submission of comments on revision of ‘annex 1 ...guidance, such as contamination control...
TRANSCRIPT

Submission of comments on Revision of ‘Annex 1: Manufacture of
Sterile Medicinal Products’
Comments from:
Name of organisation or individual
Pharmaceutical & Healthcare Sciences Society (Not-For-Profit society): PHSS Annex 1 comment platform:
Comments from Group 1: Pharmaceutical Industry and GMP consultants.
Acknowledgement: The Pharmaceutical & Healthcare Sciences Society: PHSS Annex 1 comment platform includes
contributors from the Pharmaceutical Industry, supporting GMP consultants, Pharmaceutical equipment manufacturers: Barrier
Isolator/RABS technology and Filling process machinery, suppliers: Facility monitoring system and gowning plus academics
with related research and peer reviewed publications as such provides a broad view of international stake holder interest.
The PHSS wish to acknowledge contributions with comments from the following:
PHSS Group 1 comments:
Pharmaceutical Industry: GSK, AstraZeneca, Pfizer, Merck & Co Inc, Ely Lilly France & Italy, Novartis, Allergan Westport,
Fresenius Kabi, Teva-Pliva, Alexion Ireland, Bayer. GMP Consultants: Roland Guinet (France), Gordon Farquharson (UK),
Richard Funnel (UK).
PHSS Group 2 comments:
Pharmaceutical equipment manufacturers: F Ziel GmbH (Barrier Isolator/ RABS Technology), Groninger GmbH & Bausch
Stroebel GmbH (Filling process machines), TSI & Pharmagraph (Facility monitoring systems), DuPont (Cleanroom garbing),
Sterilisation solutions (Alan Heavey), Rapid Micro Biosystems (David Jones) and Academics working in field of Good
manufacturing practice GMP: Professor Bengt Ljungqvist, Associate Professor microbiology Berit Reinmüller, Professor Matts
Ramstorp, Dr Bill Whyte.
Throughout this document, existing text is given in italics where applicable.
Where modifications have been made to existing text these areas are underlined in the proposed text change.
Where large changes have been made such that the text changes significantly there is no underlining.
The most suitable text changes considered by the PHSS are given at the start of each paragraph section.

1. General comments
General comment (1)
This draft revision of Annex 1represents an enormous change from the 2008 version, and includes a great deal of
updated information, current expectations, and focus on current technologies and practices.
However, the Pre Draft issue communication from EU Inspectors Working Group (IWG) indicated that Annex 1
would provide clear guidance to manufacturers with regard to the manufacture of sterile products so Inspectorates and
Manufacturers are not held to unclear expectations. This was understood to be of especial importance to less
technically mature organisations who currently learn via incidents or adverse regulatory inspections.
This document does not meet that expectation nor does it appear to meet current regulatory expectations as
experienced by the industry.
The draft appears to be somewhat immature with tables repeated, has conflicts in many areas with current practice,
and for example doesn’t address the EMA non-distillation WFI Q&A paper that it was supposed to replace.
The document requires improvement and clarification of technical elements, and should be subject to a further editing
process.
The use of terminology such as shall, should and must are not used consistently creating uncertainty over where
expectations are mandatory.
General Comment (2)
Note that some companies are concerned that in contrast to enabling manufacturers to take decisions relating to their
own processes using Quality Risk Management principles, some elaborated new detail is provided in the draft Annex
1 which state specific requirements in general, rather than enabling using individual risk based requirements based on
manufacturing processes.
General Comment (3)
1) The draft Annex is very prescriptive in expectations. There is risk of literal interpretations by inspectors and
industry that will add cost and complexity to sterile manufacturing operations that are not warranted for
sterility assurance. Although QRM principles are included, the statement is such that the controls must meet
or surpass the Annex, which is inconsistent with QRM concepts and subject to interpretation/local
enforcement. Industry experience has been that examples and recommendations are often interpreted by some
authorities as the mandatory requirement.
2) There is an overall emphasis on quality control and testing rather than design, validation and assurance
programs. This will add unnecessary cost and operational complexity with associated risk for burdensome
activities and risk of errors. Focusing on programs that inherently design robust sterility assurance elements
and emphasize risk reduction would provide greater levels of confidence in overall sterility assurance.
3) The draft Annex detailed requirements are based on current technology and the prescriptive methodologies
will impede implementation of new scientific, engineering and analytical methods. Although the Annex
attempts to promote modern technologies, the detailed prescriptive controls do not enable efficient, effective
use of such technology. As a result, manufacturers may be limited in new technology, testing and assurance
options for considerable time.
4) The controls outlined are often universal regardless of the technology (e.g. isolator, RABS) and therefore
manufacturers are limited in operating to maximal efficiency and assurance. Many of the controls involve
taking operations off-line limiting the time available for manufacture of product for supply. In addition, some
of the controls are invasive and may have the unintended consequence of adding risk.

Specific comments on text
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
1-4 Comment:
Document Map (Index)
1] The order of the whole document should be improved. For example, Section 5 is the first time that the cleanliness
Grades A-D are defined, yet Section 4 on personnel makes reference to them.
2] Section 8 has equipment items in it. These should be collected together in Section 6.
Proposed change:
Change the order of the sections, and rewrite Section 5 to include the equipment/processes in section 8.
Reduce the size of section 8 by having a separate section on sterilisation.
6-23 Comment:
Scope. To aid the clarity of the document, the scope should be re-written. It should be emphasised that there are two
routes in the manufacture of sterile product namely terminal sterilisation (TS) and aseptic processing. While the
former should be undertaken if feasible, it must be recognised that the growing increase in biopharma products may
increasingly preclude the option for a TS approach. This section should also include references to ICH documents for
QRM.
8-15 Existing Text
The manufacture of sterile medicinal products covers a wide range of product types, (sterile active substance through
to finished dosage form), batch sizes (single unit to multiple units), processes (from highly automated systems to
manual processes), primary packaging materials and technologies (e.g. biotechnology, classical small molecule
manufacturing and closed systems). This Annex provides general guidance that should be used for all sterile
medicinal products and sterile active substances, via adaption, using the principles of Quality Risk Management
(QRM), to ensure that microbial, particulate and pyrogen contamination associated with microbes is prevented in the
final product.
Proposed changed text:
Whenever possible products should be terminally sterilised in their primary containers as this provides the highest
level of sterility assurance based on a terminal process of known lethality. Where terminal sterilisation isn’t possible
due to the product being heat labile, the alternative approach of aseptic processing can be used. See also 8.30.
This guidance considers the manufacture of sterile medicinal products manufactured by both terminal sterilisation and
aseptic processing. It covers a wide range of product types, including sterile active ingredients (APIs) and finished
dosage forms for both classic “small molecule” and large molecule biotechnology products. In addition to product
types, guidance is also provided on batch sizes (single unit to multiple units); campaign working (single batches to
campaigns comprising sequential batches); manual processes to highly automated systems; and primary packaging
materials and certain specialised manufacturing technologies. The manufacturing environment is also considered,
including cleanrooms, RABS, and isolators, and their relationship with open and closed process systems. This Annex
provides general guidance that should be used for all sterile medicinal products and sterile active substances, using the
principles of Quality Risk Management (QRM), to ensure that microbial, particulate and pyrogenic contamination
associated with microbes is prevented in the final product, and to ensure more broadly that the products consistently
meet their quality attributes of potency, purity, sterility, and identity.
Comment:
Extended scope to include primary packaging materials and technologies including biotech and closed systems

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
17-22 Comment (1):
A definition for the Contamination Control Strategy would be very beneficial.
Existing Text
The intent of the Annex is to provide guidance for sterile medicinal products. However some of the principles and
guidance, such as contamination control strategy, room qualification, classification, monitoring and gowning, may be
used to support the manufacture of other products that are not intended to be sterile (such as certain liquids, creams,
ointments and low bioburden biological intermediates) but where the control of microbial, particulate and pyrogen
contamination, to reduce it as far as possible, is considered important.
Proposed changed text:
Important GMP principles, such as cross-contamination control, are addressed elsewhere in the GMP guidelines. The
intent of this Annex is to provide guidance for sterile medicinal products. However, some of the principles and
guidance, such as contamination control strategy, cleanroom classification, qualification, monitoring and gowning,
may be used to support the manufacture of other products that are not intended to be sterile. Examples include some
liquids, creams, ointments and low bioburden biological intermediates where the control of microbial, particulate and
pyrogenic contamination is considered important.
Comment (2)
Extended scope to include non-sterile products. This statement is too general. It needs to be more specific to eliminate
any lack of clarity. Why pyrogen contamination should be a concern for non-sterile products?
Is this statement on non-sterile products really needed in this guidance?
Proposed change: Suggest delete line 17 to 20.
Comment (3)
The proposed scope includes non-sterile medicinal products and intermediates. This revision will provide
uncertainty as to the specific application of each of the requirements of Annex 1 as they relate to the
manufacture of non-sterile products.
The governing regulatory guidelines for contamination control for the manufacture of non-sterile products
is already provided within the EU Annex 2 regulations. Within that Annex, specific cross reference to
Annex 1 is currently included.
The scope of the guideline should be unambiguous (i.e., sterile medicinal products).
If some of the principles and guidance contained in Annex 1 are to be utilised for non-sterile medicinal
products and intermediates, the relevant guidelines for those products should cross-reference the
applicable requirement in Annex 1.
Comment (4):
Distinction should be made between ‘contamination’ - microorganisms, pyrogens and particulates which represent a
direct hazard and risk to product quality and microorganisms, pyrogens and particulates which are not hazards to
product quality.
Proposed change:
However some of the principles and guidance, such as microbial and particle control strategy, room qualification,
classification, monitoring and gowning, may be used to support the manufacture of other products that are not
intended to be sterile (such as certain liquids, creams, ointments and low bioburden biological intermediates) but
where the control of microorganisms, particulates and pyrogens is considered important.
20-34 2 Principle
General Comment:
It is not clearly indicated that the direct intervention of operators in the critical grade A area should be discouraged, as
indicated by MHRA in many communications on the revision of EU GMP Annex 1.

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
This should be indicated also for the manufacturing of all open containers since at point 8.17 for the transfer of
partially stoppered containers two times the requirement is “with physical segregation from operators”.
Thus, point 8.9 is not sufficiently clear and should be “Where possible, the use of… RABS, isolators or closed
systems, should be considered in order to reduce the need for avoid direct interventions into the grade A
environment...”
Comment (1):
Existing Text
a) Facility, equipment and process design must be optimized qualified and validated according to Annex 11
and Annex 15 of EU GMP. The use of appropriate current technologies should be implemented to ensure
protection and control of the product from potential extraneous sources of particulate and microbial
contamination such as personnel, materials and the surrounding environment.
Proposed changed text:
a) Facility, equipment and process design must be optimized qualified and validated according to Annex 11
and Annex 15 of EU GMP. The use of appropriate current technologies according to EU directives 2003/94
Article 5 and 2001/83 Article 23 should be implemented to ensure protection and control of the product
from potential extraneous sources of particulate and microbial contamination such as personnel, materials
and the surrounding environment
Comment (2)
Line 26-28: Distinction should be made between ‘contamination’ - microorganisms, pyrogens and particulates which
represent a direct hazard and risk to product quality and microorganisms, pyrogens and particulates which are not
hazards to product quality.
Alternative proposed text:
The manufacture of sterile products is subject to special requirements in order to minimize risks of a microbiological
(microorganisms and pyrogens), and particulate nature. The following key areas should be considered…
Comment (3)
Line 31-34: Distinction should be made between 'contamination' - microorganisms, pyrogens and particulates which
represent a direct hazard and risk to product quality and microorganisms, pyrogens and particulates which are not
hazards to product quality. Specifically, in this context unless the transfer of microorganisms, particulates is of a
quantity or nature (e.g. pathogenic microorganisms) that exceeds the materials quality attributes at that point (i.e. at a
tolerable level) the microorganisms and particulates are not necessarily contamination.
Alternative proposed text:
The use of appropriate current technologies should be implemented to ensure protection and control of the product
from potential extraneous sources of particulate and microbial hazards such as personnel, materials and the
surrounding environment
36-38 Comment;
As “attitude” is subjective i.e. cannot be reliably measured or evaluated, it is suggested that this term is omitted.
Existing Text
Personnel must have appropriate skills, training and attitudes with a specific focus on the principles involved in the
protection of sterile product during the manufacturing, packaging and distribution processes.
Proposed changed text:
Personnel must have defined skills and training with a specific focus on the principles to ensure the safety, quality and
efficacy of sterile product(s) during the manufacturing, packaging and distribution processes.
40-42 Existing Text

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Processes and monitoring systems for sterile product manufacture must be designed, commissioned, qualified and
monitored by personnel with appropriate process, engineering and microbiological knowledge.
Proposed changed text:
Processes and monitoring systems for sterile product manufacture must be designed, commissioned, qualified and
assessed by personnel with defined process, engineering and microbiological knowledge. These systems must be
defined within the facility contamination control strategy
Comment:
Will this require evidence of qualifications of personnel who designed and commissioned systems?
44-48 Existing text
Processes, equipment, facilities and manufacturing activities should be managed in accordance with QRM principles
that provide a proactive means of identifying, scientifically evaluating and controlling potential risks to quality. Risk
assessments should be used to justify alternative approaches to those specified in this Annex only if these alternative
approaches meet or surpass the intent of this Annex.
Proposed changed text:
Processes, equipment, facilities and manufacturing activities should be managed in accordance with QRM principles
that provide a proactive means of identifying, scientifically evaluating and controlling potential risks to quality. Risk
and impact assessments should be used to identify and justify alternative approaches to those specified in this Annex
only if these alternative approaches meet or surpass the intent of this Annex.
Comment (1):
The basis of quality risk management is to establish controls commensurate with the risk. Requiring that
QRM be used, but then stating that it can only be used if the approaches meet or surpass the intent of the
annex is contradictory in nature. Furthermore, scientific rationale in addition to risk assessment should be
used to justify alternative approaches.
Proposed text change:
Processes, equipment, facilities and manufacturing activities should be managed in accordance with QRM
principles that provide a proactive means of identifying, scientifically evaluating and controlling potential
risks to quality. Risk assessments and scientific rationale should be used to justify alternative approaches
to those specified in this Annex. Comment (2):
The draft consistently advocates the use of science based risk assessments to enhance the effectiveness of the
contamination control procedures. However, there is no inclusion that the risk to the patient is dependent on the
chance that an aseptically manufactured product will support microbial growth during the shelf life following
manufacture. For example, a freeze dried product, or one which has a water activity of less than 0.6 will not support
microbial growth and so presents a greatly reduced risk to the patient than a product which does support growth1. The
design of the contamination control devices (e.g. Isolators, RABS or open workstations) and the associated
cleanrooms utilised should reflect this risk
Recommendation:
Information should be added that the risk to a patient is dependent of the likelihood that an aseptically manufactured
product that is contaminated during manufacturing will support microbial growth during the period following
manufacture prior to administration to the patient. The design of the contamination control devices (e.g. Isolators,
RABS or open workstations) and the associated cleanrooms utilised during manufacturing should reflect this risk
50-54 Existing text
Quality Assurance is particularly important, and manufacture of sterile products must strictly follow carefully
established and validated methods of manufacture and control. A contamination control strategy should be
implemented across the facility in order to assess the effectiveness of all the control and monitoring measures
employed. This assessment should lead to corrective and preventative actions being taken as necessary.
Proposed changed text:

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Quality Assurance is particularly important, and manufacture of sterile products must strictly follow carefully
established and validated methods of manufacture and control because sterility of the final product cannot be
definitively measured. A contamination control strategy should be defined and implemented across the facility in
order to assess the effectiveness of all the control and monitoring measures employed. This assessment should lead to
corrective and preventative actions being taken as necessary.
Comment (1):
Line 51: New requirement. The site is required to implement and periodically update a documented holistic
contamination control strategy.
The control strategy defined in 8.7 should be crossed referred here.
Comment (2)
Line 51-53: A single control strategy, and an appropriate strategy term is needed which includes and distinguishes
between the different approaches to controlling microorganisms, pyrogens and particulates which represent direct
hazards to product quality and microorganisms, pyrogens and particulates which do not represent a hazard to product
quality. The control strategy is required to be implemented across the facility, however controls are necessary in
systems and subsystems which may be specific to product (e.g. tests supporting assessment of controls are qualified to
ensure suitability with specific products). Clarification is suggested by changing the text and to include a description
of the term in the glossary.
Alternative proposed changed text:
A microbial and particulate control strategy should be implemented across the facility in order to assess the
effectiveness of all the facility’s control and monitoring measures employed and specific to each product.
56-57 Existing text
The strategy should consider all aspects of contamination control and its life cycle with ongoing and periodic review
and update of the strategy as appropriate.
Proposed changed text:
The strategy should consider all aspects of contamination control throughout the life cycle of the facility with ongoing
and periodic review and update of the strategy as appropriate.
Comment:
Distinction should be made between 'contamination control' of microorganisms, pyrogens and particulates which are
potential hazards to product quality (contaminants) and the control of microorganisms, pyrogens and particulates
which are not hazards to product quality. Specifically tolerable levels of microorganisms in facility areas such as
Grade D or C cleanrooms are not contaminants or contamination as long as they are a quantity and nature which is not
adverse to control or product quality.
Alternative proposed changed text
The strategy should consider all aspects of microbiological (microorganisms and pyrogens), and particulate control
and its life cycle with ongoing and periodic review and update of the strategy as appropriate.
59-62 Existing text
Contamination control and steps taken to minimise the risk of contamination from microbial and particulate sources
are a series of successively linked events or measures. These are typically assessed, controlled and monitored
individually but these many sources should be considered holistically.
Proposed changed text:
Contamination control and the proactive steps taken to minimise the risk of contamination from microbial and
particulate sources and any other extraneous material which may adulterate the product, are a series of linked events
or measures including viable and non-viable particulates counts, airflows, pressure differentials, temperature and
humidity, adherence to procedures etc. These are typically assessed, controlled and monitored individually but these
multiple sources should be considered holistically.
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Comment:
Distinction should be made between 'contamination control' of microorganisms, pyrogens and particulates which are
potential hazards to product quality (contaminants) and the control of microorganisms, pyrogens and particulates
which are not hazards to product quality.
Alternative proposed changed text
Microbial and particulate control and steps taken to minimise the risks from microbial and particulate sources are a
series of successively linked events or measures. These are typically assessed, controlled and monitored individually
but these many sources should be considered holistically
64-67 Existing text
The development of such strategies requires thorough technical and process knowledge. Potential sources of
contamination are attributable to microbiological and cellular debris (e.g. pyrogens/endotoxins) as well as
particulate matter (glass and other visible and sub-visible particles).
Proposed changed text:
The development of such contamination and control strategies requires thorough technical and process knowledge.
Potential sources of contamination are attributable to microbiological and cellular debris (e.g. pyrogens/endotoxins) as
well as particulate matter (glass and other visible and sub-visible particles).
Comment:
Distinction should be made between "contamination' - microorganisms, pyrogens and particulates which represent a
direct hazard and risk to product quality and microorganisms, pyrogens and particulates which are not hazards to
product quality.
Alternative proposed changed text
The development of such strategies requires thorough technical and process knowledge. Potential sources of hazards
are attributable to microbiological and cellular debris (e.g. pyrogens/endotoxins) as well as particulate matter (glass
and other visible and sub-visible particles).
69-70 Comment (1):
The list does not include training and qualification program. Suggest add section on training.
Comment (2):
A single control strategy, and an appropriate strategy term is needed which includes and distinguishes between the
different approaches to controlling microorganisms, pyrogens and particulates which represent direct hazards to
product quality and microorganisms, pyrogens and particulates which do not represent a hazard to product quality.
Alternative proposed changed text
Elements to be considered within such a documented microbial and particulate control strategy should include (but not
be limited to):
93-95 Comment:
Distinction should be made between 'contamination' - microorganisms, pyrogens and particulates which represent a
direct hazard and risk to product quality and microorganisms, pyrogens and particulates which are not hazards to
product quality.
Proposed changed text
Preventative maintenance - maintaining equipment and premises (planned and unplanned maintenance) to a standard
that will not add significant microbial or particulate risk
99-101 Comment:
Distinction should be made between 'contamination' - microorganisms, pyrogens and particulates which represent a
direct hazard and risk to product quality and microorganisms, pyrogens and particulates which are not hazards to
product quality. Specifically, cleanrooms and environments are expected to and normally contain a micro flora; the

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
monitoring systems need to measure this to ensure routine control and also to measure events where genuine
contamination events occur in that levels and nature of the permissible microflora are exceeded.
Proposed changed text
Monitoring systems - including an assessment of the feasibility of the introduction of scientifically sound, modern
methods that optimize the detection of environmental microbial or particulate risk
69-107 Comment:
The control strategy is suggested to include a number of listed elements. These elements are a mixture of important
and critical items within various systems and subsystems. The wording does not indicate which if any are absolutely
necessary and which are beneficially advantageous but not absolutely necessary. It is recommended that the wording
is amended to reflect systems and subsystems, specific elements therein and if these are mandatory.
Proposed changed text
Quality systems and respective elements that must he considered within the control strategy are tabulated below
3 Pharmaceutical Quality System (PQS)
General Comments:
- line 131 : “… in chapter 1 of the EU GMP, ...” should be … in chapter 1 of EU GMP Part I,...
- line 135 : “… microbial contamination ...” should be … microbial and other contamination ...
134-136 Comment (1):
No clarity regarding the definition of sterility assurance. Suggest include a definition of “sterility assurance” in the
glossary
Comment (2):
Distinction should be made between 'contamination' - microorganisms, pyrogens and particulates which represent a
direct hazard and risk to product quality and microorganisms, pyrogens and particulates which are not hazards to
product quality.
Proposed changed text:
There is an effective risk management system integrated into the product life cycle to minimize microbial and
particulate risks to ensure the safety, quality and efficacy of sterile manufactured product, including assurance of
sterility.
138-139 Existing Text
The manufacturer has sufficient knowledge and expertise in relation to the products manufactured and the
manufacturing methods employed.
Proposed changed text:
The manufacturer has defined knowledge and expertise in relation to the products manufactured and the
manufacturing methods employed and the equipment and engineering systems that have a direct impact on product
quality.
Oualitv System System Content (includes but not limited to)
Facility Desinn. traffic flows, utilities, maintenance nrcventative
and repair, cleanine. disinfection, monitorina systems
Manulaclurina Process Desicn. ccniipment. in-nrocess controls, in-process tcsls
Personnel Trainina, certification, garbina.
Procedures Vendor annroval. out sourcinc. risk assessments.
trendina. analysis, investiealional tools. CAPA.
continuous improvement
Product Raw materials, in-nrocess tests, end product tests.
containers, closures
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
145 Comment:
New requirement. The site is required to implement and periodically update a documented risk assessment. Should
this risk assessment be incorporated into the contamination control strategy described at line 51?
Proposed change (if any): Eliminate lack of clarity about the difference between the contamination control strategy
document described at line 51 and the risk assessment document described at line 145.
Comment:
There is no guidance regarding the approach to be taken to complete an effective risk assessment that can accurately
quantify the level of product microbial contamination. This requires an understanding of the fundamental risk factors
that are responsible for microbial contamination, associated with airborne deposition, surface contact or liquid
transfer, and how these factors should be combined to provide an accurate assessment of risk
Recommendation:
Include further guidance on the requirement to provide an accurate assessment of risk based upon the fundamental
mechanisms of product contamination by airborne deposition, surface contact or liquid transfer. The guidance should
reference the fundamental risk factors that relate to product (or product contacting surfaces) contamination such as
exposure area and time and the number of contacts with contaminated surfaces
152 Comment:
Chapter 1 of EU GMP vol 4-PQR section 1.10 does not include requirement for QRM. Suggest both documents are
aligned.
154-157 Comment:
Reference to transport of sterile products should be part of GDP. Suggest that this reference is removed. If it remains
then see proposed text below.
Comment:
Is this more of a GDP requirement and is this Annex the right place for this requirement ( i.e. transport)?
Existing Text
Processes associated with the finishing and transport of sterile products should not compromise the finished sterile
product in terms of container integrity or pose a risk of contamination and ensure that medicinal products are stored
and maintained in accordance with registered storage conditions.
Proposed changed text:
Processes associated with the finishing and transport of sterile products should not compromise the finished sterile
product. Aspects that should be considered include:- container integrity, risks of contamination, and avoidance of
degradation by ensuring that medicinal products are stored and maintained in accordance with their registered storage
conditions.
159-164 Existing Text
Persons responsible the quality release of sterile medicines should have appropriate access to manufacturing and
quality information and possess adequate knowledge and experience in the manufacture of sterile dosage forms and
their critical quality attributes in order to be able to ascertain that the medicines have been manufactured in
accordance with the registered specification and are of the required safety, quality and efficacy.
Proposed changed text:
Persons responsible for the certification and quality release of sterile medicines should have appropriate access to
manufacturing and quality information and possess defined knowledge and experience in the manufacture of sterile
dosage forms and their critical quality attributes in order to be able to ascertain that the medicines have been
manufactured in accordance with the registered specification and are of the required safety, quality and efficacy.
166-171 Existing Text

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
3.2 Investigations should be performed into non-conformities, such as sterility test failures or environmental
monitoring excursions or deviations from established procedures, with a specific focus regarding the potential impact
to sterility, to not only the specific batch concerned but also any other potentially impacted batch. The reasons for
including or excluding product from the scope of the investigation should be clearly recorded and justified within the
investigation.
Proposed changed text:
3.2 Investigations must be performed into non-conformities, such as sterility test failures or environmental monitoring
excursions or deviations from established procedures, with a specific focus regarding the potential impact upon
sterility. Investigations must consider not only the specific batch concerned but also any other potentially impacted
batch. The reasons for including or excluding product from the scope of the investigation should be clearly recorded
and justified within the investigation.
4. Personnel
Comment:
Before this section is introduced it would be better to describe the Premises (currently the following section) to define
Grades A-D and the concept of CNC areas. This section also requires revision to clarify and recognise best practise.
The term grade A/B cleanroom is not defined. It may be interpreted by some readers to suggest that it is acceptable for
personnel to fully enter/occupy Grade A zones, which of course is not acceptable.
175-179 Existing text:
4.1 The manufacturer should ensure that there are sufficient appropriate personnel, suitably qualified and
experienced in the manufacture and testing of sterile medicines and any of the specific manufacturing technologies
used in the site's manufacturing operations, to ensure compliance with Good Manufacturing Practice applicable to
the manufacture of sterile medicinal products.
Proposed change:
Removal of the word “appropriate”.
181-186 Existing text
4.2 Only the minimum number of personnel required should be present in cleanrooms. The maximum number of
operators in critical areas should be determined based on QRM principles, documented in the contamination control
strategy, and validated during activities such as initial qualification and aseptic process simulations, so as not to
compromise sterility assurance. This is particularly important during aseptic processing. Inspections and controls
should be conducted outside the clean areas as far as possible.
Comment:
Does this paragraph indicate that staff from the quality function should only conduct audits and inspections from
outside of the aseptic area or is it referring to the inspection of filled containers etc?
Proposed change:
Include the abbreviation (APS) after the “aseptic process simulations”.
Comment:
The document recommends that the maximum number of operators in critical areas should be determined based upon
QRM principles. By the definition of ‘critical’ areas (see Glossary, text line 2104), personnel are unlikely to be within
these areas and it is more appropriate to state that the maximum number of operators in the ‘aseptic processing room’
would be a more appropriate term.
Recommendation:
Change ‘critical area’ to aseptic processing room’
188-194 Existing text:

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
4.3 All personnel (including those performing cleaning and maintenance) employed in such areas should receive
regular training, qualification (including sampling of the operators bioburden, using methods such as contact plates,
at key locations e.g. hands arms and chest) and assessment in disciplines relevant to the correct manufacture of sterile
products. This training should include reference to hygiene, cleanroom practices, contamination control, aseptic
techniques, and potential safety implications to the patient of a loss of product sterility and in the basic elements of
microbiology.
Comment:
Monitoring requirements should be described in the monitoring section.
Proposed changed text:
4.3 All personnel, including those performing cleaning and maintenance employed in such areas should receive
regular training, qualification and assessment in disciplines relevant to the correct manufacture of sterile products.
This training should include reference to hygiene, cleanroom practices, contamination control, aseptic techniques, and
potential safety implications to the patient of a loss of product sterility and in the basic elements of microbiology.
Personnel should be monitored for microbial contamination as described in section 9.
196-208 Existing text:
4.4 The personnel working in a grade A/B cleanroom should be trained for aseptic gowning and aseptic practices.
Compliance with aseptic gowning procedures should be assessed and confirmed and this should be periodically
reassessed at least annually and should involve both visual and microbiological assessment (using additional
locations such as arms and chest). Only trained personnel who have passed the gowning assessment and have
participated in a successful aseptic process simulation (APS) test, during which they performed their normal duties,
should be authorized to enter any grade A/B area, in which aseptic operations will be conducted, or are being
conducted, whilst unsupervised. The microbial monitoring of personnel in the grade A/B area should be performed to
assess their aseptic behaviour. This monitoring should take place immediately after completion of a critical
intervention and upon each exit from the cleanroom. It should be noted that there should also be an ongoing
continuous monitoring program for personnel including some consideration of periodic monitoring under the
supervision of the quality unit.
Comment:
Use of grade A/B again. There seems to be an expectation that cleaning staff who will not be directly involved in
aseptic processing need to participate in a successful APS. This requirement has been omitted in the proposed text
below.
Proposed changed text:
4.4 As far as possible personnel should be excluded from entering Grade A zones. The personnel working in a Grade
B cleanroom and especially those supporting Grade A zones, must be trained for aseptic gowning and aseptic
practices. Compliance with aseptic gowning procedures should be assessed and confirmed and this should be
periodically reassessed at least annually and should involve both visual and microbiological assessment. Only trained
personnel who have passed the gowning assessment should be authorized to enter any grade B area, in which aseptic
operations will be conducted, or are being conducted, whilst unsupervised. Only trained personnel who have
participated in a successful aseptic process simulation (APS) test, during which they performed their normal duties,
should be authorized to undertake aseptic processing. The microbial monitoring of personnel in the grade B area
should be performed to assess their aseptic behaviour. This monitoring should take place immediately after
completion of a critical intervention and upon each exit from the cleanroom. It should be noted that there should also
be an ongoing monitoring program for personnel which will include periodic monitoring under the supervision of the
quality unit.

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Comment (1): Note some companies believe that the personnel monitoring frequency should be based upon the
activity that personnel are engaged in and should not necessarily be required on each exit.
Comment (2)
Line 199: Requalification should include visual and microbiological assessment
Proposed change (if any): suggest adding visual and microbiological assessment
Comment (3):
Line 205-206: EM on each exit of the cleanroom may be excessive. One company monitors at the end of a shift or at
the end of a campaign, at least once on each operator.
Proposed change (if any): recommend EM at each end of shift or campaign -
Define critical intervention in the glossary
Comment (4):
Line 208-208: What is the difference between critical and significant intervention?
The term “continuous monitoring” for personnel is confusing. “Routine” should be used.
-Is also gown monitoring required after a critical intervention? To be clarified. Only glove monitoring should be
required after a critical intervention, supported by risk assessment
Proposed change (if any): -Routine” should be used. Clarify if only glove monitoring is acceptable after completion of
a critical intervention, supported by risk assessment
Comment (5):
The Grade A/B cleanroom is a part of the aseptic manufacturing area in which there may be separate rooms which are
only Grade B status (contain no Grade A zones). Aseptic processing room (definition included in the Glossary) would
be a more appropriate term
Recommendation:
Change ‘Grade A/B cleanroom’ to aseptic processing room’ throughout the document.
Comment (6):
Line 200: Personnel who are validated and authorised to enter into the aseptic processing room may not perform direct
manufacturing activities e.g. to perform peripheral cleaning and disinfectant or supervisory activities, and
consequently would have no requirement to participate in a successful aseptic process simulation test.
Recommendation:
Remove the requirement for all personnel who enter into the aseptic processing room to have participated in a
successful aseptic process simulation test and specify that the requirement is specific to those personnel who have
direct manufacturing activities.
Comment (7)
Line 205: ‘Critical intervention’ is defined in the Glossary section (text line 2154) as ‘Intervention (an aseptic
manipulation or activity that occurs at the critical area)’ so the term critical is not required
There are interventions that will have been assessed to be aseptically secure, included in aseptic process simulations
and are an inherent part of the process in order for the operation to continue (e.g. replenishment of the container
closures). In these cases, it would not be appropriate to perform personnel microbial monitoring. Any such monitoring
needs to be considered for, and specific to, corrective interventions that are not included in aseptic process simulations
Recommendation:
Change ‘critical area’ to aseptic processing area
Change ‘after completion of a critical intervention’ to ‘after completion of a non validated intervention’.
Comment: (8)
Line 206: Personnel may leave the cleanroom in which the manufacturing activities are located to retrieve
consumables or related items for use during the manufacturing operation. For these activities, personnel microbial
monitoring would not be expected as personnel are returning to the cleanroom. However, when the personnel
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
subsequently leave the aseptic processing area/aseptic manufacturing area, and discard their cleanroom attire,
monitoring would be appropriate
Recommendation
Change ‘upon each exit from the cleanroom’ to ‘upon each exit from the aseptic manufacturing area’
Comment (9):
Line 205-206: Does this refer to finger sample only monitoring or gown monitoring. Gown monitoring would be
unachievable as operators would have to leave the area to re-gown mid process to avoid agar contamination from their
sterile suit.
Proposed change:
Finger sample monitoring should take place…
210-215 Existing text
4.5 There should be systems in place for disqualification of personnel from entry into cleanrooms, based on aspects
including ongoing assessment and/or the identification of an adverse trend from the personnel monitoring program.
Once disqualified, retraining and requalification is required before permitting the operator to have any further
involvement in aseptic practices. This should include consideration of participation in a successful Aseptic Process
Simulation (APS).
Proposed changed text:
4.5 There should be systems in place for disqualification of personnel from entry into cleanrooms, based on aspects
including ongoing assessment and/or the identification of an adverse trend from the personnel monitoring program.
Once disqualified, retraining and requalification is required before permitting the operator to have any further
involvement in aseptic practices. This should include participation in a successful Aseptic Process Simulation (APS)
depending upon the reasons for disqualification and its impact.
217-220 Existing text
4.6 Manufacturers should establish written procedures outlining the process by which outside staff who have not
received such training (e.g. building or maintenance contractors) need to be brought into grade A/B areas. Access by
these persons should only be given in exceptional circumstances, evaluated and recorded in accordance with the PQS.
Proposed changed text:
4.6 Manufacturers should establish written procedures outlining the process by which outside staff who have not
received such training (e.g. building or maintenance contractors) need to be brought into Grade B areas or access
Grade A zones. Access by these persons should only be given in exceptional circumstances, evaluated and recorded in
accordance with the PQS.
225-226 Comment:
“periodic health checks for such conditions should be performed.” This sentence is not clear. Who should perform the
check? Is self-assessment acceptable?
Proposed change (if any): Clarify the meaning of “periodic”.
226-228 Existing text
Actions to be taken with regard to personnel who could be introducing an undue microbiological hazard should be
described in procedures decided by a designated competent person.
Comment:
What actions can be taken/how to avoid a breach in confidentiality of personnel medical information.
227 Comment:
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Definition required for “Undue microbiological hazards”
232 Comment:
Is it problematic for Biological Quality personnel who work in the core to also work in the lab without a “rigorous,
clearly defined and effective entry procedures have been followed”?
Not clear how far you need to go to investigate this. You cannot check personal activities? What about part time
butchers, farmers? Concern? Our gowning and Cleaning and disinfection SOPs should be effective?
Proposed change (if any): More clarification of what entails a “rigorous, clearly defined entry procedure” is…
233 Comment:
The term ‘sterile product areas’ is not defined and is a new term, not used throughout the document
Proposed change:
Change ‘sterile product areas’ to ‘aseptic processing area/aseptic manufacturing area’
236-237 Existing text:
4.9 Wristwatches, make-up and jewellery and other personal items such as mobile phones should not be allowed in
clean areas.
Comment (1):
Is this referring to classified clean areas only OR CNC areas also?
Comment (2):
The definition of clean areas is not included in the glossary
The requirement to not use mobile phones in all Grades of clean rooms is too stringent.
Proposed change (if any): Include a definition of clean areas in the glossary.
Reword as follows: “Wristwatches, make-up and jewelry should not be allowed in clean areas. Other personal items
and electronic devices (e.g. mobile phones) should be allowed in clean areas, where needed, only if adequately
decontaminated and fit for purpose.
Comment (3):
More guidance on make-up is needed to suit modern culture. Make-up can now be semi-permanent, for example lip
staining.
Recommendation:
Change make up to non-permanent make-up
239-245 Existing text:
4.10 Changing and hand washing should follow a written procedure designed to minimize contamination of clean
area clothing or carry-through of contaminants to the clean areas. Garments should be visually checked for
cleanliness and integrity prior to entry to the clean room. For sterilized garments, particular attention should be
taken to ensure that garments and eye coverings have been sterilized and that their packaging is integral before use.
Reusable garments should be replaced based at a set frequency determined by qualification or if damage is
identified.
Proposed text change:
4.10 Garment changing and hand washing should follow a written procedure designed to minimize contamination of
cleanroom clothing or transfer of contaminants to the cleanrooms. Garments should be visually checked for
cleanliness and integrity prior to entry to the cleanroom. For sterilized garments, particular attention should be taken
to ensure that garments and eye coverings have been sterilized and that their packaging is integral before use.
Reusable garments should be replaced based upon a set frequency determined by qualification or when damage is
identified.
Comment:
Line 242-243: The gowning components include the garments and all other items required to complete full personnel
enclosure (e.g. garment, hood, overboots, gloves, masks, eye coverings) and all components should be confirmed to
have been sterilised with integral packaging prior to use.
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Recommendation:
Change ’particular attention should be taken to ensure that garments and eye coverings have been sterilised and that
their packaging is integral before use’ to ’particular attention should be taken to ensure that all garment components
have been sterilised and that their packaging is integral before use’
253 Comment:
‘Disinfected Shoes’ – this should not be a requirement but the type of microbial /particulate control for shoes in Grade
D should be based on the risk of contamination entering into the next stage of cleanroom.
254-260 Comment:
Disinfection of dedicated Grade D shoes should not be required. Suggest to use either overshoes or dedicated shoes
Proposed change (if any): re-word as follows: “A general protective suit and either area dedicated shoes or overshoes
should be worn.”
258-261 Comment:
The objective is to ensure appropriate measures in order not to bring contamination inside clean areas. Entry into
Grade C areas with facility dedicated shoes does not pose a risk of undue contamination. Furthermore, the
requirement for non -particle shedding garments in Grade C areas is not consistent with the non-viable particulate
limits (Grade C level classification) present in those areas. It is also not consistent with QRM principles since in those
areas there is no direct exposure of product or sterile product contact surfaces
Proposed text change:
b) Grade C: Hair, beards and moustaches should be covered. A single or two-piece trouser suit gathered at the wrists
and with high neck and appropriately disinfected or facility dedicated shoes or overshoes should be worn.
263-272 Existing text:
c) Grade A/B: Sterile headgear should totally enclose hair and facial hair; it should be tucked into the neck of the
sterile suit; a sterile face mask and sterile eye coverings should be worn to cover all facial skin and prevent the
shedding of droplets and particles. Appropriate sterilized, non-powdered rubber or plastic gloves and sterilized
footwear should be worn. Trouser-legs should be tucked inside the footwear and garment sleeves into the gloves. The
protective clothing should shed virtually no fibres or particulate matter and retain particles shed by the body.
Garments should be packed and folded in such a way as to allow operators to change into the garments with contact
to the outer surfaces of the garment reduced to a minimum.
Comment:
There is no mention of the use of gowning gloves nor under-suits which are essential in contamination control and are
used fairly ubiquitously.
Proposed text change:
b) Grade B: A dedicated single or two-piece under-suit should be worn. Sterilised outer garments (non-
shedding) must be donned for the manufacture of aseptically processed products. The single trouser suit,
gathered at the wrists and with a high neck, must provide total body coverage, such that the headgear must
fully enclose the hair and be tucked into the neck of the suit. Full face cover and eye protection must be
used to prevent the shedding of droplets. Eye protection equipment e.g. goggles, integral helmets and
glasses must be sterilised by suitable procedures. Sterilised non-powdered rubber gloves and sterilised
footwear must be worn with trouser legs tucked inside the footwear and garment sleeves into the outer
gloves (see below). The garments must be handled and donned, using a pair of sterilised gowning gloves,
such that they do not gather additional contamination and must shed virtually no fibres or particulate matter,
retaining particles shed from the body. Following gowning an additional pair of sterilised (outer) gloves
must be worn over the gowning gloves. Garments should be packed and folded in such a way as to allow
operators to change into the garments without contact to the outer surfaces of the garment and to help
prevent the garment touching the floor or other surfaces.
Comment (2)

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Line 270-272
“… with contact to the outer surfaces of the garment reduced to a minimum.” should be without contact to the outer
surfaces of the garment.
Comment (3):
Line 263: Headgear for consistency to be referred to as headwear and (as for other items of protective attire similarly
referenced throughout) it is more appropriate to refer to as being ‘sterilised’ and not as ‘sterile’ as any such sterilised
headwear is unlikely to have remained sterile during the donning process
Recommendation:
Change ‘sterile headgear’ to ‘sterilised headwear’
Comment (4):
Line 264: Data is available that confirms disinfection to be an effective process to reduce the surface concentration on
cleanroom goggles to very low levels and that the subsequent higher surface microbial concentrations recovered
during gowning qualification and on exit monitoring (from the aseptic processing area), occurs during the donning
process. Although sterilised goggles will have less surface microbial contamination than disinfected goggles, the
donning procedure will transfer more microbes than are residual on the goggles, regardless of whether they are
sterilised or disinfected. The air velocities present in cleanrooms are insufficient to remove such microbe-carrying
particles from the surface of goggles and so product contamination would only occur if product, or product contacting
surfaces, contacted the goggles. This is extremely unlikely and the risk of product contamination from microbes
present on the surface of goggles is extremely unlikely. Consequently, disinfection (with appropriate controls and
monitoring) should be considered as an acceptable method for to control surface microbial contamination on eye
coverings worn in grade A/B areas.
Recommendation
Change ‘sterile eye coverings’ to ‘sterilised or appropriately disinfected and controlled eye coverings:
Comment (5):
Line 265: The document states that face masks and eye coverings (like the other components of the gowning) should
be worn to prevent the shedding of droplets and particles. Shedding of droplets and particles will occur regardless of
the use of face masks and eye coverings but the function of these items is to control (but will not fully prevent) the
subsequent dispersal of such contaminants into the environment.
Proposed change:
Change ‘cover all facial skin and prevent the shedding of droplets and particles’ to ‘cover all facial skin and control
the dispersal of droplets and particles’.
Comment (6):
Line 266: Double gloving has been confirmed to be an effective control method to minimise the risk of product
contamination resulting from ruptured gloves. The use of double gloving should be suggested in the text.
Proposed change: Add ‘double gloving should be considered to minimise the risk of product contamination resulting
from ruptured gloves’.
275 Comment:
Dedicated socks is an excessive requirement and it would not improve contamination control.
276-279 Comment (1):
“Outdoor clothing” requires clarification as it will change depending upon the time of year.
Comment (2):
Clarification required on the purpose of the change room if cannot be used for outdoor clothing.
Comment (3):
Clarification on the use of socks e.g. what is the purpose of these and in what type room should these be donned.
Comment (4):
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Line 277: There are numerous methods that can be utilised in order to adequately control contamination in
changerooms leading to Grade B and C cleanrooms. If socks remain covered (e.g. by the use of dedicated shoes) in
such changerooms, the need for dedicated socks may not be appropriate.
Proposed change:
Change ‘It is recommended that facility suits, including dedicated socks, are worn before entry to change rooms for
Grade C and B’ to ‘It is recommended that facility suits (with consideration for dedicated socks where appropriate),
are worn before entry to change rooms for that access into Grade B and C cleanrooms’
281-284
Existing text:
4.14 For every worker in a grade A/B area, clean sterilized protective garments (including eye coverings and masks)
of an appropriate size should be provided at each work session. Gloves should be regularly disinfected during
operations. Garments and gloves should be changed at least for every working session.
Proposed text change:
4.14 For every worker in a grade B area, clean sterilized protective garments (including eye coverings and masks) of
an appropriate size as well as under garments should be provided at each work session. Gloves should be regularly
disinfected during operations. Garments and gloves should be changed at least for every working session.
286-291 Existing text:
4.15 Clean area clothing should be cleaned, handled and worn in such a way that it does not gather additional
contaminants which can later be shed. These operations should follow written procedures. Separate laundry facilities
for such clothing are desirable. Inappropriate treatment of clothing will damage fibres and may increase the risk of
shedding of particles. After washing and before sterilization, garments should be checked for integrity.
Proposed changed text:
4.15 Clean area clothing should be cleaned, handled and worn in such a way that it does not gather additional
contaminants which can later be shed. These operations should follow written procedures. Separate laundry facilities
for such clothing are required. Inappropriate treatment of clothing will damage fibres and may increase the risk of
shedding of particles. After washing and before sterilization, garments should be visually checked for wear and
damage.
293-301 Existing text:
4.16 Activities in clean areas, especially when aseptic operations are in progress, should be kept to a minimum and
movement of personnel should be controlled and methodical to avoid excessive shedding of particles and organisms
due to over-vigorous activity. Operators performing aseptic operations should adhere to strict aseptic technique at all
times. To prevent changes in air currents that introduce lower quality air, movement adjacent to the critical area
should be restricted and the obstruction of the path of the unidirectional airflow must be avoided. The ambient
temperature and humidity should be set to prevent shedding due to operators becoming too cold (leading to excessive
movement) or too hot.
Proposed changed text:
4.16 Activities in clean areas, especially when aseptic operations are in progress, should be kept to a minimum and
movement of personnel should be controlled and methodical to avoid excessive shedding of particles and micro-
organisms owing to over-vigorous activity. Operators performing aseptic operations should adhere to strict aseptic
technique at all times. To prevent adverse air currents that could introduce lower quality air into an adjacent critical
area, movement adjacent to the critical area should be restricted. The obstruction of the path of the unidirectional
airflow within Grade A zones must be avoided. The ambient temperature and relevant humidity should be specified,
maintained and monitored either to meet specific product requirements or to ensure operator comfort. Poor operator
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
comfort can lead to increased contamination dissemination and an inability of personnel to concentrate on critical
tasks.
Comment:
Line 299-300: If stated that being too cold causes excessive movement, then it should also be stated that being too hot
may increase sweating and breach of gowning
Proposed change (if any):
Suggested to change the sentence as follows “The ambient temperature and humidity should be set to prevent
shedding due to operators not being comfortable”.
5. Premises
Comment:
This section should be earlier in the document as it defines Grades A-D. The section requires modification to improve
clarity, use correct and consistent terminology, and to correct errors.
Airlocks for personnel should also be referred to as gowning rooms.
310-312 Existing text:
5.2 The various operations of component preparation, product preparation and filling should be carried out with
appropriate technical and operational separation measures within the clean area.
Proposed changed text:
5.2 The various operations of component preparation, product preparation and filling should be carried out with
appropriate technical and operational separation measures within the clean area to prevent mix-up and cross-
contamination.
314-315 Existing text:
5.3 For the manufacture of sterile medicinal products 4 grades of clean room can be distinguished.
General Comment:
Here and throughout the document the term “clean room” is used. Note cleanroom is a single word.
Proposed changed text:
5.3 For the manufacture of sterile medicinal products 4 grades of classified clean areas are defined in this guidance.
318-332 Existing text:
Grade A: The local zone for high risk operations, e.g. filling zone, stopper bowls, open ampoules and vials, making
aseptic connections. Normally, such conditions are provided by a localised air flow protection, such as laminar air
flow work stations or isolators. Unidirectional air flow systems should provide a homogeneous air speed in a range of
0.36 - 0.54 m/s (guidance value), the point at which the air speed measurement is taken should be clearly justified in
the protocol. During initial qualification and requalification air speeds may be measured either close to the terminal
air filter face or at the working height, Where ever the measurement is taken it is important to note that the key
objective is to ensure that air visualization studies should correlate with the airspeed measurement to demonstrate air
movement that supports protection of the product and open components with unidirectional air at the working height,
where high risk operations and product and components are exposed. The maintenance of unidirectional airflow
should be demonstrated and validated across the whole of the grade A area. Entry into the grade A area by operators
should be minimized by facility, process and procedural design.
Proposed changed text:
Grade A: The local zone for high risk operations, e.g. filling zone, stopper hopper and feeder bowls, open ampoules
and vials, and making aseptic connections. Normally, such conditions are provided by localised unidirectional air flow
protection, such as an uni-directional air-flow work station, RABS or isolator. Unidirectional air flow systems should
provide a homogeneous air velocity across the plane perpendicular to the airflow direction. The typical air velocity in
a traditional uni-directional airflow clean zone is an average of 0.45 m/s+/- 20%. Lower average velocities may be
advantageous in isolators and closed RABS. In some isolator applications Grade A conditions can be maintained with

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
non-unidirectional airflow. The plane at which the air velocity measurement is taken should be clearly justified in the
protocol. During initial qualification and requalification air velocities may be measured either close to the terminal air
filter face or at a defined test level. Where ever the measurement is taken it is important to note that the objective is to
ensure that air visualization studies should correlate with the air velocity measurement at a defined test plane level and
clearly demonstrate air movement that provides protection of the product and open components with unidirectional air
at the working height, where high risk operations and product and components are exposed. The maintenance of
unidirectional airflow should be demonstrated and qualified across the whole of the Grade A clean zone. Operators
must not enter the Grade A zone. Intervention access into the Grade A zone by operators should be minimized by
facility, process and procedural design
Comment (1)
Line 318: “Grade A : The local zone for high risk operations, e.g. filling zone, stopper bowls, open ampoules and
vials, making aseptic connections.” should be e.g. filling zone, stopper bowls, open primary containers (ampoules,
vials, syringes, cartridges…)....
Comment (2):
Line 321: The measurement of the unidirectional airflow (UDAF) velocity needs to be completed at a location that
when measured in the same manner will provide a consistent value that can be used to determine if there has been any
change. The UDAF passes round objects located in the working zone and creates an area that has little air movement
and so the velocity in these zones (near to product) is very low impractical to measure. Users may incorrectly specify
a higher velocity close to the exposed product which are not necessary to provide the required airflow and present
issues with airflow uniformity and increased energy costs. A distance of 15cm to 30cm from the filter face is
suggested in ISO 14644-3 (Test methods).
Alternative Proposed text change:
Unidirectional airflow should be provided in the critical area. During qualification and re-qualification the airflow
velocity should be measured at several locations to demonstrate a uniform velocity across the filter face and should be
measured 15 to 30cm from the filter face. Airflow visualisation should also be completed to demonstrate that air
movement is unidirectional.
Comment (3):
Line 330-331: Entire Grade A area must be studied with airflow visualization studies.
Clarify the meaning of …across the whole of the Grade A area”
Proposed change (if any): delete ‘whole.
Comment (4):
Line 331 & 815, 839: ‘Entry into grade A area by operators…’Regulatory authority guidance has previously
stated that the zone cannot be considered to be Grade A if personnel are present within the zone.
Alternative Proposed Change: Clarification is needed on terminology, e.g. Grade A zones that operators
enter to be termed localised unidirectional airflow (L-UDAF) or appropriate similar term.
General comment:
Throughout the document the term “laminar flow” is used. The definition of laminar airflow is provided in the
Glossary section (line 2177). According to this definition, the airflow moves in a single direction and in parallel layers
at constant velocity to the end of a straight line vector. This is not possible and the term unidirectional flow (UDAF) is
more accurate.
Proposed change: Change ‘laminar air flow’ to ‘unidirectional airflow (UDAF)’
337-338 Existing text:
Lower grades can be considered where isolator technology is used (refer to clause 5.19-5.20).
Comment:
This lacks clarity, please define precisely which background grades should be used.
343-345 Existing text:
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
5.4 In clean areas, all exposed surfaces should be smooth, impervious and unbroken in order to minimize the
shedding or accumulation of particles or micro-organisms and to permit the repeated application of cleaning agents,
and disinfectants, where used.
Proposed changed text:
5.4 In classified cleanroom areas and clean zones, all exposed surfaces should be smooth, impervious and unbroken in
order to minimize the shedding or accumulation of particles and micro-organisms and to permit the repeated
application of cleaning agents, and disinfectants, where used.
Comment:
Line 345: No description or glossary entry to explain the difference between cleaning agent and disinfectant.
Alternative Proposed Change: Include additional definitions in glossary for; Cleaning Agent (Chemical solution used
for the removal of fat based particulates), Disinfectant (Chemical agent used for the destruction of microorganisms)
347-349 Existing text:
5.5 To reduce accumulation of dust and to facilitate cleaning there should be no uncleanable recesses and a minimum
of projecting ledges, shelves, cupboards and equipment. Doors should be designed to avoid uncleanable recesses.
Proposed text change:
5.5 To reduce accumulation of dust and to facilitate cleaning there should be no uncleanable recesses and a minimum
of projecting ledges, shelves, cupboards and equipment. Doors should be designed to avoid uncleanable recesses,
open only to the higher grade and be mechanically self closing.
356-359 Existing text:
5.8 Sinks and drains should be prohibited in grade A/B areas. In other areas air breaks should be fitted between the
machine or sink and the drains. Floor drains in lower grade rooms should be fitted with traps or water seals to
prevent back flow and should be regularly cleaned and disinfected.
Proposed text change:
5.8 Sinks and drains are prohibited in Grade A and B areas. In other areas air breaks should be fitted between the
machine or sink and the drains. Drains in lower grade rooms should be fitted with water traps to prevent back flow
and should be regularly cleaned and disinfected.
361-367 Comment:
Definition needed for ‘flushed’, does this mean to remove the area from particulate?
Existing text:
5.9 Airlocks should be designed and used to provide physical separation and to minimize microbial and particulate
contamination of the different areas, and should be present for material and personnel moving from different grades,
typically airlocks used for personnel movement are separate to those used for material movement. They should be
flushed effectively with filtered air. The final stage of the airlock should, in the at-rest state, be the same grade as the
area into which it leads. The use of separate changing rooms for entering and leaving clean areas is generally
desirable.
Proposed text change:
5.9 Airlocks should be designed and used to provide physical separation between areas of different Grade, to maintain
pressure differentials and to minimize microbial and particulate contamination transfer between different areas.
Personnel airlocks (PAL or gowning room) should be separate from those used for material transfer. They should be
flushed effectively with filtered air. The final stage of the airlock should, in the at-rest state, be the same grade as the
higher graded area with which it interfaces. By first intent separate changing rooms should be used for entering and
leaving clean areas. Where this is not possible the removal of gowns must not take place in the same area where fresh
sterile gowns are put on.
369-371 Comment:
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Clarification required on cascade from D to C; this sentence details that this ‘should’ be performed; recommend that
the sentence clarifies if this is or is not required. The cascade and use of Grades should form part of site cross
contamination RA and the selected design of area should be justified.
Proposed change (if any): Recommendation is that the Eg as per section b, part iii below (lines 390-392).
Existing text:
a) Personnel airlocks. A cascade concept should be followed for personnel (e.g. from grade D to grade C to grade B).
In general hand washing facilities should be provided only in the first stage of the changing rooms.
Proposed text change:
a) Personnel airlocks. A cascade concept should be followed for personnel ( from Grade D to Grade C to Grade B). In
general hand washing facilities should be provided only in the first stage of changing rooms. Hand washing facilities
incorporating water should not be installed within Grade B PALs. Topical disinfection of gloved hands should be
used in Grade C and B PALs.
Comment:
Line 369: Jumping of grades in personnel airlock cascades seems to be not allowed
Proposed change (if any): recommend add ‘generally a cascade concept should be followed’
375-377 Existing text:
i. Pass through hatches without active filtered air supply should be avoided. If necessary, provisions and procedures
should be in place to avoid any risk of contamination (e.g. by the incoming material or by entering air).
Proposed text change:
Material airlocks and pass through hatches must be configured to prevent transfer of contamination from the less clean
to the cleaner area. The use of active filtered air supply should be considered. Provisions and procedures should be in
place to prevent contamination transfer on materials or in the air.
379-388 Existing text:
ii. For airlocks leading to grade A and B areas, only materials and equipment that have been included as part of the
qualification list should be allowed to be transferred into the grade A/B area via the air lock or pass through; the
continuity of grade A should be maintained in the aseptic core when the materials have to be transferred from grade B
to grade A areas, consideration should be given to listing these items on an authorized list. Any unapproved items that
require transfer should be an exception. Appropriate risk evaluation and mitigation strategies should be applied and
recorded as per the manufacturer's contamination control strategy and should include a specific sanitisation and
monitoring regime approved by quality assurance.
Proposed text change:
ii .For airlocks leading to Grade A and B areas, only materials and equipment that have been included on a
qualification list should be allowed to be transferred into the Grade A clean zones or Grade B cleanrooms area via an
air lock or pass through hatch. The continuity of grade A should be maintained in the Grade A aseptic core when the
materials are in transit through the Grade B cleanroom to the grade A zone. Any unapproved items that require
transfer should be an exception. Appropriate risk assessment and mitigation measures should be applied and recorded
as per the manufacturer's contamination control strategy and should include a specific sanitisation and monitoring
regime approved by quality assurance.
Comment (1):
The continuity of Grade A is an important aspect of aseptic control and is often required by regulators. This is the only
area of the entire document in which it is mentioned. It also doesn’t appear in the glossary.
Comment (2):
Line 383: “… transferred from grade B to grade A areas, ...” should be … transferred from lower grades to grade A
areas, …
Comment (3):
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Line 388: There is no definition of ‘sanitisation’ provided in the Glossary section (line 2177) and
disinfection is a more appropriate term that is included in the Glossary section.
Proposed change:
Change ‘sanitisation’ to ‘disinfection’.
390-392 Existing text:
iii. The movement of material from clean not classified (CNC) to grade C should be based on QRM principles, with
cleaning and disinfection commensurate with the risk.
Proposed text change:
iii. The movement of material from clean not classified (CNC) to Grade C cleanrooms should be based on QRM
principles, with cleaning and disinfection applied commensurate with the risk.
Comment:
Line 390: CNC is defined by the International Society for Pharmaceutical Engineering (ISPE) as ‘controlled not
classified’. To use the same abbreviation (CNC) to define an area that is ‘clean not classified’ will be confusing. It
would be more appropriate to utilise the current ISPE definition of CNC (a cGMP manufacturing area designed to
produce a consistent and controlled environment but not necessarily monitored to a given environmental
classification) or use an alternative acronym for an area that is to be defined as clean not classified.
Proposed change:
Change ‘CNC’ to controlled not classified and include the ISPE definition of CNC in the Glossary section (line 2076).
394-398 Existing text:
5.10 Both airlock doors should not be opened simultaneously. The opening of more than one door at a time should be
prevented, for airlocks leading to grade A and B an interlocking system should usually be used; for airlocks leading to
grade C and D at least a visual and/or audible warning system should be operated. Where required to maintain zone
segregation, a time delay between the closing and opening of interlocked doors should be established.
Comment (1):
Replace wording “grade C and D” with Grade C and D cleanrooms. Also remove word “usually” on Line 396.
Comment (2):
Line 397: The airborne cleanliness levels within airlocks is directly related to the rate of volume air supply into the
airlock and the rate of dispersal of contamination within the airlock. If the rate of volume air supply is sufficiently
high and the dispersal is well controlled, the operational airborne cleanliness levels will be maintained at sufficiently
low levels that may not require a time delay to prevent the transfer of contamination into the zone of highest
cleanliness.
Proposed change:
Change ‘When required to maintain zone segregation, a time delay between the closing and opening of interlocked
doors should be established’ to ‘When required to maintain zone segregation, adequate airborne cleanliness levels
should be maintained throughout operation or a time delay between the closing and opening of interlocked doors
should be established’
400-410 Existing text:
5.11 A HEPA or ULPA filtered air supply should maintain a positive pressure and an air flow relative to surrounding
areas of a lower grade under all operational conditions and should flush the area effectively. Adjacent rooms of
different grades should have a pressure differential of 10 - 15 Pascals (guidance values). Particular attention should
be paid to the protection of the zone of greatest risk, that is, the immediate environment to which a product and
cleaned components which contact the product are exposed. The recommendations regarding air supplies and
pressure differentials may need to be modified where it becomes necessary to contain some materials, e.g. pathogenic,
highly toxic, radioactive or live viral or bacterial materials or products. Decontamination of facilities, e.g. the clean
rooms and HVAC, and the treatment of air leaving a clean area may be necessary for some operations.

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Proposed text change:
5.11 A filtered air supply should maintain a positive pressure and an air flow relative to surrounding areas of a lower
grade under all operational conditions and should flush the area effectively. Terminal HEPA filters should be used for
Grade A clean zones, Grade B and C cleanrooms and should as a minimum be H14 filters to EN1822. Terminal filters
used in Grade A, B, and C areas should be periodically in-situ leak tested in accordance with ISO 14644-3. Adjacent
cleanrooms of different grades should have a pressure differential of 10 - 15 Pascals (guidance values). Particular
attention should be paid to the protection of the zone of greatest risk, that is, the immediate environment to which a
product and cleaned and sterilised components which contact the product are exposed. The recommendations
regarding air supplies and pressure differentials may need to be modified where it becomes necessary to contain some
materials, e.g. pathogenic, highly toxic, radioactive, live viral or bacterial materials or products. Decontamination of
facilities, e.g. the cleanrooms and HVAC, and the treatment of air leaving a clean area may be necessary for some
operations.
Comment:
Line 400: HEPA filters at 95 % efficiency will remove all microbe-carrying particles from supply air and a HEPA
filter (H14, reference to EN 1822, 2005) will remove all particles ≥0.5µm and so unless there is a particular
requirement, ULPA filters will achieve no extra reduction in the concentration of airborne contamination in the air
supplied to a cleanroom or workstation. ULPA filters require a much higher differential pressure, with more energy
consumption and noise and more frequent replacement.
Proposed change (if any):
Add ‘Unless there is a need to remove particles from the supply air that are smaller than ≥0.5µm, HEPA filters are
sufficient to remove all airborne contamination from the air supply, and the use of ULPA filters is not necessary.
412-421 Existing text:
5.12 It should be demonstrated that air-flow patterns do not present a contamination risk, e.g. care should be taken to
ensure that air flows do not distribute particles from a particle- generating person, operation or machine to a zone of
higher product risk. Air flow patterns should be visualised in grade A/B areas to evaluate if airflow is unidirectional.
Where unidirectional air flow is not demonstrated, corrective actions, such as design improvements, should be
implemented. In the other areas, the need to demonstrate the air flow patterns should be based on a risk assessment.
Air flow pattern studies should be performed under dynamic conditions. Video recordings of the airflow patterns are
recommended. The outcome of the air visualisation studies should be considered when establishing the facility's
environmental monitoring program.
Comment (1):
Line 415 : Change “A/B” to “Grades A and B” and change “evaluate” to “confirm”. Airflow patterns should be
carried out in both the static and dynamic condition and must include operator interventions. Video recordings must
be carried out and retained.
Comment (2):
Note some companies do not think it is possible to perform dynamic airflow visualisation studies in CRABS or
isolator lines.
Comment (3):
Line 415: “Air flow patterns should be visualised in grade A/B areas to evaluate if airflow is unidirectional...” should
include in operation including environmental monitoring such as volumetric air sampling.
Comment (4):
Line 415-418: There is no requirement to have unidirectional airflow in Grade B.
Need of risk assessment for other areas (C/D) in order not to perform smoke studies.
Proposed change (if any):
The requirement of unidirectional airflow should be for Grade A only.
Comment (5):

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Line 412-421: Not all grade A and B areas should require airflow pattern testing (as not all those areas
possess unidirectional airflow). As written the requirement would restrict the use of suitable alternatives in
the case where airflow pattern test is not physically possible (e.g. depyrogenation tunnel).
Proposed text change:
5.12 It should be demonstrated that air-flow patterns do not present a contamination risk, e.g. care should
be taken to ensure that air flows do not distribute particles from a particle generating person, operation or
machine to a zone of higher product risk. Air flow patterns should be visualised in all unidirectional
airflow areas and at the interface with areas directly adjacent to these (e.g. in critical Grade A/B
processing areas). Where unidirectional air flow is not demonstrated, corrective actions, such as design
improvements, should be implemented. In the other areas, the need to demonstrate the air flow patterns
should be based on a risk assessment. Air flow pattern studies should be performed under dynamic
conditions. Video recordings of the airflow patterns are recommended. Alternatively, simulations may be
used to demonstrate adequate airflow. The outcome of the air visualisation studies should be considered
when establishing the facility's environmental monitoring program. Comment (6):
Line 415: Grade B areas are unlikely to have unidirectional airflow and therefore assessment of unidirectional airflow
in Grade B areas is an unlikely scenario. Not all Grade A zones may have unidirectional airflow (consider a closed
isolator where non UDAF air supply can be acceptable). Proposed Change:
The evaluation of unidirectional airflow should be visualised in those zones that are supplied with unidirectional
airflow.
423-426 Existing text:
5.13 A warning system should be provided to indicate failure in the air supply and reduction of pressure differentials
below set limits. Indicators of pressure differences should be fitted between areas, based on QRM principles. These
pressure differences should be recorded regularly or otherwise documented.
Comment:
A warning system MUST be provided. Pressure differences must be monitored continuously and documented.
428-431 Existing text:
5.14 Consideration should be given to designing facilities that permit observation of activities from outside the clean
areas, e.g. through the provision of windows or remote camera access with a complete view of the area and processes
to allow observation and supervision without entry.
Proposed text change:
Facilities should be designed to permit observation of activities from outside the clean areas, e.g. through the
provision of windows or remote camera vision with a complete view of the area and processes to allow observation
and supervision without entry
434 Barrier Technologies
General comment:
Prefer the use of “bio-decontamination” rather than “disinfection”.
Comment:
The following text should be added BEFORE Line 435
Proposed text
By first intent barrier technology such as Isolators and Restricted Access Barrier Systems (RABS) should be
employed to ensure adequate separation of the Grade A zone from both operators and the surrounding environment.
This is to reduce the need for interventions into the grade A environment and to minimize the risk of contamination.
Automation of processes should also be considered to remove the risk of contamination by interventions (e.g. dry heat
tunnel, automated lyophilizer loading, SIP). The absence of such technology may be acceptable in exceptional
circumstances provided there is sufficient control and evidence of the maintenance of Grade A integrity and that the
general principles for RABS are applied

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
438-439 Comment:
“… and therefore the entry of additional materials following sterilisation should be minimized” Does that mean that
only sterilized materials can be introduced inside the RABS or isolator? This is not in accordance with filling
technologies using tubs of RTU syringes or vials since 5 to 10 tubs are introduced every minute, most of the time
without any individual integrity check of the packaging.
449-454 Existing text:
5.17 The critical zone of the RABS or isolator used for aseptic processes should meet grade A with unidirectional air
flow. Under certain circumstances turbulent airflow may be justified in a closed isolator when proven to have no
negative impact on the product. The design of the RABS and open isolators should ensure a positive airflow from the
critical zones to the surrounding areas; negative pressure isolators should only be used when containment of the
product is considered essential.
Comment (1):
The critical zone comment should cross reference section 5.3. Note that turbulent flow may be necessary for effective
fumigation of closed isolators.
Comment (2):
Line 450: Unidirectional and non-unidirectional airflow are both turbulent airflows and the term non-
UDAF is the correct term referenced in the ISO 14644 series of standards. Proposed change (if any):
Replace (throughout) ‘turbulent airflow’ with ‘non-unidirectional airflow’ (non-UDAF).
456-458 Existing text
5.18 For RABS, the background environment should meet grade B. For open RABS, or where doors may be very
rarely opened during processing, and studies should be performed to demonstrate the absence of air ingress.
Proposed text:
5.18 For RABS, the background environment must be a minimum of Grade B. For open RABS, or where doors may
be very rarely opened during processing, airflow studies should be performed to demonstrate the absence of air
ingress.
Comment:
“For open RABS, or when doors may be very rarely open during processing” what is very rarely? The doors of RABS
must never be open during processing, all interventions should be performed through the gloves of the RABS
otherwise this not a barrier technology.
460-463 Existing text:
5.19 For open, positive pressure isolators or closed isolators with decontamination by a sporicidal agent, the
surrounding area should correspond to a minimum of grade D. The disinfection regime should be included as a key
consideration when performing the risk assessment to design the contamination control strategy for an isolator.
Comment:
Minimum background for isolators should be Grade C.
465-470 Existing text:
5.20 For isolators, the required background environment can vary depending on the design of the isolator, its
application and the methods used to achieve bio-decontamination. The decision as to the supporting background
environment should be documented in a risk assessment where additional risks are identified, such as for negative
pressure isolators. Where items are introduced to the isolator after disinfection then a higher grade of background
should be considered.
Comment (1):

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Regulatory expectation should be clearly given here as to the required grade for isolators to ensure consistency of
approach. Note that for isolators used for filling operations, fresh sterilised stoppers are frequently added AFTER bio-
decontamination using RTPs.
Comment (2):
Definition required for higher grade of background.
Comment (3):
Line 465: How does this statement match the statement at paragraph 5.19 which states that “ For open, positive
pressure isolators or closed isolators with decontamination by a sporicidal agent, the surrounding area should
correspond to a minimum of grade D”?
Proposed change (if any): Ensure consistency of the 5.19 and 5.20 requirements.
Comment (4):
Line 466: Definition for bio-decontamination is required in the glossary.
472-478 Existing text:
5.21 Glove systems, as well as other parts of an isolator, are constructed of various materials that can be prone to
puncture and leakage. The materials used shall be demonstrated to have good mechanical and chemical resistance.
Integrity testing of the barrier systems and leak testing of the isolator and the glove system should be performed using
visual, mechanical and physical methods. They should be performed at defined periods, at a minimum of the
beginning and end of each batch, and following any intervention that may affect the integrity of the unit.
Proposed text change
5.21 Glove systems, as well as other parts of an isolator, are constructed of various materials that can be prone to
puncture and leakage. The materials used shall be demonstrated to have good mechanical and chemical resistance.
Integrity testing of the barrier systems and leak testing of the isolator and the glove system should be performed using
visual, mechanical and physical methods. They should be performed at defined periods, at a minimum of the
beginning and end of each batch, unless otherwise justified as in campaign processing and following any intervention
that may affect the integrity of the unit. Glove systems for RABS should be subject to visual checks to confirm their
integrity
Comment (1):
Is the leak testing at the beginning and end of processing to be applied just to gloves OR both gloves and isolator?
Comment (2):
The testing per batch should not be an absolute requirement – instead this should be based on risk to the process and
to ensure that the testing encompass all batches impacted.
Comment (3):
Line 472: Glove testing using physical methods, performed at the beginning and end of each batch, is not the common
Industry practice. Frequency be based on a risk assessment.
The difference between mechanical and physical methods is not clear. Should three checks (visual, mechanical,
physical) be performed each time?
Allow to establish the frequency of glove testing based on a risk assessment.
Clarify the difference between mechanical and physical methods and if three checks (visual, mechanical, physical)
should be performed each time
Comment (4):
There are several designs for RABS and Isolators, and while integrity testing using mechanical, physical and visual
methods is appropriate, they should not be required at the same frequency. QRM should be used to justify the
frequency for each.
Proposed text change:
5.21 Glove systems, as well as other parts of an isolator, are constructed of various materials that can be prone to
puncture and leakage. The materials used shall be demonstrated to have good mechanical and chemical resistance.
Integrity testing of the barrier systems and leak testing of the isolator and the glove system should be performed using
visual, mechanical or physical methods. They should be performed at defined periods, as determined by a quality risk
assessment of the process. A visual check should be performed at the beginning and end of each batch, and following
any intervention that may affect the integrity of the unit.
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Comment (5):
Line 476: Integrity testing of the gloves systems should confirm the integrity of batches pre and post manufacture but
the frequency of the integrity testing should be established from operational performance. This may indicate that
testing at the beginning and end of each batch may not be required with the derived testing frequency utilised to
confirm the gloves systems integrity for a number of batches.
Proposed change:
Change ‘They should be performed at defined periods, at a minimum of the beginning and end of each batch, and
following any intervention that may affect the integrity of the unit’ to
497-499 Existing text:
5.24 Clean rooms and clean air devices should be qualified in accordance with Annex 15 of EU GMP. Reference for
the classification of the clean rooms and clean air devices can be found in the ISO 14644 series of standards.
Proposed text change:
5.24 Clean rooms and clean air devices should be qualified in accordance with Annex 15 of EU GMP. Reference for
the classification of the clean rooms and clean air devices can be found in the ISO 14644 series of standards. This
standard provides the basis for determining the minimum number of sampling locations (evenly distributed), the
minimum air sample size at each location, and the method for evaluating the data. It assumes simple areas without
complexities of processes, equipment, and personnel. In order to accommodate the infinite variety of operational
cleanrooms and clean zones, the standard allows additional risk-based locations to be added.
501 Comment (1):
5µm removed for classification purposes, but left in for monitoring. From a practical perspective, it would not seem
sensible to leave it out of classification activities when it is required for routine monitoring.
Comment (2):
Line 501 & 1653: For routine monitoring, airborne particle concentrations at both >0.5μm and >5μm particle sizes is
required. The ratios of ≥0.5μm and ≥5μm particles to microbe carrying particles (MCPs) have been established by
experiment and has demonstrated that if the requirement for ≥0.5μm particles and airborne microbial counts should be
complied with, the limit required for particles ≥5μm is too low and for each cleanroom grade, the limit for ≥5μm
particles should be increased by a factor of 10. Consequently, particle generating events in cleanroom areas will
produce particle counts that, although compliant with the class limits for ≥0.5μm particles and MCPs, may exceed the
class limit for ≥5μm particles. Therefore, for classification, airborne particle concentrations at both >0.5μm and >5μm
particle sizes should be undertaken.
Proposed change (if any):
If the requirement to monitor particles at >5µm is to remain (see 1653 and 1705), replace the existing with ‘For
classification, the airborne particles equal to or greater than 0.5µm and 5µm should be measured’
505 Comment (1):
Table 1: Maximum permitted airborne particle concentration during classification, indicates that classification should
be undertaken in the “in operation” state. Clarification is needed as classification is usually performed “at rest” and
monitoring is performed “in operation”
Comment (2):
Typo in Table I: The last column header should be “ISO classification at rest/in operation” instead of “ISO
classification in operation/at rest”
Comment (3):
The recommended maximum limits for particles is not harmonised with the limits defined in the reference document
EN ISO 14644-1(2015).
Proposed change:

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Harmonise the recommended maximum limits for particles defined in Table 1 with the limits defined in EN ISO
14644-1(2015)
.
510-517 Existing Text:
5.26 For initial classification the minimum number of sampling locations can be found in ISO 14644 Part 1. However,
a higher number of samples and sample volume is typically required for the aseptic processing room and the
immediately adjacent environment (grade A/B) to include consideration of all critical processing locations such as
point of fill stopper bowls. With the exception of the aseptic processing room, the sampling locations should be
distributed evenly throughout the area of the clean room. For later stages of qualification and classification, such as
performance qualification, locations should be based on a documented risk assessment and knowledge of the process
and operations to be performed in the area.
Proposed text change:
5.26 For basic cleanroom and clean zone classification, the minimum number of sampling locations can be found in
ISO 14644 Part 1. In Grade B aseptic processing rooms, and critical Grade A zones, a higher number of sampling
locations is typically required to assess critical processing locations. For Grade B aseptic processing rooms an
example of an additional sampling location is the region immediately adjacent to open door intervention access into
Grade A clean zones or RABS; and for the critical Grade A zone the point of loading stopper hoppers, where stoppers
or other sterilised components are exposed in feeding systems, the point of fill of containers, and stopper placement
station. For other Grades of cleanroom, the sampling locations should be distributed evenly throughout the area of the
cleanroom as described in ISO 14644-1. For later stages of qualification and classification, such as performance
qualification, locations should be based on a documented risk assessment and knowledge of the process and
operations to be performed in the area
522-524 Existing text:
b) The definition of "at rest" is the room complete with all HVAC systems, utilities functioning and with
manufacturing equipment installed as specified but without personnel in the facility and the manufacturing equipment
is static.
Comment:
Add the following, “The “at rest” state provides an important performance reference datum for cleanrooms and clean
air devices”.
Comment:
Line 522-527: Add definitions of at rest and in operation to glossary section.
533-535 Existing text:
e) The particle limits given in Table 1 above for the "at rest" state should be achieved after a "clean up" period on
completion of operations. The "clean up" period should be determined during the initial classification of the rooms.
Proposed text change:
e) The particle limits given in Table 1 above for the “at rest” state should be achieved after a “clean-up” period of 10-
15 minutes (guidance value) on completion of operations. The "clean-up" period should be determined during the
qualification of the cleanrooms, and is an essential mechanism for determining the ventilation effectiveness of the
cleanroom HVAC system. ISO 14644-3, ISO 14644-4 and ISO 14644-16 contain information, guidance and test
methods on air change rate effectiveness, recovery time and its measurement.
Comment (1):
Line 534: 5.26 e) The method for the determination of the “clean up” period should be indicated such as following
ISO 14644-3 B12.

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Comment (2):
The "clean up" period should be initially determined during the initial classification of the rooms and modified as
necessary under worst case conditions
Proposed change (if any): Include a definition of “clean up period” in the glossary.
Comment (3):
Line 533 & 1655: The most appropriate scientific method to determine the ‘clean up’ rate is defined in ISO 14644-3:
2005 as the ‘Recovery Test’ (section B12.3). Further detail on the correct application of this method for EU GMP
cleanrooms has been published, including measurement at the >0.5μm particle size. No time is specified in order to
achieve clean up and so poor ventilation effectiveness is acceptable as long as the poor results are repeatable. A
minimum time must be included in the ‘clean up’ test if the test has any relevance.
Proposed change:
Include ‘An appropriate test method can be found in ISO 14644 Part 3’. A guidance time similar to the existing annex
(15 to 20 minutes) to be added
537-538 Existing text:
f) In order to meet "in operation" conditions these areas should be designed to reach certain specified air-cleanliness
levels in the "at rest" occupancy state.
Comment:
[Note: This is an error! Just meeting the “at rest” level doesn’t mean that the “in operation” will be achieved. The
ability to meet the “in operation’ level of cleanliness is determined by the ability of the unidirectional or non-
unidirectional airflow system to displace or dilute the contamination source].
[Note could “at rest occupancy state” be defined? Is it the same as “at rest”, but with people present?]
Proposed text change:
f) In order to meet “in operation” conditions these areas should be designed to reach certain specified air-cleanliness
levels in the “at rest” occupancy state, and for the unidirectional or non-unidirectional airflow system to displace or
dilute the contamination source effectively.
540 Comment:
5.27 Should be “The microbial load of the clean rooms should be determined as part of the clean room qualification
considering both environmental and human origins of micro-organisms.
544 Existing text:
Table 2: Recommended limits for microbial contamination in operation.
Comment:
Replace “limits” with “levels”.
For Grade A the value should be <1 in the table for note (b) to be valid.
The table is reproduced, more comprehensively in section 9. Suggest having full table here. The table could then be
referenced in section 9. This would also negate the need for Lines 552 to 553.
Comment:
Table 2 and line 1746 Table 6. Action limits instead of recommended maximum limits and maintain <1 instead of 1
for grade A. Add swab to contact plates and cfu/plate or swab.
- It should be indicated that the viable monitoring incubation programme should be justified as for APS in lines 1928-
1930
Comment:
Only contact plates are mentioned for qualification of microbial contamination of surfaces. It is not clear how limits
should be calculated when swabs are used.
Comment: More clarification on Table II. Do we need settle plates in the Grade D?

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Proposed change (if any): ensure consistency between 5.27 and table II. Does this refer to qualification or in
operation?
546-549 Existing text:
(a) Individual settle plates may be exposed for less than 4 hours. Where settle plates are exposed for less than 4 hours
the limits in the table should still be used, no recalculation is necessary. Settle plates should be exposed for the
duration of critical operations and changed as required after 4 hours.
Comment:
Note (a) should only apply to Grade A plates.
Comment:
Sentence asks for 4 hours but for high air change systems, plate change may be less than 4 hours but the annex does
not address this.
548 &1751 Comment:
The principles section of the draft Annex 1 encourages companies to use modern methods and apply QRM principles.
It also states that for environmental monitoring ISO14644 should be looked to for method guidance. It is therefore
inconsistent to cite a single methodology as the only appropriate solution. Furthermore, the alternative methodologies
may not be able to express results in the exact same units as cited in the Annex.
Proposed Change:
Add a note to Tables 2 and 6 in the Annex to state that “Alternative methods for sampling are acceptable, as long as
validated for the intended use and appropriate limits can be applied”.
552 Comment:
For gown qualification is it A or B limits that should be applied
558-561 Existing text:
5.29 Clean rooms should be requalified periodically and after changes to equipment, facility or processes based on
the principles of QRM. For grade A and B zones, the maximum time interval for requalification is 6 months. For
grades C and D, the maximum time interval for requalification is 12 months.
Comment (1)
The scope of the requalification testing should be clearly defined. Does it refer to cleanroom and clean zone particle
counting or standard 6 or 12 monthly filter integrity checks, air velocities and pressure differential checks?
Note these intervals may be extended, subject to comprehensive risk assessment, when system performance and
environmental monitoring systems, and historical qualification data indicate frequencies are excessive.
Comment (2)
Cleanrooms should be operated and maintained relating to process and products’ risks; the design and operation of the
cleanrooms including e.g. HVAC, cleanliness grade, maximum occupancy states, cleaning/disinfection/sanitization
regimens etc. should be covered by the contamination control strategy. Since cleanrooms are under continuous control
with data obtained from environmental monitoring programs (viable and non-viable monitoring data) and
building/facility monitoring systems (e.g. differential pressures, temperature and humidity), reliable data is already
available to ensure proper function and compliance of the cleanrooms during their operational conditions. Any changes
to the cleanrooms and processes (as e.g. introduction of new equipment) would require a re-evaluation of the cleanroom
processes per the current Quality Risk Management systems, and most often include requalification activities. When
there are no changes to the qualified status and operations in a cleanroom, compilation and trending of data is sufficient
to detect issues related to the hygienic standards and no specific requalification should be required other than the regular
integrity testing of HEPA-filters.
Many cleanrooms are uninterrupted operational, including monitoring and control. Re-qualifying the cleanrooms in
accordance to the ISO standards ‘at rest’ conditions, would not only be providing no additional insight into the status
of the cleanroom, but also lead to not justified production interruptions.
Proposed text change:
Clean rooms should be requalified after changes to equipment, facility or processes, based upon the principles of QRM.
In addition, all data obtained from continuous and regular monitoring of manufacturing cleanrooms should be
periodically compiled and trended, on a minimum of an annual basis, to provide ongoing evidence of the intended
cleanliness status.
Comment (3)

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
This section is unclear as to what is meant by clean room requalification. The section deals with both HVAC
qualification as well as cleanroom classification and the requirement is located just below the area classification
section. Clarify the sentence to indicate that it is HVAC system requalification that is intended. As the frequency of
monitoring in grade A and B is high, an additional EMPQ every six months will not provide significant additional
data for the investment required. Furthermore, Annex 1 should avoid citing a specific frequency and rely on the ISO
standard as it has in several previous sections.
Proposed Change:
5.29 Clean rooms HVAC systems should be requalified periodically and after changes to equipment, facility or
processes based on the principles of QRM and ISO14644. The rationale for the frequency of requalification should be
documented.
Comment (4):
Line 558: The tests needed to qualify and requalify a cleanroom need to be specified. In addition to classification, the
minimum tests needed to ensure that a cleanroom or workstation functions correctly are the measurements of rate of air
supply or UDAF velocity, filter system integrity and pressure differentials which should be specified.
Proposed change (if any):
Add ‘as a minimum, the following tests should be carried out: particle concentration (classification), rate of air supply
or UDAF velocity, filter system integrity and pressure differentials.
Comment (5):
Line 559: The frequency of cleanroom requalification should be risk based. Where the installation is equipped with
instrumentation for continuous or frequent monitoring (e.g. airborne particle concentrations, unidirectional airflow
velocity, room air change rate, differential pressure etc.) and operational monitoring (e.g. microbial air sampling, settle
plates, surface sampling etc.) is completed and other key test parameters (e.g. air supply velocities, room air change
rate, extract velocities, airflow visualisation etc.) are frequently measured or verified by independent testing companies,
the maximum time intervals for requalification can be extended. Guidance on testing frequencies is available in BS EN
ISO 14644 Part 2.
Proposed change:
Change ‘For grade A and B zones, the maximum time interval for requalification is 6 months. For grades C and D, the
maximum time interval for requalification is 12 months’ to ‘The frequency of requalification should consider the level
of continuous or frequent monitoring, the level of operational monitoring and other scheduled measurement of key
contamination control parameters’
563-565 Existing text:
5.30 Other characteristics, such as temperature and relative humidity, depend on the product and nature of the
operations carried out. These parameters should not interfere with the defined cleanliness standard.
Comment:
Suggest that recommended ranges for temperature and humidity are given e.g. 19+/-2°C and 45 to 50% r.h.
567 Comment:
Change title of section to “Cleaning and Disinfection”.
569-578 Comment (1):
The proposed revision clarifies that a minimum of two agents are required, and that one needs to be a
sporicidal agent – original sentence could be misconstrued as indicating 2 disinfecting agents plus a third
sporicidal agent.
Proposed Change:
5.31 The disinfection of clean areas is particularly important. They should be cleaned and disinfected
thoroughly in accordance with a written programme (for disinfection to be effective, cleaning to remove
surface contamination must be performed first)., More than one type of disinfecting agent should be
employed, one of which being a sporicidal agent to be used periodically. Disinfectants should be shown to
be effective for the duration of their in-use shelf-life taking into consideration appropriate contact time and
the manner in and surfaces on which they are utilized. Monitoring should be undertaken regularly in order
to show the effectiveness of the disinfection program and to detect the development of resistant and/or
spore forming strains. Cleaning programs should be effective in the removal of disinfectant residues.
Comment (2)
Line 572: The need to utilise more than one type of disinfectant, if a sporicidal agent is periodically used, is dependent
upon the mode of action of the disinfectant. If the mode of action is completed by bacterial cell wall/membrane, and
not by passive diffusion, resistance is unlikely.

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Proposed change:
Change ‘More than one type of disinfecting agent should be employed’ to ‘depending on the mode of action of the
disinfectant agent, more than one type of disinfection agent should be considered’
Comment (3):
Line 575: Organisms do not develop resistance to disinfectants like they do to antibiotics. The application and
chemical agent may not be effective vs this spore former.
Proposed change (if any): Recommend change “…development of resistant…” to “…Detect the appearance of new or
spore forming strains”
580 Comment:
Clarify if it is intended only for in house dilutions, not for ready to use disinfectants with a defined expiration date.
Recommended change: “Non-sterile Disinfectants and detergents used for C and D areas should be monitored for
microbial contamination…An in- use period should be determined.”
585-586 Existing text
5.33 Disinfectants should be shown to be effective when used on the specific facilities, equipment and processes that
they are used in.
Comment:
More detail on regulatory expectation is required here.
588-589 Existing text:
5.34 Fumigation or vapour disinfection of clean areas such as Vapour Hydrogen Peroxide (VHP) may be useful for
reducing microbiological contamination in inaccessible places.
Comment:
This text seems to underplay the importance of these agents in the validated, automated decontamination of properly
prepared, cleaned Grade A Isolator environments. The text may prompt readers to abandon this type of proven
technology and adopt manual processes which by their nature cannot be reliably validated. Also VHP is described as
useful for reducing microbiological contamination in accessible places - need clarification as later in the annex VHP
is described as bio-decontaminant (Conflicting definitions) bio-decontamination not referenced in the glossary
6 Equipment
General Comment:
Consider integrating equipment from other sections into this.
General improvements and clarification are required in this section.
Many will believe this relates to unitary equipment, when actually it related also to bespoke process systems
592 Comment:
The following should be added:- “In this section, the term equipment is intended to include both items of unitary
equipment, pre-engineered skid mounted systems, and in-situ assembled and build processing systems”.
593-595 Existing text:
6.1 A written, detailed description of the equipment design should be produced (including diagrams as appropriate)
and kept up to date. It should describe the product and other critical gas and fluid pathways and controls in place
Proposed text change:
6.1 A written, detailed description of the equipment design shall be produced (including diagrams as appropriate) and
kept up to date throughout the life-cycle of the equipment. It should describe the product and other critical gas and
fluid pathways and their controls.
597-598 Existing text:
6.2 Equipment monitoring requirements should be determined during qualification. Process alarm events should be
reviewed and approved and evaluated for trends
Proposed text change:
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
6.2 Equipment monitoring requirements should be defined during the design, and the operation and performance
confirmed during qualification. Process alarm events should be reviewed, and appropriate actions taken. They should
also be evaluated for trends.
600-604 Existing text:
6.3 As far as practicable equipment, fittings and services should be designed and installed so that operations,
maintenance, and repairs can be carried out outside the clean area, if maintenance has to be performed in the clean
area then precautions such as additional disinfection and additional environmental monitoring should be considered.
If sterilization is required, it should be carried out, wherever possible, after complete reassembly.
Proposed text change:
6.3 As far as practicable equipment, fittings and services should be designed and installed so that operations,
maintenance, and repairs can be carried out outside the clean area, if maintenance has to be performed in the clean
area then precautions such as issuing permits to work, clearly defined method statements, additional cleaning and
disinfection, and additional environmental monitoring should be considered. If sterilization or bio-decontamination is
required, it should be carried out, wherever possible, after complete reassembly of the equipment.
613-614 Existing text:
a) Can remove any residues that would otherwise create a barrier between the sterilizing agent and the equipment
surfaces.
Proposed text change:
a) Can remove any residues that would otherwise create a barrier between the disinfecting, bio-decontamination or
sterilizing agent and the product contact equipment surfaces.
616-617 Existing text:
b) Prevents chemical and particulate contamination of the product during the process and prior to disinfection.
Proposed text change:
c) Prevents chemical and particulate contamination of the product during the process and prior to disinfection
or bio-decontamination.
Comment:
Definition required for particulate contamination.
619 Existing text:
6.6 All critical surfaces that come into direct contact with sterile materials should be sterile.
Comment:
Change “should” for “must”.
621-623 Existing text:
6.7 All equipment such as sterilizers, air handling and filtration systems, water treatment, generation, storage and
distribution systems should be subject to qualification, monitoring and planned maintenance; their return to use
should be approved.
Proposed text change:
6.7 All equipment such as sterilizers; air handling systems (including air filtration); water treatment systems
(including water purification, storage and distribution elements) should be subject to qualification, monitoring and
planned maintenance. Upon completion of maintenance, their return to use shall be approved
Comment:
Line 621: Qualification is a typo.
There may be ambiguity on who will be approving the return to use. Approval should be by the Quality Unit.
Proposed change (if any): Correct typo.
Recommended change: “their return to use should be approved by the Quality Unit.” .
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
625-627 Existing text:
6.8 A conveyor belt should not pass through a partition between a grade A or B area and a processing area of lower
air cleanliness, unless the belt itself is continually sterilized (e.g. in a sterilizing tunnel).
Proposed text change
6.8 A conveyor belt must not pass through a partition between a Grade A clean zone or a Grade B cleanroom and a
processing area of lower air cleanliness, unless the belt itself is continually sterilized (e.g. in a sterilizing tunnel).
629-631 Existing text:
6.9 Particle counters should be qualified (including sampling tubing). Portable particle counters with a short length
of sample tubing should be used for qualification purposes. Isokinetic sample heads shall be used in unidirectional
airflow systems.
Comment:
Line 629: Portable particle counters is a new requirement. What is the definition of “short” length of tubing?
Proposed change (if any): delete ‘should’ and replace by ‘can’ in line 630
Proposed text change
6.9 Real-time particle monitoring systems using light scattering airborne particle counters (LSAPC) should be
qualified (including the sampling tubing). Portable particle counters with a short length (NMT 3 metres) of sample
tubing should be used for qualification purposes. Isokinetic sample heads shall be used in unidirectional airflow
systems. ISO 14644-2 provides guidance on real-time airborne particle monitoring.
633-635 Existing text:
6.10 Where unplanned maintenance of equipment critical to the sterility of the product is to be carried out, an
assessment of the potential impact to the sterility of the product should be performed and recorded.
Proposed text change:
6.10 Where unplanned maintenance of equipment critical to the sterility of the product is to be carried out, an
assessment of the potential impact to the sterility of the product should be performed, recorded, and evaluated as part
of the product release.
Section 7 Utilities
General Comment:
This section requires improvement, editing, and correction of errors and poor practice.
If this section is intended to replace the EMA non-Distillation WFI Q&A Paper, it is inadequate. If this paper is to be
retained, then some of the guidance is useful. The comments below are made assuming the Q&A will still apply to
EMA jurisdictions.
The WFI source water requirements are incorrect. They are dependent upon the production method.
The WFI sampling guidance is impractical, and doesn’t follow current practice.
639-640 Existing text:
7.1 The nature and amount of controls associated with utilities should be commensurate with the risk associated with
the utility determined via risk assessment.
Proposed text change
7.1 The nature and extent of controls applied to utility systems should be commensurate with the risk to product
quality associated with the utility. The impact should be determined via a documented risk assessment.
644 Existing text:
a) Directly contact product e.g. compressed gases.
Proposed text change

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
a) Directly contact product or product contact surfaces. e.g. water for washing and rinsing, compressed gases,
and steam for sterilisation.
646 Existing text:
a) Contact materials that ultimately will become part of the product.
Proposed text change
b) Materials that ultimately will become part of the product.
648 Comment:
Omit line 648 as this is covered in line 644 with text change (see above)
652-653 Existing text:
7.3 Utilities should be installed, operated and maintained in a manner to ensure the utility functions as expected.
Proposed text change
7.3 Utilities should be installed, operated, monitored and maintained in a manner so as to ensure the utility functions
as expected.
655-656 Existing text:
7.4 Results for critical parameters of the high risk utility should be subject to regular trend analysis to ensure that
system capabilities remain appropriate.
Proposed text change
7.4 Critical performance parameters of direct impact critical utilities should be monitored, and subject to regular trend
analysis to ensure that system capabilities remain appropriate.
Comment
“regular” requires definition.
658-660 Existing text:
7.5 Current drawings should be available that identify critical system attributes such as: pipeline flow, pipeline
slopes, pipeline diameter and length, tanks, valves, filters, drains and sampling points.
Proposed text change
7.5 Records of the utility installation shall be maintained throughout the system life-cycle. Such records should
include current drawings and schematic diagrams, component lists and specifications. Typical important information
includes attributes such as: pipeline flow direction, slopes, diameter and length; tank and vessel details; and valves,
filters, drains and sampling points.
Water Systems
General Comment:
The requirements for water systems are complex and detailed descriptions should be removed and reference to the
European Pharmacopoeia, or other appropriate documentation, for water systems should instead be included.
Proposed change:
Remove the requirements for water systems and refer to the European Pharmacopoeia or other specialist
documentation
670 Comment:
Replace “relevant Pharmacopoeia” with “European Pharmacopoeia”
672-676 Existing text:
7.8 Water for injections (WFI) should be produced from purified water, stored and distributed in a manner which
prevents microbial growth, for example by constant circulation at a temperature above 70oC. Where the WFI is
produced by methods other than distillation further techniques post Reverse osmosis (RO) membrane should be
considered such as nanofiltration, and ultra-filtration.
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Proposed text change
7.8 When Water for Injection (s) (WFI) is produced using distillation as the final purification step, the feed water to
the still shall be appropriate for the distillation method used. When multi-effect distillation is used, the water quality is
commonly purified water, and when vapour compression distillation is used, deionised water is suitable. Once
generated, WFI shall be stored and distributed in a manner which prevents microbial growth, for example by constant
circulation at a temperature above 70°C. When WFI is produced by non-distillation methods, the pharmacopoeias
define the required feed water quality, and provide guidance on the preferred sequence of purification steps post
reverse osmosis membrane (RO), including deionisation and ultra-filtration.
Comment:
Line 672: Specifically calls for WFI to be produced by Purified Water (i.e. final processing using PW as feed-water).
This is too restrictive.
Proposed change (if any): Suggest that it does not specify the source of the water, providing that the WFI
requirements are met once WFI is produced.
678-679 Existing text:
7.9 Water systems should be validated to maintain the appropriate levels of physical, chemical and microbial control,
taking seasonal variation into account.
Proposed text:
7.9 Water systems should be validated to demonstrate the ability to maintain the appropriate levels of physical,
chemical and microbial control, taking into account seasonal variations in raw water quality and environmental
conditions.
681 Existing text:
7.10 Water flow should remain turbulent through the pipes to prevent microbial adhesion.
Comment:
No requirement to measure flow velocity?
683-686 Existing text:
7.11 The water system should be configured to prevent the proliferation of microorganisms, e.g. sloping of piping to
provide complete drainage and the avoidance of dead legs. Where filters are included in the system, special attention
should be taken with regards to the monitoring and maintenance of these filters.
Proposed text:
7.11 The water system should be configured to prevent the proliferation of microorganisms. Techniques that should be
considered include maintenance of turbulent flow in pipelines to inhibit proliferation of microorganisms, avoidance of
dead-legs, sloping of piping to allow drainage of systems. Where filters are included in the system, special attention
should be taken with regards to the monitoring and maintenance of these filters. Sterilising grade, dead-end filters
shall not be used in purification and distribution systems as they can conceal systemic microbiological contamination.
688-689 Existing text:
7.12 Where WFI storage tanks are equipped with hydrophobic bacteria retentive vent filters the filters should be
sterilized, and the integrity of the filter tested before and after use.
Comment (1):
Should the filter be sanitised and heated?
Comment (2):
Even if generally free from microorganisms, water for injection (WFI) is not expected to be a sterile media since for
example, the European Pharmacopoeia requirements for this water quality is to contain less than 10 CFU/100ml for
microbial count, and bacterial endotoxin levels less than 0.25 IU/ml. The storage tanks, distribution systems (water
piping, loops) and points of use (taps or connections to equipment) for the WFI need to be designed, maintained,
controlled and monitored relating to the requirements for the manufacturing operations – and storage tank filters for
WFI would not need mandatory sterilization and/or integrity testing. The benefit therefore is not clear for the handling

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
of vent filters for a non-sterile medium that is continuously prepared and distributed with the same requirements as
sterilizing filters for aseptically produced product.
Proposed text change: Where WFI storage tanks are equipped with hydrophobic bacteria retentive vent filters the
filters may be sterilized, and the integrity of the filter tested before and after use, based upon Quality Risk
Management principles.
Comment (3):
Does this apply to water other than WFI?
Proposed change (if any): Applicable to both Purified Water and WFI systems regarding vent filter types.
Comment (4):
For water systems that are continuously maintained at >70oC, and with a high frequency of sampling (as indicated by
the Annex), it is unnecessary to sterilize and routinely integrity test vent filters. There are several other much more
effective and real time measures to ensure proper operation of the system (such as temperature, pressure, flow rate,
TOC and conductivity measurements of the loop), along with the routine microbial monitoring. Integrity testing
should be performed at installation and prior to removal but this was unclear in the draft Annex.
Proposed Change:
7.12 Where WFI storage tanks are equipped with hydrophobic bacteria retentive vent filters the filters should be
integrity tested after installation and prior to removal.
691-696 Existing text:
7.13 To prevent the formation of biofilms, sterilization or disinfection or regeneration of water systems should be
carried out according to a predetermined schedule and also when microbial counts exceed action and alert limits.
Disinfection of a water system with chemicals should be followed by a validated rinsing procedure. Water should be
analyzed after disinfection/regeneration; results should be approved before the start of use of the water system.
Proposed text change
7.13 Biofilms can form in ambient temperature water purification trains, and storage and distribution systems. To
prevent the formation of biofilms, sanitisation, disinfection or regeneration of water systems should be carried out
according to a predetermined schedule, and also when microbial counts exceed action levels. Disinfection of a water
system with chemicals should be followed by a validated rinsing or inactivation procedure. Water should be
monitored after disinfection, and results should be approved before the start of use of the water system.
Comment (1):
Some companies believe it is not possible to prevent the formation of biofilms and regeneration of water system
should only be undertaken in extreme cases.
Comment (2):
(Relating to Lines 691-693):
The operation of manufacturing water systems should be covered by the contamination control strategy. Since the water
systems are under continuous control related to data obtained from e.g. bioburden and bacterial endotoxin testing and
commonly in-line monitoring systems (e.g. TOC and conductivity); reliable data is available to ensure proper function
of the water systems during their operational conditions. When there are no changes to the qualified status and operations
of the water system, compilation and trending of data is sufficient to detect issues related to the hygienic standards.
Single events (i.e. exceeded alert or action limits) should not automatically render sterilization, disinfection or
regeneration of the full water system since the excursion could relate to a single point of use and not be a systemic issue
in the water system. The actions taken upon single exceeded limits from water samples should rather correlate to the
process risks, and risks of usage of water, in the manufacturing processes. Also, considerations should be given that hot
WFI water loops as such are having a self-sanitizing effect, making a pre- determined schedule for disinfection obsolete.
Proposed text change:
To prevent the formation of biofilms, sterilization or disinfection or regeneration of water systems should be carried
out based upon Quality Risk Management principles or when microbial counts or other monitoring data show a
negative trend.
Comment (3):
Line 691: Why exceeding an alert limit should require a sanitization/sterilization of the system?

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Add “routine flushing” to prevent biofilms.
Add “repeated alert limits excursions”
Proposed change (if any): Replace exceed action and alert with trending data
Comment (4)
Each of the techniques described in this section describes a way to treat biofilms when they occur. Heat is very
effective at killing biofilms, but sterilization is not required. Hot water sanitization above 65C is effective to kill
biofilms. More extreme actions such as sterilization or chemical disinfection should only be taken for-cause if
indicated by routine monitoring data and not on a routine schedule.
Proposed Change:
Replace entire section 7.13 text. Proposed wording: Periodic sanitization (thermal or chemical) of water systems
should be carried out to mitigate the formation of biofilms if the system is not kept in a continuously sanitizing
condition (temperature greater than 65oC). Other actions, such as disinfection of distribution piping or re-generation,
may also be used in response to adverse trends in the routine monitoring data. The use of chemicals for this purpose
should be followed by sampling or the use of online monitoring to ensure their removal before the start of use of the
water system
698-699 Existing text:
7.14 A suitable sampling schedule should be in place to ensure that representative water samples are obtained for
analysis on a regular basis.
Comment:
Sampling schedule should in place for both on-line and off-line testing.
Regulatory expectation for sampling schedules for purified and WFI water systems would be helpful here.
701-708 Existing text:
7.15 Regular ongoing chemical and microbial monitoring of water systems should be performed with alert limits
based on the qualification that will identify an adverse trend in the performance of the systems. Sampling should
include all outlets and user points at a specified interval. A sample from the worst case sample point, e.g. the end of
the distribution loop return, should be included each time the water is used for manufacturing and manufacturing
processes. A breach of an alert limit should trigger review and follow-up, which might include investigation and
corrective action. Any breach of an action limit should lead to a root cause investigation and risk assessment.
Comment (1):
(Lines 701-706)
The operation of manufacturing water systems should be covered by the contamination control strategy. In the strategy,
the reasoning behind sampling locations and frequencies should be established based on Quality Risk Management
principles, taking into account as a minimum, the water quality, the water system design and the use of the water in the
manufacturing process and/or in product preparation such as monitored during the water system qualification. The
requirement to sample at worst case locations at one part of a loop, whenever water is used for manufacturing processes
at other parts of the loop, needs to be clarified. On bigger WFI loop systems this sample point might be several 100metres
after the point of use, not providing any insight for the quality of water taken for the manufacturing operations.
Proposed text change (1):
Regular ongoing chemical and microbial monitoring of water systems should be performed with alert limits, sample
locations and sampling frequencies based upon the qualification that will identify an adverse trend in the performance
of the systems. Sampling should include all outlets and user points at a specified interval. A sample from the worst
case sample point, e.g. the end of the distribution loop return, should be included on a daily basis.
Alternative Proposed Text change (2):
(Lines 701-708)
7.15 Regular on-going chemical and microbial monitoring of water systems should be performed with alert levels
based on the qualification that will identify an adverse trend in the performance of the systems before action levels are
reached. Sampling should include all sample points and points-of-use at specified intervals. For WFI systems, at least
one point-of-use and the end of the distribution loop (prior to re-entry into the distribution tank) should be sampled
daily, ensuring all are sampled over a defined period of time. A breach of an alert level should initiate a review and

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
follow-up, which might include investigation and corrective action. Any breach of an action level should lead to a root
cause investigation and risk assessment.
Comment (3):
Specific sampling guidance is now included such as requiring sampling from a worst case point on the water system
each time it is used for manufacturing purposes (an example given is the return distribution header from the system).
Action limit excursions should include an assessment on the risks posed to processes that used the water.
More clarification is needed to describe the purpose of a risk assessment when an action level is reached such as
impact on process/products that used the water.
Comment (4):
The term ‘points of use’ should be used in lieu of ‘outlets’. Water systems typically have quite a number of other
“outlets” in technical areas that are used for complete system drainage or other non-processing reasons, and these
“outlets” do not typically require sampling. Also, the end of the loop may not be the worst case location. A complex
point of use will often be a worse condition than the loop return in the case of microbiological contaminants, which
are unlikely to be uniformly distributed. Clarification or rewording should also be provided about the intent of
sampling this worst case point “each time the water is used” Additionally, the terms alert and action limits are used in
this section, but the terms alert and action levels are defined in the glossary. The requirements for breach of an action
limit/level in this section are different than those imposed by the definition n lines 2023-2024 – they should be
aligned. Alert levels are initially calculated based on the qualification, but should be revised based on more recent
ongoing monitoring data. Clarification between risk assessment (done prospectively) and impact assessment
(performed after an event in order to determine impact).
Proposed Change:
7.15 Regular ongoing chemical and microbial monitoring of water systems should be performed with alert levels
based on historical results such that an adverse trend in the performance of the system would be identified. Sampling
should include all points of use at a specified interval. Risk assessment should be used to determine potential worst
case sampling locations and an appropriate sampling frequency. An exceeded alert level should trigger review and
follow-up, which might include investigation and corrective action. Any exceeded action level should lead to a root
cause investigation and impact assessment.
710-711 Existing text:
7.16 WFI systems should include continuous monitoring systems such as Total Organic Carbon (TOC) and
conductivity.
Proposed text change:
7.16 WFI systems shall be continuously monitored on-line for Total Organic Carbon (TOC), conductivity,
temperature and flow rate
Comment (1):
Continuous monitoring of TOC is a new requirement.
Interestingly only specifies WFI grade water for continuous TOC monitoring with a preference to online over offline
monitoring; other similar guidance documents such as ISPE Baseline Guide for Water & Pure/Clean Steam and USP
General Chapter <1231> still allow for either offline or online monitoring of these properties.
Proposed alternative suggestion: Offline monitoring for TOC is still an acceptable monitoring practice.
Comment (2):
The Annex should not mandate on-line continuous monitoring for TOC and Conductivity as it is not scientifically
necessary. While desirable for any newly installed systems, retrofitting of older systems would involve a very invasive
process, with high cost and high complexity and can lead to inadvertent contamination of the systems.
Proposed change:
Remove Section 7.16
Steam used for sterilization
715-716 Existing text:
7.17 Purified water, with a low level of endotoxin, should be used as the minimum quality feed water for the pure
steam generator.
Proposed text change (1):

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
7.17 Purified water, with a low level of endotoxin, shall be used as the minimum quality feed water for a pure steam
generator equipped with droplet and particle elimination. If a simple heat exchanger is used, the feed water shall be
WFI.
Comment (1):
Regulatory guidance on the expectation for low level of endotoxin would be helpful.
Comment (2):
Feed water for a pure steam generator could be derived from water that is of different quality than purified water, e.g.
from water treated with reverse osmosis. Also, the particular mention of “low level of endotoxin” does not seem to be
justified since the steam generators as such significantly reduces endotoxin levels. The quality of feed water should be
related to the performance of clean steam generator.
Proposed text change (2):
Suitably treated water, should be used as the minimum quality feed water for the pure steam generator.”
Comment (3):
Clarify what low endotoxin in feed water for pure steam generation is.
Proposed change (if any): Clarify what low endotoxin in feed water for pure steam generation is or suggest the
following wording “Pure Steam Generation systems using purified water should be qualified using water of defined
endotoxin content to ensure that the pure steam generated does not add endotoxin risk”.
Comment (4):
The statement around low level of endotoxin is subjective, and does not conform to the European Pharmacopeial
specification for Purified Water. Furthermore, well designed clean steam generators do not have an issue in handling
removal of endotoxins.
Proposed Change:
7.17 Purified water should be used as the minimum quality feed water for the pure steam generator.
718-723 Existing text
7.18 Steam used for sterilization processes should be of suitable quality and should not contain additives at a level
which could cause contamination of product or equipment. The quality of steam used for sterilization of porous loads
and for Steam-In-Place (SIP) should be assessed periodically against validated parameters. These parameters should
include consideration of the following examples: non-condensable gases, dryness value (dryness fraction), superheat
and steam condensate quality.
Proposed text change:
7.18 Steam used for sterilization processes shall be of suitable quality and should not contain additives at a level
which could cause contamination of product or equipment. When condensed, the condensate shall meet the WFI
specification. Additionally, the quality of steam used for sterilization of porous loads and for Steam-In-Place (SIP)
should be assessed periodically and whenever there is an interruption in the steam supply, against validated
parameters. These parameters include non-condensable gases, dryness value (dryness fraction), and superheat.
Reference EN285.
Comment:
Added clarification for testing the physical attributes of steam as noted within EN 285 (as well as HTM-0101). Need
clarification of what steam condensate quality is.
Proposed change (if any): Clarify that steam condensate must meet WFI compendial requirements for chemical and
endotoxin quality and should be free of microbial contamination
Comment:
Line 720: Is this necessary also for SIP systems?
Line 723: Need to clarify the expectation of steam condensate quality.
Compressed gases and vacuum systems

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
727-733 Existing text
7.19 Compressed gases that come in direct contact with the product/container primary surfaces should be of
appropriate chemical, particulate and microbiological purity, free from oil with the correct dew point specification
and, where applicable, comply with appropriate pharmacopoeial monographs. Compressed gases must be filtered
through a sterilizing filter (with a nominal pore size of a maximum of 0.22pm) at the point of use. Where used for
aseptic manufacturing, confirmation of the integrity of the final sterilization gas filter should be considered as part of
the batch release process.
Proposed text change
7.19 Compressed gases that come in direct contact with the product, product contact surfaces, or product primary
containers shall be of appropriate chemical, particulate and microbiological purity, free from oil and hydrocarbons,
with an appropriate dew point specification, and comply with appropriate pharmacopoeial monographs where
applicable. Compressed gases must be filtered through a sterilizing filter (with a nominal pore size of a maximum of
0.2μm) at the point of use. Where used for aseptic processing, confirmation of the integrity of the final sterilization
gas filter should be considered as part of the product batch release process.
Comment:
The frequency for testing the integrity of gas filter should be based upon risk assessment.
Comment:
Line 731-733: Should integrity test of gas filters be performed on a per batch basis or is a periodic testing based on
RA acceptable? If integrity testing of the gas filter is performed on a defined frequency and not by batch, what would
the batch release process requirement be?
735-736 Existing text:
7.20 There should be prevention of backflow when any vacuum or pressure system is shut off.
Proposed text change
7.20 There should be prevention of backflow when any vacuum or pressure system is shut off or activated.
738 Cooling Systems
Comment:
Change title of section to Energy utility systems
740-743 Existing text:
7.21 Major items of equipment associated with hydraulic and cooling systems should, where possible, be located
outside the filling room. Where they are located inside the filling room there should be appropriate controls to
contain any spillage and/or cross contamination associated with the hydraulics of cooling system fluids.
Proposed text change:
7.21 Major items of equipment such as those associated with vacuum, hydraulic, cooling and heating systems shall be
located outside the filling room. Where components of these systems are located inside the filling room, there should
be appropriate controls to contain any spillage and prevent cross-contamination.
745-746 Existing text:
7.22 Any leaks from the cooling system must be detectable (i.e. an indication system for leakage). In addition, there
must be adequate cooling flow within the system.
Proposed text change
7.22 Any leaks from the energy utility system must be detectable (i.e. an indication system for leakage). In addition,
there must be adequate cooling flow within the system.
748-749 Existing text:
7.23 The cooling circuit should be subject to leak testing both periodically and following any maintenance.
Proposed text change

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
7.23 Cooling and heating circuits should be subject to leak testing both periodically and following any maintenance.
751-752 Existing text:
7.24 There should be periodic cleaning/disinfection of both the vacuum system and cooling systems
Proposed text
7.24 There should be periodic cleaning/disinfection of vacuum systems.
Comment:
Add periodic “preventive maintenance”
Proposed change (if any): Cooling systems should be included in PM program.
There should be periodic preventive maintenance and cleaning/disinfection of both the vacuum system and cooling
systems.
754 8 Production and Specific Technologies
General Comment (1):
Consider moving specific equipment guidance to Section 6 and create an additional section on sterilisation. Just keep
technologies here. Many improvements to the language and clarity must be made, particularly for non-native English
speakers. Reference to relevant EN and ISO sterilisation standards must be made to ensure readers clearly understand
regulatory expectations. Some of the sterilisation lecture material is repetitive, and should be re-organised or removed.
General Comment (2):
There is no mention of vaporized hydrogen peroxide (VHP) as a sterilitant (e.g of certain isolator critical surfaces).
This is inconsistent with the position of the European Pharmacopeia. 5.1.1 Methods of Preparation of Sterile
Products, section on Gas Sterilisation (Vapor Phase Sterilisation)
Proposed Change:
Text should be added to define an acceptable approach for the use of VHP as a sterilant.
766-767 Existing text:
8.2 Filling of products for terminal sterilization should be carried out in at least a grade C environment.
Proposed text change
8.2 Filling of products for terminal sterilization should be carried out in at least a Grade C environment, with a local
Grade A air supply protecting the processing zone
769-775 Comment:
This paragraph is not required following the proposed change above for line 766 to 767.
Comment (2)
The meaning of “wide necked” is not clear. Vials are wide necked compared with ampoules. Should filling of vials
require a Grade A environment?
777-778 Existing text:
8.4 Processing of the bulk solution should include a filtration step to reduce bioburden levels and particulates prior to
filling into the final product containers.
Comment:
Process design should be handled and maintained in relation to process and product risks; the design and operations
(including any filtration steps) of processes should be based on Quality Risk Management principles and the
requirements for the specific product. Some bulk solutions as e.g. fat emulsions or suspensions cannot be filtered through
bacterial retention filters due to the physical product characteristics. Bioburden levels must be thoroughly controlled
during the process since bioburden levels cannot be reduced with the applied 10 – 20 µm pore size filters typically used
for those types of emulsion products.
Proposed text change (1)
8.4 Processing of the bulk solution should include a filtration step, employing a 0.2 micron filter where possible, to
reduce bioburden levels and particulates prior to filling into the final product containers.
Alternative text change (2)

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Processing of the bulk solution should include a filtration step to reduce bioburden levels and particulates prior to filling
into the final product containers, wherever product characteristics allow.”
785 Aseptic Preparation
787-790 Existing text:
8.6 Aseptic processing is the handling of sterile product, containers and/or devices in a controlled environment, in
which the air supply, materials and personnel are regulated to prevent microbial contamination. Additional
requirements apply to Restricted Access Barrier Systems (RABS) and isolators (refer clauses 5.15-5.22).
Proposed text change
8.6 Aseptic processing is the handling of sterile product, containers and/or devices in a controlled environment, in
which the air supply, materials and personnel are regulated to prevent microbial contamination. By first intent barrier
technology such as Isolators and Restricted Access Barrier Systems (RABS) should be employed to ensure adequate
separation of the Grade A zone from both operators and the surrounding environment. This is to reduce the need for
interventions into the grade A environment and to minimize the risk of contamination. Automation of processes
should also be considered to remove the risk of contamination by interventions (e.g. dry heat tunnel, automated
lyophilizer loading, SIP). The absence of such technology may be acceptable in exceptional circumstances provided
there is sufficient control and evidence of the maintenance of Grade A integrity.
Comment:
Note this paragraph has also been included by PHSS in Section 5 Premises under Barrier Technologies
792-797 Comment:
-Change “justifed” to “mitigated and not result in unnecessary risk to product/process’.
- Requirement for discrete aseptic process risk assessment as part of contamination control strategy
Proposed change (if any): Residual risk cannot pose unnecessary risk. Residual risks should be mitigated and
supported by a risk assessment.
805-809 Comment:
This paragraph is no longer required following proposed text change to line 787 to 790.
815 Comment (1):
The table should be modified to include under Grade A, “ Removal and cooling of unwrapped items from steriliser.
The removal of wrapped items from the sterilizer should be protected by a local Grade A air supply within the Grade
B area. There should also be a provision for a protective local Grade A air supply within the Grade D cleanroom for
the handling of components, equipment and accessories after washing prior to being sterilised.
Comment (2):
The current wording in the draft Annex 1 is (on examples of operations to be performed in grade B) “Removal of sealed
product from the grade A zone.” The mentioned example is not a good one. When product has been sealed in grade A
it can be automatically transferred under clean air (i.e. grade A air supply) into different cleanroom zones, as e.g. a grade
C zone. The product in its’ sealed container would be of no risk of being contaminated by this process design and
cleanroom grades.
Suggest that the text in Table 4: “Removal of sealed product from the Grade A zone” is omitted.
Comment (3)
Table 4 section B “Removal of sealed product from the grade A zone” is not clear since it could mean that for a vial
after capping a grade B area is required for the removal of the sealed vial.
Comment (4):
Grade A should not be required for packaged items (see also Grade B example of operations in the same table and
clause 8.41)
Proposed change (if any): Change the sentence as follows: “Grade A: Removal and cooling of unprotected (e.g.
unpackaged) items from heat sterilizers”.

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Comment (5):
Additional clarification requested to the examples in Table 4 as several of the examples can be present in several
configurations, not all of which place them in the Grade A classification category and thus can lead to confusion and
misinterpretation. Also suggest moving the requirements around aseptic connections (should be sterilized by steam in
place whenever feasible) to a more prominent section of the document. Having it placed within a table of examples
means it can easily be missed or disregarded.
Proposed change:
Table 4: Examples of operations and which grades they should be performed in
A Critical processing zone.
Aseptic assembly of filling equipment.
Aseptic connections
Aseptic compounding and mixing.
Replenishment of sterile product, containers and closures.
Removal and cooling of items from heat sterilizers (unless appropriately protected).
Staging and conveying of sterile primary packaging components (unless appropriately protected).
Aseptic filling, sealing, transfer of open or partially stoppered vials,
including interventions.
Loading and unloading (of unsealed containers) of a lyophilizer
Comment (6):
(see also 331 & 839) A Grade A environment is specified for the removal and cooling of items from heat sterilisers
and for the loading and unloading of a lyophiliser. These activities can be adequately undertaken in zones supplied
with unidirectional airflow where personnel may be present. Regulatory authority guidance has previously stated that
the zone cannot be considered to be Grade A if personnel are present within the zone.
Proposed Change:
Clarification is needed on terminology, e.g. Grade A zones that operators enter to be termed localised unidirectional
airflow (L-UDAF) or appropriate similar term
Comment (7):
Lines 839/1479 (see also 331 & 815) The document states that the transfer of partially closed containers to a
lyophilizer should be done under Grade A conditions and then clarifies this environment to be HEPA filtered positive
pressure. This activity can be adequately undertaken in zones supplied with unidirectional airflow where personnel
may be present. Regulatory authority guidance has previously stated that the zone cannot be considered to be Grade A
if personnel are present within the zone.
Proposed Change:
Clarification is needed on terminology, e.g. Grade A zones that operators enter to be termed localised unidirectional
airflow (L-UDAF) or appropriate similar term
817-819 Existing text:
Note; If Isolators are used then a risk assessment should determine the necessary background environment grade; at
least a minimum of grade D should be used. Refer clauses 5.19-5.20.
Comment:
This may indicate to the reader that Isolators are a less secure option than open processing.
821-828 Existing text:
8.11 Where the product is not subsequently sterile filtered, the preparation of equipment, components and ancillary
items and products should be done in a grade A environment with a grade B background.
8.12 Preparation and filling of sterile products such as ointments, creams, suspensions and emulsions should be
performed in a grade A environment, with a grade B background, when the product and components are exposed and
the product is not subsequently filtered or sterilized.
Proposed text change

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Where the product is not subsequently sterile filtered, the preparation of equipment, components and ancillary items
and products should be undertaken in a Grade A environment with a Grade B background, unless Isolator technology
is used. This includes the preparation and filling of sterile products such as ointments, creams, suspensions and
emulsions should be performed in a Grade A environment, with a Grade B background, when the product and
components are exposed and the product is not subsequently filtered or sterilized.
839-843 Existing text:
8.14 The transfer of partially closed containers to a lyophilizer, should be done under grade A conditions (e.g. HEPA
filtered positive pressure) at all times and, where possible, without operator intervention. Portable transfer systems
(e.g. transfer carts, portable Laminar Flow Work Stations, etc.) should ensure that the integrity of transfer system is
maintained and the process of transfer should minimize the risk of contamination.
Proposed text change
8.14 The transfer of partially closed containers to a lyophilizer, shall be undertaken under Grade A conditions at all
times, without direct operator intervention in order to minimise the risk of contamination. Technologies such as
conveyor systems, portable transfer systems (e.g. clean air transfer carts, portable unidirectional airflow workstations
should ensure that the integrity of transfer system is maintained.
845-850 Existing text:
8.15 Aseptic manipulations (including non-intrinsic aseptic connections) should be minimized using engineering
solutions such as the use of preassembled and sterilized equipment. Whenever feasible, product contact piping and
equipment should be preassembled, then cleaned and sterilized in place. The final sterile filtration should be carried
out as close as possible to the filling point and downstream of aseptic connections wherever possible
Proposed text change
8.15 Aseptic manipulations (including non-intrinsic aseptic connections) should be minimized using engineering
solutions such as the use of preassembled and sterilized equipment. Whenever feasible, product contact piping and
equipment should be preassembled, then cleaned and sterilized in place. The final sterile filtration should be carried
out as close as possible to the filling point and downstream of aseptic connections wherever possible unless the
integrity of the sterile product pathway downstream of the filter can be confirmed e.g. using integral stainless steel
piping which is subject to a SIP process and an integrity test.
855-856 Existing text:
a) Time between equipment, component, and container cleaning, drying and sterilization.
Proposed text change
The period between washing and depyrogenation of components and also their use when supplied to filling lines via
integrated depyrogenation tunnels must be limited and justified.
861-864 Existing text:
c) The time between the start of the preparation of a solution and its sterilization or filtration through a micro-
organism-retaining filter. There should be a set maximum permissible time for each product that takes into account its
composition and the prescribed method of storage.
Proposed text change
c) The time between the start of the preparation of a solution and its sterilization or filtration through a micro-
organism-retaining filter. There should be a set maximum permissible time for each product that takes into account its
composition, its susceptibility to microbial proliferation, chemical stability and the prescribed method of storage.
872-873 Existing text:
g) Maximum exposure time of sterilized containers and closures in the critical processing zone (including filling)
prior to closure.
Comment:

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Note filling needles, stopper bowls etc may be exposed for days within the processing zone when campaign filling is
undertaken.
875 Finishing of sterile products
877-881 Existing text:
8.17 Partially stoppered vials or prefilled syringes should be maintained under grade A conditions (e.g. use of
isolator technology, grade A with B background, with physical segregation from operators) or grade A LAF carts
(with suitable grade B background environment and physical segregation from operators) at all times until the
stopper is fully inserted.
Comment (1):
If the expectation is that a minimum of RABS should be employed, then this should be stated here.
Comment (2):
Since physical segregation from operators is required 2 times for partially stoppered containers this should be also
clearly indicate for open containers not partially stoppered.
883-890 Existing text:
8.18 Containers should be closed by appropriately validated methods. Containers closed by fusion, e.g. Form-Fill-
Seal Small Volume Parenteral (SVP) & Large Volume Parenteral (LVP) bags, glass or plastic ampoules, should be
subject to 100% integrity testing. Samples of other containers should be checked for integrity utilising validated
methods and in accordance with QRM, the frequency of testing should be based on the knowledge and experience of
the container and closure systems being used. A statistically valid sampling plan should be utilized. It should be noted
that visual inspection alone is not considered as an acceptable integrity test method,
Comment: (1)
Further clarification should be given on what are defined as acceptable integrity test methods i.e. microbiological or
chemical?). This should align to defined CCI methodologies given in USP1027 or state other acceptable tests.
The document should give more guidance on sampling frequencies (e.g. batch by batch testing, random batch testing)
and references to acceptable CCI tests as these vary vastly in sensitivity.
Further clarification is required on application of risk assessment as this could lead to vastly different approaches. For
example, visual inspection of a vial is limited due to occluded area under cap so this could be considered high risk in
which case 100% detection would seem appropriate. Conversely for the same container the QRM could highlight this
as a perceived low risk in which case no sampling may be required. The current position is ambiguous and could lead
to vastly different approaches.
Suggest adding 100% CCI technology should be employed when possible where there is a risk of non-detection of
defects using visual inspection processes such as in blind spots and difficult to inspect locations.
Comment (2):
LVP plastic-bags (one chamber bags and multi chamber bags), as physiochemical welded/bonded components, differ
in criticality from typically fused LVP containers (e.g. bottles produced as Blow Fill Seal from e.g. PE/PP plastic
granulates). Automated and highly controlled welding processes, together with utilising validated methods and sampling
plans for integrity testing, deliver a high assurance of package integrity for physiochemical welded/bonded plastic bags.
Welded bags can be tested in-line with vacuum/pressure in the empty bag stage rather than as filled products (after
mechanical capping), where no standardized 100% integrity tests are available. Furthermore, those bags are often
sterilized in an additional barrier over-pouch, which furthermore decreases the risk.
Proposed change (if any):
Containers should be closed by appropriately validated methods. Containers closed by fusion, e.g. Small Volume
Parenteral (SVP) & Large Volume Parenteral (LVP) manufactured from granulates, glass or plastic ampoules, should
be subjected to 100% integrity testing. Samples of other containers (including LVP plastic bags as physiochemical
welded/bonded components) should be checked for integrity utilising validated methods and in accordance with
QRM, the frequency of testing should be based on the knowledge and experience of the container and closure systems
being used. A statistically valid sampling plan should be utilized. It should be noted that visual inspection alone is not
considered as an acceptable integrity test method.”
Comment (3):

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Container closure integrity should be ensured by a robust control strategy and should leverage a series of checks and
controls throughout the process, and, as acknowledged in this section, it is this control strategy that dictates the
frequency of testing and the appropriate sample size. Sample set should always be representative of the batch.
Proposed change:
8.18 Containers should be closed by appropriately validated methods. Containers closed by fusion, e.g. Form-Fill-Seal
Small Volume Parenteral (SVP) & Large Volume Parenteral (LVP) bags, glass or plastic ampoules, should be subject
to 100% integrity testing. Samples of other containers should be checked for integrity utilising validated methods and
in accordance with QRM, the frequency of testing should be based on the knowledge and experience of the container
and closure systems being used. The sample set should be representative of the batch. It should be noted that visual
inspection alone is not considered as an acceptable integrity test method.
892-893 Existing text:
8.19 Containers sealed under vacuum should be tested for maintenance of vacuum after an appropriate, pre-
determined period and during shelf life.
Comment
Clarification is needed to confirm if this requirement is product stability (e.g. moisture ingress) or micro ingress
related – if this is specifically in products where loss of vacuum could lead to product degradation then this should be
specified.
Note - there is a concern that although vacuum levels may be lost over shelf life, this may not permit microbial or
moisture ingress. This requirement should align to the Maximum Allowable Leakage concept stated in USP1027 or
define other acceptable methodologies.
895-896 Existing text:
8.20 The container closure integrity validation should take into consideration any transportation or shipping
requirements.
Comment:
It is not clear in the document if this is CCI of primary pack after transportation and shipping (within its finished
pack) or other stages (e.g. bulk intermediate shipping) therefore further clarification should be added. Note that this
document does not actually mention requirements for the core CCI validation of the primary pack at early stages in
life cycle e.g packaging development, manufacturing and sterilisation processes.
909-913 Existing text:
8.23 In the case where capping is conducted as a clean process with grade A air supply protection, vials with missing
or displaced stoppers should be rejected prior to capping. Appropriately validated, automated methods for stopper
height detection should be in place. Microbial ingress studies (or alternative methods) should be utilized to determine
the acceptable stopper height displacement.
Comment (1):
The document should clarify that vials with missing or displaced stoppers can be inspected prior to capping but they
can be physically rejected after crimping. We assume that laser height is suitable, as well as vision, if appropriately
validated?
Suggest to reword rejection position as vials identified with displaced stoppers can be physically rejected after
capping.
This only covers micro ingress but does not consider the potential for loss of gas headspace (where critical). This
should align to the Maximum Allowable Leakage concept stated in USP1027.
Comment (2):
Line 911-913
Whenever capping is conducted as a clean process under Grade A air supply, included automated systems for missing
or displaced stopper detection, there should be no additional risk for microbial contamination, especially if in addition
the Grade A air is protected by RABS systems. Microbial ingress studies (or equivalent) would normally be performed
during container closure integrity testing (CCIT) of completely sealed finished product containers to ensure their
integrity during transport and shelf life. Performing CCIT studies of unsealed vials with different inserted stopper
heights and then setting limits based on that for a stopper positioning sensor will not add additional sterility assurance
to the product. Limits should be set conservatively and be based on full insertion.
Proposed text change:
Appropriately validated, automated methods for stopper height detection should be in place.

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Comment (3):
Should indicate “For a clean capping process where human intervention is required at the capping station...”
919-920 Existing text:
8.25 RABS and isolators may be beneficial in assuring the required conditions and minimising direct human
interventions into the capping operation.
Comment:
The statement regarding RABS/Isolators for capping should be assessed based on QRM, e.g. if the product is
sealed/closed under vacuum this may not be needed.
922-932 Existing text:
8.26 All filled containers of parenteral products should be inspected individually for extraneous contamination or
other defects. QRM principles should be used for determination of defect classification and criticality. Factors to
consider include, but are not limited, to the potential impact to the patient of the defect and the route of
administration. Different defect types should be categorized and batch performance analysed. Batches with unusual
levels of defects, when compared to routine defect levels for the process, should lead to investigation and
consideration of partial or the whole rejection of the batch concerned. A defect library should be generated and
maintained which captures all known defects. The defect library can be used as a training tool for production and
quality assurance personnel. Critical defects should not be identified during any subsequent sampling of acceptable
containers as it indicates a failure of the original inspection process.
Comment:
Line 923 to 925: Does the requirement to use QRM to assess defect criticality apply to all defect types or just
particles. Need further clarification on risk factors as this is vague.
Line 927 to 929: Need clarification on which defects types should be categorised. Typically, all critical/major should
be assessed but this is not necessary for minor cosmetic defects – a risk approach should be permissible.
Line 931 to 932: There should be a reference to applicable sampling plans (e.g. ISO2859). Additional sampling for
difficult to inspect products is not included (ref: USP790).
934-942 Existing text:
8.27 When inspection is done manually, it should be done under suitable and controlled conditions of illumination
and background. Inspection rates should be appropriately validated. Operators performing the inspection should
undergo robust visual inspection qualification (whilst wearing corrective lenses, if these are normally worn) at least
annually. The qualification should be undertaken using appropriate sample sets and taking into consideration worst
case scenarios (e.g. inspection time, line speed (where the product is transferred to the operator by a conveyor
system), component size or fatigue at the end of shift) and should include consideration of eyesight checks. Operator
distractions should be removed and frequent breaks of appropriate duration from inspection should be taken.
Comment:
Line 934 to 937: Ref: to EP 2.9.20 / USP790 manual inspection setup and method should be made for particles
inspection. Reference to the number of test set runs / passes should be made (e.g. 3 consecutive passes are required for
operator qualification).
Line 938 to 942: There are no requirements for criteria to be applied for the qualification or categories (e.g. such as
100% rejection of critical defects). Expectation would that this would be included and risk based on criticality of the
defect. Does not state any additional requirements for difficult to inspect products (e.g. change of conditions to
enhance process to inspect to extent possible).
944-946 Existing text:

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
8.28 Where automated methods of inspection are used, the process should be validated to detect known defects with
sensitivity equal to or better than manual inspection methods and the performance of the equipment checked prior to
start up and at regular intervals.
Comment:
Clarification is required at to which defects this applies to. If this covers all defects, then a comparison to manual can
lead to reduced performance for critical defects (as increased detection on minor defects can also mask poor detection
on more critical defects)..
Typically, ‘better than or equal to criteria’ is usually required for particulate defects. This draft document infers that
comparison to manual can be used for all defects (critical, major, minor). Using manual process as a baseline for all
defects can lead to a lesser standard for a poorly applied manual process. Further clarification on application of the
requirement should be made.
Additional specific criteria should apply for automated systems linking to AQL classifications e.g. 100% rejection
required for critical defects.
Clarification is also required if criteria (i.e. comparison to manual) is applicable to semi-automated inspection – this is
the typical industry requirement.
Clarification is required as to what regular means in terms of equipment checks as this is ambiguous.
No reference given to requirements for validation of re-inspection (ejects) – as USP1790 requirement “If a two-stage
inspection strategy is used, it must be validated as intended for use. Defective containers with less than a 100% PoD
will have the PoD reduced further with each stage of inspection, thus the PoD should be determined after inspection
through both stages to ensure acceptable sensitivity is maintained”.
Comment (2)
“For automated methods of inspection …the performance of the equipment checked prior to start up and at regular
intervals for a single batch” should be indicated
948-951 Existing text:
8.29 Results of the inspection should be recorded and defect types and levels trended. Reject rates for the various
defect types should also be trended. Investigations should be performed as appropriate to address adverse trends or
discovery of new defect types. Impact to product on the market should be assessed as part of this investigation.
Comment:
The requirement for analysis of batch performance is already stated in Line 926 therefore there appears to be some
duplication. Further clarification is needed on this.
953 Sterilization
Comment:
Is this section intended for product or equipment or both?
955-960 Comment (1):
Should this paragraph actually reference the EMEA document on filing new products as it is in this document that
details on how to choose the sterilization method is provided?
Comment (2):
Line 959: Pasteurisation relates to treatments for food and drink and there is no associated definition included in the
Glossary section.
Proposed change:
Remove the word ‘Pasteurisation’
Existing text:
8.30 Where possible, finished product should be terminally sterilized using a validated and controlled sterilization
process as this provides a greater assurance of sterility than a validated and controlled sterilizing filtration process
and/or aseptic processing. Where it is not possible for a product to undergo a sterilisation, consideration should be
given to using terminal bioburden reduction steps, such as heat treatments (pasteurization), combined with aseptic
processing to give improved sterility assurance.
Proposed text change
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
8.30 Where possible, finished product should be terminally sterilized using a validated and controlled sterilization
process as this provides a greater assurance of sterility than a validated and controlled sterilizing filtration process
and/or aseptic processing. Where it is not possible for a product to undergo a sterilisation, consideration should be
given to using terminal bioburden reduction steps, such as heat treatments, combined with sterile filtration and aseptic
processing to give improved sterility assurance.
967-968 Comment:
Clarification of what is considered an “atypical” sterilization cycle is required.
Please define atypical sterilization cycle in the glossary.
970-975 Existing text:
8.33 All sterilization processes should be validated. Particular attention should be given when the adopted
sterilization method is not described in the current edition of the Pharmacopoeia, or when it is used for a product
which is not a simple aqueous solution. Where possible, heat sterilization is the method of choice. Regardless, the
sterilization process must be in accordance with the registered marketing and manufacturing specifications.
Proposed text change
8.33 All sterilization processes must be validated (product and equipment, components etc). However for product
sterilisation, particular attention should be given when the adopted sterilization method is not described in the current
edition of the Pharmacopoeia, or when it is used for a product which is not a simple aqueous solution. Where possible,
heat sterilization is the method of choice. Regardless, the sterilization process must be in accordance with the
registered marketing and manufacturing specifications.
977-980 Existing text:
8.34 Before any sterilization process is adopted, its suitability for the product and equipment and its efficacy in
achieving the desired sterilizing conditions in all parts of each type of load to be processed should be demonstrated by
physical measurements and by biological indicators where appropriate.
Comment:
Include the word “also” in front of “by biological indicators”
982-985 Comment :
Clarification required on what is considered a ‘process’ e.g. does this mean the sterilization process for sterile
product?
987-989 Existing text:
8.36 For effective sterilization, the whole of the material and equipment must be subjected to the required treatment
and the process should be designed to ensure that this is achieved.
Proposed text change
8.36 For effective sterilization, the whole of the product, components, material and equipment must be subjected to
the required treatment and the process should be designed to ensure that this is achieved.
994-1003 Existing text:
8.38 Suitable biological indicators (Bis) placed at appropriate locations may be considered as an additional method
for monitoring the sterilization. Bis should be stored and used according to the manufacturer's instructions. Prior to
use of a new batch/lot of Bis, the quality of the batch/lot should be verified by confirming the viable spore count and
identity. Where Bis are used to validate and/or monitor a sterilization process (e.g. for Ethylene Oxide), positive
controls should be tested for each sterilization cycle, with strict precautions in place to avoid transferring microbial
contamination from Bis, including preventing positive control Bis from contaminating Bis exposed to the sterilization
cycle. If biological indicators are used, strict precautions should be taken to avoid transferring microbial
contamination to the manufacturing or other testing processes.
Comment:

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
BIs should be incubated for a minimum of 7 days.
1006-1010 Existing text:
Each basket, tray or other carrier of products, items of equipment or components should be clearly labelled with the
material name, its batch number and an indication of whether or not it has been sterilized. Indicators such as
autoclave tape, or irradiation indicators may be used, where appropriate, to indicate whether or not a batch (or sub-
batch) has passed through a sterilization process.
Comment:
There are other options rather than physically and individually labelling baskets, trays or other carriers of products,
equipment and components that have been sterilized to differentiate them from others which have not been sterilized.
For a double door autoclave/sterilizer that separates a fully automated pre-sterilization loading area from a fully
automated post-sterilization unloading area, there are electronic systems in place to ensure full tracking of separate
sterilizer loads; and where material names, batch numbers and sterilization records for the sterilized materials are
integrated in the validated data system.
Proposed text change:
Each basket, tray or other carrier of products, items of equipment or components should be clearly labelled or
electronically traceable with the material name, its batch number and an indication of whether or not it has been
sterilized. Indicators such as autoclave tape, or irradiation indicators may be used, where appropriate, to indicate
whether or not a batch (or sub-batch) has passed through a sterilization process.
1017-1026 Comment (1):
Suggest that this paragraph is headed with “Staging and Transfer of Sterilised Items into the Grade A zone” and re-
worded as below
Existing text:
8.41 Where possible, materials, equipment and components should be sterilized by validated methods appropriate to
the specific material. Suitable protection after sterilization should be provided to prevent recontamination. If items
sterilized "in house" are not used immediately after sterilization, these should be stored, using appropriately sealed
packaging, in at least a grade B environment, a maximum hold period should also be established. Components that
have been packaged with multiple sterile packaging layers need not be stored in grade B (where justified) if the
integrity and configuration (e.g. multiple sterile coverings that can be removed at each transfer from lower to higher
grade) of the sterile pack allows the items to be readily disinfected during transfer into the grade A zone. Where
protection is achieved by containment in sealed packaging this process should be undertaken prior to sterilisation.
Proposed text change
8.41 Materials, equipment and components must be sterilized by validated methods appropriate to the specific
material. Suitable protection after sterilization should be provided to prevent recontamination. If items sterilized "in
house", in the absence of any packaging are not used immediately after sterilization, these should be staged under
Grade A conditions using barrier technology. Items sterilised "in house" within appropriate sealed packaging, unless
utilised within Grade A isolators, should be staged within the sealed packaging and protected by a Grade A supply
located in at least a grade B environment. Where packaged items are sterilised "in house" via autoclaving for use in
Grade A isolators, the items should be staged within the sealed packaging and protected by a Grade A supply located
in at least a grade C environment. The packaging must be suitable to permit decontamination of its outer surface prior
to the exposure and removal of the sterilised contents within the Grade A environment. A maximum staging period
should be established. Components that have been packaged with multiple sterile packaging layers such as those
supplied irradiation or ethylene oxide sterilised need not be stored in grade B (where justified) if the integrity and
configuration (e.g. multiple sterile coverings that can be removed at each transfer from lower to higher grade) of the
sterile pack allows the items to be readily disinfected during transfer into the grade A zone. Where protection is
achieved by containment in sealed packaging this process should be undertaken prior to sterilisation.

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Comment (2):
Line 1019-1026
In-house sterilized items to be used for terminally sterilized products would not need to be stored in grade B, even if
only a single layer protective packaging be used. In most terminally sterilized manufacturing facilities grade B areas are
not available, although a grade A LAF-zone may be used for the filling operations in grade C, before terminal
sterilization takes place. It should be clearly stated that the requirement as mentioned in the revised Annex 1 is only
intended for aseptically produced products.
Proposed text change:
If items to be used in aseptic processing and sterilized “in house” are not used immediately after sterilization, these
should be stored, using appropriately sealed packaging, in at least a grade B environment, a maximum hold period
should also be established. Components that have been packaged with multiple sterile packaging layers need not be
stored in grade B (where justified) if the integrity and configuration (e.g. multiple sterile coverings that can be removed
at each transfer from lower to higher grade) of the sterile pack allows the items to be readily disinfected during transfer
into the grade A zone. Where protection is achieved by containment in sealed packaging this process should be
undertaken prior to sterilisation.
Comment:
Line 1022-1025: It is unclear if items must be autoclaved with multiple packaging or if it is acceptable for them to be
disinfected.
Proposed change (if any): “…need not be stored in grade B if items are packaged in multiple sterile packs or the
sterile pack allows the items to be readily disinfected during transfer…”
1028-1030 Existing text:
8.42 Transfer of materials, equipment, and components into an aseptic processing area should be via a unidirectional
process (e.g. through a double-door autoclave, a depyrogenation oven, effective transfer disinfection, or, for gaseous
or liquid materials, a bacteria-retentive filter).
Comment:
Include the words “depyrogenation tunnel”
1042-1045 Existing text:
8.44 Where materials, equipment, components and ancillary items are sterilized in sealed packaging or containers,
the integrity of the sterile protective barrier should be qualified for the maximum hold time, and the process should
include inspection of each sterile item prior to its use to ensure that the sterile protective measures have remained
integral.
Comment:
Excessive handling during inspection of each item may result in an increased contamination risk.
Comment:
Please confirm whether “qualified” refers to media fill or some other type of test e.g. physical test.
1047-1051 Existing text:
8.45 For materials, equipment, components and ancillary items that are necessary for aseptic processing but cannot
be sterilized, an effective and validated disinfection and transfer process should be in place. These items once
disinfected should be protected to prevent recontamination. These items, and others representing potential routes of
contamination, should be included in the environmental monitoring program.
Comment:
This paragraph is in the sterilisation section, yet refers to validated disinfection and transfer process?
1053-1056 Existing text:
8.46 When a depyrogenation process is used for any components or product contact equipment, validation studies
should be performed to demonstrate that the process will result in a minimum 3 log reduction in endotoxin. There is
no additional requirement to demonstrate sterilization in these cases
Proposed text change

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
8.46 When a thermal depyrogenation process is used for any components or product contact equipment, validation
studies should be performed to demonstrate that the process will provide a suitable Fh value and result in a minimum
3 log reduction in endotoxin. There is no additional requirement to demonstrate sterilization in these cases
1072-1074 Existing text:
8.48 In those cases where parametric release has been authorized, a robust system should be applied to the product
lifecycle validation and the routine monitoring of the manufacturing process. This system should be periodically
reviewed.
Comment:
Periodically requires defining.
1076-1079 Existing text:
8.49 Each heat sterilization cycle should be recorded on a time/temperature chart with a sufficiently large scale or by
other appropriate equipment with suitable accuracy and precision. Monitoring and recording systems should be
independent of the controlling system.
Comment (1):
Clarification is required here. Are independent systems required for both temperature and pressure for moist heat
sterilisation as Line 1100 to 1104 states “Time, temperature and pressure should be used to monitor the process”. Also
what is the regulatory definition of independent?
Comment (2):
Line 1078-1079: The controlling system is usually integrated with the recording systems.
Proposed change (if any): suggest a broader statement, by replacing ‘should’ with ‘can’
Comment (3):
This statement appears to have been written with much older technologies in mind. In modern facilities the equipment
utilized for sterilization is controlled by an automation system and a record of the cycle is downloaded into a historian.
With use of this kind of automation, which provides real time and much more accurate control, it is not necessary to
have separate systems for monitoring and control – only one set of instrumentation is capable of performing both tasks
accurately.
Proposed change:
8.49 Each heat sterilization cycle should be recorded by appropriate equipment with suitable accuracy and precision
1081-1084 Existing text:
8.50 The position of the temperature probes used for controlling and/or recording should have been determined
during the validation (which should include heat distribution and penetration studies), and, where applicable, also
checked against a second independent temperature probe located at the same position.
Proposed text change
8.50 The position of the temperature probes used for controlling and/or recording should have been determined during
the sterilisation cycle validation (which should include heat distribution and penetration studies). The thermometric
studies should be undertaken using independent multi-point calibrated thermocouples
1093-1096 Existing text:
8.53 After the high temperature phase of a heat sterilization cycle, precautions should be taken against contamination
of a sterilized load during cooling. Any cooling fluid or gas in contact with the product should be sterilized unless it
can be shown that any leaking container would not be approved for use.
Comment:
It is recommended that air flow visualisation studies are conducted to identify and assist in the elimination of any
adverse contaminating convection currents.
1098 Moist heat sterilization
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Comment:
Suggested additional text for clarity and guidance “Autoclaves should be controlled and maintained in accordance
with EN285. Typical sterilisation conditions within a given load are 121°C for 15 minutes and 134°C for 3.5 minutes.
Other temperatures and times may be justified when supported by scientific rationale”.
1100-1104 Existing text:
8.54 Time, temperature and pressure should be used to monitor the process. Each item sterilized should be inspected
for damage, seal and packaging material integrity and moisture on removal from the autoclave. Seal and packaging
integrity should also be inspected immediately prior to use. Any items found not to be fit for purpose should be
removed from the manufacturing area and an investigation performed.
Comment (1):
Examination of every item may lead to excessive handling and a potential for contamination
Comment (2):
Line 1102-1104: A packaged and sterilized item not passing the seal and package integrity inspection prior to use should
not automatically require a full investigation. The sterilized item would only be used in manufacturing if the sterile
package is integral, and as such a damaged package would not pose a hygienic risk to the operations and would not need
to be investigated, unless an adverse trend of defect packages is obvious.
Proposed text change:
Seal and packaging integrity should also be inspected immediately prior to use. Any items found not to be fit for
purpose should be removed from the manufacturing area
1114-1119 Existing text:
8.57 Validation should include a consideration of equilibration time, exposure time, correlation of pressure and
temperature and maximum temperature range during exposure for porous cycles and temperature, time and F0 for
fluid cycles. These critical parameters should be subject to defined limits (including appropriate tolerances) and be
confirmed as part of sterilization validation and routine cycle acceptance criteria. Revalidation should be performed
annually.
Comment:
Validation must include equilibration time, exposure time, correlation of pressure and temperature and maximum
temperature range during exposure for porous cycles and temperature, time and F0 for fluid cycles. For steam
sterilisers, guidance on equilibration time and other aspects of cycle validation can be found in EN 285 and ISO
17666-1.
Comment:
Suggest revalidation or rather requalification is directed towards worse case load as determined by QRM.
1126-1129 Existing text:
8.59 When the sterilization process includes air purging (e.g. porous autoclave loads, lyophilizer chambers) there
should be adequate assurance of air removal prior to and during sterilization. Loads to be sterilized should be
designed to support effective air removal and be free draining to prevent the build-up of condensate.
Proposed text change
8.59 When the sterilization process includes air purging (e.g. porous autoclave loads, lyophilizer chambers) there
should be adequate assurance of air removal prior to and during sterilization. Loads to be sterilized should be designed
to support effective air removal and be free draining to prevent the build-up of condensate. The fitting of an air
detector to porous load sterilisers is recommended.
1131-1134 Existing text:
8.60 The items to be sterilized, other than products in sealed containers, should be dry, wrapped in a material which
allows removal of air and penetration of steam but which prevents recontamination after sterilization. All load items
should be dry upon removal from the sterilizer. Load dryness should be confirmed as a part of sterilization process
acceptance.
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Proposed text change
8.60 The items to be sterilized, other than products in sealed containers, should be dry, wrapped in a material (unless
maintained under Grade A conditions post sterilisation) which allows removal of air and penetration of steam and
condensate removal but which prevents recontamination after sterilization. All load items should be dry upon removal
from the sterilizer. Load dryness should be confirmed as a part of sterilization process acceptance.
1140-1143 Existing text:
8.62 Care should be taken to ensure that materials or equipment are not contaminated after the sterilization exposure
phase of the cycle due to the introduction of non-sterile air into the chamber during subsequent phases; typically only
sterile filtered air would be introduced into the chamber during these phases.
Proposed text change
8.62 Care should be taken to ensure that materials or equipment are not contaminated after the sterilization exposure
phase of the cycle due to the introduction of non-sterile air into the chamber during subsequent phases; typically only
sterile filtered air would be introduced into the chamber during these phases. Moist heat sterilisers employing porous
cycles must be fitted with 0.2 micron vent filters which are capable of being sterilised and integrity tested in situ.
1145-1153 Existing text:
8.63 Where Sterilization in place (SIP) systems are used, (for example, for fixed pipework, vessels and lyophilizer
chambers), the system should be appropriately designed and validated to assure all parts of the system are subjected
to the required treatment. The system should be monitored for temperature, pressure and time at appropriate critical
locations during routine use, this is to ensure all areas are effectively and reproducibly sterilized; these critical
locations should be demonstrated as being representative, and correlated with, the slowest to heat locations during
initial and routine validation. Once a system has been sterilized by SIP it should remain integral prior to use, the
maximum duration of the hold time should be qualified.
Comment(1):
Add the following to the end of this paragraph: “Validation and requalification of SIP systems requires the use of BIs
as well as independent thermometric devices”.
There is no mention of over-pressure requirements or defined leak rate acceptance criterion?
Comment (2):
Line 1145: SIP has previously been used in the document to refer to Steam in Place, which is the correct term (not
Sterilisation in Place).
Proposed change:
Change ‘Sterilisation in Place’ to ‘Steam in Place’
1155 Dry Heat sterilization.
Comment:
Suggested additional text for clarity and guidance “Typical sterilisation conditions are 160°C for 120 minutes; Typical
depyrogenation conditions provide an Fh value equivalent to 250°C for 30 minutes. Other temperatures and times may
be justified when supported by scientific rationale”.
1161-1169 Existing text:
8.65 Dry heat sterilization or depyrogenation tunnels are typically employed to prepare components for aseptic filling
operations but may be used for other processes. Tunnels should be configured to ensure that airflow patterns protect
the integrity and performance of the sterilizing zone, by maintaining a stable pressure differential and airflow pattern
through the tunnel from the higher grade area to the lower grade area. All air supplied to the tunnel should pass
through a HEPA filter; periodic tests should be performed to demonstrate filter integrity. Any tunnel parts that come
into contact with sterilized components should be appropriately sterilized or disinfected. Critical process parameters
that should be considered during validation and/or routine processing should include, but may not be limited to:

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Comment:
It is desirable for the cooling zone to be capable of automated disinfection or sterilisation. Where located outside of
the aseptic area, the pressure differential between the cooling zone and the surrounding environment must be set at a
defined minimum value.
A HEPA filter is defined in the glossary as 99.97% efficient. Can hot air filters be reliably manufactured to this
standard?
Comment:
Line 1169: validation of depyrogenation tunnel should include a maximum exposure time in order to avoid any
damage of the filled product if the container is not sufficiently cooled down.
1185-1189 Existing text
8.67 Dry heat ovens are typically employed to sterilize or depyrogenate primary packaging components, finished
materials or APIs but may be used for other processes. They should be maintained at a positive pressure to lower
grade areas. All air entering the oven should pass through a HEPA filter. Critical process parameters that should be
considered in validation qualification and/or routine processing should include, but may not be limited to::
Comment:
Other processes include the sterilization of oil based excipients.
Periodic tests should be performed to demonstrate filter integrity.
1195 Existing text:
c) Chamber pressure.
Comment:
Also include airflow and or fan velocity
1206 Existing text:
Sterilization by radiation
Comment:
Suggested additional text for clarity and guidance “Typical sterilisation dose is 25kGy delivered over a pre-
determined time. Other doses may be justified when supported by bioburden studies and a scientific rationale”.
Reference could also be given to ISO11137.
1220 Existing text:
Sterilization with Ethylene Oxide
Comment:
Reference could be given to ISO11135.
1222-1226 Existing text:
8.72 This method should only be used when no other method is practicable. During process validation it should be
shown that there is no damaging effect on the product and that the conditions and time allowed for degassing to
reduce any residual ethylene oxide (EO) gas and reaction products to defined acceptable limits for the type of product
or material.
Proposed text change
8.72 This method should only be used when no other method is practicable. During process validation it should be
shown that there is no damaging effect on the product and that the conditions and time allowed for degassing are
sufficient to reduce any residual ethylene oxide (EO) gas and reaction products to defined acceptable limits for the
type of product or material.
Comment:
Line 1222: “8.72 This method should only be used when no other method is practicable.” This should be clarified
since many suppliers of syringes and vials in tubs and nets use this EO sterilization method.
1236-1238 Existing text:
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
8.75 Each sterilization cycle should be monitored with suitable biological indicators, using the appropriate number of
test pieces distributed throughout the load unless parametric release has been authorized by the National Competent
Authority.
Comment:
Is parametric release actually being considered for EtO sterilisation? That is BIs and sterility test not required for
articles processed by this method?
1240-1242 Existing text:
8.76 Critical process variables that should be considered as part of sterilization process validation and routine
monitoring include, but are not limited to: EO gas concentration, relative humidity, temperature and EO gas pressure
and exposure time.
Proposed text change
8.76 Critical process variables that should be considered as part of sterilization process validation and routine
monitoring include, but are not limited to: EO gas quality, EO gas quantity or concentration, relative humidity,
vacuum, temperature and EO gas pressure and exposure time.
1249 Filtration of medicinal products which cannot be sterilized in their final container
1251-1261 Existing text:
8.78 If a liquid product cannot be terminally sterilized by a microbiocidal process, it should be sterilized by filtration
through a sterile, sterilizing grade filter (with nominal pore size of 0.22 micron (or less) or with at least equivalent
micro-organism retaining properties), and subsequently aseptically filled into a previously sterilized container, the
selection of the filter used should ensure that it is compatible with the product, see 8.119.. Suitable bioburden
reduction and/or sterilizing grade filters may be used at multiple points during the manufacturing process to ensure a
low and controlled bioburden of the liquid prior to the primary sterilizing grade filter. Due to the potential additional
risks of a sterilizing filtration process as compared to other sterilization processes, a second filtration through a
sterile, sterilising grade filter (positioned as per clause 8.15), immediately prior to filling, is advisable
Proposed text change
8.78 If a liquid product cannot be terminally sterilized by a microbiocidal process, it should be sterilized by filtration
through a sterile, sterilizing grade filter (with nominal pore size of 0.22 micron (or less) or with at least equivalent
micro-organism retaining properties), and subsequently aseptically filled into a previously sterilized container, the
selection of the filter used should ensure that it is compatible with the product, see 8.119.. Suitable bioburden
reduction and/or sterilizing grade filters may be used at multiple points during the manufacturing process to ensure a
low and controlled bioburden of the liquid prior to the primary sterilizing grade filter. Due to the potential additional
risks of a sterilizing filtration process as compared to other sterilization processes, a second filtration through a sterile,
sterilising grade filter (positioned as per clause 8.15), immediately prior to filling, is advisable unless the sterile
product pathway down-stream of the filter can be demonstrated to be fully integral e.g. hard-piped stainless steel
system subject to SIP and held under positive pressure. In exceptional circumstances it may not be possible to
undertake a second sterilising filtration where process/product considerations prevent this.
Comment:
Line 1252: There are non-filterable formulations. Some biologicals such as vaccines with adjuvants cannot be
filtered, and are aseptically processed without a filtration step.
Proposed change (if any): Add it should be sterilized by filtration “when possible”
1283-1285 Existing text:
e) Allow sterilization procedures, including SIP, to be conducted as necessary. The sterilization procedures should be
validated to ensure achievement of a target sterilization assurance level (SAL) of 10"6 or better (e.g. 10 7).
Comment:
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Not sure why SAL for SIP is mentioned here OR if referring to the validation of the filtration process per se should it
read as “e) Allow sterilization procedures, including SIP, to be conducted as necessary. The sterilization by filtration
procedures should be validated to ensure achievement of a target titre reduction of 107 per cm2 or better when
challenged with a suitable test micro-organism such as B.diminuta.
1287-1289 Existing text:
f) Permit in-place integrity testing, preferably as a closed system, prior to filtration as necessary. In-place integrity
testing methods should be selected to avoid any adverse impact on the quality of the product.
Comment:
Can it be clarified if this section is for new design or does it also apply to previously designed processes where the
design did not include in line integrity testing?
1296-1299 Existing text:
8.82 Wherever possible, the product to be filtered should be used for bacterial retention testing. Where the product to
be filtered is not suitable for use in bacterial retention testing, a suitable surrogate product should be justified for use
in the test. The challenge organism used in the bacterial retention test should be justified.
Proposed text change
8.82 Wherever possible, the product to be filtered should be used for bacterial retention testing. Where the product to
be filtered is not suitable for use in bacterial retention testing e.g. as it has inherent anti-microbial properties, a suitable
surrogate product should be justified for use in the test. The challenge organism used in the bacterial retention test
should be justified.
1301-1305 Comment:
Water can be used to flush and integrity test a filter, and in this case, as long as the filter is completely dried by
filtered air, there is no need to add a product flush.
Proposed change:
8.83 Filtration parameters that should be considered in validation and routine processing should include but are not
limited to: a) If the system is flushed or integrity tested in-situ with a fluid other than the product, then flushing with
the product should be part of the process unless this fluid is WFI and the filter can be fully dried prior to use.
1311 Comment:
8.83 c) Filtration process conditions: vii indicates time taken for a know volume and pressure difference as in the
current Annex 1 but already indicated in this point 8.83 c) alineas iii, iv and v.
1328 Comment:
8.83 c) vii “ Results of these checks should be included in the batch record” should be applied to all parameters of
8.83 c) Filtration process conditions.
1331-1340 Existing text:
8.84 The integrity of the sterilized filter assembly should be verified by testing before use, in case of damage and loss
of integrity caused by processing, and should be verified by on line testing immediately after use by an appropriate
method such as a bubble point, diffusive flow, water intrusion or pressure hold test. It is recognised that for small
batch sizes, this may not be possible; in these cases an alternative approach may be taken as long as a formal risk
assessment has been performed and compliance is achieved. There should be written integrity test methods, including
acceptance criteria, and failure investigation procedures and justified conditions under which the filter integrity test
can be repeated. Results of the integrity tests (including failed and repeated tests) should be included in the batch
record.
Comment (1):
Single use system gamma irradiated filters cannot always be integrity tested prior to use. This is seen by much of
industry as a key issue. Some companies incorrectly remove filter from housing for testing, therefore is this
requirement clear enough? Note it is for product filtration only NOT air, vents, compressed gases etc.

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Comment (2)
Also there is no reference to the use of off-line testing?
Comment (3)
8.84 “It is recognised that for small batch sizes...” It should be indicated what is considered as a small batch size.:
Comment (4):
Line 131: This sentence is not clear: in case of damage and loss of integrity caused by processing,
Proposed change (if any): Delete or clarify the highlighted sentence.
Comment (5):
While in situ testing post use is useful, it should not be mandated as this may lead to much greater increased
complexity of the system and can lead to additional risk to the process. The use of off line testing will not impact the
accuracy of the filter integrity test results. Additionally, propose the removal of the examples for appropriate filter
integrity testing methods as these may evolve over time, and as long as a test is appropriate for and validated for its
intended use, the actual methodology should not be prescribed.
Proposed text change:
8.84 The integrity of the sterilized filter assembly should be verified by testing before use, in case of damage and loss
of integrity caused by processing, and should be verified immediately after use by an appropriate method. It is
recognised that for small batch sizes, this may not be possible; in these cases an alternative approach may be taken as
long as a formal risk assessment has been performed and compliance is achieved. There should be written integrity
test methods, including acceptance criteria, and failure investigation procedures and justified conditions under which
the filter integrity test can be repeated. Results of the integrity tests (including failed and repeated tests) should be
included in the batch record
1342-1344 Existing text:
8.85 The integrity of critical sterile gas and air vent filters in the filter assembly should be verified by testing after use.
The integrity of non-critical air or gas vent filters should be confirmed and recorded at appropriate intervals.
Comment:
Definition required for “after use”
Comment:
Line 1341: This requires post-use integrity testing of all critical sterile gas and vent filters.
Proposed change (if any): The term ‘critical filter’ should be defined
1346-1347 Existing text:
8.86 For gas filtration, the avoidance of unintended moistening or wetting of the filter or filter equipment is
important. This can be achieved by the use of hydrophobic filters.
Proposed text change
8.86 For gas filtration, the avoidance of unintended moistening or wetting of the filter or filter equipment is important.
For this reason, by first intent hydrophobic filters should be used.
1349 Comment:
8.87 Serial filtration: it should be indicated that the sample for bioburden must be taken before the last sterilizing filter
and the specification NMT 10 CFU/100 ml for bioburden should be indicated.
Comment:
Line 1349 &1354: Need clarification on “serial filtration” versus “redundant filter”
1354-1358 Existing text:
8.88 Where a redundant sterilizing filter is used, the additional filter does not require post- integrity testing unless the
primary sterilizing filter fails, in which case the redundant filter must then satisfactorily pass post-use integrity

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
testing. Bioburden samples should be taken prior to the first filter and the sterilizing filter, systems for taking samples
should be designed so as not to introduce contamination.
Comment:(1)
Omit wording “ the first filter and” or state that if a bioburden sample is taken prior to the first filter then it is not
necessary to take an additional sample prior to the sterilizing filter.
The meaning of redundant can vary in different parts of the world. Can the word be defined for filtration?
Comment (2)
Line 1356-1358: The requirements for product bioburden testing should be based on Quality Risk Management
principles and relate to the design of the manufacturing operations (including holding times) and product characteristics
(including growth promoting abilities). It is agreed that for aseptic manufacture a bioburden sample always should be
taken prior to the sterilizing filter, but any samples taken prior to the first filter should depend on above mentioned
aspects and can be validated. The design of the bioburden sampling regimen should be included in the overall
contamination control strategy.
Proposed text change:
Bioburden samples should be taken prior to the sterilizing filter and unless otherwise validated, before the first filter.
Systems for taking samples should be designed so as not to introduce contamination.
Comment (3):
Line 1354: 8.88 “Where a redundant sterilizing filter is used, the additional filter does not require post-integrity
testing unless...” Does that mean that the redundant filter should be tested for integrity before use even if the first filter
passed the pre-use integrity test? “Bioburden samples should be taken prior to the first filter and the sterilizing
filter...” In redundant sterilizing filtration all filters are sterilizing filters and thus bioburden should be taken only
before one sterilizing filter, the last one or the first one as explained in PICS interpretation PI 032-2 in January 2010.
The specification NMT 10 CFU/100 ml for bioburden should be indicated.
Comment (4):
Pre-filtration bioburden testing is utilized to verify the microbial load just prior to the final sterilizing filter to ensure it
is appropriate to the process. Based on the process and the product, a bioburden control strategy may suggest
additional testing at other points upstream, but that will be product and process related and should be determined by
the control strategy.
Proposed text change:
8.88 Where a redundant sterilizing filter is used, the additional filter does not require post integrity testing unless the
primary sterilizing filter fails, in which case the redundant filter must then satisfactorily pass post-use integrity testing.
Bioburden samples should be taken prior to the sterilizing filter, systems for taking samples should be designed so as
not to introduce contamination.
1360-1362 Existing text:
8.89 Liquid sterilizing filters should be discarded after the processing of a single lot. The same filter should not be
used for more than one working day unless such use has been validated.
Proposed text change (1)
8.89 Liquid sterilizing filters should be discarded after the processing of a single lot unless they are designed to be
used for multiple lots and are validated for such purposes e.g. Large scale cartridge filters for use in API manufacture
which are designed for repeated sterilisations. In all other cases the same filter should not be used for more than one
working day unless such use has been validated.
Comment (1):
Filter exchange can pose a risk to aseptic processes since closed systems need to be opened and critical areas exposed
for this operation. Exchanging the filter after every single lot would add an avoidable risk to campaign production,
where several lots of the same product are produced in a validated sequence.
The maximum usage duration, total filtration volume, number of in-line cleaning and sterilization cycles for sterilizing
filters should rather be based on a validated process and Quality Risk Management principles. The liquid sterilizing
filter should be validated related to e.g. filter conditioning, filtration time, flow rate, filtration volume, bacterial retention
testing, extractables/leachables and sterilization procedures for the filter. In-line integrity testing of the sterilizing filter
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
should be included in the validation, as well as any cleaning, flushing, drying, hold-times, etc. of the filter in-between
batches and/or after certain periods of use. The validation outcome would include provision for the maximum use of
the sterilizing filter which would be part of a standard operating procedure and/or manufacturing method. Filtration
parameters set during validation for products and/or product groups would subsequently be monitored to ensure proper
functioning of the filter during regular use.
Alternative Proposed text change (2)
Liquid sterilizing filters should be discarded after a defined and validated maximum use period.
Comment (3)
This requirement would prohibit the use of continuous manufacturing and back to back manufacturing systems from
being implemented by the industry. It is clear that filters should not be intended for re-use.
Proposed text change:
8.89 Liquid sterilizing filters should be discarded after the processing of a single lot unless such use has been
validated and is supported by the process risk assessment.
1364 Form-Fill-Seal
1366-1373 Line 1366 to 1373
Existing text:
8.90 Form-Fill-Seal (FFS) units include blow moulding from thermoplastic granulate and thermoforming from
thermoplastic film typically known as Blow-Fill-Seal (BFS) and Vertical-Form-Fill-Seal (VFFS) respectively. VFFS
process is an automated filling process, typically for terminally sterilized processes, that may utilize a single or dual
web system which constructs the primary container out of a flat roll of thermoplastic film while simultaneously filling
the formed bags with product and sealing the filled bags in a continuous process. All such containers are considered
to be sealed by fusion and, as such, fall under the requirement to perform 100% integrity testing.
Proposed text change
8.90 Form-Fill-Seal (FFS) units include blow moulding from thermoplastic granulate and thermoforming from
thermoplastic film typically known as Blow-Fill-Seal (BFS) and Vertical-Form-Fill-Seal (VFFS) respectively. VFFS
process is an automated forming/filling process, typically for terminally sterilized products, that may utilize a single or
dual web system which constructs the primary container out of a flat roll of thermoplastic film while simultaneously
filling the formed bags with product and sealing the filled bags in a continuous process. All such containers are
considered to be sealed by fusion and, as such, fall under the requirement to perform 100% integrity testing.
Comment:
Line 1372: 8.90 VFFS containers are considered to be sealed by fusion. Does that mean that BFS containers are not
considered to be sealed by fusion?
1395-1396 Existing text:
a) Determination of the "critical zone" that should be protected from contamination, and its control.
Comment:
This is open to interpretation. Please can it be stated clearly what is meant? Is it the parison forming and moulding
zone, the filling zone or the shuttle area between both?
1410-1412 Existing text:
8.95 Shuttle and Rotary-type equipment used for aseptic production which is fitted with an effective grade A air
shower should be installed in at least a grade C environment, provided that grade A/B clothing is used.
Comment:
Avoid use of term A/B. Grade B clothing should be worn.
1418-1421 Existing text:

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
8.97 For Rotary-type equipment the environment should comply with the viable and nonviable limits "at rest". It is
not normally possible to perform environmental monitoring within the parison during operation" Monitoring of the
background environment should be performed in accordance with risk management principles
Proposed text change
8.97 For Rotary-type equipment the environment should comply with the viable and nonviable limits "at rest". It is
not possible to perform environmental monitoring within the parison during operation". Controls should be in place to
assure the integrity of the parison e.g. breaches in the parison should not go undetected. Monitoring of the
background environment should be performed in accordance with risk management principles
1427-1429 Existing text:
8.99 In addition, for Shuttle-type designs, the area between parison cutting and mould sealing should be covered by a
flow of HEPA filtered or sterile air of appropriate quality to provide grade A at the critical zone.
Comment:
The sterility of formed containers in the shuttle zone is maintained by the protective heated air currents associated
with the process. Feeding this area with HEPA filtered air may well adversely impact the sterility of the process.
1434-1435 Existing text:
8.101 External particle and microbial contamination of the polymer should be prevented by appropriate design,
control, and maintenance of the polymer storage and distribution systems.
Comment:
More definition is required on the scope or minimum expectation in respect to contamination control.
1442-1444 Existing text:
8.103 Process validation should take into consideration critical operating parameters and variables of the equipment
that impact on the quality of the product, e.g. filling speed, extrusion temperature, filling times.
Proposed text change
8.103 Process validation should take into consideration critical operating parameters and variables of the equipment
that impact on the quality of the product, e.g. filling speed, extrusion temperature, mould vacuum, air supply to
mould, filling times.
1450 Lyophilisation
Comment:
The section contains no guidance for bulk lyophilisation processes as used for some API manufacturing.
1460-1461 Existing text:
8.106 The lyophilizer should be sterilized before each load. The lyophilizer should be protected from contamination
after sterilization.
Proposed text change (1)
8.106 Unless Grade A continuity has been maintained following sterilization, the lyophilizer should be cleaned and
sterilized before each load. The frequency for CIP and SIP should be determined by risk assessment.
Comment (2)
Line 1460: The sterilization processes and manufacturing equipment should be handled and maintained relating to
process and product risks; the design and operations of processes should be based on Quality Risk Management
principles. The lyophilizer use, and the characteristics and risk for the product operated in the lyophilizer, should form
the basis for control and monitoring of the lyophilizer, specifically under consideration of producing sterile APIs.
The following should be considered:
• The frequency for lyophilizer sterilization should be challenged during the regular media fills (aseptic
processing simulation) using worst case considerations.
• Historical and continuous monitoring data from the environment
• Automated loading mechanisms that minimize the risk of contamination versus manual loading mechanisms.

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
• The nature of the items to be lyophilized (e.g. finished product versus API lyophilization as Cephalosporin
and Penicillin).
• The effect on other manufacturing steps, giving the possibility for validated campaign production.
Alternative Proposed text change (2)
The lyophilizer should be sterilized in a defined and validated frequency that is challenged during aseptic process
simulations.
1471-1472 Existing text:
8.110 The integrity of the system should be monitored periodically along with consideration of the leak rate test.
Proposed text change
8.110 The integrity of the system should be monitored periodically along with the leak rate test.
1478-1479 b) Transport to the lyophilizer and loading of filled product, or other equipment into the lyophilizer should take place
under a grade A environment.
Proposed text change
c) Transport to the lyophilizer and loading of filled product, or other equipment into the lyophilizer should take
place under a grade A environment. By first intent automated systems in the absence of human intervention
should be used. Otherwise these operations should be protected by mobile Grade A RABS incorporating
glove ports to prevent direct human intervention.
Comment:
Line 1479: 8.111 b) It should be added after “… place under grade A environment.” “with physical segregation from
operators” to be in compliance with point 8.17.
1481-1483 Existing text:
c) Airflow patterns should not be adversely affected by transport devices and venting of the loading zone. Unsealed
containers should be maintained under grade A environment.
Proposed text change
c) Airflow patterns should not be adversely affected by transport devices and venting of the loading zone. Unsealed
containers should be maintained under grade A environment separated from operators by the use of barrier
technology.
1492 Closed Systems
1499 Comment:
Add physical contamination
Proposed change (if any):Contamination may be microbiological, chemical or physical
1501-1505 Existing text:
8.114 It is critical to ensure the sterility of product contact surfaces of closed systems used for aseptic processing. The
design and selection of any closed system used for aseptic processing must ensure maintenance of sterility.
Tubing/pipework that is not assembled prior to sterilization should be designed to be connected aseptically, e.g. by
intrinsic aseptic connectors or fusion systems.
Proposed text change
8.114 It is critical to ensure the sterility of all product contact surfaces used for aseptic processing, including those of
closed systems. The design and selection of any closed system used for aseptic processing must ensure maintenance of
sterility. Tubing/pipework that is not assembled prior to sterilization should be designed to be connected aseptically,
e.g. by intrinsic aseptic connectors or fusion systems.
1512-1515 Existing text:

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
8.116 The background in which closed systems are located will vary. If there is a high risk that the system will not
remain integral during processing it should be located in a grade A environment. If the system can be shown to
remain integral at every usage then lower grades, including grade D, can be considered.
Comment:
This is confusing, as it seems to suggest that you can accept a sterile system not maintaining its integrity. Surfaces in
Grade A may not be sterile, therefore if a liquid bridge occurs, there will still be a risk to contaminate the product.
While it is appreciated that the risk will be lower, this does not appear to be sound GMP.
1517 Single use Systems
1545-1546 Existing text:
i)Assessment of suppliers of disposable systems (including sterilization of these disposable systems.
Proposed text change
i) Assessment of suppliers of disposable systems, including the sterilization of the equipment. Note these systems may
consist of parts supplied from different manufacturers but received as sterilised assemblies. It is important to ensure
that the manufacturing and change control procedures are adequate and enforced.
1550-1552 Existing text:
8.119 The compatibility of materials used for product contact surfaces with the products should be ensured under the
process conditions by evaluating e.g. adsorption and reactivity to the product.
Comment:
Also need to take into account the physical dimensions of tubing etc. Non-uniform thickness of tubing may lead to
excessive wear and damage when using peristaltic pumps leading to leakage.
1580 Section 9 Viable and non-viable environmental & process monitoring:
General comment:
Overall this section is very light on detail of what regulatory expectations are. For more information and clarity it
should draw from existing peer reviewed guidance literature. As a whole the ordering of the section is a little confused
and requires re-editing.
1584 Comment:
More clarification is required for “process monitoring”.
1594-1597 Existing text:
9.3 These key elements provide information with regards to the process and facility capabilities with respect to the
maintenance of sterility assurance. The information from these systems should be used for routine batch release and
for periodic assessment during process review or investigations.
Proposed text change
9.3 These key elements as well as pressure differential, airflow, temperature and humidity data provide information
with regards to the process and facility capabilities with respect to the maintenance of sterility assurance. The
information from these systems should be used for routine batch release and for periodic assessment during process
review or investigations.
1599 Environmental monitoring
1601-1609 Existing text:
9.4 In order to establish a robust environmental monitoring program, i.e. locations, frequency of monitoring and
incubation conditions (e.g. time, temperature(s) and aerobic and or anaerobic), appropriate risk assessments should
be conducted based on detailed knowledge of the process inputs, the facility, equipment, specific processes,
operations involved and knowledge of the typical microbial flora found, consideration of other aspects such as air
visualization studies should also be included. These risk assessments should be re-evaluated at defined intervals in

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
order to confirm the effectiveness of the site's environmental monitoring program, and they should be considered in
the overall context of the trend analysis and the contamination control strategy for the site.
Comment:
A lot of this section seems to be about viable monitoring…not general monitoring requirements. Incubation
temperature is mentioned but there is no guidance regarding the possibility of selecting different temperature for
different grades.
1611-1614 Existing text:
9.5 Routine monitoring for clean rooms, clean air devices and personnel should be performed "in operation"
throughout all critical stages, including equipment set up. The locations, frequency, volume and duration of
monitoring should be determined based on the risk assessment and the results obtained during the qualification.
Comment:
This paragraph is very much open to interpretation. Routine monitoring within Grade A zones, using viable surface
sampling must only be carried out upon completion of activities. It would be useful to reference the requirements in
standards such as ISO 14644-2.
1621-1623 Existing text:
9.7 For grade A monitoring, it is important that sampling should be performed at locations posing the highest risk of
contamination to the sterile equipment surfaces, container-closures and product in order to evaluate maintenance of
aseptic conditions during critical operations.
Comment:
There is insufficient detail to determine the placement of non-viable monitoring probes. Should they be at a maximum
of 30cm from the point of interest at working height or could they be placed just under the HEPA filters because “they
would otherwise hinder routine activities”?
Proposed text change:
The monitoring of Grade A clean zones should be adequate to demonstrate the maintenance of aseptic processing
conditions during critical operations. Monitoring should be performed at locations posing the highest risk of
contamination to the sterile equipment surfaces, container-closures and product. The selection of monitoring locations
and the orientation and positioning of sampling devices should be justified and appropriate to obtain data from the
critical zones.
1625-1627 Existing text:
9.8 Appropriate alert and action limits should be set for the results of particulate and microbiological monitoring.
Alert levels should be established based on results of Performance Qualification (PQ) tests or trend data and should
be subject to periodic review.
Comment:
Should alert and action limits or alert and action levels be applied? The glossary only refers to levels.
It is unclear how non-viable alert and action levels would be set i.e. against counts per cubic foot or per cubic metre.
Could the word periodic be defined i.e. is the expectation to trend data continuously, weekly, monthly or annually etc?
1629-1631 Existing text:
9.9 The alert limits for grade B, c and D should be set based on the area performance, with the aim to have limits
lower than those specified as action limits, in order to minimise risks associated and identify potential changes that
may be detrimental to the process.
Comment:
What about non-viable alert levels in Grade A?
Comment:
Line 1629: c not in capital letter, please correct.
1633-1636 Existing text:

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
9.10 If action limits are exceeded operating procedures should prescribe a root-cause investigation followed by
corrective and preventive action. If alert limits are exceeded, operating procedures should prescribe scrutiny and
follow-up, which might include investigation and corrective action.
Comment (1):
The actions taken for both alert and action levels look almost identical and will be in most cases of Grade B viable
excursions.
Comment (2):
Line 1634: 9.10 The requirement “if alert limits are exceeded...which might include investigation...” should comply
with the point 6.9 part I of EU GMP, indicating that ” Any out of trend or out of specification data should be
addressed and subject to investigation.”
1637-1639 Existing text:
9.11 Surfaces and personnel should be monitored after critical operations. Results from monitoring should be
considered when reviewing batch documentation for finished product release.
Comment:
Again this paragraph appears to be describing viable monitoring..but it is in the general section.
1642 Non-viable monitoring
General comment:
This section would be better located within section 5 to avoid repetition and confusion. It is understood that it was
placed here to differentiate classification from monitoring, but the overall effect is not helpful.
1651-1654 Existing text:
Table 5: Recommended limits for airborne particle concentration for the monitoring of non-viable contamination
Comment (1):
The table is similar to that in section 5, Premises, but now includes at rest and in operation for 5 micron particles or
greater and reorders the limit columns i.e. “in operation” is listed before “at rest” in this table. To avoid confusion, it
would be more appropriate to have one table within the Premises section and cross refer to it from this section. The
maintenance of the 5 micron particle for monitoring is sensible.
Comment (2):
The Annex refers to ISO14644 for sampling methodology guidance and applies the ISO limits for all cases except for
the 5 micron particulates for Grade A area. There is no scientific rationale for this single discrepancy. ISO limits
should be applied in Table 5.
Proposed change:
Table 5: Recommended limits for airborne particle concentration for the monitoring of non-viable
contamination
Grade Recommended maximum limits Recommended maximum limits
for particles ≧ 0.5 μm/m3 for particles ≧ 5 μm/m3
in operation at rest in operation at rest
A 3 520 3 520 29 29
Comment (3):
Line 1653: Particle counters, which utilise laser light technology, are known to be unable to record absolute numbers
and sizes of particles and trying to count low levels of particles at ≥5μm, is subject to a high degree of uncertainty.
This is reflected in the EN ISO 14644-1(2015) standard where the monitoring of ≥5μm particles for ISO Class 5 is
excluded. It is more appropriate to record particle concentrations at one size only, specifically at the accompanying
≥0.5μm size, where the total allowable number of particles is sufficiently high to negate the inaccuracies of the
particle detection methodology.
The ratios of ≥0.5μm and ≥5μm particles to microbe carrying particles (MCPs) have been established by experiment
and has demonstrated that if the requirement for ≥0.5μm particles and airborne microbial counts (Table 6) should be

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
complied with, the limit required for particles ≥5μm is too low and for each cleanroom grade, the limit for ≥5μm
particles should be increased by a factor of 10. Consequently, particle generating events in cleanroom areas will
produce particle counts that, although compliant with the class limits for ≥0.5μm particles and MCPs, may exceed the
class limit for ≥5μm particles.
Real time monitoring, particularly within EU Grade A zones, is the most useful approach for managing excursions2.
Most particle counters operate at a flow rate of 1ft3/min where the proportioned per ft3 limit for ≥5μm particles is <1.
This anomaly causes considerable difficulties in setting a meaningful operational limit for particles ≥5μm but there are
no such issues if the single particle size of ≥0.5μm is utilised. This would also further harmonise the European
guidelines with those of the United States Food and Drug Administration. It should also be noted that the
recommended maximum limits for particles is not harmonised with the limits defined in the reference document EN
ISO 14644-1(2015).
Proposed change:
1. Measure airborne particle concentrations only at the ≥0.5μm size;
2. Harmonise the recommended maximum limits for particles defined in Table 5 with the limits defined in EN ISO
14644-1(2015);
3. If the document retains the recommendation to also monitor at the ≥5μm size, the per m3 limit for Grade A, B and C
areas should be increased to 200, 20 000 and 200 000 respectively
1655-1657 Existing text:
Note 1: The particle limits given in the table for the "at rest" state should be achieved after a short "clean up" period
defined during qualification in an unmanned state after the completion of operations (see 5.26e).
Comment:
This sentence is almost a repeat from Lines 533 to 535. Please refer to the earlier comment regarding a guidance time
to achieve clean up.
1659-1662 Existing Text:
Note 2: With regards to the monitoring of 5.0 µm, the limit of 20 is selected due to the limitations of monitoring
equipment. It should be noted that alert limits should also be set based on historical and qualification data, such that
frequent sustained recoveries below the action limit should also trigger an investigation.
Proposed text change:
Note 2: With regards to the monitoring of >=5.0 μm, the limit of 20 is selected owing to the sensitivity limitations of
the light scattering airborne particle counter (LSAPC) instrumentation. It should be noted that alert limits should also
be set based on historical and qualification data, such that frequent sustained particle counts, below the action limit
should also trigger an investigation.
Comment:
Note 2 should be “With regards of the monitoring of ≥5.0 μm ...”
Line 1661: It should be indicated what is considered as “frequent sustained recoveries” such as every batch or every
working session.
1667-1669 Existing text:
9.15 The grade A zone should be monitored continuously and with a suitable sample size (at least 28 litres (a cubic
foot) per minute) so that all interventions, transient events and any system deterioration would be captured and
alarms triggered if alert limits are exceeded.
Comment:
Some guidance is needed to understand regulatory expectations for assessing counts. Should it be against cubic foot
limits or per cubic metre limits? This is important to know as the use of the latter would clearly dilute temporarily
high counts (lasting 1 to 5 minutes for example) which would exceed cubic foot limits.

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Comment:
Line 1667: 9.15 should be “...with a suitable sample size (at least 28 litres (a cubic foot) per minute or less) so that all
interventions...”
1671-1676 Existing text:
9.16 It is recommended that a similar system be used for grade B zones although the sample frequency may be
decreased. The design of the monitoring system should be based on risk assessment and be commensurate with the
risk of the process to the product sterility assurance. The grade B zone should be monitored at such a frequency and
with suitable sample sizes that the programme captures any change in levels of contamination and system
deterioration. If alert limits are exceeded, alarms should be triggered.
Comment:
Does the wording "similar system" rule out the use of sequential monitoring devices? The wording “any change in
levels of contamination” may indicate that falls in contamination levels may warrant investigation.
1678-1681 Comments:
Table 5 gives recommended levels, however 9.17 leaves it up to the manufacturer. Please correct ambiguity.
1687-1692 Existing text:
9.19 In the case where contaminants present due to the processes involved would damage the particle counter or
present a hazard, e.g. live organisms and radiological hazards, the frequency and strategy employed should be such
as to assure the environment classification both prior to and post exposure to the risk. Additionally, monitoring
should be performed during simulated operations. Such operations should be performed at appropriately defined
intervals. The approach should be defined in the contamination control strategy.
Comment:
Can the regulatory expectation for the term “appropriately defined intervals” be more accurately defined?
1698-1701 Existing text:
9.21 The sample sizes taken for monitoring purposes using automated systems will usually be a function of the
sampling rate of the system used. It is not necessary for the sample volume to be the same as that used for formal
qualification of clean rooms and clean air devices.
Comment:
More clarity on regulatory expectation is required as different readers will interpret differently.
1703-1705 Existing text:
9.22 Although monitoring of ^ 5.0 pm particles are not required for room qualification and classification purposes, it
is required for routine monitoring purposes as they are an important diagnostic tool for early detection of machine,
equipment and HVAC failure.
Proposed text change:
9.22 Although monitoring of ≧ 5.0 μm particles are not required for room qualification and classification purposes, it
is required for routine monitoring purposes as they are considered to provide some evidence of potential microbial
contamination events; for detection of machine, equipment and HVAC deterioration; and may be indicative of poor
practice during machine set-up and routine operation.
Comment:
Line 1705: It is stated that monitoring of particles ≥5µm is ‘an important diagnostic tool for early detection of
machine equipment and HVAC failure’ This is not correct and the monitoring of particles ≥5µm provides no benefits
to the control of contamination in cleanrooms but causes numerous issues.
Proposed change:
Remove Section 9.22.
1707-1712 Existing text:

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
9.23 The occasional indication of macro particle counts, especially ^ 5.0 pm, may be considered false counts due to
electronic noise, stray light, coincidence, etc. However, consecutive or regular counting of low levels may be
indicative of a possible contamination event and should be investigated. Such events may indicate early failure of the
room air supply filtration (HVAC) system, filling equipment failure, or may also be diagnostic of poor practices
during machine set-up and routine operation.
Proposed text change:
9.23 The occasional indication of macro particle counts, (≧ 5.0 μm), may be considered false counts owing to
electronic noise, stray light, and accumulated particles shed from the sampling system. However, consecutive or
regular counting of low counts may be indicative of a possible contamination event and should be investigated.
1714-1716 Existing text:
9.24 Monitoring conditions such as frequency, sampling volume or duration, alert and action limits and corrective
action including investigation should be established in each manufacturing area based on risk assessment.
Comment:
There is an absence of guidance regarding location or orientation of sampling sensors, tube length, the avoidance of
kinks and radii requirements in tubing etc. see earlier comment re Lines 1621 to 1623.
1718 Viable monitoring:
Comment:
There are scientific papers available proving that impaction volumetric air samplers can vary markedly in their capture
efficiency depending upon their d50 value, yet there is no mention of this attribute in this document.
1720-1722 Existing text:
9.25 Where aseptic operations are performed, microbiological monitoring should be frequent using a combination of
methods such as settle plates, volumetric air, glove print and surface sampling (e.g. swabs and contact plates).
Comment (1):
The term frequent will be open to interpretation. It is suggested that reference is made to a guidance document for
more clarity.
Comment (2):
Line 1721: “...using a combination of methods such as...” should be in accordance with the PICS interpretation PI032-
2 ...using the methods indicated for each grade in Table 6
Comment (3):
This section of the Annex is specifically listing out and limiting sampling methods, whereas in lines 112-116 it
specifically stated that it did not wish to give detailed guidance on sampling methodology. Suggest making slightly
more general to ensure appropriate type of sampling is performed, without being prescriptive on the specific method
to be used.
Proposed text change:
9.25 Where aseptic operations are performed, microbiological monitoring should be frequent using a combination of
methods such as passive air, volumetric air, glove print and surface sampling.
1724-1726 Existing text:
9.26 Monitoring should include sampling of personnel at periodic intervals during the process. Particular
consideration should be given to monitoring personnel following involvement in critical interventions and on exit
from the grade A/B processing area.
Proposed text change
9.26 Monitoring of personnel should follow a defined schedule with particular consideration given following
involvement in critical interventions within the Grade A zone and on exit from the Grade B processing area. For
gowned operators working within Grade B cleanrooms locations such as gloves, arms, goggles, hood and chest must
be sampled using contact plates and/or swabs.

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
1728-1733 Existing text:
9.27 Continuous monitoring in grade A and B areas should be undertaken for the full duration of critical processing,
including equipment (aseptic set up) assembly and filling operations (i.e., an understanding of function and
interactions of each clean area). The monitoring should be performed in such a way that all interventions, transient
events and any system deterioration would be captured and any risk caused by interventions of the monitoring
operations is avoided.
Comment (1):
What is the expectation for continuous Grade B monitoring? Should it be performed using passive or active air
sampling or both? Should it be performed throughout the aseptic area (not just the processing room)? For facilities
which run campaign processing this may mean a significant increase in monitoring with areas being effectively
monitoring for 12 to 24 hours a day.
Note: no viable monitoring program is capable of capturing all risks from transient events, system deterioration etc.
Comment (2):
Line 1728-1730
Viable monitoring, as part of the environmental monitoring program should be based on Quality Risk Management
principles and included in the overall contamination control strategy. It is agreed that for aseptic production operations
viable and non-viable (total airborne) particles should be continuously monitored in critical processing areas in grade A
(with the exception of powder filling operations for which total airborne particles cannot be determined during filling
operations) – whereas the frequency of monitoring in grade B (as well as for grade C and D) should be based on a risk
assessment and product/process requirements - and be aligned with any air-visualisation/smoke studies in the aseptic
manufacturing area. Where and when a grade B area surrounding the critical processing area (grade A) poses a risk of
adding contamination into the critical area - such as during an intervention - monitoring during operations should take
place.
Proposed text change:
Continuous monitoring in the Grade A area should be undertaken for the full duration of critical processing, including
equipment (aseptic set up) assembly and filling operations (i.e., an understanding of function and interactions of each
clean area), wherever product characteristics allow.
Comment (3):
There is currently not a requirement for continuous monitoring in Grade B areas., nor is active viable air continuously
sampled in Grade A, only passive air. We recommend to be able to continue current practices.
Proposed change (if any): Reword as follows: “Continuous monitoring in Grade A area should be undertaken, and
Grade B at intervals determined by risk assessment.
Comment (4):
If the design of the areas includes critical operations or interventions with possible impact on product quality to be
performed in grade B, then its viable monitoring should be equivalent to A. Where B is only the class surrounding
class A, with no critical interventions, then there is no need for the same frequency of monitoring, since the risk and
impact are not the same. Additionally, clarify that continuous monitoring applies to passive air sampling (not
volumetric air).
Proposed text change:
9.27 Continuous passive air monitoring in grade A areas should be undertaken for the full duration of critical
processing, including equipment (aseptic set up) assembly and filling operations (i.e., an understanding of function
and interactions of each clean area). The monitoring should be performed in such a way that all interventions,
transient events and any system deterioration would be captured and any risk caused by interventions of the
monitoring operations is avoided. This could also apply to Grade B areas if surrounding critical operations or
protecting interventions, as determined by the risk assessment.
Comment (5):
Line 1728 & 1751: It is unclear what continuous (viable) monitoring in grade A and B areas relates to. It is not
feasible to continuously monitor surfaces. It is subsequently stated (line 1751) that settle plates should be exposed for
the duration of critical operations. Does exposure of settle plates throughout with supplementary air sampling meet the

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
recommendations for continuous monitoring. If a periodic volumetric air sample is taken, then particle and settle
plate sampling is more than sufficient to demonstrate that microbial contamination is under control.
Proposed change:
Define the expected approach, for settle plates and air sampling to meet the recommendations for continuous (viable)
monitoring in grade A and B areas.
1735-1736 Existing text:
9.28 Rapid microbial monitoring methods may be adopted after validation as long as they are demonstrated to be at
least equivalent to the established methodology.
Comment:
This is the only one of two references to RMMs and may indicate regulatory loss of confidence in these emerging
technologies. This absence of positive promotion will probably encourage companies to continue to use traditional
methodologies.
1738-1739 Existing text:
9.29 Sampling methods should not pose a risk of contamination to the manufacturing operations.
Comment:
Where the only intervention into the Grade zone A is to perform monitoring, can a reduced schedule be adopted to
reduce the risk of contamination? Or if continuous particle counting is carried out, and other checks and controls are
in place (validated airflows, differential pressures monitored) can viable monitoring in such areas be halted?
1741-1742 Existing text:
9.30 Additional microbiological monitoring should also be performed outside production operations, e.g. after
validation of systems, cleaning and disinfection.
Comment:
Does this apply to Grades A and B or all Grades?
1746-1766 Existing text:
Table 6: Recommended maximum limits for microbial contamination
Comment:
This table is repeated from section 5 premises but now includes gloves. Recommend that it appears in a completed
form in one section only. The same comments apply to the values and notes as per section 5. i.e. Replace “limits” with
“levels”. For Grade A the value should be <1 in the table for note (b) to be valid.
Comment (2):
Table 6 “action limits” instead of “recommended maximum limits” and for grade A <1 instead of 1 as in the previous
Annex 1. Table 6 should indicate also and swabs in the head with contact plates such as: Contact plates (diam 55
mm), Swabs, cfu/plate or swab.
As for microbiological qualification, it should be indicated that the viable monitoring incubation programme should
be justified as for APS in lines 1928-1930.- It should be indicated that after sampling the incubation should start as
soon as possible and in a delay not exceeding 4 hours. - Line 1746 Table 6 Add chest and arms with Glove print
following the requirements line 189 point 4.3 and line 199 point 4.4.
Comment (3):
Limits for gown monitoring in Grades A and B are not included
Proposed change (if any): Include limits for gown monitoring in table 6.
Comment (4):
Recommend consistent use of the word limit or level. Are they interchangeable? If so, based on the glossary
definition, recommend adding a <1 for the limits/levels to Grade A.
Proposed change (if any): Glossary definition: Action Level - An established microbial or airborne particle level that,
when exceeded, should trigger appropriate investigation and corrective action based on the investigation.
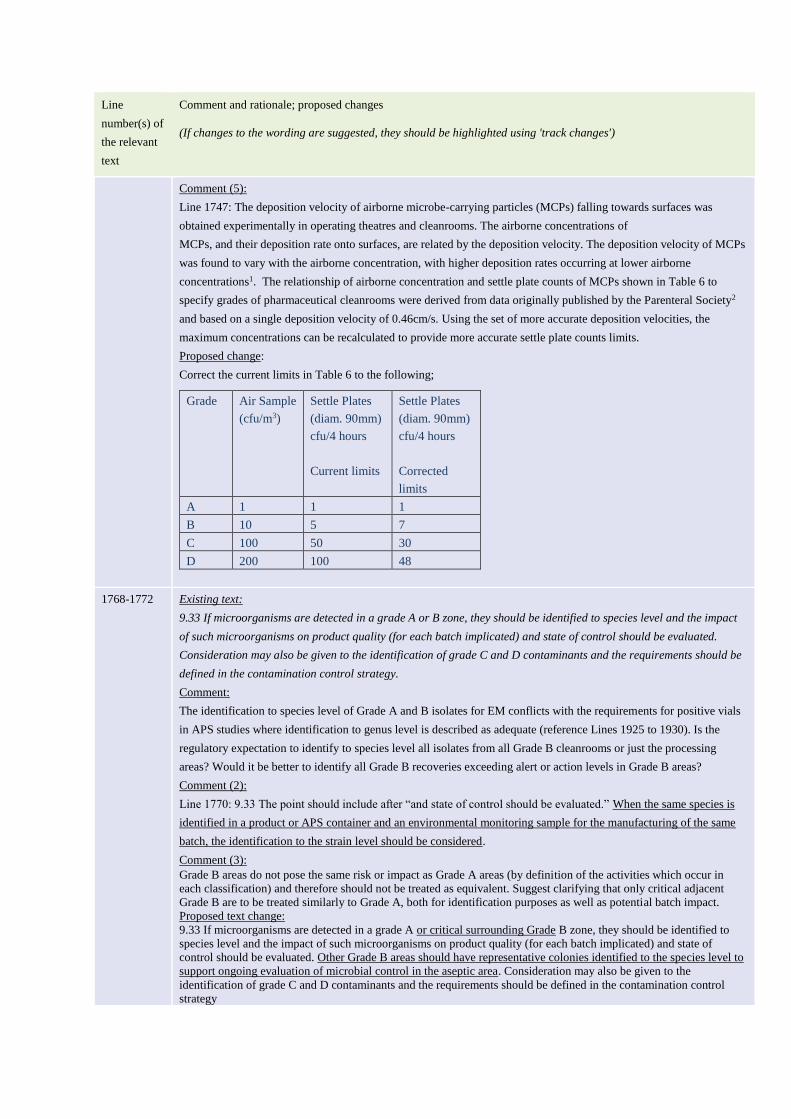
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Comment (5):
Line 1747: The deposition velocity of airborne microbe-carrying particles (MCPs) falling towards surfaces was
obtained experimentally in operating theatres and cleanrooms. The airborne concentrations of
MCPs, and their deposition rate onto surfaces, are related by the deposition velocity. The deposition velocity of MCPs
was found to vary with the airborne concentration, with higher deposition rates occurring at lower airborne
concentrations1. The relationship of airborne concentration and settle plate counts of MCPs shown in Table 6 to
specify grades of pharmaceutical cleanrooms were derived from data originally published by the Parenteral Society2
and based on a single deposition velocity of 0.46cm/s. Using the set of more accurate deposition velocities, the
maximum concentrations can be recalculated to provide more accurate settle plate counts limits.
Proposed change:
Correct the current limits in Table 6 to the following;
1768-1772 Existing text:
9.33 If microorganisms are detected in a grade A or B zone, they should be identified to species level and the impact
of such microorganisms on product quality (for each batch implicated) and state of control should be evaluated.
Consideration may also be given to the identification of grade C and D contaminants and the requirements should be
defined in the contamination control strategy.
Comment:
The identification to species level of Grade A and B isolates for EM conflicts with the requirements for positive vials
in APS studies where identification to genus level is described as adequate (reference Lines 1925 to 1930). Is the
regulatory expectation to identify to species level all isolates from all Grade B cleanrooms or just the processing
areas? Would it be better to identify all Grade B recoveries exceeding alert or action levels in Grade B areas?
Comment (2):
Line 1770: 9.33 The point should include after “and state of control should be evaluated.” When the same species is
identified in a product or APS container and an environmental monitoring sample for the manufacturing of the same
batch, the identification to the strain level should be considered.
Comment (3):
Grade B areas do not pose the same risk or impact as Grade A areas (by definition of the activities which occur in
each classification) and therefore should not be treated as equivalent. Suggest clarifying that only critical adjacent
Grade B are to be treated similarly to Grade A, both for identification purposes as well as potential batch impact.
Proposed text change:
9.33 If microorganisms are detected in a grade A or critical surrounding Grade B zone, they should be identified to
species level and the impact of such microorganisms on product quality (for each batch implicated) and state of
control should be evaluated. Other Grade B areas should have representative colonies identified to the species level to
support ongoing evaluation of microbial control in the aseptic area. Consideration may also be given to the
identification of grade C and D contaminants and the requirements should be defined in the contamination control
strategy
Grade Air Sample
(cfu/m3)
Settle Plates
(diam. 90mm)
cfu/4 hours
Current limits
Settle Plates
(diam. 90mm)
cfu/4 hours
Corrected
limits
A 1 1 1
B 10 5 7
C 100 50 30
D 200 100 48

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
1774 Aseptic process simulation (APS)1
Comment:
There is an absence of guidance for bulk manufacturing aseptic processes such as API manufacture
1776-1786 Existing text:
9.34 Periodic verification of the effectiveness of the controls in place for aseptic processing should include a process
simulation test using a sterile nutrient media and/or placebo. Selection of an appropriate nutrient media should be
made based on the ability of the media to imitate product characteristics at all processing stages. Where processing
stages may indirectly impact the viability of any introduced microbial contamination, (e.g. sterile aseptically
produced semi-solids, powders, solid materials, microspheres, liposomes and other formulations where product is
cooled or heated or lyophilized, etc.), alternative surrogate procedures that represent the operations as closely as
possible can be developed and justified. Where surrogate materials, such as buffers, are used in parts of the process
simulation, the surrogate material should not inhibit the growth of any potential contamination.
Comment (1):
The last sentence needs clarification. It is recommended that only the open stages of a bulk process should be subject
to an APS. As written there could be an expectation to seed API closed systems with media.
Comment (2)
Line 1777: and/or placebo should be deleted since it not further explained how a microbial growth can be expected
with this placebo and this is not indicated in PICS PI007, PDA TR22 or ISO 13408-1.
Comment (3):
The requirement that the media mimic product characteristics at all processing stages can be misconstrued to mean
that it should imitate characteristics such as pH, viscosity, density and others, which could make it unsuitable for the
recovery of microorganisms. Suggest clarification of the intent.
Proposed text change:
9.34 Periodic verification of the effectiveness of the controls in place for aseptic processing should include a process
simulation test using a sterile nutrient media and/or placebo. Selection of an appropriate nutrient media should be
made based on the ability of the media to recover microbiological contaminants potentially present in the processing
environment. Where processing stages may indirectly impact the viability of any introduced microbial contamination,
(e.g. sterile aseptically produced semi-solids, powders, solid materials, microspheres, liposomes and
other formulations where product is cooled or heated or lyophilized, etc.), alternative surrogate procedures that
represent the operations as closely as possible can be developed and justified. Where surrogate materials, such as
buffers, are used in parts of the process simulation, the surrogate material should not inhibit the growth of any
potential contamination.
1791-1792 Existing text:
a) Process simulation tests should assess all aseptic operations performed subsequent to the sterilisation of materials
utilised in the process.
Comment:
Same comment as above for lines 1776 to 1786.
1794 Comment
9.35 b) Should be “For non-filterable formulations or products, or after the last sterile filtration, any additional aseptic
steps should be assessed.
1796-1797 Existing text:
c) Aseptic manufacturing performed in a strict anaerobic environment should be evaluated with an anaerobic media
in addition to aerobic evaluation.

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Comment :
Processes conducted under a nitrogen atmosphere to minimise the risk of contamination will now require two media
fills: aerobic and anaerobic. This is contrary to all conventional approaches which require only an aerobic media fill.
1799-1801 Existing text:
d) Processes requiring the addition of sterile powders should employ an acceptable surrogate material in containers
identical to those utilised in the process being evaluated.
Proposed text change
Unless closed systems are employed for transfer, processes requiring the addition of sterile powders should employ an
acceptable surrogate material in containers identical to those utilised in the process being evaluated.
1803-1804 Existing text:
e) Processes involving blending, milling and subdivision of a sterile powder require similar attention.
Proposed text change
e) Processes involving open blending, open milling and open subdivision of a sterile powder, by which the powder is
exposed to the processing environment, require similar attention.
1805
footnote
Existing text:
1 For further details on the validation of aseptic processing, please refer to the PIC/S Recommendation on the Validation of Aseptic
Processing (PI 007) For PICS version only
Comment:
It is noted that this document and ISO14644 are the only guidance documents referenced in this revision.
1808-1810 Existing text:
The process simulation should duplicate the lyophilization process, with the exception of freezing and sublimation,
including partial vacuum and cycle duration and parameters as appropriate for the media.
Comment (1):
Use of the word “duplicate” could cause confusion, especially with using the term “cycle duration” in the same
sentence. The step of applying partial vacuum and vacuum release might be performed in duplicate, since those steps
are generally considered the most critical process steps in regard to possible microbial ingress. However, it must be
clear that the length of the simulated freezing and sublimation phase should be reasonable and not exceed a few hours.
The wording as currently proposed might suggest that the length of those process steps (that for some products take
several days) are to be simulated as long as the partial vacuum does not affect the media.
Proposed text change:
The process simulation should correspond as closely as possible to the regular lyophilization process, with the
exception of freezing and sublimation, using partial vacuum and parameters as appropriate for the media, and with the
exception of the total cycle duration, which may be reduced. Certain critical process steps, such as the application and
release of vacuum might be instead duplicated during the simulation.
Comment (2):
9.35 f) “...including partial vacuum and cycle duration...” cycle duration should be deleted since some lyophilization
cycles can take more than one day and there is no risk of microbial contamination inside the chamber if all the
accepted steps are simulated.
1819-1820 Existing text:
b) Corrective interventions in representative number and with the highest degree of intrusion acceptable.
Proposed text change
This should not be interpreted as validating poor practise i.e. undertaking an excessive number of intrusions in the
APS to justify the same in routine production. Passing an APS is not the same as confirming sterility, this point needs
to be made.

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
1822-1825 Existing text:
9.37 There should be an approved list of allowed interventions, both inherent and corrective, which may occur during
production and in the APS. The procedures listing the types of inherent and corrective interventions, and how to
perform them, should be updated, as necessary, to ensure consistency with the actual manufacturing activities.
Comment:
The approved list should be supported by risk assessment (FMEA) which provides confidence in the acceptability of
the intervention.
1827-1828 Comment:
Please clarify where it states “risk principles should be used” is this requiring a risk assessment?
1830-1832 Comment:
Please define “variables”.
1835 Comment:
“Bracketing or a matrix approach can be considered for initial validation of the same container/closure configuration.”
This approach could also be used for different container/closure configuration if justified.
1838-1840 Existing text:
c) The volume filled per container, which should be sufficient to ensure that the media contacts all equipment and
component surfaces that may directly contaminate the sterile product.
Proposed text change
c) The volume filled per container, which should be sufficient to ensure that the media contacts all equipment and
component surfaces that may directly contaminate the sterile product and permit the detection of growth.
1845 Existing text:
e) Ensuring that any contamination is detectable.
Comment:
No APS can ensure all contamination is detectable.
Comment:
This sentence is too general.
Proposed change (if any): Reword as follows:
“Ensuring that any contamination is detectable by performing Growth Promotion Test on the filled containers at the
APS
1847-1848 Existing text:
f) The requirement for substitution of any inert gas used in the routine aseptic manufacturing process by air, unless
anaerobic simulation is intended.
Comment:
Does this line contradict lines 1796 to 1797?
1850-1854 Existing text:
g) The duration of the process simulation filling run to ensure it is conducted over the maximum permitted filling time.
If this is not possible, then the run should be of sufficient duration to challenge the process, the operators that perform
interventions, and the capability of the processing environment to provide appropriate conditions for the manufacture
of a sterile product.
Comment:
More clarity is required. Is the regulatory expectation to carry out APS studies for the full duration of processing or be
of sufficient time to permit all interventions and critical operations to be performed?
1856-1858 Existing text:
h) Simulating normal aseptic manufacturing interruptions where the process is idle. In these cases, environmental
monitoring should be conducted to ensure that grade A conditions have been maintained.
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
Comment:
Does this simply mean carry out Grade A monitoring throughout the APS study, including when the process has
temporarily stopped? If so does this include continuous Grade A monitoring for campaign processing that are halted
over-night?
Comment:
Does continuous particulate monitoring and settle plates fulfill this requirement??
1860 Existing text:
i) The special requirements and considerations for manually intensive operations.
Comment (1):
Please clarify what is meant by the “special requirements and considerations”?
Comment (2):
“The special requirements and considerations for manually intensive operations.” Manually intensive operations
should not be considered as an acceptable aseptic process if not performed with a barrier technology.
1862-1868 Existing text:
j) Where campaign manufacturing occurs, such as in the use of barrier technologies or manufacture of sterile active
substances, consideration should be given to designing and performing the process simulation so that it simulates the
risks associated with both the beginning and the end of the campaign and demonstrating that the campaign duration
does not pose any risk. If end of production campaign APS are used, then it should be demonstrated that any residual
product does not negatively impact the recovery of any potential microbiological contamination.
Proposed text change:
Where closed systems and/or barrier technologies such as Isolators and RABS are employed then it is possible to
carry out aseptic campaign manufacturing. In such cases consideration should be given to designing and performing
the process simulation so that it simulates the risks associated with both the beginning and the end of the campaign
and demonstrating that the campaign duration does not pose any risk. If end of production campaign APS are used,
then it should be demonstrated that any residual product does not negatively impact the recovery of any potential
microbiological contamination.
Comment:
Does line 1862 to 1868 contradict lines 1850 to 1854 in that full duration of APS is not necessarily required.
1870-1873 Existing text:
k) Where barrier technologies (RABS, isolators, BPS, etc.) are used in the routine aseptic manufacturing process, the
relative risk and unique aspects of these technologies should be taken into consideration when assessing the design of
aseptic process simulation tests.
Proposed text change
k) Where barrier technologies (RABS, isolators, BPS, etc.) are used in the routine aseptic manufacturing process, the
relative reduced risk and unique aspects of these technologies, compared with open processing, should be taken into
consideration when assessing the design of aseptic process simulation tests.
1875-1880 Existing text:
9.39 For sterile active substances, batch sizes should be large enough to represent routine operation, simulate
intervention operation at the worst case, and cover potential contact surfaces. In addition, all the simulated materials
(surrogates of growth medium) should be subjected to microbiological evaluation. The recovery rate from simulation
materials should be sufficient to satisfy the evaluation of the process being simulated and should not compromise the
recovery of micro-organisms.
Comment:
Where closed systems are used which are subject to SIP and protected by integral filters, it should not be desirable to
contact these sterile surfaces with growth media.
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
1882-1889 Existing text:
9.40 Process simulation tests should be performed as initial validation, generally with three consecutive satisfactory
simulation tests per shift, and after any significant modification to the HVAC system, equipment, major facility shut
down, process and number of shifts, etc. Normally process simulation tests (periodic revalidation) should be repeated
twice a year (approximately every six months) for each aseptic process and filling line, and at least annually for each
operator. Consideration should be given to performing an APS after the last batch prior to shut down, before long
periods of inactivity or before decommissioning or relocation of a line.
Comment:
Could the regulatory definition of a shift be provided? Is it a group of persons or a period of time within a day?
Agree with performing an APS before long periods of inactivity or before decommissioning or relocation of a line, but
not prior to a planned shut-down.
1891-1894 Existing text:
9.41 Where manual filling occurs, each product, container closure, equipment train and operator should be
revalidated approximately every 6 months. The APS batch size should mimic that used in the routine aseptic
manufacturing process. An aseptic process or filling should be subject to a repeat of the initial validation when:
Comment:
There is no mention of bulk operations which involve tray drying and with manual handling and transfers.
1905-1909 Existing text:
9.42 The number of units processed (filled) for process simulation tests should be sufficient to effectively simulate all
activities that are representative of the aseptic manufacturing process; justification for the number of units to be filled
should be clearly captured in the PQS. For small batches, e.g. those under 5,000 units filled, the number of containers
for media fills should at least equal the size of the production batch.
Comment:
The minimum number of units to be filled for a valid trial is not mentioned anymore. Although specifying the
minimum and maximum number may not be totally science-based, it is helpful to align everyone to regulatory
expectations. Both regulators and manufacturers need to be clear on this important aspect.
1911-1918 Comment:
Line 1914 states the number of APS repeats should be determined using QRM principles however line 1916 states 3
successful repeats would be expected.
1920-1924 Existing text:
9.44 Filled APS units should be agitated, swirled or inverted before incubation to ensure contact of the media with all
interior surfaces in the container. Cosmetic defects, non- destructive weight checks and all other units should be
identified and incubated with the other units. Units discarded during the process simulation and not incubated should
be comparable to units discarded during a routine fill.
Comment:
There is no requirement to incubate in an inverted orientation such that the media is in contact with the container
closure. This may be unacceptable for heat sealed ampoules owing to the fragile nature of the fusion closure, but
should be possible with other dosage form presentations.
Comment (2):
Line 1924: It could be indicated that the same number of units incubated must be read at the end of the incubation.
1925-1930 Existing text:
9.45 Filled APS units should be incubated in a clear container to ensure visual detection of microbial growth.
Microorganisms isolated from contaminated units should be identified to at least the genus, and to the species level
when practical, to assist in the determination of the likely source of the contaminant. The selection of the incubation
Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
duration and temperature should be justified and appropriate for the process being simulated and the selected growth
medium.
Proposed text change
9.45 Filled APS secondary units should be incubated in a clear container to ensure visual detection of microbial
growth. It is recognised that this may not be possible for bulk API containers which will therefore require opening and
sampling at the completion of incubation to allow for the detection of growth. Microorganisms isolated from
contaminated units and containers must be identified to the species level, to assist in the determination of the likely
source of the contaminant as part of the APS investigation. The selection of the incubation duration and temperature
should be justified and appropriate for the process being simulated and the selected growth medium.
Comment:
Opaque containers are not addressed. Please include clear and/or opaque.
1952 10 Quality Control (QC)
1958-1961 Existing text:
10.2 The bioburden assay should be performed on each batch for both aseptically filled product and terminally
sterilized products and the results considered as part of the final batch review. There should be working limits on
contamination immediately before sterilization, which are related to the efficiency of the method to be used.
Comment (1):
Clarity is required here. Does the bioburden assay for an aseptically filled product refer to the pre sterile filtration
value? Also should the bioburden assay sample for a terminally sterilised product be taken immediately prior to
sterilisation or, if cold chained and controlled with adequate monitoring records, at an appropriate justified time? This
would be applicable to items sterilised by third party contractors. If the prefiltration bioburden limit is expected to be
NMT10cfu/100ml, this should be stated for clarity.
Comment (2):
Line 1958: 10.2. “The bioburden assay should be performed on each batch for both….” Does that mean that the
bioburden should be evaluated even if the product cannot be sterile filtered?
1974-1976 Existing text:
10.6 The sterility test should be performed under aseptic conditions, which are at least consistent with the standard of
clean room required for the aseptic manufacture of pharmaceutical products.
Comment:
The background environment for isolator sterility testing cabinets will be required to meet Grade C or Grade D if it is
required to be commensurate with the manufacturing environment. Please confirm if the wording,” the sterility testing
environment should be suitable to minimise the contamination risk to the test” is more acceptable?.
Comment:
Clarification is needed to ensure that Sterility testing isolators are not required to be installed in a Grade D
background environment. All material loaded into a sterility testing isolator is fully sealed and often contains
additional protection, and as a result does not require the protection of a Grade D background environment.
Proposed text change:
10.6 The sterility test should be performed under aseptic conditions, which are at least consistent with the standard of
clean room required for the aseptic manufacture of pharmaceutical products. When sterility testing isolators are used,
they can be located in an unclassified area.
1978-1983 Existing text:
10.7 Samples taken for sterility testing should be representative of the whole of the batch, but should in particular
include samples taken from parts of the batch considered to be most at risk of contamination, for example:

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
a) Products which have been filled aseptically, samples should include containers filled at the beginning and end of
the batch and after any significant intervention.
Comment:
If the sampling plan should represent the entire batch and processing conditions in the absence of intervention and
other anomalies, shouldn’t samples also be required from the middle of processing? Regulatory guidance is required
on numbers of samples or a justification for these numbers to be taken following interventions.
1985-1986 Existing text:
b) For products which have been heat sterilized in their final containers, consideration should be given to taking
samples should be taken from the potentially coolest part of the load.
Proposed text change
b) For products which have been heat sterilized in their final containers, samples should be taken from the potentially
coolest part of the load.
1988-1989 Existing text:
c) Each sterilized load should be considered as different batches and require a separate sterility test.
Comment (1)
Detailed requirements for finished product testing should be based on Quality Risk Management principles. Requiring
separate sterility tests for all sterilizer loads, regardless of type of sterilization process and/or controls in place, would
lead to significantly more sterility testing for certain terminally sterilized products compared to aseptically processed
products, which does not reflect the sterility assurance risks associated with the different processes types.
As an example: For certain flexible multi-chamber Large Volume Parenterals (LVP) bags for Total Parenteral Nutrition
(TPN), containing up to 2.5 litres in volume, the regular sterilization operations can require more than 20 individual
autoclave/sterilizer loads for a single set of preparation vessels, which are filled in less than 24 hours. The European
Pharmacopoeia’s requirement for sterility testing of LVP, i.e. units of more than 100ml container size, sets the
requirement of a minimum of 10 units per batch being tested for sterility. This will lead to 200 LVP bags part of 20
individual sterility tests for one lot of product.
Having a mandatory separate sterility test per sterilized load for terminally sterilized products would be unreasonably
more units tested compared to purely aseptically performed manufacturing processes (which are generally recognized
being higher risk processes in regard to sterility assurance).
It is agreed that each individual sterilized load must be represented in a sterility test, but the way of sampling and
whether individual or pooled sterility tests are performed should be based on control measures implemented, as e.g.:
• Appropriate design of the sterilization area with usage of double-door autoclaves that separate a (fully
automated) pre-sterilization loading area from a (fully automated) post-sterilization unloading area.
• Appropriate physical (temperature distribution and heat penetration) and microbiological validation of the
sterilization cycles for well-defined loading patterns.
• Appropriate cycle controls including independently monitoring.
• Appropriate pre-sterilization bioburden controls, including separate screening for the presence of spore
forming microorganisms (heat resistance test) where required.
Wherever a sterilization process and its control mechanisms are well established and re-validation data show consistent
and uniform heat penetration throughout autoclave loads (into all individual product units) an individual sterility test
per sterilized load does not add to the sterility assurance of the finished product.
Proposed text change:
Each sterilized load should be appropriately represented in the test for sterility.
Comment (2):
It is assumed that this only applies to TS products (which are not approved for parametric release) and not for example
vial stopper loads to be utilised in aseptic processing. There is no guidance provided for campaign filling where a
single batch may be aseptically filled over two days: Does this require a minimum of 20 units (if sufficient fill
volume/mass) to be sampled and tested per day?

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
1993-1994 Existing text:
Note: Where sterilization or lyophilization leads to separate sterility tests, consideration of performing separate
testing for other finished product tests should also be given.
Comment:
This is too prescriptive and should be part of the overall company process qualification.
1999-2000 Existing text:
10.9Media used for environmental monitoring and APS should be tested for its growth promotion capability, in
accordance with a formal written program.
Comment (1):
Guidance required for when this should be performed. Prior to use only or post use also
Comment (2):
Line 2000: It should be indicated that for settle plates used in grade A the growth promotion test should be performed
after a 4 hours exposure under grade unidirectional airflow and at the end of the incubation.
2002-2005 Comment:
Grade B areas do not pose the same risk or impact as Grade A areas (by definition of the activities which
occur in each classification) and therefore should not be treated as equivalent. Suggest clarifying that only
critical adjacent Grade B are to be treated similarly to Grade A with regards to batch review. Furthermore,
the Annex does not prescribe environmental monitoring specifications, only the recommended maximum
limits – need to maintain consistent terminology.
Proposed text change:
10.10 Environmental monitoring data generated in critical processing areas (Grade A and critical adjacent Grade B)
should be reviewed
as part of product batch release. A written plan should be available that describes the actions to be taken when data
from environmental monitoring are found out of trend or exceeding the maximum recommended limits.
2007-2009 Existing text:
10.11 The use of rapid microbial methods can also be considered. These methods should be validated for the
product(s) or processes concerned and be approved in the registered product testing specification.
Comment (1):
Only the second short reference to RMMs.
Comment (2):
It should be indicated that for sterile products with short shelf-life and used before the end of the sterility test, a rapid
microbial method must be used if possible.
Glossary Comment (1):
The glossary is very limited and requires a broad edit.
Examples:-
Action Limits are referred to throughout the document, but are called Action Levels in this section.
An intervention is described as a manipulation or activity that occurs at the critical area, but this can be less of a risk if
performed through glove ports as opposed to via an open barrier door etc. Both types of intervention require defining.
HEPA filter – should be defined as:-A High efficiency particulate air filter. An H14 filter in accordance with EN 1822
has an integral collection efficiency of 99.995%, and a local collection efficiency of 99.975% at the most penetrating
particle size (MPPS). It should be defined as per EN 1822.
Isokinetic sampling head –should be defined as:- A sampling head designed to disturb the air as little as possible by
ensuring that the air velocity entering the probe is equal to the velocity of undisturbed air at the same point.
Isolator -should be described as:- A bio-decontaminated enclosed environment supplied with Grade A (ISO 5) or
higher air quality that provides uncompromised, continuous isolation of its interior from the external environment
(e.g., surrounding cleanroom air and personnel).

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
ULPA filter should be described as:- ULPA filter - Ultra-low penetration air filter. An U15 filter in accordance with
EN 1822 has an integral collection efficiency of 99.9995%, and a local collection efficiency of 99.9975% at the most
penetrating particle size (MPPS).
Pass through hatch should be described as:- A small airlock into which only small goods can be placed.
Comment:
Line 2040:
The concentration is intended as CFU/cm2 of filter surface.
Proposed change (if any): Correct the definition, replace CFU/ ml by CFU/cm2
Comment (2):
The cleaning and EM in a CNC area should be defined by the company’s QRM principles where appropriate.
Line 2040-2042
Comment (3)
Correction: the microbial challenge should be expressed as a concentration per cm2 of the filter membrane
and not per volume to be filtered as all filter validations are performed to scale and not at actual filling
volumes.
Proposed text change:
Bacterial retention testing – This test is performed to validate that a filter can remove bacteria from a gas
or solution. The test is usually performed using a standard organism, such as Brevundimonas diminuta at a
minimum concentration of 107 Colony Forming Units/cm2.
Line 2076-2084
Existing text:
Clean Non Classified (CNC) area - An area that does not meet any of the formal predetermined grades of cleanliness
included in the Annex, i.e. grades A to D, but where a manufacturer defined level of microbial control is still required.
The area should be subject to a formal cleaning/disinfection regime and formal environmental monitoring program to
achieve the defined level of control. The level, type and frequency of both the cleaning program and the environmental
monitoring program (including contamination limits) should be based on a formal risk assessment (captured within the
wider contamination control.
strategy) and should be commensurate with the specific risks to the processes and product performed manufactured
within each CNC area.
Comment (1):
Areas not classified within the A, B, C or D grades as defined in the Annex 1 do not specifically require environmental
monitoring programs and/or limits for microbial or particulate contamination levels, which is the reason they are
controlled but not classified. The controls in place for these areas could range from temperature and/or humidity to
certain gowning requirements such as shoe covers, protective coats or safety glasses. The definition of appropriate
control measures of any CNC area should be under the responsibilities of the manufacturer and need to be aligned with
the overall contamination control strategy.
Proposed text change:
Controlled Non Classified (CNC) area - An area that does not meet any of the formal predetermined grades of cleanliness
included in the Annex, i.e. grades A to D, but where a manufacturer defined level of control is still required. The area
should be subject to a formal cleaning/disinfection regime and hygienic requirements as applicable (e.g. related to
gowning requirements) to achieve the defined level of control. The level, type and frequency of the cleaning program
and any monitoring programs (e.g. temperature, humidity) should be based on a formal risk assessment (captured within
the wider contamination control strategy) and should be commensurate with the specific risks to the processes and
product manufactured within each CNC area.”
Note: ch. 5.9 b iii in the draft Annex 1 would need to be revised with the same CNC definition, based on the proposed
change above.
Comment (2):
CNC areas should have procedures in place to control gowning, sanisation and personnel and material flow in order to
ensure the any potential contamination carried into the classified areas is minimized. It is excessive however to require
that these areas be periodically monitored to non-specific arbitrary levels. The environmental monitoring results for
the adjacent Grade D and C areas is the best indication of the effectiveness of the controls in place and are much more
frequent and reliable. Additionally, suggest rewording last sentence as there are normally no manufacturing processes

Line
number(s) of
the relevant
text
Comment and rationale; proposed changes
(If changes to the wording are suggested, they should be highlighted using 'track changes')
which take place within the CNC classified areas. Also suggest that the requirements contained within this definition
also be cited within the text of the document to ensure it is consistently applied (possibly between section 5.14 and
5.15).
Proposed text change:
Clean Non Classified (CNC) area - An area that does not meet any of the formal pre determined grades of cleanliness
included in the Annex, i.e. grades A to D, but where a level of environmental control is still required. The area should
be subject to a formal cleaning/disinfection regime and formal gowning and access control program to achieve the
defined level of control. The level, type and frequency of the cleaning program should
be based on a formal risk assessment (captured within the wider contamination control strategy) and should be
commensurate with the specific risks to the processes and product manufactured
Line 2079
Comment:
Instruction for environmental monitoring programme requirements in CNC areas where required should be included
in section 9, not the glossary
Line 2145-2147
Comment:
Suggest that the requirements contained within this definition also be cited within the text of the document to ensure it
is consistently applied (possibly between section 8.22 and 8.23).
Line 2177-2178
Comment:
The term “Laminar flow” should be removed from this document in favour of “Unidirectional flow” as it reflects the
current ISO 14644 terminology.
Proposed change:
Delete the definition and replace throughout the document
Line 2217-2220
Existing text
Sterile Product - For purposes of this guidance, sterile product refers to one or more of the elements exposed to aseptic
conditions and ultimately making up the sterile finished drug product. These elements include the containers, closures,
and components of the finished drug product.
Comment:
Sterile products can be manufactured via terminal sterilization processes of the finished product in its sealed containers.
In processing terminal sterilized products there would be no need of aseptic processing conditions.
Proposed text change:
Sterile Product - For purposes of this guidance, sterile product refers to one or more of the elements ultimately making
up the sterile finished drug product. These elements include the containers, closures, and components of the finished
drug product.