arthur c. corcoran memorial lecture -...
TRANSCRIPT

e129
Treatment-resistant hypertension (TRHT) is character-ized by persistently high arterial blood pressure (BP),
partly as a result of a dysfunctional autonomic nervous system (ANS), wherein sympathetic drive/norepinephrine spillover is increased and parasympathetic drive is decreased.1–3 The dif-ficulty in treatment of TRHT arises precisely from this partly neurogenic component because the available drug therapies do not target the central nervous system (CNS) directly. Because of this and despite recent advances in techniques such as renal denervation and carotid baroreceptor activation,4,5 successful treatment and long-term control of TRHT remain challeng-ing.6 In an attempt to develop more effective treatments, many groups are investigating specific causes of TRHT. A large body of experimental evidence implicates both genetic and environ-mental influences, such as salt sensitivity and elevated systemic renin–angiotensin system (RAS) activity7–14 in the patho-physiology of this disease. Furthermore, a majority of studies point to dysregulations in the activity within the cardiorespira-tory brain regions as a reason for increased sympathetic and decreased parasympathetic drive to the peripheral organs,14–21 resulting in end-organ damage,21–25 vascular/endothelial dys-function,26,27 and hormonal imbalance,28 which perpetuate the pathophysiology and complicate treatment strategies. Despite our increasing understanding of TRHT, the origins of this brain dysregulation remain largely unknown. Recently, the activity of the immune system29–31 and neuroimmune pathways in patients with hypertension and animal models of hypertension has been highlighted.32–36 Studies suggest that both the sympathetic and the parasympathetic arms of the ANS can exert their influence on the activity of the immune organs, tissues, and cells, and that it is the dysfunctional ANS-immune communication that may lead to hypertension and CVD.35–40 The aim of this review is to summarize the latest advances in this field and review the cur-rent understanding of connections between the autonomic and immune systems, specifically the connections between the brain and the bone marrow (BM), the largest source of hematopoietic cells in the body. In addition, we will highlight the importance of BM activity in CVD and hypertension, and propose novel bidirectional brain–BM communication hypothesis whose dys-function may have important implications in the development of therapeutic strategies for neurogenic TRHT.
Role of BM in Cardiovascular Health and Disease
BM is central in the regulation of hematopoiesis.41,42 It consti-tutes ≈4% of the total body mass in humans, produces ≈500 billion hematopoietic cells each day, and accounts for ≈90% of total hematopoiesis in the body.41,42 BM hematopoietic stem and progenitors cells (HSPCs) interact with cells of secondary lym-phoid organs such as the spleen, which regulate the HSPC dif-ferentiation and maturation.43–46 Readily available BM-derived cells are released into the circulation diurnally or in response to diverse pathophysiological cues such as vascular or tissue injury.37,47–51 The involvement of BM and HSPCs in CVD and hypertension has attracted significant attention in the last decade for several reasons. First, CVD has been linked with an overac-tive adaptive immune system. Increased inflammation has been reported in heart disease, stroke, vascular diseases, and hyper-tension in both humans and animal models.29–31,36,37,52–59 In fact, a correlation exists between elevated inflammatory responses and disease progression,31,57,60,61 and immunosupression can delay or even arrest the progression of CVD.62–67 Second, after an acute injury, the BM cells with angiogenic and reparative properties such as endothelial progenitor cells (EPCs)17,68–73 are immedi-ately recruited to the site of injury, aiding in repair of the tis-sue damage.74–78 Unfortunately, some patients with advancing CVD and other related diseases have significantly decreased pools of EPCs,79,80 and this decreased vascular potential further exacerbates the dysfunctional vasculature already seen in CVD. Third, an overactive RAS is implicated in exaggerated immune system responses in human patients as well as animal models of CVDs.14,17,81–83 In angiotensin II (Ang II)-dependent hyper-tension, increased systemic and splenic inflammatory cells and factors have a central role in initiation and maintenance of hypertension,17,35,40,82,84 presumably by conferring damage on the vasculature as a result of decreases in vascular repair–rele-vant progenitor cells. Consistent with this are observations that antihypertensive effects of the RAS inhibitors have been shown to improve the number and function of EPCs in CVD.71,85,86 Collectively, these data suggest a correlation between BM HSPCs and inflammation in development of CVD.
We have recently proposed that it is not only the increased inflammatory damage but also the imbalance in the BM-derived
Received January 6, 2014; first decision January 16, 2014; revision accepted March 7, 2014.From the Departments of Physiology and Functional Genomics (J.Z., M.M.S., M.K.R.) and Psychiatry (P.D.P., M.F.), College of Medicine; and
Department of Physiological Sciences (T.P., D.M.B., D.C.B.), College of Veterinary Medicine, University of Florida, Gainesville.Correspondence to Jasenka Zubcevic, Department of Physiology and Functional Genomics, University of Florida, 1345 Center Drive, PO BOX 100274,
M552, Gainesville FL 32610, E-mail [email protected] or Mohan K. Raizada, Department of Physiology and Functional Genomics, University of Florida, 1345 Center Drive, PO BOX 100274, M552, Gainesville FL 32610, E-mail [email protected]
Functional Neural–Bone Marrow PathwaysImplications in Hypertension and Cardiovascular Disease
Jasenka Zubcevic, Monica M. Santisteban, Teresa Pitts, David M. Baekey, Pablo D. Perez, Donald C. Bolser, Marcelo Febo, Mohan K. Raizada
(Hypertension. 2014;63:e129-e139.)© 2014 American Heart Association, Inc.
Hypertension is available at http://hyper.ahajournals.org DOI: 10.1161/HYPERTENSIONAHA.114.02440
Arthur C. Corcoran Memorial Lecture
by guest on June 30, 2018http://hyper.ahajournals.org/
Dow
nloaded from

e130 Hypertension June 2014
immune cell-dependent vascular damage and endothelial cell-driven vascular repair is central to cardiovascular patho-physiology.17,32,33,36 For example, the hypertensive phenotype of the spontaneously hypertensive rat (SHR) is characterized by chronically elevated inflammation and downregulated BM-derived EPC number and function, as evidenced by the decreased ability of the EPCs to proliferate and migrate to the site of the damage, as well as the loss of EPC angiogenic abil-ity.36 Additionally, data suggest that central effects of Ang II–induced hypertension involve an increase in the BM-derived inflammatory cells (ICs), as well as a decrease in BM-derived EPC number and function, which cannot be attributed to sec-ondary effects of systemic hypertension.14,17 Therefore, the decrease in the EPC count coupled with their decreased func-tion may perpetuate vascular damage in hypertension. One mechanism through which overactive Ang II in hypertension may affect the BM HSPC activity is by having a direct effect on the HSPC stemness, through promotion of premature dif-ferentiation.41,42 This would diminish the angiogenic and reparative abilities of HSPCs, thereby compromising vascular repair. Furthermore, our data have shown that Ang II under-mines the ability of HSPCs to home to the BM niches,87 which could further compromise their angiogenic functions. The increase in inflammatory stress can also directly reduce the function and activity of EPCs,88,89 as evidenced by clinical data demonstrating that anti-inflammatory drugs and antioxidants exert beneficial effects on the EPCs.89 Although a transient inflammatory response may in fact stimulate EPC mobiliza-tion, thereby promoting tissue repair,90 evidence suggests that chronic systemic inflammation may lead to functional impair-ment of EPCs and their depletion.91–93 In line with this is the observation that treatment with BM-derived mononuclear cells does not improve neurological recovery after stroke in the SHR, which is characterized by both increased systemic inflammation and elevated RAS, perhaps because of the high inflammation, which remained widespread in these animals.94 Taken together, it is reasonable to suggest that systemic RAS may act distinctly from its well-established classic effects to both increase ICs and decrease EPCs.
It is evident from the above discussion that production, mobilization, and release of cells from the BM is critical in maintaining regular vascular homeostasis, and that dysregula-tion at any stage in these processes leads to devastating cardio-vascular consequences. However, it is important to point out that some discrepancies exist in the literature. For example, there are studies that did not find a difference in EPCs between healthy and hypertensive humans,95 whereas others linked dysfunctional EPCs only with hypertension that was associ-ated with other comorbidities.96,97 In one such study Delva et al95 found no significant differences in EPC numbers and function between a group of carefully selected patients exhib-iting a hypertensive phenotype and control subjects. However, this patient group may have had less severe hypertension because their treatment used no more than 2 antihypertensive medications, namely RAS and beta blockers, both of which may directly affect the function of EPCs.69,71,98,99 The patients with hypertension also were treated with aspirin, known for its beneficial vascular effects65,66 including those benefiting the EPC function directly.100 These and other studies pinpoint
the complexity of the problem of TRHT, and further highlight the need for novel therapeutic targets. Some of the most rel-evant questions arising from the evidence to date are whether the BM activity is regulated by the brain, and if the dysfunc-tional brain–BM communication could account for CVD and hypertension.
Evidence of ANS Regulation of BM ActivityThe first evidence of the importance of ANS for BM cell homeostasis is derived from the circadian studies, which dem-onstrated the regulation of BM cell activity.36 It is well estab-lished that the release of BM HSPCs is rhythmically regulated in a circadian manner, for which sympathetic drive is essen-tial.36,47–50,101,102 In rodents, the increase in sympathetic drive at night is associated with the release of the surveillance immune cells from the BM, which circulate through the body in search of an infection or injury.36,47 The return of these cells to the BM signals the release of the repair cells aimed to heal and repair.47,102 However, in cases of prolonged infections or other-wise compromised immune responses, the accumulation and aging of the surveillance immune cells in the circulation leads to the loss of the trigger for release of the repair cells into the circulation.47 Clinically, this may translate into a diminished ability of the body to repair itself in times of sustained inflam-matory damage. Furthermore, loss of circadian rhythmicity of the BM cell release seems to be closely tied to any dysfunction in the sympathetic drive.36 For example, the loss of the circa-dian rhythmicity observed in diabetes mellitus–related periph-eral neuropathy is attributed to the decreased sympathetic drive to the BM and results in dysfunctional BM EPCs.102 On the contrary, our studies have demonstrated that elevated sym-pathetic drive to the BM results in a perturbed BM adrenergic receptor signaling system that is associated with loss of circa-dian rhythmicity in BM cell release.36 Therefore, we infer that the BM HSPC environment is tightly regulated, which allows it to respond quickly to an environmental change or injury, but which can also result in adaptation and plasticity of the BM cell responses, ultimately contributing to a pathological state. In the context of hypertension, we propose that the chronic elevation in the BM sympathetic drive leads to the loss of cir-cadian rhythmicity of the BM cell release, resulting in chroni-cally elevated systemic inflammatory responses and decreased EPC availability and function.36 Therefore, clinical targeting of the elevated sympathetic drive in neurogenic hypertension could have a new dimension.
The second body of evidence highlighting the importance of ANS in BM function comes from studies demonstrating increases in inflammatory cells in several disease states. Most recently, Dutta et al37 described the role of increased sympathetic drive in recruitment of BM cells after postmyo-cardial infarction. Their data showed that increased sympa-thetic drive to the BM after myocardial infarction caused the liberation of the BM HSPCs, which seeded in the spleen and boosted the production of monocytes. The increase in monocyte production and their infiltration caused the devel-opment of larger, destabilized atherosclerotic plaques with a more advanced morphology, resulting in higher incidence of recurrent myocardial events as well as increased inci-dence of stroke.37 In a similar fashion, inflammatory cells
by guest on June 30, 2018http://hyper.ahajournals.org/
Dow
nloaded from

Zubcevic et al Brain–Bone Marrow Communication in Hypertension e131
have been shown to infiltrate the brain and contribute to the neuropathology in several diseases, including CVD.103–107 Although the exact mechanism underlying the extravasation of inflammatory cells into the CNS is still an area of active research, several hypotheses are under investigation. First, an increase in chemoattractant proinflammatory molecules, such as chemokine (C-C motif) ligand 2, have been shown to be associated with the increase in BM-derived microglia and monocytes in the CNS.104,106 BM monocytes will home in on areas of the brain where the concentration of chemo-kine (C-C motif) ligand 2 is the highest. Whether this process involves the breakdown of the blood–brain barrier (BBB) is still debated. Hypoxic-ischemic brain injury is associated with BM-derived microglia infiltration into areas of the brain where the BBB had been compromised.106 However, other groups have shown that the infiltration of BM monocytes into the brain is independent of the BBB integrity.108 Therefore, it seems that although the breakdown of BBB would facilitate the infiltration of BM-derived cells into the brain, it is not necessary for this process to occur. In diabetes mellitus, for example, infiltration of BM-derived monocyte progenitors in the brain presympathetic areas contributes to the increased pool of activated inflammatory microglia and subsequent neurovascular-glial inflammatory status.104 Our preliminary data support this contention in the hypertension paradigm as well.109 Studies on the mechanism of the infiltration of BM cells into the brain of hypertensive animals are underway but could be because of a combination of BBB breakdown as described by Stern110 and increased chemoattractant, pro-inflammatory molecules.111,112 Although all the differences between BM-derived and resident microglia have yet to be elucidated, there is one fundamental difference. Resident microglia work to survey their environment become activated and help to repair acute injuries.113–115 BM-derived microg-lia have only been described to move to the CNS in chronic conditions, indicating that either their role is only important in the long-term pathophysiology of neuroinflammation or there are age-related effects on the homing of BM cells to the brain.116 The role of activated microglia in several impor-tant neurodegenerative/neuroinflammatory diseases is well established17,103–107,112,117,118; however, their influence on pre-sympathetic neurons in the context of established CVD with chronically high sympathetic drive is currently understudied.
Our recent observations in rodent models of Ang II–dependent hypertension lead us to suggest that activation of microglia in the presympathetic cardioregulatory brain areas may precede both the activation of the sympathetic drive and the increase in BP. Therefore, activation of microglia may be a crucial step in generation of high sympathetic drive to the periphery including the BM.32,33 This concept does not disregard the effects of direct Ang II action on the presym-pathetic neurons, which has been firmly established in neu-rogenic hypertension.7,14,15,119 It is plausible to suggest that the healthy cardioregulatory system is capable of regulating its own responses to sporadic Ang II challenges, until these are intensified and aided by other environmental prohypertensive insults. Consistent with this idea, an important characteristic of the microglial cell is that it exhibits characteristics of a mem-ory immune cell of the brain, meaning that it can be primed
for activation.120,121 Therefore, several prohypertensive signals may be required for microglial cell differentiation into a fully active proinflammatory stage, characterized by increased pro-liferation, migration, and release of cytokines/neurotransmit-ters that will subsequently affect the state of the surrounding presympathetic neurons. This will lead to increased sympa-thetic drive to the periphery, including to the BM, causing dys-function in BM cell activity.17 Dysfunctional BM cell activity will contribute to decreased repair and increased inflamma-tion, and consequently result in the infiltration of the immune cells to the brain, thereby perpetuating cardiovascular patho-physiology in a feed-forward manner.32
These observations support the role of increased sympathetic drive in recruitment and regulation of BM cells. Evidence also exists for a role of the parasympathetic nervous system in regulation of BM-induced inflammation. Stimulation of the vagus nerve protects from excessive cytokine production and ameliorates experimental inflammatory disease via the inflam-matory reflex mechanism. This involves activation of nicotinic acetylcholine receptor α7 (nAchRα7) on macrophages, lym-phocytes, neurons, and other cells.122 This inflammatory reflex has been described in the spleen, gut, and, recently, the BM, where it has been shown that deficiency of the BM nAchRα7 impaired vagus nerve–mediated regulation of the proinflam-matory tumor necrosis factor (TNF), in a mechanism depen-dent on the BM-derived macrophages.38,39,51,122–125 In support of these data, our recent study has demonstrated a decrease in both acetylcholine esterase and cholinergic acetyl transfer-ase in the BM of the SHR compared with the Wistar-Kyoto rat (WKY), in addition to increased BM sympathetic drive.36 As acetylcholine seems to block the acute and direct effects of norepinephrine on the release of BM ICs, this would suggest that the anti-inflammatory parasympathetic effects directly oppose the proinflammatory effects of the sympathetic drive on the BM.36 Interestingly, the vagal influence is reduced in hypertension; potentially further exacerbating the proinflam-matory state. There is no evidence of direct vagal projections to the BM, and the parasympathetic effects are most likely to be endocrine and delivered via the BM extensive vasculature (Figure 1). Alternatively, the vagus may be indirectly affecting the BM by modulating the activity of the BM postganglionic sympathetic nerves because it has been suggested in other lym-phoid tissues126 (Figure 1). Future studies should elucidate the cellular and neuronal mechanisms of the reduced parasympa-thetic drive in the hypertension-related inflammatory responses in the BM, and whether the cholinergic receptor system is as tightly regulated as the adrenergic receptors in the BM.
The evidence discussed above suggests that increased sympathetic drive to the BM, in conjunction with decreased parasympathetic influence, may initiate a cascade of signaling events culminating in dysfunctional BM cell activity and lead-ing to hypertension.36 The increase in sympathetic nerve activ-ity in prehypertensive animal models and humans supports this contention.1–3,127 However, direct measurements of BM sympathetic nerve activity in the prehypertensive state must be performed to confirm this hypothesis. Nonetheless, cumu-lative evidence supports the view that the ANS has profound influence on the BM that could be detrimental in hypertension and other CVD.
by guest on June 30, 2018http://hyper.ahajournals.org/
Dow
nloaded from

e132 Hypertension June 2014
Afferent Input From BM to the BrainIn the previous section, we have discussed the efferent brain–BM pathway and the role of the ANS in regulation of the BM cell activity in health and CVD. Our data, discussed below, sup-port the concept that an afferent neuronal input from the BM to the brain may also exist and may be involved in modification of the ANS efferent pathway to the periphery, including the BM. This bidirectional brain–BM communication hypothesis
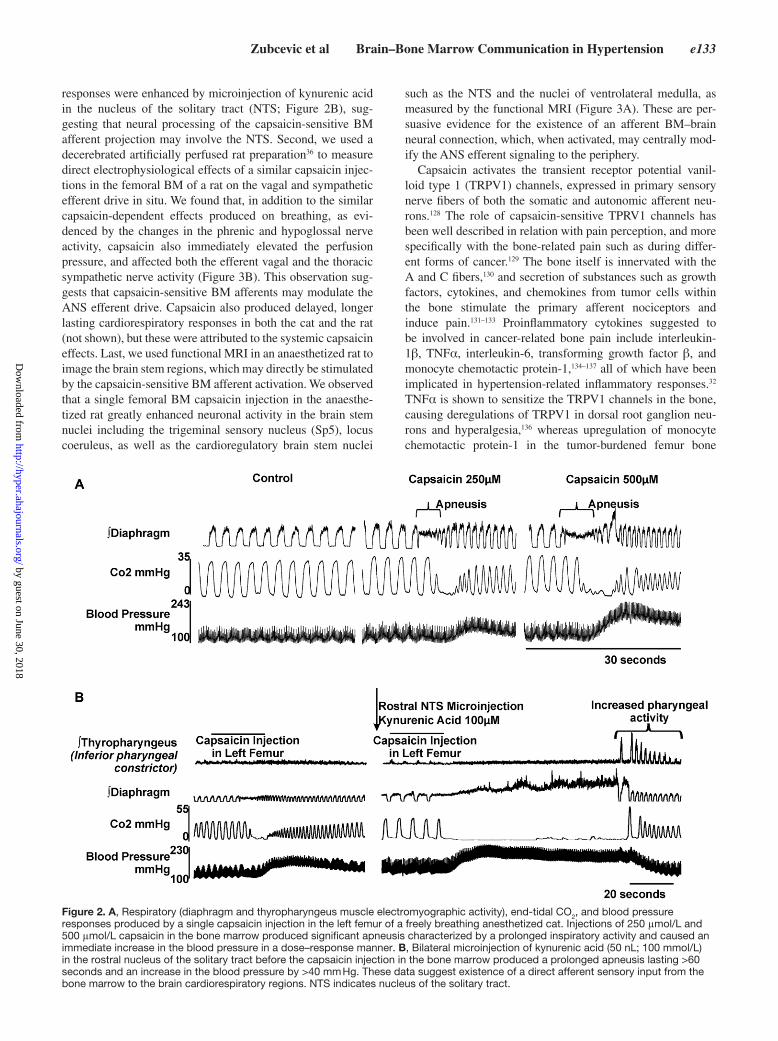
is based on the following evidence; first, we used an anaesthe-tized and nonparalyzed cat model to investigate the effects of a localized, bolus BM injection of capsaicin (0.1 mL) on car-diorespiratory variables. We observed that capsaicin injection into the BM produced an immediate dose-dependent BP eleva-tion, accompanied by apneusis, as evidence by the diaphragm and thyropharyngeus muscle electromyography recordings and measurement of end-tidal CO
2 levels (Figure 2A). These
Figure 1. Bidirectional communication between the brain and the bone marrow (BM) in cardiovascular homeostasis. Prohypertensive signals such as elevated angiotensin II (Ang II) lead to neurovascular-glial inflammation in the brain cardioregulatory areas. Dysfunctional autonomic nervous system (ANS) output is characterized by elevation in the sympathetic and a decrease in the parasympathetic drive to the periphery, including the BM. This process is coupled with elevated peripheral Ang II and causes a persistent increase in the inflammatory cell (IC) and decrease in endothelial progenitor cell (EPCs). The elevated ICs contribute to the vascular and tissue damage, whereas decreased EPC count and function contribute to the decreased repair of this damage, leading to cardiorenal pathology. The combination of extravasation of the ICs to the presympathetic brain areas and the increased somatic afferent input from the BM to the brain, via activation of the putative transient receptor potential vanilloid type 1 channels, adds to the neurovascular-glial inflammation and drives the dysfunctional ANS output to the periphery. These events perpetuate the resulting cardiovascular and renal pathophysiology and resistant hypertension. BP indicates blood pressure; PNS, parasympathetic nervous system; SNS, sympathetic nervous system.
by guest on June 30, 2018http://hyper.ahajournals.org/
Dow
nloaded from
Zubcevic et al Brain–Bone Marrow Communication in Hypertension e133
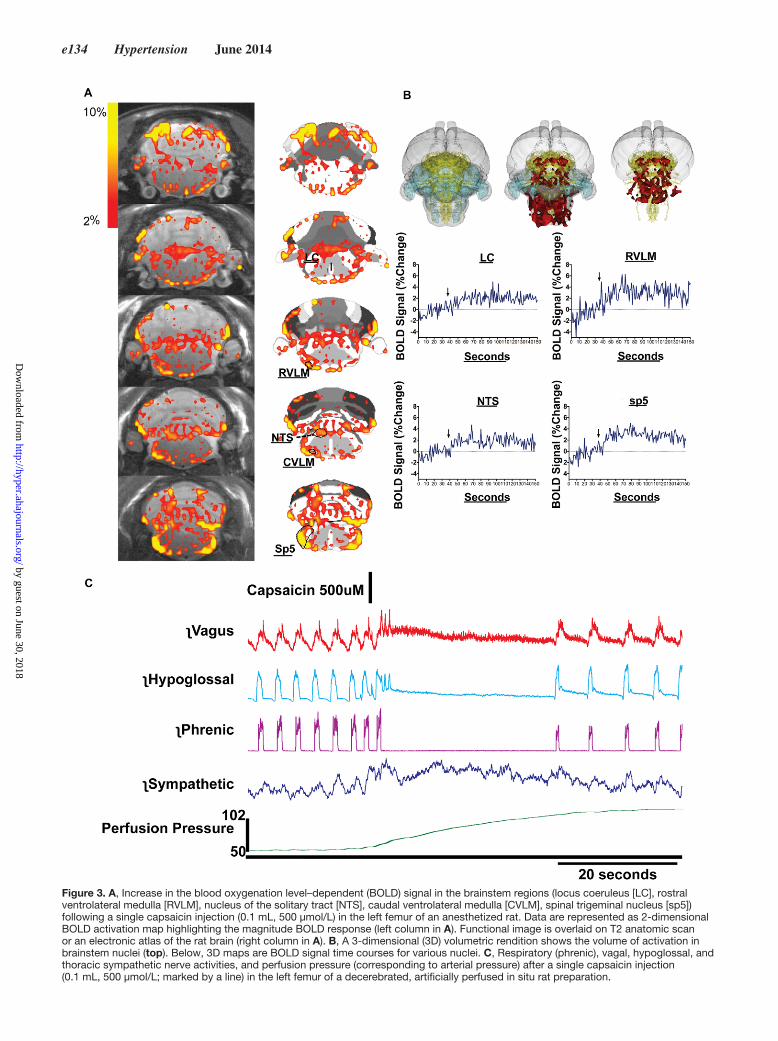
responses were enhanced by microinjection of kynurenic acid in the nucleus of the solitary tract (NTS; Figure 2B), sug-gesting that neural processing of the capsaicin-sensitive BM afferent projection may involve the NTS. Second, we used a decerebrated artificially perfused rat preparation36 to measure direct electrophysiological effects of a similar capsaicin injec-tions in the femoral BM of a rat on the vagal and sympathetic efferent drive in situ. We found that, in addition to the similar capsaicin-dependent effects produced on breathing, as evi-denced by the changes in the phrenic and hypoglossal nerve activity, capsaicin also immediately elevated the perfusion pressure, and affected both the efferent vagal and the thoracic sympathetic nerve activity (Figure 3B). This observation sug-gests that capsaicin-sensitive BM afferents may modulate the ANS efferent drive. Capsaicin also produced delayed, longer lasting cardiorespiratory responses in both the cat and the rat (not shown), but these were attributed to the systemic capsaicin effects. Last, we used functional MRI in an anaesthetized rat to image the brain stem regions, which may directly be stimulated by the capsaicin-sensitive BM afferent activation. We observed that a single femoral BM capsaicin injection in the anaesthe-tized rat greatly enhanced neuronal activity in the brain stem nuclei including the trigeminal sensory nucleus (Sp5), locus coeruleus, as well as the cardioregulatory brain stem nuclei
such as the NTS and the nuclei of ventrolateral medulla, as measured by the functional MRI (Figure 3A). These are per-suasive evidence for the existence of an afferent BM–brain neural connection, which, when activated, may centrally mod-ify the ANS efferent signaling to the periphery.
Capsaicin activates the transient receptor potential vanil-loid type 1 (TRPV1) channels, expressed in primary sensory nerve fibers of both the somatic and autonomic afferent neu-rons.128 The role of capsaicin-sensitive TPRV1 channels has been well described in relation with pain perception, and more specifically with the bone-related pain such as during differ-ent forms of cancer.129 The bone itself is innervated with the A and C fibers,130 and secretion of substances such as growth factors, cytokines, and chemokines from tumor cells within the bone stimulate the primary afferent nociceptors and induce pain.131–133 Proinflammatory cytokines suggested to be involved in cancer-related bone pain include interleukin-1β, TNFα, interleukin-6, transforming growth factor β, and monocyte chemotactic protein-1,134–137 all of which have been implicated in hypertension-related inflammatory responses.32 TNFα is shown to sensitize the TRPV1 channels in the bone, causing deregulations of TRPV1 in dorsal root ganglion neu-rons and hyperalgesia,136 whereas upregulation of monocyte chemotactic protein-1 in the tumor-burdened femur bone
Figure 2. A, Respiratory (diaphragm and thyropharyngeus muscle electromyographic activity), end-tidal CO2, and blood pressure responses produced by a single capsaicin injection in the left femur of a freely breathing anesthetized cat. Injections of 250 μmol/L and 500 μmol/L capsaicin in the bone marrow produced significant apneusis characterized by a prolonged inspiratory activity and caused an immediate increase in the blood pressure in a dose–response manner. B, Bilateral microinjection of kynurenic acid (50 nL; 100 mmol/L) in the rostral nucleus of the solitary tract before the capsaicin injection in the bone marrow produced a prolonged apneusis lasting >60 seconds and an increase in the blood pressure by >40 mm Hg. These data suggest existence of a direct afferent sensory input from the bone marrow to the brain cardiorespiratory regions. NTS indicates nucleus of the solitary tract.
by guest on June 30, 2018http://hyper.ahajournals.org/
Dow
nloaded from
e134 Hypertension June 2014
Figure 3. A, Increase in the blood oxygenation level–dependent (BOLD) signal in the brainstem regions (locus coeruleus [LC], rostral ventrolateral medulla [RVLM], nucleus of the solitary tract [NTS], caudal ventrolateral medulla [CVLM], spinal trigeminal nucleus [sp5]) following a single capsaicin injection (0.1 mL, 500 μmol/L) in the left femur of an anesthetized rat. Data are represented as 2-dimensional BOLD activation map highlighting the magnitude BOLD response (left column in A). Functional image is overlaid on T2 anatomic scan or an electronic atlas of the rat brain (right column in A). B, A 3-dimensional (3D) volumetric rendition shows the volume of activation in brainstem nuclei (top). Below, 3D maps are BOLD signal time courses for various nuclei. C, Respiratory (phrenic), vagal, hypoglossal, and thoracic sympathetic nerve activities, and perfusion pressure (corresponding to arterial pressure) after a single capsaicin injection (0.1 mL, 500 μmol/L; marked by a line) in the left femur of a decerebrated, artificially perfused in situ rat preparation.
by guest on June 30, 2018http://hyper.ahajournals.org/
Dow
nloaded from

Zubcevic et al Brain–Bone Marrow Communication in Hypertension e135
correlates with the severity of tumor progression and pain.137 Furthermore, activation of microglial C-X3-C motif receptor 1 receptors by the fractalkine-producing spinal cord neurons mediates pain during the carcinoma growth in rat tibia.138 A parallel could be drawn between the inflammatory mediators causing activation of the pain sensory afferents within the can-cerous bone, and those associated with inflammation in CVD. Activation of TRPV1 channels has profound effects on cardio-vascular homeostasis, and TRPV1 dysfunction is implicated in increased salt sensitivity, water and sodium balance in hyper-tension, and in worsening of ischemia-reperfusion injury in the heart, brain, and the liver.139–141 In view of our findings and the data from the literature, we suggest that TRPV1 in the BM may be relevant in the following manner: (1) increased proinflam-matory mediators may sensitize the TRPV1 in the BM; and (2) activation of TRPV1 may stimulate inflammatory responses in the BM cells via stimulation of the release of calcitonin related gene peptide and substance P from sensory nerves. This may be relevant in the paradigm of hypertension, which is known to be characterized by both increased BM inflammatory17 and dys-functional TRPV1 responses.140,141 Thus, sustained activation of BM TRPV1 by inflammatory factors present in hyperten-sion may exaggerate BM and systemic inflammation, contrib-uting to cardiovascular pathology in a feed-forward manner, as we have hypothesized in Figure 1. Furthermore, based on our current findings, activation of TRPV1 on BM sensory afferents may send a direct neuronal signal to the brain stem nocicep-tive- and cardioregulatory areas, thereby modifying the ANS efferent responses (Figures 2 and 3). Further experiments are needed to dissect the afferent neuronal pathways from the BM to the brain in health and CVD.
This is an attractive concept in support of bidirectional brain–BM communication. However, our functional MRI data indicate prominent activation of the sensory input-receiving nuclei such as the Sp5 brain stem nucleus and the locus coe-ruleus (Figure 3).142,143 It is highly probable that the sensory afferents originating in the BM and ending in the brain stem are of somatic origin. The NTS, an important cardioregu-latory region of the brain stem, is a site of convergence of somatic and autonomic regulatory mechanisms.144,145 In line with this, our data show that pharmacological manipulation of the NTS modified the cardiovascular responses to BM capsa-icin (Figure 2B), and that the NTS activity increases after BM capsaicin injection (Figure 3A). In hypertension, NTS activity is heavily influenced by the overactive RAS and especially Ang II, which increases the inhibitory gamma-aminobutyric acidergic influence in the NTS and dampens the vagal baro-reflex.20,146 Therefore, in the context of hypertension, high inflammation may be causing the sustained activation of the BM sensory afferents, which, together with Ang II–dependent dampened vagal responses characteristic of hypertension, could be part of a feed-forward loop resulting in even more inflammation in the periphery.
Based on this evidence, we propose the following bidirec-tional brain–BM communication hypothesis for cardiovascu-lar homeostasis (Figure 1): Prohypertensive signals such as elevated brain and systemic RAS lead to neurovascular-glial inflammation in the brain cardioregulatory areas. The resul-tant dysfunctional ANS output, characterized by elevation
in the sympathetic (and a decrease in the parasympathetic) drive to the periphery, including the BM, causes a persistent increase in the ICs and decrease in EPCs. The elevated ICs contribute to the vascular and tissue damage, while decreased EPC count and function contribute to the decreased repair of this damage, leading to cardiorenal pathology. Extravasation of the ICs to the presympathetic brain areas may add to the neurovascular-glial inflammation and drive the dysfunctional ANS output to the periphery, thereby perpetuating the result-ing cardiovascular and renal pathophysiology and resistant hypertension. The contribution of the novel somatic afferent input from the BM to the brain to our hypothesis remains to be fully investigated.
Future PerspectivesIn view of the evidence presented here, we think that the auto-nomic control of BM plays a critical role in cardiovascular homeostasis, and there seems to be a bidirectional commu-nication between the brain and BM in this process. However, many questions remain unanswered. First, what are the exact neuroanatomical sympathetic and parasympathetic pathways innervating the BM? The loss of the BM sympathetic inner-vation after denervation of the superior cervical ganglion has been shown; however, exact presympathetic and spinal neuro-nal pathways need to be elucidated to fully understand these pathways. Second, the exact nature of the BM sympathetic drive needs to be established. We have recently alluded to the respiratory coupling of the BM sympathetic nerve activ-ity, but the barosensitivity of the BM sympathetic drive and whether this is changed in hypertension remains unknown. Third, the neuroanatomical pathways and the involvement of the afferent sensory input from the BM to the brain in hyper-tension need to be established. Fourth, the specific role of the vagus in regulation of the BM cells in hypertension needs to be expounded. We think that these questions will yield important answers, which will move the field of neurogenic hypertension forward and provide novel therapeutic targets in the treatment of TRHT. One of the obvious potential thera-peutic targets would be activated microglia that influence the activity of neurons within the brain cardioregulatory areas, some of which may control the BM and affect the function of the BM HSPCs. A recent study showed that treatment with minocycline, an orally available anti-inflammatory antibiotic, which can cross the BBB and inhibit microglial activation, reduced BP and improved body weight and glucose levels in drug-resistant, morbidly obese/Type 2 diabetic patients who were also hypertensive.104 Therefore, our future investigations will focus on drug discovery of specific, orally available, BBB-crossing anti-inflammatory agents to alleviate neuro-genic hypertension.
Sources of FundingThis work was supported by the National Institutes of Health grants HL33610, HL56921, and HL102033 to M.K. Raizada. J. Zubcevic is a recipient of the American Heart Association Scientist Development Award. M.M. Santisteban is a recipient of the American Heart Association Predoctoral Fellowship.
DisclosuresNone.
by guest on June 30, 2018http://hyper.ahajournals.org/
Dow
nloaded from

e136 Hypertension June 2014
References 1. DiBona GF. Sympathetic nervous system and hypertension. Hypertension.
2013;61:556–560. 2. Esler M, Jennings G, Korner P, Willett I, Dudley F, Hasking G, Anderson
W, Lambert G. Assessment of human sympathetic nervous system activity from measurements of norepinephrine turnover. Hypertension. 1988;11:3–20.
3. Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7:335–346.
4. Esler MD, Krum H, Schlaich M, Schmieder RE, Böhm M, Sobotka PA; Symplicity HTN-2 Investigators. Renal sympathetic denervation for treat-ment of drug-resistant hypertension: one-year results from the Symplicity HTN-2 randomized, controlled trial. Circulation. 2012;126:2976–2982.
5. Bakris GL, Nadim MK, Haller H, Lovett EG, Schafer JE, Bisognano JD. Baroreflex activation therapy provides durable benefit in patients with resistant hypertension: results of long-term follow-up in the Rheos Pivotal Trial. J Am Soc Hypertens. 2012;6:152–158.
6. Calhoun DA, Jones D, Textor S, Goff DC, Murphy TP, Toto RD, White A, Cushman WC, White W, Sica D, Ferdinand K, Giles TD, Falkner B, Carey RM. Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension. 2008;51:1403–1419.
7. Bardgett ME, Holbein WW, Herrera-Rosales M, Toney GM. Ang II-Salt Hypertension Depends on Neuronal Activity in the Hypothalamic Paraventricular Nucleus but Not on Local Actions of Tumor Necrosis Factor-α. Hypertension. 2014;63:527–534.
8. Toney GM, Pedrino GR, Fink GD, Osborn JW. Does enhanced respiratory-sympathetic coupling contribute to peripheral neural mecha-nisms of angiotensin II-salt hypertension? Exp Physiol. 2010;95:587–594.
9. Huang BS, Cheung WJ, Wang H, Tan J, White RA, Leenen FH. Activation of brain renin-angiotensin-aldosterone system by central sodium in Wistar rats. Am J Physiol Heart Circ Physiol. 2006;291:H1109–H1117.
10. Huang BS, White RA, Ahmad M, Leenen FH. Role of brain corticos-terone and aldosterone in central angiotensin II-induced hypertension. Hypertension. 2013;62:564–571.
11. Yamazato M, Ferreira AJ, Yamazato Y, Diez-Freire C, Yuan L, Gillies R, Raizada MK. Gene transfer of angiotensin-converting enzyme 2 in the nucleus tractus solitarius improves baroreceptor heart rate reflex in spontaneously hypertensive rats. J Renin Angiotensin Aldosterone Syst. 2011;12:456–461.
12. Osborn JW, Fink GD, Sved AF, Toney GM, Raizada MK. Circulating angiotensin II and dietary salt: converging signals for neurogenic hyper-tension. Curr Hypertens Rep. 2007;9:228–235.
13. Yamazato M, Yamazato Y, Sun C, Diez-Freire C, Raizada MK. Overexpression of angiotensin-converting enzyme 2 in the rostral ventro-lateral medulla causes long-term decrease in blood pressure in the sponta-neously hypertensive rats. Hypertension. 2007;49:926–931.
14. Shan Z, Zubcevic J, Shi P, Jun JY, Dong Y, Murça TM, Lamont GJ, Cuadra A, Yuan W, Qi Y, Li Q, Paton JF, Katovich MJ, Sumners C, Raizada MK. Chronic knockdown of the nucleus of the solitary tract AT1 receptors increases blood inflammatory-endothelial progenitor cell ratio and exac-erbates hypertension in the spontaneously hypertensive rat. Hypertension. 2013;61:1328–1333.
15. Capone C, Faraco G, Peterson JR, Coleman C, Anrather J, Milner TA, Pickel VM, Davisson RL, Iadecola C. Central cardiovascular circuits con-tribute to the neurovascular dysfunction in angiotensin II hypertension. J Neurosci. 2012;32:4878–4886.
16. Xia H, Sriramula S, Chhabra KH, Lazartigues E. Brain angiotensin-converting enzyme type 2 shedding contributes to the devel-opment of neurogenic hypertension. Circ Res. 2013;113:1087–1096.
17. Jun JY, Zubcevic J, Qi Y, Afzal A, Carvajal JM, Thinschmidt JS, Grant MB, Mocco J, Raizada MK. Brain-mediated dysregulation of the bone marrow activity in angiotensin II-induced hypertension. Hypertension. 2012;60:1316–1323.
18. Geraldes V, Gonçalves-Rosa N, Liu B, Paton JF, Rocha I. Chronic depres-sion of hypothalamic paraventricular neuronal activity produces sustained hypotension in hypertensive rats. Exp Physiol. 2014;99:89–100.
19. Zoccal DB, Simms AE, Bonagamba LG, Braga VA, Pickering AE, Paton JF, Machado BH. Increased sympathetic outflow in juvenile rats submit-ted to chronic intermittent hypoxia correlates with enhanced expiratory activity. J Physiol. 2008;586:3253–3265.
20. Waki H, Kasparov S, Wong LF, Murphy D, Shimizu T, Paton JF. Chronic inhibition of endothelial nitric oxide synthase activity in nucleus trac-tus solitarii enhances baroreceptor reflex in conscious rats. J Physiol. 2003;546(Pt 1):233–242.
21. Marina N, Tang F, Figueiredo M, Mastitskaya S, Kasimov V, Mohamed-Ali V, Roloff E, Teschemacher AG, Gourine AV, Kasparov S. Purinergic sig-nalling in the rostral ventro-lateral medulla controls sympathetic drive and contributes to the progression of heart failure following myocardial infarction in rats. Basic Res Cardiol. 2013;108:317.
22. Llewellyn TL, Sharma NM, Zheng H, Patel KP. Effects of exercise train-ing on SFO-mediated sympathoexcitation during chronic heart failure. Am J Physiol Heart Circ Physiol. 2014;306:H121–H131.
23. Böhm M, Mahfoud F, Ukena C, et al. Rationale and design of a large registry on renal denervation: the Global SYMPLICITY registry. EuroIntervention. 2013;9:484–492.
24. Grassi G, Bertoli S, Seravalle G. Sympathetic nervous system: role in hypertension and in chronic kidney disease. Curr Opin Nephrol Hypertens. 2012;21:46–51.
25. Sobotka PA, Mahfoud F, Schlaich MP, Hoppe UC, Böhm M, Krum H. Sympatho-renal axis in chronic disease. Clin Res Cardiol. 2011;100:1049–1057.
26. Grassi G. Sympathetic overdrive and cardiovascular risk in the metabolic syndrome. Hypertens Res. 2006;29:839–847.
27. Lambert E, Straznicky N, Sari CI, Eikelis N, Hering D, Head G, Dixon J, Esler M, Schlaich M, Lambert G. Dyslipidemia is associated with sym-pathetic nervous activation and impaired endothelial function in young females. Am J Hypertens. 2013;26:250–256.
28. Littlejohn NK, Siel RB Jr, Ketsawatsomkron P, Pelham CJ, Pearson NA, Hilzendeger AM, Buehrer BA, Weidemann BJ, Li H, Davis DR, Thompson AP, Liu X, Cassell MD, Sigmund CD, Grobe JL. Hypertension in mice with transgenic activation of the brain renin-angiotensin sys-tem is vasopressin dependent. Am J Physiol Regul Integr Comp Physiol. 2013;304:R818–R828.
29. Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-alpha) and essential hypertension. J Hum Hypertens. 2005;19:149–154.
30. Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, Vinh A, Weyand CM. Inflammation, immunity, and hypertension. Hypertension. 2011;57:132–140.
31. Roifman I, Beck PL, Anderson TJ, Eisenberg MJ, Genest J. Chronic inflammatory diseases and cardiovascular risk: a systematic review. Can J Cardiol. 2011;27:174–182.
32. Santisteban MM, Zubcevic J, Baekey DM, Raizada MK. Dysfunctional brain-bone marrow communication: a paradigm shift in the pathophysiol-ogy of hypertension. Curr Hypertens Rep. 2013;15:377–389.
33. Zubcevic J, Waki H, Raizada MK, Paton JF. Autonomic-immune-vascular interaction: an emerging concept for neurogenic hypertension. Hypertension. 2011;57:1026–1033.
34. Abboud FM, Harwani SC, Chapleau MW. Autonomic neural regulation of the immune system: implications for hypertension and cardiovascular disease. Hypertension. 2012;59:755–762.
35. Harwani SC, Chapleau MW, Legge KL, Ballas ZK, Abboud FM. Neurohormonal modulation of the innate immune system is proinflam-matory in the prehypertensive spontaneously hypertensive rat, a genetic model of essential hypertension. Circ Res. 2012;111:1190–1197.
36. Zubcevic J, Jun JY, Kim S, Perez PD, Afzal A, Shan Z, Li W, Santisteban MM, Yuan W, Febo M, Mocco J, Feng Y, Scott E, Baekey DM, Raizada MK. Altered inflammatory response is associated with an impaired auto-nomic input to the bone marrow in the spontaneously hypertensive rat. Hypertension. 2014;63:542–550.
37. Dutta P, Courties G, Wei Y, et al. Myocardial infarction accelerates athero-sclerosis. Nature. 2012;487:325–329.
38. Matteoli G, Gomez-Pinilla PJ, Nemethova A, Di Giovangiulio M, Cailotto C, van Bree SH, Michel K, Tracey KJ, Schemann M, Boesmans W, Vanden Berghe P, Boeckxstaens GE. A distinct vagal anti-inflammatory pathway modulates intestinal muscularis resident macrophages independent of the spleen. Gut. 2013; doi: 10.1136/gutjnl-2013–304676.
39. Ji H, Rabbi MF, Labis B, Pavlov VA, Tracey KJ, Ghia JE. Central cholin-ergic activation of a vagus nerve-to-spleen circuit alleviates experimental colitis. Mucosal Immunol. 2014;7:335–347.
40. Ganta CK, Lu N, Helwig BG, Blecha F, Ganta RR, Zheng L, Ross CR, Musch TI, Fels RJ, Kenney MJ. Central angiotensin II-enhanced splenic cytokine gene expression is mediated by the sympathetic nervous system. Am J Physiol Heart Circ Physiol. 2005;289:H1683–H1691.
41. Rodgers KE, Dizerega GS. Contribution of the Local RAS to Hematopoietic Function: A Novel Therapeutic Target. Front Endocrinol (Lausanne). 2013;4:157.
42. Haznedaroglu IC, Beyazit Y. Local bone marrow renin-angiotensin system in primitive, definitive and neoplastic haematopoiesis. Clin Sci (Lond). 2013;124:307–323.
by guest on June 30, 2018http://hyper.ahajournals.org/
Dow
nloaded from

Zubcevic et al Brain–Bone Marrow Communication in Hypertension e137
43. Tan JK, O’Neill HC. Concise review: Dendritic cell development in the context of the spleen microenvironment. Stem Cells. 2007;25:2139–2145.
44. Malhotra D, Fletcher AL, Turley SJ. Stromal and hematopoietic cells in secondary lymphoid organs: partners in immunity. Immunol Rev. 2013;251:160–176.
45. van der Laan AM, Ter Horst EN, Delewi R, Begieneman MP, Krijnen PA, Hirsch A, Lavaei M, Nahrendorf M, Horrevoets AJ, Niessen HW, Piek JJ. Monocyte subset accumulation in the human heart following acute myocardial infarction and the role of the spleen as monocyte reservoir. Eur Heart J. 2014;35:376–385.
46. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616.
47. Casanova-Acebes M, Pitaval C, Weiss LA, Nombela-Arrieta C, Chèvre R, A-González N, Kunisaki Y, Zhang D, van Rooijen N, Silberstein LE, Weber C, Nagasawa T, Frenette PS, Castrillo A, Hidalgo A. Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell. 2013;153:1025–1035.
48. Scheiermann C, Kunisaki Y, Lucas D, Chow A, Jang JE, Zhang D, Hashimoto D, Merad M, Frenette PS. Adrenergic nerves govern circadian leukocyte recruitment to tissues. Immunity. 2012;37:290–301.
49. Méndez-Ferrer S, Battista M, Frenette PS. Cooperation of beta(2)- and beta(3)-adrenergic receptors in hematopoietic progenitor cell mobiliza-tion. Ann N Y Acad Sci. 2010;1192:139–144.
50. Scheiermann C, Kunisaki Y, Frenette PS. Circadian control of the immune system. Nat Rev Immunol. 2013;13:190–198.
51. Olofsson PS, Katz DA, Rosas-Ballina M, Levine YA, Ochani M, Valdes-Ferrer SI, Pavlov VA, Tracey KJ, Chavan SS. Alpha7 nico-tinic acetylcholine receptor (alpha7nAChR) expression in bone marrow-derived non-t cells is required for the inflammatory reflex. Mol Med. 2012;18:539–543.
52. Nagareddy P, Smyth SS. Inflammation and thrombosis in cardiovascular disease. Curr Opin Hematol. 2013;20:457–463.
53. Crowson CS, Liao KP, Davis JM 3rd, Solomon DH, Matteson EL, Knutson KL, Hlatky MA, Gabriel SE. Rheumatoid arthritis and cardio-vascular disease. Am Heart J. 2013;166:622–628.
54. Ghattas A, Griffiths HR, Devitt A, Lip GY, Shantsila E. Monocytes in coronary artery disease and atherosclerosis: where are we now? J Am Coll Cardiol. 2013;62:1541–1551.
55. Courties G, Moskowitz MA, Nahrendorf M. The innate immune system after ischemic injury: lessons to be learned from the heart and brain. JAMA Neurol. 2014;71:233–236.
56. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808.
57. Sesso HD, Buring JE, Rifai N, Blake GJ, Gaziano JM, Ridker PM. C-reactive protein and the risk of developing hypertension. JAMA. 2003;290:2945–2951.
58. Harrison DG, Marvar PJ, Titze JM. Vascular inflammatory cells in hyper-tension. Front Physiol. 2012;3:128.
59. Zubcevic J, Jun JY, Lamont G, Murça TM, Shi P, Yuan W, Lin F, Carvajal JM, Li Q, Sumners C, Raizada MK, Shan Z. Nucleus of the solitary tract (pro)renin receptor-mediated antihypertensive effect involves nuclear factor-κB-cytokine signaling in the spontaneously hypertensive rat. Hypertension. 2013;61:622–627.
60. Koenig W. High-sensitivity C-reactive protein and atherosclerotic dis-ease: from improved risk prediction to risk-guided therapy. Int J Cardiol. 2013;168:5126–5134.
61. Murea M, Register TC, Divers J, Bowden DW, Carr JJ, Hightower CR, Xu J, Smith SC, Hruska KA, Langefeld CD, Freedman BI. Relationships between serum MCP-1 and subclinical kidney disease: African American-Diabetes Heart Study. BMC Nephrol. 2012;13:148.
62. van Diepen JA, Berbée JF, Havekes LM, Rensen PC. Interactions between inflammation and lipid metabolism: relevance for efficacy of anti-inflammatory drugs in the treatment of atherosclerosis. Atherosclerosis. 2013;228:306–315.
63. Ba D, Takeichi N, Kodama T, Kobayashi H. Restoration of T cell depression and suppression of blood pressure in spontaneously hypertensive rats (SHR) by thymus grafts or thymus extracts. J Immunol. 1982;128:1211–1216.
64. Khraibi AA. Association between disturbances in the immune system and hypertension. Am J Hypertens. 1991;4(7 Pt 1):635–641.
65. Baigent C, Blackwell L, Collins R, Emberson J, Godwin J, Peto R, Buring J, Hennekens C, Kearney P, Meade T, Patrono C, Roncaglioni MC, Zanchetti A. Aspirin in the primary and secondary prevention of vascular
disease: Collaborative meta-analysis of individual participant data from randomised trials. Lancet. 2009;373:1849–1860.
66. Rodríguez LA, Cea-Soriano L, Martín-Merino E, Johansson S. Discontinuation of low dose aspirin and risk of myocardial infarction: case-control study in UK primary care. BMJ. 2011;343:d4094.
67. Soubrier M, Rosenbaum D, Tatar Z, Lahaye C, Dubost JJ, Mathieu S. Vascular effects of nonsteroidal antiinflammatory drugs. Joint Bone Spine. 2013;80:358–362.
68. Tsai TH, Chai HT, Sun CK, Yen CH, Leu S, Chen YL, Chung SY, Ko SF, Chang HW, Wu CJ, Yip HK. Obesity suppresses circulating level and function of endothelial progenitor cells and heart function. J Transl Med. 2012;10:137.
69. Calò LA, Dal Maso L, Pagnin E, Ravarotto V, Facco M, Boscaro E, Maiolino G, Pessina AC, Rossi GP. Effect of olmesartan medoxomil on number and survival of circulating endothelial progenitor cells and calcitonin gene related peptide in hypertensive patients. J Hypertens. 2014;32:193–199.
70. Liu HF, Qi XW, Ma LL, Yao DK, Wang L. Atorvastatin improves endothe-lial progenitor cell function and reduces pulmonary hypertension in patients with chronic pulmonary heart disease. Exp Clin Cardiol. 2013;18:e40–e43.
71. Suzuki R, Fukuda N, Katakawa M, Tsunemi A, Tahira Y, Matsumoto T, Ueno T, Soma M. Effects of an angiotensin ii receptor blocker on the impaired function of endothelial progenitor cells in patients with essential hypertension. Am J Hypertens. 2013; doi: 10.1093/ajh/hpt208.
72. Imanishi T, Moriwaki C, Hano T, Nishio I. Endothelial progenitor cell senescence is accelerated in both experimental hypertensive rats and patients with essential hypertension. J Hypertens. 2005;23:1831–1837.
73. Giannotti G, Doerries C, Mocharla PS, Mueller MF, Bahlmann FH, Horvàth T, Jiang H, Sorrentino SA, Steenken N, Manes C, Marzilli M, Rudolph KL, Lüscher TF, Drexler H, Landmesser U. Impaired endothelial repair capacity of early endothelial progenitor cells in prehypertension: relation to endothelial dysfunction. Hypertension. 2010;55:1389–1397.
74. Liu J, Wang Y, Akamatsu Y, Lee CC, Stetler RA, Lawton MT, Yang GY. Vascular remodeling after ischemic stroke: Mechanisms and therapeutic potentials. Prog Neurobiol. 2013; doi: 10.1016/j.pneurobio.2013.11.004.
75. Shinozuka K, Dailey T, Tajiri N, Ishikawa H, Kim DW, Pabon M, Acosta S, Kaneko Y, Borlongan CV. Stem Cells for Neurovascular Repair in Stroke. J Stem Cell Res Ther. 2013;4:12912.
76. Mackie AR, Losordo DW. CD34-positive stem cells: in the treat-ment of heart and vascular disease in human beings. Tex Heart Inst J. 2011;38:474–485.
77. Krishnamurthy P, Thal M, Verma S, Hoxha E, Lambers E, Ramirez V, Qin G, Losordo D, Kishore R. Interleukin-10 deficiency impairs bone marrow-derived endothelial progenitor cell survival and function in isch-emic myocardium. Circ Res. 2011;109:1280–1289.
78. Tongers J, Losordo DW, Landmesser U. Stem and progenitor cell-based therapy in ischaemic heart disease: promise, uncertainties, and challenges. Eur Heart J. 2011;32:1197–1206.
79. Valgimigli M, Rigolin GM, Fucili A, Porta MD, Soukhomovskaia O, Malagutti P, Bugli AM, Bragotti LZ, Francolini G, Mauro E, Castoldi G, Ferrari R. CD34+ and endothelial progenitor cells in patients with various degrees of congestive heart failure. Circulation. 2004;110:1209–1212.
80. Fadini GP, de Kreutzenberg SV, Coracina A, Baesso I, Agostini C, Tiengo A, Avogaro A. Circulating CD34+ cells, metabolic syndrome, and cardio-vascular risk. Eur Heart J. 2006;27:2247–2255.
81. Schiffrin EL. Immune mechanisms in hypertension and vascular injury. Clin Sci (Lond). 2014;126:267–274.
82. Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C, Gordon FJ, Harrison DG. Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angio-tensin II-induced hypertension. Circ Res. 2010;107:263–270.
83. Aroor AR, Demarco VG, Jia G, Sun Z, Nistala R, Meininger GA, Sowers JR. The Role of Tissue Renin-Angiotensin-Aldosterone System in the Development of Endothelial Dysfunction and Arterial Stiffness. Front Endocrinol (Lausanne). 2013;4:161.
84. Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460.
85. Yao EH, Fukuda N, Matsumoto T, Kobayashi N, Katakawa M, Yamamoto C, Tsunemi A, Suzuki R, Ueno T, Matsumoto K. Losartan improves the impaired function of endothelial progenitor cells in hypertension via an antioxidant effect. Hypertens Res. 2007;30:1119–1128.
86. Cacciatore F, Bruzzese G, Vitale DF, Liguori A, de Nigris F, Fiorito C, Infante T, Donatelli F, Minucci PB, Ignarro LJ, Napoli C. Effects of ACE inhibition on circulating endothelial progenitor cells, vascular damage,
by guest on June 30, 2018http://hyper.ahajournals.org/
Dow
nloaded from

e138 Hypertension June 2014
and oxidative stress in hypertensive patients. Eur J Clin Pharmacol. 2011;67:877–883.
87. Kim S, Scott EW, Raizada, Mohan K. Angiotensin II regulates hemato-poietic stem cell proliferation, differentiation, and engraftment efficacy. Hypertension. 2013;62:A156.
88. Stenvinkel P, Pecoits-Filho R, Lindholm B. Coronary artery disease in end-stage renal disease: no longer a simple plumbing problem. J Am Soc Nephrol. 2003;14:1927–1939.
89. Tousoulis D, Andreou I, Antoniades C, Tentolouris C, Stefanadis C. Role of inflammation and oxidative stress in endothelial progenitor cell func-tion and mobilization: therapeutic implications for cardiovascular dis-eases. Atherosclerosis. 2008;201:236–247.
90. Rabelink TJ, de Boer HC, de Koning EJ, van Zonneveld AJ. Endothelial progenitor cells: more than an inflammatory response? Arterioscler Thromb Vasc Biol. 2004;24:834–838.
91. Heeschen C, Lehmann R, Honold J, Assmus B, Aicher A, Walter DH, Martin H, Zeiher AM, Dimmeler S. Profoundly reduced neovasculariza-tion capacity of bone marrow mononuclear cells derived from patients with chronic ischemic heart disease. Circulation. 2004;109:1615–1622.
92. Reynolds JA, Robertson AC, Bruce IN, Alexander MY. Improving car-diovascular outcomes in rheumatic diseases: Therapeutic potential of circulating endothelial progenitor cells. Pharmacol Ther. 2013; doi: 10.1016/j.pharmthera.2013.12.008.
93. Ablin JN, Boguslavski V, Aloush V, Elkayam O, Paran D, Caspi D, George J. Effect of anti-TNFalpha treatment on circulating endothelial progeni-tor cells (EPCs) in rheumatoid arthritis. Life Sci. 2006;79:2364–2369.
94. Minnerup J, Wagner DC, Strecker JK, Pösel C, Sevimli-Abdis S, Schmidt A, Schilling M, Boltze J, Diederich K, Schäbitz WR. Bone marrow-derived mononuclear cells do not exert acute neuroprotection after stroke in spon-taneously hypertensive rats. Front Cell Neurosci. 2014;7:288.
95. Delva P, Degan M, Vallerio P, Arosio E, Minuz P, Amen G, Di Chio M, Lechi A. Endothelial progenitor cells in patients with essential hyperten-sion. J Hypertens. 2007;25:127–132.
96. Rossi F, Bertone C, Michelon E, Bianco MJ, Santiemma V. High-density lipoprotein cholesterol affects early endothelial progenitor cell num-ber and endothelial function in obese women. Obesity (Silver Spring). 2013;21:2356–2361.
97. De Ciuceis C, Pilu A, Cappelli C, Porteri E, Zani F, Santoro A, Gandossi E, Boari GE, Rizzardi N, Castellano M, Rizzoni D, Agabiti Rosei E. Decreased number of circulating endothelial progenitor cells in patients with Graves’ hyperthyroidism. J Endocrinol Invest. 2011;34:335–339.
98. Besler C, Doerries C, Giannotti G, Lüscher TF, Landmesser U. Pharmacological approaches to improve endothelial repair mechanisms. Expert Rev Cardiovasc Ther. 2008;6:1071–1082.
99. Yao EH, Fukuda N, Matsumoto T, Katakawa M, Yamamoto C, Han Y, Ueno T, Kobayashi N, Matsumoto K. Effects of the antioxidative beta-blocker celiprolol on endothelial progenitor cells in hypertensive rats. Am J Hypertens. 2008;21:1062–1068.
100. Hu Z, Zhang F, Yang Z, Zhang J, Zhang D, Yang N, Zhang Y, Cao K. Low-dose aspirin promotes endothelial progenitor cell migration and adhesion and prevents senescence. Cell Biol Int. 2008;32:761–768.
101. Méndez-Ferrer S, Lucas D, Battista M, Frenette PS. Haematopoietic stem cell release is regulated by circadian oscillations. Nature. 2008;452:442–447.
102. Busik JV, Tikhonenko M, Bhatwadekar A, et al. Diabetic retinopathy is associated with bone marrow neuropathy and a depressed peripheral clock. J Exp Med. 2009;206:2897–2906.
103. Vacas S, Degos V, Tracey KJ, Maze M. High-mobility group box 1 protein initiates postoperative cognitive decline by engaging bone marrow-derived macrophages. Anesthesiology. 2013; doi: 10.1097/ALN.0000000000000045.
104. Hu P, Thinschmidt JS, Yan Y, et al. CNS inflammation and bone marrow neuropathy in type 1 diabetes. Am J Pathol. 2013;183:1608–1620.
105. Chen SK, Tvrdik P, Peden E, Cho S, Wu S, Spangrude G, Capecchi MR. Hematopoietic origin of pathological grooming in Hoxb8 mutant mice. Cell. 2010;141:775–785.
106. Lampron A, Pimentel-Coelho PM, Rivest S. Migration of bone marrow-derived cells into the central nervous system in models of neu-rodegeneration. J Comp Neurol. 2013;521:3863–3876.
107. Rodriguez M, Alvarez-Erviti L, Blesa FJ, Rodríguez-Oroz MC, Arina A, Melero I, Ramos LI, Obeso JA. Bone-marrow-derived cell differentia-tion into microglia: a study in a progressive mouse model of Parkinson’s disease. Neurobiol Dis. 2007;28:316–325.
108. Shaftel SS, Carlson TJ, Olschowka JA, Kyrkanides S, Matousek SB, O’Banion MK. Chronic interleukin-1beta expression in mouse brain
leads to leukocyte infiltration and neutrophil-independent blood brain barrier permeability without overt neurodegeneration. J Neurosci. 2007;27:9301–9309.
109. Santisteban MM, Zubcevic J, Kim S, Marulanda Carvajal J, Zingler MB, Joseph J, McCarter MA, Raizada MK. Reconstitution of bone marrow with wky cells lowers central/peripheral inflammation and blood pres-sure in the SHR. Hypertension. 2013;62:A606.
110. Biancardi VC, Son SJ, Ahmadi S, Filosa JA, Stern JE. Circulating Angiotensin II Gains Access to the Hypothalamus and Brain Stem During Hypertension via Breakdown of the Blood-Brain Barrier. Hypertension. 2014;63:572–579.
111. Shi P, Raizada MK, Sumners C. Brain cytokines as neuromodulators in cardiovascular control. Clin Exp Pharmacol Physiol. 2010;37:e52–e57.
112. Shi P, Diez-Freire C, Jun JY, Qi Y, Katovich MJ, Li Q, Sriramula S, Francis J, Sumners C, Raizada MK. Brain microglial cytokines in neuro-genic hypertension. Hypertension. 2010;56:297–303.
113. Cunningham C. Microglia and neurodegeneration: the role of systemic inflammation. Glia. 2013;61:71–90.
114. Benarroch EE. Microglia: Multiple roles in surveillance, circuit shaping, and response to injury. Neurology. 2013;81:1079–1088.
115. Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science. 2013;339:156–161.
116. Soulet D, Rivest S. Bone-marrow-derived microglia: myth or reality? Curr Opin Pharmacol. 2008;8:508–518.
117. Yenari MA, Xu L, Tang XN, Qiao Y, Giffard RG. Microglia potentiate damage to blood-brain barrier constituents: improvement by minocy-cline in vivo and in vitro. Stroke. 2006;37:1087–1093.
118. Liu B. Modulation of microglial pro-inflammatory and neurotoxic activ-ity for the treatment of Parkinson’s disease. AAPS J. 2006;8:E606–E621.
119. Wang G, Coleman CG, Chan J, Faraco G, Marques-Lopes J, Milner TA, Guruju MR, Anrather J, Davisson RL, Iadecola C, Pickel VM. Angiotensin II slow-pressor hypertension enhances NMDA currents and NOX2-dependent superoxide production in hypothalamic paraventricular neurons. Am J Physiol Regul Integr Comp Physiol. 2013;304:R1096–R1106.
120. Perry VH, Teeling J. Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin Immunopathol. 2013;35:601–612.
121. Norden DM, Godbout JP. Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol Appl Neurobiol. 2013;39:19–34.
122. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflam-mation. Nature. 2003;421:384–388.
123. Czura CJ, Schultz A, Kaipel M, Khadem A, Huston JM, Pavlov VA, Redl H, Tracey KJ. Vagus nerve stimulation regulates hemostasis in swine. Shock. 2010;33:608–613.
124. Pavlov VA, Tracey KJ. The vagus nerve and the inflammatory reflex–linking immunity and metabolism. Nat Rev Endocrinol. 2012;8:743–754.
125. Rosas-Ballina M, Olofsson PS, Ochani M, Valdés-Ferrer SI, Levine YA, Reardon C, Tusche MW, Pavlov VA, Andersson U, Chavan S, Mak TW, Tracey KJ. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. 2011;334:98–101.
126. Rosas-Ballina M, Ochani M, Parrish WR, Ochani K, Harris YT, Huston JM, Chavan S, Tracey KJ. Splenic nerve is required for cholinergic anti-inflammatory pathway control of TNF in endotoxemia. Proc Natl Acad Sci U S A. 2008;105:11008–11013.
127. Grassi G, Cattaneo BM, Seravalle G, Lanfranchi A, Mancia G. Baroreflex control of sympathetic nerve activity in essential and secondary hyper-tension. Hypertension. 1998;31:68–72.
128. Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313.
129. Lozano-Ondoua AN, Symons-Liguori AM, Vanderah TW. Cancer-induced bone pain: Mechanisms and models. Neurosci Lett. 2013;557 Pt A:52–59.
130. Mach DB, Rogers SD, Sabino MC, Luger NM, Schwei MJ, Pomonis JD, Keyser CP, Clohisy DR, Adams DJ, O’Leary P, Mantyh PW. Origins of skeletal pain: sensory and sympathetic innervation of the mouse femur. Neuroscience. 2002;113:155–166.
131. Mundy GR. Pathophysiology of cancer-associated hypercalcemia. Semin Oncol. 1990;17(2 Suppl 5):10–15.
132. Schwei MJ, Honore P, Rogers SD, Salak-Johnson JL, Finke MP, Ramnaraine ML, Clohisy DR, Mantyh PW. Neurochemical and cellular reorganization of the spinal cord in a murine model of bone cancer pain. J Neurosci. 1999;19:10886–10897.
by guest on June 30, 2018http://hyper.ahajournals.org/
Dow
nloaded from

Zubcevic et al Brain–Bone Marrow Communication in Hypertension e139
133. Watkins LR, Wiertelak EP, Goehler LE, Smith KP, Martin D, Maier SF. Characterization of cytokine-induced hyperalgesia. Brain Res. 1994;654:15–26.
134. Baamonde A, Curto-Reyes V, Juárez L, Meana A, Hidalgo A, Menéndez L. Antihyperalgesic effects induced by the IL-1 recep-tor antagonist anakinra and increased IL-1beta levels in inflamed and osteosarcoma-bearing mice. Life Sci. 2007;81:673–682.
135. Geis C, Graulich M, Wissmann A, Hagenacker T, Thomale J, Sommer C, Schäfers M. Evoked pain behavior and spinal glia activation is depen-dent on tumor necrosis factor receptor 1 and 2 in a mouse model of bone cancer pain. Neuroscience. 2010;169:463–474.
136. Constantin CE, Mair N, Sailer CA, Andratsch M, Xu ZZ, Blumer MJ, Scherbakov N, Davis JB, Bluethmann H, Ji RR, Kress M. Endogenous tumor necrosis factor alpha (TNFalpha) requires TNF receptor type 2 to generate heat hyperalgesia in a mouse cancer model. J Neurosci. 2008;28:5072–5081.
137. Lozano-Ondoua AN, Hanlon KE, Symons-Liguori AM, et al. Disease modification of breast cancer-induced bone remodeling by cannabinoid 2 receptor agonists. J Bone Miner Res. 2013;28:92–107.
138. Hu JH, Yang JP, Liu L, Li CF, Wang LN, Ji FH, Cheng H. Involvement of CX3CR1 in bone cancer pain through the activation of microglia p38 MAPK pathway in the spinal cord. Brain Res. 2012;1465:1–9.
139. Yu SQ, Wang DH. Enhanced salt sensitivity following shRNA silenc-ing of neuronal TRPV1 in rat spinal cord. Acta Pharmacol Sin. 2011;32:845–852.
140. Hollis M, Wang DH. Transient receptor potential vanilloid in blood pres-sure regulation. Curr Opin Nephrol Hypertens. 2013;22:170–176.
141. Wang Y, Wang DH. Neural control of blood pressure: focusing on capsaicin-sensitive sensory nerves. Cardiovasc Hematol Disord Drug Targets. 2007;7:37–46.
142. Cortelli P, Pierangeli G. Chronic pain-autonomic interactions. Neurol Sci. 2003;24 Suppl 2:S68–S70.
143. Jaggi AS, Singh N. Role of different brain areas in peripheral nerve injury-induced neuropathic pain. Brain Res. 2011;1381:187–201.
144. Boscan P, Pickering AE, Paton JF. The nucleus of the solitary tract: an integrating station for nociceptive and cardiorespiratory afferents. Exp Physiol. 2002;87:259–266.
145. Potts JT, Paton JF, Mitchell JH, Garry MG, Kline G, Anguelov PT, Lee SM. Contraction-sensitive skeletal muscle afferents inhibit arterial baro-receptor signalling in the nucleus of the solitary tract: role of intrinsic GABA interneurons. Neuroscience. 2003;119:201–214.
146. Wong LF, Polson JW, Murphy D, Paton JF, Kasparov S. Genetic and phar-macological dissection of pathways involved in the angiotensin II-mediated depression of baroreflex function. FASEB J. 2002;16:1595–1601.
by guest on June 30, 2018http://hyper.ahajournals.org/
Dow
nloaded from

Donald C. Bolser, Marcelo Febo and Mohan K. RaizadaJasenka Zubcevic, Monica M. Santisteban, Teresa Pitts, David M. Baekey, Pablo D. Perez,
Cardiovascular DiseaseBone Marrow Pathways: Implications in Hypertension and−Functional Neural
Print ISSN: 0194-911X. Online ISSN: 1524-4563 Copyright © 2014 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Hypertension doi: 10.1161/HYPERTENSIONAHA.114.02440
2014;63:e129-e139; originally published online March 31, 2014;Hypertension.
http://hyper.ahajournals.org/content/63/6/e129World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://hyper.ahajournals.org//subscriptions/
is online at: Hypertension Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information aboutOffice. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialHypertensionin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on June 30, 2018http://hyper.ahajournals.org/
Dow
nloaded from