targetingthe c-met pathway potentiates glioblastoma ... · targetingthe c-met pathway potentiates...
TRANSCRIPT

Targeting the c-Met Pathway Potentiates GlioblastomaResponses to ;-RadiationBachchu Lal,1 Shuli Xia,1Roger Abounader,1andJohn Laterra1,2,3
Abstract Purpose: Resistance to current cytotoxic therapies limits the treatment of most solid malignan-cies. This results, in part, from the overactivation of receptor tyrosine kinases and their down-stream pathways in tumor cells and their associated vasculature. In this report, we ask if targetingthe multifunctional mitogenic, cytoprotective, and angiogenic scatter factor/hepatocyte growthfactor (SF/HGF)/c-Met pathway potentiates antitumor responses to g-radiation.Experimental Design: Endogenous expression of SF/HGF and c-Met was targeted in U87MGhuman malignant glioma cells and xenografts using chimeric U1/ribozymes. The effects ofU1/ribozymes F g-radiation on glioma cell proliferation, apoptosis, xenograft growth, and ani-mal survival were examined.Results: U1/ribozymes knocked down SF/HGF and c-Met mRNA and protein levels, sensitizedcells to g-radiation (P < 0.005), and enhanced radiation-induced caspase-dependent cytotoxicityin vitro (P < 0.005). Intravenous U1/ribozyme therapy as liposome/DNA complexes or radiationalonemodestlyandtransientlyinhibitedthegrowthofs.c.U87xenografts.Combiningthetherapiescausedtumorregressionanda40%tumorcurerate. Inanimalsbearingintracranialxenografts, long-termsurvivalwas0%inresponsetoradiation,20%inresponsetointratumoraladenoviral-basedU1/ribozyme delivery, and 80% (P < 0.0005) in response to combining U1/ribozymes with radiation.Thisapparentsynergisticantitumor responsewasassociatedwithaf70%decreaseincellprolifer-ation (P < 0.001)andaf14-to40-foldincrease inapoptosis (P <0.0001)withinxenografts.Conclusions: Targeting the SF/HGF/c-Met pathway markedly potentiates the antigliomaresponse to g-radiation. Clinical trials using novel SF/HGF/c-Met pathway inhibitors in gliomaand other malignancies associated with c-Met activation should ultimate include concurrent radi-ation and potentially other cytotoxic therapeutics.
Expression of the transmembraneous receptor tyrosine kinasec-Met and its secreted ligand scatter factor/hepatocyte growthfactor (SF/HGF) is associated with malignant progressionand poor survival in glioma and many other solid malignancies(1–5). SF/HGF and c-Met are increasingly seen as promisingtherapeutic targets due to these clinical associations and to theirmultifunctional autocrine and paracrine tumor-promotingactivities. c-Met activation by tumor-derived SF/HGF stimulatestumor cell migration/invasion, proliferation, and resistance tovarious cytotoxic stimuli (3, 6, 7). SF/HGF is also a potentphysiologic and tumor-associated endothelial cell survivalfactor and angiogenic factor (8, 9) that correlates with tumorangiogenesis and potentiates the angiogenic effects of otherangiogenic factors (10, 11).
SF/HGF inhibits apoptotic mechanisms and enhances cellsurvival in various normal and malignant cell types and inmultiple contexts. In Madin-Darby canine kidney cell epithe-lial cells, SF/HGF inhibits apoptosis induced by detachment(12) and a constitutively activated truncated c-Met rendersnormal hepatocytes resistant to apoptotic cell death (13). Werecently found that SF/HGF protects neonatal cerebellargranule cells from excitotoxic death (14). In malignant celllines such as mammary carcinoma and malignant glioma, wefound that SF/HGF protects against death induced by a varietyof DNA damaging agents, including the topoisomeraseinhibitors Adriamycin and camptothecin and ionizing radia-tion both in vitro and in vivo (15–17). Whereas the bulk ofevidence points to predominantly cytoprotective and anti-apoptotic effects of c-Met activation, other studies have foundthat c-Met promotes apoptosis in certain sarcoma andhepatoma cell lines (18, 19). Recently, Tulasne et al. foundthat the c-Met tyrosine kinase can induce epithelial cellapoptosis in a SF/HGF-independent manner in response toin vitro cell stress (20).
We have used gain-of-function and loss-of-functionapproaches to determine how endogenous SF/HGF/c-Metsignaling contributes to glioma malignancy. SF/HGF genetransfer to experimental rat and human glioma enhancestumorigenicity, tumor growth rates, and tumor-associatedangiogenesis (9, 21). Stable and near-complete knockdownof endogenous SF/HGF or c-Met gene expression potently
Cancer Therapy: Preclinical
Authors’Affiliations: 1Departments of Neurology, 2Oncology and 3Neuroscience,The Johns Hopkins University School of Medicine and The Kennedy KriegerResearch Institute, Baltimore, MarylandReceived1/24/05; revised 3/18/05; accepted 3/29/05.Grant support: NIH grants R01NS32148 (J. Laterra), R01NS043987 (J. Laterra),and R01NS045209 (R. Abounader).The costs of publication of this article were defrayed in part by the payment of pagecharges.This article must therefore be hereby marked advertisement in accordancewith18 U.S.C. Section1734 solely to indicate this fact.Requests for reprints: John Laterra, Kennedy Krieger Research Institute, 707North Broadway, Baltimore, MD 21205. Phone: 443-923-2679; Fax: 443-923-2695; E-mail: [email protected].
F2005 American Association for Cancer Research.
www.aacrjournals.org Clin Cancer Res 2005;11(12) June15, 20054479
Research. on May 31, 2020. © 2005 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

inhibits the tumorigenicity of malignant glioma cells thatexpress autocrine/paracrine SF/HGF/c-Met loops, a characteris-tic of the majority of human malignant gliomas (22).Delivering anti-SF/HGF and anti-Met U1/ribozymes to prees-tablished glioma xenografts has produced statistically andbiologically significant tumor growth inhibition of a moremodest degree (23). In these previous studies, targeting the SF/HGF/c-Met pathway in glioma xenografts had antiangiogenicand proapoptotic effects suggesting that targeting this pathwaywould enhance antiglioma responses to traditional cytotoxictreatment modalities.
In this present study, we show that targeting the SF/HGF/c-Met pathway in vivo substantially enhances the response ofpreestablished human glioma xenografts to relatively low dosesof hypofractionated g-radiation. Furthermore, we show that theapparent synergistic antitumor response occurs through multi-ple mechanisms including glioma cell growth arrest andactivation of glioma cell apoptosis pathways. Our preclinicalfindings establish a basis for combining strategies that target theSF/HGF/c-Met pathway with g-radiation in the treatment ofmalignant glioma and possibly other cancers associated withc-Met pathway activation.
Materials and Methods
U1/ribozymes and expression vectors. Anti-SF/HGF and anti-c-MetU1snRNA/ribozyme/antisense chimeric transgenes (U1/ribozymes)and their expression vectors were synthesized and extensivelycharacterized as previously described (22, 23). Endotoxin-free anti-SF/HGF, anti-Met, and control U1/ribozyme expression plasmids(designated pU1/SF, pU1/Met, or pU1/control, respectively) werecomplexed with liposomes containing the cationic lipid DOTIM (1-[2-[9(Z)-octadecenoy]-ethyl][-2-]([8](Z)-heptadecenyl]-3-[hydroxyethyl]-imidazolinium chloride) and cholesterol (kindly provided by Dr. GaryKoe, Valentis, Inc., Burlingame, CA) for i.v. delivery to animalsbearing s.c. tumors as previously described (23). Replication-defectiveadenoviruses expressing anti-SF/HGF and anti-Met U1/ribozymes orcontrol U1 sequences (designated Ad-U1/SF and Ad-U1/Met and Ad-U1/control, respectively) were constructed and produced as previouslydescribed (23). Viral preparations purified by ultracentrifugation anddialysis and titered by plaque forming assays at f1012 plaque-forming unit/mL were kindly provided by Dr. Andrea Gambotto(NHL Vector Core Facility, Human Gene Therapy Center, Universityof Pittsburgh).
Northern hybridization and immunoblottting. U87 MG humanmalignant glioma cells were cultured in Eagle’s MEM (CellgroMediatech, Washington, DC) containing 10% fetal bovine serum(Cellgro Mediatech), 0.15% sodium bicarbonate (Life Technologies,Rockville, MD), 1 mmol/L sodium pyruvate (Life Technologies), 0.1mol/L nonessential amino acids (Life Technologies), and 500 Ag/mLpenicillin-streptomycin (Life Technologies) at 37jC, 5% CO2/95% air.Subconfluent cell monolayers received Ad-U1 control, Ad-U1/SF, or Ad-U1/Met (f100 multiplicity of infection). Approximately 48 hours later,cells were harvested and total cellular RNA was purified as previouslydescribed (22). Northern analysis using 32P-labeled cDNA probesspecific for either human SF/HGF, human c-Met, or glyceraldehyde-3-phosphate dehydrogenase was done as previously described (22). Foranalysis of SF/HGF and c-Met protein, cells received adenovirus twice24 hours apart and total protein was isolated in the presence of proteaseinhibitors 48 hours later. Western immunoblot analysis of total cellularprotein (30 Ag per lane) using anti-SF/HGF and anti-c-Met antibodies(Santa Cruz, Inc., Santa Cruz, CA) was done as previously described(22, 23).
In vitro cell viability. U87 MG glioblastoma cells were seeded at adensity of 2,000 cells per well in 12-well tissue culture plates. After
24 hours, adherent cells were treated twice at 24 hours intervals with
either Ad-U1/control or Ad-U1/ribozyme (f100 multiplicity of
infection). At 72 hours following the first adenoviral treatment, cells
were subjected to 20 Gy g-radiation (radiation source = 132Cesium
GammaCell Irradiator; Atomic Energy of Canada, Ltd., Mississauga,
Ontario, Canada) or mock irradiated. In a subset of experiments,
cultures received the caspase inhibitor Z-VAD-FMK (Sigma, St.
Louis, MO) to a final concentration of 50 Amol/L (or buffer only as
control) 30 minutes before the first adenoviral vector treatment and
again 30 minutes before radiation treatment. At the times indicated,
adherent cells were suspended by trypsinization and counted using a
Beckman Coulter Counter (Fullerton, CA). Terminal deoxynucleotidyl
transferase–mediated nick-end labeling (TUNEL) immunocytochemis-
try was done on the pooled populations of adherent and floating cells
as previously described (15).Tumor implantation and animal treatments. For s.c. tumors, U87
MG cells (4 � 106 per animal) were implanted in the flanks of
athymic nude mice (n = 10). When the tumors reached a size of f25
mm3, the animals began treatment with liposomes complexed with
pU1/SF + pU1/Met ribozymes (1:1 mixture) or pU1/control (30 Ag
total DNA in 100 AL) with or without radiation (300 cGy/dose)
delivered by a 132Cesium irradiator. Liposome/DNA complexes were
delivered by tail vein injection on days 5, 15, and 25 following tumor
implantation as previously described (23). Tumors were irradiated on
days 7, 12, 17, 22, and 27 following tumor implantation. Tumor sizes
were measured every 5 days with calipers and volumes were calculated
using the formula: volume = (length � width2) / 2 (9). Animals were
sacrificed if tumor volume reached f200 mm3. For intracranial
tumors, SCID/beige mice were implanted with transcranial cannulae
(Plastics One, Inc., Roanoke, VA) with the proximal end within right
caudate/putamen. Cannulae were used for tumor implantation and
adenovirus delivery to insure precise and replicable delivery of
adenoviruses directly into the tumors (24). U87 MG cells (1 � 105
per animal) were implanted (n = 20) 7 days after cannulae placement.
The animals subsequently received intratumoral injections of Ad-U1/
SF + Ad-U1/Met (1:1 mixture) or Ad-U1/control (5 � 107 plaque-
forming unit in 5 AL) with or without radiation (300 cGy/dose).
Fig. 1. Chimeric U1/ribozymes inhibit glioma cell expression of SF/HGF and c-Met.U87MGhuman glioblastoma cells were treated with replication-defectiveadenovirus coding for anti-SF/HGFU1/ribozyme (Ad-U1/SF), anti-Met U1/ribozyme(Ad-U1/Met), or control (Ad-U1).Total cellular RNA and protein were isolated andsubjected to Northern andWestern analyses, respectively, using specific cDNAprobes and antibodies for human SF/HGF and c-Met as described in Materials andMethods. Densitometric analyses (data not shown) show that U1/ribozymesreduced cell levels of the targeted mRNA byf80% to 95% relative toglyceraldehyde-3-phosphate dehydrogenase (GAPDH). SF/HGF and c-Metproteins were reduced byf80% andf50% to 60%, respectively.
Cancer Therapy: Preclinical
www.aacrjournals.orgClin Cancer Res 2005;11(12) June15, 2005 4480
Research. on May 31, 2020. © 2005 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
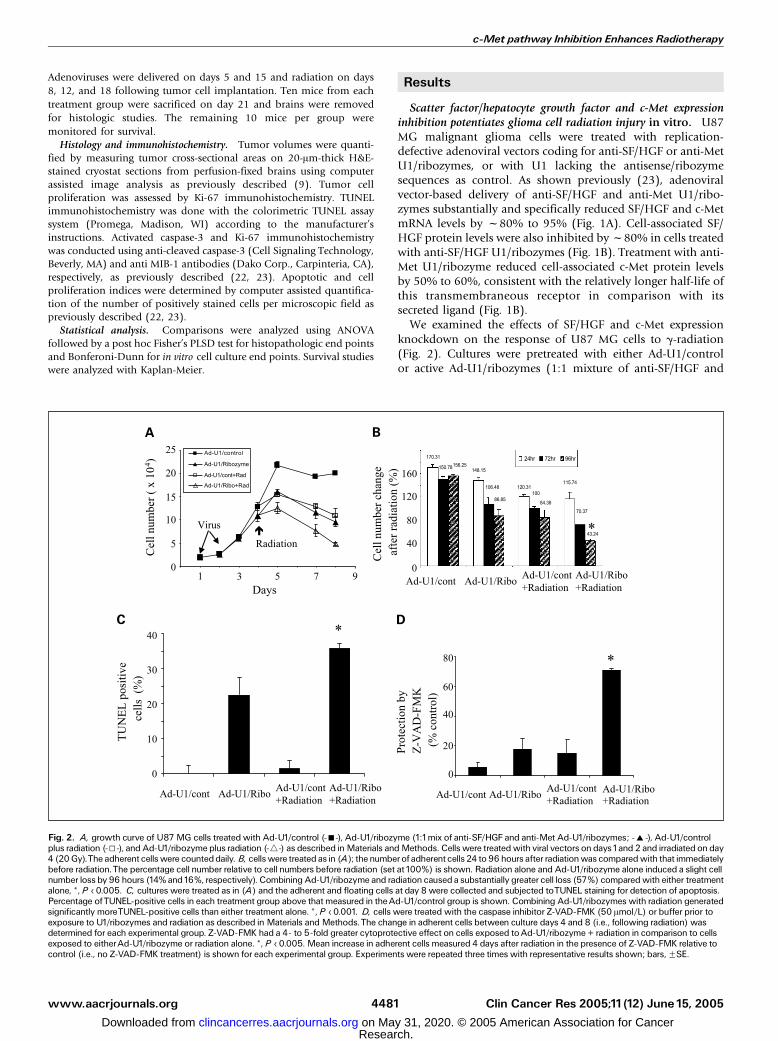
Adenoviruses were delivered on days 5 and 15 and radiation on days
8, 12, and 18 following tumor cell implantation. Ten mice from each
treatment group were sacrificed on day 21 and brains were removed
for histologic studies. The remaining 10 mice per group were
monitored for survival.Histology and immunohistochemistry. Tumor volumes were quanti-
fied by measuring tumor cross-sectional areas on 20-Am-thick H&E-stained cryostat sections from perfusion-fixed brains using computerassisted image analysis as previously described (9). Tumor cellproliferation was assessed by Ki-67 immunohistochemistry. TUNELimmunohistochemistry was done with the colorimetric TUNEL assaysystem (Promega, Madison, WI) according to the manufacturer’sinstructions. Activated caspase-3 and Ki-67 immunohistochemistrywas conducted using anti-cleaved caspase-3 (Cell Signaling Technology,Beverly, MA) and anti MIB-1 antibodies (Dako Corp., Carpinteria, CA),respectively, as previously described (22, 23). Apoptotic and cellproliferation indices were determined by computer assisted quantifica-tion of the number of positively stained cells per microscopic field aspreviously described (22, 23).
Statistical analysis. Comparisons were analyzed using ANOVAfollowed by a post hoc Fisher’s PLSD test for histopathologic end pointsand Bonferoni-Dunn for in vitro cell culture end points. Survival studieswere analyzed with Kaplan-Meier.
Results
Scatter factor/hepatocyte growth factor and c-Met expressioninhibition potentiates glioma cell radiation injury in vitro. U87MG malignant glioma cells were treated with replication-defective adenoviral vectors coding for anti-SF/HGF or anti-MetU1/ribozymes, or with U1 lacking the antisense/ribozymesequences as control. As shown previously (23), adenoviralvector-based delivery of anti-SF/HGF and anti-Met U1/ribo-zymes substantially and specifically reduced SF/HGF and c-MetmRNA levels by f80% to 95% (Fig. 1A). Cell-associated SF/HGF protein levels were also inhibited by f80% in cells treatedwith anti-SF/HGF U1/ribozymes (Fig. 1B). Treatment with anti-Met U1/ribozyme reduced cell-associated c-Met protein levelsby 50% to 60%, consistent with the relatively longer half-life ofthis transmembraneous receptor in comparison with itssecreted ligand (Fig. 1B).
We examined the effects of SF/HGF and c-Met expressionknockdown on the response of U87 MG cells to g-radiation(Fig. 2). Cultures were pretreated with either Ad-U1/controlor active Ad-U1/ribozymes (1:1 mixture of anti-SF/HGF and
Fig. 2. A, growth curve of U87MG cells treatedwith Ad-U1/control (-n-), Ad-U1/ribozyme (1:1mix of anti-SF/HGF and anti-Met Ad-U1/ribozymes; -E-), Ad-U1/controlplus radiation (-5-), and Ad-U1/ribozyme plus radiation (-4-) as described inMaterials andMethods. Cells were treatedwith viral vectors on days1and 2 and irradiated on day4 (20Gy).The adherent cellswere counteddaily. B, cellswere treated as in (A); thenumberof adherent cells 24 to 96 hours after radiationwas comparedwith that immediatelybefore radiation.The percentage cell number relative to cell numbers before radiation (set at100%) is shown. Radiation alone and Ad-U1/ribozyme alone induced a slight cellnumber loss by 96 hours (14% and16%, respectively). Combining Ad-U1/ribozyme and radiation causeda substantially greater cell loss (57%) comparedwith either treatmentalone, *, P < 0.005. C, cultures were treated as in (A) and the adherent and floating cells at day 8 were collected and subjected toTUNEL staining for detection of apoptosis.Percentage ofTUNEL-positive cells in each treatment group above that measured in theAd-U1/control group is shown. Combining Ad-U1/ribozymes with radiation generatedsignificantly moreTUNEL-positive cells than either treatment alone. *, P < 0.001. D, cells were treated with the caspase inhibitor Z-VAD-FMK (50 Amol/L) or buffer prior toexposure to U1/ribozymes and radiation as described in Materials andMethods.The change in adherent cells between culture days 4 and 8 (i.e., following radiation) wasdetermined for each experimental group. Z-VAD-FMK had a 4- to 5-fold greater cytoprotective effect on cells exposed toAd-U1/ribozyme + radiation in comparison to cellsexposed to eitherAd-U1/ribozyme or radiation alone. *, P < 0.005. Mean increase in adherent cells measured 4 days after radiation in the presence of Z-VAD-FMK relative tocontrol (i.e., no Z-VAD-FMK treatment) is shown for each experimental group. Experiments were repeated three times with representative results shown; bars,FSE.
c-Met pathway Inhibition Enhances Radiotherapy
www.aacrjournals.org Clin Cancer Res 2005;11(12) June15, 20054481
Research. on May 31, 2020. © 2005 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
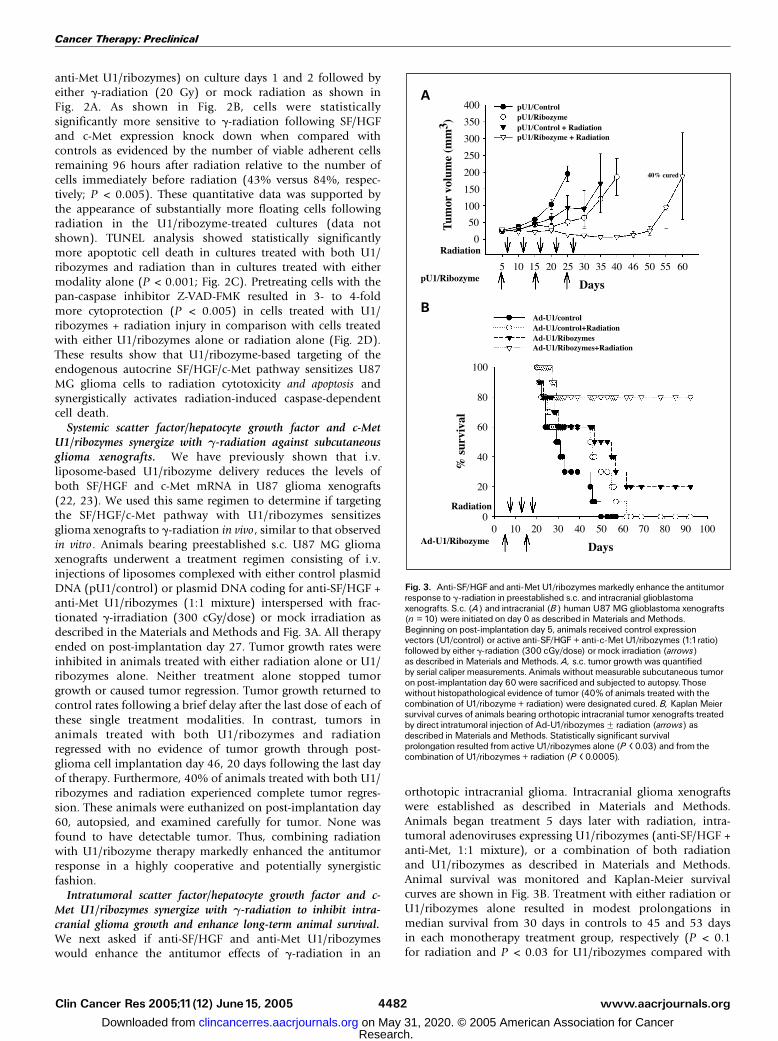
anti-Met U1/ribozymes) on culture days 1 and 2 followed byeither g-radiation (20 Gy) or mock radiation as shown inFig. 2A. As shown in Fig. 2B, cells were statisticallysignificantly more sensitive to g-radiation following SF/HGFand c-Met expression knock down when compared withcontrols as evidenced by the number of viable adherent cellsremaining 96 hours after radiation relative to the number ofcells immediately before radiation (43% versus 84%, respec-tively; P < 0.005). These quantitative data was supported bythe appearance of substantially more floating cells followingradiation in the U1/ribozyme-treated cultures (data notshown). TUNEL analysis showed statistically significantlymore apoptotic cell death in cultures treated with both U1/ribozymes and radiation than in cultures treated with eithermodality alone (P < 0.001; Fig. 2C). Pretreating cells with thepan-caspase inhibitor Z-VAD-FMK resulted in 3- to 4-foldmore cytoprotection (P < 0.005) in cells treated with U1/ribozymes + radiation injury in comparison with cells treatedwith either U1/ribozymes alone or radiation alone (Fig. 2D).These results show that U1/ribozyme-based targeting of theendogenous autocrine SF/HGF/c-Met pathway sensitizes U87MG glioma cells to radiation cytotoxicity and apoptosis andsynergistically activates radiation-induced caspase-dependentcell death.
Systemic scatter factor/hepatocyte growth factor and c-MetU1/ribozymes synergize with g-radiation against subcutaneousglioma xenografts. We have previously shown that i.v.liposome-based U1/ribozyme delivery reduces the levels ofboth SF/HGF and c-Met mRNA in U87 glioma xenografts(22, 23). We used this same regimen to determine if targetingthe SF/HGF/c-Met pathway with U1/ribozymes sensitizesglioma xenografts to g-radiation in vivo , similar to that observedin vitro. Animals bearing preestablished s.c. U87 MG gliomaxenografts underwent a treatment regimen consisting of i.v.injections of liposomes complexed with either control plasmidDNA (pU1/control) or plasmid DNA coding for anti-SF/HGF +anti-Met U1/ribozymes (1:1 mixture) interspersed with frac-tionated g-irradiation (300 cGy/dose) or mock irradiation asdescribed in the Materials and Methods and Fig. 3A. All therapyended on post-implantation day 27. Tumor growth rates wereinhibited in animals treated with either radiation alone or U1/ribozymes alone. Neither treatment alone stopped tumorgrowth or caused tumor regression. Tumor growth returned tocontrol rates following a brief delay after the last dose of each ofthese single treatment modalities. In contrast, tumors inanimals treated with both U1/ribozymes and radiationregressed with no evidence of tumor growth through post-glioma cell implantation day 46, 20 days following the last dayof therapy. Furthermore, 40% of animals treated with both U1/ribozymes and radiation experienced complete tumor regres-sion. These animals were euthanized on post-implantation day60, autopsied, and examined carefully for tumor. None wasfound to have detectable tumor. Thus, combining radiationwith U1/ribozyme therapy markedly enhanced the antitumorresponse in a highly cooperative and potentially synergisticfashion.
Intratumoral scatter factor/hepatocyte growth factor and c-Met U1/ribozymes synergize with g-radiation to inhibit intra-cranial glioma growth and enhance long-term animal survival.We next asked if anti-SF/HGF and anti-Met U1/ribozymeswould enhance the antitumor effects of g-radiation in an
orthotopic intracranial glioma. Intracranial glioma xenograftswere established as described in Materials and Methods.Animals began treatment 5 days later with radiation, intra-tumoral adenoviruses expressing U1/ribozymes (anti-SF/HGF +anti-Met, 1:1 mixture), or a combination of both radiationand U1/ribozymes as described in Materials and Methods.Animal survival was monitored and Kaplan-Meier survivalcurves are shown in Fig. 3B. Treatment with either radiation orU1/ribozymes alone resulted in modest prolongations inmedian survival from 30 days in controls to 45 and 53 daysin each monotherapy treatment group, respectively (P < 0.1for radiation and P < 0.03 for U1/ribozymes compared with
Fig. 3. Anti-SF/HGF and anti-Met U1/ribozymes markedly enhance the antitumorresponse to g-radiation in preestablished s.c. and intracranial glioblastomaxenografts. S.c. (A) and intracranial (B) human U87MG glioblastoma xenografts(n = 10) were initiated on day 0 as described in Materials andMethods.Beginning on post-implantation day 5, animals received control expressionvectors (U1/control) or active anti-SF/HGF + anti-c-Met U1/ribozymes (1:1ratio)followed by either g-radiation (300 cGy/dose) or mock irradiation (arrows)as described in Materials andMethods. A, s.c. tumor growth was quantifiedby serial caliper measurements. Animals without measurable subcutaneous tumoron post-implantation day 60 were sacrificed and subjected to autopsy.Thosewithout histopathological evidence of tumor (40% of animals treated with thecombination of U1/ribozyme + radiation) were designated cured. B, KaplanMeiersurvival curves of animals bearing orthotopic intracranial tumor xenografts treatedby direct intratumoral injection of Ad-U1/ribozymesF radiation (arrows) asdescribed in Materials andMethods. Statistically significant survivalprolongation resulted from active U1/ribozymes alone (P < 0.03) and from thecombination of U1/ribozymes + radiation (P < 0.0005).
Cancer Therapy: Preclinical
www.aacrjournals.orgClin Cancer Res 2005;11(12) June15, 2005 4482
Research. on May 31, 2020. © 2005 American Association for Cancerclincancerres.aacrjournals.org Downloaded from
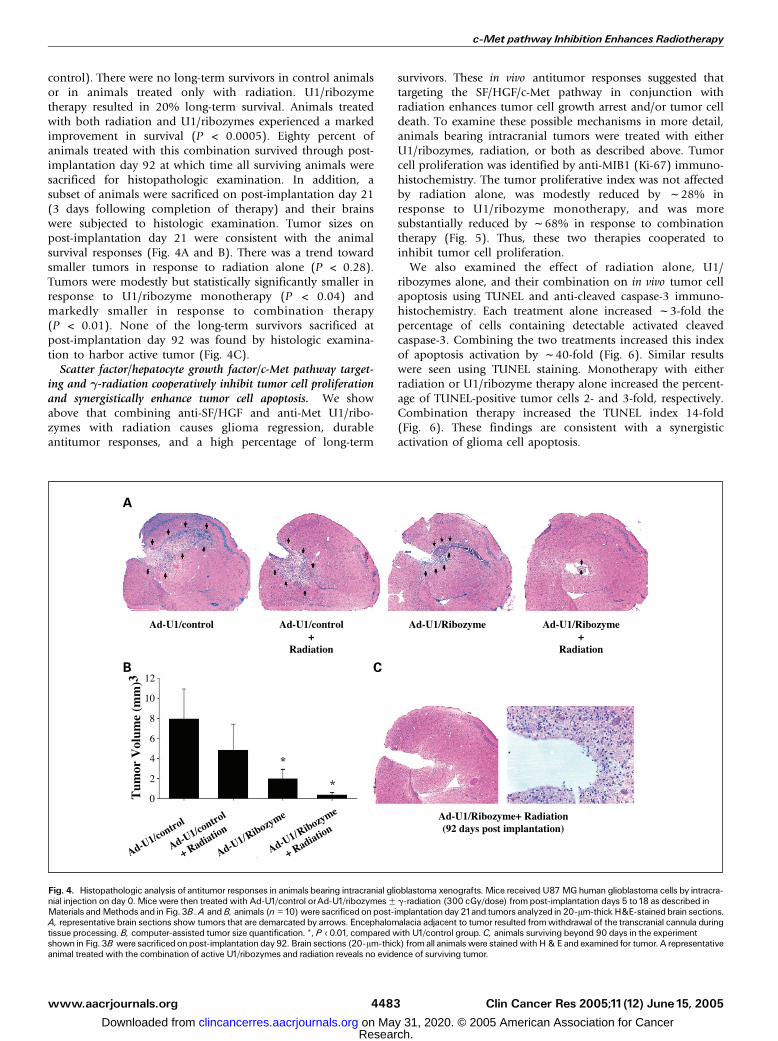
control). There were no long-term survivors in control animalsor in animals treated only with radiation. U1/ribozymetherapy resulted in 20% long-term survival. Animals treatedwith both radiation and U1/ribozymes experienced a markedimprovement in survival (P < 0.0005). Eighty percent ofanimals treated with this combination survived through post-implantation day 92 at which time all surviving animals weresacrificed for histopathologic examination. In addition, asubset of animals were sacrificed on post-implantation day 21(3 days following completion of therapy) and their brainswere subjected to histologic examination. Tumor sizes onpost-implantation day 21 were consistent with the animalsurvival responses (Fig. 4A and B). There was a trend towardsmaller tumors in response to radiation alone (P < 0.28).Tumors were modestly but statistically significantly smaller inresponse to U1/ribozyme monotherapy (P < 0.04) andmarkedly smaller in response to combination therapy(P < 0.01). None of the long-term survivors sacrificed atpost-implantation day 92 was found by histologic examina-tion to harbor active tumor (Fig. 4C).
Scatter factor/hepatocyte growth factor/c-Met pathway target-ing and g-radiation cooperatively inhibit tumor cell proliferationand synergistically enhance tumor cell apoptosis. We showabove that combining anti-SF/HGF and anti-Met U1/ribo-zymes with radiation causes glioma regression, durableantitumor responses, and a high percentage of long-term
survivors. These in vivo antitumor responses suggested thattargeting the SF/HGF/c-Met pathway in conjunction withradiation enhances tumor cell growth arrest and/or tumor celldeath. To examine these possible mechanisms in more detail,animals bearing intracranial tumors were treated with eitherU1/ribozymes, radiation, or both as described above. Tumorcell proliferation was identified by anti-MIB1 (Ki-67) immuno-histochemistry. The tumor proliferative index was not affectedby radiation alone, was modestly reduced by f28% inresponse to U1/ribozyme monotherapy, and was moresubstantially reduced by f68% in response to combinationtherapy (Fig. 5). Thus, these two therapies cooperated toinhibit tumor cell proliferation.
We also examined the effect of radiation alone, U1/ribozymes alone, and their combination on in vivo tumor cellapoptosis using TUNEL and anti-cleaved caspase-3 immuno-histochemistry. Each treatment alone increased f3-fold thepercentage of cells containing detectable activated cleavedcaspase-3. Combining the two treatments increased this indexof apoptosis activation by f40-fold (Fig. 6). Similar resultswere seen using TUNEL staining. Monotherapy with eitherradiation or U1/ribozyme therapy alone increased the percent-age of TUNEL-positive tumor cells 2- and 3-fold, respectively.Combination therapy increased the TUNEL index 14-fold(Fig. 6). These findings are consistent with a synergisticactivation of glioma cell apoptosis.
Fig. 4. Histopathologic analysis of antitumor responses in animals bearing intracranial glioblastoma xenografts. Mice received U87MGhuman glioblastoma cells by intracra-nial injection on day 0. Mice were then treated with Ad-U1/control orAd-U1/ribozymesF g-radiation (300 cGy/dose) frompost-implantation days 5 to18 as described inMaterials andMethods and in Fig. 3B.A andB, animals (n =10)were sacrificedonpost-implantationday 21and tumors analyzed in 20-Am-thick H&E-stainedbrain sections.A, representative brain sections show tumors that are demarcated by arrows. Encephalomalacia adjacent to tumor resulted fromwithdrawal of the transcranial cannula duringtissue processing. B, computer-assisted tumor size quantification. *, P < 0.01, compared with U1/control group. C, animals surviving beyond 90 days in the experimentshown in Fig. 3B were sacrificedonpost-implantationday 92. Brain sections (20-Am-thick) from all animalswere stainedwithH&E and examined for tumor. A representativeanimal treated with the combination of active U1/ribozymes and radiation reveals no evidence of surviving tumor.
c-Met pathway Inhibition Enhances Radiotherapy
www.aacrjournals.org Clin Cancer Res 2005;11(12) June15, 20054483
Research. on May 31, 2020. © 2005 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

Discussion
There are likely multiple mechanisms for the enhancedtumor cell growth arrest and death observed in vivo in thisstudy. Glioma cell–derived SF/HGF activates c-Met receptorspresent on the tumor cells in an autocrine fashion and on hostparenchymal cells (e.g., endothelial cells), in a paracrinefashion. Interfering with the autocrine loop is expected todirectly down-regulate glioma cell cycle and cytoprotectivepathways (6). The in vitro antiproliferative and cell deathresponses of U87 MG cells to autocrine loop inhibition usingU1/ribozymes in this present study essentially confirm thatsuch direct antitumor effects can occur. These results arealso consistent with our previous findings that knocking downc-Met receptor expression in glioma cells lacking an autocrineloop blocks the ability of recombinant SF/HGF to protectagainst chemotherapy-induced cytotoxicity (5). Inhibiting theparacrine SF/HGF/c-Met loop in tumor xenografts includingU87 MG glioma generates an antiangiogenic response that canalso enhance tumor responses to cytotoxic therapeutics in vivoas originally proposed by Teicher et al. and recently confirmedin clinical trial (25 – 28). The relative contributions ofautocrine and paracrine pathway inhibition to the potentiationof g-radiation by SF/HGF/c-Met pathway targeting have notbeen specifically defined and may vary between tumor types,organ sites, and mode of pathway inhibition (i.e., systemicversus direct intratumoral).
We found that anti-SF/HGF and anti-Met U1/ribozymesenhance antitumor radiation responses in both s.c. andintracranial tumor xenografts, tissue sites with distinctenvironmental features. These distinctions include vascularphenotype, immunologic environment, extracellular matrix,and the nature of peritumoral mesenchymal cell types(e.g., glial cells being unique to the central nervous system).Thus, the radiosensitizing response to SF/HGF/c-Met pathwayinhibition in other c-Met-dependent tumors, many of whichcommonly metastasize to multiple organ sites, is not likely tobe organ specific. Our findings also show that the therapeuticresponses to the U1/ribozymes are independent of deliveryroute (direct intratumoral or systemic) and specific deliverysystem (plasmid DNA/liposome complexes versus adenovi-rus). Therefore, nonspecific affects such as those potentiallyseen from vector-specific host interactions are not likelyto account for the antitumor responses observed in ourexperiments.
The molecular mechanisms by which SF/HGF directlyprotects against DNA damaging agents have been examinedin multiple malignant and nonneoplastic cell lines (15–17).Cytoprotection partially requires signaling through phospha-tidylinositol-3V-kinase and the downstream serine/threoninekinase c-Akt, a pathway potently activated by c-Met, inbreast carcinoma, glioblastoma, and nonneoplastic Madin-Darby canine kidney cell epithelial cells. Activation of thispathway is accentuated in cells lacking the dual phos-phatase MMAC1/PTEN (mutated in multiple aggressivecancers/phosphatase and tensin homologue), a tumorsuppressor frequently absent in glioblastoma multiformeand absent in the U87 MG cell line used in this study.Fan et al. (29) have recently found that nuclear factor-nBdownstream of c-Akt- and p21-activated kinase (Pak-1) canbe additional mediators of SF/HGF-dependent cytoprotec-
tion. This recent study further implicated nuclear factor-nB–mediated transcription of at least two genes, TRAF-2 andcIAP-2 , in tumor cytoprotection by SF/HGF. A comprehen-sive understanding of c-Met-dependent cytoprotection,
Fig. 5. SF/HGF/c-Met targeting potentiates the effect of g-radiation on tumor cellproliferation inhibition.Mice receivedU87MGcells by intracranial injectiononday 0.They were then treated with control vectors or active U1/ribozymesF radiation andsacrificed on post-implantation day 21as described in Fig. 3B.Tumors were thenexamined for cell proliferation using Ki-67 immunohistochemistry as described inMaterials andMethods (A).The number of labeled cells per high-poweredmicroscopic fieldwas quantified by computer-assisted image analysis. *,P < 0.0001compared with Ad-U1/control. +, P < 0.001compared with eitherAd-U1/ribozymeorAd-U1/ribozyme + radiation.
Cancer Therapy: Preclinical
www.aacrjournals.orgClin Cancer Res 2005;11(12) June15, 2005 4484
Research. on May 31, 2020. © 2005 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

particularly the transcriptional-dependent aspects, remains tobe fully determined.
In summary, we show that a molecular therapeutic strategythat targets SF/HGF and c-Met generates potent antitumoractivity when combined with g-radiation. End pointssupporting the cooperative, and by certain criteria, synergisticantitumor response from combining these two modalitiesinclude the regression and apparent cure of s.c. gliomaxenografts, the long-term survival and apparent cure ofanimals bearing intracranial glioma xenografts, and histologicevidence of tumor cell growth arrest and apoptosis. Thepronounced cooperative therapeutic responses presented hereare significant and timely in light of the expectation that
pharmacologic inhibitors of c-Met activation, such as smallmolecule c-Met kinase inhibitors, receptor antagonists such astruncated SF/HGF proteins (e.g., NK4), decoy solublechimeric c-Met receptors, or humanized neutralizing anti-SF/HGF and anti-c-Met antibodies, will soon be available forclinical testing (30–33). Our results highlight the need toultimately evaluate the clinical efficacy of such reagents inconjunction with other cytotoxic regimens such as ionizingradiation therapy and chemotherapy. These would beparticularly practical treatment strategies in patients withnewly diagnosed malignant glioma, essentially all of whoreceive radiation often with chemotherapy as first-linetherapy (34).
Fig. 6. SF/HGF/c-Met targeting and g-radiation synergistically enhance tumor cell apoptosis. Intracranial U87 MG xenografts were established and treated as in Fig. 5.Post-implantation day 21tumors were then examined for cell apoptosis by imunohistochemistry for activated caspase-3 (A) andTUNEL staining (C). Cell labeling wasquantified by computer-assisted image analysis (B andD). Differences in blue-green staining between panels in (A) represent variations in methyl green counterstaining. Asexpected anti ^ activated caspase-3 generates a predominantly cytoplasmic stain relative toTUNEL that labels condensed/fragmented nuclei. *, P < 0.0001.
c-Met pathway Inhibition Enhances Radiotherapy
www.aacrjournals.org Clin Cancer Res 2005;11(12) June15, 20054485
Research. on May 31, 2020. © 2005 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

References1. Birchmeier C, Birchmeier W, Gherardi E, VandeWoude GF. Met, metastasis, motility and more. NatRevMol Cell Biol 2003;4:915^25.
2. Koochekpour S, Jeffers M, Rulong S, et al. Met andhepatocyte growth factor/scatter factor expression inhuman gliomas. Cancer Res1997;57:5391^8.
3. Lamszus K, Liang J, Laterra J, et al. Scatter factorpromotes motility of human glioma and neuromi-crovascular endothelial cells. Int J Cancer 1998;75:19^28.
4. Freije WA, Castro-Vargas FE, Fang Z, et al. Geneexpression profiling of gliomas strongly predicts sur-vival. Cancer Res 2004;64:6503^10.
5. Rosen EM, Laterra J, Joseph A, et al. Scatter factorexpression and regulation in human glial tumors. Int JCancer1996;67:248^55.
6. Bowers DC, Fan S, Rosen EM, LaterraJ. Hepatocytegrowth factor inhibits chemotherapy and radiationinduced cell apoptosis in U373 human glioblastomacell lines. Proc AmAssoc Cancer Res1999;40:325.
7.Walter KA,HossainMA, LuddyC,GoelN, ReznickTE,Laterra J. Scatter factor/hepatocyte growth factorstimulation of glioblastoma cell cycle progressionthrough G1 is c-Myc dependent, and independent ofp27 suppression, cdk2 activation, or E2F1-dependenttranscription. Mol Cell Biol 2002;22:2703^15.
8. Rosen EM, Goldberg ID. Regulation of angiogenesisby scatter factor. EXS1997;79:193^208.
9. Laterra J, Nam M, Rosen E, Rao JS, Johnston P.Scatter factor/hepatocyte growth factor gene transferto 9L glioma cells enhances glioma growth and angio-genesis in vivo. Lab Invest1997;76:565^77.
10. Schmidt MO,Westphal M, Hagel C, et al. Levels ofvascular endothelial growth factor, hepatocyte growthfactor/scatter factor andbasic fibroblast growth factorin human gliomas and their relationship to angiogene-sis. IntJCancer1999;84:10^8.
11. Gerritsen ME, Tomlinson JE, Zlot C, Ziman M,Hwang S. Using gene expression profiling to identifythe molecular basis of the synergistic actions ofhepatocyte growth factor and vascular endothelialgrowth factor in human endothelial cells. Br J Phar-macol 2003;140:595^610.
12. Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induce apoptosis. JCell Biol 1994;124:619^26.
13. Amicone L, Spagnoli FM, Spath G, et al. Transgenic
expression in the liverof truncatedMet blocksapopto-sis andpermits immortalizationof hepatocytes. EMBOJ1997;16:495^503.
14. HossainMA, BailoneJC,GomesR, LaterraJ.Neuro-protection by scatter factor/hepatocyte growth factorand FGF-1in cerebellar granule neurons is phosphati-dylinositol 3-kinase/Akt-dependent and MAPK/CREB-independent. JNeurochem 2002;81:365^78.
15. Bowers DC, Fan S,Walter K, et al. Scatter factor/hepatocyte growth factor activates AKTand protectsagainst cytotoxic death in human glioblastoma viaPI3-kinase and AKT-dependent pathways. CancerRes 2000;60:4277^83.
16. Fan S,WangJ-A,Yuan R-Q, et al. Scatter factor pro-tects epithelial and carcinoma cells against apoptosisinduced by DNA damaging agents. Oncogene 1998;17:131^41.
17. Fan S, Ma YX,Wang J, et al. The cytokine scatterfactor inhibits apoptosis and enhances DNA repair bya common mechanism involving signaling throughphosphatidyl inositol 3V kinase. Oncogene 2000;19:2212^23.
18. Gohda E, Okauchi H, Iwao M,Yamamoto I. Induc-tion of apoptosis by hepatocyte growth factor/scatterfactor and its augmentation by phorbol esters in MethA cells. Biochem Biophys Res Commun 1998;245:278^83.
19. Arakaki N, KaziJA, Kazihara T, Ohnishi T, DaikuharaY. Hepatocyte growth factor/scatter factor activatesthe apoptosis signaling pathway by increasing cas-pase-3 activity in sarcoma180 cells. BiochemBiophysRes Commun1998;245:211^5.
20. Tulasne D, Deheuninck J, Lourenco FC, et al. Proa-poptotic function of the MET tyrosine kinase receptorthrough caspase cleavage. Mol Cell Biol 2004;24:10328^39.
21. Laterra J, Rosen EM, Nam M, Ranganathan S,Fielding K, Johnston P. Scatter factor/hepatocytegrowth factor expression enhances human glioblasto-ma tumorigenicity and growth. Biochem Biophys ResComm1997;235:743^7.
22. Abounader R, Ranganathan S, Lal B, et al. Rever-sion of human glioblastoma malignancy by U1smallnuclear RNA/ribozyme targeting of scatter factor/hepatocyte growth factor and c-met expression.JNatl Cancer Inst1999;91:1548^56.
23. Abounader R, Lal B, Luddy C, et al. In vivo targeting
of SF/HGF and c-met expression via U1snRNA/ribo-zymes inhibits glioma growth and angiogenesis andpromotes apoptosis. FASEB J,16:108 ^ 110, 2002(published on-line November 29, 2001; http://www.fasebj.org/cgi/doi/10.1096/fj.01-0421fje).
24. Lal S, Lacroix M,Tofilon P, Fuller GN, Sawaya R,Lang FF. An implantable guide-screw system for braintumor studies in small animals. JNeurosurg 2000;92:326^33.
25. Teicher BA, Holden SA, Ara G, KorbutT, Menon K.Comparison of several antiangiogenic regimens aloneandwith cytotoxic therapies in the Lewis lungcarcino-ma. Cancer Chemother Pharmacol1996;38:169^77.
26. Hess C,VuongV, Hegyi I, et al. Effect ofVEGF recep-tor inhibitor PTK787/ZK222584 [correction ofZK222548] combined with ionizing radiation on en-dothelial cells and tumour growth. Br J Cancer 2001;85:2010^6.
27. Teicher BA. A systems approach to cancer therapy.(Antioncogenics + standard cytotoxics ^mecha-nism(s) of interaction). Cancer Metastasis Rev 1996;15:247^72.
28. Mayer RJ. Two steps forward in the treatment ofcolorectal cancer. NEnglJMed 2004;350:2406^8.
29. Fan S, GaoM,Meng Q, et al. Role of NF-KB signal-ing inhepatocyte growth factor/scatter factormediat-ed cell protection. Oncogene 2005;24:1749^66.
30. Christensen JG, Schreck R, Burrows J, et al. Aselective small molecule inhibitor of c-Met kinaseinhibits c-Met-dependent phenotypes in vitro andexhibits cytoreductive antitumor activity in vivo.Cancer Res 2003;63:7345^55.
31. Date K, Matsumoto K, Kuba K, Shimura H,TanakaM, NakamuraT. Inhibition of tumor growth and inva-sion by a four-kringle antagonist (HGF/NK4) for hepa-tocyte growth factor. Oncogene1998;17:3045^54.
32. Michieli P, Mazzone M, Basilico C, et al. Targetingthe tumor and its microenvironment by a dual-functiondecoyMet receptor. Cancer Cell 2004;6:61^73.
33. Cao B, SuY, OskarssonM, et al. Neutralizingmono-clonal antibodies to hepatocyte growth factor/scatterfactor (HGF/SF) display antitumor activity in animalmodels. Proc Natl Acad Sci US A 2001;98:7443^8.
34. Chang SM, Lamborn KR, Malec M, et al. Phase IIstudy of temozolomide and thalidomide with radiationtherapy for newly diagnosed glioblastomamultiforme.IntJRadiat Oncol Biol Phys 2004;60:353^7.
Cancer Therapy: Preclinical
www.aacrjournals.orgClin Cancer Res 2005;11(12) June15, 2005 4486
Research. on May 31, 2020. © 2005 American Association for Cancerclincancerres.aacrjournals.org Downloaded from

2005;11:4479-4486. Clin Cancer Res Bachchu Lal, Shuli Xia, Roger Abounader, et al.
-RadiationγResponses to Targeting the c-Met Pathway Potentiates Glioblastoma
Updated version
http://clincancerres.aacrjournals.org/content/11/12/4479
Access the most recent version of this article at:
Cited articles
http://clincancerres.aacrjournals.org/content/11/12/4479.full#ref-list-1
This article cites 33 articles, 9 of which you can access for free at:
Citing articles
http://clincancerres.aacrjournals.org/content/11/12/4479.full#related-urls
This article has been cited by 16 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://clincancerres.aacrjournals.org/content/11/12/4479To request permission to re-use all or part of this article, use this link
Research. on May 31, 2020. © 2005 American Association for Cancerclincancerres.aacrjournals.org Downloaded from