original article - homepage |...
TRANSCRIPT

901
The renin-angiotensin system is widely accepted as a central mediator of chronic kidney disease (CKD) and is the main
target of therapies aiming to slow down or ameliorate the pro-gression of the disease.1 Aliskiren is a novel, direct inhibitor of renin activation that has been added to angiotensin-converting enzyme blockers and angiotensin II receptor 1 inhibitors and, at least theoretically, may avoid angiotensin II escape that is observed after treatment with drugs from the other 2 classes.2,3
Although the beneficial effect of aliskiren in normalizing blood pressure and proteinuria has been shown in both humans4,5 and animal models,6,7 data regarding its anti-inflammatory and, most importantly, its antifibrotic capacity in models of CKD are still missing. To evaluate the effect of aliskiren on regres-sion of renal fibrosis, we used a heterozygous transgenic mouse model that expresses an artificial mouse renin gene (RenTg) in the liver at constantly high levels8 against which aliskiren has been proven to be effective.9,10 These transgenic mice are characterized by elevated blood pressure and a progressive increase in proteinuria, which, at later stages, is accompanied by established lesions typical of CKD, such as perivascular and
periglomerular inflammation, glomerulosclerosis, mesangial expansion, tubular dilation, and cardiac fibrosis.11 The main advantage of this model of hypertension-induced renal disease is that, because of its slow progression, it better resembles the kinetics and characteristics of human pathology.
We administered aliskiren in 10-month-old RenTg mice (RenTg ASK), an age that lesions in the kidneys are well established. Our results indicate that renin inhibition induced a shift in the balance between profibrotic and antifibrotic mediators and pathways leading to the regression of renal fibrosis and restoration of normal tissue structure and function independent of blood pressure normalization.
Materials and MethodsAnimals and Experimental DesignBriefly, experiments were performed in RenTg mice on the 129SV background.8 Aliskiren (25 mg/kg per day) was administered through osmotic minipumps and hydralazine (40 mg/kg per day) orally, both for 28 days. Please see the online-only Data Supplement for detailed methods.
Abstract—Aliskiren, a direct renin inhibitor, is a novel antihypertensive drug. To study whether aliskiren can reverse chronic kidney disease, we administered it to renin transgenic mice, a strain characterized by elevated blood pressure and a slow decline of renal function, mimicking well the progression of hypertensive chronic kidney disease. Ten-month-old transgenic mice were treated either with aliskiren or placebo for 28 days. Age-matched wild-type mice treated or not with aliskiren were considered as normotensive controls. Aliskiren reduced blood pressure to wild-type levels from as early as day 14. Proteinuria and cardiac hypertrophy and fibrosis were also normalized. Renal interstitial fibrosis and inflammation were significantly ameliorated in aliskiren-treated mice (shown by the decrease of proinflammatory and profibrotic markers), and the phenotypes of tubular epithelial cells and podocytes were restored as evidenced by the reappearance of cellular proteins characteristic of normal phenotype of these cells. Profibrotic p38 and Erk mitogen-activated protein kinases were highly activated in placebo-treated transgenic animals. Aliskiren treatment cancelled this activation. This nephroprotection was not attributed to the antihypertensive activity of aliskiren, because blood pressure normalization after treatment with hydralazine failed to induce the regression of renal fibrosis. Direct inhibition of renin can restore renal function and structure in aged hypertensive animals with existing proteinuria. This finding suggests that, in addition to antihypertensive action, aliskiren can be also used to treat chronic kidney disease. (Hypertension. 2013;61:901-907.) • Online Data Supplement
Key Words: aliskiren ■ fibrosis ■ renal disease regression ■ renin
Received November 15, 2012; first decision January 24, 2013; revision accepted January 25, 2013.From the Institut National de la Santé et de la Recherche Médicale Unité Mixte de Recherche S702, Hôpital Tenon, Paris, France (P.K., L.W., A.B.A.,
J.-C.D., C.C.); and Novartis Pharmaceuticals Corp, East Hanover, NJ (D.L.F.).The online-only Data Supplement is available with this article at http://hyper.ahajournals.org/lookup/suppl/doi:10.1161/HYPERTENSIONAHA.
111.00639/-/DC1.Correspondence to Christos Chatziantoniou, Institut National de la Santé et de la Recherche Médicale U702, Tenon Hospital, 4 Rue de la Chine, 75020
Paris, France. E-mail [email protected]
Renin Inhibition Reverses Renal Disease in Transgenic Mice by Shifting the Balance Between
Profibrotic and Antifibrotic AgentsPanagiotis Kavvadas, Lise Weis, Ahmed B. Abed, David L. Feldman, Jean-Claude Dussaule,
Christos Chatziantoniou
© 2013 American Heart Association, Inc.
Hypertension is available at http://hyper.ahajournals.org DOI: 10.1161/HYPERTENSIONAHA.111.00639
Original Article
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from

902 Hypertension April 2013
ResultsAliskiren Normalizes Systolic Blood Pressure, Albuminuria, Glomerular Filtration Rate, and Cardiac Hypertrophy in RenTg MiceTen-month-old RenTg mice displayed elevated systolic blood pressure (141±6 vs 104±6 mm Hg for age-matched wild-type mice; P<0.05) and albuminuria (83.4±6.0 vs 2.8±1.0 mg/mmol of creatinine; P<0.05). After 14 days of treatment with aliskiren, blood pressure of the RenTg ASK animals was already normalized (100±7 mm Hg, P<0.05 versus RenTg placebo-treated [PBS], RenTg PBS; Figure S1A, available in the online-only Data Supplement). Albuminuria followed a similar pattern, being reduced at day 14 to 13.8±5.0 mg/mmol of creatinine and reaching control levels (4.8±3 mg/mmol of creatinine, P<0.05 versus RenTg PBS) at day 28. To further evaluate the effect of aliskiren on renal function, glomerular filtration rate was measured in a second group of age-matched RenTg mice (n=5) before and after aliskiren treatment. Our results showed a significant improvement in response to aliskiren, because RenTg mice had significantly decreased glomerular filtration rate (132±8 vs 220±12 μL/min for the wild-type mice; P<0.05) before aliskiren treat-ment that was practically normalized with aliskiren (189±4 vs 205±7 for the wild-type at day 28; P value not significant). In addition, RenTg PBS mice showed a statistically significant increase in cardiac mass (7.53±0.40 vs 5.60±0.36 mg/g for wild-type PBS; P<0.05), which was fully abolished by aliski-ren. Aliskiren administration in wild-type mice did not affect the above parameters.
To examine whether regression of renal fibrosis by aliskiren is simply the outcome of blood pressure normalization, we treated RenTg animals with hydralazine. Hydralazine normal-ized blood pressure from as early as day 14 (98±6 at day 14 vs 146±5 mm Hg at day 0; P<0.05), showing the same pattern with aliskiren (Figure S1B). However, blood pressure normal-ization was not accompanied by improvement in albuminuria (61.20±12.30 mg/mmol of creatinine at day 14 with a further minimal reduction to 50.10±5.24 mg/mmol of creatinine by day 28) and cardiac hypertrophy (7.07±0.16 for hydralazine-treated RenTg vs 5.09±0.30 mg/kg for wild-type hydralazine; P<0.05). Taken together, these results indicate that the thera-peutic effect of aliskiren is not just related to its antihyper-tensive activity. The effects of aliskiren and hydralazine on systolic blood pressure were further verified using radiotelem-etry measurements in separate experiments (Figure S1C and S1D).
Aliskiren Ameliorates Renal InflammationRenin-overexpressing mice are characterized by severe peri-vascular inflammatory lesions.9 To evaluate the effect of aliskiren on renal inflammation, we performed immunohis-tochemistry for the macrophage marker F4/80 (Figure 1A). The excessive F4/80 staining observed in RenTg PBS ani-mals was absent in the RenTg ASK animals, and the tissue resembled that of the wild-type animals. To extend this result we evaluated the expression of inflammatory markers vascular cell adhesion molecule, tumor necrosis factor-α, and mono-cyte chemoattractant protein 1 (Figure 1B through 1D). All of
these markers followed a similar pattern of expression; they were upregulated in RenTg PBS animals (300%) and returned to wild-type levels during aliskiren, indicating that renin inhi-bition ameliorates renal inflammation almost to a complete extent. Treatment with hydralazine had no beneficial effect on inflammation, as shown by real-time polymerase chain reac-tion for the same markers (Figure S2).
Figure 1. Effect of aliskiren on renal inflammation. Immunostaining for the macrophage marker F4/80 revealed a significant reduction in macrophage infiltration in aliskiren-treated animals (A). Real-time polymerase chain reaction (PCR) for the inflammatory markers vascular cell adhesion molecule (VCAM), tumor necrosis factor (TNF)-α, and monocyte chemoattractant protein (MCP) 1 showed that the upregulation of these markers in heterozygous transgenic mouse model that expresses an artificial mouse renin gene (RenTg) placebo-treated (PBS) animals was fully attenuated by aliskiren (B through D). *P<0.05 vs wild-type (WT) PBS, #P<0.05 vs RenTg PBS.
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from

Kavvadas et al Aliskiren Reverses Hypertensive Renal Disease 903
Aliskiren Reduces Renal FibrosisRenal interstitial fibrosis was evaluated with Masson trichrome (Figure S3A and S3B) and Sirius red stains (Figure 2A). Masson staining revealed massive perivascular cellular infil-trations, protein load inside tubular lumens, and interstitial fibrosis in RenTg animals. Quantification of total collagen with Sirius red showed that ≈20% of the interstitium was covered by collagen (versus ≈5% in wild-type). Treatment with aliskiren resulted in a statistically significant resolu-tion of cellular infiltrates and a decrease in collagen depos-its (Figures 2A and S4A). Reverse-transcription polymerase chain reaction for collagens I and III further confirmed Sirius red results (Figure 2C and 2D). Moreover, immunofluores-cence for the myofibroblast marker α-smooth muscle actin showed that treatment with aliskiren diminished interstitial staining for α-smooth muscle actin, which was restricted to vascular smooth muscle cells (Figure S4). mRNA levels of plasminogen activator inhibitor 1, a major mediator of fibrosis and inhibitor of fibrinolysis, were also normalized in response to aliskiren (data not shown). Hydralazine treatment did not improve renal damage (Figure S5) or decrease mRNA levels
of profibrotic mediators such as transforming growth factor (TGF)-β and connective tissue growth factor (Figure S3).
Aliskiren Restores the Podocyte Phenotype and Activates Tubular Cell ProliferationThe increase of proteinuria in RenTg mice is accompanied by decreased expression of nephrin (Figure 3D). Treatment with aliskiren resulted in a 20% increase in nephrin expression, indicating restoration of the slit diaphragm in the glomerular filtration barrier. Proximal tubular cells of RenTg PBS animals displayed significantly decreased proliferation versus wild-type mice, as shown by Ki67 staining (Figure 3A and 3B). Tubular cell cycle arrest, which is known to be induced by angiotensin II12 and is proposed to promote renal fibrosis,13 was reversed after treatment with aliskiren. Moreover, the high levels of mRNA for kidney injury molecule 1, a marker for tubular damage, observed in RenTg PBS mice were significantly reduced in RenTg ASK (Figure 3C). Taken together, these results indicate that aliskiren induced a beneficial effect on the integrity of tubular epithelium.
Aliskiren Shifts the Balance Between Profibrotic and Antifibrotic Mechanisms/PathwaysNext we investigated the effect of aliskiren on profibrotic mediators in the kidneys of RenTg mice. Reverse-transcription polymerase chain reaction for TGF-β and connective tissue growth factor, considered as main mediators of renal fibrosis, revealed a statistically significant increase in the expressions of both genes in RenTg PBS animals (Figure 4A and 4B). This upregulation was almost completely reversed in aliskiren-treated mice. Mitogen-activated protein kinases Erk and P38, which also mediate many profibrotic actions in response to angiotensin II, were highly activated in RenTg PBS mice, as shown by Western blot (Figure 4C and 4D). In contrast, RenTg ASK mice showed statistically significant reductions in the levels of phosphorylation compared with the RenTg mice. Bone morphogenetic proteins 4 and 7, which are considered to exert antifibrotic actions and to antagonize the effect of TGF-β, were highly upregulated after aliskiren treatment, indicating activation of antifibrotic mechanisms (Figure S6A and S6B). Because bone morphogenetic proteins exert their antifibrotic actions through phosphorylation of SMAD1/5/8, we evaluated SMAD phosphorylation levels with Western blotting. Our results showed that SMAD1/5/8 phosphorylation levels tended to decrease in RenTg PBS animals (borderline significance) but returned to wild-type levels in aliskiren-treated animals (Figure S6C).
Aliskiren Effect on Novel Profibrotic MechanismsRecently, hypoxia, endoplasmic reticulum stress, and DNA methylation have been proposed to participate in the progression of renal fibrosis. Therefore, we assessed these parameters in the kidneys of RenTg mice. Our results showed that endoplasmic reticulum stress, assessed by binding immunoglobulin protein, increased in RenTg animals, but aliskiren did not alter this increase (Figure S7A). In contrast, aliskiren reduced to normal levels the upregulation of hypoxia-inducible factor-1α (Figure S7B). Finally, levels of DNMT1, an enzyme that catalyzes DNA methylation and is proposed to be involved in the progression of fibrosis, showed a slight,
Figure 2. Effect of aliskiren on renal fibrosis. Sirius red staining (A) revealed that aliskiren significantly reduced collagen deposits. Staining was quantified by measuring the area of red staining and expressed as collagen area/total tissue area % (B). Real-time polymerase chain reaction (PCR) for collagen III further confirmed Sirius red staining results (C). *P<0.05 vs wild-type (WT) placebo-treated (PBS), #P<0.05 vs heterozygous transgenic mouse model that expresses an artificial mouse renin gene (RenTg) PBS.
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from

904 Hypertension April 2013
nonstatistically significant increase in RenTg mice, which was normalized by aliskiren (Figure S7C).
Aliskiren Reduces Cardiac FibrosisThe renin-angiotensin system plays a key role in the regu-lation of cardiovascular function, with angiotensin II being
involved in the pathophysiology of cardiac disease and fibro-sis. To evaluate collagen deposition and extracellular matrix accumulation in cardiac tissue, we performed Masson tri-chrome staining on hearts from RenTg mice (Figure 5A and 5B). Quantification of the extracellular matrix area in these hearts showed that the significant increase observed in RenTg
Figure 3. Effect of aliskiren on tubular epithelium and podocytes. Staining for the proliferation marker Ki67 showed that aliskiren reactivated cell cycle progression in proximal tubules (A and B). Real-time polymerase chain reaction (PCR) for the tubular injury marker kidney injury molecule (KIM) 1 further showed that aliskiren promotes restoration of tubular epithelium structure (C). Nephrin protein levels (D) were increased after treatment with aliskiren. *P<0.05 vs wild-type (WT) placebo-treated (PBS), #P<0.05 vs heterozygous transgenic mouse model that expresses an artificial mouse renin gene (RenTg) PBS.
Figure 4. Effect of aliskiren on profibrotic pathways/mediators. Real-time polymerase chain reaction (PCR) for transforming growth factor (TGF)-β (A) and connective tissue growth factor (CTGF; B) showed that treatment with aliskiren completely reversed the upregulation observed in the heterozygous transgenic mouse model that expresses an artificial mouse renin gene (RenTg) placebo-treated (PBS) animals. Profibrotic Erk (C) and P38 (D) phosphorylation was increased sharply in RenTg vs wild-type animals. Treatment with aliskiren significantly reduced phosphorylation levels. *P<0.05 vs wild-type (WT) PBS, #P<0.05 vs RenTg PBS.
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from

Kavvadas et al Aliskiren Reverses Hypertensive Renal Disease 905
PBS animals was fully reversed after treatment with aliski-ren. Moreover, atrial natriuretic peptide expression, a com-mon marker of cardiac dysfunction, was highly upregulated in RenTg PBS mice (Figure 5C), whereas treatment with aliskiren reduced atrial natriuretic peptide levels, indicating improvement of cardiac function. In sharp contrast, treatment with hydralazine did not significantly decrease extracellular matrix deposition (Figure S8).
DiscussionIn the present study we used a renin-overexpressing mouse model, characterized by hypertension-induced chronic renal disease, to study the renoprotective mechanisms of the direct renin inhibitor aliskiren. The advantage of this transgenic model is that renal disease progresses more slowly than in many other angiotensin II models and, thus, more closely mimics the kinetics of hypertension-induced CKD in humans. Our previous results11 have revealed that 10- to 11-month-old RenTg mice display albuminuria levels >80 mg/mmol of creatinine and established lesions of renal inflammation and fibrosis. Accordingly, the current work studied whether renin inhibition can produce a curative effect in the setting of existing, progressive renal damage and, if true, attempted to identify the mechanisms involved in the progression and regression of the disease.
RenTg animals were hypertensive, proteinuric, had compromised renal function, and showed established lesions typical of chronic renal disease, such as perivascular and periglomerular inflammation, glomerulosclerosis, mesangial expansion, and tubular dilation, as well as cardiac hypertrophy and fibrosis. In aliskiren-treated animals, systolic blood pressure and albuminuria were normalized from as early as day 14 of treatment, whereas heart:body weight ratio and glomerular filtration rate were also normalized to wild-type
levels. These results are in agreement with previous studies about the efficacy of aliskiren in lowering blood pressure and normalizing albuminuria.6,7 An additional important finding was that renin inhibition suppressed fibrillar collagen expression and reduced to normal the exaggerated accumulation of the extracellular matrix in kidneys of RenTg mice. Because 10- to 11-month-old RenTg mice have existing renal fibrosis and albuminuria, our findings strongly suggest that renin inhibition reversed well-established renal damage. In agreement with this antifibrotic action, aliskiren prevented the development of renal fibrosis in nonhypertensive models of renal disease, such as in unilateral ureteral obstruction,10,14 a model of rapid tubulointerstitial inflammation, and in Col4α3–/– mice,15 a model mimicking Alport syndrome. To our knowledge, our study is the first report in mice showing that aliskiren can reverse (and not just prevent) the progression of renal fibrosis associated with hypertension-induced CKD, at least in the renin-overexpressing model. Renin inhibition also exhibited a beneficial effect on cardiac fibrosis, as evidenced by an impressive reduction in extracellular matrix accumulation, in agreement with previous studies on the effect of aliskiren on cardiac injury and fibrosis.16–18
Diminished renal fibrosis was accompanied by almost complete absence of cellular infiltrates and protein in tubu-lar lumens. The marker of tubular epithelial injury, kidney injury molecule 1, was normalized after treatment with aliski-ren, whereas a reactivation of tubular cell proliferation was induced, thus reversing one potential mechanism of promot-ing renal disease, as described recently.13 The podocyte marker nephrin was also upregulated after treatment with aliskiren, suggesting that renin inhibition exerts a beneficial effect on both the tubular and glomerular compartments of the kidney.
A new and important finding of this work is that regression of renal inflammation and fibrosis was paralleled by a shift in
Figure 5. Effect of aliskiren on cardiac fibrosis. Masson trichrome staining showed that the heterozygous transgenic mouse model that expresses an artificial mouse renin gene (RenTg) placebo-treated (PBS) cardiac tissue is characterized by large extracellular matrix (ECM) deposits that were almost fully diminished after treatment with aliskiren (A). Staining was quantified by measuring extracellular matrix (ECM)-covered area/total tissue area (B). Real-time polymerase chain reaction (PCR) for atrial natriuretic peptide (ANP), a common marker of cardiac disease, showed that aliskiren induces a downregulation of this gene (C). *P<0.05 vs wild-type (WT) PBS.
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from

906 Hypertension April 2013
the balance between profibrotic and antifibrotic mediators and pathways. The profibrotic growth factors TGF-β and connec-tive tissue growth factor were upregulated in RenTg animals, whereas Erk and P38, which are believed to be important targets of angiotensin II, were highly phosphorylated and, therefore, activated.19 Treatment with aliskiren not only sig-nificantly reduced the gene expression or activation of these profibrotic mediators but also increased gene expression of bone morphogenetic proteins 4 and 7, which, in turn, resulted in enhanced phosphorylation and activation of the antifibrotic SMAD1/5/8 pathway. Moreover, hepatocyte growth factor, a growth factor believed to exert antifibrotic actions by counter-acting actions of angiotensin II and TGF-β,20 was also upregu-lated in aliskiren-treated mice (data not shown).
Endoplasmic reticulum stress,21 hypoxia,22 and methyla-tion23 are 3 new mechanisms proposed to be involved in the progression of renal fibrosis, at least in specific models of renal disease. Our results showed that treatment with aliski-ren did not induce normalization of endoplasmic reticulum stress in RenTg mice, indicating that this mechanism is not of great significance in regression of fibrosis, at least in this model. Levels of hypoxia-inducible factor-1α were normal-ized after treatment with aliskiren. This may reflect inhibition of angiotensin II–induced hypoxia (eg, via vasoconstriction) that results in tubular and capillary damage. Methylation of fibroblast genes has recently been proposed as a novel mecha-nism for maintaining fibroblasts in an active state. Levels of DNMT1, the encoding gene for the main methyltransferase involved in renal disease, showed the anticipated pattern; they were increased in RenTg PBS animals and normalized after aliskiren treatment. However, the observed differences were rather small (20%). The fact that we used the whole renal cortex for DNMT1 quantification and not isolated renal fibro-blasts could be an explanation for the small differences.
Studies on the regression of fibrosis in models of hyper-tension-induced disease must address whether the observed regression and tissue regeneration are exclusively attributed to blood pressure normalization. Hydralazine is a known antihypertensive drug with poor antifibrotic capacity.23 Our results showed that hydralazine normalized systolic blood pressure exhibiting similar kinetics to aliskiren. However, hydralazine treatment did not protect renal structure and func-tion, as evidenced by the persistence of albuminuria and his-tological lesions. Moreover, treatment with hydralazine had no beneficial effect on renal inflammation and fibrosis or in cardiac fibrosis. These results indicate that normalizing blood pressure alone is not sufficient to regress renal and cardiac fibrosis. These observations are in agreement with our pre-vious results in the l-NG-nitroarginine methyl ester model of renal disease.24,25 Moreover, our previous results with RenTg mice treated with an angiotensin II receptor blocker have shown that regression of renal fibrosis and restoration of renal structure can occur without normalization of systolic blood pressure.11 It is possible that the contribution of blood pressure in hypertensive models of renal injury is model and renal compartment dependent. Thus, the contribution of per-fusion pressure in the renal injury was minimal in the outer cortical region but important in the juxtamedullary glomeruli in angiotensin II–infused rats,26 whereas the elevations of
perfusion pressure contributed significantly to the renal fibro-sis in the Dahl salt-sensitive rats.27 In contrast, renal injury was independent of elevated blood pressure in the model of angiotensin II + l-NG-nitroarginine methyl ester–induced hypertension.28
PerspectivesOur study provides evidence that renin inhibition is an effi-cient curative treatment of CKD. Aliskiren normalized blood pressure, proteinuria, glomerular filtration rate, cardiac hyper-trophy, and fibrosis. Moreover, it produced regression of renal inflammation and fibrosis accompanied by restoration of nor-mal tissue architecture. This improvement was accompanied by a shift in the balance between profibrotic and antifibrotic mechanisms and pathways of the TGF-β superfamily. This shift of balance in favor of the antifibrotic mechanisms and parameters and the renoprotective and cardioprotective effects in general is not exclusively attributed to blood pressure nor-malization. These results indicate that blockade of renin pro-tects from all of the angiotensin II deleterious actions, both in plasma and locally in the kidneys, while avoiding angiotensin II escape that has been observed with other renin-angiotensin system blockers and inhibitors, further supporting the consid-eration of studies to reverse renal fibrosis in humans.
AcknowledgmentsRenin transgenic mice were kindly provided by Oliver Smithies. Telemetry measurements were performed in the department of bio-chemistry of Hôpital Europeen Georges Pompidou (Paris, France) by Veronique Baudrie.
Sources of FundingP.K. was a fellow of the European Renal Association-European Dialysis Transplantation Association and of the European Molecular Biology Organization. The project was financially supported by Institut National de la Santé et de la Recherche Médicale, University Pierre and Marie Curie, and Novartis Pharmaceuticals Corp.
DisclosuresDr Feldman is a full-time employee of Novartis Institutes for Biomedical Research, Novartis Pharmaceuticals Corp.
References 1. Remuzzi G, Ruggenenti P, Perico N. Chronic renal diseases: renopro-
tective benefits of renin-angiotensin system inhibition. Ann Intern Med. 2002;136:604–615.
2. Rahuel J, Rasetti V, Maibaum J, Rüeger H, Göschke R, Cohen NC, Stutz S, Cumin F, Fuhrer W, Wood JM, Grütter MG. Structure-based drug design: the discovery of novel nonpeptide orally active inhibitors of human renin. Chem Biol. 2000;7:493–504.
3. Wiggins KJ, Kelly DJ. Aliskiren: a novel renoprotective agent or simply an alternative to ACE inhibitors? Kidney Int. 2009;76:23–31.
4. Siddiqi L, Oey PL, Blankestijn PJ. Aliskiren reduces sympathetic nerve activity and blood pressure in chronic kidney disease patients. Nephrol Dial Transplant. 2011;26:2930–2934.
5. Moriyama T, Tsuruta Y, Kojima C, Itabashi M, Sugiura H, Takei T, Ogawa T, Uchida K, Tsuchiya K, Nitta K. Beneficial effect of aliskiren combined with olmesartan in reducing urinary protein excretion in patients with chronic kidney disease. Int Urol Nephrol. 2012;44:841–845.
6. Tain YL, Hsu CN, Lin CY, Huang LT, Lau YT. Aliskiren prevents hyper-tension and reduces asymmetric dimethylarginine in young spontaneously hypertensive rats. Eur J Pharmacol. 2011;670:561–565.
7. Feldman DL, Jin L, Xuan H, Contrepas A, Zhou Y, Webb RL, Mueller DN, Feldt S, Cumin F, Maniara W, Persohn E, Schuetz H, Jan Danser AH, Nguyen G. Effects of aliskiren on blood pressure, albuminuria, and (pro)
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from

Kavvadas et al Aliskiren Reverses Hypertensive Renal Disease 907
renin receptor expression in diabetic TG(mRen-2)27 rats. Hypertension. 2008;52:130–136.
8. Caron KM, James LR, Kim HS, Morham SG, Sequeira Lopez ML, Gomez RA, Reudelhuber TL, Smithies O. A genetically clamped renin transgene for the induction of hypertension. Proc Natl Acad Sci USA. 2002;99:8248–8252.
9. Lu H, Rateri DL, Feldman DL, Jr RJ, Fukamizu A, Ishida J, Oesterling EG, Cassis LA, Daugherty A. Renin inhibition reduces hypercholesterol-emia-induced atherosclerosis in mice. J Clin Invest. 2008;118:984–993.
10. Choi DE, Jeong JY, Lim BJ, Chang YK, Na KR, Shin YT, Lee KW. Aliskiren ameliorates renal inflammation and fibrosis induced by unilat-eral ureteral obstruction in mice. J Urol. 2011;186:694–701.
11. Huby AC, Rastaldi MP, Caron K, Smithies O, Dussaule JC, Chatziantoniou C. Restoration of podocyte structure and improvement of chronic renal dis-ease in transgenic mice overexpressing renin. PLoS ONE. 2009;4:e6721.
12. Hannken T, Schroeder R, Zahner G, Stahl RA, Wolf G. Reactive oxy-gen species stimulate p44/42 mitogen-activated protein kinase and induce p27(Kip1): role in angiotensin II-mediated hypertrophy of proximal tubu-lar cells. J Am Soc Nephrol. 2000;11:1387–1397.
13. Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16:535–43, 1p following 143.
14. Wu WP, Chang CH, Chiu YT, Ku CL, Wen MC, Shu KH, Wu MJ. A reduction of unilateral ureteral obstruction-induced renal fibrosis by a therapy combining valsartan with aliskiren. Am J Physiol Renal Physiol. 2010;299:F929–F941.
15. Gross O, Girgert R, Rubel D, Temme J, Theissen S, Müller GA. Renal protective effects of aliskiren beyond its antihypertensive property in a mouse model of progressive fibrosis. Am J Hypertens. 2011;24:355–361.
16. Pilz B, Shagdarsuren E, Wellner M, Fiebeler A, Dechend R, Gratze P, Meiners S, Feldman DL, Webb RL, Garrelds IM, Jan Danser AH, Luft FC, Müller DN. Aliskiren, a human renin inhibitor, ameliorates cardiac and renal damage in double-transgenic rats. Hypertension. 2005;46:569–576.
17. Fischer R, Dechend R, Qadri F, et al. Dietary n-3 polyunsaturated fatty acids and direct renin inhibition improve electrical remodeling in a model of high human renin hypertension. Hypertension. 2008;51:540–546.
18. Singh VP, Le B, Khode R, Baker KM, Kumar R. Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes. 2008;57:3297–3306.
19. de Borst MH, Navis G, de Boer RA, Huitema S, Vis LM, van Gilst WH, van Goor H. Specific MAP-kinase blockade protects against renal damage in homozygous TGR(mRen2)27 rats. Lab Invest. 2003;83:1761–1770.
20. Iekushi K, Taniyama Y, Kusunoki H, Azuma J, Sanada F, Okayama K, Koibuchi N, Iwabayashi M, Rakugi H, Morishita R. Hepatocyte growth factor attenuates transforming growth factor-β-angiotensin II crosstalk through inhibition of the PTEN/Akt pathway. Hypertension. 2011;58:190–196.
21. Chiang CK, Hsu SP, Wu CT, Huang JW, Cheng HT, Chang YW, Hung KY, Wu KD, Liu SH. Endoplasmic reticulum stress implicated in the develop-ment of renal fibrosis. Mol Med. 2011;17:1295–1305.
22. Nangaku M. Chronic hypoxia and tubulointerstitial injury: a final common pathway to end-stage renal failure. J Am Soc Nephrol. 2006;17:17–25.
23. Bechtel W, McGoohan S, Zeisberg EM, Müller GA, Kalbacher H, Salant DJ, Müller CA, Kalluri R, Zeisberg M. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med. 2010;16:544–550.
24. Boffa JJ, Lu Y, Placier S, Stefanski A, Dussaule JC, Chatziantoniou C. Regression of renal vascular and glomerular fibrosis: role of angiotensin II receptor antagonism and matrix metalloproteinases. J Am Soc Nephrol. 2003;14:1132–1144.
25. Helle F, Iversen BM, Chatziantoniou C. Losartan increases NO release in afferent arterioles during regression of L-NAME-induced renal damage. Am J Physiol Renal Physiol. 2010;298:F1170–F1177.
26. Mori T, Cowley AW Jr. Role of pressure in angiotensin II-induced renal injury: chronic servo-control of renal perfusion pressure in rats. Hypertension. 2004;43:752–759.
27. Mori T, Polichnowski A, Glocka P, Kaldunski M, Ohsaki Y, Liang M, Cowley AW Jr. High perfusion pressure accelerates renal injury in salt-sensitive hypertension. J Am Soc Nephrol. 2008;19:1472–1482.
28. Polichnowski AJ, Lu L, Cowley AW Jr. Renal injury in angiotensin II+L-NAME-induced hypertensive rats is independent of elevated blood pres-sure. Am J Physiol Renal Physiol. 2011;300:F1008–F1016.
What Is New?•Our study is among the first experimental studies document-
ing the efficacy of direct renin inhibition to reverse hypertensive renal disease and describing the mechanisms involved in this process.
What Is Relevant?• CKD is an incurable-to-date pathology that progresses with age to end-
stage renal Disease. Hypertension is among the leading causes, and
there is an urgent need to develop therapeutic strategies that can arrest (or, even better, reverse) the decline of renal function.
Summary
We found that aliskiren administration to aged, proteinuric mice displaying a constantly activated renin-angiotensin system was ca-pable of improving the renal function and structure through a shift of the balance between profibrotic and antifibrotic members of the TGF-β/bone morphogenetic proteins superfamily.
Novelty and Significance
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from

and Christos ChatziantoniouPanagiotis Kavvadas, Lise Weis, Ahmed B. Abed, David L. Feldman, Jean-Claude Dussaule
Between Profibrotic and Antifibrotic AgentsRenin Inhibition Reverses Renal Disease in Transgenic Mice by Shifting the Balance
Print ISSN: 0194-911X. Online ISSN: 1524-4563 Copyright © 2013 American Heart Association, Inc. All rights reserved.
is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Hypertension doi: 10.1161/HYPERTENSIONAHA.111.00639
2013;61:901-907; originally published online February 25, 2013;Hypertension.
http://hyper.ahajournals.org/content/61/4/901World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://hyper.ahajournals.org/content/suppl/2013/02/25/HYPERTENSIONAHA.111.00639.DC1Data Supplement (unedited) at:
http://hyper.ahajournals.org//subscriptions/
is online at: Hypertension Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer this process is available in the
click Request Permissions in the middle column of the Web page under Services. Further information aboutOffice. Once the online version of the published article for which permission is being requested is located,
can be obtained via RightsLink, a service of the Copyright Clearance Center, not the EditorialHypertensionin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on June 1, 2018http://hyper.ahajournals.org/
Dow
nloaded from

On line supplement
RENIN INHIBITION REVERSES RENAL DISEASE IN TRANSGENIC MICE BY SHIFTING THE BALANCE BETWEEN PRO AND ANTI-FIBROTIC AGENTS
Panagiotis Kavvadas, PhD1, Lise Weis, MD1, Ahmed B. Abed MD1, David L.
Feldman, PhD2, Jean-Claude Dussaule, MD, PhD1, Christos Chatziantoniou,
PhD1
1: INSERM UMR S702, Hopital Tenon, Paris, France
2: Novartis Pharamaceuticals Corp., East Hanover, New Jersey, USA
Short title: Aliskiren reverses hypertensive renal disease
Corresponding author: Christos Chatziantoniou, PhD, Inserm U702, Tenon
Hospital, 4 Rue De la Chine, 75021, Paris, France
Tel. +33 1 56016653 – Fax. +33 1 56016659
e-mail: [email protected]

Materials and methods Animals and experimental design Experiments were performed in Ren Tg on the 129SV background as described elsewhere8. This strain expresses mouse renin in the liver at a constantly high level, leading to elevated levels of prorenin and active renin. For the aliskiren protocol, osmotic mini-pumps (Alzet 2004) containing either PBS or the renin inhibitor aliskiren (25mg/kg/day) were implanted subcutaneously in ten months old heterozygous Ren Tg (n=7 for both groups) and wild type (n=4 for both groups) mice for 28 days. Mice used in our experimental protocol were chosen based on their microalbuminuria levels. Briefly, we used 9-10 month old RenTg mice with albuminuria ≥ 70mg/mmol. Such mice exhibit excessive inflammation accompanied with significant tissue damage and fibrosis1. For the hydralazine protocol, hydralazine (40mg/kg/day, Sigma Aldrich, Saint-Louis, MO, USA) was administered for 28 days to 7 RenTg and 4 wild type mice. In addition, 5 RenTg and 4 wild type were used for the GFR measurements and 8 RenTg mice for monitoring blood pressure with telemetry. A total of 34 RenTg and 16 wild type mice were used. All animals were handled in strict accordance with good animal practice as defined by the relevant national animal welfare bodies of France and all animal work was approved by the appropriate committee of Inserm and the University Pierre et Marie Curie, Paris. Blood pressure measurement Systolic blood pressure was measured every 7 days with the tail-cuff (Kent Scientific Corporation, Vancouver, BC, Canada) and the telemetry methods. For tail-cuff, animals were conscious, slightly warmed-up and acclimated to the measurement procedure for several days before measurements. Measurements were always taken at the same time of day (10:00-11:30 am). A mean value was determined from a minimum of 10 measurements per animal. For telemetry, mice were anesthetized initially with 5% isoflurane in an oxygen stream and maintained on 2–3% isoflurane. Mice were kept on a heating pad throughout implantation of the BP telemeter (modelTA11PA-C10; Data Sciences International, St. Paul, MN, Department of biochemistry, Hôpital Européen Georges Pompidou Paris, France). The catheter was inserted into the left femoral artery and the telemetric transmitter probe was positioned subcutaneously on the left flank. Animals were housed in individual cages placed on top of the telemetric receivers in a light-dark cycled recording room (7h00 to 19h00) for a 1-wk recovery period before the initiation of the experiment. Four mice were subcutaneously implanted with mini-pump containing aliskiren (25/mg/kg/day) and another four received hydralazine through drinking water (40mg/kg/day). Measurements were done daily for the first two weeks and on days 21 and 28 thereafter as described elsewhere3. Urinary albumin excretion Urine samples were collected every 7 days. Before the initial collection all mice were acclimated in metabolic cages with free access to water for 16-hour urine collection. Microalbuminaria was measured using the Olympus System Reagent (OSR6167) and an Olympus AU400 apparatus (Laboratory of Biochemistry, UFR de Medecine Paris 7 – Site Bichat, Paris). Urinary albumin concentration was normalized to urinary creatinine concentration and expressed as mg of albumin/mmol of creatinine. Glomerular filtration rate

Glomerular filtration rate (GFR) was measured using the single bolus injection method of FITC-inulin as described elsewhere2. Briefly, RenTg mice (n=5) and their wild type littermates (n=4) were injected retro-orbitally, under light aesthesia (isoflurane) with 3.74µl/g of body weight 5% FITC-inulin (Sigma-Aldrich) solution. Blood samples were collected every 15 minutes for a period of 90 minutes and plasma was isolated after centrifugation. GFR was estimated and expresses in µl/min ± SEM. The following day, the RenTg mice were implanted with osmotic minipumps containing aliskiren as described above. After 28 days of treatment, GFR was measured again for both groups. Renal and cardiac morphology and fibrosis Formalin-fixed renal and cardiac tissues were processed by routine histological methods and stained with Sirius red and Masson trichrome stains. Four photomicrographs from random, non-overlapping fields were captured using a 20x objective lens. The images were quantified by image analysis and results were expressed as % red or blue stained tissue /total tissue area ± SEM. Photomicrographs are representative of the histological appearances of the respective groups. RNA extraction and quantitative Real-Time PCR Total RNA was extracted from renal tissue using TRI reagent (Molecular Research Center, Cincinnati, OH, USA) according to the manufacturer’s instructions. RNA quality was evaluated by the optical density at 260 and 280 nm. cDNA was synthesized from 1µg RNA using the Fermentas H Minus First Strand cDNA Synthesis kit according to the manufacturer’s instructions. Real Time PCR was performed with the Roche Light Cycler 480 sequence Detection System using SYBR Green PCR Master Mix (Qiagen). GUSB was used as housekeeping gene. All samples were tested in duplicates. Results are expressed as % change vs wild type PBS animals ± SEM. Primers are listed in Table S1. Immunohistochemistry and immunofluorescence Immunohistochemistry was performed on 4µm sections from paraffin embedded tissue for F4/80 (1/200, AbD Serotec, Oxford, UK) and Ki67 (1:200, Millipore, Billerica, MA, USA). Immunofluorescence for α-SMA (1/500, Sigma-Aldrich) was performed on 4µm thick, 4% paraformaldehyde-fixed cryosections. Protein extraction and western blot Renal tissue was homogenized in RIPA lysis buffer (Santa Cruz, Santa-Cruz, CA, USA), kept on ice for 30 min and then centrifuged. Total protein concentrations were measured with the Bradford assay (Biorad). 30µg of total protein were loaded in 4-12% polyacrylamide gels (Invitrogen) and then transferred to nitrocellulose membrane (Millipore). Membranes with incubated with anti-pP38, P38, pERK1/2, ERK1/2 (Santa Cruz), pSMAD1/5/8 (Cell Signaling, Boston, MA, USA), nephrin (Santa Cruz) and β-actin (Sigma-Aldrich). The bound antibodies were labeled with horseradish peroxidase-conjugated secondary antibodies (GE Healthcare, UK) and detected with the ECL Plus Detection System (GE Healthcare). A minimum of 4 animals per group were used for every experiment. Western blots were scanned with the GS-800 calibrated densitometer, and images were analyzed with Quantity One image-processing software (Bio-Rad). If not stated otherwise, β-Actin was used as loading control. Data were expressed as mean values ± SEM of optical density.

Statistical Analysis Values are expressed as mean ± SEM. Data were analyzed using one-way ANOVA followed by Protected Least Significance Difference Fisher’s test. Results with p<0.05 were considered statistical significant. References
1. Huby AC, Rastaldi MP, Caron K, Smithies O, Dussaule JC, Chatziantoniou C. Restoration of podocyte structure and improvement of chronic renal diseases in transgenic mice overexpressing renin. Plos One. 2009;4:e6721.
2. Qi Z, Whitt I, Mehta A, Jin J, Zhao M, Harris RC, Fogo AB, Breyer MD. Serial determination of glomerular filtration rate in conscious mice using FITC-inulin clearance. Am J Physiol Renal Physiol. 2003;286:F590-F596.
3. Bergaya S, Faure S, Baudrie V, Rio M, Escoubet B, Bonnin P, Henrion D, Loirand G, Achard JM, Jeunemaitre X, Hadchouel J. WNK1 regulates vasoconstriction and blood pressure response to α 1-adrenergic stimulation. Hypertension. 2011;58:439-45.

Table S1 - List of primers GENE SENSE ANTISENSE
VCAM TGGTGAAATGGAATCTGAACC CCCAGATGGTGGTTTCCTT
TNF‐α TCTTCTCATTCCTGCTTGTGG ATGAGAGGGAGGCCATTTG
MCP‐1 GTTGGCTCAGCCAGATGCA AGCCTACTCATTGGGATCATCTTG
Collagen I GCAGGTTCACCTACTCTGTCCT CTTGCCCCATTCATTTGTCT
Collagen III TCCCCTGGAATCTGTGAATC TGAGTCGAATTGGGGAGAAT
PAI‐1 AGGATCGAGGTAAACGAGAGC GCGGGCTGAGATGACAAA
Nephrin ACTACGCCCTCTTCAAATGCA TCGAGGGCCTCATACCTGAT
Podocin CCATCTGGTTCTGCATAAAGG CCAGGACCTTTGGCTCTTC
KIM‐1 CCAACATCAATCAGAGTCTCTACC TGTCTCATGGGGACAAAATG
BMP4 ACCAATGGAGCCATTCCGTAG CTCATGTAATCCGGAATGACGG
BMP7 CCTGGGCTTACAGCTCTCTG GGTGGCGTTCATGTAGGAGT
TGF‐β TGGAGCAACATGTGGAACTC GTCAGCAGCCGGTTACCA
CTGF TGACCTGGAGGAAAACATTAAGA AGCCCTGTATGTCTTCACACTG
ANP GAGAAGATGCCGGTAGAAGA AAGCACTGCCGTCTCTCAGA
BiP CTGAGGCGTATTTGGGAAAG TCATGACATTCAGTCCAGCAA
HIF-1α GCACTAGACAAAGTTCACCTGAGA CGCTATCCACATCAAAGCAA
DNMT1 CAAATAGATCCCCAAGATCCAG CGGAACTAGGTGAAGTTTCAAAAA
GUSB CTCTGGTGGCCTTACCTGAT CAGTTGTTGTCACCTTCACCTC

S1
****
###
60
80
100
120
140
160
0 14 21 28
mmHg
DAYS
Systolic blood pressure
WT PBS
WT ASK
RenTg PBS
RenTg ASK
A
60
80
100
120
140
160
0 7 14 21 28
mmHg
Systolic Blood Pressure
WT Hyd
Ren Tg Hyd
*
*
B
50
70
90
110
130
150
170
PC 7 14 21 28
mm
Hg
days
Telemetry systolic blood pressure (day)
ask
hyd
C

S1: Time course of systolic blood pressure in the aliskiren (A) and hydralazine (B) treated mice as measured with the tail cuff method. * P < 0.05 vs WT PBS, # P < 0.05 vs RenTg PBS. Radiotelemetry measurements of systolic blood pressure during day (07:00-19:00h, C) and night (19:00-07:00h, D) cycles further verified the anti-hypertensive effect of aliskiren and hydralazine. Data are presents as mean values ± SEM.
50
70
90
110
130
150
170
190
PC 7 14 21 28
mm
Hg
days
Telemetry systolic blood pressure (night)
ask
hyd
D
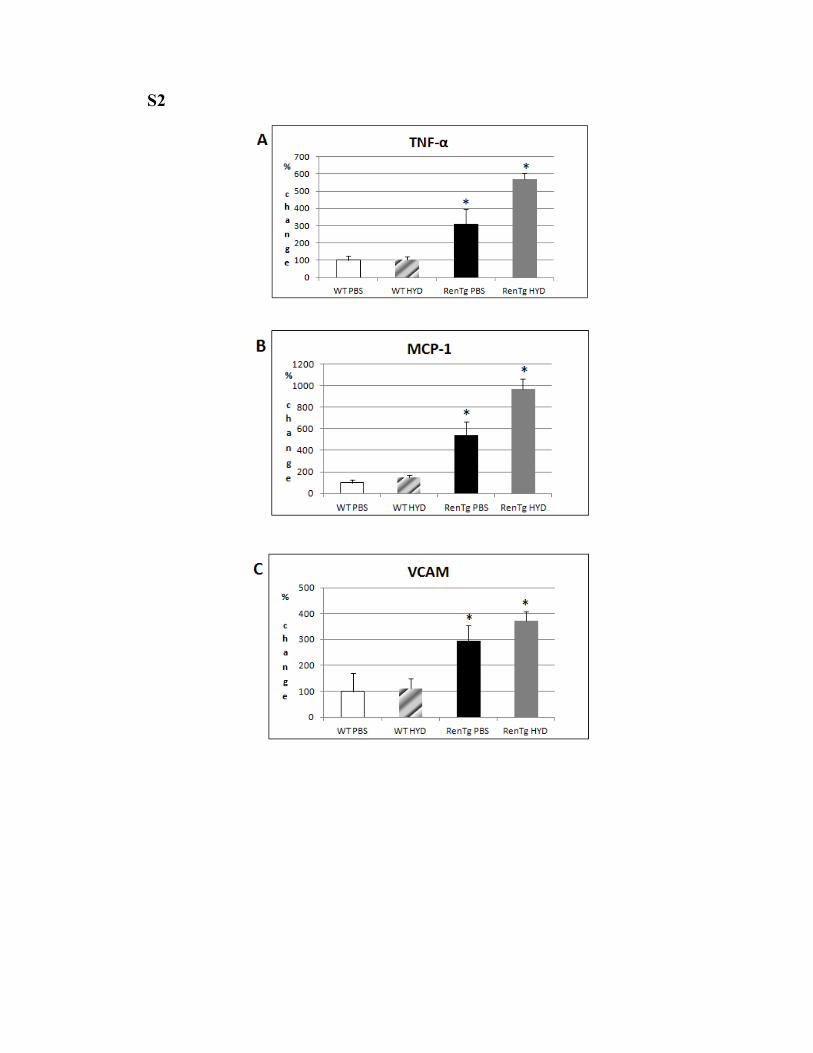
S2

S2: Effect of hydralazine on inflammatory and fibrotic markers. mRNA levels of TNF-α (Α), MCP-1 (B), VCAM (C), TGF-β (D) and CTGF (E) remained elevated after treatment with hydralazine. * P < 0.05 vs WT PBS.
S3

S3: Effect of aliskiren on renal fibrosis. Masson trichrome staining showed that RenTg PBS renal tissue is characterized by huge cellular infiltrates (white arrow), proteinuria (white asterisk) and perivascular and interstitial fibrosis. Treatment with aliskiren significantly improved all the above mentioned parameters (A). Fibrotic foci quantification verified aliskiren beneficial effect on regression of fibrosis (B). * = P < 0.05 vs WT PBS, # = P < 0.05 vs RenTg PBS.
WT ASK
RenTg ASK RenTg PBS
WT PBS
A
*
B
0
2
4
6
8
10
WT PBS WT ASK RenTg PBS RenTg ASK
fibrotic
foci
Interstitial fibrotic foci
#

S4
S4: Effect of aliskiren on fibrosis-related parameters. Myofibroblasts marker α-SMA is expressed in vascular smooth muscle cells in WT animals. In RenTg PBS mice, staining was observed in the interstitium indicating the presence of myofibroblasts. Treatment with aliskiren diminishes interstitial staining for α-SMA.

S5
S5: Effect of hydralazine on renal fibrosis. Hydralazine did not improve renal histology in RenTg mice since perivascular inflammation and ECM deposits were still evident with Masson’s staining (A). Quantification of the fibrotic foci (B) further verified the histological observations. * P < 0.05 vs WT PBS.

S6
S6: mRNA of the anti-fibrotic mediators BMP4 (A) and BMP7 (B) was highly upregulated after treatment with aliskiren. In addition, anti-fibrotic Smad’s1/5/8 activity was reduced in RenTg PBS mice and restored after treatment with aliskiren (C).
0
50
100
150
200
250
300
WT PBS WT ASK RenTg PBS RenTg ASK
%
change
BMP4
* #
A
0
200
400
600
800
1000
1200
WT PBS WT ASK RenTg PBS RenTg ASK
%
change
BMP7*
*
* #
B
Actin
WT PBS
WT ASK
RenTgPBS
RenTgASK
pSMAD1/5/8
C
0
50
100
150
200
WT PBS WT ASK RenTg PBS RenTg ASK
%
Densit
y
pSMAD1/5/8 /Actin

S7
S7: Effect of aliskiren on markers of novel pro-fibrotic mechanisms. mRNA levels of BiP (A), HIF-1α (Β) and DNMT1 (C) were evaluated with Real Time PCR. * = P < 0.05 vs WT PBS, # P < 0.05 vs RenTg PBS.

S8
S8: Hydralazine effect on cardiac fibrosis. Masson’s staining (A) revealed extensive ECM deposits in the tissue of the hydralazine-treated RenTg mice. Quantification of the ECM covered area (B) showed a non-significant decrease in the hydralazine treated animals compared to the RenTg PBS ones. * = P < 0.05 vs WT PBS.