determine if there was a
direct relationship between the covalent binding and the rash. This work led to the innate sensing
mechanisms of cell damage by the keratinocyte NLRP3 inflammasome, an extension of the core
project, which will be a key future directive. Each of the most meaningful findings from the
aforementioned five overall objectives based on this hypothesis will be addressed here.

150
5.2 Discussion and Limitations
Although direct oxidation and covalent binding of the quinone methide metabolite was
observed in the liver, we were not able to produce a mouse model of NVP-induced liver injury.
This is most likely due to the fact that the dominant response in the liver is immune tolerance, and
despite using knockout mice, which should have impaired immune tolerance, I was not able to
produce an animal model of NVP-induced liver injury similar to the injury that occurs in humans.
In addition, attempts to use immune stimulation via TLR 3 and TLR 7 agonists (poly(I:C) and
imiquimod, respectively) failed, as did attempts to deplete circulating regulatory T-cells through
irradiation studies. These studies demonstrated that the presence of covalent binding is
insufficient to sustain an immune response. The results with other drugs that are known to cause
idiosyncratic liver injury in humans and have been studied in our laboratory have been similar.
This is consistent with the fact that the dominant immune response in the liver is tolerance, and
the majority of patients who develop mild and transient increases in ALT eventually adapt;
however, it is still surprising that nothing seems to work to break the presumed tolerance. The
interplay of a variety of a tolerogenic factors and molecules in the liver must be further
understood if we are to succeed in developing a model of NVP-induced liver injury.
The hepatic studies allowed us to show that direct oxidation of NVP is capable of
producing a reactive quinone methide metabolite, which can adduct hepatic proteins and
expressed P450’s in humans and rodents. We were also able to show binding is associated with an
increase in certain pro-inflammatory cytokines (IL-18 in C57 male mice, unpublished, and IFN-γ,
published74), which precedes immune cell infiltrate in the liver of susceptible mouse strains.
Importantly, we found that 12-OH-NVP and DNVP were not able to produce substantial binding,
giving credence to the direct oxidation of NVP leading to a reactive metabolite. Further work is
however required to produce a valid animal model of liver injury.
The major finding of this thesis is that the NVP-induced skin rash is definitively caused by
a reactive metabolite produced in the skin. This supports the role of reactive metabolites in
inducing IDRs. Earlier studies performed over the years suggested that the sulfate metabolite was
not responsible for the rash. Specifically, the sulfate was found to have very low reactivity
towards model nucleophiles even in the presence of base,11
and in vivo, inhibition of sulfate
formation by depleting the cofactor did not prevent the rash.39
However, at the time these studies
were performed, we had not produced an anti-serum and had no way of identifying what occurred

151
within the skin. This is important for three reasons: first, because the skin is capable of metabolic
biotransformation and possesses the necessary SULT enzyme to form 12-OH-NVP sulfate;
second, because 12-OH-NVP is not reactive and could easily arrive in the skin and undergo
sulfation there; and third, because nucleophilic groups behave very differently when part of an
entire protein molecule. Therefore the physiological relevance had to be examined by examination
of the skin itself.
Numerous methods were attempted to identify covalent adducts in the skin. Whole skin
revealed the presence of an artifact band on the western blot, which interfered with detection of
true binding. When a method was found to separate the dermis and epidermis, the artifact band
was determined to originate from the dermis and the NVP binding was found in the epidermis.
This is not surprising given that the epidermis is highly cellular and composed primarily of
keratinocytes that possess sulfotransferase enzymes. We found in later studies that the 12-OH-
NVP sulfate covalently binds to cultured keratinocytes (unpublished results). This binding may
cause cell damage leading to the activation of innate pattern recognition receptors (PRRs) such as
NOD-like receptors (NLR’s) in the skin; this has become the focus of ongoing studies. An
important question is how do the keratinocytes sense cell stress or damage; is it mediated by
covalent binding or does 12-OH-NVP sulfate directly activate NLR’s such as the NLRP3
inflammasome? In order for the Langerhans cells to migrate to regional lymph nodes, they must
dissociate from suprabasal epidermal layers, and this is suggested to occur through the down
regulation of E-cadherin adhesions. The exact signal(s) sent by keratinocytes in situations of cell
stress are not known and could be useful biomarkers if they could be identified.
The pathological findings in the skin formed an interesting and novel portion of the study.
Sub-maximal epidermal binding was determined to occur by day 10 of NVP treatment, and
correlated with pathological epidermal changes. These changes, observed histologically, occurred
much earlier for 12-OH-NVP than for NVP itself. This supports the finding that 12-OH-NVP
sulfate, formed in the skin, is responsible for the skin rash. The primary epidermal pathology
observed was acanthosis, which is defined as diffuse epidermal hyperplasia. It has been shown
that the cytokine, IL-22, mediates acanthosis in inflammatory skin conditions. IL-22 is a TH17
cytokine that acts through IL-23 to induce dermal inflammation and thickening of the stratum
spinosum.105
The IL-22 cytokine is best characterized in the context of skin inflammation, and its
involvement has been observed in both psoriasis and atopic dermatitis.106,107
Specifically, IL-22

152
functions in epidermal remodeling, keratinocyte proliferation, and hyperplasia.107
IL-22 deficient
mice were shown to produce decreased IL-23-mediated dermal inflammation.105
Additionally,
direct injection of IL-23 into the skin of mice was found to produce IL-22-dependent dermal
inflammation and acanthosis.108
In turn, IL-23 is a key upstream regulator of IL-17, and both
cytokines combined induce inflammatory reactions. IL-22 is increasingly being studied in the
context of epithelial innate immunity, and it appears to be important in very early stages of skin
inflammation. It may be a key cytokine in drug-induced skin rash of NVP and other drugs,
especially in the initial stages initiating skin rash. One limitation of our work was the lack of
broad screening or characterization of early-acting cytokines, which may be important for
induction of the rash. It would be interesting to test the involvement of IL-22, IL-23, or IL-17 in
the pathogenesis of NVP-induced skin rash.
Although both humans and female BN rats develop acanthosis and skin rash in response to
NVP, numerous mouse strains were found not to. We later found that mice simply do not sulfate
the 12-OH-NVP compound. There exists multiple reports of SULT mRNA present in skin of mice
(i.e. 2B1b > 2B1a); however, there is no evidence that these SULT isoforms express similar
substrate specificity to human or rat SULTs found within skin.109
For example, the mouse 2B1b
isoform expresses high catalytic activity towards cholesterol metabolism in skin, but there is no
evidence that its substrate specificity is similar to that of human skin SULT isoforms for other
compounds. Furthermore, another epidermal mouse SULT, St2b2, was found to have the highest
expression level in epidermis from 3-day-old mice, which then decreased before maturation.110
This suggests sulfotransferases may not be widely expressed in older mice; 8 - 10 week old mice
were used in our studies and their age may therefore be a reason for their lack of development of
rash. Additionally this St2b2 isoform in mice is thought to control epidermal cell differentiation
by controlling cholesterol sulfate levels in the cells, but little is known about its ability to
metabolize foreign compounds. Of note is the turnover of keratinocytes in different species.
Humans turnover KCs every 40 – 56 days111
while mice possess a turnover rate of only 8 – 10
days112
; this large difference may also account for lack of rash in mice because they may turn over
the cells and get rid of abnormal, adducted cellular proteins much faster than humans. An
exhaustive PubMed search failed to yield any information about KC turnover rate in rat skin.
In conclusion, the major finding of the work concerning the NVP-induced skin rash is that
12-OH-NVP sulfate, produced in the skin, is responsible for inducing the rash. All three

153
objectives were met: there is clearly covalent binding of a NVP metabolite in the skin as
demonstrated with both in vitro and in vivo studies; determination that 12-OH-NVP sulfate is the
major metabolite responsible for covalent binding in skin; and the skin rash was prevented by
using a topical sulfotransferase inhibitor that also blocked the formation of covalent skin
adducts.74,113
An inhibitor of hepatic sulfation, salicylamide, did not prevent covalent binding in
the skin, presumably because it was unable to deplete PAPS in the epidermis due to slow
turnover, whereas topical inhibition of sulfotransferase by 1-phenyl-1-hexanol was successful in
preventing covalent binding and the rash.113
A 73% reduction in hepatic PAPs was found to occur
2 hours following administration of 2 mmol/kg salicylamide, the same dose used in our work.72 In
addition, SULT enzymes exhibit different organ-specific expression; therefore, the enzyme that
sulfates salicylamide may be absent from the skin. The focus now turns to the response of
keratinocytes to covalent adducts: how do they signal stress, what do they activate, and how does
this, in turn, lead to an immune response, which in rare cases progress to such a severe adverse
reaction as SJS-TEN? The answer to this may lie in the inflammasome, discussed in the next
section.
5.3 Future Directions
This project has opened an entirely new area for the study of drug-induced skin rashes.
Sulfotransferase enzymes are unique in being one of the few drug metabolizing enzymes that has
significant activity in the skin (other enzymes such as flavin monoxygenases may also be
important).71
Our group was the first to show that a reactive sulfate formed within the skin can
cause a skin rash. We are also the first group to study the early immune response to covalent
epidermal adducts, specifically how this early immune activation could lead to the ultimate rash.
It is likely that there are a variety of other drugs that can form reactive benzylic sulfates in the
skin and cause a rash through this mechanism. Trimethoprim, a bacteriostatic antibiotic, is one
example that will be studied in the future by the Uetrecht group. Sulfation is also a high affinity,
low capacity pathway, and sulfation reactions predominate at lower concentrations. Because the
doses used in humans are typically far lower than in animals, sulfation represents an important
route of both clearance and bioactivation in humans. Although sulfation has been studied as a
route of drug clearance, this is primarily in the context of hepatic metabolism, and its role in skin
metabolism may have been overlooked. While we do not have concrete data to suggest sulfation

154
is significant in the clearance of drugs in the skin, KCs are rich in this enzyme class. In addition,
many new drug compounds are being developed that have greater efficacy at lower dosages;
sulfation may become an important route of metabolism for these drugs as well and could be
studied as part of a future project.
It appears that an essential part of the mechanism of the NVP-induced skin rash involves
activation of inflammasomes by the reactive sulfate metabolite. We have obtained preliminary
data showing an increase in IL-1β in both the dermis and epidermis of rats that develop a skin
rash. We have also been able to show that blocking IL-1β via the IL-1 receptor antagonist
Anakinra blocks the development of full skin rash despite the presence of epidermal adducts
(manuscript in preparation). This has provided a sound basis for pursuing the NLRP3
inflammasome studies, which we are currently pursuing using human keratinocytes. Other drugs
causing skin rash that are intrinsically reactive such as penicillin and telaprevir may directly
activate inflammasomes, and this may be the mechanism by which they cause skin rashes. If this
is true, it may represent a way to screen some types of drug candidates for their potential to cause
skin rashes. In fact, drugs that work by irreversibly binding to targets are becoming increasingly
popular, and many of these drugs are associated with a high incidence of skin rash. If they also
covalently bind in the skin, this may be responsible for activating NLRP3 and induction of skin
rash. This will be examined in the future by the Uetrecht group.
Inflammasome complexes encompass a wide variety of multimeric protein forms and their
expression differs in various organs. Although NLR expression in the liver is in fact significantly
different than in the skin, this may represent a general mechanism of IDR induction by a
mechanism for innate immune instruction of adaptive immunity. Numerous cells of the body are
known to express various inflammasome machinery (neutrophils, monocyte-derived cells,
macrophages, Kupffer cells, etc.), and a number of these cells have been shown to be involved in
idiosyncratic drug reactions (agranulocytosis, drug-induced liver injury, etc.). Thus the study of
the inflammasome and innate immune sensing in NVP-induced skin rash extends far beyond the
NVP model itself. Our ultimate goal is to use the study of the NLRP3 found in keratinocytes as a
model system to develop biomarkers for other cell types involved in various idiosyncratic drug
reactions. The keratinocyte work may allow us to develop screening tools or protocols to test for
early immune activation that is applicable to several other organ-specific toxicities. If our
hypothesis involving activation of the NLRP3 inflammasome leading to skin rash is supported by

155
future studies, we will attempt to produce a method to screen other drugs that are capable of
haptenating skin proteins for their ability to up-regulate IL-1β and IL-18.
Additionally, severe forms of skin rash (Stevens Johnson Syndrome or toxic epidermal
necrolysis) involve the release of granzyme A/porin, which leads to detachment of skin.
Granzyme A has been shown capable of activating IL-1β to its mature form via cleavage of its
pro-peptide. Our work will aid in the further study of IL-1β and its role in drug-induced
detachment of skin. This relationship of granzyme A and IL-1β has never before been examined
in the context of drug-induced skin rash in response to cutaneous covalent adducts, and it is hoped
that our work will open a door to study this important relationship.
Implications of this research extend beyond the NVP-induced IDR. Our skin rash model
has allowed us to test not only specific, but also general hypotheses of idiosyncratic drug
reactions, which would otherwise be impossible to test any other way. These general findings may
eventually be applicable to other type B adverse toxicities, ultimately leading to production of
safer drugs.

156
Bibliography
(1) Lazarou, J., Pomeranz, B. H., and Corey, P. N. (1998) Incidence of adverse drug
reactions in hospitalized patients: A meta-analysis of prospective studies. JAMA 279,
1200-1205.
(2) Lasser, K. E., Allen, P. D., Woolhandler, S. J., Himmelstein, D. U., Wolfe, S. M., and
Bor, D. H. (2002) Timing of new black box warnings and withdrawals for prescription
medications. JAMA 287, 2215-2220.
(3) Edwards, I. R., and Aronson, J. K. (2000) Adverse drug reactions: definitions,
diagnosis, and management. Lancet 356, 1255-1259.
(4) Rawlins, M., and Thompson, J. Textbook of adverse drug reactions; Oxford
University Press: Oxford, 1991.
(5) Jick, H. (1984) Adverse drug reactions: the magnitude of the problem. J Allergy Clin
Immunol 74, 555-557.
(6) Uetrecht, J. (2007) Idiosyncratic drug reactions: current understanding. Annu Rev
Pharmacol Toxicol 47, 513-539.
(7) Uetrecht, J., and Naisbitt, D. J. (2013) Idiosyncratic adverse drug reactions: current
concepts. Pharmacol Rev 65, 779-808.
(8) Park, B., Pirmohamed, M., and Kitteringham, N. (1998) Role of drug disposition in
drug hypersensitivity: a chemical, molecular, and clinical perspective. Chemical Research
in Toxicology 11, 969.
(9) Roujeau, J. C., and Stern, R. S. (1994) Severe Adverse Cutaneous Reactions to Drugs.
New England Journal of Medicine 331, 1272-1285.
(10) Yawalkar, N., Egli, F., Hari, Y., Nievergelt, H., Braathen, L., and Pichler, W. J.
(2000) Infiltration of cytotoxic T cells in drug-induced cutaneous eruptions. Clinical &
Experimental Allergy 30, 847-855.
(11) Mannargudi, M. B., University of Toronto, 2009.
(12) Moeschlin, S., and Wagner, K. (1952) Agranulocytosis due to the Occurrence of
Leukocyte-Agglutinins. Acta Haematol 8, 29–41.
(13) Gardner, I., Leeder, J. S., Chin, T., Zahid, N., and Uetrecht, J. P. (1998) A
comparison of the covalent binding of clozapine and olanzapine to human neutrophils in
vitro and in vivo. Mol Pharmacol 53, 999-1008.
(14) Marchesi, C., Paini, M., Tamborini, S., Ampollini, P., and Maggini, C. Recurrence of
clozapine-induced agranulocytosis; J Clin Psychopharmacol. 2005 Jun;25(3):276-7.
(15) Uetrecht, J. P. (1992) The role of leukocyte-generated reactive metabolites in the
pathogenesis of idiosyncratic drug reactions. Drug Metab Rev 24, 299-366.
(16) Uetrecht, J. (2007) Idiosyncratic Drug Reactions: Past, Present, and Future. Chem
Res Toxicol 21, 84-92.
(17) Kitteringham, N. R., Kenna, J. G., and Park, B. K. (1995) Detection of
autoantibodies directed against human hepatic endoplasmic reticulum in sera from patients
with halothane-associated hepatitis. Br J Clin Pharmacol 40, 379-386.
(18) Uetrecht, J. (2008) Immune-Mediated Adverse Drug Reactions. Chem Res Toxicol
22, 24-34.
(19) Mallal, S., Nolan, D., Witt, C., Masel, G., Martin, A. M., Moore, C., Sayer, D.,
Castley, A., Mamotte, C., Maxwell, D., James, I., and Christiansen, F. T. (2002)

157
Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and
hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. The Lancet 359, 727-
732.
(20) Martin, A. M., Nolan, D., James, I., Cameron, P., Keller, J., Moore, C., Phillips, E.,
Christiansen, F. T., and Mallal, S. (2005) Predisposition to nevirapine hypersensitivity
associated with HLA-DRB1*0101 and abrogated by low CD4 T-cell counts. Aids 19, 97-
99.
(21) Martin, A., Nolan, D., Almeida, C. A., Rauch, A., and Mallal, S. (2006) Predicting
and diagnosing abacavir and nevirapine drug hypersensitivity: from bedside to bench and
back again. Pharmacogenomics 7, 15-23.
(22) Landsteiner, K., and Jacobs, J. (1936) Studies on the sensitization of animals with
simple chemical compounds. II. . The Journal of Experimental Medicine 64, 625-639.
(23) Chipinda, I., Hettick, J. M., and Siegel, P. D. (2011) Haptenation: Chemical
Reactivity and Protein Binding. Journal of Allergy 2011.
(24) Vergani, D., Mieli-Vergani, G., Alberti, A., Neuberger, J., Eddleston, A. L., Davis,
M., and Williams, R. (1980) Antibodies to the surface of halothane-altered rabbit
hepatocytes in patients with severe halothane-associated hepatitis. N Engl J Med 303, 66-
71.
(25) Lecoeur, S., André, C., and Beaune, P. H. (1996) Tienilic acid-induced autoimmune
hepatitis: anti-liver and-kidney microsomal type 2 autoantibodies recognize a three-site
conformational epitope on cytochrome P4502C9. Molecular Pharmacology 50, 326-333.
(26) Matzinger, P. (1994) Tolerance, danger, and the extended family. Annu Rev Immunol
12, 991-1045.
(27) Seguin, B., and Uetrecht, J. (2003) The danger hypothesis applied to idiosyncratic
drug reactions. Curr Opin Allergy Clin Immunol 3, 235-242.
(28) Li, J., and Uetrecht, J. P. (2010) The danger hypothesis applied to idiosyncratic drug
reactions. Handb Exp Pharmacol 196, 493-509.
(29) Zanni, M. P., von Greyerz, S., Schnyder, B., Brander, K. A., Frutig, K., Hari, Y.,
Valitutti, S., and Pichler, W. J. (1998) HLA-restricted, processing-and metabolism-
independent pathway of drug recognition by human alpha beta T lymphocytes. Journal of
Clinical Investigation 102, 1591.
(30) Gerber, B. O., and Pichler, W. J. (2006) Noncovalent interactions of drugs with
immune receptors may mediate drug-induced hypersensitivity reactions. The AAPS
journal 8, E160-E165.
(31) Schnyder, B., Burkhart, C., Schnyder-Frutig, K., von Greyerz, S., Naisbitt, D. J.,
Pirmohamed, M., Park, B. K., and Pichler, W. J. (2000) Recognition of Sulfamethoxazole
and Its Reactive Metabolites by Drug-Specific CD4+ T Cells from Allergic Individuals.
The Journal of Immunology 164, 6647-6654.
(32) Lavergne, S. N., Whitaker, P., Peckham, D., Conway, S., Park, B. K., and Naisbitt,
D. J. (2010) Drug metabolite-specific lymphocyte responses in sulfamethoxazole allergic
patients with cystic fibrosis. Chem Res Toxicol 23, 1009-1011.
(33) Kindmark, A., Jawaid, A., Harbron, C. G., Barratt, B. J., Bengtsson, O. F.,
Andersson, T. B., Carlsson, S., Cederbrant, K. E., Gibson, N. J., Armstrong, M.,
Lagerstrom-Fermer, M. E., Dellsen, A., Brown, E. M., Thornton, M., Dukes, C., Jenkins,
S. C., Firth, M. A., Harrod, G. O., Pinel, T. H., Billing-Clason, S. M. E., Cardon, L. R.,
and March, R. E. (2007) Genome-wide pharmacogenetic investigation of a hepatic adverse

158
event without clinical signs of immunopathology suggests an underlying immune
pathogenesis. Pharmacogenomics J 8, 186-195.
(34) Klaassen, C. D. Toxicology The Basic Science of Poisons; 7th ed.; McGraw-Hill
Companies, 2008.
(35) Nelson, D. R. (2003) Comparison of P450s from human and fugu: 420 million years
of vertebrate P450 evolution. Archives of Biochemistry and Biophysics 409, 18-24.
(36) Guengerich, F. P. (2006) Cytochrome P450s and other enzymes in drug metabolism
and toxicity. Aaps J 8, E101-111.
(37) Josephy, P. D., and Mannervik, B.; Second ed.; Oxford University Press, 2006.
(38) Uetrecht, J., and Trager, W. Drug Metabolism Chemical and Enzymatic Aspects
Informa Healthcare, 2007.
(39) Novalen, M., University of Toronto, 2010.
(40) Kaplowitz, N. (2004) Drug-Induced Liver Injury. Clinical Infectious Diseases 38,
S44-S48.
(41) Lau, A. H., de Creus, A., Lu, L., and Thomson, A. W. (2003) Liver tolerance
mediated by antigen presenting cells: fact or fiction? Gut 52, 1075-1078.
(42) Gimmi, C. D., Freeman, G. J., Gribben, J. G., Gray, G., and Nadler, L. M. (1993)
Human T-cell clonal anergy is induced by antigen presentation in the absence of B7
costimulation. Proc Natl Acad Sci U S A 90, 6586-6590.
(43) Holt, M. P., and Ju, C. (2006) Mechanisms of drug-induced liver injury. AAPS J 8,
E48-54.
(44) Chan, L. S. Animal Models of Human Inflammatory Skin Diseases; CRC Press LLC,
2004.
(45) Svensson, C. K. (2009) Biotransformation of drugs in human skin. Drug Metab
Dispos 37, 247-253.
(46) Gutowska-Owsiak, D., and Ogg, G. S. (2012) The epidermis as an adjuvant. J Invest
Dermatol 132, 940-948.
(47) Nestle, F. O., Di Meglio, P., Qin, J.-Z., and Nickoloff, B. J. (2009) Skin immune
sentinels in health and disease. Nat Rev Immunol 9, 679-691.
(48) Schenten, D., and Medzhitov, R. (2011) The control of adaptive immune responses
by the innate immune system. Adv Immunol 109, 87-124.
(49) Watanabe, H., Gaide, O., Petrilli, V., Martinon, F., Contassot, E., Roques, S.,
Kummer, J. A., Tschopp, J., and French, L. E. (2007) Activation of the IL-1beta-
processing inflammasome is involved in contact hypersensitivity. J Invest Dermatol 127,
1956-1963.
(50) Gutowska-Owsiak, D., and Ogg, G. S. (2012) The Epidermis as an Adjuvant. J
Invest Dermatol 132, 940-948.
(51) Wang, B., Amerio, P., and Sauder, D. N. (1999) Role of cytokines in epidermal
Langerhans cell migration. J Leukoc Biol 66, 33-39.
(52) Feldmeyer, L., Werner, S., French, L. E., and Beer, H.-D. (2010) Interleukin-1,
inflammasomes and the skin. European Journal of Cell Biology 89, 638-644.
(53) Nickoloff, B. J. (2008) Saving the skin from drug-induced detachment. Nat Med 14,
1311-1313.
(54) Chung, W. H., Hung, S. I., Yang, J. Y., Su, S. C., Huang, S. P., Wei, C. Y., Chin, S.
W., Chiou, C. C., Chu, S. C., Ho, H. C., Yang, C. H., Lu, C. F., Wu, J. Y., Liao, Y. D., and
Chen, Y. T. (2008) Granulysin is a key mediator for disseminated keratinocyte death in
Stevens-Johnson syndrome and toxic epidermal necrolysis. Nat Med 14, 1343-1350.

159
(55) Usui, T., Mise, M., Hashizume, T., Yabuki, M., and Komuro, S. (2009) Evaluation of
the Potential for Drug-Induced Liver Injury Based on in Vitro Covalent Binding to Human
Liver Proteins. Drug Metabolism and Disposition 37, 2383-2392.
(56) Oesch, F., Fabian, E., Oesch-Bartlomowicz, B., Werner, C., and Landsiedel, R.
(2007) Drug-Metabolizing Enzymes in the Skin of Man, Rat, and Pig. Drug Metabolism
Reviews 39, 659-698.
(57) Chapman, E., Best, M. D., Hanson, S. R., and Wong, C. H. (2004) Sulfotransferases:
structure, mechanism, biological activity, inhibition, and synthetic utility. Angew Chem Int
Ed Engl 43, 3526-3548.
(58) Funk, C., Pantze, M., Jehle, L., Ponelle, C., Scheuermann, G., Lazendic, M., and
Gasser, R. (2001) Troglitazone-induced intrahepatic cholestasis by an interference with
the hepatobiliary export of bile acids in male and female rats. Correlation with the gender
difference in troglitazone sulfate formation and the inhibition of the canalicular bile salt
export pump (Bsep) by troglitazone and troglitazone sulfate. Toxicology 167, 83-98.
(59) Pollard, R. B., Robinson, P., and Dransfield, K. (1998) Safety profile of nevirapine, a
nonnucleoside reverse transcriptase inhibitor for the treatment of human
immunodeficiency virus infection. Clinical Therapeutics 20, 1071-1092.
(60) Hall, D. B., and Macgregor, T. R. (2007) Case-control exploration of relationships
between early rash or liver toxicity and plasma concentrations of nevirapine and primary
metabolites. HIV Clin Trials 8, 391-399.
(61) Patel, S. M., Johnson, S., Belknap, S. M., Chan, J., Sha, B. E., and Bennett, C. (2004)
Serious Adverse Cutaneous and Hepatic Toxicities Associated With Nevirapine Use by
Non-HIV-Infected Individuals. JAIDS Journal of Acquired Immune Deficiency Syndromes
35, 120-125.
(62) Yuan, J., Guo, S., Hall, D., Cammett, A. M., Jayadev, S., Distel, M., Storfer, S.,
Huang, Z., Mootsikapun, P., Ruxrungtham, K., Podzamczer, D., and Haas, D. W. (2011)
Toxicogenomics of nevirapine-associated cutaneous and hepatic adverse events among
populations of African, Asian, and European descent. Aids 25, 1271-1280.
(63) Mallal, S., Nolan, D., Witt, C., Masel, G., Martin, A. M., Moore, C., Sayer, D.,
Castley, A., Mamotte, C., Maxwell, D., James, I., and Christiansen, F. T. (2002)
Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and
hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet 359, 727-732.
(64) Popovic, M., Shenton, J., Chen, J., Baban, A., Tharmanathan, T., Mannargudi, B.,
Abdulla, D., and Uetrecht, J. In Adverse Drug Reactions; Springer, 2010.
(65) Shenton, J. M., Teranishi, M., Abu-Asab, M. S., Yager, J. A., and Uetrecht, J. P.
(2003) Characterization of a potential animal model of an idiosyncratic drug reaction:
nevirapine-induced skin rash in the rat. Chem Res Toxicol 16, 1078-1089.
(66) Shenton, J. M., Popovic, M., Chen, J., Masson, M. J., and Uetrecht, J. P. (2005)
Evidence of an immune-mediated mechanism for an idiosyncratic nevirapine-induced
reaction in the female Brown Norway rat. Chem Res Toxicol 18, 1799-1813.
(67) Havlir, D., Cheeseman, S. H., Mclaughlin, M., Murphy, R., Erice, A., Spector, S. A.,
Greenough, T. C., Sullivan, J. L., Hall, D., Myers, M., Lamson, M., and Richman, D. D.
(1995) High-Dose Nevirapine: Safety, Pharmacokinetics, And Antiviral Effect In Patients
With Human Immunodeficiency Virus Infection. Journal of Infectious Diseases 171, 537-
545.

160
(68) Chen, J., Mannargudi, B. M., Xu, L., and Uetrecht, J. (2008) Demonstration of the
metabolic pathway responsible for nevirapine-induced skin rash. Chem Res Toxicol 21,
1862-1870.
(69) Riska, P., Lamson, M., MacGregor, T., Sabo, J., Hattox, S., Pav, J., and Keirns, J.
(1999) Disposition and biotransformation of the antiretroviral drug nevirapine in humans.
Drug Metab Dispos 27, 895-901.
(70) Merk, H. F. (2009) Drug skin metabolites and allergic drug reactions. Curr Opin
Allergy Clin Immunol 9, 311-315.
(71) Svensson, C. K. (2009) Biotransformation of Drugs in Human Skin. Drug
Metabolism and Disposition 37, 247-253.
(72) Kim, H. J., Cho, J. H., and Klaassen, C. D. (1995) Depletion of hepatic 3'-
phosphoadenosine 5'-phosphosulfate (PAPS) and sulfate in rats by xenobiotics that are
sulfated. Journal of Pharmacology and Experimental Therapeutics 275, 654-658.
(73) Oguro, T., Gregus, Z., Madhu, C., Liu, L., and Klaassen, C. D. (1994) Molybdate
depletes hepatic 3-phosphoadenosine 5-phosphosulfate and impairs the sulfation of
acetaminophen in rats. J Pharmacol Exp Ther 270, 1145-1151.
(74) Sharma, A. M., Li, Y., Novalen, M., Hayes, M. A., and Uetrecht, J. (2012)
Bioactivation of Nevirapine to a Reactive Quinone Methide: Implications for Liver Injury.
Chem Res Toxicol 25, 1708-1719.
(75) Mueller, D. L. (2004) E3 ubiquitin ligases as T cell anergy factors. Nat Immunol 5,
883-890.
(76) Sharpe, A. H., Wherry, E. J., Ahmed, R., and Freeman, G. J. (2007) The function of
programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat
Immunol 8, 239-245.
(77) MacGregor, T. R., and Hall, D. B. (2007) Case-control exploration of relationships
between early rash or liver toxicity and plasma concentrations of nevirapine and primary
metabolites. HIV Clin. Trials 8, 391-399.
(78) Wen, B., Chen, Y., and Fitch, W. L. (2009) Metabolic activation of nevirapine in
human liver microsomes: dehydrogenation and inactivation of cytochrome P450 3A4.
Drug Metab Dispos 37, 1557-1562.
(79) Srivastava, A., Lian, L.-Y., Maggs, J. L., Chaponda, M., Pirmohamed, M., Williams,
D. P., and Park, B. K. (2010) Quantifying the Metabolic Activation of Nevirapine in
Patients by Integrated Applications of NMR and Mass Spectrometries. Drug Metabolism
and Disposition 38, 122-132.
(80) Zimmermann, C., van Waterschoot, R. A. B., Harmsen, S., Maier, A., Gutmann, H.,
and Schinkel, A. H. (2009) PXR-mediated induction of human CYP3A4 and mouse
Cyp3a11 by the glucocorticoid budesonide. European Journal of Pharmaceutical Sciences
36, 565-571.
(81) Grozinger, K. G., Fuchs, V., Hargrave, K. D., Mauldin, S., Vitous, J., Campbell, S.,
and Adams, J. (1995) Synthesis of nevirapine and its major metabolite. Journal of
Heterocyclic Chemistry 32, 259-263.
(82) Kelly, T. A., Proudfoot, J. R., McNeil, D. W., Patel, U. R., David, E., Hargrave, K.
D., Grob, P. M., Cardozo, M., Agarwal, A., and Adams, J. (1995) Novel Non-nucleoside
Inhibitors of Human Immunodeficiency Virus Type 1 Reverse Transcriptase. 5. 4-
Substituted and 2,4-Disubstituted Analogs of Nevirapine. J Med Chem 38, 4839-4847.

161
(83) Chauret, N., Gauthier, A., and Nicoll-Griffith, D. A. (1998) Effect of Common
Organic Solvents on in Vitro Cytochrome P450-Mediated Metabolic Activities in Human
Liver Microsomes. Drug Metabolism and Disposition 26, 1-4.
(84) Reid, M. J., Lamé, M. W., Morin, D., Wilson, D. W., and Segall, H. J. (1998)
Involvement of cytochrome P450 3A in the metabolism and covalent binding of 14C-
monocrotaline in rat liver microsomes. Journal of Biochemical and Molecular Toxicology
12, 157-166.
(85) Omiecinski, C. J., Hassett, C., and Costa, P. (1990) Developmental expression and in
situ localization of the phenobarbital-inducible rat hepatic mRNAs for cytochromes
CYP2B1, CYP2B2, CYP2C6, and CYP3A1. Molecular Pharmacology 38, 462-470.
(86) Pampori, N. A., and Shapiro, B. H. (1994) Over-expression of CYP2C11, the major
male-specific form of hepatic cytochrome P450, in the presence of nominal pulses of
circulating growth hormone in adult male rats neonatally exposed to low levels of
monosodium glutamate. Journal of Pharmacology and Experimental Therapeutics 271,
1067-1073.
(87) Kato, R., and Yamazoe, Y. (1992) Sex-specific cytochrome P450 as a cause of sex-
and species-related differences in drug toxicity. Toxicology Letters 64-65, 661-667.
(88) Yano, J. K., Wester, M. R., Schoch, G. A., Griffin, K. J., Stout, C. D., and Johnson,
E. F. (2004) The Structure of Human Microsomal Cytochrome P450 3A4 Determined by
X-ray Crystallography to 2.05-A Resolution. J. Biol. Chem. 279, 38091-38094.
(89) Sharma, A. M., Klarskov, K., and Uetrecht, J. (2013) Nevirapine Bioactivation and
Covalent Binding in the Skin. Chemical Research in Toxicology 26, 410-421.
(90) Keane, N., et al. Proceedings of the 4th International AIDS Society Conference on
HIV Pathogenesis, Treatment and Prevention, Sydney, Australia, 2007.
(91) Shenton, J. M., Chen, J., and Uetrecht, J. P. (2004) Animal models of idiosyncratic
drug reactions. Chemico-biological interactions 150, 53-70.
(92) Bersoff-Matcha, S. J., Miller, W. C., Aberg, J. A., van der Horst, C., Hamrick, H. J.,
Powderly, W. G., and Mundy, L. M. (2001) Sex Differences in Nevirapine Rash. Clinical
Infectious Diseases 32, 124-129.
(93) Chen, X., Tharmanathan, T., Mannargudi, B., Gou, H., and Uetrecht, J. P. (2009) A
study of the specificity of lymphocytes in nevirapine-induced skin rash. J Pharmacol Exp
Ther 331, 836-841.
(94) Banoglu, E., and Duffel, M. W. (1997) Studies on the Interactions of Chiral
Secondary Alcohols with Rat Hydroxysteroid Sulfotransferase STa. Drug Metabolism and
Disposition 25, 1304-1310.
(95) Oesch, F., Fabian, E., Oesch-Bartlomowicz, B., Werner, C., and Landsiedel, R.
(2007) Drug-metabolizing enzymes in the skin of man, rat, and pig. Drug Metab Rev 39,
659-698.
(96) Lichti, U., Anders, J., and Yuspa, S. H. (2008) Isolation and short-term culture of
primary keratinocytes, hair follicle populations and dermal cells from newborn mice and
keratinocytes from adult mice for in vitro analysis and for grafting to immunodeficient
mice. Nat. Protocols 3, 799-810.
(97) Pichler, W. J. (2003) Drug-induced autoimmunity. Current Opinion in Allergy and
Clinical Immunology 3, 249-253.
(98) “Divisions of AIDS Table for Grading Severity of Adult Adverse Experiences. AIDS
Clinical Trial Group Protocol Management Handbook. Divisions of AIDS (DAIDS)
Regulatory Operations Center (ROC) Titled Serious Adverse Experience (SAE) Reporting

162
Manual for: AIDS Clinical Trail Groups (AACTG and PACTG), Community Programs
for Clinical Research on AIDS (CPCRA), Intramural Research Programs (IRP). ,” Frontier
Science, 1998.
(99) Park, K., Williams, D. P., Naisbitt, D. J., Kitteringham, N. R., and Pirmohamed, M.
(2005) Investigation of toxic metabolites during drug development. Toxicology and
Applied Pharmacology 207, 425-434.
(100) Kalgutkar, A. S., Gardner, I., Obach, R. S., Shaffer, C. L., Callegari, E., Henne, K.
R., Mutlib, A. E., Dalvie, D. K., Lee, J. S., Nakai, Y., O'Donnell, J. P., Boer, J., and
Harriman, S. P. (2005) A comprehensive listing of bioactivation pathways of organic
functional groups. Curr Drug Metab 6, 161-225.
(101) Pichler, W. J. (2002) Pharmacological interaction of drugs with antigen-specific
immune receptors: the p-i concept. Current Opinion in Allergy and Clinical Immunology
2, 301-305.
(102) Pavek, P., and Dvorak, Z. (2008) Xenobiotic-induced transcriptional regulation of
xenobiotic metabolizing enzymes of the cytochrome P450 superfamily in human
extrahepatic tissues. Curr Drug Metab 9, 129-143.
(103) Nagar, S., Walther, S., and Blanchard, R. L. (2006) Sulfotransferase (SULT) 1A1
Polymorphic Variants *1, *2, and *3 Are Associated with Altered Enzymatic Activity,
Cellular Phenotype, and Protein Degradation. Molecular Pharmacology 69, 2084-2092.
(104) You, Q., Cheng, L., Kedl, R. M., and Ju, C. (2008) Mechanism of T cell tolerance
induction by murine hepatic Kupffer cells. Hepatology 48, 978-990.
(105) Zheng, Y., Danilenko, D. M., Valdez, P., Kasman, I., Eastham-Anderson, J., Wu, J.,
and Ouyang, W. (2007) Interleukin-22, a TH17 cytokine, mediates IL-23-induced dermal
inflammation and acanthosis. Nature 445, 648-651.
(106) Bernard, F., #xe7, ois-Xavier, Morel, F., Camus, M., Pedretti, N., Barrault, C.,
Garnier, J., and Lecron, J.-C. (2012) Keratinocytes under Fire of Proinflammatory
Cytokines: Bona Fide Innate Immune Cells Involved in the Physiopathology of Chronic
Atopic Dermatitis and Psoriasis. Journal of Allergy 2012, 10.
(107) Zenewicz, L. A., and Flavell, R. A. (2011) Recent advances in IL-22 biology.
International Immunology 23, 159-163.
(108) Bromley, S. K., Larson, R. P., Ziegler, S. F., and Luster, A. D. (2013) IL-23 induces
atopic dermatitis-like inflammation instead of psoriasis-like inflammation in CCR2-
deficient mice. PLoS One 8, 5.
(109) Shimizu, C., Fuda, H., Yanai, H., and Strott, C. A. (2003) Conservation of the
Hydroxysteroid Sulfotransferase SULT2B1 Gene Structure in the Mouse: Pre- and
Postnatal Expression, Kinetic Analysis of Isoforms, and Comparison with Prototypical
SULT2A1. Endocrinology 144, 1186-1193.
(110) Shimada, M., Matsuda, T., Sato, A., Akase, T., Matsubara, T., Nagata, K., and
Yamazoe, Y. (2008) Expression of a skin cholesterol sulfotransferase, St2b2, is a trigger
of epidermal cell differentiation. Xenobiotica 38, 1487-1499.
(111) Halprin, K. M. (1972) Epidermal "turnover time"--a re-examination. Br J Dermatol
86, 14-19.
(112) Potten, C. S., Saffhill, R., and Maibach, H. I. (1987) Measurement of the transit
time for cells through the epidermis and stratum corneum of the mouse and guinea-pig.
Cell Tissue Kinet 20, 461-472.

163
(113) Sharma, A. M., Novalen, M., Tanino, T., and Uetrecht, J. P. (2013) 12-OH-
Nevirapine Sulfate, Formed in the Skin, Is Responsible for Nevirapine-Induced Skin Rash.
Chem Res Toxicol 26, 817-827.

164
‘I have no special talent. I am only passionately curious’
- Albert Einstein