congenital neuroblastoma
TRANSCRIPT

J. clin. Path. (1965), 18, 54
Congenital neuroblastomaA. R. EVANS
From the Group Laboratory, the Royal Infirmary, Wigan
SYNOPSiS The clinical histories and post-mortem findings in five cases of neuroblastoma are
described, and an account given of the microscopic characteristics of the tumours. In four of thecases the tumour was present at birth and was probably so in the fifth case. In only one case was
the presence of the malignant tumour a significant factor in causing death. The differential diagnosisof such tumours is discussed.The accumulated evidence of many recorded cases suggests that neuroblastoma, becoming
manifest in the early months or weeks of life, and congenital tumour, would be included in such agroup, and has an appreciably better prognosis than has this same tumour when it becomes manifestin later childhood. The literature is briefly reviewed to illustrate this aspect of prognosis and possiblereasons for it are indicated.
With the possible exception of retinoblastoma,congenital malignant tumours are not common.
Wells (1940), in an extensive and critical reviewof the available literature to determine the occurrenceand significance of malignant tumours present at orbefore birth, accepted 66 reported cases as establishedbeyond doubt. Of these tumours 33 were sarcomataand 23 neuroblastoma. The neuroblastomataincluded four cases encountered by the reviewer in aseries of 3,000 necropsies on newborn and stillborninfants at the University of Chicago Hospitals. Insummary Wells emphasized that while half of theestablished congenital tumours were sarcomata,there were very nearly as many cases of neuro-blastoma and that a vastly larger proportion of allrecorded neuroblastoma (about 275 cases had beenreported in the literature up to 1938) was present atbirth as compared with the relatively small pro-portion of congenital sarcomata.
Instances of congenital neuroblastoma recordedsince this review by Wells include the cases describedby Hepler (1943), Larimer (1949), Beck and Howard(1951), and Haber and Bennington (1963).
Five further cases, in four of which the tumour wascertainly present at birth, and was probably so in thefifth, are described in this report. Three of themformed part of a total of 59 cases of neuroblastomaregistered at the Children's Tumour Registry of theUniversity of Manchester in the nine-year periodSeptember 1953 to September 1962. I observed theremaining two cases in a series of 28 necropsies onstillborn and newborn infants attheBillinge Hospital,Orrell, during the year 1963.Received for publication 8 April 1964.
CASE 1
The mother was a primigravida aged 25 years. Pregnancywas complicated by mild toxaemia but terminated in anormal labour with the birth of a male child weighing7 lb. 3 oz. The child was 'slow to gasp' and, despiteclearance of airways of mucus andoxygenadministration,respiration remained impaired. Clinical examinationrevealed a tense swelling in the upper abdomen. Thishad a firm lower edge palpable just below the level ofthe umbilicus. Peripheral blood examination showed ahaemoglobin level of 13-3 g./100 ml.; leucocytes 18,100/c.mm. (neutrophils 60%, lymphocytes 37%, monocytes2%, eosinophils 1 %), The film showed no abnormalwhite cells, and a degree of polychromasia with occasionallate normoblasts which was considered to be within thenormal limits for a child of this age. The blood urea levelwas 36 mg./100 ml. An intravenous pyelogram outlinedan apparently normal renal tract. Respiratory distressbecame progressive, despite continuous oxygen admini-stration and other restorative measures, and the generalcondition deteriorated rapidly. Death occurred 34 hoursafter birth.
NECROPSY
There was no evidence of birth trauma. The faceand finger nail beds were deeply cyanosed.The lungs were congested and showed widespread
patchy atalectatic areas.The left adrenal was occupied by a soft rounded
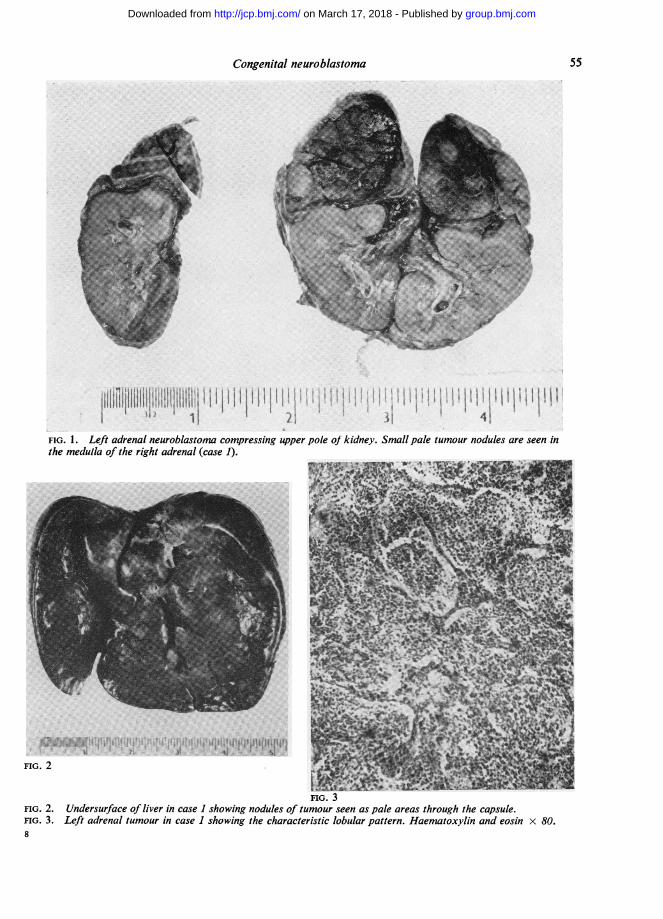
lobulated vascular medullary tumour 3 x 2 x1-5 cm. The cut surface was dark red, mottled withlighter pink and yellow areas and showed anincomplete rim of recognizable cortex. The tumourwas well demarcated, compressing but not infiltrat-ing the upper pole of the left kidney (Fig. 1).
54
group.bmj.com on March 17, 2018 - Published by http://jcp.bmj.com/Downloaded from
Congenital neuroblastoma
?7ttIiS..;1*1T5: 41
FIG. 1. Left adrenal neuroblastona compressing upper pole of kidney. Small pale tumour nodules are seen inthe medulla of the right adrenal (case 1).
¼%:~.~-:".: ~-~:,.~ ~ .I ii~.. 'A
FIG. 2
FIG. 3FIG. 2. Undersurface of liver in case 1 showing nodules of tumour seen as pale areas through the capsule.FIG. 3. Left adrenal tumour in case 1 showing the characteristic lobular pattern. Haematoxylin and eosin x 80.
55
I. I.!.Mh
group.bmj.com on March 17, 2018 - Published by http://jcp.bmj.com/Downloaded from
A. R. Evans
*e~~~~~~~~~~~~~~~~~~~~~~~~~~~~~..D-*..X
't VA
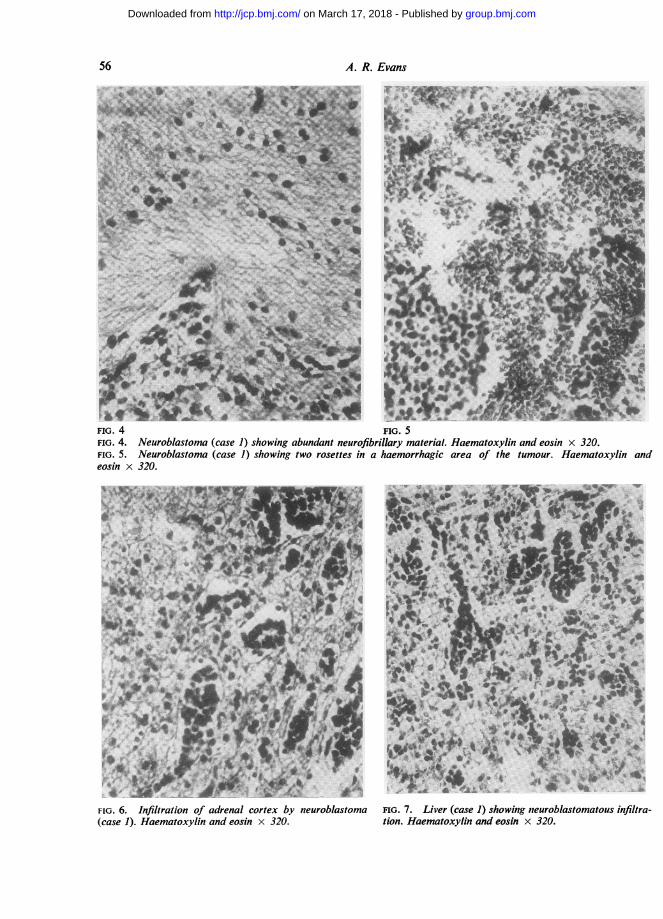
FIG. 4 FIG. 5FIG. 4. Neuroblastoma (case 1) showing abundant neurofibrillary material. Haematoxylin and eosin x 320.FIG. 5. Neuroblastoma (case 1) showing two rosettes in a haemorrhagic area of the tumour. Haematoxylin andeosin x 320.
" -j p ri.^ r v r;.*-0 sre ;0 c.:7so-4A-
FIG. 6. Infiltration of adrenal cortex by neuroblastoma FIG. 7. Liver (case 1) showing neuroblastomatous infiltra-(case 1). Haematoxylin and eosin x 320. tion. Haematoxylin and eosin x 320.
56
group.bmj.com on March 17, 2018 - Published by http://jcp.bmj.com/Downloaded from

Congenital neuroblastoma
The right adrenal appeared normal in size andshape but the cut surface showed a few small greytumour nodules in the medulla, each measuring1 to 2 mm. in diameter.The liver was large, weighing 600 g. The edges
were smooth and rounded and the whole of thesurface was mottled by numerous low nodules ofgreyish yellow tumour tissue varying in diameterfrom 1 to 5 mm. The cut surface showed a similarpattern of tumour infiltration (Fig. 2).A single para-aortic lymph node was enlarged by
tumour. The remaining organs showed no abnormal-ity apart from varying degrees of congestion.
HISTOLOGY
LEFT ADRENAL Sections showed a neuroblastoma.The tumour cells were small with darkly stainingnuclei showing rather dense chromatin and obscureintranuclear detail. Nuclear diameter varied fromabout 8,u to 14[s. In the smallest cells the nucleiwere deeply and evenly staining with only aninconspicuous rim of scanty cytoplasm. Suchlymphocyte-like cells have been called sympatho-gonia from their resemblance to those cells ofneuroectodermal origin which are the formativecells of the sympathetic nervous system (Blacklock,1934). Most of the tumour cells were somewhatlarger having a rather more open coarsely vesicular,nuclear chromatin pattern and sometimes showingmore plentiful cytoplasm. The larger cells have beendescribed as sympathoblasts, again from theirresemblance to those cells in developing sympatheticnervous tissue which are derived from sympatho-gonia and represent a first step in differentiationtowards mature ganglion cells.
Architecturally the tumour presented a lobulatedpattern (Fig. 3) with sheets of tumour cells dividedby a vascular supporting stroma into acinar areasof varying diameter. The stroma varied in densityfrom broad bands of mature fibrous tissue in someareas to delicate strands of capillaries with littleconnective tissue in others. Within the lobules thetumour cells showed a characteristic 'loosely packed'pattern with an abundance of eosinophilic fibrillarymaterial arranged as a loose meshwork or as parallelbundles of fibrils lying between the cells and cellgroups (Fig. 4). Occasional individual fibrils wereseen arising as extensions from the cytoplasm oftumour cells and some small groups of cells showedball-like clumping and sometimes a circular arrange-ment around a central core of fibrillary or granularmaterial (Fig. 5). Such rosettes, often described as acharacteristic feature of neuroblastoma, were illformed and scarce.Numerous areas of haemorrhage were seen
throughout the tumour and the incomplete rim ofrather stretched cortex was extensively infiltrated bytumour (Fig. 6).
RIGHT ADRENAL The medulla contained areas ofneuroblastoma and cortical infiltration was evident.
LIVER Normal hepatic architecture was obliteratedby extensive tumour infiltration (Fig. 7). Short cordsof liver cells with occasional haemopoietic foci werewidely separated from one another by strands andsheets of tumour cells, and in some areas tumourcells were seen lying within portal vessels. Thetumour cells were similar to those seen in theprimary growth but fibrillary substances was notplentiful and of course the lobular pattern of theprimary tumour was not apparent.
CASE 2
The mother was 40 years of age and had had threeprevious pregnancies, all normal, and all three of thechildren were alive and well. The current pregnancy wasnormal but terminated with premature rupture ofmembranes followed by breech delivery of a male child(5 lb. 10 oz.) some 55 hours later. Satisfactory normalrespiration was never established and the child diedwithin 30 minutes of birth.
NECROPSY
There was no evidence of birth trauma. The faceand nail beds were deeply cyanosed.The lungs showed extensive atalectasis and were
congested with a few small areas of pulmonaryhaemorrhage.The left adrenal contained a soft, rounded,
demarcated medullary tumour measuring 3 x 3 x
Itllillllllllt!l6lIIttIit I I I IjI|tt I I tI I Ii IFIG.8. Lf adreatIIIIIIIIIm1` u I 2)i
FIG. 8. Left adrenal tumour (case 2).
57
group.bmj.com on March 17, 2018 - Published by http://jcp.bmj.com/Downloaded from
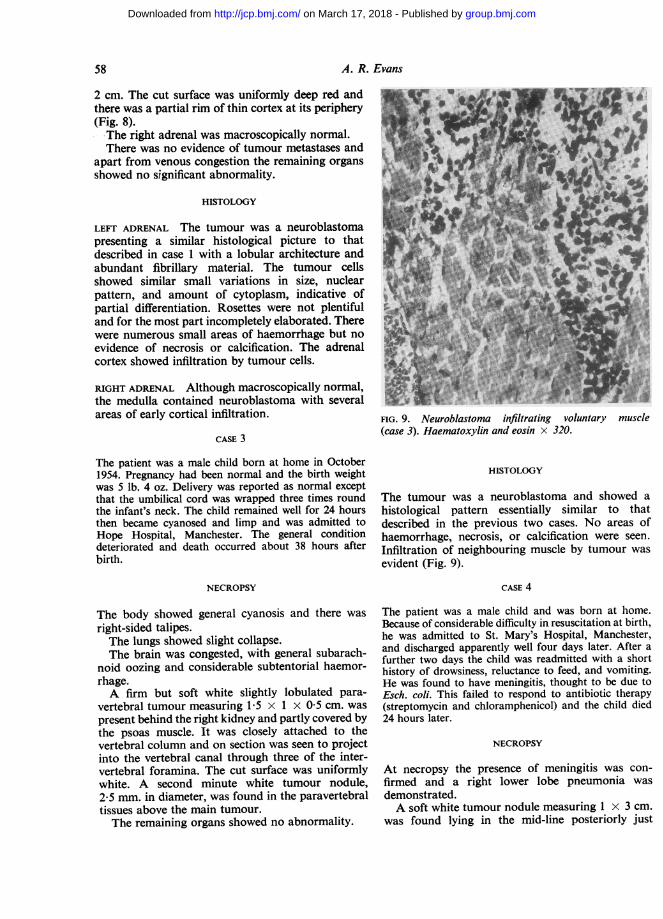
2 cm. The cut surface was uniformly deep red andthere was a partial rim of thin cortex at its periphery(Fig. 8).The right adrenal was macroscopically normal.There was no evidence of tumour metastases and
apart from venous congestion the remaining organsshowed no significant abnormality.
HISTOLOGY
LEFT ADRENAL The tumour was a neuroblastomapresenting a similar histological picture to thatdescribed in case 1 with a lobular architecture andabundant fibrillary material. The tumour cellsshowed similar small variations in size, nuclearpattern, and amount of cytoplasm, indicative ofpartial differentiation. Rosettes were not plentifuland for the most part incompletely elaborated. Therewere numerous small areas of haemorrhage but noevidence of necrosis or calcification. The adrenalcortex showed infiltration by tumour cells.
RIGHT ADRENAL Although macroscopically normal,the medulla contained neuroblastoma with severalareas of early cortical infiltration.
CASE 3
The patient was a male child born at home in October1954. Pregnancy had been normal and the birth weightwas 5 lb. 4 oz. Delivery was reported as normal exceptthat the umbilical cord was wrapped three times roundthe infant's neck. The child remained well for 24 hoursthen became cyanosed and limp and was admitted toHope Hospital, Manchester. The general conditiondeteriorated and death occurred about 38 hours afterbirth.
NECROPSY
The body showed general cyanosis and there was
right-sided talipes.The lungs showed slight collapse.
The brain was congested, with general subarach-noid oozing and considerable subtentorial haemor-rhage.A firm but soft white slightly lobulated para-
vertebral tumour measuring 1-5 x 1 x 05 cm. was
present behind the right kidney and partly covered bythe psoas muscle. It was closely attached to thevertebral column and on section was seen to projectinto the vertebral canal through three of the inter-vertebral foramina. The cut surface was uniformlywhite. A second minute white tumour nodule,2-5 mm. in diameter, was found in the paravertebraltissues above the main tumour.The remaining organs showed no abnormality.
FIG. 9. Neuroblastoma infiltrating voluntary muscle(case 3). Haematoxylin and eosin x 320.
HISTOLOGY
The tumour was a neuroblastoma and showed ahistological pattern essentially similar to thatdescribed in the previous two cases. No areas ofhaemorrhage, necrosis, or calcification were seen.Infiltration of neighbouring muscle by tumour wasevident (Fig. 9).
CASE 4
The patient was a male child and was born at home.Because of considerable difficulty in resuscitation at birth,he was admitted to St. Mary's Hospital, Manchester,and discharged apparently well four days later. After afurther two days the child was readmitted with a shorthistory of drowsiness, reluctance to feed, and vomiting.He was found to have meningitis, thought to be due toEsch. coli. This failed to respond to antibiotic therapy(streptomycin and chloramphenicol) and the child died24 hours later.
NECROPSY
At necropsy the presence of meningitis was con-firmed and a right lower lobe pneumonia wasdemonstrated.A soft white tumour nodule measuring 1 x 3 cm.
was found lying in the mid-line posteriorly just
58 A. R. Evans
group.bmj.com on March 17, 2018 - Published by http://jcp.bmj.com/Downloaded from
Congenital neuroblastoma
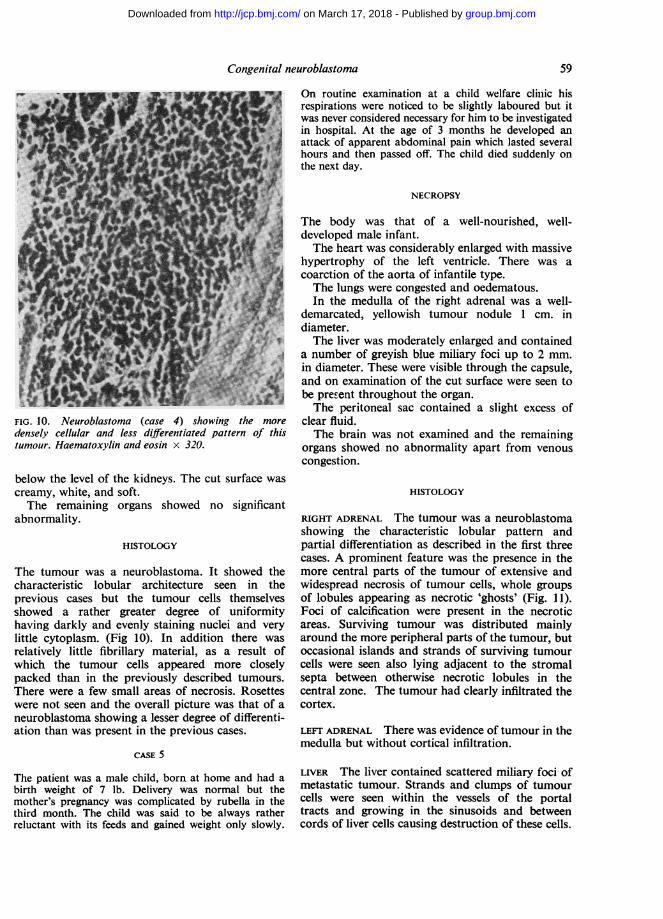
FIG. 10. Neuroblastoma (case 4) showing the more
densely cellular and less differentiated pattern of thistumour. Haematoxylin and eosin x 320.
below the level of the kidneys. The cut surface was
creamy, white, and soft.The remaining organs showed no significant
abnormality.
HISTOLOGY
The tumour was a neuroblastoma. It showed thecharacteristic lobular architecture seen in theprevious cases but the tumour cells themselvesshowed a rather greater degree of uniformityhaving darkly and evenly staining nuclei and verylittle cytoplasm. (Fig 10). In addition there was
relatively little fibrillary material, as a result ofwhich the tumour cells appeared more closelypacked than in the previously described tumours.There were a few small areas of necrosis. Rosetteswere not seen and the overall picture was that of aneuroblastoma showing a lesser degree of differenti-ation than was present in the previous cases.
CASE 5
The patient was a male child, born at home and had abirth weight of 7 lb. Delivery was normal but themother's pregnancy was complicated by rubella in thethird month. The child was said to be always ratherreluctant with its feeds and gained weight only slowly.
On routine examination at a child welfare cliniic hisrespirations were noticed to be slightly laboured but itwas never considered necessary for him to be investigatedin hospital. At the age of 3 months he developed anattack of apparent abdominal pain which lasted severalhours and then passed off. The child died suddenly onthe next day.
NECROPSY
The body was that of a well-nourished, well-developed male infant.The heart was considerably enlarged with massive
hypertrophy of the left ventricle. There was acoarction of the aorta of infantile type.The lungs were congested and oedematous.In the medulla of the right adrenal was a well-
demarcated, yellowish tumour nodule 1 cm. indiameter.The liver was moderately enlarged and contained
a number of greyish blue miliary foci up to 2 mm.in diameter. These were visible through the capsule,and on examination of the cut surface were seen tobe prerent throughout the organ.The peritoneal sac contained a slight excess of
clear fluid.The brain was not examined and the remaining
organs showed no abnormality apart from venouscongestion.
HISTOLOGY
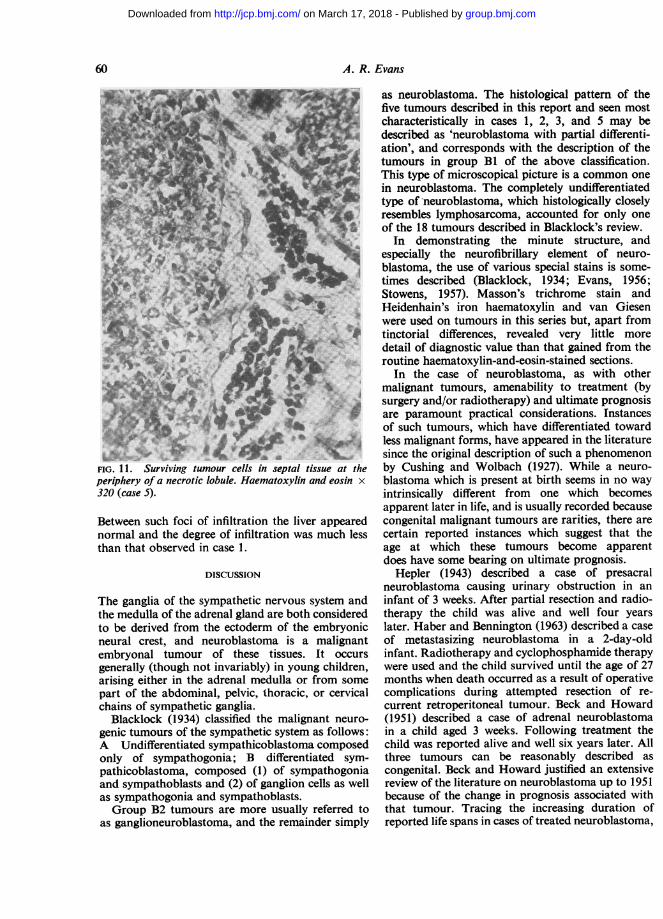
RIGHT ADRENAL The tumour was a neuroblastomashowing the characteristic lobular pattern andpartial differentiation as described in the first threecases. A prominent feature was the presence in themore central parts of the tumour of extensive andwidespread necrosis of tumour cells, whole groupsof lobules appearing as necrotic 'ghosts' (Fig. 11).Foci of calcification were present in the necroticareas. Surviving tumour was distributed mainlyaround the more peripheral parts of the tumour, butoccasional islands and strands of surviving tumourcells were seen also lying adjacent to the stromalsepta between otherwise necrotic lobules in thecentral zone. The tumour had clearly infiltrated thecortex.
LEFT ADRENAL There was evidence of tumour in themedulla but without cortical infiltration.
LIVER The liver contained scattered miliary foci ofmetastatic tumour. Strands and clumps of tumourcells were seen within the vessels of the portaltracts and growing in the sinusoids and betweencords of liver cells causing destruction of these cells.
59
group.bmj.com on March 17, 2018 - Published by http://jcp.bmj.com/Downloaded from
A. R. Evans
At6 ..
FIG. 11. Surviving tumour cells in septal tissue at theperiphery of a necrotic lobule. Haematoxylin and eosin x320 (case 5).
Between such foci of infiltration the liver appearednormal and the degree of infiltration was much lessthan that observed in case 1.
DISCUSSION
The ganglia of the sympathetic nervous system andthe medulla of the adrenal gland are both consideredto be derived from the ectoderm of the embryonicneural crest, and neuroblastoma is a malignantembryonal tumour of these tissues. It occursgenerally (though not invariably) in young children,arising either in the adrenal medulla or from some
part of the abdominal, pelvic, thoracic, or cervicalchains of sympathetic ganglia.
Blacklock (1934) classified the malignant neuro-
genic tumours of the sympathetic system as follows:A Undifferentiated sympathicoblastoma composedonly of sympathogonia; B differentiated sym-pathicoblastoma, composed (1) of sympathogoniaand sympathoblasts and (2) of ganglion cells as wellas sympathogonia and sympathoblasts.Group B2 tumours are more usually referred to
as ganglioneuroblastoma, and the remainder simply
as neuroblastoma. The histological pattern of thefive tumours described in this report and seen mostcharacteristically in cases 1, 2, 3, and 5 may bedescribed as 'neuroblastoma with partial differenti-ation', and corresponds with the description of thetumours in group Bi of the above classification.This type of microscopical picture is a common onein neuroblastoma. The completely undifferentiatedtype of neuroblastoma, which histologically closelyresembles lymphosarcoma, accounted for only oneof the 18 tumours described in Blacklock's review.
In demonstrating the minute structure, andespecially the neurofibrillary element of neuro-blastoma, the use of various special stains is some-times described (Blacklock, 1934; Evans, 1956;Stowens, 1957). Masson's trichrome stain andHeidenhain's iron haematoxylin and van Giesenwere used on tumours in this series but, apart fromtinctorial differences, revealed very little moredetail of diagnostic value than that gained from theroutine haematoxylin-and-eosin-stained sections.
In the case of neuroblastoma, as with othermalignant tumours, amenability to treatment (bysurgery and/or radiotherapy) and ultimate prognosisare paramount practical considerations. Instancesof such tumours, which have differentiated towardless malignant forms, have appeared in the literaturesince the original description of such a phenomenonby Cushing and Wolbach (1927). While a neuro-blastoma which is present at birth seems in no wayintrinsically different from one which becomesapparent later in life, and is usually recorded becausecongenital malignant tumours are rarities, there arecertain reported instances which suggest that theage at which these tumours become apparentdoes have some bearing on ultimate prognosis.
Hepler (1943) described a case of presacralneuroblastoma causing urinary obstruction in aninfant of 3 weeks. After partial resection and radio-therapy the child was alive and well four yearslater. Haber and Bennington (1963) described a caseof metastasizing neuroblastoma in a 2-day-oldinfant. Radiotherapy and cyclophosphamide therapywere used and the child survived until the age of 27months when death occurred as a result of operativecomplications during attempted resection of re-current retroperitoneal tumour. Beck and Howard(1951) described a case of adrenal neuroblastomain a child aged 3 weeks. Following treatment thechild was reported alive and well six years later. Allthree tumours can be reasonably described ascongenital. Beck and Howard justified an extensivereview of the literature on neuroblastoma up to 1951because of the change in prognosis associated withthat tumour. Tracing the increasing duration ofreported life spans in cases of treated neuroblastoma,
60
group.bmj.com on March 17, 2018 - Published by http://jcp.bmj.com/Downloaded from

Congenital neuroblastoma
they stressed that early diagnosis and treatment hadbecome of considerably increased value, and pointedout that of a total of 475 cases of neuroblastomareported up to that time, 47 (about 10%) hadbeen recorded as cures. These figures reflected a farmore favourable outlook than that given byBlacklock in 1934, when, from his studies of neuro-blastoma in patients at the Royal Hospital for SickChildren at Glasgow, he concluded that the averageduration of life was eight or nine weeks from thefirst symptoms.
Stowens (1957), in a review of neuroblastoma andrelated tumours, drew attention to the relationshipbetween prognosis and the age at which the tumourbecame apparent. Drawing his material from thefiles of the Armed Forces Institute of Pathology, heincluded in his review a series of 105 cases of neuro-blastoma for which complete information wasavailable, and, defining a survivor as one who livedat least 18 months without recurrence or metastases,found that 14 (13 %) of these cases were survivors.Forty-five of the 105 cases were in the first year oflife when diagnosed and of these 13 (29%) weresurvivors. The remaining survivor was one of 27cases which fell into the age group 2 to 4 years attime of diagnosis, giving a survival rate of 4% forthis age group. There were no survivors in theremaining 33 patients whose ages at diagnosisranged from 5 years upwards and included onepatient aged over 50 years. Basing his conclusionson the differences in biological behaviour of thesetumours, including the disproportionately highsurvival rate in cases where the tumour becamemanifest in the first year of life, Stowens suggestedthat tumours, which on histological grounds areclassified as neuroblastoma, actually comprise twogroups. 1 A group of tumours of congenital originresulting from disruption of normal embryogenesisof the sympathetic nervous system, which he called'congenital neuroblastoma' and considered themalways to become manifest in the first few years oflife, and that about 30% of them were amenable tosurgical cure and might even undergo spontaneousregression. 2 A group which resulted frommalignant degeneration of fully mature elements ofthe sympathetic nervous system. These, he suggested,be designated 'neuroblastic sarcoma'. Whileanatomically indistinguishable from 'congenitalneuroblastoma', 'neurogenic sarcoma' was con-sidered to occur with random frequency at all agesand to be invariably fatal.Such a hypothesis is somewhat at variance with
the more usually accepted concept of neuroblastoma,as for example as defined by Evans (1956), whodescribed neuroblastoma as a malignant tumour ofembryonic tissue which is composed of sympatho-
gonia and sympathoblasts, and the continued growthof which is dependent on the proliferation of thesecells. While agreeing that some neuroblastomata arecongenital in origin, Stowens (1957) considered thatsuch a postulate failed to explain the origin of thistumour when occurring in the older age groups. Insuch cases it would be necessary to assume that thetumours are capable of either lying dormant forconsiderable periods of time or having vastlydifferent growth rates. Neither assumption seemedto him probable.However this may be, it seems possible that the
disproportionately high survival rate noticed incases of neuroblastoma diagnosed and treated in thefirst year of life is not necessarily due to any intrinsicdifference in tumours which happen to be present ator shortly after birth but to the fact that routinephysical examinations, for one reason or another,are relatively frequent just at this age. If one suchexamination should reveal the presence of a palpableabdominal mass in a child possibly otherwiseasymptomatic, it could lead to the rapid diagnosisand treatment of a neuroblastoma arising in theadrenal or some part of the abdominal sympatheticchain, both of which are common sites of origin forthis tumour. Further, such a more or less chancefinding, when it occurs before actual symptomsreferable to the tumour or its metastases becomemanifest, may initiate treatment at a stage when itschances of success are relatively high. In thisconnexion it is interesting to note that in eight ofStowen's survivors, diagnosis and early surgeryfollowed the more or less accidental finding of apalpable abdominal mass on routine physicalexamination in otherwise asymptomatic infants(Stowens, 1957). Again, in two of the cases alreadymentioned (Beck and Howard, 1951; Haber andBennington, 1963) early treatment of neuroblastomafollowed similar chance discoveries of palpableabdominal masses on clinical examination, andsomewhat similar patterns of diagnosis followed bysuccessful treatment are seen in cases described byWyatt and Farber (1941) and by Koop, Kiesewetter,and Horn (1955). Schaffer (1960), commenting on theincreasingly favourable outlook in neuroblastoma,and quoting from the reviews of Farber (1940), ofWittenborg (1950), and of Koop et al. (1955),concluded that there is general agreement that theyounger the child is when the disease becomesmanifest the better the prognosis. Whatever thereason, be it an intrinsic property of the congenitaltumour, a tendency towards maturation to lessmalignant forms with the possibility of spontaneousregression, or merely that an abdominal mass is morelikely to be promptly discovered in an infant orneonate than in an older child, it is apparent that
61
group.bmj.com on March 17, 2018 - Published by http://jcp.bmj.com/Downloaded from

neuroblastoma, present at birth, has a surprisinglyfavourable prognosis. In at least four of the fivecases described in this report death was not directlydue to a malignant tumour.
Since neuroblastoma, becoming manifest in thefirst year of life and particularly in the neonatalperiod, commonly presents as an abdominal masswhich may be either the primary tumour or fre-quently a liver enlarged by metastases, the problemin diagnosis lies in distinguishing it from othercauses of palpable abdominal tumour. Conditionswhich require consideration in the differentialdiagnosis include: 1 Renal enlargements not dueto tumour, such as hydronephrosis and cystickidney; 2 the enlarged livers of haemolyticdisease and congenital syphilis, and the congestedliver of cardiac failure seen in some cases of con-genital heart lesion. The additional clinical features,serological tests for syphilis, and demonstrationof rhesus or other incompatibility, are obviousaids in distinguishing these conditions. It maybe mentioned that the occasional occurrenceof jaundice and, more frequently, a moderate degreeof anaemia in congenital neuroblastoma may causeinitial confusion, especially when it is rememberedthat the peripheral blood film of a newborn mayshow considerable polychromasia together withnumbers of normoblasts which may be misinter-preted as evidence of haemolytic disease when seenin association with the above-mentioned features.3 Other abdominal tumours of infancy, includingWilms' tumour, lymphosarcoma, and the leuk-aemias, may be congenital, for example, fivecongenital malignant renal tumours recorded byWells (1940), but the fact of the tumour beingpresentat or shortly after birth is more in favour of itsbeing a neuroblastoma. Blood and marrow examin-ation will help to differentiate the leukaemias.Wilms' tumour is probably the commonest of thesethree tumours to present as an abdominal massshortly after birth and is usually unilaterally situated.Intravenous pyelography may aid diagnosis byrevealing pelvic and calyceal distortion by intra-renal tumour or downward displacement of thekidney by adrenal neuroblastoma. Examination ofurinary catecholamines may demonstrate the alter-ation in cathecholamine metabolism which has beenfound in association with neuroblastoma but not intumours which are not of neural crest origin(Voorhess and Gardner, 1962). Also it seems thatsuch estimations of urinary catecholamine, itsprecursors and metabolites, may be of value infollowing up cases of treated neuroblastoma anddetermining the effects of therapy.
In most cases confirmation of diagnosis will followsurgical exploration when the site of the tumour
will become evident. Neuroblastoma commonlyarises from one or other adrenal, or from sympatheticganglionic tissue in the vicinity of the kidneys,although, of course, it may arise from such gangli-onic tissue at practically any site. Ultimate micro-scopic diagnosis from examination of the removedor biopsied tumour will present little difficultyprovided it is possible to demonstrate such charac-teristic features as cellular variation with somedifferentiation, presence of neurofibrillary materialand perhaps rosette formation. If, however, thetumour is of the undifferentiated variety, moredifficulty in microscopic assessment is to be expected.Such undifferentiated forms are composed entirelyof sympathogonia (Blacklock, 1934), which aresmall round cells with darkly staining nuclei offairly uniform size and, in the absence of much orany fibrillary material, would present a fairlyuniform picture of closely packed cells very muchresembling that seen in lymphosarcoma. In describ-ing such a tumour, Blacklock drew attention to thetendency for cells at the growing margin to becomeclumped into ball-like clusters similar to thosefound in developing sympathetic nervous tissue, andconsidered this a valuable feature in differentiatingsuch a tumour from lymphosarcoma. Probably manyneuroblastomata of this type have been described assarcoma, lymphosarcoma, or round cell tumour ofthe adrenal, especially in reports antedating the des-cription of the distinguishing characteristics ofneuro-blastoma by Wright (1910).
I wish to thank Dr. J. K. Steward of the Children'sTumour Registry, University of Manchester, for provid-ing material for histological examination and for accessto records for three cases; Drs. H. B. Marsden and G.J. Crawford for permission to use their post-mortemreports on two cases; Drs. R. M. Forrester and G. M.Komrower for allowing me to publish cases sometime intheir care; Dr. J. Schrager for helpful advice; and Mr. J.Molyneux for the photographs.
REFERENCES
Beck, S. M. Jr., and Howard, P. J. (1951). Amer. J. Dis. Child., 82,325.
Blacklock, J. W. S. (1934). J. Path. Bact., 39, 27.Cushing, H., and Wolbach, S. B. (1927). Amer. J. Path., 3, 203.Evans, R. W. (1956). Histological Appearances of Tumours. Living-
stone, Edinburgh and London.Farber, S. (1940). Amer. J. Dis. Child., 60, 749.Haber, S. L., and Bennington, J. L. (1963). Arch. Path., 76, 121.Hepler, A. B. (1943). J. Urol., 49, 777.Koop, C. E., Kiesewetter, W. B., and Horn, R. C. (1955). Pediatrics,
16, 652.Larimer, R. C. (1949). J. Pediat., 34, 365.Schaffer, A. J. (1960). Diseases of the Newborn. Saunders, Philadelphia
and London.Stowens, D. (1957). Arch. Path., 63, 451.Voorhess, M. L., and Gardner, L. I. (1962). J. clin. Endocr., 22, 126.Wells, H. G. (1940). Arch. Path., 30, 535.Wittenborg, M. H. (1950). Radiology, 54, 679.Wright, J. H. (1910). J. exp. Med., 12, 556.Wyatt, G. M., and Farber, S. (1941). Amer. J. Roentgenol., 46, 485.
62 A. R. Evans
group.bmj.com on March 17, 2018 - Published by http://jcp.bmj.com/Downloaded from

Congenital neuroblastoma
A. R. Evans
doi: 10.1136/jcp.18.1.541965 18: 54-62 J Clin Pathol
http://jcp.bmj.com/content/18/1/54Updated information and services can be found at:
These include:
serviceEmail alerting
the online article. article. Sign up in the box at the top right corner of Receive free email alerts when new articles cite this
Notes
http://group.bmj.com/group/rights-licensing/permissionsTo request permissions go to:
http://journals.bmj.com/cgi/reprintformTo order reprints go to:
http://group.bmj.com/subscribe/To subscribe to BMJ go to:
group.bmj.com on March 17, 2018 - Published by http://jcp.bmj.com/Downloaded from