unlocking the secrets of fda's orange book: an … ©2007 foley & lardner llp 1 11...
TRANSCRIPT

1©2007 Foley & Lardner LLP
1
11
Unlocking the Secrets of FDA's Orange Book:An Introduction to Therapeutic Equivalence, Drug Patents, Exclusivities, and More
Co-sponsored by FOI Services, Inc. and Reedfax
David L. Rosen, B.S. Pharm., J.D.
Foley & Lardner LLP (202) 672-5430 phoneWashington Harbour (202) 672-5599 fax3000 K St., NW, Suite 500 [email protected], DC
February 2007
2
22

2©2007 Foley & Lardner LLP
3
33
Approved Drug Products List with Therapeutic Equivalence Evaluations
Commonly referred to as the FDA's “Orange Book”
First published in January 1979 in conjunction with the FTC’s Model Drug Product Selection Act to assist states and facilitate generic substitution
The authoritative reference source for drugs products that that have been approved by FDA under the federal Food, Drug and Cosmetic Act.
The FDA's Orange Book lists all products that have beenapproved by FDA for safety and effectiveness, alphabetically by ingredient(s) in the product.
It also lists a therapeutic equivalence code for all multi-source products
4
44
FDA Orange Book Overview (cont.)
With the passage of the Drug Price Competition Act of 1984, (the Hatch Waxman Act), the FDA's Orange Book took on additional significance
– Repository and source Patents
Periods of marketing exclusivity
associated with reference listed drugs.
Searchable on version available at
– www.fda.gov/cder/orange/default.htm

3©2007 Foley & Lardner LLP
5
55
Topics for Discussion
FDA's Orange Book history and trivia
Organization of the Orange Book and how it is maintained;
Therapeutic equivalence codes – what they are and why they are important
Types of patents can be listed and codes associated with those patents;
Market exclusivity codes associated with products.
My goal is that you will leave this web conference with
– a through understanding and appreciation of the FDA's Orange Book
– how to use it to provide high level strategic advice to advance the goals and objectives of your company.
6
66
Hot FDA Topics
Increased focus on drug/device safety– Response to IOM critique– FDA pilot re-evaluation of New Molecular Entity safety – Separate safety center for drug post marketing issues– Strengthen science to improve device safety over product lifecycle
Reauthorization of the Prescription Drug User Fee Act– User fees for ANDAs– Increased resources for drug safety
New proposed legislation– Block authorized generics– Follow on Protein Products
Marketing unapproved new drugs– FDA is trying to clean up the marketplace
Improve export certification programConsolidation of FDA to White Oak campusStrengthen food safety
4©2007 Foley & Lardner LLP
7
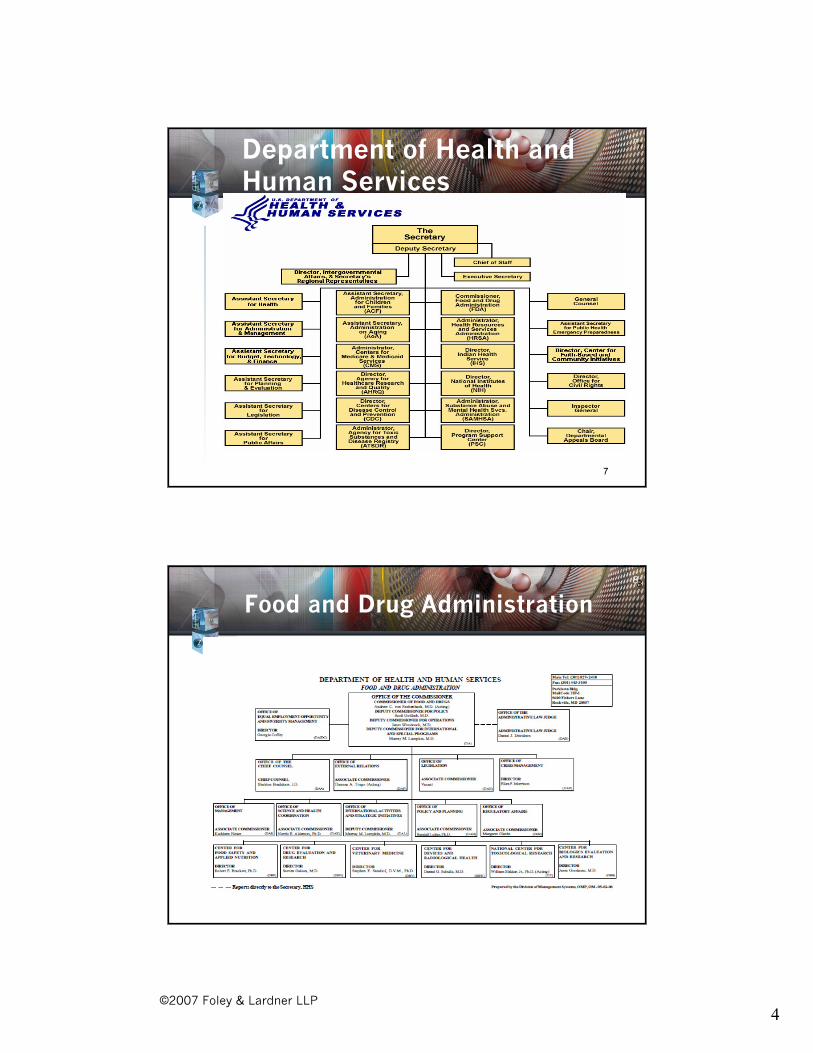
77Department of Health and Human Services
8
88
Food and Drug Administration

5©2007 Foley & Lardner LLP
9
99
©2006 Foley & Lardner LLP

6©2007 Foley & Lardner LLP
11
1111
Important Statutory Milestones - Drug Law1900 1910 1920 1930 1940 1950 1960 1970 1980 1990
2003
A. Pure Food and Drug Act of 1906
B. Federal Food, Drug and Cosmetic Act of 1938
C. Durham-Humphrey Amendments of 1955
D. Kefauver-Harris Amendments of 1962
E. Orphan Drug Act -1983; Amendments of 1985 & 1988
F. Drug Price Competition and Patent Term Restoration Act of 1984
G. Prescription Drug Marketing Act of 1988
H. Prescription Drug User Fee Act of 1992
I. Generic Drug Enforcement Act of 1992
J. Food and Drug Modernization Act of 1997
K. Best Pharmaceuticals for Children’s Act of 2002
L. Access to Affordable Pharmaceuticals – part of the Medicare Modernization Act 0f 2003
12
1212The New Drug Development Process

7©2007 Foley & Lardner LLP
13
1313Full New Drug ApplicationSection 505(b)(1) of the FD&C Act
Full reports of investigations demonstrating safety and effectiveness
Components of the drug
Composition of the drug
Full description of the methods, facilities and controls used for the manufacturing processing and packaging of the drug
Samples, as required
Labeling
Patent number and expiration date which claims the drug or method of using the drug
14
1414New Drug ApplicationSection 505(b)(2) of the FD&C Act
An application where the investigations relied upon by the applicant for approval
were not conducted by or for the applicantapplicant has not obtained a right of reference from the person by or for whom the investigations were conducted
Must include a patent certification
Can list Patents in the Orange Book

8©2007 Foley & Lardner LLP
15
1515
Examples of 505(b)(2) Applications
Dosage form
Strength
Route of administration
Substituted active ingredient in combo
Formulation
Active Ingredient
Intentional bioinequivalence
Combo of individually approved products
New indication
Rx/OTC switch
Different product than in OTC monograph
Naturally derived or recombinant ingredient
New molecular entity
16
1616Abbreviated New Drug ApplicationSection 505(j) of the FD&C Act
An applicant must provide information regarding the generic drug as compared to the reference listed drug
Conditions of use prescribed, recommended or suggested in the labeling have been previously approved
Active ingredient(s) is the same
Route of administration, dosage form and strength are the same
Product is bioequivalent
Labeling is the same
An applicant must also provide a certification with respect toeach patent that claims reference listed drug

9©2007 Foley & Lardner LLP
17
1717
Petition Submitted If Different In:
Route of Administration
Dosage Form
Strength
or
API* Substituted for API in a listed Combination Drug
API = Active Pharmaceutical Ingredient
18
1818
Paragraph IVCites Section 505(j)(2)(B)(ii)
Contains Required Bioavailability and/or Bioequivalence Data
ANDA Number
Established Name
API, Strength, Dosage Form of Proposed Drug Product
Patent Number and Expiration Date of Each Patent
Detailed Statement of the Factual and Legal Basis for Non-infringement, unenforceability and/or invalidity

10©2007 Foley & Lardner LLP
19
1919
Exclusivity Protection Available for a NDA Application
Three year exclusivity
– if one or more of the clinical investigations, other than BA/BE studies, was essential to approval and conducted by or for the sponsor
Five year exclusivity
– for a new chemical entity.
Pediatric exclusivity (6 months)
Orphan Drug
20
2020
Near the end of30 month stay, NDA holderis granted a new patent andsubmits to Orange Book
within 30 days.No metabolite, intermediates
or packaging patents.
ANDA holder submitsp IV cert. to
“new” listed patent
NDA holder sues on newpatent,
No second 30 monthstay
180 day Exclusivity is “triggered”.•Exclusivity Can Be Triggered by
• Commercial Marketing• But can also be Forfeited!
Patent grantedfor drug productexpires in 2009.
No metabolite, intermediates or packaging patents
New Drug Application(NDA) submitted. NDA holder submits
patent to Orange Book
Abbreviated New DrugApplication (ANDA) submitted 1st ANDA
with p IV certification
ANDA is acceptedfor filing
NDA Holder bringssuit w/45 days,
Automatic 30-MonthStay of Approval
Only One 30 Month Stay
ANDA applicant notifiesNDA holder of p IV
certification. NDA holderhas 45 days to bring suit.
NDA Approvedw/5 years of NCE
Exclusivity
[ANDA cannot be submitted for 5 years unlessit includes a p IV & then it is 4 years]
ANDA applicant wins atDistrict Court level
ANDA Holder is potentially liable for treble damages for willful patent infringement if they lose at the appellate level.
Patent holder appeals decision to Circuit Court. [6-12 months]
Current Law and FDA Regulations Including Changes from Medicare Modernization Act of 2003

11©2007 Foley & Lardner LLP
21
2121
A Brief Primer on Patents
22
2222

12©2007 Foley & Lardner LLP
23
2323
Legal Authority
Constitution, Art. 1, Section 8: “The Congress shall have the power… to promote the progress of science and useful arts, by securing for limited times to authors and inventors the exclusive right to their respective writings and discoveries.”
Basic philosophy is to encourage inventors to disclose their inventions to the public in return for the limited right to exclude others from practicing the invention.
24
2424
What is a Patent?
A limited grant from the U.S. government giving the inventor the right to exclude others from making, using, offering to sell, or selling the patented invention in the U.S.
The right to exclude is measured by how the claims of the patent define the invention
“Negative Right”--Not a right to do anything except exclude others from practicing invention for a limited period of time.

13©2007 Foley & Lardner LLP
25
2525
Patents
Protect practical applications of ideas
Right to exclude others from– Making– Using– Offering for sale– Selling– ImportingInvention in the U.S.
26
2626
Types of PatentsUtility—processes, machines, articles of manufacture, compositions of matter, methods of manufacture, methods of use, business methods
Design—ornamental aspects of an invention
Plant—plants that can be asexually reproduced (except plants that are not tuber-propagated or found in an uncultivated states)—right to exclude others from asexually reproducing the plant or selling or using the plant if it is asexually reproduced
Plant Variety—novel varieties of sexually reproduced plants (Dep’t of Agriculture)

14©2007 Foley & Lardner LLP
27
2727
Patent Duration
Utility patent term is 20 years from date application filed– Pre-1995 patent term 17 years from
date patent issued
Design patent term is 14 years
28
2828
One-Year Statutory Bar
Must apply for patent within one year after you use it or offer it for sale in the U.S. or you disclose it in a publication
Part of Give and Take for Patent

15©2007 Foley & Lardner LLP
29
2929
– New – It has not already been invented by someone else
– Non-obvious – It is not an obvious variation of a prior invention
– Useful – It has a useful purpose– Disclosure – Patent specification must
describe the invention in detail and enable one of skill in the art how to make and use it (cf. Trade secret)
– Time Bars
Patentability:
30
3030
1984: Roche v. Bolar
Roche v. Bolar, 733 F2d 858 (Fed. Cir. 1984).
– In 1983 Bolar Pharmaceuticals was interested in making a generic of Dalmane (Flurazepam HCl), a psychotropic drug manufactured by Hoffmann-La Roche.
Roche’s Composition of Matter Patent 3,299,053 expired in 1984.
– Prior to expiration of the patent
Bolar brought into the U.S. 5 kgs of flurazepam for use in product development (formulation, bioequivalence, dissolution testing) .
– Roche learned of this and brought suit for infringement.

16©2007 Foley & Lardner LLP
31
3131
1984: Roche v. Bolar
– In April 1984, the Fed. Cir. held:
Bolar’s use was infringing, and
it was up to Congress, not the Courts, to create an exception specifically geared to drug development.
– Soon afterwards, Congress passed the Drug Price Competition and Patent Term Restoration Act of 1984 (“Hatch-Waxman”) which included § 271(e).
– This specifically overruled Roche v. Bolar
– Permitted generic companies to begin product development prior to patent expiration
Act of infringement – the submission of the ANDA
32
3232
35 U.S.C. § 271(e)
“It shall not be an act of infringement to make, use, or sell a patented invention (other than a new animal drug or veterinary biological product (as those terms are used in the Federal Food, Drug, and Cosmetic Act ... )) solely for uses reasonably related to the development and submission of information under a Federal law which regulates the manufacture, use or sale of drugs.”

17©2007 Foley & Lardner LLP
33
3333
Why Get Patents?
1. To protect the investment in research and development of your product or process and prevent others from copying your invention
2. To license your invention to others and thereby generate licensing revenues
3. To have bargaining chips for cross licensing
4. To increase your company’s value
34
3434
Listing of Patents in FDA’s Orange Book
Patent holder provided with opportunity to delay FDA approval of ANDA for a generic version– “lists” patents covering an approved product in the
FDA’s Orange BookNo metabolite patentsNo intermediate patentsNo packaging patents
– Must identify – by patent claim - specific methods of uses covered by patent that are in the approved labeling
– listing enables patent holder to file suit under 271(e)(2) to bar immediate FDA approval of the generic drug

18©2007 Foley & Lardner LLP
35
3535
Limits to Orange Book Listed Patents
Patents having only method of making (process patents) claims are not listable in the Orange Book
Proprietors of biological agents unable to list a patent in the Orange Book - may be loath to have a regulatory pathway allowing approval of a generic biologic, based on data they generated
No comparable Orange Book for Biologics at present– List of products approved by CBER– www.fda.gov/cber/products.htm
36
3636
Intellectual Property – Biologics
IP for Biologics
– for some biologics, only “method of making” patent claims are available
– naturally occurring biological agent and its activity (i.e., uses) may be known
– Patentable invention was developing a manufacturing process for commercial scale up

19©2007 Foley & Lardner LLP
37
3737Current Remedies Available to Biologic Patent Holder
Patent holder would still be able to sue a generic biologic in district court if they infringe
But “method of making” patents are more easily designed around
So much of the value in the biologic may be the cost and effort invested in the FDA approval process
38
3838
Patent Term RestorationTo be eligible for PTR, a patent must satisfy all of
the following conditions:– An application for extension has been filed within 60
days of FDA approval;
– the term of the patent has not yet expired;
– the patent has not previously been extended;
– the product was subject to an FDA regulatory review period before its commercial marketing or use; and
– the product represents the first permitted commercial marketing or use of the product after regulatory review.

20©2007 Foley & Lardner LLP
39
3939
PTR: Length of Extension
Maximum patent term extension is five years.
Total patent life for the product with the patent extension cannot exceed 14 years from the
product’s approval date.
40
4040
PTR: Calculating Length of Extension
Regulatory review period is divided into two phases: Testing and Approval Phase
– Testing Phase = IND → Submission of NDA– Approval Phase = Submission of NDA → Approval
Half credit for testing phaseFull credit for approval phase

21©2007 Foley & Lardner LLP
41
4141
42
4242
Historical Development of the List of Approved Drugs
1. Maximum allowable cost – estimated acquisition cost for drugs purchased by the government
2. Repeal of anti-substitution laws
3. States sought advice from FDA on which products were FDA approved – but FDA did not have an organized, verified data base (1975-1978)

22©2007 Foley & Lardner LLP
43
4343
Historical Development of the List (cont.)
4. Lack of bioequivalence information was a roadblock to generic substitution
5. U.S. Congress Office of Technology Assessment (OTA) 1974 Report on Bioequivalence –recommendation that FDA prepare official list of interchangeable drugs
6. FDA proposed bioavailability/ bioequivalence regulations – June 1975
44
4444
Historical Development of the List (cont.)
7. FDA list of holders of approved drug applications for drugs presenting actual or potential bioequivalence problems (the “Blue Book”) 1976
8. FDA final bioavailability/bioequivalence regulations – 1977
9. FDA agreed to help New York prepare drug substitution Iist (Green Book) – later FDA agrees to help Illinois prepare a similar list (Illinois Formulary)
10. FDA realizes it can't serve individual state needs and prepares first Orange Book in Jan 1979

23©2007 Foley & Lardner LLP
45
4545
Orange Book Trivia Questions
What was the color of the cover of FDA’s first Orange Book?
Who was the FDA Commissioner at the time the FDA’s Orange Book was first published?
Name a person who is still at FDA that was one of the chief architects of the Orange Book.
46
4646
Contents of the Orange Book
1. Introduction defining terms, listings, therapeutic equivalence codes, and FDA policies
2. Key on how to use the lists
3. Lists of RX and OTC drugs that have been approved under full New Drug Applications (NDAs) and Abbreviated New Drug Applications (ANDAs)

24©2007 Foley & Lardner LLP
47
4747
Contents of the Orange Book (cont.)
4. List of drug product that are regulated by the center for biologics evaluation and research (CBER) – these include drugs that are used in processing blood and blood products
5. List of discontinued products6. List of orphan drug designations7. List of drugs that must demonstrate in vivo
bioequivalence only if they fail dissolution
48
4848
Contents of the Orange Book (cont.)
8. Appendices – product listing by
- trade name- generic name- application holder;
List of uniform terms dosage forms and routes of administration
9. List of patent and exclusivity information- Searchable by patent number on line

25©2007 Foley & Lardner LLP
49
4949
Important Definitions
1. Pharmaceutical Equivalents – same active ingredient(s); dosage form; strength or concentration; and route of administration
2. Pharmaceutical Alternatives – drugs that contain the same therapeutic moiety but are different salts, esters, or complexes of the moiety or are different dosage forms or strengths
3. Therapeutic Equivalents – drugs which an (1) approved by FDA as safe and effective; (2) pharmaceutical equivalents; (3) bioequivalent; (4) adequately labeled; and (5) manufactured in compliance with current good manufacturing practices
50
5050
Important Definitions (Cont.)
4. Bioequivalencea. The rate and extent of absorption of the test drug
are not significantly different from that of the reference product when administered under similar experimental conditions
b. The extent of absorption of the test drug is notsignificantly different from that of the reference product when administered under similar experimental conditions; the difference in rate is intentional, reflected in the labeling, is not essential to the attainment of effective concentrations, and is considered medical insignificant for the drug

26©2007 Foley & Lardner LLP
51
5151
Important Definitions (Cont.)
Bioequivalence Statistical method for establishing BE
Confidence limits for PK parameters AUC and Cmax
Analysis of Variancetest product average/reference product average lower bound reference product average/test product average upper bound
Calculate the 90% Confidence Limit bounds of 80-125
No more than a 5% statistical chance that a generic product thatis not truly equivalenct to the reference product will be approved
52
5252
Important Definitions (Cont.)
Reference listed drug (RLD)– Listed drug identified by FDA (with a “+”
symbol) in the Orange Book which an applicant relies upon in seeking approval of its ANDA or 505(b)(2) NDA
– RLD is the basis of comparison for use in in vivobioequivalence and in vitro dissolution
– Applicants must address patent and exclusivity issues associated with the RLD that are listed in the Orange Book
NOTE – must check OB frequently (often multiple times daily) for new patents and exclusivities – this can hold up FDA approval

27©2007 Foley & Lardner LLP
53
5353
Important Definitions (Cont.)
Practitioner / User ResponsibilitiesEvaluations of therapeutic equivalence are based upon FDA’s scientific and medical evaluations
Products can be expected to have the equivalent clinical effect and no difference in their potential in adverse effects
Products may vary in shape, scoring configuration, release mechanisms, packaging, excipients (colors, flavors, preservatives) expiration dates and labeling (in certain situations - can exclude items that are are protected by patent/exclusivity)
54
5454
So the last time you went to the pharmacy to pick up a prescription…
Did you get or request a generic version, if it was available??
Why or why not???

28©2007 Foley & Lardner LLP
55
5555
Organization of Listings
Drug product list arranged alphabetically by active ingredient; each listing contains:– Dosage form– Route of administration– Product name– Applicant name– Strength– NDA/ANDA number– Approval date for products approved on or after
January 1, 1982– Effective approval date where applicable
56
5656
Definition of Therapeutic Equivalence Codes
Codes which denote therapeutic equivalence ratings assigned by FDA
"A" codes – drug products that FDA considers to be therapeutically equivalent to other pharmaceutically equivalent products
"B" codes – drug products that FDA considers NOT to be therapeutically equivalent to other pharmaceutically equivalent products

29©2007 Foley & Lardner LLP
57
5757
Explanation of Therapeutic Equivalence Codes
AA Products in conventional dosage forms not presenting bioequilvalence problems
AN Solutions and powders for aerosolization
AO Injectable oil solutions
AP Injectable aqueous solutions
AT Topical products
“A” Products for which actual or potential bioequivalence problems have been resolved with adequate in vivo or in vitro evidence supporting bioequivalence are designated as follows:
AB Products meeting necessary bioequivalence requirements
58
5858
Explanation of Therapeutic Equivalence Codes (cont.)
“B” products for which actual or potential bioequivalence problems have not been resolved by adequate evidence of bioequivalence:
These products often have such a problem with specific dosage forms rather than with the active ingredients.
BC Extended-release dosage forms (tablets, capsules, injectables)
BD Active ingredients and dosage forms with documented bioequivalence problems
BE Delayed-release, enteric coated oral dosage forms
BN Products in aerosol-nebulizer drug delivery systems

30©2007 Foley & Lardner LLP
59
5959
Explanation of Therapeutic Equivalence Codes (cont.)
BP Active ingredients and dosage forms with potential bioequivalence problems
BR Suppositives or enemas that deliver drugs for systemic absorptions
BS Products having drug-standard deficiencies
BT Topical products with bioequivalence issues
BX Drug products for which the data are insufficient to determine therapeutic equivalence
60
6060
Basis of Therapeutic Equivalence Determinations
1. Drugs products must be approved as both safe and effective
2. They must be pharmaceutical equivalents having identical amounts of active ingredient(s) in the same dosage form and route of administration, and must meet USP or other applicable standards of identity, strength, quality, and purity.

31©2007 Foley & Lardner LLP
61
6161
Basis of Therapuetic Equivalence Determinations (cont.)
3. They are bioequivalent in that the drugs do not present a known or potential bioequivalence problem and meet an acceptable in vitro standard; or there is data demonstrating in vivo bioequivalence.
4. The drug products are adequately labeled.
5. The drug products are manufactured in compliance with current good manufacturing practices.
62
6262
Products Rated by FDA as NOT Therapeutically Equivalent
1. Certain applications approved before passage of the Drug Price Competition and Patent Term Restoration Act in 1984
2. Products on the market without FDA approval
3. In the absence of data, FDA is NOT willing nor able to make therapeutic equivalence determinations

32©2007 Foley & Lardner LLP
63
6363
64
6464

33©2007 Foley & Lardner LLP
65
6565
Example of a “B” Rated Product
Albuterol
AB Armstrong Pharms 0.09MG/INH N73273 001 Aug 14, 1996AB Genpharm 0.09MG/INH N73045 001 Aug 19, 1997AB Ivax Pharms 0.09MG/INH N73272 001 Dec. 28, 1995AB Pliva 0.09MG/INH N74072 001 Aug 01, 1996
BN Proventil 0.09MG/INH N17559 001
Schering
VentolinAB + Glaxo Wellcome 0.09MG/INH N18473 001
AlbuterolAerosol, Metered, Inhalation
66
6666Examples of Pre-1938 Drug Products Which Are Not Listed in FDA’s Orange Book
Certain single entity narcotic pain relief agents– Codeine Phosphate
Certain immediate release potassium replacement therapiesCertain sedative/hypnotic agents– Barbiturates

34©2007 Foley & Lardner LLP
67
6767
What is an “Authorized Generic”
Before or at the patent expiration
At the time of introduction of the first generic version into the market– either just before or at the same time
Brand Pharma entered into a manufacturing and distribution agreement with a generic company
Generic company or subsidiary of the Brand company sells a version of the drug manufactured under the NDA as a “generic” version– Attempt to preempt and capture a share of the generic
market – Sometime stem from out of court patent law suit settlements
68
6868
Examples of Products Marketed by Two Different Manufacturers Under the Same New Drug Application
Authorized Generic Issues
Proventil – Schering Albuterol Sulfate – Warrick
Z-Pak - Pfizer Azithromycin – Greenstone
Paxil – GSK Paroxetine – Par Pharm
Procardia – Pfizer Nifedipine - Mylan

35©2007 Foley & Lardner LLP
69
6969Debate Regarding the Substitution of Generic for Brand Name NTI Drug Products
Is an FDA approved generic version of an NTI drug readily substitutable for the brand name drug?Should NTI drugs be subject to stricter bioequivalence standards?What about NTI drugs that have not gone through the FDA approval process?
70
7070
When I was in pharmacy school, I was taught the following products could present issues when switching patients from one company’s product to another…
DigoxinLevothyroxine sodiumWarfarin sodiumPhentoin sodiumTheophylline

36©2007 Foley & Lardner LLP
71
7171
Definition
A narrow therapeutic ratio drug is defined by the FDA– Less than 2 fold difference in the median lethal
dose LD50 and effective dose; or– Less than 2 fold difference in the minimum
toxic concentrations and minimum effective concentrations in the blood, and
– Safe and effective use of the drug product requires careful titration and safe monitoring
72
7272
Access to Life-Saving Medicines (ALSMA)
Introduced Sept. 2006 by Rep. Waxman, Sen. Schumer, and Sen. Clinton.

37©2007 Foley & Lardner LLP
73
7373
Summary of ALSMA Provisions
– Amends section 351 of the PHS Act to authorize FDA to approve abbreviated applications for biological products that are “comparable” to previously approved (“reference”) biological products.
– A comparable biological product application must demonstrate that:
there are no clinically meaningful differences between the two products; the new product shares the “principal molecular structural features” of the reference product and the same mechanism(s) of action, if known.
– FDA has discretion on a case-by-case basis to determine what studies are necessary to establish comparability, and may require a clinical study or studies, but only if necessary.
– A comparable biological product application will be subject to user fees.
74
7474
Summary of ALSMA Provisions
– An applicant for a comparable biological product may elect, but is not required, to establish interchangeability.
tax credits for the cost of studies demonstrating interchangeabilitygrants the first applicant to obtain approval of an interchangeable version of a biological product a period of exclusive marketing during which no other interchangeable version of the product may be approved.
– Applicants for comparable biological products may elect to ask the holder of the reference product:
for a list of patents on the product;and may elect to notify the reference product holder and owner of one or more of the patents identified that the applicant has filed a comparable biological product application.If the applicant sends a notice, it must contain a detailed statement explaining why the identified patent(s) is invalid, unenforceable, or not infringed.If the reference product holder fails to disclose a relevant patent, it may not enforce that patent against that applicant.If no patent infringement action is brought within 45 days of notice of a
challenge, the remedy in any later action to enforce that patent against that applicant is limited to a reasonable royalty.

38©2007 Foley & Lardner LLP
75
7575
Definitions Under ALSMA
A biological product is “comparable” to a previously approved (“reference”) biological product if there are no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity and potency of the product, based on non-clinical studies and clinical studies, as necessary.
An “interchangeable product” is a product that can be expected to produce the same clinical result(s) as the reference product in any given patient.
76
7676Submission of Comparable Biological Product Applications.
– An abbreviated application for a comparable biological product to must contain information showing that, among other things:
the product is comparable to the reference product; the two products have comparable principal structural features; the two products have the same mechanism of action, if known; the proposed product label carries one or more of the approved indications for the reference product; and the route of administration, dosage form, and strength, are the same as the reference product.
– Can submit an application for a product that differs from, or incorporates a change to, the reference product, if the application contains sufficient information to show that the new product is safe, pure, and potent.

39©2007 Foley & Lardner LLP
77
7777
FDA Review of Comparable Biological Product Applications
Contains provisions that track the requirements of Hatch-Waxman for abbreviated application review procedures
Encourages FDA to meet with sponsors of comparable biological products and reach written agreements with them on the design and size of studies.
78
7878
Approval of Comparable Biological Applications
FDA must approve a comparable biological application unless:
There is insufficient information to show that the product is “comparable” to the reference product,for the condition(s) of use in the proposed labeling.
– If the mechanism of action of the reference product is known, the applicant must demonstrate comparability for at least one proposed condition of use. If the mechanism of action is unknown, the applicant must demonstrate comparability for each proposed condition of use.
There is insufficient information to show that product and reference product have comparable principal molecular structural features; There is insufficient information to show that the two products have the same mechanism(s) of action, if known;

40©2007 Foley & Lardner LLP
79
7979
Approval of Comparable Biological Applications (cont.)
The new product differs from the reference product in route of administration, dosage form, or strength; The inactive ingredients used in, or the composition of, the new product are unsafe; The controls used in manufacturing the product are inadequate to assure identity, strength, quality, and purity; The reference product has been, or is being, withdrawn for safety or effectiveness reasons; or
The application contains an untrue statement of fact.
The Secretary may also approve an enhanced version of a reference product if the application contains sufficient information to establish safety and efficacy.
80
8080Interchangeability Determinations, Labeling, and Exclusivity
The bill acknowledges that Interchangeable biological products would generate the greatest cost savings:– there currently is no equivalent of a simple “bioequivalence” study for
biologics; – significantly more costly and difficult to produce than comparable products
The bill therefore provides incentives for the development of interchangeable products, but does not require that each comparable biologic be interchangeable.
– Applicant may request an interchangeability determination. If adetermination is made before approval, FDA must publish a therapeutic comparability evaluation code for the product at the time of approval.
– Tax credits are made available for studies conducted to establish that a comparable biological product is interchangeable with the reference product.
– If FDA determines that a comparable biological product is interchangeable with the reference product, the bill permits the label of the product to state that the product is interchangeable with the reference product for the approved conditions of use.

41©2007 Foley & Lardner LLP
81
8181Interchangeability Determinations, Labeling, and Exclusivity
– If an applicant is the first to establish that its product is interchangeable with the reference product, the bill prevents FDA from approving a subsequent application for an interchangeable version of the reference drug,until the earlier of:
180 days from first commercial marketing;1 year after a final court decision or dismissal with prejudice of all patent infringement cases instituted under this subsection;36 months after approval if such patent litigation is ongoing; 1 year after approval, if no such patent litigation was instituted.
– The bill also prohibits the marketing of a “rebrandedinterchangeable product” distributed with the authorization of the reference product holder during the exclusive marketing period.
82
8282
Final Action on ApplicationsFDA must approve or disapprove an application for a comparable biological product eight months after submission, or 180 days after the application is accepted for filing by FDA, whichever is earlier, unless the final action date is extended by joint agreement of the applicant and FDA. Provisions to prevent frivolous petitions from delaying the approval of comparable biologics. – FDA must not fail or refuse to take action by the final action date on
the ground that a third party has made such a request, nor may acourt enjoin FDA from taking final action or staying an approvalexcept by permanent injunction.
– A permanent injunction may not be issued unless the person seeking the injunction demonstrates an injury of more than irrecoverableeconomic loss.
– Requires a company who files a citizen petition to delay approval of acomparable biologic to do so at least 110 days before the effective approval date. A company may not file a lawsuit concerning a late-filed petition until 180 days after it was filed.
FDA must report to Congress any extension of the final action date and any failure to meet the final action date.

42©2007 Foley & Lardner LLP
83
8383
Patent Provisions
Early resolution of patent disputes is essential to ensuring that irrelevant or invalid patents do not delay
competition in the marketplace.
– Patent Requests. Applicants for comparable biological products may elect to ask the holder of the reference product for a list of patents related to the product.
The holder must respond within 60 days with a list of all related patents, including process patents, owned by or licensed to the holder, and may demand payment up to $1,000. For a period of twoyears, the holder must update the list within 30 days of the issuance
a new related patent or license.
84
8484
Patent Provisions
– Patent Notifications. The comparable biological product applicant may elect to notify the reference product holder and patent owner that it intends to challenge one or more patents from the list provided.Any notice must contain a detailed statement of the factual and legal bases for the claim of invalidity or non-infringement and identify a judicial district in which the applicant consents to be sued.
– Patent Remedies. The patent laws are amended by providing that if a patent is not disclosed in response to a request, that patent may not be enforced against that applicant.
If a patent is disclosed and is the subject of a notice, but no patent infringement action is brought within 45 days of notice in the judicial district identified in the notice, or is not maintained through a final decision or dismissal with prejudice, the remedies in any later action to enforce that patent against the submitter of the notice are limited to reasonable royalties. The federal law governing declaratory judgments is amended to prohibit reference product holders from bringing actions for a declaration of patent infringement, validity, or enforceability with respect to patents that were not subject to a notice.

43©2007 Foley & Lardner LLP
85
8585
Contact Information
David L. Rosen, B.S. Pharm, J.D.
Foley & Lardner LLP (202) 672-5430 phoneWashington Harbour (202) 672-5599 fax3000 K St., NW, Suite 500 [email protected], DC