the mechanism of action of histidase: amino-enzyme ... · the mechanism of action of histidase:...
TRANSCRIPT

THE JOURNAL osB~omo~ca~ CHEMISTRY Vol. 237, No. 3, March 1962
Printed in U.S.A
The Mechanism of Action of Histidase: Amino-enzyme
Formation and Partial Reactions
ALAN PETERKOFSKY
From the National Institute of Arthritis and Metabolic Diseases, National Institutes of Health, United States Public Health Service, Bethesda 14, Maryland
(Received for publication, September 18, 1961)
Histidase catalyzes the irreversible nonoxidative deamination of histidine to urocanic acid. This reaction (Equation 1) has been studied in mammalian and bacterial preparations (1). Although enzymes which carry out similar deaminations of other amino acids (notably aspartic acid) have been described, to date no detailed studies of the mechanism of the process of nonoxida- tive deamination have been reported.
H H
I -z-CooH -NH,t - -C=C-COOH
rlHH
HN 4
NH2 HN 4
(I)
In the present work, some properties of histidase purified from Pseudomonas jluorescens are described. It is demonstrated that the enzyme catalyzes an exchange of urocanic acid and of hy- drogen ion into histidine but that no ammonia exchange into histidine occurs. A mechanism for the histidase reaction based on these observations is presented. A preliminary report of some of these findings has been published (2).
EXPERIMENTAL PROCEDURE
Materials and Methods
n-Histidine monohydrochloride was a product of Nutritional Biochemicals Company. nn-H&dine-2-C’* was obtained from the Nuclear-Chicago Company. Urocanic acid was prepared enzymatically by the method of Mehler, Tabor, and Hayaishi (3). Urocanic acid-Cl4 was prepared as described in the text. Cyanomethylimidazole and imidazolelact,ic acid were gifts from Dr. Hugo Bauer. TtO was purchased from Tracerlab, Inc. DEAE-cellulose (4) was a product of Eastman Organic Chemi- cals. Sephadex G-50 was purchased from Pharmacia, Uppsala, Sweden. Dowex 50W was obtained from The Baker Chemical Company. 2-Mercaptoethanol was purchased from Eastman Organic Chemicals. 2,5-Diphenyloxazole and 1,4-bis-2’(5’- phenyloxazolyl)-benzene were obtained from Tracerlab, Inc., and Hyaminer hydroxide in methanol was supplied by The Packard Instrument Company. Histidine decarboxylase was a commercial preparation from the Worthington Biochemical Company. Diamine oxidase and aldehyde dehydrogenase were prepared by the methods of Tabor (5) and Weissbach, Redfield, and Udenfriend (6). Pseudomonas jtuorescens was grown and harvest~ed as previously described (7).
Protein was determined by the method of Lowry et al. (8).
1 The trade mark of Rohm and Haas for p-(diisobutylcresoxy- ethoxyethyl)dimethyl benzyl ammonium hydroxide.
Imidazole compounds were detected on paper chromatograms (9) and quantitatively determined in solution (10) by the Pauly test. Histidase was determined by a spectrophotometric assay (11). Tritium counting was done in a Packard Tri-Carb liquid scintillation spectrometer. Carbon-14 was estimated, after drying samples on stainless steel planchets, in a Nuclear-Chicago gas flow counter. A Beckman model DU spectrophotometer was used for spectrophotometric determinations. N15 was de- termined in a mass spectrometer.
Separation of Histidine and Urocanic Acid for Analyses of Exchange Experiments
Reaction mixtures containing histidine and urocanic acid were neutralized with NaOH and applied to columns of Dowex 5OW- H+ (8% cross-linked 200 to 400 mesh) (1 X 12 cm). The columns were washed with 10 ml of water, then with 200 ml of 1.1 N HCl to elute the urocanic acid. Further elution with 20 ml of 6 N HCl quantitatively removed the histidine. The histidine eluates were evaporated to dryness on the steam bath to remove excess HCl. They could now be assayed by the Pauly test (lo), and their radioactivity, either tritium or CY4, could be determined.
Enzymatic Preparation of CY- Urocanic Acid-Urocanic acid-Cl4 was prepared for use in exchange experiments by the histidase- catalyzed deamination of histidine-2-Ci4. A preparation of 53 pmoles of n-histidine-2-C4 (3.96 mc per mmole), 250 pmoles of sodium carbonate buffer (pH 9.2), 30 pmoles of mercaptoethanol, and 2 ml of histidase (0.37 mg per ml, DEAE-cellulose fraction) in a total volume of 2.8 ml was incubated at 36” for 4 hours, at which time the histidine was essentially completely converted to urocanic acid. The incubation mixture was acidified to pH 5 with HCl, then applied to a column of Dowex 5OW-H+ (0.8 x 10 cm). The column was washed with 10 ml of water, then eluted with 200 ml of 1 .l N HCI. Radioactive urocanic acid was eluted between 100 and 160 ml. This fraction was dried in a vacuum several times to drive off excess hydrochloric acid. A total of 40 rmoles of radioactive urocanic acid was recovered. Paper chromatography of this product in the solvent of Meister, Sober, and Tice (12) indicated only one ultraviolet-absorbing area which contained all the radioactivity and the RF of which was identical with that of standard urocanic acid.
Preparation of Enzymatically Labeled Tritiated H&dine-A preparation of 600 pmoles of histidine (adjusted to pH 8 with NaOH), 600 pmoles of sodium carbonate buffer (pH 9.2), 73 pmoles of mercaptoethanol, 25 mc of TSO, and 60-fold purified histidase (0.6 mg) in a total volume of 7.2 ml was incubated at
787
by guest on August 13, 2018
http://ww
w.jbc.org/
Dow
nloaded from
788 Mechanism of Histidase Vol. 237, No. 3
25”. The rate of formation of urocanic acid was followed by its absorption at 277 rnp. When the optical density of a 1: 1,000 dilution had reached 0.800 (l-cm light path), corresponding to about 50 To conversion of the histidine to urocanic acid, the reac- tion mixture was frozen and lyophilized. Absolute ethanol (10 ml) was added to destroy the enzyme, after which the mix- ture was again evaporated to dryness in a vacuum. Repeated evaporations served to remove the last traces of exchangeable tritium. The residue was dissolved in water, streaked on What- man No. 3 paper and chromatographed in the t-butanol-formic acid-water solvent of Meister et al. (12). The histidine was eluted with water. In this manner, 240 pmoles of labeled histi- dine was recovered. Determination of the radioactivity of the preparation indicated that 0.14 patom of tritium had been in- corporated per micromole of histidine.
RESULTS
Purification of Enzyme
Pseudomonas Jluorescens (ATCC 11,299) was cultured in a medium containing hiitidine, harvested, and washed as described by Tabor and Mehler (7). From this point, all manipulations were performed at 2-5”. A total of 35 g (wet weight) of frozen cells was suspended in 0.05 M potassium phosphate (pH 7.2) to a volume of 110 ml and disrupted for 20 minutes in a Raytheon 10 kc sonic oscillator cooled to 3”. The sonically treated prep- aration was dispensed into test tubes (20 X 190 mm) and held in a water bath at 80” for 15 minutes, then cooled in an ice bath and centrifuged for 20 minutes at 20,000 X g. To the superna- tant solution (45 ml) was slowly added with stirring an equal volume of ammonium sulfate solution (saturated at 0”). After stirring for 5 minutes, the precipitate was sedimented by cen- trifugation for 20 minutes at 20,000 X g. The precipitate was dissolved in 0.05 M sodium pyrophosphate, pH 9.2, to a volume of 10 ml and applied to a column of Sephadex G-50 (2 X 20 cm), previously primed with 0.1 M Tris-acetate, pH 8.0. The protein was washed through the column with additional 0.1 M Tris- acetate, pH 8.0. A yellow band of impurity remained on the column. Those fractions containing protein were pooled and applied to a column of DEAE-cellulose (4) (2 x 12 cm) which had previously been adjusted to pH 8.0 and washed with 0.1 M
TABLE I Purification of pseudomonas histidase
Fraction Volume
Crude extract Heated extract Ammonium sulfate pre-
cipitate DEAE-cellulose frac-
tions 12-16
nZ1
85 50 10
41
Total units”
Total protein
2,377,500 2,860 1,400,000 300 1,400,000 116
1,148,OOO 15
Specific activity
mits/mg protein
940
4670 12,100
75.000
a One unit is defined as that amount of enzyme resulting in a change in optical density of 0.001 per minute at 25” in the follow- ing assay (11). Sodium pyrophosphate (30 pmoles), mercapto- ethanol (15 rmoles), histidine (sodium salt, 10 pmoles), and en- zyme were incubated in a total volume of 3 ml and the change of optical density at 277 rnp was followed in quartz cuvettes of l-cm light path until the rate was constant.
r I I I I 1 I 1 0 50 too 150 200 250 300
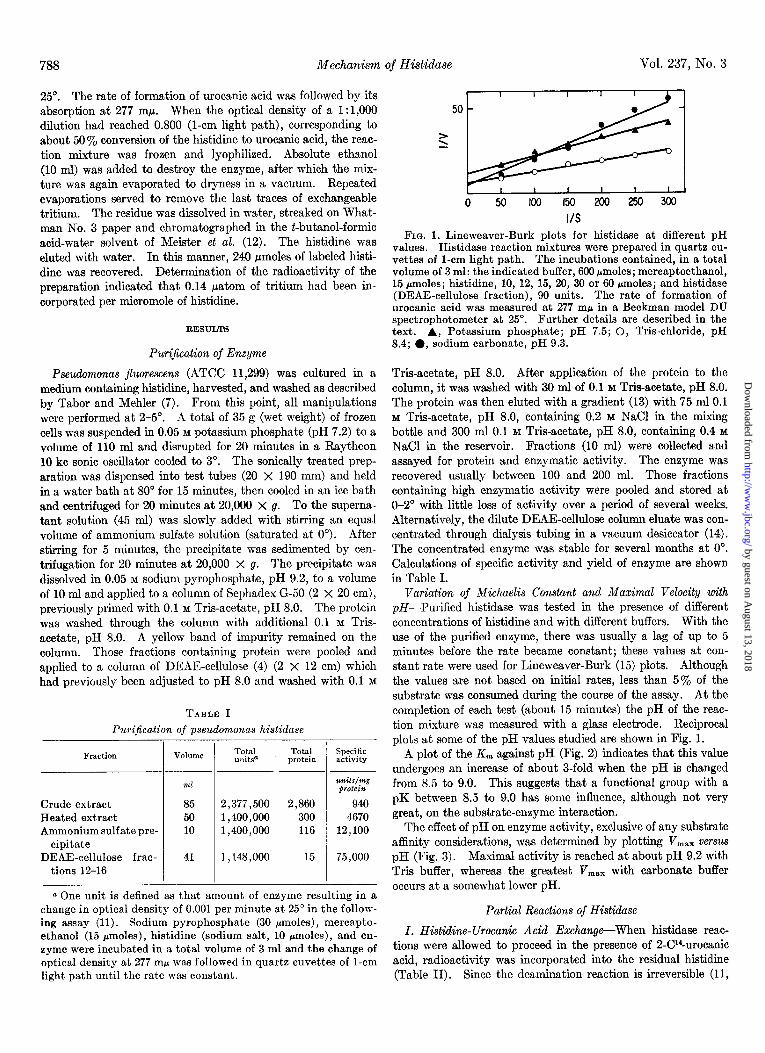
I/S FIG. 1. Lineweaver-Burk plots for histidase at different pH
values. Histidase reaction mixtures were prepared in quartz cu- vettes of l-cm light path. The incubations contained, in a total volume of 3 ml: the indicated buffer, 660 Mmoles; mercaptoethanol, 15 rmoles; histidine, 10, 12, 15, 20, 30 or 66 rmoles; and histidase (DEAE-cellulose fraction), 90 units. The rate of formation of urocanic acid was measured at 277 rnh in a Beckman model DU spectrophotometer at 25”. Further details are described in the text. A, Potassium phosphate; pH 7.5; 0, Tris-chloride, pH 8.4; 0, sodium carbonate, pH 9.3.
Tris-acetate, pH 8.0. After application of the protein to the column, it was washed with 30 ml of 0.1 M Tris-acetate, pH 8.0. The protein was then eluted with a gradient (13) with 75 ml 0.1 M Tris-acetate, pH 8.0, containing 0.2 M NaCl in the mixing bottle and 300 ml 0.1 M Tris-acetate, pH 8.0, containing 0.4 M
NaCl in the reservoir. Fractions (10 ml) were collected and assayed for protein and enzymatic activity. The enzyme was recovered usually between 100 and 200 ml. Those fractions containing high enzymatic activity were pooled and stored at O-2” with little loss of activity over a period of several weeks. Alternatively, the dilute DEAE-cellulose column eluate was con- centrated through dialysis tubing in a vacuum desiccator (14). The concentrated enzyme was stable for several months at 0”. Calculations of specific activity and yield of enzyme are shown in Table I.
Variation of Michaelis Constant and Maximal velocity with pH-Purified histidase was tested in the presence of different concentrations of histidine and with different buffers. With the use of the purified enzyme, there was usually a lag of up to 5 minutes before the rate became constant; these values at con- stant rate were used for Lineweaver-Burk (15) plots. Although the values are not based on initial rates, less than 5% of the substrate was consumed during the course of the assay. At the completion of each test (about 15 minutes) the pH of the reac- tion mixture was measured with a glass electrode. Reciprocal plots at some of the pH values studied are shown in Fig. 1.
A plot of the K, against pH (Fig. 2) indicates that this value undergoes an increase of about 3-fold when the pH is changed from 8.5 to 9.0. This suggests that a functional group with a pK between 8.5 to 9.0 has some influence, although not very great, on the substrate-enzyme interaction.
The effect of pH on enzyme activity, exclusive of any substrate affinity considerations, was determined by plotting Vmax versus pH (Fig. 3). Maximal activity is reached at about pH 9.2 with Tris buffer, whereas the greatest Vmax with carbonate buffer occurs at a somewhat lower pH.
Partial Reactions of Histidase
I. Histidine- Urocanic Acid Exchange-When histidase reac- tions were allowed to proceed in the presence of 2-CY4-urocanic acid, radioactivity was incorporated into the residual histidine (Table II). Since the deamination reaction is irreversible (11,
by guest on August 13, 2018
http://ww
w.jbc.org/
Dow
nloaded from

March 1962 A. Pete&of&y 789
16), the incorporation is not due to the condensation of free ammonia with urocanic acid (incubation No. 6). These data are consistent with a mechanism for the histidase reaction involv- ing the reversible formation of an amino-enzyme complex derived from histiclme with the release of free urocanic acid. Recom- bination of urocanic acid with the amino-enzyme leads to re- synthesis of histidine (the urocanic acid exchange reaction). The irreversible decomposition of the amino-enzyme compound leads to the formation of free ammonia and regeneration of the enzyme. That the amino-enzyme can be formed from histidme but not from free ammonia accounts for the irreversibility of the reaction.
Enzyme + histidine *
urocanic acid + H+ + amino-enzyme (1)
Hf + amino-enzyme -+ enzyme + NH, (2)
II. Histidine-Hydrogen Ion Exchange-The formulation of the histidase reaction mechanism, shown above, as involving reversi- ble amino-enzyme formation requires (a) urocanic acid exchange and (b) exchange of hydrogen ion between the medium and the B carbon of histidine. As shown in Table III, the enzyme cata- lyzes the incorporation of solvent tritium into a form that is stable to repeated drying. Both the complete histidase reaction
4 I I I I
q:. . * 27:
:
I I I I 6 7 8 9 IO
PH
FIG. 2. Variation of K, with pH. Experimental conditions are those described for Fig. 1. A, Potassium phosphate; 0, Tris- chloride; 0, sodium carbonate.
I I I I
PH
FIG. 3. Variation of V,,, with pH. Experimental details are ident,ical with those for Fig. 1.
TABLE II Histidase-catalyzed incorporation of C”-urocanic acid into hi&dine
The complete system contained, in a total volume of 0.5 ml: sodium carbonate buffer (pH 9.2), 50 pmoles; histidine (sodium salt), 10 pmoles; mercaptoethanol, 10 pmoles; sodium urocanate- 04 (3,280 c.p.m. per pmole),a 5 rmoles; histidase (80X purified), 0.074 mg. Where indicated, 5 pmoles of NH&I were added.
The reactions were terminated by the addition of 0.5 ml of 12% trichloroacetic acid. Residual histidine was purified on Dowex 50-H+ and assayed calorimetrically by a modified diazo test (10). Urocanic acid formation was estimated spectrophotometrically (11).
1ncub;tion Omissions Time
None None None Enzyme Mercaptoethanol Histidine (ammonia
added)
rzin pVdeS pm&
0 5.3 0.03 15 6.2 0.24 60 8.3 0.48 60 5.5 0.02 60 7.3 0.18 60 5.4 0
T
Urocanic cid at end o experiment
Urocanic acid-C” in- corporated
Into histidin@
D Urocanic acid-Cl4 was prepared biosynthetically by the action of histidase on 2-Cl4 (ring-labeled) histidine, as described in “Materials and Methods.”
b The incorporation of radioactivity from CY-urocanic acid into histidine was roughly corrected for the change in specific activity of urocanic acid during the course of the reaction due to the pro- duction of additional urocanic acid derived from histidine. The specific activity of the urocanic acid used for the calculation of incorporation in any experiment was taken as the average of the specific activities at the beginning and end of the incubation.
and the tritium incorporation show a dependency on mercapto- ethanol. No tritium is incorporated in the presence of urocanic acid and ammonia, when histidine is absent. This suggests that the incorporation is dependent on the preliminary formation of amino-enzyme from histidine, but is not due to a net synthesis of histidine by reversal of the histidase reaction.
Demonstration that Solvent T&urn Is Incorporated into Histidine and Not into Urocanic Acid
A complete histidase incubation (as described in Table III) was carried out in the presence of solvent T,O. After repeated lyophilizations to remove exchangeable tritium, the incubation mixture was dissolved in water and streaked on Whatman No. 3 paper. Descending chromatography was run in a solvent of t-butanol-formic acid-water (70 : 15 : 15) (12). Urocanic acid (RF 0.72) was located by its ultraviolet absorption and reaction in the Pauly test, whereas histidine (Rp 0.31) was located by its reaction with the Pauly reagent. The areas corresponding to histidine and urocanic acid were eluted with water, dried, and prepared for scintillation counting. The histidine recovered (25 pmoles) contained 1,410 c.p.m. corresponding to an incorporation of 6.10 patoms of tritium. On the other hand, the recovered urocanic acid (20 pmoles) contained 7 c.p.m., indicating an incorporation of not more than 0.03 patom of tritium. The association of the tritium label with histidine and not with urocanic acid suggests a stereospecific tritiation of histidine. These data are consistent with the idea that the hydrogen atom
by guest on August 13, 2018
http://ww
w.jbc.org/
Dow
nloaded from

790 Mechanism of Histidase Vol. 237, No. 3
TABLE III
Incorporation of solvent tritium during histidase reaction in TtO
The complete system contained in a total volume of 1.55 ml: sodium carbonate buffer (pH 9.2), 100 pmoles; histidine (sodium salt), 100 pmoles (except incubation No. 4,50 rmoles); mercapto- ethanol, 15 pmoles; 4 mc of TzO; histidase (60X purified), 0.1 mg. Where indicated, sodium urocanate (50 rmoles) and NHaOH (50 pmoles) were included.
After incubation, the reaction mixtures were frozen and lyo- philized, then treated with absolute ethanol to destroy the enzyme. Repeated addition of water and lyophilization were con- tinued to remove residual exchangeable tritium. The dried sam- ples were dissolved in a mixture of 2 ml of 1 M Hyamine hydroxide in methanol, 2 ml of toluene containing 1.5oj, 2,5-diphenyloxazole and 0.005% 1,4-bis-2’(5’-phenyloxazolyl)-benzene and 13 ml of toluene. The samples were counted in a Packard Tri-Carb liquid scintillation spectrometer, model 314. The extent of urocanic acid formation at the end of the incubation period was estimated spectrophotometrica~ly (11).
Incubation No.
-
-_
Omissions Time
None None None Enzyme (urocanic
acid + NH, added) Mercaptoethanol Histidine (urocanic
acid + NH, added)
man /.4mo2es patoms 0 1.5 0.35
15 19 0.92 60 47 4.73 60 68 0.32
60 13 0.58 60 61 0.23
Urocanic :id at end c experiment
-
If il
-
Tritium ocorporated
of histidine that participates in the exchange reaction is identical with the hydrogen atom that is lost in the conversion of histidine to urocanic acid. Such a thesis is substantiated further by the experiments subsequently described.
Localization of Tritium Label on 0 Carbon of Histidine
To determine the position of the histidine molecule which had become labeled in the enzymatic reaction, it was felt desirable to degrade the amino acid to imidazoleacetic acid. Retention of the radioactivity from histidine in imidazoleacetic acid would eliminate the a: carbon of histidine as being the labeled site. Such a result would leave only the /? carbon and the imidazole ring hydrogens to be considered.
Chemical Degradation of Histidine to Imidazoleacetic Acid
I. Nitric Acid Degradation-It has been reported (17) that L-histidine-hydrochloride may be oxidized with nitric acid to form imidazoleacetic acid. The identification of the product was based on an RF essentially identical to that of imidazole-
acetic acid in a propanol-ammonia solvent. However, on compar- ison of this compound with imidazoleacetic acid in several other solvent systems2 it became apparent that the product of nitric acid oxidation was, in fact, imidazolelactic acid.3 Therefore, this method could not be utilized for our purposes.
II. Silver Oxide Degradation-Sprinson and Rittenberg (18),
2 A. Peterkofsky and H. Tabor, unpublished results. 3 Solvent System 1: n-propanol, 75; 28yo NH*OH, 1.5; water,
23.5. Solvent System 2: n-propanol, 75; glacial acetic acid, 1.5; water, 23.5. Solvent System 3: ethanol, 77; water, 23. RF values in these solvents are reported in (10).
in a study of the metabolism of the branched chain amino acids, have used silver oxide to convert these amino acids to the corre- sponding carboxylic acid with one carbon less. We attempted to utilize this procedure for the degradation of histidine to imidazoleacetic acid. However, it appeared that histidine was resistant to this oxidation; examination of the reaction mixture by paper chromatography gave no evidence for the formation of new Pauly-reactive compounds.
III. Sodium Hypochlorite Degradation-Bauer and Tabor (19) have presented the details for the oxidation of histidine to cyano- methylimidazole by sodium hypochlorite. Subsequent hy- drolysis of the cyanomethylimidazole resulted in a good yield of imidazoleacetic acid. For a specific chemical degradation to be useful in the localization of the radioactive hydrogen label in a compound, it must be rigorously demonstrated that none of the intermediates in such a degradation are subject to nonspecific exchange reactions. To test this possibility, histidine was con- verted to imidazoleacetic acid in a medium of T,O under the conditions described by Bauer and Tabor. The isolated imid- azoleacetic acid had incorporated a significant amount of ti-itium. Since Reitz (20) and Sprinson and Rittenberg (18) have amply demonstrated the lability of the CY hydrogen of nitriles, it was apparent that the cyanomethylimidazole formed in the histidine degradation underwent such an exchange reac- tion. On the basis of this observation, the conditions for this degradation were modified to take full advantage of the exchange reaction. Histidine labeled with tritium by the histidase reac- tion was converted to cyanomethylimidazole by incubation for 6 hours with sodium hypochlorite. The NaOH concentration was then adjusted to 0.3 N and incubation was continued over- night at room temperature. The long exposure of cyanomethyl- imidazole to alkaline conditions is necessary for the complete exchange of carbon-bound hydrogen adjacent to the cyano group. Subsequent hydrolysis of the nitrile (2 hours at loo’, 1 N NaOH) led to unlabeled imidazoleacetic acid. This result is consistent with the idea that the histidine was originally labeled in the /3 position of the side chain, and that the label was removed in the alkali-catalyzed exchange reaction of cyanomethylimidazole, but does not exclude the possibility that it was the cx carbon or a position on the imidazole ring which carried the label.
An attempt was made to exclude the imidazole ring hydrogens as the site of labeling in the enzyme-catalyzed hydrogen exchange reaction. Incubation of cyanomethylimidazole in 0.3 N NaOH in a medium containing TzO results in significant incorporation of tritium, but similar treatment of histidine or urocanic acid leads to no labeling. These data indicate that imidazole ring hydrogens are not labile to alkali. Since the enzymatically labeled histidine loses its radioactivity after conversion to cyano- methylimidazole and subsequent exposure to alkali, it seems likely that the imidazole ring hydrogens are not involved.
Enzymatic Degradation of Histidine to Imidazoleacetic Acid
To substantiate further that histidine was labeled on the /3 carbon and to eliminate the possibility that it was labeled on the CY carbon, an enzymatic degradation to imidazoleacetic acid, with retention of the label, was carried out. Labeled histidine was first converted in essentially quantitative yield to histamine by the action of crude histidine decarboxylase according to the procedure of Gale (21). The specific activity of the resultant histamine was 82 y0 that of the starting histidine. The histamine was then essentially completely oxidized to imidazoleacetic acid
by guest on August 13, 2018
http://ww
w.jbc.org/
Dow
nloaded from
March 1962 A. Pete&of&y 791
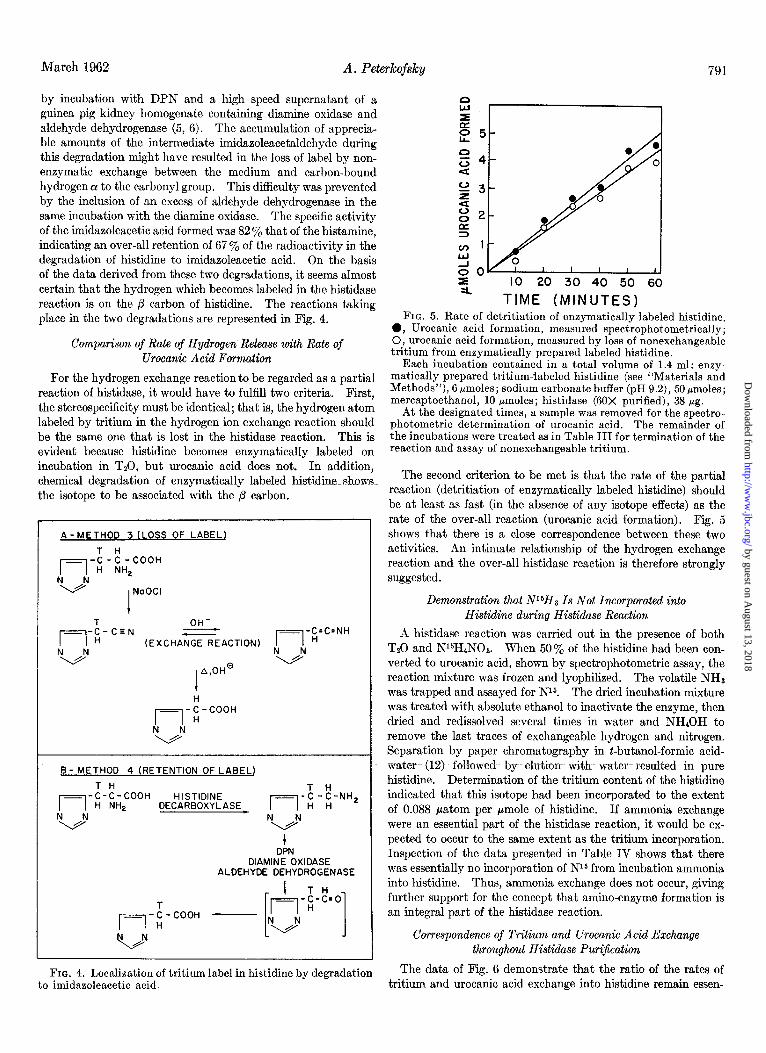
by incubation with DPN and a high speed supernatant of a guinea pig kidney homogenate containing diamine oxidase and aldehyde dehydrogenase (5, 6). The accumulation of apprecia- ble amounts of the intermediate imidazoleacetaldehyde during this degradation might have resulted in the loss of label by non- enzymatic exchange between the medium and carbon-bound hydrogen QI to the carbonyl group. This difficulty was prevented by the inclusion of an excess of aldehyde dehydrogenase in the same incubation with the diamine oxidase. The specific activity of the imidazoleacetic acid formed was 82 y. that of the histamine, indicating an over-all retention of 67 Y. of the radioactivity in the degradation of histidine to imidazoleacetic acid. On the basis of the data derived from these two degradations, it seems almost certain that the hydrogen which becomes labeled in the histidase reaction is on the ,B carbon of histidine. The reactions taking place in the two degradations are represented in Fig. 4.
Comparison of Rate of Hydrogen Release with Rate of Urocanic Acid Formation
For the hydrogen exchange reaction to be regarded as a partial reaction of histidase, it would have to fulfill two criteria. First, the stereospecificity must be identical; that is, the hydrogen atom labeled by tritium in the hydrogen ion exchange reaction should be the same one that is lost in the histidase reaction. This is evident because histidine becomes enzymatically labeled on incubation in TzO, but urocanic acid does not. In addition, chemical degradation of enzymatically labeled histidine shows the isotope to be associated with the p carbon.
A -METHOD 3 (LOSS OF LABEL)
,mN-; - $COOH
w NaOCl
(EXCHA=EACTION)
I3 - METHOD 4 (RETENTION OF LABEL)
Nm-$;OOH HISTIDINE DECARBOXYLASE
-F-:-NH,
N c IHH
v Nd
t DPN
DIAMINE OXIDASE ALDEHYDE DEHYDROGENASE I TN-“-cooH - [ 1 Nm-E-z=o 4 /
=L 20 30 40 50 60
TIME (MINUTES) FIG. 5. Rate of detritiation of enzymatically labeled histidine.
0, Urocanic acid formation, measured spectrophotometrically; 0, urocanic acid formation, measured by loss of nonexchangeable tritium from enzymatically prepared labeled histidine.
Each incubation contained in a total volume of 1.4 ml: enzy- matically prepared tritiumiabeled histidine (see “Materials and Methods”), 6 pmoles; sodium carbonate buffer (pH 9.2), 50pmoles; mercaptoethanol, 10 pmoles; histidase (60X purified), 38 pg.
At the designated times, a sample was removed for the spectro- photometric determination of urocanic acid. The remainder of the incubations were treated as in Table III for termination of the reaction and assay of nonexchangeable tritium.
The second criterion to be met is that the rate of the partial reaction (detritiation of enzymatically labeled histidine) should be at least as fast (in the absence of any isotope effects) as the rate of the over-all reaction (urocanic acid formation). Fig. 5 shows that there is a close correspondence between these two activities. An intimate relationship of the hydrogen exchange reaction and the over-all histidase reaction is therefore strongly suggested.
Demonstration that N15H3 Is Not Incorporated into Hi&dine during Histidase Reaction
A histidase reaction was carried out in the presence of both TzO and N15H4N03. When 50% of the histidine had been con- verted to urocanic acid, shown by spectrophotometric assay, the reaction mixture was frozen and lyophilized. The volatile NH3 was trapped and assayed for N15. The dried incubation mixture was treated with absolute ethanol to inactivate the enzyme, then dried and redissolved several times in water and NH40H to remove the last traces of exchangeable hydrogen and nitrogen. Separation by paper chromatography in t-butanol-formic acid- -water (12) followed by- elution -with- -water- resulttied in pure histidine. Determination of the tritium content of the histidine indicated that this isotope had been incorporated to the extent of 0.088 patom per pmole of histidine. If ammonia exchange were an essential part of the histidase reaction, it would be ex- pected to occur to the same extent as the tritium incorporation. Inspection of the data presented in Table IV shows that there was essentially no incorporation of Nr5 from incubation ammonia into histidine. Thus, ammonia exchange does not occur, giving further support for the concept that amino-enzyme formation is an integral part of the histidase reaction.
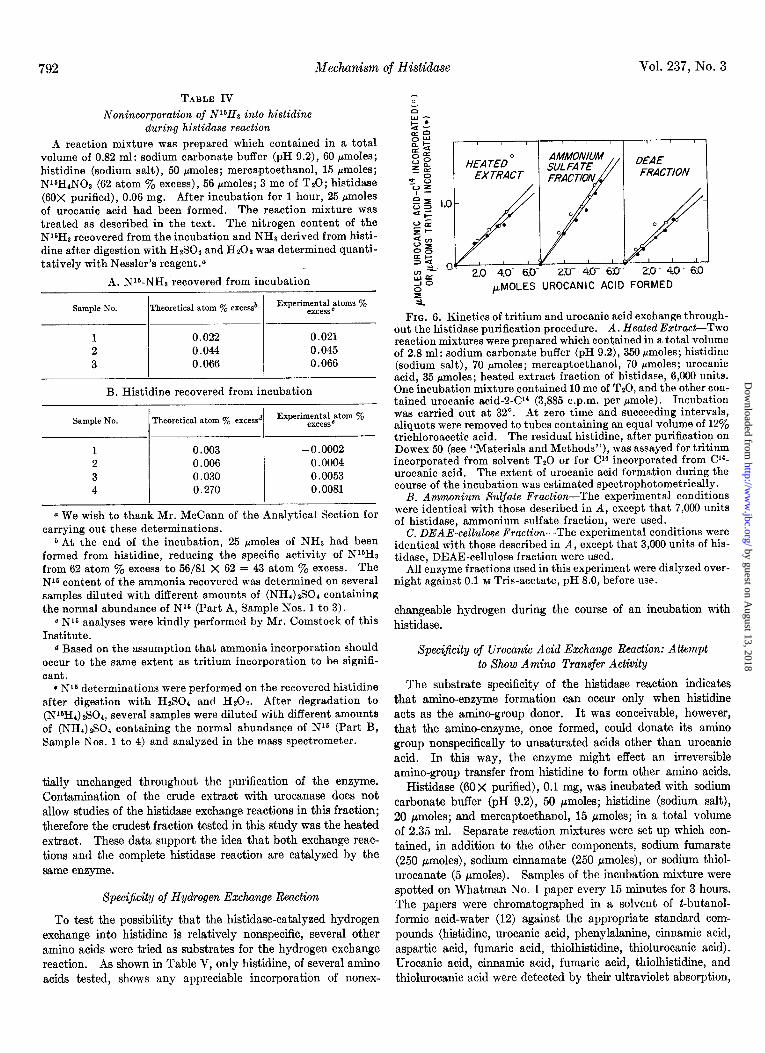
Correspondence of T&urn and Urocanic Acid Exchange throughout Histidase Purijcation
The data of Fig. 6 demonstrate that the ratio of the rates of FIG. 4. Localization of tritium label in histidine by degradation to imidazoleacetic acid. tritium and urocanic acid exchange into histidine remain essen-
by guest on August 13, 2018
http://ww
w.jbc.org/
Dow
nloaded from
792 Mechanism of Histidase Vol. 237, No. 3
TABLE IV Non&corporation of N16H8 into hi&dine
during histidase reaction A reaction mixture was prepared which contained in a total
volume of 0.82 ml: sodium carbonate buffer (pH 9.2), 60 pmoles; histidine (sodium salt), 50 pmoles; mercaptoethanol, 15 pmoles; N16H4N0s (62 atom ‘% excess), 56 pmoles; 3 mc of TYzO; histidase (60X purified), 0.06 mg. After incubation for 1 hour, 25 pmoles of urocanic acid had been formed. The reaction mixture was treated as described in the text. The nitrogen content of the NIbHa recovered from the incubation and NH, derived from histi- dine after digestion with HzS04 and HZOZ was determined quanti- tatively with Nessler’s reagent.”
A. N16-NH8 recovered from incubation
Sample No. Theoretical atom y. excessb Experimental atoms % excess0
1 0.022 0.021 2 0.044 0.045 3 0.066 0.066
B. Histidine recovered from incubation
Sample No. Theoretical atom y. excessJ Experimental atom % excess’
1 0.003 -0.0002 2 0.006 0.0004 3 0.030 0.0053 4 0.270 0.0081
a We wish to thank Mr. McCann of the Analytical Section for carrying out these determinations.
b At the end of the incubation, 25 pmoles of NH3 had been formed from histidine, reducing the specific activity of N16Hs from 62 atom y. excess to 56/81 X 62 = 43 atom To excess. The Nl6 content of the ammonia recovered was determined on several samples diluted with different amounts of (NHa)&Oa containing the normal abundance of N” (Part A, Sample Nos. 1 to 3).
c N’s analyses were kindly performed by Mr. Comstock of this Institute.
d Based on the assumption that ammonia incorporation should occur to the same extent as tritium incorporation to be signifi- cant.
6 N’s determinations were performed on the recovered histidine after digestion with Hz’S04 and Hz02. After degradation to (N16Hl)2S04, several samples were diluted with different amounts of (NH4)2S04 containing the normal abundance of N16 (Part B, Sample Nos. 1 to 4) and analyzed in the mass spectrometer.
tially unchanged throughout the purification of the enzyme. Contamination of the crude extract with urocanase does not allow studies of the histidase exchange reactions in this fraction; therefore the crudest fraction tested in this study was the heated extract. These data support the idea that both exchange reac- tions and the complete histidase reaction are catalyzed by the same enzyme.
Specificity of Hydrogen Exchange Reaction
To test the possibility that the histidase-catalyzed hydrogen exchange into histiclme is relatively nonspecific, several other amino acids were tried as substrates for the hydrogen exchange reaction. As shown in Table V, only histidine, of several amino acids tested, shows any appreciable incorporation of nonex-
HEATED ’ EXTRACT
L
2x7 4.0--
6” I
pMOLES UROCANIC ACID FORMED
=L
FIQ. 6. Kinetics of tritium and urocanio acid exchange through- out the histidase purification procedure. A. Heated Extract-Two reaction mixtures were prepared which contained in a total volume of 2.8 ml: sodium carbonate buffer (pH 9.2), 350 pmoles; histidine (sodium salt), 70 rmoles; mercaptoethanol, 70 pmoles; urocanic acid, 35 pmoles; heated extract fraction of histidase, 6,000 units. One incubation mixture contained 10 mc of TsO, and the other con- tained urocanic acid-2-Cl4 (3,885 c.p.m. per pmole). Incubation was carried out at 32”. At zero time and succeeding intervals, aliquots were removed to tubes containing an equal volume of 12% trichloroacetic acid. The residual histidine, after purification on Dowex 50 (see “Materials and Methods”), was assayed for tritium incorporated from solvent TzO or for Cl4 incorporated from C14- urocanic acid. The extent of urocanic acid formation during the course of the incubation was estimated spectrophotometrically.
B. Ammonium Sulfate Fraction-The experimental conditions were identical with those described in A, except that 7,000 units of histidase, ammonium sulfate fraction, were used.
C. DEAE-cellulose Fraction-The experimental conditions were identical with those described in A, except that 3,000 units of his- tidase, DEAE-cellulose fraction were used.
All enzyme fractions used in this experiment were dialyzed over- night against 0.1 M Tris-acetate, pH 8.0, before use.
changeable hydrogen during the course of an incubation with histidase.
Specijkity of Urocank Acid Exchange Reaction: Attempt to Show Amino Transfer Activity
The substrate specificity of the histidase reaction indicates that amino-enzyme formation can occur only when histidine acts as the amino-group donor. It was conceivable, however, that the amino-enzyme, once formed, could donate its amino group nonspecifically to unsaturated acids other than urocanic acid. In this way, the enzyme might effect an irreversible amino-group transfer from histidine to form other amino acids.
Histidase (60x purified), 0.1 mg, was incubated with sodium carbonate buffer (pH 9.2), 50 /J,moles; histidine (sodium salt), 20 pmoles; and mercaptoethanol, 15 pmoles; in a total volume of 2.35 ml. Separate reaction mixtures were set up which con- tained, in addition to the other components, sodium fumarate (250 pmoles), sodium cinnamate (250 pmoles), or sodium thiol- urocanate (5 pmoles). Samples of the incubation mixture were spotted on Whatman No. 1 paper every 15 minutes for 3 hours. The papers were chromatographed in a solvent of t-butanol- formic acid-water (12) against the appropriate standard com- pounds (histidine, urocanic acid, phenylalanine, cinnamic acid, aspartic acid, fumaric acid, thiolhistidine, thiolurocanic acid). Urocanic acid, cinnamic acid, fumaric acid, thiolhistidine, and thiolurocanic acid were detected by their ultraviolet absorption,
by guest on August 13, 2018
http://ww
w.jbc.org/
Dow
nloaded from

March 1962 A. Pete&of&y 793
whereas the other compounds were visualized after spraying with the ninhydrin reagent (22). There was no evidence for the appearance of new amino acids in any of the incubation mixtures. Thus, the specificity of olefin acceptor does not appear to allow for the transfer of the amino group of histidine to unsaturated compounds other than urocanic acid.
Effect of pH on Partial Reactions of Histiduse
The labiliiation of a proton as an essential step of the histidase mechanism suggests that relative rates of one or the other partial reactions compared to the complete reaction might be markedly affected by pH changes. With this purpose in mind, a study was made of the effect of diierent pH values and buffers on the sev- eral reactions characteristic of this system.
Histidase reactions were carried out for 1 hour at 35” in a total volume of 0.6 ml and contained: histidine (sodium salt), 10 pmoles; mercaptoethanol, 10 pmoles; urocanic acid, 5 pmoles; the specified buffer, 100 pmoles; and 280 units of histidase (DEAE-cellulose fraction). One set of incubations contained 2 mc of solvent TzO, and the other, otherwise identical, contained urocanic acid-04 (18,000 c.p.m.). After 1 hour, the amount of urocanic acid formed was determined spectrophotometrically and the reaction was then terminated by the addition of an equal volume of 12% trichloroacetic acid. The remaining histidine was purified and assayed for the amount of tritium or Cl4 incorporated.
The results of such an experiment are shown in Fig. 7. It is
TABLE V Test of amino acids as substrates for the histidase-catalyzed
hydrogen exchange reaction
A preparation of 10 pmoles of the designated amino acid (as the sodium salt), 50 pmoles of sodium carbonate buffer (pH 9.2), 10 pmoles of mercaptoethanol, 1.2 mc of TzO, and histidase (0.074 mg) m a total volume of 0.47 ml, was incubated at 37” for the time intervals shown. At the end of the incubation period, 0.5 ml of 12y0 trichloroacetic acid was added to the reactions. They were then neutralized and applied to Dowex 50-H+ columns (1 X 15 cm). After washing the columns with 100 ml of water, the amino acids were eluted with 20 ml of 6 N HCl. The HCl eluates were evaporated on the steam bath and the dried samples prepared for tritium counting.
Substrate
Histidine
Incubation time
hr
0
Phenylalanine
Lysine
Leucine
Alanine
Aspartic acid
0 1
0 1
0 1
0 1
0 1
-
_-
-
Solvent tritium incorporated
c.$bn.
22 169
5 4
14 9
11 4
4 7
3 18
PH FIG. 7. Resolution of the tritium and urocanic acid exchange
reactions from the breakdown of the amino-enzyme complex. Experimental details are described in the text. The buffers were: pH 5.0 sodium succinate; pH 6.0 potassium phosphate; pH 7.0 and 8.0 Tris-acetate; pH 9.2 and 9.8 sodium carbonate. The enzyme used in this experiment was previously dialyzed against 0.001 M Tris-acetate, pH 8.0.
evident from these data that at lower pH the tritium and urocanic acid exchange reactions increase in rate relative to the break- down of the amino-enzyme complex (measured by the net forma- tion of urocanic acid). Conversely, as the pH increases, the relative rate of destruction of the amino-enzyme complex in- creases. The exchange reactions are essentially unaffected by changing the pH from 8 to 7, whereas the net reaction is 650/, inhibited by changing the pH from 8 to 7. This indicates a relative stabilization of the amino-enzyme compound at the lower pH. Additional support is given to the idea that amino- enzyme formation takes place by a concerted process by the observation that the tritium and urocanic acid exchange reac- tions occur at essentially identical rates throughout the entire pH range tested.
It should be pointed out that the data of Fig. 7 do not repre- sent a legitimate pH optimal curve for the enzyme. Not only were the experiments performed at substrate concentrations too low to saturate the enzyme, but the experiments at the different pH values were also done with different buffers. A marked influence on the enzyme activity by different buffers was demon- strated in Fig. 3.
E$ect of Urocanic Acid Concentration on Tritium and Urocanic Acid Exchange Reactions
The rates of exchange of tritium and of urocanic acid show essentially the same response to different concentrations of urocanic acid. In addition to the other evidence, this supports the proposal that the tritium and urocanic acid exchange reac- tions reflect a single reaction rather than a sequence of two reac- tions. The measurement of the dependence of the two exchange reactions on the concentration of urocanic acid provides a means for the determination of the Km for urocanic acid (Fig. 8). The Km for urocanic acid, 7 X lO+ M, is of the same order as that for histidine.
by guest on August 13, 2018
http://ww
w.jbc.org/
Dow
nloaded from

794 Mechanism of Histidase Vol. 237, No. 3
I/ I l TRIT/UM EXCHANGE 1
30 A UROCANIC ACID
$(M) FIG. 8. Variation of tritium and urocanic acid exchange reac-
tions with urocanic acid concentration. Data plotted according to Lineweaver and Burk (15). Incubations were carried out at 36” for 70 minutes in a total volume of 0.66 ml and contained: Tris-acetate (nH 9.0). 209 umoles: histidine (sodium salt), 10
~Z I I
pmoles; mercaptoethanol, lO.pmoles; DEAE-cellulose fraction of histidase, previously dialyzed against 0.001 M Tris-acetate (pH 8.0), 300 units; and urocanic acid, 2, 5, 10, 14, 20, or 30 Mmoles. One set of incubations contained 2 mc of solvent TzO, and the other set contained C-labeled urocanic acid (5,400 c.p.m. per rmole). The reactions were terminated by the addition of 0.65 ml of 12% trichloroacetic acid. Residual histidine was purified on Dowex 50 as described in “Materials and Methods” and as- sayed for the incorporation of either tritium or CY4-urocanic acid. I’ = microatoms tritium or micromoles urocanic acid-Cl4 incor- porated per rmole of residual histidine during the incubation.
DISCUSSION
Although this study has provided insufficient data to formu- late a detailed picture of the process of biological deamination, it may be useful to compare the enzymatic reaction with the “&elimination” reactions of organic compounds (23). Base catalysis is characteristic of both the chemical and enzymatic reaction (the optimal pH for histidase is 9.2). Secondly, strongly electron-attracting groups are essential for effective elimination in the model reaction. The indication that some functional group on the enzyme becomes bound to the amino group of histidine during the deamination process (as evidenced by amino- enzyme formation) is consistent with the idea that increased electron withdrawal on that nitrogen is an essential step. Fi- nally, the p-elimination generally occurs as a concerted, rather than a stepwise, process. Since under no set of experimental conditions used in this study was there an indication for the separation of tritium and urocanic acid exchange, a concerted mechanism is also favored in the enzymatic reaction. Thus, although other possible mechanisms are not excluded, it seems reasonable to suggest a tentative scheme for the enzymatic process which embodies all the features of the chemical reactions as shown in Equation 2.
g* HI
Im\ ‘1 P */c- c\ 1 COOH
= H,O + Im, P
C y2
f”,= “COOH (2)
I 2
inzyme
Im= lmidazole
knzyme 1 H2O
NH,+ Enzyme + OH’
The mode of linkage of ammonia to the protein in the amino- enzyme remains obscure. Using the techniques of Backer et al. for the isolation of enzyme-substrate compounds (24, 25), it might be possible to isolate this intermediate. The relative stability of the amino-enzyme complex at pH 6 to 7 might be useful in the accumulation and isolation of this compound. In addition, since the decomposition of the amino-enzyme com- pound is visualized as a hydrolytic process, the stability of this complex may be increased by carrying out the reaction in a nonaqueous medium.
In addition to the binding of the amino group of histidine to the enzyme, a different part of the substrate, most likely the imidazole ring, also specifically interacts with the protein. This specificity of binding may account for the inability of amino acids other than histidine to participate in the enzymatic hydrogen exchange reaction or for unsaturated acids other than urocanic acid to serve as acceptors of the amino group of histidine.
@Methylaspartase, the enzyme (from Clostridium tetano-
morphum) that converts fl-methylaspartic acid to mesaconic acid, has been crystallized (26) and its mechanism studied (27). Experiments carried out in D20 indicate that deuterium ex- changes into @-methylaspartate more rapidly than the amino acid is transformed into mesaconate. On the basis of these data, Bright and Ingraham suggested that the enzymatic reaction proceeds via an anion resulting from the release of a proton to the medium. These experiments are consistent with a reaction mechanism involving an amino-enzyme. However, more con- clusive tests would have to be made to substantiate such a hypothesis.
Several other enzymes catalyzing nonoxidative deamination reactions have been described. The substrates for these include aspartic acid (28)) P-alanyl-CoA (29), ergothioneine (30) 14 phenyl- alanine (31), and tyrosine.5 In contrast with the studies on @-methylaspartase, deuterium exchange experiments with aspartase (29) indicate that the rate of incorporation of deuterium exactly parallels the rate of amination of fumarate. These data have been used to argue against a reaction mechanism involving a preliminary proton release. More definitive tests are also required in this case to rule out the amino-enzyme type of mecha- nism. To date, no mechanism studies have been carried out on the other enzymes mentioned. Thus, it is premature to specu- late on the importance of the amino-enzyme mechanism in other deamination reactions.
SUMMARY
1. Several properties of purified Pseudomonas histidase have been studied. The K, for histidine varies between 9 X 10m3 M
and 2.4 X 10d2 M. The K, for urocanic acid at pH 9.0 is 7 X lo+ M.
2. In addition to the irreversible deamination reaction, the purified enzyme catalyzes two partial reactions, a hydrogen and a urocanic acid exchange into histidine. Specificity studies on the exchange reactions show that amino acids other than histidine do not undergo hydrogen exchange and that unsaturated acids other than urocanic acid will not accept the amino group of histidine.
3. Specific degradations of histidine labeled with tritium in the hydrogen exchange reaction show that the labilized hydrogen is on the ,L3 carbon of histidine.
4 J. B. Wolff, to be published. 5 A. C. Niesh, personal communication.
by guest on August 13, 2018
http://ww
w.jbc.org/
Dow
nloaded from

March 1962 A. Pete&of&y 795
4. Based on the observed exchange reactions and the lack of exchange of ammonia into histidine, a mechanism is proposed involving the formation of an amino-enzyme, which can either reversibly form histidine or irreversibly be degraded to ammonia and free enzyme.
Acknowledgments-The continued interest of Dr. Herbert Tabor in this problem is greatly appreciated. I am also indebted to Dr. E. Backer for suggesting the urocanic acid exchange experiments.
REFERENCES
1. TABOR, H., Pharmacol. Revs., 6,299 (1954). 2. PETERKOFSKY, A., Biochem. and Biophys. Research Communs.,
3, 445 (1960). 3. MEHLER, A. H., TABOR, H., AND HAYAISHI, O., in W. W. WES-
TERFIELD (Editor), Biochemical preparations, Vol. IV, John Wiley and Sons, Inc., New York, 1955, p. 50.
4. PETERSON, E. A., AND SOBER, H. A., J. Am. Chem. Sot., 76, 751 (1956).
5. TABOR, H., J. Biol. Chem., 188, 125 (1951). 6. WEISSBACH, H., REDFIELD, B. G., AND UDENFRIEND, S., J.
Biol. Chem., 229, 953 (1957). 7. TABOR, H., AND MEHLER, A. II., in S. P. COLOWICK AND N. 0.
KAPLAN (Editors), Methods in enzymology, Vol. II, Academic Press, Inc., New York, 1955, p. 22s. --.
8. LOWRY, 0. H.. ROSEBROUGH. N. J.. FARR. A. L.. AND RANDALL. R. J.; J. Biol. Chem., 193; 265 (1951).’ ’
9. AMES, B., AND MITCHELL, H. K., J. Am. Chem. Sot., 74, 252 (1952).
27. BRIGHT, H. J., AND INGRAAAM, L. L., Abstr. Meeting of Am. Chem. Sot., March, 1961, p. 9C.
28. ENGLARD, S., J. Biol. Chek., 233, 1003 (1958). 29. VAGELOS. P. R.. EARL. J. M.. AND STADTMAN. E. R.. J. Biol.
Chem.,‘234, 4iO (1959). ’ 10. TABOR, H., in S. P. COLOWICH AND N. 0. KAPLAN (Editors), Methods in enzymology, Vol. III, Academic Press Inc., New 30. YANASUGONDHA, D., AND APPLEMAN, M. D., J. Bacterial., 74, York, 1957, p. 623. 381 (1957).
Il. MEHLER, A. H., AND TABOR, H., J. Biol. Chem., 201,775 (1953). 31. KOUKOL, J., AND CONN, E. E., Federation Proc., 20, 373 (1961)
12. MEISTER, A., SOBER, H., AND TICE, S., J. Biol. Chem., 189, 577 (1951).
13. BUSCH, H., HURLBERT, R. B., AND POTTER, V. R., J. Biol. Chem., 196, 717 (1952).
14. SOBER, H. A., GUTTER, F. J., WYCKOFF, M. M., AND PETERSON, E. A., J. Am. Chem. Sot., 78,756 (1956).
15. LINEWEAVER, H., AND BURK, D., J. Am. Chem. Sot., 66, 658 (1934).
16. YOSHIOKA, Y., &aka Daigaku Igaku Zassi, 7, 377 (1955). 17. AMES, B. N., MITCHELL, H. K., AND MITCHELL, M. B., J. Am.
Chem. SOL, 76,1015 (1953). 18. SPRINSON, D. B., AND RITTENBERG, D., J. Biol. Chem., 164,
405 (1950). 19. BAUER, H., AND TABOR, H., in D. SHEMIN (Editor), Biochemi-
cal preparations, Vol. V, John Wilev and Sons, Inc., New York, i957, p. 9i.
20. REITZ, O., 2. physik. Chem. (Leipzig) Abt. A, 183, 371 (1939). 21. GALE, E. F., in D. GLICK (Editor). Methods of biochemical
analysis, Vol. IV, Inters&&e P&lishers, New York, 1954, p. 285.
22. STEPKA, W., in S. P. COLOWICK AND N. 0. KAPLAN (Editors), Methods in enzymology, Vol. III, Academic Press, Inc., New York, 1957, p. 504.
23. GOULD, E. S., Mechanism and structure in organic chemistry,
Henry Holt and Co., New York, 1960, p. 478. 24. KRIMSKY, I., AND RACKER, E., Science,.i22, 319 (1955). 25. DATTA. A. G.. AND RACKER. E.. J. BioE. Chem.. 236.624 (1961). 26. BRIGH;, H. j., AND INGR~HA& L. L., Biochim. et Bibphys.
Acta, 44, 586 (1960).
by guest on August 13, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Alan PeterkofskyReactions
The Mechanism of Action of Histidase: Amino-enzyme Formation and Partial
1962, 237:787-795.J. Biol. Chem.
http://www.jbc.org/content/237/3/787.citation
Access the most updated version of this article at
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/237/3/787.citation.full.html#ref-list-1
This article cites 0 references, 0 of which can be accessed free at
by guest on August 13, 2018
http://ww
w.jbc.org/
Dow
nloaded from