discoidin domain receptor 1 null mice are protected...
TRANSCRIPT

Discoidin Domain Receptor 1 Null Mice Are Protectedagainst Hypertension-Induced Renal Disease
Martin Flamant,* Sandrine Placier,* Anita Rodenas,† Cyrile Anne Curat,‡
Wolfgang F. Vogel,§ Christos Chatziantoniou,* and Jean-Claude Dussaule*�
*INSERM U702, Tenon Hospital, Pierre et Marie Curie University, †Department of Pathology, Tenon Hospital, and�AP-HP, Department of Physiology, School of Medicine St. Antoine, Pierre et Marie Curie University, Paris, France;‡Institute of Cardiovascular Physiology, J.W. Goethe University, Frankfurt, Germany; and §Department of LaboratoryMedicine and Pathobiology, University of Toronto, Toronto, Ontario, Canada
A frequent complication of hypertension is the development of chronic renal failure. This pathology usually is initiated byinflammatory events and is characterized by the abnormal accumulation of collagens within the renal tissue. The purpose ofthis study was to investigate the role of discoidin domain receptor 1 (DDR1), a nonintegrin collagen receptor that displaystyrosine-kinase activity, in the development of renal fibrosis. To this end, hypertension was induced with angiotensin in micethat were genetically deficient of DDR1 and in wild-type controls. After 4 or 6 wk of angiotensin II administration, wild-typemice developed hypertension that was associated with perivascular inflammation, glomerular sclerosis, and proteinuria.Systolic pressure increase was similar in the DDR1-deficient mice, but the histologic lesions of glomerular fibrosis andinflammation were significantly blunted and proteinuria was markedly prevented. Immunostaining for lymphocytes, mac-rophages, and collagens I and IV was prominent in the renal cortex of wild-type mice but substantially reduced in DDR1 nullmice. In separate experiments, renal cortical slices of DDR1 null mice showed a blunted response of chemokines to LPS thatwas accompanied by a considerable protection against the LPS-induced mortality. These results indicate the importance ofDDR1 in mediating inflammation and fibrosis. Use of DDR1 inhibitors could provide a completely novel therapeuticapproach against diseases that have these combined pathologies.
J Am Soc Nephrol 17: 3374–3381, 2006. doi: 10.1681/ASN.2006060677
H ypertension frequently is complicated by the devel-opment of chronic renal failure, a complex pathologythat is initiated by inflammatory events that evolve
to increased synthesis and accumulation of extracellular matrix(ECM; mainly collagens) within the renal tissue and lead overtime to loss of function and ESRD. To date, no efficient treat-ment that can stop or, even more desirable, reverse the declineof renal function exists. Therefore, the understanding of thesystems and/or mechanisms that are involved in the develop-ment of renal vascular inflammation and fibrosis will providevaluable information to design specific pharmacologic targetsto treat this incurable disease.
Important advancements have been made regarding themechanisms that are involved in the development of chronicrenal failure. These studies focused mainly in the systems oragents that promote ECM synthesis and progression of renaldisease. We and other investigators, for instance, clearly iden-tified and characterized the signaling pathways that vasocon-strictor peptides are using to activate collagen synthesis (1–4).Less is known about the mechanisms regarding the postsyn-
thesis regulation of ECM, such as matrix anchoring and inter-actions with the cell membrane.
Among the systems that interact with the ECM are the dis-coidin domain receptors (DDR). They are the first identifiedreceptor tyrosine kinases that bind directly to the ECM (5).DDR1 binds all types of collagens and is widely expressed in avariety of tissues, including vascular smooth muscle, mesan-gial, and renal epithelial cells and macrophages (6–8). Aorticsmooth muscle cells that were cultivated from DDR1 null miceshowed decreased proliferation, collagen attachment, migra-tion and, matrix metalloproteinase-2/9 activity compared withcell cultures from wild-type controls (6,9). In addition, neointi-mal development was severalfold reduced in DDR1 null micecompared with wild-type controls after vascular injury in ca-rotids. It is interesting that an important part of this reductionwas due to a dramatic decrease of collagen deposition in theneointima (6).
On the basis of these results, we hypothesized that DDR1could be involved in the mechanisms of the hypertension-associated renal fibrosis. To test this hypothesis, we examinedthe development and the severity of renal vascular and glomer-ular lesions in DDR1 null mice and compared them with wild-type controls, using an experimental model of hypertension-induced renal disease (angiotensin II [AngII] infusion). Wefound that DDR1 null mice are protected against the develop-ment of renal failure because of negligible perivascular and
Received June 29, 2006. Accepted September 12, 2006.
Published online ahead of print. Publication date available at www.jasn.org.
Address correspondence to: Dr. Christos Chatziantoniou, INSERM U702, HopitalTenon, 4 rue de la Chine, Paris 75020, France. Phone: �331-5601-6653; Fax:�331-4364-5448; E-mail: [email protected]
Copyright © 2006 by the American Society of Nephrology ISSN: 1046-6673/1712-3374

glomerular infiltration accompanied by reduced levels of theabnormal accumulation of collagens I and IV.
Materials and MethodsTreatment
Male transgenic mice that weighed 30 to 35 g (4 to 6 mo of age) at thetime of the experiments were fed high-NaCl (5%) mouse food withwater available ad libitum. The higher-than-normal salt diet acceleratesthe development of hypertension and aggravates the degree of therenal and vascular lesions. The generation and genotyping of mice wasdescribed previously (6,10). The original background of the DDR1-nullmice was a mix of 129/Sv with CD1. These mice have been backcrossedfive times to 129/Sv. No difference of the genetic background wasfound between DDR1�/� and wild-type controls after microsatelliteanalysis of DNA samples from 26 mice (13 DDR1�/� and 13 wildtype). The breeding couples that were used in our protocol wereheterozygotes, and experiments were performed using DDR1-null miceand wild-type littermates. All animal procedures were in accordancewith the European Union Guidelines for the Care and use of LaboratoryAnimals.
AngII (Sigma Chemical, St. Louis, MO) was infused subcutaneously(1 �g/kg per min) using osmotic minipumps (Model 1004; Alzet,Cupertino, CA) for 4 or 6 wk. No mortality was observed in DDR1-nulland wild-type littermates for these periods of time. In preliminaryexperiments, we established that this infusion rate of AngII was grad-ually increasing BP (from day 3) and was producing glomerular andvascular lesions (from day 14). A total of 74 DDR1 null and 76 wild-typecontrol mice were used.
Systolic BP was measured twice per week by the tail-cuff methodadapted to the mouse as described previously using the Chart moduleof the MacLab software (1,2). To avoid variations in BP as a result ofday cycle, all measurements were carried out between 9 and 11 a.m.Eight measurements from each mouse were taken at 2-min intervals,and a mean value was determined.
Isolation of Renal Cortical SlicesThe technique to isolate renal cortical slices from mouse kidney was
similar to that previously described (1,2). The cortical tissue was usedfor morphology, immunocytochemistry, or cytokine evaluation accord-ing to the different protocols described next.
Renal HistologyKidneys from at least 10 mice from each group were immersed in
Dubosq solution. After fixation, cortical slices of each kidney wereembedded in paraffin after conventional processing (alcohol dehydra-tion), and 3-�m-thick sections were stained with Masson trichromicsolution for staining of ECM proteins.
Morphologic EvaluationSections of kidneys were examined on a blinded basis for the level of
glomerular ischemia, glomerular sclerosis, and periglomerular andperivascular infiltration using a 0 to 4� injury scale as describedpreviously (1–3). At least 200 glomeruli were scored to estimate thesclerotic index of an animal.
Immunohistochemistry for DDR1, Collagens I and IV, CD3,and F4–80
Four-microgram-thick cryostat sections of renal cortex were fixedwith acetone for 7 min. After blockade of endogenous peroxidase, theywere immunostained with an anti–collagen I or anti–collagen IV (bothat 10 �g/ml; Chemicon, Temecula, CA) or anti-DDR1 (4 �g/ml; Santa
Cruz Biotechnology, Santa Cruz, CA) and the Envision kit (Dako,Carpinteria, CA) was applied for 30 min at room temperature. Stainingwas revealed by applying DAB kit (Dako), hematoxylin QS (Vector,Burlingame, CA), and Permanent Mounting Media Aqueous based(Innovex, Richmond, VA).
For staining of inflammatory cells, 4-�m-thick sections of paraffin-embedded kidneys were dewaxed, heated in citric acid solution, andincubated with a polyclonal rabbit anti-human CD3 (Dako) or a biotin-ylated polyclonal rat anti-mouse F4–80 (Serotec, Oxford, UK) at aconcentration of 3 and 10 �g/ml, respectively. For CD3 immunostain-ing, sections first were treated with biotinylated anti-rabbit IgG, fol-lowed by AB solution treatment. The development was performedusing 3,3-diaminobenzidine–glucose oxidase and light counterstainingwith hematoxylin.
The double-stain experiments with monocytes/macrophages wereperformed using frozen sections fixed to acetone for 7 min and thenwashed with PBS using anti-DDR1 (C-20; Santa Cruz Biotechnology),anti-IgG rabbit TRITC (Jackson Immunoresearch, West Grove, PA), andrat anti-mouse F4–80–FITC (Serotec). Immunofluorescence micro-graphs were obtained using an Olympus BX 51 camera DP70 (Olym-pus, Rungis, France).
Blood Cell CountWhite blood cells were counted and identified using the ADVIA 120
Hematology System (Bayer Diagnostics, Puteaux, France), a technologythat uses peroxidase staining and is based on cytochemical light scatterand light absorption measurements.
Measurement of Urinary Albumin ExcretionThe day before the mice were killed, they were transferred into
metabolic cages and urine samples were collected for a 24-h period.Measurements of microalbuminuria were performed using the Olym-pus System Reagent (ref OSR6167) and an Olympus AU 400 apparatus.Urinary albumin concentration was normalized to urinary creatinineconcentration, and values were expressed as mg albumin/�mol creat-inine.
LPS AdministrationEndotoxemic shock was produced in DDR1�/� and wild-type con-
trols (n � 10 per strain) by intraperitoneal injections of LPS (10 mg/kg).Survival curves were established for a 36-h period. In additional in vitroexperiments, renal cortical slices that were freshly isolated from bothstrains of mice were stimulated by LPS at the concentrations of 100 and1000 ng/ml. Incubation lasted 4 h in RPMI at 37°C, and monocytechemoattractant protein-1 (MCP-1) concentration was measured in thesupernatants using a commercial ELISA kit (R&D System, Minneapolis,MN).
Statistical AnalysesStatistical analyses were performed using ANOVA followed by Pro-
tected Least Significance Difference Fisher test of the Statview softwarepackage. Results with P � 0.05 were considered statistically significant.All values are means � SE.
ResultsDDR1 Protein Expression Is Increased in Renal Cortex andIs Accompanied by Severe Nephroangio- andGlomerulosclerosis during AngII-Induced Hypertension
Continuous perfusion of AngII gradually increased BP inwild-type controls (Figure 1). At the same period, DDR1 ex-pression was increased in renal vessels and within glomeruli as
J Am Soc Nephrol 17: 3374–3381, 2006 DDR1 in Renal Inflammation and Fibrosis 3375

evidenced by immunocytochemistry using an antibody thatwas specific to DDR1 (Figure 2A). In untreated mice, however,DDR1 immunostaining was present at a lesser degree in renalvessels and was almost negligible in glomeruli (Figure 2B). Inagreement with the literature, AngII produced severe vascularand glomerular lesions and profoundly altered the structure ofthe renal vasculature. These alterations were characterizedmainly by the appearance of sclerotic glomeruli as evidencedby the abnormal deposition of ECM, the presence of periglo-merular and perivascular infiltrates, the formation of fibrinwithin the vascular wall, and the deposition of protein aggre-gates in tubular lumen (Figures 3, A, D, and E, and 4). At least50% of vessels showed fibrin-like lesions (Figure 3E). Almost allinflammatory cells were positive to anti-CD3 antibody (specificmarker of lymphocytes) and were massively localized arounddamaged vessels and glomeruli (Figure 5A). A minor fractionof these infiltrating cells were positive for anti-F4–80 antibodystaining, a specific marker of monocytes and macrophages(Figure 5C). Very little staining was observed in or around thetubular interstitium. In contrast, control tissue showed a verysmall number of positive cells (Figure 5, E and F).
To test whether infiltrating cells were DDR1 positive, weperformed experiments to examine whether there was co-local-ization of DDR1 expression with leukocytes in the renal corticaltissue of wild-type mice. As shown in Figure 5, G through I,inflammatory cells did not stain with anti-DDR1 antibody,whereas DDR1 was expressed mainly on smooth muscle cellsof renal vessels. These morphologic alterations were accompa-nied by the appearance of albuminuria (Figure 6).
DDR1 Null Mice Showed Reduced Structural andFunctional Alterations during AngII-Induced Hypertension
Baseline systolic pressure of DDR1 null mice was not differ-ent from that of wild-type controls and increased during AngIItreatment to a similar level compared with hypertensive wild-type mice (Figure 1). In addition, levels of leukocytes were notdifferent between DDR1�/� and wild-type mice under controlconditions (Table 1). Despite the similarity of the pressureresponse, the AngII-induced renal lesions were markedly de-creased in DDR1 null mice (Figures 3, B and F, and 4). Inaddition, immunostaining for lymphocytes or macrophages
Figure 1. Systolic BP increase after angiotensin II (AngII) infu-sion for 42 d in wild-type and discoidin domain receptor 1(DDR1) null mice. Values are means � SEM; n � 10; **P � 0.01versus control.
Figure 2. Representative examples of DDR1 expression revealedby immunocytochemistry in the renal cortex of mice that re-ceived a continuous infusion of AngII (A) or placebo (B) for28 d. G, glomerulus; V, renal vessel. Note the increased expres-sion of DDR1 within glomeruli and renal vessels in the AngII-treated group. Bar � 20 �m.
Figure 3. Representative examples of renal cortical morphology(bar � 20 �m) revealed by Masson’s trichrome stain in wild-type (A, D, and E) or DDR1 null mice (B and F) that weretreated for 28 d with AngII. (C) Wild-type mouse infused withplacebo for 28 d. In A through C the magnification is lower toallow an overall view of the renal cortex. Note the lower levelsof extracellular matrix deposition (green), formation of intratu-bular protein precipitation (red), and the quasi-absence of in-filtrating cells in DDR1 null mice (B and F). The cellular infil-trates are mainly perivascular and periglomerular in the wild-type mice (A and D, arrows); the arrows in E show fibrin-likeformation within renal vessel walls.
3376 Journal of the American Society of Nephrology J Am Soc Nephrol 17: 3374–3381, 2006
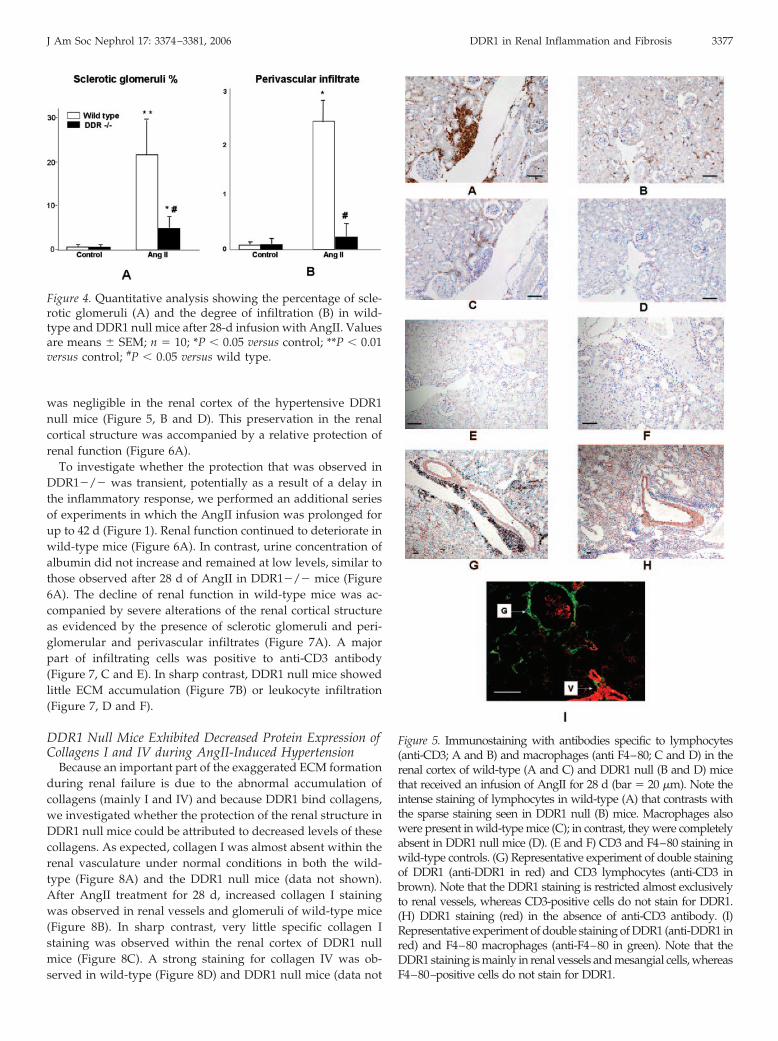
was negligible in the renal cortex of the hypertensive DDR1null mice (Figure 5, B and D). This preservation in the renalcortical structure was accompanied by a relative protection ofrenal function (Figure 6A).
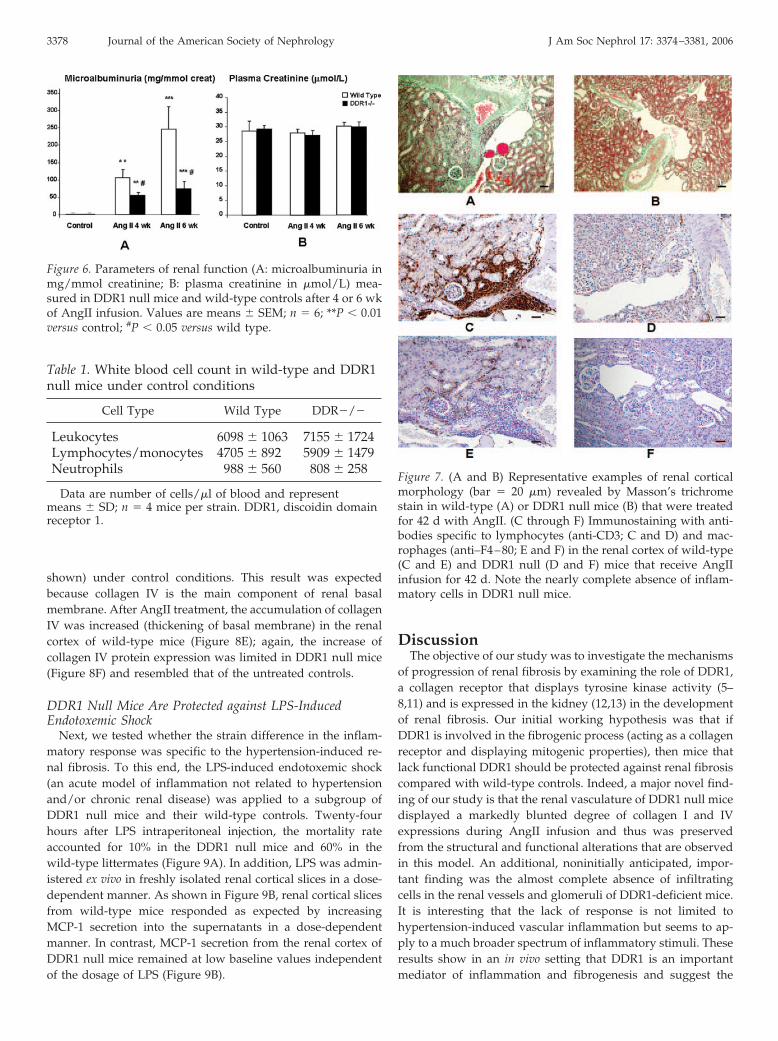
To investigate whether the protection that was observed inDDR1�/� was transient, potentially as a result of a delay inthe inflammatory response, we performed an additional seriesof experiments in which the AngII infusion was prolonged forup to 42 d (Figure 1). Renal function continued to deteriorate inwild-type mice (Figure 6A). In contrast, urine concentration ofalbumin did not increase and remained at low levels, similar tothose observed after 28 d of AngII in DDR1�/� mice (Figure6A). The decline of renal function in wild-type mice was ac-companied by severe alterations of the renal cortical structureas evidenced by the presence of sclerotic glomeruli and peri-glomerular and perivascular infiltrates (Figure 7A). A majorpart of infiltrating cells was positive to anti-CD3 antibody(Figure 7, C and E). In sharp contrast, DDR1 null mice showedlittle ECM accumulation (Figure 7B) or leukocyte infiltration(Figure 7, D and F).
DDR1 Null Mice Exhibited Decreased Protein Expression ofCollagens I and IV during AngII-Induced Hypertension
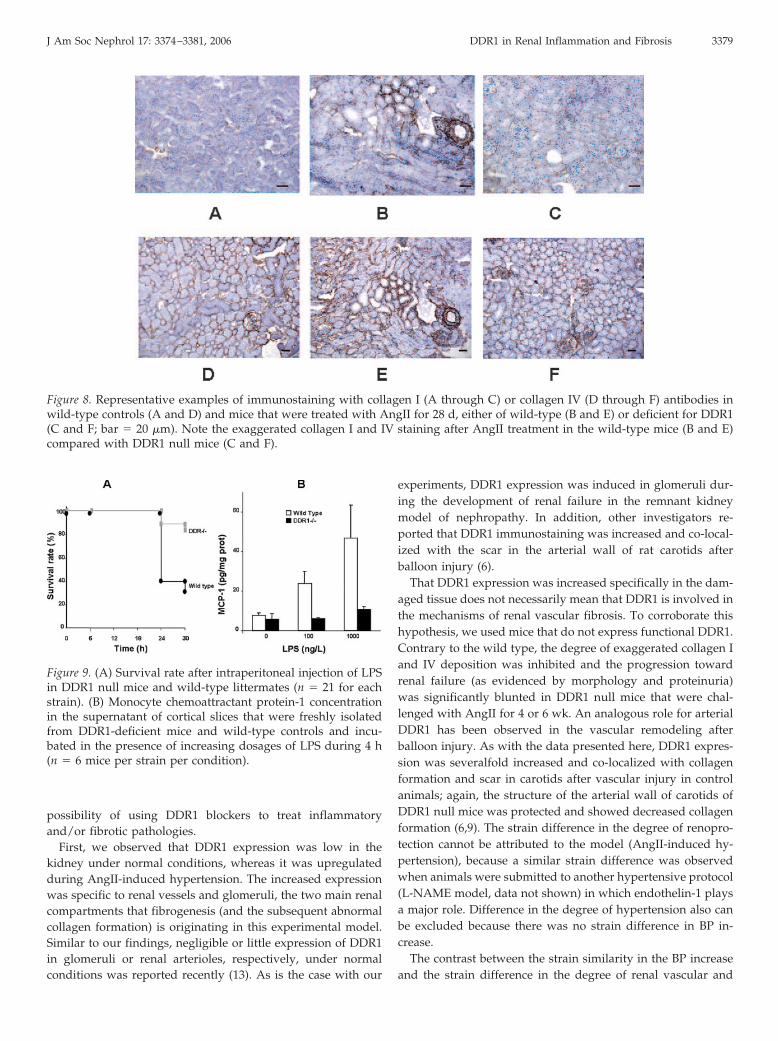
Because an important part of the exaggerated ECM formationduring renal failure is due to the abnormal accumulation ofcollagens (mainly I and IV) and because DDR1 bind collagens,we investigated whether the protection of the renal structure inDDR1 null mice could be attributed to decreased levels of thesecollagens. As expected, collagen I was almost absent within therenal vasculature under normal conditions in both the wild-type (Figure 8A) and the DDR1 null mice (data not shown).After AngII treatment for 28 d, increased collagen I stainingwas observed in renal vessels and glomeruli of wild-type mice(Figure 8B). In sharp contrast, very little specific collagen Istaining was observed within the renal cortex of DDR1 nullmice (Figure 8C). A strong staining for collagen IV was ob-served in wild-type (Figure 8D) and DDR1 null mice (data not
Figure 4. Quantitative analysis showing the percentage of scle-rotic glomeruli (A) and the degree of infiltration (B) in wild-type and DDR1 null mice after 28-d infusion with AngII. Valuesare means � SEM; n � 10; *P � 0.05 versus control; **P � 0.01versus control; #P � 0.05 versus wild type.
Figure 5. Immunostaining with antibodies specific to lymphocytes(anti-CD3; A and B) and macrophages (anti F4–80; C and D) in therenal cortex of wild-type (A and C) and DDR1 null (B and D) micethat received an infusion of AngII for 28 d (bar � 20 �m). Note theintense staining of lymphocytes in wild-type (A) that contrasts withthe sparse staining seen in DDR1 null (B) mice. Macrophages alsowere present in wild-type mice (C); in contrast, they were completelyabsent in DDR1 null mice (D). (E and F) CD3 and F4–80 staining inwild-type controls. (G) Representative experiment of double stainingof DDR1 (anti-DDR1 in red) and CD3 lymphocytes (anti-CD3 inbrown). Note that the DDR1 staining is restricted almost exclusivelyto renal vessels, whereas CD3-positive cells do not stain for DDR1.(H) DDR1 staining (red) in the absence of anti-CD3 antibody. (I)Representative experiment of double staining of DDR1 (anti-DDR1 inred) and F4–80 macrophages (anti-F4–80 in green). Note that theDDR1 staining is mainly in renal vessels and mesangial cells, whereasF4–80–positive cells do not stain for DDR1.
J Am Soc Nephrol 17: 3374–3381, 2006 DDR1 in Renal Inflammation and Fibrosis 3377
shown) under control conditions. This result was expectedbecause collagen IV is the main component of renal basalmembrane. After AngII treatment, the accumulation of collagenIV was increased (thickening of basal membrane) in the renalcortex of wild-type mice (Figure 8E); again, the increase ofcollagen IV protein expression was limited in DDR1 null mice(Figure 8F) and resembled that of the untreated controls.
DDR1 Null Mice Are Protected against LPS-InducedEndotoxemic Shock
Next, we tested whether the strain difference in the inflam-matory response was specific to the hypertension-induced re-nal fibrosis. To this end, the LPS-induced endotoxemic shock(an acute model of inflammation not related to hypertensionand/or chronic renal disease) was applied to a subgroup ofDDR1 null mice and their wild-type controls. Twenty-fourhours after LPS intraperitoneal injection, the mortality rateaccounted for 10% in the DDR1 null mice and 60% in thewild-type littermates (Figure 9A). In addition, LPS was admin-istered ex vivo in freshly isolated renal cortical slices in a dose-dependent manner. As shown in Figure 9B, renal cortical slicesfrom wild-type mice responded as expected by increasingMCP-1 secretion into the supernatants in a dose-dependentmanner. In contrast, MCP-1 secretion from the renal cortex ofDDR1 null mice remained at low baseline values independentof the dosage of LPS (Figure 9B).
DiscussionThe objective of our study was to investigate the mechanisms
of progression of renal fibrosis by examining the role of DDR1,a collagen receptor that displays tyrosine kinase activity (5–8,11) and is expressed in the kidney (12,13) in the developmentof renal fibrosis. Our initial working hypothesis was that ifDDR1 is involved in the fibrogenic process (acting as a collagenreceptor and displaying mitogenic properties), then mice thatlack functional DDR1 should be protected against renal fibrosiscompared with wild-type controls. Indeed, a major novel find-ing of our study is that the renal vasculature of DDR1 null micedisplayed a markedly blunted degree of collagen I and IVexpressions during AngII infusion and thus was preservedfrom the structural and functional alterations that are observedin this model. An additional, noninitially anticipated, impor-tant finding was the almost complete absence of infiltratingcells in the renal vessels and glomeruli of DDR1-deficient mice.It is interesting that the lack of response is not limited tohypertension-induced vascular inflammation but seems to ap-ply to a much broader spectrum of inflammatory stimuli. Theseresults show in an in vivo setting that DDR1 is an importantmediator of inflammation and fibrogenesis and suggest the
Figure 6. Parameters of renal function (A: microalbuminuria inmg/mmol creatinine; B: plasma creatinine in �mol/L) mea-sured in DDR1 null mice and wild-type controls after 4 or 6 wkof AngII infusion. Values are means � SEM; n � 6; **P � 0.01versus control; #P � 0.05 versus wild type.
Table 1. White blood cell count in wild-type and DDR1null mice under control conditions
Cell Type Wild Type DDR�/�
Leukocytes 6098 � 1063 7155 � 1724Lymphocytes/monocytes 4705 � 892 5909 � 1479Neutrophils 988 � 560 808 � 258
Data are number of cells/�l of blood and representmeans � SD; n � 4 mice per strain. DDR1, discoidin domainreceptor 1.
Figure 7. (A and B) Representative examples of renal corticalmorphology (bar � 20 �m) revealed by Masson’s trichromestain in wild-type (A) or DDR1 null mice (B) that were treatedfor 42 d with AngII. (C through F) Immunostaining with anti-bodies specific to lymphocytes (anti-CD3; C and D) and mac-rophages (anti–F4–80; E and F) in the renal cortex of wild-type(C and E) and DDR1 null (D and F) mice that receive AngIIinfusion for 42 d. Note the nearly complete absence of inflam-matory cells in DDR1 null mice.
3378 Journal of the American Society of Nephrology J Am Soc Nephrol 17: 3374–3381, 2006
possibility of using DDR1 blockers to treat inflammatoryand/or fibrotic pathologies.
First, we observed that DDR1 expression was low in thekidney under normal conditions, whereas it was upregulatedduring AngII-induced hypertension. The increased expressionwas specific to renal vessels and glomeruli, the two main renalcompartments that fibrogenesis (and the subsequent abnormalcollagen formation) is originating in this experimental model.Similar to our findings, negligible or little expression of DDR1in glomeruli or renal arterioles, respectively, under normalconditions was reported recently (13). As is the case with our
experiments, DDR1 expression was induced in glomeruli dur-ing the development of renal failure in the remnant kidneymodel of nephropathy. In addition, other investigators re-ported that DDR1 immunostaining was increased and co-local-ized with the scar in the arterial wall of rat carotids afterballoon injury (6).
That DDR1 expression was increased specifically in the dam-aged tissue does not necessarily mean that DDR1 is involved inthe mechanisms of renal vascular fibrosis. To corroborate thishypothesis, we used mice that do not express functional DDR1.Contrary to the wild type, the degree of exaggerated collagen Iand IV deposition was inhibited and the progression towardrenal failure (as evidenced by morphology and proteinuria)was significantly blunted in DDR1 null mice that were chal-lenged with AngII for 4 or 6 wk. An analogous role for arterialDDR1 has been observed in the vascular remodeling afterballoon injury. As with the data presented here, DDR1 expres-sion was severalfold increased and co-localized with collagenformation and scar in carotids after vascular injury in controlanimals; again, the structure of the arterial wall of carotids ofDDR1 null mice was protected and showed decreased collagenformation (6,9). The strain difference in the degree of renopro-tection cannot be attributed to the model (AngII-induced hy-pertension), because a similar strain difference was observedwhen animals were submitted to another hypertensive protocol(L-NAME model, data not shown) in which endothelin-1 playsa major role. Difference in the degree of hypertension also canbe excluded because there was no strain difference in BP in-crease.
The contrast between the strain similarity in the BP increaseand the strain difference in the degree of renal vascular and
Figure 8. Representative examples of immunostaining with collagen I (A through C) or collagen IV (D through F) antibodies inwild-type controls (A and D) and mice that were treated with AngII for 28 d, either of wild-type (B and E) or deficient for DDR1(C and F; bar � 20 �m). Note the exaggerated collagen I and IV staining after AngII treatment in the wild-type mice (B and E)compared with DDR1 null mice (C and F).
Figure 9. (A) Survival rate after intraperitoneal injection of LPSin DDR1 null mice and wild-type littermates (n � 21 for eachstrain). (B) Monocyte chemoattractant protein-1 concentrationin the supernatant of cortical slices that were freshly isolatedfrom DDR1-deficient mice and wild-type controls and incu-bated in the presence of increasing dosages of LPS during 4 h(n � 6 mice per strain per condition).
J Am Soc Nephrol 17: 3374–3381, 2006 DDR1 in Renal Inflammation and Fibrosis 3379

glomerular damage suggests that systemic pressure increaseand development of renal fibrosis are not always associated.This result adds to our previous observations in which thedevelopment or prevention of renal structure was independentof the BP increase during hypertension (1,2,14,15). There is,however, a new element that distinguishes the present from theprevious studies: The prevention and/or protection seen pre-viously was observed during pharmacologic antagonism ofendothelin or AngII and was attributed to the difference of thelocal-renal versus systemic activation of the vasoconstrictor’sreceptor. This is not the case in these studies. A possible expla-nation is that collagen and DDR1 interact in a positive feedbackmanner to amplify the fibrogenic effect of AngII. In agreementwith this hypothesis, DDR1 is activated in vascular smoothmuscle cells (VSMC) that grow in collagen substrate, andVSMC that display functional DDR1 proliferate in a collagensubstrate faster than cells that are deficient of DDR (6,9,16). Inaddition, we found that AngII and endothelin-1 activate colla-gen I and IV genes and induce fibrosis in the renal vasculature(1,2,17). Therefore, we propose that AngII promotes collagen,which in turn upregulates DDR1 expression in the renal vas-culature; once activated, DDR1 promotes remodeling, cellularproliferation, and excessive matrix deposition. When DDR1 isabsent, the feedback is broken at the step of vasoconstrictor-induced collagen formation. This hypothesis also provides anexplanation for why the exaggerated formation of collagenswas blunted but not completely normalized in DDR1 null mice.Alternatively, AngII could transactivate DDR1 independent ofcollagen formation, as it does with some other families oftyrosine kinase receptors such as PDGF and EGF receptors(18,19).
The other major phenotypic difference in our study concernsthe inflammatory response. The prolonged AngII action onvessels frequently is accompanied by the recruitment of infil-trating cells and the induction of vascular inflammation (20,21).Therefore, it was not surprising to see infiltrating cells aroundrenal vessels and glomeruli in the wild-type mice. This con-trasted with the almost complete absence of inflammatory cellsin the renal vasculature of mice that lacked DDR1. This differ-ence cannot be secondary to impaired basal levels of leukocytesbecause no strain difference was observed in the white cellcount under normal conditions. Furthermore, that DDR1 nullmice showed no cell infiltration even at prolonged time ofAngII administration (6 wk) in which renal function worsenedfurther in wild-type controls indicates that DDR1 deficiencyprevented rather than delayed leukocyte infiltration. The dou-ble-stain experiments suggest that the interaction betweenDDR1 and cell infiltration is mediated by the DDR1 expressedon the VSMC of renal vessels in the model of hypertensivenephropathy.
Several in vitro studies have suggested that DDR1 could beinvolved in the inflammatory response. DDR1 is expressedduring differentiation of monocytes to macrophages, and thisdifferentiation is facilitated by collagens (22). A monocyte cellline that was transfected with DDR1 underwent differentiationand responded to inflammatory stimuli, whereas it remainedundifferentiated and nonresponsive to inflammation in the ab-
sence of DDR1 expression (8,23). An amplifier role for DDR1was proposed: Proinflammatory agents induce expression ofDDR1 in the scarred tissue; interaction of DDR1 with collagenof the ECM in turn promotes differentiation of monocytes tomacrophages and upregulation of cytokine secretion through asignaling cascade involving p38 mitogen-activated protein ki-nase and NF-�B (8,22–24). These in vitro studies suggested apossible role of DDR1 in mediating inflammatory responses.Our study is among the first reports to show that DDR1 indeedis a major mediator of the inflammation, and its absence isaccompanied by a deficient inflammatory response in an in vivopathology. In agreement with our results, a recent study ob-served that short-term administration of small interferenceRNA against DDR1 significantly inhibited expression of DDR1in bronchoepithelial cells and protected animals from the de-velopment of bleomycin-induced lung damage (25). It thereforeseems that the role of DDR1 as mediator of inflammatoryresponse can be extended to a more generalized inflammatoryevent, because it applies in pathologies that range from chronicmodels of hypertension to acute models of inflammation (bleo-mycin, LPS). We propose that DDR1 participates in fibrosis asan amplifier of the AngII-induced collagen synthesis and ininflammation as an attractant or facilitator of cellular infiltra-tion and cytokine secretion. When these two physiopathologicmechanisms are met (as is the case with the renal chronicfailure), DDR1 becomes a crucial factor of the progression of thedisease.
ConclusionThis is among the first in vivo studies to investigate the role
of DDR1, a collagen receptor, in the physiopathologic mecha-nism(s) of renal fibrotic disease. Mice that lacked DDR1 showeddecreased collagen formation and an absence of inflammatorycell infiltration and were protected against the development ofhypertension-associated chronic renal failure. Development ofinhibitors or blockers of systems that, like DDR1, mediate bothfibrosis and inflammation could provide a completely noveltherapeutic approach against diseases with these combinedpathologies.
AcknowledgmentsThis work was financially supported by the “Institut National de la
Sante et de la Recherche Medicale,” the “Faculte de Medecine Pierre etMarie Curie,” and an ACI grant from the “Ministere de la Recherche.”
M.F. was research fellow of INSERM.
References1. Chatziantoniou C, Boffa JJ, Ardaillou R, Dussaule JC: Ni-
tric oxide inhibition induces early activation of type I col-lagen gene in renal resistance vessels and glomeruli intransgenic mice: Role of endothelin. J Clin Invest 101: 2780–2789, 1998
2. Boffa JJ, Tharaux PL, Placier S, Ardaillou R, Dussaule JC,Chatziantoniou C: Angiotensin II activates collagen type Igene in the renal vasculature of transgenic mice duringinhibition of nitric oxide synthesis: Evidence for an endo-
3380 Journal of the American Society of Nephrology J Am Soc Nephrol 17: 3374–3381, 2006

thelin-mediated mechanism. Circulation 100: 1901–1908,1999
3. Francois H, Placier S, Flamant M, Tharaux PL, Chansel D,Dussaule JC, Chatziantoniou C: Prevention of renal vascu-lar and glomerular fibrosis by epidermal growth factorreceptor inhibition. FASEB J 18: 926–928, 2004
4. Chatziantoniou C, Dussaule JC: Insights in the mecha-nisms of renal fibrosis: Is it possible to achieve regression?Am J Physiol Renal Physiol 289: F227–F234, 2005
5. Vogel W, Gish GD, Alves F, Pawson T: The discoidindomain receptor tyrosine kinases are activated by collagen.Mol Cell 1: 13–23, 1997
6. Hou G, Vogel W, Bendeck MP: The discoidin domainreceptor tyrosine kinase DDR1 in arterial wound repair.J Clin Invest 107: 727–735, 2001
7. Curat AC, Vogel WF: Discoidin domain receptor 1 controlsgrowth and adhesion of mesangial cells. J Am Soc Nephrol13: 2648–2656, 2002
8. Matsuyama W, Wang L, Farrar WL, Faure M, YoshimuraT: Activation of discoidin domain receptor 1 isoform bwith collagen up-regulates chemokine production in hu-man macrophages: Role of p38 mitogen-activated proteinkinase and NF-kappa B. J Immunol 172: 2332–2340, 2004
9. Hou G, Vogel WF, Bendeck MP: Tyrosine kinase activity ofdiscoidin domain receptor 1 is necessary for smooth mus-cle cell migration and matrix metalloproteinase expression.Circ Res 90: 1147–1149, 2002
10. Vogel WF, Aszodi A, Alves F, Pawson T: Discoidin domainreceptor 1 tyrosine kinase has an essential role in mam-mary gland development. Mol Cell Biol 21: 2906–2917, 2001
11. Shrivastava A, Radziejewski C, Campbell E, Kovac L,McGlynn M, Ryan TE, Davis S, Goldfarb MP, Glass DJ,Lemke G, Yancopoulos GD: An orphan receptor tyrosinekinase family whose members serve as nonintegrin colla-gen receptors. Mol Cell 1: 25–34, 1997
12. Gross O, Beirowski B, Harvey SJ, McFadden C, Chen D,Tam S, Thorner PS, Smyth N, Addicks K, Bloch W, Ni-nomiya Y, Sado Y, Weber M, Vogel WF: DDR1-deficientmice show localized subepithelial GBM thickening withfocal loss of slit diaphragms and proteinuria. Kidney Int 66:102–111, 2004
13. Lee R, Eidman KE, Kren SM, Hostetter TH, Segal Y: Local-ization of discoidin domain receptors in rat kidney.Nephron Exp Nephrol 97: e62–e70, 2004
14. Ying L, Flamant M, Vandermeersch S, Boffa JJ, Chatzianto-niou C, Dussaule JC, Chansel D: Renal effects of omapat-rilat and captopril in salt-loaded nitric oxide deficient rats.Hypertension 42: 937–944, 2003
15. Boffa JJ, Tharaux PL, Dussaule JC, Chatziantoniou C: Re-gression of renal vascular fibrosis by endothelin receptorantagonism. Hypertension 37: 490–496, 2001
16. Vogel WF: Discoidin domain receptors: Structural relationsand functional implications. FASEB J 13[Suppl]: S77–S82,1999
17. Boffa JJ, Ying L, Placier S, Stefanski A, Dussaule JC, Chat-ziantoniou C: Regression of renal vascular and glomerularfibrosis: Role of angiotensin II receptor antagonism andmetalloproteinases. J Am Soc Nephrol 14: 1132–1144, 2003
18. Inagami T, Eguchi S, Numaguchi K, Motley ED, Tang H,Matsumoto T, Yamakawa T: Cross-talk between angioten-sin II receptors and the tyrosine kinases and phosphatases.J Am Soc Nephrol 10[Suppl 11]: S57–S61, 1999
19. Saito Y, Berk BC: Transactivation: A novel signaling path-way from angiotensin II to tyrosine kinase receptors. J MolCell Cardiol 33: 3–7, 2001
20. Fliser D, Buchholz K, Haller H; EUropean Trial on Olme-sartan and Pravastatin in Inflammation and Atherosclero-sis (EUTOPIA) Investigators: Anti-inflammatory effects ofangiotensin II subtype 1 receptor blockade in hypertensivepatients with microinflammation. Circulation 110: 1103–1107, 2004
21. Ruiz-Ortega M, Lorenzo O, Suzuki Y, Ruperez M, Egido J:Proinflammatory actions of angiotensins. Curr Opin Neph-rol Hypertens 10: 321–329, 2001
22. Kamohara H, Yamashiro S, Galligan C, Yoshimura T: Dis-coidin domain receptor 1 isoform-alpha (DDR1alpha) pro-motes migration of leukocytes in three-dimensional colla-gen lattices. FASEB J 15: 2724–2726, 2001
23. Matsuyama W, Faure M, Yoshimura T: Activation of dis-coidin domain receptor 1 facilitates the maturation of hu-man monocyte-derived dendritic cells through the TNFreceptor associated factor 6/TGF-beta-activated protein ki-nase 1 binding protein 1 beta/p38 alpha mitogen-activatedprotein kinase signaling cascade. J Immunol 171: 3520–3532,2003
24. Matsuyama W, Kamohara H, Galligan C, Faure M, Yo-shimura T: Interaction of discoidin domain receptor 1 iso-form beta (DDR1beta) with collagen activates p38 mitogen-activated protein kinase and promotes differentiation ofmacrophages. FASEB J 17: 1286–1288, 2003
25. Matsuyama W, Watanabe M, Shirahama Y, Hirano R, Mit-suyama H, Higashimoto I, Osame M, Arimura K: Suppres-sion of discoidin domain receptor 1 by RNA interferenceattenuates lung inflammation. J Immunol 176: 1928–1936,2006
J Am Soc Nephrol 17: 3374–3381, 2006 DDR1 in Renal Inflammation and Fibrosis 3381