inprocess as per usp ip bp injection
TRANSCRIPT

INJECTIONGeneral chapter

Introduction• Parenteral Preparations are sterile products intended for
administration by injection, infusion or implantation into the body.
• They may be preparations intended for direct parenteral administration or they may be parenteral products for constituting or diluting prior to administration.
• There are five main types of Parenteral reparations, namely, Injections, Infusions, Powders for Injection, Concentrated Solutions for Injection and Implants.
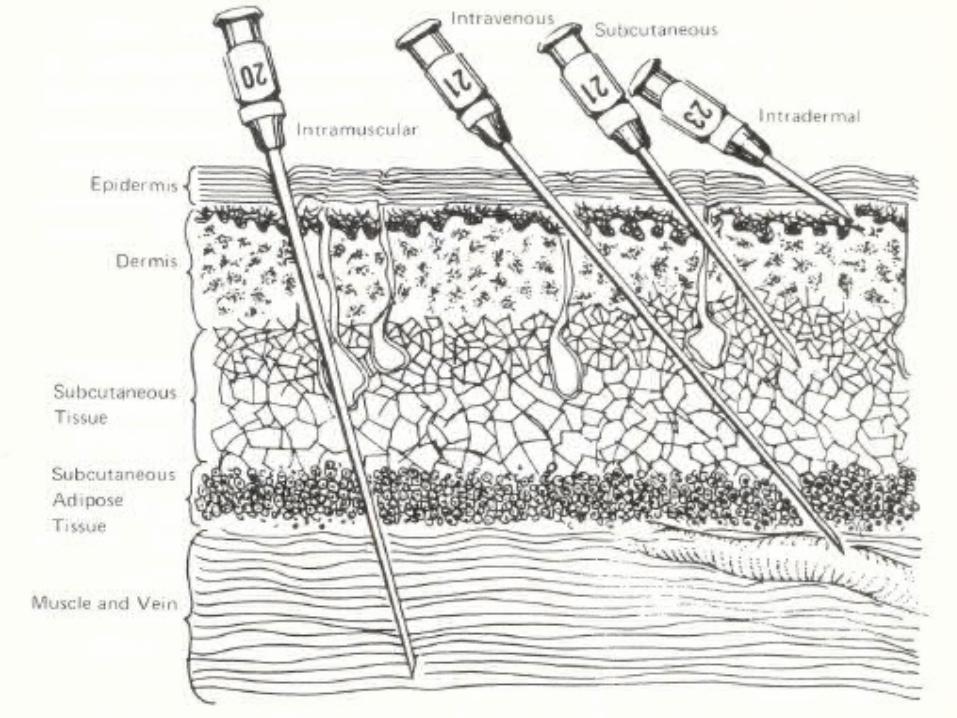

Parenteral Routes• Intradermal– Small volumes 0.1ml– Absorption is slow slow onset of action
• Subcutaneous– <2ml volumes– Much more rapid absorption than ID
• Intramuscular– 2 – 5ml volumes– More rapid absorption than by SC– Can formulate a delayed response
• Intravenous– Small to large volumes– No absorption of drug directly in vein
Sterile Dosage Forms: S. Turco, R. King

Pharmacokinetics for Absorption• IV route immediate & total access to active
drug molecules• IM, SC, ID require an absorption step
– The vascularity of the IM route is greater than the SC route absorption
Sterile Dosage Forms: S. Turco, R. King

Container• Containers for Parenteral Preparations are made
as far as possible from materials that • 1) are sufficiently transparent to permit visual inspection of the
contents, except for implants; • 2) do not adversely affect the quality of the preparation under the
ordinary conditions of handling, shipment, storage, sale and use; • 3) do not permit diffusion into or across the walls of the container
or yield foreign substances into the preparation.
• Parenteral Preparations may be supplied in glass ampoules, vials or bottles or in other containers such as plastic bottles or bags or in prefilled syringes the integrity of which is ensured by suitable means.

Tests• Particulate matter• Uniformity of content• Extractable volume• Sterility • Pyrogens• Bacterial endotoxin
• Leak test
Pharmacopoial
Non- Pharmacopoial

Particulate Contamination (IP/BP/USP)• For the purpose of this test, particulate contamination is defined
as the unintentional presence in injections and infusions, of extraneous, mobile, undissolved substances, other than gas bubbles.
• Parenteral preparations including solutions constituted from sterile solids are expected to be free from particles of approximately 50 μm or more that can be observed by inspection with the unaided eye
• However, parenteral preparations in containers that are labelled as containing 100 ml or more of a single-dose large volume injection intended for administration by intravenous infusion should comply with the limits of sub-visible particles prescribed in this test

• The test does not apply to multiple dose injections, to single dose small volume parenteral preparations and to parenteral solutions constituted from sterile solids.
• For these preparations the following method of visual assessment is adequate.
• For visible particles• Apparatus. A viewing station comprising:
-A matt black panel of suitable size kept in a vertical position,-A non-glare white panel of the same size kept next to the black panel, and- an adjustable lamp holder fitted with a light diffuser (such as two 13W fluorescent tubes, each about 52.5 cm in length); the intensity of illumination is kept at 2000 to 3750 lux, or higher for plastic and colored glass containers.
• Method. Remove any labels on the container, wash and dry the outside. Gently invert the container or swirl it, ensuring that air bubbles are not formed, and observe for about 5 seconds in front of the white panel. Repeat the procedure in front of the black panel. Note the presence of any particles.

For sub-visible particles• Two methods are specified, • one involving the counting of particles viewed under a
microscope and • the other based on the count of particles causing light
obscuration.

Method 1. Microscopic particle count test• This method is suitable for revealing the presence of particles the
longest axis or effective linear dimension of which is 10 μm or more.• Apparatus. A suitable binocular microscope, filter assembly and
membrane filter for retention of particles.

Method 2. Light obscuration particle count test• This method is not suitable for preparations with reduced clarity or
increased viscosity such as emulsions, colloids.• liposomal preparations and products that produce air or gas
bubbles when drawn into the sensor.• Precautions during testing. The same general precautions as• for Method 1 apply.• Apparatus. A suitable particle counter based on the principle of
light blockage and capable of automatic counting and sizing of particles. The manufacturer’s instructions for installation and operation should be followed.

(USP/BP limit)

Uniformity of content (IP/BP)• Unless otherwise stated in the individual monograph, suspensions for
injection that are presented in single dose containers and that contain less than 10 mg or less than 10 per cent of active ingredient (IP) comply with the following test.
• less than 25 mg or less than 25 per cent of active ingredient (BP/USP)• For suspensions for injection containing more than one active
ingredient carry out the test for each active ingredient that corresponds to the above conditions.
• The test for Uniformity of content should be carried out only after the content of active ingredient(s) in a pooled sample of the preparation has been shown to be within accepted limits of the stated content.
• Determine the content of active ingredient(s) of each of 10 containers taken at random, using the method given in the monograph or by any other suitable analytical method of equivalent accuracy and precision.
As per BP
Unless otherwise prescribed or justified and authorized, powders for injections or infusions with a content of active substance less than 2 mg or less than 2 per cent of the total mass, or with a unit mass equal to or less than 40 mg comply with test A for uniformity of content of single-dose preparations. If the preparation contains more than one active substance, the requirement applies only to those substances that correspond to the above conditions.

• The preparation under examination complies with the test if the individual values thus obtained are all between 85 and 115 per cent of the average value.
• The preparation under examination fails to comply with the test if more than one individual value is outside the limits 85 to 115 per cent of the average value or if any one individual value is outside the limits 75 to 125 per cent of the average value.
• If one individual value is outside the limits 85 to 115 per cent but within the limits 75 to 125 per cent of the average value, repeat the determination using another 20 containers taken at random.
• The preparation under examination complies with the test if in the total sample of 30 containers not more than one individual value is outside the limits 85 to 115 per cent and none is outside the limits 75 to 125 per cent of the average value.
• NOTE — the test for Uniformity of content is not applicable to suspensions for injection containing multivitamins and trace elements.

Extractable volume (IP)• Where the nominal volume does not exceed 5 ml, the containers
comply with the requirements of Method 1 and where the nominal volume is greater than 5 ml, the containers comply with the requirements of Method 2. Suspensions should be shaken before the contents are withdrawn; oily injections may be warmed but should be cooled to 25º C before carrying out the test.
• Method 1- Use 6 containers, 5 for the test and 1 for rinsing the syringe used.
• Inspect the 5 containers to be used in the test visually and ensure that each contains approximately the same volume of the preparation.
• Using a syringe with a capacity not exceeding twice the volume to be measured and fitted with a suitable needle, take up a small quantity of the liquid under examination from the container reserved for rinsing the syringe, and discharge it from the syringe whilst the needle is pointing upwards so as to expel any air.

• Withdraw as much as possible the contents of one of the containers reserved for the test and transfer, without emptying the needle, to a dry graduated cylinder of such capacity that the total combined volume to be measured occupies not less than 40 per cent of the nominal volume of the cylinder. Repeat the procedure until the contents of the 5 containers have been transferred and measure the volume. The average content of the 5 containers is not less than the nominal volume and not more than 115 per cent of the nominal volume.
• Method 2 — Transfer the contents of not less than 3 containers separately to dry graduated cylinders such that the volume to be measured occupies not less than 40 per cent of the nominal volume of the cylinder and measure the volume transferred.
• The contents of each container are not less than the nominal volume and not more than 110 per cent of the nominal volume
• Multiple dose containers labelled to yield a specific number of doses shall contain a sufficient excess to permit the withdrawal of the designated number of doses.

Extractable volume (BP)• Single dose container: • Select 1 container if the nominal volume is 10 ml or more, • 3 containers if the nominal volume is more than 3 ml and less than 10 ml, • 5 containers if the nominal volume is 3 ml or less
• Take up individually the total contents of each container selected into a dry syringe of a capacity not exceeding 3 times the volume to be measured, and fitted with a 21-gauge needle not less than 2.5 cm in length
• Take up individually the total contents of each container selected into a dry syringe of a capacity not exceeding 3 times the volume to be measured, and fitted with a 21-gauge needle not less than 2.5 cm in length. Expel any air bubbles from the syringe and needle, then discharge the contents of the syringe without emptying the needle into a standardized dry cylinder (graduated to contain rather than to deliver the designated volumes) of such size that the volume to be measured occupies at least 40 per cent of its graduated volume. Alternatively, the volume of the contents in millilitres may be calculated as the mass in grams divided by the density.

• For containers with a nominal volume of 2 ml or less, the contents of a sufficient number of containers may be pooled to obtain the volume required for the measurement provided that a separate, dry syringe assembly is used for each container. The contents of containers holding 10 mL or more may be determined by opening them and emptying the contents directly into the graduated cylinder or tared beaker.
• • The volume is not less than the nominal volume in case
of containers examined individually, or, in case of containers with a nominal volume of 2 ml or less, is not less than the sum of the nominal volumes of the containers taken collectively.

Multi-dose container: (BP)• For injections in multi-dose containers labelled to yield a specific
number of doses of a stated volume, select one container and proceed as directed for single-dose containers using the same number of separate syringe assemblies as the number of doses specified.
• The volume is such that each syringe delivers not less than the stated dose

Sterility (IP/BP/USP)• The test for sterility is applied to pharmacopoial articles that are
required according to the Pharmacopoeia to be sterile.• However, a satisfactory result only indicates that no contaminating
viable micro-organisms have been found in the sample examined in the conditions of the test. If the number of micro-organisms present in a given amount of the article under examination is large, the probability of detecting them increases.
• Very low levels of contamination cannot be detected on the basis of random sampling of a lot. Moreover, if contamination is not uniform throughout the lot, random sampling cannot detect contamination with any certainty.
• Compliance with the test for sterility alone cannot therefore provide absolute assurance of freedom from microbial contamination. Greater assurance of sterility must come from reliable manufacturing procedures and compliance with good manufacturing practices
• The test must be carried out under aseptic conditions designed to avoid accidental contamination of the product during testing. For achieving these conditions, a grade A laminar airflow cabinet or an isolator is recommended.
• minimum number of items recommended to be tested in relation to the number of items in the batch on the assumption that the preparation has been manufactured under conditions designed to exclude contamination.

Culture Media• Fluid Thio-glycollate Medium-For use with clear fluid Products.
Use fluid thio-glycollate medium by incubating it at 30°C to 35°C.
• Alternative Thio-glycollate Medium — For use with turbid and viscid products.
• Use alternative thio-glycollate medium in a manner that will• assure anaerobic conditions for the duration of the incubation at
30°C to 35°C. • Soyabean-casein Digest Medium: Use soyabean-casein digest
medium by incubating it at 20°C to 25°C under aerobic conditions.• Suitability of Media:• Sterility• Growth Promotion Test

• The media are suitable if a clearly visible growth of the microorganisms occurs. The tests may be conducted simultaneously with any test for sterility done using the same lot of media. However, such tests will be considered invalid if the test media show inadequate growth response.

Test procedure• Either of the following methods, • Method A – Membrane Filtration or • Method B – Direct Inoculation, may be followed.
• Method A is to be preferred where the substance under examination is • a) an oil, • b) an ointment that can be put into solution, • c) a non-bacteriostatic solid not readily soluble in the culture medium, and d) a
soluble powder or a liquid that possesses bacteriostatic and/or fungistatic properties.
• For liquid products where the volume in a container is 100 ml or more, Method A should be used.

Quantities of Sample to be used• For parenteral preparations. Whenever possible use the• whole contents of the container, but in any case not less than
the quantities prescribed in Table 3, diluting where necessary to about 100 ml with a suitable diluents such as fluid A.
• For ophthalmic and other non-parenteral preparations. Take an amount within the range prescribed in column (A) of Table 4, if necessary, using the contents of more than one container, and mix thoroughly. For each medium use the amount specified in column (B) of Table 4, taken from the mixed sample.


Method of Test• For aqueous solutions.• For liquids immiscible with aqueous vehicles, and
suspensions.• For oils and oily solutions.• For ointments and creams.• For soluble solids.• For solids for injection other than antibiotics.• For antibiotic solids, bulks, and blends.• For antibiotics in packages of 5 g or less.• For sterile devices

Method B – Direct Inoculation Method of Test
• For aqueous solutions and suspensions.• For oils and oily solutions.• For ointments and creams.• For solids.• For surgical dressings and related articles.• For sterile devices.

Observation and Interpretation of Results• At intervals during the incubation period and at its conclusion,
examine the media for macroscopic evidence of microbial growth. • If the material being tested renders the medium turbid so that the
presence or absence of microbial growth cannot be easily determined by visual examination, 14 days after the beginning of incubation, transfer portions (each not less than 1 ml) of the medium to fresh vessels of the same medium and then incubate the original and transfer vessels for not less than 4 days.
• If no evidence of microbial growth is found, the preparation under examination complies with the test for sterility.
• If evidence of microbial growth is found, the preparation under examination does not comply with the test for sterility.
• Do not repeat the test unless it can be clearly shown that the test was invalid for causes unrelated to the preparation under examination.
• The test may be considered invalid only when one or more of the following conditions are fulfilled:

• a) Microbial growth is found in the negative controls;• b) Data on microbial monitoring of the sterility testing facility
show a fault;• c) A review of the testing procedure used for the test in question
reveals a fault;• d) After identifying the micro-organisms isolated from the
containers showing microbial growth, the growth may be ascribed without any doubt to faults with respect to the materials and/or technique used in conducting the test procedure.
• If the test is declared to be invalid, repeat with the same number of units as in the original test. If no evidence of microbial growth is found in the repeat test, the preparation under examination complies with the test for sterility. If microbial growth is found in the repeat test and confirmed microscopically, the preparation under examination does not comply with the test for sterility.
a) The data of the microbiological monitoring of the sterility testing facility show a fault; b) A review of the testing procedure used during the test in question reveals a fault; c) Microbial growth is found in the negative controls; d) After determination of the identity of the micro-organisms isolated from the test, the growth of this species or these species may be ascribed unequivocally to faults with respect to the material and/or the technique used in conducting the sterility test procedure.

Pyrogens (IP/BP/USP)• The test involves measurement of the rise in body temperature of
rabbits following the intravenous injection of a sterile solution of the substance under examination. It is designed for products that can be tolerated by the test rabbit in a dose not exceeding 10 ml per kg injected intravenously within a period of not more than 10 minutes.
• Test Animals• Use healthy, adult rabbits of either sex, preferably of the same variety,
weighing not less than 1.5 kg, fed on a complete and balanced diet and not showing loss of body weight during the week preceding the test. House the animals individually in an area of uniform temperature (± 2º), preferably with uniform humidity, and free from disturbances likely to excite them. Do not use animals for pyrogen tests more frequently than once every 48 hours.
• After a pyrogen test in the course of which a rabbit’s temperature has risen by 0.6º or more, or after a rabbit has been given a test substance that was adjudged pyrogenic, at least 2 weeks must be allowed to elapse before the animals is used again.

• Materials• All glassware, syringes and needles must be thoroughly washed
with water for injections and heated in a hot air oven at 250º for 30 minutes or at 200º for 1 hour. Treat all diluents and solutions for washing and rinsing of devices in a manner that will assure that they are sterile and pyrogen-free.
• The retaining boxes for rabbits in which the temperature is being measured by electrical device should be made in such a way that the animals are retained only by loosely-fitting neck stocks and the rest of the body remains relatively free so that the rabbits may sit in a normal position. The animals must be put in the boxes 1 hour before the test and remain in them throughout the test. Ensure that the room temperature where the test is carried out is within 3º of that of the rabbits living quarters or in which the rabbits have been kept for at least 18 hours before the test. Withhold food from the animals overnight and until the test is completed; withhold water during the test.

• Recording of Temperature• Use an accurate temperature-sensing device such as a clinical thermometer or
thermistor or other suitable probes that have been calibrated to assure an accuracy of 0.1º and have been tested to determine that a maximum reading is reached in less than 5 minutes. Insert the thermometer or temperature-sensing probe into the rectum of the test rabbit to a depth of about 5 cm.
• The depth of insertion is constant for any one rabbit in any one test. If an electrical device is used, it should be inserted in the rectum of the rabbit 90 minutes before the injection of the solution being examined and left in position throughout the test. After a period of time not less than that previously determined as sufficient, record the rabbit’s body temperature.
• Preliminary Test (Sham Test)• If animals are used for the first time in a pyrogen test or have not been used
during the 2 previous weeks, condition them 1 to 3 days before testing the substance under examination by injecting intravenously into them 10 ml per kg of body weight of a pyrogen-free saline solution warmed to about 38.5º.
• Record the temperatures of the animals, beginning at least 90 minutes before injection and continuing for 3 hours after injection of the solution being examined. Any animal showing a temperature variation of 0.6º or more must not be used in the main test.

• Recording of Temperature• Use an accurate temperature-sensing device such as a clinical thermometer or
thermistor or other suitable probes that have been calibrated to assure an accuracy of 0.1º and have been tested to determine that a maximum reading is reached in less than 5 minutes. Insert the thermometer or temperature-sensing probe into the rectum of the test rabbit to a depth of about 5 cm.
• The depth of insertion is constant for any one rabbit in any one test. If an electrical device is used, it should be inserted in the rectum of the rabbit 90 minutes before the injection of the solution being examined and left in position throughout the test. After a period of time not less than that previously determined as sufficient, record the rabbit’s body temperature.
• Preliminary Test (Sham Test)• If animals are used for the first time in a pyrogen test or have not been used
during the 2 previous weeks, condition them 1 to 3 days before testing the substance under examination by injecting intravenously into them 10 ml per kg of body weight of a pyrogen-free saline solution warmed to about 38.5º.
• Record the temperatures of the animals, beginning at least 90 minutes before injection and continuing for 3 hours after injection of the solution being examined. Any animal showing a temperature variation of 0.6º or more must not be used in the main test.

• Main Test• Carry out the test using a group of three rabbits.• Preparation of the sample: Dissolve the substance under examination in, or
dilute with, pyrogen-free saline solution or other solution prescribed in the monograph. Warm the liquid under examination to approximately 38.5º before injection procedure.
• Record the temperature of each animal at intervals of not more than 30 minutes, beginning at least 90 minutes before the injection of the solution under examination and continuing for 3 hours after the injection.
• Not more than 40 minutes immediately preceding the injection of the test dose, record the “initial temperature” of each rabbit, which is the mean of two temperatures recorded for that rabbit at an interval of 30 minutes in the 40-minute period.
• Rabbits showing a temperature variation greater than 0.2º between two successive readings in the determination of “initial temperature” should not be used for the test.
• In any one group of test animals, use only those animals whose “initial temperatures” do not vary by more than 1º from each other, and do not use any rabbit having a temperature higher than 39.8º and lower than 38º.

• Inject the solution under examination slowly into the marginal vein of the ear of each rabbit over a period not exceeding 4 minutes, unless otherwise prescribed in the monograph. The amount of sample to be injected varies according to the preparation under examination and is prescribed in the individual monograph. The volume of injection is not less than 0.5 ml per kg and not more than 10 ml per kg of body weight. Record the temperature of each animal at half-hourly intervals for 3 hours after the injection. The difference between the “initial temperature” and the “maximum temperature” which is the highest temperature recorded for a rabbit is taken to be its response. When this difference is negative, the results are counted as a zero response.
• Interpretation of results: If the sum of the responses of the group of three rabbits does not exceed 1.4º and if the response of any individual rabbit is less than 0.6º, the preparation under examination passes the test.
• If the response of any rabbit is 0.6º or more, or if the sum of the response of the three rabbits exceeds 1.4º, continue the test using five other rabbits.
• If not more than three of the eight rabbits show individual responses of 0.6º or more, and if the sum of responses of the group of eight rabbits does not exceed 3.7º, the preparation under examination passes the test.


Endotoxin Testing (IP/BP)
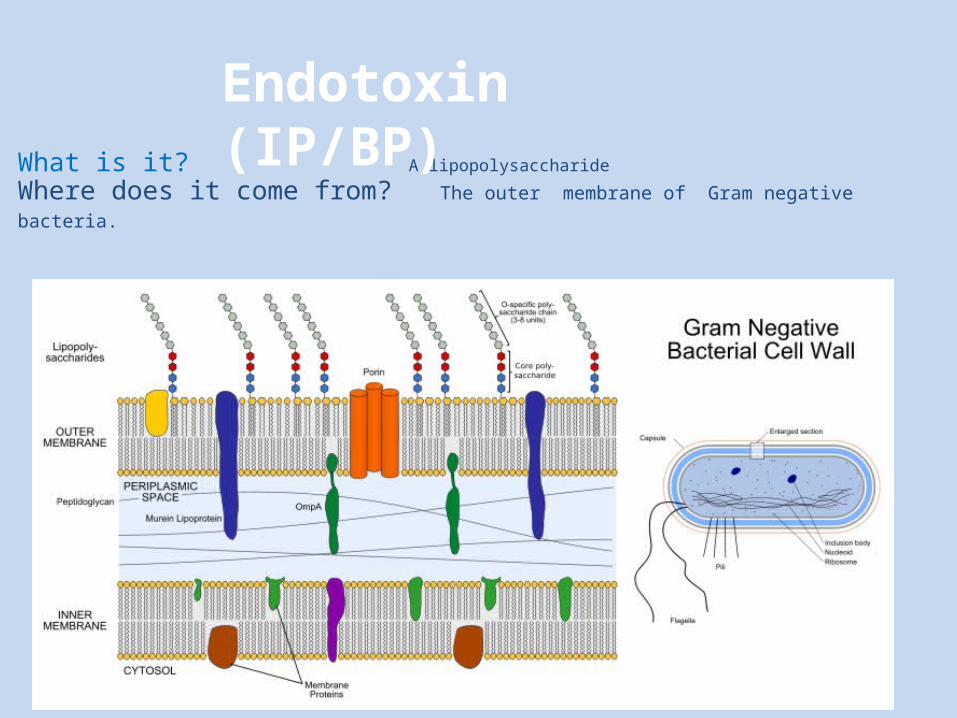
What is it? A lipopolysaccharideWhere does it come from? The outer membrane of Gram negative bacteria.
Endotoxin (IP/BP)

• The test for bacterial endotoxins (BET) measures the concentration of bacterial endotoxins that may be present in the sample or on the article to which the test is applied using a lysate derived from the hemolymph cells or amoebocytes of the horseshoe crab, Limulus polyphemus and other species.
• The addition of a solution containing endotoxins to a solution of the lysate produces turbidity, precipitation or gelation of the mixture.
• However, addition of a chromogenic substrate to a solution of the lysate results in development of color due to release of chromophore from the substrate upon activation by the endotoxin present in the solution.
• The rate of reaction depends on the concentration of endotoxin, the pH and the temperature.
• The reaction requires the presence of certain bivalent cations, a clotting cascade enzyme system and clottable protein, all of which are provided by the lysate.
• The following methods can be used to monitor the endotoxins concentration in a product official in the Pharmacopoeia and to determine whether the product complies with the limit specified in the monograph.
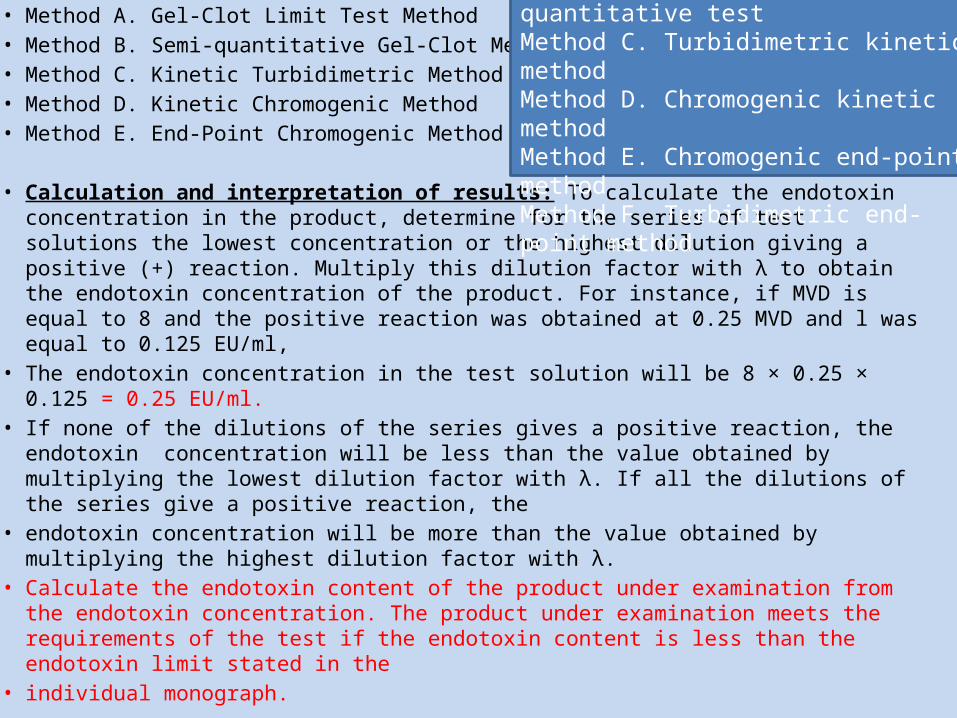
• Method A. Gel-Clot Limit Test Method• Method B. Semi-quantitative Gel-Clot Method• Method C. Kinetic Turbidimetric Method• Method D. Kinetic Chromogenic Method• Method E. End-Point Chromogenic Method
• Calculation and interpretation of results: To calculate the endotoxin concentration in the product, determine for the series of test solutions the lowest concentration or the highest dilution giving a positive (+) reaction. Multiply this dilution factor with λ to obtain the endotoxin concentration of the product. For instance, if MVD is equal to 8 and the positive reaction was obtained at 0.25 MVD and l was equal to 0.125 EU/ml,
• The endotoxin concentration in the test solution will be 8 × 0.25 × 0.125 = 0.25 EU/ml.• If none of the dilutions of the series gives a positive reaction, the endotoxin
concentration will be less than the value obtained by multiplying the lowest dilution factor with λ. If all the dilutions of the series give a positive reaction, the
• endotoxin concentration will be more than the value obtained by multiplying the highest dilution factor with λ.
• Calculate the endotoxin content of the product under examination from the endotoxin concentration. The product under examination meets the requirements of the test if the endotoxin content is less than the endotoxin limit stated in the
• individual monograph.
Method A. Gel-clot method: limit test Method B. Gel-clot method: quantitative test Method C. Turbidimetric kinetic method Method D. Chromogenic kinetic method Method E. Chromogenic end-point method Method F. Turbidimetric end-point method

Powder for injection
• Powders for injection are sterile, solid substances (including freeze-dried materials) which are distributed in their final containers and which, when shaken with the prescribed volume of the appropriate sterile liquid, rapidly form clear and practically particle-free solutions or uniform suspensions.

Tests (IP/BP)• Particulate matter• Uniformity of content• Uniformity of weight • Clarity of solution • Sterility • Bacterial Endotoxin• Pyrogens •
Pharmacopoial


Uniformity of content (IP)• Unless otherwise stated in the individual monograph, Powders for injection
that contain 10 mg or less than 10 mg or less than 10 per cent of active ingredient or that have a unit weight equal to or less than 50 mg comply with the test for Uniformity of content described under Injections.
• For Powders for injection containing more than one active ingredient carry out the test for each active ingredient that corresponds to the above conditions.
• The test is not applicable to Powders for injection containing multivitamins and trace elements.
• The test for Uniformity of content should be carried out only after the content of active ingredient(s) in a pooled sample of the preparation has been shown to be within accepted limits of the stated content.
As per BP
Unless otherwise prescribed or justified and authorized, powders for injections or infusions with a content of active substance less than 2 mg or less than 2 per cent of the total mass, or with a unit mass equal to or less than 40 mg comply with test A for uniformity of content of single-dose preparations. If the preparation contains more than one active substance, the requirement applies only to those substances that correspond to the above conditions.
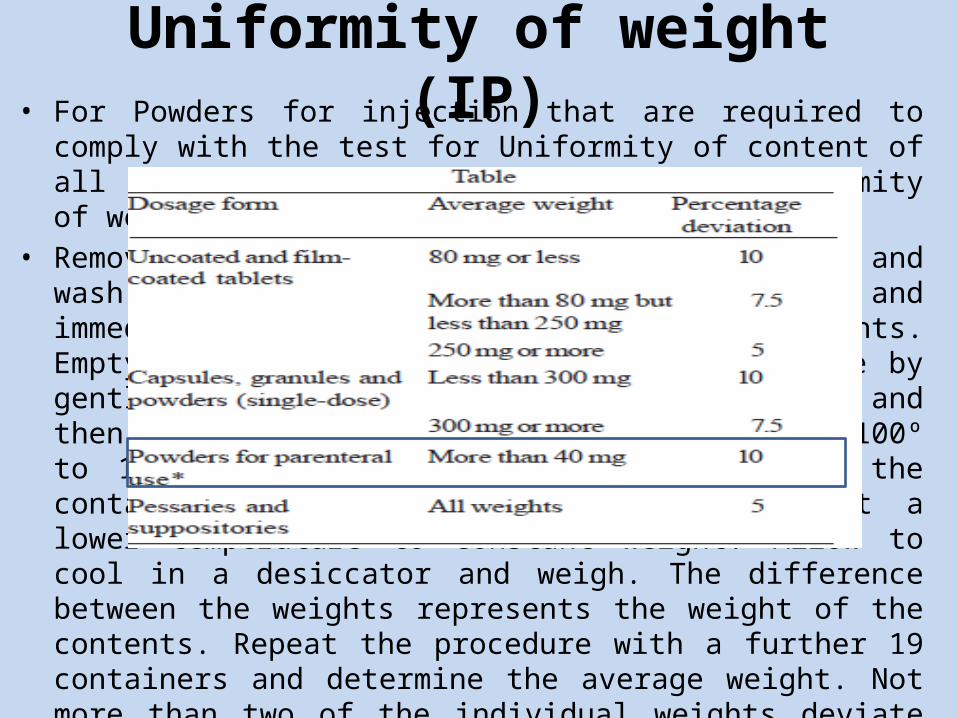
Uniformity of weight (IP)• For Powders for injection that are required to comply with the test for
Uniformity of content of all active ingredients, the test for Uniformity of weight is not required.
• Remove any adherent labels from a container and wash and dry the outside. Open the container and immediately weigh the container and its contents. Empty the container as completely as possible by gentle tapping, rinse if necessary with water and then with ethanol (95 per cent) and dry at 100º to 105º for 1 hour or, if the nature of the container precludes such treatment, dry at a lower temperature to constant weight. Allow to cool in a desiccator and weigh. The difference between the weights represents the weight of the contents. Repeat the procedure with a further 19 containers and determine the average weight. Not more than two of the individual weights deviate from the average weight by more than 10 per cent and none deviates by more than 20 per cent.
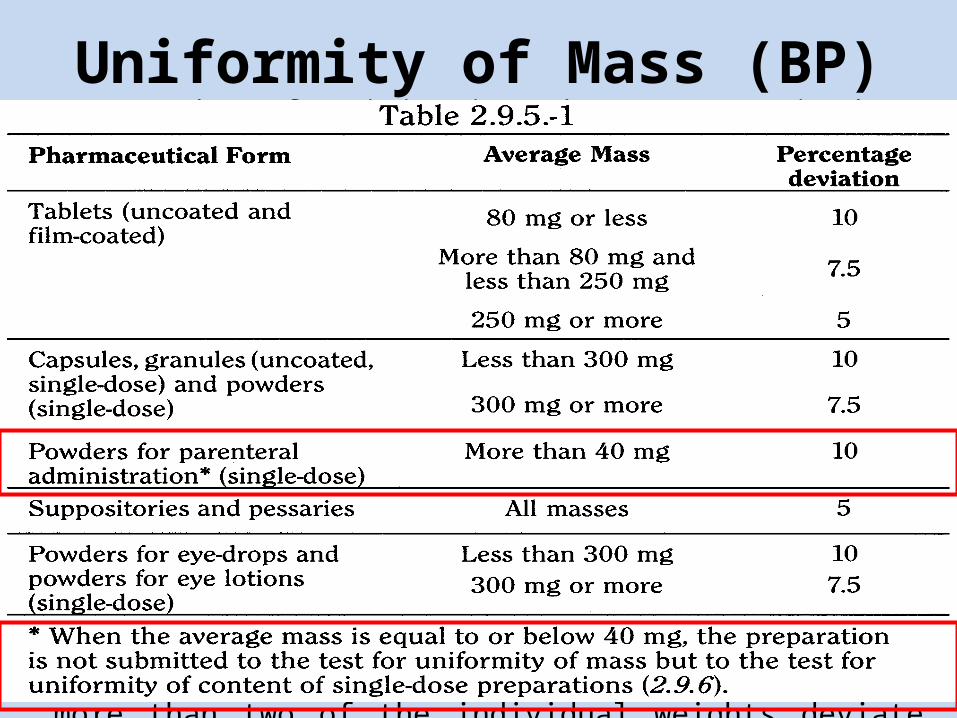
Uniformity of Mass (BP) • For Powders for injection that are required to comply with the test for
Uniformity of content of all active ingredients, the test for Uniformity of weight is not required.
• Remove any adherent labels from a container and wash and dry the outside. Open the container and immediately weigh the container and its contents. Empty the container as completely as possible by gentle tapping, rinse if necessary with water and then with ethanol (95 per cent) and dry at 100º to 105º for 1 hour or, if the nature of the container precludes such treatment, dry at a lower temperature to constant weight. Allow to cool in a desiccator and weigh. The difference between the weights represents the weight of the contents. Repeat the procedure with a further 19 containers and determine the average weight. Not more than two of the individual weights deviate from the average weight by more than 10 per cent and none deviates by more than 20 per cent.

Clarity of solution
• Constitute the injection as directed on the label.
• a) The solid dissolves completely, leaving no visible residue as undissolved matter.
• b) The constituted injection is not significantly less clear than an equal volume of the diluents or of water for injections contained in a similar container and examined in the same manner.

• Particulate matter. Constitute the injection as directed on the label; the solution is essentially free from particles of foreign matter that can be seen on visual inspection.
• Sterility Powders for injection comply with the test for sterility.