introduction - inflibnetshodhganga.inflibnet.ac.in/bitstream/10603/13077/7/07_chapter...
TRANSCRIPT

Introduction
Page 1
1.1 Introduction
Catalysis plays a vital role in providing a society with fuels, commodity and fine
chemicals, pharmaceuticals and means to protect the environment. It is clear that catalysis
has a multidimensional impact on society. The chemical industry largely depends on
catalysis and it is estimated that 80% of the industrial chemical processes in the 21st
century will be based on catalytic processes [1].
Catalysis is a phenomenon by which chemical reactions are accelerated by small quantity
of foreign substances called catalysts. A catalyst is a surface active material i.e. catalysis
occurs at the surface of the material and hence the activity of the catalyst depends very
much on the nature of its surface. Recognizing the exact nature of these surface species
and fine tuning them for still better catalytic performance are the main objectives of
catalyst research.
There are different types of catalysts. They range from a proton, H+ through Lewis acids,
organometallic complexes, organic and inorganic polymers, all the way to enzymes.
Catalysts are divided into three categories: Bio(Enzyme) catalysis, Homogeneous and
Heterogeneous catalysis [2-3].
In biocatalysis enzymes or microorganisms catalyze various biochemical reactions.
Prominent examples of biochemical reactions are isomerization of glucose to fructose by
using enzymes such as glucoamylase immobilized on silicon dioxide. Conversion of
acrylonitrile to acrylamide by coryne bacteria entrapped in polyacrylamide gel. The
classic dupont route to adipic acid and frosts biosynthetic route is followed using a
genetically modified E.Coli cell.
A reaction is called homogeneously catalyzed when the catalysts and the reactants or the
solution form a same physical phase. Typical examples of homogeneous catalysts and the
reaction catalyzed by them are as given below.
Homogeneous catalysts Reactions catalyzed
Organometallic complexes Wilkinson olefin polymerization catalyst
Carbonyls of Fe, Co and Rh Hydroformylation of olefins to corresponding

Introduction
Page 2
aldehydes by carbonyls of Co or Rh.
Metal salts of organic acids Oxidation of toluene to benzoic acid in presence
of Co and Mn benzoates,
Heterogneous catalysis
Heterogeneous catalysis involves systems in which catalyst and reactants form separate
physical phases. In many instances the catalysts are in solid phase whereas, the reactants
are either in vapor or liquid phase. Examples of major industrial process using
heterogeneous catalysts are given below:
Heterogeneous catalysts Reactions catalyzed
Metals Polymerization of olefins by Ti- Zeigler-Natta catalyst
Metals oxides Oxidation of xylene to phthalic acids by Vanadium
oxide catalyst
Supported metal/metal oxides Hydrogenation of propene to propane in the presence of
supported metal catalysts.
Dehydrogenation of alkanes to alkenes by Pt/Al2O3
catalyst.
Zeolites Isomerization of xylenes and toluene’s to p-xylenes by
H-ZSM-5 zeolite.
Clays Cracking of long chain alkanes to alkanes and
alkylation of C3-C5 alkanes to C7-C9 isoalkanes using
clays
The special categories of heterogeneous catalysis are photo catalysis, electro catalysis
and environmental catalysis. In photo catalysis, light is adsorbed by the catalysis or a
reactant during a reaction. One example is utilization of semiconductor catalysts
(titanium, zinc and iron oxides) for photochemical degradation of organic substances on
self-cleaning surfaces. The main aim of environmental catalysis is environment
protection. Examples are reduction of NOx in stack gases with ammonia on V2O5-TiO2
catalyst and the removal of NOx, CO and hydrocarbons from automobile exhaust gases
by using the so called three- way-catalysts consisting of Rh-Pt-CeO2-Al2O3 deposited on

Introduction
Page 3
ceramic honey combs. Electro catalysis involves oxidation/reduction by transfer of
electrons. Example: the use of catalytically active electrodes in electrolysis processes
such as chlor-alkali electrolysis and in fuel cells [2].
One of the key objectives of green chemistry is the waste minimization. Further a
sustainable process is one that optimizes the use of resources, while still leaving
sufficient resources for future generations. Heterogeneous catalysis is an important tool in
both the cases. In fact, as far as chemistry is concerned heterogeneous catalysis is a key to
sustainability.
1.1.1 Heterogeneous catalysts
The important categories of heterogeneous catalysts, also known as solid catalysts are
metal oxides, supported metal oxides, zeolites, clays, AlPO4 [4-9]. These materials may
be acidic/basic or oxidation/ reduction properties which catalyze a number of organic
transformations. Thus, in principle by suitable selection of a solid catalyst any type of
organic transformation can be catalyzed for the production of bulk and fine chemicals.
Heterogeneous metal catalysis is highlighted, in particular those associated with the
designing and characterization of catalytic materials, the surface science of catalysis,
catalyst testing and green chemistry.
The efficient use of heterogeneous solid catalysts offers many advantages over
conventional homogeneous liquid catalysts. Product isolation is simplified and reactions
often run under milder conditions and give higher selectivity. The atom efficiency of the
reaction is improved, the processes is simplified, precious raw materials used in the
manufacturing of the catalyst are given increased lifetime (through reuse) and the volume
of waste is significantly reduced [10, 11].
1.1.2 Solid acid catalysts
Metal oxides, supported metal oxides, zeolites, AlPO4 possess surface acid sites, and
hence are known as solid acids. The acid sites may be Bronsted or Lewis types. The
concentration and strength of these acid sites depend on texture and structure of the
Introduction
Page 4
solidacids which intern depend on their pre and post synthetic modifications. Types of
solid acids and examples are shown in Table 1[12].
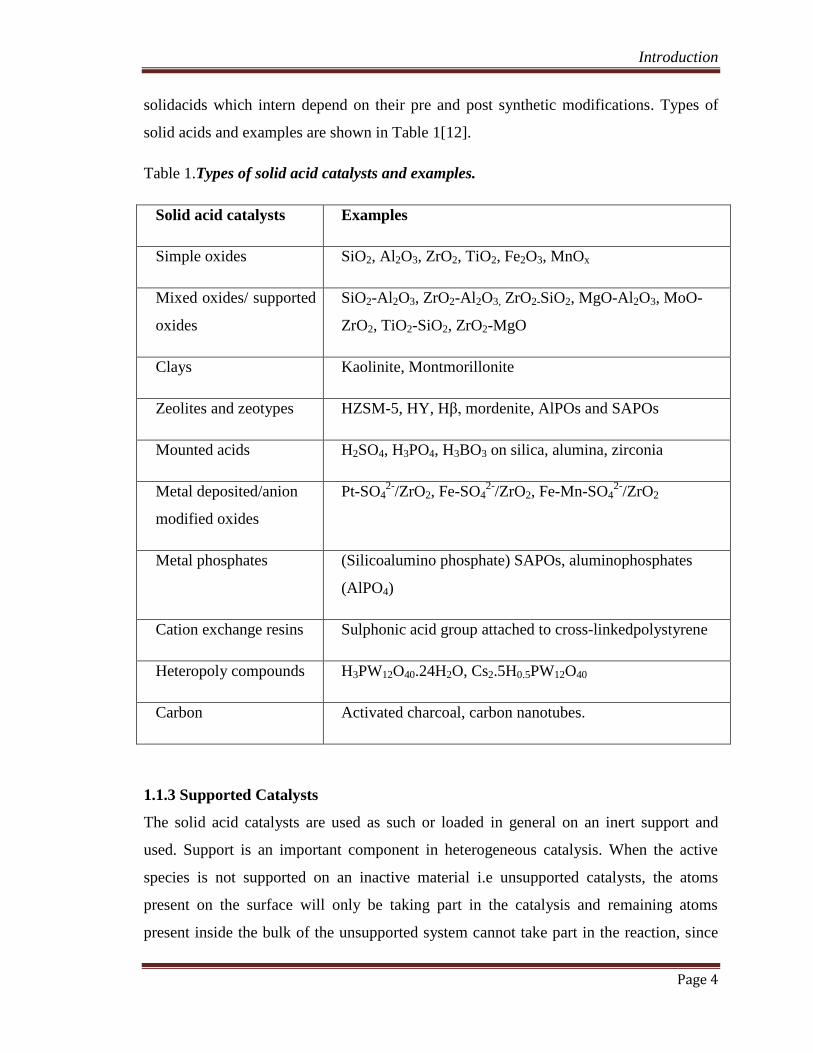
Table 1.Types of solid acid catalysts and examples.
Solid acid catalysts Examples
Simple oxides SiO2, Al2O3, ZrO2, TiO2, Fe2O3, MnOx
Mixed oxides/ supported
oxides
SiO2-Al2O3, ZrO2-Al2O3, ZrO2-SiO2, MgO-Al2O3, MoO-
ZrO2, TiO2-SiO2, ZrO2-MgO
Clays Kaolinite, Montmorillonite
Zeolites and zeotypes HZSM-5, HY, Hβ, mordenite, AlPOs and SAPOs
Mounted acids H2SO4, H3PO4, H3BO3 on silica, alumina, zirconia
Metal deposited/anion
modified oxides
Pt-SO42-
/ZrO2, Fe-SO42-
/ZrO2, Fe-Mn-SO42-
/ZrO2
Metal phosphates (Silicoalumino phosphate) SAPOs, aluminophosphates
(AlPO4)
Cation exchange resins Sulphonic acid group attached to cross-linkedpolystyrene
Heteropoly compounds H3PW12O40.24H2O, Cs2.5H0.5PW12O40
Carbon Activated charcoal, carbon nanotubes.
1.1.3 Supported Catalysts
The solid acid catalysts are used as such or loaded in general on an inert support and
used. Support is an important component in heterogeneous catalysis. When the active
species is not supported on an inactive material i.e unsupported catalysts, the atoms
present on the surface will only be taking part in the catalysis and remaining atoms
present inside the bulk of the unsupported system cannot take part in the reaction, since

Introduction
Page 5
these atomic or molecular species remain inaccessible. In order to overcome this
problem, the active materials are often loaded on a support surface [13, 14].
Some of the important characteristic features of a support are:
Supports provide optimal dispersion of the active component, good acceleribility and
stability. Hence, a porous material which has high surface area is preferred as a
support. The porous nature of the support may also control the transport of the
reactant and the product molecules affecting the overall conversion.
The support diminishes the amount of the active component needed and increases the
effective surface area of the catalyst.
The support holds on its surface the microcrystalline particles of the active
component and prevents its sintering. Hence metal-support interactions are an
important in supported catalysis.
The support may interact with the active component deposited on it and form a new
complex which may have better catalytic activity and selectivity than that of the
support or the active component.
Non porous
Porous
Supported catalysts
1.1.4 Metal oxides in catalysis
Metal oxides make up a large and important class of catalytically active materials.
Their surface properties and chemistry is determined by their composition and structure,
the bonding character, the co-ordination of surface atoms and hydroxyl groups in exposed
terminating crystallographic faces. Metal oxides may be acidic or basic and also exhibit
redox properties. They may have simple composition like in simple and binary oxides,
but many technologically important oxide catalysts are complex and multicomponent

Introduction
Page 6
materials. Metal oxides form an important class of industrial catalysts due to their
versatile nature [15]. Among the metal oxides which are widely employed as catalysts
and supports, the prominent ones are alumina, silica, zirconia and titania (Al2O3, SiO2,
ZrO2 and TiO2) [16].
These oxides form a set of catalysts encompassing a wide spectrum of catalytic activity
because of their acid-base and redox properties in their unmodified forms. Modification
of these simple oxides have opened up new vistas in the field of catalysis and
revolutionized the chemical industry giving rise to even solid super acids.
Realizing the boundless potential that has remained yet unexplored in the domain of
metal oxides, a lot of work is being done over alumina and zirconia catalyst systems. A
brief description of preparation, properties and catalytic applications of alumina, and
zirconia, their modified forms are given in the following section.
1.2 Alumina as catalyst and catalyst support
1.2.1 Preparation of alumina (Al2O3): The most important methods in practice for
laboratory preparation of alumina [17, 18] are
(a) Hydrolysis of an aluminum salt usually the nitrate using aqueous ammonia
Al(NO3)3 + Aq NH3→Al(OH)3+ NH4NO3
(b) Hydrolysis of an aluminum isopropoxide by water
3Al(OC3H7)3+ 3H2O →3Al(OH)3+3 C3H7OH
(c) Thermal decomposition of aluminum isopropoxide and
∆
Al(OC3H7)3→Al2O3
(d) Decomposition of aqueous Na/K-aluminate by CO2 etc.
2NaAlO2+ CO2→ Al2O3 + Na2CO3
1.2.2 Crystalline phases of alumina
Generally, the starting material for aluminum oxide is a precipitated hydroxide-
gibbsite or bayerite, both having the composition Al(OH)3. The hydroxide may be

Introduction
Page 7
crystalline or amorphous. When the hydroxide is subjected to calcination at different
temperatures various phases of alumina are formed. The α-phase is the only
thermodynamically stable oxide of aluminium and is the final product of the calcination
process which follows the bayer treatment. The nature of these phase transformations has
now been studied for many years, and the pathways involved in the calcination include:
Gibbsite → boehmite (γ-AlOOH) → γ-alumina (γ-Al2O3) → δ-alumina (δ-Al2O3) → θ-
alumina (θ-Al2O3) → α-alumina. [19, 20] (Figure 1).
Figure 1. Thermal transformation sequence of aluminum hydroxides.
Each transition phase exhibits a distinct powder X-ray diffraction pattern. These phase
transformations are of fundamental importance in determining their catalytic activity
[21]. Thus the catalytic activity of alumina is influenced by the conditions at which it is
prepared such as the source from which alumina is obtained, precipitation and aging of
the hydroxide, preheating time and temperature and final calcinations temperature [22-
24].
1.2.3 Acidic and basic properties of alumina
Precipitated aluminum hydroxide has surface hydroxyl groups, on dehydroxylation
defects are generated. Defects may also be caused by anion addition. These defects are
potential sites of catalytic activity. The defect sites strongly influences the chemical

Introduction
Page 8
properties of the remaining hydroxyls on the surface. Little is known about the electronic
and geometric structure of these defects or the specifics and their influence on the
reactivity of working catalyst. There are five different types of hydroxyl groups in
alumina as observed by Khozinger and P. Rathnaswamy (figure 2).The occurrence and
number of each type depends on the relative contribution of specific crystal faces. The
various OH groups are expected to have varying chemical properties. Type III should
exhibit the highest acidity, while types IA and IB should be the most basic.Lewis and
Bronsted acidity in aluminum hydroxide may be generated by subjecting it to heat
treatment figure 3. [25].
Figure 2. Types of OH groups and planes of alumina as observed by H. Knozinger and P.
Rathnaswamy.
Figure 3. Co-ordination of alumina.

Introduction
Page 9
1.2.4 Catalytic applications of alumina
The catalytic applications of alumina are based on its surface acidic and basic properties,
these applications have been extensively reviewed and reported [13]. Metal oxide and
anion modified alumina are used in many catalytic applications like opening of aliphatic
epoxides [26], as auto-exhaust catalyst [27], nitration of aromatic compounds [28],
solvent free esterification of carboxylic acid [29], selective catalytic oxidation of
ammonia in gasified biomass [30], isomerization of alkenes, dehydration of alcohols,
hydrogenation of aromatic compounds, dehydrogenation, isomerization, oxidation,
reduction, esterification and trans-esterification reactions [31-38].
1.3 Zirconia as catalyst and catalyst support
1.3.1 Preparation of zirconia (ZrO2): Different routes, such as chemical precipitation,
hydrothermal, gas-condensation, sonochemical, and sol-gel processes are employed for
the synthesis of zirconium hydroxide which in turn is converted into zirconia by
calcination [39-43].
1.3.2 Crystalline phases of Zirconia
Figure 4. Thermal transformation sequence of Zirconia.
ZrO2 exist in three polymorphic forms: these are
Cubic (c) which has a fluorite structure in which Zr atoms are coordinated to
eight oxygen atoms
Monoclinic (m) it sometimes referred to as the baddeleyite structure and
Tetragonal (t) which has distorted fluorite structure whose diffraction patterns
can be indexed to a face centered tetragonal cell.
< 1170 °C 1170°C-2370 °C >2370
Monoclinic Tetragonal Cubic

Introduction
Page 10
A schematic crystal structure of the above three forms are showed in figure 5.
Figure 5. Crystal structure of monoclinic (a), tetragonal (b) and cubic zirconia(c). Source:
[44].
1.3.3 Phase transformation in zirconia:
Tetragonal phase of zirconia has been found to be catalytically active. Several
attempts have been made to stabilize this phase. Zirconia has been found to change
phases under certain temperatures and pressures. At normal atmospheric pressure the
oxide is monoclinic at room temperature. It then transforms to the tetragonal and cubic
phases with an increase in temperature. The monoclinic-tetragonal phase transformation
has been extensively studied due to its theoretical and practical importance [45-50]. The
monoclinic to tetragonal phase transformation rate depends on the particle size of the
zirconia powder and process of powder preparation [51, 52]. The larger the particle size
of the prepared zirconia, the faster the phase transformation occurs.
Addition of sulfate anions can also play an important role in phase
transformation. Bridging sulfate ions stabilize the structure of zirconia since it can retard
the formation of oxo-bonds between zirconium atoms and oxygen atoms (figure 6). This
will prevent sintering at high temperature and hence, prevent rapid phase transformation
and will stabilize the surface area [53, 54]. Factors like precursor, pH, and aging time
influence the phase transformation [55]. The sample precipitated at low pH exhibited fast
phase transition from tetragonal to monoclinic. Furthermore, the phase transformation
occurred more rapidly in an oxygen environment than in an inert gas atmosphere.

Introduction
Page 11
Figure 6. Structure of sulphated zirconia [56]
1.3.4. Acidic and base properties of zirconia
Zirconia is amphoteric oxides which possess strong acid-base properties. The
existence of co-coordinately unsaturated cations is responsible for Lewis acidity.
Adsorption of water molecules results in the reversible transformation of Lewis acid sites
into Bronsted acid sites. Bronsted acid sites and Lewis acid sites are the probable forms
of acidity at lower and higher temperatures respectively [57]. Its application has however
been limited by the presence of mild acidic and basic sites on its surface. In this regard
recent years, there are many investigations on increasing the acid strength of zirconia by
surface as well as structural modification. The most prominent among them is anchoring
catalytically active metal oxides, addition of cationic or anionic substances such as WO3,
SO42-
and MoO3 at sub-monolayer level to generate newer acidic sites.
The results achieved in recent years are quite remarkable considering the fact that
strength of the order of 100% H2SO4 can be achieved by these modifications which is
rarely found in any heterogeneous catalyst [58]. The grafting of sulfated species on ZrO2,
strong acidic sites are developed on their surfaces which are termed as “super acidic.
Tungsten oxide species dispersed on zirconia supports (WOx-ZrO2) comprise another
interesting class of solid acids [59]. The strong acid sites originates on these materials when
zirconia oxyhydroxide (ZrOx(OH)4-2x) is impregnated with solutions containing tungstate
anions and then oxidized at high temperatures.
1.3.5 Catalytic applications of zirconia
ZrO2 has been used as an acid-base catalyst and also as an catalyst support for numerous
organic reactions in heterogeneous catalysis. Abundant literature is available in catalysis
where zirconia based solid acids are employed in organic transformations [60-64].

Introduction
Page 12
1.4 Oxides of manganese and its catalytic applications
Manganese oxides represent a group of transition metal (TM) oxides which hold
promising properties for a range of chemical reactions. Due to the variable valence of
manganese cations, the chemistry of its compounds is very rich. This holds true for
manganese oxides as well. The common manganese oxides stable at ambient conditions
are: Mn3O4, Mn2O3 and MnO2 and MnO.
The thermodynamic stability of these oxides increases in the given order with increasing
temperature and decreasing partial pressure, a fact which can be made use of while
preparing the different oxides [65]. Manganese oxide has been used as a substitute for the
noble metal catalysis, but, because of the lower surface area the catalytic activity was
often disturbed [66, 67]. Therefore, there has been tremendous research interest on the
laboratory synthesis of manganese oxides with various structures to improve the surface
area and catalytic ability. The surface area of manganese oxide is not very high,
especially at high temperatures. So manganese oxides mixed with or supported on other
high surface area materials are extensively investigated. MnOx-ZrO2 system has been
shown to have a better reducibility compared to MnOx supported on other materials, such
as alumina, titania, and silica [68].
1.4.1 Effect of calcination on manganese oxide
As the calcination temperature increases, the surface area decreases and the phase
also changes (the Mn ions transform from higher oxidation state to lower state). The heat
treatment at various temperatures leads to the decomposition of the manganese oxide.
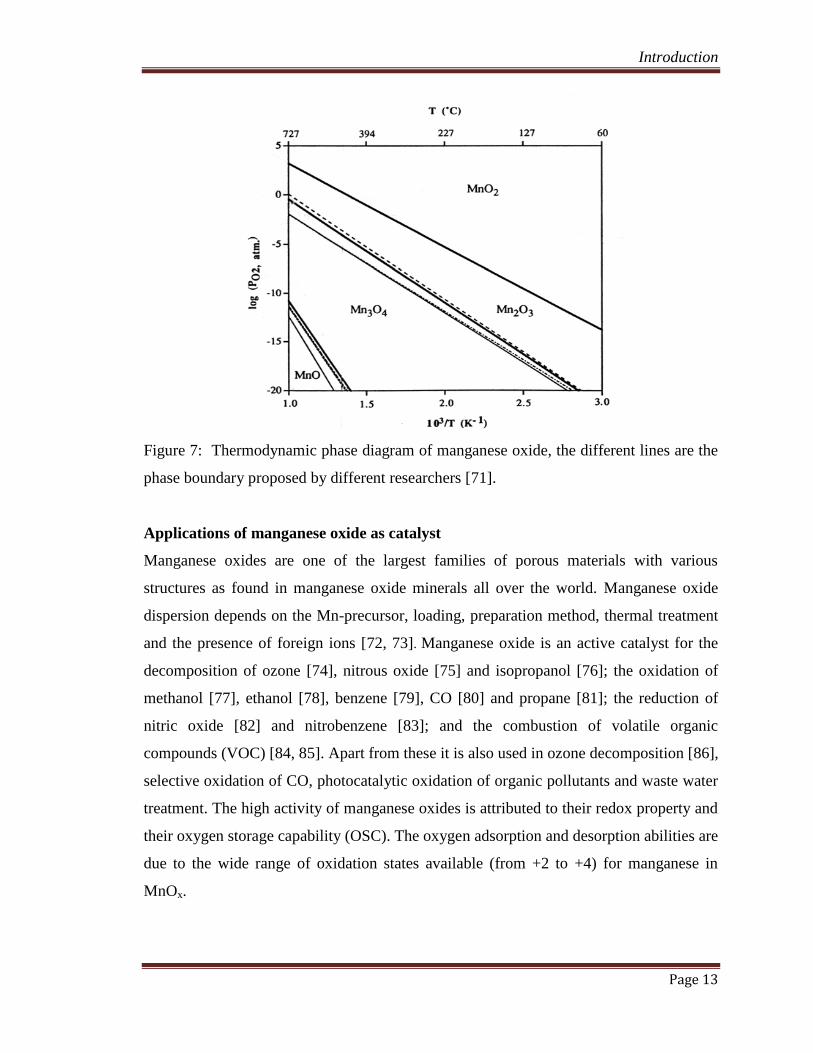
According to the phase diagram (Figure 7.) the occurrence of phase depends on the
temperature and oxygen partial pressure. In the ambient pressure, from about room
temperature to 400oC, the stable phase is MnO2 between 400 and 700
oC, the stable phase
is Mn2O3. Mn2O3 further decomposes to Mn3O4 and then MnO above 700oC. The
literature data showed that the crystalline manganese oxide was MnO2 or Mn2O3 after
synthesis and low temperature calcination (~300oC. And indeed the crystalline phases
changed with calcination temperatures and atmosphere for pure manganese oxide or
supported manganese oxide [69-71].
Introduction
Page 13
Figure 7: Thermodynamic phase diagram of manganese oxide, the different lines are the
phase boundary proposed by different researchers [71].
Applications of manganese oxide as catalyst
Manganese oxides are one of the largest families of porous materials with various
structures as found in manganese oxide minerals all over the world. Manganese oxide
dispersion depends on the Mn-precursor, loading, preparation method, thermal treatment
and the presence of foreign ions [72, 73]. Manganese oxide is an active catalyst for the
decomposition of ozone [74], nitrous oxide [75] and isopropanol [76]; the oxidation of
methanol [77], ethanol [78], benzene [79], CO [80] and propane [81]; the reduction of
nitric oxide [82] and nitrobenzene [83]; and the combustion of volatile organic
compounds (VOC) [84, 85]. Apart from these it is also used in ozone decomposition [86],
selective oxidation of CO, photocatalytic oxidation of organic pollutants and waste water
treatment. The high activity of manganese oxides is attributed to their redox property and
their oxygen storage capability (OSC). The oxygen adsorption and desorption abilities are
due to the wide range of oxidation states available (from +2 to +4) for manganese in
MnOx.

Introduction
Page 14
1.5 Experimental aspects of catalysis
One of the important aspects of catalysis research is the identification of catalytic activity
and structure relationship of the catalyst. Up to date in most cases this remains as an
unsolved problem. A rational design of a catalyst is governed by, in general three
interconnected principles: preparation, characterization and reactivity. A triangular
relationship between these three principles has been shown by many authors in the design
of the catalyst for a specific application. (Figure 8)
Figure 8. The scheme of interconnectivity of catalyst design on synthesis characterization
and reactivity [1]
The above diagram shows how preparation of a catalyst is related to characteristics and
reactivity and reactivity depends on both preparation and characteristics. The following
aspects of catalyst design are briefly described:
Preparation of heterogeneous catalyst
Characterization techniques
Catalytic activity studies.
1.5.1 Preparation of catalysts
The preparation method of catalysts has a strong influence on their final properties [87].
Various method of preparation of heterogeneous catalysts, principles of the methods with
Reactivity
Preparation characterization
Catalyst design

Introduction
Page 15
specific examples has been published by Selim Senkan [88]. The preparation parameters
that influence the physico-chemical properties of the precipitate are shown in the figure 9.
Figure 9. Parameters affecting the properties of the precipitate and the main properties
influenced.
Precipitation-impregnation and co-precipitation are the most commonly employed
methods for the preparation of heterogeneous catalysts.
Precipitation
In the precipitation method the metal salts are dissolved in water and the precipitation is
carried out by slow addition of required amount of precipitating agent like ammonium
hydroxide or sodium carbonate at room temperature until the pH reaches a required
value. The precipitate thus obtained is washed with distilled water to remove all anions
present, dried overnight at 110-120ºC. After precipitation/impregnation, the catalyst mass
is normally subjected to drying and calcination at desired temperature.

Introduction
Page 16
Impregnation
In this procedure a certain volume of solution containing known amount of the precursor
of active phase is contacted with a previously weighed solid support. The amount of
metal ions that interactively remain bound to the support surface depends on the
adsorption capacity of the support, the period of impregnation and temperature of
impregnation. Wet impregnation and incipient wetness impregnation are the two methods
used in catalyst preparation, which can be distinguished depending on the volume of the
solution. In wet impregnation excess solution is generally used. After a certain time, the
solid is separated and the excess solvent is removed by evaporation. In the incipient
wetness impregnation the volume of solution of appropriate concentration is equal to or
slightly less than the pore volume of the support.
Co-precipitation
In Co-precipitation method two or more metal salts are completely dissolved in water and
the precipitation is carried out by slow addition of required amount of precipitating agent
until pH reaches required value. The precipitate thus obtained is aged for required
duration, washed with distilled water to remove all anions present, dried overnight at
100-120ºC. The catalyst mass are finely ground and subjected to calcinations at desired
temperature depending on the catalyst material prepared.
1.5.2 Catalyst characterization techniques
Characterization is an important field in catalysis. Spectroscopy, microscopy, diffractions
and methods based on adsorption or desorption and bulk reactions, all offer tools to
investigate the nature of an active catalyst. With such information we can understand the
catalyst better, so that modify them or even design new catalyst.
For the development of active, selective and stable catalyst we need to identify the
structural property that discriminates efficient from less efficient catalyst.

Introduction
Page 17
Various spectroscopic and non-spectroscopic techniques have been employed to
characterize the solid acid catalysts include:
Fourier Transform infrared (FT-IR) spectroscopy
Powder X-ray diffraction studies (P-XRD)
N2 adsorption for BET surface area
Temperature programmed desorption of ammonia (TPD-NH3) and
n-butyl amine back titration method
Temperature programmed reduction (TPR)
Scanning electron microscopy (SEM)
Transmission electron microscopy (TEM)
Inductively coupling plasma (ICP).
A detail description of the principles and applications of the above techniques employed
in catalyst characterization is published by I. Chorkendorff et al. [3]
1.5.3 Catalytic activity determination
The activity of a catalyst in a chosen organic transformation is determined mainly by two
methods: Liquid phase and vapour phase catalytic activity studies.
In liquid phase studies, the reactant/s is/are taken in a suitable reaction vessel is mixed
with the catalyst to be evaluated. The reaction mixture is analyzed for the progress of the
reaction using a suitable analytical technique. In vapour phase studies, pre-heated
reactant or a mixture of reactants is passed over a solid catalyst bed maintained at a
definite temperature. The reaction products are condensed and analyzed using a suitable
analytical technique.
The degree of activity of a catalyst is described by the turn over number (TON) and the
catalytic efficiency by the turnover frequency (TOF) [1-3].
Turnover Number (TON): The turnover number specifies the maximum use that can be
made of a catalyst for a special reaction under defined conditions by a number of

Introduction
Page 18
molecular reactions or reaction cycles occurring at the reactive center up to the decay of
activity.
The relationship between TOF and TON is:
TON = TOF [time–1
] .Lifetime of the catalyst [time] [–]
For industrial applications the TON is in the range 106-10
7.
Turnover Frequency (TOF): The turnover frequency TOF quantifies the specific
activity of a catalytic center for a special reaction under defined reaction conditions by
the number of molecular reactions or catalytic cycles occurring at the center per unit
time. For heterogeneous catalysts the number of active centers is derived usually from
sorption methods.
For most relevant industrial applications the TOF is in the range 10–2
- 102S
–1
(enzymes103-10
7S
-1).
1.6 Objectives and the scope of the proposed research work
It is evident from the reported literature that alumina, zirconia, modified forms of these
materials (as supported oxides as well as their anion modified forms) are extensively
investigated for their catalytic activity. However the reports on manganese oxide
modified alumina and zirconia as catalytic materials in organic transformations are
scarce. Application of the catalyst with respect to particular phase of the materials is less
i.e application of oxy-hydroxy phase of alumina (Boehmite) in organic transformations
and stabilization of the tetragonal phase of zirconia which is highly unstable and

Introduction
Page 19
catalytically active could be achieved by modification by loading with some metal
oxides. Further the correlation between catalytic activity and the texture of the material
prepared need to be clearly understood. Thus this topic of study is still a rewarding and
demanding.
The main objectives of the present work are:
To prepare manganese oxide modified alumina and zirconia catalysts with different
percentage of manganese oxide.
To investigate the surface and bulk properties of the materials by suitable
characterization techniques.
To understand the precise role of manganese oxide on acid-base properties of
alumina and zirconia support.
To develop recyclable, ecofriendly and selective solid acid catalysts to synthesize a
few pharmacologically important organic fine chemicals.
To examine the structure-catalytic activity relationship if any.
To achieve the above objectives, the following methodology was adopted.
1.7 Methodology
Preparation alumina and zirconia supports and those containing different percentages
of manganese using precipitation-impregnation method and co-precipitation method
using ammonia as precipitating agent.
Physico-chemical characterization of these catalysts by various techniques such as
BET surface area, TPD-NH3, XRD, FT-IR, ICP, SEM and TEM.
Evaluation of the catalytic activity in organic transformations which involves
synthesis of benzimidazoles, benzodiazepines, bis(indolyl)methane,
dihydropyrimidinone, biphenylurea and quinoxalines.
Comparison of the catalytic activity and selectivity of these catalysts with their
physico-chemical properties.

Introduction
Page 20
1.8 Organization of the thesis
The work carried out in the present doctoral work has been organized into five chapters.
Chapter I Introduction
Chapter II Preparation and characterization of alumina, zirconia,
manganese oxide modified alumina and zirconia catalysts and
their catalytic activity in the synthesis of Benzimidazoles and
Benzodiazepines
Chapter III Section A: Selective synthesis of Bis(indolyl)methanes using
Mn/Al2O3 and Mn/ZrO2 catalysts: The role of surface acidity
and particle morphology
Section B: Synthesis of Dihydropyrimidinones using modified
alumina and zirconia as catalysts
Chapter IV Synthesis of Quinoxalines: Role of catalyst acidity of various
solid acids
Chapter V Non phosgene method for the synthesis of Biphenyl urea using
alumina based catalysts prepared by different methods
Summary and Conclusion

Introduction
Page 21
References
1. Catalysis, selected applications, Ed. B. Viswanathan, Narosa publishing house, New
Delhi, 2009.
2. Heterogeneous catalysis and solid catalysts. Olaf. Deutschmann et al., 2009, Wiley-
VCH VerlagGmbh and Co. KGaA. Weinheim
3. I. Chorkendorff, J.W. Niemantsverdriet. Concepts of modern catalysis and kinetics,
WILRY-VCH GmbH and Co. KGaA. 2003.
4. A.F. Bedilo, M.A. Plotnikov, N.V. Mezentseva, A.M. Volodin, G.M.
Zhidomirov, I.M. Rybkina, K.J. Klabundeb. Phys. Chem. Chem. Phys.7 (2005)
3059.
5. D.W. Breck. Zeolite molecular sieves, structure chemistry and use. John Wiley and
Sons, New York, 1974.
6. J. Weitkamp, Solid State Ionics 131 (2000) 175.
7. Hartmann, L. Kevan. Chemical Reviews, 99 (1999) 635.
8. G. Nagendrappa, Appl. Clay Sci. 53 (2011) 106.
9. M.B. Gawande, R.K. Pandey, R.V. Jayaram. Catal. Sci. Technol. 2 (2012) 1113.
10. J.H. Clark. Acc. Chem. Res. 35 (2002) 791.
11. T. Okuhara, Chem. Rev. 102 (2002) 3641.
12. K. Tanabe. "Solid Acid and Bases", Academic Press, New York (1990) 136.
13. M. Trueba, S.P. Trasatti. Euro. J. Inorg. Chem. (2005) 3393
14. David C. Sherrington, A.P. Kybett. “Supported Catalysts and their
Applications”.Angewandte. Chemie. 41(2002) 520.
15. J.K. Acres, A.J. Bird, J.W. Jenkins, F. King. Catalysis (1981) 1.
16. I. Chorkendorff, J.W. Niemantsverdriet. Concepts of modern catalysis and kinetics.
Wiley-VHC, (2003).
17. P. Kim, J.B. Joo, H. Kim, W. Kim, Y. Kim, I.K. Song, J. Yi. Catal. Let. 104 (2005)
181.
18. T. Lopez, A. Romero, A. Chavela, L. Razo, R. Gomez. React. Kinet. Catal. Lett. 43
(1991) 307.
19. J.B. Peri, R.B. Hannan. Ibid. 64 (1960) 1526.
20. H. Knozinger. P. Ratnaswamy. Catal. Rev. 17 (1978) 31

Introduction
Page 22
21. G. Paglia, Doctoral thesis, Curtin. University of Technology (February 2004).
22. M. Jayamani. B. Viswanathan. C.N. Pillai. J. Catal, 89 (1984) 560.
23. H. Pines, W.O. Haag. J. Amer. Chem. Soc. 82(1960) 2471.
24. H. Sonntag, K.Z. Rodel, An org. Allg. Chem. 343(1966) 131.
25. M. Trueba, S.P. Trasatti. Euro. J. Inorg. Chem. (2005) 3393
26. G.H. Posner, D.Z. Rogers. J. Am. Chem. Soc. (1977) 8214.
27. N.K Labhsetwar, A. Watanabe, R.B. Biniwale, R. Kumar, T. Mitsuhashi, App Catal
B: Envi. 33 (2001)165.
28. M. Hosseini-sarvari, M. Tavakolian and S. Ashenagar, Iranian Journal of Science &
Technology.34 (2010) 215.
29. H. Sharghi, M. HosseiniSarvari, R. Eskandari. J. Chem. Res. (2005) 482.
30. S. Nassos, E.E. Svensson, M. Boutonnet, S.G. Jaras,Appl. Catal. B: Environ. 74
(2007) 92.
31. G.H. Posner, Angew. Chem, 90 (1978) 527.
32. J.M. Campelo, R. Chakraborty, J.M. Marinas, A.A. Romero. Catal. Lett. 54 (1998)
91.
33. S. Narayanan, R.P Unnikrishnan, J. Chem. Soc, 93 (1997) 2009.
34. S. Narayanan, R.P Unnikrishnan, Appl. Catal, 145 (1996) 231.
35. T.H.E. Nabarawy, Adsorpt. Sci. Technol, 15 (1997) 25.
36. J. Jansson. J. Catal, 194 (2000) 55.
37. H. Praliaud, S. Mikhailenko, Z. Chajar, M. Primet. Appl. Catal:B, 16 (1998) 359.
38. N. Nagaraju, G. Kuriakose, J. Mol. Catal. A, 223 (2004) 155.
39. Z. Wang, D. Tao, G. Guo, S. Jin, F. Wei, W. Qian, S. Hong, J. Guo, Materials
Letters, 60 (2006), 3104.
40. A.C. Dodd, P.G. Mc Cormick, J. Eur. Ceram. Soc. 22 (2002) 1823
41. J. Liang, X. Jiang, G. Liu, Z. Deng, J. Zhuang, Fuli Li and Yadong Li, Mater. Res.
Bull.38 (2003) 161.
42. L. Li, W. Wang, Sol. State Comm. 127 (2003) 639.
43. Y.V. Kolenko, V.D. Maximov, A.A. Burukhin, V.A. Muhanov, B.R. Churagulov,
Mater. Sci. Eng.23 (2003) 1033.
44. R.H.J. Hannink, P.M. Kelly, B.C. Muddle. J. Am. Ceram. Soc. 83 (2000) 461.

Introduction
Page 23
45. F. Heshmatpour, R.B. Aghakhanpour. Powder Technology 205 (2011) 193
46. F. Zhang, C-H. Chen. J. Am. Chem. Soc. 89 (2006) 1028.
47. J. Hong, D. De La Torre, L. Gao, K. Miyamoto and H. Miyamoto .J. Mater. Sci.
Lett.17 (1998) 1313.
48. R. Srinivasan. R.J. De Angelis, G. Ice, B.H. Davis. J. Mater. Res. 6 (1991) 1287.
49. V. Santos. M. Zeni, C.P. Bergmann, J.M. Hohemberger. Rev.Adv.Mater.Sci. 17
(2008) 62.
50. F. Kazemi, A. Saberi, S. Malek-ahmadi, S. Sohrabi, H.R. Rezaie, M. Tahriri.
Ceramics – Silikaty. 55 (2011) 26.
51. K. Ishid, K. Hirotu, O. Yagamuchi. J. Am. Ceram. Soc.77 (1994) 1391.
52. M. Trunec. Ceramics – Silikaty. 52 (2008) 165.
53. C. Norman, P. Goulding and I. Mc Alpine.Catal. Today, 20 (1994) 313.
54. C. Norman, P. Goulding, P. Moles.Stud. Surf. Sci. Catal. 90 (1994) 269.
55. R. Srinivasan, M. Harris, S. Simpson, R. De Angelis, B. Davis. J. Mater. Res.3
(1988) 787.
56. A.F. Bedilo, M.A. Plotnikov, N.V. Mezentseva, A.M. Volodin, G.M.
Zhidomirov, I.M. Rybkina, K.J. Klabundeb. Phys. Chem. Chem.Phys.7 (2005) 3059.
57. K. Tanabe. "Solid Acid and Bases", Academic Press, New York (1990) 136.
58. B.M. Reddy, M.K. Patil. Chem. Rev. 109 (2009) 2185.
59. B.G. Mishra et al., Colloid and Surfaces. A 317 (2008) 234.
60. T. Yamaguchi, Catal. Today, 20 (1994) 199.
61. R.H.J.Hannink, P.M.Kelly, B.C. Muddle. J. Am. Ceram. Soc. 83 (2000)461.
62. D. Smith, H. Newkirk, Acta. Cryst. 18 (1965) 983.
63. M. Finnis, T. Paxton. Phys. Rev. Lett.81 (1998) 5149.
64. J. Mc Cullough, K. Trueblood. Acta. Cryst. 12 (1959)507
65. W. Feltknecht, Pure Appl Chem. 9 (1964) 423.
66. C.E. Langley, C. Biljana, C.E. Banks, R.G. Compton. Jpn. Soc. Anal. Chem.23
(2007) 165.
67. C. Xia, W .Ning, G. Lin, Sens. Actuators B. 137 (2009) 710.
68. F. Arena, T. Torre, C. Raimondo, A. Parmaliana. Phys. Chem. Chem. Phys. 3 (2001)
1911.

Introduction
Page 24
69. L.M. Fei, Z. Bo, Y.X. Xin. Journal of Rare Earths, 12 (1994) 259.
70. C. Kappenstein, T. Wahdan, D. Duprez, M.I. Zaki, D. Brands, E. Poels, A. Bliek,
Preparation of Catalysis VI, (1995) 699.
71. S. Fritsch, A. Navrotsky. J. Am. Ceram. Soc. 79 (1996) 1761.
72. I.R. Craciun, N. Dulamita. Catal. Lett. 46 (1997) 229.
73. F. Kapteijn, F. van Langeveld, D. Moulijn, J.A. Andreini, A. vuurman, M.A. Turek,
A.M. Jehng, J.M. Wachs, J. Catal. 150 (1994) 94.
74. B. Dhandapani, S.T. Oyama, Appl. Catal. B: Environmental. 11 (1997) 129.
75. T. Yamashita, A. Vannice, J. Catal. 161 (1996) 254.
76. J. Ma, G.K. Chuah, S. Jaenicke, R. Gopalakrishnan, K.L. Tan, Phys. Chem. 100
(1995) 585.
77. M.A. Baltanas, A.B Stiles, J.R. Katzer. Appl. Catal. 28 (1986) 13.
78. L.J. Pettersson, A.M. Wahlberg, S.G. Jaras, surface science and catalysis. 116 (1998)
465.
79. A. Naydenov, D. Mehandjiev. Appl. Catal. A: General. 97 (1993) 17.
80. R. Craciun, B. Nentwick, K. Hadjiivanov, H. Knözinger. Appl. Catal. A: General.
243 (2003) 67.
81. M. Baldi, E. Finochhio, C. Pistarino, G. Busca. Appl. Catal. A: General, 173 (1998)
61.
82. F. Kapteijn, L. Singoredjo, A. Andreini, J.A. Moulijn. Appl. Catal. B: Environ. 3
(1994) 173.
83. A. Maltha, L. Favre, F.T. Kist, H.F. Zuur, A. P. Ponec, J. Catal. 149 (1994) 364.
84. M. Baldi, V.S. Escribano, J.M. Gallardo-Amores, F. Milella, G. Busca. Appl. Catal.
B: Environ. 17 (1998) L175.
85. M. Baldi, E. Finocchio, F. Milella, G. Busca, Appl. Catal. B : Environ. 16 (1998) 43.
86. R. Radhakrishna, S.T. Oyama. J. Catal. 196 (2001) 182.
87. M. Shimokawabe, R. Furuichi, T. Ishii, Thermochim Acta 21 (1977) 373.
88. S. Senkan. Angew. Chem. Int. Ed. 40 (2001) 312.