cancer a disease of the genome – genomic instability a disease of the cell cycle – checkpoint...
TRANSCRIPT


Cancer
• A disease of the genome– Genomic instability
• A disease of the cell cycle– Checkpoint failures
• A disease of the aged
• Benign vs. Malign• What makes a cancer cell so special?
– Interaction with basal lamina– Interactions with other cells– Blood Supply

DysplasticACF
EarlyAdenoma
IntermediateAdenoma
LateAdenoma Carcinoma MetastasisNormal
Epithelium
APCother changes?
DCCDPC4/JV18?
K-Ras
MMRDeficiency

Cancer Progression Example: Colorectal Cancer
• APC mutations initiate the neoplastic process.• Patients with FAP inherit APC mutations and develop numerous dysplastic aberrant
crypt foci ACF, which then progress• MMR deficiency speeds up this process• K-RAS is an oncogene that requires only one genetic event for its activation.• The other specific genes indicated are tumor suppressor genes that require two
genetic events (one in each allele) for their inactivation• Chromosome 18q21 may contain several different tumor suppressor genes
involved in colorectal neoplasia, with DCC, DPC4, and JV18–1 genes proposed as candidates

Cancer
• The pathway to carcinogenesis consists of 4-7 rate-limiting events.
• Tumor development proceeds by a process analogous to Darwinian evolution.
• The multiple lines of cellular defense may explain why cancer is not more frequent during an average human lifetime.
• If you live long enough, you will develop cancer.
Cell, Vol. 100, 57–70, January, 2000

Cancer• The pathway to carcinogenesis consists of 4-7 rate-
limiting events.– Recently, as little as 2: activation of a pro-growth (c-myc)
factor and a survival factor (Bcl-XL) or suppression of a proapoptotic p53 or p19ARF
• Tumor development proceeds by a process analogous to Darwinian evolution.
• The multiple lines of cellular defense and the maintainence of DNA fidelity may explain why cancer is not more frequent during an average human lifetime.
• If you live long enough, you will develop cancer.
Cell, Vol. 100, 57–70, January, 2000

Cell, Vol. 100, 57–70, January, 2000
General Charactistics of Cancer
• All cancers must acquire several of the same six hallmark capabilities
• Means and order of acquisition vary significantly.
The catalyst of acquiring these is Genomic instability.

Cancer: A disease of the Genome
Genomic Stability• Given the standard mutation rate in dividing cells,
coincident with the fidelity of DNA replication, the time it would take to achieve a sufficiently mutated state for cancer would far surpass the human lifespan.– A normal error frequency of 1 base-pair change in roughly 109
base pairs for each cell generation (1 in a billion).– A single gene that encodes an average-sized protein (~103 base
pairs) suffers a mutation once in about 106 cell generations. – This number is roughly consistent with the evolutionary estimate -
one mutation appears in an average gene in the germ line every 200,000 years.

Genomic Instability
DNA damage/mutation Apoptosis
Mitosis
ArrestRepair
AttemptExtensiv
e
damage
CorrectionNormal Cell
Damage toApoptoticPathway
MitosisDamage
Cancer Cell
Inability toSense Damage
DamagedRepair
Mechanisms
Inability toarrest
Rapid accumulationof genomic damage.
leads to accumulation of more key cancer cellcharacteristics
Perpetuation oferrors to daughter
cells.
DNA damage/mutation

Environmental Causes of Genomic Instability and Aneuploidy
• Oncogenic Viruses• Chemical carcinogens• Ionizing radiation
Loss of checkpoints +
circumvention of apoptosis =
genomic instability

Cell, Vol. 100, 57–70, January, 2000
General Charactistics of Cancer
• All cancers must acquire the same six hallmark capabilities
• Means and order of acquisition vary significantly.
• The catalyst of acquiring these is Genomic instability.

Characteristics of Tumor Cells
Genome Instability
• Given the normal mutation rate in dividing cells, the time it would take to achieve a sufficiently mutated state for cancer would far surpass the human lifespan.
• A normal error frequency of 1 base-pair change in roughly 109 base pairs for each cell generation (1 in 1,000,000,000, or one in a billion).
• A single gene that encodes an average-sized protein (~103 base pairs) suffers a mutation once in about 106 cell generations.
• This number is roughly consistent with the evolutionary estimate - one mutation appears in an average gene in the germ line every 200,000 years.

Cancer: A Disease of the Genome
Genomic Stability• Given the standard mutation rate in dividing cells, coincident
with the fidelity of DNA replication, the time it would take to achieve a sufficiently mutated state for cancer would far surpass the human lifespan
– A normal error frequency of 1 base-pair change in roughly 109 base pairs for each cell generation (1 in a billion)
– A single gene that encodes an average-sized protein (~103 base pairs) suffers a mutation once in about 106 cell generations
– This number is roughly consistent with the evolutionary estimate – one mutation appears in an average gene in the germ line every 200,000 years.

How is Fidelity Normally Maintained?
• Protein level: Wobble Effect.• Genetic level: Distinct DNA repair mechanisms.– DNA polymerase exonuclease activity– post-replicative recognition, excision, and repair of
mismatches
• Other strategy Affecting stability:• 5% exons• Extensive introns

Genomic Instability
DNA damage/mutation Apoptosis
Mitosis
Arrest RepairAttempt
Extensive
damage
CorrectionNormal Cell
Damage toApoptoticPathway
MitosisDamage
Cancer Cell
Inability toSense Damage
DamagedRepairMechanisms
Inability toarrest
Rapid accumulationof genomic damage.
leads to accumulation of more key cancer cellcharacteristics
Perpetuation oferrors to daughtercells.

DNA Damage
Arrest
Normal Cell
Cancer Cell
RepairDamage
Apoptosis
Mitosis
DamagedRepair
Mechanism
InabilityTo SenseDamage
Inability to Arrest
DNADamage
Damage to Apoptotic Pathway
Mitosis Rapid accumulationof genetic damage
Perpetuation ofErrors to
Daughter Cells
Genomic Instability
DNA Damage

Aneuploidy• Euploidy: Having a chromosome number that is an exact multiple
of the monoploid number.• Aneuploidy: Having a chromosome number that is not an exact
multiple of the usually haploid number.• Develops from defects in the process of chromosome segregation.• Most benign tumors are diploid.• All malignant tumors are aneuploid; this is one of the identifying
characteristics of malignant tumor cells.• Is required for cell immortalization; it is a critical rate-limiting step
of tumorigenesis – therefore, it develops early in the progression.
• Caused by the same factors which cause genomic instability.

Causes of Genomic Instability and Aneuploidy
• Oncogenic Viruses• Chemical carcinogens• Ionizing radiation• Spontaneous occurrence in p53-/- cell lines
derived from Li-Fraumeni patients (germline mutation in the p53 gene)

Once genomic instability is in effect, cancer cells then follow a quicker path towards the accumulation of necessary
mutations required to reach full-blown carcinogenesis.

Cell 2000 Jan 7;100(1):57-70

Acquired Characteristics of Tumor Cells
1. Self-Sufficiency in Growth Signals2. Insensitivity to Antigrowth Signals3. Evading Apoptosis4. Limitless Replicative Potential
(Immortalization)5. Sustained Angiogenesis6. Tissue Invasion and Metastasis

Characteristics of Tumor Cells
I: Self-Sufficiency in Growth Signals• many oncogenes mimick normal growth signaling.
• 3 strategies: – 1: Alteration of extracellular growth signals (Autocrine stimulation)
• PDGF (platelet-derived growth factor) • TGFa (tumor growth factor alpha)
– 2: Signal Transducer alteration• A: wild-type GF receptors are overexpressed: hypersensitivity
– EGF-R/erbB• B: Structural alteration of receptors can result in ligand-independent signaling
– EGF receptor lacking most of the cytoplasmic domain fire constitutively – 3: Intracellular circuits
• RAS - 25% of human tumors, constitutive mutants
Cell, Vol. 100, 57–70, January, 2000

II: Insensitivity to Antigrowth Signals
• Normal tissue: – Antiproliferative signals operate to maintain
cellular quiescence and tissue homeostasis.• Soluble growth inhibitors • Immobilized inhibitors embedded
– the extracellular matrix – the surfaces of nearby cells
– These growth-inhibitory signals, like their positively acting counterparts, are received by transmembrane cell surface receptors coupled to intracellular signaling circuits.

E Cadhedrin
CAMs
antigrowth signals
b-catenin
Lef/Tcf transcription factor
N-CAM
Wilms tumor, neuroblastoma, and small cell lung cancer
Epithelial Cancers
antigrowth signals
Growth Inhibitory Signal via binding
Extracellular Matrix
Integrins
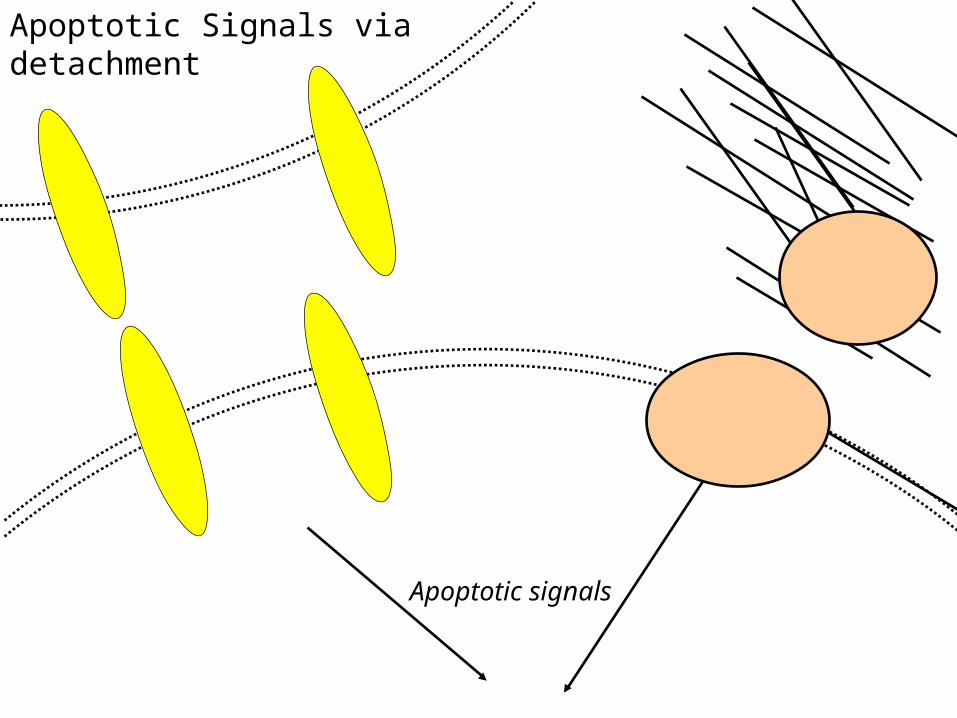
Apoptotic signals
Apoptotic Signals via detachment

III: Evading Apoptosis: future lecture• Bcl-2: Oncogene that doesn’t promote growth, but inhibits death. usually
found mutated in conjunction with c-myc, which is a powerful activator of the cell cycle.
• p53: 50% of human tumors have this gene mutated.• Decoy death receptors• etc.

DR-4/5
TRAIL
DcR
TRAIL
Decoy Receptor
Can still bind ligand, but unable to transduce signal
DD

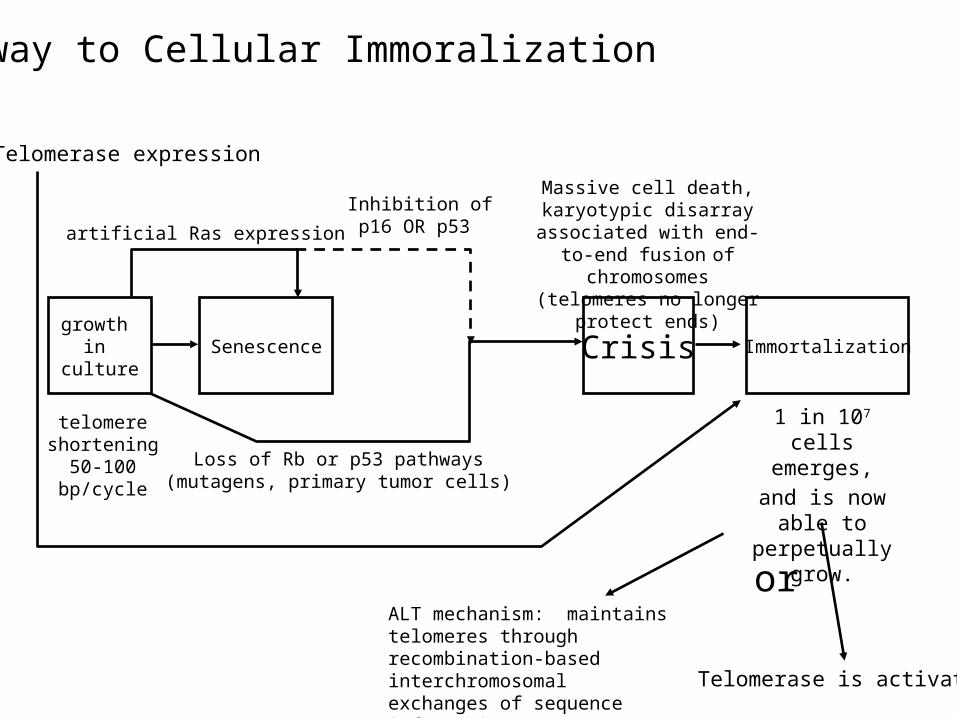
IV: Limitless Replicative Potential: Immortalization
• Even cells which are free to divide at their whim eventually stop growing.
• This is due to telomere shortening.• How does a cell arrive at immortality?

growth in culture
Senescence
Loss of Rb or p53 pathways(mutagens, primary tumor cells)
ImmortalizationCrisis
Massive cell death, karyotypic disarray associated with end-to-end fusion of chromosomes (telomeres no longer protect ends)
1 in 107 cells emerges,and is now able to perpetually grow.
Pathway to Cellular Immoralization
ALT mechanism: maintains telomeres through recombination-based interchromosomal exchanges of sequence information Telomerase is activated
or
Telomerase expression
artificial Ras expression
Inhibition of p16 OR p53
telomere shortening50-100 bp/cycle
growth in
cultureSenescence
Loss of Rb or p53 pathways(mutagens, primary tumor cells)
ImmortalizationCrisis
Massive cell death, karyotypic disarray
associated with end-to-end fusion of chromosomes
(telomeres no longer protect ends)
1 in 107 cells emerges,
and is now able to perpetually grow.
Pathway to Cellular Immoralization
ALT mechanism: maintains telomeres through recombination-based interchromosomal exchanges of sequence information Telomerase is activated
or
Telomerase expression
artificial Ras expression
Inhibition of p16 OR p53
telomere shortening
50-100 bp/cycle

HDACs & HATS: Chromatin Remodelling Overview
B

Pathways to Cellular Immortalization
GrowthIn
cultureSenescence
Many divisionsCRISIS Immortalization
Ectopic Ras expressionInhibition of p16 or p53
Massive cell death; karyotypic disarray associated with end-to-end fusion of chromosomes (telomeres no longer protect ends)
1 in 107 cellsEmerges
And is now able toPerpetually grow
Telomerase isActivated
ALT Mechanism: MaintainsTelomeres through recombination-based interchromasomal exchanges of sequence information
Telomerase expression
Loss of Rb / p53 pathways

V: Sustained Angiogenesis (growth of new blood vessels)
• All cells, normal and cancer alike, must reside within 100 µm of a capillary blood vessel.
• A tumor which fails to activate angiogenesis can only grow to ~2mm in diameter.

5. Sustained Angiogenesis
• All cells, normal and cancer alike, must reside within 100 mm of a capillary blood vessel.
• A tumor which fails to activate angiogenesis can only grow to ~2mm in diameter

Simplistic model of TumorAngiogenesis
endothelial cells

VI: Tissue Invasion and Metastasis
• 90% of human cancer deaths are due to metastatic tumors.
• Invasion and metastasis are exceedingly complex processes, and their genetic and biochemical determinants remain incompletely understood.

Molecular Defects in Cancer:Two Paradigms
Tumor SuppressorsLoss of function• p53• p16• p53• p16• Rb• pTEN• p27• etc. etc.
OncogenesGain of function• c-myc• Ras• Cyclin D• CDK4• Bcl-2• Survivin• Oncogenic Viral Proteins
Each protein is resopnsible in evading one or more of the core six characteristics necessary for tumorigenesis

Fighting Cancer

Chemotherapy• The idea behind chemotherapy is simple: kill cells which are
proliferating. • Chemotherpeutic agents induce apoptosis (specific to the
cycling cells) and necrosis (nonspecific and therefore less effective)
• The Failure of Chemotherapy– unfortunately, many cancers have damaged apoptotic response
pathways, leading to ineffective treatment– 50% of cancers have a mutation in p53, which is crucial for
apoptotic response.– Many normal cells proliferate at the same rate (if not faster) than
tumor cells– Many tumor cells don’t necessarily grow faster as they simply just
don’t die.

Chemotherapy• The idea behind chemotherapy is simple: kill cells that are proliferating
through inducing death pathways.• Chemotherapeutic agents induce apoptosis (specific to the cycling cells)
and necrosis (nonspecific and therefore less effective)
• The failure of chemotherapy:– unfortunately, many cancers have damaged apoptotic response pathways,
leading to ineffective treatment.– 50% of cancers have mutated p53, crucial for the apoptotic response.– Many normal cells proliferate at the same rate (if not faster) than tumor cells– Many tumor cells don’t necessarily grow faster as they simply just don’t die
– Strategy: Reactivate death pathways, then hit them with a toxic stress (synergy between chemotherapy and a sensitizing agent).

Targeting Apoptosis in Cancer Therapy
• Cancer can be distilled to two fundamental genetic lesions: – excessive proliferation– constitutively active survival signaling pathway
• To compensate, pro-growth signals are also capable of inducing pro-apoptotic signals (c-myc)– Provides a failsafe mechanism to offset oncogenic capacity– irony: creates a pressure for tumor cell selection.
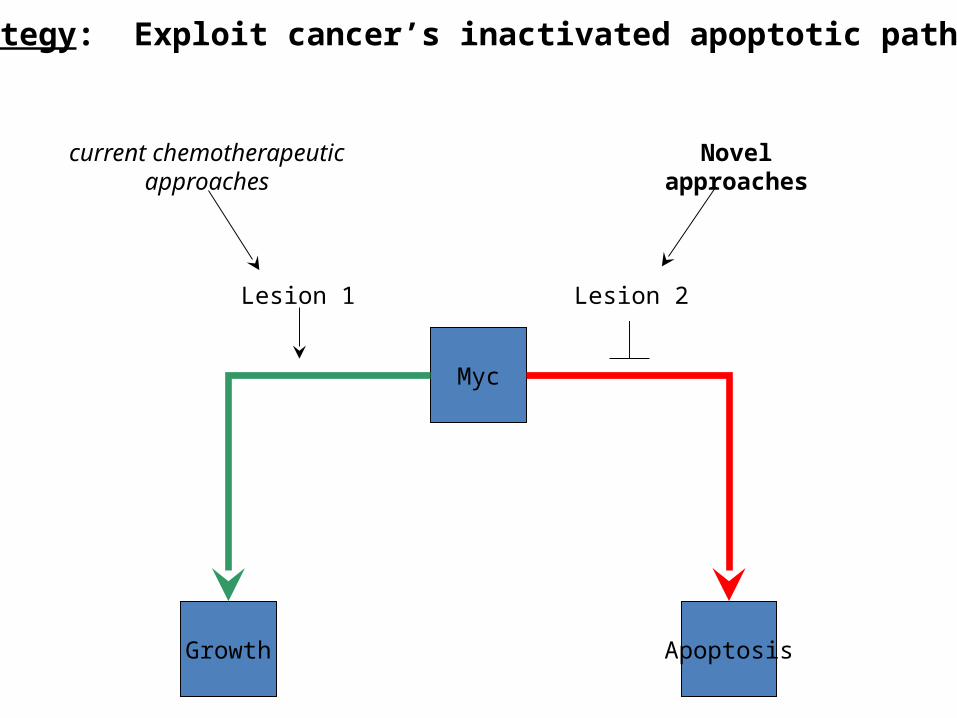
Myc
Growth Apoptosis
Lesion 1 Lesion 2
current chemotherapeuticapproaches
Novelapproaches
Strategy: Exploit cancer’s inactivated apoptotic pathways
