bioinformatica t3-scoring matrices
TRANSCRIPT


FBW
08-10-2012
Wim Van Criekinge

Inhoud Lessen: Bioinformatica
GEEN LES


Ove
rvie
w
• Introduction
– Short recap on databases
– Definitions
• Scoring Matrices
– Theoretical
– Empirial
• PAM (pam-simulator.pl)
• BLOSUM
• Pairwise alignment
– Dot-plots (dotplot-simulator.pl)
Overview

Major sites
NCBI - The National Center for Biotechnology Information
http://www.ncbi.nlm.nih.gov/
The National Center for Biotechnology Information (NCBI) at
the National Library of Medicine (NLM), a part of the National
Institutes of Health (NIH).
ExPASy - Molecular Biology Server
http://expasy.hcuge.ch/www/
Molecular biology WWW server of the Swiss Institute of
Bioinformatics (SIB). This server is dedicated to the analysis of
protein sequences and structures as well as 2-D PAGE
EBI - European Bioinformatics Institute
http://www.ebi.ac.uk/

Anno 2002 Anno 2003

Anno 2004

Anno 2005

Anno 2006

Anno 2007

Anno 2009

Anno 2010
Anno 2010

Anno 2011

Anno 2012

Anno 2012

Ove
rvie
w
• Introduction
– Short recap on databases
– Definitions
• Scoring Matrices
– Theoretical
– Empirial
• PAM (pam-simulator.pl)
• BLOSUM
• Pairwise alignment
– Dot-plots (dotplot-simulator.pl)
Overview

IdentityThe extent to which two (nucleotide or amino acid)
sequences are invariant.
HomologySimilarity attributed to descent from a common ancestor.
Definitions
RBP: 26 RVKENFDKARFSGTWYAMAKKDPEGLFLQDNIVAEFSVDETGQMSATAKGRVRLLNNWD- 84
+ K ++ + + GTW++MA+ L + A V T + +L+ W+
glycodelin: 23 QTKQDLELPKLAGTWHSMAMA-TNNISLMATLKAPLRVHITSLLPTPEDNLEIVLHRWEN 81

OrthologousHomologous sequences in different species
that arose from a common ancestral gene
during speciation; may or may not be responsible
for a similar function.
ParalogousHomologous sequences within a single species
that arose by gene duplication.
Definitions

speciation
duplication

fly GAKKVIISAP SAD.APM..F VCGVNLDAYK PDMKVVSNAS CTTNCLAPLA
human GAKRVIISAP SAD.APM..F VMGVNHEKYD NSLKIISNAS CTTNCLAPLA
plant GAKKVIISAP SAD.APM..F VVGVNEHTYQ PNMDIVSNAS CTTNCLAPLA
bacterium GAKKVVMTGP SKDNTPM..F VKGANFDKY. AGQDIVSNAS CTTNCLAPLA
yeast GAKKVVITAP SS.TAPM..F VMGVNEEKYT SDLKIVSNAS CTTNCLAPLA
archaeon GADKVLISAP PKGDEPVKQL VYGVNHDEYD GE.DVVSNAS CTTNSITPVA
fly KVINDNFEIV EGLMTTVHAT TATQKTVDGP SGKLWRDGRG AAQNIIPAST
human KVIHDNFGIV EGLMTTVHAI TATQKTVDGP SGKLWRDGRG ALQNIIPAST
plant KVVHEEFGIL EGLMTTVHAT TATQKTVDGP SMKDWRGGRG ASQNIIPSST
bacterium KVINDNFGII EGLMTTVHAT TATQKTVDGP SHKDWRGGRG ASQNIIPSST
yeast KVINDAFGIE EGLMTTVHSL TATQKTVDGP SHKDWRGGRT ASGNIIPSST
archaeon KVLDEEFGIN AGQLTTVHAY TGSQNLMDGP NGKP.RRRRA AAENIIPTST
fly GAAKAVGKVI PALNGKLTGM AFRVPTPNVS VVDLTVRLGK GASYDEIKAK
human GAAKAVGKVI PELNGKLTGM AFRVPTANVS VVDLTCRLEK PAKYDDIKKV
plant GAAKAVGKVL PELNGKLTGM AFRVPTSNVS VVDLTCRLEK GASYEDVKAA
bacterium GAAKAVGKVL PELNGKLTGM AFRVPTPNVS VVDLTVRLEK AATYEQIKAA
yeast GAAKAVGKVL PELQGKLTGM AFRVPTVDVS VVDLTVKLNK ETTYDEIKKV
archaeon GAAQAATEVL PELEGKLDGM AIRVPVPNGS ITEFVVDLDD DVTESDVNAA
Multiple sequence alignment of
glyceraldehyde- 3-phsophate dehydrogenases

This power of sequence alignments
• empirical finding: if two biological sequences are sufficiently similar, almost invariably they have similar biological functions and will be descended from a common ancestor.
• (i) function is encoded into sequence, this means: the sequence provides the syntax and
• (ii) there is a redundancy in the encoding, many positions in the sequence may be changed without perceptible changes in the function, thus the semantics of the encoding is robust.

Ove
rvie
w
• Introduction
– Short recap on databases
– Definitions
• Scoring Matrices
– Theoretical
– Empirial
• PAM (pam-simulator.pl)
• BLOSUM
• Pairwise alignment
– Dot-plots (dotplot-simulator.pl)
Overview

A metric …
It is very important to realize, that all subsequent results depend critically on just how this is done and what model lies at the basis for the construction of a specific scoring matrix.
A scoring matrix is a tool to quantify how well a certain model is represented in the alignment of two sequences, and any result obtained by its application is meaningful exclusively in the context of that model.

Scoring matrices appear in all analysis
involving sequence comparison.
The choice of matrix can strongly influence
the outcome of the analysis.
Scoring matrices implicitly represent a
particular theory of evolution.
Understanding theories underlying a given
scoring matrix can aid in making proper
choice.
• Nucleic acid and Protein Scoring Matrices
Importance of scoring matrices

• Identity matrix (similarity) BLAST matrix (similarity) A T C G A T C G
A 1 0 0 0 A 5 -4 -4 -4
T 0 1 0 0 T -4 5 -4 -4
C 0 0 1 0 C -4 -4 5 -4
G 0 0 0 1 G -4 -4 -4 5
• Transition/Transversion Matrix A T C G
A 0 5 5 1T 5 0 1 5C 5 1 0 5G 1 5 5 0
Nucleic Acid Scoring Matrices
G and C
purine-pyrimidine
A and T
purine -pyrimidine

• Nucleotide bases fall into two categories depending on the ring structure of the base. Purines (Adenine and Guanine) are two ring bases, pyrimidines (Cytosine and Thymine) are single ring bases. Mutations in DNA are changes in which one base is replaced by another.
• A mutation that conserves the ring number is called a transition (e.g., A -> G or C -> T) a mutation that changes the ring number are called transversions. (e.g. A -> C or A -> T and so on).
A T C G
A 0 5 5 1
T 5 0 1 5
C 5 1 0 5
G 1 5 5 0
Transition/Transversion Matrix

• Although there are more ways to create a transversion, the number of transitions observed to occur in nature (i.e., when comparing related DNA sequences) is much greater. Since the likelihood of transitions is greater, it is sometimes desireable to create a weight matrix which takes this propensity into account when comparing two DNA sequences.
• Use of a Transition/Transversion Matrix reduces noise in comparisons of distantly related sequences.
Transition/Transversion Matrix
A T C G
A 0 5 5 1
T 5 0 1 5
C 5 1 0 5
G 1 5 5 0

The Genome Chose Its Alphabet With Care
• Of all the nucleotide bases available, why did nature pick the four we know as A, T, G, and C for the genomic alphabet ?
• The choice of A, T, G, and C incorporates a tactic for minimizing the occurrence of errors in the pairing of bases, in the same way that error-coding systems are incorporated into ISBNs on books, credit card numbers, bank accounts, and airline tickets.

• In the error-coding theory first developed in 1950 by Bell Telephone Laboratories researcher Richard Hamming, a so-called parity bit is added to the end of digital numbers to make the digits add up to an even number. For example, when transmitting the number 100110, you would add an extra 1 onto the end (100110,1), and the number 100001 would have a zero added (100001,0). The most likely transmission error is a single digit changed from 1 to 0 or vice versa. Such a change would cause the sum of the digits to be odd, and the recipient of that number can assume that it was incorrectly transmitted.
The Genome Chose Its Alphabet With Care

• Represent each nucleotide as a four-digit binary number.
• The first three digits represent the three bonding sites that each nucleotide presents to its partner. Each site is either a hydrogen donor or acceptor; a nucleotide offering donor-acceptor-acceptor sites would be represented as 100 and would bond only with an acceptor-donor-donor nucleotide, or 011.
• The fourth digit is 1 if the nucleotide is a single-ringed pyrimidine type and 0 if it is a double-ringed purine type.
• Nucleotides readily bond with members of the other type.
The Genome Chose Its Alphabet With Care

• The final digit acted as a parity bit: The four digits of A, T, G, and C all add up to an even number.
• Nature restricted its choice to nucleotides of even parity because "alphabets composed of nucleotides of mixed parity would have catastrophic error rates.
• For example, nucleotide C (100,1) binds naturally to nucleotide G (011,0), but it might accidentally bind to the odd parity nucleotide X (010,0), because there is just one mismatch. Such a bond would be weak compared to C-G but not impossible. However, C is highly unlikely to bond to any other even-parity nucleotides, such as the idealized amino-adenine (101,0), because there are two mismatches
• So, nature has avoided such mistakes by banishing all odd-parity nucleotides from the DNA alphabet.
The Genome Chose Its Alphabet With Care

• The simplest metric in use is the
identity metric.
• If two amino acids are the same,
they are given one score, if they are
not, they are given a different score -
regardless, of what the replacement
is.
• One may give a score of 1 for
matches and 0 for mismatches - this
leads to the frequently used unitary
matrix
Protein Scoring Matrices: Unitary Matrix

Protein Scoring Matrices: Unitary Matrix
A R N D C Q E G H I L K M F P S T W Y V
A 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
R 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
N 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
D 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
C 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
Q 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0
E 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0
G 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0
H 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0
I 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0
L 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0
K 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0
M 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0
F 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0
P 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0
S 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0
T 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0
W 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0
Y 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0
V 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1

Protein Scoring Matrices: Unitary Matrix
• The simplest matrix:
– High scores for Identities
– Low scores for non-identities
• Works for closely related proteins
• Or one could assign +6 for a match and -1 for a mismatch, this would be a matrix useful for local alignment procedures, where a negative expectation value for randomly aligned sequences is required to ensure that the score will not grow simply from extending the alignment in a random way.

A very crude model of an evolutionary
relationship could be implemented in a
scoring matrix in the following way: since
all point-mutations arise from nucleotide
changes, the probability that an observed
amino acid pair is related by chance,
rather than inheritance should depend on
the number of point mutations necessary
to transform one codon into the other.
A metric resulting from this model would
define the distance between two amino
acids by the minimal number of nucleotide
changes required.
Genetic Code Matrix

A S G L K V T P E D N I Q R F Y C H M W Z B X
Ala = A O 1 1 2 2 1 1 1 1 1 2 2 2 2 2 2 2 2 2 2 2 2 2
Ser = S 1 O 1 1 2 2 1 1 2 2 1 1 2 1 1 1 1 2 2 1 2 2 2
Gly = G 1 1 0 2 2 1 2 2 1 1 2 2 2 1 2 2 1 2 2 1 2 2 2
Leu = L 2 1 2 0 2 1 2 1 2 2 2 1 1 1 1 2 2 1 1 1 2 2 2
Lys = K 2 2 2 2 0 2 1 2 1 2 1 1 1 1 2 2 2 2 1 2 1 2 2
Val = V 1 2 1 1 2 0 2 2 1 1 2 1 2 2 1 2 2 2 1 2 2 2 2
Thr = T 1 1 2 2 1 2 0 1 2 2 1 1 2 1 2 2 2 2 1 2 2 2 2
Pro = P 1 1 2 1 2 2 1 0 2 2 2 2 1 1 2 2 2 1 2 2 2 2 2
Glu - E 1 2 1 2 1 1 2 2 0 1 2 2 1 2 2 2 2 2 2 2 1 2 2
Asp = D 1 2 1 2 2 1 2 2 1 O 1 2 2 2 2 1 2 1 2 2 2 1 2
Asn = N 2 1 2 2 1 2 1 2 2 1 O 1 2 2 2 1 2 1 2 2 2 1 2
Ile = I 2 1 2 1 1 1 1 2 2 2 1 0 2 1 1 2 2 2 1 2 2 2 2
Gln = Q 2 2 2 1 1 2 2 1 1 2 2 2 0 1 2 2 2 1 2 2 1 2 2
Arg = R 2 1 1 1 1 2 1 1 2 2 2 1 1 0 2 2 1 1 1 1 2 2 2
Phe = F 2 1 2 1 2 1 2 2 2 2 2 1 2 2 0 1 1 2 2 2 2 2 2
Tyr = Y 2 1 2 2 2 2 2 2 2 1 1 2 2 2 1 O 1 1 3 2 2 1 2
Cys = C 2 1 1 2 2 2 2 2 2 2 2 2 2 1 1 1 0 2 2 1 2 2 2
His = H 2 2 2 1 2 2 2 1 2 1 1 2 1 1 2 1 2 0 2 2 2 1 2
Met = M 2 2 2 1 1 1 1 2 2 2 2 1 2 1 2 3 2 2 0 2 2 2 2
Trp = W 2 1 1 1 2 2 2 2 2 2 2 2 2 1 2 2 1 2 2 0 2 2 2
Glx = Z 2 2 2 2 1 2 2 2 1 2 2 2 1 2 2 2 2 2 2 2 1 2 2
Asx = B 2 2 2 2 2 2 2 2 2 1 1 2 2 2 2 1 2 1 2 2 2 1 2
??? = X 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2
The table is generated by calculating the minimum number of base changes required to
convert an amino acid in row i to an amino acid in column j.
Note Met->Tyr is the only change that requires all 3 codon positions to change.
Genetic Code Matrix

This genetic code matrix already
improves sensitivity and specificity
of alignments from the identity
matrix.
The fact that the genetic code matrix
works to align related proteins, in
the same way that matrices derived
from amino-acid properties work
says something very interesting
about the genetic code: namely that
it appears to have evolved to
minimize the effects of point
mutations.
Genetic Code Matrix

Genetic Code Matrix

• Simple identity, which scores only identical amino
acids as a match.
• Genetic code changes, which scores the
minimum number of nucieotide changes to change
a codon for one amino acid into a codon for the
other.
• Chemical similarity of amino acid side chains,
which scores as a match two amino acids which
have a similar side chain, such as hydrophobic,
charged and polar amino acid groups.
Overview

All proteins are polymers of the 20 naturally occuring amino acids. They are listed here along with their
abbreviations :-Alanine Ala A
Cysteine Cys C
Aspartic AciD Asp D
Glutamic Acid Glu E
Phenylalanine Phe F
Glycine Gly G
Histidine His H
Isoleucine Ile I
Lysine Lys K
Leucine Leu L
Methionine Met M
AsparagiNe Asn N
Proline Pro P
Glutamine Gln Q
ARginine Arg R
Serine Ser S
Threonine Thr T
Valine Val V
Tryptophan Trp W
TYrosine Tyr Y
Amino Acid Residues
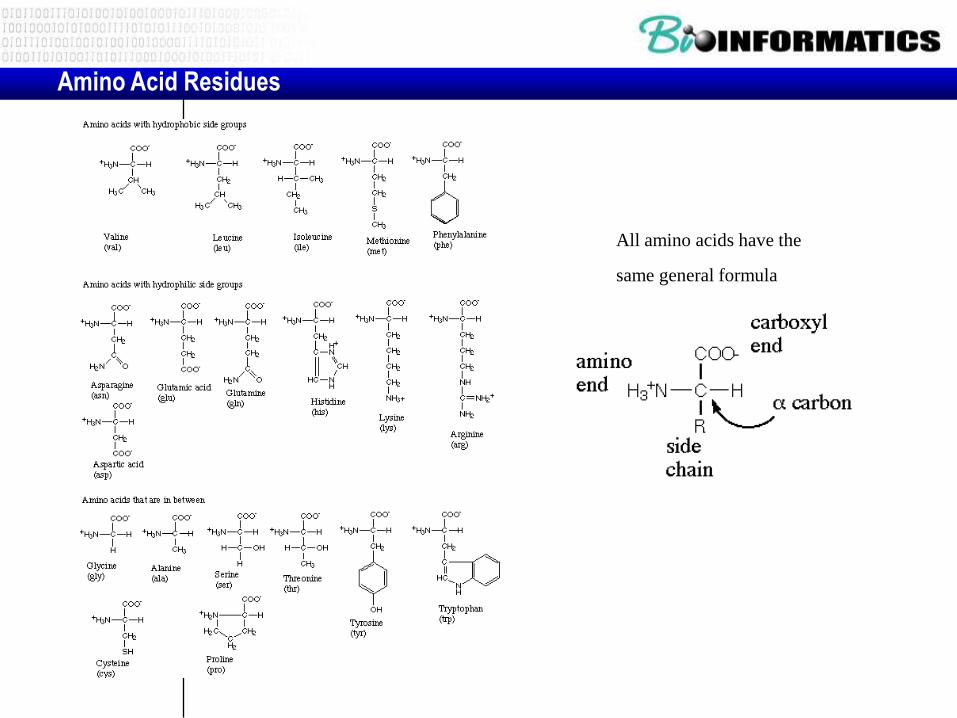
All amino acids have the
same general formula
Amino Acid Residues

• Hydrophobic-aliphatic amino acids: Their side chains consist of non-polar methyl- or methylene-groups.
– These amino acids are usually located on the interior of the protein as they are hydrophobic in nature.
– All except for alanine are bifurcated. In the cases of Val and Ile the bifurcation is close to the main chain and can therefore restrict the conformation of the polypeptide by steric hindrance.
– red and blue atoms represent polar main chain groups
Amino Acid Residues

Amino Acid Residues

• Hydrophobic-aromatic: Only
phenylalanine is entirely non-polar.
Tyrosine's phenolic side chain has a
hydroxyl substituent and tryptophan
has a nitrogen atom in its indole ring
sytem.
– These residues are nearly always found
to be largely buried in the hydrophobic
interior of a proteins as they are
prdeominantly non-polar in nature.
– However, the polar atoms of tyrosine
and tryptophan allow hydrogen bonding
interactions to be made with other
residues or even solvent molecules
Amino Acid Residues

Amino Acid Residues

Neutral-polar side chains: a number of
small aliphatic side chains containing polar
groups which cannot ionize readily.
– Serine and threonine possess hydroxyl groups in
their side chains and as these polar groups are
close to the main chain they can form hydrogen
bonds with it. This can influence the local
conformation of the polypeptide,
– Residues such as serine and asparagine are
known to adopt conformations which most other
amino acids cannot.
– The amino acids asparagine and glutamine
posses amide groups in their side chains which
are usually hydrogen-bonded whenever they
occur in the interior of a protein.
Amino Acid Residues

Amino Acid Residues

• Acidic amino acids: Aspartate and glutamate have carboxyl side chains and are therefore negatively charged at physiological pH (around neutral).
– The strongly polar nature of these residues means that they are most often found on the surface of globular proteins where they can interact favourably with solvent molecules.
– These residues can also take part in electrostatic interactions with positively charged basic amino acids.
– Aspartate and glutamate also can take on catalytic roles in the active sites of enzymes and are well known for their metal ion binding abilities
Amino Acid Residues

Amino Acid Residues

• Basic amino acids:
– histidine has the lowest pKa (around 6) and is
therefore neutral at around physiological pH.
• This amino acid occurs very frequently in enzyme
active sites as it can function as a very efficient
general acid-base catalyst.
• It also acts as a metal ion ligand in numerous
protein families.
– Lysine and arginine are more strongly basic and
are positively charged at physiological pH's. They
are generally solvated but do occasionally occur
in the interior of a protein where they are usually
involved in electrostatic interactions with
negatively charged groups such as Asp or Glu.
• Lys and Arg have important roles in anion-binding
proteins as they can interact electrostatically with
the ligand.
Amino Acid Residues

Amino Acid Residues

Conformationally important residues: Glycine and
proline are unique amino acids. They appear to
influence the conformation of the polypeptide.
• Glycine essentially lacks a side chain and therefore
can adopt conformations which are sterically
forbidden for other amino acids. This confers a high
degree of local flexibility on the polypeptide.
– Accordingly, glycine residues are frequently found in
turn regions of proteins where the backbone has to
make a sharp turn.
– Glycine occurs abundantly in certain fibrous proteins
due to its flexibility and because its small size allows
adjacent polypeptide chains to pack together closely.
• In contrast, proline is the most rigid of the twenty
naturally occurring amino acids since its side chain
is covalently linked with the main chain nitrogen
Amino Acid Residues

Amino Acid Residues

Here is one list where amino acids are grouped according to the characteristics of the side chains:
Aliphatic - alanine, glycine, isoleucine, leucine, proline, valine,
Aromatic - phenylalanine, tryptophan, tyrosine,
Acidic - aspartic acid, glutamic acid,
Basic - arginine, histidine, lysine,
Hydroxylic - serine, threonine
Sulphur-containing - cysteine, methionine
Amidic (containing amide group) -asparagine, glutamine
Amino Acid Residues

R K D E B Z S N Q G X T H A C M P V L I Y F W
Arg = R 10 10 9 9 8 8 6 6 6 5 5 5 5 5 4 3 3 3 3 3 2 1 0
Lys = K 10 10 9 9 8 8 6 6 6 5 5 5 5 5 4 3 3 3 3 3 2 1 0
Asp = D 9 9 10 10 8 8 7 6 6 6 5 5 5 5 5 4 4 4 3 3 3 2 1
Glu = E 9 9 10 10 8 8 7 6 6 6 5 5 5 5 5 4 4 4 3 3 3 2 1
Asx = B 8 8 8 8 10 10 8 8 8 8 7 7 7 7 6 6 6 5 5 5 4 4 3
Glx = Z 8 8 8 8 10 10 8 8 8 8 7 7 7 7 6 6 6 5 5 5 4 4 3
Ser = S 6 6 7 7 8 8 10 10 10 10 9 9 9 9 8 8 7 7 7 7 6 6 4
Asn = N 6 6 6 6 8 8 10 10 10 10 9 9 9 9 8 8 8 7 7 7 6 6 4
Gln = Q 6 6 6 6 8 8 10 10 10 10 9 9 9 9 8 8 8 7 7 7 6 6 4
Gly = G 5 5 6 6 8 8 10 10 10 10 9 9 9 9 8 8 8 8 7 7 6 6 5
??? = X 5 5 5 5 7 7 9 9 9 9 10 10 10 10 9 9 8 8 8 8 7 7 5
Thr = T 5 5 5 5 7 7 9 9 9 9 10 10 10 10 9 9 8 8 8 8 7 7 5
His = H 5 5 5 5 7 7 9 9 9 9 10 10 10 10 9 9 9 8 8 8 7 7 5
Ala = A 5 5 5 5 7 7 9 9 9 9 10 10 10 10 9 9 9 8 8 8 7 7 5
Cys = C 4 4 5 5 6 6 8 8 8 8 9 9 9 9 10 10 9 9 9 9 8 8 5
Met = M 3 3 4 4 6 6 8 8 8 8 9 9 9 9 10 10 10 10 9 9 8 8 7
Pro = P 3 3 4 4 6 6 7 8 8 8 8 8 9 9 9 10 10 10 9 9 9 8 7
Val = V 3 3 4 4 5 5 7 7 7 8 8 8 8 8 9 10 10 10 10 10 9 8 7
Leu = L 3 3 3 3 5 5 7 7 7 7 8 8 8 8 9 9 9 10 10 10 9 9 8
Ile = I 3 3 3 3 5 5 7 7 7 7 8 8 8 8 9 9 9 10 10 10 9 9 8
Tyr = Y 2 2 3 3 4 4 6 6 6 6 7 7 7 7 8 8 9 9 9 9 10 10 8
Phe = F 1 1 2 2 4 4 6 6 6 6 7 7 7 7 8 8 8 8 9 9 10 10 9
Trp = W 0 0 1 1 3 3 4 4 4 5 5 5 5 5 6 7 7 7 8 8 8 9 10
Hydrophobicity matrix
•Physical/Chemical characteristics: Attempt to quantify some physical or chemical attribute of
•the residues and arbitrarily assign weights based on similarities of the residues in this chosen property.

Other similarity scoring matrices might be constructed from
any property of amino acids that can be quantified
- partition coefficients between hydrophobic and hydrophilic phases
- charge
- molecular volume
Unfortunately, …

AAindex
Amino acid indices and similarity matrices (http://www.genome.ad.jp/dbget/aaindex.html)
List of 494 Amino Acid Indices in AAindex ver.6.0
• ANDN920101 alpha-CH chemical shifts (Andersen et al., 1992)
• ARGP820101 Hydrophobicity index (Argos et al., 1982)
• ARGP820102 Signal sequence helical potential (Argos et al., 1982)
• ARGP820103 Membrane-buried preference parameters (Argos et al., 1982)
• BEGF750101 Conformational parameter of inner helix (Beghin-Dirkx, 1975)
• BEGF750102 Conformational parameter of beta-structure (Beghin-Dirkx, 1975)
• BEGF750103 Conformational parameter of beta-turn (Beghin-Dirkx, 1975)
• BHAR880101 Average flexibility indices (Bhaskaran-Ponnuswamy, 1988)
• BIGC670101 Residue volume (Bigelow, 1967)
• BIOV880101 Information value for accessibility; average fraction 35% (Biou et al., 1988)
• BIOV880102 Information value for accessibility; average fraction 23% (Biou et al., 1988)
• BROC820101 Retention coefficient in TFA (Browne et al., 1982)
• BROC820102 Retention coefficient in HFBA (Browne et al., 1982)
• BULH740101 Transfer free energy to surface (Bull-Breese, 1974)
• BULH740102 Apparent partial specific volume (Bull-Breese, 1974)

Protein Eng. 1996 Jan;9(1):27-36.

• Simple identity, which scores only identical amino
acids as a match.
• Genetic code changes, which scores the
minimum number of nucieotide changes to change
a codon for one amino acid into a codon for the
other.
• Chemical similarity of amino acid side chains,
which scores as a match two amino acids which
have a similar side chain, such as hydrophobic,
charged and polar amino acid groups.
• The Dayhoff percent accepted mutation (PAM)
family of matrices, which scores amino acid pairs
on the basis of the expected frequency of
substitution of one amino acid for the other during
protein evolution.
Overview

• In the absence of a valid model derived from first principles, an empirical approachseems more appropriate to score amino acid similarity.
• This approach is based on the assumption that once the evolutionary relationship of two sequences is established, the residues that did exchange are similar.
Dayhoff Matrix

Model of Evolution:
“Proteins evolve through a succesion of
independent point mutations, that are
accepted in a population and
subsequently can be observed in the
sequence pool.”
Definition:
The evolutionary distance between two
sequences is the (minimal) number of
point mutations that was necessary to
evolve one sequence into the other
Overview

• The model used here states that
proteins evolve through a succesion of
independent point mutations, that are
accepted in a population and
subsequently can be observed in the
sequence pool.
• We can define an evolutionary
distance between two sequences as
the number of point mutations that was
necessary to evolve one sequence into
the other.
Principle

• M.O. Dayhoff and colleagues
introduced the term "accepted point
mutation" for a mutation that is stably
fixed in the gene pool in the course
of evolution. Thus a measure of
evolutionary distance between two
sequences can be defined:
• A PAM (Percent accepted mutation)
is one accepted point mutation on
the path between two sequences,
per 100 residues.
Overview

First step: finding “accepted mutations”
In order to identify accepted point
mutations, a complete phylogenetic
tree including all ancestral sequences
has to be constructed. To avoid a
large degree of ambiguities in this
step, Dayhoff and colleagues
restricted their analysis to sequence
families with more than 85% identity.
Principles of Scoring Matrix Construction

Identification of accepted point mutations:
•Collection of correct (manual) alignments
• 1300 sequences in 72 families
• closely related in order not to get multiply
changes at the same position
• Construct a complete phylogenetic tree including all
ancestral sequences.
• Dayhoff et al restricted their analysis to
sequence families with more than 85%
identity.
• Tabulate into a 20x20 matrix the amino acid pair
exchanges for each of the observed and inferred
sequences.
Overview

ACGH DBGH ADIJ CBIJ
\ / \ /
\ / \ /
B - C \ / A - D B - D \ / A - C
\ / \ /
\/ \/
ABGH ABIJ
\ /
\ I - G /
\ J - H /
\ /
\ /
|
|
|
Overview

Dayhoff’s PAM1 mutation probability matrix (Transition Matrix)
A
Ala
R
Arg
N
Asn
D
Asp
C
Cys
Q
Gln
E
Glu
G
Gly
H
His
I
Ile
A 9867 2 9 10 3 8 17 21 2 6
R 1 9913 1 0 1 10 0 0 10 3
N 4 1 9822 36 0 4 6 6 21 3
D 6 0 42 9859 0 6 53 6 4 1
C 1 1 0 0 9973 0 0 0 1 1
Q 3 9 4 5 0 9876 27 1 23 1
E 10 0 7 56 0 35 9865 4 2 3
G 21 1 12 11 1 3 7 9935 1 0
H 1 8 18 3 1 20 1 0 9912 0
I 2 2 3 1 2 1 2 0 0 9872
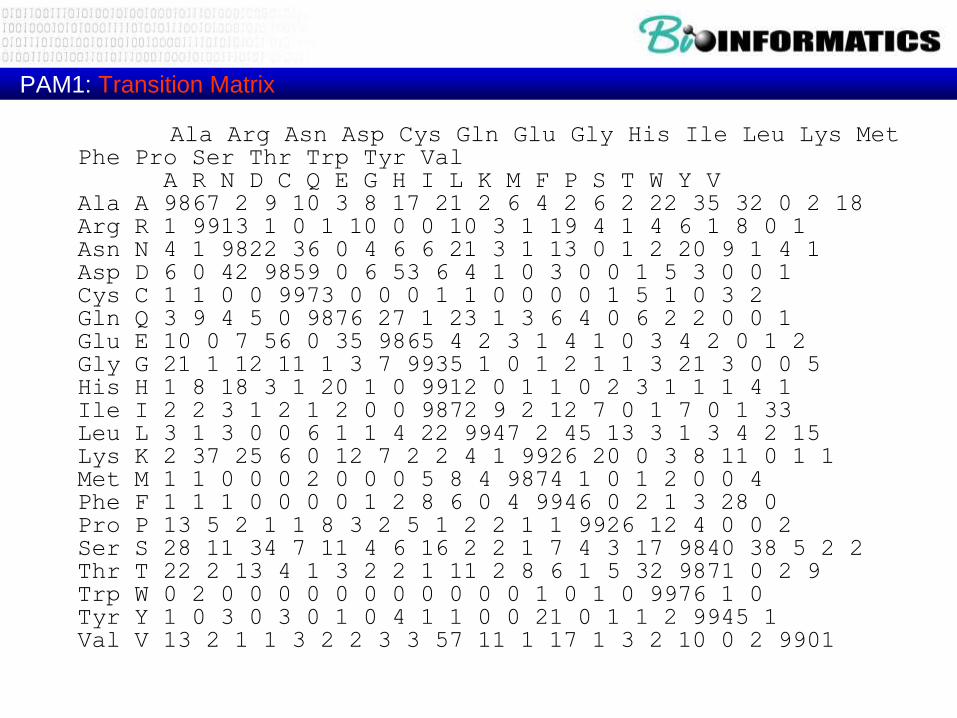
PAM1: Transition Matrix
Ala Arg Asn Asp Cys Gln Glu Gly His Ile Leu Lys Met Phe Pro Ser Thr Trp Tyr Val
A R N D C Q E G H I L K M F P S T W Y VAla A 9867 2 9 10 3 8 17 21 2 6 4 2 6 2 22 35 32 0 2 18Arg R 1 9913 1 0 1 10 0 0 10 3 1 19 4 1 4 6 1 8 0 1Asn N 4 1 9822 36 0 4 6 6 21 3 1 13 0 1 2 20 9 1 4 1Asp D 6 0 42 9859 0 6 53 6 4 1 0 3 0 0 1 5 3 0 0 1Cys C 1 1 0 0 9973 0 0 0 1 1 0 0 0 0 1 5 1 0 3 2Gln Q 3 9 4 5 0 9876 27 1 23 1 3 6 4 0 6 2 2 0 0 1Glu E 10 0 7 56 0 35 9865 4 2 3 1 4 1 0 3 4 2 0 1 2Gly G 21 1 12 11 1 3 7 9935 1 0 1 2 1 1 3 21 3 0 0 5His H 1 8 18 3 1 20 1 0 9912 0 1 1 0 2 3 1 1 1 4 1Ile I 2 2 3 1 2 1 2 0 0 9872 9 2 12 7 0 1 7 0 1 33Leu L 3 1 3 0 0 6 1 1 4 22 9947 2 45 13 3 1 3 4 2 15Lys K 2 37 25 6 0 12 7 2 2 4 1 9926 20 0 3 8 11 0 1 1Met M 1 1 0 0 0 2 0 0 0 5 8 4 9874 1 0 1 2 0 0 4Phe F 1 1 1 0 0 0 0 1 2 8 6 0 4 9946 0 2 1 3 28 0Pro P 13 5 2 1 1 8 3 2 5 1 2 2 1 1 9926 12 4 0 0 2Ser S 28 11 34 7 11 4 6 16 2 2 1 7 4 3 17 9840 38 5 2 2Thr T 22 2 13 4 1 3 2 2 1 11 2 8 6 1 5 32 9871 0 2 9Trp W 0 2 0 0 0 0 0 0 0 0 0 0 0 1 0 1 0 9976 1 0Tyr Y 1 0 3 0 3 0 1 0 4 1 1 0 0 21 0 1 1 2 9945 1Val V 13 2 1 1 3 2 2 3 3 57 11 1 17 1 3 2 10 0 2 9901

Numbers of accepted point mutations (x10)
accumulated from closely related
sequences.
Fractional exchanges result when ancestral
sequences are ambiguous: the
probabilities are distributed equally
among all possibilities.
The total number of exchanges tallied was
1,572. Note that 36 exchanges were
never observed.
The Asp-Glu pair had the largest number of
exchanges
PAM1: Transition Matrix

Second step: Frequencies of Occurence
If the properties of amino acids differ and if
they occur with different frequencies, all
statements we can make about the average
properties of sequences will depend on the
frequencies of occurence of the individual
amino acids. These frequencies of
occurence are approximated by the
frequencies of observation. They are the
number of occurences of a given amino acid
divided by the number of amino-acids
observed.
The sum of all is one.
Principles of Scoring Matrix Construction

Amino acid frequencies
1978 1991
L 0.085 0.091
A 0.087 0.077
G 0.089 0.074
S 0.070 0.069
V 0.065 0.066
E 0.050 0.062
T 0.058 0.059
K 0.081 0.059
I 0.037 0.053
D 0.047 0.052
R 0.041 0.051
P 0.051 0.051
N 0.040 0.043
Q 0.038 0.041
F 0.040 0.040
Y 0.030 0.032
M 0.015 0.024
H 0.034 0.023
C 0.033 0.020
W 0.010 0.014
Second step: Frequencies of Occurence

Third step: Relative Mutabilities
• To obtain a complete picture of the mutational process, the amino-acids that do not mutate must be taken into account too.
• We need to know: what is the chance, on average, that a given amino acid will mutate at all. This is the relative mutability of the amino acid.
• It is obtained by multiplying the number of observed changes by the amino acids frequency of occurence.
Principles of Scoring Matrix Construction

Compute amino acid mutability, mj, i.e., the propability
of a given amino acid, j, to be replaced.
Aligned A D A
Sequences A D B
Amino Acids A B D
Observed Changes 1 1 0
Frequency of Occurence 3 1 2
(Total Composition)
Relative Mutability .33 1 0
Overview

1978 1991A 100 100C 20 44D 106 86E 102 77F 41 51G 49 50H 66 91I 96 103K 56 72L 40 54M 94 93N 134 104P 56 58Q 93 84R 65 83S 120 117T 97 107V 74 98W 18 25Y 41 50
Principles of Scoring Matrix Construction

Fourth step: Mutation Probability Matrix
• With these data the probability that an amino acid in
row i of the matrix will replace the amino acid in
column j can be calculated: it is the mutability of amino
acid j, multiplied by the relative pair exchange
frequency (the pair exchange frequency for ij divided
by the sum of all pair exchange frequencies for amino
acid i).
Mij= The mutation probability matrix gives the
probability, that an amino acid i will replace an amino
acid of type j in a given evolutionary interval, in two
related sequences
Principles of Scoring Matrix Construction
ADB
ADA
A D B
A
D
B
i
j

Fifth step: The Evolutionary Distance
• Since the represent the probabilites
for amino acids to remain
conserved, if we scale all cells of our
matrix by a constant factor we can
scale the matrix to reflect a specific
overall probability of change. We
may chose so that the expected
number of changes is 1 %, this
gives the matrix for the evolutionary
distance of 1 PAM.
Principles of Scoring Matrix Construction

6. Relatedness Odds
• By comparison, the probability that
that same event is observed by
random chance is simply given by
the frequency of occurence of
amino acid i
• Rij = probability that j replaces i in
related proteins
• Piran = probability that j replaces I by
chance (eg unrelated proteins)
• Piran = fi = the frequency of
occurance of amino acid i
Principles of Scoring Matrix Construction

Last step: the log-odds matrix• Since multiplication is a computationally
expensive process, it is preferrable to add
the logarithms of the matrix elements. This
matrix, the log odds matrix, is the
foundation of quantitative sequence
comparisons under an evolutionary model.
• Since the Dayhoff matrix was taken as the
log to base 10, a value of +1 would mean
that the corresponding pair has been
observed 10 times more frequently than
expected by chance. A value of -0.2 would
mean that the observed pair was observed
1.6 times less frequently than chance
would predict.
Principles of Scoring Matrix Construction

• http://www.bio.brandeis.edu/InterpGenes/Proj
ect/align12.htm

A B C D E F G H I K L M N P Q R S T V W Y Z
0.4 0.0 -0.4 0.0 0.0 -0.8 0.2 -0.2 -0.2 -0.2 -0.4 -0.2 0.0 0.2 0.0 -0.4 0.2 0.2 0.0 -1.2 -0.6 0.0 A
0.5 -0.9 0.6 0.4 -1.0 0.1 0.3 -0.4 0.1 -0.7 -0.5 0.4 -0.2 0.3 -0.1 0.1 0.0 -0.4 -1.1 -0.6 0.4 B
2.4 -1.0 -1.0 -0.8 -0.6 -0.6 -0.4 -1.0 -1.2 -1.0 -0.8 -0.6 -1.0 -0.8 0.0 -0.4 -0.4 -1.6 0.0 -1.0 C
0.8 0.6 -1.2 0.2 0.2 -0.4 0.0 -0.8 -0.6 0.4 -0.2 0.4 -0.2 0.0 0.0 -0.4 -1.4 -0.8 0.5 D
0.8 -1.0 0.0 0.2 -0.4 0.0 -0.6 -0.4 0.2 -0.2 0.4 -0.2 0.0 0.0 -0.4 -1.4 -0.8 0.6 E
1.8 -1.0 -0.4 0.2 -1.0 0.4 0.0 -0.8 -1.0 -1.0 -0.8 -0.6 -0.6 -0.2 0.0 1.4 -1.0 F
1.0 -0.4 -0.6 -0.4 -0.8 -0.6 0.0 -0.2 -0.2 -0.6 0.2 0.0 -0.2 -1.4 -1.0 -0.1 G
1.2 -0.4 0.0 -0.4 -0.4 0.4 0.0 0.6 0.4 -0.2 -0.2 -0.4 -0.6 0.0 -0.4 H
1.0 -0.4 0.4 0.4 -0.4 -0.4 -0.4 -0.4 -0.2 0.0 0.8 -1.0 -0.2 -0.4 I
1.0 -0.6 0.0 0.2 -0.2 0.2 0.6 0.0 0.0 -0.4 -0.6 -0.8 0.1 K
1.2 0.8 -0.6 -0.6 -0.4 -0.6 -0.6 -0.4 0.4 -0.4 -0.2 -0.5 L
1.2 -0.4 -0.4 -0.2 0.0 -0.4 -0.2 0.4 -0.8 -0.4 -0.3 M
0.4 -0.2 0.2 0.0 0.2 0.0 -0.4 -0.8 -0.4 0.2 N
1.2 0.0 0.0 0.2 0.0 -0.2 -1.2 -1.0 -0.1 P
0.8 0.2 -0.2 -0.2 -0.4 -1.0 -0.8 0.6 Q
1.2 0.0 -0.2 -0.4 0.4 -0.8 0.6 R
0.4 0.2 -0.2 -0.4 -0.6 -0.1 S
0.6 0.0 -1.0 -0.6 -0.1 T
0.8 -1.2 -0.4 -0.4 V
3.4 0.0 -1.2 W
2.0 -0.8 Y
0.6 Z
PAM 1 Scoring Matrix

• Some of the properties go into the makeup of PAM matrices are - amino acid residue size, shape, local concentrations of electric charge, van der Waals surface, ability to form salt bridges, hydrophobic interactions, and hydrogen bonds. – These patterns are imposed principally
by natural selection and only secondarily by the constraints of the genetic code.
– Coming up with one’s own matrix of weights based on some logical features may not be very successful because your logical features may have been over-written by other more important considerations.
Overview

• Two aspects of this process cause the
evolutionary distance to be unequal in
general to the number of observed
differences between the sequences:
– First, there is a chance that a certain
residue may have mutated, than reverted,
hiding the effect of the mutation.
– Second, specific residues may have
mutated more than once, thus the number
of point mutations is likely to be larger
than the number of differences between
the two sequences..
Principles of Scoring Matrix Construction

Similarity ve. distance

• Initialize:
– Generate Random protein (1000 aa)
• Simulate evolution (eg 250 for PAM250)
– Apply PAM1 Transition matrix to each amino
acid
– Use Weighted Random Selection
• Iterate
– Measure difference to orginal protein
Experiment: pam-simulator.pl
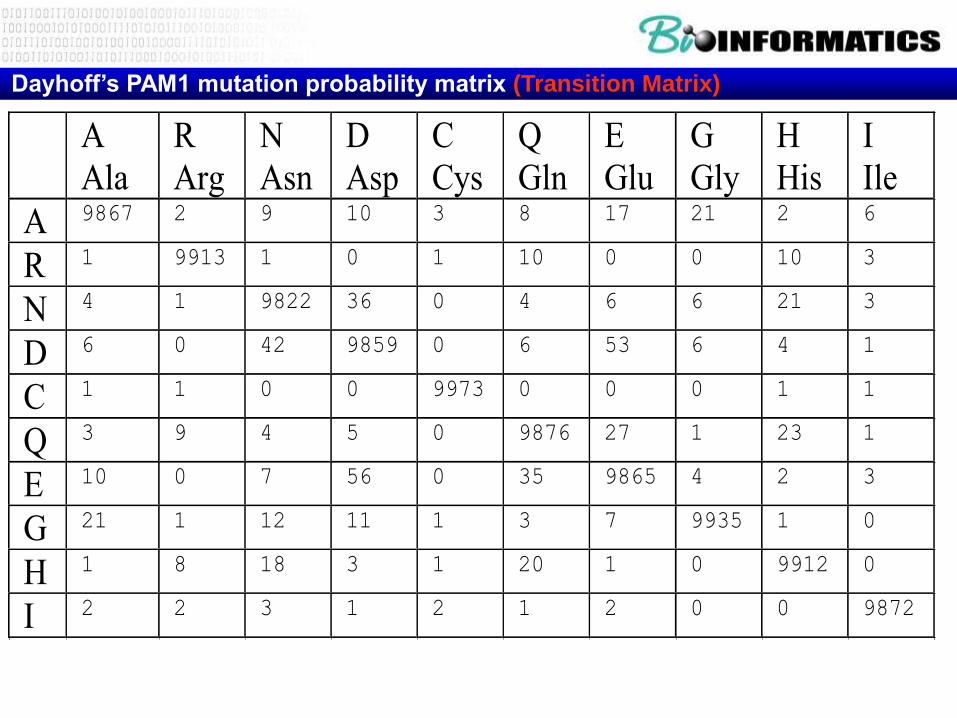
Dayhoff’s PAM1 mutation probability matrix (Transition Matrix)
A
Ala
R
Arg
N
Asn
D
Asp
C
Cys
Q
Gln
E
Glu
G
Gly
H
His
I
Ile
A 9867 2 9 10 3 8 17 21 2 6
R 1 9913 1 0 1 10 0 0 10 3
N 4 1 9822 36 0 4 6 6 21 3
D 6 0 42 9859 0 6 53 6 4 1
C 1 1 0 0 9973 0 0 0 1 1
Q 3 9 4 5 0 9876 27 1 23 1
E 10 0 7 56 0 35 9865 4 2 3
G 21 1 12 11 1 3 7 9935 1 0
H 1 8 18 3 1 20 1 0 9912 0
I 2 2 3 1 2 1 2 0 0 9872

Weighted Random Selection
• Ala => Xxx (%)
A
R
N
D
C
Q
E
G
H
I
L
K
M
F
P
S
T
W
Y
V

PAM-Simulator
PAM-simulator
0
20
40
60
80
100
120
0 50 100 150 200 250 300
PAM
%id
en
tity

PAM-Simulator
PAM-Simulator
0
10
20
30
40
50
60
70
80
90
100
0 200 400 600 800 1000 1200 1400 1600 1800 2000
PAM
% i
den
tity

PAM Value Distance(%)
80 50
100 60
200 75
250 85 <- Twilight zone
300 92
(From Doolittle, 1987, Of URFs and ORFs,
University Science Books)
Some PAM values and their corresponding observed distances
•When the PAM distance value between two distantly related proteins nears the value 250 it becomes difficult to tell whether the two proteins are homologous, or that they are two at randomly taken proteins that can be aligned by chance. In that case we speak of the 'twilight zone'.
•The relation between the observed percentage in distance of two sequences versus PAM value. Two randomly diverging sequences change in a negatively exponential fashion. After the insertion of gaps to two random sequences, it can be expected that they will be 80 - 90 % dissimilar (from Doolittle, 1987 ).

• Creation of a pam series from evolutionary
simulations
• pam2=pam1^2
• pam3=pam1^3
• And so on…
• pam30,60,90,120,250,300
• low pam - closely related sequences
– high scores for identity and low scores for
substitutions - closer to the identity matrix
• high pam - distant sequences
– at pam2000 all information is degenerate except
for cysteins
• pam250 is the most popular and general
– one amino acid in five remains unchanged
(mutability varies among the amino acids)
Overview


250 PAM evolutionary distance
A R N D C Q E G H I L K M F P
Ala A 13 6 9 9 5 8 9 12 6 8 6 7 7 4 11Arg R 3 17 4 3 2 5 3 2 6 3 2 9 4 1 4 Asn N 4 4 6 7 2 5 6 4 6 3 2 5 3 2 4Asp D 5 4 8 11 1 7 10 5 6 3 2 5 3 1 4 Cys C 2 1 1 1 52 1 1 2 2 2 1 1 1 1 2Gln Q 3 5 5 6 1 10 7 3 7 2 3 5 3 1 4Glu E 5 4 7 11 1 9 12 5 6 3 2 5 3 1 4Gly G 12 5 10 10 4 7 9 27 5 5 4 6 5 3 8 His H 2 5 5 4 2 7 4 2 15 2 2 3 2 2 3 Ile I 3 2 2 2 2 2 2 2 2 10 6 2 6 5 2 Leu L 6 4 4 3 2 6 4 3 5 15 34 4 20 13 5 Lys K 6 18 10 8 2 10 8 5 8 5 4 24 9 2 6 Met M 1 1 1 1 0 1 1 1 1 2 3 2 6 2 1 Phe F 2 1 2 1 1 1 1 1 3 5 6 1 4 32 1 Pro P 7 5 5 4 3 5 4 5 5 3 3 4 3 2 20 Ser S 9 6 8 7 7 6 7 9 6 5 4 7 5 3 9 Thr T 8 5 6 6 4 5 5 6 4 6 4 6 5 3 6 Trp W 0 2 0 0 0 0 0 0 1 0 1 0 0 1 0 Tyr Y 1 1 2 1 3 1 1 1 3 2 2 1 2 15 1 Val V 7 4 4 4 4 4 4 4 5 4 15 10 4 10 5[column on left represents the replacement amino acid]
Mutation probability matrix for the evolutionary distance of 250 PAMs. To simplify the appearance, the elements are shown multiplied by 100.
In comparing two sequences of average amino acid frequency at this evolutionary distance, there is a 13% probability that a position containing Ala in the first sequence will contain Ala in the second. There is a 3% chance that it will contain Arg, and so forth.
Overview

4 3 2 1 0
A brief history of time (BYA)
Origin of
life
Origin of
eukaryotes insectsFungi/animal
Plant/animal
Earliest
fossils
BYA

Margaret Dayhoff’s 34 protein superfamilies
Protein PAMs per 100 million years
Ig kappa chain 37
Kappa casein 33
Lactalbumin 27
Hemoglobin 12
Myoglobin 8.9
Insulin 4.4
Histone H4 0.10
Ubiquitin 0.00

Many sequences depart from average composition.
Rare replacements were observed too infrequently to resolve relative probabilities accurately (for 36 pairs no replacements were observed!).
Errors in 1PAM are magnified in the extrapolation to 250PAM.
Distantly related sequences usually have islands (blocks) of conserved residues. This implies that replacement is not equally probable over entire sequence.
Sources of error

• Simple identity, which scores only identical amino acids as a match.
• Genetic code changes, which scores the minimum number of nucieotide changes to change a codon for one amino acid into a codon for the other.
• Chemical similarity of amino acid side chains, which scores as a match two amino acids which have a similar side chain, such as hydrophobic, charged and polar amino acid groups.
• The Dayhoff percent accepted mutation (PAM) family of matrices, which scores amino acid pairs on the basis of the expected frequency of substitution of one amino acid for the other during protein evolution.
• The blocks substitution matrix (BLOSUM) amino acid substitution tables, which scores amino acid pairs based on the frequency of amino acid substitutions in aligned sequence motifs called blocks which are found in protein families
Overview

• Henikoff & Henikoff (Henikoff, S. &
Henikoff J.G. (1992) PNAS 89:10915-
10919)
• asking about the relatedness of distantly
related amino acid sequences ?
• They use blocks of sequence fragments
from different protein families which can
be aligned without the introduction of
gaps. These sequence blocks correspond
to the more highly conserved regions.
BLOSUM: Blocks Substitution Matrix

BLOSUM (BLOck – SUM) scoring
DDNAAV
DNAVDD
NNVAVV
Block = ungapped alignent
Eg. Amino Acids D N V A
a b c d e f
1
2
3
S = 3 sequences
W = 6 aa
N= (W*S*(S-1))/2 = 18 pairs

A. Observed pairs
DDNAAV
DNAVDD
NNVAVV
a b c d e f
1
2
3
D N A V
D
N
A
V
1
4
1
3
1
1
1
1
4 1
f fij
D N A V
D
N
A
V
.056
.222
.056
.167
.056
.056
.056
.056
.222 .056
gij
/18
Relative frequency table
Probability of obtaining a pair
if randomly choosing pairs
from block

AB. Expected pairs
DDDDD
NNNN
AAAA
VVVVV
DDNAAV
DNAVDD
NNVAVV
Pi
5/18
4/18
4/18
5/18
P{Draw DN pair}= P{Draw D, then N or Draw M, then D}
P{Draw DN pair}= PDPN + PNPD = 2 * (5/18)*(4/18) = .123
D N A V
D
N
A
V
.077
.123
.154
.123
.049
.123
.099
.049
.123 .049
eijRandom rel. frequency table
Probability of obtaining a pair of
each amino acid drawn
independently from block

C. Summary (A/B)
sij = log2 gij/eij
(sij) is basic BLOSUM score matrix
Notes:
• Observed pairs in blocks contain information about
relationships at all levels of evolutionary distance
simultaneously (Cf: Dayhoffs’s close relationships)
• Actual algorithm generates observed + expected pair
distributions by accumalution over a set of approx. 2000
ungapped blocks of varrying with (w) + depth (s)

• blosum30,35,40,45,50,55,60,62,65,70,75,80,85,90
• transition frequencies observed directly by identifying blocks that are at least
– 45% identical (BLOSUM45)
– 50% identical (BLOSUM50)
– 62% identical (BLOSUM62) etc.
• No extrapolation made
• High blosum - closely related sequences
• Low blosum - distant sequences
• blosum45 pam250
• blosum62 pam160
• blosum62 is the most popular matrix
The BLOSUM Series

Overview

• Church of the Flying Spaghetti Monster
• http://www.venganza.org/about/open-letter

• Which matrix should I use?
– Matrices derived from observed substitution data
(e.g. the Dayhoff or BLOSUM matrices) are
superior to identity, genetic code or physical
property matrices.
– Schwartz and Dayhoff recommended a mutation
data matrix for the distance of 250 PAMs as a
result of a study using a dynamic programming
procedure to compare a variety of proteins known
to be distantly related.
• The 250 PAM matrix was selected since in Monte
Carlo studies matrices reflecting this evolutionary
distance gave a consistently higher significance
score than other matrices in the range 0.750 PAM.
The matrix also gave better scores when compared
to the genetic code matrix and identity scoring.
Overview

• When comparing sequences that were not known in advance to be related, for example when database scanning:
– default scoring matrix used is the
BLOSUM62 matrix
– if one is restricted to using
only PAM scoring matrices, then
the PAM120 is recommended for
general protein similarity searches
• When using a local alignment method, Altschul suggests that three matrices should ideally be used: PAM40, PAM120 and PAM250, the lower PAM matrices will tend to find short alignments of highly similar sequences, while higher PAM matrices will find longer, weaker local alignments.
Which matrix should I use?

Rat versus
mouse RBP
Rat versus
bacterial
lipocalin

– Henikoff and Henikoff have compared the
BLOSUM matrices to PAM by evaluating how
effectively the matrices can detect known members
of a protein family from a database when searching
with the ungapped local alignment program
BLAST. They conclude that overall the BLOSUM
62 matrix is the most effective.
• However, all the substitution matrices investigated
perform better than BLOSUM 62 for a proportion of
the families. This suggests that no single matrix is
the complete answer for all sequence comparisons.
• It is probably best to compliment the BLOSUM 62
matrix with comparisons using 250 PAMS, and
Overington structurally derived matrices.
– It seems likely that as more protein three
dimensional structures are determined, substitution
tables derived from structure comparison will give
the most reliable data.
Overview

Overv
iew
• Introduction
– Short recap on databases
– Definitions
• Scoring Matrices
– Theoretical
– Empirial
• PAM (pam-simulator.pl)
• BLOSUM
• Pairwise alignment
– Dot-plots (dotplot-simulator.pl)
Overview

Dotplots
• What is it ?
– Graphical representation using two orthogonal
axes and “dots” for regions of similarity.
– In a bioinformatics context two sequence are
used on the axes and dots are plotted when a
given treshold is met in a given window.
• Dot-plotting is the best way to see all of the
structures in common between two
sequences or to visualize all of the repeated
or inverted repeated structures in one
sequence

Dot Plot References
Gibbs, A. J. & McIntyre, G. A. (1970).
The diagram method for comparing sequences. its use with
amino acid and nucleotide sequences.
Eur. J. Biochem. 16, 1-11.
Staden, R. (1982).
An interactive graphics program for comparing and aligning
nucleic-acid and amino-acid sequences.
Nucl. Acid. Res. 10 (9), 2951-2961.

Visual Alignments (Dot Plots)
• Matrix
– Rows: Characters in one sequence
– Columns: Characters in second sequence
• Filling
– Loop through each row; if character in row, col match, fill
in the cell
– Continue until all cells have been examined

Dotplot-simulator.pl
print " $seq1\n";
for(my $teller=0;$teller<=$seq2_length;$teller++){
print substr($seq2,$teller,1);
$w2=substr($seq2,$teller,$window);
for(my $teller2=0;$teller2<=$seq_length;$teller2++){
$w1=substr($seq1,$teller2,$window);
if($w1 eq $w2){print "*";}else{print " ";}
}
print"\n";
}

Overview
Window size = 1, stringency 100%

Noise in Dot Plots
• Nucleic Acids (DNA, RNA)
– 1 out of 4 bases matches at random
• Stringency
– Window size is considered
– Percentage of bases matching in the window is
set as threshold

Reduction of Dot Plot Noise
Self alignment of ACCTGAGCTCACCTGAGTTA

Dotplot-simulator.pl
Example: ZK822 Genomic and cDNA
Gene prediction:
How many exons ?
Confirm donor and aceptor sites ?
Remember to check the reverse complement !

Chromosome Y self comparison

• Regions of similarity appear
as diagonal runs of dots
• Reverse diagonals
(perpendicular to diagonal)
indicate inversions
• Reverse diagonals crossing
diagonals (Xs) indicate
palindromes
• A gap is introduced by each
vertical or horizontal skip
Overview

• Window size changes with goal
of analysis
– size of average exon
– size of average protein structural
element
– size of gene promoter
– size of enzyme active site
Overview

Rules of thumb
Don't get too many points, about 3-5 times the length of the sequence is about right (1-2%)
Window size about 20 for distant proteins 12 for nucleic acid
Check sequence vs. itself
Check sequence vs. sequence
Anticipate results
(e.g. “in-house” sequence vs genomic, question)
Overview

Available Dot Plot Programs
Dotlet (Java Applet)
http://www.isrec.isb-
sib.ch/java/dotlet/Dotlet.
html

Available Dot Plot Programs
Dotter (http://www.cgr.ki.se/cgr/groups/sonnhammer/Dotter.html)

Available Dot Plot Programs
EMBOSS DotMatcher, DotPath,DotUp

Weblems
• W3.1: Why does 2 PAM, i.e. 1 PAM multiplied with itself, not correspond to exactly 2% of the amino acids having mutated, but a little less than 2% ? Or, in other words, why does a 250 PAM matrix not correspond to 250% accepted mutations ?
• W3.2: Is it biologically plausible that the C-C and W-W entries in the scoring matrices are the most prominent ? Which entries (or groups of entries) are the least prominent ?
• W3.3: What is OMIM ? How many entries are there ? What percentage of OMIM listed diseases has no known (gene) cause ?
• W3.4: Pick one disease mapped to chromosome Y from OMIM where only a mapping region is known. How many candidate genes can you find in the locus using ENSEMBL ? Can you link ontology terms for the candidates to the disease phenotype ?