sirt6 overexpression potentiates apoptosis evasion in ... · sirt6 overexpression potentiates...
TRANSCRIPT

Biology of Human Tumors
SIRT6 Overexpression Potentiates ApoptosisEvasion in Hepatocellular Carcinoma viaBCL2-Associated X Protein–DependentApoptotic PathwayLong-Kuan Ran1,2, Yong Chen3, Zhen-Zhen Zhang4, Na-Na Tao1, Ji-Hua Ren1, Li Zhou5,Hua Tang1, Xiang Chen1, Ke Chen1,Wan-Yu Li1, Ai-Long Huang1,2, and Juan Chen1
Abstract
Purpose: To characterize the functional role of SIRT6 in hepa-tocellular carcinoma (HCC).
Experimental Design: The expression of SIRT6 in 60 pairedparaffin-embedded HCC tissues and adjacent nontumoral livertissues was examined by immunohistochemistry. The expressionof SIRT6 in 101 paired frozen HCC tissues and adjacent non-tumoral liver tissues was analyzed by Western blotting analysisand qPCR. The biologic consequences of overexpression andknockdown of SIRT6 in HCC cell lines were studied in vitro andin vivo.
Results: SIRT6 expression was frequently upregulated in clin-ical HCC samples, and its expression was highly associated withtumor grade (P¼ 0.02), tumor size (P ¼ 0.02), vascular invasion(P¼ 0.004), and shorter survival (P¼ 0.024). Depletion of SIRT6from multiple liver cancer cell lines inhibited their growth and
induced apoptosis in vitro. At themolecular level,weobserved thatthe activation of the BCL2-associated X protein (Bax) signalingpathway, a major pathway that determines cancer cell apoptosis,is regulated by SIRT6 via its deacetylase activity. SIRT6 wasrecruited to the promoter of Bax, where it deacetylated histone3 lysine 9 and suppressed its promoter activity. Binding oftranscription factors (p53 and E2F-1) to Bax promoter was alsogenerally increased in SIRT6-depleted cells. In mouse xenografts,SIRT6 suppression inhibited tumor growth and induced apopto-sis. Finally, there is a negative correlation between SIRT6 and BaxmRNA expressions in human HCC samples.
Conclusions: SIRT6 is an important protumorigenic factor inliver carcinogenesis. Thus, the therapeutic targeting of SIRT6may offer options for HCC treatment. Clin Cancer Res; 22(13);3372–82. �2016 AACR.
IntroductionHepatocellular carcinoma (HCC) is the third most frequent
cause of cancer-related deaths worldwide, and its incidence rate isincreasing (1). Despite progress in the diagnosis and treatment ofHCC, its biology remains poorly understood, limiting patientoutcome overall. Although this disease is biologically heteroge-
neous, the dysregulation of cellular proliferation and of apoptosisoccurs frequently and contributes to the malignant phenotype(2). Defects in and the disruption of the death regulator pathwayin cancer cells contribute to resistance to anticancer therapies (3).Therefore, greater understanding of how cancer cells evade deathstimuli is urgently needed for the development of new therapeutictargets for HCC treatment.
The sirtuin (SIRT) family of NADþ-dependent protein deace-tylases has been implicated in life-span regulation in yeast,worms, and flies (4). Seven members of the SIRT family(SIRT1–SIRT7) have been identified in mammals. SIRT6 is anuclear NADþ-dependent deacetylase that regulates multiplemolecular pathways to modulate DNA repair, telomere integrity,and aging. SIRT6 regulates double-strand break (DSB) repair byrecruiting SNF2H to DNA break sites (5), deacetylating the DSB-resection protein CtIP (6), and activating PARP1 under oxidativestress (7). SIRT6 specifically deacetylates histone H3 lysine 9(H3K9) on telomeric chromatin, which is required for telomeremaintenance (8). SIRT6 also attenuates NF-kB signaling viaH3K9deacetylation, which is an important regulator of aging-relatedcellular processes (9). Emerging evidence has also implicatedSIRT6 in the development of various cancers, including breastcancer (10), skin cancer (11), prostate cancer (12), lung cancer(13), pancreatic cancer (14), and gliomas (15). SIRT6 regulatestumorigenesis by directly interacting with oncogenic proteins orby modulating the acetylation of H3K9 in oncogene promoters(15–18). However, the role of SIRT6 in HCC remains unclear.
1Key LaboratoryofMolecular Biology for Infectious Diseases (Ministryof Education), Institute for Viral Hepatitis, Department of InfectiousDiseases, The Second Affiliated Hospital, Chongqing Medical Univer-sity,Chongqing,China. 2Collaborative InnovationCenter forDiagnosisand Treatment of Infectious Diseases, Zhejiang University, Zhejiang,China. 3Department of Hepatobiliary Surgery, First Affiliated Hospital,Chongqing Medical University, Chongqing, China. 4Department ofInfectious Diseases, The Children’s Hospital of Chongqing MedicalUniversity, Chongqing, China. 5Department of Epidemiology, Schoolof Public Health and Management, Chongqing Medical University,Chongqing, China.
L.-K. Ran, Y. Chen, and Z.-Z. Zhang contributed equally to this article.
Note: Supplementary data for this article are available at Clinical CancerResearch Online (http://clincancerres.aacrjournals.org/).
Corresponding Authors: Juan Chen, The Second Affiliated Hospital, 1# MedicalRoad, Yuzhong District, Chongqing 40016, China. Phone: 86-23-68815112; Fax:86-23-68486780; E-mail: [email protected]; or Ai-Long Huang, Phone:86-23-68818112; Fax: 86-23-68486780; Email: [email protected]
doi: 10.1158/1078-0432.CCR-15-1638
�2016 American Association for Cancer Research.
ClinicalCancerResearch
Clin Cancer Res; 22(13) July 1, 20163372
on April 30, 2020. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 9, 2016; DOI: 10.1158/1078-0432.CCR-15-1638

We previously reported that SIRT1 and SIRT2 are critical parti-cipants in HCC development. SIRT1 is essential for proliferationand telomere maintenance (19), whereas SIRT2 mediates theepithelial–mesenchymal transition (EMT) of cancer cells (20).In the present study, we found that SIRT6 was upregulated in asubset of HCC tissues and cell lines. SIRT6 overexpression inprimary HCC tumors correlated with tumor size and grade.Furthermore, we discovered an oncogenic function of SIRT6 inHCC that involved the inhibition of cell apoptosis via the Bax-dependent signaling pathway in vitro and in vivo.
Materials and MethodsPlasmids and antibodies
SIRT6 short hairpin RNA (shSIRT6-1 and shSIRT6-2) or non-targeting shRNA (shCont) were purchased from Shanghai Gen-echemCompany Limited. Sequences of shSIRT6-1 and shSIRT6-2are 50-GCTACGTTGACGAGGTCATGA-30 and 50-GCCTCTGAC-TTGCTGTGTTGT-30. Sequence of shCont is 50-GCAACAAGAT-GAAGAGCACCAA-30. Anti-SIRT6 (NB100-2522) and Anti-Bax(NBP1-88682) were obtained from Novus Biologicals. Anti-Bax(#5023S), anti-COX IV (#4850), anti-Smac (#2954S), anti-His-toneH3(#3638), anti-HistoneH4(#07-327), anti-Bcl-2 (#2870),anti-Cytochrome C (#4280S), anti-PARP (#9542), anti-cleavedPARP (#5625) and anti-caspase 9 (#9502), anti-E2F-1 (#3742),anti-p53 (#2527), anti–Acetyl-Lyine (#9441), anti-caspase 3(#9665), and anti-cleaved caspase 3(#9664) were obtained fromCell Signaling Technology. Anti–acetyl-HistoneH3 (Ac-Lys9) (07-352) and anti–acetyl-Histone H4 (Ac-Lys16) (07-329) were fromMillipore. Anti–b-actin (sc-1616) and anti-GAPDH (sc-365062)were purchased from Santa Cruz Biotechnology. The plenti-c-Myc-DDK-SIRT6 expression vector was obtained from OriGene.siRNAs targeting Bax and siRNAs with scrambled sequences wereobtained from Invitrogen.
Cell cultureHepG2, PLC/PRF/5, SK-Hep-1, and Hep3B cells were obtained
from the American Type Culture Collection. Huh-7 cell line was
acquired from the Health Science Research Resource Bank. MIHAcell linewas obtained fromProfessor BenC.B. Ko (TheHongKongPolytechnic University), and SMMC-7721 cell line was obtainedfrom Professor Ni Tang (Chong Qing medical University).HepG2, PLC/PRF/5, SK-Hep-1, Huh-7, SMMC-7721, Hep3B, andMIHA cell lines were cultivated in DMEM containing 10% FBS(Gibco BRL). HepG2 was maintained in MEM with 10% FBS.Primary human hepatocytes (PHH) were purchased from Scien-cell Research laboratories and cultured in hepatocyte medium(Sciencell). All cells were cultivated at 37�C in 5%CO2. Cells wereauthenticated by short-tandem repeat (STR) fingerprinting byBeijing Microread Genetics Company Limited recently.
HCC specimensA set of 101 pairs of primary and corresponding adjacent
nontumoral liver tissues were collected from the First AffiliatedHospital of Chongqing Medical University. The study protocolconformed to the ethical guidelines of the 1975 Declaration ofHelsinki and was approved by the Clinical Research Ethics Com-mittee of Chongqing Medical University. Total RNA and proteinswere obtained from these specimens.
Coimmunoprecipitation assay and Western blot analysisCellswere harvested and lysedwith RIPA lysis buffer containing
a protease inhibitor cocktail (Roche Diagnostics). Then, thelysates were precipitated using protein magnetic beads (Milli-pore). ForWestern blotting analysis, the lysates were separated bySDS-PAGE and transferred onto a nitrocellulose membrane. Themembrane was then blocked with 5% nonfat milk for 1 hour andincubated with relevant primary antibodies overnight at 4�C. Theblots were developed using ECL Western blotting reagents (Milli-pore). Band intensities of Western blot were quantified by ImageJv1.37 software.
Real-time qPCRTotal RNA was extracted using TRIzol Reagent (Invitrogen).
cDNA was synthesized from 1 mg of total RNA using an iScriptcDNA Synthesis kit (Bio-Rad). Relative gene expression levelsfollowing different treatments were determined using FastStartUniversal SYBR Green Master Mix (Roche Diagnostics) withb-actin mRNA as an endogenous control. Values represent themean � the SDs of three independent experiments. The expres-sion values of target genes were calculated by using the 2�DDCt
method. The primers were designed by our group and are listed inSupplementary Table S1.
Chromatin immunoprecipitation assayChromatin immunoprecipitation (ChIP) assays were per-
formed with genomic DNA samples from cross-linked cells usinga specific antibody according to the manufacturer's protocol(Millipore). A region of the Bax promoter was amplified fromthe immunoprecipitated DNA samples by PCR using the senseprimer F 50-TAAAAATTAACCAGGGGCGG-30 and the antisenseprimer R 50-TCACTGTGTTGCCCAGGCTG-30.
Cell proliferation assayCell proliferation in response to SIRT6 silencing or overexpres-
sion was determined by a trypan blue exclusion assay (ThermoFisher Scientific). DNA synthesis was examined using a Click-iTEdU Imaging kit (Invitrogen) according to the manufacturer'sinstructions.
Translational Relevance
Optimal therapeutic strategies for hepatocellular carcinoma(HCC) patients are still challenging due to its high recurrencerate after surgical resection and chemotherapy resistance.Growing evidence shows that genetic and epigenetic altera-tions are involved in HCC progression; however, the under-lying molecular mechanisms have not been fully elucidated.Here, we found that SIRT6 was significantly upregulated inHCC tissues, and upregulation of SIRT6 was significantlyassociated with increased tumor grade, tumor size, vascularinvasion, and shorter survival. Our in vitro and in vivo studiesshowed that gene silencing of SIRT6 suppressed cell prolifer-ation and promoted cellular apoptosis by activating the Bax-dependent apoptotic signal pathway in HCC cells. Further-more, SIRT6 knockdown could increase liver cancer cell sen-sitivity to chemotherapy drug doxorubicin. Collectively, thenewly identified SIRT6–Bax axis partially illustrates themolec-ular mechanism of HCC progression and represents a novelpotential therapeutic target for HCC treatment.
The Role of SIRT6 in HCC
www.aacrjournals.org Clin Cancer Res; 22(13) July 1, 2016 3373
on April 30, 2020. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 9, 2016; DOI: 10.1158/1078-0432.CCR-15-1638

Colony formation assay and soft-agar assayThe colony formation assay and soft-agar assay have been
described previously (19).
Flow cytometryCell apoptosis was analyzed by fluorescence-activated cell
sorting analysis as described previously (21).
Extraction of mitochondria and nuclear proteinsNuclear proteins were purified using NE-PER Nuclear and
Cytoplasmic Extraction Reagents (Thermo Fisher Scientific)according to the manufacturer's instructions. Mitochondrial pro-teins were extracted using the Mitochondria/Cytosol Fraction Kit(Abcam) according to the manufacturer's instructions.
Immunofluorescence and immunohistochemistrySK-Hep-1 cells infected with lentivirus expressing shCont or
shSIRT6 were seeded on coverslips, fixed in 4% paraformalde-hyde, and permeabilized using 0.5% Triton X-100. Then, the cellswere incubated with rabbit anti-Bax antibody (Novus Biologicals;dilution 1:100) and a FITC-coupled anti-rabbit secondary anti-body. The cells were counterstained with DAPI to label the nucleiand examined by fluorescence microscopy (Leica TCS SP2).
Immunohistochemistry was performed on paraffin-embeddedsections. The tissue sections were deparaffinized, rehydrated, andmicrowaved-heated in sodium citrate buffer (10mmol/L, pH6.0)for antigen retrieval. Then, the slides were incubated with primaryantibody (anti-Bax, 1:50; anti-Ki67, 1:100; anti-SIRT6, 1:200;anti-cleaved PARP, 1:200). Diaminobenzidine (DAB) stainingwas used for detecting immunoreactivity (Dako). Counterstain-ing was performed using hematoxylin. The scoring of SIRT6 inHCC tissues was carried out by two independent pathologistsaccording to the proportion of tumor cells with positive nuclearstaining: 0 (<10%); 1 (10%–30%); 2 (30%–50%); and 3 (<50%).HScore takes into consideration the intensity of the staining andthe percentage of positive cells per the formula: HScore¼ 1� (%light staining) þ 2 � (% moderate staining) þ 3 � (%strongstaining). HScores range from 0 to 300 (22).
Luciferase reporter assayDistinct lengths of the Bax promoter fragment were subcloned
into the pGL3-basic vector to produce pGL3-982, pGL3-803,pGL3-588, pGL3-389, andpGL3-213. These different vectorswerecotransfected with shCont or shSIRT6. pRL-TK was cotransfectedto normalize the transfection efficiency. Luciferase activity wasmeasured using the dual-luciferase Reporter Assay System (Pro-mega) according to the manufacturer's instructions.
Animal modelMale BALB/c nude mice (4 weeks of age) received single
subcutaneous flank injection of SK-Hep-1 cells suspended in200 mL DMEM/Matrigel (1:1 mixture). Tumor growth was mon-itored by bidimensional measurements using a caliper. Tumor-bearing mice were sacrificed 4 weeks after inoculation, and thetumors were removed for further study.
Statistical analysisThe SIRT6 expression levels in HCC and nontumoral liver
tissueswere comparedusing the Student paired t test. Correlationsbetween SIRT6 and individual clinicopathologic parameters wereevaluated using a nonparametric c2 test and Spearman's s rank
test. The Kaplan–Meier method was used to estimate the survivalrates for SIRT6 expression. Equivalences of the survival curveswere tested by log-rank statistics. All statistical analyses wereperformed using SPSS 19.0 software (IBM Corporation).
ResultsSIRT6 expression in human HCC
First, we analyzed SIRT6 expression patterns in eight cell lines,including six HCC cell lines (Huh-7, HepG2, PLC/PRF/5, SMMC-7721, Hep3B, and SK-Hep-1), one immortalized liver cell line(MIHA), and PHH. SIRT6 was almost undetectable in PHH,whereas high-level expression of SIRT6 was observed in the sixHCC cell lines at both the mRNA and protein levels (Fig. 1Aand B).
Next, we determined SIRT6 expression in 60 paired paraffin-embedded HCC tissues and adjacent nontumoral livers by usingimmunohistochemistry. SIRT6 immunoreactivity was graded asnegative (score 0), low (scores 1–2), and high (score 3). In the 60cases examined, positive SIRT6 expression was detected in 25 of60 (41.6%) HCC tissues (Fig. 1C). In contrast, SIRT6 staining wasonly detected in 9 of 60 (15%) adjacent nontumoral liver. Amongthe 25 positive HCC samples, 15 showed low staining and 10showed strong staining of SIRT6. SIRT6 expression was furtheranalyzed in 101 pairs of frozen HCC and adjacent nontumoralliver tissues by usingWestern blotting analysis. SIRT6 overexpres-sion was detected in 67 of 101 (66%) of HCCs compared withtheir adjacent nontumoral liver tissues (Fig. 1D and Supplemen-tary Fig. S1). Furthermore, the average SIRT6 protein levels weresignificantly higher in tumor tissues relative to nontumoral livertissues, indicating that SIRT6 was frequently overexpressed inHCC (Fig. 1E). We also compared SIRT6 mRNA levels in thesesamples by qPCR. SIRT6 mRNA levels were significantly upregu-lated inHCC tissues comparedwith nontumoral liver tissues (Fig.1F), suggesting that SIRT6 overexpression inHCC is regulated in atranscription-dependent manner. Correlative analysis of SIRT6protein levels with clinicopathologic features suggested signifi-cant association between increased SIRT6 expression and tumorsize (P¼ 0.02), tumor grade (P¼ 0.02), and vascular invasion (P¼ 0.004; Table 1). Among these 101 HCCs, detailed survivalinformation was available for 53 cases. The Kaplan–Meier anal-ysis revealed that patients with high SIRT6 expression levels intissues had significantly shorter overall survival rates than thosewith low SIRT6 expression (P ¼ 0.024; Fig. 1G).
SIRT6 depletion in HCC cells inhibited proliferation andinduced apoptosis
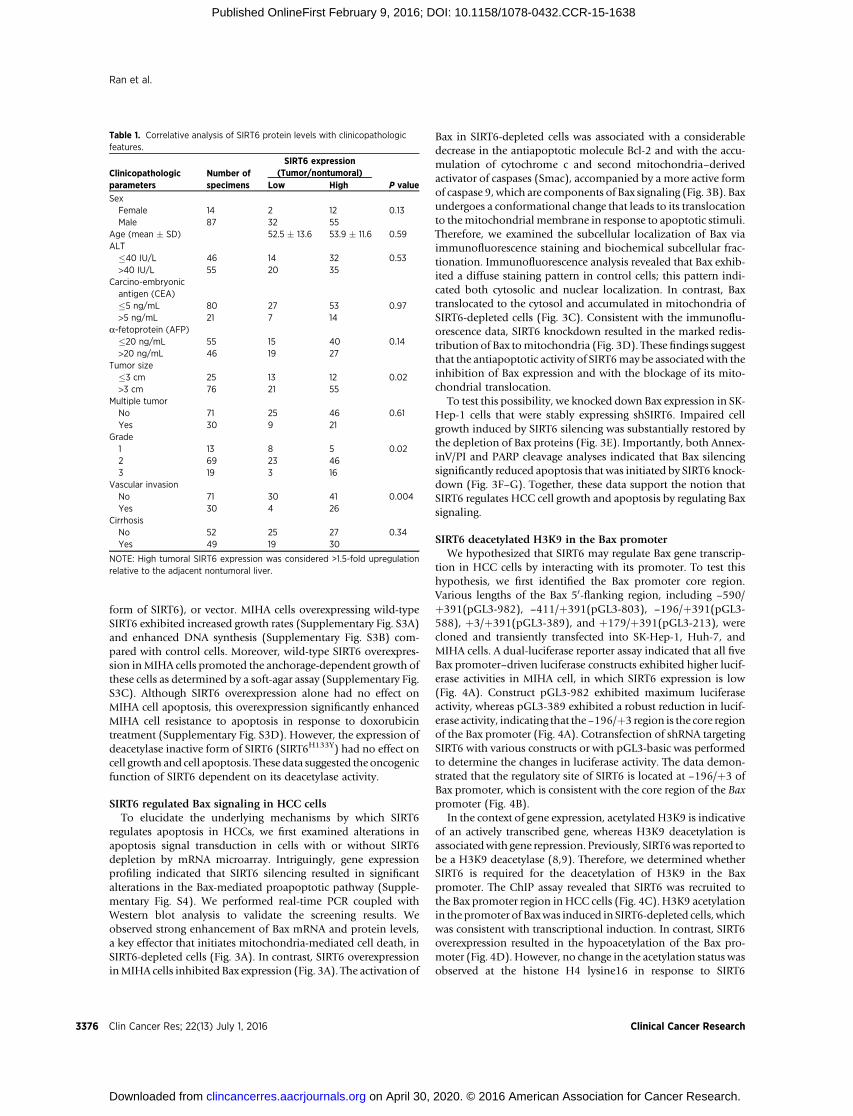
We analyzed the cellular loss-of-function phenotype via lenti-virus-mediated shRNA interference to address the functionalimportance of SIRT6 in HCC development. Two independentshRNAs (shSIRT6-1 and shSIRT6-2) induced efficient SIRT6knockdown in four HCC cell lines (PLC/PRF/5, SMMC-7721,Huh-7, and SK-Hep-1) compared with scrambled shRNA(shCont)–infected cells (Fig. 2A). The loss of SIRT6 significantlydecreased the proliferation rate of all HCC cell lines examined(Fig. 2B). Furthermore, SIRT6 knockdown reduced the numbersand sizes of SK-Hep-1 cell colonies as determined by a colonyformation assay (Fig. 2C). EdU staining also indicated that theloss of SIRT6 dramatically inhibited DNA synthesis (Fig. 2D).
Next, we determined if SIRT6 plays a role in the regulation ofcell death. AnnexinV/PI assays indicated a dramatic increase in the
Ran et al.
Clin Cancer Res; 22(13) July 1, 2016 Clinical Cancer Research3374
on April 30, 2020. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 9, 2016; DOI: 10.1158/1078-0432.CCR-15-1638

apoptosis rate in SIRT6-depleted cells comparedwith their respec-tive controls (Fig. 2E). SIRT6 depletion–induced apoptosis wasfurther evidenced by enhanced PARP cleavage and caspase 3cleavage (Fig. 2F). In addition, we further examined the potentialrole of SIRT6 in resistance to the DNA-damaging agent doxoru-bicin. SIRT6 silencing promoted doxorubicin-induced apoptosisin Huh-7 and SK-Hep-1 cells, as evidenced by the AnnexinV/PIassays and by PARP cleavage (Supplementary Fig. S2A and S2B).
Together, these data suggest that SIRT6 may play a role in theregulation of HCC proliferation and apoptosis.
SIRT6 overexpression in immortalized liver cell line promotedcell proliferation
To further elucidate the oncogenic role of SIRT6 in HCCtumorigenesis, we infected immortalized liver cell line (MIHA)with lentivirus expressing SIRT6, SIRT6H133Y (deacetylase inactive
MIH
A
MIH
A
Huh-7
Huh-7
PLC/P
RF/5
PLC/P
RF/5
SMM
C-772
1
SMM
C-772
1
Hep3B
Hep3B
HepG2
HepG2
SK-Hep
-1
SK-Hep
-1
PHH
PHH
SIRT6
Negative
A
C
E
G
F
B
D
HCC
AdjacentNontumoral liver
Adjacentnontumoral liver
Overall survival
Time (months)
Per
cen
t su
rviv
al (
%)
TumorTumor
050
60
70
80
90
100
110
50 100 150 200
P = 0.024
SIRT6 not overexpressed (n = 20)
SIRT6 overexpressed (n = 33)
Adjacentnontumoral liver
0.01
0.1
Rel
ativ
e S
IRT
6 m
RN
Aex
pres
sion
Rel
ativ
e S
IRT
6 pr
otei
nex
pres
sion
1
10
100
0.1
1
10
100
Low High
0
2
4
6
8
Rel
ativ
e m
RN
A le
vel o
f SIR
T6
β-Actin
SIRT6
HCC1
T
0.01 0.01 6.21 0.01 0.30 0.01 2.20 0.01 1.32 0.33 0.01 0.01
4.01 1.43 3.66 1.54 0.01 0.01 1.60 0.01 1.49 0.01 0.01 0.12
5.21 0.10 0.01 0.95 0.06 0.01 3.11 0.01 3.88 0.50 0.69 3.21
0.01 0.01 4.51 0.26 3.45 0.45 4.41 0.95 3.59 1.51 1.36 0.93
N T N T N T N T N T N
T N T N T N T N T N T N
T N T N T N T N T N T N
T N T N T N T N T N T N
HCC2 HCC3 HCC4 HCC5 HCC6
HCC7 HCC8 HCC9 HCC10 HCC11 HCC12
HCC13 HCC14 HCC15 HCC16 HCC17 HCC18
HCC19 HCC20 HCC21 HCC22 HCC23 HCC24
β-Actin
SIRT6
β-Actin
SIRT6
β-Actin
SIRT6
β-Actin
Figure 1.Differential expression of SIRT6 in HCC samples. A and B, SIRT6 mRNA and protein expression in six liver cancer cell lines, the immortalized liver cell line (MIHA),and PHH. b-Actin (43 kDa) was used as a reference gene for real-time PCR and as a loading control for Western blot analysis. C, immunohistochemicalexamination of SIRT6 in 60paired primary HCC tissues and adjacent nontumoral tissues. Magnification,�400. D,Western blot analysis of SIRT6 (37 kDa) in 101 pairedfrozen HCC tissues (T) and adjacent nontumor liver tissues (N). b-Actin was used as a loading control. E, quantitative analysis of SIRT6 protein levels in 101paired HCC tissues. � , P < 0.01. F, real-time PCR analysis of SIRT6 mRNA levels in 101 paired HCC tissues and adjacent nontumoral tissues. b-Actin mRNA expressionwas used as an internal control. � , P < 0.01. G, Kaplan–Meier analysis of overall survival in 53 HCC patients based on SIRT6 expression.
The Role of SIRT6 in HCC
www.aacrjournals.org Clin Cancer Res; 22(13) July 1, 2016 3375
on April 30, 2020. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 9, 2016; DOI: 10.1158/1078-0432.CCR-15-1638
form of SIRT6), or vector. MIHA cells overexpressing wild-typeSIRT6 exhibited increased growth rates (Supplementary Fig. S3A)and enhanced DNA synthesis (Supplementary Fig. S3B) com-pared with control cells. Moreover, wild-type SIRT6 overexpres-sion inMIHA cells promoted the anchorage-dependent growth ofthese cells as determined by a soft-agar assay (Supplementary Fig.S3C). Although SIRT6 overexpression alone had no effect onMIHA cell apoptosis, this overexpression significantly enhancedMIHA cell resistance to apoptosis in response to doxorubicintreatment (Supplementary Fig. S3D). However, the expression ofdeacetylase inactive form of SIRT6 (SIRT6H133Y) had no effect oncell growth and cell apoptosis. These data suggested the oncogenicfunction of SIRT6 dependent on its deacetylase activity.
SIRT6 regulated Bax signaling in HCC cellsTo elucidate the underlying mechanisms by which SIRT6
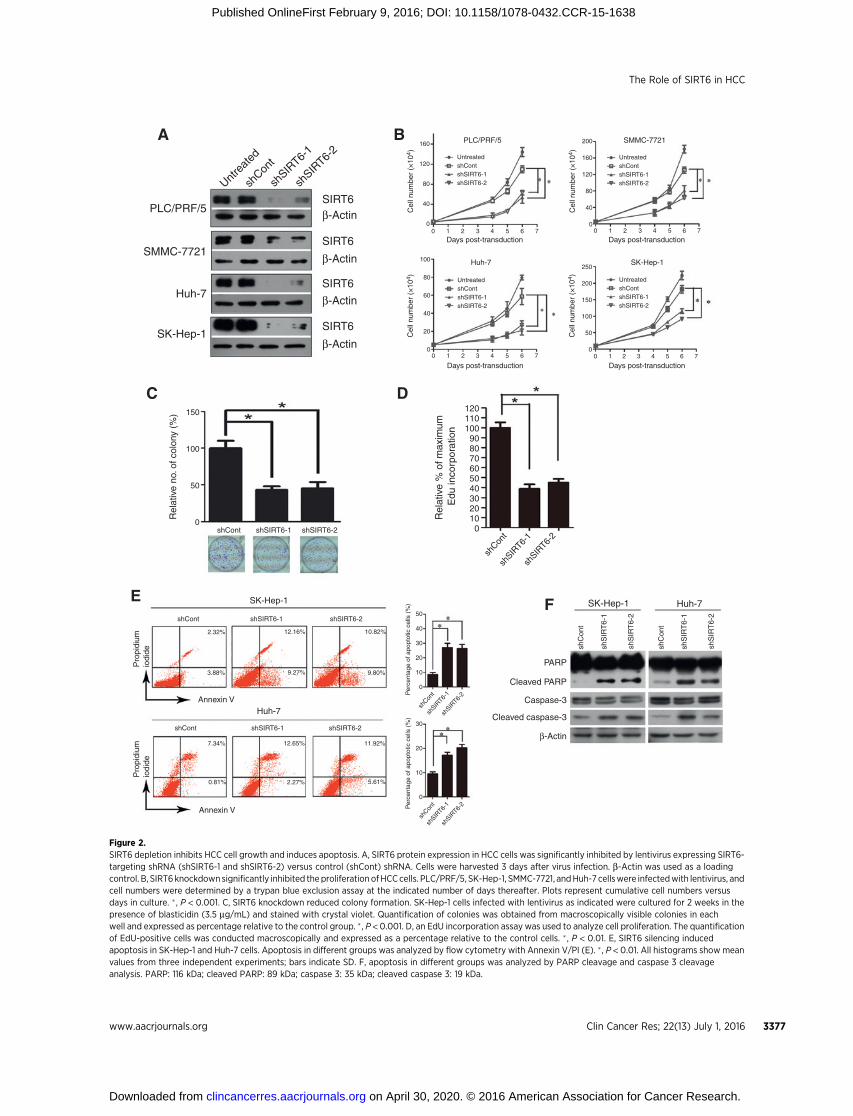
regulates apoptosis in HCCs, we first examined alterations inapoptosis signal transduction in cells with or without SIRT6depletion by mRNA microarray. Intriguingly, gene expressionprofiling indicated that SIRT6 silencing resulted in significantalterations in the Bax-mediated proapoptotic pathway (Supple-mentary Fig. S4). We performed real-time PCR coupled withWestern blot analysis to validate the screening results. Weobserved strong enhancement of Bax mRNA and protein levels,a key effector that initiates mitochondria-mediated cell death, inSIRT6-depleted cells (Fig. 3A). In contrast, SIRT6 overexpressioninMIHA cells inhibited Bax expression (Fig. 3A). The activation of
Bax in SIRT6-depleted cells was associated with a considerabledecrease in the antiapoptotic molecule Bcl-2 and with the accu-mulation of cytochrome c and second mitochondria–derivedactivator of caspases (Smac), accompanied by a more active formof caspase 9, which are components of Bax signaling (Fig. 3B). Baxundergoes a conformational change that leads to its translocationto themitochondrial membrane in response to apoptotic stimuli.Therefore, we examined the subcellular localization of Bax viaimmunofluorescence staining and biochemical subcellular frac-tionation. Immunofluorescence analysis revealed that Bax exhib-ited a diffuse staining pattern in control cells; this pattern indi-cated both cytosolic and nuclear localization. In contrast, Baxtranslocated to the cytosol and accumulated in mitochondria ofSIRT6-depleted cells (Fig. 3C). Consistent with the immunoflu-orescence data, SIRT6 knockdown resulted in the marked redis-tribution of Bax tomitochondria (Fig. 3D). These findings suggestthat the antiapoptotic activity of SIRT6may be associatedwith theinhibition of Bax expression and with the blockage of its mito-chondrial translocation.
To test this possibility, we knocked down Bax expression in SK-Hep-1 cells that were stably expressing shSIRT6. Impaired cellgrowth induced by SIRT6 silencing was substantially restored bythe depletion of Bax proteins (Fig. 3E). Importantly, both Annex-inV/PI and PARP cleavage analyses indicated that Bax silencingsignificantly reduced apoptosis that was initiated by SIRT6 knock-down (Fig. 3F–G). Together, these data support the notion thatSIRT6 regulates HCC cell growth and apoptosis by regulating Baxsignaling.
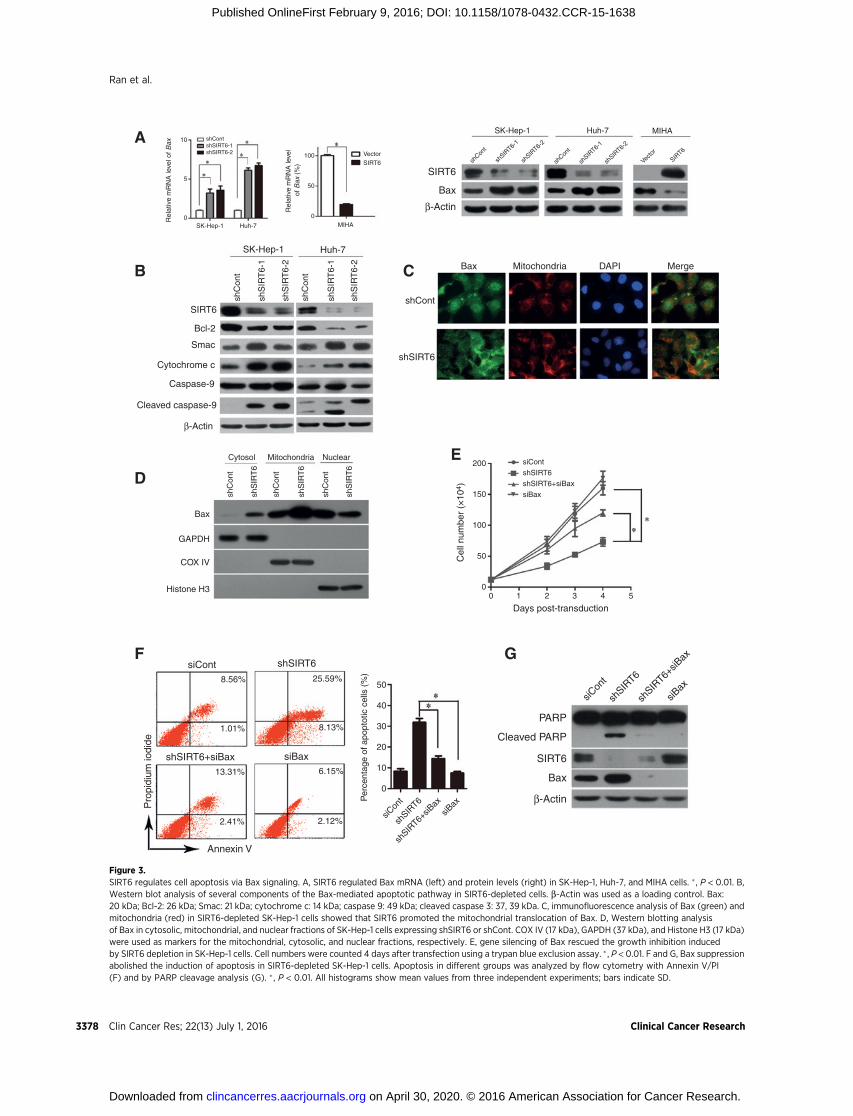
SIRT6 deacetylated H3K9 in the Bax promoterWe hypothesized that SIRT6 may regulate Bax gene transcrip-
tion in HCC cells by interacting with its promoter. To test thishypothesis, we first identified the Bax promoter core region.Various lengths of the Bax 50-flanking region, including –590/þ391(pGL3-982), –411/þ391(pGL3-803), –196/þ391(pGL3-588), þ3/þ391(pGL3-389), and þ179/þ391(pGL3-213), werecloned and transiently transfected into SK-Hep-1, Huh-7, andMIHA cells. A dual-luciferase reporter assay indicated that all fiveBax promoter–driven luciferase constructs exhibited higher lucif-erase activities in MIHA cell, in which SIRT6 expression is low(Fig. 4A). Construct pGL3-982 exhibited maximum luciferaseactivity, whereas pGL3-389 exhibited a robust reduction in lucif-erase activity, indicating that the–196/þ3 region is the core regionof the Bax promoter (Fig. 4A). Cotransfection of shRNA targetingSIRT6 with various constructs or with pGL3-basic was performedto determine the changes in luciferase activity. The data demon-strated that the regulatory site of SIRT6 is located at –196/þ3 ofBax promoter, which is consistent with the core region of the Baxpromoter (Fig. 4B).
In the context of gene expression, acetylated H3K9 is indicativeof an actively transcribed gene, whereas H3K9 deacetylation isassociatedwith gene repression. Previously, SIRT6was reported tobe a H3K9 deacetylase (8,9). Therefore, we determined whetherSIRT6 is required for the deacetylation of H3K9 in the Baxpromoter. The ChIP assay revealed that SIRT6 was recruited tothe Bax promoter region in HCC cells (Fig. 4C). H3K9 acetylationin the promoter of Baxwas induced in SIRT6-depleted cells, whichwas consistent with transcriptional induction. In contrast, SIRT6overexpression resulted in the hypoacetylation of the Bax pro-moter (Fig. 4D). However, no change in the acetylation status wasobserved at the histone H4 lysine16 in response to SIRT6
Table 1. Correlative analysis of SIRT6 protein levels with clinicopathologicfeatures.
SIRT6 expression(Tumor/nontumoral)Clinicopathologic
parametersNumber ofspecimens Low High P value
SexFemale 14 2 12 0.13Male 87 32 55
Age (mean � SD) 52.5 � 13.6 53.9 � 11.6 0.59ALT�40 IU/L 46 14 32 0.53>40 IU/L 55 20 35
Carcino-embryonicantigen (CEA)�5 ng/mL 80 27 53 0.97>5 ng/mL 21 7 14
a-fetoprotein (AFP)�20 ng/mL 55 15 40 0.14>20 ng/mL 46 19 27
Tumor size�3 cm 25 13 12 0.02>3 cm 76 21 55
Multiple tumorNo 71 25 46 0.61Yes 30 9 21
Grade1 13 8 5 0.022 69 23 463 19 3 16
Vascular invasionNo 71 30 41 0.004Yes 30 4 26
CirrhosisNo 52 25 27 0.34Yes 49 19 30
NOTE: High tumoral SIRT6 expression was considered >1.5-fold upregulationrelative to the adjacent nontumoral liver.
Ran et al.
Clin Cancer Res; 22(13) July 1, 2016 Clinical Cancer Research3376
on April 30, 2020. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 9, 2016; DOI: 10.1158/1078-0432.CCR-15-1638
Untrea
ted
shCon
t
shSIR
T6-1
shSIR
T6-2
PLC/PRF/5
SMMC-7721
Huh-7
SK-Hep-1
SIRT6β-Actin
SIRT6
SIRT6
SIRT6
β-Actin
β-Actin
β-Actin
β-Actin
PARP
Cleaved PARP
Caspase-3
Cleaved caspase-3
SK-Hep-1 Huh-7
shC
ont
shC
ont
shS
IRT
6-1
shS
IRT
6-1
shS
IRT
6-2
shS
IRT
6-2
shCont shSIRT6-1 shSIRT6-2
150
100
50
0
Rel
ativ
e no
. of c
olon
y (%
)
PLC/PRF/5
Huh-7 SK-Hep-1
UntreatedshContshSIRT6-1shSIRT6-2
UntreatedshContshSIRT6-1shSIRT6-2
UntreatedshContshSIRT6-1shSIRT6-2
UntreatedshContshSIRT6-1shSIRT6-2
SMMC-7721
Days post-transduction
Days post-transduction Days post-transduction
Days post-transduction
Cel
l num
ber
(×10
4 )C
ell n
umbe
r (×
104 )
Cel
l num
ber
(×10
4 )C
ell n
umbe
r (×
104 )
160
120
80
40
0
200
160
120
80
40
0
100
80
60
40
20
0
250
200
150
100
50
0
0 1 2 3 4 5 6 7 0 1 2 3 4 5 6 7
0 1 2 3 4 5 6 70 1 2 3 4 5 6 7
120110100
908070605040302010
0
shCon
tsh
SIRT6-
1sh
SIRT6-
2
shCon
tsh
SIRT6-
1sh
SIRT6-
2
shCon
tsh
SIRT6-
1sh
SIRT6-
2
Rel
ativ
e %
of m
axim
um
Edu
inco
rpor
atio
n
SK-Hep-1
Huh-7
shCont shSIRT6-1 shSIRT6-2
shCont shSIRT6-1 shSIRT6-2
Annexin V
Annexin V
Pro
pidi
umio
dide
Pro
pidi
umio
dide
2.32%
3.88%
7.34% 12.65% 11.92%
5.61%2.27%0.81%
9.27%
12.16% 10.82%
9.80%
50
40
30
20
10
0
30
20
10
0
Per
cent
age
of a
popt
otic
cel
ls (
%)
Per
cent
age
of a
popt
otic
cel
ls (
%)
A
C
E F
D
B
Figure 2.SIRT6 depletion inhibits HCC cell growth and induces apoptosis. A, SIRT6 protein expression in HCC cells was significantly inhibited by lentivirus expressing SIRT6-targeting shRNA (shSIRT6-1 and shSIRT6-2) versus control (shCont) shRNA. Cells were harvested 3 days after virus infection. b-Actin was used as a loadingcontrol. B, SIRT6knockdownsignificantly inhibited the proliferation of HCCcells. PLC/PRF/5, SK-Hep-1, SMMC-7721, andHuh-7 cellswere infectedwith lentivirus, andcell numbers were determined by a trypan blue exclusion assay at the indicated number of days thereafter. Plots represent cumulative cell numbers versusdays in culture. � , P < 0.001. C, SIRT6 knockdown reduced colony formation. SK-Hep-1 cells infected with lentivirus as indicated were cultured for 2 weeks in thepresence of blasticidin (3.5 mg/mL) and stained with crystal violet. Quantification of colonies was obtained from macroscopically visible colonies in eachwell and expressed as percentage relative to the control group. � , P < 0.001. D, an EdU incorporation assay was used to analyze cell proliferation. The quantificationof EdU-positive cells was conducted macroscopically and expressed as a percentage relative to the control cells. � , P < 0.01. E, SIRT6 silencing inducedapoptosis in SK-Hep-1 and Huh-7 cells. Apoptosis in different groups was analyzed by flow cytometry with Annexin V/PI (E). � , P < 0.01. All histograms showmeanvalues from three independent experiments; bars indicate SD. F, apoptosis in different groups was analyzed by PARP cleavage and caspase 3 cleavageanalysis. PARP: 116 kDa; cleaved PARP: 89 kDa; caspase 3: 35 kDa; cleaved caspase 3: 19 kDa.
The Role of SIRT6 in HCC
www.aacrjournals.org Clin Cancer Res; 22(13) July 1, 2016 3377
on April 30, 2020. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 9, 2016; DOI: 10.1158/1078-0432.CCR-15-1638
10
5
0SK-Hep-1 Huh-7
100
50
0MIHA
VectorSIRT6
shContshSIRT6-1shSIRT6-2
Rel
ativ
e m
RN
A le
vel o
f Bax
Rel
ativ
e m
RN
A le
vel
of B
ax (
%)
SK-Hep-1 Huh-7 MIHA
SIRT6
Bax
β-Actin
shCon
t
shCon
t
shSIR
T6-1
shSIR
T6-1
shSIR
T6-2
shSIR
T6-2
SIRT6
Vector
SK-Hep-1 Huh-7
shCont
shSIRT6
Bax Mitochondria DAPI Merge
SIRT6
Bcl-2
Smac
Cytochrome c
Caspase-9
Cleaved caspase-9
β-Actin
shS
IRT
6-1
shS
IRT
6-1
shS
IRT
6-2
shS
IRT
6-2
shC
ont
shC
ont
Cytosol Mitochondria Nuclear
shC
ont
shC
ont
shC
ont
shS
IRT
6
shS
IRT
6
shS
IRT
6
Bax
GAPDH
COX IV
Histone H3
200
150
100
50
00 1 2 3 4 5
Days post-transduction
Cel
l num
ber
(×10
4 )siCont
shSIRT6
shSIRT6+siBax
siBax
siCont shSIRT6
shSIRT6+siBax siBax
Annexin V
Pro
pidi
um io
dide
8.56%
1.01%
13.31%
2.41% 2.12%
6.15%
8.13%
25.59%50
40
30
20
10
0
siCon
t
siCon
t
shSIR
T6
shSIR
T6
shSIR
T6+siB
ax
shSIR
T6+siB
ax
siBax
siBax
Per
cent
age
of a
popt
otic
cel
ls (
%)
PARP
Cleaved PARP
SIRT6
Bax
β-Actin
A
B
D
F G
E
C
Figure 3.SIRT6 regulates cell apoptosis via Bax signaling. A, SIRT6 regulated Bax mRNA (left) and protein levels (right) in SK-Hep-1, Huh-7, and MIHA cells. � , P < 0.01. B,Western blot analysis of several components of the Bax-mediated apoptotic pathway in SIRT6-depleted cells. b-Actin was used as a loading control. Bax:20 kDa; Bcl-2: 26 kDa; Smac: 21 kDa; cytochrome c: 14 kDa; caspase 9: 49 kDa; cleaved caspase 3: 37, 39 kDa. C, immunofluorescence analysis of Bax (green) andmitochondria (red) in SIRT6-depleted SK-Hep-1 cells showed that SIRT6 promoted the mitochondrial translocation of Bax. D, Western blotting analysisof Bax in cytosolic, mitochondrial, and nuclear fractions of SK-Hep-1 cells expressing shSIRT6 or shCont. COX IV (17 kDa), GAPDH (37 kDa), and Histone H3 (17 kDa)were used as markers for the mitochondrial, cytosolic, and nuclear fractions, respectively. E, gene silencing of Bax rescued the growth inhibition inducedby SIRT6 depletion in SK-Hep-1 cells. Cell numbers were counted 4 days after transfection using a trypan blue exclusion assay. � , P < 0.01. F and G, Bax suppressionabolished the induction of apoptosis in SIRT6-depleted SK-Hep-1 cells. Apoptosis in different groups was analyzed by flow cytometry with Annexin V/PI(F) and by PARP cleavage analysis (G). � , P < 0.01. All histograms show mean values from three independent experiments; bars indicate SD.
Ran et al.
Clin Cancer Res; 22(13) July 1, 2016 Clinical Cancer Research3378
on April 30, 2020. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 9, 2016; DOI: 10.1158/1078-0432.CCR-15-1638

Figure 4.SIRT6 regulates Bax gene expression via the deacetylation of H3 lysine 9. A, the promoter activity of the Bax gene was measured using a dual-luciferase reporterassay. SK-Hep-1, Huh-7, and MIHA cells were transfected with pGL3-basic or reporter constructs containing various lengths of the 50-flanking region of theBax gene as indicated. The data are presented as the mean � SD of three independent experiments. B, SIRT6 depletion activated the Bax promoter. SK-Hep-1 andHuh-7 cells were transfected with pGL3-982, pGL3-803, pGL3-588, pGL3-389, and pGL3-213 after infection with lentivirus expressing shSIRT6. The data arepresented as the mean � SD of three independent experiments. �, P < 0.01. C, ChIP assay was performed to confirm the interaction between SIRT6 and thepromoter region of Bax. D, SIRT6 is required for H3K9 deacetylation at the Bax promoter. ChIP assay with anti-H3K9Ac or anti-H4K16Ac was performed inSK-Hep-1 cells infected with lentivirus expressing shSIRT6 or shCont; H3K9 or H4K16 acetylation at the Bax promoter (mean � SD) is shown relative to input.� ,P<0.01. E, SIRT6 silencing promoted transcription factor p53 andE2F-1 occupancy at the promoter of Bax gene. ChIP assaywith anti-p53 or E2F-1was performed inSK-Hep-1 cells. p53 or E2F-1 occupancy (mean � SD) at promoter in SIRT6-depleted cells relative to input is shown.� , P < 0.01. All histograms show meanvalues from three independent experiments; bars indicate SD.
The Role of SIRT6 in HCC
www.aacrjournals.org Clin Cancer Res; 22(13) July 1, 2016 3379
on April 30, 2020. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 9, 2016; DOI: 10.1158/1078-0432.CCR-15-1638
depletion or overexpression (Fig. 4D). Then, we asked whetherH3K9 hyperacetylation of the Bax promoter in SIRT6-depletedcells affected its accessibility to DNA-binding factor. The tran-
scription factors p53 and E2F1 were selected based on publisheddata. TheChIP assay revealed that the occupancy of p53 andE2F-1in the Bax promoter was significantly enhanced in SIRT6-depleted
1,200
900
600
300
00 7 14 21 28
shCont
shSIRT6
shSIRT6+shBax
shContshSIRT6shSIRT6+shBax
shCont shSIRT6 shSIRT6+shBax
shCont
shSIR
T6
shSIR
T6+sh
Bax
shCon
t
shSIR
T6
shSIR
T6+sh
Bax
Days after injection
Tum
or v
olum
e (m
m3 )
Tum
or v
olum
e (m
m3 )
SIRT6
Bax
β-Actin
shCont
shSIRT6
shSIRT6+shBax
1,500
1,000
500
0
1.4
1.2
1.0
0.8
0.6
0.4
0.2
0.0Tu
mor
wei
ght (
g)
Anti-SIRT6
Anti-Ki67
Anti-cleaved
Anti-Bax
-PARP
shCont shSIRT6 shSIRT6+shBax
300
200
100
0
300
200
100
0
300
200
100
0
300
200
100
0
HS
core
sH
Sco
res
HS
core
sH
Sco
res
Rel
ativ
e B
ax e
xpre
ssio
n
5
4
3
2
1
00 1 2 3 4 5
Relative SIRT6 expression
Spearman's rank = –0.39P < 0.0001
A
B
D
E
C
Figure 5.SIRT6depletion inhibits HCCgrowth in vivo. SK-Hep-1 cells infectedwith lentivirus expressing shSIRT6, shSIRT6þshBax, or shContwere injected subcutaneously into4-week-old nude mice. At 4 weeks after implantation, the animals were sacrificed, and the tumor masses were excised. A, analysis of tumor volumeover a 4-week time course. The data represent the mean � SD. � , P < 0.001. B, representative images of tumors formed by the implantation of SK-Hep-1 cellsexpressing shSIRT6, shSIRT6þshBax, or shCont. C, tumor weights in different groups of nude mice. Data represent the mean � SD. Bars indicate SD;� ,P <0.001. D, representative images of immunohistochemical staining of SIRT6, Bax, cleavedPARP, andKi67 in tumor xenograftswith hematoxylin counterstaining.Magnification, �400. Quantification of positive cells was calculated based on HScore formula. � , P < 0.01. E, correlation analysis SIRT6 mRNA and BaxmRNA in 101 frozen HCC tissues. SIRT6 and Bax mRNA was first normalized by the expression level of b-actin, and the induction of SIRT6 and Bax in HCC overnontumoral liver in each patients was calculated. Correlation analysis was analyzed by Spearman s rank test.
Clin Cancer Res; 22(13) July 1, 2016 Clinical Cancer Research3380
Ran et al.
on April 30, 2020. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 9, 2016; DOI: 10.1158/1078-0432.CCR-15-1638

cells. Together, these data suggest that SIRT6 is recruited to the Baxpromoter to deacetylate H3K9, affecting its accessibility to tran-scription factors (Fig. 4E).
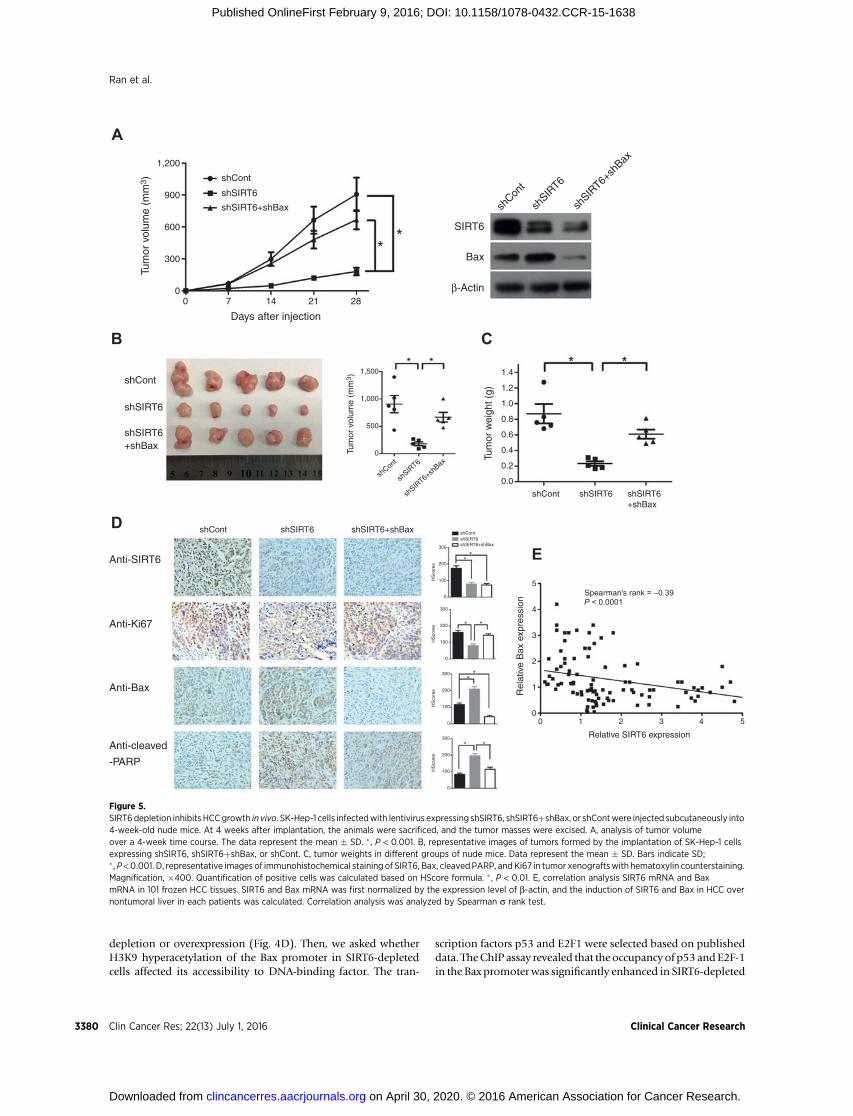
SIRT6 knockdown inhibits tumor growth and inducedapoptosis in vivo
To investigate whether SIRT6 knockdown in HCC cells sup-presses tumorigenicity in vivo, SK-Hep-1 cells infected with len-tivirus expressing vector, shSIRT6, and shSIRT6þshBax werexenografted into nude mice subcutaneously. The growth rate oftumors derived from shSIRT6-infected cells was reduced signifi-cantly compared with vector-infected cells (Fig. 5A). Similarly,SIRT6 knockdown significantly inhibited both the size andweightof tumors, whereas the effect was attenuated by the Bax suppres-sion (Fig. 5B and C). SIRT6 knockdown in tumors tissues wasassociated with increased levels of cleaved PARP and decreasedlevel of the cell proliferation marker Ki-67, and the effect waspartially reversed by Bax knockdown (Fig. 5D). These data sug-gested that the SIRT6 knockdown in tumor cells can suppresstumor growth and induce apoptosis in vivo. Finally, we alsoexamined SIRT6 and Bax mRNA in 101 HCC tissues that hasbeen used to determine the relevance of SIRT6-regulated Baxpathway in human subjects. Correlative analysis further revealeda negative correlation between SIRT6mRNAandBaxmRNA levels(Spearman rank¼ –0.39, P<0.001). These data suggested that theSIRT6–Bax regulator axis might exist in vivo (Fig. 5E).
DiscussionThe role of SIRT6 in tumor development is controversial. A loss
of SIRT6 in mouse embryonic fibroblasts led to tumor formationindependent of oncogene activation, suggesting that SIRT6 maybe a tumor suppressor (18). Furthermore, SIRT6 downregulationwas observed in human pancreatic ductal adenocarcinomas (18),colorectal carcinomas, and head and neck cancers (23). However,SIRT6 has been implicated as an oncogene in skin cancer (11) andprostate cancer (12). SIRT6 is upregulated in human skin cancer,where it promotes skin tumorigenesis by regulating COX-2expression (11). SIRT6 is overexpressed in prostate cancer, andthe inhibition of SIRT6 inprostate cancer reduces cell viability andincreases sensitivity to chemotherapeutics (12). Taken together,thesefindings suggest that SIRT6 is both tumor suppressive and anoncogenic depending on the context and gene dose.
Regarding the role of SIRT6 in HCC, discrepancies still existbetween the findings of our study and of other groups. Marquardtand colleagues noted a significant reduction of SIRT6 in HCCspecimens based on analysis of a publicly available cancer micro-array database (24). These researchers further observed a reduc-tion in SIRT6 mRNA expression in 45% (24/53) of HCC speci-mens from their recently published HCC database (24,25). Incontrast with their findings, by analyzing SIRT6 expression in 101paired frozen HCC tissues and 60 paired paraffin-embeddedsections, we convincingly showed that both SIRT6 mRNA andprotein levels were significantly upregulated in the majority ofHCC tissues compared with adjacent nontumoral liver tissues.Interestingly, high SIRT6 expressionwas associatedwith increasedtumor grade, tumor size, and vascular invasion. HCC patientswith high SIRT6 expression showed significant correlation withpoor overall survival rate. These clinical data suggest that SIRT6may act as an oncogene in HCC development. Unfortunately, theMarquardt study did not determine the expression of the SIRT6 in
HCC clinical samples, which precluded a direct comparisonbetween the two studies. We speculate that the different findingsin these two studies may be due to different HCC population indifferent region. We also found that SIRT6 expression was low inthree normal liver tissues and in PHH cells. SIRT6 suppressioninhibited the growth of HCC cells in vitro and in vivo. These datafurther support the protumorigenic function of SIRT6 in HCCdevelopment.
The dysregulation or blockade of apoptosis machinery incancer cells represents a potential mechanism of HCC carcino-genesis (26). Defects in the apoptosis pathway in cancer cellscontribute to resistance to anticancer therapies (27). In terms offunction, we found that SIRT6 suppression induced apoptosis inHCC cells and increased cell sensitivity to doxorubicin treatment.Concordantly, SIRT6 knockdown in human prostate cancer cellsled to increased apoptosis and enhanced chemotherapeutic sen-sitivity (12). However, according to Meter and colleagues, SIRT6overexpression induces massive apoptosis in cancer cells, includ-ing fibrosarcoma, cervical carcinoma, primary breast tumor, andmetastatic breast tumor cell lines (28). This cell death requires themono-ADP-ribosyltransferase but not the deacetylase activity ofSIRT6 and is mediated by the activation of both the p53 and p73apoptotic signaling cascades in cancer cells (28). Unfortunately,the study byMeter and colleagues did not include any liver cancercell lines. Therefore, the function of SIRT6 in cancer may dependon tissue-specific molecular profiles.
We further revealed that SIRT6 contributes to the apoptosisprocess in hepatoma cells partially via Bax regulation. SIRT6silencing also induced the expression of Smac, cytochrome C,and cleaved caspase 9, which are downstream effectors of Baxactivation, suggesting that the Bax signaling pathway is active inSIRT6-depleted cells. Next, we observed that SIRT6 negativelyregulates Bax transcription via H3K9 deacetylation. Our findingsuggests a novelmechanism for SIRT6 in the epigenetic regulationof Bax inHCC cells. To our knowledge, thisfinding constitutes thefirst demonstration of a role for SIRT6 in gene expression via themodulation of chromatin in HCC. In the context of gene expres-sion, acetylated H3K9 is associated with actively transcribedgenes. In SIRT6-depleted cells, the hyperacetylation of H3K9 atthe Bax promoter indeed enhanced the accessibility of transcrip-tion factors such as p53 to chromatin. Transcription factor p53 is adirect transcriptional activator of Bax gene (29,30). In HCC cells,SIRT6 is recruited to promoter of Bax gene, deacetylates histoneH3K9, decreases the accessibility of transcription factors such asp53 to chromatin, and thereby contributes to the termination ofBax transcription. However, whether SIRT6 physically interactedwith transcription factors p53 needs our further investigation.
In summary, we demonstrated that SIRT6 acts as an oncogenein HCC development by blocking Bax expression and mitochon-drial translocation. SIRT6 suppression also increased the sensi-tivity of liver cancer cells to chemotherapeutics. Our findingshighlight the importance of the SIRT family in HCC carcinogen-esis and provide useful tools for the development of mechanism-based cancer prevention strategies.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: A.-L. Huang, J. ChenDevelopment of methodology: Z.-Z. Zhang
www.aacrjournals.org Clin Cancer Res; 22(13) July 1, 2016 3381
The Role of SIRT6 in HCC
on April 30, 2020. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 9, 2016; DOI: 10.1158/1078-0432.CCR-15-1638

Acquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): L.-K. Ran, Y. Chen, N.-N. Tao, L. Zhou, W.-Y. LiAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): H. Tang, X. ChenWriting, review, and/or revision of the manuscript: W.-Y. Li, A.-L. HuangAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): N.-N. Tao, J.-H. Ren, K. Chen
Grant SupportThis study was supported by the National Natural Science Foundation of
China (81472271 and 81270559), the National Science and Technology Major
Project (2013ZX10002002), the Chongqing Natural Science Foundation(cstc2012jjA10047), and theMajor project of Chongqing Science & TechnologyCommission (cstc2013jcyjC10002, to A.-L. Huang).
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received July 14, 2015; revised January 11, 2016; accepted January 17, 2016;published OnlineFirst February 9, 2016.
References1. Shiraha H, Yamamoto K, Namba M. Human hepatocyte carcinogenesis
(review). Int J Oncol 2013;42:1133–8.2. Schattenberg JM, Schuchmann M, Galle PR. Cell death and hepatocarci-
nogenesis: Dysregulation of apoptosis signaling pathways. J GastroenterolHHepatol 2011;26 Suppl 1:213–9.
3. Kelly GL, Strasser A. The essential role of evasion from cell death in cancer.Adv Cancer Res 2011;111:39–96.
4. Haigis MC, Guarente LP. Mammalian sirtuins–emerging roles in physiol-ogy, aging, and calorie restriction. Genes Dev 2006;20:2913–21.
5. Toiber D, Erdel F, Bouazoune K, SilbermanDM, Zhong L, Mulligan P, et al.SIRT6 recruits SNF2H to DNA break sites, preventing genomic instabilitythrough chromatin remodeling. Mol Cell 2013;51:454–68.
6. Kaidi A, Weinert BT, Choudhary C, Jackson SP. Human SIRT6 promotesDNA end resection through CtIP deacetylation. Science 2010;329:1348–53.
7. Mao Z, Hine C, Tian X, VanMeter M, AuM, Vaidya A, et al. SIRT6 promotesDNA repair under stress by activating PARP1. Science 2011;332:1443–6.
8. Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M,et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomericchromatin. Nature 2008;452:492–6.
9. Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, et al.SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependentgene expression and organismal life span. Cell 2009;136:62–74.
10. Khongkow M, Olmos Y, Gong C, Gomes AR, Monteiro LJ, Yague E, et al.SIRT6modulates paclitaxel and epirubicin resistance and survival in breastcancer. Carcinogenesis 2013;34:1476–86.
11. Ming M, HanW, Zhao B, Sundaresan NR, Deng CX, Gupta MP, et al. SIRT6Promotes COX-2 Expression and Acts as an Oncogene in Skin Cancer.Cancer Res 2014;74:5925–33.
12. Liu Y, Xie QR, Wang B, Shao J, Zhang T, Liu T, et al. Inhibition of SIRT6 inprostate cancer reduces cell viability and increases sensitivity to chemother-apeutics. Prot Cell 2013Aug 27. [Epub ahead of print].
13. Han Z, Liu L, Liu Y, Li S. Sirtuin SIRT6 suppresses cell proliferation throughinhibition of Twist1 expression in non-small cell lung cancer. Int J Clin ExpPathol 2014;7:4774–81.
14. Bauer I, Grozio A, Lasiglie D, Basile G, Sturla L, Magnone M, et al. TheNADþ-dependent histone deacetylase SIRT6 promotes cytokine produc-tion andmigration in pancreatic cancer cells by regulatingCa2þ responses.J Biol Chem 2012;287:40924–37.
15. Chen X, Hao B, Liu Y, Dai D, Han G, Li Y, et al. The histone deacetylaseSIRT6 suppresses the expression of the RNA-binding protein PCBP2 inglioma. Biochem Biophys Res Commun 2014;446:364–9.
16. Min L, Ji Y, Bakiri L, Qiu Z, Cen J, Chen X, et al. Liver cancer initiation iscontrolled by AP-1 through SIRT6-dependent inhibition of survivin. NatCell Biol 2012;14:1203–11.
17. Lefort K, Brooks Y, Ostano P, Cario-Andre M, Calpini V, Guinea-Viniegra J,et al. A miR-34a-SIRT6 axis in the squamous cell differentiation network.EMBO J 2013;32:2248–63.
18. Sebastian C, Zwaans BM, Silberman DM, Gymrek M, Goren A, Zhong L,et al. The histone deacetylase SIRT6 is a tumor suppressor that controlscancer metabolism. Cell 2012;151:1185–99.
19. Chen J, Zhang B, Wong N, Lo AW, To KF, Chan AW, et al. Sirtuin 1 isupregulated in a subset of hepatocellular carcinomas where it is essentialfor telomere maintenance and tumor cell growth. Cancer Res 2011;71:4138–49.
20. Chen J, Chan AW, To KF, Chen W, Zhang Z, Ren J, et al. SIRT2 over-expression in hepatocellular carcinoma mediates epithelial to mesenchy-mal transition by protein kinase B/glycogen synthase kinase-3beta/beta-catenin signaling. Hepatology 2013;57:2287–98.
21. Law BY, Wang M, Ma DL, Al-Mousa F, Michelangeli F, Cheng SH, et al.Alisol B, a novel inhibitor of the sarcoplasmic/endoplasmic reticulum Ca(2þ) ATPase pump, induces autophagy, endoplasmic reticulum stress, andapoptosis. Mol Cancer Therap 2010;9:718–30.
22. Lavorato-Rocha AM, Anjos LG, Cunha IW, Vassallo J, Soares FA, Rocha RM.Immunohistochemical assessment of PTEN in vulvar cancer: best practicesfor tissue staining, evaluation, and clinical association. Methods 2015;77–78:20–4.
23. Lai CC, Lin PM, Lin SF, Hsu CH, LinHC,HuML, et al. Altered expression ofSIRT gene family in head and neck squamous cell carcinoma. Tumour Biol2013;34:1847–54.
24. Marquardt JU, Fischer K, Baus K, Kashyap A,Ma S, KruppM, et al. Sirtuin-6-dependent genetic and epigenetic alterations are associated with poorclinical outcome in hepatocellular carcinoma patients. Hepatology 2013;58:1054–64.
25. Andersen JB, Factor VM, Marquardt JU, Raggi C, Lee YH, Seo D, et al. Anintegrated genomic and epigenomic approach predicts therapeuticresponse to zebularine in human liver cancer. Sci Translat Med 2010;2:54ra77.
26. Fabregat I, Roncero C, FernandezM. Survival and apoptosis: a dysregulatedbalance in liver cancer. Liver Int 2007;27:155–62.
27. Tsuruo T, Naito M, Tomida A, Fujita N, Mashima T, Sakamoto H, et al.Molecular targeting therapy of cancer: drug resistance, apoptosis andsurvival signal. Cancer Sci 2003;94:15–21.
28. Van Meter M, Mao Z, Gorbunova V, Seluanov A. SIRT6 overexpressioninducesmassive apoptosis in cancer cells but not in normal cells. Cell Cycle2011;10:3153–8.
29. Moll UM, Wolff S, Speidel D, Deppert W. Transcription-independent pro-apoptotic functions of p53. Curr Opin Cell Biol 2005;17:631–6.
30. Speidel D. Transcription-independent p53 apoptosis: an alternative routeto death. Trends Cell Biol 2010;20:14–24.
Clin Cancer Res; 22(13) July 1, 2016 Clinical Cancer Research3382
Ran et al.
on April 30, 2020. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 9, 2016; DOI: 10.1158/1078-0432.CCR-15-1638

2016;22:3372-3382. Published OnlineFirst February 9, 2016.Clin Cancer Res Long-Kuan Ran, Yong Chen, Zhen-Zhen Zhang, et al. Dependent Apoptotic Pathway
−Hepatocellular Carcinoma via BCL2-Associated X Protein SIRT6 Overexpression Potentiates Apoptosis Evasion in
Updated version
10.1158/1078-0432.CCR-15-1638doi:
Access the most recent version of this article at:
Material
Supplementary
http://clincancerres.aacrjournals.org/content/suppl/2016/02/09/1078-0432.CCR-15-1638.DC1
Access the most recent supplemental material at:
Cited articles
http://clincancerres.aacrjournals.org/content/22/13/3372.full#ref-list-1
This article cites 29 articles, 7 of which you can access for free at:
Citing articles
http://clincancerres.aacrjournals.org/content/22/13/3372.full#related-urls
This article has been cited by 3 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://clincancerres.aacrjournals.org/content/22/13/3372To request permission to re-use all or part of this article, use this link
on April 30, 2020. © 2016 American Association for Cancer Research. clincancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 9, 2016; DOI: 10.1158/1078-0432.CCR-15-1638