tutorial in molecular biology
DESCRIPTION
An introductory articleTRANSCRIPT

CLINICAL ORTHOPAEDICS AND RELATED RESEARCH Number 320, pp 247-278 0 1995 Lippincott-Raven Publishers
SECTION 111 REGULAR AND SPECIAL FEATURES
Tutorial Molecular Biology for the Clinician Part II. Tools of Molecular Biology
Eileen M. Shore, PhD*; and Frederick S. Kaplan, MD*.**
This is the second of a series of tutorials on mol- ecular biology for the clinician. Part I intro- duced general principles of nucleic acid and gene structure. This tutorial will build on those general principles and describe some of the commonly used techniques employed in re- search and diagnosis to examine genes and gene function. The tools of molecular biology include recombinant DNA techniques that have revolu- tionized the study of DNA and genetic informa- tion. Recombinant DNA technology allows the isolation of specific regions of chromosomal DNA and the recovery of unlimited quantities of that DNA for analyses, including determination of the nucleotide sequence of genes, examina- tion of the mechanisms of gene regulation, and the identification of gene products (RNA and protein).
From *Department of Orthopaedic Surgery, University of Pennsylvania School of Medicine, Philadelphia, PA. **Division of Metabolic Bone Disease, Department of Orthopaedic Surgery, Hospital of the University of Pennsylvania, Philadelphia, PA. Reprint requests to Eileen M. Shore, PhD, University of Pennsylvania, Department of Orthopaedics, 424 Stemmler Hall, 36th and Hamilton Walk, Philadelphia, PA19104-6081.
Historically, the practice of medicine began as the treatment of symptoms (such as fever, skin rash). Later, physicians began to search for and treat the causes of these symptoms (such as bacterial and viral infections, poor nutrition). Later still, physicians used this knowledge to cure or prevent the diseases themselves. However, some diseases could not be prevented by the treatment of exoge- nous factors, because the diseases were not external to the individual, but rather an in- nate part of an individual’s genetic makeup.
Modern clinical and human genetics strive to identify gene alterations that lead to dis- ease. Gene mutations can lead to the synthesis of a nonfunctional gene product (ribonucleic acid [RNA] or protein) or to the dysregulated production of an RNAor protein. Understand- ing gene structure and function has led to the development of improved methods of diagno- sis and treatment of many diseases. Identifica- tion of the gene that is responsible for a dis- ease is often the first step in identifying the protein and the biochemical pathway that is perturbed in that disease. Molecular biology technology provides the tools that are being used to determine the structure of genes, to
247

24% Shore and Kaplan Clinical Orthopaedics and Related Research
understand the regulation and expression of gene activity, and to design therapies that will lead to the effective treatment of diseases caused by the malfunction of genes.
INTRODUCTION TO CLONING AND RECOMBINANT DNA TECHNOLOGY
The analysis of a gene would be impossible if only a single molecule of that gene was avail- able for study. Recombinant deoxyribonucleic acid (DNA) technology and molecular cloning provide the means to recover multiple copies of a segment of DNA, producing large amounts of material for analysis.
The human genome contains 50,000 to 100,000 genes encoded by hundreds of mil- lions of base pairs of DNA. Molecular cloning is the isolation of a specific segment of DNA (such as a gene or part of a gene) and the generation of many identical copies, or clones, of that segment of DNA. An iso- lated segment of DNA cannot replicate itself. This limitation is overcome by joining (or ligat- ing) the DNA segment to a vector (or carrier) DNA molecule which is capable of its own replication and which also can replicate the DNA segment that has been ligated to it. This new combination of the DNA segment of interest with the vector DNA is a recombi- nant DNA molecule. To provide the neces- sary enzymes and other cellular machinery required for DNA replication, recombinant DNA molecules are inserted into host cells, typically bacteria such as Escherichia coli, which become factories to reproduce large quantities of the recombinant DNA mole- cule. This procedure is summarized in Figure 1 and described in detail in the following sections.
The authors describe the molecular biology techniques that are used to construct a recom- binant DNA molecule and some of the tech- nology that is used to characterize the struc- ture and function of cloned genes. The last sections introduce methods of RNA and pro- tein analysis and the analysis of transcrip-
tional regulation and gene expression. This article is not intended to provide protocols for these techniques, but to present a general overview that will aid the reader in under- standing the principles behind the technology.
ISOLATION AND CHARACTERIZATION OF SPECIFIC GENES
Restriction Endonucleases One of the most important discoveries lead- ing to the isolation and manipulation of spe- cific genes was identifying restriction en- donucleases. These enzymes are synthesized by many bacterial species as part of a de- fense system to degrade the DNA of invad- ing viruses. Each restriction endonuclease (also referred to as a restriction enzyme) recog- nizes and binds a specific sequence of DNA nu- cleotides and cuts double-stranded DNA within or nearby the recognition sequence (restriction site), breaking the phosphodiester bonds of the nucleotide chain. Recognition sites for restriction endonucleases occur in the DNA of the bacteria as well; however, the bacteria contain enzymes that modify its DNA by methylation of nucleotides that contain ade- nine (A) or cytosine ( C ) bases, blocking cleavage of its DNA by the restriction en- donuclease. The unmethylated vital DNA will be unprotected and will be cut or re- stricted (prevented from functioning) when it enters the bacterial cell.
Double-stranded DNA will be broken only if phosphodiester bonds, which form the backbone of a polynucleotide chain, are cut in each strand. Restriction endonuclease recognition sites are typically 4 to 8 nu- cleotides long and occur as inverted palin- dromes: The same sequence occurs in both strands in reverse order (Fig 2). The enzyme then can recognize and cut the same se- quence on both strands. Some restriction en- zymes cut both DNA strands at the center of symmetry, generating fragments of DNA with blunt ends. Others cut each DNAstrand at the same sites on each strand, but offset

Number 320 November, 1995 Tools of Molecular Biology 249
Nucleus
+ Isolate DNA
from cell
Eukaryotic Cell
segments
Transformation u Bacteria, into host cells host cell t o replicate 0
Recombinant DNA molecules
I Replication of
recombinant DNA
/ n
OW Ligation to join DNA segments
with vector DNAS Vector
DNA molecules
Fig 1. Recombinant DNA. In eukary- otic cells, DNA resides in the chromo- somes within the cell nucleus. The DNA is extracted from the cells and enzymatically cut into small pieces. These pieces of DNA are ligated to vector DNAs, self-replicating DNA molecules that will facilitate the syn- thesis of many copies of any piece of DNA inserted into them. A commonly used vector is plasmid DNA, small cir- cular molecules of double-stranded DNA that are found in bacterial cells. The joining of plasmid vector DNA with eukaryotic cellular DNA (DNAs from 2 different sources) forms recom- binant DNA molecules. The recombi- nant DNA molecules are introduced into bacterial host cells by a process known as transformation, under con- ditions that favor the entry of a sinale
recombinant molecule per host cell. Each transformed host cell becomes a factory for the synthesis-of many copies of a single recombinant molecule (see also Fig 5A).
from the center of symmetry, generating fragments with single stranded ends (Fig 2).
The isolation and commercial manufac- ture of the restriction endonucleases have provided a set of highly specific molecular scalpels that allow DNAto be cut into highly specific and reproducible pieces. More than 150 different restriction enzymes, most of which recognize different nucleotide se- quences, are available commercially. Re- striction endonucleases have been purified from many species of bacteria. The enzymes are named by a 3- to 4-letter abbreviation that identifies the bacterial species from which the enzyme was isolated, followed by Roman numerals to distinguish enzymes that have the same bacterial origin. For example, EcoRI was the first restriction endonuclease purified from Escherichia coli; Hind I11 was the third restriction enzyme isolated from Haemophilus influenza Rd; and PvuI and PvuII were recovered from Proteus vulgaris.
A specific restriction endonuclease will cut, or digest, any (unmethylated) double- stranded DNA molecule into specific seg- ments of DNA because the enzyme will cut the DNA reproducibly where there are
recognition sites for that enzyme. Such a di- gestion of DNA results in the production of a set of DNA segments called restriction fragments.
A specific region of DNA can be cut singly or in combination with several restric- tion enzymes, and a restriction map of that region of DNA, showing the position of each restriction enzyme recognition site relative to the others, can be constructed (Fig 3) . Be- cause each restriction endonuclease recog- nizes a short DNA sequence, a restriction map provides some DNA sequence informa- tion about the DNAregion without determin- ing the complete nucleotide sequence. Com- parison of restriction maps for a particular gene region among individuals or species can indicate the degree of similarity among them. Restriction maps also provide impor- tant information about a gene region that is used in gene cloning and genetic engineering (to be discussed in detail in later sections).
DNA Cloning A detailed analysis of a gene or a specific segment of DNArequires large amounts of a specific DNA fragment. This can be accom-

250 Shore and Kaplan Clinical Orthopaedics and Related Research
Restriction RecognitionlCut Endonuclease S i t e Ends Produced
ECO RI S ' G ~ A A T T C S ' 5 'G 3' 5' A A T T C 3' 3 ' 6 5' 3' C T T A AAG 5' 3 ' C T T A AS'
Hind 111 5' A ~ A G c T T 3' 3 ' 1 T C G AAA 5'
5' A 3' 3 ' T T C G A S '
5 ' A G C T T 3 ' 3 ' A 5'
Taq I 5' TVC G A 3' 5 ' T 3' 5' C G A 3' 3 ' A G CAT 5' 3 ' A G C 5 ' 3 ' 1 5'
3 ' G G G A C C C 5 ' 3 'G G G 5' 3 ' C C C S ' Sma I 5' C C CVG G G 3' 5' c c c 3' 5 'G G G 3'
P"" I S ' C G A Y C G 3 ' 5 ' C G A T 3 ' 5 ' C G 3' 3 ' G C A T A G C S ' 3' G C 5' 3 ' T A G C s '
Fig 2. Restriction endonuclease recognition and cut sites. A restriction endonuclease binds to a specific sequence of nucleotides in double- stranded DNA. The recognition sequences for restriction endonucleases are inverted palin- dromes: reading in the 5' to 3' direction, both strands of the recognition site have the same se- quence. Most commonly used restriction en- donucleases recognize sequences of 4 to 8 nucleotides and cut within the recognition site. Cuts in the DNA strands are made at symmetri- cal positions on each DNA strand, as indicated by black arrowheads in the examples shown. Some restriction endonucleases, such as Sma I, cut the double-stranded DNA at the center of symmetry, generating fragments of DNA with flush (or blunt) ends. Other enzymes will cut at offset (or staggered) sites, generating single stranded tails, or overhangs. Eco RI, Hind 111, and Taq I generate 5' overhangs; Pvu I creates a 3' overhang. Most of the examples (Eco RI, Hind Ill, Sma I, Pvu I) recognize sequences of 6 nucleotides and commonly are referred to as 6- cutters.Taq I is a 4-cutter.
plished by using recombinant DNA mole- cules produced by DNA cloning.
Ligation Many restriction endonucleases cleave dou- ble-stranded DNA by staggered cuts that gen- erate single-stranded tails at the cut site (Figs 2,4). These ends of DNA are described as co- hesive ends, or sticky ends, because the single strands can form complementary base pairs with single-stranded tails on other DNAmole- cules that have been generated by cutting with the same enzyme. Any 2 DNAmolecules with complementary single-stranded tails can be joined together by complementary base pair-
~
2 0 kb molecule of DNA
I
A and 8
- 8 kb 1 5 k b 12 kb
- 5 kb
- 5 kb - 7 kb - 8 kb
2 0 kb A 15 kb I , l Z k b $1 8 kb I
A+8 I 5 kb 7 kb (1 8 kb 1
J A B
Restr ict ion Map: L Fig 3. Restriction map.A restriction map shows the position of restriction sites within a segment of DNA. For the linear 20 kb of DNA molecule depicted, the positions of restriction sites for re- striction enzymes A and B can be determined by cutting the DNA with these 2 enzymes in sin- gle digests with each enzyme and by a double digest using both enzymes together.The resulting fragments will be <20 kb if the enzyme has cut within the 20-kb molecule. The positions of cut sites that are consistent with the restric- tion fragment sizes generated can be deter- mined, and a restriction map for those sites can be constructed.
ing followed by reformation of the phospho- diester bonds on each DNA strand by the en- zyme DNA ligase (Fig 4). Note that some re- striction enzymes generate blunt ends, with no single-stranded tail, rather than sticky ends (Fig 2). Blunt ends also can be ligated to- gether by DNA ligase. The ligation of 2 DNA molecules from different sources creates a re- combinant DNA molecuIe.
Vector DNAs To generate many copies of a DNA molecule, the DNA must have the ability to replicate it- self. Most isolated pieces of DNAdo not have this ability, and therefore require a carrier DNA molecule, or vector. The vectors that commonly are used in molecular biology studies can self-replicate in bacterial or yeast

Number 320 November, 1995 Tools of Molecular Biology 251
I
Digestion with Restriction Enzyme
(EcoRI)
I Ligation with DNA Ligase
5' -
3' -
Fig 4A-C. Ligation of DNA. (A) A double- stranded segment of DNA with an Eco RI recognition site is depicted. The nucleotides of the inverted palindrome recognition site (5' GAATTC 3' on both strands) are represented by the boxes containing the base (A = adenine; C = cytosine; G = guanine;T = thymine) of each nucleotide. The hydrogen bonds between base pairs of the 2 DNA strands are indicated by striped lines: 2 hydrogen bonds between A-T pairs and 3 between G-C pairs. Arrows indicate the cutting site of the enzyme. (8) On digestion with Eco RI, the phosphodiester bonds be- tween the nucleotides at the cut site are bro- ken, leaving a phosphate group (P) at free 5' ends and a hydroxyl group (OH) at free 3' ends. After disruption of the phosphodiester bonds, the hydrogen bonds between base pairs are not sufficient to hold the double strands to- gether and the fragments of double stranded DNA to either side of the cut site will separate. Because Eco RI cuts at staggered sites, the cut ends of the DNA fragments will have single- stranded tails, or overhangs. (C) The overhangs that result from staggered cuts commonly are referred to as sticky ends because the single- stranded ends can reform hvdroaen bonds be-
tween A-T and G-C pairs. If the single-stranded overhangs contain complementary base sequence to allow formation of hydrogen bonds between base pairs, they are described as being compatible. (A fragment generated with cutting by Eco RI and 1 generated by Hind Ill would not be compatible because their single-stranded overhangs could not form complementary base pairs [see Fig 21). The enzyme DNA ligase catalyzes the formation of phosphodiester bonds and is used to join frag- ments of DNA having compatible sticky ends, for example in the formation of recombinant DNAs.
host cells. Recombinant DNA molecules that are formed using restriction enzymes and DNA ligase to join a segment of human DNA and vector DNA can be introduced into the host cells and replicated to generate large quantities of the recombinant molecule. Be- cause the host cells can be grown indefinitely in the laboratory, unlimited amounts of the re- combinant DNA can be obtained.
Plasmids and phagemids are commonly used vectors for the cloning of short (up to 10 kb) segments of DNA. Plasmids, which occur in nature, are circular double-stranded DNA molecules found in bacteria and replicate inde- pendently of the host chromosome. Phagemids are genetically altered, or engineered, plas- mids to which regions of bacteriophage M13 (a bacteria virus) have been added. The M13 genome can be replicated into double-stranded
or single-stranded forms. By inserting the in- formation that encodes single-strand synthesis into a plasmid, the plasmid can be induced to generate single-stranded DNA, a property that especially has been useful for DNA sequenc- ing technology (see below).
Plasmids and phagemids have been engi- neered to meet the specific needs of molecu- lar cloning. They contain an origin of repli- cation to allow propagation in bacteria, 1 or more selectable markers (to identify bacteria that carry the plasmid or phagemid), and multiple recognition sites for restriction en- donucleases for ease of insertion of a foreign DNA segment. The method of propagation of plasmid DNA is shown schematically in Figure 5A.
Larger fragments of DNA can be cloned into other types of vectors (Table 1). Bacte-

252 Shore and Kaplan Clinical Orthopaedics and Related Research
A Plasmid and phagemid growth
plasmid DNA 0
ooo 0" Transform Bacteria
Cel ls
J colonies
B Bacteriophage 1 growth
0 '2 DNA
Packaw I DNi I @0
I Transfect Bacteria
Cel ls
f-
Phage Growth i assembly
-DNA replication -phage particle
Bacterial Cell Lysis
Fig 5A-B. Growth of phage and plasmid DNAs. (A) Plasmids are small circular double- stranded DNAs. Plasmid DNA molecules are depicted here as small circles; the heavy line within each circle represents a segment of DNA inserted within the plasmid DNA. In the labora- tory, bacteria (Escherichia coli) cells can be made permeable to the uptake of DNA. The plasmid enters these competent cells in a process known as transformation, under condi- tions that favor the entry of a single plasmid molecule per cell. Plasmid DNA will replicate in each host bacterial cell, generating many copies of the plasmid within the cell. A single cell with its replicated plasmid DNA is shown in the figure. After transformation, the bacterial cell mixture is plated on agar containing a se- lective antibiotic, and only transformed cells that contain an antibiotic resistance gene car- ried within the plasmid DNA will grow; most plasmids used in recombinant DNA techniques contain an antibiotic resistance gene. Because the bacterial cells are spread across the agar surface at a low density, the plasmid-containing cells will be well isolated from each other. As each of these bacteria cells grow and divide, its plasmid DNA will be distributed to its daughter cells in which the Dlasmid DNA continues to
replicate. Each transformed bacterial cell will form a colony of cells (a cluster of cells, all of which are descended from a single parental cell) containing the plasmid DNA. Cells that carry no plasmid, and therefore no antibiotic resistance gene, have been plated on the agar surface as well, but are not seen because they cannot grow and form visible colonies. (B) Bacteriophage h (lambda) DNA is double-stranded and can exist as a linear or circular form. Phage 3, DNA molecules are depicted here as circles; the heavy line represents a segment of DNA inserted within the phage DNA.To enter bacterial host cells (Escherichia coli), h DNA must be packaged into a protein coat to form infectious phage particles.The phage particles insert the h DNA into the bacteria by a process known as trans- fection. The h DNA enters the host cell in a linear form and then circularizes within the host cell to replicate itself. Generally, only a single phage particle will infect a given cell. In the host cell, the h DNA replicates and new phage particles form. A single cell containing replicated and packaged h DNA is de- picted in the figure.Over time, the phage growth will cause the bacterial cell to lyse, or burst, releasing the phage particles to infect additional host cells. In the laboratory, phage can be grown on an agar surface in a petri dish.The phage are mixed with bacterial cells then plated at a low density such that an infected cell is well isolated from other infected cells on the dish. As the phage grows and is released by cell lysis, the phage particles reinfect additional host cells which in turn are lysed.The cell lysis eventu- ally results in a cleared area against an as yet uninfected bacterial lawn. (Bacteriophage MI3 is a fil- amentous bacteriophage that is not packaged into the same type of protein coat as bacteriophage h; MI3 phage does lyse cells and form plaques.)
riophage lambda (k) , a bacterial virus (or replicates like plasmids within the host cells phage), can accomodate up to 23 kb of DNA. (Fig 5). Very large pieces of chromosomal Fragments of approximately 45 kb can be in- DNA (range, 100-1500 kb) can be recovered serted into cosmid vectors. Cosmids are hy- in yeast artificial chromosomes (YACs). brid molecules derived from plasmid and Yeast artificial chromosomes have been con- phage DNAs. The DNA of these hybrid mol- structed to contain centromeres and telomeres ecules is packaged into phage particles but to allow replication and segregation during

Number 320 November, 1995 Tools of Molecular Biology 253
TABLE 1. Cloning Vectors
Cloning Vector Size of Insert
Plasmid and phagemid < I 0 kb
Bacteriophage h <23 kb
Cosmid <45 kb
Yeast artificial chromosome (YAC) 100-1500 kb
yeast cell division like normal yeast chromo- somes.
DNA Libraries The discussion thus far has described how pieces of DNA are copied or cloned. The next sections will begin to describe how a specific gene of interest may be recovered.
The first step in isolating a particular DNA sequence is to construct a set of recombinant DNA molecules from a source that contains the sequence of interest. This set of clones is called a library and ideally contains all se- quences contained in the original source (ge- nomic DNA or messenger ribonucleic acid [mRNA]). Once a library is constructed, it is screened to identify the specific clone con- taining the DNA sequence of interest.
Genomic Libraries A genomic DNA library includes all DNA se- quences contained in the chromosomes of a cell (genomic DNA). Typically, purified ge- nomic DNA is digested with a restriction en- zyme in a brief incubation, so that the enzyme is not given sufficient time to cut at all the recognition sites for that enzyme. Assuming that the limited number of cuts that are made occur without bias for specific positions along the DNAmolecules, a set of large overlapping fragments will be obtained. These fragments are ligated into bacteriophage DNA, and the recombinant bacteriophage DNA molecules are packaged into infectious phage (viral) par- ticles. These viral particles are incubated with Escherichia coli cells, some of which will be infected by a single phage particle. The phage DNA will replicate in its host bacterial cell,
each bacterium producing DNA from a single recombinant clone. When the phage-cell mix- ture is plated on agar in a petri dish, clear areas of cell lysis (called plaques) appear against a lawn of uninfected bacterial growth (Fig 5B). Each plaque is derived from a single infecting phage particle: The initially infected bac- terium is lysed after the production of multiple new phage particles, releasing the newly syn- thesized phage to infect additional cells.
Complementary DNA (cDNA) Libraries Only a portion (estimated to be approxi- mately 10%) of the total DNAin the human genome contains protein coding gene se- quences. The function of most of the remain- ing DNA sequences of the genome is not un- derstood. However, some nonprotein coding sequences contain gene regulatory se- quences or are transcribed sequences that are removed from the primary RNA transcript (Fig 6). After splicing to remove the introns, the resulting mRNA contains a continuous protein coding region flanked on either side by untranslated regions (UTRs). Messenger ribonucleic acid (mRNA) for a particular gene only will be present in cells that tran- scribe that gene. Some genes are transcribed (or expressed) in all cell types; these fre- quently are referred to as housekeeping genes because they are necessary for general cell maintenance. Other genes are regulated specifically to be expressed in only certain cell types (for example, hemoglobin in red blood cells) or at very specific times (for ex- ample, during development or tissue regen- eration as in fracture healing). Such specific gene regulation is what makes 1 type of cell functionally different from another type.
A genomic DNA library contains the en- tire DNA content of a cell. A complementary DNA (cDNA) library represents the entire RNA content of a cell or tissue. In other words, a cDNAlibrary contains only the gene sequences that are transcribed in the cells from which the library was made. Because any given cell type expresses only a subset of its genes, the cDNA library used as a source for

254 Shore and Kaplan Clinical Orthopaedics and Related Research
Fig 6. Gene and mRNA structure. Genes, located on double-stranded
Chromo~ma, .............. ............ DNA within chromosomes, are the DNA ........ fundamental physical and functional
units of heredity. A gene is an or- dered sequence of nucleotides that contains the coding information for an RNA transcript and the regulatory information that controls the synthe- sis of that transcript. The top portion of the figure depicts a segment of chromosomal DNA with the tran- scribed region of a hypothetical gene shown as boxes (exons: white boxes; introns: striped boxes). By conven-
tion, a gene is depicted with its transcription start site (the beginning of exon 1) to the left.Transcrip- tional regulatory sequences, including the site for RNA polymerase complex binding, occur immediately upstream of the transcriptional start site (to the left of the transcript start site in the fig- ure) in a region called the promoter. (Nontranscribed DNA sequences adjacent to the transcribed re- gion are indicated by dashed lines.) Additional regulatory sequences (not shown) can occur further upstream, downstream, or even within the transcribed portion of the gene. Ribonucleic acid poly- merase synthesizes a single-stranded primary RNA transcript that is colinear with the gene se- quences. For most genes, the primary RNA transcript contains intron and exon regions. The exons (numbered, white boxes) include all the coding sequences for the protein product of the gene, plus nonprotein coding sequences upstream from the initiation codon (start site of translation) and down- stream from the termination codon (end of translation) that are known as UTRs (untranslated re- gions). The introns (striped boxes) are sequences that interrupt the coding sequences of a gene. lntrons are transcribed into RNA, but are removed from the primary RNA transcript when it is processed to form the mature messenger RNA (mRNA).Two other RNA processing steps also occur to RNAs that encode proteins.The first is a modification of the 5‘ end (or start) of the RNA molecule; this occurs as soon as transcription begins and is found on both primary RNA transcripts and mRNA.The other is the addition of a poly(A) tail to the 3’ end of the mRNA.The primary RNA tran- script extends beyond what will be the end of the mRNA; it is trimmed back to a specific site and the poly(A) tail is added. Note that a genomic DNA clone of a gene, which is derived directly from the chromosomal DNA, can contain intron and exon sequences, and regulatory sequences that flank the transcribed regions. In contrast, a cDNA clone is obtained by synthesizing a DNA copy of mRNA; this cDNA will therefore only contain exon sequences.
Start of trandmon (initiation codon) (termination mdon)
End 01 translation
..........
ftr~=ript
Primary RNA transcript
‘paly(A)tall
Stan of translation End Of translation WAAMAAAA mRNA
a specific gene must be derived from cells that transcribe that gene.
This is a very important distinction be- tween these 2 libraries. For example, a hu- man genomic DNA library will contain the same DNA sequences regardless of the cells or tissue from which the DNA is derived. This is because all the cells, with few excep- tions, of an organism have the same DNA content. In contrast, a cDNA library is de- rived from the mRNA contained in a cell or tissue. Since the mRNA content of each tis- sue is different, because of different cellular functions, cDNA libraries will reflect only the genes that are being actively transcribed by the cells of that tissue.
Complementary DNA libraries are con- structed from DNA copies of mRNA. A DNA copy of an RNA will reflect the same informa- tion (nucleotide sequence) contained in the RNA, while giving the additional advantage of manipulations (such as cloning) that are possi- ble with a DNA molecule but not RNA. To construct a cDNA library, total RNA is iso- lated from the cells or tissue of interest and then processed to obtain a fraction enriched for mRNAs. Most eukaryotic mRNAs contain a series of adenine nucleotide residues at their 3’ ends. This run of adenines (which are not encoded within the gene, but are added to the mRNA after transcription) is known as a poly(A) tail (Fig 6). The cellular functions of

Number 320 November, 1995 Tools of Molecular Biology 255
oligo(dT) ceW+ .... .....I.... ... ........ . .... \.. . .. ................. . ... . . . ... . .... .... ...... . . . . . . . . ..:;.:.:..
only mRNA with poly(A) tail will bind lo oligo(dT) celluloSe
\ rRNA and tRNA flow through the column low sah solution
poly(A) mRNA is eluted from the oligo(dT) column wlth a
A A low salt buffer
I
Fig 7. Poly(A) RNA selection. To construct a cDNA library, total RNA is processed to obtain a fraction enriched for mRNAs. Most eukaryotic mRNAs contain a series of adenine nucleotide residues at their 3' ends. This run of adenines (which are not encoded within the gene, but are added to the mRNA after transcription) is known as a poly(A) tail. The cellular functions of the poly(A) tail are not well understood; however, it serves as a useful way to separate mRNA (1 Y0-2Y0 of the total RNA) away from tRNAs and rRNAs. Short oligonucleotides containing only deoxythymidine residues [oligo(dT)] are at- tached to cellulose beads [oligo(dT) cellulose] that are packed into a small column. When total RNA in solution is poured into the column, the poly(A) tails of the mRNA (represented by As in the figure) will hybridize to the oligo(dT) causing the mRNA to bind to the column. Ribosomal RNAs and tRNAs will not bind and are washed away. The poly(A) mRNA can be eluted easily from the column using a low salt buffer that is un- favorable to hybridization of poly(A) to oligo(dT).
the poly(A) tail are not well understood; how- ever, it serves as a useful way to separate &A (1%-2% of the total FWA) away from tRNAs and rRNAs (Fig 7). After the enrich- ment for poly(A) mRNA, an enzyme called re-
mRNA AAAAA 3'
Synthesize cDNA strand from mRNA with reverse transcriptase
AAAAA 3' m 5'
i i
5' First strand 3' <
Remove mRNA and synthesize second DNA strand
cDNA
Double
cDNA 3' stranded ' > y 5'
Fig 8. Complimentary DNA synthesis. Comple- mentary DNA (cDNA) is a DNA copy of an RNA molecule. Most messenger RNAs (mRNAs) are synthesized with a poly(A) tail at the 3' end of the RNA. A short oligonucleotide of poly(dT) can hybridize (complementary base pair) to the poly(A) tail and serve as a primer (provide a 3' OH from which nucleic acid synthesis can ex- tend) for DNA synthesis. Reverse transcriptase is an RNA-dependent DNA polymerase that is able to use an RNA template for the synthesis of a complementary strand of DNA. To form a double-stranded cDNA molecule, the RNA template is removed enzymatically and the second strand of DNA can be synthesized by DNA polymerase using the first cDNA strand as
verse transcriptase (an RNA-dependent DNA polymerase) is used to synthesize cDNA from RNA templates (Fig 8). Reverse transcriptase requires a primer (starting nucleotides with a free 3' hydroxyl group) to initiate synthesis of DNA: either oligo(dT) primers, which will anneal to the poly(A) tail of mRNAs, or random primers, which anneal within the RNA sequence, are used. Through a series of enzymatic steps, the RNA strands of the RNA:cDNA hybrid molecules are removed and replaced by DNA. The double-stranded cDNA molecules are ligated into a cloning vector to create a cDNAlibrary that represents all the transcripts in the starting cells or tissue. Because most cDNAs are only a few kilobases in length, they can be cloned into either a bac- teriophage or a plasmid vector.
Cloned DNA in a plasmid vector is in- serted into bacterial cells for replication through a process known as transformation (Fig 5A). Bacterial cells must first be treated to make their cell walls permeable to the up- take of DNA; these cells are described as be-

Clinical Orthopaedics 256 Shore and Kaplan and Related Research
ing competent. Once the bacteria have been transformed with the recombinant DNA, the cells are grown under selective conditions, usually with an antibiotic that will allow only the cells containing plasmid DNA to grow. When the cells are plated at low den- sity on agar under selective conditions, the cells containing plasmid DNA will grow and replicate forming a colony of cells on the agar surface. Because only a single DNA molecule typically enters a given cell, each colony is synthesizing the DNAof a single recombinant molecule.
At the start of this section, a distinction was made between the gene content of ge- nomic DNA libraries (contain all genes and DNA sequences) and cDNA libraries (con- tain only the expressed gene sequences for a given cell type or tissue). The differences be- tween a cDNA clone and a genomic DNA clone for a given gene also need to be em- phasized (Fig 6). Because a cDNAclone was derived by copying an mRNA, the only gene sequences represented in a cDNA clone are the exons contained in the processed or ma- ture mRNAof the gene. However, a genomic DNA clone for the same gene will contain, in addition to the exon regions, the introns and the sequences flanking the transcribed re- gion which typically contain information that regulates gene expression.
The 2 types of clones, cDNA and genomic DNA, provide useful but different information in gene analysis. For example, the amino acid sequence of a protein can be determined easily from a cloned cDNAbecause its nucleotide se- quence is not interrupted by introns as it is for genomic DNA. The protein and cDNA se- quences can be compared with databases of known sequences to provide clues to the func- tion of the gene. Complementary DNAs fre- quently are used to identify mutations that cause disease or to study structure-function relationships of the protein product. However, other questions can be answered only by iso- lating genes from a genomic DNA library, for example, the examination of the gene se- quences that regulate gene expression (such as
promoter regions) or the analysis of the struc- tural organization of a cluster of genes. Ge- nomic DNA contains intron and exon se- quences, allowing an analysis of mutations that affect splicing.
Screening a DNA Library Once a phage or plasmid DNA library has been plated to separate individual recombinant DNA clones on an agar plate, a method of screening, or probing, for a specific clone con- taining the DNAsequence of interest is needed. The first step in screening a library is to transfer the cell colonies or phage plaques to a solid sup- port, usually a nitrocellulose or nylon filter. A filter disk is placed on the agar surface contain- ing the cells or phage and then carefully lifted off, carrying some of the cells or phage in an exact replica of the position each clone has on the agar surface (Fig 9). The filters are treated to lyse the cells and/or phage and re- lease their DNAs. The DNA then is dena- tured to single strands and fixed to adhere to the filter. The filters containing the recombi- nant DNAs now are ready to be hybridized with a labeled probe to detect specific DNA sequences.
Nucleic Acid Hybridization and Probes The most commonly used screening method is nucleic acid hybridization. Hybridization is the formation of hydrogen bonds between base pairs of complementary strands of DNA or RNA. Under the appropriate condi- tions, single-stranded DNAs can anneal, or hybridize, to one another if their DNA se- qences are complementary (Fig 10). Con- versely, a double-stranded DNA molecule can be denatured to separate the strands into 2 single-stranded DNA molecules.
A probe is a DNA or RNA sequence that carries a tag or label so that it readily can be recognized and detected. Incorporation of a radiolabeled molecule, such as 32P, is used frequently to tag the probe DNAor RNA, al- though nonradioactive methods also can be used. Standard labeling methods for DNAin- clude end-labeling, nick translation, and ran-

Number 320 November, 1995 Tools of Molecular Biology 257
A filter disk is placed on surface of plate
Phage stick a I .
to filter
Filter is lifted from I agar plate ,,
on agar plate
DNA on filter is denatured and hybridized with labeled probe
Hybridized filter is exposed to Xray film to identify ps i t ive clone
Positive clone can be selected from phage plaques on original plate
Fig 9. Screening a recombinant DNA library. Cloned DNAs can be screened for the pres- ence of a specific sequence. In the example shown, recombinant DNA molecules in a phage vector are plated and grown as plaques on agar plates (see Fig 5B). When a filter disk made of nitrocellulose or nylon is placed over the plaques, some of the phage particles will stick to the filter when the filter is lifted from the plate. The filter, called a plaque lift, is a replica of the plaque positions as they occur on the agar surface. The phage particles on the plaque lift are treated to release the phage DNA, and the DNA is denatured to single strands and fixed, or adhered, to the filter.The filter is incubated with a labeled probe specific for the DNA sequence of interest under conditions that promote the hybridization of the probe with phage DNA through the formation of base pairs between complementary sequences (see Fig 10). After hybridization, the filter is washed to remove un- bound probe molecules and then exposed to xray film. Recombinant DNAs containing se- quence complementary to the probe are recog- nized by positive (dark) spots on the film where probe DNA has bound. A positive clone is iden- tified by matching the autoradiogram with the plaque lift and with the original agar plate con- taining the plaques. The plaque corresponding
to the positive signal can be selected for further analysis of its recombinant DNA. This screening method also can be applied to recombinant DNAs in plasmid vectors. After growing colonies on plates (see Fig 5A), colony lifts can be made and processed similarly as plaque lifts.
dom priming (Fig 11). Labeled RNAs are synthesized by in vitro transcription. When a single-stranded probe DNA or RNA is incu- bated with filters containing clones from a recombinant DNA library, the probe will hy- bridize to the recombinant clone because the DNA sequences are complementary to one another. After hybridization and washing to remove unhybridized probe, the filter is ex- posed to xray film. Energy emitted from the radioactive probe (now hybridized to its com- plementary recombinant clone) will cause a dark spot to appear on the film at the position of the positive clone. The film and the origi- nal agar plate containing the recombinant clones are aligned with each other, and the recombinant cell colony or phage plaque containing the positive recombinant DNA can be selected (Fig 9).
Where do probes come from? When no information about the gene of interest is available, the protein of interest is purified and a portion of its amino acid sequence is determined. The genetic code, which relates a single amino acid to its several encoding nucleotide triplet codons, is used to anticipate the putative nucleotide sequence of the gene that encodes the protein. Oligonucleotides (short sequences of nucleotides, Fig 10) cor- responding to the several possible gene se- quences are synthesized to be used as probes for screening a cDNA library. Once a cDNA clone for the gene of interest has been re- covered, the cDNA itself can be labeled and used as a probe to screen a genomic DNA li- brary. As discussed above, cDNA and ge- nomic DNA clones provide important, but different, information for gene analysis. Be-

258 Shore and Kaplan Clinical Orthopaedics
and Related Research
oligonucleotide /
3'C AT A A C G G AT C C G TAT A G G A 5' 5'---- ATG A A ATG C ACT G T ATTG C C T AG G C AT ATC C TAG C G A T----3'
\ target DNA
Fig 10. Nucleic acid hybridization. A target sequence is shown in the 5' to 3' orientation. Nu- cleotides are represented by their base content (A, C, G, T). Above the target sequence, a short se- quence of nucleotides (an oligonucleotide) with a complementary sequence is shown in the 3' to 5' direction (in the antiparallel orientation with respect to the target strand). This oligonucleotide of 20 nucleotides can be referred to as a 20-mer. Two nucleic acids that have complementary nucleotide sequences can form base pairs between the complementary bases, resulting in a region of double- stranded DNA. This process is known as hybridization. If the oligonucleotide is labeled with a de- tectable tag, such as a radioisotope, it can be used to identify a piece of DNA containing a complementary sequence. Hybridization probes can be synthetically made oligonucleotides or se- quences that are up to a few kilobases in length such as a cDNA.
cause of the greater complexity of genomic DNA, it is often difficult to use an oligonucleotide probe to identify a clone of interest from a genomic DNA library. A cDNA clone, however, is used easily for this purpose.
An alternative to screening directly for gene sequences with a nucleic acid probe is to screen for the protein encoded by the gene. In this case, a cDNA library is con- structed using a special type of vector known as an expression vector, which facilitates the translation of the cDNA into a protein. An expression library can be screened using an antibody to the protein product. Expression vectors typically contain bacterial promoters that allow the transcription of the cloned cDNA in Escherichia coli cells. The host cell is capable of protein synthesis from these transcripts. The expression library is plated as has been described above, and then screened by incubating filter lifts of the plated library with an antibody specific for the protein of interest. Detection methods, similar to those used for Western blotting and described in a later section, are used to identify the clone that expressed the protein. The positive clone is recovered, and the se-
quence of the DNA can be readily deter- mined for gene analysis.
Gel Electrophoresis Gel electrophoresis is a technique that sepa- rates DNAmolecules by size and is used to visualize fragments of DNA. Different gel compositions and electrophoretic conditions are used depending on the type of separation required. Apolyacrylamide gel is used to sepa- rate DNA molecules of <500 base pairs. An agarose gel is used to separate DNAmolecules of 300 to 10,000 nucleotide pairs. Pulsed-field gel electrophoresis is a variation of agarose gel electrophoresis that is used to separate very large DNA molecules including entire bac- terial and yeast chromosomes.
For gel electrophoresis, the DNA sample is placed in a well at 1 end of a gel. Because each nucleotide in a nucleic acid molecule carries a single negative charge, when an electric current is applied the DNA migrates toward the positive electrode. The agarose (or acrylamide) matrix acts like a sieve for the DNA, allowing smaller DNA molecules to move more quickly through the matrix and thereby separating the DNA fragments according to their size (molecular weight).

Number 320 November, 1995 Tools of Molecular Biology 259
A Nlck Translation
double stranded DNA treat DNA with DNase to produce single stranded DNA synthesis,
nicks extending from the DNA nick, incorporates labeled dNTPs
B Random Priming
c *.***- 3'
+ 5' *****- 5' 3'
--9 + -***** -***** :: + 3' 5' + 3' 5' 5' 3'
double stranded DNA denature DNA to single strands DNA synthesis, extending from oligonucleotide primers, incorporates labeled dNTPs
Fig 11A-6. Labeling of double-stranded DNA to generate probes for nucleic acid hybridization.Two methods for uniform labeling of double-stranded DNA molecules are shown. In the nick translation method (A), double-stranded DNA is treated with an enzyme (DNase I) that produces single- stranded nicks in the DNA strands. These nicks break the phosphodiester bond between 2 adjacent nucleotides on the same DNA strand. The 3' OH that is now present at the nick site can serve as a primer from which DNA polymerase can synthesize DNA (displacing the existing unlabeled DNA) using the complementary DNA strand as a template. Labeled (such as with 32P) deoxynucleoside triphosphates (dNTPs) are included in the DNA synthesis reaction so that the newly synthesized DNA will contain a detectable tag. (Arrows show the direction of DNA synthesis-always 5' to 3'- and asterisks (*) represent the newly synthesized labeled DNA.) In the random priming method (B), double-stranded DNA is denatured (either by heat or alkali) to single-stranded DNA molecules. The single strands are incubated with short oligonucleotides (typically 6-9 nucleotides) of random se- quences under conditions that favor hybridization of the oligonucleotides to the single-stranded DNAs. Deoxyribonucleic acid polymerase, in the presence of labeled dNTPs, synthesizes DNA ex- tending from the 3' OH of the primers and using the single-stranded DNA molecules as the template to direct the incorporation of complementary nucleotides. In recent years, random priming has be- come more commonly used to label probes for filter hybridization (see Figs 9, 13) than nick transla- tion. In addition to these 2 methods, there are several other ways to generate labeled nucleic acid probes. For example, single-stranded RNA probes can be generated by the transcription of DNA se- quences that have been cloned into a vector that contains a phage RNA polymerase promoter. A phage RNA polymerase is used to synthesize uniformly labeled probes to detect homologous DNA or RNA sequences. Unlike probes that are generated from double-stranded DNA molecules, single- stranded probes have the advantage of avoiding the problem of hybridization competition between the complementary strands of the probe DNA and the target DNA or RNA. In addition to the uniform labeling methods that have been described, DNA molecules also can be tagged by end labeling techniques. End labeling with polynucleotide kinase, which transfers a labeled phosphate to the 5' end of the DNA, is typically the preferred method for labeling oligonucleotides.
All DNA fragments of a given size will mi- dye ethidium bromide. Molecules of ethid- grate at the same rate through the gel. ium bromide insert between the stacked
Deoxyribonucleic acid fragments sepa- bases of a DNA double helix, and when the rated by gel electrophoresis can be visual- stained gel is exposed to ultraviolet light the ized by staining the gel with the fluorescent ethidium-DNA will fluoresce. The length of
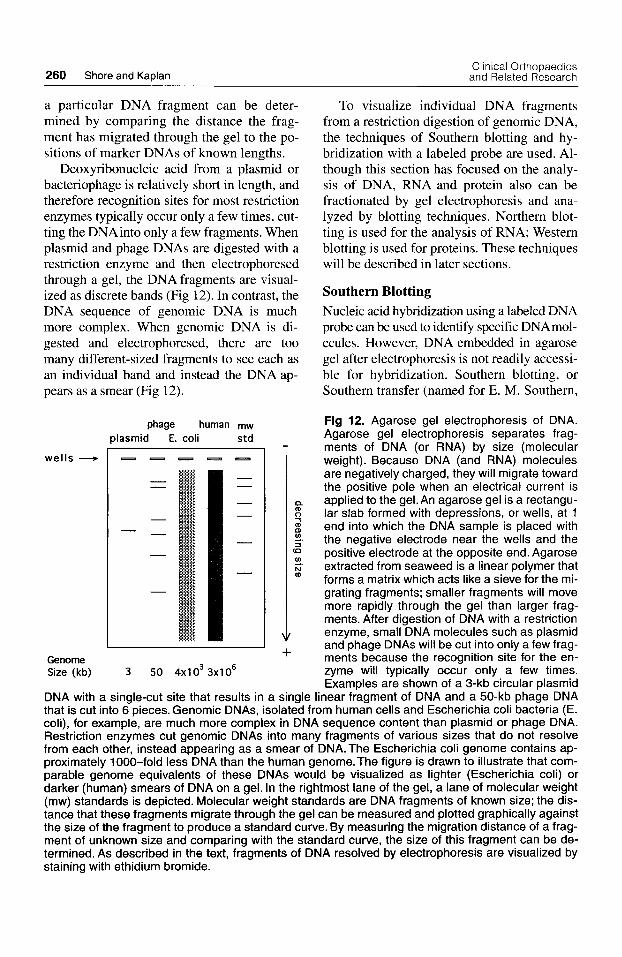
260 Shore and Kaplan Clinical Orthopaedics and Related Research
a particular DNA fragment can be deter- mined by comparing the distance the frag- ment has migrated through the gel to the po- sitions of marker DNAs of known lengths.
Deoxyribonucleic acid from a plasmid or bacteriophage is relatively short in length, and therefore recognition sites for most restriction enzymes typically occur only a few times, cut- ting the DNAinto only a few fragments. When plasmid and phage DNAs are digested with a restriction enzyme and then electrophoresed through a gel, the DNA fragments are visual- ized as discrete bands (Fig 12). In contrast, the DNA sequence of genomic DNA is much more complex. When genomic DNA is di- gested and electrophoresed, there are too many different-sized fragments to see each as an individual band and instead the DNA ap- pears as a smear (Fig 12).
wells -
Genome Size (kb)
phage human mw dasmid E. coli std
a (D
n 1 (D LD
3 (CI
4.
2 N m
+ 3 50 4x103 3x106
To visualize individual DNA fragments from a restriction digestion of genomic DNA, the techniques of Southern blotting and hy- bridization with a labeled probe are used. Al- though this section has focused on the analy- sis of DNA, RNA and protein also can be fractionated by gel electrophoresis and ana- lyzed by blotting techniques. Northern blot- ting is used for the analysis of RNA; Western blotting is used for proteins. These techniques will be described in later sections.
Southern Blotting Nucleic acid hybridization using a labeled DNA probe can be used to identify specific DNAmol- ecules. However, DNA embedded in agarose gel after electrophoresis is not readily accessi- ble for hybridization. Southern blotting, or Southern transfer (named for E. M. Southern,
Fig 12. Agarose gel electrophoresis of DNA. Agarose gel electrophoresis separates frag- ments of DNA (or RNA) by size (molecular weight). Because DNA (and RNA) molecules are negatively charged, they will migrate toward the positive pole when an electrical current is applied to the gel. An agarose gel is a rectangu- lar slab formed with depressions, or wells, at 1 end into which the DNA sample is placed with the negative electrode near the wells and the positive electrode at the opposite end. Agarose extracted from seaweed is a linear polymer that forms a matrix which acts like a sieve for the mi- grating fragments; smaller fragments will move more rapidly through the gel than larger frag- ments. After digestion of DNA with a restriction enzyme, small DNA molecules such as plasmid and phage DNAs will be cut into only a few frag- ments because the recognition site for the en- zyme will typically occur only a few times. ExamDles are shown of a 3-kb circular Dlasmid
DNA with a single-cut site that results in a single linear fiagment of DNA and a 50-kb phage DNA that is cut into 6 pieces. Genomic DNAs, isolated from human cells and Escherichia coli bacteria (E. coli), for example, are much more complex in DNA sequence content than plasmid or phage DNA. Restriction enzymes cut genomic DNAs into many fragments of various sizes that do not resolve from each other, instead appearing as a smear of DNA. The Escherichia coli genome contains ap- proximately 1000-fold less DNA than the human genome. The figure is drawn to illustrate that com- parable genome equivalents of these DNAs would be visualized as lighter (Escherichia coli) or darker (human) smears of DNA on a gel. In the rightmost lane of the gel, a lane of molecular weight (mw) standards is depicted. Molecular weight standards are DNA fragments of known size; the dis- tance that these fragments migrate through the gel can be measured and plotted graphically against the size of the fragment to produce a standard curve. By measuring the migration distance of a frag- ment of unknown size and comparing with the standard curve, the size of this fragment can be de- termined. As described in the text, fragments of DNA resolved by electrophoresis are visualized by staining with ethidium bromide.

Number 320 November. 1995 Tools of Molecular Biology 261
the developer of the technique), is a technique of transferring DNA that has been elec- trophoresed through an agarose gel to a solid support for hybridization.
After restriction digestion and electrophore- sis, the gel containing the size-fractionated DNA is incubated in a solution that contains a strong base (sodium hydroxide) to denature the DNA to separate the complementary strands of the double-stranded DNA fragments. The now single-stranded DNA molecules are trans- ferred to a piece of nitrocellulose or nylon filter by blotting and capillary action (Fig 13A). This process preserves the relative po- sitions of each of the DNAfragments, result- ing in a filter replica of the gel. (The forma- tion of colony and plaque lifts, described above for screening a DNAlibrary, are varia- tions of the Southern blot.) To identify a spe- cific DNA fragment, a labeled probe con- taining the sequences of interest is hybridized with the Southern blot filter. Af- ter washing to remove excess probe, the fil- ter is exposed to xray film. Bands of DNA that have hybridized to the labeled probe will produce dark bands on the developed film. The sizes of the hybridized bands can be de- termined by their positions relative to the po- sitions of molecular size standards.
Southern blotting has become a standard procedure for medical diagnostics. The DNA of a patient can be examined as to whether a particular gene is present, deleted, or altered in that individual. For example, by examining the pattern of restriction fragments (restric- tion map), it can be determined if the gene ap- pears to have a grossly normal structure or if portions of the gene are deleted or rearranged.
DNA Sequencing The information encoded by DNA is con- tained in the sequence of nucleotides of the DNA. Although several methods for deter- mining DNA sequence have been developed, the most commonly used method is dideoxy chain termination.
Dideoxy chain termination is an enzymatic sequencing method that uses a DNA poly- merase to read the sequence of nucleotides
from a single-stranded DNA molecule (tem- plate). The segment of DNA to be sequenced usually is cloned into a vector, allowing suffi- cient quantities of DNA for the sequencing re- action to be obtained. Single-stranded template DNA can be synthesized as single-stranded molecules by cloning the target DNA into a bacteriophage M13 or phagemid vector; alter- natively, single-stranded template can be ob- tained by denaturation of double-stranded DNA that has been cloned into a plasmid phagemid vector.
Deoxyribonucleic acid synthesis by DNA polymerase only can occur by extending the newly synthesized strand from a 3’ OH (hy- droxyl) group. A short sequence of nucleotides that is complementary in sequence to the tem- plate DNA can serve to prime the DNA syn- thesis (Fig 14). No prior knowledge of the tar- get DNA sequence is necessary, because an oligonucleotide primer that has sequence com- plementary to vector DNA can be used to ex- tend DNA synthesis into the target region.
The chain terminators used in this sequenc- ing method are dideoxy forms of the 4 DNA nucleotides (dNTPs: dATP, dCTP, dGTP, dTTP). Dideoxy nucleotides (ddNTPs) can be incorporated into a growing nucleotide chain, but they will block the addition of any further nucleotides caused by the absence of the 3’ hydroxyl (OH) needed for phosphodiester bond formation (Fig 15). When a small amount of 1 ddNTP is included with the 4 dNTPs in a DNA synthesis reaction, there is competition between extension and termina- tion. The result is a set of DNA molecules whose lengths are determined by the distance between the primer used to initiate synthesis and the site of ddNTP termination (Fig 16).
For each template DNA and primer, 4 re- actions are done, each contains the 4 dNTPs (deoxy nucleotide triphosphates) and a chain terminating nucleotide specific for 1 of the 4 DNA nucleotides. The 4 reactions will pro- duce a set of newly synthesized DNA mole- cules that terminate at positions occupied by every nucleotide in the template strand (Fig 16). Asequencing reaction contains some la- beled (usually 35S or 32P) nucleotide so that
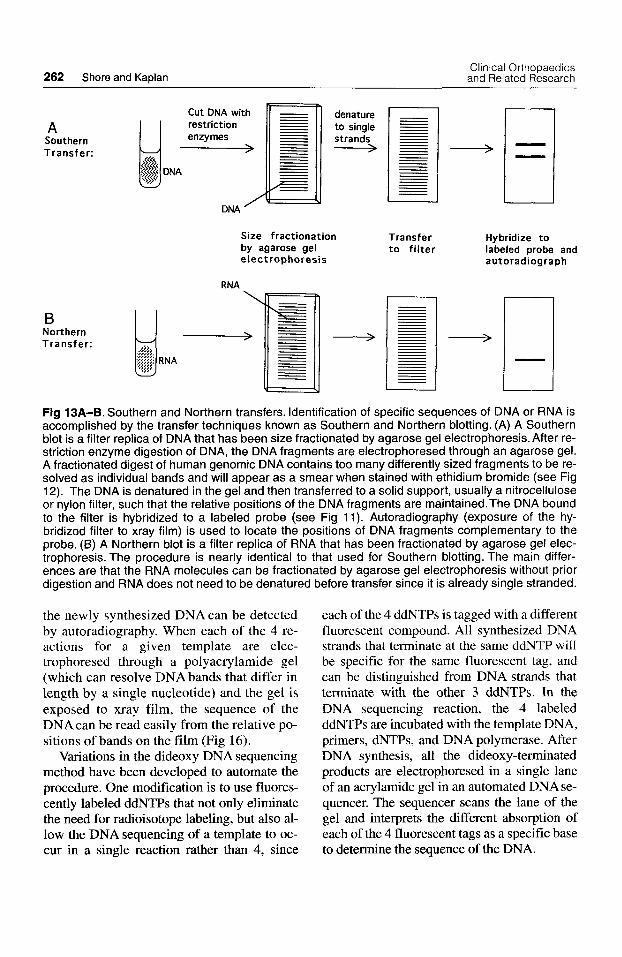
262 Shore and Kaplan Clinical Orthopaedics and Related Research
Size fractionation Transfer Hybridize to by agarose gel to filter labeled probe and electrophoresis autoradiograph
RNA I
Ll Fig 13A-B. Southern and Northern transfers. Identification of specific sequences of DNA or RNA is accomplished by the transfer techniques known as Southern and Northern blotting. (A) A Southern blot is a filter replica of DNA that has been size fractionated by agarose gel electrophoresis. After re- striction enzyme digestion of DNA, the DNA fragments are electrophoresed through an agarose gel. A fractionated digest of human genomic DNA contains too many differently sized fragments to be re- solved as individual bands and will appear as a smear when stained with ethidium bromide (see Fig 12). The DNA is denatured in the gel and then transferred to a solid support, usually a nitrocellulose or nylon filter, such that the relative positions of the DNA fragments are maintained.The DNA bound to the filter is hybridized to a labeled probe (see Fig 11). Autoradiography (exposure of the hy- bridized filter to xray film) is used to locate the positions of DNA fragments complementary to the probe. (6) A Northern blot is a filter replica of RNA that has been fractionated by agarose gel elec- trophoresis. The procedure is nearly identical to that used for Southern blotting. The main differ- ences are that the RNA molecules can be fractionated by agarose gel electrophoresis without prior digestion and RNA does not need to be denatured before transfer since it is already single stranded.
the newly synthesized DNA can be detected by autoradiography. When each of the 4 re- actions for a given template are elec- trophoresed through a polyacrylamide gel (which can resolve DNA bands that differ in length by a single nucleotide) and the gel is exposed to xray film, the sequence of the DNA can be read easily from the relative po- sitions of bands on the film (Fig 16).
Variations in the dideoxy DNA sequencing method have been developed to automate the procedure. One modification is to use fluores- cently labeled ddNTPs that not only eliminate the need for radioisotope labeling, but also al- low the DNA sequencing of a template to oc- cur in a single reaction rather than 4, since
each of the 4 ddNTPs is tagged with a different fluorescent compound. All synthesized DNA strands that terminate at the same ddNTP will be specific for the same fluorescent tag, and can be distinguished from DNA strands that terminate with the other 3 ddNTPs. In the DNA sequencing reaction, the 4 labeled ddNTPs are incubated with the template DNA, primers, dNTPs, and DNA polymerase. After DNA synthesis, all the dideoxy-terminated products are electrophoresed in a single lane of an acrylamide gel in an automated DNA se- quencer. The sequencer scans the lane of the gel and interprets the different absorption of each of the 4 fluorescent tags as a specific base to determine the sequence of the DNA.

Number 320 November, 1995 Tools of Molecular Biology 263
target DNA
5'---- A T G A A AT G C A C T G T A T T G C C T A G G C AT A T C C T A G C G AT----3' < OH 3' A T C C G T A T A G G A T C G C T A 5 , ,
\ direction of DNA synthesis
oligonucleotide primer
DNA polymerase dNTPs (dATP, dCTP, dGTP, dTTP) I
5'---- A T G A A A T G C A C T G T A T T G C C T A G G C AT A T C C T A G C G AT---- 3' 3 1 T A C T T T A C G T G A C A T A A C G G A T C C G T A T A G G A T C G CTA5t
newly-synthesized DNA Fig 14. Primer extension. Deoxyribonucleic acid synthesis by DNA polymerase occurs through the sequential addition of deoxynucleotides to free 3' hydroxyl (OH) groups. A short sequence of nu- cleotides (an oligonucleotide primer) provides the 3' OH to start, or prime, DNA synthesis. For a DNA sequencing reaction, the oligonucleotide is designed and in vitro synthesized to have a nu- cleotide sequence that is complementary to the template DNA strand. Alternatively, the primer can be complementary to vector sequences at a site adjacent to the target sequence. The primer hy- bridizes to the template through complementary base pairing and positions the primer, and therefore the start of DNA synthesis, at a specific location along the template. The specific nucleotide that is added (A, C, G, or T) is determined by the template DNA that directs the addition of a complemen- tary nucleotide. Deoxynucleoside triphosphates (dNTPs: dATP, dCTP, dGTP, dTTP) are the sub- strates used by DNA polymerase for DNA synthesis (see also Fig 15). The direction of DNA synthesis is described as occurring in the 5' to 3' direction.
Eventually, the DNA sequence of the ap- proximately 6 billion nucleotides that en- code the entire human genome will be deter- mined. This information will be invaluable for predicting the amino acid sequence of the protein product of newly identified genes, for providing the background to compare and identify gene mutations in individual pa- tients, and for designing probes and primers to be used for clinical diagnostics.
Polymerase Chain Reaction The polymerase chain reaction (PCR) is a powerful technique that has revolutionized molecular genetics during the past 10 years.
Polymerase chain reaction is used to amplify (make many copies of) a specific DNA or RNA sequence (Fig 17), even if the target DNA is present in very small amounts in a mixture of DNA.
Polymerase chain reaction uses a DNA polymerase to synthesize DNAfrom 2 primers (short oligonucIeotides of typically 18-30 nu- cleotides) that hybridize to opposite strands of the template (or target) DNA. The primers are arranged such that the primer extension reac- tion from each strand directs the synthesis of DNA toward the other primer. In this way, 1 primer synthesizes a DNA strand that can be primed by the other strand. Both the double

264 Shore and Kaplan Clinical Orthopaedics
and Related Research
Deoxyribonucleotide Dideoxyribonucleotide
(dNTP) (ddNTP)
Fig 15. Deoxynucleotides and dideoxynu- cleotides. Deoxyribonucleic acid molecules are chains of deoxynucleotides. Synthesis of DNA occurs through t h e removal of 2 phosphate groups from a deoxynucleoside triphosphate (dNTP) and the addition of the resulting de- oxynucleoside monophosphate to a 3' hydroxyl (OH) group of the growing nucleotide chain.The 3' OH of 1 nucleotide is used for phosphodi- ester bond formation with the next nucleotide. Because a dideoxynucleotide contains no 3' OH group, it acts as a chain terminator for DNA synthesis when incorporated into a growing nu- cleotide chain.
strands of DNA in the target sequence will be replicated, synthesizing a segment of DNAde- fined by the 2 primers (Fig 17).
The DNA polymerase used in a poly- merase chain reaction reaction has been iso- lated from a heat-resistant strain of bacteria. (Several heat stable DNA polymerases cur- rently are available for polymerase chain re- action. The first to be used, Taq polymerase, was derived from the bacteria Thermus aquaticus.) A heat-resistant polymerase will retain its enzymatic activity through the high temperatures, needed to denature the double strands of the DNA template to single strands that can hybridize with the oligonu- cleotide primers. The stability of the poly- merase allows the DNA synthesis reaction to go through many cycles of strand denatura- tion and DNA synthesis, resulting in an ex- ponential amplification of the target DNA.
A polymerase chain reaction reaction con- tains the template DNA, a heat stable DNA polymerase, dNTPs (dATP, dCTP, dGTP, dTIT), the 2 primers, and the appropriate buffer. Each polymerase chain reaction cycle has 3 steps: (1) denaturation (at approximately 95" C) to separate the double-stranded DNA
template to single strands; (2) annealing (at approximately 55" C, depending on the primers used) to allow hybriduation of the primers to the single strands of the template; and (3) primer ex- tension (at approximately 72" C ) to synthesize DNA by extending from the 3' end of the primer (Fig 17). Repetition of these 3 steps over 25 to 30 cycles results in an amplifica- tion of the number of copies of the target se- quence by approximately 1 million-fold (less than predicted with exponential amplifica- tion of 100% efficiency, but a substantial in- crease of the starting DNA nevertheless). With use of polymerase chain reaction ma- chines or thermal cyclers, an amplification cycle takes only approximately 10 minutes.
One major advantage of polymerase chain reaction is the small amount of starting mate- rial necessary for an amplification reaction. Polymerase chain reaction can selectively amplify the DNA or RNA from a few cells, or even a single cell, generating sufficient amounts of DNA to be used, for example, for DNA sequence analysis or as a hybridization probe. In many cases polymerase chain reac- tion eliminates the need to clone a DNA fragment before these analyses; however, polymerase chain reaction does not eliminate the necessity for the initial cloning of a gene because it is necessary to know enough DNA sequence information about the target se- quence to be able to design the primers that are used in the amplification reaction.
When polymerase chain reaction is used to examine an mRNA of interest, a DNA copy first must be synthesized using the same enzyme, reverse transcriptase, that is used to prepare cDNA libraries (Fig 8). Primers and DNA polymerase then are used to synthesize the second DNA strand, fol- lowed by several cycles of amplification as in DNA polymerase chain reaction. This technique is called reverse transcriptase polymerase chain reaction, or RT-PCR.
Polymerase chain reaction is used exten- sively in the molecular analysis of genetic disease, molecular diagnosis, and forensic medicine. Polymerase chain reaction can be

Number 320 November, 1995 Tools of Molecular Biology 265
Fig 16. Deoxyribonucleic acid se- quencing by dideoxy chain termina- tion. Using a single-stranded DNA template, a complementary primer with a 3' OH group, and all 4 nucleoside triphosphates (dATP, dCTP, dGTP, dlTP), DNA polymerase will catalyze chain extension from the primer, and synthesize a DNA strand complemen- tary to the template (see also Fig 14). (top). The template DNA is the target DNA whose sequence will be deter-
*ATATajGTA - TCGTA- W A - IWA- mined. The primer hybridizes to the template through complementary
A ~ T A T ~ T - A T G A T A T ~ T - ATE~TATAQx~T- ATcMA~xAT- base pairing and positions the primer, and the start of DNA synthesis, at a
A - T A T ~ T - A ~ T A T ~ T - specific location along the template. Therefore, the first complementary nu- cleotide that is added to the primer will be the same for every newly synthe- sized DNA molecule generated from that primer and a given DNA template. In the example shown, the first nu- cleotide added is an A, the second is a T, and so on. If DNA synthesis is stopped at intervals, all fragments that are the same length will end in the same
- nucleotide (middle). For the dideoxy chain termination sequencing method,
- dideoxyncleotides (ddNTPs) are used - - G T to terminate DNA synthesis at each of
arate reactions. In the figure, DNAs in each of the 4 reactions are depicted:The top strand shows the nu- cleotide sequence of a portion of the template adjacent to the site of primer binding, and the bottom strand represents the newly synthesized DNA that extends from the primer and is complementary to the template strand. Insertion of a ddNTP is represented by *A, *C, *G, *T. In the A reaction, for example, a combination of dATP and ddATP is included and for each position where an A can be incorporated, ei- ther a dATP or a ddATP can be added. If ddATP (*A) is inserted the chain will terminate, but if dATP (A) is used DNA synthesis will continue incorporating the other dNTPs. When the next A is to be inserted, again DNA synthesis may or may not be terminated (bottom). Electrophoresis and autoradiography is used to detect the DNA extended from the primer. A small amount of a tagged (usually with 32P or 35S) dNTP is included in the sequencing reactions to label the newly synthesized DNA.The fragment lengths of the newly synthesized DNA molecules are determined for each of the 4 reactions by electrophoresis through a high resolution polyacrylamide gel which resolves DNA fragments that differ in length by a single nucleotide. Each DNA fragment synthesized from the A reaction must terminate at an A, and so on.The shortest fragments will have terminated after the addition of only a few nucleotides to the primer. By size ordering the fragments generated from each of the 4 reactions, the DNA sequence of the newly synthesized DNA is determined. The sequence is read from the bottom of the gel (the shortest frag- ments, closest to the primer) to the top of the gel (longer fragments extending from the primer) in the 5' to 3' direction.This sequence is complementary to the template strand.
DNA
S'---ATCGATATAGCCATAGCTGTGATCCGTAATGTAC---3'
3,TCGACACTAGGCATTACATG ' 'Primer I
dATP, dCTP, dGTP, dTTP labeled dNTP
DNA polymerase
+ddATP v +ddCTP +ddGTP + d m P
A m T A T m T - ATGATATAWXT - ATOXTATAGXAT- ATE4TATAQD.T -
%TA - IAGCTATATCGGTA - 'A- TTATATCGGTA-
I A T W A -
IATAmA- %TajGTA- \ 7; ATGATATA-T-
AlO3TATAKO.T- *AGCTATATCGGTA -
ATGATATAKCAT - ATGATATWT-
7 A -
l A C G T
I.1 - - -
5' the nucleotides (A, C, G, or T) in 4 sep-
used in the analysis of specific genes or gene segments from the DNA of individual pa- tients for the analysis of mutation or varia- tions in DNA sequence. Before the availabil- ity of polymerase chain reaction technology,
this was possible only by constructing a cDNA or genomic DNA library from each individual patient and then screening the li- brary for the gene of interest, a procedure that takes many days. Using polymerase

Clinical Orthopaedics and Related Research 266 Shore and Kaplan
4 denature
- 4 anned primers - I - - -
4 DNA synthesis (extenson)
z P 4 denature
- - J anneal primers -
_3 - - - - - - - -
double stranded 5' 3q /DNA template 3' 5'
5' 3' I 4 denature
Cycle '2
Cycle3 etc
3' 5' I
P - - xe
J anneal primers
5' 3'--5' 3' 3' 5'-3' 5 ' 4 DNA synthesis (extension)
Cycle 1
Fig 17. Polymerase chain reaction (PCR). Poly- merase chain reaction is used for the amplifica- tion of a specific region of DNA. Any double stranded DNA can be amplified by this method providing that the sequence flanking the region to be amplified has been determined. This se- quence information is used to design 2 oligonu- cleotides, 1 complementary to each strand of the target template DNA (bold lines), that will serve as primers for DNA synthesis across the region to be amplified. One primer is shown as a white box and the other primer as a striped box. Three basic steps occur in each polymerase chain reaction cycle: denaturation of double- stranded DNA, annealing of primers, and exten- sion of DNA synthesis. In the first cycle, double-stranded template DNA is heated to de- nature, or separate, the double strands. Subse- quent cooling of the DNA in the presence of a large excess of the 2 oligonucleotide primers al- lows the hybridization of the primers to their complementary sequences in the template DNA. Deoxyribonucleic acid polymerase, with the 4 nucleoside triphosphates, extend DNA synthesis from the 3' ends of the 2 primers.The cycle of denaturation, annealing, and extension is repeated, usually 25 to 30 times, to synthesize many copies of the DNA region between the 2 primers. Many of the products of the first few rounds of amplification are heterogeneous in size because extension of the DNA strands can continue past the primer site when copying di- rectly from the template DNA strands. However,
as more strands that end at the primer site are synthesized, the portion of DNA between the primers will be preferentially amplified and becomes the dominant product of the reaction.
chain reaction, the targeted gene region can be amplified (using primers designed from the known sequence of a normal copy of the gene) in a single day, and the DNAs from many patients can be easily analyzed simul- taneously. After polymerase chain reaction, the amplified DNA can be cloned for further analysis or the polymerase chain reaction product can be analyzed directly. In addition to screening for mutations, polymerase chain reaction is also useful to detect viral infec- tions such as acquired immunodeficiency syndrome (AIDS). Even if the virus is pre- sent in low abundance, as long as its nu- cleotide sequence is known, the viral DNA can be detected by polymerase chain reac- tion amplification. Polymerase chain reac-
tion also is becoming of increasing impor- tance in forensics. Although the DNA of all people is very similar, each individual's DNAis slightly different. These differences can be detected through polymerase chain reaction and used to generate a unique DNA profile, analogous to a person's fin- gerprints.
ANALYSIS OF GENE STRUCTURE
Many techniques for analyzing DNA have been described above: gene cloning, gel elec- trophoresis, Southern blotting, DNA sequenc- ing, and chain reaction amplification. In this section, several additional applications of re- combinant DNA technology are described.

Number 320 November. 1995 Tools of Molecular Biology 267
A Restriction Map
0 1 2 3 4 5 6 7 8 9 1 0 k b I
Allele A
0 Probe
H3 RI
Allele B 3* 0 Probe
B Southern blot of EcoRl digest
AA AB BB
6 kb
2 kb
Restriction Mapping and Restriction Fragment Length Polymorphisms
A restriction endonuclease will cut a double- stranded DNA molecule into pieces of DNA known as restriction fragments. The recogni- tion site of each enzyme is specific for a partic- ular short nucleotide sequence (Fig 2). When a sample of the same DNAis digested with a dif- ferent restriction enzyme, cleavage will occur at different sites within the DNA; the sizes and numbers of fragments from the 2 digestions will differ. By using several enzymes and com- binations of these enzymes, a restriction map of the DNA can be constructed (Fig 3). This map shows the positions of each restriction site relative to the others. Restriction maps are use- ful in the process of gene cloning (see above). Specific restriction fragments can be visual- ized after gel electrophoresis and staining with ethidium bromide for cloned DNAs, or by Southern blotting and hybridization with a spe- cific probe for genomic DNA.
Fig 18A-6. Restriction fragment length poly- morphisms (RFLPs). (A) Restriction maps for 2 alleles are shown. Both alleles have recogni- tion sites for Hind Ill (H3), Eco RI (RI), and Xho I (X) at the same locations, with the ex- ception of an Eco RI site that is present in al- lele A (at 5 kb from the leftmost Hind Ill site) but is absent in allele B. (B) Genomic DNA from individuals that are homozygous for al- lele A (AA), heterozygous for alleles A and B (AB), or homozygous for allele B (BB) is di- gested with Eco RI, electrophoresed through an agarose gel, and Southern blotted (see Fig 13A). The blot is probed with the 0.75 kb Xho I-Eco RI fragment from the region of interest (shown in [A] beneath each restriction map, at positions 6.25 to 7 kb). The probe detects a single Eco RI fragment of 2 kb in AA homozy- gotes. This fragment is the result of Eco RI sites at positions 5 kb and 7 kb, as shown in the allele A map above. The probe detects a single Eco RI fragment of 6 kb in BB homozy- gotes. This fragment is the result of Eco RI sites at positions 1 kb and 7 kb, as shown in the allele B map above. The probe detects 2 Eco RI fragments in AB heterozygotes: 1 of 2 kb generated from the A allele and 1 of 6 kb generated from the B allele.
Because restriction sites characterize spe- cific regions of DNA sequence, restriction maps also serve as a means of comparing the same region of DNA from several sources (such as among individuals or between species) without determining a complete DNA sequence. Differences in the size (length of the DNA fragment) and/or number of restriction fragments generated from a DNA region when the DNAs are cut with the same restriction en- donuclease are known as restriction fragment length polymorphisms (RFLPs)(Fig 18). The variation in a DNAregion detected by an re- striction fragment length polymorphism re- flects differences in the DNA sequences being compared. This difference may be attributable to a nucleotide alteration that changes the re- striction endonuclease recognition site itself, or can reflect a mutation such as an insertion or deletion of DNA sequence in the restriction fragment that is generated.
Southern blotting and restriction fragment length polymorphism analysis are used to

Clinical Orthopaedics 268 Shore and Kaplan and Related Research
examine variation among individuals of a family, leading to the recognition of poly- morphisms that represent either normal vari- ation in the population or variation that may be associated with a particular disease phe- notype (Fig 18). In many cases an restriction fragment length polymorphism is a marker that is linked, or coinherited, with the dis- ease allele.
Genetic Linkage Analysis and Positional Cloning Genetic mapping through genetic linkage analysis and positional cloning have become very important in medical genetics. These are the only methods that allow the identifi- cation of some genes, such as certain disease genes, for which the gene product has not been identified yet. Functional cloning, which has been the traditional strategy for cloning genes, begins by identifying and pu- rifying the protein product of the gene, fol- lowed by determining a partial amino acid sequence of the protein and using that infor- mation to design oligonucleotide probes that can be used to screen a library and recover the gene that encodes the protein of interest.
In contrast, positional cloning relies only on the chromosomal location of the gene. Genetic linkage analysis maps the disease gene to a specific chromosomal position. Genes in that chromosome region then are identified, cloned, and examined for correla- tion to the disease in question. The RNA and protein products of genes identified in this manner can be characterized and examined to reveal the cellular pathways involved in the disease condition.
When 2 genes are located on different chromosomes, they are not linked, and the probability that any allele of the first gene will be coinherited with a specific allele of the second gene is random, or 50%. This also is true for 2 genetic loci that are located far apart on the same chromosome, because a crossover event will almost always occur be- tween these loci. The closer that 2 genes or genetic markers are located on the same
chromosome, the greater the probability that specific alleles for these genes will be inher- ited together or show linkage.
In genetic linkage analysis, polymorphic markers distributed throughout the chromo- somes are tested for coinheritance with a particular disease phenotype within a family.
Linkage analysis is facilitated greatly by large numbers of polymorphic markers spaced at small intervals along the DNA of the chromosomes, and the efforts of many laboratories during the past few years have re- sulted in a catalog of such markers. Many of the useful markers are restriction fragment length polymorphisms, but the most of the markers are derived from polymerase chain reaction amplification of short tandemly re- peated sequences.
Scattered throughout the human genome are many loci that contain a variable number of identical sequences joined together in tan- dem. These variable number tandem repeat (VNTR) sequences are 2 to 100 nucleotides long and occur in 3 to 60 or more copies. Mi- crosatellites, or short tandem repeat polymor- phisms (STRPs), form a subgroup of variable number tandem repeat sequences that consists of 1 to 5 nucleotides repeated in a tandem arrangement. At a specific chromosomal lo- cus, the number of repeats in an short tandem repeat polymorphism is highly variable among individuals, such that only individuals who are related to one another will tend to contain a spe- cific short tandem repeat polymorphism with the same number of repeats. Because short tandem repeat polymorphisms are very poly- morphic, they are extremely useful as genetic linkage markers. The number of repeats within an short tandem repeat polymorphism at a specific chromosomal locus can be de- tected in individuals through polymerase chain reaction methodology. Polymerase chain reaction primers are designed from the sequence flanking the repeat region, and used to amplify the repeat region from an individual’s genomic DNA. The poly- merase chain reaction products are elec- trophoresed through polyacrylamide gels that

Number 320 November, 1995 Tools of Molecular Biology 269
can resolve amplified DNAs that differ in as little as 1 nucleotide in length. By comparing the inheritance pattern of the marker among affected and unaffected members of a family with the presence of the disease, linkage can be established or excluded.
In addition to their use as genetic linkage markers, variable number tandem repeat se- quences have important uses in forensic med- icine and identification. Some variable num- ber tandem repeat sequences, such as short tandem repeat polymorphisms, are detected readily by polymerase chain reaction tech- niques as described above. Other variable number tandem repeat sequences are detected through restriction digestion and Southern blotting of genomic DNA (Fig 19). If the blot is incubated with a probe that will hybridize only to a single locus containing the variable number tandem repeat sequence (for exam- ple, the probe contains unique sequence adja- cent to the repeat region), the 2 alleles for that locus in an individual can be visualized. An example of the inheritance of allelic poly- morphisms at a single locus is shown in Fig- ure 19. The same blot also can be incubated with a probe that will hybridize to the re- peated sequence. This method will not iden- tify a specific variable number tandem repeat sequence at a single chromosomal locus, but instead will identify multiple loci that con- tain that repeated sequence. The pattern of DNA fragments generated from the DNA of a single individual is essentially unique for that person. Use of variable number tandem repeat sequences to compare the identities of 2 people in this manner is known as DNA fingerprinting.
Physical Mapping of Genes Cloned DNAs (cDNA or genomic) can be used to identify the chromosomal location of a gene. This can be important in associating a specific gene with a disease, for example, when a disease has been genetically mapped to a chromosomal locus or the disease is asso- ciated with a chromosome translocation. Two methods for identifying the chromosome on
which a gene is located are fluorescence in situ hybridization (FISH), which hybridizes a probe labeled with a fluorescent tag to chro- mosomes and determines the gene location microscopically, and hybridization to a so- matic cell hybrid mapping panel, which uses restriction enzyme digestion of DNA, South- ern blotting, and hybridization to correlate probe hybridization with a specific human chromosome or chromosome region.
METHODS OF RNA ANALYSIS
Many of the techniques applied to DNAanalysis can also be applied with slight variations to RNA analysis, such as gel electrophoresis, blotting to a membrane, and hybridization. Other methodolo- gies, such as restriction endonuclease diges- tion, cloning, and sequencing, can be applied only to DNA. One way to circumvent these limitations is to synthesize a complementary DNA copy (cDNA) of the RNA of interest (Fig 8) and then analyze the cDNA.
To understand gene function and regula- tion, it is necessary to examine the cells and tissues in which a particular gene is active and determine the abundance of the RNA transcript and the timing of its expression. Several methods are available for the detec- tion of RNA and are described briefly.
Northern Analysis
Northern analysis is very similar in procedure to Southern analysis (see above), except that in Northern analysis RNA is analyzed (Fig 13B). After isolation of RNAfrom cells or tis- sue, the RNA is size fractionated by agarose gel electrophoresis. Unlike DNA that is di- gested with restriction enzymes before elec- trophoresis, the lengths of RNAs are typically in the size range that will resolve easily on an agarose gel. A replica of the gel is made by transferring, or blotting, the RNAfrom the gel to a piece of nitrocellulose or nylon mem- brane. Specific RNAs are detected by hy- bridization with a labeled probe, followed by au toradiography.

270 Shore and Kaplan Clinical Orthopaedics and Related Research
Allele
1
2
3 4
5
6
7
8
Fig 19. Detection of allelic polymorphisms. A hypothetical example of the inheritance of alle- les for a single locus in a family is described. The top portion of the figure shows the family pedigree. Circles are females and squares are males. The parents (5 and 6) and grandparents (1 and 2; 3 and 4) of 6 children (7-12) are rep- resented. Genomic DNA from each individual can be obtained easily from the white blood cells in a peripheral blood sample. In this exam- ple, it has been previously determined that the DNA at the locus of interest shows 8 polymor- phic variants, or alleles, when the DNA has been cut with a particular restriction endonucle- ase. The polymorphic variants are restriction fragments of unique lengths that can be re- solved by agarose gel electrophoresis (see also Fig 18). After digestion of the DNA from each individual with the restriction endonuclease, the DNA is electrophoresed through an agarose gel. The gel is Southern blotted and the filter is hy- bridized with a labeled probe.The probe is comple- mentary to DNA sequences that occur at a single site, or locus, of interest within the chromosomal DNA. After probe hybridization, the filter is exposed to xray film, and the resulting autoradiogram is shown below the pedigree. Each lane of the au- toradiogram corresponds to the DNA of the indi- vidual in the pedigree with which it lines up. Each individual is found to contain 2 different al- leles for the locus examined, and the inheri- tance pattern of the alleles can be followed. For example, individual 5 has inherited a ’1’ allele from his father and a ‘5’ allele from his mother; individual 6 has inherited a ‘7’allele from her fa- ther and a ‘8’ allele from her mother. This type of analysis can be used to examine family rela- tionships, or used as a diagnostic tool if a spe- cific allele is found to be associated with a disease phenotype.
Northern analysis can be used to deter- mine the relative abundance of a particular mRNA, for example, in comparing the RNA from a normal tissue with that from a disease tissue. Northern analysis also will reveal the size of the detected RNA, indicating, for ex- ample, if a mutation has caused a shortened form of the mRNA to be synthesized.
The RNA that is applied to the gel may be either total cellular RNA or the poly(A) frac- tion of the total RNA. Poly(A) RNA, or polyadenylated mRNA, is the RNA fraction that is translated during protein synthesis. Abundant mRNAs can be detected by North- em analysis of total RNA; however, less abun- dant mRNAs are detected more easily when enriched in the poly(A) fraction (Figs 6,7).
Ribonuclease Protection Assay An alternate method for the detection of a specific RNA is the ribonuclease protection assay. In this method, a short labeled RNA probe (typically 100-500 nucleotides) spe- cific for the mRNAof interest is hybridized in solution to the isolated cellular RNA. The hy- bridization reaction is treated enzymatically to preferentially digest single stranded RNAs (unhybridized molecules and single stranded ends on hybrid molecules). The resulting RNA-RNA hybrids then are electrophoresed through a high resolution polyacrylamide gel for identification and quantification.
The ribonuclease protection assay is more sensitive than Northern analysis in detecting very low amounts of a specific mRNA. This method also allows for quantitation of the RNA. It cannot, however, determine the size of the synthesized mRNA because only the protected fragment of RNA corresponding to the size of the probe will be detected.
In Situ Hybridization When RNA is extracted from cells or tissue, the information about the localization of the RNAis lost. In situ hybridization is used to identify the distribution of specific RNAs in intact cells and tissues. Cells or sectioned tis- sue are prepared on microscope slides and

Number 320 November, 1995 Tools of Molecular Biology 271
treated using methods that retain the in- tegrity of the RNA and favor hybridization of a labeled probe to the RNA. The probes that are used for in situ hybridization can be DNAor RNAprobes, and can be tagged with radioactive, histochemical, or fluorescent la- bels. The same hybridization principles that apply to Northern and Southern analysis ap- ply to in situ hybridization as well. After hy- bridization, the distribution of the target RNA is determined microscopically. (Note that the term in situ hybridization also is used to describe the hybridization of a la- beled probe to the DNA in chromosomes.)
METHODS OF PROTEIN ANALYSIS
Much information about normal and abnor- mal gene function can be understood by the examination of gene structure. However, to understand the effect that a gene defect has on its function the protein products need to be examined.
Western Analysis Western blotting is analogous to Southern and Northern blotting but applies to the analysis of proteins rather than nucleic acids. Western blotting is used to determine the size and relative abundance of proteins. As with Northern analysis, this methodology can provide information on the differential expression of the gene product in a patient with a genetic disease.
Proteins are extracted from cells or tissue and size-fractionated by electrophoresis through a polyacrylamide gel. The gel typi- cally contains sodium dodecyl sulfate that binds to proteins to give them a negative charge, allowing them to migrate to the posi- tive electrode during electrophoresis. Be- cause of the small pore size of a polyacry- lamide gel, transfer of the proteins to a nylon membrane is facilitated by electrophoretic transfer (rather than capillary action which is typically used for nucleic acid transfer from agarose gels). The filter containing the frac- tionated proteins is incubated with antibod-
ies that will specifically bind the target pro- tein. This antibody-protein complex is de- tected by incubation with a labeled sec- ondary antibody detected by a radiographic, fluorescent, or histochemical signal.
Immunohistochemistry Immunohistochemistry methods are analo- gous to in situ hybridization for nucleic acids and are used to detect the distribution of a specific protein within a cell or tissue. The localization of a protein to specific cells of a tissue or to specific sites within the cell (such as the cell membrane, nucleus, or cy- toplasm) provides important information about the function or misfunction of the pro- tein. As in Western blotting, a specific anti- body serves as the probe to detect the pro- tein of interest.
ANALYSIS OF TRANSCRIPTIONAL REGULATION AND GENE EXPRESSION
Many genetic defects cause disease because an incorrect form of the gene product is syn- thesized. However, disease also can be the result of the over or under abundance of a gene product, or the synthesis of that product at an inappropriate place or time. In these in- stances, the genetic defect affects the regula- tion of the gene expression, often at the tran- scriptional level. Analysis of gene regulation requires the identification and characteriza- tion of the regulatory regions of the gene: the regions of a gene that encode binding sites for the RNA polymerase complex and for other promoter DNA binding proteins that activate or repress the synthesis of the RNA transcript by RNA polymerase.
The promoter is the gene regulatory region that determines the position of transcription initiation by directing the site of RNA poly- merase complex binding to the gene. It is lo- cated upstream (away from the direction of transcription) of the mRNA start site that codes for the 5’ end of the mRNA(Fig 6). Ad- ditional DNA sequences (or elements) within

272 Shore and Kaplan Clinical Orthopaedics and Related Research
the promoter region can direct the rate and tis- sue specificity of mRNA synthesis.
By mapping the precise location of the RNA start site of an mRNA, the position of the promoter region can be determined. Sev- eral techniques currently are used to map the 5’ ends of mRNAs, including primer exten- sion, S 1 nuclease digestion, and rapid ampli- fication of cDNAends (RACE) analysis.
Once a promoter region has been identi- fied and cloned, analyses to identify the DNA binding sites for transcriptional regula- tory proteins can be done and sequence spe- cific DNA binding proteins can be isolated and identified. DNA sequencing of the pro- moter will reveal DNA sequence elements that have homology to known transcription factor binding sites; however, actual use of these sites must be verified either by evi- dence of specific protein binding or by a functional assay showing the influence of the sites on transcription. Techniques such as DNA footprinting and the gel mobility shift assay are used to confirm protein binding to a specific promoter sequence. Promoter re- gions can be tested functionally in reporter gene expression assays.
Gel Mobility Shift Assay Age1 mobility shift assay (also known as a gel retardation assay) is a means of showing the binding of proteins to a promoter region. In this assay, a specific segment of a pro- moter region is labeled with a radioisotope tag and incubated with a nuclear protein ex- tract. (Because transcription occurs within the nucleus of a eukaryotic cell, the tran- scriptional regulatory proteins can be recov- ered in a nuclear extract.) The mixture is electrophoresed through a polyacrylamide gel. If proteins bind to the promoter DNA, the resulting DNA:protein complex will mi- grate more slowly through the gel than DNA alone. The identity and specificity of binding can be determined by competition of protein binding using oligonucleotides.
Gel shift assays can be used to identify binding of previously characterized regula-
tory proteins. However, novel transcription factors can be identified by this method as well. The DNA:protein complex can be re- covered from the gel and the proteins purified and analyzed. This methodology will help to identify the nuclear proteins that regulate the transcriptional activation of a gene of interest.
DNA Footprinting In DNAfootprinting, a specific segment of a promoter region is labeled with a radioisotope tag and incubated with a specific transcription regulatory protein. The DNA:protein complex is partially digested with DNase I. The DNA regions that are protected from digestion due to protein binding are identified after elec- trophoresis through a high resolution polyacry- lamide gel. This technique helps determine the exact region of DNA:protein interaction in- volved in the regulation of gene activity.
Reporter Gene Expression Assays To study the function of putative gene regula- tory regions, these regions can be cloned into an expression vector containing a reporter gene. The inserted DNA fragment sits adja- cent to a gene that encodes a protein that read- ily can be detected in an assay (reporter gene). The cloned DNA is transfected into mam- malian cells in culture. If the DNAcloned into the expression vector contains sequences that promote transcriptional activation, an mRNA will be synthesized from the reporter gene and the protein product will be made and detected by an appropriate assay. Several reporter sys- tems are currently in use, including chloram- phenicol acetyltransferase and firefly lu- ciferase. In a typical experimental approach, regions of DNA sequence in the promoter are tested systematically to determine their effect on the level of reporter transcript that can be synthesized. In this way, functional regulatory sequences can be identified.
Transgenic Mice Analysis of gene expression in cell culture (in vitro) can provide much information about the regulation of gene expression. However,

Number 320 November, 1995 Tools of Molecular Biology 273
many questions, such as the genetic control of mammalian development, require an animal (in vivo) system. In recent years, technical ad- vances have provided the ability to introduce foreign genes into mice (forming transgenic mice) and to examine the pattern of expres- sion and the phenotypic effects of the intro- duced gene (called a transgene).
Transgenic mice are produced by mi- croinjecting several hundred copies of the cloned gene into fertilized mouse eggs. The eggs then are transferred to a foster mother where some of the injected eggs will develop to term and contain the foreign DNA inte- grated into the mouse chromosomes. If the transgene is integrated within the chromo- somes of the germ line cells, the transgene will be stably inherited by the progeny of the transgenic mouse.
Although the site of DNA integration ap- pears to be random regarding chromosomal location, the transgene often is expressed in a tissue-specific pattern similar to that of the endogenous copy of the gene. This allows the examination of expression of the gene or vari- ants of the transgene to be examined. The in- sertion site of a transgene also can be within a chromosomal location that contains a gene which, now interrupted, results in a mutant phenotype. Characterization of the disrupted locus has led to the identification of previ- ously unknown genes. Transgenes also can be targeted to be expressed in specific tissues: Cloning the gene of interest with a tissue spe- cific promoter will direct its expression to oc- cur only in tissues which can activate that pro- moter. The timing of transgene expression also can be regulated by cloning the gene of interest with an inducible promoter that can be activated at specific times.
Valuable information on protein function also can be gained through observation of the effects of the absence of a specific gene product. Knockout mice are a type of trans- genic mouse which results in a null mutation for the gene of interest. Through homolo- gous recombination, which targets the trans- gene to the site of the endogenous copy of
the gene in the mouse chromosomes, the en- dogenous mouse gene can be disrupted. Mice homozygous for the disrupted gene are bred and examined.
SUMMARY
The techniques described in this article have been developed during the past 25 years and provide a powerful set of tools that have rev- olutionized the understanding of gene struc- ture and function in health and disease. All these tools readily are applicable to a more fundamental understanding of the genes in- volved in the embryogenesis, maintenance, repair, regeneration, and aging of the muscu- loskeletal system. Many of these techniques are being used daily in medical research lab- oratories, in medical diagnostic laboratories, and in forensic medicine laboratories. This article presented the basic tools and princi- ples of molecular biology, but only touched on the many variations and applications of this technology. It is incumbent on the in- formed physician to understand the princi- ples behind these methods, their possible use, and their inherent limitations. They will con- tinue to transform our world, enlighten our un- derstanding of the biologic basis of our exis- tence, and enhance our ability to prevent and treat human disease.
Further Reading Alberts B, Bray D, Lewis J, et al: Molecular Biology of
the Cell. Ed 3. New York, Garland Publishing, Inc 1994.
Drlica K: Understanding DNA and Gene Cloning: A Guide for the Curious. Ed 2. New York, J Wiley and Sons, Inc 1992.
Lewin B: Genes V. New York, Oxford University Press 1994.
Sambrook J, Fritsch EF, Maniatis T Molecular Cloning: ALaboratory Manual. Ed 2. New York, Cold Spring Harbor Laboratory Press 1989.
Shore EM, Kaplan FS: Molecular Biology for the Clini- cian. Part 1 General Principles. Clin Orthop 306:264-283, 1994.
Thompson MW, McInnes RR, Willard HF: Genetics in Medicine. Ed 5. Philadelphia, WB Saunders Com- pany 1991.
Watson JD, Gilman M, Witkowski J, Zoller M: Recom- binant DNA, Ed 2. New York, WH Freeman and Company 1992.

274 Shore and Kaplan Clinical Orthopaedics and Related Research
GLOSSARY
5‘ and 3’ Nucleic acids have 5’ (5 prime) and 3’ (3 prime) ends, designating the orientation of the ri- bose (in RNA) or deoxyribose (in DNA) sugar- phosphate backbone of the polynucleotide. Synthesis of DNAor RNAalways occurs by the ad- dition of a nucleotide to the 3’ end of a polynu- cleotide; synthesis is described as occurring in the 5’ to 3’ direction. By convention, the end of a gene where the promoter region is located (and therefore the position of the transcription start site) is called the 5’, or upstream, region; the opposite end is the 3‘, or downstream, region.
A,C,G,T Abbreviations for the 4 types of nucleotides in DNArepresenting the base contained in each nu- cleotide: A (adenine), C (cytosine), G (guanine), or T (thymine). In a double-stranded DNA molecule, the 2 polynucleotide chains are held together by hy- drogen bonds between pairs of bases: Aalways pairs with T C always pairs with G. (Also see: nucleotide, base) Abbreviation for the nucleotide base uracil, which replaces thymine (T) in RNA.
Alleles are alternate forms of a genetic locus. For each locus, a single allele is inherited from each parent.
A set of related sequences, each about 300 bp long, that are dispersed throughout the human genome. The named was derived from the Alu I cleavage site that each element contains.
amino acids The units, or building blocks, of proteins. antisense strand of DNA The strand of double
stranded DNA that is complementary to mRNA. This strand is the transcribed DNA strand, serving as the template for RNAsynthesis, sometimes referred to as the noncoding DNA strand (Also see sense strand).
The 2 polynucleotide strands of the DNA double helix associate with each other in opposite orientations: the 5‘ end of 1 strand is aligned with the 3’ end of the other.
Achromosome that is not 1 of the sex (X or Y) chromosomes.
A bacteria virus. Bacteriophage DNAs are used as cloning vectors. Two types of bacterio- phage commonly used are bacteriophage lambda (h) and bacteriophage M13.
The nitrogenous base that is part of the nucleotide building blocks of DNA and RNA. Frequently, the name of the base is used as a shorthand term for the nucleotide because the base is the distinctive component of each nucleotide. When referring to double-stranded DNA, the term base often is used instead of base pair.
Two complementary nucleotide bases joined by hydrogen bonds. The 2 strands of double stranded DNA are held together through the hydro- gen bonds of base pairing. C always pairs with G; A pairs with T (in DNA) or U (in RNA).
A highly conserved DNA sequence or secondary (folded) structure of DNA. For example, the TATA box, which is recognized and hound by the RNA polymerase I1 complex.
U
allele
Alu family
antiparallel
autosome
bacteriophage
base
base pair (bp)
box
cap A modified nucleotide (7-methyl-guanosine) that is added to the 5’ end of mRNAs. Appears necessary for processing, stability, and translation.
cDNA Complementary DNAor copy DNAis synthetic DNA that has been copied from an mRNA template through the activity of the enzyme reverse transcrip- tase. cDNA, distinct from genomic DNA, will in- clude only those regions of the genome that are contained in mature RNA (exons).
chromatin The compact structure of double-stranded DNA and associated proteins that compose the chro- mosomes.
chromosome An autonomous unit of the genome that contains many genes. Each chromosome contains a single long molecule of double-stranded DNA that is complexed with proteins to form chromatin.
This term is used to describe a DNA molecule produced by recombinant DNA technology. Most accurately, a clone is a large number of identical cells or molecules that are derived from a single an- cestral cell or molecule.
clone
coding DNA strand coding region The RNA coding region of a gene is
the DNA segment that is transcribed to form an RNA. The protein coding region is the segment of mRNA that is translated into the amino acid se- quence of the protein product of a gene. Note that although the protein coding region is always con- tained within the mRNA coding region, the mRNA coding region will include segments that are not used for protein coding.
codon A sequence of 3 consecutive bases (a triplet) in a DNA or RNA polynucleotide that specifies an amino acid or a stop signal for translation. (Also see degeneracy.)
Ends of double stranded DNA mole- cules in which 1 of the strands extends further than the other. The protruding strand is single stranded and therefore has the capacity to base pair with an- other single stranded end that contains a comple- mentary base pair sequence. This forms a basis for joining 2 DNA molecules together. Cohesive ends are also known as sticky ends.
colinearity The linear array of nucleotide base se- quence of DNA (and the RNA transcribed from that DNA) directly corresponds to the sequence of amino acids in the protein encoded by that DNA (or RNA).
colony A visible cluster of bacterial cells, usually grown on an agar surface. All cells within a colony are derived from a single parent cell; therefore, all the cells are identical. The colony often is referred to as a clone.
An idealized sequence of nu- cleotides derived from comparison of several DNA sequence elements with similar function. The nu- cleotide at each position in the consensus sequence is the 1 most frequently found at that position. For example, a novel DNAsequence can be screened for similarity to the consensus sequence of the TATA box to predict potential sites of RNA polymerase I1 complex binding.
An artificially constructed cloning vector that has been derived from plasmid and phage DNAs. Cosmids contain the cos gene of bacteriophage
See sense strand of DNA.
cohesive ends
consensus sequence
cosmid

Number 320 November. 1995 Tools of Molecular Biology 275
lambda and can be packaged into lambda phage par- ticles for transfection into Escherichia coli cells, but are able to replicate like plasmids within the host cells. Cosmids facilitate the cloning of larger frag- ments than can be obtained with plasmid vectors.
degeneracy The genetic code is described as degener- ate because of the redundancy in coding specificity: Most of the 20 amino acids are encoded by more than 1 of the 64 codons.
The loss of a portion of DNA; it can be a sin- gle base or a large segment of a chromosome.
An enzymatic DNA se- quencing method that depends on termination of DNA synthesis at specific nucleotides.
Nucleotides that con- tain no hydroxyl group at either the 2‘ or 3’ position of the ribose. DNA synthesis cannot be extended from a ddNTP by DNA polymerase because there is no 3’ hydroxyl group with which to form a phospho- diester bond.
A cell that contains 2 homologs of each chromosome. A human diploid cell contains 46 chromosomes: 2 copies of each of the 22 autosomes plus 2 sex chromosomes.
DNA (deoxyribonucleic acid) DNA is a polynu- cleotide that contains the code of information (genes) that specifies the cellular functions of all liv- ing organisms (with the exception of some viruses that use RNAas their genetic material).
The process of using highly poly- morphic DNA markers, such as restriction fragment length polymorphisms and variable number tandem repeats, to compare the identities of individuals.
DNA ligase An enzyme that joins the ends of 2 DNA molecules together by catalyzing the formation of phosphodiester bonds between 3’OH and 5’P ends.
DNA polymerase An enzyme that synthesizes DNA. DNA synthesis DNA synthesis always occurs in the
5’ to 3’ direction and requires an existing DNA strand to serve as a template. The newly synthe- sized DNA strand has a complementary base se- quence relative to the template strand, and therefore is able to hydrogen bond through complementary base pairing to the template DNAstrand.
downstream A term used to describe sequences pro- ceeding further in the direction of expression of a gene. For example, the coding region is downstream of the transcriptional start site. It also is used to refer to the noncoding region flanking the 3‘ end of the RNAcoding region of a gene.
enhancer Transcriptional regulatory DNA sequences (or elements) that can function from upstream or downstream of the transcribed region of a gene and from distances of several kilobases. Enhancers only function in cis; therefore, they only can affect genes on the same chromosome.
Atranscribed region of a gene that is present in a mature (postsplicing) RNA. An exon can contain translated regions and untranslated regions (UTRs).
A set of DNA fragments (typically cDNAs) that have been cloned into a vector that fa- cilitates the translation of the cDNA into a protein.
FISH (fluorescence in situ hybridization) A hy- bridization technique that uses fluorescently labeled
deletion
dideoxy chain termination
dideoxy nucleotides (ddNTPs)
diploid cell
DNA fingerprinting
exon
expression library
probes to locate complementary molecules in chro- mosomes or tissue sections.
DNA sequences that are upstream or downstream of the transcribed region of a gene.
A deletion or insertion in DNA that is not a multiple of 3 nucleotides. This changes the reading frame (the selection of triplet bases that form codons), resulting in a protein that contains an altered amino acid sequence, typically of an incor- rect length.
functional cloning Gene cloning that is accomplished by first identifying and partially characterizing the protein product of the gene.
gel electrophoresis A method of separating molecules (DNA, RNA, or protein) based on their sizes and electrical charge. The molecules are drawn through a gel matrix (such as agarose or acrylamide) by an electrical field. Small molecules will migrate through the gel faster than larger molecules (assum- ing charge and conformation have neutral effects).
A unit of heredity. In molecular biology, a seg- ment of genomic (chromosomal) DNA that is re- quired for production of a functional product (protein or RNA). A gene contains coding and regulatory re- gions.
Describes the process by which the DNAsequences of a gene are converted into RNAor RNA and protein.
genetic code The triplet codons that specify the 20 amino acids. The genetic code interprets nucleotide (DNAand RNA) language into protein.
Chromosomal loci (genes or DNA markers) on the same chromosome show link- age if they have a tendency to be inherited together. Linkage analysis within families is used to show linkage of 1 or more chromosomal loci with a partic- ular disease phenotype. In this way, disease genes can be identified through positional cloning.
genome The entire genetic information contained within the complete DNA sequence of the chromo- somes in a cell, an individual, a population, or a species.
genomic DNA DNA that has been isolated directly from cells or chromosomes or the cloned copies of all or part of that DNA. This DNA includes DNA se- quences from coding and noncoding regions of the genome.
Positively charged (basic) proteins that asso- ciate with DNA in the chromosomes. Histones pro- vide the structural components for the primary level of chromatin compaction and have been highly con- served through evolution.
homologous chromosomes (homologs) Chromo- somes that contain the same gene loci in the same or- der. In a diploid cell, which contains 2 copies of each homolog, 1 homolog is inherited from each parent.
The process of matching complemen- tary strands of DNA or RNA or both to form a dou- ble stranded molecule.
immunohistochemistry Analogous to in situ hy- bridization for the localization of nucleic acids, im- munohistochemistry techniques are used to localize a specific protein within cells or tissue through anti- body binding.
flanking regions
frameshift mutation
gene
gene expression
genetic linkage analysis
histones
hybridization

276 Shore and Kaplan Clinical Orthopaedics and Related Research
in situ hybridization Hybridization of a DNAor RNA probe to a target molecule that has not been ex- tracted from its cellular location, for example, within a chromosome or in a fixed tissue section.
independent assortment The random inheritance of chromosomes and unlinked genes.
insertion The addition of nucleotides into a region of DNA; it can be a single base or a large segment of a chromosome.
intron A transcribed region of a gene that is removed from the primary RNA transcript.
inversion A rearrangement of chromosomal DNA that flips a region of DNAend to end.
kilobase (kb) One thousand nucleotide bases in a DNA or RNA sequence. When refemng to doubled- stranded DNA, the term is used as shorthand for kilobase pairs.
library A collection of clones (cloned DNA) from a particular source such as an organism or specific tis- sue.
ligation The joining of 2 DNA molecules through the formation of phosphodiester bonds (see also: DNA ligase).
marker An identifiable locus on a chromosome (such as a gene, a restriction endonuclease recogni- tion site, or a VNTR) whose inheritance can be fol- lowed. Markers can be within or outside of genes. The most useful markers are those that have many allelic forms (are highly polymorphic) within the population.
meiosis The type of cell division that produces 4 hap- loid gamete cells from a diploid cell.
meiotic recombination The reciprocal (equal) ex- change of portions of chromosomes that occurs be- tween chromatids of homologous chromosomes. This recombination, also called crossing over, oc- curs during the first prophase of meiosis.
An RNA that contains pro- tein coding information. Eukaryotic mRNAs are transcribed from “Class 11” genes by RNA poly- merase 11.
This typically involves the modification of a cytosine base in DNA by the addition of a methyl group. Methylation appears to play an im- portant, though not well understood, role in the reg- ulation of gene expression.
missense mutation Changes of I or more nucleotides in a codon that alters the amino acid specified by that codon.
molecular cloning The isolation of a specific segment of DNA (such as a gene or part of a gene) and the generation of many identical copies, or clones, of that segment of DNA.
A heritable change in the genomic DNA se- quence. A mutation may be neutral, deleterious, or simply provide genetic variability. Mutations (gene changes) are the foundation of evolution.
noncoding DNA strand See antisense strand of DNA. nonsense mutation A base substitution in DNA that
changes an amino acid codon to a termination codon. Northern transfer The process of transferring RNA
that has been electrophoresed through an agarose gel to a solid filter support for hybridization.
nucleic acid hybridization See hybridization.
messenger RNA (mRNA)
methylation
mutation
nucleosome The basic unit of chromatin structure which consists of -200 bp of DNAand a histone oc- tamer (146 base pairs of DNAwrapped around a his- tone octamer corn plus a segment of linker DNA which associates with histone H1).
A molecule composed of a nitrogenous base, a 5-carbon sugar, and a phosphate group. Anu- cleic acid (such as DNA) is a polymer of many nu- cleotides. A nucleoside is composed of a nitrogenous base and a 5-carbon sugar (no phosphate groups). The 4 nucleosides in DNA are deoxyadenosine, de- oxycytidine, deoxyguanosine, and deoxythymidine; in RNA they are adenosine, cytidine, guanosine, and uridine (uridine replaces thymidine). Nucleotides are named by their nucleoside and the number of added phosphate groups, for example, adenosine triphos- phate (ATP). (See also A, C, G, T, U).
nucleotide
oligonucleotide phage See bacteriophage. plaque
A short sequence of nucleotides.
A clear area in a lawn of bacteria on an agar plate. The clearing contains bacteriophage particles that have been released when cells infected with phage were lysed, or killed, by the viral infection. Released phage can infect additional bacterial cells that will be lysed subsequently, increasing the plaque size.
A genetically engineered plasmid to which regions of bacteriophage M13 have been added for increased versatility as a cloning vector.
A small, circular double-stranded DNAmole- cule that is found in bacteria and replicates indepen- dently of the host cell chromosome. Plasmids commonly are used as vectors in molecular cloning.
Achange in a single nucleotide of DNA. A DNA synthesis
reaction in which a specific region of DNA is copied, or amplified, many times.
polymorphism A detectable difference in a DNA se- quence among individuals at a specific locus.
polypeptide Achain of amino acids. Aprotein may be composed of 1 or more polypeptides.
positional cloning Gene cloning that is based on the chromosomal location of the gene as identified through genetic linkage analysis.
primer A short oligonucleotide to which nucleotides can be added by DNApolymerase.
probe A DNA or RNA molecule that is labeled, or tagged, and used to locate a complementary DNA or RNA molecule through hybridization.
promoter DNA sequences, usually located in the re- gion flanking the 5’ end of the RNA coding region of a gene, that determine the site of initiation of tran- scription and the quantity and sometimes the tissue specificity of the mRNA. The promoter directs which of the DNA double strands will be the tem- plate strand for transcription. The promoter is part of the regulatory region of a gene.
A DNA molecule that contains segments of DNA from different origins (for example, a piece of human DNA that has been joined to a plasmid DNA).
regulatory gene A gene that encodes a product, either RNA or protein, that regulates the expression of other genes.
phagemid
plasmid
point mutation polymerase chain reaction (PCR)
recombinant DNA molecule

Number 320 November, 1995 Tools of Molecular Biology 277
restriction endonucleases Enzymes that recognize and cut double-stranded DNA at specific nucleotide sequences (restriction sites). In bacterial cells, from which these enzymes have been isolated, these en- zymes function to protect the cell against infection by foreign DNA.
restriction fragment Asegment of DNAthat is gener- ated when a larger DNA molecule is cut with a re- striction enzyme. For example, a 3-kb linear DNA molecule could be cut by the restriction endonucle- ase EcoRI to generate 2 restriction fragments: 1 that is 1 kb and 1 that is 2 kb.
restriction fragment length polymorphism (RFLP) Cleavage of DNA from 2 sources (for example, from 2 individuals) may generate restriction frag- ments of different lengths in a specific region of the DNA. This occurs if there are differences (or poly- morphisms) in the DNA sequence (such as a single nucleotide difference) that creates or removes a recognition site for a specific restriction endonucle- ase or an insertion or deletion of DNA). Note that there are many DNA sequence differences among individuals and not all differences cause a gene de- fect or functional alteration.
restriction map A map of a segment of DNA that shows the relative positions of restriction endonu- clease recognition sites.
An RNA-dependent DNA pol y- merase that is used to synthesize cDNA from RNA templates.
In eukaryotic cells, a pri- mary rRNA transcript is synthesized by RNA poly- merase I and processed to yield 28S, 18S, and 5.8s rRNA. 18s rRNA complexes with proteins to fonn the small ribosomal subunit; 28S, 5.8S, and 5s (syn- thesized by RNA polymerase 111) rRNAs complex with proteins to form the large ribosomal subunit.
Cellular organelles that mediate the inter- action between mRNA and tRNAs during transla- tion of the mRNA to protein. Ribosomes are composed of 2 distinct subunits, both of which con- tain rRNAand proteins.
A polynucleotide synthesized from a DNA template. RNA structure is similar to DNA structure, except that the sugar component is a ribose instead of a deoxyribose and the base uracil replaces thymine.
ribonuclease protection assay A highly sensitive method for the detection of RNAmolecules. This is the preferred method for quantitation of RNAs but, unlike Northern analysis, cannot be used to deter- mine the size of a particular RNAmolecule.
A PCR reac- tion in which the starting template to be amplified is an RNAinstead of a DNAmolecule. AcDNAis first synthesized from the RNA using reverse transcrip- tase, followed by polymerase chain reaction amplifi- cation of the double-stranded cDNA.
The process of identifying a fragment of DNAcontaining a specific DNAsequence among all of the clones in a DNA library.
semiconservative replication The mode by which double-stranded DNA is copied to produce 2 iden- tical DNA double-stranded DNA molecules. The 2
reverse transcriptase
ribosomal RNA (rRNA)
ribosomes
RNA (ribonucleic acid)
RT-PCR (reverse transcriptase PCR)
screening
strands of the double helix separate (hydrogen bonds between base pairs are broken) and each strand serves as a template for the synthesis of a complementary strand. The process is semiconser- vative because each of the resulting double- stranded molecules contains a newly synthesized DNA strand and a strand from the original DNA molecule.
The strand of double-stranded DNA that has the same 5’ to 3‘ sequence (or sense) as mRNA; also called the coding strand. This strand does not serve as the template strand for transcrip- tion. (Also see antisense strand.)
Refers to the nucleotide sequence, or order, of a given segment of DNA or RNA. Each nu- cleotide in a DNA sequence is identified by the base which that nucleotide contains: adenine (A), thymine (T), guanine (G), or cytosine (C). In RNA, uracil (U) replaces T.
A change in the DNA sequence that has no discernible effect.
The process of transferring DNA that has been electrophoresed through an agarose gel to a solid filter support for hybridization.
splice site The donor splice site is the boundary be- tween the 3‘ end of an exon and the 5’ end of the ad- jacent intron. The acceptor splice site is the boundary between the 3’ end of an intron and the 5’ end of the adjacent exon.
The removal (splicing out) of introns and the joining (splicing together) of exons to form mature mRNA from the primary transcript.
sense strand of DNA
sequence
silent mutation
Southern transfer
splicing
stop codon See termination codon. short tandem repeat polymorphisms (STRPs) Also
called microsatellites; these repeated sequences are variable number tandem repeats that contain 5 or fewer nucleotides repeated in a tandem arrange- ment.
structural gene A gene that codes for an RNA or pro- tein product.
TATA box A conserved AT-rich sequence found in the promoter region of many eukaryotic genes. This sequence element, located -25 base pairs upstream of the transcription start site, determines the site of transcription initiation.
A DNA strand that directs the sequence of newly synthesized DNA or RNA through comple- mentary base pairing. See antisense strand of DNA and semiconservative replication.
One of the 3 codons that specify the end of polypeptide synthesis; also called a stop codon.
transcript The RNA product synthesized by an RNA polymerase enzyme.
transcription The synthesis of RNA by an RNApoly- merase. This synthesis requires a DNA template.
transcription factor A protein that is needed for tran- scription to occur, but is not part of RNA poly- merase itself. May bind directly to a DNA sequence element or to other proteins in promoter and en- hancer regions of a gene.
The process of a bacteriophage, or bacte- rial virus, infecting a bacterial cell.
template
termination codon
transfection
transgene See transgenic mice.

278 Shore and Kaplan Clinical Orthopaedics and Related Research
transgenic mice
transformation
Mice that carry a foreign gene (or transgene) within their genomic DNA.
The process by which a bacterial cell takes up DNA molecules such as plasmid DNA. (Note that the term transformation also is used to de- scribe the conversion of a normal cell into a tumor cell.)
translocation The transfer of a portion of 1 chromo- some to another chromosome.
upstream A term used to describe sequences opposite to the direction of gene expression. For example, the ini- tiation codon is upstream from the termination codon. Also used to describe the noncoding region flanking the 5’ end of the RNAcoding region of a gene.
A segment of a gene that is transcribed and contained within an exon but is not translated into amino acid sequence. Untranslated re- gions commonly occur at the 3’ and 5’ ends of a mRNA.
A DNAmolecule, such as a plasmid or bacterio- phage, that is used as a carrier molecule for cloned
untranslated region (UTR)
vector
DNAs. The vector contains information that will al- low recombinant molecules to be replicated in host bacterial cells.
variable number tandem repeats (VNTRs) The hu- man genome contains many loci that contain multi- ple copies of short DNAsequences. These repeated sequences can occur within or outside of genes. Variable number tandem repeats loci are highly polymorphic within the population. Variable number tandem repeats are used as polymorphic markers in genetic linkage analyses and in the identification of individuals such as in forensics and paternity testing.
Western transfer The process of transferring proteins that have been electrophoresed through an acry- lamide gel to a solid filter support for detection of a specific protein by antibody binding.
cloning vectors that contain the centromere and telomere sequences of yeast chromosomes, and are used to clone very large (>lo00 kb) DNAfragments.
Yeast artificial chromosome (YAC)