sciencesearch.defra.gov.uksciencesearch.defra.gov.uk/document.aspx?document=vm... · web viewthe...
TRANSCRIPT

Preliminary evaluation of the potential risks to consumers of animal-derived food products following non-authorised veterinary use of chemicals FINAL REPORT
March 2007

The Institute of Environment and Health was established at Cranfield University in November 2005. The Institute is principally funded by UK Government Departments and Agencies by way of specific research and consultancy contracts.
The views expressed here do not necessarily represent those of any Government Department or Agency.
This document is a Final Report by the Institute of Environment and Health and the Central Science Laboratory for the Department for Environment Food and Rural Affairs.
Prepared by P Holmes1, LS Levy1, L Assem1, A Boxall3, M Litchfield2, Jane Cotterill3, Qasim Chaundhry3 & M Taylor3
1 Institute of Environment and Health, Cranfield Health, Cranfield University2 Consultant3 Central Science Laboratory
The authors gratefully acknowledge the contributions made to the conduct of the project, and to the preparation of early drafts of the report, by E Stutt, J Massey, L Shuker, P Rumsby and C Courage while at the MRC Institute for Environment and Health
Institute of Environment and Health, 2007
Institute of Environment and HealthCranfield HealthCranfield UniversitySilsoeBedfordshireMK45 4DTUK
Central Science LaboratorySand HuttonYorkYO41 1LZ

ContentsPage No.
Executive Summary_____________________________________________________________________11 Introduction_________________________________________________________________________32 Methodology________________________________________________________________________6
2.1 Exposure and risk assessment_________________________________________________62.2 Hazard assessment____________________________________________________________9
3 Evaluations_________________________________________________________________________123.1 Chloramphenicol______________________________________________________________123.2 Clenbuterol___________________________________________________________________133.3 Dimetridazole________________________________________________________________133.4 Enrofloxacin__________________________________________________________________143.5 17-Estradiol_________________________________________________________________143.6 Flavomycin___________________________________________________________________153.7 Furazolidone__________________________________________________________________153.8 Lasalocid sodium_____________________________________________________________163.9 Malachite green______________________________________________________________163.10 Nalidixic acid________________________________________________________________173.11 Nandrolone__________________________________________________________________173.12 Narasin______________________________________________________________________183.13 Phenylbutazone_____________________________________________________________183.14 Progesterone________________________________________________________________183.15 Salbutamol__________________________________________________________________193.16 Streptomycin________________________________________________________________193.17 Testosterone________________________________________________________________20
4 Conclusions________________________________________________________________________294.1 Discussion and conclusions___________________________________________________294.2 Recommendations____________________________________________________________31
5 References_________________________________________________________________________32Annex 1 Chloramphenicol___________________________________________________________34Annex 2 Clenbuterol_________________________________________________________________41Annex 3 Dimetridazole______________________________________________________________46Annex 4 Enrofloxacin________________________________________________________________53Annex 5 17β-Estradiol_______________________________________________________________62Annex 6 Flavomycin_________________________________________________________________73Annex 7 Furazolidone_______________________________________________________________79

Contents continuedPage No.
Annex 8 Lasalocid Sodium__________________________________________________________90Annex 9 Malachite Green___________________________________________________________98Annex 10 Nalidixic Acid____________________________________________________________108Annex 11 Nandrolone______________________________________________________________113Annex 12 Narasin__________________________________________________________________121Annex 13 Phenylbutazone_________________________________________________________128Annex 14 Progesterone____________________________________________________________137Annex 15 Salbutamol______________________________________________________________145Annex 16 Streptomycin____________________________________________________________150Annex 17 Testosterone____________________________________________________________159Annex 18 Toxicity Searches___________________________________________________________166

Executive SummaryThis preliminary (pilot) evaluation assesses the potential risks to consumers from exposure to chemical residues following theoretical non-authorised veterinary use of 17 selected chemicals in food-producing animals on the basis of a series of simplistic worst case assumptions.
A detailed literature review of published toxicological data was conducted by the Institute of Environment and Health (IEH) for each of the 17 selected chemicals. In addition, Central Science Laboratory (CSL) compiled information on the uses of the substances and undertook structure–activity relationship (SAR) modelling for one chemical for which the toxicological database was inadequate for evaluation. Total daily intakes of meat, fish and eggs, but not diary products, for toddlers and adults were estimated, using recognised dietary surveys. Worst-case estimates of residue concentrations in each of these food types were made, using combinations of residue data reported by Veterinary Residues Committee (VRC), experimentally derived residue data and/or where measured residue data were not available, crude calculations. The experimentally-derived data were compiled by CSL who also determined the calculated estimates, where they were required. Daily dietary intakes and estimated worst-case residue concentrations were then combined, to estimate a theoretical total exposure from animal foodstuffs for each chemical. Once the potential worst-case intakes had been estimated, comparisons were made with Acceptable Daily Intake (ADI) values or, where these were not available, relevant toxicological data, in order to make a statement regarding the potential for adverse health effects among consumers, following such intakes.
It should be emphasised that there is no evidence that the theoretical residue concentrations used to calculate the dietary intakes have ever been reached or indeed are ever likely to be reached, for any of the substances discussed in this evaluation. In addition, the food intake values derived from dietary surveys also, at best, represent “worst-case” scenarios. For these reasons, any results of this exercise that might be seen as an apparent cause for concern, based on dietary intake, should be seen rather as triggers for further enquiry, not as a need for risk reduction measures at this stage.
Based on the limited, conservative evaluation conducted and presented herein, 8 of the 17 selected chemicals — 17-estradiol, flavomycin, naladixic acid, nandrolone, narasin, progesterone, streptomycin and testosterone —were considered not to be a potential health risk to consumers following non-authorised veterinary use in food-producing animals. However, in the case of flavomycin, further consideration of its toxicological properties may be appropriate.
Owing to the nature of their toxic properties (e.g. genotoxic carcinogens), a zero tolerance approach was taken for several of the chemicals evaluated; that is, any contamination of foodstuffs by such chemicals would constitute a theoretical risk, albeit miniscule, to consumer health. The worst-case estimates of exposure via animal food products calculated in this evaluation would, therefore, be unacceptable if they were actually to occur; although, in reality, the actual impacts on human health and their significance are not known. Chemicals falling into this category were chloramphenicol, furazolidone, malachite green and phenylbutazone. Of these, only chloramphenicol and malachite green have been detected in food samples in the UK but at much lower concentrations than have been estimated in this conservative1, preliminary risk assessment exercise. Phenylbutazone and furazolidone have not been detected in food samples, which provides considerable reassurance as to the scale of any health risk to the public although a chemical A0Z which is a potential, but not specific, marker for furazolidone exposure, has occasionally been noted in a few samples. Worst-case intakes exceeding the ADI were also calculated for enrofloxacin, particularly in the case of toddlers. However, this was in large part a reflection of the use of a microbiologically-, rather than
1 Throughout this document, the term “conservative” is taken to mean a precautionary approach, based upon assumptions of “worst case” exposure scenarios
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
1

toxicologically-, based ADI. Considerable reassurance as to the level of risk posed to the public is provided by the finding that only one sample of poultry meat contamination with this compound has been identified in UK surveillance programmes in the period 2001–2003
The worst-case intakes calculated for clenbuterol significantly exceed the ADI for toddler, and 97.5% adult consumers. Such exposures would be unacceptable given that, while considered not genotoxic or carcinogenic, it is pharmacologically active and has reproductive toxic potential. However, analysis of food samples under UK surveillance schemes has not identified contamination by β-agonists, such as clenbuterol, which provides considerable reassurance as to the scale of risk to the public.
The worst-case intakes calculated for lasalocid sodium significantly exceeded the proposed ADI and were between up to 20% of the established experimental No-Observed-Effect Level (NOEL). Hence this could represent an erosion of the margin of safety for consumers; however, the impact of this is uncertain, as the toxicological dataset for lasalocid sodium is limited.
No ADI has been recommended for either dimetridazole, owing to inadequacies in the toxicological dataset, or salbutamol, as there are no licensed veterinary products. Therefore, for the purposes of this pilot evaluation, project-specific de novo ‘Intake of Concern’ (IOC) values were derived from the limited data available. A conservative NOEL was selected for each substance, and an uncertainty factor applied, depending on the source of the NOEL and uncertainties in the respective datasets. Comparison of calculated worst-case salbutamol intake values with the extremely conservative project-specific IOC identified exceedence of 20% of the value for toddlers but UK surveillance results indicate that there is little cause for concern about the potential for any consumer health impact following unauthorised veterinary use of this substance. The calculated worst-case intake values for dimetridazole were up to five times the project-specific IOC. Such high intakes together with the uncertain genotoxic potential of this substance would raise concern if the calculated intakes were to be realised. However, the UK National Surveillance scheme andr the Non-Statutory Surveillance Scheme, (VRC, 2001; VRC, 2002a; VRC, 2003) have found only one isolated instance of dimetridazole contamination.
In order to improve the accuracy of future risk assessments of this nature, it is recommended that attention be given to obtaining improved measures of likely ‘real world’ residue concentrations and to obtaining improved toxicokinetic and toxicodynamic datasets for those chemicals considered to pose the greatest potential risk.
Where, for some of the substances, available data have not been sufficient to reach a secure conclusion regarding, in particular, carcinogenicity and/or genotoxicty, as is notably the case for phenylbutazone and flavomycin, it would be appropriate to seek further advice from the relevant government expert committees, such as the Committee on Carcinogenicity of Chemicals in Food, Consumer Products and the Environment and Committee on Mutagenicity of Chemicals in Food, Consumer Products and the Environment.
The preliminary evaluations made herein may be useful in assisting the VRC and other surveillance schemes to target appropriate and cost-effective sampling strategies for detecting non-authorised veterinary use of chemicals.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
2

1 IntroductionVeterinary medicines play an important part in maintaining the health and well-being of both food-producing and companion animals in the UK, as elsewhere in the world. However, in order to protect consumers, some veterinary medicines are no longer authorised for use in food-producing animals or their use is restricted in some way. In addition, there are chemicals that could possibly be used in food-producing animals that have never been authorised for veterinary use. Evidence for such misuse is demonstrated by the detection of some of these chemicals in food products, in residue detection programmes, as exemplified by reports by the Veterinary Residues Committee (VRC, 2001; VRC, 2002a; VRC, 2003). This has caused some concern and, as a result, the MRC Institute for Environment and Health, together with the Central Science Laboratory (CSL), was commissioned by the Department for Environment Food and Rural Affairs (Defra) to investigate potential health risks to consumers of 17 selected chemicals following unauthorised veterinary use in food-producing animals (see Table 1.1). Following the closure of the MRC Institute for Environment and Health in October 2005, the key Institute scientists, now at the Institute of Environment and Health (IEH) at Cranfield Univeristy, were commissioned by the Medical Research Council (MRC) to complete the risk evaluation and produce the final report.
This project was a preliminary pilot exercise to assess the likely extent of any problem. The assessment was generally restricted to consumption of meat, fish or eggs, except for some instances where the chemical under consideration had been identified in other foods in a UK monitoring programme. Exposure arising from consumption of diary produce was specifically excluded from the scope of this preliminary exercise. The substances included in this assessment were selected by Defra, and fall into the following groups.
Hormones, stilbenes and β-agonists, the use of which, for growth promotion in food-producing animals, is specifically prohibited under Council Directive 96/22/EC. However, a small number of these substances do have authorisations that permit therapeutic use under very limited circumstances, for example clenbuterol (a β-agonist), which is used in tocolysis for bovines.
The small number of substances that are listed in Annex IV of Commission Regulation 2377/90/EC, which means that they must not be used in food-producing animals. Under the Maximum Residue Limit (MRL) controls, all authorised therapeutics must have MRLs listed in Annexes I-III of the Regulation. Examples of Annex IV substances include chloramphenicol and the nitrofurans. Substances are in Annex IV because it was not possible to assign a more satisfactory classification when they were submitted for determination of an MRL. The reasons for this might be based on toxicological concerns or missing data that the companies making the MRL application were not able to provide.
Substances not authorised for use as veterinary medicines but detected in residue surveillance programmes in the UK or elsewhere. Examples are malachite green in fish and chloramphenicol in imported chicken.
Permitted pharmaceutical products that might be used in non-permitted ways; for example, enrofloxacin, lasalocid sodium, narasin, flavomycin and streptomycin.
Council Directive 96/23/EC commits EU Member States to analyse food samples for residues of veterinary medicines to ensure that their home-produced foods of animal origin are safe. In the UK the National Surveillance Scheme covers red meat, poultry meat, wild and farmed game, farmed fish (salmon and trout), eggs, milk and honey. Authorised officers of the different government agencies collect samples on behalf of Defra’s Veterinary Medicines Directorate. All of the results that are
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
3

above the relevant MRL or ‘Action Level’2 are passed to the Food Standards Agency, where toxicologists give a scientific opinion on the relevance of the residues for human health. A follow-up investigation is conducted for every sample with a residue above the MRL or ‘Action Level’.
The Non-Statutory Surveillance Scheme was set up to complement the National Surveillance Scheme. It mainly looks for the use of banned or unauthorised substances imported meat, farmed fish and honey.
Section 2 of this report describes how, for each of the 17 selected chemicals, total daily intakes of meat, fish and eggs for toddlers and adults were estimated, using recognised dietary surveys, and combined with highly conservative calculations or estimates, or measurements of residue concentrations, provided by the Central Science Laboratory (CSL), to estimate exposures from animal foodstuffs (Section 2.1). Once the potential exposures had been estimated, the potential health effects that could occur were assessed (Section 2.2). Summary data on use and exposure, toxicity and any current guideline values, for all 17 selected chemicals, are provided in Annexes 1–17. Finally, given the pilot nature of this project, the risks of health effects occurring at the estimated intakes for each chemical were assessed assuming a series of simplistic “worst case” scenarios, and these are reported in Section 3.
2 For many veterinary residues an MRL is not set or may not be relevant for a number of reasons: substances banned from use in food animals; analysis of tissues and substances not normally eaten; substances in the surveillance scheme that are not veterinary medicines; feed additives, which are not classed as veterinary medicines. The Action Level is usually any confirmed residue, and is based on the limitations of the analytical methodology, so might not indicate health concerns if exceeded (VRC, 2003).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
4

Table 1.1 Chemicals with potential for non-authorised veterinary use included in this evaluationChemical name Use Note on illegal use in UK
Chloramphenicol Antibiotic No MRL. Prohibited for use in food-producing animals.
Clenbuterol Bronchodilator Prohibited for use as a growth-promoting agent, but authorised for use for other purposes in animals intended for human consumption.
Dimetridazole Antiparasitic agent No MRL. Prohibited for use in food-producing animals.
Enrofloxacin Antibiotic Authorised for use in animals intended for human consumption, but with withdrawal periods.
17-Estradiol Estrogenic hormone No MRL. Hormonal growth promoters prohibited in farm animals.
Flavomycin(Flavophospholipol; Bambermycins)
Antimicrobial No MRL
Furazolidone Antimicrobial agent No MRL. Prohibited for use in food-producing animals.
Lasalocid sodium Coccidiostat Not permitted for use in laying hens.
Malachite green Antimicrobial agent No MRL. Prohibited in farmed fish.
Nalidixic acid Antibacterial agent No MRL. Limited usefulness; replaced with more efficacious quinolones. No authorised veterinary medicines.
Nandrolone Anabolic steroid No MRL. Prohibited for use in food-producing animals.
Narasin Coccidiostat Authorised but with withdrawal periods
Phenylbutazone Anti-inflammatory No MRL. Prohibited for use in food-producing animals.
Progesterone Steroid hormone No MRL could be set.
Salbutamol(Albuterol)
Bronchodilator No MRL. Prohibited for use as a growth-promoting agent.
Streptomycin Antibiotic Authorised for use in animals intended for human consumption, but with withdrawal periods.
Testosterone Steroid hormone No MRL. Prohibited for use in food-producing animals.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
5

2 Methodology2.1 Exposure and risk assessment Theoretical “worst case” consumer exposures resulting from the non-authorised veterinary use of the 17 selected chemicals were estimated from the measured or calculated concentrations of these compounds in meat and meat-derived foodstuffs and the amount of such foodstuffs consumed by an ‘average’ and ‘high-level’ consumer, on a long-term basis. Consideration of intakes that might arise from the consumption of dairy produce derived from exposed animals was, because of the limited preliminary nature of this excerise, specifically excluded from consideration. The potential maximum intakes were estimated for both adults and toddlers.
In estimating exposure, measured concentrations of the chemicals in animal products were used if they were readily available from authoritative sources, such as the annual reports of the VRC (2001; 2002a; 2003). In some cases experimentally-derived data on measured concentrations in meat products were available from studies conducted previously at CSL. Results from the CSL studies, which took the form of concentrations determined in animal tissues following experimental administration of the chemical, are reported in Annexes 1–17 (Sections 2.1). For each compound, the highest value reported from amongst the various sources was used in the risk assessment.
If measured data were not available, likely “worst-case” concentrations in meat products were estimated for those animal species in which there was believed to be a potential for the substance to be used. Estimates were made using Equation 1:
Equation 1M = D × T × Fabs × (1 – Fexc)
where M is the predicted concentration in meat (mg/kg); D is the dose of substance (mg/kg/day); T is the duration of the treatment (d); Fabs is the fraction of compound absorbed; and Fexc is the fraction of compound excreted.
Values generated using Equation 1 would be anticipated to be significant overestimates, as conservative values were selected for input and conservative assumptions were made where complete data were not available (e.g. for the proportions of the chemicals absorbed or excreted).
The amount of the residues of the selected chemicals to which a consumer might be exposed in their diet was calculated, based on estimates of the quantities of animal products consumed by populations in the UK and Europe, using the National Diet and Nutrition Survey (NDNS; ONS & MRC Human Nutrition Research, 2002) and the World Health Organization Global Environment Monitoring System/Food Contamination Monitoring and Assessment Programme (GEMS/Food; WHO, 2003) datasets, respectively. The potential daily intake of each chemical (in µg/day) from any single category of animal product was estimated by multiplying the concentrations of the residue in each product by the amount of the product consumed per day. The total daily intake per person for each substance was calculated by summing the intakes resulting from each product that might contain that particular residue. Calculations of intake per person on a body weight basis (µg/kg bw/day) used a typical adult bodyweight of 60 kg (WHO, 1997) and a selected bodyweight of 11 kg for toddlers3
(children aged 1–2 years). In order to ensure a highly conservative (worst-case) scenario was considered, it was assumed that all relevant foodstuffs consumed in the diet would contain residues at the maximum estimated concentration on a long-term basis.3Defra and Environment Agency (2002) give a typical bodyweight of 11 kg for children aged 1–2 years and 14 kg for those aged 2–3 years
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
6

Average daily consumption values of animal produce for the ‘typical European consumer’ were taken from the GEMS/Food Regional Diet for Europe (WHO, 2003). GEMS/Food has developed five regional diets for predicting the dietary intakes of pesticides and other contaminants in food. The GEMS/Food Regional Diets are based on Food Balance Sheet (FBS) data compiled by the Food and Agriculture Organization (FAO) of the United Nations. Data on the consumption of those animal products used in this assessment are presented below in Table 2.1.
Table 2.1 Consumption data from GEMS/Food European Regional Diet Animal product Consumption per person (g/day)
Eggs 37.5Chicken 49.4Chicken liver 0.3Turkey 8.3Poultry 58.4Beef 63.9Beef (cattle) offal 6.0Pork 75.8Pig offal 5.0Sheep/lamb 10.3Sheep offal 1.3Salmon & trout 33.9Prawns 3.0Honey 1.3
From WHO (2003) The selected value for chicken (49.4 g) comprises the European regional diet value for chicken meat (44.0 g), poultry fats (5.3 g) and poultry skin (0.1 g). The selected value for turkey (8.3 g) is made up of the GEMS/Food value for turkey meat (7.3 g) and a value of 1 g allocated for the proportion of skin and fat likely to come from this source. The selected value for poultry combines the published values for poultry meat (53.0 g), poultry fats (5.3 g) and poultry skin (0.1 g). The European regional values for offal were used for the consumption of liver and kidney. Consumption values for marine and freshwater fish were combined to give a conservative estimate for consumption of salmon and trout. The European regional value for ‘crustaceans (fresh/frozen)’ was used for prawns.
UK dietary information for adults was derived from the most recent NDNS (ONS & MRC Human Nutrition Research, 2002). This is a survey of the diet and nutrition of adults aged 19 to 64 years living in private households in Great Britain, and was conducted between July 2000 and June 2001. Data for average (median) and high-level (97.5th percentile from the NDNS dataset, provided by the Food Standards Agency) consumers of animal products were used in the risk assessment and are presented in Table 2.2.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
7

Table 2.2 Median and high-level (97.5th percentile) consumption data for adults in Great BritainAnimal product Median consumption per
person (g/day)97.5th percentile consumption (g/day)1
Eggs and egg dishes 37.9 75Poultry 70.9 123 Beef 68.6 86Pork 47.0 119Sheep/lamb 36.0 60Liver, liver products & dishes 12.3 36Salmon & trout (oily fish) 19.6 66Prawns (shellfish) 11.6 32Honey 4.02 231 All 97.5th centile data rounded to the nearest whole number2 Mean daily consumption rate (median value unavailable)Median consumption data: The selected value for poultry comprises the NSDS daily consumption values for ‘coated chicken and turkey’ (23.9 g) and ‘chicken and turkey dishes’ (47.0 g). The selected value for beef comprises the NDNS values for ‘beef, veal and dishes’ (45.7 g), 50% of the value for ‘burgers and kebabs’ (29.1 g; 14.55 g allocated to beef) and 50% of the value for ‘sausages’ (16.7 g; 8.35 g allocated to beef). The selected value for pork comprises the NDNS values for ‘bacon and ham’ (16.7 g), ‘pork and dishes’ (21.9 g) and 50% of the value for ‘sausages’ (16.7 g; 8.35 g allocated to pork). The selected value for sheep/lamb comprises the NSDS values for ‘lamb and dishes’ (21 4 g) and 50% of the value for ‘burgers and kebabs’ (29.1 g; 14.55 g allocated to sheep/lamb).
Information on median food intakes consumed by toddlers was taken from the NDNS survey conducted between July 1992 and June 1993 (OPCS & MRC Research Council Dunn Nutrition Unit, 1995). Data on high-level consumption for toddlers were obtained from the 2000–2001 dataset, which was provided by the Food Standards Agency.
Table 2.3 Median and high-level (97.5th percentile) consumption data for toddlers in Great BritainAnimal product Median consumption per
person (g/day)97.5th percentile consumption (g/day)1
Eggs and egg dishes 12.3 50a
Poultry 16.5 44 Beef 26.8 58Pork 14.9 53Sheep/lamb 9.4 32Liver, liver products & dishes 6.7 35Salmon & trout (oily fish) 6.4 22Prawns (shellfish) 6.3 19Honey 2.02 81 All 97.5th centile data rounded to the nearest whole number2 Mean daily consumption rate (median value unavailable)Median consumption data: The selected value for poultry comprises the NSDS daily consumption values for ‘coated chicken and turkey’ (9.6 g) and ‘chicken and turkey dishes’ (6.9 g). The selected value for beef comprises the NDNS values for ‘beef, veal and dishes’ (16.3 g), 50% of the value for ‘burgers and kebabs’ (10 g; 5 g allocated to beef) and 50% of
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
8

the value for ‘sausages’ (11 g; 5.5 g allocated to beef). The selected value for pork comprises the NDNS values for ‘bacon and ham’ (4.4 g), ‘pork and dishes’ (5 g) and 50% of the value for ‘sausages’ (1 g; 5.5 g allocated to pork). The selected value for sheep/lamb comprises the NSDS values for ‘lamb and dishes’ (4.4 g) and 50% of the value for ‘burgers and kebabs’ (10 g; 5 g allocated to sheep/lamb).
Where possible, intakes were calculated by direct combination of the measured or estimated concentrations in animal products and the daily consumption of that product. Conservative assumptions were employed where this was not possible. For example, poultry consumption data were used in conjunction with measured concentrations in chicken meat, for enrofloxacin and dimetridazole, as tissue residue concentrations were not available for turkey and it is possible that these two substances may be used for the treatment of turkeys. Additionally, separate consumption data for chicken and turkey were not available from the NDNS dataset so, where tissue concentrations were available for both birds, the highest concentration value was used in conjunction with the overall consumption for poultry. It was also assumed that all liver and kidney consumed contained the highest concentration measured or estimated for a particular substance. In the case of the high-level consumer from the NDNS dataset, all fish were assumed to be salmon and trout and the highest concentration of malachite green measured in these fish was used to assess the potential maximum intake.
Worst-case intakes determined from worst-case residue concentrations in meat products and conservative estimates of consumption patterns for such food products, as described above, are presented in Section 3.
The potential for risks posed by these worst-case exposures was characterised, for each of the 17 selected chemicals, by calculating the proportion of the Acceptable Daily Intake (ADI) that was contributed by these intakes. Where no ADI was available, for the purposes of this pilot evaluation, a project-specific de novo ‘Intake of Concern’ (IOC) was derived from the toxicologically limited data available. To do this a conservative NOEL was selected for each substance, and an uncertainty factor applied, depending on the source of the NOEL and uncertainties in the respective datasets. The nature of the potential health hazard(s) posed by a compound was also considered at this stage, in order to estimate the likelihood that adverse effects might occur and to assess their potential severity. Risk assessments are reported in Section 3.
The National and Non-Statutory Surveillance Scheme results for the years 2001-2003 were consulted and, where appropriate, findings are reported in this assessment, in order to put the calculated residue concentrations and hence the worst-case risk assessments into perspective.
2.2 Hazard assessment2.2.1 Toxicological profileA toxicological profile was prepared for each of the selected chemicals using, primarily, reviews produced by authoritative organisations (see Section 2.2.2). In many cases reliance on such sources for a complete dataset was not possible, so full literature searches were conducted (as described in Annex 18).
The aim of the literature review was to produce a toxicological profile sufficient to highlight the key toxicological endpoints of concern for each substance; the intention was not to produce a fully comprehensive review of all the available literature. Indeed, while it is recognised that a considerable amount of data on these chemicals may be held by regulatory authorities, much of this is unpublished and, potentially, considered commercially sensitive, and therefore is not publically available for use in reviews or assessments such as this.
Where available, recent published data summarised by the WHO/FAO Joint Expert Committee on Food Additives (JECFA) and the EU European Medicines Agency (EMEA) Committee on Veterinary
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
9

Medicinal Products (CVMP) were used in preference to data from other sources; WHO/FAO Joint Meeting on Pesticide Residues (JMPR) reports were also used. However, in general, original articles cited in such expert committee reviews were not retrieved and reviewed. It should be noted that the EMEA CVMP reports are, unfortunately, not referenced; thus the provenance of original studies cannot be identified from these summaries and, furthermore, the WHO/FAO reports may include information from unpublished sources.
The toxicological profiles for each selected chemical are presented in Annexes 1–17.
2.2.2 Databanks, review sources and toxicity searchesThe ToxNet, InChem, JECFA, CVMP and Agency for Toxic Substances and Disease Registry (ATSDR) web sites were either searched or browsed in order to identify databank records or reviews for each of the 17 selected chemicals. Results from these searches are given in Table 2.4. Where data sources identified as described above were not adequate to provide a suitable database for the present exercise, additional searches were undertaken. Additional toxicity search strategies are outlined in Annex 18.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
10

Table 2.4 Databank records and major review articles identified1
Chemical Name(CAS No)
TOXNET EMEA InChem
JECFA ATSDR
ChemID HSDB
IRIS
GeneTox
CCRIS
Chloramphenicol56-75-7
x x
Clenbuterol (Clembuterol)37148-27-9(hydrochloride)21898-19-1
x x x x x x
Dimetridazole551-92-8
x x x x
Enrofloxacin93106-60-6
x x
x x
17β-Estradiol50-28-2
x x x x
Flavomycin(Flavophospholipol;Bambermycins)11015-37-5
x x x x x x x x
Furazodilone67-45-8
x x
Lasalocid sodium25999-20-6
x x x x x x x x
Malachite green569-64-2
x x x x x x
Nalidixic acid389-08-2
x x x x x
Nandrolone434-22-0
x x x x x x
Narasin55134-13-9
x x x x x x x x
Phenylbutazone50-33-9
x x x x
Progesterone57-83-0
x x x
Salbutamol(Albuterol)18559-94-9
x x x x x x x x
Streptomycin57-92-1
x x x
Testosterone58-22-0
x x x
1The number of ticks indicates the number of records
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
11

3 EvaluationsTheoretical worst-case dietary intake values, for each of the 17 selected chemicals, determined from worst-case estimates of concentrations in meat products and conservative estimates of consumption patterns for such food products, as described above, are presented in Tables 3.1–3.3 at the end of this section. Theoretical intake values based on GEMS/Food data for Europe are presented in Table 3.1. Theoretical intake values for adults and toddlers based on the NDNS datasets are presented in Tables 3.2 and 3.3, respectively. By comparing these values with the ADI (or IOC) for each chemical and taking into account the nature of any potential hazard, a summary risk evaluation was made for each of the 17 selected compounds, as described below.
3.1 ChloramphenicolThe use of the antibiotic chloramphenicol in food-producing animals is prohibited. No ADI and therefore no MRLs have been set for this substance owing to the lack of a No-Observed-Adverse-Effect Level (NOAEL) for the induction of aplastic anaemia in experimental animals and humans and also concerns about potential genotoxicity and the lack of adequate data for carcinogenicity and fetotoxicity. All residues in foodstuffs should therefore, be treated as unacceptable. A minimum required performance limit (MRPL) of 0.3μg/kg has been established for residue analysis (EC, 2003; see Annex 1).
Use of GEMS/Food dietary intake data for Europe (WHO, 2003) gave a theoretical mean chloramphenicol intake of approximately 41 g/kg bw/day (Table 3.1). Using the NDNS dataset (OPCS & MRC Research Council Dunn Nutrition Unit, 1995; ONS & MRC Human Nutrition Research, 2002), the theoretical median chloramphenicol intakes for adults (Table 3.2) and toddlers (Table 3.3) were calculated to be approximately 37 and 76 g/kg bw/day, respectively. Based on 97.5th percentile consumption data from the NDNS dataset, theoretical chloramphenicol intakes would be 58 and 185 g/kg bw/day for adults (Table 3.2) and toddlers (Table 3.3), respectively.
Oral bioavailability of this substance is high, and rates of clearance in humans vary with age and renal function (see Annex 1). Therefore, for the purpose of this assessment, it has been assumed that most of any ingested dose of chloramphenicol, should it occur, would be systemically available, and that the very young and individuals with renal insufficiencies might be more susceptible to its effects. It should be noted that although a NOAEL for aplastic anaemia is not available, this is partly a reflection of the use of relatively high doses in experimental animal studies and in human clinical practice. Clinical experience, from administration of relatively high doses (50–100 mg/kg bw/day) of chloramphenicol to humans of all ages, has demonstrated that this disorder only occurs rarely (1 in 10 000 to 1 in several 100 000 up to a year after completion of a course of 8-80 g total intake). Grey Baby Syndrome is also associated with doses in excess of 25 mg/kg bw/day. However, it is recommended that patients are closely monitored and repeated courses of treatment are avoided. None of the experimental animal studies tested employed dosages as low as the theoretical worst-case dietary intakes calculated above. Thus, while there is a need to eliminate residues of chloramphenicol from foodstuffs, even the worst-case exposures estimated in this exercise are approximately 1000 times less than the therapeutic doses used in human medicine; such exposure would not, therefore, be expected to pose a significant risk to the UK consumer and, in practice, the chance of exposure of the population to appreciable quantities of this compound through the routes considered herein would appear remote.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
12

3.2 ClenbuterolClenbuterol is authorised for use in cattle and horses for a limited range of conditions. The EMEA CVMP has established a toxicologically-based ADI of 0.15 μg/kg bw/day and a pharmacologically-based ADI of 0.0042 μg/kg bw/day. The lower, pharmacologically-based, value derived from data from humans was used in this assessment.
Using the GEMS/Food data, the theoretical intake for clenbuterol from beef-based foods (0.0034 μg/kg bw/day) was approximately 81 % of the ADI (Table 3.1). Using the NDNS dataset, the theoretical median intakes were approximately 87 % (0.0037 μg/kg bw/day) and 186 % (0.0078 μg/kg bw/day) of the ADI for adults and toddlers respectively, and the 97.5 th percentile clenbuterol intakes were approximately 109 % (0.0046 μg/kg bw/day) and 402 % (0.0169 μg/kg bw/day) of the ADI for adults and toddlers respectively (see Tables 3.2 and 3.3).
Thus, even using the highly conservative scenarios based on consumers with mean or median diets calculated clenbuterol intakes for adults, while in some instances approaching the level of the ADI, do not exceed this value. A marginal exceedance of the ADI was only found, in adults, in the case of a 97.5th % consumer (109% ADI). However, calculated intakes in toddlers exceed that ADI by a considerable margin even for those on a mean/median diet, and given its biological activity, exposure of toddlers to such levels would be of concern. As recognised by the VRC (VRC, 2001; VRC, 2002) there exists a potential for misuse of this and other β-agonists at high dosages as growth promoters. However, it must be stressed that the theoretical maximum intakes for toddlers calculated here assume that all beef-derived foodstuffs the child consumes are contaminated at the maximum calculated residues of clenbuterol. Reassurance that this would not occur in practice is given by the fact that β-agonist contamination has not been identified in food samples analysed under the UK National Surveillance scheme or the Non-Statutory Surveillance scheme during the period 2001-2003 (VRC, 2001; 2002a; 2003).
3.3 DimetridazoleDimetridazole is not authorised for use in food-producing animals. No ADI or MRLs have been set for this substance. The data for genotoxicity and carcinogenicity were considered inadequate by the EMEA CVMP, which concluded that genotoxic activity could not be excluded for this substance. JECFA was unable to recommend an ADI owing to the lack of data, but did note the NOAEL of 4 mg/kg bw/day for a rat carcinogenicity study (see Annex 3).
In the light of the limited level of concern regarding the potential genotoxicity of dimetridazole, it was considered appropriate to derive a project-specific ‘Intake of Concern’ (IOC) of 40μg/kg bw/day (based upon a low-fold uncertainity factor of 100) to use as a comparative value for the purpose of this exercise only. Use of GEMS/Food dietary intake data gave a calculated theoretical intake of approximately 48 μg/kg bw/day or 121 % of the IOC (Table 1). When the NDNS dataset was used, the calculated theoretical intake, based upon median consumption values, were approximately 32 and 56 μg/kg bw/day for adults and toddlers, respectively; the 97.5th percentable intakes were approximately 78 and 199 μg/kg bw/day for adults and toddlers, respectively (Tables 3.2 & 3.3). Therefore, with the exception of the median consumer values for the NDNS dataset, all theoretical exposures exceeded the project specific IOC of 40 μg/kg bw/day with up to a 5-fold exceedance for the worst-case scenarios in toddlers receiving a 97.5th percentile diet
Irrespective of the above discussion, there would be concern regarding human health if dimetridazole were to enter human diets through unauthorised use in food-producing animals, because it is not possible to discount the possibility that it could be a non-threshold genotoxin. Reassurance is provided since dimetridazole has not been detected in samples analysed under the UK National Surveillance scheme. However, it has been detected in 6/40 samples of UK quail eggs at concentrations of 3-41 g/kg (6800 g/kg used in exposure calculation), during the Non-Statutory
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
13

Surveillance Scheme sampling of 2002 (VRC, 2001; VRC, 2002a; VRC, 2003). Following an investigation of the mill concerned, it was concluded that the contamination could have arisen from cross-contamination of feed, rather than as a result of deliberate unauthorised use of dimetridazole (VRC, 2002a).
3.4 EnrofloxacinEnrofloxacin is an antibiotic authorised for use in food-producing animals. However, it is not authorised for use in hens from which eggs are intended to be produced for human consumption. The toxicological ADI recommended by the EMEA CVMP is 30 g/kg bw/day, based upon a NOEL of 3 mg/kg bw/day for arthropathy in dogs. However, a lower microbiologically-based value of 6.2 g/kg bw/day has also been established on which a range of MRLs have been set for tissues of several species including pigs, cattle and poultry. A lower microbiologically-based ADI of 0-2 μg/kg bw/day was, however, proposed by JECFA, and it is this low microbiologically-based ADI of 2 μg/kg bw/day that has been used in the assessment (see Annex 4).
Theoretical daily enrofloxacin intakes calculated using European food survey data from GEMS/Food (WHO, 2003) and experimentally derived residue concentrations accounted for approximately 258 % of the ADI (approximately 5.17 g/kg bw/day; see Table 3.1). Similarly, use of the NDNS dataset (OPCS & MRC Research Council Dunn Nutrition Unit, 1995; ONS & MRC Human Nutrition Research, 2002) gave theoretical median enrofloxacin intakes of approximately 289 % of the ADI (5.78 g/kg bw/day) for adults, and approximately 482 % of ADI (9.63 g/kg bw/day) for toddlers. When the theoretical 97.5th percentile residue intakes were considered, marked exceedence of the ADI was noted for both adults (550 %) and toddlers (1745 %; see Tables 3.2 & 3.3). Although representing a potential risk to health, particularly in the case of toddlers, it should be noted that, were the toxicologically-based ADI to be considered, the only exceedence of ADI would be for the theoretical 97.5th percentile residue intake of toddlers, and then only by a very small amount (16 %).
In interpreting these findings, it must be stressed that the calculated theoretical intakes represent an absolute worst-case and assume that all of the meat and eggs consumed would be contaminated with the maximum calculated residues of enrofloxacin, and that the experimentally-derived tissue concentrations used to calculate daily intakes were approximately 10 times the MRL established by the EMEA CVMP; such a situation is highly unlikely to occur in practice. A high degree of reassurance that this situation does not apply within the UK is provided by the finding that only one sample of poultry analysed either under the UK National Surveillance scheme or the Non-Statutory Surveillance scheme during the period 2001-2003 (VRC, 2001; VRC, 2002; VRC, 2003) was found to be contaminated with enrofloxacin. It should also be noted that a significant proportion of the estimated intakes (approximately 40-50 %) is attributable to eggs. These should not constitute a source when enrofloxacin is administered according to its authorised uses, and no evidence of such contamination of eggs has been noted in the UK surveillance schemes.
3.5 17-Estradiol17-Estradiol is prohibited from use as a growth promoter in farm animals and can only be used in food-producing animals as a veterinary treatment under strict veterinary control. Given these restrictions, the EMEA CVMP has not established an ADI or set MRLs. However, JECFA has established an ADI of 0-50 ng/kg bw/day on the basis of changes in several hormone-dependent parameters in a study of healthy post-menopausal women, for which an NOEL of 5 g/kg bw/day was determined (see Annex 5). The ADI recommended by JECFA has, therefore, been employed for this assessment.
Using the GEMS/Food data, the theoretical 17-estradiol intake (0.02 g/kg bw/day) was approximately 43 % of the ADI (Table 3.1). Using the NDNS dataset the theoretical median 17-estradiol intakes were approximately 46 % (0.02 g/kg bw/day) and 97 % (0.06 g/kg bw/day) of the
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
14

ADI for adults and toddlers, respectively, and the 97.5 th percentile 17-estradiol intakes were approximately 57 % (0.03g/kg bw/day) and 211 % (0.11 g/kg bw/day) of the ADI for adults and toddlers, respectively (see Tables 3.2 & 3.3). There are a number of reasons why the apparent high fraction, or exceedence, of the ADI value should not raise particular concern for consumer health. Firstly, tissue concentrations of 17-estradiol in cattle following treatment with estradiol in an intravaginal device were calculated using assumptions of 100 % bioavailability and zero excretion. These assumptions are likely to be significant over- and under-estimates, respectively; thus the calculated tissue concentration applied in this assessment is unrealistically large and an extreme worst-case. Secondly, the intake has been calculated based on the assumption that all of the beef/beef products eaten by consumers are contaminated by 17-estradiol from unauthorised use. In reality, unauthorised use is likely to be sporadic and would not affect all sources of beef. Finally, it should be noted that no allowance has been made in these calculations for the oral bioavailability of the residues in humans. However, oral bioavailability of 17-estradiol in humans is known to be poor owing to hepatic first pass metabolism. Therefore, only a small fraction of any ingested estradiol would be expected to become systemically available. Given these considerations, it is concluded that there is no appreciable risk to consumers. However, should values for measured concentrations in meat from cattle become available, it would be appropriate to revisit this assessment using more realistic exposure scenarios. There is further reassurance in the fact that 17-estradiol has not been detected in samples analysed under the UK National Surveillance scheme or the Non-Statutory Surveillance Scheme during the period 2001-2003 (VRC, 2001; VRC, 2002a; VRC, 2003).
3.6 FlavomycinFlavomycin (also known as flavophospholipol or bambermycins; Annex 6), while not a veteninary medicine per se, was used as an antimicrobial growth promoter in feed. However, in Europe its use in cattle, pigs and poultry was licensed only until December 2005, after which its use in food-producing animals has been prohibited. Therefore, there is currently no valid European-derived value suitable for consideration in the assessment of risk. It is however noted that the Australian government has published an ADI of 0.3 μg/kg/day (Australian Government, 2005).
Owing to its heteropolar behaviour, flavomycin tends to form complexes, so absorption from the gut is virtually zero, and no appreciable tissue residues would be expected to occur in exposed animals. The absence of residues and the low systemic availability of flavomycin mean that no attempt to estimate ‘worst case’ dietary exposures of the UK population was made, and there is considered to be negligible risk to consumer health effects following consumption of meat or meat-derived products.
It should be noted that the published database on the toxicity of flavomycin is sparse and clearly inadequate for a detailed risk evaluation. Use of the in silico predictive model DERECK suggests it is plausible that flavomycin has genotoxic properties. It may, therefore, be appropriate to consider additional data gathering on the toxicodynamic properties of this substance.
3.7 FurazolidoneFurazolidone is an antibiotic that is no longer authorised for use in veterinary medicine and, therefore, in food-producing animals. ADI and MRL values have not been recommended as the parent compound and its main metabolite, 3-amino-oxazolidone-2, are genotoxic carcinogens (see Annex 7). A minium required performance limit (MRLP) of 1 μg/kg analysticl standard has been established for residue analysis (EC, 2005; see Annex 7).
Using GEMS/Food European dietary survey data (WHO, 2003), the theoretical total intake of furazolidone was 83 µg/kg bw/day. Using the NDNS dataset (OPCS & MRC Research Council Dunn Nutrition Unit, 1995; ONS & MRC Human Nutrition Research, 2002), the theoretical median intakes for adults and toddlers were approximately 99 and 138 µg/kg bw/day, respectively, and the 97.5th percentile values were approximately 166 and 359 µg/kg bw/day, respectively (Tables 3.1, 3.2 & 3.3).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
15

For comparison purposes, only, these worst-case intakes could be considered in relation to the pre-2003 Australian/New Zealand ADI of 0.4 µg/kg bw/day, which is no longer recognised by the regulatory authorities in Australia and New Zealand. In all cases, the calculated exposures exceed this ‘nominal’ ADI by significant amounts. Furthermore, since it is assumed that there is no threshold for the carcinogenic properties of furazolidone, even minimal exposure to this substance would be unacceptable. Therefore, exposure to the calculated theoretical worst-case intakes, above, would pose an appreciable risk to consumer health. However, such intakes are likely to be highly unrealistical owing to the extremely conservative calculations used in deriving them. Residues of the marker metabolite 3-amino-oxazolidone-2 (AOZ) have been detected in a small number of samples of imported chicken, honey and warm water prawns in 2002 or 2003 under the UK National Surveillance Scheme and the Non-Statutory Surveillance Scheme during the period 2001-2004 (VRC, 2001; VRC, 2002a; VRC, 2003 VRC, 2004). One incident of AOZ contamination was also identified in UK-bred chicken tissue in 2004, which was subsequently found to be due to contamination of old water tanks in a poultry house with furazolidone, arising from historic (then legal) use of the substance (VRC, 2004). Such isolated instances of contamination are unlikely to represent a significant risk to public health. Hwever, this underlines the need for continued monitoring for this contaminant. It should however, be noted that furazolidone is not the only potential source of AOZ residues, since AOX may also arise from other sources (EFSA, 2005).
3.8 Lasalocid sodiumWhile lasalocid sodium is approved for use as a poultry coccidiostat and an ADI and MRLs have been set for poultry by the EMEA, its use in laying hens is prohibited. It is authorised for use as a growth promoter for cattle in some countries, such as the USA. EMEA has proposed an ADI of 2.5 g/kg bw/day for lasalocid sodium. The EFSA identified a possible ADI of 5 μg/kg bw/day based on a 2-year chronic oral toxicity study in rats and maternal toxicity in pregnant rabbits but didn’t recommend this because of a number of other concerns (see Annex 8).. A lower ADI of 1 µg/kg bw/day, based on a NOEL of 2 µg/kg bw/day, has been established by the Australian Government (Australian Government, 2005), However, the EMEA ADI values has been used in this assessment.
Using GEMS/Food dietary survey data for a standard European diet (WHO, 2003), the theoretical total intake of lasalocid sodium was approximately 13 g/kg bw/day or 512 % of the ADI (Table 3.1). Using the NDNS dataset (OPCS & MRC Research Council Dunn Nutrition Unit, 1995; ONS & MRC Human Nutrition Research, 2002), the theoretical median intakes for adults and toddlers were approximately 1094 % and 1443 % of the ADI (27 and 36 g/kg bw/day), respectively, and the theoretical 97.5th percentile values were approximately 1912 % and 4053 % of the ADI (48 and 101 g/kg bw/day), respectively (Tables 3.2 & 3.3).
While the calculated intake values are based on worst-case contamination of dietary constituents, given that they are substantially greater than the ADI and up to approximately 20 % of the NOAEL in experimental animals (500 g/kg bw/day), there would be an obvious erosion of the margin of safety with these exposures. However, the potential significance to consumer health is unclear, particularly for the majority of the population whose poultry, beef and egg intake is very unlikely to contain such worst-case residue concentrations. It is also noted that the calculated tissue concentrations are far in excess of the MRLs for poultry.
3.9 Malachite greenNo ADI or MRLs exist for malachite green, as its use in farmed fish is prohibited because of mutagenic and potential carcinogenic properties. However, malachite green is recognised as a highly effective treatment against infections in fish and is inexpensive. Therefore there is some reason to suspect that unauthorised use in fish farms may occur on occasions (see Annex 9). A minimum required performance limit (MRLP) of 2 g/kg has been established for residue analysis (EC, 2004).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
16

In 2003 as part of the UK National Surveillance Scheme (VRC, 2003), malachite green was detected at tissue concentrations of 5 and 8 g/kg in 2/84 samples from salmon and trout farms. The VRC (2002a) also reported that malachite green was detected at a concentration of 12 µg/kg in 1/67 samples of trout analysed during 2002. The total theoretical intake of malachite green when GEMS food survey data were used was 0.06 g/kg bw/day (Table 3.1). When the NDNS dataset was used, the theoretical median intakes were 0.04 and 0.07 g/kg bw/day for adults and toddlers, respectively, and, for the 97.5th percentile, intakes were 0.13 and 0.24 g/kg bw/day for adults and toddlers, respectively (Tables 3.2 & 3.3).
The toxicological properties of malachite green mean that it poses a potentially serious risk to the health of consumers who eat contaminated salmon and trout. This is reflected in the regulatory position that no residue should be tolerated. Indeed, Defra takes the use of malachite green extremely seriously. For example, following the detection of malachite green in the fish samples in 2003, fish movement restrictions were placed on the farms involved and Defra conducted investigations into its illegal use (VRC, 2003).
3.10 Nalidixic acidNalidixic acid is used in human medicine to treat kidney infections, and it requires high doses in order to reach therapeutic concentrations in the kidney. Even at the high doses of nalidixic acid employed, therapeutically active concentrations are not attained in any other tissue; hence, its clinical usefulness is restricted to renal infection. There are no licensed veterinary medicines that contain nalidixic acid (see Annex 10), and no ADI and, therefore, no MRLs have been established.
It is not anticipated that unauthorised veterinary use of medicines containing nalidixic acid should occur as there are no recommended dose rates for animals, and use of formulations intended for human medicine would be expected to be prohibitively expensive. It is important to note that in the extremely unlikely event of consumer exposure via meat products, tissue concentrations would be unlikely to reach levels at which effects would be observed. It is not anticipated that unauthorised use of nalidixic acid will occur and therefore exposure calculations were not performed for this substance.
3.11 NandroloneNo ADI has been recommended for nandrolone and there are no licensed products for use in food-producing animals, so MRLs have not been established (see Annex 11).
Using the GEMS/Food dietary survey data for a standard European diet (WHO, 2003), the theoretical nandrolone intakes were 0.26 g/kg bw/day (Table 3.1). Using the NDNS dataset (OPCS & MRC Research Council Dunn Nutrition Unit, 1995; ONS & MRC Human Nutrition Research, 2002), the theoretical median intakes of nandrolone were 0.25 g/kg bw/day and 0.47 g/kg bw/day for adults and toddlers, respectively. Using the 97.5th percentile dietary survey data, the intakes were 0.44 g/kg bw/day and 1.30 g/kg bw/day for adults and toddlers, respectively (Tables 3.2 & 3.3). It should be noted that all of these calculated intakes are significantly below (approximately 100-500 fold) the lowest Lowest-Observed-Adverse-Effect Level (LOAEL; 140 g/kg bw/day) observed in animal studies, in which nandrolone was administered subcutaneously and effects on craniofacial growth and behaviour (higher doses only) were observed. In addition, nandrolone is known to have poor oral bioavailability so it is expected that only a small proportion of any dietary intake would be systemically available, which further reduces the probability that effects on consumer health could occur.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
17

3.12 NarasinAn ADI for the antibiotic narasin of 5 g/kg bw/day and an MRL of 50 g/kg for all tissues have been established by the EFSA (see Annex 12).
Using food intake data produced by GEMS/Food (WHO, 2003) to calculate possible daily intake of narasin from contaminated meat, the theoretical mean daily intake of narasin would be only approximately 13 % of the ADI (0.65 g/kg bw/day; Table 3.1). Similarly, the theoretical median intakes of narasin for adults and toddlers, calculated using UK dietary survey results (OPCS & MRC Research Council Dunn Nutrition Unit, 1995; ONS & MRC Human Nutrition Research, 2002) , were 0.74 and 1.01 g/kg bw/day, respectively; that is, 20 % or less of the ADI. The 97.5th percentile intakes of narasin were calculated as 1.40 and 2.92 g/kg bw/day, or 28 % and 58 % of the ADI for adults and toddlers, respectively (Tables 3.2 & 3.3). The latter finding indicates the potential for an appreciable exposure among children. However, the calculation assumes that all meat products consumed by an individual would contain narasin at a constant high (worst-case) concentration. This is not expected to occur in reality and, therefore, the intakes used in this assessment represent the absolute worst-case scenario. On this basis, it is not anticipated that consumption of meat and meat products would result in ingestion of narasin to the extent that adverse consumer health effects would result. Narasin has not been detected in samples analysed under the UK National Surveillance scheme during 2001-2003 or the Non-Statutory Surveillance Scheme during the period 1998-2003 (VRC, 2001; VRC, 2002a; VRC, 2002b; VRC, 2002c; VRC, 2003).
3.13 PhenylbutazonePhenylbutazone is a non-steroidal anti-inflammatory drug, which is prohibited for use in food-producing animals owing to its potential to cause blood dyscrasias and its possible carcinogenicity in humans. Due to the nature of the effects of phenylbutazone, no ADI has been established or MRL values set (see Annex 13).
Illegal and extra-label use of phenylbutazone in cattle may occasionally occur. Therefore, tissue concentrations for cattle were calculated using the realistic, therapeutically relevant oral doses that would be expected to be employed and worst-case assumptions for absorption and excretion kinetics. The derived tissue residue concentration was combined with dietary survey data for beef to calculate worst-case phenylbutazone intakes for adults and toddlers (Tables 3.1, 3.2 & 3.3). Using the GEMS/Food dietary survey results (WHO, 2003), the worst-case theoretical phenylbutazone intake was 85 g/kg bw/day. Using the NDNS dataset (OPCS & MRC Research Council Dunn Nutrition Unit, 1995; ONS & MRC Human Nutrition Research, 2002) gave theoretical median phenylbutazone intakes of 91 and 195 g/kg bw/day for adults and toddlers, respectively. Calculations using the 97.5 th
percentile Food Standards Agency dietary data gave phenylbutazone intakes of 115 and 422 g/kg bw/day for adults and toddlers, respectively. Since there is no NOAEL for the effects of phenylbutazone on blood, all of the above intakes would be considered unacceptable and a potential risk to consumer health.
3.14 ProgesteroneProgesterone is a steroid hormone, which is licensed in the UK to control oestrus and improve synchronisation of oestrus in cows. As progesterone is an endogenous hormone in mammals, it must be regarded as a natural constituent of food of animal origin. The EMEA has not recommended an ADI or set MRLs for progesterone. For the purposes of this assessment, the ADI (30 g/kg bw/day) recommended by JECFA was used. This was based on a Lowest-Observed-Effect Level (LOEL) of 3.3 mg/kg bw, which led to concentrations in blood similar to those found during the luteal phase of the human ovulatory cycle (see Annex 14).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
18

The most likely source of exposure to progesterone is following veterinary use in beef cattle. Therefore, in the absence of measured values, animal tissue concentrations were calculated using realistic veterinary doses and worst-case assumptions for absorption and excretion kinetics. None of the derived theoretical worst-case intake values exceeded 1% of the ADI; therefore, they were not considered to represent any risk to consumer health.
3.15 SalbutamolThere are no licensed veterinary products that contain salbutamol in the EU. Because of its β2-agonistic activity, salbutamol is prohibited for use as a growth-promoting agent, and there are no recommended ADI or MRL values (see Annex 15). The data for oral toxicity are extremely limited for salbutamol. However, for the purposes of this risk assessment, a NOAEL of 17 mg/kg bw/day for cardiotoxicity from a one-month oral study in rats was used. To allow an assessment of the calculated salbutamol intakes, a project-specific IOC of 17 g/kg bw/day was derived by dividing the NOAEL by a total uncertainty factor of 1000 (to allow for intra- and inter-species variation and the limited nature of the toxicity database for salbutamol).
Using the GEMS/Food dietary survey results for Europe (WHO, 2003) gave theoretical worst-case salbutamol intakes of 1.87 g/kg bw/day (Table 3.1). Using the NDNS dataset (OPCS & MRC Research Council Dunn Nutrition Unit, 1995; ONS & MRC Human Nutrition Research, 2002) gave theoretical median salbutamol intakes of 2.03 and 4.31 g/kg bw/day for adults and toddlers, respectively. Calculations using the 97.5th percentile NDNS data gave salbutamol intakes of 2.55 and 9.33 g/kg bw/day for adults and toddlers, respectively (Tables 3.2 & 3.3). These intake values are less than 20 % of the project-specific IOC, except for toddlers, where intakes of up to 55% have been calculated, suggesting the potential for an appreciable exposure in this subpopulation. However, considering the extremely conservative calculation for tissue concentrations in cattle and the clearly unrealistic assumption that all consumed beef and poultry are contaminated with salbutamol, this is not considered of particular concern. Indeed, further reassurance is provided by the results of surveillance programmes in the UK. The only recent cases of detection of salbutamol residues related to low levels detected in a small number of broiler turkey and cattle samples. Further investigation however found this to be attributed to use of medication containing salbutamol prior to sample collection by the collecting officer rather than through illegal use in animals (VRC, 2003a).
3.16 StreptomycinThe toxicological ADI for the antibiotic streptomycin (see Annex 16) is 25 g/kg bw/day, and MRLs have been derived using this ADI value.
Streptomycin was detected in imported honey during 2002 (VRC, 2003). The highest concentration found, 180 µg/kg, was used in the worst-case risk scenario as the basis for this potential source of dietary exposure. Based on GEMS/Food data (WHO, 2003), the theoretical intake of streptomycin was approximately 3.28 g/kg bw/day (13 % of the ADI; Table 3.1). Based on NDNS data (OPCS & MRC Research Council Dunn Nutrition Unit, 1995; ONS & MRC Human Nutrition Research, 2002), adult and toddler theoretical median intakes are 2.57 (10% of ADI) and 5.26 (21% of ADI) g/kg bw/day. The 97.5th percentile intake for adults is 7.38 g/kg bw/day or 30 % of the ADI and for toddlers is 25 μg/kg bw/day or 103 % of the ADI (Tables 3.2 & 3.3). The intake data assume that all meat products consumed by an individual contain streptomycin at a constant concentration, but in reality this is very unlikely, and streptomycin is poorly absorbed when ingested. Therefore, it is highly unlikely that the calculated intakes will be systemically available, and the intakes calculated represent an absolute worst-case scenario. Overall, it is not anticipated that inappropriate veterinary use of streptomycin would pose a significant risk to the health of consumers of animal products in the UK.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
19

3.17 TestosteroneTestosterone is an androgenic steroid hormone; it is prohibited for use in farm animals and is also prohibited for use as a fattening agent. Owing to the restrictions in place, the EMEA CVMP has not recommended an ADI or set MRLs for testosterone. JECFA has recommended an ADI of 0-2 g/kg bw/day for testosterone, based on a study in human eunuchs. The JECFA ADI has been used for the following assessment (see Annex 17).
Using the GEMS/Food data (WHO, 2003), the theoretical testosterone intake was approximately 7 % of the ADI (0.14 g/kg bw/day; Table 3.1). Using the NDNS data, which is relevant to the diet in Great Britain (OPCS & MRC Research Council Dunn Nutrition Unit, 1995; ONS & MRC Human Nutrition Research, 2002), the theoretical median testosterone intakes were approximately 0.15 and 0.32 g/kg bw/day or 7 % and 16 % of the ADI for adults and toddlers, respectively. The 97.5 th
percentile testosterone intakes were approximately 0.19 and 0.69 g/kg bw/day or 9% and 34% of the ADI for adults and toddlers, respectively (Tables 3.2 & 3.3). The bioavailability of oral testosterone has not been reported but it is generally considered to be negligible, and any that is absorbed would be expected to be cleared rapidly (see Annex 17). Since the calculated worst-case intakes of testosterone from beef/beef products were no more than 34 % of the ADI, and only a small proportion of this intake value would in any case be expected to be absorbed, there is no apparent health concern for consumers of beef that contains testosterone up to the concentrations used in this assessment.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
20

Table 3.1 Theoretical intake values resulting from worst-case non-authorised veterinary use of chemicals, based on GEMS/Food data for EuropeVeterniary Medicine (VM)
ADI (µg/kg bw/day)
Animal/product Estimated Product Concentration (µg/kg)
Estimated Product Intake (kg/day) GEMS
VM intake (µg/kg bw/day)
% ADI
Chloramphenicol No ADI Cattle (beef) 25000 0.06 26.63 Pigs (pork/bacon) 11000 0.08 13.90Prawns 0.9 0.00 0.00Honey 1.4 0.00 0.00
Clenbuterol 0.0042 Cattle (beef) 3.2 0.06 0.0034 81.142
Dimetridazole
Project specific IOC Chicken 1600 0.06 1.56 3.8940 Eggs 6800 0.04 4.25 10.63
Pigs (pork/bacon) 33600 0.08 42.45 106.1248.26 120.641
Enrofloxacin 2 Cattle (beef) 700 0.06 0.75 37.28(microbial) Beef kidney/liver 1300 0.01 0.13 6.50
Chicken 2050 0.06 2.00 99.77Chicken liver 5750 0.00 0.03 1.44Eggs 3630 0.04 2.27 113.44Pigs (pork/bacon) 0.079 0.08 0.00 0.00
5.17 258.421
17β-Estradiol 0.05 Cattle (beef) 20 0.06 0.02 42.602
Flavomycin (flavophospholipol; bambermycins)
300 - - - - -
Furazolidone No ADI Pigs (pork/bacon) 250 0.08 0.32 Pig kidney/liver 1800 0.01 0.15Calves (beef) 11900 0.06 12.67Poultry 71400 0.06 69.50
Lasalocid sodium 2.5 Chicken 9000 0.05 7.41 296.40Turkey 21000 0.01 2.91 116.20Cattle (beef) 310 0.06 0.33 13.21Eggs 3450 0.04 2.16 86.25
12.80 512.061
Malachite green No ADI Salmon 120 0.03 0.06 Trout 35 0.00 0.00
Nalidixic acid No ADINandrolone No ADI Cattle (beef) 100 0.06 0.11
Sheep 100 0.01 0.02Pigs (pork/bacon) 100 0.08 0.13
Narasin 5 Chicken 500 0.05 0.41 8.23Pigs (pork/bacon) 190 0.08 0.24 4.80
0.65 13.03Phenylbutazone No ADI Cattle (beef) 80000 0.06 85.20Progesterone 30 Cattle (beef) 1.5 0.06 0.00 0.01
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
21

Table 3.1 continued Veterniary Medicine (VM)
ADI (µg/kg bw/day)
Animal/product Estimated Product Concentration (µg/kg)
Estimated Product Intake (kg/day) GEMS
VM intake (µg/kg bw/day)
% ADI
Salbutamol
Project specific IOC Poultry 13 0.06 0.01 0.0717 Cattle (beef) 1760 0.06 1.87 11.03
1.89 11.10Streptomycin 25 Cattle (beef) 250 0.06 0.27 1.07
Beef kidney/liver 2800 0.01 0.28 1.12Sheep 200 0.01 0.03 0.14Sheep liver/kidney 938 0.00 0.02 0.08Pigs (pork/bacon) 2000 0.08 2.53 10.11Pig kidney/liver 1756 0.01 0.15 0.59Honey 380 0.00 0.01 0.03
3.28 13.13Testosterone 2 Cattle (beef) 130 0.06 0.14 6.92
1 Greater than 100% of ADI/IOC2 Greater than 20% of ADI/IOC
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
22

Table 3.2 Theoretical intake values for adults resulting from worst-case non-authorised veterinary use of chemicals, based on National Diet and Nutrition Surveys from Great BritainVeterinary Medicine (VM)
ADI (µg/kg bw/day)
Animal/Product
Estimated Product Concentration (µg/kg)
Estimated Product Intake (kg/day) 19-64 yrs median
VM intake (µg/kg bw/day) ; Median Consumer
% ADI Product Intake (kg/day) 97.5th %
VM intake (µg/kg bw/day): 97.5th % Consumer
% ADI
Chloramphenicol No ADI Cattle (beef) 25000 0.07 28.58 0.09 35.83 Pigs (pork/bacon) 11000 0.05 8.61 0.12 21.82Prawns 0.9 0.01 0.00 0.03 0.00Honey 1.4 0.00 0.00 0.02 0.00
Clenbuterol 0.0042 Cattle (beef) 3.2 0.07 0.00 87.112 0.09 0.00 109.211
Dimetridazole Project specific IOC
Chicken 1600 0.07 1.89 4.73 0.12 3.28 8.20
40 Eggs 6800 0.04 4.30 10.74 0.08 8.50 21.25Pigs 33600 0.05 26.29 65.73 0.12 66.64 166.60
32.48 81.202 78.42 196.051
Enrofloxacin 2 Cattle (beef) 700 0.07 0.80 40.02 0.09 1.00 50.17Beef kidney/liver 1300 0.01 0.27 13.33 0.06 1.26 62.83Chicken 2050 0.07 2.42 121.12 0.12 4.20 210.13Chicken liver 5750Eggs 3630 0.04 2.29 114.65 0.08 4.54 226.88Pigs (pork/bacon) 0.079 0.05 0.00 0.00 0.12 0.00 0.01
5.78 289.111 11.00 550.011
17β-Estradiol 0.05 Cattle (beef) 20 0.07 0.02 45.732 0.09 0.03 57.332
Flavomycin (flavophospholipol; bambermycins)
300 - - - - - - - -
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
23

Table 3.2 continuedVeterinary Medicine (VM)
ADI (µg/kg bw/day)
Animal/Product
Estimated Product Concentration (µg/kg)
Estimated Product Intake (kg/day) 19-64 yrs median
VM intake (µg/kg bw/day) ; Median Consumer
% ADI Product Intake (kg/day) 97.5th %
VM intake (µg/kg bw/day): 97.5th % Consumer
% ADI
Furazolidone No ADI Pigs (pork/bacon) 250 0.05 0.20 0.12 0.50 Pig kidney/liver 1800 0.01 0.37 0.06 1.74Calves (beef) 11900 0.07 13.61 0.09 17.06Poultry 71400 0.07 84.37 0.12 146.37
Lasalocid sodium 2.5 Chicken 9000Turkey 21000 0.07 24.82 992.60 0.12 43.05 1722.00Cattle (beef) 310 0.07 0.35 14.18 0.09 0.44 17.77Eggs 3450 0.04 2.18 87.17 0.08 4.31 172.50
27.35 1093.951 47.81 1912.271
Malachite green No ADI Salmon 120 0.02 0.04 0.07 0.13 Trout 35
Nalidixic acid No ADINandrolone No ADI Cattle (beef) 100 0.07 0.11 0.09 0.14
Sheep 100 0.04 0.06 0.06 0.10Pigs (pork/bacon) 100 0.05 0.08 0.12 0.20
Narasin 5 Chicken 500 0.07 0.59 11.82 0.12 1.03 20.50Pigs (pork/bacon) 190 0.05 0.15 2.97 0.12 0.38 7.54
0.74 14.79 1.40 28.042
Phenylbutazone No ADI Cattle (beef) 80000 0.07 91.47 0.09 114.67Progesterone 30 Cattle (beef) 1.5 0.07 0.00 0.01 0.09 0.00 0.01
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
24

Table 3.2 continuedVeterinary Medicine (VM)
ADI (µg/kg bw/day)
Animal/Product
Estimated Product Concentration (µg/kg)
Estimated Product Intake (kg/day) 19-64 yrs median
VM intake (µg/kg bw/day) ; Median Consumer
% ADI Product Intake (kg/day) 97.5th %
VM intake (µg/kg bw/day): 97.5th % Consumer
% ADI
Salbutamol Project specific IOC
Poultry 13 0.07 0.02 0.09 0.12 0.03 0.16
17 Cattle (beef) 1760 0.07 2.01 11.84 0.09 2.52 14.842.03 11.93 2.55 15.00
Streptomycin 25 Cattle (beef) 250 0.07 0.29 1.14 0.09 0.36 1.43Beef kidney/liver 2800 0.01 0.57 2.30 0.06 2.71 10.83Sheep 200 0.04 0.12 0.48 0.06 0.20 0.80Sheep liver/kidney 938Pigs (pork/bacon) 2000 0.05 1.57 6.26 0.12 3.97 15.87Pig kidney/liver 1756Honey 380 0.00 0.03 0.10 0.02 0.15 0.58
2.57 10.28 7.38 29.512
Testosterone 2 Beef 130 0.07 0.15 7.43 0.09 0.19 9.321 Greater than 100% of ADI/IOC2 Greater than 20% of ADI/IOC
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
25

Table 3.3 Theoretical intake values for toddlers resulting from worst-case non-authorised veterinary use of chemicals, based on National Diet and Nutrition Surveys from Great BritainVeterinary Medicine (VM)
ADI (µg/kg bw/day)
Animal/Product
Estimated Product Concentration (µg/kg)
Product Intake (kg/day) 1.5-2.5 yrs median
VM intake (µg/kg bw/day) : Median Consumer
% ADI Estimated Product Intake (kg/day) 97.5th %
VM intake (µg/kg bw/day) :97.5th % Consumer
% ADI
Chloramphenicol No ADI Cattle (beef) 25000 0.03 60.91 0.06 131.82 Pigs (pork/bacon) 11000 0.02 14.90 0.05 53.00Prawns 0.9 0.00 0.00 0.02 0.00Honey 1.4 0.00 0.00 0.01 0.00
Clenbuterol 0.0042 Cattle (beef) 3.2 0.03 0.0078 185.631 0.06 0.0169 401.731
Dimetridazole Project specific IOC
Chicken 1600 0.02 2.40 6.00 0.04 6.40 16.00
40 Eggs 6800 0.01 7.60 19.01 0.05 30.91 77.27Pigs 33600 0.01 45.51 113.78 0.05 161.89 404.73
55.52 138.791 199.20 498.001
Enrofloxacin 2 Cattle (beef) 700 0.03 1.71 85.27 0.06 3.69 184.55Beef kidney/liver 1300 0.01 0.79 39.59 0.06 6.50 325.00Chicken 2050 0.02 3.08 153.75 0.04 8.20 410.00Chicken liver 5750Eggs 3630 0.01 4.06 202.95 0.05 16.50 825.00Pigs (pork/bacon) 0.079 0.01 0.00 0.01 0.05 0.00 0.02
9.63 481.571 34.89 1744.561
17β-Estradiol 0.05 Cattle (beef) 20 0.03 0.05 97.4522 0.06 0.11 210.911Flavomycin (flavophospholipol; bambermycins)
300 - - - - - - - -
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
26

Table 3.3 continuedVeterinary Medicine (VM)
ADI (µg/kg bw/day)
Animal/Product
Estimated Product Concentration (µg/kg)
Product Intake (kg/day) 1.5-2.5 yrs median
VM intake (µg/kg bw/day) : Median Consumer
% ADI Estimated Product Intake (kg/day) 97.5th %
VM intake (µg/kg bw/day) :97.5th % Consumer
% ADI
Furazolidone No ADI Pigs (pork/bacon) 250 0.01 0.34 0.05 1.20 Pig kidney/liver 1800 0.01 1.10 0.06 9.00Calves (beef) 11900 0.03 28.99 0.06 62.75Poultry 71400 0.02 107.10 0.04 285.60
Lasalocid sodium 2.5 Chicken 9000 Turkey 21000 0.02 31.50 1260.00 0.04 84.00 3360.00Cattle (beef) 310 0.03 0.76 30.21 0.06 1.63 65.38Eggs 3450 0.01 3.86 154.31 0.05 15.68 627.27
36.11 1444.521 101.32 4052.651
Malachite green No ADI Salmon 120 0.01 0.07 0.02 0.24 Trout 35
Nalidixic acid No ADI No exposureNandrolone No ADI Cattle (beef) 100 0.03 0.24 0.06 0.53
Sheep 100 0.01 0.09 0.03 0.29Pigs (pork/bacon) 100 0.01 0.14 0.05 0.48
Narasin 5 Chicken 500 0.02 0.75 15.00 0.04 2.00 40.00Pigs (pork/bacon) 190 0.01 0.26 5.15 0.05 0.92 18.31
1.01 20.152 2.92 58.312
Phenylbutazone No ADI Cattle (beef) 80000 0.03 194.91 0.06 421.82Progesterone 30 Cattle (beef) 1.5 0.03 0.00 0.01 0.06 0.01 0.03
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
27

Table 3.3 continuedVeterinary Medicine (VM)
ADI (µg/kg bw/day)
Animal/Product
Estimated Product Concentration (µg/kg)
Product Intake (kg/day) 1.5-2.5 yrs median
VM intake (µg/kg bw/day) : Median Consumer
% ADI Estimated Product Intake (kg/day) 97.5th %
VM intake (µg/kg bw/day) :97.5th % Consumer
% ADI
Salbutamol Project specific IOC
Poultry 13 0.02 0.02 0.11 0.04 0.05 0.31
17 Cattle (beef) 1760 0.03 4.29 25.22 0.06 9.28 54.594.31 25.342 9.33 54.892
Streptomycin 25 Cattle (beef) 250 0.03 0.61 2.44 0.06 1.32 5.27Beef kidney/liver 2800 0.01 1.71 6.82 0.06 14.00 56.00Sheep 200 0.01 0.17 0.68 0.03 0.58 2.33Sheep liver/kidney 938Pigs (pork/bacon) 2000 0.01 2.71 10.84 0.05 9.64 38.55Pig kidney/liver 1756Honey 380 0.00 0.07 0.28 0.01 0.28 1.11
5.26 21.052 25.81 103.251
Testosterone 2 Beef 130 0.03 0.32 15.84 0.06 0.69 34.272
1 Greater than 100% of ADI/IOC2 Greater than 20% of ADI/IOCNB Honey consumption for toddlers is based on mean rather median value
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
28

4 Conclusions4.1 Discussion and conclusionsGiven the preliminary nature of this exercise and the limited datasets available for the 17 chemicals reviewed herein and uncertainties about their use, the animal tissue and foodstuffs residue concentrations employed throughout this risk assessment are based upon theoretical, worst-case values and scenarios. This has, of necessity, resulted in the derived estimates of potential human intake being unrealistically high. Factors that make a particularly important contribution to the overestimation are as follows.
Calculations of tissue concentrations in meat were often based on poor or incomplete toxicokinetic datasets, particularly with regard to the food-producing animals concerned.
For experimentally-derived tissue concentrations in meat, it was generally unclear whether the withdrawal period before sacrifice was realistic. In most cases it appeared that only a minimal period was used. Therefore, the tissue concentrations obtained are probably significant exaggerations of those that would be experienced in ‘real’ situations. Conversely, it must be accepted that where a substance is used in an unauthorised way, the recommended withdrawal periods might be ignored.
In the calculation of daily intake values from the dietary survey data, it was assumed that the total dietary intake of the potentially contaminated meats or meat products would contain residues at the worst-case concentration. However, people obtain their foodstuffs from a variety of sources, and the assumption that all foodstuff would contain these worst-case residue concentrations all of the time is clearly most unlikely.
It is emphasised that there is no evidence that the theoretical residue concentrations used to calculate the dietary intakes have ever been reached or indeed are ever likely to be reached, for any of the substances discussed in this evaluation. In addition, the food intake values derived from dietary surveys also represent worst-case scenarios. For these reasons, results of this exercise that might be seen as an apparent reason for concern, should be seen rather as triggers for further enquiry, and not as a trigger for considering the need for risk reduction measures at this stage.
The findings from the risk assessments are summarised in Table 4.1, and discussed in more detail below.
Based on the limited, conservative evaluation conducted, for 8 of the 17 selected chemicals —17-estradiol, flavomycin, naladixic acid, nandrolone, narasin, progesterone, streptomycin and testosterone — there was considered to be no potential health risk to consumers following non-authorised veterinary use in food-producing animals, although further consideration of the toxic properties of flavomycin may be appropriate.
Owing to the nature of their toxic properties (e.g. genotoxic carcinogens), a zero tolerance approach was taken for several of the chemicals evaluated; that is, any contamination of foodstuffs by such chemicals would constitute a theoretical risk to consumer health. The worst-case estimates of exposure via animal food products calculated in this evaluation would, therefore, be unacceptable if they were actually to occur. In reality, the actual impacts on human health and their significance are not known. The chemicals falling into this category were chloramphenicol, furazolidone, malachite green and phenylbutazone; the biological properties of clenbuterol also suggest that human exposure should be avoided. Chloramphenicol and malachite green have been detected in food samples in the UK but at much lower concentrations than have been estimated in this preliminary risk assessment exercise, while only one sample of poultry analysed had an elevated level of clenbuterol.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
29

Table 4.1 Summary of risk assessment of non-authorised veterinary useChemical name Calculated intake Overall assessment
(Comment)>20% ADI/
IOC>100% ADI/ IOC
Adult
Toddler
Adult Toddler
Chloramphenicol No exposure considered acceptable (No established ADI)
Clenbuterol Y Y* Y Potential concern were evidence of exposure identified
Dimetridazole Y Y Y Potential concern were evidence of exposure identified (Project specific IOC)
Enrofloxacin Y Y Potential concern where evidence of exposure identified
17-Estradiol Y Y Y* Not considered a significant risk to health of public
Flavomycin,(Flavophospholipol; Bambermycins)
No anticipated exposure; Not considered a significant risk to health of public
Furazolidone No exposure considered acceptable (No ADI)
Lasalocid sodium Y Y Calculated exposure would lead to erosion of margin of safety
Malachite green No exposure considered acceptable (No ADI)
Nalidixic acid Not considered a significant risk to health of public (No ADI)
Nandrolone Not considered a significant risk to health of public (No ADI)
Narasin Y* Y Not considered a significant risk to health of public
Phenylbutazone No exposure considered acceptable (No ADI)
Progesterone Not considered a significant risk to health of public
Salbutamol(Albuterol)
Y Not considered a significant risk to health of public (Project specific IOC)
Streptomycin Y* Y Y* Not considered a significant risk to health of public
Testosterone Y* Not considered a significant risk to health of public
ADI, acceptable daily intake; IOC, intake of concern; Y, exceedance at mean/median intake level, Y*, exceedance at worst case (97.5%) level only
Furazolidone and phenylbutazone have not themselves been detected in any food samples (VRC, 2001; VRC, 2002a; VRC, 2003). While a marker metabolite of furazolidone, AOZ, has occasionally been identified in a few samples of (mainly imported) foodstuffs, it is not firmly established if this represents cases of misuse of furazolidone or if it is due to contamination from other materials that can form AOZ on breakdown. However, overall, the findings suggest that there is a considerable degree of reassurance as to the scale of any health risk to the public.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
30

In the assessment for lasalocid sodium, based on a combination of calculated worst-case tissue concentrations and concentrations reported by VRC (2002a), the overall intake applied to the risk assessment was considered to represent a suitably extreme scenario to constitute a worst-case assessment. The worst-case intakes calculated for lasalocid sodium significantly exceeded the ADI proposed by the EFSA or the Australian government, and were between 2 and 18% (GEMS and 97.5 th
percentile for children, respectively) of the established experimental NOEL. Hence there is an erosion of the margin of safety for consumers. However, the impact of this is uncertain, as the toxicological dataset for lasalocid is limited.
No ADIs have been recommended for dimetridazole (owing to inadequacies in the toxicological dataset) and salbutamol (as there are no licensed veterinary products). Thus, for the purposes of this pilot evaluation, project-specific de novo IOCs, which were based on a conservative NOEL for each substance and an appropriate uncertainty factor, were derived as comparators. Comparison of calculated worst-case salbutamol intake values with the extremely conservative project-specific IOC derived for salbutamol only showed exceedence of 20% of the IOC for toddlers. However, in the light of the findings from the surveillance schemes, there was considered to be little cause for concern with regard to the potential impacts on consumer health following its unauthorised veterinary use. The calculated worst-case intake values for dimetridazole were up to five - times the project-specific IOC. Such high potential intakes, together with the uncertain genotoxicity of this substance, would raise concern if the calculated intakes were to be realised. However, to date, only one instance of contamination of quails eggs has been detected under the Non-Statutory Surveillance Scheme (VRC, 2001; VRC, 2002a; VRC, 2003); and this was attributed to cross-contamination rather than deliberate unauthorised use.
4.2 Recommendations In order to improve the accuracy of future risk assessments of this nature, it is recommended
that attention be given to obtaining improved measures of likely ‘real world’ residue concentrations (where there is, suggested or established, non-authorised veterinary use of a given chemical) and to obtaining improved toxicokinetic and toxicodynamic datasets for those chemicals identified as posing the greatest potential risk. Such data might be available from currently unpublished sources (e.g. industry data). Alternatively, the use of predictive models or ‘read-across’ between chemicals of similar structure or properties might provide suitable surrogate data.
Where, for some of the substances, available published data have not been sufficient to reach a secure conclusion regarding, in particular, carcinogenicity and/or genotoxicty, as is notably the case for phenylbutazone and flavomycin, it would be appropriate to seek further advice from the relevant government expert committees, such as the Committee on Carcinogenicity of Chemicals in Food, Consumer Products and the Environment and the Committee on Mutagenicity of Chemicals in Food, Consumer Products and the Environment
The approach used in making these evaluations in this pilot exercise may be useful in assisting the VRC and other surveillance schemes, through identifying chemicals of particular concern, to target appropriate and cost-effective sampling strategies for detecting non-authorised veterinary use of chemicals.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
31

5 References
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
32

Annexes
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
33

Annex 1 Chloramphenicol1 IntroductionChloramphenicol (2,2-dichloro-N-[2-hydroxy-1-(hydroxymethyl)-2-(4-nitrophenyl)ethyl] acetamide; CAS No. 56-75-7) is a broad-spectrum bacteriostatic antibacterial. It is active against rickettsial and Chlamydophila infections, the majority of obligate anaerobes, most Gram-positive aerobes, and non-enteric aerobes including Actinobacillus, Bordetella, Haemophilus, Pasteurella multocida, and Mannheimia haemolytica. Enterobacteriaceae, including Escherichia and Salmonella spp., are intrinsically susceptible but plasmid-mediated resistance is widespread (GENE-TOX, 1995; EMEA, 1996; JECFA, 2001).
2 Uses and exposureChloramphenicol is used in the treatment of human Salmonella typhi infection (typhoid) and as over the counter (OTC) eye drops formulations (Bishop, 2004). In veterinary medicine, the use of chloramphenicol is restricted to companion animals (dogs and cats) and horses and as such would be used for treating individual animals rather than a group. The only licensed product in the UK is for the treatment of bacterial eye infections with eye drops (Chloromycetin V RedidropsTM) containing 0.5% chloramphenicol. Human preparations containing chloramphenicol may also be prescribed for animal use under the ‘cascade’ system and include Chloramphenicol (non-proprietary) eye drops and eye ointment; Chloramphenicol capsules (non-proprietary) and KemicetineTM powder for reconstitution and injection (Bishop, 2004).
The drug is included in Annex IV of Regulation 2377/90/EEC which prohibits its use in food-producing animals and it is no longer available in any licensed products for use in these species in the UK and EU countries. However, prior to its inclusion in Annex IV, it was available as licensed veterinary preparations for use in cattle and pigs (ChloramphenicolTM) and cattle (IntramycetinTM, SensicolTM) in a form for oral administration or as an injectable broad spectrum antibiotic. The dose rates were 10–25 mg/kg bw 1–2 times daily for cattle and 11 mg/kg bw daily for pigs. In dogs, the dose rate was 50 mg/kg bw 1–2 times daily and in cats, 25 mg/kg bw 1–2 times daily (Bishop, 2004).
The recommended therapeutic dose of chloramphenicol in infants over one month of age and in adults is 50–100 mg/kg bw/day. A lower dose of 25–50 mg/kg bw/day is recommended for neonates (<1 month of age) due to their greater susceptibility. Nonetheless, in view of its established toxicities, close monitoring of patients is recommended, which should include regular monitoring of the achieved systemic exposure level in the young. It has also been suggested that chloramphenicol should never be employed for diseases that are treatable with other agents. In addition, it is recommended that repeated courses of treatment should be avoided where possible (JECFA, 1994; HSDB, 2003).
Other potential sources of human exposure include ingestion of animal products containing illegal drug residues. However, no products for use in food-producing animals containing chloramphenicol are legally available in the UK and, given the existing uses and formulations available, the possibility of indirect exposure of the general population would appear remote.
2.1 Exposure concentrations in meatUsing information on usage of chloramphenicol and on absorption and excretion, likely ‘worst case’ concentrations in meat products were estimated using Equation 1:
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
34

Equation 1M = D × T × Fabs × (1 – Fexc)
where M is the predicted concentration in meat (mg/kg); D is the dose of substance (mg/kg bw/d); T is the duration of the treatment (d); Fabs is the fraction of compound absorbed; and Fexc is the fraction of compound excreted.
For cattle, it was assumed that chloramphenicol is given by injection (i.e. Fabs = 1.0) at a dose rate of 25 mg/kg bw/d for three days and that the fraction of drug excreted was 0.70 (based on a rate study). Using these input values, a ‘worst case’ concentration of 25 mg/kg was obtained.
For pigs, it was assumed that chloramphenicol is given by injection (i.e. F abs = 1.0) at a dose rate of 11 mg/kg bw/d for three days and that the fraction of drug excreted was 0.70. Using these input values, a ‘worst case’ concentration of 11 mg/kg was obtained.
Chloramphenicol has been detected in 2/45 imported prawn samples at concentrations of 0.7 and 0.9 µg/kg during 2001 (VRC, 2002) and at 0.8 µg/kg during 2002 in 1/113 samples (VRC, 2003). The highest measured concentration of 0.9 µg/kg was used in considering a ‘worst case’ risk scenario for dietary exposure. The same compound was also detected in imported honey during 2002 at concentrations of 0.3–1.4 µg/kg in 5/200 samples (VRC, 2002; VRC, 2003). The highest concentration of 1.4 µg/kg was used in considering a ‘worst case’ risk scenario for dietary exposure. Chloramphenicol has not been found in foods produced within the UK and sampled under the UK National Surveillance scheme (VRC, 2003) during the period 2001-2003 (2004 results are not yet published).
3 ToxicokineticsA number of analytical methods have been developed for detecting chloramphenicol in tissues to levels of 1 µg/kg (EMEA, 1996).
In adult humans oral absorption of chloramphenicol has been shown to be rapid, with serum levels of 20–40 mg/ℓ achieved after administration of 29 mg/kg and 40–60 mg/ℓ after 57 mg/kg. It is also well absorbed via the oral route by infants and neonates, with serum levels peaking at 20–24 mg/ℓ following administration of 40 mg/kg. There is also evidence suggesting that, although limited, dermal absorption does occur (JECFA, 1988; EMEA, 1996; HSDB, 2003).
In animals, chloramphenicol is rapidly absorbed following oral or parenteral administration, with maximum blood levels generally occurring between 1 and 5 hours post dose (EMEA, 1996). Absorption was rapid following oral dosing of dogs with 50 mg/kg, and resulted in plasma levels of 16.5 g/ℓ two hours later. Similar results were reported for rabbits following oral dosing at 16 mg/kg (JECFA, 1988).
In all the species studied, distribution of chloramphenicol following absorption was rapid and widespread throughout the body (JECFA, 1988; EMEA, 1996). In humans, regardless of dose route, rapid distribution occurs; the volume of distribution is approximately 0.7–1.4 ℓ/kg; highest concentrations have been noted after intravenous (i.v.) or oral administration and extensive protein binding occurs. Chloramphenicol has been shown to cross the placenta and has been detected in seminal fluid and saliva (JECFA, 1988).
Interspecies differences in metabolism have been identified (EMEA, 1996). In humans, 93% of an oral dose has been shown to be excreted via the urine, with the major metabolite being the glucuronide. In one study, 48% of the administered dose was excreted, of which only 6% was as the parent and 4% as the base (JECFA, 1988). While the rate of glucuronide conjugation has been shown
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
35

to be lower in those with hepatic insufficiency, the metabolism of the drug remained essentially normal overall (HSDB, 2003).
In rats, the major metabolite is also the glucuronide, although the oxamic acid, alcohol and base derivatives are also detected. In the dog, however, the base and glucuronide are the major metabolites and, in the goat, the glucuronide, oxamic, acetylarylamine, arylamine and base have all been identified (JECFA, 1988).
In humans, 90% of excretion is via the kidney, and glomerular excretion is considered the primary route. Only 15% is excreted in the form of the parent compound; the majority is in the form of a number of metabolites, including conjugated forms. Rates of clearance vary with age: clearance rates in neonates are 0.46–9.76 ℓ/hour and in infants 1.8–2.1 ℓ/hour. Values of 6.03 and 9.59 ℓ/hr have been reported in two children over 2.5 years of age. There is some evidence that plasma clearance is impaired in patients showing chloramphenicol-induced bone marrow suppression, when compared with those that showed no toxic response. Clearance is also known to be reduced in individuals with renal insufficiency (JECFA, 1988).
When other species are considered, the kidney is also the principal route of elimination (EMEA, 1996). In rats up to 70% of the administered dose may be excreted via the urine after oral dosing, and only 0.4% of the dose was detected in bile 4 hours after an intramuscular dose of 40 mg/kg (JECFA, 1988). In newborn pigs, the majority of an i.v. dose was excreted in the urine, with only a small quantity being detected in the bile (JECFA, 1988). As noted above, other bodily fluids may also act as vehicles for excretion. Intramuscular administration of 10 mg/kg to cattle resulted in a concentration of 1 mg/ℓ in milk six hours after dosing, and up to 1.3% of a dose has been shown to be excreted in the milk in humans (JECFA, 1988).
4 Toxicity profile4.1 Acute toxicityThere are a few studies in humans that report that allergic contact dermatitis may be associated with the topical use of chloramphenicol (JECFA, 1988); most cases are associated with the use of eye drops (JECFA, 1988). There is also evidence that systemic administration can elicit a reaction at sites previously exposed and sensitised to the drug (JECFA, 1988).
Chloramphenicol is considered moderately toxic to mice (EMEA, 1996), with LD50 values by the intravenous route of 1530 mg/kg bw in non-pregnant and 1210 mg/kg bw in pregnant mice (JECFA, 1988). There is also evidence from dogs, supported by an in vitro study, that minor haematological changes may be elicited by a single dose of chloramphenicol (see Table 4.1).
Table 4.1 Haematological effects following single dose of chloramphenicol Species Treatment Effect
Dogs Chloramphenicol Decrease in protein synthesis in platelets
In vitro on marrow cells (species not specified)
Chloramphenicol Inhibition of erythroid and granulocyte colonies in one out of two studies
Data from JECFA (1988)
4.2 Repeat dose toxicityIn humans, chloramphenicol treatment is associated with a low incidence of blood dyscrasias, including aplastic anaemia (EMEA, 1996). While cases of chloramphenicol-induced aplastic anaemia
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
36

are generally irreversible and often fatal, the other types of bone marrow suppression that can be elicited generally show reversibility following withdrawal of treatment (JECFA, 1988; JECFA, 1994).
The mechanism of toxicity that underlies development of aplastic anaemia has not been defined with any certainty, although it has been suggested that a metabolite, nitrosochloroamphenicol, may be the causal agent. It has been estimated that the condition may develop (at an incidence of approx. 1:10 000 to 1:several 100 000) several months or even a year after completion of a course of treatment involving total intakes of 8 to 80 g; however, the minimum dose associated with development of the condition is uncertain (EMEA, 1996). The WHO estimated the incidence of this disease in those treated with chloramphenicol to be approximately 1:30 000 (JECFA, 1994). Cases have usually involved oral or parenteral administration. However, there are a small number of cases reporting the development of this disease following ophthalmic, dermal or inhalation exposure (JECFA, 1988; JECFA, 1994). The EMEA considered that recent epidemiological studies demonstrated that ophthalmic use was not associated with aplastic anaemia (EMEA, 1996), a view supported by WHO, which also concluded that there was also no relationship between the disease and veterinary use of chloramphenicol (JECFA, 1994). In any event, since the level of systemic exposure arising from ophthalmic use has not been established, the EMEA concluded that it was not possible to establish a threshold effect level on the basis of the evidence from ophthalmic use (EMEA, 1996).
It has been suggested that hepatic toxicity is evident in at least some patients who then go on to develop aplastic anaemia, and it is possible that there may be a genetic component to susceptibility (JECFA, 1994).
Reversible suppression of the bone marrow erythroid series shortly after treatment is also associated with chloramphenicol treatment in some patients. Effects are thought to be due to reduced haem synthesis, possibly as a result of interference with mitochondrial protein synthesis, but are unlikely to occur at plasma levels below 20 mg/ℓ (JECFA, 1988).
Chloramphenicol treatment has also been associated with the ‘Grey baby syndrome’, in which cardiovascular collapse occurs within 2 to 9 days of the start of dosing of some newborn or premature infants. The symptoms include inappetance, vomiting, abdominal distension, cyanosis, flaccidity, shock and lowered body temperature. The syndrome is generally associated with dosages exceeding 25 mg/kg bw/day (or plasma levels greater than 30 µg/mℓ), and death occurs in approximately 60% of cases (JECFA, 1988; JECFA, 1994). Although not known with any certainty, there are some experimental data suggesting that mitochondrial mechanisms may be important (JECFA, 1988).
Experimentally, there has been extensive investigation of the potential for chloramphenicol to cause haematological effects (Table 4.2; JECFA, 1988). Although results are variable, there is evidence of effects on haematological profiles in at least some instances. Effects on a number of enzymes and on mitochondrial function have been established. Chloramphenicol has been shown to act as a suicide substrate for cytochrome P450 (particularly phenobarbitome-inducible forms), possibly by binding of the oxamic acid metabolite with a lysine residue of the cytochrome (JECFA, 1988). This activity has been shown to affect the metabolism and clearance rates of other chemicals, including pharmaceuticals (JECFA, 1988). Other metabolic effects include inhibition of hepatic N-demethylase, glucose-6-phosphate dehydrogenase and carboxylesterase, and disruption of mitochondrial protein synthesis (possibly through a range of actions, including inhibition of DNA polymerase, ATP synthesis and entry of NAD-linked substrates into mitochondria; JECFA, 1988).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
37

Table 4.2 Haematological effects following repeated doses of chloramphenicolSpecies Treatment Effect
Mice X-irradiation+/-chloramphenicol Reduced recovery of bone marrow following X-ray irradiation in chloramphenicol treated mice
Mice Busulphan+/-chloramphenicol Treatment with either chemical alone had no effect, but treatment with both led to progressive fall in stem cell and granulocyte precursor cell numbers. Separate study using similar regimen found no effects of treatment
Mice and dogs Lethal X-ray dosing+/- chloramphenicol
Higher doses of chloramphenicol resulted in decreased erythropoiesis and granulocyte formation in irradiated dogs
Neonatal calves Chloramphenicol No effect on haematology or bone marrow cellularity in two separate studies
Calves Chloramphenicol Partial aplasia of marrow
Data from JECFA (1988)
Chloramphenicol has also been associated experimentally with ototoxicity in guinea pigs and rats; the effect involves the hair and supporting cells of the Organ of Corti and the cochlea (JECFA, 1988). However, studies on the potential of chloroamphenicol to cause ocular toxicity were negative (JECFA, 1988).
4.3 Carcinogenicity and mutagenicityA number of case reports have described the development of leukaemia in patients following chloramphenicol-induced aplastic anaemia. A follow-up study showed three cases of leukaemia in 126 patients with bone-marrow depression following chloramphenicol treatment (IARC, 1987).
The administration of 2.5 mg chloramphenicol, 5 days/week for 5 weeks, to mice previously treated with busulphan or vehicle alone, resulted in an increase in the incidence of lymphoma in those mice given both chloramphenicol and busulphan and a slight increase in those given chloramphenicol alone (JECFA, 1988). In one inadequately reported study, administration of chloramphenicol via the drinking water was reported to result in increased incidence of lymphomas in two strains of mice and hepatocellular carcinoma in one of the mouse strains (EMEA, 1996). However, another study suggested that cloramphenicol was protective to mice for the cirrhotic and hepatic carcinogenic effects of N-2-fluorenyldiacetamide (JECFA, 1988).
A variable pattern of response has been noted with various in vitro assays for genotoxicity or mutagenicity (JECFA, 1988).
DNA fragmentation was reported for Chinese hamster V79 cell and rat hepatocytes (JECFA, 1994; EMEA, 1996) and a weak positive response for DNA strand breaks was noted in bacterial and rat hepatic cells (JECFA, 1988; JECFA, 1994). Positive results have also been reported for DNA repair assays on cultured human and rat hepatocytes (JECFA, 1994; EMEA, 1996), and there was a significant increase in the frequency of 6-thioguanine resistant clones in V79 cells exposed to chloramphenicol (JECFA, 1994; EMEA, 1996). Inhibition of DNA synthesis has been noted in lymphocytes and in a phage of E. coli (JECFA, 1988).
In the earlier of the WHO reports (JECFA, 1988), it was noted that, in general, negative results were found in bacterial reverse mutation assays, bacterial DNA repair assays, a HGPRT test in Chinese Hamster Ovary cells, in a test for sister chromatid exchange in human lymphocytes, a micronucleus test, and a test for DNA binding. Negative results in Ames tests were attributed to its bacterial toxicity (JECFA, 1988). However, the subsequent WHO review reported positive or weakly positive findings in sister chromatid exchange assays in human lymphocytes, V79 cells and bovine fibroblasts (JECFA,
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
38

1994). It should also be noted that three metabolites of chloramphenicol (nitroso-chloramphenicol, dehydro-chloramphenicol and dehydro-chloramphenicol-base) are clearly mutagenic in vitro (EMEA, 1996).
WHO report that chromosomal aberration tests give generally positive results (JECFA, 1988). An increase in chromosomal aberration was noted in cultured human lymphocytes, in mouse bone marrow, and in F1-mouse hepatocytes (JECFA, 1988). Increased sister chromatid exchange was noted in cultured human lymphocytes, V79 cells and bovine fibroblasts (EMEA, 1996).
In vivo, chromosomal abnormalities were induced in bone marrow cells taken following oral administration at 50 or 100 mg/kg to mice. However, a single oral dose of 1250 mg/kg to rats caused no effect on the incidence of micronuclei in rat bone marrow cells (EMEA, 1996). WHO state that results are generally negative in dominant lethal tests in rodents or D. melanogaster (JECFA, 1988).
Overall, WHO concluded that chloramphenicol has only a very low genotoxic capacity, and that, when considering the metabolites with greater activity, 3 to 4 logs of concentration separate their possible genotoxic serum concentrations from that which might result from ingestion of residues from foods of animal origin (JECFA, 1994). IARC concluded that there was limited evidence for the carcinogenicity of chloramphenicol in humans, that experimental data were inadequate, and therefore classified chloramphenicol as possibly carcinogenic to human (Group 2B; IARC, 1987).
4.4 Reproductive and developmental toxicityThe WHO reported on a single case study of a 61-year old man who died following chloramphenicol treatment and the subsequent development of aplastic anaemia. Necropsy showed that there was loss of testicular germinal epithelium (but not Sertoli) cells. However, a definitive link with chloramphenicol treatment could not be established since the individual had also previously received treatment with ampicillin (JECFA, 1988).
In a small (599 cases) epidemiology study, treatment of pregnant women with chloramphenicol and/or tetracyclines has been associated with development of cleft-lip in their offspring. However, because of methodological limitations, it was not possible to determine whether tetracycline or chloramphenicol was the causal agent (JECFA, 1988). While IARC report a relative risk of 1.17 for malformation in a large study conducted between 1959 and 1965, two other smaller studies were noted not to have shown any effect (IARC, 1990).
Experimentally, treatment with chloramphenicol is associated with increased rates of fetal death and fetal growth impairment in rats, mice and rabbits. A NOEL of 500 mg/kg/day was established in the rabbits but no NOEL was established in rats or mice for which effects were seen at the lowest dose (500 mg/kg/day) tested (JECFA, 1988; JECFA, 1994; EMEA, 1996).
Teratology studies in Macaca mulatto and rabbits were negative. However, increased incidences of umbilical hernia and delayed ossification have been reported in rats and, in some studies, ossification delays and a low incidence of fused sternebrae were observed in mice exposed in utero. Functional changes to the nervous system have also been reported in rodents following gestational or neonatal exposure. In rats, learning deficits were noted in the offspring of treated dams and in 60-day old rats that had been injected with chloramphenicol on each of the first 3 days after birth. In mice, reduced learning ability, increased threshold to electroshock seizure, and impaired performance in an open-field test were noted in animals following exposure on days 5 to 7 of gestation. Chick embryos have also shown a number of abnormalities (including cardiovascular anomalies and neural tube defects) following exposure to chloramphenicol (JECFA, 1988; JECFA, 1994). Evidence of potential teratogenic activity has also been noted in the mouse embryo limb bud and the rat embryo midbrain and limb bud assays in vitro (JECFA, 1988).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
39

5 Guidelines and standardsJECFA has noted that human exposure to chloramphenicol residues in food would be expected to be of the same order as the systemic exposure that might arise from ophthalmic use and, as such, would not be expected to cause a demonstrable change in the incidence of plastic anaemia. Nonetheless, JECFA did not establish an ADI because of the lack of information on the carcinogenicity of chloramphenicol and its established role in aplastic anaemia (JECFA, 1994).
EMEA CVMP have also concluded that it is not possible to establish an ADI for chloramphenicol because of the inability to establish a threshold for the haematological effects, its genotoxicity, lack of adequate carcinogenicity study, lack of a NOEL for fetotoxicity, and the absence of adequate reproductive toxicity data. Given this, and the lack of adequate information as to the metabolic residues of toxicological concern, it was recommended that no MRL could be determined and chloramphenicol was therefore included in Annex IV of Council Regulation (EEC) No. 2377/90 (EMEA, 1996). Nonetheless, in line with Council Directive 2002/657 EC, a non-toxicologically based Minimum Required Performance Limit (MRPL) of 0.3 g/kg analytical performance standard has been established for chloramphenical, for use in analysis of samples of meat, eggs, milk, urine, aquaculture products and honey (EC, 2003).
6 ReferencesBishop Y, ed (2004) The Veterinary National Formulary (Sixth Edition), London, UK, Royal Pharmaceutical Society of Great Britain/British Veterinary Association
EC (2003) Commission Decision of 13 March 2003 amending Decision 2002/657/EC as regards the setting of minimum required performance limits (MRLPs) for certain residues in food of animal origin (notified under document number C(2003)764) (2003/181/EC) Official Journal of the European Union, L71, 17-18 Available July 2005 at: http://europa.eu.int/eur-lex/pri/en/oj/dat/2003/l_071/l_07120030315en00170018.pdf
EMEA (undated) Chloramphenicol. Summary Report, London, UK, European Agency for the Evaluation of Medicinal Products, Available [May 2005] at http://www.emea.europa.eu/pdfs/vet/mrls/chloramphenicol.pdf
GENE-TOX (1995) Chloramphenicol. From: Genetic Toxicology (Mutagenicity) Databank, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [November 2003] at http://http://toxnet.nlm.nih.gov
HSDB (2003) Chloramphenicol. From: Hazardous Substances Databank, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [November 2003] at http://http://toxnet.nlm.nih.gov
IARC (1987) IARC Monographs on Evaluation of Carcinogenic Risks of Chemicals to Humans, Vol Supplement 7, Overall Evaluations of Carcinogenicity: An Updating of IARC Monograph Volumes 1-42, Lyon, France, International Agency for Cancer Research
IARC (1990) Summaries and Evaluations, Vol 50, Chloramphenicol, Lyon, France, International Agency for Cancer Research
JECFA (1988) Chloramphenicol (Food Additives Series 23), Geneva, Switzerland, World Health Organization, Available [November 2003] at; http://www.inchem.org/documents/jecfa/jecmono/v23je03.htm
JECFA (1994) Chloramphenicol (Food Additives Series 33), Geneva, Switzerland, World Health Organization, Available [November 2003] at; http://www.inchem.org/documents/jecfa/jecmono/v33je03.htm
JECFA (2001) Chloramphenicol (Summary of Evaluations performed by the Joint FAO/WHO Expert Committee on Food Additives), Geneva, Switzerland, World Health Organization, Available [November 2003] at; http://www.inchem.org/documents/jecfa/jeceval/jec_326.htm
VRC (2002) Annual Report on Surveillance for Veterinary Residues in Food in the UK, 2002, Addlestone, UK, The Veterinary Residues Committee
VRC (2003) Annual Report on Surveillance for Veterinary Residues in Food in the UK, 2003, Addlestone, UK, The Veterinary Residues Committee
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
40

Annex 2 Clenbuterol1 IntroductionClenbuterol (4-amino-3,5-dichloro-[[(1,1-dimethylethyl)amino]methyl]-3,5-dichlorobenzyl alcohol, CAS no. 37148-27-9) is a direct-acting 2-sympathomimetric agent, which is usually administered as a hydrochloride salt. It is manufactured as a 50:50 racemic mixture and most of the pharmacological activity is associated with the L-form. It exerts a potent bronchiolytic effect by preferential action on 2-adrenoceptors in smooth muscle, resulting in the relaxation of bronchial smooth muscle and a decrease in airway resistance. It also has a similar effect on uterine smooth muscle cell membrane, resulting in the relaxation of the uterus, that is tocolysis (JECFA, 1991; EMEA, 2000)..
2 Uses and exposureClenbuterol is used as a bronchodilator for horses and is administered orally or parenterally with a recommended treatment regime of 0.8 g/kg, twice daily, for up to 10 days (JECFA, 1991). It may also be administered by intramuscular or intravenous routes. Clenbuterol is also used as a tocolytic in cattle and horses (EMEA, 2000).
Clenbuterol is also a human medicine used as for the treatment of chronic obstructive airway disease, with a recommended dose of 10 to 20 g, twice a day (JECFA, 1991; EMEA, 2000).
2.1 Exposure concentrations in meatUsing information on usage of clenbuterol and on absorption and excretion, likely ‘worst case’ concentrations in meat products were estimated using Equation 1:
Equation 1M = D × T × Fabs × (1 – Fexc)
where M is the predicted concentration in meat (mg/kg); D is the dose of substance (mg/kg bw/d); T is the duration of the treatment (d); Fabs is the fraction of compound absorbed; and Fexc is the fraction of compound excreted.
It was assumed that all of the clenbuterol is absorbed following administration (i.e. F abs = 1.0) at a dose rate of 1.6 g/kg bw/d for ten days and that the fraction of drug excreted was 0.8 (i.e. 60% in the urine and 20% in the faeces). Using these input values, a ‘worst case’ concentration of 3.2 g/kg was obtained and used to calculate theoretical intakes from consumption of beef and beef products.
3 ToxicokineticsClenbuterol is well absorbed after oral administration to laboratory animals and humans. In most species peak blood concentrations are achieved 2 to 3 hours after oral dosing. Clenbuterol is widely distributed to the tissues and has been shown to cross the placenta in pregnant rats, dogs, baboons and cows. In rats, 60% of a radioactive dose was recovered in the urine after 48 hours with a further 20% being recovered in faeces. Approximately 7% appeared in the bile. Levels in the tissues were relatively low, except for the liver, kidneys and lungs (EMEA, 2000).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
41

In all species, clenbuterol is excreted in the urine, predominantly unmetabolised, with five minor metabolites detected (EMEA, 2000).
4 Toxicity profile4.1 Acute toxicityMany of the toxicological studies are old and have not been conducted according to recent regulatory protocols and reporting standards.
Clenbuterol is of moderate to high acute toxicity. The acute oral LD50 value for the racemic mixture ranged from 80 to 180 mg/kg bw. It is more toxic when administered parenterally. The L-form is more toxic than the D-form, with the acute intravenous LD50 values in the mouse being 23.8 and 50 mg/kg bw, respectively. Acute toxicity includes lethargy, increased heart rates and tonic-clonic convulsions (JECFA, 1991; EMEA, 2000).
Clenbuterol was not a sensitiser in the Maximisation test or when tested by the Buhler test (EMEA, 2000).
4.2 Repeat dose toxicityA number of clinical trials have been carried out with clenbuterol in healthy human volunteers, pregnant women with premature labour pains, patients with coronary heart disease and chronic obstructive airway disease. From these studies, a pharmacological NOAEL in humans of 2.5 g/day was derived (EMEA, 2000).
Several repeated dose studies have been conducted in dogs, using oral and parenteral routes of administration. Cardiotoxicity was the main observed toxicity. In a 3-month study with oral doses of 0, 0.4, 4 and 20 mg/kg bw/day, a dose-related tachycardia was observed at the lowest dose, while in other studies, myocardial necrosis was apparent at 2.5 mg/kg bw/day and above. NOAELs have not been established in the dog (JECFA, 1991; EMEA, 2000).
Repeat dose studies have been carried out in the rat for durations from 1–18 months, using oral and parenteral administration. Increased respiration and heart rate were observed following intravenous dosing at 16 mg/kg bw/day. Dose-related myocardial lesions were found in several studies and hepatotoxicity with increased liver function enzymes and focal necrosis occurred following intravenous dosing at 16 mg/kg bw/day. Myocardial lesions were also found in a 6-month study at 1 mg/kg bw/day, the lowest dose tested. Inconsistent results were obtained in an 18-month study; bradycardia was observed rather than tachycardia and, upon histochemical examination of the myocardium, a reduction in enzyme activity was observed in both the left and right ventricle walls; the effect was greatest at the lowest concentration, 0.1 mg/kg bw/day (JECFA, 1991).
In an incomplete 1-month mouse study, increased liver weight was observed following oral dosing at 12.5 mg/kg bw/day with a NOAEL of 2.5 mg/kg bw/day (EMEA, 2000).
Inhalation studies have been carried out but are not considered reliable owing to methodological problems (EMEA, 2000).
4.3 Carcinogenicity and mutagenicityIn a 2-year carcinogenicity study in the mouse at doses 0, 0.1, 1 and 25 mg/kg bw/day, administered in drinking water, increased heart rates and heart weights were observed but there was no evidence of carcinogenicity (JECFA, 1991; EMEA, 2000).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
42

A carcinogenicity study was conducted on two strains of rats, Chbb:THOM (dose levels of 0, 6.25, 12.5 or 25 mg/kg bw/day) and Sprague-Dawley (0, 25 mg/kg bw/day). The dose was administered in the feed for 20 weeks then in drinking water for the remaining time owing to contamination problems. There was a reduction in weight. No increase in tumour incidence was observed apart from an increase in mesovarian leiomyomas in the treated Sprague-Dawley rats. Several -agonists have been shown to induce such tumours in certain species of rats, and there is evidence from a study on salbutamol that co-administration with a -antagonist (such as propranol) abolishes these tumours. The induction of mesovarian leiomyomas appears to be a function of adrenergic stimulation. Human epidemiology indicated that mesovarian leiomyomas were not increased in women treated with adrenergic agents. Therefore it was concluded that clenbuterol was not carcinogenic (JECFA, 1991; EMEA, 2000).
In incomplete studies not carried out to modern standards, clenbuterol was negative in two bacterial assays for gene mutation and a HGPRT mutation assay in V79 cells. In a well-conducted mouse lymphoma assay, clenbuterol was negative in the absence of metabolic activation but there was a small increase in mutant frequency at the highest doses of 700 and 800 g/mℓ in the presence of metabolic activation; however, these results were not reproducible. In an in vitro cytogenetic assay in human lymphocytes, there were occasional significant increased numbers of aberrant cells but the effects were not reproducible or dose-dependent (JECFA, 1991; EMEA, 2000).
An in vivo micronucleus test gave negative results but the maximum tolerated dose was not achieved and the polychromatic to normochromatic erythrocyte ratio was not calculated. There was no evidence that clenbuterol induced chromosomal aberration in Chinese hamster bone marrow following sub-acute (5-day) oral dosing at 50% of the LD50 value (JECFA, 1991; EMEA, 2000).
In conclusion, although the studies are incomplete and not of the most recent standards of conduct and reporting, the evidence suggests that clenbuterol is not genotoxic or carcinogenic.
4.4 Reproductive and developmental toxicityIn a study to investigate perinatal toxicity, groups of pregnant female Chbb:THOM rats were given oral doses of clenbuterol (0, 1, 7 or 50 mg/kg bw/day) from day 15 of gestation to day 21 post-partum. Maternal food consumption was reduced in all treated groups and the number of pups born dead was increased in a dose-dependent manner. Post-natal mortality also increased in all treatment groups with all pups in the highest dose group dead by day 7 of lactation (JECFA, 1991; EMEA, 2000).
In a reproductive toxicity study, groups of Chbb:THOM rats were given oral doses of clenbuterol (0, 1, 7 or 50 mg/kg bw/day). Treatment of males started 10 weeks prior to mating, while females were treated 2 weeks prior to mating and treatment continued through gestation and lactation. On day 14 of gestation 50% of the females were killed and the uterine contents were examined. The remainder were allowed to deliver normally and to rear the offspring to weaning. To investigate the high pup mortality in the perinatal toxicity study, the litters of six control dams were exchanged with those from dams given 50 mg/kg bw. There was no effect on fertility, corpora lutea, implantations or resorption rates. There was evidence of maternal toxicity (reduced bodyweight gain) at 50 mg/kg bw. Pup weight at birth was significantly reduced in all treatment groups and there was a decrease in the number of viable pups in the 7 and 50 mg/kg bw groups. All the pups in the 50 mg/kg bw group died during the first day of lactation; it made no difference whether the pups were suckled by their own mothers or those from the untreated control group. The majority of the pups from the control group who were suckled by treated mothers survived to weaning (JECFA, 1991; EMEA, 2000).
The previous study was repeated using lower dose levels (0, 1.5, 7.5 or 15 g/kg bw/day). There was no effect on fertility, gestation length, number of corpora lutea, implantation rate, incidence of resorption, teratogenicity or fetotoxicity at any dose level. Pup survival, bodyweight gain and performance in behavioural tests were unaffected by treatment. The NOAEL was considered to be greater than 15 g/kg bw/day (JECFA, 1991; EMEA, 2000).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
43

Three teratogenicity studies have been conducted in rats using oral administration. In the first study, with doses between 0–1 mg/kg bw/day given on days 6 to 15 of gestation, there was no evidence of maternal toxicity, teratogenicity or fetotoxicity. In the second study the dams were given daily doses of 0, 0.01, 1, 10 or 100 mg/kg bw. At 10 or 100 mg/kg bw, maternal toxicity, increased resorption, reduced live births, and an increase in malformations were observed. These malformations included hydrocephalus, ansarka, anophthalmia, rib malformation and splintering of the vertebrae. The third study was conducted using the same doses and including a littering phase with 5 dams/dose. Again dose-response increases in malformations were seen at the highest two doses and in the littering phase, pup development was retarded and malformations were seen at the highest two doses. The oral NOAEL was again established as 1 mg/kg bw (JECFA, 1991; EMEA, 2000).
In a number of studies in rabbits that were poorly conducted and reported, teratogenicity was observed at maternally toxic dose levels and an oral dose of 1 mg/kg bw was a NOAEL for maternal toxicity (reduced body weight again and food consumption), fetotoxicity (reduced mean litter and fetal weight) and teratogenicity (cleft palate and synostosis; JECFA, 1991; EMEA, 2000).
5 Guidelines and standardsEMEA CVMP (EMEA, 2000) has established a toxicological ADI of 0.15 g/kg bw, by applying a safety factor of 100 to a NOAEL of 15 g/kg bw/day from the reproductive toxicity study in rats, taking into account reduced pup weight in related studies. This is similar to the ADI of 0.2 g/kg bw calculated by applying a safety factor of 500 to the dose level of 0.4 mg/kg bw/day, the lowest dose in the 3-month repeat dose dog study. The safety factor was considered justified as this dose was not a NOAEL and caused tachycardia but no other effects.
EMEA CVMP has also established a pharmacological ADI of 0.25 g for a 60 kg adult (i.e. 0.0042 g/kg bw), by applying a safety factor of 10 to the human NOAEL of 2.5 µg/day. This safety factor was justified because asthmatics are especially sensitive to the bronchodilatory effects of -agonists (EMEA, 2000; VRC, 2001; VRC, 2002; VRC, 2003).
It is considered that the pharmacological ADI derived from human data was the most relevant ADI for risk assessment (EMEA, 2000).
In accordance with the provisions of Council Directive 96/22/EC concerning the prohibition on the use in stock farming of certain substances having hormonal or thyrostatic action and of -agonists, clenbuterol was listed in Annex I of Council Regulation (EEC) No. 2377/90. Table 5.1 summarises the MRLs derived by EMEA (2000).
Table 5.1 MRLs for clenbuterolPharmacologically active substance
Marker residue
Animal species
MRL Target tissue
Clenbuterol hydrochloride
Clenbuterol Bovine 0.1 g/kg Muscle0.5 g/kg Liver0.5 g/kg Kidney0.05 g/kg Milk
Equidae 0.1 g/kg Muscle0.5 g/kg Liver 0.5 g/kg Kidney
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
44

6 ReferencesEMEA (2000) Clenbuterol. Summary Report (2) (EMEA/MRL/723/99-Final), London, UK, European Agency for the Evaluation of Medicinal Products, Available [May 2005] at http://www.emea.europa.eu/pdfs/vet/mrls/072399en.pdf
JECFA (1991) Clenbuterol (Food Additives Series 38), Geneva, Switzerland, World Health Organization, Available [May 2005] at; http://www.inchem.org/documents/jecfa/jecmono/v38je02.htm
VRC (2001) Annual Report on Surveillance for Veterinary Residues in 2001, Addlestone, UK, Veterinary Residues Committee
VRC (2002) Annual Report on Surveillance for Veterinary Residues in Food in the UK, 2002, Addlestone, UK, Veterinary Residues Committee
VRC (2003) Annual Report on Surveillance for Veterinary Residues in Food in the UK, 2003, Addlestone, UK, Veterinary Residues Committee
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
45

Annex 3 Dimetridazole 1 IntroductionDimetridazole (1,2-dimethyl-5-nitroimidazole; CAS No.586-84-5) is a drug with antiparasitic activity used for the prevention and treatment of histomoniasis in turkeys, the treatment of trichomoniasis in pigeons, genital trichomoniasis in cattle and the prevention and treatment of haemorrhagic enteritis in pigs (EMEA, 1994). All products were voluntarily withdrawn from the market in 2003.
2 Uses and exposureDimetridazole was available as EmtrylTM in many countries, and was used for treatment of anaerobic bacteria in pigs, notably swine dysentery (Serpulina/Treponema); the protozoal infections, blackhead (Histomonas), in turkeys, chickens and gamebirds (pheasants, partridge) and guinea fowl; hexamitosis (Hexamita/Spironucleus) in game birds; and trichomoniosis (Trichomonas) in gamebirds and pigeons. It is currently only licensed and available for treatment of trichomoniosis in pigeons not intended for human consumption (Harkanker SolubleTM; Taylor, 2000).
For turkeys, chickens, gamebirds and pigs, dimetridazole was available either as a feed additive or for medication via the drinking water (Taylor, 2004).
Premix for use as feed additives (Emtryl PremixTM, Emtryl PremixTM, Lutrizol SwineTM) contained 22.5% dimetridazole w/w. The dose rate for inclusion in the diet for the prevention of blackhead and histomoniosis in turkeys was 450–900 g/tonne of premix equivalent to 100–200 g dimetridazole/tonne of feed medicated continuously to within 28 days of slaughter. For the prevention of blackhead and histomoniosis in pheasants, partridges and guinea fowl, the inclusion rate was 560–670 g/tonne of premix equivalent to 125–150 g dimetridazole /tonne of feed, to be fed continuously to within 28 days of slaughter. For the treatment of blackhead and histomoniosis in turkeys, chickens and gamebirds the dose rate for inclusion was 2.25 kg dimetridazole/tonne of feed for 7–14 days followed by the recommended inclusion rate for prevention according to the host species. In pigs for the treatment of swine dysentery, premix (Emtryl Presciption PureTM) containing 40% w/w dimetridazole, was administered as a feed additive at 200 g/tonne equivalent to 200 g/dimetridazole/tonne throughout the period of risk or until 28 days prior to slaughter (Taylor, 2000).
Dimetridazole was also available as a 40% w/w powder (Emtryl SolubleTM, Emtryl Prescription SolubleTM) for medication of drinking water. For the treatment of blackhead in turkeys, chickens and gamebirds, the dose rate was 26.7 g/100 ℓ of drinking water for 12 days. In rapidly spreading outbreaks the recommendation was 53.4 g/100 ℓ for 3–5 days, followed by 26.7 g/ℓ for 7–9 days; thereafter provision of medicated feed at the dose rate recommended above. For prophylaxis, the recommended dose rate was 13.3 g/100 ℓ throughout the period of susceptibility or to within the recommended withdrawal period prior to slaughter. In pigs, the dose rate was 26.7 g/100 ℓ for 3–7 days for treatment of 13.3 g/100 ℓ for prophylaxis (Taylor, 2000).
For use in the treatment of trichomoniosis in pigeons, dimetridazole is available as an oral powder in a 3 g soluble sachet at 400 mg/g w/w dimetridazole for medication of the drinking water. The dose rate is 1.2 g/4.5 ℓ of powder in the drinking water (Taylor, 2004).
In some countries, dimetridazole has been recommended for the treatment of venereal trichomoniosis (Tritrichomonas foetus) in cattle by preputial lavage. Dimetridazole has never been recommended for this purpose in the UK as the disease has been eradicated. As such, there are no recommended dose rates for use in cattle.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
46

For treatment of pigs, dimetridazole is applied either in feed or in solution at a dose of 200 g/tonne (equivalent to 10 mg/kg bodyweight). Animals are typically treated for between 3 and 14 days (NRA, 2002).
Dimetridazole in included in Annex IV of Regulation 2377/90/EEC, which prohibits its use in food producing animals in Europe. Products containing dimetridazole have been voluntarily suspended by the manufacturers and will become unavailable once existing stocks are depleted. The gamebird industry in the UK has expressed great concern over the loss of this compound as no alternatives exist for the treatment of blackhead and hexamitosis in pheasants and partridge. It is thought that many gamekeepers have stockpiled products containing dimetridazole as a short-term solution to the control of these diseases. As a consequence there is the potential for human exposure via ingestion of animal products containing drug residues.
2.1 Exposure concentrations in meatDimetridazole is absorbed from the gut in the target species; about 88% of the parent drug is eliminated from turkeys in about 3 days, and 76% from the pig in about 7 days. Residue depletion studies indicate that dimetridazole and its major, hydroxylated metabolite could be detected in the skin and fat of pigs after 9 days and in turkeys after 12 days (EMEA, 1994). Prior to voluntary removal from the market in 2003, withdrawal periods for meat products were set at 28 days for all species.
Experimentally-derived data are available on concentrations of dimetridazole in meat products. Chickens were treated with dimetridazole in drinking water following typical treatment regimes (concentration in water 260 mg/ℓ for 3 months). Following treatment, the chickens were slaughtered and concentrations of dimetridazole and the major metabolite, 2-hydroxydimetridazole were then measured in meat material and in eggs (Table 2.1). Maximum concentrations of the parent compound were observed in egg samples.
Table 2.1 Concentrations of dimetridazole and its metabolite in chicken meat and eggs obtained from animals dosed following typical treatment regimes
Dimetridazole (mg/kg)
2-hydroxydimetridazole (mg/kg)
Chicken muscle 1.6 3.9Chicken liver 0.072 0.033Chicken eggs 6.8 1.3
No measured data were available for dimetridazole in pigs. Information on usage of dimetridazole and on absorption and excretion were therefore used with Equation 1 to estimate likely ‘worst case’ concentrations in pig meat:
Equation 1M = D × T × Fabs × (1–Fexc)
where M is the predicted concentration in meat (mg/kg); D is the dose of substance (mg/kg bw/d); T is the duration of the treatment (d); Fabs is the fraction of compound absorbed; and Fexc is the fraction of compound excreted.
Assuming that dimetridazole was given at a dose of 10 mg/kg bw/d for 14 days and that all of this is absorbed (i.e. Fabs = 1.0) and that the fraction of drug excreted was 0.76, a ‘worst case’ concentration of 33.6 mg/kg was obtained.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
47

3 ToxicokineticsRoutine analytical methods are inadequate because the toxicological and analytical significance of the metabolites measured (dimetridazole and its hydroxylated derivative) are not clear. No validated analytical methods for assaying the residues in edible tissue are available (EMEA, 1994).
Dimetridazole is absorbed from the gastrointestinal tract in both laboratory and target species. About 88% of the administered dose is eliminated from turkeys within 3 days and about 76% is eliminated from pigs within 7 days. In both turkeys and pigs, the predominant oxidative metabolite found in urine is 2-hydroxymethyl-1-methyl-5-nitroimidazole. Although there was inadequate information on the biotransformation of dimetridazole in the target species and it was not possible to establish the ratio between the hydroxylated derivative and total residues, this metabolite has been recognised by JECFA as the major one in tissues (EMEA, 1994).
The quantity of bound residues was evaluated from two depletion studies performed on turkeys and pigs with 14C-dimetridazole following oral dosing. In both species, about 50% of the total radioactivity was not extracted and the nature of the bound residues was unclear. In more recent studies, in pigs and turkeys at therapeutic doses, dimetridazole and its major hydroxylated metabolite could be detected in the skin and fat of pigs until 9 days and in turkeys until 12 days after treatment (EMEA, 1994).
4 Toxicity profile4.1 Acute toxicityOral LD50 values have been determined in mice and rats as shown in Table 4.1 (JECFA, 1990).
Table 4.1 Oral LD50s for dimetridazole in mice and ratsSpecies Sex LD50 (mg/kg bw)
Mouse M, F 1790M, F 1790a–2000
60a–290Rat M, F 1600a–2500
70a
From JECFA, 1990aAdministered as ‘emtryl soluble’, which contains dimetridazole (40%), potassium dihydrogen phosphate (22%) and potassium sulphate (38%)
4.2 Repeat dose toxicityDiets containing 0, 0.2, 0.4, 0.6, 0.8 or 1% dimetridazole were fed to groups of 10 Simonsen Albino (SPF) male and female rats for 13 weeks. Data, submitted in summary form, showed that 3 females in the top dose group did not survive the full 13 weeks of study. The deaths occurred about 4 weeks after the first signs of ataxia, tilted head, anaemic appearance, excitation and convulsion, which occurred after 5 weeks of treatment. Testicular atrophy and degeneration occurred in all dimetridazole-treated male rats. These changes involved severe atrophy of seminiferous tubules with spermatogenic arrest of primary and secondary spermatocytes. A decrease in the numbers of primary follicles and increased degeneration of follicular epithelium were also noted in the ovaries of female rats treated with dimetridazole. Gastritis was observed in rats from each group except the control and 0.6% groups. Minimal focal infiltrations of leucocytes and occasional degenerative myocardial fibres were observed in 3 rats in each of the 0.6 and 0.8% groups (1 in the control group). This was considered to be suggestive of some myocardial toxicity (JECFA, 1990). A 2-month study in male and female rats after oral administration of dimetridazole at 100, 2000 or 5000 ppm revealed that the progesterone levels in
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
48

females increased at 2000 ppm (+112%) and at 5000 ppm (+167%). Progesterone levels were not affected by treatment in male rats (EMEA, 1994).
Diets containing 0.36 or 1.08% dimetridazole were fed to groups of one beagle dog of each sex for 4 weeks. There was no control group. Results, submitted in summary form, indicated that food consumption was markedly reduced in dogs in the 1.08% group when compared to the 0.36% group. Two weeks after treatment started, the female dog from the 1.08% group exhibited ataxia, predominant in the hindquarters. The male in this group also showed the same signs 3 days later. This condition became worse in both dogs until the study was terminated. No toxic signs were noted in the animals from the 0.36% group. Mild nephrosis, haemorrhagic and petechial haemorrhages and nephrosis of the kidney, haemorrhages of heart and spleen, central lobular cirrhosis and haemorrhages of liver were seen in dogs of the high dose group. Kidneys from animals in this group showed moderate cloudy swelling in the cells lining convoluted tubules and tubules comprising the medullary ray. The reaction was less in degree in the 0.36% group. Mild atrophy of the seminiferous tubules with no mature spermatocytes present and moderate degeneration of spermatids were observed in the testes of the male dog from the 1.08 % group. Very mild degenerative changes in spermatids and a reduced number of spermatocytes were also noted in the testes of the male dog from the 0.36% group. These changes were suggestive of dimetridazole-related effects (JECFA, 1990).
Groups of 2 male and 2 female beagle dogs, approximately 12–30 weeks old were given dimetridazole orally at dose levels of 0, 16, 33, 66 and 132 mg/kg bw/day for 13 weeks. Bodyweight gain and food consumption of all dimetridazole groups were less than that of the control group, particularly at the two highest dose levels. The dogs at 0, 16 and 33 mg/kg bw/day remained in relatively good health throughout the study. Anorexia, ataxia, convulsions and opisthotonos were seen in dogs at 66 and 132 mg/kg bw/day in a dose-related intensity. The study terminated early at 40 days for these 2 groups. In another 13 week study, groups of 4 male and 4 female beagle dogs were given dimetridazole orally at 0, 5, 10, 20 or 40 mg/kg bw/day. No unusual clinical signs were observed in any of the dogs at any time during the study. There were no drug-related effects on bodyweight, food consumption, urinalysis, haematology, biochemistry, organ weight or histopathology. Neither ophthalmological nor neurological examinations revealed any changes attributable to dimetridazole. It was concluded that daily doses of up to 40 mg/kg bw/day dimetridazole were well tolerated by the dogs over the period of 13 weeks (JECFA, 1990).
4.3 Carcinogenicity and mutagenicityThirty-five female Sprague–Dawley rats were fed 0.2% dimetridazole in diet (equal to 200 mg/kg bw/day) for 46 weeks, followed by control diet for an additional 20 weeks. Another group of female rats were fed control diet for 66 weeks. At 66 weeks there was a clear increase of benign mammary gland tumours in treated rats (25/35) compared with controls (4/35). The mean number of mammary tumours per rat was also increased in treated rats (1.7) compared to controls (1.0). Malignant mammary tumours did not occur in either group. It was not certain, in this study of only 66 weeks, if dimetridazole caused an actual increase in the tumour incidence or decreased the time for development of tumours that occurred spontaneously (JECFA, 1990).
In another study, diets containing 0, 100, 400 or 2000 ppm dimetridazole were fed to groups of 50 CFY rats of each sex for 122 weeks. The approximate daily intake of dimetridazole over the period of the study was 0, 3.8, 15.1 and 77.7 mg/kg bw/day in males and 0, 4.6, 18.3 and 94.1 mg/kg bw/day in females. At the termination of the study, the survival rates were: control males 30%, females 46%; 100 ppm males 38%, females 42%; 400 ppm males 28%, females 36%; 2000 ppm males 20%, females 14%. No adverse changes in bodyweight were noted for treated males during the study. In the females, except for the first 20 weeks of study, there was a tendency for the group mean bodyweights of all the treated groups to be slightly lower than those of the control group. Food consumption was unaffected by treatment. Necropsies were performed on all rats that died during the study or were sacrificed at the end of the study and microscopic examinations were undertaken. A significant increase in benign tumours (adenoma, fibroadenoma, fibroma) of the mammary gland occurred in
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
49

male and female rats from the 2000 ppm groups and a smaller increase was observed in the 400 ppm females. An increase in tumour multiplicity was observed at this site in the mid- and high-dose level females. Malignant mammary gland tumours were not increased in treated rats and there was no significant increase for any tumour type in other tissues. The NOEL for benign mammary gland tumours was considered to be 100 ppm dimetridazole (JECFA, 1990). In an addition to this study, using a similar experimental protocol, groups of 50 CYF rats of each sex were fed diets containing 0 or 10 ppm dimetridazole for 128 weeks. The approximate daily intakes of dimetridazole over the study period were 0.45 mg/kg bw for males and 0.57 mg/kg bw for females. At the end of the study the survival rates were: control males 32%, females 20%; 10 ppm males 12%, females 22%. Treatment with dimetridazole had no effect on group mean bodyweight or food consumption and there were no clinical signs due to treatment. There was no increase of benign or malignant mammary tumours in treated rats of either sex. However, at interim sacrifices, more tumour-bearing rats were found in dimetridazole treated rats than in the controls (JECFA, 1990).
CVMP suggested that the increase in benign mammary tumours in rats and the induction of elevated progesterone levels seen in the 2-month study in rats (see Section 4.2), could be coincidental rather than causally related. Progesterone levels were raised only in female rats but the tumours occurred in rats of both sexes. The Committee recognised that carcinogenicity studies with other nitroimidazoles indicate that these substances can cause malignant tumours in mice. No carcinogenic assessment was available for dimetridazole in mice (EMEA, 1994).
Both the CVMP and WHO Committees considered that the carcinogenicity studies on dimetridazole to be inadequate. The WHO Committee noted that three long-term rat studies were reported between 1973 and 1977. While meeting the requirements for that period, they were not conducted in accordance with present day standards for carcinogenicity studies.
The genotoxic potential of dimetridazole has been evaluated in several in vitro and in vivo studies, as summarised in Tables 4.2 and 4.3.
Table 4.2 Results of in vitro genotoxicity assays on dimetridazole Test system Conc. Dimetridazole Result
S. typhimurium, 4strains 50–200 µg/plate (± S9) Positivea
S. typhimurium, 4 strains 0.03 mM (- S9) PositiveS. typhimurium, 4 strains 0.01 µg/ml (± S9) PositiveS. typhimurium, nitroreductase deficient
100 µg/ml Negative
D. melanogaster 1.4 mM NegativeS. cerevisiae D4 0.05% (w/v) PositiveChinese hamster, CHO/HGPRT 820–2800 µg/ml NegativeChinese hamster lung fibroblast, UDS
200 µg/ml Negative
From JECFA (1990)aCCRIS (1994)
Although dimetridazole showed mutagenic activity in several bacterial assays, it was shown that this activity was linked to the enzyme activity of nitroreductases of the bacteria used in the tests. The compound gave negative results with nitroreductase deficient strains (JECFA, 1990; EMEA, 1994). This reasoning weakens the case for dimetridazole being a mammalian genotoxin.
Table 4.3 Results of in vivo genotoxicity tests on dimetridazole Test system Dose (mg/kg bw) Result
Mouse, dominant lethal 1000 NegativeMouse, micronucleus test 980 Negative
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
50

Rat, unscheduled DNA synthesis 1000 Negative
From JECFA (1990)
A WHO Committee noted that there was a lack of mutagenic effect for dimetridazole in in vitro and in vivo mammalian systems and considered that the mechanism for the production of benign mammary tumours in the rat was unlikely to be genotoxic (JECFA, 1990). However, a CVMP Committee noted that other nitroimidazoles had induced chromosomal aberrations in human lymphocytes in vitro and in vivo at high therapeutic dose levels. Therefore, the possibility that dimetridazole might be genotoxic could not be excluded (EMEA, 1994).
4.4 Reproductive and developmental toxicityGroups of 10 male and 20 female weanling CFY rats, comprising the F0 generation, were maintained on diets containing 0, 100 or 2000 ppm dimetridazole for approximately 80 days prior to the first mating and throughout the production of 3 generations. Dimetridazole markedly reduced the weight gain and food intake of the F0 males at the 2000 ppm dose level. During each of the six whelping phases, the fertility, viability and length of gestation period were comparable for the control and dimetridazole-treated groups. The numbers of pups dying in the F1b offspring from both matings were markedly and often significantly increased in both treated groups compared with the control group. This was due almost entirely to the increased number of dams that ceased lactating. The possibility of drug-induced, non-lactation in the F1b dams could not be excluded, but since similar effects were not observed in either the F0 or F2b rats, this was most unlikely. It was concluded that although some of the results obtained were contradictory, dimetridazole did not adversely affect the reproductive performance in the rat (JECFA, 1990).
Dimetridazole was administered by gavage to 4 groups of 23 pregnant New Zealand White rabbits on days 6–18 of gestation at dose levels of 0, 30, 60 or 120 mg/kg bw/day. On day 29 of gestation, the animals were killed for examination of uterine contents. Dose-related maternal toxicity, shown by a reduction in food intake and bodyweight gain and abortion, was observed in all dimetridazole treated groups. Death and total litter resorption were seen at the highest dose level. Although there was evidence of a slight reduction in fetal and placental weight, it was concluded that morphological development of the fetuses was unaffected by dimetridazole treatment (JECFA, 1990).
5 Guidelines and standardsJECFA could not establish an ADI for dimetridazole solely on the basis of a NOEL of 100 ppm in the diet, equal to 4 mg/kg bw/day, reported from a long-term rat study (see Section 4.3), in the absence of results from a carcinogenicity study in a second species. No MRL was determined (JECFA, 1989).
Similarly, EMEA CVMP could not derive a NOEL, which could serve as a basis for establishment of an ADI, from the toxicity studies conducted, since treatment related effects were observed at the lowest dose rates used.
There was inadequate information on the transformation of dimetridazole in the target species and it was not possible to identify a marker residue or target tissues. No MRLs could be established for dimetridazole in tissues of pigs and turkeys. In addition the parent drug has the potential to be mutagenic and carcinogenic; dimetridazole was, therefore, added to Annex IV of EU 2377/90 (EMEA, 1994).
6 ReferencesCCRIS (1994) Dimetridazole. From: Chemical Carcinogenesis Research Information System, Bethesda MD, USA, Specialized Information Services, National Library of Medicinehttp://http://toxnet.nlm.nih.gov
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
51

EMEA (1994) Dimetridazole (3) Summary Report, London, UK, European Agency for the Evaluation of Medicinal Products, Available [March 2004] at http://www.emea.europa.eu/pdfs/vet/mrls/dimetridazole3.pdf
JECFA (1989) Dimetridazole (Joint FAO/WHO Expert Committee on Food Additives), Geneva, Switzerland, World Health Organization, Available [January 2004] at; http://www.inchem.org/documents/jecfa/jeceval/jec_504.htm
JECFA (1990) Dimetridazole (Food Additives Series 25), Geneva, Switzerland, World Health Organization, Available [June 2003] at; http://www.inchem.org/documents/jecfa/jecmono/v25je03.htm
NRA (2002) Dimetridazole - Review Scope Document, Canberra, Australia, National Registration Authority for Agricultural and Veterinary Medicines
Taylor MA (2000) Antiprotozoals. In: Bishop Y, ed, The Veterinary National Formulary (Fifth Edition), London, UK, Royal Pharmaceutical Society of Great Britain/British Veterinary Association, pp 173–185
Taylor MA (2004) Antiprotozoals. In: Bishop Y, ed, The Veterinary National Formulary (Sixth Edition), London, UK, Royal Pharmaceutical Society of Great Britain/British Veterinary Association, pp 171–179
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
52

Annex 4 Enrofloxacin 1 IntroductionEnrofloxacin (1,4-dihydro-1-cyclopropyl-7-(4-ethyl-1-piperazinyl)-6-fluoro-4-oxo-3-quinoline-carboxylic acid; CAS No. 93106-60-6) is a fluoroquinolone antibiotic; it acts by inhibition of bacterial DNA gyrase in Gram-negative bacteria. It is administered by subcutaneous (s.c.) injection to cattle, by intramuscular (i.m.) injection to pigs and orally to cattle, pigs, turkeys and chickens for treatment of infections of the respiratory and alimentary tract (EMEA, 1998c). In some countries, enrofloxacin is authorised for use in sheep, goats and rabbits (EMEA, 1999).
Ciprofloxacin, a major metabolite of enrofloxacin, is used in human medicine by oral administration for the treatment of a wide variety of infectious diseases (EMEA, 1999).
2 Uses and exposureIn veterinary medicine in the UK, enrofloxacin is used for treating enrofloxacin-sensitive infections in cattle, pigs, chickens, turkeys, dogs, cats, and exotic species such as small mammals, reptiles and birds, for which a range of licensed products are available. There are concerns over the use of fluoroquinolones in food-producing animals due to the emergence of strains of Campylobacter and Salmonella with reduced susceptibilities to this group of compounds and the potential impact on human health (Bishop, 2004).
Cattle and pigs are treated by s.c. or i.m. injection, respectively, at a dose rate of 2.5 mg/kg daily for 3 days with either Baytril 5%TMor 10%TM (50 or 100 mg/mℓ enrofloxacin w/v). The dose rate may be increased to 5 mg/kg for 5 days for salmonellosis or treatment of complicated respiratory disease. A single, depot injection containing 100 mg/mℓ enrofloxacin is available for use in cattle (Baytril MaxTM). An oral solution containing 25 mg/mℓ is available for the treatment of calves at 2.5 mg/kg (Baytril 2.5% Oral SolutionTM). Piglets up to 10 kg bw may be treated orally with 1–2 mℓ of an oral solution containing 5 mg/mℓ enrofloxacin at a dose rate of approximately 1.5 mg/kg (Baytril Piglet DoserTM). Withdrawal periods for cattle are 14 days for meat and 84 hours for milk and for pigs 10 days for meat (NOAH, 2004).
Chickens and turkeys are medicated by addition to the drinking water at a dose rate of 10 mg/kg daily for 3–10 days (Baytril Oral 10% solution — 100 mg/ml enrofloxacin w/v). Treated birds should not be slaughtered within 8 days and the drug should not be given to birds within 14 days of commencement of laying (Bishop, 2004).
Dogs and cats are treated at 5 mg/kg with either Baytril 2.5%TMor 5%TM (25 or 50 mg/ml enrofloxacin w/v) once daily or twice daily by s.c. injection, or by oral tablets (Baytril TabletsTM) containing either 15, 50 or 150 mg enrofloxacin, according to weight and size (Bishop, 2004).
Potentially, humans might be exposed via ingestion of animal products containing drug residues.
2.1 Exposure concentrations in meatEnrofloxacin is well absorbed following oral administration in all target species and is widely distributed to all tissues with the highest concentrations in liver and kidney and rapid excretion via faeces and urine. The main metabolite, ciprofloxacin, is also readily absorbed. Enrofloxacin binds strongly to cattle faeces, but less so to poultry excreta.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
53

Experimentally-derived data are available on concentrations of enrofloxacin in meat products. Cattle were treated with enrofloxacin at 2.5 mg/kg bw/d for five days. Chickens were treated following typical treatment regimes (10 mg/kg bw/d for 10 d). Following treatment, the cattle were slaughtered and concentrations of enrofloxacin and the major metabolite, ciprofloxacin, were then measured in meat material. Enrofloxacin concentrations ranged from 0.7–5.75 mg/kg whereas ciprofloxacin concentrations ranged from 0.07–3.55 mg/kg (Table 2.1). Maximum concentrations of both enrofloxacin and ciprofloxacin were detected in the liver of both species.
Data are also available on the concentrations of enrofloxacin in eggs (Heitzman, 1997). Laying hens were administered enrofloxacin and concentrations of both enrofloxacin and ciprofloxacin were measured in the egg white and the egg yolk. The results are shown in Table 2.1.
Table 2.1 Concentrations of enrofloxacin and its metabolite, ciprofloxacin, in meat from cattle and chickens
Concentration of enrofloxacin (mg/kg)
Concentration of ciprofloxacin (mg/kg)
Beef – muscle 0.7 0.86 Beef – kidney 1.3 3.25 Beef – liver 1.31 3.55 Chicken – muscle 2.05 0.07 Chicken – liver 5.75 2.1 Laying hen – egg white (1 d withdrawal) 3.63 0.200Laying hen – egg yolk (1 d withdrawal) 3.14 0.21
3 ToxicokineticsAn analytical method is available for the determination of the combined residues of enrofloxacin and ciprofloxacin (the marker residue) in sheep tissues. The method involves extraction of the residues followed by column clean-up and analysis by HPLC with fluorescence detection. The limits of quantification were 25 µg/kg for enrofloxacin and for ciprofloxacin for ovine muscle and fat and 50 µg/kg for ovine liver and kidney (EMEA, 1999).
Enrofloxacin was well absorbed after oral administration to rats and the target species. In rats, the bioavailability of enrofloxacin was estimated to be 75% after a single oral dose of 5 mg/kg bw of radiolabelled enrofloxacin. The compound was widely distributed to all tissues with the highest concentrations in the liver and kidney. Elimination was rapid via urine and faeces, with most of the administered radioactivity excreted during the first 24 h after administration. Rat urine, after dosing with enrofloxacin (5 × 5 mg/kg bw/day), contained the following compounds: enrofloxacin 17%, ciprofloxacin 31%, oxociprofloxacin 5%, enrofloxacin amide 23%, dioxociprofloxacin 9%, desethylene ciprofloxacin 3%, desethylene enrofloxacin 2%, N-formyl ciprofloxacin <2%, oxoenrofloxacin <2%, and hydroxy oxoenrofloxacin 3% (EMEA, 1998b).
The main metabolite of enrofloxacin, ciprofloxacin, is well absorbed. In humans, the bioavailability of orally administered ciprofloxacin was 63–69%. The main sites of absorption in humans were the duodenum and jejenum. Ciprofloxacin was eliminated by both renal and non-renal routes. It was calculated that about 14% of orally administered ciprofloxacin was excreted in human faeces. In humans, oxociprofloxacin was the main metabolite of ciprofloxacin found in urine (EMEA, 1998b).
Enrofloxacin was well absorbed in rabbits after oral, i.m. and s.c. administration of 5 mg/kg bw. Cmax
values of 0.45, 3.04 and 2.07 µg/mℓ, respectively, were attained 1–2 hours after administration. The respective bioavailabilities were estimated to be 61%, 92% and 72%. In an in vitro study using fractions of rabbit liver, the metabolism of enrofloxacin was shown to be similar to that of the rat,
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
54

with ciprofloxacin as the main metabolite (12–15% of radioactivity). Rabbits were dosed orally, in drinking water, with 13 mg enrofloxacin/kg bw/day for 7 consecutive days. Residues of enrofloxalin in liver and muscle were detectable only in samples taken one day after treatment (<10–20 µg/kg). No residues were detected in fat. In kidney, residues of enrofloxacin declined from 13–100 µg/kg, one day after treatment, to <10–30 µg/kg, 4 days after the end of treatment (EMEA, 1998a). Rabbits were given a single oral dose of 5 mg/kg enrofloxacin and killed at intervals up to 12 h later. The mean tissue levels (enrofloxacin + ciprofloxacin) at 1 h were: liver 1411 µg/kg; kidney 1683 µg/kg, muscle 543 µg/kg and lung 151 µg/kg. These tissue levels declined rapidly: at 12 hours the respective values were 179 and 225 µg/kg for liver and kidney and were non-detectable for muscle and lung (Cagnardi et al., 2003).
The pharmacokinetics of enrofloxacin and ciprofloxacin were studied in dogs after an oral tablet of 5 mg/kg enrofloxacin and intravenous (i.v.) injections of 5 mg/kg enrofloxacin and ciprofloxacin. A model was used to examine enrofloxacin metabolism and fitted parameters were used to calculate plasma clearance: 0.729 ℓ/h/kg for enrofloxacin, 0.468 ℓ/h/kg for ciprofloxacin; distribution volume: 2.45 ℓ/kg for enrofloxacin, 1.92 ℓ/kg for ciprofloxacin; mean residence time: 3.47 hours for enrofloxacin, 4.2 hours for ciprofloxacin. Enrofloxacin was mainly metabolised to ciprofloxacin. The fraction of metabolised drug was similar after i.v. and oral doses, the hepatic first pass effect being low at 7.5% (HSDB, 2002).
Enrofloxacin was administered to goats by a single i.m. injection of 2.5 mg/kg bw. The peak plasma concentrations of enrofloxacin (1.13 µg/ml) and ciprofloxacin (0.24 µg/ml) were found at 0.8 and 1.2 h, respectively. The elimination half-life, volume of distribution, total body clearance and mean residence time of enrofloxacin were 0.74 hours, 1.42 ℓ/kg, 1329 mℓ/h/kg and 1.54 hours respectively. The metabolic conversion of enrofloxacin to ciprofloxacin was appreciable (36%) and the sum of the plasma concentrations of enrofloxacin and ciprofloxacin was maintained at or above 0.1 µg/ml for up to 4 hours. Enrofloxacin was given by single i.v. and i.m. administrations of 5 mg/kg bw to 5 adult Angora goats. Pharmacokinetics were best described by a two-compartment open model. The elimination half-life and volume of distribution after i.v. and i.m administrations were similar at 4.0–4.7 h and 1.2–1.5 ℓ/kg, respectively. Enrofloxacin was rapidly (t½, 0.25 h) and almost completely absorbed (90%) after i.m. administration (HSDB, 2002).
Serum concentrations and pharmacokinetics of enrofloxacin were studied in 6 mares after i.v. and (intragastric) i.g. administration at a single dose of 7.5 mg/kg bw. At 5 minutes after the i.v. injection, the mean serum concentration was 9.04 µg/mℓ, decreasing to 0.09 µg/mℓ by 24 hours. The elimination half-life was 5.33 ± 1.05 h and the area under the serum concentration versus time curve (AUC) was 21.03 ± 5.19 mg.h/ℓ. After the i.g. dose the mean peak serum concentration was 0.94 ± 0.97 µg/mℓ at 4 hours, declining to 0.29 ± 0.12 µg/mℓ by 24 hours (HSDB, 2002).
Four Jersey cows, 3–4 years old, 370 ± 20 kg weight were injected with enrofloxacin i.v. at a single dose of 5 mg/kg. Blood samples were collected at intervals up to 30 hours post-injection. The plasma concentration of enrofloxacin was 2.7 µg/mℓ at 2 minutes after dosing. The therapeutic concentration (0.1 µg/ml) of enrofloxacin and ciprofloxacin remained in the plasma for more than 12 and 8 hours, respectively. The plasma concentration time profile following the single i.v. dose was adequately fitted to a two-compartment open model. The elimination rate constant was 0.282/h with an elimination half-life of 2.61 hours. Enrofloxacin was widely distributed in the extravascular compartments. The clearance of enrofloxacin in this study was comparable to that seen in dairy cows of 21 mℓ/kg/min. The AUC of enrofloxacin in plasma was 4.4 µg.h/mℓ. Ciprofloxacin, the active metabolite, appeared within 2 minutes of the enrofloxacin injection at a concentration of 0.26 µg/mℓ with a peak plasma level of 1 µg/mℓ at 30 minutes. Its elimination half-life was 3.1 h and the AUC was 3.7 µg.h/ml (Varma et al., 2003).
The pharmacokinetics of i.v. and orally administered enrofloxacin were investigated in 6 adult sheep in single dose and multiple dose regimes. Single dose studies included an i.v. bolus of 5 mg/kg enrofloxacin and an oral suspension of 10 mg/kg crushed 68 mg enrofloxacin tablets. The multiple
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
55

dose regimen consisted of a mixture of 10 mg/kg enrofloxacin solution and grain per 24 hours for 7 days. The results for the single i.v. dose were: t1/2 = 4.76 ± 2.71 h, clearance = 210 ± 0.25 mℓ/kg/h and volume of distribution = 0.40 ± 0.06 ℓ/kg. The mean AUC was 23.23 ± 12.60 µg.h/mℓ. For the oral dose, the parameters were: t1/2 = 14.99 ± 1.14 h, the mean AUC was 29.92 ± 11.75 µg.h/mℓ and Cmax = 1.24 ± 0.98 µg/mℓ. For the multiple dosing, the mean Cmax was 3.11 ± 1.82 µg/mℓ. The amount of enrofloxacin metabolised to ciprofloxacin was <20% in all the studies. Enrofloxacin administered orally to sheep has a prolonged t1/2 and acts as a sustained release product (Bermingham & Papich, 2000). The pharmacokinetics of enrofloxacin were also examined after its i.v. and i.m. administration in 6 lactating sheep. After i.v. injection, as a bolus, the elimination half-life, the volume of distribution and the AUC were 3.30 ± 0.36 h, 2.91 ± 0.17 ℓ/kg and 4.19 ± 0.18 µg.h/ml, respectively. The maximum milk concentrations of enrofloxacin (Cmax) and the AUC were 2.38 ± 0.14 µg/mℓ and 23.76 ± 2.21 µg.h/mℓ, respectively. After i.m. administration, the elimination half-life, Cmax, time of Cmax
and bioavailability were 3.87 ± 0.10 h, 0.74 ± 0.07 µg/mℓ, 0.83 ± 0.12 h and 75.35% respectively. The Cmax and AUC for milk were 1.94 ± 0.13 µg/mℓ and 24.81 ± 2.25 µg.h/mℓ, respectively (Haritova et al., 2003).
Eight male pigs were used to study the pharmacokinetics of enrofloxacin after single i.v. and i.m. administrations at 2.5 mg/kg bw. Twelve pigs were used to study tissue residue distribution; they were given daily doses of enrofloxacin at 2.5 mg/kg i.m. for 3 days. The mean elimination half-life and mean residence time (MRT) of enrofloxacin in plasma were 9.64 ± 1.49 and 12.77 ± 2.15 h, respectively, after i.v. administration and 12.06 ± 0.68 and 17.15 ± 1.04 h, respectively, after i.m. administration. The bioavailability of enrofloxacin after i.m. administration was 74.53 ± 5.20%; the maximal plasma concentration of 1.17 ± 0.23 µg/ml was detected 1.81 ± 0.23 h after i.m. administration. After i.m. dosing, the metabolite, ciprofloxacin, was 51.5% of the parent drug plasma concentrations. The peak ciprofloxacin plasma level of 0.71 ± 0.14 µg/ml occurred at 1.75 ± 0.63 h after enrofloxacin administration. Mean concentrations of enrofloxacin and ciprofloxacin ranging between 0.029 and 0.079 µg/ml were detected 5 days after the last injection in muscle, liver , kidney and adipose tissue. After 10 days, ciprofloxacin was not detected in any tissue and enrofloxacin was detected only in liver (0.02 µg/g) and kidney (0.01 µg/g; Anadón et al., 1999).
Plasma enrofloxacin concentrations after single i.v. dosing to healthy and E. coli-infected broilers were best described by a two-compartment model. The disposition kinetics in the healthy and infected birds were as follows: elimination half-life 4.75 vs 3.63 h; apparent volume of the central compartment 1.11 vs 1.57 ℓ/kg; rate constant for transfer from peripheral to central compartment 1.15 vs 1.4 ℓ/h; and total body clearance 0.35 vs 0.53 ℓ/h/kg. After oral administration, the absorption half-life in infected birds was significantly longer than in healthy birds, while the elimination half-life and MRT were significantly shorter. Bioavailability was higher in infected birds at 72.5% compared with healthy birds at 69.8% (HSDB, 2002).
Several studies have been undertaken to quantify and characterise tissue residues in target species. When pigs were injected s.c. with 5 mg 14C-enrofloxacin/kg bw/day for 5 consecutive days, mean total residue levels (3 animals) in kidney, liver, muscle and fat fell from 2080, 1790, 862 and 148 µg equivalents/kg, respectively, at one day after the last dose, to 141, 277, 28 and 10 µg equivalents/kg, respectively, at 5 days. The residues, from 1 gilt and 1 barrow killed 12 hours after dosing, were characterised as approximately 80, 80, 90 and 99% as enrofloxacin in liver, muscle, kidney and fat; some ciprofloxacin was also present. When cattle were injected s.c. with 5 mg 14C-enrofloxacin/kg bw/day for 5 consecutive days, total residue levels in liver, kidney, muscle and fat fell from 10 050, 7470, 1540 and 625 µg/kg, respectively, at 8 hours after the last dose to 639, 66, 4 and 6 µg/kg, respectively, at 14 days. In the residues of tissues from 1 steer and 1 heifer killed 8 hours after the last dose, enrofloxacin accounted for 21–26%, 27–31%, 55–56% and 35–59% in the liver, kidney, muscle and fat respectively. Ciprofloxacin accounted for 38–44%, 50–55%, 33–34% and 5–7% of the residues in these same tissues. Oxoenrofloxacin was the major component of the residues in fat. When 3 lactating cows were dosed i.v. with 5 mg 14C-enrofloxacin/kg bw/day for 5 consecutive days, the total residue levels in milk samples fell from approximately 3267 µg/ℓ at 6 hours after dosing to <17–140 µg/kg at 57 hours. About 80% of the residues in milk samples taken 6–24 hours
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
56

after dosing were ciprofloxacin and 8% were enrofloxacin. In neonatal sheep given a single oral dose of 7.5 mg enrofloxacin/kg bw, the mean residue levels of enrofloxacin, 2 days after dosing, in liver, kidney, muscle and fat were 520, 1370, 993 and 1370 µg/kg, respectively, and these declined to 48, 13, 13 and 13 µg/kg 4 days after dosing. Over the same time period, mean residues of ciprofloxacin in these tissues declined from 483, 175, 265 and 175 µg/kg in liver, kidney, muscle and fat to 53, 13, 13 and 13 µg/kg. When poultry were dosed orally with 10 mg 14C enrofloxacin/kg bw for 7 days, the total residue levels in liver samples taken over 6–24 h after the last dose declined from 4890 µg equivalents/kg to 89 µg/kg in chickens and from 8240 µg equivalents/kg to 2662 µg/kg in turkeys. In chickens, 61–66%, 51%, 53–62% and 85% of the total residues present in kidney, liver, fat and muscle, 6 h after dosing, were composed of enrofloxacin. The comparative values for turkeys were 58–60%, 94–97%, and 99% for liver, kidney and muscle, respectively (EMEA, 1998a; EMEA, 1998b).
In an in vitro experiment, the inhibition of P-450 reductase by enrofloxacin was assessed by measuring the NADPH-cytochrome c reductase activity and the inhibition of P-450 1A1, 1A2 and 11B in rat liver microsomes. NADPH-cytochrome c reductase was not affected. Enrofloxacin induced a strong concentration-dependent inhibition of P-450 1A1 and 1A2. In an in vivo study, the effects of five administrations of 5, 25, or 100 mg enrofloxacin/kg bw/day were assessed in rats. A slight induction of P-450 11B1 and 11B2 expression and activity (140% of controls) was present only at the 5 mg/kg bw/day dose level (Vancutsem & Babish, 1996). Enrofloxacin was administered orally to 25-day old commercial broiler chickens at a dosage of 10 mg/kg bw daily for a 3 day period. Enrofloxacin was found significantly to inhibit microsomal cytochrome P-450 monooxygenases up to 9 days post-treatment (Shlosberg et al., 1997).
4 Toxicity profile4.1 Acute toxicityAlthough there appear to be no conventional acute toxicity tests for enrofloxacin, the compound is considered to be of low acute toxicity on the basis of its reactions in other types of toxicity testing. For example, acute signs were not seen at high dose levels (up to about 1000 mg/kg bw) of enrofloxacin in a 13-week dietary study in rats (EMEA, 1998c).
The main metabolite of enrofloxacin, ciprofloxacin, has been widely used in human medicine for several years; side effects reported include gastrointestinal disturbances, some hypersensitivity reactions and crystalluria (EMEA, 1998c).
4.2 Repeat dose toxicityIn a 13-week study in rats, enrofloxacin was administered in the feed at doses up to 7500 mg/kg. Reduced bodyweight gain was observed in both sexes receiving the top dose. Caecal distension occurred with no corresponding microscopic alterations, a common finding in rats given large doses of antibiotics. Histopathological changes were observed in the knee joints in 3 out of 30 animals receiving the top dose and, in male rats at this dose level, round or oval cells in the epididymides (12/15 rats) and in the seminiferous tubules (5/15 rats). These cells probably represented a degenerating or necrotic ‘cap phase’ spermatid. The no-effect level for the study was considered to be 300 mg/kg enrofloxacin in diet (equal to 40 mg/kg bw). In order to examine the testicular effects further, a study incorporating recovery periods was undertaken. In rats at the lowest dietary dose administered (125 mg/kg = 10 mg/kg bw/day), no abnormal spermatozoa were found. Neither was abnormal spermatozoa found in the seminiferous tubules of any rat at the end of a 90-day recovery phase and recovery of the epididymides was also apparent. The effects on rat testes were therefore reversible. Degenerative changes, typical of those induced by floroquinolone antibiotics, were found in the articular cartilage after administration of enrofloxacin to immature animals such as calves, piglets and puppies. Dogs were the most sensitive to this effect and a NOEL of 3 mg/kg bw/day was established for arthropathy in a 13-week repeat dose study in which the dogs were 3 months old at the
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
57

start (EMEA, 1998c). Following a re-evaluation of the results of the 13-week repeat dose toxicity studies in rats and dogs, it was concluded that effects seen on the testes of dogs were inconsistent, not dose related and were of a different type from those seen in rats. The toxicological ADI for enrofloxacin should therefore be based on the NOEL of 3 mg/kg bw/day for arthropathy in juvenile dogs (EMEA, 1998b).
Ten Turkish Shepherd dogs, 24–30 months old, were given enrofloxacin i.m. at 5 mg/kg bw daily for 14 days and blood samples were taken regularly during the study period for investigation of biochemical, haematological and blood gas parameters. Acidosis and temporary increases in indirect bilirubin, sodium, partial pressure of CO2 and mean corpuscular volume and decreased levels of inorganic phosphorus, ionised calcium, potassium, partial pressure of O2 and standard bicarbonate were observed (Tras et al., 2001).
The possible relationship between the administration of parenteral enrofloxacin and the onset of acute retinal degeneration in cats was evaluated in a retrospective clinical study. The cats identified were all of the domestic shorthair breed; 7 were females and 10 were males. Ages ranged from 3 to 16 years. The daily and total dosage of enrofloxacin and number of days of administration were highly variable. The clinical signs were most often mydriasis and acute blindness. All cats had diffuse retinal degeneration as evidenced by increased tapetal reflectivity and retinal vascular attenuation. Vision returned in a few cats, but retinal degeneration persisted or even progressed. Histopathology of 2 eyes revealed primarily outer retinal degeneration with diffuse loss of outer nuclear and photoreceptor layers and hypertrophy and proliferation of the retinal pigment epithelium (Gelatt et al., 2001).
No evidence of leg weakness was observed in poultry treated at the recommended rate of enrofloxacin. No evidence of arthropathy was observed in calves and pigs treated according to the recommended dosage regime. However, oral administration of 30 mg/kg bw/day for 14 days to calves and 50 mg/kg bw/day to pigs produced histopathological evidence of arthropathy. No adverse effects were observed in neonatal lambs treated orally with 5–7 mg/kg bw enrofloxacin and the lambs grew normally over the first 3 months of life (EMEA, 1998a). Toxicity problems associated with cartilage and bone development have occurred with enrofloxacin use in young horses and it is therefore not recommended for use in this species (Vancutsem et al., 1990).
A frequency of 0.5–5% of nervous manifestations have been described after a dose of 250 mg ciprofloxacin in humans (= 3.6 mg/kg bw). The intensity of these manifestations is diverse and includes headaches, dizziness, tremors, hallucinations, psychosis and seizures (Vancutsem et al., 1990).
4.3 Carcinogenicity and mutagenicity In a rat carcinogenicity study on enrofloxacin, the incidence of endocardial tumours was elevated in females at the top dose (level not stated). When the incidence of endocardial tumours was combined with the incidence of proliferative lesions, the difference from the controls was significant in both sexes at the top dose. Also, the incidence of bile duct hyperplasia was increased in a dose-related manner. However, an independent review of the histopathology indicated that both the endocardial neoplasms and the Schwann cell-like hyperplasia were not associated with the administration of enrofloxacin. In a second carcinogenicity study in the same strain of rat, no neoplastic change attributable to enrofloxacin treatment was observed. However, because evidence of bile duct hyperplasia was apparent at the lowest dose level, a third study was carried out. Histopathological examinations after 12 months showed no evidence of a dose-related increase in bile duct hyperplasia at dose levels of up to 50 mg/kg enrofloxacin in the diet. Although the incidence of malignant lymphoma was increased in treated groups in a carcinogenicity study in the mouse with enrofloxacin, the incidence was within the historical control range. It was concluded from these studies that enrofloxacin was not carcinogenic (EMEA, 1998c).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
58

Several in vitro mutagenicity assays have been reported for enrofloxacin. The compound produced no increase in revertants in a bacterial assay for gene mutation though only very low drug concentrations could be used because of the high toxicity shown towards the tester strains of bacteria. Sporadic increases in mutant frequency were observed in a CHO HGPRT forward mutation assay but with no evidence of a dose–response and an in vitro UDS assay gave negative results (EMEA, 1998c). Other reported studies included a S. typhimurium assay with the TA102 tester strain with enrofloxacin at 0.015 µg/plate (- S9) which gave a positive result and a mouse lymphoma L5178Y (TK+/TK-) test (- S9) with enrofloxacin at 500 µg/mℓ which also gave a positive result. Another mouse lymphoma L5178Y (HPRT) 6-thioguanine test gave a negative result. A Chinese hamster V-79 assay (- S9) with enrofloxacin at 600 µg/mℓ was also negative (CCRIS, 2000). Chromosomal aberrations were evaluated in cultures of human peripheral lymphocytes exposed to enrofloxacin or ciprofloxacin. The control analysis resulted in 3.6 ± 0.6 chromosomal aberrations/100 cells while the treated cultures resulted in 8.3 ± 0.8 and 9.6 ± 1.2 aberrations/100 cells at 5 and 50 µg/mℓ enrofloxacin and 5.6 ± 1.3 and 7.7 ± 3.5 aberrations/100 cells at 5 and 25 µg/mℓ ciprofloxacin. Some cytotoxic effect was noted with enrofloxacin at 50 µg/ml. These results indicate a genotoxic effect for these two compounds in this system (Gorla et al., 1999).
There was a small but dose-related increase in sister chromatid exchanges per metaphase in an in vivo assay in hamster bone marrow but the increase was not statistically significant and no evidence of mutagenicity was found in either an in vivo mouse micronucleus test nor in an in vivo cytogenetics assay in rat bone marrow (EMEA, 1998c).
4.4 Reproductive and developmental toxicityEffects on the male rat epididymides and seminiferous tubules have been described in a 13-week study in Section 4.2. Similar effects on the male reproductive tract due to enrofloxacin were observed in several other studies in rats including a multigeneration study in which reproductive performance was impaired at the top dose level of 7500 mg/kg but not at 500 mg/kg. These effects on spermatogenesis were not observed in repeat dose studies in other species. Enrofloxacin was not teratogenic in either the rat or the chinchilla and NOELs for fetotoxicity of 50 and 25 mg/kg bw/day, respectively, were established (EMEA, 1998c). Treatment of rabbits according to the recommended dosage regime had no adverse effects on general health, reproductive performance or development of the offspring (EMEA, 1998a).
The placental transfer of enrofloxacin and ciprofloxacin was evaluated in a two-step infusion programme to establish steady state maternal plasma concentrations for these drugs. For each compound, the placenta in 5 rabbits was perfused for 200 minutes with Earle’s enriched bicarbonate buffer at a flow rate of 1.5 mℓ/min. Plasma protein binding estimation indicated no difference between the drugs. Placental clearance of the drugs was significantly different: 0.88 ± 0.13 mℓ/min for enrofloxacin and 0.06 ± 0.02 mℓ/min for ciprofloxacin. These values accounted for 81 and 5%, respectively, of the placental clearance found for antipyrene, a commonly used indicator of placental exchange (HSDB, 2002).
5 Guidelines and standardsThe EMEA CVMP has based a toxicological ADI for enrofloxacin on the NOEL of 3 mg/kg bw/day for arthropathy in juvenile dogs (see Section 4.2). By applying a safety factor of 100 the toxicological ADI for enrofloxacin was determined to be 30 µg/kg bw (EMEA, 1998b)
A microbiological ADI for enrofloxacin has also been calculated, using a formula recommended by the CVMP involving a daily faecal bolus of 150 g. The calculated microbiological ADI was shown to be 6.2 µg/kg bw, that is 372 µg/person (EMEA, 1998b).
The CVMP recommended the inclusion of enrofloxacin in Annex 1 of Council Regulation (EEC) No. 2377/90 in accordance with the MRLs shown in Table 5.1. In coming to these recommendations, the
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
59

Committee took into account that a microbiological ADI of 372 µg/person had been established for enrofloxacin (EMEA, 2002).
In 1997, JECFA established a microbiological ADI of 0-2 g/kg bw/day on the basis of data on the antimicrobial activity of enrofloxacin against 10 strains of the most sensitive relevant genus isolated from the human gastrointestinal tract (JECFA, 1997).
Table 5.1 MRLs for enrofloxacinMarker residue Animal species MRL
(μg/kg)
Target tissue
Sum of enrofloxacin & ciprofloxacin Bovine, ovine, caprine 100 Muscle100 Fat300 Liver200 Kidney100 Milk
Porcine, rabbits 100 Muscle100 Fat200 Liver300 Kidney
Poultrya 100 Muscle100 Skin+fat200 Liver300 Kidney
All food-producing 100 Musclespecies except the above 100 Fat
200 Liver200 Kidney
aNot for use in animals from which eggs are produced for human consumption
It was estimated that extending the MRLs to all food-producing species, as proposed above, would result in a consumer intake not exceeding 74% of the CVMP microbiologically-based ADI (EMEA, 2002).
The US FDA has set a tolerance of 0.3 ppm for residues of enrofloxacin (marker residue) in muscle (target tissue) of chickens and turkeys. For cattle, a tolerance of 0.1 ppm for desethylene ciprofloxacin (marker residue) has been established in liver, the target tissue (HSDB, 2002).
6 ReferencesAnadón A, Martinez-Larrañaga MR, Díaz MJ, Fernández-Cruz ML, Martínez MA, Frejo MT, Martínez M, Iturbe J & Tafur M (1999) Pharmacokinetic variables and tissue residues of enrofloxacin and ciprofloxacin in healthy pigs. Am J Vet Res, 60, 1377-1382
Bishop Y, ed (2004) The Veterinary National Formulary (Sixth Edition), London, UK, Royal Pharmaceutical Society of Great Britain/British Veterinary Association
Bermingham EC & Papich MG (2000) The pharmacokinetics of intravenous and oral enrofloxacin in sheep. [Abstract] J Vet Int Med, 14, 332
Cagnardi P, Villa R, Sonzogni O & Carli S (2003) Tissue distribution and depletion of enrofloxacin in rabbit. J Vet Pharmacol Ther, 26 (Suppl 1), 284
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
60

CCRIS (2000) Enrofloxacin. From: Chemical Carcinogenesis Research Information System, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [January 2004] at http://toxnet.nlm.nih.gov
Cerniglia CE & Kotarski S (1999) Evaluation of veterinary drug residues in food for their potential to affect human intestinal microflora. Regul Toxicol Pharmacol, 29, 238-261
EMEA (1998a) Enrofloxacin (extension to sheep, rabbits and lactating cows) Summary Report (3) (EMEA/MRL/389/98-Final), London, UK, European Agency for the Evaluation of Medicinal Products, Available [March 2004a] at http://www.emea.europa.eu/pdfs/vet/mrls/038998en.pdf
EMEA (1998b) Enrofloxacin (modification for bovine, porcine and poultry) Summary Report (2) (EMEA/MRL/388/98-Final), London, UK, European Agency for the Evaluation of Medicinal Products, Available [March 2004b] at http://www.emea.europa.eu/pdfs/vet/mrls/038898en.pdf
EMEA (1998c) Enrofloxacin. Summary Report (1), London, UK, European Agency for the Evaluation of Medicinal Products, Available [March 2004c] at http://www.emea.europa.eu/pdfs/vet/mrls/enro01en.pdf
EMEA (1999) Enrofloxacin (extension to sheep) Summary Report (4) (EMEA/MRL/574/99-Final), London, UK, European Agency for the Evaluation of Medicinal Products, Available [March 2004] at http://www.emea.europa.eu/pdfs/vet/mrls/057499en.pdf
EMEA (2002) Enrofloxacin (extension to all food producing species) Summary Report (5) (EMEA/MRL/820/02-Final), London, UK, European Agency for the Evaluation of Medicinal Products, Available [March 2004] at http://www.emea.europa.eu/pdfs/vet/mrls/082002en.pdf
Gelatt KN, van der Woerdt A, Ketring KL, Andrew SE, Brooks DE, Biros DJ, Denis HM & Cutler TJ (2001) Enrofloxacin-associated retinal degeneration in cats. Vet Ophthalmol, 4, 99-106
Gorla N, García-Ovando H & Larripa I (1999) Chromosomal aberrations in human lymphocytes exposed in vitro to enrofloxacin and ciprofloxacin. Toxicol Letts, 104, 43-48
Haritova A, Lashev L & Pashov D (2003) Pharmacokinetics of enrofloxacin in lactating sheep. Res Vet Sci, 74, 241-245
Heitzman RJ (1997) Veterinary Drug Residue: Enrofloxacin (Report to the Joint FAO/WHO Expert Committee on Food Additives)
HSDB (2002) Enrofloxacin. From: Hazardous Substances Databank, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [June 2003] at http://toxnet.nlm.nih.gov
NOAH (2004) Compendium of Data Sheets for Animal Medicines 2005, Enfield, UK, National Office of Animal Health Ltd
JECFA (1997) Toxicological Evaluation of Certain Veterinary Drug Residues in Food (Food Additives Series 39), Geneva, Switzerland, World Health Organization, Available [July 2004] at http://www.inchem.org/documents/jecfa/jecmono/v39je06.htm
Shlosberg A, Ershov E, Bellaiche M, Hanji V, Weisman Y & Soback S (1997) The inhibitory effects of the fluoroquinolone antimicrobials norfloxacin and enrofloxacin on hepatic microsomal cytochrome P-450 monooxygenases in broiler chickens. Drug Metab Drug Interact, 14, 109-122
Tras B, Maden M, Bas AL, Elmas M, Yazar E & Civelek T (2001) Investigation of biochemical and haematological side-effects of enrofloxacin in dogs. J Vet Med A, 48, 59-63
Vancutsem PM & Babish JG (1996) In vitro and in vivo study of the effects of enrofloxacin on hepatic cytochrome P-450: Potential for drug interactions. Vet Hum Toxicol, 38, 254-259
Vancutsem PM, Babish JG & Schwark WS (1990) The fluoroquinolone antimicrobials: Structure, antimicrobial activity, pharmacokinetics, clinical use in domestic animals and toxicity. Cornell Vet, 80, 173-186
Varma R, Ahmad AH, Sharma LD, Aggarwal P & Ahuja V (2003) Pharmacokinetics of enrofloxacin and its active metabolite ciprofloxacin in cows following single dose intravenous administration. J Vet Pharmacol Ther, 26, 303-305
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
61

Annex 5 17β-Estradiol1 Introduction17β-Estradiol (estradiol; 17β-estra-1,3,5(10)-triene-3,17-diol; CAS No. 50-28-2) is the most active naturally occurring estrogenic hormone. It has been used in human medicine for the treatment of the climacteric, particularly for vasomotor and psychological disturbances. It has also been used for local treatment of atropic vaginitis, for the chemotherapy of advanced prostatic cancer and for the prevention of post-partum breast engorgement. Other uses include the treatment of primary amenorrhoea, delayed onset of puberty and chemotherapy of breast neoplasms in postmenopausal women. It has been used in veterinary medicine for estrogenic hormone therapy, administered by injection to mares and cows (1–5 mg), ewes (0.3–2.5 mg) and bitches (0.1–1 mg; IARC, 1979).
Estradiol is now normally used for medical treatment in the form of its semi-synthetic esters such as the valerate and cypionate. These salts are used for the treatment of menopausal symptoms, female hypogonadism and primary ovarian failure (HSDB, 2002). Estradiol is commercially available as micronised tablets, as topical transdermal patches, as a vaginal cream and as an extended-release vaginal insert. Estradiol salts are commercially available as injectable suspensions in oil for parenteral administration (IARC, 1999).
2 Uses and exposureEstradiol is an estrogenic hormone licensed currently for use in veterinary medicine in the UK for treating misalliance in the bitch (unwanted pregnancy) and in the male dog, excessive libido, anal adenoma and prostate hyperplasia. EU Directives 96/22/EC and 2003/74/EC prohibit the use of substances having a hormonal action for growth promotion in farm animals and drastically reduces the circumstances under which estradiol may be administered for other purposes to food producing animals. There are currently three uses permissible on a transitional basis and under strict veterinary control. These are treatment of fetal mummification, pyometra in cattle and estrus induction in cattle, horses, sheep and goats. The latter use was to be phased out by September 2006. Steps have to be taken to ensure that the treated animal and its products are not used for human or animal consumption (Bishop, 2004).
There are currently only two products containing estradiol licensed for use in veterinary medicine in the UK. MesalinTM containing estradiol benzoate at 200 g/ml is indicated for misalliance in dogs at 10 g/kg on 2–3 occasions after mating. PridTM, an intravaginal device containing 10 mg estradiol benzoate and 1.55 g progesterone is used to stimulate ovarian activity in anovulatory and subestrus cows and for the synchronisation of estrus to allowed predetermined insemination. The device is normally inserted for 12 days (Bishop, 2004).
Estradiol benzoateTM was available, until recently, as an oily suspension containing 5 mg/ml estradiol benzoate for use in cattle for treatment of endometritis and pyometra at a dose rate of 3 mg/500 kg repeated at 7-day intervals if required. However, the product is no longer available in the UK (Bishop, 1996). Potential sources of human exposure are extremely limited but could conceivably occur through ingestion of animal products containing drug residues following use in food animals. Treatment of the food animals would, howver, only be expected to occur through use of existing, (declining) stocks and then generally only for individual or small numbers of animals. The intravaginal device form requires a nil milk withdrawal period and animals must not be slaughtered for 24 hours after insertion or 6 hours after removal. When available, the withdrawal periods for injectable products were set at 15 days for meat and nil for milk in cattle. Use of pre-existing
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
62

injectable preparations for cattle remains permissible but use ie expected to continue to decline as stocks become exhausted.
The US FDA requirements for the implantation or injectable dosage form new animal drugs are: Conditions of use: for implantation in steers and heifers; Indications for use: for increased rate of weight gain in suckling and pastured growing steers; for improved feed efficiency and increased rate of weight gain in confined steers and heifers (HSDB, 2002).
2.1 Exposure concentrations in meatUsing information on usage of estradiol and on absorption and excretion, likely ‘worst case’ concentrations in meat products were estimated using Equation 1:
Equation 1M = D × T × Fabs × (1–Fexc)
where M is the predicted concentration in meat (mg/kg); D is the dose of substance (mg/kg bw/day); T is the duration of the treatment (days); Fabs is the fraction of compound absorbed; and Fexc is the fraction of compound excreted.
For cattle it was assumed that the total of 10 mg is applied to 450 kg cattle giving a dose of 0.02 mg/kg bw . Assuming 100% bioavailability and no excretion, a ‘worst case’ concentration of 0.02 mg/kg was calculated.
There was insufficient information to estimate concentrations of estradiol in sheep.
3 ToxicokineticsThe absorption of the various estradiol preparations differs, even when the route of exposure remains the same. For example, crystalline estradiol applied dermally as a cream diffuses more readily through the skin to the systemic circulation than esterified estradiol because it is more lipophilic than its ester derivative. Similarly, micronised estradiol is absorbed more rapidly than crystalline estradiol because of its small particle size. The absorption of estradiol in humans has been studied extensively; however, the results are difficult to compare as different preparations of estradiol and different routes of administration have been used. Some examples are described in the following passages. The oral administration of a first estradiol tablet, 2 mg micronised estradiol, to 32 healthy post-menopausal women resulted in a maximal plasma estradiol concentration of 1084 pg/ml, 49 minutes after administration, which decreased rapidly in the subsequent 3 hours. After the fifth tablet, the average concentration of estradiol was about 418 pg/ml, which was 12 times greater than that found when a transdermal patch was used. A comparison of the pharmacokinetic parameters of oral and sublingual administration of micronised estradiol to post-menopausal women revealed that the time to the maximal concentration of estradiol was significantly different by the two routes, being 1 hour or less for sublingual and 6.5–7.6 hours for oral administration. Transdermal estradiol provides physiological levels of estradiol at a constant rate; the transdermal route avoids loss of drug by the hepatic first-pass effect and minimally affects hepatic protein metabolism. Maximal serum concentrations of estradiol are reached within 2–8 hours of application of a transdermal system. When 50 µg/day transdermal therapy is used on a long-term basis, the mean steady-state serum concentrations can be 20–50 pg/ml. Pellets of 25 and 50 mg estradiol produced serum concentrations of estradiol of approximately 50–70 pg/ml and 100–120 pg/ml respectively, for up to several months. Relatively constant serum concentrations of approximately 150 pg/ml estradiol and estrone were achieved for 21 days from a vaginal ring containing 400 mg estradiol. The bioavailability of estradiol from vaginal rings has been reported to be 13 ± 7 % (range, 7–27 %) in post-menopausal women (IARC, 1999).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
63

Most of the available data on distribution are based on intravenous (i.v.) administration. After i.v. administration of estradiol to post-menopausal women, a high clearance (1.8 ± 0.6 ℓ/min) and a low distribution volume (51 ± 28 ℓ) were found. Estrogens circulate in the blood bound to albumin (about 60 %) and sex hormone binding globulin, SHBG, (about 38 %). Estradiol binds weakly to albumin (low affinity/high capacity; plasma concentration, 40 g/ℓ), about one-third is tightly bound to SHBG (high affinity/low capacity) and a small fraction (<3 %) is ‘free’. After i.v. administration, the distribution volume of estradiol at steady state was only about 70 ℓ, representing 1.5–2 times the total body water in fertile women. Its low level of distribution is consistent with its high level of binding to plasma proteins. Because of the high concentration of albumin (about 5 orders of magnitude higher than SHBG) and its rapid dissociation, albumin may serve a more important regulating role (IARC, 1999).
Limited experimental studies are available on the absorption, distribution and excretion of estradiol. Estradiol represented only 6 % of the total estrogen detected in the hepatic portal vein after estradiol was placed in the stomach of a prepubertal pig; thus, most of the estradiol was converted or conjugated before entering the hepatic portal vein. The blood concentrations of estradiol glucuronide, estrone glucuronide and estrone sulphate but not that of estradiol or estrone in the jugular vein rose and remained elevated for several hours, indicating that estradiol and estrone are completely converted and/or removed by the liver. In a comparison of the serum and tissue concentrations of estradiol in fertile female and in castrated male Syrian golden hamsters, estradiol pellets (20 mg) were implanted into the shoulder region of groups of 4–6 hamsters every 45 days to maintain the hormone concentration, and the animals were killed after 15 days and at 30-day intervals. The average serum estradiol level in the cycling female hamsters was 79 pg/mℓ on days 1–2 and 311 pg/mℓ on days 3–4, attaining a maximum of 358 pg/mℓ on day 4 of the cycle. The levels on days 3–4 of the cycle were 3-fold higher than those on day 1 in uterine tissue, 2-fold higher in renal tissue and 2.6-fold higher in hepatic tissue. As expected, the serum estradiol concentrations of untreated castrated male hamsters did not vary appreciably over the 6 months of the study and the average was about 32 pg/mℓ. A 10 mg dose of crystalline estradiol placed in the rectum of prepubertal gilts resulted in increased concentrations of estradiol, estrone, estradiol glucuronide, estrone glucuronide and estrone sulphate in the hepatic portal vein within 30 minutes and the levels remained elevated for several hours. After estradiol was placed in the stomach of the prepubertal gilts, the levels of estradiol, estrone, estradiol glucuronide, estrone glucuronide and estrone sulphate in the hepatic portal vein rose within 5 minutes and remained elevated for several hours. Most of the conjugated metabolites in liver and kidney of cattle are glucuronides (85–95 %). In pregnant rhesus monkeys, estradiol was eliminated from the maternal circulation principally by conversion to glucuronide conjugates. After pulse injection of 3H-estrone sulphate to adult rhesus monkeys, the initial volume of distribution was 4.6 ± 0.9 ℓ and the metabolic clearance rate was 42 ± 2.9 ℓ/day. After infusion, estrone sulphate is cleared slowly and is converted to both estrone and estradiol (IARC, 1999).
The distribution of estradiol in female Wistar rats was measured in heart, liver, kidney, brain and plasma by radioimmunoassay for 24 h after i.v. administration of 0.1 mg/kg bw or after intragastric (i.g.) administration of 10 mg/kg bw. The concentration of estradiol in liver was 20 times higher after i.g. than after i.v. administration when equivalent plasma levels of hormone were evaluated. Negligible differences were seen in the estradiol concentrations in other tissues. The tissue concentrations of estradiol were higher than those in plasma at all times. The absolute bioavailability, as measured by comparison of the dose-corrected values for the area under the integrated concentration–time curve (AUC), was 8.3 % after an i.g. dose of 10 mg/kg bw. The total clearance was 154 mℓ/min/kg bw. The half-life of estradiol in liver was 2.6 h. Fourteen young women received a single dose of 2, 4 or 8 mg estradiol orally or 0.3 mg i.v. The 8 mg dose resulted in incomplete absorption. The absolute bioavailability of the 4 mg dose was calculated to be 5%. The mean ratio of free estrone:estradiol was 1 after i.v. injection and 20 after oral administration. In a two-compartment model, the AUC for young women given the 0.3 mg dose i.v. was 4000 pg-h/mℓ; total clearance was 22 mℓ/min/kg bw. The terminal plasma half-life of estradiol after i.v. administration to humans was 27 min, the volume of distribution was calculated to be 0.082 ℓ/kg bw (JECFA, 2000).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
64

The metabolic disposition of estrogens includes oxidative reaction, mainly hydroxylation, and conjugative reactions by glucuronidation, sulphonation and/or O-methylation. Estradiol is converted to estrone by a 17β-hydroxysteroid dehydrogenase; the estrone produced is further metabolised to 16 α-hydroxyestrone and then to estriol. Hydroxylation of estriol at the 2 position is a major metabolic pathway in the liver. There are large inter-individual differences in estradiol 2-hydroxylation in human liver samples, which may reflect differences in estrogenic action. 4-Hydroxylation of estradiol to a catechol is a minor pathway (usually <15 % of 2-hydroxylation) in the liver. 4-Hydroxylation of estradiol is the dominant pathway of catechol estrogen formation in human breast and uterus. In humans, 4-hydroxylation of estradiol is catalysed by the cytochrome P-450 enzyme CYP1B1. Estradiol and estrone hydroxylates can undergo metabolic redox cycling in vitro to generate free radicals such as superoxide and chemically reactive estrogen semiquinone/quinone intermediates. In the presence of fatty acid acyl-coenzyme A, estradiol can be converted at the C-17 position to the very lipophilic estrogen fatty acid esters by enzymes present in liver and in estrogen target organs such as breast and placenta. Estradiol, estrone and estriol are excreted in the bile as glucuronides and undergo enterohepatic recirculation. Their glucuronides are hydrolysed in the intestine and unconjugated estradiol or estrone is reabsorbed from the intestine by enterohepatic cycling. After administration of estradiol to rats, glucuronides and sulphates of 16-keto-estradiol and of 2- and 3-methyl esters of 2-hydroxyestriol and 2-hydroxy-16-ketoestradiol were excreted in the bile (IARC, 1999).
Lysosomes from male Syrian hamster livers and kidneys can catalyse the deconjugation of estradiol and estrone glucuronides. The rates of deconjugation were higher in the kidney than in the liver, by 56 % for estrone and 34 % for estradiol. Treatment of hamsters for 9 days with subcutaneous (s.c.) implants containing 25 mg estradiol (releasing 61 µg/day) increased lysosomal estrone and estradiol 3 β-glucuronidase activity in kidney by 15 and 25 %, respectively, and by about 100% in liver. Human liver microsomal sulphatases convert estrone sulphate to estrone before 16 α-hydroxylation (JECFA, 2000).
The metabolism of estradiol and estrone is similar in rats and in humans, in that both species transform these steroids mainly by (aromatic) 2-hydroxylation and also by 16 α-hydroxylation. Glucuronides of the various metabolites are excreted in the bile. Differences in the metabolism of estrogens by humans and rats lie mostly in the type of conjugation. When estriol is administered to rats, glucuronides and, to a lesser extent, sulphates of 16-ketoestradiol and of 2- and 3- methyl ethers of 2-hydroxyestriol and 2-hydroxy-16-ketoestradiol are excreted in the bile. In contrast, hydroxylations at C-6 or C-7 of ring B of estradiol and estrone are a minor pathway in rats. In rabbit liver, the presence of two enzymes unique to this organ leads to a metabolite pattern that cannot be compared with the human situation. Rabbit liver microsomes contain glucosyl and N-acetylglucosaminyl transferases, which transfer glucose from UDP-glucose to the 3-hydroxy group of estrone and estradiol or N-acetylglucosamine from the UDP form to the 17 α-hydroxy group of estradiol-17 alpha. In addition, a 17 α-hydroxysteroid dehydrogenase is found in rabbit liver cytosol. Hence, the major metabolite of estrone and estradiol in rabbits is 17 α-estradiol, which is mainly conjugated to estradiol-17 α-N-acetylglucosaminide, or to estradiol-3-glucuronide-17 α-N-acetylglucosaminide. These metabolic pathways do not occur in man. Sulphates are the major conjugates in guinea pigs and metabolism by 16 α-hydroxylation is thought to take place on the estrone 3-sulphate. Glucuronidisation, if any, plays only a minor role, in contrast to the human situation. Canine metabolism of estrogens also differs from that of humans. Only small amounts of labelled estradiol are converted to estriol; the bulk of the radioactive dose is excreted in urine as conjugates of estradiol and estrone. After administration of labelled estriol to dogs, a unique pattern of conjugates was found in bile and urine, probably including polyglucuronides. Furthermore, no significant enterohepaptic circulation occurs in contrast to the distribution of estrogenic hormones in most other species. 17 α-Estradiol is a major metabolite in species such as the domestic fowl, bulls and sheep. Liver tissues of pigs contain significant amounts of 17 β-hydroxysteroid dehydrogenase and glucuronyl transferase; in minipigs, oxidoreduction at C-17 is the predominant reaction, whereas in contrast to humans, hydroxylation of estrogens plays only a minor role. On the basis of the urinary and faecal excretion of estrogens, non-human primates resemble humans much more closely than
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
65

rodents or dogs. The conjugates of estrogens are similar in rhesus monkeys and in humans. Baboons also show some similarities to humans in estrogen metabolism (IARC, 1979).
Thus, esters of estradiol are rapidly cleaved to estradiol in vivo, and may also be considered to be endogenous substances, since the residues are structurally identical to estradiol produced in humans and other mammals after hydrolysis (JECFA, 2000).
4 Toxicity profile4.1 Acute toxicityThere appear to be no conventional acute toxicity studies on estradiol. However, estradiol is considered to be rather inactive when administered orally, due to gastrointestinal and/or hepatic inactivation (JECFA, 2000).
The therapeutic dose of fine-particle estrogen given orally is 0.5–2 mg/day. No adverse effects were reported in children after accidental ingestion of large doses of estrogen-containing oral contraceptives. An electroencephalogram of a young woman who took 160 mg estradiol valerate (80 tablets of 2 mg), showed traces typical of subcortical disturbance on the first day; however, the recording was normal one week later (JECFA, 2000).
In clinical studies, the most common adverse effect reported with transdermal estradiol therapy was erythema and irritation at the application site, which occurred to some degree in up to 31% of patients and required discontinuance in about 2–7 % of patients. The irritation generally resolved completely within a day or so (HSDB, 2002).
4.2 Repeat dose toxicityEstradiol implant overdose (200 mg estradiol by implant every 3 weeks) led to fluid retention, facial swelling and pitting oedema of the thighs. Serum estradiol levels were elevated and fell over several years after the implants were discontinued. In 2 other patients, repeated implants of estradiol over a 1–2 year period led to hypertension, flushing, fatigue and nasal congestion (HSDB, 2002). Other side effects from the use of estrogens for contraception or hormone replacement include thromboembolic and other vascular diseases, breakthrough bleeding, gall bladder disorders, nausea, migraine and mood changes (JECFA, 2000).
In a study of 23 healthy postmenopausal women receiving 0.3 mg/day of fine particle estradiol, no effect was seen on serum concentrations of follicle-stimulating hormone, angiotensinogen, SHBG or CBG. Thus, 0.3 mg/day (5 µg/kg bw/day) was the NOEL for these hormonal effects. This was used as the basis for setting the ADI for estradiol (see Section 5, JECFA, 2000).
Estradiol was administered in the diet to female Crl:CD BR rats at doses equal to 0, 0.003, 0.17, 0.69 or 4.1 mg/kg bw/day for 90 days. Administration of doses 0.17 mg/kg bw produced dose dependent increases in bodyweight, food consumption and feed efficiency. At 0.69 and 4.1 mg/kg bw, minimal to mild non-regenerative anaemia, lymphopenia, decreased serum cholesterol and altered splenic lymphocyte subtypes were observed. Changes in the weights of several organs were noted. Evidence of ovarian malfunction was found at doses 0.17 mg/kg bw. Pathological changes in males and females fed 0.69 or 4.1 mg/kg bw included centrilobular hepatocellular hypertrophy, diffuse hyperplasia of the pituitary gland, feminisation of the male mammary gland, mammary gland hyperplasia in females, cystic ovarian follicles, hypertrophy of the endometrium and endometrial glands in the uterus, degeneration of the seminiferous epithelium and atrophy of the testes and accessory sex glands (JECFA, 2000).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
66

Total doses of 575–825 mg estradiol were planted s.c. at intervals of 5–6 weeks over a 24–28 month period in 5 female rhesus monkeys. Cystic hyperplasia of the mammary gland was evident but no tumours were found (HSDB, 2002).
4.3 Carcinogenicity and mutagenicityA number of studies on the carcinogenicity of estradiol have been carried out in several animal species using a variety of dose routes. As early as 1979, an IARC Working Group summarised the available data as follows: ‘Estradiol and its esters were tested in mice, rats, hamsters, guinea pigs and monkeys by s.c. injection or implantation and in mice by oral administration. Its s.c. administration resulted in increased incidences of mammary, pituitary, uterine, cervical, vaginal and lymphoid tumours and interstitial cell tumours of the testis in mice. In rats, there was an increased incidence of mammary and/or pituitary tumours. In hamsters, a high incidence of malignant kidney tumours occurred in intact and castrated males and in ovariectomised females, but not in intact females. In guinea pigs, diffuse fibromyomatous uterine and abdominal lesions were observed. Oral administration of estradiol in mice led to an increased mammary tumour incidence. The s.c. injections in neonatal mice resulted in precancerous and cancerous cervical and vaginal lesions in later life and an increased incidence of mammary tumours’ (IARC, 1979).
Since 1979 several more animal studies have added to the evidence of the carcinogenic potential of estradiol as indicated by the following examples. Groups of 200–227 female C3H/HeJ mice, 6 weeks old, with a high titre of antibodies to the mouse mammary tumour virus (MTV+) factor were fed diets containing 0, 100, 1000 or 5000 µg/kg (ppb) estradiol for 104 weeks. At that time, the incidence of cervical adenosis was increased in 8/20 mice at 1000 ppb estradiol and 3/6 at 5000 ppb, and the incidence of uterine adenocarcinomas was increased in the latter group (5/207 versus 0/227 controls). Mammary hyperplastic alveolar nodules were increased by this dose, from 0/57 in controls to 5/78 at weeks 40–65 and 6/50 in controls to 6/17 at weeks 92–105; the time to development of mammary adenocarcinomas was also shortened. Groups of 2–16 female Fischer 344 rats, 7 weeks old, were each injected s.c. with 5 mg estradiol dipropionate once every 2 weeks for 13 weeks. Ten untreated female rats were used as controls. No pituitary tumour was observed in the control rats, but pituitary adenomas were observed in 11/12 treated animals killed at week 7 and carcinomas were observed in 6/6 rats killed at week 9 and 16/16 at week 13. Male Syrian golden hamsters were orchidectomised at 7 weeks of age, then 4 weeks later, they received implants every 3 months of pellets containing 20 mg estradiol. After 5.3 months, renal cell dysplasia and infiltrating and non-infiltrating renal carcinoma were observed in 5/5 estradiol treated animals. No tumour was observed in untreated control hamsters (IARC, 1999).
In its evaluation of the available data, an IARC Working Group considered that there was ‘sufficient’ evidence for the carcinogenicity of estradiol and estrone in experimental animals (IARC, 1999). In a review of the long-term studies on the carcinogenicity of estradiol in animals, a WHO Committee noted that oral and parenteral administration of estradiol increased the incidences of tumours only in hormone-dependent tissues, including the kidneys of male Syrian hamsters. The Committee concluded that the carcinogenicity of estradiol is most probably a result of its interaction with hormonal receptors (JECFA, 2000).
In its evaluation of the human data, an IARC Working Group noted that there were no case reports or epidemiological studies on estradiol alone. In the absence of adequate data in humans, the Working Group considered it reasonable, for practical purposes, to regard estradiol as if it presented a carcinogenic risk to humans. Studies in humans strongly indicate that the administration of estrogens is causally related to an increased incidence of endometrial carcinoma; there was no evidence that estradiol is different from other estrogens in this respect (IARC, 1979). This view was echoed at a later date by a WHO Committee, which concluded that the data on the use of estrogens for postmenopausal hormone replacement therapy were appropriate for evaluating the safety of estradiol. Epidemiological studies on women who took estrogens either alone or in combination with progestogens and androgens, showed that the risks of cancers at most sites were unaffected; the risks
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
67

for cancers of the endometrium and breast were increased (JECFA, 2000). In their more recent evaluation, an IARC Working Group concluded that there was ‘sufficient evidence’ in humans for the carcinogenicity of postmenopausal estrogen therapy (IARC, 1999).
In mice, estradiol enhanced the incidences of endometrial adenomatous hyperplasia, atypical hyperplasia and adenocarcinomas induced by N-methyl-N-nitrosourea and N-ethyl-N-nitrosourea. A continuously high serum estradiol concentration and a low progesterone concentration appeared to be important for the development of endometrial carcinomas in mice. Estradiol suppressed the development of uterine cervical carcinomas induced by 3-methylcholanthrene. In rats, large doses of estradiol suppressed the development of mammary carcinomas induced by N-methyl-N-nitrosourea. Combined treatment of ovariectomised rats with estradiol and N-methyl-N-nitrosourea induced vaginal polyps. Estradiol did not show promoting effects in the livers of rats initiated with N-nitrosodiethylamine. Estradiol did not affect mammary tumour development in intact or ovariectomised female rats treated with 7,12-dimethylbenz(a)anthracene (IARC, 1999). In a number of tumour inhibition studies in rats, estradiol was shown to inhibit tumour formation due to N-ethyl-N-nitrosourea or N-methyl-N-nitrosourea in kidney, mammary gland or liver (CCRIS, 2003).
Although the major route of metabolism for estrogens inactivates them and facilitates their excretion, a minor metabolic pathway activates a small proportion of estrogen to catechol intermediates, with significant potential for damaging DNA. At higher concentrations, which may or may not involve receptor mediation, estrogens have been reported to induce changes in DNA and chromosomes (IARC, 1999).
A large number of in vitro genotoxicity studies have been undertaken on estradiol encompassing a variety of end-points (GENE-TOX, 1998; IARC, 1999; JECFA, 2000; CCRIS, 2003). A representative selection of these assays is shown in Table 4.1 (IARC, 1999).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
68

Table 4.1 In vitro genotoxicity assays with estradiolTest system ± S9 Dose(LED/HID)a Result
S. typhimurium, 5 strains ± S9 2500 µg/plate NegativeS. typhimurium, 3 strains ± S9 500 µg/plate NegativeDNA strand breaks, hamster V79 cells ± S9 816 µg/ml NegativeDNA strand breaks, rat hepatocytes - S9 82 µg/ml NegativeGene mutation, hamster lung V79 cells ± S9 27.2 µg/ml NegativeSCE, mouse kidney cells - S9 2.7 µg/ml NegativeMicronucleus, hamster embryo cells - S9 2.7 µg/ml PositiveChromosomal aberrations, hamster embryo cells
- S9 8.17 µg/ml Negative
Aneuploidy, hamster DON cells - S9 13.6 µg/ml PositiveAneuploidy, hamster embryo cells - S9 0.82 µg/ml PositiveCell transformation, C3H 10T1/2 cells - S9 0.27 µg/ml PositiveCell transformation, hamster embryo cells - S9 2.72 µg/ml PositiveCell transformation, mouse mammary cells
- S9 0.2 µg/ml Negative
SCE, human lymphocytes ± S9 27.2 µg/ml NegativeMicronucleus, human lymphocytes -S9 1.3 µg/ml PositiveChromosomal aberrations, human lymphocytes
± S9 27.2 µg/ml Negative
Aneuploidy, human lymphocytes -S9 13.6 µg/ml Weak positive
From IARC (1999). SCE, sister chromatid exchangeaLED, lowest effective dose; HID, highest ineffective dose
The above results and other in vitro genotoxicity data have been summarised as follows. Gene mutations were not induced in S. typhimurium by estradiol. DNA strand breaks were not induced by estradiol in rat hepatocytes or hamster ovary cells in the absence or presence of a metabolic activation system. Estradiol weakly induced DNA breakage in mouse brain cells. It did not induce DNA repair in a mouse mammary cell line and did not give rise to unscheduled DNA synthesis in hamster embryo cells. It did not induce gene mutations at either the hprt or Na+/K+ ATPase loci, or sister chromatid exchange (SCE) or chromosomal aberrations in hamster embryo cells, whereas the formation of micronuclei was increased in these cells in a single study. In several studies, aneuploidy was induced in hamster embryo cells, male hamster cells and human foreskin fibroblasts. In rodent cells, estradiol was shown to cause cell transformation in 5 studies with different experimental designs, but it gave negative results in 2 studies. In hamster cells, it gave rise to the formation of DNA adducts, but it did not cause oxidative DNA damage. In human lymphocytes, estradiol caused micronucleus formation but no chromosomal aberrations or SCE; it weakly induced aneuploidy (IARC, 1999).
A number of in vivo genotoxicity studies to a variety of end-points have also been undertaken with estradiol (IARC, 1999; JECFA, 2000). A selection of these studies is shown in Table 4.2 (IARC, 1999).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
69

Table 4.2 In vivo genotoxicity studies with estradiol Test system Dose (LED or HID)a Result
DNA strand breaks, hamster kidney 250 µg/day, s.c. implant × 1, 7 days PositiveDNA strand breaks, hamster liver 250 µg/day, s.c. implant × 1, 7 days NegativeSCE, mouse uterine cervix/horn 5 µg s.c. × 1 PositiveSCE, female mouse kidney 5 µg s.c. × 1 NegativeChromosomal aberrations, hamster kidney 125 µg/day, s.c. implant × 1, 5 months PositiveAneuploidy, mouse kidney 5 µg s.c. × 1 NegativeAneuploidy, hamster kidney 20 mg s.c. implant × 1, 3.5 months PositiveBinding (covalent) to DNA, rat liver 0.3 mg/kg bw/day, orally × 1 NegativeBinding (covalent) to DNA, hamster kidney 22.5 mg s.c. implant × 1, 7 months Positive
From IARC (1999). SCE, sister chromatid exchangeaLED, lowest effective dose; HID, highest ineffective dose
The above results and those from other in vivo genotoxicity studies have been summarised as follows: DNA strand breakage was induced in the kidneys but not in the livers of hamsters after s.c. implantation of estradiol at low doses and in both liver and kidneys after a much higher dose; an even higher dose of estradiol administered to male hamsters i.p. had no effect on either kidneys or liver. Estradiol induced SCE in mouse uterine cells but not in kidney cells and it did not cause aneuploidy in either cell type. It gave rise to chromosomal aberrations and aneuploidy in renal cells of hamsters (IARC, 1999).
In a study on adduct formation, male Syrian hamsters were given 2, 10, 50 or 150 mg/kg bw estradiol by intraperitoneal (i.p.) injection and sacrificed 4 hours later; or with 100 mg/kg bw and sacrificed 1–8 hours later. Their livers and kidneys were examined for 8-hydroxy-2'-deoxyguanosine (8-OHdG) as a marker of hydroxyl radical interaction with DNA. Four hours after dosing with 50 mg/kg bw, the renal 8-OHdG levels were double those of controls. No dose dependence was observed. The levels of hepatic DNA adducts in treated animals were similar to those in controls. In animals injected with 100 mg/kg bw estradiol, the number of hepatic DNA adducts was increased 1 and 2 h after dosing (JECFA, 2000).
The genotoxic potential of the metabolites of estradiol has also been examined. DNA strand breakage was demonstrated in kidney cells of male hamsters treated s.c. with 4-hydroxyestradiol. In hamster cells in vitro, 4-hydroxyestradiol caused cell transformation and formation of DNA adducts in the presence of exogenous metabolic activation. Induction of oxidative damage in male hamster liver DNA and binding to calf thymus DNA were seen after in vitro treatment with 4-hydroxyestradiol and similar results were observed in vivo in mammary cells of rats treated with the compound. DNA strand breakage was not demonstrated in kidney cells from male hamsters treated s.c. with 2-hydroxyestradiol and this compound did not bind to liver DNA of hamsters in vitro in one study. In hamster cells, 2-hydroxyestradiol caused cell transformation and formation of DNA adducts in the presence of exogenous metabolic activation. Estradiol-3,4-quinone bound to DNA both in vitro and in vivo in rat mammary cells. Estrone did not cause gene mutation at various loci in hamster ovary cells. It did not induce oxidative damage in hamster liver DNA, nor did it bind to kidney or liver DNA of male hamsters or to liver DNA of rats treated in vivo. In vitro, estrone-3,4-quinone induced DNA strand breaks in human MCF-7 cells and bound weakly to calf thymus DNA (IARC, 1999).
A WHO Committee made an overall assessment of the genotoxic potential of estradiol. It noted that the compound did not cause gene mutations in vitro. In some other assays, sporadic but unconfirmed positive results were obtained. There was more consistent evidence for the induction of micronuclei in vitro, aneuploidy in vitro, cell transformation in vitro, oxidative damage to DNA in vivo and DNA single-stranded breakage in vivo by estradiol. The Committee concluded that estradiol has genotoxic potential (JECFA, 2000).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
70

4.4 Reproductive and developmental toxicityICR mouse fetuses from the 15th or 17th day of gestation were injected s.c. with 50 µg estradiol. Irreversible cornification or stratification of the vaginal epithelium was seen at birth in 85% of those treated on day 17 and in 66% of those treated on day 15. When examined at the age of 3 months, corpora lutea were absent in 4/12 treated on day 17 and in 5/6 mice treated on day 15. Administration of 50 µg estradiol on the day of birth or 3 days later to ICR mice resulted in the absence of corpora lutea. The changes in their vaginal epithelium were less marked. Pregnant rats were given s.c. injections of 0.8–35 mg estradiol daily from day 12–13 until day 18–21 of gestation. Only 12/28 estradiol treated rats carried to term; litter size was reduced and post-partum mortality was 66%. In female offspring, abnormalities of the reproductive tract and absence of corpora lutea were observed. In male offspring, 3–6 pairs of well-developed nipples, undescended testes and impairment of Wolffian-derived tissues occurred. Rabbits were injected i.m. with 15 or 30 µg estradiol for 3–5 consecutive days during different periods of gestation, starting on the 5th day. Administration of 15 µg terminated pregnancy when given before day 21 of gestation; later, 30 µg were required to terminate pregnancy. In reviewing this and other evidence, an IARC Working Group concluded that estradiol has teratogenic actions on the genital tract and possibly on other organs and impairs fertility (IARC, 1979). Ten milligrams estradiol was implanted s.c. in 5 female Sprague–Dawley rats on day 10 of pregnancy. Control rats were given dextran by the same method. The treatment with estradiol resulted in complete resorption of embryos in all test animals (JECFA, 2000).
Estradiol was administered in the feed to female Crl:CD BR rats at doses equal to 0, 0.003, 0.17, 0.69 or 4.1 mg/kg bw/day in a 90-day, one generation study. The animals in the two highest dose groups produced no pups. The mean daily intakes of the F1 females were 0, 0.005 and 0.27 mg/kg bw/day, respectively. In the F0 generation, doses of >0.17 mg/kg bw produced a dose-dependent increase in serum estradiol concentration and all doses produced a dose-dependent decrease in serum progesterone concentration on test day 90 which correlated with ovarian atrophy and lack of corpora lutea. The serum concentration of luteinising hormone was decreased at all times at >0.69 mg/kg bw and at 0.17 mg/kg bw on test day 90. In the F1 generation on postnatal day 28, the serum estradiol level was increased and that of progesterone decreased at 0.27 mg/kg bw. No change in serum prolactin, follicle-stimulating hormone or luteinising hormone levels was noted. Dietary estradiol caused marked effects on the estrus cycle at 0.17 mg/kg bw (F0), at 0.27 mg/kg bw (F1) and at 0.69 and 4.1 mg/kg bw (F0). The weight of the pups from the 0.003 and 0.17 mg/kg bw dose groups decreased relative to the control group; the weights in the former group recovered after birth and remained similar to those of controls throughout the study. The bodyweights of the animals at 0.27 mg/kg bw remained below control levels throughout the study. Parental administration of estradiol did not affect the anogenital distance in male or female pups. Onset of sexual maturity as measured by prepubertal separation in males and vaginal opening in females, was delayed. Some of the histopathological findings were more severe in the F1 generation than in the parent generation (JECFA, 2000).
The embryotoxicity of steroidal estrogens, including estradiol, was examined in cultured whole embryos obtained from Sprague–Dawley rats. Preliminary experiments resulted in a steep dose-response curve ranging from 0.05 to 0.5 mmol/ℓ. The concentrations tested resulted in low embryo lethality (2–20%). The commonest effect observed was hypoplasia of the prosencephalon. Estradiol and estrone were markedly and statistically significantly more toxic in the presence of metabolic activation. In this system, estradiol was more efficiently converted to catechol estrogens in male liver (JECFA, 2000).
A review of the birth certificates and hospital records of 7723 infants whose mothers had reported using oral contraceptives indicated that these compounds presented no major teratogenic hazard (JECFA, 2000).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
71

5 Guidelines and standardsJECFA established an ADI of 0–0.05 µg/kg bw on the basis of the NOEL of 0.3 mg/day (equivalent to 5 µg/kg bw/day) in studies of changes in several hormone-dependent parameters in postmenopausal women (see Section 4.2). A safety factor of 10 was used to account for normal variation among individuals and an additional factor of 10 was adopted to protect sensitive populations (JECFA, 2000).
The EMEA CVMP has not established an ADI for residues of any of the estrogenic hormones in meat, and no MRLs have been established. As a consequence of this, estradiol was added to Annex II of EU 2377/90. Products containing this compound are now only licensed for use in dogs.
6 References Bishop Y, ed (1996) The Veterinary National Formulary (Third Edition), London, UK, Royal Pharmaceutical Society of Great Britain/British Veterinary Association
Bishop Y, ed (2004) The Veterinary National Formulary (Sixth Edition), London, UK, Royal Pharmaceutical Society of Great Britain/British Veterinary Association
CCRIS (2003) 17-Estradiol. From: Chemical Carcinogenesis Research Information System, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [January 2004] at http://http://toxnet.nlm.nih.gov
GENE-TOX (1998) -Estradiol. From: Genetic Toxicology (Mutagenicity) Databank, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [January 2004] at http://http://toxnet.nlm.nih.gov
HSDB (2002) Estradiol. From: Hazardous Substances Databank, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [January 2004] at http://http://toxnet.nlm.nih.gov
IARC (1979) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol 21, Sex Hormones (II): Oestradiol-17, Oestradiol 3-Benzoate, Oestradiol Dipropionate, Oestradiol-17-Valerate and Polyoestradiol Phosphate, Lyon, France, International Agency for Cancer Research
IARC (1999) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol 72, Hormonal Contraception and Post-Menopausal Hormonal Therapy, Lyon, France, International Agency for Research on Cancer
JECFA (2000) Toxicological Evaluation of Certain Veterinary Drug Residues in Food (Food Additives Series 43), Geneva, Switzerland, World Health Organization, available [January 2004] at http://www.inchem.org/documents/jecfa/jecmono/v43je05.htm
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
72

Annex 6 Flavomycin1.1 IntroductionFlavomycin, otherwise known as flavophospholipol or bambermycins (CAS No. 11015-37-5), is a complex antibiotic comprising at least four active components. It is obtained from cultures of Streptomyces bambergiensis, S. ghanaensis, S. ederensis, S. geysirensis and related strains (Merck, 2001).
The structure of flavomycin, as used in the predictive model, is presented in Figure 1
Figure 1 Flavomycin structure
2 Uses and exposureFlavomycin is a phosphorus-containing glycolipid antibacterial and was available in the UK as a zootechnical feed additive used as a production enhancer in cattle, pigs, laying hens, broiler chickens, turkeys, and rabbits. Two products containing flavomycin were licensed in the UK (Flaveco 40TM and Flavomycin 80TM; Bishop, 2004), but were only permitted for use as antimicrobial growth promoting substances until 31 December 2005 within the EU (EC, 2003).
Both Flaveco 40 TM and Flavomycin 80TM contain 8% w/w flavomycin (80g/kg premix) and were available as a premix for addition to the feed.
In calves up to 26 weeks of age, the dose rate for inclusion of flavomycin in the diet was 150–400 g premix, equivalent to 6–16 g/tonne in feed, or 200–400 g premix equivalent to 8–16 g/tonne in milk replacer. In fattening cattle, the inclusion rate was 50–250 g premix, equivalent
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
73

to 2–10 g/tonne, added to completed supplementary feed or free-access minerals (100 kg bw — maximum daily dose 40 mg; >100 kg bw — 40 mg plus 1.5 mg per additional 10 kg bw).
In pigs, dose rates for inclusion in the diet were: up to 6 months of age,25–500 g premix, equivalent to 1–20 g/tonne of feed; and up to 3 months of age,250–500 g premix, equivalent to 10–20 g/tonne of milk replacer or creep feed.
In poultry, dose rates for inclusion in the diet were: in broiler chickens up to 16 weeks of age, 25–500 g premix, equivalent to 1–20 g/tonne of feed; in laying hens,50–125 g premix, equivalent to 2–5 g/tonne of feed; and in turkeys up to 26 weeks of age, 25–500 g premix, equivalent to 1–20 g/tonne of feed.
In rabbits, the dose rate for inclusion in the diet was 50–100 g premix, equivalent to 2–4 g/tonne of feed.
3 ToxicokineticsFlavomycin is a macromolecule that, tends to form complexes owing to its heteropolar behaviour. Absorption and bioavailability from the gut would, therefore, be expected to be extremely low and no tissue residues would be predicted to result. Indeed, no evidence of tissue residues were found in chickens fed diet containing flavomycin at 20ppm (Mulder and van der Hulst-Van Arkel et al., 1976). For all species, there was a nil withdrawal period. Excretion is via the faeces (almost 100%) in the form of an intact microbiologically-active molecule and the subsequent half-life in faeces is 21 days; (Eco Animal Health, 2006).
4 Toxicity profileSearches were made for publicly available information on the toxicity of flavomycin, and supplemented by use of the online program ChemIDPlus (SIS NLM, 2003), but the available toxicity database identified for flavomycin was very limited. Additional information was, therefore, supplied by an ‘in-silico’ evaluation of the potential toxicity of flavomycin, which was conducted using an expert software system DEREK (Deductive Estimation of Risk from Existing Knowledge, Lhasa Ltd).
For this, the two-dimensional chemical structure of flavomycin was downloaded as a ‘.mol file’, using the online program ChemIDPlus, and imported into DEREK for analysis.
The expert software DEREK (for Windows version 8.0.1; Lhasa Ltd) works by matching structural entities in a query structure with structural alerts that are associated with different toxicity endpoints (toxicophores). A structure alert is the set of structural features in a molecule that signals a possibility that the substance may show a particular toxic effect. This is similar to the definition of a toxicophore (a structural feature believed to be responsible for its toxic effect) and a pharmacophore (a structural feature believed to be responsible for a useful pharmacological effect), but alerts and toxicophores are not always identical. An alert may include information about additional features that increase or decrease the effectiveness of a toxicophore. In total the programme uses 482 structural alerts associated with different endpoints.
For species that include bacteria (Salmonella typhimurium), guinea pig, hamster, human, mammal, mouse, primate, rat and rodent, the following toxicity endpoints are predicted:
Carcinogenicity, such as photocarcinogenicity
Irritation, such as irritation of the eye and the gastrointestinal tract
Miscellaneous endpoints, such as anaphylaxis and anticholinesterase activity
Genotoxicity, such as mutagenicity and chromosome damage
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
74

Respiratory sensitisation, such as occupational asthma
Skin sensitisation, such as photoallergenicity
Thyroid toxicity.
DEREK provides an indication of the likelihood of each predicted adverse effect using a specified terminology ranging from ‘certain’ — there is proof that the proposition is true, through a series of statements, such as ‘plausible’ — the weight of evidence supports the proposition, to ‘contradicted’ — there is proof both that the proposition is true and that it is false.
DEREK was used to predict different endpoints and their likelihoods in a range of species. Results were compiled using the standard DEREK reporting format. Summaries of the positive alerts identified are tabulated below. Other structural alerts were either not identified for this molecule or were insignificant on the probability scale. Thus, based on current toxicological knowledge, it is considered that flavomycin is unlikely to exhibit any of the toxic activity assessed by DEREK other than those described below.
4.1 Acute toxicityData on acute toxicity identified from DEREK and ChemIDPlus are summarised in Tables 4.1 and 4.2.
Table 4.1 Structural alerts (acute toxicity) identified in flavomycin by DEREK
DEREK Alert
Description of Alert
Endpoint Species Location in the molecule (shown in bold)
420 1,3-Diketone.Number of matches = 1
Skin sensitization Guinea pigHamsterHumanMammalMousePrimateRat Rodent
O
R1 R4
O
R2 R3
R1, R4 = CR2, R3 = any
424 Aldehyde precursor. Number of matches = 2
Skin sensitization Guinea pigHamsterHumanMammalMousePrimateRat Rodent
O O
R1 H
R3R2
R1 = C, H
R2, R3 = any
The analysis by DEREK concluded that the presence of the structural alerts in the molecule indicated that:
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
75

it is considered plausible (i.e. there is a weight of evidence) that flavomycin will exhibit skin sensitisation in some animals (guinea pig, hamster, mouse, rat and rodent); it is considered doubted (i.e. the weight of evidence opposes the proposition) that flavomycin will exhibit skin sensitisation in the human and the evidence is equivocal (there is an equal weight of evidence for and against the proposition) in mammals and primates. This endpoint is predicted because the compound is an aldehyde precursor and also a tautomer of flavomycin contains a 1,3-diketone group.
Thus, in summary, the predictions obtained using DEREK indicate that it is possible (there is a weight of evidence) that flavomycin will cause skin sensitisation in some animals although probably not in humans.
Table 4.2 Toxicity data for flavomycin obtained using ChemIDPlus
Organism Test Type RouteReported Dose (Normalized)
Effect Reference
Dog LDLo Intravenous 600 mg/kg
Autonomic nervous system (smooth muscle relaxant), behavioural wakefulness, and depressed somnolence
SIS NLM (2004)
Mouse LD50 Intravenous 200 mg/kg SIS NLM (2004)
Mouse LD50 Subcutaneous 500 mg/kg SIS NLM (2004)
The acute toxicity of flavomycin in mammals by parenteral routes appears to be limited. However, no data on acute oral toxicity were identified.
4.2 Repeat dose toxicityBased on current toxicological knowledge, analysis by DEREK indicated no endpoints related to repeat dose toxicity.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
76

4.3 Carcinogenicity and mutagenicityTable 4.3 Structural alerts (mutagenicity and chromosome damage) identified in flavomycin by DEREKDEREK Alert
Description of Alert
Endpoint Species Location in the molecule (shown in bold)
305 Alkyl ester of phosphoric or phosphonic acid.. Number of matches = 1
Mutagenicity Bacteria, e.g Salmonella typhimurium PR1
R1
R2
R2
R1 = O, S
R2 = O, S, N
R3 = C, H
PR1
R1
R3
R2
(I) (II)
1 1
2 2
3
309 Substituted vinyl ketone. Number of matches = 1
Chromosome damage.
Guinea pigHamsterHumanMammalMousePrimateRat Rodent
R2
R1
R3
OR4
R1-R3 = any but at least one not H
R4 = C
The analysis by DEREK concluded that the presence of the structural alerts in the molecule indicated that:
it is considered plausible (i.e. there is a weight of evidence) that flavomycin will exhibit mutagenicity in vitro in bacteria owing to the presence of an alkyl ester of a phosphoric acid group;
it is considered plausible (i.e. there is a weight of evidence) that flavomycin will cause chromosomal damage in the guinea pig, hamster, human, mammal, mouse, primate, rat and rodent owing to the presence of a substituted vinyl ketone group.
Thus, the predictions obtained using DEREK indicate that it is plausible (there is a weight of evidence) that flavomycin will exhibit mutagenicity in bacteria and will cause chromosomal damage in mammals.
4.4 Reproductive and developmental toxictyBased on current toxicological knowledge, analysis by DEREK indicated no endpoints related to reproductive or developmental toxicity.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
77

5 Guidelines and standardsAn ADI of 0.3 mg/kg bw/day has been published by the Australian Government (Australian Government, 2005). Although permitted for use in Europe as antimicrobial growth promoters until recenty, this authorisation lapsed on 31 December 2005.
6 ReferencesAustralian Government (2005) ADI List - Acceptable Daily Intakes for Agricultural and Veterinary Chemicals , Canberra, Australia, Australian Government, Department of Health and Ageing, Available [June 2005] at; http://www.tga.gov.au/docs/html/adi.htm
Bishop, Y (2004) The veterinary formulary: 5th edition. BVA and Associated Pharmaceutical Press, London.
EC (2003) Commission Regulation (EC) No 1831/2003 of 22 September 2003 on additives for use in animal nutrition Official Journal of the European Union, L269, 29-43. Available [July 2006] at: http://europa.eu.int/eur-lex/pri/en/oj/dat/2003/l_268/l_26820031018en00290043.pdf
Eco Animal Health (2006) Flaveco: Brand Flavophospholipol. Available at: http://www.ecoanimalhealth.com/g/g_flaveco.html, Accessed [2006, 17 August]
Merck (2001) The Merck Index (Thirteenth Edition), Rahway NJ, USA, Merck and Co Ltd ONS & MRC Human Nutrition Research (2002) The National Diet and Nutrition Survey: Adults Aged 19-64
Mulder RW & van der Hulst-Van Arkel MC (1976) Antibiotic residues in organs and muscle tissue of broilers. I. Bacitracin, flavomycin, spiramycin and viriniamycin residues following administration of diets containing low levels of these antibiotics (Dutch) Tijdscher Diergeneeskd, 101, 1194-1198
SIS NLM (2003) Flavomycin. From: ChemIDplus, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [June 2003] at http://chem.sis.nlm.nih.gov/chemidplus/
SIS NLM (2004) Bambermycins. From: ChemIDplus, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [September 2005] at http://chem.sis.nlm.nih.gov/chemidplus/
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
78

Annex 7 Furazolidone 1 IntroductionFurazolidone (3-[[(5-nitro-2-furanyl) methylene]-amino]-2-oxazolidinone; CAS No. 67-45-8) is a nitrofuran derivative, which was used in the prophylactic and therapeutic treatment of infections caused by bacteria or protozoa in poultry, cattle (calves), pigs, rabbits and fish. It acts by gradual inhibition of monoamine oxidase (JECFA, 1992; EMEA, 1997; SIS NLM, 2004).
Furazolidone is used in the treatment of cholera when anti-infective therapy is indicated as an adjunct to fluid and electrolyte replacement. It is also used for the specific and symptomatic treatment of diarrhoea and enteritis caused by susceptible bacteria or protozoa. It has been prescribed to treat giardiasis in children (HSDB, 2002).
2 Uses and exposureFurazolidone was available under various trade names throughout the world (e.g Furazolidone BPTM, Furazolidone-250TM, Furazolidone SuspendibleTM, FuroxTM, FuroxaneTM, FuroxoneTM, MicrodoneTM, NeftinTM, NifulidoneTM in the UK) for the treatment of bacterial infections in poultry, pigs and calves. The drug was also used in the treatment of protozoal infections notably histomoniosis or blackhead (Histomonas meleagridis) and hexamitosis (Hexamita/Spironucleus spp) in turkeys. Historically, it was also used as an anticoccidial agent in combination with Nitrofurazone (BifuranTM) for the treatment of coccidiosis in poultry in the 1950s (Taylor, personal data).
Furazolidone was placed in Annex IV of Regulation 2377/90/EEC, which prohibits use in medicinal products for food-producing species and is no longer available in any licensed products in the UK and EU countries.
Furazolidone was available in several formulations.
As a feed additive, products containing 20% w/w furazolidone (e.g. MicroBio Furazolidone PremixTM, Furazolidone BPTM, Furazolidone 250TM, Pharmsure Furazolidone 20%TM, Neftin 200TM) were used in chickens and turkeys for the treatment of salmonellosis, blackhead, bacterial infections complicating respiratory disease, colisepticaemias, and in turkeys, Arizona Disease (Salmonella arizona syn. Arizona hinshawii) and hexamitosis at a dose rate of 400 g/tonne of feed for 10 days (or 14 days for blackhead and hexamitosis in turkeys). In weaner pigs it was used to treat salmonellosis and other bacterial diseases at a rate of 300 g/tonne of feed for 7 days and in unweaned pigs 600 g/tonne of creep feed. In ducks it was used to treat salmonellosis and E. coli infections, and in rabbits to treat bacterial enteritis, both at 100 g/tonne of feed for 10 days (Bishop, 1994).
Furazolidone was also available as water-based suspendable preparations containing 6% w/v (Micro-Bio Furazolidone Suspendable TM, Furoxone TM) for dosing livestock at 15–25 g/ℓ for 7–14 days; as a 7.5% w/v suspension for dosing individual calves (Neftin Calf Doser TM) for salmonellosis and scours caused by E. coli and other enteric bacteria at 10 mg/kg daily (227 mℓ dose applicator giving 258 mg/dose); or as 5% suspension for dosing individual piglets for the treatment of bacterial scours at 50–200 mg/daily (100 mℓ dose applicator giving 50 mg/dose; Bishop, 2004).
No products containing furazolidone are legally available in the UK. Exposure to humans is likely to be very low, the exception being the illegal use of the substance in food animals and subsequent ingestion of animal products containing residues.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
79

2.1 Exposure concentrations in meatFurazolidone is partially metabolised in the gastrointestinal tract. The parent drug and its metabolites are well absorbed and extensively metabolised in the tissues and the major route of excretion is via the urine. The concentration of total residues of furazolidone in pigs (given 300 mg/kg in the diet for 10–14 days) and chickens (given 220 mg/kg for 4 days) was still in the mg/kg range after 14 and 3 days withdrawal periods, respectively (Mitch Kelly, CSL, unpublished data).
Experimentally-derived data are available on concentrations of furazolidone in meat products. Pigs were treated continuously with furazolidone in feed at a rate of 400 g/tonne of feed for five days. Immediately following treatment, the pigs were slaughtered and concentrations of furazolidone and its free and bound metabolites were obtained. Measured concentrations of furazolidone in kidney, liver and muscle were 1.5, 1.8 and 0.25 mg/kg, respectively.
Experimental data were not available for furazolidone in poultry and calves. Information on usage of furazolidone and on absorption and excretion were therefore used with Equation 1 to estimate likely ‘worst case’ concentrations in calf and poultry meat:
Equation 1M = D × T × Fabs × (1–Fexc)
where M is the predicted concentration in meat (mg/kg); D is the dose of substance (mg/kg bw/d); T is the duration of the treatment (d); Fabs is the fraction of compound absorbed; and Fexc is the fraction of compound excreted.
For calves it was assumed that furazolidone is given at a dose of 10 mg/kg bw/d for 14 d, that 85% of this is absorbed (i.e. Fabs = 0.85) and that the fraction of drug excreted was 0.9. This gave a ‘worst case’ concentration in calf meat of 11.9 mg/kg.
For poultry, it was assumed that the dose rate was 400 g/tonne of feed (corresponding to a dose rate of approximately 30 mg/kg bw/d) and that animals were treated for 4 weeks. Assuming that 85% of this is absorbed and that the fraction of the drug excreted is 90%, then the ‘worst case’ concentration in chicken meat is 71.4 mg/kg.
3 ToxicokineticsThe absorption of radioactivity after administration of 14C-furazolidone in PEG 200 to rats, calculated from the total recovered in urine, bile and tissues, was 87% of the dose. The extent of absorption of radioactivity after administration of 14C-furazolidone by diet or muscle was 90 and 96% of the dose, respectively (EMEA, 1997).
Groups of rats were dosed with 14C-furazolidone labelled either at the aldehyde carbon on the nitrofuran ring or at the methylene carbons of the oxazolidine ring. At 48 h after dosing, rats treated with aldehyde-labelled furazolidone excreted the major part (45.9%) via the urine, with 38.2% via the faeces and 2.1% exhaled as 14CO2. The major route of the excreted methylene-labelled furazolidone was via the urine (71.6%), with 34.5% via the faeces and 3.6% in expired air. After both treatments, the main radioactivity in tissues was present in liver and kidney with smaller amounts in heart, muscle and testes. In a separate study, rats received a diet containing methylene-labelled 14C-furazolidone for 10 days, after pre-treatment with unlabelled furazolidone for 19 days. Excretion of radioactivity in urine and faeces was 62.2 and 25%, respectively. 14C-labelled tissue concentrations were in the order liver>kidney>heart>blood>muscle>testes>fat. Elimination was biphasic with a fast and slow component. The half-lives were: liver 0.9 and 4.8 days; kidney 0.7 and 7.5 days; heart 1.1 and 14.1 days; blood 0.8 and 14.8 days; muscle 1.0 and 29.0 days and testes 0.7 and 8.2 days. Four cannulated bile duct rats were administered, by intubation, 16.5 mg 14C-furazolidone (methylene- and formyl-
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
80

labelled)/kg bw in PEG-200. Rats were sacrificed after 72 h. Excretion was 46.6% of the absorbed dose in urine and 36.5% in bile. Residual radioactivity in tissues was 3.7% (JECFA, 1993).
Formyl-labelled 14C-furazolidone was administered to chickens at a dose of 220 mg/kg in the feed for 4 days following a 21 day feeding period with unlabelled furazolidone at the same rate. Maximum levels of radioactivity in liver and kidney (15.6 and 11.7 mg-equivalents/kg, respectively) were about 5–8 times the radioactivity present in fat and muscle. Radioactivity in these tissues was 0.31–0.49 mg-equivalents/kg 16 days after dosing. Depletion of radioactivity was a biphasic process, with a fast component half-life of 1.5 days and a slower component half-life of about 4 days. In another study, white leghorn chickens, fed furazolidone at 220 mg/kg in feed for 3 weeks were treated with 14C-furazolidone (methylene-labelled) for 4 days. Depletion was biphasic with a rapidly excreted component half-life of about 0.4 days and a slower component half-life of about 4 days. At 11 days after dosing, levels of radioactivity in liver, kidney, muscle and fat were 0.867, 0.576, 0.466 and 1.46 mg-equivalents/kg, respectively. Chickens were administered 220 mg formyl-labelled 14C-furazolidone/kg in feed for 10 days. Radioactivity was measured daily in urine, faeces and expired air and the chickens were sacrificed on day 11. Recovery of radioactivity in urine and faeces was 92% of the total dose, with 0.6% in expired air. Total recovery in tissue was 2.8% of administered 14C, with the greater proportions in liver, kidney and the crop and its contents (JECFA, 1993).
A Yorkshire barrow, maintained on feed containing 330 mg/kg furazolidone for 3 weeks was administered 1.25 mg 14C-furazolidone (formyl-labelled)/kg bw orally. Urine and faeces were collected and the pig sacrificed 48 h after dosing. The total 14C-recovery in urine and faeces was 90%. Most of the radioactivity in tissues was found in kidney, liver and thyroid with lesser amounts in bile, blood, muscle and fat. One female pig was treated with formyl-labelled 14C-furazolidone and, another, was treated with methylene-labelled 14C-furazolidone at a rate of 5 mg/kg bw/day for 5 days. A total of 42% and 47%, respectively, of applied radioactivity was excreted via the urine. Radioactivity in edible tissue was highest in liver at 5.15 and 7.80 mg-equivalents/kg, respectively and lowest in muscle at 1.00 and 1.05 mg/equivalents/kg, respectively . In urine, at least 15 metabolites were found with <5% comprising the parent compound. In another study, 4 male pigs were treated with 14C-furazolidone (labelled in both the formyl and methylene position) in the feed for 5 consecutive days at a rate of 5 mg/kg bw/day. Pigs were sacrificed 5 or 14 days after the last dose. Total excretion in urine was 51–56% of the administered dose. At least 11 metabolites were present in the urine but no parent compound was found. Piglets were dosed orally with 12 mg/kg bw 14C-furazolidone (methylene-labelled) for 10 days. Peak blood and plasma levels of the parent compound were reached within 30 minutes at 835 and 955 ng/mℓ, respectively; no detectable level was found 3–4 hours after the last dose. The half-life in blood was about 60 minutes. One day after the last dose, all the radioactivity was associated with plasma proteins. Excretion via the urine and faeces totalled 61 and 18% of the administered radioactive dose, respectively. For residual radioactivity in tissue, 2 h after sacrifice, 9, 14 and 35% could not be extracted from kidney, liver and muscle, respectively ; 14 days later, these amounts were 31, 27 and 56%, respectively (JECFA, 1993).
A fast conversion of 14C-furazolidone (methylene-labelled) was observed in liver microsomes of 3-MC induced rats incubated under aerobic and anaerobic conditions. The two major metabolites formed were 3-(4-cyano-2-oxobutylid-ene-amino)-2-oxazolidone and 2,3-dihydro-3-cyanomethyl-2-hydroxy-5-nitro-2-di (2-oxo-oxazolidin-3-yl) iminomethyl-furo [2,3-β] furan, both accounting for about 16.5% of the total extractable radioactivity. The metabolism of 14C-furazolidone (methylene-labelled) was also studied in swine liver microsomes under aerobic and anearobic conditions. 3-(4-cyano-2-oxobutyli-deneamino)-2-oxazolidone and 2,3-dihydro-3-cyanomethyl-2-hydroxyl-5-nitro-1α, 2-di(2-oxo-oxoazolidin-3-yl) iminomethyl-furo[2,3-β] furan were the major ethylacetate extractable metabolites, formed via the open chain acrylonitrile derivative of furazolidone. Another metabolite, formed by microsomes in the presence of mercaptoethanol was identified as a mercaptoethanol conjugate and, in the presence of GSH, the metabolite formed was shown to be a GSH conjugate of furazolidone. Furazolidone was rapidly transformed by porcine hepatocyte cultures resulting in the formation of 3-(4-cyano-2-oxobutylidene amino)-2-oxazo-lidone which amounted to 15% of the total metabolites (JECFA, 1993).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
81

Male Wistar rats were given 100 mg 14C-furazolidone (formyl-labelled)/kg bw orally. N-(4-carboxy-2-oxobutylideneamino)-2-oxazolidone, α-ketoglutaric acid, 3-(4-cyano-2-oxobutylidenamino)-2-oxazolidone and N-(5-acetoamido-2-furfurylidene)-3-amino-2-oxazolidone were identified in urine collected from the rats (JECFA, 1993).
The metabolites of aldehyde-labelled 14C-furazolidone in pig urine were investigated and more than 30 radiolabelled metabolites were found. Three primary metabolites of furazolidone were identified as 5-nitro-2-furoic acid, an orange coloured ‘415’ chromogen and a yellow ‘415’ chromogen, with half-lives of 30, 22 and 19 min, respectively. The orange ‘415’ metabolite contains an intact 5-nitro-2-furfural moiety and is the most abundant furazolidone-related metabolite found in pig urine. Analysis of urine from a pig treated with 14C-furazolidone, also labelled with 15N in the 5-nitro position, showed 14 metabolites. Eleven of them degraded to two or three degradation products which could not be identified. The presence of 15N was detected in one minor metabolite. It was concluded that during the metabolism of furazolidone, the nitro group was removed extensively from the molecule (JECFA, 1993).
Four human subjects were given a single dose of 400 mg furazolidone/day, administered in a tablet or capsule. The 24 h urine samples collected after administration, contained 0.003–0.16% unchanged furazolidone when analysed by HPLC. By using a more specific complexation method, no furazolidone was detected, (<2 µg/mℓ). Furazolidone was given to 10 human adults as 2 daily doses of 200 mg each for 21 days. Plasma levels, analysed by HPLC (detection limit 0.002 µg/mℓ) ranged from trace amounts to 0.489 µg/mℓ (JECFA, 1993).
A 14C-furazolidone residue depletion study was undertaken in pigs subjected to 0, 21 and 45 day withdrawal times following a 14-day oral treatment at 16.5 mg/kg bw/day. The 14C furazolidone was labelled in the oxazolidinone ring and in the imine carbon adjacent to the nitrofuran ring. The depletion values over the 45 day withdrawal period are shown in Table 3.1 (EMEA, 1997).
Table 3.1 Radioactive residues (mg/kg) in pig tissues during the 45 day withdrawal periodTissue 0 day 21 days 45 days
Liver 41.1 4.4 2.1Kidney 34.4 3.4 2.0Muscle 13.2 3.3 2.4Fat 6.2 3.0 1.9
3-Amino-oxazolidone-2 (AOZ) has been proposed as the marker residue. The ratio of released 3-amino-oxazolidone-2 to total bound residues was determined in the pig livers from the residue depletion study. 3-amino-oxazolidone-2 could be released from 18% of the bound residues at day 0 and this fraction decreased to 8 and 6%, respectively after the 21 and 45 day withdrawals. An analytical method for the detection of 3-amino-oxazolidone-2, released from protein bound residues of furazolidone in pig livers, was validated with spiked and incurred tissues (EMEA, 1997). However, it is important to note that furazolidone is not the only potential source of this metabolite, since it may also be derived from commercially-derived blowing agents used in production of plastic sealing gaskets (EFSA, 2005).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
82

4 Toxicity profile4.1 Acute toxicityOral LD50 values of furazolidone in rats have been determined to be 1508 and 2336 mg/kg bw and, in mice, as 1110 and 1782 mg/kg bw. The mouse intraperitoneal (i.p.) LD50 has been determined to be 300 mg/kg bw (JECFA, 1993; HSDB, 2002).
A dose of 500 mg furazolidone, moistened with 0.5 mℓ 0.5% saline, caused slight erythema in rabbits when applied under semi-occlusive conditions to the clipped skin of 3 male and 3 female New Zealand White rabbits. Nine New Zealand White rabbits received 30 mg furazolidone in the conjunctival sac of the right eye and 3/9 eyes were washed 20 seconds after treatment. After 24 hours, slight conjunctival irritation was observed in both the washed and unwashed eyes. Corneal ulcerations were observed in 2/6 unwashed eyes. All eyes were normal by day 7 (JECFA, 1993).
Allergic contact eczema has been reported in subjects handling animal feed containing furazolidone. Hypersensitivity reactions to oral furazolidone have occurred in a small number of patients and generally subside when the drug is discontinued (HSDB, 2002).
4.2 Repeat dose toxicityRats were fed furazolidone at dose levels of 10, 50 and 100 mg/kg in diet, corresponding to 1, 4 and 8.5 mg/kg bw/day, respectively , for 3 months. Hepatocellular vacuolisation was seen at the 100 mg/kg dose level. Decreases in food consumption and bodyweight were recorded in the animals in the 50 and 100 mg/kg dose groups. An anaemic effect was also seen in these two dose groups. The NOEL for this study was 10 mg/kg in diet (= 1 mg/kg bw/day; EMEA, 1997).
Male Wistar rats, 6/group, were fed diets containing 0, 10, 100 or 200 mg/kg furazolidone for 13 months (= 0, 0.5, 5 or 10 mg/kg bw/day). No effects were observed for a number of experimental parameters except for a slight increase in relative liver and spleen weight at the highest dose. Slight hypertrophy of the liver cells was noted at all dose levels. In another study, Carworth rats, 20/sex/group, were fed furazolidone in diet at 1000 mg/kg (= 50 mg/kg bw/day) for 45 weeks. After 5 weeks treatment and at termination, the rats showed a significant decrease in bodyweight gain, food consumption and efficiency. A significantly increased incidence of palpable mammary tumours was observed in female rats. At microscopy, most of these tumours were found to be adenomas, fibromas and fibroadenomas. Two were adenocarcinomas (JECFA, 1993).
In two poorly reported studies, furazolidone was administered orally to groups of beagle dogs. In the first study, male and female dogs were given doses of 7.5, 25 or 50 mg/kg bw/day for varying periods over 6 months. In all treated dogs, neurological symptoms and histopathological changes in the region of the basal ganglia, decreased sperm and tubular testicular degeneration were reported. These changes appeared to be reversible. In the other study, 4 dogs/sex/group were administered furazolidone at 0, 1, 5 or 15 mg/kg bw/day for 2 years. Neurological symptoms and cataracts were observed in dogs at the highest dose group. Decreased sperm motility, abnormal sperm and decreased testes weight were observed in the mid- and high dose group males. Increased relative kidney weight was seen in high dose females (JECFA, 1993).
A 3-month dietary toxicity study was undertaken in dogs with the metabolite, 3-amino-oxazolidone-2, at dose levels of 1, 3 or 6 mg/kg bw/day. Anaemia, prolongation of thrombin time and increased serum hepatic enzyme activity were seen at the mid- and high dose levels. Males at the highest dose had increased serum triglyceride levels and increased liver weight. A dose dependent cholestatis was observed in all treatment groups. Therefore a NOEL could not be established for this study (EMEA, 1997).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
83

The effect of furazolidone on enzyme activity has been examined in several species. After single oral doses of 2–100 mg/kg bw, a decrease in rat liver mitochondrial monoamine oxidase (MAO) was observed in about 4 hours. Maximal inhibition occurred in 16–24 hours and enzyme activity returned to normal after 21 days. In chickens fed 400 mg furazolidone/kg in feed for 10 days, MAO activity in the brain, heart and alimentary tract was inhibited. The treatment increased the amount of 5-hydroxy-tryptamine in the brain and potentiated the vasodepressor action of tyramine. Significant diamine oxidase inhibition was observed in plasma, duodenal mucosa, liver, heart and brain of rabbits given 50 mg furazolidone/kg bw orally for 5 days. Recovery was complete in these tissues 14 days after the last dose (JECFA, 1993).
Furazolidone, administered to patients with essential hypertension, produced marked supersensitivity to tyramine and amphetamine, inhibition of intestinal MAO and increased urinary excretion of tryptamine. Continued administration produced cumulative inhibition of MAO activity (JECFA, 1993).
4.3 Carcinogenicity and mutagenicityGroups of Swiss MBR/ICR mice (50/sex/group) were fed diets containing 0, 75, 150 or 300 mg/kg furazolidone for 13 months (= 12, 24 or 47 mg/kg bw/day). The mice were maintained on normal diet for a further 10 months. Survival was decreased in the mid- and high dose groups. The incidence of bronchial adenocarcinomas was significantly increased in the mid- and high dose groups in both sexes (male incidences: 13/49, 19/48, 26/50, 37/50; female: 15/50, 18/50, 20/47, 30/48 for control, low, mid and high doses, respectively ). The incidence of lymphosarcomas was significantly increased in the mid and high dose males (1/49, 7/48, 10/50, 10/50 for control, low, mid and high dose, respectively ) (JECFA, 1993).
Groups of Sprague–Dawley rats, 35/sex/group, were fed diets containing furazolidone for 2 years. The consumption of test compound per group was 0, 0.7, 4 or 10 mg/kg bw/day for males and 0, 0.8, 4.3 or 14 mg/kg bw/day for females. At termination, mid and high dose female rats showed dose-related decreased erythrocyte, haemoglobin and haematocrit counts and an increased neutrophil count. Relative liver weight was increased in high dose males and there was an increase in parathyroid hyperplasia without ‘renal ricketts’. In males and females, there was an increase in adrenal cortical hyperplasia at the highest dose level. High dose females showed an increase in thyroid atrophy. Females exhibited increased mammary tumour incidence as shown in Table 4.1. The mean onset time of mammary neoplasms was approximately 2 months earlier in the mid and high dose females than in other groups (JECFA, 1993).
Table 4.1 Mammary tumour incidences in female Sprague–Dawley ratsMammary tumour type
Control Low Mid High
No. animals examined 34 35 33 35Multiple 3 6 11 19Malignant 1 3 3 5Fibroadenoma 0 6 2 10Adenocarcinoma 1 2 2 3Carcinosarcoma 0 1 2 1
Groups of Fischer 344 rats (50/sex/group) were fed diets containing 0, 250, 500 or 1000 mg/kg furazolidone (= 0, 12.5, 25 or 50 mg/kg bw/day) for 20 months. The surviving rats were maintained on control diet for at least 4 months or until 90% of the rats had died. Extensive histopathological examinations were performed on all moribund and sacrificed rats. At the mid (males only) and highest dose the mortality rate was increased; it was 90% at the highest dose at 24 months. At this time, bodyweight gain was significantly decreased at the 500 and 1000 mg/kg levels. At the highest dose
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
84

level, haemoglobin, haematocrit and the number of erythrocytes were significantly decreased in male rats and an increase in ‘non-renal rickets’ was observed. The incidence of testicular atrophy was increased in mid and high dose males and the incidence of adrenal cortical hyperplasia, lipolysis, congestion and haemorrhage was increased in high dose males only. In high dose females, a significant increase in the incidence of mammary gland adenocarcinomas was observed. An increased incidence of sebaceous gland adenomas and thyroid adenomas was observed in both sexes at mid and high dose and of basal-cell epithelioma and carcinomas in males of the high dose group. The results for these tumour incidences are shown in Table 4.2 (JECFA, 1993).
Table 4.2 Tumour incidences in male and female Fischer 344 ratsMales Control Low Mid High
No. rats examined 49 50 50 49Sebaceous gland adenomas 0 0 8 11Basal cell epithelioma 0 2 4 8Basal cell carcinoma 0 0 0 2Thyroid adenoma 1 2 12 19
FemalesNo. rats examined 49 50 50 50Sebaceous gland adenomas 1 2 6 10Mammary neoplasms 11 29 40 30Mammary adenocarcinomas 0 0 0 6Thyroid adenomas 0 1 12 7
Sprague–Dawley rats (50/sex/group) were fed diets containing 0, 250, 500 or 1000 mg furazolidone/kg (= 0, 12.5, 25, or 50 mg/kg bw/day) for 20 months following the same protocol as for the Fischer 344 rat study above. Mortality for the control, low, mid and high dose males was 4/50, 11/50, 17/50 and 30/50 and, for females, 9/50, 12/50, 8/50 and 29/50, respectively. Bodyweight gain was significantly decreased in mid and high dose males and high dose females. At the end of treatment, the number of erythrocytes was decreased in females at mid and high dose levels; high dose males showed an increased neutrophil/lymphocyte ratio and a decreased lymphocyte count. An increased incidence of hepatic necrosis was seen in all treated rats, especially high dose females. Testicular atrophy was seen in mid and high dose males. A dose-related increase in adrenal cortical hyperplasia was observed in females at all dose levels. In the high dose group, significantly increased incidences were recorded for mammary adenocarcinomas in females and for neural astrocytomas in males. Female rats showed a significant increase in the incidence of mammary neoplasms (benign and malignant combined) at all dose levels, but without a dose–response relationship (JECFA, 1993).
A large number of in vitro genotoxicity studies has been undertaken on furazolidone to a variety of end-points (JECFA, 1993; GENE-TOX, 1998; CCRIS, 2002). Many have shown positive results. A representative selection of the results of these assays is shown in Table 4.3 (JECFA, 1993).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
85

Table 4.3 Results for in vitro genotoxicity assays on furazolidoneTest system Concentration Result
S. typhimuriuma, TA98 (±S9) 0.1–2.5 µg/plate PositiveE. coli, WP2uvraA, Spot test 50 µg/plate PositiveE. gracilis, reverse mutation 25, 50, 100 µg/mℓ PositiveCHO cells, gene mutation (±S9) Up to 125 µg/mℓ PositiveE. coli, WP2, SOS function/mutation 0.8, 8.0 µM PositiveD. melanogaster, S-LRL test 0.5 mM PositiveHuman lymphocytes, chromosome aberrations 0.2, 2.0, 20.0 µg/mℓ PositiveHuman lymphocytes, chromosome aberrations 0.5–100 µM NegativeHuman lymphocytes, SCE 0.2, 2.0, 20 mg/mℓ PositiveE. coli, WP2uvrA, DNA repair 1.0–10 µM PositiveHuman lymphocytes, UDS 5–100 µM NegativeRat hepatocytes, UDS 0.5–1000 nM/mℓ PositiveaNitroreductase deficient
Two in vivo micronucleus tests with furazolidone have been undertaken in mice. In one test, mice were dosed the test compound in methylcellulose i.p. at 300 mg/kg bw. In the other test, the mice were dosed orally at 100 or 500 mg/kg bw. The result after i.p. dosing was negative and, after oral dosing, it was equivocal (JECFA, 1993).
Some of the metabolites of furazolidone have also been assessed with in vitro genotoxicity assays as shown in Table 4.4 (JECFA, 1993).
Table 4.4 Results of in vitro genotoxicity assays with furazolidone metabolitesMetabolite Test system Concentration ResultaA S. typhimurium, 2 strains 100–5000 µg/plate NegativebB S. typhimurium, 2 strains 0.1mg/plate NegativecC E. gracilis, rev. mutation 2.5, 7.5 µg/mℓ PositivedD DNA repair, rodent hepat. 10-3 M, 10-5 M NegativeaA = 3-Amino-oxazolidinon-2; bB = 3-(4-cyano-2-oxobutylideneamino)-2-oxazolidone; cC = 5-nitro-2-furaldehyde; dD = 2-hydroxyethylhydrazine
The marker residue metabolite, 3-amino-oxazolidone-2, has been assessed in a battery of in vitro and in vivo assays. In the S. typhimurium assay, negative results were obtained with tester strains TA98 and TA1537 (+S9) and in TA100 (-S9). Positive results were obtained with tester strains TA1535 (±S9) and TA100 (+S9). Slightly positive results were obtained in an E. coli plate test in the presence or absence of S9. The metabolite produced a statistically significant increase in chromosomal aberrations using human peripheral lymphocytes without S9. In the presence of S9, the test gave a negative result. Two micronucleus tests in mice were performed. In one test, male and female mice were treated with a single i.p. dose of 500 mg/kg bw and 1000 mg/kg bw, respectively. A significant increase in micronucleated polychromatic erythrocytes was seen in animals of both sexes. In the other test, males received single i.p. doses of 32–500 mg/kg bw and, females, 250–1500 mg/kg bw. A statistically significant increase in the number of micronucleated polychromatic erythrocytes was observed in males of the highest dose group at 48 hours but not at 24 hours post administration. The compound induced an increase in the frequency of micronuclated polychromatic erythrocytes in individual animals of both sexes in all dose groups. The Committee for Veterinary Medicinal Products considered that 3-amino-oxazolidone-2 was mutagenic in all these test systems (EMEA, 1997).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
86

On the basis of the positive results of furazolidone in genotoxicity tests in vitro and the increased incidence of malignant tumours in mice and rats, a WHO committee concluded that the compound was a genotoxic carcinogen (JECFA, 1993).
4.4 Reproductive and developmental toxicityGroups of Sprague–Dawley rats (20/sex) were assessed in a three-generation reproduction study. Female rats, only, were administered 500 mg furazolidone/kg in diet. This dose level was lowered to 400 mg/kg by day 16 and then to 250 mg/kg by day 37 because of growth depression effects. Three matings per generation were performed. Pups from the first litter were sacrificed 21 days after birth. Pups from the second litter were used for the second generation and pups from the third litter were given a teratological examination. No treatment-related effects were found for reproductive parameters, resulting in a NOEL equivalent to 12.5 mg/kg bw/day (JECFA, 1993).
Groups of albino C strain mice were administered furazolidone at doses up to 2 g/kg during pregnancy varying from day 1–11. Abortions or fetal deaths occurred in all mice treated with doses 1 g/kg when treatment started before day 8 of pregnancy. When treatment was started at day 10, abortions or fetal death occurred in only 2/9 mice. Litter weight was decreased in a dose-related manner but no congenital abnormalities were observed. Groups of 10 pregnant New Zealand White rabbits were orally administered 30 mg furazolidone/kg bw/day on days 7–15 of pregnancy. On day 29 of pregnancy, the dams were sacrificed and the fetuses delivered by caesarean section. Food consumption and bodyweight gain were significantly decreased. No embryotoxicity or teratogenicity was observed (JECFA, 1993).
Five male rats/group received diets containing 330 or 660 mg furazolidone/kg for 14 weeks and 3 male rats/group were given the same doses for 12 weeks (equivalent to 16 or 33 mg/kg bw/day) with a 2 week recovery period. A control group of 4 rats was used. No effects were seen in rats of the low dose group. Testes weight was markedly decreased in high dose rats. The testes in these rats showed oedema of the interstitium and atrophy and degeneration of the sperm producing tubules. These effects were still present after the recovery period. In a follow-up study, male rats were fed a diet containing 660 mg furazolidone/kg and histopathology was performed on animals killed 5 days or 6 weeks after withdrawal from the test diet. Testes weight was decreased in 4/6 rats and in 3/6 rats atrophic seminiferous tubules and abundant intertubular transudate were seen. In 2 of these rats, the epididimi had sperm stasis. All prostates were normal. Two males, which had been retained for 14 weeks after withdrawal of diet, were each placed with 2 untreated females. These males showed normal libido and fecundated the females but the litters produced had slightly fewer young than those from litters sired by control males. In another study, groups of 3 Donryu male rats were fed 0 or 100 mg furazolidone/kg bw for 7 days, with the rats being sacrificed on day 8. Relative testes weights were slightly decreased. The number of mature spermatozoa was decreased and degenerative changes in the seminiferous tubules (sloughing of spermatocytes and multinucleated cells) were observed (JECFA, 1993).
Reprotoxicity studies have also been carried out on other species. Groups of New Hampshire chickens were fed diets containing 55, 110 or 220 mg furazolidone/kg feed for 4–16 weeks. No effects were observed on bodyweight and egg production, hatchability of eggs and shell quality. Thyroid and adrenal weights were reduced. A group of Hampshire and Duroc pigs (9 sows and 12 gilts) were fed a diet containing 300 mg/kg furazolidone for 2 weeks at breeding and 150 mg/kg for 3 weeks at farrowing. Another group acted as control. There was no difference in the number of pigs born, the number of pigs weaned or weight gain of the pigs. Pup weight at weaning was slightly higher in the treated group. Male Nubian goats received furazolidone at a dose of 10 or 40 mg/kg bw. At both doses, ejaculate volume, the number of motile spermatozoa per ejaculate and the number of live spermatozoa per ejaculate were significantly decreased (JECFA, 1993).
Studies on endocrine toxicity have been carried out on rats. Groups of ovariectomised Sprague–Dawley, Fischer 344 and Long Evans rats received single oral doses varying from 50 to 500 mg
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
87

furazolidone/kg bw. After a 2 week recovery period, positive dose-related lordosis (becoming sexually perceptive) behaviour was observed in the Sprague–Dawley rats only. Groups of immature superovulated Sprague–Dawley rats were dosed orally with 100 or 500 mg furazolidone/kg bw. In all groups, ovulation was initiated about 24 h earlier than the control group. The same shift in ovulation time was observed with a positive control group treated with progesterone (JECFA, 1993).
The feeding of 1000 mg furazolidone/kg diet for 30, 60 or 90 days produced a marked inhibition of the conversion of progesterone into corticosterone (11-hydroxylation) in the adrenals of Sprague–Dawley female rats. A considerably lesser effect was found in Fischer 344 females. The oral administration of 100 or 500 mg furazolidone/kg bw to mature female rats produced a significant decrease in serum prolactin during morning proestrus but not in the afternoon proestrus. Administration of 500 mg/kg bw during the morning estrus resulted in a slight decrease in serum prolactin levels. A diet containing 500, 750 or 1000 mg furazolidone/kg administered to female rats for 30 days, resulted in a significant increase in serum testosterone at the highest dose (JECFA, 1993).
Male mature chickens were fed a diet containing 400 or 800 mg furazolidone/kg feed for 10 days. Testes weight was decreased in both groups. At 800 mg/kg, there was a significant reduction in the concentration of testosterone in plasma and testes. Bolus administration of doses of 40 or 80 mg/kg bw for 5 days caused the same effect. The size of the testes, wattles and combs were significantly reduced and the MAO activity in the testes was also inhibited by furazolidone. The testicular concentrations of 5-hydroxytryptamine were significantly raised in all treated birds, except for those fed 400 mg/kg for 10 days (JECFA, 1993).
5 Guidelines and standardsJECFA concluded that furazolidone was a genotoxic carcinogen (see Section 4.3). The Committee also noted that few of the furazolidone metabolites had been either identified or quantified in rats and pigs. JECFA also concluded that insufficient data were available on the nature and toxic potential of compounds released from the bound residues. Because of these conclusions JECFA was unable to establish an ADI or MRLs (JECFA, 1992; JECFA, 1993). Nonetheless, in line with Council Directive 2002/657 EC, a non-toxicologically based Minimum Required Performance Limit (MRPL) of 1 g/kg analytical performance standard has been established for furazolidone (and its metabolites), for use in analysis of samples of poultry meat and aquaculture products (EC, 2003).
During use, withdrawal periods for meat products were set at 7 days for all species. EMEA CVMP could derive no NOEL that could serve as a basis for establishment of an ADI from toxicity studies conducted since treatment related effects were observed at the lowest dose rates used; no MRLs have been established for furazolidone in food; and products containing this compound are no longer in use. In the light of the above and as the parent drug and the main metabolite (3-amino-oxazolidone-2) have been shown to be carcinogenic and mutagenic, furazolidone was added to Annex IV of EU 2377/90 and all products withdrawn from the market in 1997 (EMEA, 1997).
The US FDA established a tolerance of zero for residues of furazolidone in the uncooked edible tissues of swine (HSDB, 2002).
Prior to 1992, Furazolidone was registered for use in Australia, and an ADI of 0.4 g/kg bw/day established based on a NOEL of 0.75 mg/kg bw/day in a long-term study in Sprague-Dawley rats, using a 2000-fold uncertainty factor (see Section 4.3). This ADI was withdrawn in 2003 as furazolidone was no longer in use following a ban of use in food-producing animals (FSANZ, 2004).
6 ReferencesBishop Y, ed (1994) The Veterinary National Formulary (Second Edition), London, UK, Royal Pharmaceutical Society of Great Britain/British Veterinary Association
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
88

Bishop Y, ed (2004) The Veterinary National Formulary (Sixth Edition), London, UK, Royal Pharmaceutical Society of Great Britain/British Veterinary Association
CCRIS (2002) Furazolidone. From: Chemical Carcinogenesis Research Information System, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [March 2004] at http://http://toxnet.nlm.nih.gov
EC (2003) Commission Decision of 13 March 2003 amending Decision 2002/657/EC as regards the setting of minimum required performance limits (MRLPs) for certain residues in food of animal origin (notified under document number C(2003)764) (2003/181/EC) Official Journal of the European Union, L71, 17-18 Available July 2005 at: http://europa.eu.int/eur-lex/pri/en/oj/dat/2003/l_071/l_07120030315en00170018.pdf
EFSA (2005) Opinion of the Scientific Panel on Food Additives, Flavourings, Processing Aids and Materials in Contact with Food on a request from the Commisson related to Semicarbazide in Food Question number EFSA-2003-235 Adopted on 21 June 2005 by written procedure. European Food Standards Agency. The EFSA Journal, 219, 1-36
EMEA (1997) Furazolidone Summary Report, London, UK, European Agency for the Evaluation of Medicinal Products, Available [March 2004] at http://www.emea.europa.eu/pdfs/vet/mrls/Furazolidone.pdf
FSANZ (2004) Nitrofurans in Prawns: A Toxicological Review and Risk Assessment. Food Standards Australia New Zealand, Canberra, Australia
GENE-TOX (1998) Furazolidone. From: Genetic Toxicology (Mutagenicity) Databank, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [June 2004] at http://http://toxnet.nlm.nih.gov
HSDB (2002) Furazolidone. From: Hazardous Substances Databank, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [June 2003] at http://http://toxnet.nlm.nih.gov
JECFA (1992) Furazolidone (Joint FAO/WHO Expert Committee on Food Additives), Geneva, Switzerland, World Health Organization, Available [January 2004] at http://www.inchem.org/documents/jecfa/jeceval/jec_661.htm
JECFA (1993) Furazolidone (Food Additives Series 31), Geneva, Switzerland, World Health Organization, Available [January 2004] at http://www.inchem.org/documents/jecfa/jecmono/v31je06.htm
SIS NLM (2004) Furazolidone. From: ChemIDplus, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [January 2004] at http://http://chem.sis.nlm.nih.gov/chemidplus/
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
89

Annex 8 Lasalocid Sodium 1 IntroductionLasalocid sodium (6-(7-(5-ethyl-5-(5-ethyltetrahydro-5-hydroxy-6-methyl-2H-pyran-2-yl)tetrahydro-3-methyl-2-furanyl)-4-hydroxy-3,5-dimethyl-6-oxononyl)-2-hydroxy-3-methyl-benzoic acid, monosodium salt; CAS No. 25999-20-6) is an ionophore capable of combining with monovalent and bivalent ions and transporting them through lipophilic biological membranes. Lasalocid was first introduced into animal agriculture as a poultry coccidiostat, marketed as AVATEC. This was approved for use in broiler rations at 75–125 ppm. Another lasalocid product, BOVATEC, is used for feedlot cattle to improve feed efficiency and rate of weight gain. It was approved for use in beef cattle at 30 g/ton. Lasalocid has also been helpful in preventing acidosis in high grain-fed cattle (Galitzer & Oehme, 1984).
In fowl, the increase in productivity due to lasalocid is derived primarily through control of coccidia capable of adversely affecting animal health. Increases in ruminants’ productivity results, most importantly, from the increase in feed conversion due to shifts in rumen microflora populations. Lasalocid prevents acidosis in feedlot cattle by inhibiting major lactic acid producing bacteria, except Selenomonas, but not affecting rumen bacteria that ferment lactic acid. In addition, it exerts favourable effects on ruminal function by enhancing propionate formation and inhibiting methane production (Oehme & Pickrell, 1999).
2 Uses and exposureLasalocid sodium, available as AvatecTM in many countries, is classified as a zootechnical feed additive used for the prophylaxis of coccidiosis in chickens, turkeys and gamebirds (pheasants, partridges). AvatecTM contains 15% w/w lasalocid sodium (150 g/kg premix) in a corncob carrier (Taylor, 2004).
For chickens, the dose rate for inclusion in the diet is 500–830 g/tonne of premix equivalent to 75–125 g lasalocid sodium /tonne of feed. For broiler chickens reared in the UK, the usual dose rate is 600 g of premix equivalent to 90 g/tonne lasalocid sodium (90 mg/kg = 0.009%). This may be fed in the diet continually from 1-day old, to 5 days before slaughter (approximately 6 weeks of age) for broiler chickens, or 16 weeks of age for layer replacement chickens. As an alternative, the drug may be fed in the diet for a shorter period of 1–3 weeks as part of a ‘rotation’, ‘shuttle’ or ‘sandwich’ control programme aimed at limiting the development of resistance, or increasing anticoccidial performance (Taylor, 2004).
The withdrawal period in broiler chickens prior to slaughter is 5 days. The product is contraindicated in layer replacement chickens over 16 weeks of age (Taylor, 2004).
In turkeys, the dose rate for inclusion in the diet is 600–830 g/tonne of premix equivalent to 90–125 g lasalocid sodium/tonne of feed, to be fed continuously from 1 day of age to a maximum age of 12 weeks. The withdrawal period in turkeys prior to slaughter is 5 days (Taylor, 2004).
For pheasants and partridges, the inclusion rate is 600–800 g/tonne of premix equivalent to 90–120 g lasalocid sodium/tonne of feed, to be fed continuously from 1 day of age to a maximum age of 12 weeks. The withdrawal period in gamebirds prior to slaughter is 7 days (Taylor, 2004).
In some countries, such as USA and Canada, lasalocid is available as a medicated premix (Bovatec TM) for use as a growth promoter by improving feed efficiency and increasing weight gain in beef cattle raised in confinement. The dose rate for inclusion in the diet is 36 mg/kg (0.0036%) or 240 g/tonne
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
90

of Premix (150 g/kg) equivalent to 250 mg/animal/day for those weighing up to 300 kg and 350 mg/animal/day for animals greater than 300 kg (Taylor, personal data).
In Australia and New Zealand Lasalocid is licensed only for use in broiler chickens and in layer replacements up to the age of 16 weeks and is therefore not legally permitted in foodstuffs used for laying hens. However, cross-contamination at the feed mill and mistakes with feed batches have led to detectable residues in eggs. No withdrawal period has been set for layer replacement birds that come into lay before 16 weeks of age [Reference to be supplied by CSL].
Humans are potentially exposed to lasalocid sodium via ingestion of animal products containing drug residues.
2.1 Exposure concentrations in meatUsing information on usage of lasalocid and on absorption and excretion, likely ‘worst case’ concentrations in meat products were estimated using Equation 1:
Equation 1M = D × T × Fabs × (1–Fexc)
where M is the predicted concentration in meat (mg/kg); D is the dose of substance (mg/kg bw/d); T is the duration of the treatment (d); Fabs is the fraction of compound absorbed; and Fexc is the fraction of compound excreted.
For chickens, it was assumed that lasalocid is given in feed for 40 days at a dose rate of 90 mg/kg bw//d. The bioavailability of lasalocid is reported to be low and the metabolism is reported to be high, values of 5% absorption and 95% excretion were therefore used. Using these input values, a ‘worst case’ concentration of 9 mg/kg was obtained.
For turkeys, it was assumed that lasalocid is given in feed for 84 days at a dose rate of 100 mg/kg bw/d. The bioavailability of lasalocid is reported to be low and the metabolism is reported to be high; values of 5% absorption and 95% excretion were therefore used. Using these input values, a ‘worst case’ concentration of 21 mg/kg was obtained.
For cattle it was assumed that lasalocid is given to cattle (average weight 200 kg) at a dose rate of 1.25 mg/kg bw/d for 100 d. The bioavailability of lasalocid is reported to be low and the metabolism is reported to be high, values of 5% absorption and 95% excretion were therefore used. Using these input values, a ‘worst case’ concentration of 0.31 mg/kg bw was obtained.
In addition, the Veterinary Residues Committee (VRC, 2002) reported that lasalocid concentrations of 50-3450 µg/kg were detected in eggs during 2001, with 18/255 samples exceeding the MRL. The highest detected concentration of 3450 µg/kg was used in the worst-case risk scenarios for dietary exposure.
3 ToxicokineticsTotal distribution and elimination studies with 14C-lasalocid in beef cattle showed that the liver was the only edible tissue that contained substantial quantities of lasalocid-derived radioactivity at day 0 withdrawal time. The levels in the liver of cattle dosed at 1 mg/kg bw were about 8 ppm. These levels were of concern regarding their possible harmful inotropic effects for some consumers, e.g. people with ischaemic coronary arteries. However, efforts to isolate cattle liver metabolites were hampered by several factors. Because of its complex structure, 14C-labelling of lasalocid could not be carried out by synthetic methods and, because of the problems of synthesis, potential metabolites were not available for comparison purposes. Moreover, there appeared to be a very large number of metabolites
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
91

produced; approximately 12% of the liver residues at Day 0 withdrawal was intact parent compound, the remainder consisted of a multitude of minor metabolites, none of which constituted >3% of the total liver residues. About 80% of the total liver radioactivity remained unidentified (Weiss, 1990).
Because of the above difficulties, other approaches were taken to evaluate possible metabolic consequences of the breakdown of lasalocid. The cattle liver radioactivity was separated into an ethanol-soluble and an ethanol-insoluble fraction and the bioavailabilities of these fractions were evaluated in the bile-cannulated rat model. This showed that the ethanol-insoluble fraction, which was about 18% of the liver radioactivity, was essentially non-bioavailable. About 40% of the ethanol-soluble fraction, which represented about 82% of the activity, was bioavailable. The inotropic activity of the lasalocid metabolites was evaluated using an in vitro isolated guinea pig atrial muscle system. In preliminary experiments, it was determined that metabolite concentrations of about 900 µg/ml in DMSO would be required. Because of the low residue levels in the liver, it was decided to test the metabolites in the faeces as well. The faeces were subjected to a solvent extraction procedure designed to separate most of the intact lasalocid from metabolites. Solvent fractions were dissolved in DMSO to give a concentration of about 900 µg/ml lasalocid equivalents. The results from the isolated guinea pig trial muscle tests for both cattle faeces and liver extracts indicated that the metabolites produced by the cattle are not significantly more inotropic than lasalocid (Weiss, 1990).
4 Toxicity profile4.1 Acute toxicityAcute LD50 values have been determined for lasalocid in a number of species as shown in Table 4.1.
Table 4.1 Acute LD50 values for lasalocidSpecies Route LD50 (mg/kg bw) Reference
Rat Oral 122 Galitzer & Oehme (1984)Rat Oral >130 Gad et al. (1985)Mouse Oral 146 Galitzer & Oehme (1984)Mouse Oral >100 Gad et al. (1985)Rabbit Oral 40 Galitzer & Oehme (1984)Horse Oral 21.5a Hanson et al. (1981)Poultry Oral 71.5 Galitzer & Oehme (1984)Rat I.P.b 8 Gad et al. (1985)Mouse I.P. 40 Gad et al. (1985)Rabbit Dermal >50 Gad et al. (1985)aValue disputed by Kronfeld (2002), see belowbIntraperitoneal
A range of acute toxic effects has been described for lasalocid in rodents. These consisted of aggressive behaviour and other neurotoxic effects due to action on the central nervous system and peripheral nervous system. There was also marked cardiotoxicity and adrenergic stimulation (Gad et al., 1985). Cattle given lasalocid at single oral doses of 50 mg/kg bw or more, showed muscle tremors in the flank, increased heart and respiratory rates and anorexia, all appearing within the first 24 h; the effects were transient at 50 mg/kg bw (Galitzer & Oehme, 1984).
Lasalocid was given to horses in a series of sequentially increasing single oral doses ranging between 5 and 30 mg/kg bw, with appropriate recovery periods between treatments. The clinical signs of toxicity were progressive and included depression, ataxia, paresis and paralysis with partial anorexia. The lowest dose at which a horse died was 15 mg/kg bw. The LD50 was estimated using a method which was conservative yet appropriate for the small number of animals used and for the re-dosing procedure employed; the LD50 was calculated to be 21.5 mg/kg bw as indicated in Table 4.1 (Hanson
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
92

et al., 1981). The estimated LD50 for lasalocid in horses was challenged by Kronfeld (2002), who pointed out that it is based on an analysis of data from 4 horses that died at dose levels of 15, 21, 22 and 26 mg/kg bw. This analysis neglects data from another 6 horses that survived at dose levels of 5, 10, 14, 18, 19, 25, 29 and 30 mg/kg bw and was therefore biased by selection of data. In view of this, the 21.5 mg/kg bw estimate for the LD50 for lasalocid in horses should not be perpetuated without question and needs to be better quantified (Kronfeld, 2002).
Lasalocid was evaluated in primary dermal and eye irritation tests. In dermal testing, 0.1 g of the material was placed on 1-inch squares on the shaved backs of six New Zealand White rabbits and occluded for 24 hours. The reactions on the test sites were scored by the Draize scale at 24, 48 and 72 h after the removal of the patches and a primary dermal irritation index (PII) calculated from these results. Ocular irritation potential was evaluated by placing a single drop of material into the left eye of one rabbit, the other eye was untreated and acted as control. The resulting reaction was graded and scored, according to the Draize procedure, at 1, 2, 3, 4, 7 and 10 days post installation. For lasalocid, the PII was 1.1 and the eye irritation score was moderate (Gad et al., 1985).
4.2 Repeat dose toxicityLasalocid causes a variety of toxic effects when given by repeated dose administration to animals at elevated levels or due to misformulation. In cattle, lasalocid is reported to be toxic to cardiac and skeletal muscle. Cardiotoxicity in cattle is expressed as respiratory distress, oedema and hydrothorax. In horses, cardiac muscle is most affected, leading to tachycardia and cardiac muscle damage. Avian hepatopathies and skeletal weakness have been reported for lasalocid administration (Oehme & Pickrell, 1999). Neurotoxic effects such as tremors, convulsions and reduced seizure thresholds have been observed for lasalocid in dogs, rats and man (Gad & Gad, 2003).
No observable effect levels (NOELs) for lasalocid have been established in long-term studies with mice, rats and dogs as shown in Table 4.2 (Oehme & Pickrell, 1999).
Table 4.2 NOELs established for lasalocid in long-term (2 year) animal studiesSpecies NOEL
mg/kg bw/d Dietary (ppm)
Mouse 2.4 120Rat 2.4 120Dog 1.0 –
Apart from the NOEL values, no other information was found for these long-term studies.
The lasalocid product, BOVATEC, produced no adverse effects when fed to beef cattle at the recommended rate of 30 g/ton (0.97 mg/kg bw). Feeding cattle up to five times this dose for 252 days produced no adverse clinical signs except for transient diarrhoea on days 4–7. In another study, cattle fed five times the recommended rate for 182 days showed no significant changes in haematology or serum chemistry except for increases in globulin and aminotransferase activity and a decrease in uric acid. Swine have been fed lasalocid at 3.78 mg/kg bw with no adverse effects. Ingestion at 21 mg/kg bw for 2 days also caused no effects. Transient muscle weakness occurred at 35 mg/kgbw and death occurred at 58 mg/kgbw when fed for 1 day (Galitzer & Oehme, 1984). When 3 horses were fed pelleted broiler feed containing 125 g lasalocid/metric ton, there was a limited intake of lasalocid at 1.52–1.88 mg/kg bw/day for 11 days. None of these horses showed clinical signs of lasalocid toxicity (Hanson et al., 1981).
Feeding trials with chickens demonstrated that lasalocid, at high dose levels (300–400 mg/kg in feed), caused irreversible damage to axons in the spinal cord. Clinically, birds were mainly unaffected for 4–7 days but then rapidly developed inappetance, depression and a preference to sit. Later, birds
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
93

rocked on their hocks and those that could stand were ataxic and unsure of foot placement. More chronic stages were walking on tip-toe or partial paralysis. Mortality was usually low despite severe weight loss. Histopathology in affected birds revealed consistent abnormalities, only in the spinal cord, in which swollen, degenerating and fragmented myelin sheaths and axons were seen in the ventral columns of the thoracic cord, later manifest as vacuolisation of these areas (Shlosberg et al., 1985).
The effects of accidental feeding of lasalocid sodium to broiler breeder chickens on two farms have been described. In one farm, the flock was 29 weeks of age when an obvious slowing down in the rate of increase of egg production was first observed. At the second farm, the flock was 44 weeks old when a drop in egg production was observed within a period of 15 days. At the same time a few cockerels showed clinical signs characterised by leg weakness and partial ataxia. Analysis of feed samples from both farms revealed levels of lasalocid ranging from 115–150 ppm. A direct correlation between the length of time the flock received the contaminated feed and the decrease in fertility and hatchability was observed. The level of lasalocid found in egg yolk increased from 2.5 mg/kg in eggs collected after 3 days of lasalocid administration up to 18.5 mg/kg in eggs collected after 14 days administration. The fertility and hatchability returned to normal within about 2 weeks, after the contaminated feed had been replaced. Post mortem examination of the cockerels with leg weakness and ataxia did not reveal any macroscopic lesions. Microscopic examination showed severe pathological changes in the muscles of the affected cockerels. The predominant lesions included loss of striation and hyalinisation of the muscle fibres; vacuolation of the sarcoplasm with enlarged and occasionally centrally located nuclei were frequently observed. Foci of axonal swelling and fragmentation in the sciatic nerve and signs of slight testicular degeneration, characterised by sloughing into the lumen of cells of the germinal epithelium associated with the presence of giant cells, were also observed in some samples taken from the affected cockerels (Perelman et al., 1993). Further information on effects on fertility and affected offspring, in this study, are described in Section 4.4.
Some studies have been undertaken to examine the mechanism of lasalocid’s toxic action. Beagle dogs were used to study the effect of lasalocid on ventricular function, total body haemodynamics and blood volume. Animals were anaesthetised with a combination of barbital and pentobarbital. After a 30 min period of stabilisation, lasalocid was administered intravenously (i.v.) at 1 mg/kg bw. The parameters measured included heart rate, myocardial contractility, cardiac index, stroke index, right atrial pressure, arterial blood pressure and total peripheral resistance. Lasalocid produced a significant increase in cardiac index and pressure gradient for venous return and a non-significant decrease in resistance to venous return and right atrial pressure. There was a significant increase in heart rate and arterial pressure and a significant decrease in total peripheral resistance and a non-significant decrease in stroke volume index. These results and those for other parameters are shown in Table 4.3 (Osborne et al., 1977).
Table 4.3 Cardiac function results for dogs given lasalocid at 1 mg/kg bw (i.v.)Parameter Control Lasalocid P valueHeart rate (beats/min) 153 ± 9 194 ± 7 0.05Cardiac index 3636 ± 248 5175 ± 300 0.01Stroke volume index 23.8 ± 1.3 27.0 ± 2.2 NSRight atrial pressure (mmHg) 1.0 ± 0.6 0.4 ± 0.6 NSMean arterial bp 137.4 ± 7.1 160.4 ± 8.9 0.05Total peripheral resistance 7.4 ± 0.5 6.1 ± 0.5 0.05Mean circulatory pressure 10.3 ± 0.7 12.1 ± 0.9 0.005Pressure gradient for venous return 9.4 ± 1.1 11.7 ± 1.3 0.005Resistance to venous return 0.26 ± 0.04 0.23 ± 0.03 NSThe accumulated experimental results indicate that the cardiovascular effect of lasalocid embodies two distinct mechanisms — an increase in tension of all cardiovascular tissue and a dynamic alteration in cardiovascular homeostasis secondary to the increase in tension. The mechanism of the
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
94

lasalocid effect on myocardial contractility appears to be complex. The results demonstrate that all cardiovascular tissue is involved in expressing the effects produced by lasalocid. A role for an ionophore mediated calcium perturbation in the contraction-coupling process of cardiovascular tissue myofibrils should be considered as a possible mechanism (Osborne et al., 1977).
Similar results for the effects of lasalocid on the dog’s cardiovascular system were also obtained by Schwartz (1976). A single injection of lasalocid into a catheter placed in the right atrium or into the femoral vein of an anaesthetised animal caused a prolonged increase in mean blood pressure and aortic cardiac output as well as a direct positive inotropic effect. An explanation for the action of lasalocid was put forward; that it energises a membrane-mediated calcium–proton or perhaps a calcium–sodium exchange that leads to an increased availability of calcium. This would include vascular smooth muscle as well.
The mechanisms of the neurotoxic action of lasalocid have been examined in vitro. Dispersed cerebral cells prepared from 15–16 day mouse fetuses were cultured for 7 days and exposed to lasalocid for periods of 4, 18 or 48 hours. Cultures were examined by phase contrast microscopy and processed for scanning electron microscopy. Lasalocid (1 and 2 µM) induced neurotoxic effects that were evident at 4 hours, including swelling of perikarya followed by cytolysis of most neurons present in the cultures. In the next 18 hours, virtually the entire neuronal population degenerated. Cultures exposed to 0.5 µM lasalocid for 48 hours showed only mild damage to some neurons and, at 0.2 µM, no damage was observed. Glial and other non-neuronal cells were not damaged by exposure to 2 µM lasalocid and cell division resumed on returning the cultures to regular growth media. Lasalocid (2 µM) was not toxic to cultured rat astrocytes. Lasalocid (1 µM) induced 45Ca2+ influx to 40% above control values. The voltage-sensitive calcium channel antagonists nimodipine and D-600 did not inhibit lasalocid-mediated 45Ca2+ influx, whereas MK-801, a non-competitive N-methyl-D-aspartate (NMDA) receptor/channel antagonist, exclusively blocked this influx and prevented cytotoxic damage. Conversely, NMDA, which by itself mediated 45Ca2+ influx in these cultures, potentiated lasalocid-induced 45Ca2+ influx and cellular damage. Similar results were obtained using cerebral cultures prepared from prenatal 15–16 day rat fetuses. These observations demonstrate the selective neurotoxicity of lasalocid to cultured cerebral neurons and may imply the involvement of the NMDA receptor/channel in lasalocid-mediated neurotoxicity (Safran et al., 1993; Safran et al., 1996).
4.3 Carcinogenicity and mutagenicityLong-term (2 year) rat and mouse dietary toxicity studies have been undertaken with lasalocid (Oehme & Pickrell, 1999). However, apart from reporting NOELs of 120 ppm lasalocid (2.4 mg/kg bw), no other information regarding study design, toxic effects or histopathology was given.
No information could be found for mutagenic assessments of lasalocid. However, the European Food Safety Authority (EFSA, 2004) concluded that lasalocid sodium is not genotoxic and that there was no evidence of carcinogenicity. The studies on which this conclusion was reached were not given.
4.4 Reproductive and developmental toxicityLasalocid supplementation (0.15 or 0.23 kg; 70 mg/hd/day) increased the percentage of lambs born/ewe (P <0.05) and kg of lamb weaned (productivity) because of improved lambing percentage (Oehme & Pickrell, 1999).
A slight decrease in egg production was reported at two broiler breeder farms receiving feed from the same mill. On the first farm, the flock was 29 weeks of age when an obvious slowing down of egg production occurred. At the second farm, the flock was 44 weeks old when a drop in egg production of between 4% and 8% was observed within a period of 15 days. At the same time, a few cockerels showed clinical signs characterised by leg weakness and partial ataxia. Analysis of feed samples revealed levels of lasalocid of 115–150 ppm. A decrease of 10% in the fertility and hatchability was
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
95

observed in eggs collected 6 days after the introduction of contaminated feed; a decrease of more than 60% was seen in birds that had been fed the ration for 12 days. The fertility and hatchability returned to normal levels about 2 weeks after the contaminated feed was replaced. An examination of eggs, which failed to hatch, showed a large number of infertile eggs (29–34%), an increase in early embryonic death (5.4–6.6%) and many fully developed piping chicks that were unable to hatch (20.4–24.0%). More than 10% of the chicks hatched from these eggs developed leg weakness and ataxia. Post mortem of one-day-old chicks with leg weakness and piping chicks unable to hatch did not show any macroscopic lesions. Microscopic examination revealed muscular lesions in the one-day-old chicks which consisted of occasional foci of myofibres with basophilic sarcoplasm and enlarged nuclei. Similar changes were observed in the muscles of piping chicks unable to hatch (Perelman et al., 1993).
5 Guidelines and standardsIn 2004, EMEA CVMP (EMEA, 2004), based upon a toxicologically-based ADI of 2.5 ug/kg/day (i.e. 150 μg/60kg bw/day), recommended the establishment of MRLs in accordance with Council regulation (EEC) No. 2377/90 for lasalocid in poultry species. These MRLs are summarised in Table 5.1.
Table 5.1 MRLs for lasalocid Substance
Marker residue
Animal species
MRL Target tissue
Other provisions
Lasalocid Lasalocid A Poultry 20 µg/kg100 µg/kg100 µg/kg50 µg/kg
MuscleFat+skinLiverKidney
Not for use in animals from which eggs are produced for human consumption
The EFSA concluded that the lowest NOEL was 0.5 mg/kg bw/day, established in a 2-year chronic oral toxicity study in rats and the maternal toxicity study in rabbits (EFSA, 2004). However, it was noted that it was not possible to establish a NOEL for reproductive endpoints because of study limitations. Applying an uncertainty factor of 100 to the chronic toxicity NOEL would lead to an ADI of 5 µg/kg bw/day. However, the similarity between the metabolic profiles of lasalocid sodium in the laboratory animals and chickens has not been thoroughly established. Therefore, concern remains regarding the adequacy of the evaluation of residues in chicken tissues and no ADI or MRL was set.
The Australian Government have based their standard on a NOEL of 2 mg/kg bw/day, although the specific study and endpoints on which this was based was not specified and, on the basis of this NOEL, an ADI of 1 µg/kg bw/day was established (Australian Government, 2005).
6 ReferencesAustralian Government (2005) ADI List - Acceptable Daily Intakes for Agricultural and Veterinary Chemicals, Canberra, Australia, Australian Government, Department of Health and Ageing, Available [June 2005] at; http://www.tga.gov.au/docs/html/adi.htm
EMEA (2004) Lasalocid: Summary Opinion of the Committee for Medicinal Products for Veterinary Use on the Establishment of Maximum Residue Limits (EMEA/CVMP/962/04), London, UK, European Agency for the Evaluation of Medicinal Products, Available [May 2005] at http://www.emea.europa.eu/pdfs/vet/mrls/mrlopinions/096204en.pdf
EFSA (2004) Opinion of the Scientific Panel on Additives and Products or Substances used in Animal Feed on the Reevaluation of Coccidiostat Avatec in Accordance with article 9G of Council Directive 70/524/EEC (EFSA-Q-2003-042), Brussels, Belgium, European Food Safety Authority, available [May 2005] at http://www.efsa.eu.int/science/feedap/feedap_opinions/423_en.html
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
96

Gad SC & Gad SE (2003) A functional observational battery for use in canine toxicity studies: Development and validation. Int J Toxicol, 22, 415-422
Gad SC, Reilly C, Siino K, Gavigan FA & Witz G (1985) Thirteen cationic ionophores: Their acute toxicity, neurobehavioral and membrane effects. Drug Chem Toxicol, 8, 451-468
Galitzer SJ & Oehme FW (1984) A literature review on the toxicity of lasalocid, a polyether antibiotic. Vet Hum Toxicol, 26, 322-326
Hanson LJ, Eisenbeis HG & Givens SV (1981) Toxic effects of lasalocid in horses. Am J Vet Res, 42, 456-461
Kronfeld DS (2002) Lasalocid toxicosis is inadequately quantified for horses. Vet Hum Toxicol, 44, 245-247
Oehme FW & Pickrell JA (1999) An analysis of the chronic oral toxicity of polyether ionophore antibiotics in animals. Vet Hum Toxicol, 41, 251-257
Osborne MW, Wenger JJ & Zanko MT (1977) The cardiovascular pharmacology of the antibiotic ionophore Ro 2-2985 (X537A). J Pharmacol Exp Ther, 200, 195-206
Perelman B, Pirak M & Smith B (1993) Effects of the accidental feeding of lasalocid sodium to broiler breeder chickens. Vet Record, 132, 271-273
Safran N, Haring R, Gurwltz D, Shainberg A, Halili I, Levy A, Bogin E & Shahar A (1996) Selective neurotoxicity induced by the ionophore lasalocid in rat dissociated cerebral cultures, involvement of the NMDA receptor/channel. Neurotoxicology, 17, 883-896
Safran N, Shainberg A, Haring R, Gurwitz D & Shahar A (1993) Selective neurotoxicity induced by lasalocid in dissociated cerebral cultures. Toxicol In Vitro, 7, 345-352
Schwartz A (1976) Cellular and molecular mechanisms involved in cardiac cell function: Effects of an antibiotic ionophore. Acta Med Scand, 199 (Suppl 587), 71-82
Shlosberg A, Weisman Y, Klopfer U & Perl S (1985) Neurotoxic action of lasalocid at high doses (letter). Vet Record, 117, 394
Taylor MA (2004) Antiprotozoals. In: Bishop Y, ed, The Veterinary National Formulary (Sixth Edition), London, UK, Royal Pharmaceutical Society of Great Britain/British Veterinary Association, pp 171-179
Weiss G (1990) The integration of pharmacological and toxicological testing of tissue residues in the evaluation of their human food safety. Drug Metab Rev, 22, 829-848
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
97

Annex 9 Malachite Green 1 IntroductionMalachite green (ammonium, (4-(p-(dimethylamino)--phenylbenzylidene)-2-5-cyclohexadien-1-ylidene)-dimethyl-, chloride; CAS No. 569-64-2)is a N-methylated diaminotriphenylmethane dye used in the fish and dye industries. Its powerful antimicrobial activity has been attributed to inhibition of intracellular enzymes, intercalcation into DNA and/or interaction with cellular membranes. Although not approved for use in aquaculture in the USA, malachite green has been used as an antifungal treatment for fish since the 1930s. Random sampling of fish from markets in the UK indicated the continued use of malachite green in the aquaculture industry (Culp et al., 1999).
2 Uses and exposureMalachite green is available for the treatment of fungal infections and some ectoparasitic infections of fish in the UK. It is available as a range of products for ornamental fish on the UK market either on its own (Aquarium Care FungusTM; Koi Care MalachiteTM; Malachite; Malachite Green SolutionTM; Nishicare Anti-Fungus TreatmentTM); in combination with potassium iodide (CostapurTM); more usually with formaldehyde (FormalachiteTM; IchcideTM; Koi CareTM; ParapureTM; Pond Aid EradickTM; Fungus ControlTM; ProtobanTM; Tetrafin Goldfish DiseaseTM; Tetrafin GoldmedTM; TetraMedica ContraSpotTM; TetraPond MedifinTM; White Spot ControlTM; White Spot TerminatorTM) often referred to as Leteux-Meyer mixture; or with acriflavine and quinine sulphate (Pond Prode No3TM) for the treatment of external fungal (Saprolegnia) and protozoal (Icthyophthirius, Icthyobodo, Chilodonella), and with some products for fluke infections (Gyrodactylus, Dactylogyrus) (Bishop, 2004).
Malachite green had been used for decades in fish farming for the treatment of fish and eggs against Saprolegnia and juvenile fish against white spot (Icthyophthirius). Similarly, many preparations have been used, and continue to remain in use, for the treatment of ornamental fish, despite many products not having a marketing authorisation. As a consequence, many of the recommendations for its use have not been substantiated. For farmed fish intended for human consumption, there are a number of chemical treatments for ectoparasitic and fungal infections available. Many are considered as non-medicinal curative substances although consumer groups have expressed reservations and any potential risks for these substances remain unproven. Compounds used include formaldehyde, chloramine and sodium chloride. The use of malachite green in farmed fish is prohibited under EC Directive 2377/90 because of potential human health risks, although illegal and continued use in the aquaculture industry is apparently occurring as shown by random sampling at fish markets (Culp et al., 1999).
Treatment with malachite green can be given as a dip for individual fish at 50–60 ppm (50–60 mℓ/1000 ℓ) for 10–30 seconds; by bath at 1–2 ppm (1–2 mℓ/1000 ℓ) for 30–60 minutes or for larger numbers of fish in aquariums or ponds by prolonged immersion at 0.1 ppm (0.1 mℓ/1000 ℓ) for 30–96 hours. Eggs are treated for fungal infection by bath at 0.5 ppm for 1 hour. Care should be taken with regard to the source of malachite green, as some grades containing zinc may be lethal to fish. Low doses should be used at low pH and higher doses used at high pH. Residues of this chemical persist in tissues and it may only be used in ornamental fish or eggs not intended for human consumption (Bishop, 2004).
Leteux-Meyer mixture contains 3.3 g/ℓ in 35–40% formaldehyde solution. This combination is used in ornamental fish as an ectoparasiticide in particular for white spot (Icthyophthirius) and slime disease (Icthyobodo, Chilodonella) and is effective against secondary fungal overgrowth. It is given by bath at 25 ppm (25 ml/1000 ℓ) for 1 hour or by prolonged immersion at 15 ppm (15 mℓ/1000 ℓ) at
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
98

3–4 day intervals for 3 treatments for white spot. When treating ornamental fish, the volume of diluted chemical required may be small and difficult to titrate correctly leading to overdose and possible toxicity (Taylor, personal communication).
In ponds or aquaria containing ornamental fish, water change is required following treatment to reduce chemical concentrations to acceptable levels. In farmed fish, treatment is recommended in quarantine tanks or in commercial re-circulating systems, within isolated individual tanks with the water discharged to waste. Disposal of medicated wastewater from fish farming enterprises requires a Discharge consent from the Environment Agency or SEPA (Bishop, 2004).
With the exception of a US Fish and Wildlife Service investigational new animal drug exemption for treating specific threatened aquatic species, malachite green is not approved for use on any aquatic species by the US FDA or EPA (Culp & Beland, 1996).
As malachite green is a highly effective and cheap fungicide and ectoparasiticide when used in fish, there is likely to be a high probability of abuse. This could lead to potential human exposure by ingestion of animal products containing drug residues.
2.1 Exposure concentrations in meatHuman exposure to illegal use of malachite green due to consumption of treated fish has been reported with residues of up to 35 µg/kg malachite green measured in random samples of trout sold in UK markets. Residue levels of the metabolite, leucomalachite green (9–96 µg/kg) have been detected in trout marketed for sale, which are 30-fold higher than residues of the parent compound (0.4–3.4 µg/kg) (Culp et al., 1999). A 1 mg/ℓ bath for 1 hour can produce a blood serum level of up to 14 mg/kg at temperate water temperatures. Its elimination is extremely slow and in excess of 2000 degree days (200 days at 10 oC) are required for the residues of one treatment to be eliminated (Alderman & Clifton-Hadley, 1993).
Experimentally derived data are available on concentrations of malachite green in meat products. Salmon were treated with malachite green using a 10 mg/ℓ bath treatment. Fish were slaughtered either 4 hours post treatment or 24 hours post treatment and concentrations of malachite green in muscle and skin were obtained. In samples taken 4 hours post treatment the concentration was 0.35 mg/kg whereas in samples taken 24 hours post treatment concentrations were 0.12 mg/kg.
3 ToxicokineticsThe carbinol form of malachite green is much more lipophilic than the cation from which it is derived and it is most likely that in this form malachite green enters cells. In the body, malachite green appears to be reduced, to some extent, to leucomalachite green. Leucomalachite green has been detected in the liver, kidneys, heart, lungs and muscles of rats 2 hours after an intravenous (i.v.) injection of malachite green and in Ehrlich’s ascitic tumour cells 3 hours after intraperitoneal (i.p.) injection (Culp & Beland, 1996).
Rainbow trout were exposed to a bath treatment of 1.6 ppm malachite green for 40 minutes, with water temperatures of 8o or 16 oC and a pH of 7.6. In these conditions, the dye–carbinol equilibrium was such that 95% of the malachite green was in the carbinol form. The maximum concentrations of malachite green in serum, liver and kidney were detected immediately after exposure, with levels ranging from 7.8 to 34.0 ppm. A peak concentration of 10.8 ppm was reached in muscle at 90–120 minutes after exposure. The highest concentration of malachite green, 34 ppm, was found in kidneys at 16 oC. It was calculated that at a constant 16 oC temperature, malachite green residues would fall below 0.1 ppb after 615 days. The high level in kidney was confirmed in a study with fingerling trout exposed to 2 ppm 14C-malachite green for 1 hour. The maximum level in kidney, about 9 ppm, was reached by 1 hour after exposure and persisted for the remainder of the 21 day study. 14C-Levels in other tissues such as liver, muscle and skin decreased over the time period. Analysis of methylene
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
99

chloride extracts of kidney showed the presence of high levels of chromatic- and leuco-malachite green (Culp & Beland, 1996).
The preferential concentration of malachite green in the kidney was not found in rodents. Male and female Sprague–Dawley rats were administered 2 mg 14C-malachite green/kg bw by gavage, and kidney, liver, muscle and skin were analysed after 1 week. Approximately twice the amount of radioactivity was found in the liver as in each of the other tissues, but the radioactivity was <10 % of the maximum found in trout extracts. Approximately 96 % of the dose was excreted in the urine and faeces by 7 days after dosing (Culp & Beland, 1996).
Male F344 rats and female B6C3F1 mice were fed 0, 100 or 600 ppm malachite green or similar molar equivalents of leucomalachite green (0, 96 or 580 ppm) for 28 days. At the end of the feeding period, DNA was isolated from the livers and adduct levels measured using a 32P-postlabeling assay. The results of this assay indicated the formation of a DNA adduct or co-eluting adducts, that increased with increasing dose, in the rats and mice fed malachite green or leucomalachite green. In addition, a series of desmethyl derivatives were observed by HPLC/APCI/MS analysis of liver extracts from both species. These results indicate that malachite green may undergo a reduction to leucomalachite green or, a cytochrome P-450 catalysed N-demethylation to mono- and di-desmethyl malachite green. Leucomalachite green could also undergo a similar N-demethylation by cytochrome P-450 (Culp & Beland, 1996).
Malachite green, used to treat and prevent fungal and parasitic infections, is reduced to leucomalachite green and accumulates in the tissues of exposed fish. Malachite green residues have also been detected in eggs and fry of rainbow trout and Atlantic salmon (Srivastava et al., 2004). Human exposure to malachite green, due to consumption of treated fish, has been documented. Residues of up to 35 µg/kg malachite green have been measured in random samples of trout sold in UK markets. Leucomalachite green residues, 9–96 µg/kg, have been detected in trout, marketed for sale, at levels 30-fold higher than malachite green residues, 0.4–3.4 µg/kg (Culp et al., 1999).
4 Toxicity profile4.1 Acute toxicityMalachite green oxalate was dosed orally to groups of Wistar rats (5/sex/group) as a 12 mℓ aqueous solution/kg bw. The animals were observed 1, 2, 3 and 5 h after dosing and then each day for 14 days. Reduced motor activity, diarrhoea and piloerection were observed during the first day. The major findings were hyperemia and atonia of the intestinal walls, often in conjunction with dilatation of the gastro-intestinal tract. Survivors were free of toxic signs after 2 days. The LD 50 was calculated to be 275 (223–341) mg/kg bw. Mice appear to be more sensitive than rats, as a range-finding study showed that approximately half of the animals died at a dose of 50 mg/kg bw (Clemmensen et al., 1984).
The acute oral toxicity was also assessed in female Sprague–Dawley rats. They were administered malachite green at doses of 300, 450, 600 or 750 mg/kg bw, presumably by gavage. The 24 h-LD50
was determined to be 520 mg/kg bw. Effects observed included depression, prostration, emaciation and coma (Culp & Beland, 1996). An acute oral LD50 of 80 mg/kg bw and an i.p. LD50 of 4.2 mg/kg bw have been reported for malachite green in mice (HSDB, 2003).
No signs of systemic toxicity were observed after occlusive dermal application of malachite green oxalate at a dose of 2000 mg/kg bw. A 20 % suspension of malachite green did not produce visible erythema or oedema on either rat or guinea pig skin. Within the conditions of a guinea pig maximisation test, no effects could be seen on control or treated animals. Instillation of an 8% aqueous solution of malachite green produced marked oedema, substantial discharge and slight hyperemia of the conjunctiva, which disappeared after 24 hours, in the eyes of 2/3 rabbits. No effect on the cornea or iris was observed and all changes were fully resolved after 8 days (Clemmensen et
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
100

al., 1984). In one patient, an attempt to treat conjunctivitis with a 1 % solution of malachite green rsulted in destructive keratitis with hypopyon and terminated in bilateral blindness due to corneal opacification (HSDB, 2003).
Eleven patients with clinical signs of contact sensitivity to therapeutically used triphenylmethane dyes (eczema localised mainly on the legs), were patch tested with malachite green (2 % in water). The patches were on for 20–24 hours and were evaluated for 6–7 days. Positive reactions were observed in 6/11 of the investigated patients. This shows the possibility of cross sensitisation between the dyes (Bielicky & Novák, 1969).
4.2 Repeat dose toxicityGroups of Wistar rats, 8/sex/group, were fed 0, 10, 100 or 1000 ppm malachite green in the diet for 28 days. All animals were autopsied and selected tissues weighed and examined microscopically. No clinical effects were observed in the dosed animals except for apparent hyperactive behaviour of rats at 1000 ppm. The animals in this group also showed a significant reduction in weight gain and a reduced food intake. In the highest dose females, an increase in lymphocytes and a concomitant decrease in neutrophils were seen, together with a slight but significant decrease in packed cell volume. The males in the highest dose group showed a significant increase in plasma urea (Clemmensen et al., 1984).
Comprehensive 28-day toxicity studies have been undertaken in rats and mice for both malachite green and leucomalachite green. Groups of B6C3F1 mice and F344 rats (8/sex/group) were fed malachite green at concentrations of 0, 25, 100, 300, 600 and 1200 ppm in diet for 28 days. Separate groups of male rats and female mice, 8/group, were fed diets of leucomalachite green at levels of 0, 290, 580 and 1160 ppm. Additional groups of male and female rats were fed 0 or 1200 ppm malachite green for 4 or 21 days for triiodothyronine (T3), thyroxine (T4) and thyroid stimulating hormone (TSH) analyses. Male rats were fed untreated diet or diet containing 1160 ppm leucomalachite green for the same purpose (Culp et al., 1999).
For malachite green, the mean body weights of female rats at 1200 ppm were decreased during weeks 1–4 of administration. A significant increase in liver:bodyweight ratio was seen in female rats at 300, 600 and 1200 ppm and , in males, at 600 and 1200 ppm. There was an increasing trend in γ-glutamyl transferase (γ-GT) activity in rats of both sexes with the value in the 1200 ppm females being 4.2 times greater than the control group. The male rats had a slight but significant decrease in mean haemoglobin values at 300, 600 and 1200 ppm. The T3 levels were significantly higher in female rats fed 1200 ppm malachite green compared with the control group on day 21. The T4 levels were significantly lower on days 4 and 21 in this group. Minimal-mild hepatocyte vacuolisation was observed in 7/8 female rats fed 1200 ppm and in 1/8 males fed 600 ppm and 4/8 males fed 1200 ppm malachite green (Culp et al., 1999).
For leucomalachite green, male rats fed 1160 ppm had significantly lower body weights at weeks 2, 3 and 4 and significant decreases in bodyweight in the 580 ppm group at weeks 3 and 4. The liver: bodyweight ratio was significantly increased in all 3 dose groups and γ-GT activity was 2.2-fold higher than control in the top dose males. The erythrocyte count, haemoglobin and haematocrit levels showed slight but significant decreases from controls at 1160 ppm. There was a significant decrease in T4 and increase in TSH levels on days 4 and 21 at 1160 ppm compared with the controls. In male rats fed leucomalachite green, hepatocyte vacuolisation, primarily mid-zonal and centrilobular in location, was seen in 7/8 rats fed 1160 ppm, 5/8 fed 580 ppm and 2/8 fed 290 ppm. Apoptotic follicular epithelial cells in the thyroid gland were observed in 2/8 rats fed 1160 ppm and 2/8 fed 580 ppm (Culp et al., 1999).
In the 28-day mice studies, the females at 1200 ppm malachite green had significantly lower body weights at weeks 3 and 4. Slight haematological changes occurred at 600 and 1200 ppm in females and in males at 1200 ppm. There were no adverse histopathological changes due to treatment. In the
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
101

mice fed leucomalachite green, females at 1160 ppm had significantly lower body weights at week 4. All females in this dose group had scattered dead or degenerate cells in the transitional epithelium of the urinary bladder. Examination of thin sections revealed that many apparent apoptotic cells were contained within phagocytic vacuoles inside viable epithelial cells. The cytoplasm of the apparently apoptotic cells was moderately positive for the presence of DNA fragments and condensed nuclei stained intensely for DNA fragmentation (Culp et al., 1999).
Overall, the results of these 28-day studies in rats and mice showed that leucomalachite green has the potential to cause a greater number and more severe effects than malachite green (Culp et al., 1999).
Malachite green infused into the renal arteries of dogs, produced marked increases in the excretion of water, sodium, potassium, chloride, calcium and phosphate. The dye localised primarily in the renal cortex, indicating proximal or distal tubular uptake. It also appeared to cause a direct vasoconstriction of the renal arterioles. It has been reported that non-pregnant New Zealand White rabbits were able to tolerate 13 consecutive daily doses of 50 mg malachite green/kg bw (Culp & Beland, 1996).
4.3 Carcinogenicity and mutagenicityIn 2004, the UK Committee on Mutagenicity of Chemicals in Food, Consumer Products and the Environment (COM) and the Committee on Carcinogenicity of Chemicals in Food, Consumer Products and the Environment (COC) issued a joint statement on the mutagenicity and carcinogenicity of malachite green and leucomalachite green (COM & COC, 2004). Where appropriate, the conclusions of these committees have been incorporated in this section, and the overall conclusion that malachite green should be regarded as an in vivo mutagen has been used in the risk assessment.
The main study on which the COC based their conclusion for the carcinogenic potential of malachite green was conducted by the UK National Toxicology Programme (NTP). They considered that this study provided the only available bioassay on which any sort of assessment could be based. Female F344 and female B6C3F1 mice (female considered to be most sensitive gender), were fed a diet containing malachite green for 104 weeks. The rats were given doses of approximately 0, 1, 21 and 43 mg/kg bw/day. Survival was unaffected by treatment, and there was a slight reduction in body weight gain in the two highest dose levels. At autopsy it was noted that relative liver weights were increased at the highest dose. There was equivocal evidence of carcinogenicity in the rats, based on an increase in thyroid gland tumours (adenoma and carcinoma combined), hepatocellular adenomas and mammary gland carcinomas. The mice were given doses of approximately 0, 15, 33 and 67 mg/kg bw/day. There was a slight reduction in body weight gain at the highest dose, but survival was unaffected. There was no evidence of carcinogenic activity in the mice. Overall, the COC concluded that there is no convincing evidence for any carcinogenic effect with malachite green in these studies (COM & COC, 2004).
Similarly, NTP also studied the carcinogenic potential of leucomalachite green in male and female F344 rats and female mice following exposure via the diet for 104 weeks. The rats were fed diets containing approximately 0, 5, 15 or 30 mg/kg bw/day for males and 0, 6, 17 or 35 mg/kg bw/day for the females. Survival was unaffected by the treatment but there was a reduction in body weight gain at the highest dose for males and females. At autopsy liver weights were increased at the two highest doses. There was equivocal evidence of carcinogenic activity in male rats based on an increase in interstitialcell adenoma of the testes and the occurrence of thyroid gland follicular cell adenoma and carcinoma (combined). There was also equivocal evidence in the female rats, based on an increased incidence of hepatocellular adenoma or carcinoma (combined). The mice were fed diets containing approximately 0, 13, 31 or 63 mg/kg bw/day. Survival was unaffected by the treatment. There was evidence of carcinogenic activity based on an increase in hepatocellular adenoma or carcinoma combined. Based on these study results, the COC concluded that it would be prudent to consider leucomalachite green as a carcinogen.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
102

The longer term toxicity of malachite green has been reported briefly for 3 rodent studies. CD mice were given 0, 5 or 20 ppm malachite green in drinking water 5 days a week for 52 weeks. All of the mice given the 20 ppm diet were reported to have sluggish movements. A slight dose-related increase was seen in kidney weight in treated males, as well as an increase in the incidence of hydronephrosis. No significant differences were found in tumorigenicity between treated and control mice. Rats were fed diets containing 0, 0.03, 0.3 or 3.0 % malachite green and all animals in the 2 higher doses died within 1 week. The remaining rats were killed after 64 weeks of treatment. Histopathological examination did not reveal any toxicologically significant changes. In the third study, an increased number of tumours was reported in Wistar rats fed malachite green for a long but unspecified period at doses 20–33% of the lethal dose (about 30–35 mg/kg bw; Culp & Beland, 1996).
Male Wistar rats were administered 200 ppm diethylnitrosamine (DEN) in drinking water for 4 weeks. Then, after a 2-week interval untreated, 25 ppm malachite green was administered to the animals in drinking water for 10 weeks. A significant increase in the size and number of total γ-GT positive foci was observed in rats treated with DEN and a further increase in these hepatic pre-neoplastic lesions was observed after the treatment with malachite green. No morphological changes were observed in rats treated only with malachite green (Culp & Beland, 1996).
Groups of Wistar rats were treated as follows: an untreated control group; a group given 100 ppm malachite green in drinking water for 4 months; and a group administered 200 ppm DEN in water for 1 month followed by water for a 15 day recovery period and then, 100 ppm malachite green in water for 4 months. At termination, the livers from the animals in each group were removed, weighed and examined for markers of cell proliferation. The rats administered malachite green subsequent to DEN treatment showed an increase in liver weight/100 g bw, whereas malachite green alone showed no effect. Immunohistochemical staining showed an elevated level of proliferating cell nuclear antigen (PCNA) in animals administered malachite green subsequent to DEN treatment. Western and Northern blot hybridisation showed an increased expression of both cyclin D1 and its associated kinase cdk4 and cyclin B1 and its associated kinase cdc2 in livers of rats administered malachite green after administration of DEN as compared with untreated or DEN controls. The increased level of mRNA was due to the increased rate of transcription in these genes as studied by run-on transcription assay. These data provide evidence for a link between dysregulation of the two critical checkpoints of the cell cycle as one of the possible mechanisms during tumour promotion by malachite green (Gupta et al., 2003).
An in vitro investigation of tumour promotion was carried out using hepatocytes isolated from male Wistar rats. The effect of malachite green on DNA synthesis in primary cultures of the hepatocytes maintained under fully defined conditions was studied. The rate of DNA synthesis in both untreated and hepatocytes treated with epidermal growth factor, EGF, (10 ng/mℓ) were inhibited by malachite green at concentrations of 0.025–0.4 µg/mℓ. These inhibitory effects were concomitant with an extensive release of lactate dehydrogenase which began after 24 hours. Malachite green inhibited DNA synthesis when added after only 16 hours to hepatocytes either primed or not primed with EFG. This indicates that the target site may be other than the EFG-receptor or EFG-mediated early events involving signal transduction. This then indicates that cytotoxic and mitoinhibitory properties of malachite green possibly play an important role during tumour promotion (Rao, 1995).
The toxicological mechanisms of malachite green were investigated further by studies on free radical formation, its malignant transforming potential on Syrian hamster embryo (SHE) cells and the characterisation of these cells in relation to cell proliferation and antiapoptotic markers and for tumour induction. Electron spin resonance analysis showed formation of reactive free radicals during exposure of malachite green to SHE cells. The compound induced the formation of Type II and Type III morphologically transformed foci in a dose-related manner using the SHE cell transformation assay. Immortal cell lines were established and one of these showed enhanced DNA synthesis and increased presence of PCNA. When these immortal cells were injected subcutaneously (s.c.) into nude mice, they developed tumours which were transplantable and were, histopathologically, sarcomas (Mahudawala et al., 1999).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
103

Possible mechanisms of genotoxic action were also investigated for leucomalachite green. Big Blue female rats, 6/group, were fed leucomalachite green in diet at levels of up to 543 ppm for up to 32 weeks. At the end of the treatment period, rats were sacrificed and their livers removed for lacl mutational analysis. The spleen and bone marrow were also removed for lymphocyte Hprt and micronucleus assays, respectively. No significant increases in the frequency of micronuclei or Hprt mutants were observed for any of the doses or time points assayed. When corrected for clonality, the 16-week lacl mutation frequency (36 ± 10 × 10-6) in treated rats was not significantly different from the clonally corrected control frequency (17 ± 9 × 10-6; p = 0.06). The lacl mutational spectrum in treated rats, also, was not significantly different from that found for control rats (p = 0.09). Therefore the authors considered that this isolated positive was an artefact due to the disproportionate expansion of spontaneous lacl mutations. Taken together, these data indicate that the DNA adducts produced by leucomalachite green in rats (see Culp et al., 1999 in Section 3) do not result in detectable levels of genotoxicity (Manjanatha et al., 2004). COM (COM & COC, 2004) noted that this study did not optimise the chances of detecting a mutagenic response.
Studies to investigate cll mutations induced by leucomalachite green in the liver of Big Blue mice gave a positive result. Following administration of diet containing 408 ppm leucomalachite green, a statistically significant (p<0.05) increase in mutations was noted. A spectrum of mutations that was distinct from that of the controls was found. In similar studies using rats, such effects were not observed (COM & COC, 2004).
The results of several in vitro mutagenicity assays, mainly bacterial, for malachite green are shown in Table 4.1 (Clemmensen et al., 1984; Fessard et al., 1999).
Table 4.1 In vitro mutagenicity assays for malachite greenTest system Concentration ResultS. typhimurium, TA1535 (±S9) 0.05–160 µg/plate NegativeS. typhimurium, TA1537 (±S9) 0.05–160 µg/plate NegativeS. typhimurium, TA100 (±S9) 0.05–160 µg/plate NegativeS. typhimurium, TA 98 (-S9) 0.05–160 µg/plate NegativeS. typhimurium, TA 98 (+S9) 0.05–160 µg/plate PositiveS. typhimurium, TA 97A (±S9) 0.01–10 µg/plate NegativeS. typhimurium, TA 98 (±S9) 0.01–10 µg/plate NegativeS. typhimurium, TA 100 (±S9) 0.01–10 µg/plate NegativeS. typhimurium, TA 102 (±S9) 0.01–10 µg/plate NegativeCHO/HGPRT (CHO-K1) (±S9) 0.001–1 µg/ml Negative
It should be noted, for the above assays, that malachite green was very cytotoxic to the bacterial and mammalian cells, although the addition of S9 decreased this effect. For example, in the CHO/HGPRT assay, the mutagenic potential of the dye could be evaluated only at the lower concentrations of 0.001–0.05 µg/mℓ without S9 but up to 1 µg/mℓ with S9. The COM mentioned, but did not describe, a study in which there was some evidence of clastogenicity in Chinese Hamster Lung cells (COM & COC, 2004).
The major metabolite of malachite green, leucomalachite green, was also assayed in bacterial and mammalian cell in vitro mutagenicity assays, with results shown in Table 4.2 (Fessard et al., 1999). The potential DNA damaging effect of malachite green was evaluated using the single cell gel electrophoresis (Comet) assay on CHO cells in the presence or absence of S9. The compound induced DNA damage only at cytotoxic doses. Loss of cell viability was observed for doses of 3 µg/ml, with parallel increase in DNA alterations. After metabolic activation, however, DNA damage was observed at doses of 15–20 µg/mℓ with only low cytotoxicity. Leucomalachite green did not have any effect on cell viability or DNA damage at doses up to 500 µg/mℓ, in the absence or presence of metabolic
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
104

activation, in the Comet assay (Fessard et al., 1999). The COM decided that, owing to the limitations of the above Salmonella, CHO/HGPRT and Comet assays, and the fact that no data were available on clastogenicity, it was not possible to make an adequate assessment of the mutagenic potential of leucomalachite green (COM & COC, 2004).
Table 4.2 In vitro mutagenicity assays for leucomalachite greenTest system Concentration ResultS. typhimurium, TA97A (±S9) 10–2000 µg/plate NegativeS. typhimurium, TA98 (±S9) 10–2000 µg/plate NegativeS. typhimurium, TA100 (±S9) 10–2000 µg/plate NegativeS. typhimurium, TA102 (±S9) 10–2000 µg/plate NegativeCHO/HGPRT, (±S9) 5–100 µg/ml Negative
An in vivo micronucleus test for malachite green was carried out on groups of 5 mice. They were given the maximum tolerated dose, 37.5 mg/kg bw, by gavage and killed 24, 42 or 66 hours after dosing. Bone marrow smears were prepared from each animal, stained and 1000 polychromatic erythrocytes counted. No significant increase in the number of micronuclei was found in any of the malachite green treated groups at any of the sampling times. A positive control, cyclophosphamide, produced the expected increases in micronuclei (Clemmensen et al., 1984). In 2004, COM agreed that the dose level used in this study was adequate, but that it was difficult to assess the value of the negative result in the absence of appropriate information on bone marrow toxicity or data to show that malachite green and/or its metabolites reached the bone marrow (COM & COC, 2004).
NTP has conducted studies to investigate DNA adduct formation in the liver of rats and mice following 28 day repeated dose exposure to malachite green and leucomalachite green. Male F344 rats and female B6C3F1 mice were fed 0, 9, 100 or 600 ppm malachite green, or 0, 96 or 580 ppm leucomalachite green in their diets for 28 days. 32P post-labelling analysis of liver DNA indicated a single adduct, or co-eluting adducts, with both compounds. In both cases the level of adducts increased significantly with increasing dose. In rats, the level of adducts was similar for malachite green and leucomalachite green. In mice, malachite green gave a clear dose–response, which was slightly lower than that observed in rats, whereas leucomalachite green produced very low levels of adducts, the significance of which was doubtful. Therefore no conclusion regarding leucomalachite green could be drawn from this study. Later studies in female Big Blue rats and mice produced evidence of DNA adduct formation in the rat for malachite green only (COM & COC, 2004).
Neither compound induced micronuclei in peripheral lymphocytes or hprt mutations in splenic lymphocytes in the above 28 day study. However, the COM noted that the studies did not optimise the chances of obtaining a positive response and no definite conclusion could be drawn (COM & COC, 2004).
Overall, the COM concluded that “in view of the demonstration of DNA adduct formation in samples from both rats and mice, malachite green should be regarded as an in vivo mutagen”. For leucomalachite green the conclusion was “in view of the demonstration of the induction of mutations in liver DNA of female B6C3F1 mice, leucomalachite green should be regarded as an in vivo mutagen” (COM & COC, 2004). The COC also concluded that although there was no convincing evidence for a carcinogenic effect for malachite green, there was some evidence for leucomalachite green (hepatocellular adenoma or carcinoma in mice) and therefore it would be prudent to regard leucomalachite green as a genotoxic carcinogen.
4.4 Reproductive and developmental toxicitySignificant teratological effects were observed in New Zealand White rabbits administered 0, 5, 10 or 20 mg malachite green/kg bw by gavage on days 6–18 of gestation. At all 3 dose levels there were significant increases in pre-implantation losses, due to early resorption of the fetus and decreases in
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
105

the number of living fetuses. The bodyweights of the progeny were less than those of the controls with the differences being significant in the 5 and 20 mg/kg bw groups. Developmental anomalies were observed in all treated groups. Skeletal deviations were the most common abnormality and included incomplete ossification of vertebrae and phalanges and malformed skulls. Enlargement of the liver, heart and abdominal cavity was also observed. The abnormalities were not strictly dose-related, with progeny having 18.5 %, 38.0 %, 33.9 % and 47.0 % deviations in the 0, 5, 10 and 20 mg/kg bw treatment groups, respectively. Thalidomide, used as a positive control, caused similar types of changes in 94 % of the offspring. Maternal toxicity findings in the malachite green animals were not available (Culp & Beland, 1996).
In a preliminary investigation, groups of female hooded rats, 20/group, were either given an intramuscular injection with malachite green at a dose 1/5 of the lethal dose (LD) for 30 days and then, after coitus, ¼ LD for 5 days or, they were injected the compound i.p. at ¼ LD. In both cases the treatment partly or completely prevented the production of offspring. In the few cases when progeny were produced, alopecia on the back was the notable abnormality (Sokolowska-Pituchowa et al., 1965).
5 Guidelines and standardsNo NOEL for malachite green has been established which could serve as a basis for establishment of an ADI, and no MRLs have been set in fish or fish products. Nonetheless, in line with Council Directive 2002/657 EC, a non-toxicologically based Minimum Required Performance Limit (MRPL) of 2 g/kg analytical performance standard has been established for malachite green and leucomalachite green, for use in analysis of samples of meat from aquaculture products (EC, 2004).
6 ReferencesAlderman DJ & Clifton-Hadley RS (1993) Malachite green: A pharmacokinetic study in rainbow trout, Oncorhynchus mykiss (Walbaum). J Fish Dis, 16, 297-311
Bishop Y, ed (2004) The Veterinary National Formulary (Sixth Edition), London, UK, Royal Pharmaceutical Society of Great Britain/British Veterinary Association
Bielicky T & Novák M (1969) Contact-group sensitization to triphenylmethane dyes. Gentian violet, brilliant green, and malachite green. Arch Dermatol, 100, 540-543
Clemmensen S, Jensen JC, Jensen NJ, Meyer O, Olsen P & Würtzen G (1984) Toxicological studies on malachite green: A triphenylmethane dye. Arch Toxicol, 56, 43-45
COM & COC (2004) Joint COM and COC Statement on the Mutagenicity and Carcinogenicity of Malachite Green (MG) and Leucomalachite Green (LMG) (COM/04/S4 and COC/04/S7 - December 2004), London, UK, Department of Health, Committees on Mutagenicity and Carcinogenicity of Chemicals in Food, Consumer Products and the Environment, Available [May 2005] at http://www.advisorybodies.doh.gov.uk/com/mglmg.htm
Culp SJ & Beland FA (1996) Malachite green: A toxicological review. J Am Coll Toxicol, 15, 219-238
Culp SJ, Blankenship LR, Kusewitt DF, Doerge DR, Mulligan LT & Beland FA (1999) Toxicity and metabolism of malachite green and leucomalachite green during short-term feeding to Fischer 344 rats and B6C3F1 mice. Chem Biol Interact, 122, 153-170
EC (2004) Commission Decision of 22 December 2003 amending Decision 2002/657/EC as regards the setting of minimum required performance limits (MRLPs) for certain residues in food of animal origin (notified under document number C(2003)764) (2004/25/EC) Official Journal of the European Union, L6, 38-39. Available July 2006 at http://europa.eu.int/eur-lex/pri/en/oj/dat/2004/l_006/l_00620040110en00380039.pdf
Fessard V, Godard T, Huet S, Mourot A & Poul JM (1999) Mutagenicity of malachite green and leucomalachite green in in vitro tests. J Appl Toxicol, 19, 421-430
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
106

Gupta S, Sundarrajan M & Rao KVK (2003) Tumor promotion by metanil yellow and malachite green during rat hepatocarcinogenesis is associated with dysregulated expression of cell cycle regulatory proteins. Teratog Carcinog Mutagen, Suppl 1, 301-312
HSDB (2003) Malachite green. From: Hazardous Substances Databank, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [January 2004] at http://toxnet.nlm.nih.gov
Mahudawala DM, Redkar AA, Wagh A, Gladstone B & Rao KVK (1999) Malignant transformation of Syrian hamster embryo (SHE) cells in culture by malachite green: An agent of environmental importance. Indian J Exp Biol, 37, 904-918
Manjanatha MG, Shelton SD, Bishop M, Shaddock JG, Dobrovolsky VN, Heflich RH, Webb PJ, Blankenship LR, Beland FA, Greenlees KJ & Culp SJ (2004) Analysis of mutations and bone marrow micronuclei in Big Blue® rats fed leucomalachite green. Mutat Res, 547, 5-18
Rao KV (1995) Inhibition of DNA synthesis in primary rat hepatocyte cultures by malachite green: A new liver tumor promoter. Toxicol Letts, 81, 107-113
Sokolowska-Pituchowa J, Kowalczykowa J, Kus J, Piotrowski J & Sawicki B (1965) Teratogenic effect of malachite green in experimental animals. Preliminary report. Folia Biol, 13, 311-315
Srivastava S, Sinha R & Roy D (2004) Toxicological effects of malachite green. Aquat Toxicol, 66, 319-329
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
107

Annex 10 Nalidixic Acid 1 IntroductionNalidixic acid (1-ethyl-1,4-dihydro-7-methyl-4-oxo-1,8-naphthyridine-3-carboxylic acid) is a quinolone antibacterial that can attain concentrations in urine sufficient to treat urinary tract infections, in humans, caused by most Gram-negative aerobic organisms. It acts by rapidly inhibiting DNA synthesis by promoting cleavage of bacterial DNA in the DNA–enzyme complexes of DNA gyrase and type IV topoisomerase, resulting in rapid bacterial death. However, modest and variable serum and tissue concentrations, coupled with minimum inhibitory concentrations, have limited the usefulness of nalidixic acid in treating systemic infections. Nalidixic acid has been replaced, in recent years, by the more effective 2nd and 3rd generation quinolones (Siporin, 1989; Oliphant & Green, 2002).
2 Uses and ExposureThere are no products containing nalidixic acid licensed for use in either domestic livestock or companion animals in the UK. Given the wide availability of quinolone derivatives (fluoroquinolones) it is extremely unlikely that nalidixic acid will be used in domestic livestock.
Several preparations containing nalidixic acid are licensed for human use in the UK (NegramTM, UribenTM). The principal route of excretion is via urine and because of this nalidixic acid is prescribed for the treatment of urinary tract infections in humans. Drug concentrations in other tissues are insufficient for therapeutic effects. Under the Medicines (restrictions on the Administration of Veterinary Medicinal Products) Regulations 1994, where no authorised veterinary medicine exists for a condition in a particular species, a veterinarian may prescribe for an individual animal, or a small number of animals kept on the same premises, an authorised human medicine under the ‘cascade’ system. Urinary tract infections are generally diagnosed in cattle and companion animals only and require antibiotic treatment. Only individual animals are usually involved and there exist several licensed antibiotics available for the treatment of drug-sensitive urinary tract infections in these species. There would be no indication for use of nalidixic acid, given the availability of alternative medicines, unless under exceptional circumstances where, for example, antibiotic sensitivity tests indicated antibiotic resistance to other compounds and sensitivity to nalidixic acid.
There is no information on pharmacokinetics or excretion of nalidixic acid in food animals, and given that there are no licensed veterinary medicines, no MRLs have been established. There is therefore little or no perceived risk of human exposure to nalidixic acid from residues in meat, or from other potential uses in animals.
3 ToxicokineticsIn rats and mice, oral doses of nalidixic acid are rapidly absorbed with peak blood concentrations after about 1 hour. Elimination is via the kidneys, peaking at about 6 hours after administration. Eighty per cent of the dose is eliminated in the first 8 hours. In dogs, highly effective therapeutic concentrations appear in urine within 2–3 hours after oral administration. Approximately 96% of nalidixic acid administered orally to humans is absorbed. Plasma concentrations of 20–50 µg/mℓ can be achieved, with 93–97% of the acid bound to plasma proteins. The plasma half-life is 8 hours (HSDB, 2002).
Nalidixic acid is partially metabolised in the liver to hydroxynalidixic acid and to the glucuronic acid conjugates of nalidixic acid and hydroxynalidixic acid. The drug is also partially metabolised to the
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
108

dicarboxylic acid derivative and there is some evidence indicating that this metabolite is formed in the kidney. Renal excretion is rapid and almost complete within 24 hours of a dose of nalidixic acid; 2–3% is excreted unchanged, 13% as the active metabolite and >80% as inactive metabolites. The active entity does not accumulate in patients with impaired renal function, but inactive metabolites accumulate and may cause a toxic reaction (HSDB, 2002).
Orally administered nalidixic acid and its active metabolite are distributed to most tissues, particularly the kidneys. Traces of the drug cross the placenta. It is excreted in breast milk. The drug does not penetrate into prostatic fluid (HSDB, 2002).
4 Toxicity profile4.1 Acute toxicityAcute toxicity of nalidixic acid in humans may be manifest by toxic psychoses, convulsions, increased intracranial pressure or metabolic acidosis. Vomiting, nausea and lethargy may also occur. Because of the rapid excretion of nalidixic acid, such reactions are usually short-lived, persisting only 2–3 hours. A 19-year-old took 28 g nalidixic acid and suffered coma and other serious symptoms but survived. Seizures have been reported in children receiving 50 mg/kg/day or greater of nalidixic acid. Allergic reactions to nalidixic acid commonly include rash and urticaria. Eosinophilia, pruritus and photosensitivity occur occasionally (HSDB, 2002).
Oral, subcutaneous and intravenous LD50 values for nalidixic acid in mice have been reported as 3.3 g/kg bw, 0.5 g/kg bw and 0.176 g/kg bw, respectively. A rat oral LD50 of 1160 mg/kg bw has also been reported (HSDB, 2002).
4.2 Repeat dose toxicityThe most frequent adverse effects of quinolones in human clinical use are gastrointestinal reactions such as nausea, vomiting, diarrhoea, anorexia, dyspepsia and heartburn.
Visual disturbances, including overbrightness of lights, blurred vision, difficulty in focusing, decreased visual acuity, double vision and alteration of colour perception have been reported in patients receiving nalidixic acid. Nystagmus and eye pain or burning have also been reported (HSDB, 2002).
Owing to induction of toxic lesions in the articular cartilage of juvenile but not adult animals (see below) the use of quinolones has been restricted in children and growing adolescents. Adverse drug reactions in the skeletal system, including arthralgia, joint swelling, arthropathy and arthritis, have been reported at low incidences in patients after treatment with nalidixic acid. One study showed the absence of joint abnormalities in 279 children receiving nalidixic acid.
Vomiting was also observed in dogs after oral administration of nalidixic acid at a dose of 200 mg/kg bw. Oral and intraperitoneal nalidixic acid induced vomiting in cats. The effects on gastrointestinal transit and gastric emptying rate have also been studied. Mouse intestinal transit was reported to be reduced by nalidixic acid. It was also reported that the drug decreased gastric and intestinal motility in rabbits (Takayama et al., 1995).
Nalidixic acid causes lameness in immature dogs due to permanent damage of the cartilage of weight-bearing joints. Prolonged use of the drug in dogs and cats has caused retinal degeneration leading to blindness in some cases (HSDB, 2002).
Nalidixic acid has shown a slight excitatory EEG pattern in rats and seizure discharges in cats. It has been found to decrease spontaneous motor activity in mice at high doses. The drug has also been reported to increase the activity at lower doses (Takayama et al., 1995).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
109

When administered intravenously, nalidixic acid produces hypotension and bradycardia in animals, especially in anaesthetised dogs and cats. Intravenous doses of 5–10 mg/kg bw i.v. in dogs and 10–30 mg/kg bw in cats caused decreases in blood pressure. Since blood pressure is a function of cardiac output and total peripheral vascular resistance, the hypotension induced by nalidixic acid should result from cardiac depression and/or peripheral vasodilatation. The drug was reported to decrease cardiac contractility in isolated guinea pig atria. Using Langendorf preparations, it was found that nalidixic acid at extremely high concentrations had negative inotropic activity in isolated guinea pig heart but positive inotropic activity on isolated rat heart. The effect on vascular tone has also been studied. Nalidixic acid slightly increased flow in isolated rabbit ear vessels but potentiated the contractile response of isolated rabbit aorta to epinephrine (Takayama et al., 1995).
Nalidixic acid causes chondrotoxicity in laboratory animals, inducing toxic lesions in the articular cartilage of juvenile, but not adult animals.
The induction of another lesion, osteochondrosis, occurs after the oral administration of nalidixic acid to rats, in the distal femur, a predilection site of blistering. Blistering was induced on the ventral aspect of the femoral condyle after single or several repeated administrations, while osteochondrotic lesions were noted on the caudal aspect after subchronic treatment. It is uncertain whether adverse drug reactions of quinolones in humans are similar to blister formation observed in animals (Takayama et al., 1995).
Nalidixic acid is a well known photosensitiser that induces skin lesions. The phototoxicity of quinolones was compared in albino BALB/c mice using a method in which doses of the compounds and UVA irradiation, which induced no change in the auricle when given alone, induced erythema and thickening of the auricle upon single oral administration of the quinolone followed by irradiation for 4 hours. By this method, the order of toxic potential was lomefloxacin > enoxacin, nalidixic acid > ofloxacin, ciprofloxacin. Most quinolones produce no direct toxicity to the eyes of laboratory animals; however, nalidixic acid causes retinal degeneration in cats. The first step in a phototoxic reaction is reported to be the absorption of light by a phototoxic compound in a tissue and then the compound or a breakdown product becomes a chromophore. However, nalidixic acid induces photohaemolysis in vitro without undergoing toxic photoproduct conversion (Takayama et al., 1995).
4.3 Carcinogenicity and mutagenicityTwo-year studies have been conducted by feeding diets containing 0, 2000 or 4000 ppm nalidixic acid to groups of 50 male, 50 female F344/N rats and to 50 male, 50 female B6C3F1 mice. There was evidence of carcinogenic activity of nalidixic acid in the F344/N rats as indicated by increased incidences of preputial gland neoplasms in males and clitoral gland neoplasms in females. There was equivocal evidence of carcinogenic activity for male mice as indicated by marginally increased incidences of subcutaneous tissue neoplasms. There was no evidence of carcinogenic action in the female mice (HSDB, 2002). The preputial gland of male rats is anatomically and histologically similar to the clitoral gland of female rats, appearing as holocrine branched tubulo-alveolar glands derived from sebaceous glands. The structure and pheromonal activity of the glands are regulated by both pituitary and sex hormones, including testosterone, estrogen and progesterone (Takayama et al., 1995). The tumour response induced by nalidixic acid was considered unusual and suggestive of a non-genotoxic hormonal mechanism of carcinogenicity. Coincident with the above increases in tumour incidence in rats, there were also marked decreases in tumour incidence at other sites such as the pituitary gland in males and the haemopoietic system and mammary gland in females (Tennant & Ashby, 1991).
A number of in vitro mutagenicity studies have been carried out on nalidixic acid. The bacterial tests were mainly negative, other end-points showed both negative and positive results. In vitro assays with adequate description of test conditions are shown in Table 4.1 (CCRIS, 1998).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
110

Table 4.1 In vitro mutagenicity assays for nalidixic acid Test system Concentration Result
S. typhimurium TA100 (-S9) 0.03–3.3 µg/plate NegativeS. typhimurium TA1535 (-S9) 0.03–3.3 µg/plate NegativeS. typhimurium TA97 (-S9) 0.03–3.3 µg/plate NegativeS. typhimurium TA98 (-S9) 0.03–3.3 µg/plate NegativeS. typhimurium TA100 (+S9) 0.1–10 µg/plate NegativeS. typhimurium TA100 (+S9) 0.1–33 µg/plate NegativeS. typhimurium TA1535 (+S9) 0.1–10 µg/plate NegativeS. typhimurium TA97 (+S9) 0.1–10 µg/plate NegativeS. typhimurium TA98 (+S9) 0.1–10 µg/plate NegativeS. typhimurium TA100 (+S9) 0.1–10 µg/plate NegativeS. typhimurium TA102 (-S9) 3 µg/plate or 8 µg/mℓ PositiveS. typhimurium TA102 (-S9) 0.625–10 µg/plate PositiveS. typhimurium TA2638 (-S9) 0.625–10 µg/plate NegativeMouse lymphocytes L5178Y,TK+/TK-(+S9) 30–100 µg/mℓ NegativeMouse lymphocytes L5178Y,HPRT (-S9) 200 µg/mℓ NegativeMouse lymphocytes L5178Y,TK+/TK-(-S9) 200 µg/mℓ NegativeChinese hamster V79 (-S9) 160 µg/mℓ NegativeE. coli WP2 (-S9) 0.625-10 µg/plate PositiveE. coli WP2 UVRA (-S9) 0.625-10 µg/plate Positive
From CCRIS (1998)
Results for other in vitro assays for nalidixic acid have been reported but without details of the study conditions, as listed in Table 4.2 (Fort, 1992; Takayama et al., 1995).
For reported in vivo studies, nalidixic acid was mutagenic in the sex-linked recessive lethal test in Drosophila, but was negative in the mouse dominant lethal test. Sister chromatid exchange frequency was increased in peripheral lymphocytes from children treated therapeutically with nalidixic acid (Fort, 1992).
In summary, on the basis of the available data, the most likely mechanism for the positive genotoxicity results with nalidixic acid is via interference with topoisomerase action rather than a direct effect on DNA (Fort, 1992).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
111

Table 4.2 Results for other in vitro mutagenicity assays for nalidixic acidEnd-point ResultDNA damageBacterial PositiveMammalian UDS PositiveMammalian UDS NegativeBreakage PositiveRepair (B. subtilis, rec) PositiveRepair (B. subtilis) NegativeRepair (CHO) NegativeSCE (Human lymphocytes) PositiveChromosomal aberrationCultured human lymphocyte cells PositiveGene mutationMultigene sporulation PositivePhage induction PositiveChinese hamster V79 NegativeMouse lymphoma L5178Y Positive
4.4 Reproductive and developmental toxicityIn rats receiving 1600 ppm nalidixic acid for 13 weeks, there was a degeneration of the germinal epithelium of the seminiferous tubules of the testes, indicating that the compound may affect hormone synthesis in the testis (Takayama et al., 1995).
5 Guidelines and standardsNo information available.
6 ReferencesCCRIS (1998) Nalidixic acid. From: Chemical Carcinogenesis Research Information System, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [March 2004] at http://toxnet.nlm.nih.gov
Fort FL (1992) Mutagenicity of quinolone antibacterials. Drug Saf, 7, 214-222
HSDB (2002) Nalidixic acid. From: Hazardous Substances Databank, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [March 2004] at http://toxnet.nlm.nih.gov
Oliphant CM & Green GM (2002) Quinolones: A comprehensive review. Am Fam Physician, 65, 455-464
Siporin C (1989) The evolution of fluorinated quinolones: Pharmacology, microbiological activity, clinical uses and toxicities. Ann Rev Microbiol, 43, 601-627
Takayama S, Hirohashi M, Kato M & Shimada H (1995) Toxicity of quinolone antimicrobial agents. J Toxicol Environ Health, 45, 1-45
Tennant RW & Ashby J (1991) Classification according to chemical structure, mutagenicity to Salmonella and level of carcinogenicity of a further 39 chemicals tested for carcinogenicity by the U.S. National Toxicology Progran. Mutat Res, 257, 209-227
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
112

Annex 11 Nandrolone 1 IntroductionNandrolone (19-nortestosterone) (17β-hydroxy-estr-4-en-3-one; CAS No. 434-22-0) is a steroid with androgenic and anabolic properties. It has been used therapeutically to treat various ailments, as for other anabolic steroids. Nandrolone has been used in the treatment of chronic wasting diseases under conditions in which negative nitrogen balance exists. Although nandrolone phenpropionate is less androgenic than testosterone, in doses that exert anabolic actions, virilisation may occur after high doses or during chronic administration. The androgenic virilising action is required in treatment with this agent for inoperable breast cancer in women. Nandrolone deconoate (50 mg by injection every 3 weeks) increases peripheral and axial bone mass without side effects in osteoporotic women. Because of their anabolic and androgenic effects on performance and physique, nandrolone and other androgens have been misused by athletes, bodybuilders and weight lifters. Although not approved in the USA, nandrolone has been used as a veterinary agent for long acting repository effects, particularly for racing greyhounds (HSDB, 2002).
2 Uses and exposureNandrolone is an anabolic steroid, which is prohibited for use in animals intended for human consumption and in animals used in competitions (e.g. horses, racing greyhounds). Anabolic steroids stimulate appetite, increase muscle mass and increase production of erythrocytes and may be used as an adjunct to the treatment of chronic renal failure, in debilitating diseases, convalescence and to promote tissue repair. They are also indicated in the management of hypoplastic anaemia and anaemia due to uraemia and neoplasia (Bishop, 2004).
Nandrolone is now only licensed in the UK for use in dogs and cats and is available in several formulations as oily, depot injections (LaurobolinTM — nandrolone laurate 25 or 50 mg/mℓ; NandrolinTM — nandrolone phenylpropionate 25 or 50 mg/mℓ; RetarbolinTM — nandrolone cyclohexylproprionate 10 mg/mℓ; Bishop, 2004).
Nandrolone, as the above formulations, was available for use in cattle, sheep, goats, pigs and horses until 1997, for its anabolic and supportive therapy of a variety of medical and surgical situations, generally on an individual basis. Nandrolone laurate, available in preparations of 25 or 50 mg/mℓ, was administered at doses of up to 200 mg (1 mg/kg) to adult cattle and horses; up to 100 mg (1 mg/kg) to calves, sheep, goats and pigs; and up to 40 mg (1 mg/kg) to lambs and piglets, all by single injection with the option of repeating at 3 week intervals. Nandrolone phenylpropionate, available in preparations of 25 or 50 mg/ml, was administered at doses of up to 400 mg (1 mg/kg) to adult cattle and horses; up to 100 mg (1 mg/kg) to calves, sheep, goats and pigs; and up to 40 mg (1 mg/kg) to lambs and piglets, all by single injection with the option of repeating at 7–10 week intervals. Nandrolone cyclohexylproprionate, available in preparations of 10 or 50 mg/ml, was administered at doses of up to 50–100 mg (1 mg/kg) to adult cattle and horses; 50 mg (1 mg/kg) to calves, sheep, goats and pigs, all by single injection. Withdrawal periods were 14–28 days depending on the product and formulation (Bishop, 1994).
A potential source of human exposure to nandrolone is via ingestion of animal products containing drug residues following illegal use in food animals. Treatment of food animals would only occur through illegal use and would thus be expected to occur in individual or small numbers of animals only.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
113

2.1 Exposure concentrations in meatUsing information on usage of nandrolone and on absorption and excretion, likely ‘worst case’ concentrations in meat products were estimated using Equation 1:
Equation 1M = D × T × Fabs × (1–Fexc)
where M is the predicted concentration in meat (mg/kg); D is the dose of substance (mg/kg bw/d); T is the duration of the treatment (d); Fabs is the fraction of compound absorbed; and Fexc is the fraction of compound excreted.
It was assumed that nandrolone is used illegally as a growth promoter in cattle at a dose of 1 mg/kg bw and that it is applied by injection (i.e. Fabs = 1.0) on one occasion and that the fraction of drug excreted was 0.9. Using these input values, a ‘worst case’ concentration of 0.1 mg/kg was obtained. As the dose rate for sheep and pigs is also 1 mg/kg, illegal use of nandrolone in these animals will also result in exposure ‘worst case’ concentrations of 0.1 mg/kg.
3 ToxicokineticsIn metabolic studies with 14C-nandrolone in mice and calves, the administered radioactivity was quickly eliminated, mainly in urine. Ten weeks after administration, the residual levels in tissues were low. Isomers of 3-hydroxyestran-17-one estrane-3, 17-diol were identified by GLC-mass spectrometry in the urine of a crossbred horse given nandrolone by intramuscular injection (i.m.; HSDB, 2002).
Isolation and characterisation of metabolites from beagle dog and rat urine following administration of STS 557, a compound structurally related to nandrolone, revealed the following biotransformation pathways: hydroxylation in different positions of the steroid molecule; aromatisation of ring A; hydrogenation of a double bond; simultaneous hydroxylation and hydrogenation; and alteration of the 17α-side chain with loss of nitrogen (Schubert et al., 1983).
Nandrolone decanoate was injected into healthy volunteers. One group of females received one injection of 100 mg and 3 groups of males received one injection of 200 mg, 2 repeat injections of 100 mg or 4 repeat injections of 50 mg, respectively. The serum levels of nandrolone were used to estimate pharmacokinetic parameters. There was a mean half-life of 6 days for the release of the ester from the muscular injection depot into the general circulation. For the combined processes of hydrolysis of nandrolone deconoate and the distribution and elimination of nandrolone, the mean half-life was 4.3 h. The mean nandrolone serum clearance was 1.55 ℓ/h/kg . The half-life of hydrolysis of nandrolone deconoate in serum was of the order of 1 hour or less. The data are consistent with linear kinetics (Wijnand et al., 1985).
Equimolar concentrations of the 7α-methyl-derivative of nandrolone (7α-MN) and its acetate were administered to cynomolgus monkeys as a subcutaneous (s.c.) infusion. Serum levels of 7α-MN were measured by radioimmunoassay in blood samples collected daily for 4 days during steady state. The serum 7α-MN levels were not significantly different in the 2 groups (11.3 ± 1.6 vs 13.1 ± 1.2 nmol/ℓ). This indicated that 7α-MN acetate was rapidly converted to 7α-MN in circulation. The hydrolysis of 7α-MN acetate to 7α-MN was confirmed by the in vitro incubation of 7α-MN acetate with blood or plasma. A single intravenous (i.v.) bolus of 500 µg 7α-MN in each of 9 healthy men (22–39 years old) led to peak serum levels 3 min after dosing, followed by an exponential decline, reaching undetectable levels by 180 min (LOD = 28 pg/mℓ). The average terminal half-life and the metabolic clearance rate were calculated to be 40 min and 2360 ℓ/day respectively. It was also shown that 7α-MN did not bind to sex hormone binding globulin (Kumar et al., 1997).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
114

The metabolism and receptor binding of nandrolone and testosterone were studied under in vitro and in vivo conditions. The results from both the incubation studies with 3H-nandrolone and 3H-testosterone in rat tissue homogenates and infusion studies with 3H-nandrolone and 3H-testosterone in conscious rats showed the importance of the enzymes 5α-reductase and 3α/β-hydroxysteroidoxido-reductases in the prostate and of 17β-hydroxysteroid dehydrogenase in the kidney for the effects on these tissues. The in vitro and in vivo studies showed that nandrolone and testosterone were relatively stable in the spleen, thymus and muscular tissue. In vitro binding studies with the androgen receptor in intact human cells show that 5α-reduction increases the affinity of testosterone and decreases the affinity of nandrolone. The results help to explain the relatively strong effect of nandrolone compared with that of testosterone on tissues devoid of 5α-reductase activity, for example muscular tissue, and that there may be a strong effect of nandrolone on tissues that in addition, have a high 17β-hydroxysteroid dehydrogenase activity, for example kidney (Bergink et al., 1985).
Nandrolone and its 17α-cyanomethyl-derivative were incubated with liver microsomes from female rats for 60 minutes in the presence of NADPH. Steroid extracts from the incubation mixtures were separated by TLC. Both compounds were almost completely transformed, by >95% (Schubert et al., 1983).
4 Toxicity profile4.1 Acute toxicityAcute dosage can produce nausea and gastrointestinal upset in humans. Patients are expected to recover after acute over-dosage, but there are few data (PIMS, 1998).
Male and female Sprague–Dawley rats weighing 120–200 g were treated i.m. with various doses of nandrolone homofarnesate. The compound was administered in olive oil in a constant volume of 5 ml/kg. After dosing, the animals were observed for 8 days. The results showed that the compound was well tolerated up to 500 mg/kg bw (Coppi et al., 1973).
Single intracutaneous injections of micronised crystalline suspensions of nandrolone phenyl-propionate induced local thickening of the epidermis of female hairless hamsters accompanied by a marked increase in sebaceous gland volume over 7 days (HSDB, 2002).
Nandrolone decanoate was administered by intraperitoneal (i.p.) injection to male ICR mice in the dose range of 50 to 500 mg/kg bw and behavioural effects were measured between 5 minutes and 3 hours. The compound did not alter spontaneous locomotor or forced (rotarod) activity. In terms of an indirect effect, a 10 minute nandrolone pre-treatment reduced, by 30 %, the dose of s.c. pentobarbital necessary to induce loss of righting-reflex, without altering the duration of the loss. It appears that the relatively non-toxic anabolic steroid may augment the behavioural toxicity produced by other types of drugs (Compton, 1993).
4.2 Repeat dose toxicityMale and female Sprague–Dawley rats, 140–160 g body weight were dosed i.m. once weekly for 14 weeks with nandrolone homofarsenate. Groups of rats, 15 males 15 females/group, were dosed the test compound at 0, 12.5, 25 or 50 mg/kg. Haematology assays were carried out before and at the end of treatment and renal function tests at the end of treatment. At termination, gross post mortem examination was undertaken on all animals, organ weights measured and histopathological examination performed on a range of tissues. There was a significant increase in kidney weights in rats of both sexes at dose levels of 25 and 50 mg/kg. There were decreases in adrenal, brain and ovary weights in females at all dose levels. There was also a non-dose related decrease in testis weight in the males. Treated male rats grew normally but the females showed increased growth compared with controls from 4 weeks of treatment onwards. There was no effect on urinary function. The total serum
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
115

protein values of the 25 and 50 mg/kg females were reduced due to treatment. The histopathology of the liver, kidney, heart, spleen thyroid and lungs was not altered by treatment with the test substance. In the thymus, adrenals, testes and ovaries there was an indication of endocrine effects (Coppi et al., 1973).
A two part study was performed to determine the effects of high doses of nandrolone deconoate. Groups of 15 male and 15 female Wistar rats were dosed i.m. weekly with either physiological saline or approximately 100 mg/kg nandrolone deconoate. After 6 doses, the rats were sacrificed and a range of organs taken for microscopic examination. Treated female body weights were comparable to controls but treated males were significantly lighter than controls. Treated male livers had less lipid than control males. The uteri of treated females displayed abnormal vacuolisation, stromal oedema and peliosis. In the second study, groups of 6 male rats were treated with saline or nandrolone deconoate as in the first study. The nandrolone rats had lower weights and ate less than controls. The kidneys were heavier and the testes and liver were lighter than the controls. Roentgenograms taken at 0, 3 and 6 weeks of the study, revealed no significant differences in tibial length between groups at any time point (Yu-Yahiro et al., 1989).
The effect of nandrolone was evaluated in male castrate Greyface lambs. Two groups of 5 lambs were used, one as control and the other administered nandrolone. The compound was given as s.c. implants, 50 mg each time, 100 days and 37 days before slaughter. Nandrolone produced a small increase in body weight (23.5 kg versus 21.7 kg in controls) and decreased thymus weight (1.13 g/kg versus 1.89 g/kg bw in controls). The binding capacity of skeletal muscle for the synthetic glucocorticoid dexamethasone was increased by nandrolone treatment (Galbraith et al., 1986).
Nandrolone phenpropionate was administered to groups of Sprague-Dawley rats to study the effects on craniofacial growth. A control group consisted of 10 males and 10 females, a low dose nandrolone group had 14 males and 16 females and received s.c. doses of 1 mg/kg bw/week, a high dose nandrolone group had 16 males and 14 females, dosed 10 mg/kg bw/week; dosing continued for 9 weeks. At termination, direct millimetric measurements of the skeletodental variables were obtained using electronic sliding dial callipers. Fourteen linear dimensions were measured on each skull. Nandrolone treatment significantly increased all measures of the craniofacial complex, of the order of 3–5 %, except for some precocious calvarial dimensions. Significant alterations also occurred in facial morphology. The low dose group exhibited proportionate increases in most craniofacial dimensions but the high dose produced overt shape changes, notably a maxillomandibular, anteroposterior jaw discrepancy due to maxillary excess. Females were more sensitive to the nandrolone phenpropionate effects than males (Barrett & Harris, 1993).
Five week old female Sprague–Dawley rats were divided into 2 groups with 60 animals in each group. The experimental group rats were injected s.c. with 1 mg nandrolone phenylpropionate in the interscapsular region on alternate days. Rats were sacrificed at 60 and 120 days of age. There was about a 20 % increase in body weight in the treated animals. Cephalometric analysis of soft X-ray cephalograms showed that administration of the steroid caused an increase in total skull length, elongation of the maxillary and mandibular incisors, an increase in the depth of the antegonial notch and downward-forward growth of the viscerocranium against the neurocranium. These results indicate that nandrolone phenylpropionate may accelerate craniofacial growth and/or induce high functional activity of the masticatory muscles in female rats (Noda et al., 1994).
Spontaneously hypertensive male rats, 7 weeks old, were assigned to a control group and a nandrolone deconoate group, with 6 rats/group. In the treated group, rats received the test substance s.c. at 20 mg/kg bw/day for 6 weeks. Metabolic, blood pressure and heart rate measurements were performed on all rats every 2 weeks. After 6 weeks of treatment, blood pressure was higher in treated animals than in controls. At termination, certain tissues were removed and weighed. The weights of kidney, levator ani muscle and seminal vesicle (with prostate glands) were higher in the treated rats than in controls. Hearts were fixed in 10 % buffered formaldehyde and myocardial dimension measurements made. The nandrolone animals showed increased vertical ventricular diameters and
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
116

displayed higher vertical ventricular circumferences when compared with controls. Myocardial inflammatory and fibrotic changes were significantly more evident in the nandrolone treated rats (Tseng et al., 1994).
Two studies were undertaken to evaluate the effect of nandrolone deconoate on rabbit arteries and aorta. In both studies, male New Zealand rabbits were treated with nandrolone deconoate at 10 mg/kg bw/week i.m. for 4, 8 and 12 weeks. In each study, control and treated rabbits were sacrificed and the thoracic aorta and mesenteric and femoral arteries isolated and divided into segments. The nandrolone treatment in the first study reduced the contraction elicited by noradrenaline, 5-hydroxytryptamine (5-HT) and angiotensin in the aorta and those caused by 5-HT in mesenteric arteries. In the aorta, the contractions elicited by phorbol 12,13-dibutyrate, an activator of protein kinase C, were reduced by nandrolone treatment. These results indicated that the thoracic aorta is the most affected by nandrolone treatment and that the reduction of contractile responses appears to be due to changes in protein kinase C activity and/or in a mechanism situated beyond protein C activation (Ferrer et al., 1994b). In the second study, the blood pressure of the animals was unaltered by nandrolone treatment and averaged 83.0 ± 4.0 in controls and 84.3 ± 3.2 mmHg in rabbits treated for 12 weeks. The treatment abolished endothelium-dependent relaxation caused by acetylcholine and the Ca2+-ionophore A23187 in the thoracic aorta and reduced endothelium-independent relaxation induced by exogenous nitric oxide (NO) or sodium nitroprusside. In contrast, relaxation induced by acetylcholine, NO or sodium nitroprusside in mesenteric and femoral arteries was unaltered by nandrolone treatment. Bioassay experiments using donor segments and endothelium-denuded bioassay rings from thoracic aorta show that acetylcholine applied either through control or treated (12 weeks) donor segments produced similar relaxation in bioassay rings from control rabbits, but this relaxation was markedly reduced in rings from treated rabbits. The increases of cyclic GMP levels induced by acetylcholine or sodium nitroprusside in segments from thoracic aorta were abolished by nandrolone treatment. These results indicated that the treatment with nandrolone reduces NO-mediated relaxation only in the thoracic aorta by inhibition of guanylate cyclase; endothelial NO production and vasodilator machinery being unaltered (Ferrer et al., 1994a).
Behavioural animal studies with nandrolone have been the subject of some repeated dose investigations. In a study with male Long Evans rats, gonadally intact animals were administered nandrolone at 5 mg/kg bw s.c. 5 days/week for 12 weeks. Each rat was then tested for aggression, 4.5 hours after injections. Treated rats were paired with either gonadally intact or castrated opponents. Aggression was tested during tail pinch of the subject rat and during tail pinch of the opponent rat. Tail pinch did not increase aggression in nandrolone treated rats, whereas it was significantly increased in rats treated similarly with testosterone propionate (McGinnis et al., 2002). In another study, intact and castrated male Wistar rats were injected i.m. with a 0.2 ml suspensions of 0, 10 or 50 mg nandrolone decanoate in cottonseed oil once/week for 8 weeks. After the sixth injection, locomotor activity was measured in an open-field and the acquisition of lever press behaviour was assessed in an autoshaping procedure. Locomotor activity decreased for all groups of rats with continued exposure to the open-field, with no difference between treated and controls. Rats treated with the highest dose spent more time in the margin of the open-field. There was no significant difference between groups on any of the learning measures. The high dose steroid administration inhibited growth in intact and castrated rats and resulted in heavier kidneys and lighter testes. These results indicate that high dose steroids produce observable physiological changes and that they may also interfere with behaviour other than aggressive behaviour, although the evidence from this study is not overwhelming (Minkin et al., 1993).
An investigation was made to see whether there was a relationship between nandrolone deconoate and voluntary ethanol intake in rats. Male Wistar rats (20/group) were divided into a control group and a group receiving daily injections of nandrolone deconoate s.c. at 15 mg/kg bw for 14 days. One subgroup of treated animals (10 rats) was tested for voluntary alcohol intake 1 week after the end of the treatment period and another subgroup received alcohol 3 weeks after the end of nandrolone treatment. Assessment of defensive behaviours and immunoreactivity (ir) levels of the brain opioid peptides dynorphin B and Metenkephalin-Arg-Phe (MEAP) were performed. The nandrolone
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
117

deconoate treated rats were significantly more aggressive and showed lower fleeing and freezing reaction than the controls. Treatment with the steroid enhanced voluntary alcohol intake at 1 and 3 weeks after the end of the treatment period. These animals had a decreased dynophin B-ir activity in the nucleus accumbens, decreased levels of MEAP-ir in the periaquaductal gray and higher levels of MEAP-ir in the hypothalamus compared with controls. This indicates that the altered dynorphin B-ir activity may promote the rewarding effects of ethanol and thereby increasing alcohol intake, whereas MEAP-ir may be associated with the ability to control the aggressive reaction (Johansson et al., 2000). In order to obtain information on the brain regions affected by anabolic steroids, the distribution of neurons containing c-Fos, the protein product of immediate early gene c-fos and Fos-related antigens, was studied after treatment of guinea pigs with nandrolone deconoate i.m. at 15 mg/kg bw/day for 14 days. The behaviour of the animals was monitored for 1 h each day. Animals treated with nandrolone deconoate exhibited a significantly greater incidence of biting behaviour during the 14-day treatment period than controls. A significantly greater density of c-Fos and Fos- related antigen-positive neurons was found in the central nucleus of the amygdala and of Fos-related antigen-positive neurons in the frontal cortex, the shell of the nucleus accumbens and the supraoptic nucleus in nandrolone treated animals than in controls. Therefore, nandrolone induced Fos in brain regions involved in stress, behavioural responses and reward (Johansson-Steensland et al., 2002).
The effects of androgenic anabolic steroids (AAS) on hypothalamic–pituitary–adrenal axis function or corticotropin releasing factor, which may be involved in mediating some of the psychiatric symptoms associated with AAS abuse, were evaluated in male Sprague–Dawley rats. The animals received one daily i.m. injection of nandrolone deconoate at 15 mg/kg bw for 3 days. The rats were sacrificed either 1 hour or 24 hours after the last injection, brain regions dissected and trunk blood collected. One hour after the last injection, nandrolone significantly increased circulating levels of both corticosterone and ACTH levels. In the amygdala, CRF mRNA levels were unchanged 1 hour after the last injection of nandrolone but were significantly reduced at 24 hours. The same was found for hypothalamic proopio-melanocortin (POMC). No significant effects were observed on hypothalamic CRF mRNA, POMC m RNA in the amygdala or CRF R1 mRNA in the anterior pituitary (Schlussman et al., 2000).
The behavioural studies described above have been undertaken to understand better the adverse effects and sites of action in the CNS of androgenic anabolic steroids (AAS). This is in response to the problems posed by the abuse of AAS among bodybuilders, athletes and others to enhance performance and muscle mass. Behavioural changes observed in humans under the influence of AAS include increased irritability, aggression and sensitivity to provocation. Nandrolone has been linked clinically to mood changes, hostility and aggression (Johansson-Steensland et al., 2002).
4.3 Carcinogenicity and mutagenicityNo information was found for carcinogenicity and mutagenicity.
4.4 Reproductive and developmental toxicityFifteen embryonated SPF chicken eggs were incubated at 37 oC and dipped for 5 seconds into ethanolic solutions containing various concentrations of nandrolone. The dipping was done on the 3rd day of incubation (day 19). After hatching (day 0), the chickens were maintained in tradition cages and, at day 10, the birds were weighed, sacrificed, checked for gross abnormalities and the weight of the bursa of Fabricius determined. In an associated study, eggs were dipped into an ethanolic solution containing 10 mg/mℓ nandrolone. Birds were killed 10 days after hatching and samples of bursa and thymus were processed for histopathological examination. The effect of various concentrations of nandrolone on bursa weights is shown in Table 4.1.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
118

Table 4.1 The effect of various concentrations of nandrolone on bursa weightNandrolone Conc. (mg/ml)
10 3 1 0.3 Placebo
Bursa weight (g) Exp 1 0.046 - 0.214 - 0.261Bursa weight (g) Exp 2 0.05 0.161 0.223 0.245 0.225
In the associated study, the number of follicles in the bursa of 10 day old chickens was markedly inhibited by the nandrolone treatment. Also, the thymus size was reduced. The bursa inhibiting capacity of the steroid does not appear to correlate with its endocrine properties (Verheul et al., 1986).
5 Guidelines and standardsNo NOEL which could serve as a basis for establishment of an ADI has been derived, and no MRL has been established for nandrolone. As a consequence of this, nandrolone was added to Annex IV of EU 2377/90. Products remain registered but preclude use in all food-producing animals. Products containing nandrolone are now only licensed for use in dogs and cats.
6 ReferencesBarrett RL & Harris EF (1993) Anabolic steroids and craniofacial growth in the rat. Angle Orthod, 63, 289-298
Bergink EW, Geelen JAA & Turpijn EW (1985) Metabolism and receptor binding of nandrolone and testosterone under in vitro and in vivo conditions. Acta Endocrinol, 110 (Suppl 271), 31-37
Bishop Y, ed (1994) The Veterinary National Formulary (Second Edition), London, UK, Royal Pharmaceutical Society of Great Britain/British Veterinary Association
Bishop Y, ed (2004) The Veterinary National Formulary (Sixth Edition), London, UK, Royal Pharmaceutical Society of Great Britain/British Veterinary Association
Compton DR (1993) Anabolic steroids: Evidence in mice for the production of indirectly-mediated behavioral effects at doses relevant to human abuse patterns. NIDA Research Monograph Series, 132, 378
Coppi G, Gaetani M & Bonardi G (1973) Pharmacological and toxicological properties of 19-nortestosterone-homofarnesate (DA 1979), a new anabolic agent. Arzneimittelforschung, 23, 693-700
Ferrer M, Encabo A, Marín J & Balfagón G (1994a) Chronic treatment with the anabolic steroid, nandrolone, inhibits vasodilator responses in rabbit aorta. Eur J Pharmacol, 252, 233-241
Ferrer M, Encabo A, Marín J & Balfagón G (1994b) Treatment with the anabolic steroid, nandrolone, reduces vasoconstrictor responses in rabbit arteries. Eur J Pharmacol, 258, 103-110
Galbraith H, Berry AD, Henderson GD & Jessiman C (1986) Effect of naturally occurring and synthetic androgens on growth, body composition and muscle glucocorticoid receptors in wether lambs. [Abstract] Anim Prod, 42, 433
HSDB (2002) Nandrolone. From: Hazardous Substances Databank, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [January 2004] at http://toxnet.nlm.nih.gov
Johansson P, Lindqvist A-S, Nyberg F & Fahlke C (2000) Anabolic androgenic steroids affects alcohol intake, defensive behaviors and brain opioid peptides in the rat. Pharmacol Biochem Behav, 67, 271-279
Johansson-Steensland P, Nyberg F & Chahl L (2002) The anabolic androgenic steroid, nandrolone decanoate, increases the density of Fos-like immunoreactive neurons in limbic regions of guinea-pig brain. Eur J Neurosci, 15, 539-544
Kumar N, Suvisaari J, Tsong Y-Y, Aguillaume C, Bardin CW, Lähteenmaki P & Sundaram K (1997) Pharmacokinetics of 7-methyl-19-nortestosterone in men and cynomolgus monkeys. J Androl, 18, 352-358
McGinnis MY, Lumia AR, Breuer ME & Possidente B (2002) Physical provocation potentiates aggression in male rats receiving anabolic androgenic steroids. Horm Behav, 41, 101-110
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
119

Minkin DM, Meyer ME & Van Haaren F (1993) Behavioral effects of long-term administration of an anabolic steroid in intact and castrated male Wistar rats. Pharmacol Biochem Behav, 44, 959-963
Noda K, Chang H-P, Takahashi I, Kinoshita Z & Kawamoto T (1994) Effects of the anabolic steroid nandrolone phenylpropionate on craniofacial growth in rats. J Morphol, 220, 25-33
PIMS (1998) Nandrolone. From: Poisons Information Monographs, Geneva, Switzerland, World Health Organization, International Programme on Chemical Safety, available [January 2004] at http://www.inchem.org/documents/pims/pharm/pim910.htm
Schlussman SD, Zhou Y, Johansson P, Kiuru A, Ho A, Nyberg F & Kreek MJ (2000) Effects of the androgenic anabolic steroid, nandrolone decanoate, on adrenocorticotropin hormone, corticosterone and proopiomelanocortin, corticotropin releasing factor (CRF) and CRF receptor1 mRNA levels in the hypothalamus, pituitary and amygdala of the rat. Neurosci Letts, 284, 190-194
Schubert K, Hobe G, Kaufmann G, Schumann G, Wehrberger K & Hörhold C (1983) Studies on biotransformation of STS 557. Exp Clin Endocrinol, 81, 168-174
Tseng YT, Rockhold RW, Hoskins B & Ho IK (1994) Cardiovascular toxicities of nandrolone and cocaine in spontaneously hypertensive rats. Fundam Appl Toxicol, 22, 113-121
Verheul HAM, Tittes E, V, Kelder J & Schuurs AHWM (1986) Effects of steroids with different endocrine profiles on the development, morphology and function of the bursa of fabricius in chickens. J Steroid Biochem, 25, 665-675
Wijnand HP, Bosch AMG & Donker CW (1985) Pharmacokinetic parameters of nandrolone (19-nortestosterone) after intramuscular administration of nandrolone decanoate (Deca-Durabolin®) to healthy volunteers. Acta Endocrinol, 110 (Suppl 271), 19-30
Yu-Yahiro JA, Michael RH, Nasrallah DV & Schofield B (1989) Morphologic and histologic abnormalities in female and male rats treated with anabolic steroids. Am J Sports Med, 17, 686-689
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
120

Annex 12 Narasin1 IntroductionNarasin (alpha-ethyl-6-(5-(2-(5-ethyltetrahydro-5-hydroxy-6-methyl-2H-pyran-2-yl)-15-hydroxy-2,10,12- trimethyl-1,6,8-trioxadispiro(4.1.5.3)pentadec-13-en-9-yl)-2-hydroxy-1,3-dimethyl-4-oxoheptyl) tetrahydro-3,5-methyl-2H-pyran-2-acetic acid; CAS No. 55134-13-9) is a member of the group of polyether carboxylic ionophore antibiotics that are used as coccidiostats for poultry and as growth promoters for pigs. Narasin forms electrically neutral complexes with monovalent cations (Na+, K+), which promote an exchange-diffusion transport of the cations across biological membranes. This facilitated transport produces effects on cellular function and metabolism and is the basis of the known physiological and toxicological effects of narasin (Novilla et al., 1994).
In toxicology studies, the mycelial and, when appropriate, the crystalline forms of narasin were tested and the doses used were based on similar analytical narasin activity (Novilla et al., 1994).
2 Uses and exposureNarasin, available as MontebanTM and MontebanTM in the UK, is classified as a zootechnical feed additive used for the prophylaxis of coccidiosis in chickens. In some countries it is also approved for use as a growth promoter in growing and finishing pigs. Narasin is also active against Gram-positive bacteria and is active against necrotic enteritis (Clostridium perfringens; Bishop, 2004).
In the UK, Monteban G100TM contains 10 % w/w narasin (100 g/kg premix) and is available for use in broiler chickens, only, for the treatment of coccidiosis. Narasin is also available in combination with nicarbazin as MaxibanTM. Maxiban G160TM is also used for the prophylaxis of coccidiosis in chickens and contains 8 % w/w narasin (80 g/kg premix) and 8 % w/w nicarbazin (80 g/kg premix). The combination product is aimed at limiting the development of resistance, by synergistically increasing anticoccidial performance (Bishop, 2004).
The dose rate for inclusion of Monteban G100TM in the diet is 700 g/tonne of premix, equivalent to 70g narasin/tonne of feed (0.007 %w/w). The dose rate for inclusion of Maxiban G160 TM in the diet is 500–625 g/tonne of feed equivalent to 40–50 g narasin /tonne and 40–50 g nicarbazin/tonne of feed (50 mg/kg = 0.005 % each). Both may be fed in the diet continually from 1 day old, to 5 days prior to slaughter (approximately 6 weeks of age; Bishop, 2004).
The withdrawal period in broiler chickens prior to slaughter is 5 days. The products are contraindicated in layer replacement chickens, turkeys and guinea fowl. Narasin is extremely toxic to other species and has proved fatal in horses and dogs. Narasin should not be used within 7 days of tiamulin medication (Taylor, 2004).
The authors are aware that in some countries, such as the USA and Canada, narasin is available as a medicated premix (Monteban 70 PremixTM), containing 7 % w/w narasin (70 g/kg premix) for use as follows: for the treatment of coccidiosis in chickens; for the prevention of necrotic enteritis in chickens; and as a growth promoter by improving feed efficiency and increasing weight gain in growing and finishing pigs. In chickens, the dose rate is 70 mg narasin/kg complete feed (0.007 %). To improve weight gain in pigs the dose rate is 15 mg/kg complete feed (0.0015 %). In the USA, narasin (Monteban) is approved in combination with bacitracin zinc (Albac) for the prevention of coccidiosis and to improve weight gain and food efficiency in broiler chickens at concentrations of 54–70 g naracin/ton and 4–50 g bacitracin zinc/ton of feed.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
121

Potential sources of human exposure include ingestion of animal products containing drug residues. However, no contamination with narasin has been detected in samples of food monitored under the VRC non-statutory monitoring programme between 1998 and 2001 (VRC, 2002).
2.1 Exposure concentrations in meatUsing information on usage of narasin and on absorption and excretion, likely ‘worst case’ concentrations in meat products were estimated using Equation 1:
Equation 1M = D × T × Fabs × (1–Fexc)
where M is the predicted concentration in meat (mg/kg); D is the dose of substance (mg/kg/d); T is the duration of the treatment (d); Fabs is the fraction of compound absorbed; and Fexc is the fraction of compound excreted.
Narasin is absorbed to an unknown extent and excreted rapidly by the chicken. The excretion routes are not established in the chicken but the main metabolic pathway involves oxidative processes leading to the formation of metabolites in tissues and excreta, with the liver the target tissue. Unchanged narasin is not detectable in tissues after 6 hours withdrawal, with the exception of the skin/fat (until 12 h) where it represents the major fraction. For chickens, it was assumed that narasin is given in feed for 40 days at 70 g/tonne of feed (corresponding to a dose rate of 5 mg/kg bw/d). The bioavailability of lasalocid is reported to be low and the metabolism is reported to be high; values of 5 % absorption and 95 % excretion were therefore used. Using these input values, a ‘worst case’ concentration of 0.5 mg/kg was obtained.
For pigs, it was assumed that narasin is given in feed for 100 days at 15 g/tonne of feed (corresponding to a dose rate of approximately 0.75 mg/kg bw/d). The bioavailability of narasin is reported to be low and the metabolism is reported to be high; values of 5 % absorption and 95 % excretion were therefore used. Using these input values, a ‘worst case’ concentration of 0.19 mg/kg was obtained.
3 ToxicokineticsNo information was found.
4 Toxicity profile4.1 Acute toxicityNarasin was administered orally as single doses to experimental animals and intravenous (i.v.) to rats. The animals used were ICR mice, Harlan Wistar rats, New Zealand White rabbits and beagle dogs. All animals were fasted overnight prior to treatment. Animals were observed for signs of toxicity for 14 days and a median LD50 was calculated. Consistent signs of toxicity included anorexia, hypoactivity, leg weakness, ataxia, depression and diarrhoea.
The LD50 values are shown in Table 4.1 (Novilla et al., 1994).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
122

Table 4.1 The acute oral toxicity (LD50s) of narasin in experimental animalsSpecies Sex LD50 ± SE (mg/kg bw)
Mycelial narasin
Crystalline narasin
Mouse M 15.8 ± 2.6 22.8 ± 2.9F 16.7 ± 2.1 36.7 ± 4.3
Rat M 21.1 ± 2.9 40.8 ± 4F 18.5 ± 2.3 33.8 ± 6F 1.96 ± 0.14 (i.v.)
Rabbit M, F >10.75 15.5 ± 3.9Dog M, F >10 >10
Dermal toxicity and irritation were evaluated in New Zealand white rabbits (3/sex/group) by topical application of mycelial narasin on to the skin of the back. The treatment sites were occluded for 24 hours and the animals were observed for 14 days. There was no obvious toxicity or irritation at a single dose of 250 mg narasin/kg bw (Novilla et al., 1994). [NOTE: There appears to be some confusion about the dose used in this study. In the Materials & Methods it is stated to be 10.75 mg/kg bw; in the Results, it is shown as 250 mg/kg.]
An acute inhalation study was conducted with a group of 5 male and 5 female Wistar rats. The animals were exposed head only for 30 minutes to an atmospheric concentration of mycelial narasin equivalent to 9.72 mg narasin/m3 air. The mycelial preparation was difficult to disperse and it was difficult to attain an air concentration of 0.4 mg/ℓ. The mass median aerodynamic diameter of the particles was 7.75 µm. The median lethal concentration (LC50) of mycelial narasin was >0.4 mg/ℓ (Novilla et al., 1994). [NOTE: Again, there is a discrepancy between the exposure value of 9.72 mg/m3 in the ‘Methods’ section and 0.4 mg/ℓ (400 mg/m3) in the ‘Results’ section.]
For the evaluation of ocular irritation, a group of 5 male and 4 female rabbits were used. One eye of each animal was instilled with 0.1 mℓ mycelial narasin into the conjunctival sac. Two minutes after treatment, 3 treated eyes were rinsed for 2 minutes using 300 mℓ sterile physiological saline solution. Ocular response was graded after 1 hour and 1, 2, 3, 7, 14 and 21 days. Direct exposure caused severe corneal, iridal and conjunctival irritation. Two of the 6 rabbits developed pannus, indicative of permanent eye damage. In the other 3 rabbits, rinsing eyes immediately after exposure was effective in reducing the degree of irritation and eyes returned to normal within 2 days (Novilla et al., 1994).
The skin sensitisation of narasin was evaluated in a group of 10 female albino guinea pigs. A series of 10 intracutaneous injections of 0.25% crystalline narasin in sunflower oil was given to each animal over a 3 week period. The first injection was 0.05 mℓ and the remaining 9 were 0.1 mℓ each. Two weeks after the tenth injection, a challenge was made using 0.05 mℓ solution for each animal. The irritation wheal diameters, measured 24 hours after each injection, were compared. There was no indication of delayed hypersensitivity in guinea pigs in this study (Novilla et al., 1994).
4.2 Repeat dose toxicityGroups of 15 male and 15 female Wistar rats, 4–5 weeks old, were fed diets containing 0, 15, 40 or 100 ppm crystalline narasin in one study and 0, 15, 30 or 60 ppm mycelial narasin in another study. All animals were examined daily for clinical signs of toxicity and body weight and food consumption were measured weekly. Blood samples were collected from surviving animals for haematological and blood chemistry evaluations. The rats were necropsied and pathological examinations made on major tissues. In the first study, rats at the middle and top dose levels had decreased body weight gain and food utilisation. In top dose rats, anorexia was observed early in 1 male and in all females throughout the study. In the second study, decreased body weight gain occurred at the 30 and 60 ppm dose levels. The no effect level was determined to be 15 ppm in both studies (Novilla et al., 1994).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
123

Groups of 15 male and female B6C3F1 mice were fed diets containing 0, 10, 20 or 40 ppm narasin in one study and 0, 60, 80 or 100 ppm in another study. Animals were examined daily for clinical signs of toxicity and body weights were measured weekly. Haematological and blood chemistry values were obtained before necropsy and pathological examination was made on major tissues at necrosy. There were no changes of toxicological significance in the first study. In the second study, the only effect was growth impairment in males at 80 and 100 ppm and in females at 100 ppm (Novilla et al., 1994).
Three 3-month studies were undertaken in dogs. In the first study in mature beagles, groups of 4 males and 4 females, 9–14 months old, were given crystalline narasin by capsule daily at doses of 0, 0.75 or 2.25 mg/kg bw (2 doses) or 6.0 mg/kg bw (1 dose). After the first dose, the mid- and top dose animals presented severe weakness, ataxia, prostration and vomiting. Therefore, dosing was stopped and following a 2–3 week recovery period, the narasin dosage was changed to 0.25 (formerly 6.0), 0.75 or 1.5 (formerly 2.25) mg/kg bw/day for 3 months. In the second study, mycelial narasin was administered by capsule daily to groups of 4 male and 4 female beagles, 13–16 months old, at doses of 0, 0.5, 1.0 or 2.0 mg/kg bw. In a third study, groups of young beagles, 4 months old, were administered 0, 0.5, 1.0 or 2.0 mg mycelial narasin/kg bw or 2.0 mg crystalline narasin/kg bw/day for 3 months. Clinical signs were observed daily and body weights were measured weekly. Haematological and biochemical assays were performed periodically during the study, Electrocardiograms (ECG) were conducted on all young dogs before treatment and 2 hours after dosing at 1, 2 and 3 months. At necropsy, pathological examination was made on major tissues. Crystalline narasin at doses up to 1.5 mg/kg bw/day was without effect on the parameters measured in mature dogs. Mycelial narasin at a dose of 2.0 mg/kg bw/day induced leg weakness and incoordination in 5/8 dogs; this lasted for 1 day to 3 weeks in 4 dogs and for 3 months in another dog. Some of these dogs had abnormal ECGs. In this study, there were no effects at 0.5 or 1.0 mg/kg bw/day. Adverse clinical signs were observed in the study with young dogs at the top dose of 2.0 mg/kg bw/day including leg weakness, ataxia, recumbency, laboured breathing, excessive salivation and anorexia. These signs were more pronounced in dogs given crystalline narasin than in those given mycelial narasin. Three dogs in each of these 2.0 mg/kg bw/day groups had treatment related lesions consisting of focal degeneration and regeneration of skeletal muscles including the diaphragm. Focal degeneration of intramuscular nerves was observed in the 3 crystalline narasin treated dogs and in 1 mycelial narasin treated dog with skeletal muscle lesions. No effects were seen at 0.5 or 1.0 mg/kg bw/day in young dogs (Novilla et al., 1994).
Repeat exposure inhalation studies were conducted in dogs in 9 m3 inhalation chambers. In a 14-day study (10 exposures), groups of 2 male and 2 female beagles, 11–14 months old, were exposed to 0, 0.113, 0.276 or 1.057 mg narasin/m3 in air. In a 3-month study (65 exposures) groups of 4 male and 4 female dogs, 9–10 months old, were exposed to 0, 0.114, 0.276 or 0.874 mg/m3 in air. For the 3 exposure groups, the median equivalent aerodynamic diameters of the aerosols were 11.44–12.87 µm in the 14-day study and 16.69–17.33 µm in the 3-month study. Clinical observations were made daily and body weight, haematology, blood chemistry, pulmonary function and ECGs were measured at appropriate times before and during the studies. All animals were necropsied and pathological examinations undertaken on major tissues. All dogs survived in the 14-day study. Treatment related effects occurred in the top dose animals as ptyalism, ataxia, limb paresis, tremors, dyspnea and ocular irritation. Tissue effects in top dose dogs consisted of severe necrosis and moderate regeneration of skeletal muscle, with slight degeneration of the sciatic nerve in a female, moderate degeneration of the sciatic nerve in a male and slight necrosis and regeneration of skeletal muscle in another female. No effects, except for mild ocular irritation in 1 female, were seen in the mid- and low exposure groups. In the 3-month study, 1 male died after 40 exposures at the top dose. No effects were found in low exposure dogs. In dogs exposed to 0.276 and 0.874 mg/m3, signs of toxicity included ptyalism, ocular irritation, dyspnea, hypotonic abdominal muscle and limb paresis. Mild to severe inflammatory or degenerative lesions occurred in the trachea, lung, cornea, skeletal muscle and sciatic nerve in dogs in these 2 groups in a dose related manner (Novilla et al., 1994).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
124

Groups of beagle dogs weighing 8–15 kg were surgically prepared for the determination of blood pressure in the left femoral artery and blood flow in the left anterior descending coronary artery. In an i.v. study, 4 anaesthetised and 4 conscious dogs received successive doses of narasin dissolved in 0.2 mℓ ethanol and diluted with 5% dextrose in water by bolus injection into the cannulated left femoral vein every 10 minutes to give cumulative doses of 0.0076, 0.0153, 0,0382, 0.076 and 0.153 mg/kg bw. Control anaesthetised or conscious dogs were given vehicle only. In an oral study, 4 conscious dogs were administered, by gavage, 1.53 mg narasin/kg bw dissolved in 0.2% ethanol and suspended in 10% acacia. Control dogs received 15 mℓ 10% acacia. Coronary flow, mean blood pressure and heart rate were measured after each dose in the i.v. study and at 10, 20, 30, 40, 50, 60, 90 and 120 minutes after dosing in the oral study. Narasin increased coronary flow following i.v. dosing in both anaesthetised and conscious dogs. Doses estimated to increase coronary flow by 100% were 0.0458 mg/kg bw and 0.040 mg/kg bw in anaesthetised and conscious dogs, respectively. Doses of 0.076 and 0.153 mg/kg bw i.v. in anaesthetised dogs and 0.153mg/kg bw i.v. in conscious dogs, increased mean blood pressure. The dose of 0.153 mg/kg bw i.v. increased heart rates in both anaesthetised and conscious dogs. There was no effect on coronary flow, mean blood pressure or heart rate following oral dosing of narasin (Novilla et al., 1994).
Chronic toxicity dietary studies (6 months to 1 year) were undertaken in rats or dogs. In the 1 year rat study, groups of 15 Harlan Wistar rats/sex/group, 4–6 weeks old, were derived from parents that had been maintained on diets containing 0, 7.5, 15 or 30 ppm narasin. The young rats were placed on similar diets for 1 year that provided average daily intakes equivalent to 0.24–0.74, 0.51–1.41 or 0.98–2.84 mg/kg bw for males and 0.35–0.79, 0.77–1.58 or 1.55–4.89 mg/kg bw in females, respectively. The rats were examined daily for clinical signs of toxicity. Body weights, haematology and clinical chemistry parameters were determined during the test period. Animals that died during the study and those at study termination were subjected to gross and microscopic examination. Survival was not affected by treatment. The only treatment related effect was reduced body weight gain in female rats at the top dose level, supporting a no effect level of 15 ppm narasin in the diet (Novilla et al., 1994).
Six-month and 1-year studies were carried out on beagle dogs. In the 6-month study, groups of 4 males and 4 females, 12–16 months old, were administered 0, 0.5, 1.0 or 1.5 mg mycelial narasin/kg bw/day orally. In the 1-year study, 5 month old dogs, 4/sex/group, were given daily oral doses of 0, 0.5, 1.0 or 2.0 mycelial narasin/kg bw. Another group was treated with 2.0 mg crystalline narasin/kg bw/day. The animals were observed daily and body weights, haematology and clinical chemistry parameters were determined at specified intervals before and during the 6-month and 1-year studies. Ophthalmoscopic and ECG examinations were conducted before during and at the end of a study. All dogs were necropsied and major tissues evaluated histopathologically. Except for 1 abnormal ECG at 1 month in 1 female given 1.5 mg mycelial narasin/kg bw/day, the other dogs were unaffected by the test material in the 6-month study. In the 1-year study, dose-related signs of toxicity were observed for dogs at 1.0 and 2.0 mg/kg bw/day. These consisted of excessive salivation, leg weakness, decreased appetite, laboured breathing, hypoactivity and recumbence, loss of body weight and degenerative and/or regenerative changes in cardiac and skeletal muscles and peripheral nerves. Mycelial narasin had a greater effect than crystalline narasin. A no effect level of 0.5 mg mycelial narasin/kg bw/day was supported by the 1 year study (Novilla et al., 1994).
4.3 Carcinogenicity and mutagenicityGroups of Wistar rats (40/sex/dose), derived from parents maintained on diets containing 0, 7.5, 15 or 30 ppm narasin (from multigeneration study, see Section 4.4), were placed on identical dietary dose levels for 2 years. Groups of 5–6 week old B6 C3 F1 mice, 30/sex/dose, were fed diets containing 0, 5, 15 or 50 ppm mycelial narasin or 50 ppm crystalline narasin for 2 years. The animals were observed daily and body weights, haematological and clinical chemistry values were determined at intervals during the study period. Animals dying during the study and those surviving to the end of the study were necropsied and the tissues submitted to histopathological examination. In the 2-year rat study, narasin did not induce cardiac or skeletal muscle lesions, other non-neoplastic alterations or
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
125

neoplasms. Similar results were obtained in the 2-year mouse studies with either the mycelial or crystalline forms of narasin. Therefore, these rodent bioassays demonstrated that narasin did not give rise to cumulative toxicity or carcinogenicity (Novilla et al., 1994).
A battery of in vitro tests was performed to evaluate the potential genotoxicity of narasin. Crystalline narasin was used in these assays. Bacterial mutation was assessed in a gradient plate assay using 8 histidine auxotrophs of S. typhimurium and 2 tryptophan auxotrophs of E. coli exposed to narasin concentrations of 0.1–1000 µg/mℓ. Bacterial assays were also conducted with S. typhimurium strains TA98 and TA100 at narasin concentrations of 1–1000 µg/plate with and without metabolic activation. Assays for DNA damage were conducted in primary cultures of adult rat hepatocytes. In 2 studies, cell cultures were incubated for 20 hours with concentrations of 0.5–1000 nmol/mℓ narasin. A forward mutation test using L5178Y TK+/- mouse lymphoma cells was conducted with narasin concentrations of 0.1–10 µg/mℓ, with and without metabolic activation. The results of these in vitro tests showed that narasin was not mutagenic in bacterial or mammalian cells and that it did not induce DNA repair (Novilla et al., 1994).
Potential chromosomal damage was assessed in vivo in Chinese hamsters treated orally with narasin at doses of 1, 5, 10, 15, 20, 25, 30, 35, 45 or 50 mg/kg bw. The frequency of sister chromatid exchanges (SCE) was measured in bone marrow cells obtained at necropsy. The results showed that narasin did not induce SCEs in this test (Novilla et al., 1994).
4.4 Reproductive and developmental toxicityIn a rat multigeneration study, mycelial narasin was fed to 3 parental generations and their offspring. Initially, the F0 parents, 45/sex/group, were fed narasin in their diet at 0, 15, 30 or 60 ppm. However, the animals in the 60 ppm group had significantly lower body weights, progeny weights and survival. Thereafter, the study was conducted with dietary levels of 0, 7.5, 15 or 30 ppm narasin using the F1b offspring with 22/sex/dose to produce the next generation. The F1a offspring were assigned to the 2-year study (see Section 4.3). The F1c offspring were submitted to a teratology assessment. In the next generation, the F2a offspring, 1/sex/litter, were given a gross internal examination. In the final generation, gross internal examination was carried out on all F3a progeny with histopathology on 1/sex/litter and progeny from the F3b litter were evaluated for teratogenic defects by macroscopic and microscopic examinations for visceral or skeletal abnormalities. The reproductive performance of the parents was evaluated throughout the study. Narasin, at any dose level, did not affect reproductive performance, including fertility, litter size, gestation length and survival, progeny survival and sex distribution. Parental and progeny weights were significantly depressed at the 30 ppm narasin dose level. The teratology evaluations undertaken during the study showed that narasin was without effect on the developing rat fetus (Novilla et al., 1994).
Groups of 15 pregnant Dutch Belted rabbits, 3–4 months old, were given oral doses of narasin daily at 0, 0.6, 1.2 or 1.8 mg/kg bw on gestation days 6–18. On gestation day 28, the dams were killed and evaluated for reproductive performance and the fetuses examined for abnormalities. Maternal toxicity was seen at the 1.2 and 1.8 mg/kg bw dose levels as evidenced by leg weakness, incoordination and abortion in some of the animals. A slight increase in resorptions was recorded at the top dose. However, there was no evidence for a teratogenic effect due to narasin in this study (Novilla et al., 1994).
5 Guidelines and standardsThe lowest NOEL identified in oral toxicity studies was 0.5 mg/kg bw/day, and the ADI has been set at 5 µg/kg bw (equal to 300 µg narasin/day for a person of 60 kg bodyweight). A uniform MRL for all tissues was proposed as 50 µg narasin/kg (0.05 ppm; EFSA, 2004).
Based on a stated NOEL of 1.5 mg/kg bw/day for an unspecified study, a slightly higher ADI of 10 µg/kg bw/day has been proposed by the Australian Government (Australian Government, 2005).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
126

6 ReferencesAustralian Government (2005) ADI List- Acceptable Daily Intakes for Agricultural and Veterinary Chemicals , Canberra, Australia, Australian Government, Department of Health and Ageing, Available [June 2005] at http://www.tga.gov.au/docs/html/adi.htm
Bishop Y, ed (2004) The Veterinary National Formulary (Sixth Edition), London, UK, Royal Pharmaceutical Society of Great Britain/British Veterinary Association
EFSA (2004) Opinion of the Scientific Panel on Additives and Products or Substances used in Animal Feed on a Request from the Commission on the Re-evaluation of Efficacy and Safety of the Coccidiostat Monteban® G100 in Accordance with Article 9G of Council Directive 70/524/EEC (Question No. EFSA-2003-046), Brussels, Belgium, European Food Safety Authority, available [July 2005] at http://www.efsa.eu.int/science/feedap/feedap_opinions/518_en.html
Novilla MN, Owen NV & Todd GC (1994) The comparative toxicology of narasin in laboratory animals. Vet Hum Toxicol, 36, 318-323
Taylor MA (2004) Antiprotozoals. In: Bishop Y, ed, The Veterinary National Formulary (Sixth Edition), London, UK, Royal Pharmaceutical Society of Great Britain/British Veterinary Association, pp 171-179
VRC (2002) Non-Statutory Programme – Proposals for the 2003 Programme (VRC/02/39) Veterinary Residues Committee, Addelstone, UK. Available [July 2006] at: http://www.vet-residues-committee.gov.uk/
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
127

Annex 13 Phenylbutazone1 IntroductionPhenylbutazone (4-butyl-1,2-diphenyl-3,5-pyrazolidinedione) is a non-steroidal anti-inflammatory drug (NSAID) drug that has anti-inflammatory, antipyretic and analgesic activities. It has been effective in the treatment of ankylosing spondylitis in humans. It has also been useful for rheumatoid arthritis and Reiter’s syndrome. Although phenylbutazone is effective for gouty arthritis, risk/benefit considerations indicate that the drug should not be employed for this disease (SIS NLM, 2004). Because of potentially serious adverse haematological effects, phenylbutazone should now be used only when other less toxic NSAIDs are ineffective. It is not recommended for use in individuals under 15 or over 60 years of age. In addition, the drug should be administered only to carefully selected patients who are under close medical supervision (HSDB, 2002).
Oxyphenbutazone, the main metabolite of phenylbutazone, is also produced commercially and has a similar therapeutic and toxicological profile (IARC, 1977; Drugs.com®, 2004).
Table 1.1 Synonyms and trade names for phenylbutazoneAlindor; Alkabutazona; Alqoverin; Anerval; Anpuzone; Antadol; Anuspiramin; Arthrizin; Artrizin; Artrizone; Artropan; Azdid; Azobutil; Benzone; Betazed; Bizolin 200; B.T.Z.; Butacote; Butacompren; Butadion; Butadiona; Butadione; Butagesic; Butalgina; Butalan; Butalidon; Butaluy; Butaphen; Buta-Phen; Butapirazol; Butapyrazole; Butarecbon; Butartril; Butartrina; Butazina; Butazolidin; Butazolidine; Butazona; Butazone; Butidiona; Butiwas-simple; Butone; Butoz; 4-Butyl-1,2-diphenyl-pyrazolidine-3,5-dione; 3,5-Dioxo-1,2-diphenyl-4-n-butylpyrazolidine; Butylpyrin; Buvetzone; Buzon; Chembutazone; Digibutina; Diossidone; Diozol; Diphebuzol; Diphenylbutazone; 1,2-Diphenyl-4-butyl-3,5-pyrazolidinedione; 1,2-Diphenyl-3,5-dioxo-4-butylpyrazolidine; 1,2-Diphenyl-2,3-dioxo-4-n-butylpyrazolidine; Ecobutazone; Elmedal; Equi Bute; Eributazone; 'Esteve'; Febuzina; Fenartil; Fenibutasan; Fenibutazona; Fenylbutazon; Fenilbutazona; Fenilbutina; Fenilbutine; Fenibutol; Fenilidina; Fenotone; Flexazone; G 13,871; IA-But; Intalbut; Intrabutazone; Ipsoflame; Kadol; Lingel; Malgesic; Mephabutazone; Merizone; Nadazone; Nadozone; Neo-Zoline; Novophenyl; Phebuzin; Phebuzine; Phen-Buta-Vet; Phenbutazol; Phenopyrine; Phenylbetazone; Phenylbutaz; Phenylbutazonum; Phenyl-Mobuzon; Pirarreumol 'B'; Praecirheumin; Pyrabutol; Pyrazolidin; Rectofasa; Reudo; Reudox; Reumasyl; Reumazin; Reumazol; Reumune; Reupolar; Robizon-V; Rubatone; Scanbutazone; Schemergin; Shigrodin; Tazone; Tetnor; Tevcodyne; Therazone; Ticinil; Todalgil; Uzone; VAC-10; Wescozone; Zolaphen; Zolidinum
2 Uses and exposurePhenylbutazone is an anti-inflammatory agent with analgesic, antipyretic and mild uricosuric activity although its precise mechanism of action is uncertain (HSDB, 2002). It has been used in humans for the symptomatic treatment of acute gout, rheumatoid arthritis, rheumatoid spondylitis, osteoarthritis, psoriatic arthritis, acute superficial thrombophlebitis and for several other severe acute local inflammatory conditions (IARC, 1977) although it is poorly tolerated in many patients with some side effects occurring in 10–45% of patients (HSDB, 2002). Human therapeutic dosages vary, for example, from 300–600 mg/day for the treatment of rheumatoid arthritis to 200 mg every 4 hours for up to 4 days for the treatment of gout. In many countries, phenylbutazone is approved only for the treatment of severe ankylosing spondylitis that is unresponsive to other NSAIDs, while the use of phenylbutazone to relieve the pain and inflammation of acute painful shoulder (i.e. peritendinitis, capsulitis, or bursitis of that joint) is no longer FDA-approved. It has been strongly recommended that use of phenylbutazone is restricted to short-term treatment of severe flares of rheumatic disease, gout, or calcium pyrophosphate deposition disease (Drugs.com®, 2004).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
128

Phenylbutazone is still authorised for human use in Europe (Bishop, 2004). Phenylbutazone is no longer approved for use as a human medicine in the USA (FDA, 2003). Under US FDA requirements it is included in the Approved Drug Products with Therapeutic Equivalence Evaluations List (FDA, 1992; HSDB, 2002).
Medical authorities in the USA and other countries have recommended a series of restrictions in the use of phenylbutazone. These include the following warnings. a) The drug should be used with caution in pregnant women and nursing mothers, b) the drug is contraindicated in patients with gastrointestinal tract problems, c) the drug is contraindicated in children under 14 years of age, d) it is inadvisable to use the drug in elderly patients, e) the drug should be used with caution in patients with severe cardiac or renal disease, f) because of potentially serious haematological effects, phenylbutazone should be used only when other less toxic non-steroidal anti-inflammatory agents are ineffective (HSDB, 2002).
The use of phenylbutazone in food producing animals is effectively banned by EMEA CVMP, as an application to set MRLs could not be progressed (EMEA, 1999; EMEA, 2000). However, it may still be employed in non-food species (including horses) not intended for food use and in food-species in exceptional circumstances provided specified prolonged withdrawal periods are employed (FVE, 2002).
Under US-FDA requirements phenylbutazone can be used in dogs and horses (HSDB, 2002). The FDA has recently specifically prohibited extra-label use of phenylbutazone in female dairy cattle aged 20 months or older (FDA, 2003).
Phenylbutazone is used for the treatment of inflammation and pain associated with the musculoskeletal system in dogs, cats, and arthritis and lameness in horses. In companion animals (dogs and cats), the drug is administered as oral tablets containing 25, 100 or 200 mg phenylbutazone at a dose rate of 6-20 mg/kg daily for 7 days usually in divided doses 2 or 3 times daily (Companazone 25mgTM; Phenylbutazone 100mg, 200mgTM). In horses, in-feed (Equipalazone powderTM, Pro-Dynam PowderTM), oral paste (Equipalazone Paste E-PPTM) and injectable formulations (Equipalazone InjectionTM) are available. The dose rate for oral formulations is 4.4 mg/kg twice daily on day 1; thereafter 2.2 mg/kg twice daily for 2-4 days followed by 2.2 mg/kg daily or on alternate days. In ponies the dose rate is 4.4 mg/kg on alternate days. Injection is given as a single dose at 4.4 mg/kg and may then be followed by oral treatment as above (Bishop, 2004).
No MRLs have been established for meat and meat products and no NOEL, which could serve as a basis for establishment of an ADI, has been set. As a consequence of this, and its carcinogenic potential, phenylbutazone is rarely used in human medicine and banned for use in food producing animals. It may only be used in horses not intended for human consumption (Bishop, 2004).
2.1 Exposure concentrations in meatIllegal and extra-label use of phenylbutazone in cattle may occasionally occur and has been reported in some countries.
Limited information is available on metabolism and excretion in cattle. Following intravenous injection of 4.4 mg/kg the mean elimination half-life (t1/2) was shown to be 35.9 hours. Intramuscular and oral routes resulted in a much longer half-life (t1/2) due to slow absorption from the gastrointestinal tract and depot formation and sequestration at the intramuscular site. When given orally, there may be several peaks in plasma concentration possibly due to adsorption onto rumen contents. Two hydroxylated metabolites of phenylbutazone, oxyphenbutazone and -hydroxyphenbutozone were detected in plasma and urine at levels which suggested poor re-absorption and renal excretion and that these derivatives were probably formed more slowly in cattle than in horses (Bishop, 2004).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
129

Using information on usage of phenylbutazone and on absorption and excretion, likely ‘worst case’ concentrations in meat products were estimated using Equation 1:
Equation 1 M = D × T × Fabs × (1–Fexc)
where M is the predicted concentration in meat (mg/kg); D is the dose of substance (mg/kg bw/d); T is the duration of the treatment (d); Fabs is the fraction of compound absorbed; and Fexc is the fraction of compound excreted.
Phenylbutazone is likely to be used to treat cattle with chronic lameness; it was assumed that phenylbutazone is given in feed for 365 days at a dose rate of 4.4 mg/kg bw/d, that it is completely absorbed and that 90% is excreted. Using these input values, a ‘worst case’ concentration of 80 mg/kg was obtained.
3 ToxicokineticsMethods of analysis identified by the International Agency for Research on Cancer (IARC, 1977) included, for phenylbutazone, titration and spectrophotometry and, for oxyphenutazone, titration and UV spectrophotometry, while plasma phenylbutazone and oxyphenylbutazone can be detected using HPLC or GLC. More recently, it has been reported that sensitive analytical techniques available include an HPLC with photodiode-array detector method with detection and quantification limits of 0.016 and 0.029 µg/mℓ, respectively, and an EU-validated GC–MS-based method for analysis in plasma (Gonzalez-Martin et al., 2002; Hines et al., 2004).
Phenylbutazone appears to be rapidly and completely absorbed from the gastrointestinal tract. Approximately 60 % of a single 400 mg oral dose of radiolabelled phenylbutazone is excreted in urine in 21 days; 27 % of the dose was excreted in faeces. Phenylbutazone binds avidly to serum proteins and is found in negligible quantities in saliva. After administration of a single 400 mg dose to humans, the plasma concentration of unaltered drug is characterised by an early maximum of 36 µg/mℓ at 3 hours and by slow decay between 7 and 336 hours, corresponding to an elimination half-life of 88 hours. Following oral administration of a single 300 mg dose to healthy fasting men, peak plasma phenylbutazone concentrations, averaging 43.3 µg/mℓ, were reached within 2.5 hours. Administration of 300–400 mg phenylbutazone daily to patients with rheumatoid arthritis usually produces steady state plasma levels averaging about 95 µg/mℓ in 3–4 days. The plasma half-life was reported to be shorter in children than adults; in one study, it was about 40 hours in children 1–7 years of age. Plasma half-lives may be somewhat longer in geriatric patients than in younger adults (HSDB, 2002).
Phenylbutazone is extensively metabolised before excretion. Studies involving humans revealed that no detectable parent compound was excreted in the urine. Phenylbutazone is biotransformed by the hepatic microsomal system. Major metabolites include oxyphenbutazone (ring hydroxylation), -hydroxyphenylbutazone (side-chain hydroxylation), -hydroxyoxyphenbutazone (dihydroxy metabolite) and 4-hydroxyphenylbutazone. The formation of the lactone form of -hydroxyphenylbutazone has been shown to be an insignificant reaction in humans. In addition to the primary metabolites, glucuronide and sulphate conjugates of these metabolites have been detected in varying proportions. In humans, conjugates represent about 50% of urinary metabolites. After a single 400 mg oral dose of radiolabelled phenylbutazone, about 40% of the total urinary excretion occurred as the C-glucuronide of phenylbutazone, 12 % as the C-glucuronide of -hydroxyphenylbutazone, 10 % as the sum of unconjugated metabolites and <1 % as unchanged phenylbutazone. The plasma half-life of oxyphenbutazone is similar to that of the parent compound at 50–100 hours. In a multiple dose study in patients with rheumatoid arthritis, plasma levels of total oxyphenbutazone decreased with increasing phenylbutazone dose. This indicated that increased
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
130

chronic doses of phenylbutazone might stimulate elimination of oxyphenbutazone or inhibit oxyphenbutazone formation (HSDB, 2002).
Phenylbutazone and oxyphenbutazone both cross the placenta, and both are distributed into milk (HSDB, 2002).
In mice, administration of radiolabelled phenylbutazone has demonstrated that the highest levels occur in the blood with liver, heart, lungs and kidneys also showing radioactivity; lower amounts are found in the CNS. The plasma half-life of phenylbutazone is shorter in animals than humans, being approximately 6 hours in dogs, 5 hours in guinea-pigs and 3 hours in rabbits (IARC, 1977). Studies in horses and rats indicate that less than 3 % of an administered dose of phenylbutazone is excreted unchanged in the urine. In rats and horses γ-hydroxyphenylbutazone represents a major metabolite (approx. 35 %) and is found in two interchangeable forms, a lactone and a straight-chain. In rats approximately 35–40 % of the metabolites occur as conjugated forms in the urine, including glucuronides and sulphate conjugates. However, glucuronidation does not occur in the horse (HSDB, 2002).
4 Toxicity profile4.1 Acute toxicityIn humans, serious symptoms have developed in adults after ingestions of 4 to 40 g of phenylbutazone. A 2 g ingestion in a 1-year old resulted in death (HSDB, 2002). Severe phenylbutazone overdose is characterised by multi-organ failure, including cardiogenic shock and asystole; sinus tachycardia has also been noted. Fatalities may occur, associated with the appearance of a range of symptoms including gastrointestinal disturbance, convulsions, metabolic acidosis, hepatic necrosis and renal failure; other symptoms of phenylbutazone overdose may include: nausea, omitting, epigastric pain, excessive perspiration, euphoria, psychosis, headache, vertigo and giddiness, nystigmus, insomnia, tinnitus, hearing impairment, oedema from sodium retention, hypertension, cyanosis, respiratory depression, hallucinations, drowsiness and stupor. (HSDB, 2002).
The oral LD50 values of phenylbutazone are 680 mg/kg bw in mice, 1000 mg/kg bw in rats and 146 mg/kg bw in rabbits. The intravenous (i.v.) LD50 values are 120 mg/kg bw in mice and 150 mg/kg bw in rats. The oral LD50 of oxyphenbutazone in dogs is 575 mg/kg bw and its i.v. LD50 in rats is 105 mg/kg bw (IARC, 1977).
In rabbits, a 20 % solution of phenylbutazone was found to be too irritating to administer sub-conjunctivally (HSDB, 2002).
No information on dermal or eye irritancy or sensitization potential has been identified.
4.2 Repeat dose toxicityIn humans, recognised adverse effects of treatment with phenylbutazone included development or exacerbation of peptic ulcer, hepatitis, interstitial nephritis and effects on the bone marrow (leukopenia, agranulocytosis, and rarely aplastic anaemia) and thrombocytopenia; other lesser symptoms include diarrhoea, vertigo, insomnia, euphoria, nervousness and oedema (IARC, 1977; HSDB, 2002; Silva, 2004).
In addition, Type 3 (serum sickness) hypersensitivity has developed following phenylbutazone treatment (IARC, 1977; HSDB, 2002), and it has also been shown to reduce thyroidal iodine uptake (apparently as a secondary effect to the inhibition of microsomal enzyme functions) and may also cause epidermal necrolysis (with ocular involvement; HSDB, 2002). The toxicity profile of the metabolite oxyphenbutazone is similar to that of the parent compound (IARC, 1977).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
131

Limited data are available on the toxicity of phenylbutazone in experimental animals. In a poorly described account of a repeated dose study in dogs, IARC noted that treatment of dogs by intramuscular administration with 3.6 to 35.2 g of phenylbutazone over 10 to 103 days resulted in gastric ulceration (IARC, 1977).
In a 19-day study in which F344/n rats were given phenylbutazone by oral gavage, 4/5 males and 5/5 females given 600 mg/kg/day, 2/5 females given 300 mg/kg/day and 1/5 females given 150 mg/kg/day died during the study period. Impaired growth was also noted in rats given 300 mg/kg/day or above. However in an associated study in B6C3F1 mice using a similar protocol, no effects of treatment were detected (NTP, 1990).
In 13-week repeated dose studies in which F344/n rats were given dosages of up to 300 mg/kg/day, and B6C3F1 mice were given dosages of up to 600 mg/kg/day, deaths were noted during the treatment period in 7/10 male and 8/10 female rats given 300 mg/kg/day, 1/10 male and 2/10 female rats given 200 mg/kg/day, and 5/10 male and 4/10 female mice given 400 mg/kg/day. The adverse responses to treatment in rats included the following: clinical signs, such as diarrhoea, lack of grooming and inactivity; impaired growth at 200 mg/kg/day or above; and dose-related renal changes (including renal papillary necrosis, papillary oedema, and multifocal mineralization). Testicular degeneration was also noted in a few rats at 200 mg/kg/day, and lymphoid depletion was seen in some early decedents given 200 mg/kg/day or above. In mice the effects of treatment were less severe, with only an increase in body weight-relative liver weights being noted for males given 300 or 600 mg/kg/day (NTP, 1990).
The NTP has also conducted 2-year carcinogenicity studies in rats and mice (see Section 4.3 for study details). A reduction in body weight gain was noted in the rats given the high dosage of 100 mg/kg/day. Pathological examination of the kidneys showed a dosage-related increase in nephropathy, papillary necrosis, mineralization and acute inflammation of the proximal convoluted tubules. Stomach ulcers were also noted in rats of either sex given 100 mg/kg/day, while females in this group also showed acanthosis, hyperkeratosis and basal cell hyperplasia of the stomach, adrenal medullary hyperplasia and pulmonary histiocytic cellular inflammation. In contrast, male mice showed dosage-related hepatic pathology, comprising cytomegaly (mainly centrilobular), karyomegaly, hepatocellular degeneration and necrosis, fatty change and peliosis hepatis (NTP, 1990).
4.3 Carcinogenicity and mutagenicitySeveral cases of leukaemia have been reported by IARC in patients treated with phenylbutazone. The more notable cases are described as follows. A male aged 69 years was treated with 600 mg and then 400 mg phenylbutazone daily for 3 weeks, when he developed a mild anaemia, leucocytosis and lymphadenopathy in the neck and axillae. He died 20 months later and post mortem showed myeloid leukaemia. A 63-year old male had received a course of fifty 100 mg phenylbutazone tablets approximately 18 months before admission to hospital for severe angina. Three weeks after admission, his blood picture was suggestive of myeloid leukaemia, confirmed at necropsy 6 days later. A case was reported of acute leukaemia in a 71-year old man who, 3 months earlier, had been treated with twenty 200 mg phenylbutazone tablets over 6 days. A 56-year old female was treated with 300 mg phenylbutazone daily for 17 days. After 2 weeks, she became seriously ill and was found to have an aplastic anaemia that was accompanied by extensive bruising; she died a week later. Autopsy showed massive cerebral haemorrhage and diagnosis was of an acute leukaemia. The onset of a fatal myelomonocytic leukaemia was described in a 44-year old woman treated daily with doses of 100–300 mg phenylbutazone for consecutive periods of 12, 8 and 19 months for rheumatoid arthritis. A total of 8 cases of acute leukaemia (5 men, 3 women) following phenylbutazone therapy, were observed in Australia in the years 1959–1963. The doses ranged from approximately 3–100 g and the duration of treatment lasted from 1 week to 4 years continuously or 7 years intermittently. The first symptoms related to leukaemia were observed from a few weeks to 1 year after cessation of phenylbutazone treatment. Three cases of acute leukaemia following phenylbutazone therapy, out of
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
132

50 cases of acute leukaemia, were recorded in Scandinavia between 1959 and 1964. Acute leukaemia or a leukaemoid reaction has also been observed after treatment with oxyphenbutazone. One case concerned a 47-year old man who, 13 months before admission with a temperature, had been given 1.9 g oxyphenbutazone over a 2-week period. On admission, he was given 0.6 g of the drug; blood examination showed that he had a leukaemia, of which he died approximately 4 months later (IARC, 1977).
A follow-up study was conducted on 25 patients who had developed bone marrow depression following phenylbutazone therapy. Eighteen patients recovered within 2 weeks to 4 years, four were still under care at the time of the report and three died within 2 weeks to 2 years, two of aplastic anaemia and one of a myeloproliferative disorder, probably a leukaemia (IARC, 1977).
When evaluating the data in humans, an IARC Working Group considered that the evidence for carcinogenicity to humans was inadequate. The significance of the cases of leukaemia that had been reported in patients following phenylbutazone therapy could not be evaluated, given the widespread use of phenylbutazone. No significant excess of leukaemia or other malignancy was observed during 1969–1976 among 3660 members of a prepaid health plan prescribed phenylbutazone during 1969–1973. In a case–control study of patients with leukaemia or lymphoma and a subset with myelocytic leukaemia, there was no clear association between the amount or duration of phenylbutazone therapy and risk of leukaemia (IARC, 1987). In conclusion, IARC, while not having any animal data available to them in 1987 (see below), concluded that the evidence for carcinogenicity to humans was inadequate (Group 3).
Long-term studies with phenylbutazone have been conducted in rats and mice under the aegis of the US NTP. Groups of 50 F344/N rats/sex were administered phenylbutazone in corn oil by gavage at 0, 50 or 100 mg/kg bw, 5 days/week for 103 weeks. Mean body weights of high dose rats were generally 6–11% lower than those of vehicle controls. Survival in all groups was similar although it was significantly lower in the low dose males and non-significantly lower in the top dose females compared with the controls. There was equivocal evidence for carcinogenic activity of phenylbutazone in this study as shown by the occurrence of small numbers of renal tubular cell adenomas and carcinomas in treated male rats. There was also some evidence of carcinogenic activity in the females shown by the presence of two rare transitional cell carcinomas in the top dose group. None had ever been seen in untreated females previously. Tubular cell adenomas in the females may also have been associated with the administration of phenylbutazone. The compound was nephrotoxic to rats as shown by the dose-related increase in the severity of age-related nephropathy, necrosis of the renal papilla and mineralisation of the collecting ducts in the papilla. Groups of 50 B6C3F1 mice/sex were administered phenylbutazone in corn oil by gavage at 0, 150 or 300 mg/kg bw, 5 days/week for 103 weeks. Mean body weights were similar among the groups except for the high dose female mice, which weighed 4–11 % less than vehicle controls. There was some evidence of carcinogenicity in treated male mice as shown by the increased incidence of hepatocellular adenomas or carcinomas (combined). There was no evidence of carcinogenicity in the females (HSDB, 2002).
The long-term toxicity and carcinogenic potential of phenylbutazone was also investigated in a dietary study in DONRYU rats. The animals were fed diets containing 0, 1250 or 2500 ppm phenylbutazone for 2 years. The control group contained 100 rats of each sex and the treated groups 50 rats of each sex. Dose-dependent positive trends were seen in the occurrence of leukaemia, neoplastic nodules of the liver and pheochromocytomas of the adrenal glands (HSDB, 2002).
The NTP has subsequently reported on 2-year carcinogenicity studies in F344/n rats and B6C3F1 mice given dosages of up to 100 or 300 mg/kg/day, respectively, 5 days a week, for 103 weeks. In these studies, the occurrence of a small number of renal tubular cell adenoma and carcinoma in male rats was considered to be equivocal evidence of carcinogenicity but in female rats the detection of a small number of very rare transitional cell carcinomas in high dosage animals and the increased incidence of tubular cell adenomas, was considered to provide some evidence of carcinogenicity. For mice, some evidence of carcinogenicity was provided by an increase in the combined incidence of
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
133

hepatocellular adenoma and carcinoma but no evidence of carcinogenicity was noted in female mice (NTP, 1990).
The NTP has also reported on studies of the promotional potential of phenylbutazone in rats pre-exposed to either N-ethyl-N-nitrosourea or N-propyl-N-nitrosourea and subsequently maintained on either basal diet or a diet containing 200 mg/kg phenylbutazone for 104 weeks. A slight potentiation of tumorigenicity was noted in the case of renal and thyroid tumours but not for a number of other tumour types that were induced by either of the nitroso-ureas (NTP, 1990).
Evidence for the mutagenic potential of phenylbutazone is conflicting.
IARC has reported on genotoxic reactions in patients receiving phenylbutazone treatment. A significant increase in chromosome abnormalities was recorded in cultured human peripheral leucocytes from patients treated with phenylbutazone for rheumatic disorders for at least 3 months with doses ranging from 100–500 mg/day. In the same study, a few of the patients showed negative results for chromosome damage in bone marrow cells. Negative results have also been reported for chromosome damage in bone marrow cells from humans receiving 400 mg/day phenylbutazone for 1 week (IARC, 1977).
Phenylbutazone was tested for mutagenicity in the Salmonella/microsome preincubation assay using a standard protocol approved by the US NTP. The compound was tested at doses of 0.033, 0.10, 0.33, 1.0, 3.3 and 10 mg/plate using strains TA1535, TA1537, TA97, TA98 and TA100 in the presence and absence of rat or hamster liver S9. Phenylbutazone was negative in these assays. It was toxic at 3.3 mg/plate in strain TA100 (HSDB, 2002; CCRIS, 2003).
Further in vitro mutagenicity tests were undertaken using a variety of techniques. No growth inhibition due to DNA damage was observed in B. subtilis rec-/rec+, with or without S9. Induction of chromosomal aberrations by phenylbutazone was reported in cultured hamster lung fibroblasts in the absence of S9, in Chinese hamster ovary cells in the presence of S9 and in human lymphocyte cultures, but not in human fibroblast cultures. The frequency of sister chromatid exchanges was not increased in Chinese hamster ovary cells nor in human fibroblasts treated with phenylbutazone (HSDB, 2002). The compound showed a positive result in the mouse lymphoma L5178Y test system using the suspension plate method. The dose range for phenylbutazone (dissolved in acetone) was 200–800 µg/mℓ (CCRIS, 2003). Phenylbutazone did not induce non-disjunction or crossing over in A. nidulans in the absence of metabolic activation; its metabolite, oxyphenbutazone, had a weak but significant activity (IARC, 1977).
Phenylbutazone has been tested in several in vivo assays for clastogenicity. A positive result has been reported for the induction of micronuclei in bone marrow cells of Swiss albino mice given phenylbutazone orally or by intraperitoneal (i.p.) injection. The total doses were 75–200 mg/kg bw, administered in 2 equal portions at 24 hour intervals; mice were killed 6 hours after the second administration. Other studies have shown negative outcomes. A micronucleus test in the bone marrow of male BALB/c mice administered phenylbutazone at 400 mg/kg bw as a single i.p. injection gave a negative result. No induction of chromosomal aberrations was observed in bone marrow cells of Chinese hamsters after oral administration or of rats after i.p. administration of phenylbutazone, nor in germinal cells of male mice after oral administration. Spermatocytes from male mice administered phenylbutazone did not demonstrate an increase in chromosomal aberrations. No induction of dominant lethal mutations was detected in the germ cells of CLFP male mice given i.p. injection of 100 mg/kg bw phenylbutazone, in male BALB/c mice given i.p. injections of 400 mg/kg bw or in the ovum of female mice administered 400 mg/kg bw phenylbutazone orally at the time of estrus (HSDB, 2002).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
134

4.4 Reproductive and developmental toxicityIt is believed that in humans, treatment of nursing mothers may result in severe effects in their offspring, including blood dyscrasias (Drugs.com®, 2004).
No impairment of fertility has been identified in studies on mice, Chinese hamsters and rats at dosages of up to 33 times the maximum human daily dose.
Although showing no teratogenic potential, phenylbutazone has been shown to cause a slight decrease in litter size in rats and rabbits at doses of 16 times the maximum human therapeutic dose (FDA Pregnancy category C4) (Drugs.com®, 2004).
The experimental evidence on the embryotoxicity of phenylbutazone is inconclusive, although it has been reported that inhibitors of prostaglandin synthesis can adversely effect the fetal cardiovascular system, suggesting a potential cause for concern with this compound (HSDB, 2002).
5 Guidelines and standardsNo NOEL was identified that could serve as a basis for an ADI value. The EMEA CVMP has not established MRLs for meat and meat products.
6 ReferencesBishop Y, ed (2004) The Veterinary National Formulary (Sixth Edition), London, UK, Royal Pharmaceutical Society of Great Britain/British Veterinary Association
CCRIS (2003) Phenylbutazone. From: Chemical Carcinogenesis Research Information System, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [January 2004] at http://http://toxnet.nlm.nih.gov
Drugs.com® (2004) Phenylbutazone. From: Drugs.com®: Drugs Information Online - Prescription Drug Information for Consumers and Professionals, available [October 2004] at http://www.drugs.com/MMX/Phenylbutazone.html
EMEA (1999) Position Paper on Availability of Veterinary Medicines Agreed on 17 March 1999 (EMEA/CVMP/151/99-FINAL), London, UK, European Agency for the Evaluation of Medicinal Products, available at http://www.emea.europa.eu/pdfs/vet/press/pp/015199en.pdf
EMEA (2000) Update of the Position Paper on Availability of Veterinary Medicines Agreed on 21 June 2000 (EMEA/CVMP/411/00-FINAL), London, UK, European Agency for the Evaluation of Medicinal Products, available at http://www.emea.europa.eu/pdfs/vet/press/pp/041100en.pdf
FDA (1992) Approved Drug Products with Therapeutic Equivalence Evaluations List, Rockville MD, USA, US Food and Drug Administration, Department of Health and Human Services
FDA (2003) FDA Veterinarian Newsletter, Volume XVIII, No. II, Rockville MD, USA, US Food and Drug Administration, Department of Health and Human Services, available [October 2004] at http://www.fda.gov/cvm/index/fdavet/2003/Mar-Apr03.htm
FVE (2002) Veterinary Medicinal Products: FVE Suggestions Endorsed by European Parliament (FVE/02/127), Brussels, Belgium, Federation of Veterinarians of Europe
Gonzalez-Martin MI, Sanchez-Gonzalez CI, Jimenez-Hernandez A, Garcia-Cachan MD, Castro de Cabo MJ & Garzon-Cuadrado AL (2002) Determination by high-performance liquid chromatography of phenylbutazone in samples of plasma from fighting bulls. J Chromatogr B, 769, 119–126
4 Requires use of labelling phrase ‘There have been no adequate, well-controlled studies in women, but studies using animals have shown a harmful effect on the fetus, or there haven't been any studies in either women or animals. Caution is advised, but the benefits of the medication may outweigh the potential risks’
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
135

Hines S, Pearce C, Bright J & Teale P (2004) Development and validation of a quantitative gas chromatography - mass spectrometry confirmatory method for phenylbutazone in equine plasma. Chromatographia, 59 (Suppl), S109–S114
HSDB (2002) Phenylbutazone. From: Hazardous Substances Databank, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [January 2004] at http://http://toxnet.nlm.nih.gov
IARC (1977) IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Man. Summaries and Evaluations, Vol 13, Phenylbutazone and Oxyphenbutazone, Lyon, France, International Agency for Research on Cancer
IARC (1987) IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Man. Summaries and Evaluations, Vol Suppl 7, Phenylbutazone (Group 3), Lyon, France, International Agency for Research on Cancer
NTP (1990) Toxicology and Carcinogenesis Studies of Phenylbutazone in F344/N Rats and B6C3F1 Mice (Gavage Studies) (NIH Publication No. 90-2822), USA, National Institutes of Health
Silva FG (2004) Chemical-induced nephropathy: A review of the renal tubulointerstitial lesions in humans. Toxic Pathol, 32 (Suppl 2), 71–84
SIS NLM (2004) Phenylbutazone. From: ChemIDplus, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [January 2004] at http://http://chem.sis.nlm.nih.gov/chemidplus/
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
136

Annex 14 Progesterone1 IntroductionProgesterone (pregn-4-ene-3, 20-dione; CAS No. 57-83-0) is the principal progestational hormone of the body, secreted by the corpus luteum, adrenal cortex and placenta. Its chief function is to prepare the uterus for the reception and development of the fertilised ovum (SIS NLM, 2004). Progesterone has been used in human medicine for the treatment of secondary amenorrhoea and dysfunctional uterine bleeding. Progesterone-delivering devices are used for contraception and they are also suitable for post-menopausal hormone therapy (IARC, 1999).
2 Uses and exposureProgesterone is a naturally occurring steroid hormone used therapeutically for disorders of the reproductive system and zootechnically for estrous synchronisation and preparation of animals for embryo transfer. It is licensed in the UK to control estrus and improve synchronisation of estrus in cows, either alone, or in combination with estradiol benzoate. Ezi-Breed CIDRTM contains 1.9 g progesterone, for intravaginal administration for 7–12 days. PridTM, an intravaginal device containing 10 mg estradiol benzoate and 1.55 g progesterone and is normally inserted for 12 days (Bishop, 2004).
Natural progesterone levels in plasma show a large variation, depending on the species, sex, age and physiological status. Physiological plasma concentrations in cattle and horses during estrus are less than 0.2 to 8 ng/mℓ and less than 0.3 to 22 ng/mℓ, respectively, and during pregnancy less than 8 to 12 ng/mℓ and less than 7 to 25 ng/mℓ, respectively. Pharmacokinetic studies with commercial products at the recommended dosages show that after intravenous, intramuscular or intravaginal treatment of cattle, the plasma progesterone concentrations are elevated only within the first few hours after treatment, but never higher than the values observed under physiological conditions (EMEA, 1999).
In cattle, the highest average physiological progesterone concentrations are found in fat (2.5–16.7 g/kg in fattening stock and heifers and 239–360.2 g/kg in pregnant cattle). Lower concentrations are found in liver, kidney and muscle.
In milk and milk products progesterone levels are correlated with the percentage of fat: average physiological levels up to 4.6, 6.5, 72.7 µg/ℓ and 300 g/kg are found in skim milk, buttermilk, cream and butter, respectively. In milk itself physiological progesterone levels relate to the stage of the estrus cycle, with minimal concentrations at estrus (less than 0.2 to 0.92 µg/ℓ) and highest levels during the luteal phase (0.2 to 30 µg/ℓ) and pregnancy (20 to 35.7 g/ℓ). Milk residue experiments with the commercial products at the recommended dosages show that after intravenous, intramuscular or intravaginal treatment of cattle, the milk progesterone levels are highly correlated with the plasma progesterone levels: the milk progesterone concentrations are elevated only in the first few milkings after treatment, but never higher than the values observed under physiological conditions. As the administration of the commercial products containing progesterone failed to induce progesterone blood concentrations exceeding the physiological levels, no specific residue studies in edible tissues other than fat and injection site have been carried out.
Despite being licensed for use in the UK, the hormonal activity of progesterone prohibits its use as a growth promoting agent in food-producing animals.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
137

No residues of progesterone are permitted by US-FDA in excess of the following increments above the concentrations of progesterone naturally present in untreated animals: In uncooked edible tissues of steers and calves: 3 ppb for muscle; 12 ppb for fat; 9 ppb for kidney and 6 ppb for liver; In uncooked edible tissues of lambs: 3 ppb for muscle and 15 ppb for fat, kidney and liver (HSDB, 2003).
2.1 Exposure concentrations in meatUsing information on usage of progesterone and on absorption and excretion, likely ‘worst case’ concentrations in meat products were estimated using Equation 1:
Equation 1M = D × T × Fabs × (1–Fexc)
where M is the predicted concentration in meat (mg/kg); D is the dose of substance (mg/kg bw/d); T is the duration of the treatment (d); Fabs is the fraction of compound absorbed; and Fexc is the fraction of compound excreted.
It was assumed that 1.55 ng of progesterone is given to cattle in the form of an intravaginal sponge to treat estrus. Using an animal weight of 450 kg, this gives a dose rate of 0.003 ng/kg. Assuming 50 % bioavailability and no excretion a ‘worst case’ concentration of 0.0015 mg/kg was obtained. Following intravenous injection of progesterone to cows, 50 % of the dose is recovered in the faeces and only 3 % in the urine. However, it should be noted that progesterone accumulates in fatty tissue, owing to its lipophilic properties, and in tissues/organs containing progesterone receptors.
3 ToxicokineticsAbsorption from the gut is rapid. However, progesterone does not have appreciable activity by the oral route due to extensive first pass metabolism in the liver and gastrointestinal tract, such that the oral bioavailability of exogenously administered progesterone is less than 10 %. Progesterone is available in an oil solution for intramuscular (i.m.) administration but the preparation can cause significant local irritation when given by this route. Dosing via certain fine particle forms has been studied for possible use in contraception or in hormone replacement therapy (JECFA, 2000; HSDB, 2003). Progesterone is bound to serum corticosteroid-binding globulin (CBG) and albumin. Approximately 17 % of serum progesterone is bound to CBG and 80 % to albumin; 2.5 % is in the free form (JECFA, 2000).
Elimination of progesterone after intravenous (i.v.) administration of 500 mg/kg bw to ovariectomised rats was described by a two-compartment model, with distribution and elimination half-lives of 0.13 ± 0.024 and 1.2 ± 0.21 h, respectively, and a volume of distribution of 2.4 ℓ/kg bw. In 5 ovariectomised gilts given 47.4 nmol progesterone, bi-exponential clearance was observed, with mean half-lives of 2.5 and 34 minutes. The half-life of the first compartment indicated that the liver is the principal organ of clearance. Six adult anestrous bitches were given progesterone i.m. at 2 mg/kg bw; a peak serum concentration of the compound of 34 ng/mℓ was reached 1.8 hours after dosing. By 72 hours, the serum concentrations had reached the pre-injection level of 0.9 ng/ml. The mean half-life of absorption was 0.5 hours and the mean half-life of elimination was 12 hours (JECFA, 2000).
The i.v. administration of 3H-progesterone to cynomolgus monkeys resulted in the total disappearance of the hormone from the circulation within 3 hours; 0.5–1.75 hours later, about 5% of the initial maximal concentration of the hormone reappeared, perhaps as a result of delayed release from tissue stores. In female cynomolgus monkeys, progesterone has a volume of distribution of 1.75 ℓ/kg bw and a plasma clearance of 0.06 ℓ/kg bw per min. In comparison with humans, plasma progesterone binding is greater and progesterone clearance is slower in cynomolgus monkeys (IARC, 1999). The
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
138

percutaneous absorption of progesterone was studied in the Mexican hairless dog and compared with that observed in humans. Total absorption and maximal absorption rates were greater in humans than the hairless dog. Progesterone persisted on the dog skin much longer than on human skin (HSDB, 2003).
Seven subjects consumed several oral formulations containing 200 mg progesterone and blood samples were collected for up to 6 hours later. A mean peak serum concentration of 30 ng/mℓ progesterone was achieved with a fine-particle formulation in oil 2 hours after administration; the mean peak concentrations achieved with the other formulations were 9.6 ng/mℓ at 4 hours with plain-milled, 13 ng/mℓ at 3.2 hours with fine-particle, 11 ng/mℓ at 4 hours with plain-milled in oil and 11 ng/mℓ at 4.1 hours with fine-particle enteric coated capsules. The bioavailability of 50–200 mg fine-particle progesterone was studied in normally menstruating women treated by sublingual, oral (capsule and tablet), vaginal or rectal administration during the follicular phase. Increased serum concentrations persisted for over 8 hours in women given >100 mg progesterone vaginally or rectally and for over 24 hours in those given 200 mg tablets orally. After oral intake, absorption is enhanced about 2-fold by the presence of food but the bioavailability appears to be low, the integrated area under the curve of concentration versus time after i.m. injection of progesterone being about 10 times larger than after oral intake (JECFA, 2000).
Progesterone is converted, for subsequent elimination, by hydroxylation to more polar and less active compounds, particularly at C21, C6 and C16. Cytochromes P450 and other enzymes in the hepatic and extrahepatic tissues catalyse the hydroxylation. Tissues and species differ in the metabolism of progesterone. For example, human but not rat kidneys possess 6β-hydroxylase activity. Steroid 21-hydroxylase in liver is provided primarily by CYP2C5 in rabbits and by CYP2C6 in rats, which are P450 isozymes that have not been found in human liver. Progesterone is eliminated via the urine after conversion, in the liver, to pregnane-3 α, 20 α-diol (preganediol) and conjugation to glucuronide at C3. Pregnanediol is the major urinary metabolite of progesterone and serves as an index of progesterone secretion. The metabolic clearance rate for progesterone is approximately 2200 ℓ/day. Progesterone can be metabolised to 17 α-hydroxyprogesterone by P450 17 -hydroxylase and further to androstenedione. Cells that do not express this enzyme cannot metabolise progesterone to androgenic or estrogenic hormones (JECFA, 2000).
Five post-menopausal women received 100 mg progesterone per day orally for 5 days. The peak plasma progesterone concentrations, 7–11 ng/ml, were reached within 4 hours after the last dose and the levels remained raised for 96 hours. The concentration of the metabolite 20 α-dihydroprogesterone was similar to that of progesterone at 3–5.1 ng/mℓ. Other metabolites in the plasma included pregnaediol-3 α-glucuronide (550–1000 ng/mℓ) and 17-hydroxyprogesterone (1.4–3.2 ng/mℓ) (JECFA, 2000). Female chimpanzees, injected with progesterone i.v., produced preganediol as the major urinary metabolite at up to 50 % of the dose. Pregnanolone (2 %) and pregnanestriol (2 %) were also isolated from the urine (HSDB, 2003).
4 Toxicity profile4.1 Acute toxicityNo conventional studies in animals treated with progesterone were noted for acute and repeat dose toxicity and few other studies were available. Because progesterone binds specifically and with high affinity to its receptor, the hormonal effects are the most sensitive toxicological end-points (JECFA, 2000).
After determining the bupivacaine AD50 (the concentration of bupivacaine that caused 50% of all beating rat heart myocyte cultures to become arrhythmic) the effect of 1 hour progesterone hydrogen chloride exposure on myocyte contractile rhythm was determined. Each concentration of progesterone (6.25, 12.5, 25 and 50 µg/mℓ) caused a significant and concentration dependent reduction in the AD50
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
139

for bupivacaine. The progesterone effects on bupivacaine arrhythmogenicity were not potentiated by epinephrine (HSDB, 2003).
4.2 Repeat dose toxicityChanges in endometrial morphology were investigated in 43 patients without ovaries who were given progesterone i.m., intravaginally or orally. The response to i.m. administration in oil was heterogeneous, whereas the endometrial morphology seen after intravaginal administration of fine-particle progesterone closely matched that observed in the natural cycle and pregnancies were supported in 2 women. Orally administered progesterone did not affect endometrial morphology. Two groups of 6 healthy postmenopausal women were given 300 mg of fine-particle progesterone either once or twice a day on days 1–14 after estrogen priming for 30 days. Endometrial biopsy samples were then taken for examination and compared with biopsy samples obtained from the women before therapy. The group given progesterone once a day showed incomplete secretory conversion, whereas the group receiving 2 doses a day showed full secretory conversion with suppression of mitotic activity and the presence of a predecidual reaction. Staining for glycogen was significantly increased in both groups. Ribosomal RNA was decreased by 50 and 73 % in the 2 groups, respectively. The nuclear receptor content decreased in every region of the endometrium in both groups (JECFA, 2000).
Sixty women with oligomenorrhea or amenorrhoea, received a 10 day course of 200 or 300 mg fine-particle progesterone orally. Withdrawal bleeding occurred in 90 % of the women receiving 300 mg progesterone, 58 % of those receiving 200 mg and 29 % of those given a placebo. Their lipid concentrations were unchanged. Side-effects in women receiving fine-particle progesterone orally as part of hormone replacement therapy included minor changes in plasma lipoprotein profile in some patients but no effect on haemostatic parameters such as platelet aggregation and fibronolytic capacity (JECFA, 2000).
Progesterone may have depressant and hypnotic actions in the central nervous system, which may account for reports of drowsiness after hormone administration (HSDB, 2003).
Progesterone pre-treatment (5 mg/kg bw/day for 21 days) caused a significant increase in bupivacaine arrhythmogenicity in intact pentobarbital anaesthetised rats. There was a significant decrease in the time to onset of arrhythmia compared with control non-progesterone treated rats (6.2 ± 1.3 versus 30.8 ± 2.5 min). The results of this study and the one above indicate that progesterone can potentiate bupivacaine arrhythmogenicity in vivo and in vitro (HSDB, 2003).
Progesterone given by subcutaneous (s.c.) injection to female beagle dogs for a total of 74 weeks at an increasing dosage from 0.08 to 22.5 mg/day caused endometrial hyperplasia. No tumours were found in animals killed 24 hours after the last dose but fibro-adenomatous nodules occurred in 2/5 dogs given the highest doses of progesterone (HSDB, 2003).
Female rabbits were treated with large doses of progesterone for up to 763 days and hydroxyprogesterone caproate was given i.m. every other week at an average dose of 13 mg/rabbit to 19 animals. The rabbits developed numerous cysts of the endometrium, sometimes associated with atypical hyperplasia. Active mammary secretion also occurred. Other tissues, including the ovary, adrenal, thyroid and pituitary glands, were not significantly affected. No pre-cancerous endometrial changes or cancers were found (JECFA, 2000).
4.3 Carcinogenicity and mutagenicityThe results from long-term studies with progesterone administered by s.c. or i.m. injections to mice, rabbits or dogs have been extensively reviewed by IARC. Progesterone increased the incidences of ovarian, uterine and mammary tumours in mice and mammary gland tumours in dogs. Neonatal treatment increased the occurrence of pre-cancerous and cancerous lesions of the genital tract and mammary tumorigenesis in female mice. The IARC Working Group considered that the evidence for carcinogenicity of progesterone to animals was ‘sufficient’ (Group 2B; IARC, 1987).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
140

In a review of the role of hormones, including progesterone, in mammary neoplasia in rodents and their relevance to human risk assessment, it was noted that rodent models mimic some but not all of the complex external and endogenous factors involved in initiation, promotion and progression of carcinogenesis. Tumour type and incidence are influenced by the age, reproductive history and endocrine milieu of the host at the time of exposure. In rats, most spontaneously developed neoplasias, with the exception of leukaemia, are of endocrine organs or organs under endocrine control. It was concluded that a mechanism-based approach is not sufficient for human risk assessment and that this approach should be coupled to and validated by traditional long-term bioassays (JECFA, 2000).
No pre-cancerous endometrial changes or cancers were found in female rabbits treated with large doses of progesterone for up to 763 days, with hydroxyprogesterone caproate given i.m. every other week at an average dose of 13 mg/rabbit to 19 animals (JECFA, 2000).
Implanted high doses of progesterone (14 mg in pellets every 28 days over 104 weeks) s.c. in 59 female C3HXA Hybrid mice (with mouse tumour virus) produced breast cancers at a significantly earlier age and higher incidence (88 % at 70 weeks) than in 58 untreated control mice (62 % at 93 weeks). No mammary tumours were seen in 27 progesterone treated intact males. After 18 months treatment with 59–900 µg/day progesterone (s.c. implantation), so-called ovarian granulosa cell tumours were found in 27/83 BALB/C mice, most of them measuring <0.5 mm diameter. One microscopic tumour occurred among 33 control mice killed after 18 months. Following absorption from s.c. pellets of 18–900 µg/day progesterone, uterine sarcomas were observed in 15/142 mice after a period of 18 months; most of these tumours were very small. No tumours were found in 33 controls (HSDB, 2003).
A dose of 2.5 mg progesterone, in peanut oil, given s.c. 5 times/week for 19 weeks, increased the incidence of breast tumours induced by 3-methylcholanthrene (3-MC) in C3H female mice (with MTV) to 23/23, compared with 5/24 for 3-MC alone and 2/25 for progesterone alone. Progesterone given to 27 C3H mice, without the mouse tumour virus, did not induce tumours within 42 weeks. AXC rats were implanted with pellets containing 20 mg progesterone/100 g bw and observed for 40 weeks. There was an increase in the incidence of liver cell carcinomas induced by N-2- fluorenyldiacetamide in intact male rats (11/11 v 7/12), in castrated males (5/13 v 1/9) and in ovariectomised females (4/12 v 0/14) (HSDB, 2003).
The possible influence of progesterone as an inhibitor of tumour formation was assessed in rat studies. Sprague–Dawley female rats, 20/group, were put on diets containing 0 or 2560 mg/kg progesterone for 187 days. The study started 7 days prior to the single i.v. injection of 50 mg/kg bw N-methyl-N-nitrosourea. The incidence of mammary gland tumours due to the carcinogen, 100 %, was not significantly affected (incidence 95%) by the presence of progesterone. In another study with female Wistar rats, 11/group, progesterone was dosed s.c. at 0 or 1 mg/100 g bw 3/week for 150 days after carcinogen treatment. The carcinogen, 7, 12-dimethylbenz(a)anthracene, had been injected into exposed mammary tissue of hyperprolactinemic rats at a single dose of 4 mg in 0.1 ml sesame oil when the rats were 73 days old. The incidence of mammary gland adenocarcinoma due to the carcinogen, 73 %, was significantly reduced (incidence, 45 %) by the action of progesterone (CCRIS, 1999).
A group of 291 female BALB/cfC3H/Crgl mice, bearing the mouse mammary tumour virus, received daily s.c. injections of 100 µg progesterone, 5 µg estradiol and 100 µg progesterone or 20 µg estradiol and 100 µg progesterone for 5 days beginning 36 hours after birth. One half the mice were ovariectomised on day 40. The mice were killed and autopsied at the time of onset of mammary tumours or at 1 year of age. The results for mammary tumour formation are shown in Table 4.1. The administration of progesterone resulted in an earlier age of onset and a higher incidence of mammary tumours; this also occurred for the estrogen + progesterone treatments. In treated mice, ovariectomised on day 40, normal mammary development did not occur and mammary tumours failed to appear, regardless of neonatal treatment. It was concluded that progesterone causes ovary
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
141

dependent, persistent vaginal cornification and hyperplastic changes in the vaginal and cervical epithelium and significantly increases the incidence of mammary tumours in mammary tumour virus bearing mice (JECFA, 2000).
Table 4.1 Incidence of mammary tumours in mice treated neonatallyNeonatal treatment (µg) No. bearing tumours/
No. examined (%) Age at appearance (months)
Estradiol Progesterone
0 100 23/32 (72) 9.35 100 20/32 (63) 9.320 100 33/44 (75) 9.60 0 5/17 (29) 11.3
Progesterone has been the subject of numerous in vitro mutagenicity assays. Results in a number of bacterial assays using S. typhimurium and E. coli test systems were negative; see Table 4.2 (CCRIS, 1999).
Table 4.2 In vitro mutagenicity studies with progesterone, bacterial systemsTest system/strain + or – S9 Test conc.
(µg/plate)Result
S. typhimurium/TA100 -S9 100–10 000 NegativeS. typhimurium/TA100 +S9a 100–10 000 NegativeS. typhimurium/TA1535 -S9 100–10 000 NegativeS. typhimurium/TA1535 +S9a 100–10 000 NegativeS. typhimurium/TA97 -S9 100–10 000 NegativeS. typhimurium/TA97 +S9a 100–10 000 NegativeS. typhimurium/TA98 -S9 100–10 000 NegativeS. typhimurium/TA98 +S9a 100–10 000 NegativeE. coli/WP2 UVRA -S9 0.3–333.3 NegativeE. coli/WP2 UVRA +S9b 0.3–333.3 NegativeaRat or hamster liver S9bRat or mouse or hamster liver S9
Other in vitro assays have been reported but with minimal description of the assay conditions. The results are shown in Table 4.3 (GENE-TOX, 1998; HSDB, 2003).
The mutagenic potential of progesterone was also evaluated by in vivo studies. It was administered by gavage to female rats as a single dose of 100 mg/kg bw 3 days before a two-thirds hepatectomy and the rats were killed for cell sampling 2 days after the hepatectomy. The frequency of micronucleated hepatocytes was 3.5-fold higher in progesterone treated rats than in control animals. No increase in the frequency of binucleated hepatocytes was observed. A dose of 100 mg/kg bw progesterone administered by gavage weekly for 6 weeks, followed by partial hepatectomy, did not increase the number of -glutamyltranspeptidase positive foci in liver. Progesterone did not induce dominant lethal mutations in mice or chromosomal aberrations in rat bone marrow in vivo. In 3 male and 3 female Han:Wistar rats given progesterone at 100 mg/kg bw/day intragastric (i.g.) as an aqueous microcrystalline suspension for 14 days, no progesterone specific adducts were observed in the liver, as determined by 32P-postlabelling (JECFA, 2000).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
142

Table 4.3 Other in vitro mutagenicity assays with progesteroneAssay Cell type Result
Cell transformationa Mouse, BALB/c-3T3 NegativeCell transformationa (clonal) Syrian hamster embryo NegativeCell transformationb Syrian hamster embryo PositiveCell transformationa (viral enhanced) Rat embryo, RLV/1706 PositiveCell transformationb (viral enhanced) Baby rat kidney PositiveDNA repaira E. coli polA, W3119 v P3478 InconclusiveDNA repairb Rat liver NegativeSperm morphologya Human (male) PositiveSperm morphologyb Mouse (male) NegativeaGENE-TOX (1998)2HSDB (2003)
An IARC working group, reporting in 1987, summarised the available information as follows: ‘Progesterone did not induce dominant lethal mutations in mice or chromosomal aberrations in rats treated in vivo. It did not induce chromosomal aberrations or SCEs in cultured human cells, nor chromosomal aberrations or DNA strand breaks in rodent cells. Studies on transformation of rodent cells, in vitro were inconclusive. Progesterone was not mutagenic in bacteria’. At a later date, a WHO Committee concluded that, on balance, progesterone has no genotoxic potential (JECFA, 2000).
4.4 Reproductive and developmental toxicityThe pregnancies of 42 women, treated with progesterone suppositories (average total dose of 2236 mg), resulted in 28% spontaneous abortions but no malformations. Another 45 pregnancies, treated with progesterone i.m. (average total dose 1009 mg), resulted in 5.8 % spontaneous abortions and 2 malformations (a unilateral undescended testis and a meningomyelocele; HSDB, 2003).
Six pregnant rhesus monkeys were treated with progesterone in oil i.m. at a dose of 50 mg/day for 5 days each week. Treatment was begun on days 24–28 of gestation and was maintained until delivery. The offspring of the animals treated with progesterone (3 males and 3 females) were delivered at about term and no abnormalities were noted. It was concluded that progesterone, in comparatively large doses, produces no abnormalities and does not interfere with the normal conclusion of pregnancy in rhesus monkeys. Progesterone at doses of 5–200 mg was administered daily by s.c. injection to 22 pregnant Long–Evans rats on days 15–20 of gestation. The pups were surgically removed from the dams on day 22 and killed after 20 days. No difference in the anogenital distance was observed between treated and control rats and sex was easily determined (JECFA, 2000).
In 1977 the US-FDA restricted the use of progesterone in pregnancy based upon reports of cardiac malformations, CNS defects, masculinisation of female fetuses and limb defects. However, re-evaluation of these studies failed to show an association between these defects and progesterone (HSDB, 2003). A WHO Committee noted that no multigeneration study for reproductive toxicity on progesterone was available. It noted that developmental toxicity was not seen in studies in rats and rhesus monkeys. Exogenous progesterone has been used to maintain pregnancy, with no evidence of toxicity and with no effect on the normal outcome of pregnancy (JECFA, 2000).
5 Guidelines and standardsIn 2000, JECFA noted that the oral administration of a single dose of 200 mg of fine-particle progesterone (equivalent to 3.3 mg/kg bw) to women provided concentrations in blood similar to those found during the luteal phase of the ovulatory cycle. This dose was considered to be the lowest observed effect level (LOEL) in humans. The Committee established an ADI of 0–30 µg/kg bw for progesterone on the basis of the LOEL of 3.3 mg/kg bw. A safety factor of 100 was used to allow for
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
143

extrapolation from a LOEL to a NOEL, and to account for normal variations among individuals (JECFA, 2000).
6 ReferencesBishop Y, ed (2004) The Veterinary National Formulary (Sixth Edition), London, UK, Royal Pharmaceutical Society of Great Britain/British Veterinary Association
CCRIS (1999) Progesterone. From: Chemical Carcinogenesis Research Information System, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [January 2004] at http://http://toxnet.nlm.nih.gov
EMEA (1999). Progesterone: Summary Report (EMEA/MRL/146/96-FINAL), London, UK, European Agency for the Evaluation of Medicinal Products, available at http://www.emea.eu.int/pdfs/vet/mrls/014696en.pdf
GENE-TOX (1998) Progesterone. From: Genetic Toxicology (Mutagenicity) Databank, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [January 2004] at http://http://toxnet.nlm.nih.gov
HSDB (2003) Progesterone. From: Hazardous Substances Databank, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [January 2004] at http://http://toxnet.nlm.nih.gov
IARC (1987) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Supplement 7, Overall Evaluations of Carcinogenicity: An Updating of IARC Monographs Volumes 1 to 42, Oestrogens, progestins and combinations, Lyon, France, International Agency for Research on Cancer
IARC (1999) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol 72, Hormonal Contraception and Post-Menopausal Hormonal Therapy, Lyon, France, International Agency for Research on Cancer
JECFA (2000) Toxicological Evaluation of Certain Veterinary Drug Residues in Food (Food Additives Series 43), Geneva, Switzerland, World Health Organization, Available [January 2004] at http://www.inchem.org/documents/jecfa/jecmono/v43je05.htm
SIS NLM (2004) Progesterone. From: ChemIDplus, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [January 2004] at http://http://chem.sis.nlm.nih.gov/chemidplus/
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
144

Annex 15 Salbutamol 1 IntroductionSalbutamol, Albuterol (α1-(((1,1-dimethylethyl)amino)methyl)-4-hydroxy-1,3-benzene-dimethanol; CAS No. 18559-94-9) is a non-catecholamine β2-adrenoreceptor stimulant with minimal β1 activity. It is a relatively selective β2-adrenoreceptor stimulant. Salbutamol has rapid, potent and long-lasting bronchodilator activity and only minor inotropic or chronotropic effects. Its main clinical use is for relief of asthma in adults and children. It is administered either by inhalation or orally (Libretto, 1994).
Salbutamol (Albuterol) exists in the (R)- and (S)- isomer forms, with the (R)-isomer exhibiting a 100-fold greater affinity for the β-adrenergic receptor than the (S)-isomer. Racemic salbutamol is the entity used for therapy (Handley, 1999).
2 Uses and exposureThere are no products containing salbutamol licensed for use in either domestic livestock, or companion animals in the UK. Instances of salbutamol contamination of a small number of chicken, turkey and cattle samples was reported in 2002 in the UK, but further investigation showed this to have arisen from the use of medication containing salbutamol by the collecting officer prior to collection of samples, rathen than to illegal use in animals (VRC, 2002)
Several human preparations containing salbutamol are licensed in the UK as aerosolised formulations for the relief of asthma in adults and children (SalbutamolTM, VentolinTM). Under the Medicines (restrictions on the Administration of Veterinary Medicinal Products) Regulations 1994, where no authorised veterinary medicine exists for a condition in a particular species, a veterinarian may prescribe for an individual animal, or a small number of animals kept on the same premises, an authorised human medicine under the ‘cascade’ system. Salbutamol is used in veterinary medicine under the cascade system as a ‘rescue therapy’ for horses with respiratory distress due to allergic respiratory disease. Administration is by inhalation using proprietary human applicators, as an alternative to oral or intravenous clenbuterol (VentipulminTM), also a β2-adrenoceptor stimulant, licensed as a veterinary medicine for use in horses (Bishop, 2004).
β2 Agonists have anabolic properties, and salbutamol taken at high doses, is potentially open to abuse as a performance enhancer and growth promoter particularly in beef animals, to produce leaner carcases. All β2 agonists are prohibited in the EU for use as growth-enhancing agents. As clenbuterol is the only licensed β2 agonist in veterinary medicine, illicit use of this drug, rather than salbutamol, has occurred in some European countries, resulting in detection of residues in meat products such as liver.
The likelihood of consumer exposure resulting from illegal use in fattening animals is unquantifiable, although appears to be decreasing due to monitoring and enforcement of EU legislation.
2.1 Exposure concentrations in meatThere is no information on pharmacokinetics or excretion of salbutamol in food animals. In Canada, detection limits have been set at 300 mg/kg.
Experimentally derived data are available on concentrations of salbutamol in meat products. Poultry were treated with salbutamol at a dose of 150 µg/kg bw/d for 10 days. Following treatment, the
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
145

animals were slaughtered and the meat analysed. Concentrations of approximately 13 µg/kg were detected in the meat.
No measured data were available for salbutamol in cattle. Information on usage of salbutamol and on absorption and excretion was therefore used with Equation 1 to estimate likely ‘worst case’ concentrations in pig meat:
Equation 1M = D × T × Fabs × (1–Fexc)
where M is the predicted concentration in meat (mg/kg); D is the dose of substance (mg/kg bw/d); T is the duration of the treatment (d); Fabs is the fraction of compound absorbed; and Fexc is the fraction of compound excreted.
Assuming that salbutamol is given in feed at 1 g/tonne (corresponding to a dose of 0.05 mg/kg/d) for 175 days and that all of this is absorbed (i.e. Fabs = 1.0) and 80% of the drug excreted (i.e. Fexc = 0.8) a ‘worst case’ concentration of 1.76 mg/kg was obtained.
3 ToxicokineticsIsomeric assays developed to quantitate (R)- and (S)- salbutamol show that after oral or inhalation administration of racemic salbutamol to man, the area under the curve for plasma levels of (S)-salbutamol is 6-fold that of (R)-salbutamol. It would have been expected that the plasma concentrations of both isomers would be equivalent. However, many tissues such as human lung, liver, platelets and other cells have the preferred capacity to sulphate (R)-salbutamol. Also, (S)-salbutamol, the dominant plasma isomer, is poorly metabolised compared with (R)-salbutamol. Since (S)-salbutamol does not exhibit bronchodilation, this has led to difficulties in correlating plasma levels with the onset and duration of racemic salbutamol drug action (Handley, 1999).
HSDB (2005) states that 69–90 % is excreted renally (60 % as metabolite) and 4 % faecally; elimination half-life is 3.8–6 hours (species unspecified; presumably human data). EMEA report 81 % excretion in urine for clenbuterol, a similar β-agonist, in cows (EMEA, 2000).
4 Toxicity profile4.1 Acute toxicitySalbutamol has been known to induce asthma-like side effects in patients, seen as paradoxic bronchospasm. It exhibits moderate to severe paradoxic bronchospasm in about 15.4% of patients. Many cases of paradoxical bronchospasm to salbutamol are life threatening and have been determined to be independent of route of administration or formulation. Also, patients with severe asthma who are treated with salbutamol may not exhibit any benefit, although inflammation may be involved (Handley, 1999).
Acute oral, intravenous (i.v.) and intraperitoneal (i.p.) studies have been carried out with salbutamol on rats and mice. The LD50 results obtained from these studies are shown in Table 4.1 (Libretto, 1994).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
146

Table 4.1 Acute LD50 values for salbutamolSpecies/route Sex LD50 (mg/kg bw)Mouse; i.v. M 70.5Mouse; i.v. F 75.3Rat; i.v. M 61.5Rat; i.v. F 59Young rat; oral M, F >1000Mice/rats; oral - >2000Fed 8-week old rats; oral M 8600Fasted 8-week old rats; oral M 2900Fed 8-week old rats; oral F 7600Fasted 8-week old rats; oral F 410010 day old rats; i.p. M 21320 day old rats; i.p. M 37210 day old rats; i.p. F 16920 day old rats; i.p. F 415
Eight days after administration, there was no effect on the mean body weights of 10- and 20-day old Sprague–Dawley rats given single 1000 mg/kg bw oral doses of salbutamol. Some statistically significant differences were found in mean organ weights of treated animals compared with controls including increases of up to 9.5% in mean heart weights in males.
In several toxicology models, the lethal doses for rats by i.v. or oral administration of (S)- and (R)-salbutamol were nearly equivalent. Thus, (S)-salbutamol, which does not bind to the β-adrenergic receptor nor contribute to efficacy, caused acute lethality at doses similar to the racemic drug. In other studies in mice, (S)-salbutamol had i.v. toxicity comparable to racemic salbutamol (Handley, 1999).
4.2 Repeat dose toxicityNo signs of toxicity were observed in rats given daily oral doses of 50 mg salbutamol/kg bw for 4 months or in rats which inhaled about 3.3 mg/kg bw over a period of 3 hours every day for 6 weeks. In a 1 month oral toxicity study, 2 female Sprague–Dawley rats died after receiving 150 mg salbutamol/kg bw/day for 3–4 weeks. However, no adverse changes in ECGs, haematology or clinical chemistry were noted in any animals throughout the study. There was no toxicity to dogs following administration of 0.15 mg salbutamol/kg bw/day by aerosol for 12 weeks. However, dogs that had received oral doses ranging between 0.02 and 25 mg salbutamol/kg bw/day for 4 months showed dose-related tachycardia that was most marked 1 hour after administration. Flushing of the skin was also observed in high dose animals (Libretto, 1994).
Rabbits were given 150 mg salbutamol/kg bw/day orally or 3 mg/kg bw/day subcutaneously (s.c.) for 9 days. No histological changes were seen in the hearts of these animals at doses greater than those used clinically. Salbutamol also had no cardiotoxic effects on dogs given forty 100 µg doses by metered inhalers (Libretto, 1994).
Heart weights increased significantly, by up to 27 %, in rats dosed orally for 1 month with 17, 50 or 150 mg salbutamol/kg bw/day. There were no associated changes in ECGs. Congestion, interstitial oedema, hypertrophy of muscle fibres, focal myocardial necrosis and fibrosis were seen at histopathological examination. These changes were not evident 1 month after cessation of treatment. Significant increases in lung weight were also found. Similarly, increases in heart weight were also found when Sprague–Dawley rats were given s.c. doses of salbutamol at 50 mg/kg bw/day for 3 days. Slight myocardial necrosis, characterised by regression and inflammatory changes, occurred with doses from 0.5–50 mg/kg bw/day; doses of 0.1 mg/kg bw/day did not result in myocardial necrosis. The increase in heart weight was a result of the oedema associated with the necrosis (Libretto, 1994).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
147

Salbutamol was investigated in a number of studies by a variety of routes in a range of species. Overall, the findings were unexceptional apart from four findings in the rat when the test substance was given orally. These were growth of the salivary gland, enlargement of the Harderian gland, and an increase in colloid in the pituitary and mesovarian leiomyomas. The leiomyomas were produced after prolonged administration and are discussed further in Section 4.3 (Libretto, 1994).
4.3 Carcinogenicity and mutagenicityAdministration of salbutamol sulphate orally to Long–Evans or Sprague–Dawley rats at 20 mg/kg bw/day for 96 weeks resulted in mesovarian leiomyomas in about 25 % of the Sprague–Dawley rats and 5–10 % of the Long–Evans rats. Mesovarian leiomyomas were also found in long-term studies in which Charles River CD rats were treated with doses of up to 50 mg/kg bw/day, the maximum tolerated dose. Salbutamol did not induce mesovarian leiomyomas in mice, hamsters, dogs or non-human primates. Also, the mesovarian leiomyomas appear to be only fully expressed in one strain of rats, the Sprague–Dawley. No such tumours were found in the CFE strain and, in the Long–Evans rats, they occurred at a low frequency (Libretto, 1994).
Salbutamol was evaluated in an oral toxicity 2-year study in rats to test the hypothesis that mesovarian leiomyomas were an expression of excessive pharmacological activity. Mesovarian leiomyomas or smooth muscle hyperplasia occurred in the rats and the incidence of the leiomyomas was dose related. Furthermore, the induction of the mesovarian leiomyomas was completely prevented by the concurrent administration with salbutamol of propanol, a specific β-receptor antagonist. This confirmed that the formation of these tumours was a consequence of β-adrenergic stimulation (Libretto, 1994).
Mesovarian smooth muscle cells have β2-adrenoreceptors, which mediate relaxation. The proliferation of mesovarian muscle caused by β-stimulants in the rat may be an adaptive physiological response to continuous relaxation of the muscle. Any such stimulant at a sufficient dosage will therefore induce leiomyomas. The consensus view of leiomyomas induced in rats by salbutamol is that the tumours are benign with no sign of malignancy. The relevance of the findings to humans is not known because rat ovaries are anatomically very different from those of women. Leiomyoma of the ovary or mesovarium in women is rare. Any substantial increase in their incidence that resulted from the use of β-stimulant drugs would therefore be obvious. Such lesions have not been reported in association with the use of salbutamol in bronchial asthma for over 20 years (Libretto, 1994).
When tested at concentrations up to one million times its effective concentration on β2-adrenoreceptors, salbutamol was not mutagenic in in vitro tests involving four different microorganisms with and without metabolic activation with liver microsomes. No mutagenic activity was found for salbutamol using Ames reversion mutation and repair tests, Kada’s reversion test and recombination assay, nor with Zimmerman’s reversion and mitotic gene conversion tests (Libretto, 1994).
4.4 Reproductive and developmental toxicityDaily doses of up to 100 mg salbutamol/kg bw to pregnant rabbits and up to 50 mg/kg bw to pregnant rats, did not induce any abnormalities in fetuses. The possible effects of salbutamol on fetal development were studied in mice, rats and rabbits. Doses ranging from small multiples to several thousand times the human therapeutic dose were administered by various routes during early stages of differentiation and advanced organogenesis. Fetal malformations, mainly in the form of cleft palate, were produced in the mouse particularly when the drug was administered s.c. at high levels. The total incidence of malformations was 1 % in controls and 10 % in salbutamol-treated animals. The occurrence of cleft palate varied between trials and, within some trials, was statistically comparable with control incidences. Subcutaneous doses of salbutamol to pregnant rats and rabbits did not cause an increase in cleft palate. However, doses amounting to 20 000–40 000 times that prescribed in therapy resulted in other types of unspecified malformations in rabbits (Libretto, 1994).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
148

5 Guidelines and standardGiven that there are no licensed veterinary medicines, no MRLs have been established for food-producing species.
In 1992, the US-FDA guidelines recognised racemic drugs as combination drugs and required both isomers to be evaluated experimentally, clinically and toxicologically. This approach to guidelines that encourage single isomer drugs above racemic mixtures has also been included in similar guidelines in Europe, Canada and Japan (Handley, 1999). The impact of these guidelines on salbutamol could not be ascertained, nor was any other information found on the subject of guidelines and standards regarding salbutamol.
6 ReferencesBishop Y, ed (2004) The Veterinary National Formulary (Sixth Edition), London, UK, Royal Pharmaceutical Society of Great Britain/British Veterinary Association
EMEA (2000) Clenbuterol. Summary Report (2) (EMEA/MRL/723/99-Final), London, UK, European Agency for the Evaluation of Medicinal Products, Available [May 2005] at http://www.emea.europa.eu/pdfs/vet/mrls/072399en.pdf
Handley D (1999) The asthma-like pharmacology of (S)-isomers of agonists. J Allergy Clin Immunol, 104, S69–S76
HSDB (2005) Albuterol. From: Hazardous Substances Databank, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [June 2005] at http://http://toxnet.nlm.nih.gov
Libretto SE (1994) A review of the toxicology of a salbutamol (albuterol). Arch Toxicol, 68, 213–216
VRC (2002) Annual Report on Surveillance for Veterinary Residues in Food in the UK, 2002, Addlestone, UK, The Veterinary Residues Committee
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
149

Annex 16 Streptomycin1 IntroductionStreptomycin (0-2-deoxy-(methylamino)-alpha-L-glucopyranosyl-(1-2)-O-5-deoxy-3C-formyl-alpha-L-lyxofuranosyl-(1-4)-N,N-bis(aminoiminomethyl)-D-streptamine; CAS No. 57-92-1) is an aminoglycoside antibiotic used in veterinary medicine to treat bacterial diseases in cattle, pigs, sheep and poultry. Streptomycin was the first clinically effective drug to become available for the treatment of tuberculosis in humans (HSDB, 2002).
2 Uses and exposureStreptomycin (and dihydrostreptomycin) belongs to the aminoglycoside group of antibiotics, which are bactericidal and active against a wide range of Gram-negative and some Gram-positive bacterial organisms. The chemical structure and biological activity of streptomycin and dihydrostreptomycin are similar and safety evaluations for the two substances are usually carried out together. The oral route is required for the treatment of gastrointestinal infections and the treatment of systemic infections requires that the drug be administered by injection.
The recommended therapeutic doses of streptomycin for the treatment of bacterial diseases in cattle, pigs, sheep and poultry range from 5–10 mg/kg bw/day for 3–5days by the parenteral route or from 25–100 mg/kg bw/day for 3–5 days via drinking water (EMEA, 2002). Streptomycin was the first clinically effective drug to become available for the treatment of tuberculosis in humans.
Streptomycin is only available as injectable formulations (Devomycin or Devomycin-DTM) in the UK for use against streptomycin-sensitive systemic infections in horses, cattle, sheep, goats, dogs and cats. Streptomycin, available as the sulphate salt, is administered either on its own (1 mℓ streptomycin sulphate/25 kg bw at 250mg/mℓ w/v) or in combination with dihydrostreptomycin sulphate (1 mℓ streptomycin sulphate and dihydrostreptomycin sulphate/30 kg b. at 150 mg/mℓ w/v for each active ingredient) equivalent to 10 mg active ingredients/kg bw, by intramuscular injection (i.m.) to horses, cattle, sheep and goats. In dogs and cats, the dose rates are increased to 25 mg/kg bw (1 mℓ/10 kg bw streptomycin sulphate at 250 mg/mℓ w/v and 1 mℓ/12 kg bw streptomycin sulphate and dihydrostreptomycin sulphate both at 150 mg/mℓ w/v).
Dihydrostreptomycin is also formulated in combination with procaine benzylpenicillin to provide broad-spectrum anthelmintic activity against Gram-positive and Gram-negative bacterial organisms and may be synergistic. These preparations are available for use in cattle, sheep, pigs and horses in the UK by i.m. injection (Depomycin ForteTM, Duphapen + StrepTM, Pen & StrepTM, StreptacareTM, StreptopenTM) and by subcutaneous (s.c.) injection in dogs and cats ((Depomycin ForteTM). All preparations contain 250 mg/mℓ dihydrostreptomycin sulphate and 200 mg/mℓ procaine benzylpenicillin. The recommended dose rates are 0.04–0.05 mℓ/kg (10 mℓ/kg of each active ingredient) in horses, cattle, sheep and pigs and 0.1 mℓ/kg in dogs and cats. Withdrawal periods are between 18–28 days for meat in cattle, 18–35 days in sheep, and 18–28 days in pigs. Products should not be used in horses intended for human consumption. Milk withdrawal periods are 2.5 days in cattle, and should not be used in sheep that produce milk for human consumption.
Dihydrostreptomycin is also used in combination with procaine penicillin (Streptopen Milking CowTM) framycetin sulphate, penethamate hydroiodide and prednisolone (Leo Yellow Milking CowTM), or neomycin sulphate, novobiocin sodium, prednisolone, procaine benzylpenicillin (Tetra-DeltaTM) as intramammary pastes for the treatment of mastitis in lactating cattle, by infusion into the teat canal. Treatment is one dose per infected ‘quarter’ (mammary gland); the dose of
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
150

dihydrostreptomycin sulphate varies between 100–500 mg according to the product used and may, under exceptional circumstances, increase to 4-fold these levels if all four ‘quarters’ are infected. Combination products for mastitis control containing dihydrostreptomycin sulphate are also used in dry (non-lactating) cow therapy (Nafpenzal DCTM, Streptopen Dry CowTM). The dose is 400–2000 mg per animal, given as one infusion by ‘quarter’ at drying off.
Potential sources of human exposure are ingestion of animal products containing drug residues and exposure to animal wastes containing the compound. The formulations do not predispose to operator exposure although certain compound preparations containing penicillins may cause sensitisation following accidental injection, inhalation, ingestion or skin contact.
2.1 Exposure concentrations in meatExperimentally derived data are available on concentrations of streptomycin in meat products. Pigs were treated with streptomycin at 5 mg/kg/day following typical treatment regimes. Samples of meat were then taken either within 1 hour of dosing or within 24 hours and analysed. The concentration 1 hour after dosing was 53.7 mg/kg and this had dropped to 0.18–2 mg/kg 24 hours after dosing.
Streptomycin was detected in imported honey during 2002 (VRC, 2003); 13/200 samples that exceeded the MRL/‘action level’ contained concentrations of 23–180 µg/kg.
3 ToxicokineticsAn analytical method based on HPLC with fluorescence detection is available for the determination of streptomycin residues in animal tissues and in bovine and ovine milk. The limits of quantification (LOQ) were 200 µg/kg for edible tissue and 50 µg/kg for milk (EMEA, 2002). In animals and humans, streptomycin is poorly absorbed from the gastrointestinal tract and the majority of an oral dose is recovered in faeces. After parenteral administration of the drug to farm animals, the majority of the dose is excreted unchanged in the urine. Since only a very small proportion of potential residues in farm animals is likely to be in metabolite form, the parent compound was identified as the marker residue (EMEA, 2002).
Residue data in tissues have been obtained from cattle, sheep or pigs treated with streptomycin. Twelve cattle were treated with streptomycin sulphate by deep i.m. injection at a dose of 10 mg/kg bw/day for 3 consecutive days. Animals were killed at 3, 5 or 7 days after the final dose. Residues of streptomycin, below the LOQ (250 µg/kg), were recorded in muscle, liver and fat, 7 days after the final dose. Levels in kidney ranged from 978–2800 µg/kg. Two days after the last of repeated i.m. injections of streptomycin in sheep at 10 mg/kg bw/day for 3 days, the concentrations of residues in muscle and fat were below the LOQ (200 µg/kg). In liver and kidney the mean streptomycin concentrations were 938 and 886 µg/kg, respectively. A group of four pigs received a combination of streptomycin and dihydrostreptomycin sulphate (10 mg/kg bw of each), by the i.m. route, once daily for 3 days. Two days after the last injection, the concentrations of residues in muscle and fat were below the LOQ. In liver and kidney the mean residue values were 472 and 1756 µg/kg respectively (EMEA, 2002).
Residue data were also obtained for bovine and ovine milk. Eight lactating cows were treated by i.m. injection with a combination of streptomycin sulphate and dihydrostreptomycin sulphate at a dose of 10 mg/kg bw /day for 3 consecutive days. Milk samples were taken at 12, 24, 36, 48, 60, 72, 84 and 96 hours after the final dose. After 12 hours, all milk samples had detectable levels of streptomycin ranging from 173–265 µg/kg. At 36 hours, three samples had streptomycin levels <LOQ (50 µg/kg) and five samples contained levels just above the LOQ at 51.6–61.7μg/kg. At the subsequent sampling times, the levels of streptomycin were below the LOQ of 50 µg/kg. In a similar study, eight lactating sheep were treated by i.m. injection with a combination of streptomycin sulphate and dihydrostreptomycin sulphate at a dose of 10 mg/kg bw/ day for 3 consecutive days. Twelve hours after the final dose the mean streptomycin concentration in milk was 241.6 µg/kg. By 48 hours, the
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
151
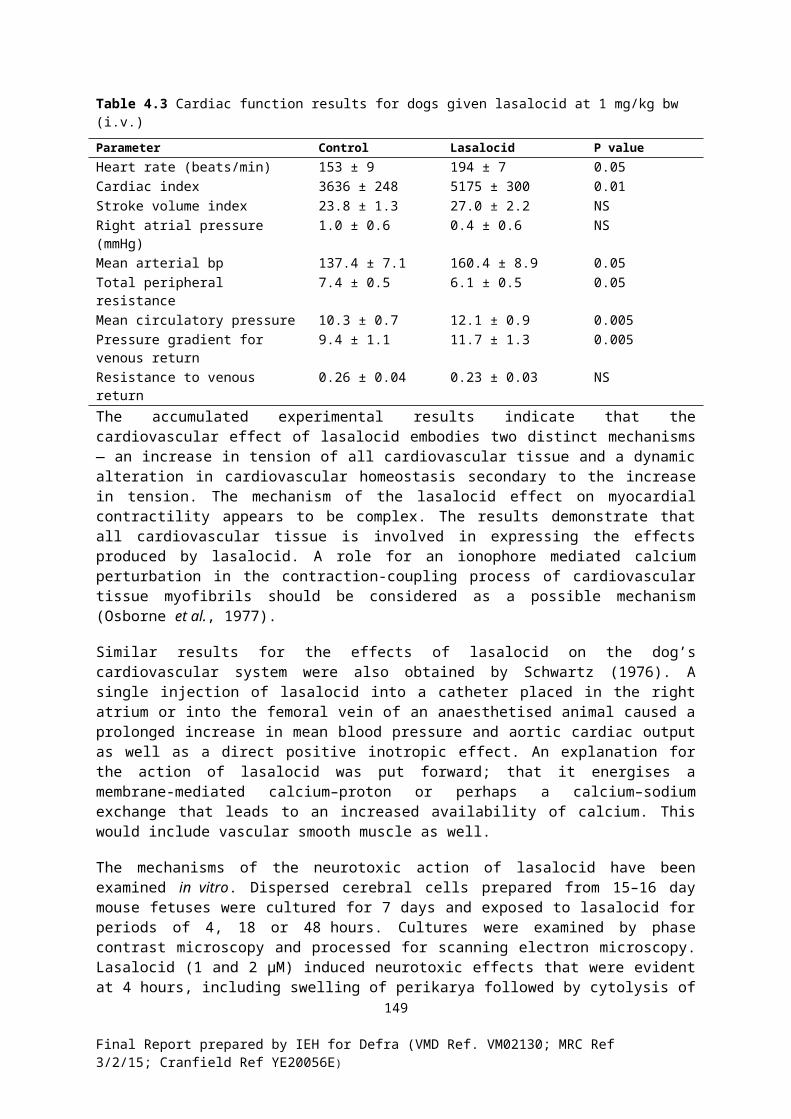
mean concentration had decreased to 72.4 µg/kg and from then on the streptomycin concentrations in subsequent milk samples were less than the LOQ of 50 µg/kg (EMEA, 2002).
The elimination of streptomycin from rodents, rabbits, dogs and monkeys was assessed in a series of studies reported in 1945 and 1948. After s.c. injection of 5000–50 000 units of streptomycin/kg bw in mice, the peak blood concentration of 6.5 units/mℓ was reached in approximately 15 minutes. After oral doses of 200 000 units/kg bw, the peak blood level of 2 units/mℓ was reached at 45–60 minutes. After administration of a single intravenous (i.v.) dose of 5000 units/kg bw to rabbits, 5–10% of the dose was recovered in bile over 8 hours after injection. After oral administration of 420 000 units streptomycin to dogs, no drug was detected in plasma, but up to 3.9% of the dose was recovered from urine. After oral administration to dogs of 100 000–200 000 units/kg bw streptomycin, no drug was detected in bile and 5–10% was recovered in urine. Twenty-four hours after the administration, 60–80% of the drug was recovered unabsorbed from the gastrointestinal tract. The volume of distribution in dogs after injection of 20 mg/kg bw streptomycin was 23–36% of bodyweight, corresponding to extracellular fluid volume. Renal clearance appeared to be by glomerular filtration alone and was 34–59 mℓ plasma/minute. After i.m. injection of 10 000 or 50 000 units streptomycin/kg bw to groups of 2 monkeys, 61–69% and 38–42% of the dose was excreted in urine, respectively, after 1 day (JECFA, 1995).
The pharmacokinetic parameters in humans following i.m. adminstration are summarised in Table 3.1 (JECFA, 1995).
Table 3.1 Pharmacokinetic parameters for streptomycin in humansParameter Data
Adult daily therapeutic dose 15–25 mg/kg bw/dayRoute of administration IntramuscularNormal dosage interval 12 hoursPeak serum level 25–30 µg/mℓHalf-life
Normal 2.5 hoursAneuric 50–110 hours
Elderly 9 hoursPremature/newborn infants 7 hoursVolume of distribution
Well nourished patients 95.9 ± 19.5 litresMalnurourished patients 66.3 ± 7.4 litres
Plasma protein binding 35%
After oral administration of streptomycin to humans, 60–100 % of the drug was recovered unchanged in faeces. Approximately 20 % of a parenteral dose of streptomycin to humans could not be accounted for in urine. No metabolites were identified. Approximately 1 % was excreted in bile. The aminoglycosides are reported not to be metabolised in humans and are excreted in their active forms by glomerular filtration. Streptomycin is poorly absorbed by inhalation (JECFA, 1995).
4 Toxicity Profile4.1 Acute toxicityIn humans, hypersensitivity reactions may occur in response to streptomycin treatment. Skin reactions are reported to occur in 5 % of patients. Severe exfoliative dermatitis and anaphylaxis have occurred. Sensitisation is common among those handling streptomycin occupationally.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
152

In mice, the oral LD50 values ranged from 15 000 to 30 000 mg/kg bw, the s.c. LD50 was 500–600 mg/kg bw, the i.v. LD50 was 85–111mg/kg bw and the intraperitoneal (i.p.) LD50 was 610–575 mg /kg bw. Clinical signs of toxicity included restlessness, respiratory depression, loss of balance, unconciousness, motor paralysis and coma, following all routes of administration. At autopsy, the only anatomical changes were haemorrhagic lesions in the gastrointestinal tract of mice dosed orally (JECFA, 1995).
The oral LD50 of streptomycin sulphate in hamsters was 400 mg/kg bw. The LD50 after s.c. administration was >500 mg/kg bw. Some animals died up to 2 weeks after dosing. Clinical signs of toxicity included listlessness, ruffled fur, decreased food intake and diarrhoea. The s.c. LD50 for streptomycin in guinea pigs was 400 mg/kg bw (JECFA, 1995).
The i.v. injection of 10 mg/kg bw ‘pure’ streptomycin to cats had no effect on blood pressure; 20 mg/kg bw gradually depressed blood pressure, which subsequently returned to normal. High doses of 120–375 mg/kg bw caused vasomotor and respiratory paralysis for several hours if artificial respiration was maintained. Out of 12 cats receiving a parenteral dose of 250–350 mg/kg bw streptomycin, four developed respiratory failure and thee died (JECFA, 1995).
The i.v. injection of streptomycin at doses of 220–440 mg/kg bw in dogs caused an irreversible depression of blood pressure. Respiration was stimulated by low i.v. doses but paralysed by high (165 mg/kg bw) i.v. doses. Intravenous and s.c. administration of 30–70 mg/kg bw streptomycin to monkeys caused marked respiratory depression, which sometimes required artificial respiration. (JECFA, 1995).
An aqueous solution of streptomycin (8 mg base/mℓ) and an ophthalmic ointment containing 1 mg/g streptomycin were applied to the conjunctival sac of lightly anaesthetised rabbits for 30 minutes. Occasional redness of the conjunctiva was observed, which persisted up to 12 hours after application. Application of streptomycin as either an aqueous solution (8 mg/mℓ) or as an ointment (1 mg/g) to the buccal membrane of dogs for 15 minutes had no adverse effects. The intradermal. injection of 0.1–0.4 mg streptomycin in the abdominal skin of guinea pigs produced slight reddening followed, occasionally, by blister formation. Groups of rabbits given intrapleural injections of streptomycin at 1, 10 or 100 mg/kg bw showed dose-related increases in pleural fluid and congestion of the diaphragm 4 days after dosing (JECFA, 1995).
4.2 Repeat dose toxicitySpecial attention has been devoted to the ototoxic effects seen in several species due to streptomycin administration. Streptomycin damages the hair cells of the Organ of Corti in the cochlea and the hair cells of the vestibular apparatus, which are found in the macula of the saccule, the macula of the utricule and the ampullae of the three semicircular canals. It does not damage the eighth cranial nerve.
In humans, the incidence of vestibular toxicity is particularly high in patients receiving streptomycin; nearly 20 % of individuals who received 500 mg twice daily for 4 weeks developed clinically detectable, irreversible vestibular damage. Up to 75 % of patients who received 2 g streptomycin for more than 60 days showed evidence of nystagmus or postural imbalance (HSDB, 2002). Patients presenting with ototoxic side effects after treatment with streptomycin were studied retrospectively. Vertigo was reported by the end of the first week of treatment in 25/26 patients receiving streptomycin at doses of 0.25–2 g/person/day (3–36 mg/kg bw/day). The route of administration was not stated but was probably i.m. injection. The total doses were 4–71 g streptomycin. In most cases (13/19 patients) treatment lasted from 1 week to 4 months. In 6/19 patients the symptoms were still present at examinations performed 1.5–6 years after their first appearance. In 13/18 patients, functional impairment of renal clearance was reported. These tended to be in patients treated for longer periods (9–19 days). At lower doses (15 mg/kg bw/day) for about 7 days, only 1 case of vestibular damage was reported when over 1000 patients were treated with streptomycin (JECFA, 1995).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
153

Evidence of minor renal tubular dysfunction, such as urinary casts and a minor degree of albuminuria, is not uncommon in humans treated with streptomycin. Severe renal damage is rare and renal effects are usually reversible on cessation of therapy. Streptomycin has been reported to cause toxic neuritis of the branches of the trigeminal nerve, resulting in numbness, tingling or burning sensations in the mouth or face of patients. The administration of the drug may produce dysfuntion of the optic nerve. Scotomas, presenting as enlargement of the blind spot, have been associated with treatment. CNS depression characterised by stupor and flaccidity and, in some cases, by coma and respiratory depression, has been reported in infants receiving high doses of streptomycin (JECFA, 1995; HSDB, 2002).
Mice given streptomycin by s.c. injection at 150mg/kg bw/day, in three equally divided doses for 6 days, or 1000 mg/kg bw/day, in five divided doses s.c. for 6 days, or 150, 300, 600 or 1500 mg/kg bw/day, orally in the diet, remained clinically normal during treatment and for the following 10-day observation period. No adverse gross pathology was observed. Groups of rats injected s.c. with 100 mg/kg bw/day streptomycin in three divided doses for 72 days showed no clinical signs of toxicity or any treatment related histopathological effects. In an escalating dose study, hamsters were fed, initially, 2 mg/kg bw/day streptomycin in diet. The dose was then doubled every 2 weeks until the dose reached 64 mg/kg bw/day at 3 months. All animals survived. Histopathological examination revealed damage of the intestine, caecum and liver in some of the animals. Groups of guinea pigs were treated with 20, 30, 40 or 60 mg/kg bw/day streptomycin s.c. in 3 divided doses for 6–8 weeks. Two animals at 40 mg/kg bw/day died of unknown causes on day 15. The other animals remained clinically normal and no gross abnormal findings were noted at post-mortem (JECFA, 1995).
Ten cats (5/sex) were treated daily with streptomycin at 200 mg/kg bw/day s.c. for 90 days. All the cats showed clinical signs of toxicity after 2 weeks of treatment with disturbed vestibular function manifested by ataxia, loss of righting reflex and head oscillations, salivation and decreased body weight. The streptomycin dose was then decreased to the maximum tolerated dose based on food and water intake (between 25 and 100 mg/kg bw/day). The vestibular dysfunction persisted at this lower dose. Five dogs were injected s.c. or i.m. with streptomycin at 50 or 100 mg/kg bw/day in 3 divided doses for 20 days. All animals developed proteinuria at 1–2 weeks. Casts, epithelial cells and leucocytes were observed in urine. At necropsy, 1 dog in the high dose group had liver and kidney changes. Three dogs developed a change in gait and posture suggesting a labyrinthine or cerebellar disturbance. Auditory impairment was noted based on failure of these dogs to respond normally to sudden noises. Dogs treated daily by i.m. injections of 44 mg/kg bw/day streptomycin for 14 days developed vestibular dysfunction. Dogs treated with 187 mg/kg bw/day for 28 days developed bilateral liquefaction necrosis of the ventral cochlear nuclei (JECFA, 1995).
A series of studies were undertaken on monkeys using s.c., i.v. or i.m. routes of administration of streptomycin. Most animals were either unaffected or had minimal effects, apart from fatty metamorphosis in the liver and, less often, in the kidney, in monkeys dosed at 25 mg/kg bw/day or more. The reversibility of the fatty changes was examined in monkeys given 25 mg/kg bw/day i.v. for 5 days. At 10 days after the last dose, a large amount of fat was present in the liver and a slight amount was present in the kidney. At 66 days, no pathological changes were observed (JECFA, 1995).
It was reported that guinea pigs treated for 21–60 days with 200–400 mg/kg bw/day streptomycin parenterally showed changes in both the peripheral and central sides of the vestibular and cochlear systems. It was noted that the vestibular system was more severely affected than the cochlear system (JECFA, 2002). In 90-day studies in guinea pigs and cats, the NOEL for ototoxic effects was determined to be 40 mg/kg bw/day (EMEA, 2002).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
154

4.3 Carcinogenicity and mutagenicityA long-term study in rodents has not been undertaken on streptomycin. A 2-year study in rats has been carried out on the closely related compound dihydrostreptomycin. The drug was administered in the diet to groups of 35 rats/sex at dose levels of 1,5 or 10 mg/kg bw/day. Interim sacrifices of 5 rats/sex/group were made at 6 and 12 months; the remaining animals continued on the treated diet for up to 2 years. At 18 months and 2 years, bodyweights were slightly decreased in males at the top dose. At 2 years, the incidence of tumours in treated groups was no higher than in the control group. The NOEL for the study was 5 mg/kg bw/day based upon the decreased bodyweights in males at the high dose. Although the study does not meet current standards, the JECFA Committee concluded that it represented an adequate test of the carcinogenic potential of the compound. Since the chemical structure, pharmacokinetic properties and toxicity profile of the two compounds are almost identical, the JECFA Committee concluded that the carcinogenic potential of streptomycin had been satisfactorily assessed in the 2-year oral toxicity study with dihydrostreptomycin (JECFA, 1995).
Streptomycin binds to and alters the configuration of the 30S subunit of ribosomes, thus inhibiting protein synthesis and causing misreading of the genetic code. RNA and DNA syntheses are unaffected. The results of genotoxicity assays with streptomycin are shown in Table 4.2 (GENE-TOX, 1991; CCRIS, 1994; JECFA, 1995).
Table 4.2 Results of genotoxicity assays on streptomycinTest system Test object Concentration Results
In vitroCytogenetics Human lymphocytes 4.7–13.7 mg/mℓ EquivocalCytogenetics Human lymphocytes 50–2000 µg/mℓ NegativeCytogenetics Human lymphocytes 50–300 µg/mℓ Inconclusive1
Cytogenetics Mammalian cells 10–20 mM PositiveBacterial S. typhimurium (TA102, +S9) 3.125–25 µg/plate PositiveBacterial S. typhimurium (TA102, -S9) 3.125–25 µg/plate PositiveChromosome aberr. Vicia faba Not stated PositiveDNA repair B. subtilis (H17 v M45T) Not stated InconclusiveDNA repair E. coli polA (W3119 v P3478) Not stated NegativeIn vivoCytogenetics Human lymphocytes2 0.75–1.0 g/day NegativeCytogenetics Human lymphocytes2 Not stated Negative1 Streptomycin tested in combination with penicillin.2 Human lymphocytes obtained from treated tuberculosis patients.
4.4 Reproductive and developmental toxicity The incidence of congenital malformations in new-borns was examined in 1619 mothers who had received treatment for tuberculosis with streptomycin, hydrasid and p-amino-salicylic acid. The results were compared with those from a control group of 2711 healthy pregnant women. The incidence of congenital malformations was 2.34 % in the treated subjects and 2.56 % in the control group. No difference was observed in the pattern of malformations in the 2 groups, although the nature of these malformations was not specified and the dose and time of treatment were not stated (JECFA, 1995). A review was made of the pregnancy outcomes of women receiving streptomycin for the treatment of tuberculosis. The doses administered ranged from 15 to 30 mg/kg bw, twice weekly, for all or part of their pregnancy. Ear defects were observed in the children, consisting of vestibular dysfunction and varying degrees of hearing loss. No other adverse effects were reported (EMEA, 2002).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
155

Groups of 6–7 pregnant Swiss mice were given single s.c. injections of streptomycin at doses of 0.025 or 0.25 µg/kg bw on day 14 of gestation. There was no effect on litter size and no malformations were observed in any of the fetuses. Female offspring showed a temporary decrease in bodyweight gain at the low dose. In the high dose group, both females and males had reduced bodyweight gain up to day 35 day 17, respectively. Seminal vesicle and adrenal weights were reduced at the low dose in both sexes. At the high dose, all organ weights were reduced except for liver.
Pregnant C57BL mice were given i.m. injections of streptomycin at 250 mg/kg bw, twice daily. The timing of the treatment in relation to gestation was not stated. There was no effect on litter size, no external malformation and no gross malformation of the brain or cranial segments of the cervical medulla. Streptomycin crossed the placental barrier and was identified by microbiological evaluation of tissue fluids of embryos from treated dams. Streptomycin was given by s.c. injection to 14 pregnant mice at 400 µg/kg bw/day on days 9, 10 and 11 of pregnancy. The number of implants was reduced in treated mice (179 v 351 in controls). The percentages of fetal deaths and live fetuses were similar in treated and control animals. Bodyweights of the offspring were significantly reduced compared to controls. No malformations were observed in fetuses in the treated group. In a further study, ICR mice were treated i.p. during days 12–16 of gestation with 250 mg/kg bw/day streptomycin. Offspring were examined by behavioural tests and the morphology of the inner ear was examined by electron microscopy. Bodyweight increase, activity and functional development, such as grooming, were unaffected by treatment. Vestibular function was reduced compared with controls. Morphological changes included degeneration and polyp-like cytoplasmic extrusions of the inner hair cells (JECFA, 1995). No teratogenic effects occurred in guinea pigs given up to 200 mg/kg bw/day streptomycin i.m. or in rabbits given 5 or 10 mg/kg bw/day of dihydrostreptomycin orally on days 6–18 of gestation. Streptomycin was regarded as not teratogenic as a result of these studies (EMEA, 2002).
No adverse effects on the reproductive performance of farm animals, due to streptomycin, have been discovered as a result of literature reviews on the subject or from the assessment of field data. Streptomycin did not affect sperm quality, fertility or reproductive performance and did not induce adverse developmental effects among offspring. It was concluded that the use of streptomycin in food-producing animals treated in accordance with good practice in the use of veterinary drugs does not present a risk to peri- and post-natal development in these animals (EMEA, 2002).
The JECFA Committee considered that the data in animals and humans indicated that the effects of streptomycin on the middle ear of fetuses were a manifestation of fetotoxicity. The Committee concluded that the compound was not a teratogen (JECFA, 1995).
5 Guidelines and standardsJECFA confirmed that the appropriate NOEL to establish the ADI for streptomycin is that derived from the 2-year oral toxicity study in rats on dihydrostreptomycin (see Section 4.3). Applying a safety factor of 100, a group ADI of 0–50 µg/kg bw for the combined residues of streptomycin and dihydrostreptomycin was established (EMEA, 2002; JECFA, 2002). This recommendation was also accepted by the Australian Government (Australian Government, 2005).
As the pharmacokinetics, pharmacodynamics and toxicological profiles of both streptomycin and dihydrostreptomycin are similar, EMEA CVMP established an ADI of 25 µg/kg bw for both streptomycin and dihydrostreptomycin, using the NOEL from the 2-year rat study for dihydrostreptomycin. They applied a safety factor of 200 owing to the limited data on reproductive toxicity (EMEA, 2002).
A microbiological ADI has also been calculated using a formula recommended by the EMEA CVMP, involving a daily faecal bolus of 150 g. The calculated microbiological ADI was 0–80 µg/kg bw. JECFA modified the calculation by replacing the faecal bolus value (150 g) with a value for a colonic
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
156

content of 220 g. This increases the ADI, based on microbiological activity, for the combined residues of streptomycin and dihydrostreptomycin to 0–120 µg/kg bw (EMEA, 2002).
More recently, JECFA recommended definitive MRLs for edible tissues of cattle, pigs, sheep and chickens as follows: 600 µg/kg for muscle, fat and liver, 1000 µg/kg for kidney and a temporary MRL of 200 µg/kg for bovine milk. The marker residue was identified as the sum of streptomycin and dihydrostreptomycin (EMEA, 2002; JECFA, 2002). The EMEA CVMP, further to the consideration of the grounds for appeal, recommends the inclusion of streptomycin in Annex I of Council Regulation (EEC) No.2377/90 in accordance with Table 5.1.
Table 5.1 MRLs for streptomycinA ctive substance
Marker residue Animal species MRLs Target tissues
Streptomycin Streptomycin Bovine, ovine 500 µg/kg Muscle500 µg/kg Fat500 µg/kg Liver1000 µg/kg Kidney200 µg/kg Milk
Porcine 500 µg/kg Muscle500 µg/kg Skin+fat500 µg/kg Liver1000 µg/kg Kidney
Based on these MRL values, the daily intake will represent approximately 40% of the toxicological ADI (EMEA, 2002).
The US-FDA established a tolerance of zero for residues of streptomycin in the uncooked tissues of chickens, turkeys and swine and in eggs (HSDB, 2002).
6 ReferencesAustralian Government (2005) ADI List - Acceptable Daily Intakes for Agricultural and Veterinary Chemicals, Canberra, Australia, Australian Government, Department of Health and Ageing, Available [June 2005] at; http://www.tga.gov.au/docs/html/adi.htm
CCRIS (1994) Streptomycin. From: Chemical Carcinogenesis Research Information System, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [January 2004] at http://toxnet.nlm.nih.gov
EMEA (2002) Streptomycin. Summary Report (3) (EMEA/MRL/809/01-FINAL), London, UK, European Agency for the Evaluation of Medicinal Products, available [May 2005] at http://www.emea.europa.eu/pdfs/vet/mrls/080901en.pdf
GENE-TOX (1991) Streptomycin. From: Genetic Toxicology (Mutagenicity) Databank, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [January 2004] at http://toxnet.nlm.nih.gov
HSDB (2002) Streptomycin. From: Hazardous Substances Databank, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [January 2004] at http://toxnet.nlm.nih.gov
JECFA (1995) Streptomycin (Food Additives Series 34), Geneva, Switzerland, World Health Organization, available [January 2004] at http://www.inchem.org/documents/jecfa/jecmono/v34je04.htm
JECFA (2002) Streptomycin (Joint FAO/WHO Expert Committee on Food Additives), Geneva, Switzerland, World Health Organization, available [July 2005] at http://www.inchem.org/documents/jecfa/jeceval/jec_1911.htm
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
157

VRC (2002) Annual Report on Surveillance for Veterinary Residues in Food in the UK, 2002 , Addlestone, UK, The Veterinary Residues Committee
VRC (2003) Annual Report on Surveillance for Veterinary Residues in Food in the UK, 2003, Addlestone, UK, The Veterinary Residues Committee
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
158

Annex 17 Testosterone1 IntroductionTestosterone (17 -hydroxyandrost-4-en-3-one; CAS No. 58-22-0) is a naturally occurring 19-carbon steroid that has potent androgenic properties, including promotion and maintenance of primary and secondary anatomical and physiological male sexual characteristics (JECFA, 2000).
In men, testosterone is used for the management of congenital or acquired primary hypogonadism, resulting from orchidectomy or from testicular failure caused by cryptorchidism, bilateral torsion, orchitis or vanishing testis syndrome. Testosterone is also used in males for the management of congenital or acquired hypogonadotropic hypogonadism. If any of these conditions occur before puberty, androgen replacement therapy will be necessary during adolescence for the development of secondary sexual characteristics. In women, testosterone has been used for the palliative treatment of androgen-responsive, advanced, inoperable, metastatic (skeletal) carcinoma of the breast in females who are 1–5 years postmenopausal. Short-acting androgen preparations are preferred, particularly during the early stages of androgen therapy, since androgens occasionally appear to accelerate the disease. Because of their anabolic and androgenic effects on performance and physique, androgens have been misused and abused by athletes, bodybuilders and weight lifters (HSDB, 2002).
2 Uses and exposureTestosterone propionate (200 mg) in combination with estradiol benzoate (20 mg) is administered to cattle as an ear implant to increase the rate of weight gain (growth promotion) and to improve feed efficiency. Testosterone propionate is rapidly cleaved to testosterone in vivo and is thus considered an ‘endogenous substance’, as the residues are structurally identical to testosterone produced by humans and other mammalian species (JECFA, 2000).
Testosterone is administered to animals as either testosterone esters, or methyltestosterone, for deficient libido, reversion of feminisation after testicular tumour removal in male dogs; treatment of estrogen-dependent mammary tumours, suppression of estrus, pseudopregnancy in bitches; and hormonal alopecias in both sexes. DuratestoneTM is licensed in the UK as an oily depot for use in dogs. It comprises a mixture of testosterone esters (testosterone decanoate 20 mg/mℓ, testosterone isocaproate12 mg/mℓ, testosterone phenylproprionate 12 mg/mℓ, testosterone proprionate 6 mg/mℓ) at 0.05–0.1 mℓ/kg. It may also be given to cats at the same dose rate to treat alopecias (Bishop, 2004).
Methyltestosterone is available as a 5 mg tablet formulation (OrandroneTM) and is given to dogs at a dose rate of 0.5 mg/kg (Bishop, 2004).
Potential sources of human exposure include ingestion of animal products containing drug residues following illegal use in food animals. Treatment of food animals would only occur through illegal use. The production rate of endogenous testosterone and hence natural testosterone levels in plasma show large variations, depending on species, sex, age and physiological status. EU Directive 81/602/EEC prohibits the administration to farm animals of substances having an androgenic effect and the marketing and processing of meat from these animals. This is further extended for testosterone by 88/146/EEC; its use for fattening purposes is prevented and use is limited only to zootechnic and therapeutic purposes, as prescribed by a veterinarian. As a consequence, MRLs for muscle, liver, kidney and fat in have not been specified in food animals.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
159

2.1 Exposure concentrations in meatUsing information on usage of testosterone and on absorption and excretion, likely ‘worst case’ concentrations in meat products were estimated using Equation 1:
Equation 1M = D × T × Fabs × (1–Fexc)
where M is the predicted concentration in meat (mg/kg); D is the dose of substance (mg/kg bw/d); T is the duration of the treatment (d); Fabs is the fraction of compound absorbed; and Fexc is the fraction of compound excreted.
Testosterone is likely to be given to cattle of around 450 kg weight in the form of a subcutaneous pellet injection containing 600 mg of active ingredient. This gives a dose rate of 1.3 mg/kg. It was assumed that bioavailability was 100 % and that 90 % of the substance is excreted in the urine. Using these input values a ‘worst case’ concentration of testosterone in meat products of 0.13 mg/kg is obtained.
3 ToxicokineticsTestosterone is generally considered to be inactive when given by the oral route owing to gastrointestinal and/or hepatic inactivation. Maintenance of physiological concentrations after injection of testosterone is also difficult because of its rapid clearance (JECFA, 2000). The esters of testosterone, such as the propionate, cypionate and enanthate, are partially cleaved in vivo to release the parent compound (HSDB, 2002).
When radiolabelled testosterone is administered intragastric (i.g.) to rats, about 25 % appears in bile within 12 hours as glucuronide and sulphate conjugates, indicating enterohepatic circulation. About 90 % of an administered dose of radiolabelled testosterone is found in urine and about 6 % undergoes enterohepatic circulation and appears in faeces. The plasma half-life of testosterone after intravenous (i.v.) administration was about 10 minutes. The plasma concentrations of testosterone were measured by radioimmunoassay after oral administration of 25 mg of the compound to young women and compared with that in subjects receiving testosterone at 1.5 µg/kg bw by i.v. injection. Bioavailable testosterone represented 3.6 % of administered compound. The low bioavailability of orally administered testosterone is attributed to its high metabolic clearance rate of 25 mℓ/min/kg. The estimated bioavailability provides justification for the high doses (120–140 mg/day) considered to be necessary to replace daily production (5–7 mg) (JECFA, 2000).
In normal men, circulating androgens are bound to sex hormone binding globulin (SHBG) and, to a lesser extent, to serum albumin. Only 1–2 % of the testosterone in circulation is unbound; 44 % is bound to SHBG and 54 % is bound to albumin and other proteins. Plasma SHBG is secreted from the liver, but adult rodent livers do not produce the secretory form of SHBG (JECFA, 2000).
Testosterone is inactivated primarily in the liver. Metabolism to androstanedione involves oxidation of 17-OH groups; 5 α-reduction of ring A of androstanedione and reduction of the 3-keto group leads to the formation of androsterone. Alternatively, androstenedione can be reduced in the 5-β position and can undergo 3-keto reduction to form etiocholanolone. Dihydrotestosterone is converted in the liver to androsterone, androstanedione and androstanediol. It is transformed to 5 α-dihydrotestosterone in target organs such as the prostate, sebaceous glands and seminal vesicles; only the latter compound binds to androgen receptor sites in these target organs. Testosterone and its metabolites are conjugated with glucuronic and sulphuric acids and are excreted in urine and faeces. Approximately 90% of a dose of testosterone is excreted in urine in this way (HSDB, 2002).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
160

Significant differences occur between the metabolic pathways of testosterone in rodents and humans. Hepatic rat testosterone 7 α-hydroxylase (P450 2A1) and mouse testosterone 15 α-hydroxylase (P450 2A4) are not found in human liver. Testosterone is converted to its 6 β form by human liver CYP3A4. The major pathways for inactivation of testosterone in female mouse liver are hydroxylation at the 16 β-, 6 α- and 16 α-positions followed by conjugation. Metabolism of testosterone in rat liver produces 7 α-, 6 β- 16 α- and 2 α-hydroxytestosterones. No 16 β-hydroxylase (CYP2B1) activity was detected in rat liver. The rat small intestine is capable of oxidising testosterone, the enzyme is present in homogenates of rat gastric and duodenal mucosa at higher levels than in the jejunum. The major portal vein metabolite was androstenedione, indicating that the gut rather than the liver is primarily responsible for the metabolism of orally administered testosterone. Testosterone is a precursor of other physiologically relevant steroids. The active metabolite, dihydrotestosterone (DHT), formed through the action of 5 -reductase, is an important physiological substrate for the androgen receptor and is considered to be the main cellular mediator of androgen activity in some tissues (JECFA, 2000).
4 Toxicity profile4.1 Acute toxicityAdverse effects associated with testosterone in humans include acne, flushing of the skin, increased or decreased libido, habituation and oedema. Other adverse effects associated with testosterone therapy include nausea, bladder irritability, sleeplessness, headache, anxiety and mental depression. Hypersensitivity reactions, such as skin manifestations or anaphylactoid reactions have occurred rarely with testosterone (HSDB, 2002).
There do not appear to be any conventional acute toxicity studies for testosterone in animals (PIMS, 1998). Because testosterone is rapidly inactivated after oral administration, it is likely to have low acute toxicity by this route (JECFA, 2000).
4.2 Repeat dose toxicityThere do not appear to be any conventional repeat dose toxicity studies for testosterone in animals (PIMS, 1998).
Six intact adult male baboons received weekly intramuscular (i.m.) injections of 200 mg testosterone enanthate (equivalent to 8 mg/kg bw) for up to 28 weeks, while two control animals had weekly injections of the vehicle only. Quantitative increases in the weight and volume of both prostatic lobes were seen after 15 weeks of treatment and, by week 28, there was an increase in stromal tissue with papillary in-growth or invagination of glandular epithelium in the caudal lobe of the prostate. The serum concentrations of testosterone and DHT were significantly elevated, from 10 and 2–3 ng/mℓ to 30-40 and 5–6 ng/mℓ, respectively. The androstenedione concentrations were increased by 3–4 times and that of estradiol from 20 to 80–90 pg/mℓ. It was concluded that these steroids play a direct role in inducing early benign prostate hypertrophy in baboons and that the observations are similar to those in human benign prostate hypertrophy (JECFA, 2000).
A significant increase in prostatic weight occurred after 6 weeks treatment of castrated rats with testosterone. Histopathologically, glandular hyperplasia of the prostate was noted and the number of bromodeoxyuridine-positive cells showed an increase (HSDB, 2002).
Testosterone ethanate was given i.m. every other week at an average dose of 15 mg to 21 female rabbits for up to 763 days. The treatment induced two adenomatous polyps of the endometrium in one animal but one control animal developed similar polyps. No other tissues such as the ovary, adrenal, thyroid or pituitary gland was altered significantly (JECFA, 2000).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
161

4.3 Carcinogenicity and mutagenicityAn IARC Working Group, reviewing the carcinogenicity to humans of the androgenic (anabolic) steroids, considered the evidence to be ‘limited’. Hepatocellular carcinomas, cholangiocarcinomas and adenomas have been reported after extended treatment of patients with methyltestosterone, testosterone enanthate and nandrolone decanoate for hypogonadadism, hypopituitarism, chronic renal failure and generalised weakness. The fact that castration palliates prostatic cancer indicates that testosterone may be involved in the genesis of these tumours and a number of epidemiological observations suggest that increased testosterone levels may increase the risk for prostatic cancer. In addition, patients with cirrhosis, who have depressed testosterone levels, have low rates of prostatic cancer and prostatic cancer is seemingly unknown among castrates (IARC, 1987).
Male rats received s.c. implants of 1–3 10 mg pellets of testosterone propionate:cholesterol in the ratio of 9:1, replaced at 6–8 week intervals for up to 91 weeks. Prostatic carcinoma occurred in 0/13, 5/30 and 11/55 rats receiving 1, 2 or 3 pellets, respectively. The incidence in untreated animals was 0.48%. Twenty female rats were given 0.5 mg testosterone propionate in arachis oil s.c. weekly from the age of 3 days; the doses were then increased to 1 mg when the rats were 21 days old and then to 2.5 mg when they were 6 months old. Three out of ten rats surviving to 16 months had theca cell ovarian tumours and with marked epithelial hyperplasia in the uteri of 5/10 rats. No ovarian or uterine tumours were seen in control animals. Cervical uterine tumours were found in 26/42 (C57BL×DBA)F1 mice implanted with 1–2 mg pellets of testosterone propionate twice weekly for their lifespan. The tumours were infiltrating and metastasised to the lungs in 10 mice. In female BALB/CCRGL mice injected s.c. with 25 µg testosterone in water daily for the first 5 days after birth, 7/9 developed hyperplastic epithelial lesions resembling epidermoid carcinomas (vaginal sqamous-cell tumours) at about 71 weeks of age. Daily s.c. injections to female BALB/CFC3H mice (with mouse tumour virus) of 5 or 20 µg testosterone in 0.02 ml sesame oil for the first 5 days of postnatal life, resulted in increased mammary tumour incidence, by 16 months of age, of 42/49 and 22/35 respectively (HSDB, 2002).
In female Sprague–Dawley rats, single s.c. injection of 1 mg testosterone propionate in 0.05 mℓ sesame oil at 2 days of age enhanced the incidence of auditory sebaceous gland tumours induced by DMBA administered i.g. at 50 days of age. The tumour incidence, 250 days after DMBA administration, was 8/32 and 1/20 in the treated and control groups respectively. The s.c. implantation of 50 mg testosterone:cholesterol at 1:1, enhanced the incidence of bladder tumours in nitrosoamine treated ovariectomised Wistar rats to 8/11. Testosterone treatment on its own induced no tumours (HSDB, 2002). Fischer 344 rats were given 3,2'-dimethyl-4-aminobiphenyl (a prostate carcinogen) at 50 mg/kg bw 10 times at 2 week intervals and then, from week 20, testosterone propionate by s.c. Silastic implant for 40 weeks, as 7 cycles of 30 days treatment and 10 days withdrawal. The administration of testosterone resulted in suppression of the development of ventral prostate adenocarcinomas and slight (non-significant) increases in the incidences of invasive carcinomas of the lateral prostate seminal vesicles (JECFA, 2000).
An IARC Working Group summarised the evidence for the carcinogenicity to animals due to testosterone. Testosterone propionate, tested for carcinogenicity in mice and rats by s.c. implantation, produced cervical uterine tumours in female mice and prostatic adenocarcinomas in male rats. Neonatal treatment of female mice by s.c. injection of testosterone induced hyperplastic epithelial lesions of the genital tract and increased the incidence of mammary tumours. Dihydrotestosterone also increased the incidence of mammary tumours in mice when given neonatally by s.c. injection. The s.c. administration of testosterone propionate, following i.v. treatment with N-methyl-N-nitrosourea, produced a high incidence of prostatic adenocarcinoma not seen with the individual compounds alone. The Working Group considered that the evidence for carcinogenicity to animals was ‘sufficient’ for testosterone (IARC, 1987). A WHO Committee affirmed that the increased rate of prostatic cancer detected in the rat studies, is consistent with the hormonally mediated effects of testosterone and its metabolites (JECFA, 2000).
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
162

The overall evaluation of IARC (1987) is that androgenic (anabolic) steroids are probably carcinogenic to humans (Group 2A).
A few genotoxicity studies have been reported for testosterone. Testosterone at concentrations of 1–100 µg/mℓ had weak transforming activity, but with no dose–response relationship, in Syrian hamster embryo cells. In another study, the growth of Syrian hamster embryo cells was reduced by incubation with testosterone or testosterone propionate at 10–30 µg/mℓ in a dose-dependent manner and the two compounds were equipotent in producing morphological transformation. At 30 µg/ml, the transformation frequency induced by testosterone and the propionate was one half that induced by 1 µg/mℓ benzo(a)pyrene. Neither steroid significantly increased the frequency of chromosomal aberrations or aneuploidy. Testosterone did not induce gene mutations at the hprt or Na+/K+ ATPase locus. When testosterone was added at a final concentration of 2 mmol/ℓ to DNA obtained from human surgical resections, rat liver, HepG2 cells and calf thymus, it did not form adducts with the naked DNA. No adducts were observed in DNA isolated from HepG2 cells incubated with 10–100 µmol/ℓ testosterone for 24 h (JECFA, 2000). A sperm morphology assay with male human cells was reported to give a positive result, while a similar assay with male mouse cells was reported to be negative (GENE-TOX, 1991).
In vivo, DNA strand breaks were observed in the dorsolateral prostate of Nobel rats treated with a combination of testosterone and estradiol but not in those treated with testosterone alone. NBL/Cr rats received two Silastic implants s.c., one of which contained testosterone and the other estradiol at the age of 8–9 weeks and were killed 8, 16 or 24 weeks after the start of treatment. The quantity of steroid in the implant was not stated but the release rate was predicted to increase the estradiol concentrations by about 14-fold while maintaining normal plasma testosterone concentrations. The prostate glands were found to contain 2 major endogenous adducts and several minor spots irrespective of treatment but, in treated animals, the relative levels of the 2 adducts were decreased from 1 and 10 × 109 to 0.5 and 3 × 109 in the ventral but not in the dorsolateral or anterior prostate. A major adduct spot was observed in the dorsolateral prostate of the animals treated with both hormones for 16 and 20 weeks but not in those treated for 8 weeks or in control animals. The presence of this adduct coincided with the occurrence of dysplastic lesions in the dorsolateral prostate. It was suggested that testosterone enhances prostatic carcinogenesis, perhaps by generating estradiol through peripheral conversion (JECFA, 2000).
4.4 Reproductive and developmental toxicityMasculinisation of the fetal external genitalia is a basic biological phenomenon with the administration of testosterone. Side effects of testosterone administration in humans include hirsutism, deepening of voice, precocious puberty, epiphyseal closure in immature males, increased libido, priapism, oligospermia and enlargement of clitoris in females. Oligospermia and decreased ejaculatory volume may occur in males receiving excessive dosage or prolonged administration of the drug. The administration of 25 mg testosterone propionate daily for 6 weeks in males caused a decrease in spermatogenesis. Amenorrhea and other menstrual irregularities and inhibition of gonadotropin secretion occur commonly in females with administration of the drug. Testosterone may cause fetal harm when administered to pregnant women. Androgenic effects include clitoral hypertrophy, labial fusion of the external genital fold to form a scrotal-like structure, abnormal vaginal development and persistence of urogenital sinus in female offspring of women who were given androgens during pregnancy (HSDB, 2002).
Increasing the plasma concentration of testosterone to above physiological levels results in decreased secretion of follicle-stimulating and luteinising hormones and decreased testicular volume and sperm production. The effectiveness of orally administered testosterone was assessed in 5 eunuchs in a trial in which the controls received a placebo. Initial studies indicated that a dose of 25 mg testosterone had no effect on any clinical parameter including sexual desire, erection, ejaculation and general well-being. In a second study, a dose of 100 mg/day did not alter the clinical parameters, whereas 400 mg/day (100 mg, 4 times/day) was fully effective. A WHO Committee considered the dose of
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
163

100 mg/day (equivalent to 1.7 mg/kg bw/day) to be the NOEL for this study and used it as the basis for setting the ADI for testosterone (see Section 5). Oral administration of testosterone at 30 mg/day did not restore sexual function to castrates, while 100 mg/day had a slight effect; 20 mg testosterone propionate injected twice a week had moderate potency (JECFA, 2000).
Complete resorption of embryos was seen in 7 female Sprague–Dawley rats that received 10 mg testosterone as a s.c. implant on day 10 of gestation. No effect was seen in control pregnant rats given dextran by the same route (JECFA, 2000).
Daily s.c. injections for 4–8 days to give total doses of 1–55 mg testosterone propionate to rats between days 12–19 of gestation resulted in resorptions, necrosis, lethality, post-partum mortality and various degrees of masculinisation in female offspring. The effects were correlated directly with dose and period of administration. The injection of 100 mg testosterone propionate in rats on day 14 of gestation, induced small or absent mammary glands in offspring of both sexes and the absence of nipples in females only. Single s.c. injection of 4 mg/kg bw testosterone to rats between days 5–8 of gestation, prevented implantation in 11/16 animals; injections on days 9–11 produced fetal loss or delayed parturition in 9/13 rats. Injection of 20 mg/kg bw on days 1, 5 or 9 led to fetal loss in all treated animals. Testosterone propionate or sesame oil vehicle were given to rats during the last week of pregnancy. Testosterone significantly increased anogenital distance and delayed vaginal opening of progeny. When females that had been exposed to testosterone in utero were tested for breeding capacity, a significantly smaller number mated than in the control group. Pregnant female guinea pigs were treated with testosterone or vehicle from days 28–65 of gestation. The offspring were gonadectomised and tested as adults for lordosis and androgen-activated mounting behaviour. Prenatal testosterone treatments altered the hormonal requirements for androgen-activated mounting in females such that they resembled normal males and did not require aromatisation as adults (HSDB, 2002).
5 Guidelines and standardsJECFA established an ADI of 0–2 µg/kg bw for testosterone on the basis of the NOEL of 100 mg/day (equivalent to 1.7 mg/kg bw/d) in the study of eunuchs (see Section 4.4) and using a safety factor of 1000. The large safety factor was used in order to protect sensitive populations and because of the small number of subjects in the study from which the NOEL was identified (JECFA, 2000). MRLs for muscle, liver, kidney and fat (cattle), were not specified by JECFA (1999).
EU Directive 81/602/EEC prohibits the administration to farm animals of substances having an androgenic effect and the marketing and processing of meat from these animals. This is further extended for testosterone by 88/146/EEC, preventing its use for fattening purposes and limiting use to zootechnic and therapeutic purposes, as prescribed by a veterinarian. As a consequence, MRLs for muscle, liver, kidney and fat have not been specified for food animals by EMEA CVMP.
The US-FDA Approved Drug Products with Therapeutic Equivalence Evaluations List identifies currently marketed prescription drug products, including testosterone, approved on the basis of safety and effectiveness by FDA under sections 505 and 507 of the Federal Food, Drug and Cosmetic Act (HSDB, 2002).
6 ReferencesBishop Y, ed (2004) The Veterinary National Formulary (Sixth Edition), London, UK, Royal Pharmaceutical Society of Great Britain/British Veterinary Association
GENE-TOX (1991) Testosterone. From: Genetic Toxicology (Mutagenicity) Databank, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [January 2004] at http://http://toxnet.nlm.nih.gov
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
164

HSDB (2002) Testosterone. From: Hazardous Substances Databank, Bethesda MD, USA, Specialized Information Services, National Library of Medicine, available [January 2004] at http://http://toxnet.nlm.nih.gov
IARC (1987) IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Man. Summaries and Evaluations, Vol Suppl 7, Testosterone, Lyon, France, International Agency for Research on Cancer
JECFA (1999) Testosterone (Joint FAO/WHO Expert Committee on Food Additives), Geneva, Switzerland, World Health Organization, Available [January 2004] at http://www.inchem.org/documents/jecfa/jeceval/jec_1624.htm
JECFA (2000) Toxicological Evaluation of Certain Veterinary Drug Residues in Food (Food Additives Series 43), Geneva, Switzerland, World Health Organization, Available [January 2004] at http://www.inchem.org/documents/jecfa/jecmono/v43je05.htm
PIMS (1998) Testosterone. From: Poisons Information Monographs, Geneva, Switzerland, World Health Organization, International Programme on Chemical Safety, available [January 2004] at http://http://www.inchem.org/documents/pims/pharm/pim519.htm
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
165

Annex 18 Toxicity SearchesWhere data sources identified as described in Section 2 were not adequate to provide a suitable database to prepare toxicological profiles for any of the 17 selected chemicals, additional searches were undertaken. Searches to identify additional data on toxicity, carcinogenicity or reproductive effects were performed on the host Datastar in the major biomedical databases (see Table I for summary). The type of search performed for each chemical is summarised in Table II.
Table I Toxicity search database summaryDatabase Database producer Database
labelCoverage
Medline National Library of Medicine MEZZ 1951-presentMEDL 1996-present
ToxFile Dialog Corporation AG1 TOXL 1965-presentCancerlit National Cancer Institute CANC 1967-2003Embase Elsevier B.V. EMZZ 1974-present
EMED 1996-presentBiosis Biosis BIZZ 1969-present
BIOL 1996-present1 Using data provided by the National Library of Medicine
A summary of the toxicity terms used is shown in Table III. In all databases, phrases and descriptors were combined using the ‘and’ operator with synonyms and CAS numbers for the individual chemicals. In Medline, ToxFile and Embase, the ‘with’ command was used when a specific descriptor was available for a chemical in order to use specific descriptor subheadings.
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
166

Table II Type of searches performedChemical Date
performed
Search type Database Yearlimit
Reviews Only
Medline Toxfile
CancerLit Embase Biosis
Bambermycins 1/4/2004 Standard toxicity MEZZ x EMZZ BIZZ None xBambermycins 17/6/2004 Standard toxicity MEZZ x EMED BIZZ None xEnrofloxacin 1/4/2004 Standard toxicity
ADI/TDI/Guideline valuesMEZZ x EMZZ BIZZ None x
Lasalocid acid 1/4/2004 Standard toxicity MEZZ x EMZZ BIZZ None xMalachite green 1/4/2004 Standard toxicity MEZZ x EMZZ BIZZ None xNalidixic acid 2/4/2004 Standard toxicity MEZZ EMZZ BIZZ None Nandrolone 2/4/2004 Standard toxicity MEZZ EMZZ BIZZ None Nandrolone 9/6/2004 Standard toxicity MEZZ EMZZ BIZZ None xNarasin 2/4/2004 Standard toxicity MEZZ EMZZ BIZZ None Phenylbutazone 13/1/2005 Targeted Reproductive
Toxicity/CarcinogenicityMEDL EMED BIOL 2000+ x
Salbutamol 2/4/2004 Standard toxicity MEZZ EMZZ BIZZ None
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
167

Table III Summary of the standard toxicity search strategyOperator Search Phrase/Descriptor
Medline
Veterinary medicine.ti,de,ab.1
CAS number.rn.AND2 Health adj2 effect$13.ti,ab,de.
Adverse adj effect$1.ti,ab.Toxicology#4
Toxicity-tests#Toxic$8.ti,ab.Carcinogen$5.ti,de,ab.Teratogen$5.ti,de,ab.Mutagen$5.ti,de,ab.Mutagenicity-tests#Neurotoxic$8.ti,de,ab.Cytotoxic$8.ti,de,ab.Genotoxic$8.ti,de,ab.Poison$3.ti,ab.
Veterinary medicine.de. WITH2 Toxicity.de.Adverse adj effect$1.de.Poisoning.de.
Embase
Veterinary medicine.ti,de,ab.CAS number.rn.
AND Health adj effect$1.ti,de,ab.Adverse adj effect$1.ti,de,ab.Toxic$8.ti,ab.Toxicity#Toxicity-testing#Carcinogen$5.ti,de,ab.Mutagen$5.ti,de,ab.Teratogen$5.ti,de,ab.Neurotoxic$8.ti,de,ab.Cytotoxic$8.ti,de,ab.Genotoxic$8.ti,de,ab.Poison$3.ti,de,ab.
Veterinary medicine.de. WITH Drug adj toxicity.de.Biosis
Veterinary medicine.ti,de,ab.CAS number.rn.
AND Health adj effect$1.ti,de,ab. Adverse adj effect$1.ti,de,ab. 225#5
toxic$8.ti,ab. Carcinogen$5.ti,de,ab. Teratogen$5.ti,de,ab. Mutagen$5.ti,de,ab. Neurotoxic$8.ti,de,ab.Cytotoxic$8.ti,de,ab. Genotoxic$8.ti,de,ab. Poison$3.ti,de,ab.
Veterinary medicine.de. WITH toxic$8.de.1Letters indicate the fields searched .de. = descriptors, .ti. = title, .ab. = abstract, .rn. = registry number2Proximity operators: ADJ = adjacent to; WITH = in the same sentence, words in any order; AND = in the same paragraph3$1 – Truncation symbol, a number indicates the number of characters allowed4# - Term was exploded to include all terms below in the hierarchical descriptor tree5Concept code for toxicity
Final Report prepared by IEH for Defra (VMD Ref. VM02130; MRC Ref 3/2/15; Cranfield Ref YE20056E)
168