studies related to identification of toxic metabolites of
TRANSCRIPT

Studies Related to Identification of Toxic Metabolites of Benzene and
Their Chemical Reactivity
SUMMARY SUBMITTED TO THE
DEPARTMENT OF BIOCHEMISTRY (Faculty of Life Sciences)
ALIGARH MUSLIM UNIVERSITY, ALIGARH FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY IN BIOCHEMISTRY
By
PETROLEUM TOXICITY DIVISION
Industrial Toxicology Research Centre Lucknow (India)
m

Summary
Small amounts of a large variety of substances normally regarded as foreign to the
body are always existent in the environment. These are often toxic and occur frequently.
In the modern world, man is increasingly dependent upon the use of synthetic chemicals
and other domestic products in larger number.
Benzene is frequently used as an industrial solvent of the modem time. Its excellent
solvent properties and its use as a starting material for wide ranging chemicals and
domestic products of commercial importance has made it indispensable. Its value in our
modern society is great, but due to its toxic nature, the use of t)enzene would undoubtedly
have an impact on our way of life. The use of benzene has been on the rise and
obviously the risk of exposure is also increasing. Though the exposure levels to benzene
can be brought down to acceptable levels but can not be eliminated.
Benzene is a well known genotoxic and carcinogenic agent. But the precise
mechanism of its genotoxicity and carcinogenicity are yet to be known. Therefore, in this
study an attempt has been made to explain the probable metabolite(s) of benzene to
induce haematotoxicity and role of transition metal ions (iron and copper) to explain the

Summary 2
possible biochemical mechanisms of genoloxicity and carcinogenicity expressed during
benzene exposure.
Release of iron from ferritin by 1,2,4-benzenetriol
Iron is a vital metallic cofactor of several important biomolecules. It is normally
localized in ferritin (main iron storage protein) or transferrin (iron transport protein). But
this iron, when it gets decompartmentalized manifests lethally by triggering off free radical
reactions by means of Fenton and Haber-Weiss type of reactions:
Fe(lll) + Reductant > Fe(ll) + Oxidized Reductant
2Fe(ll) + O2+ 2H* > 2Fe(lll) + H A
Fe(ll) + HjOj -— > Fe(lll) +-OH + OH"
Large human prospective studies suggest that excess body free iron is associated
v/ith carcinogenesis, such as primary hepatocellular carcinoma, colon cancer, lung cancer,
bladder cancer, acute lymphocytic leukemia and neuroblastoma. Bleomycin detectable
iron has been shown to accumulate in significant concentrations in the bone marrow of
benzene exposed rats. Polyphenolic metabolite of benzene, particularly 1,2,4-
benzenetriol (a Irihydroxy benzene) was found to release significant amount of iron from
horse spleen ferritin under aerobic and anaerobic conditions. The release of iron from
ferritin during autooxidation of 1,2,4-benzenetriol in wvo may result in increased
intracellular concentrations of free iron. Although, the physiological mechanism of iron
mobilization from ferritin is pooriy understood, the observed iron release from ferritin by
1,2,4-benzenetriol could occur through the direct reduction of iron by the hydroxy
hydroquinone form or via its autooxidation intermediates i.e., superoxide radical and
semiquinone It was observed that the iron released from ferritin in the presence of
1,2,4-benzenelriol was capable of inducing lipid peroxidation and catalyzing
bleomycin-dependenl calf thymus DNA degradation. The mechanism by which iron is
released from bone marrow cells and whether it acts as a prooxidant during benzene
toxicity is not known. However, the present observation offer a new mechanism that the
iron released from ferntin by 1,2,4-benzenetriol could contribute to the better

Summary 3
Polyphenol-iron complex: A probable toxic metabolite of benzene
An attempt was made to examine in vitro formation of polyphenoi:iron complex.
Complex formation was fractionated on Sephadex G-10 column chromatography. Results
clearly show the formation of polyphenoUron complex under in vitro condition. Polyphenol
iron complex formation was also observed during the release of iron from ferritin in the
presence of 1,2,4-benzenetriol. Although, benzene exposure leads to accumulation of
iron as well as polyphenolic metabolites of benzene in the bone marrow against the
concentration gradient, the formation of such complex in vivo is yet to be identified.
We evaluated the haematotoxicity of hydroquinone:iron complex as a possible
haematotoxic intermediate during benzene exposure. Results indicate that hydroquinone
iron chelate showed marginal increase in haematological parameters in experimental
animals But Ihe precise mechanism of marginal, yet significant increase in haema
tological indices is not clear, Atleast, some of the effects observed in hydroquinone:iron
treated group showed similarities to some extent with what has been shown to occur
following exposure of benzene to mice.
Glutathionyl hydroquinone: A possible toxic metabolite of benzene
Glutathionyl hydroquinone has been isolated as one of the urinary metabolites of
hydroquinone, indicating that glutathionyl hydroquinone formation occurs in vivo and
autooxidized at several fold higher than the parent hydroquinone to glutathionyl
benzoquinone We noticed that glutathionyl hydroquinone is a potent prooxidant to
damage both calf thymus as well as plasmid pUC 18 DNA and this observation may be
of toxicological importance in relation to benzene induced genotoxicity.
Haematotoxicity of glutathionyl hydroquinone was studied in male mice at dose 100
mg/kg body weight, intraperitoneally for one month. Increased pattern of relative liver and
spleen weight and WBC were noticed, but these results were not conclusive to explain
GHQ as potent haematotoxic metabolite of benzene The pharmacokinetics and
distribution of administered GHQ Is not known. Whether GHQ administered I.p. reaches

Summary 4
understanding of the toxicity of benzene. Further, elucidation of the mechanisms of
iron-mediated oxidative damage may be important for the prevention of carcinogenesis.
Prooxidant and antioxidant properties of iron: polyphenol complex
Polyphenolic metabolites of benzene viz, hydroquinone and 1,2,4- benzenetriol have
been shown to be toxic and the suggested mechanism includes free radical fonnation via
superoxide during their autooxidation and the covalent binding of semiquinone to DNA,
RNA and other cellular macromolecules. Benzene exposure also leads to an
accumulation of bleomycin sensitive iron in bone marrow. 1,2.4-Benzenetriol, a trihydroxy
metabolite of benzene was capable of releasing iron from horse spleen ferritin, which may
lead to accumulation of iron in bone marrow during benzene exposure, as the
polyphenolic metabolites are accumulated against the concentration gradient in the bone
marrow. The ferrous iron may be chelated to the polyhydroxy metabolites of benzene viz.
hydroquinone and 1,2,4-benzenetriol.
Studies carried out with hydroquinone/1,2,4-benzenetriol and iron showed several
fold increase in iron-catalyzed bleomycin- dependent degradation of calf thymus DNA as
compared to iron alone. Complex formation some how increase the availability of iron to
degrade DNA. It is also possible that the hydroquinone and 1,2,4-benzenetriol possess
combined iron-chelating and iron ion reducing properties to stimulate DNA degradation
in the presence of bleomycin. Further, iron:polyphenol complex appears to be stable and
intact as the recovery from bone marrow lysate was complete. On the other hand,
iron polyphenol complexes have shown an antioxidant activity by inhibiting of
iron-dependent lipid peroxidation in rat brain homogenate and glutamate degradation.
This is similar to the antioxidant properties exhibited by flavonoids due to their ability to
chelate iron. Thus the polyphenolic chelates limiting the availability of iron for lipid
peroxidation and at the same time facilitate bleomycin- dependent degradation of DNA.

Summmy
the target organ i.e. bone marrow is yet to be evaluated. Other reason may be due to
difference in sensitivity of species towards different benzene metabolites.
Role of transition metal ions (Fe.Cu) during benzene toxicity
Earlier studies have provided enough evidence that transition metals including iron
and copper at e capable of directly mediating the activation or metabolism of several
xenobiotics via a metal redox mechanism, which leads to the formation of more reactive
oxygen and other organic free radicals. We observed that exposure to benzene leads to
increase in total iron content in liver and isolated rat liver nuclei. Benzene metabolites
induced the DNA damage particularly in the presence of iron and copper. This study
would help in better understanding of the chromosomal aberrations/abnormalities
associated with benzene exposure. Copper which is associated with DNA, may play a
potential role in nuclear DNA damage in presence of hydroquinone. However, the
molecular mechanism of its damage is not well understood. Significant increase in iron
content in liver and in isolated liver nuclei during benzene exposure and the copper which
is associated with DNA may have toxicological implications for cytotoxicity of liver cells
and liver nuclear DNA damage in wVo.
In the light of our investigations following conclusions can be made;
• 1,2,4-Benzenetriol is a potent reductant of ferric iron of ferritin and releases iron from
ferritin core. The release of iron from bone marrow lysate by 1,2,4-benzenetriol may
be of toxicological significance during benzene exposure as this could lead to
disruption of intracellular iron homeostasis in bone marrow cells.
• The iron:polyphenol chelate, on one hand, is a more potent DNA cleaving agent in
the presence of bleomycin, and on the other hand, it is a less effective free radical
generator as compared to iron alone. However, the formation of such a complex in
VIVO IS not known. Further studies are needed to characterize such complexes in
vivo and evaluate their toxicological significance during benzene exposure.
B Polyphenol forms complex with iron under in vitro condition and may be a possible
h««»rri8t!iit©i«iQ interma^iista during benzene exposure.

Siunmary
The present in vitro studies revealed that glutathionyl hydroquinone is a potent toxic
metabolite of benzene and acts as a prooxidant due to its faster autooxidation to
glutathionyl benzoquinone and oxyradicals which may induce DNA damage.
Exposure to benzene resulted in significant increase in total iron content in liver
tissue and in isolated liver nuclei.
Benzene metabolites (particularly hydroquinone) induced genotoxic effect in
presence of copper which is associated with the nuclear DNA. However, further
studies are needed to explain the precise mechanism of DNA damage and
chromosomal aberration in wVo during benzene exposure and effect of transition
metal ions in benzene induced genotoxicity and human leukemia.

Studies Related to Identification of Toxic Metabolites of Benzene and
Their Chemical Reactivity
THESIS SUBMITTED TO THE
DEPARTMENT OF BIOCHEMISTRY (Faculty of Life Sciences)
ALIGARH MUSLIM UNIVERSITY, ALIGARH FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY IN BIOCHEMISTRY
By
PETROLEUM TOXICITY DIVISION
Industrial Toxicology Research Centre Lucknow (India)
1996

, r , . ^ ^ . M , , g ^ ^
'^^^.
' • / Ace No. ^
« * / , . , ••'•^ UNIVtR^^"^^
T4873

DEPARTMENT OF BIOCHEMISTRY F A C U L T Y O F L I F E S C I E N C E S
ALIGARH MUSLIM UNIVERSITY ALIGARH—202002 (INDIA)
TELEPHONE : 4 00 74 1 TELEX: 564-230 AMU IN
Ref. No- Dated-
Dr. Jawaid Iqbal Lecturer Department of Biochemistry
CERTIFICATE
This is to certify that the work embodied in this thesis entitled "Studies related to identification of toxic metabolites of benzene and their chemical reactivity" has been carried out by Mr. Sarfaraz Ahmad under my supervision.
He has fulfilled all the requirements of the Aligarh Muslim University, Aligarh regarding the nature and period prescribed for investigational work for the degree of Doctor of Philosophy in Biochemistry.
The work included in this thesis is original unless stated othenwise and has not been submitted for any other degree.
Dated: T^ov. c^fh , /'-f7^ (Jawaid Iqbal)

Industrial Toxicology Research Centre
F M NO. - 0522-278227 Telex -0535-2456 ^JJLM M A H A T M A GANDHI MARC
12 2 0 1 0 7 y S ^ ^ POST BOX No. 80
2 i 7 5 8 6 3 ^ ^ a » L U C K N O W-226001 T e l e g r a n i - I N T O X r ^ — ^ U.P.INDIA
Dr. G.S. Rao Assistant Director Petroleum Toxicity Division
CERTIFICATE
This is to certify that the work embodied in this thesis entitled "Studies related to identification of toxic metabolites of benzene and their chemical reactivity" has been carried out by Mr. Sarfaraz Ahmad under my supervision
He has fulfilled all the requirements of the Aligarh Muslim University, Aligarh regarding the nature and period prescribed for investigational work for the degree of Doctor of Philosophy in Biochemistry
The work included in this thesis is onginal unless stated otherwise and has not been submitted for any other degree. .
Dated: | ^ "^ ^ ' (Dr. G.S. Rao)

'a
£< <yu£^

^^^4^jed /He ^fune^tde fzJeadu^ /a 'ieci^im4^ Jee^ decide o^a^ta/i^uJe (uu^
^^/u^e^leJiHeM ^ ^-i. (/.S. /?ao^ Scue/t-^cd^^-/, /^eAo/e^i^*t '/o^t^c^ ^^Md^oj^,
-Z^sc^^^^v 2>ef2a^^He^o^^/ocAe/fud^, ^acu/^ ^-^^ S(ue/iced, /?/i^a^
yfvudu/H 'Unioe^d^^, //uaoyiJi wAode u/u^auicia^ au/c£a^tce, //icedda^
e/u:au^Ui^e/*ie^ a^ia a^!^c^io^taJe oe/uwiou^ a/A/cA Aad MOUGA/^HU 'iedeoyicA
J^a/n ^<nJeA^Ax 2>^. /^.(?. <^4/tf*taA, ^i^^iecAn, ^ne^^oAuoA %^t^caAi^
y uUdA Aa exfz^edd /fu^ di/tce^ AAta^tAd Aa P'ioA. J^. <^(uee/*uii^c^ui,
(?Aiu^Ufui^n, 2>epa^tA/He^c^^/ocAe4^fudAi4^, '^acu/A^ o^J?ii^ <^cu&^fced.^ /7.AA.^.
//Aa^G^Aoi At^ /inAe^edA<2^ AieAa. -ie/ioe^ec^ du^^/t^ AA[e Ae/iu^ oA AA(^ ata^.
^AWOUAC^AB co/idAiuecAad a 'te/*udd OH m4(^ pa^iA^y AUAAa 'teco^^^^ J2^-i.
/^cuu <^Aa/iAe^, 3^. (/.,^.2). ^ufzAd, 2)^. S.A^. (/oeA, AAt. /^Oi^ £^^iaAa^AAt.
/7A<A^A7^^ An AAe^ d^^ce^te AceAi a^ta cuHi^iAAe ax^^e^i^^^AuJiH.
y 'ieca^ /H4^ d^/tce^ AA^i^nAd Ax A^4o/. S.M JAaJ/, 3^. AA/:. ^^du/^
a ^ 3^. j2a4^4^uM t^adaiH ^nAa ^le^^eoAJ Aa /ne AAe ^ad^u/uiAi/i^ / ^ ^ ^
/^locAe^fudA^.
^ oAda acA^^UiJiAecAae AAe dAi/HuAoA^^ co/Hpa/uiUidAi^ a/iit iuuuoAAe
coop.e'UiAiOH oAoAA/fu^ caAAea^^<ed edpec^oAAi^ 3^. VoA^-A, ^a44uAA, <oAcoeA,
<^Aui^:AaA, ^a^a^, SAtoAeeA^ SAa/nd cuuz /Ad^^f.
AA<(^ Aeoyt^eAAAAa^Ad Aa Af'^. J^oAdAjfu A^a^^ Aid dufze^ Ai^fii/t^ o^AAid
AAedid a^AAi-. /^a/H ciA^ mic40fiA.aAo^4af2At4^.
^(^tJAuAAcx e^tfi^iedd /Hi^ cAeefz de^de O^AUJ^ CUUA^iedfiecA Aa /H4^ pO'tenAd,
AioAAeM oJuA didAe4^ Aa^ e^t^^JiH^ AAei^ /HO^ d^ippoiA a ^ ma<tiM^*/H

coofze^iauo^ a^^^^uuu ^z/A.icA cMea/H O/^HU ut^^/tHo. /i^ud, zAed^ uMuliz HW Jui-cte
/Hd'/e^uciu^^, y (uda ac^^taa/la^^ /ace OH^ oMec^ui^ ^ *H^ 'ie/a^^/ed OHtz

CONTENTS
Page 1
i i i
Abbreviations
Preface
Chapter -1 Benzene an Overview 1-25
Chapter - U Materials and Methods 26-40
Chapter - HI Release of Iron from Ferritin by 1,2,4-benzenetriol 41-54
Chapter - IV Prooxidant and Antioxidant Properties of Polyphenol-iron 55-64 Complex: an Implication for Benzene Toxicity
Chapter - V Polyphenol - iron Complex: a Probable Toxic Metabolite 65-78 of Benzene.
Chapter - VI Glutathionyl Hydroquinone: a Possible Toxic Metabolite 79-97 of Benzene.
Chapter - VII Role of Transition Metal Ions (Fe, Cu) During Benzene Toxicity 98-114
Summary 115-120
Bibliography 121-149
List of Publications 150
IIII till till It

8-ALA
AML
BQ
BSA
BT
b.w.
CML
dl
DLC
DNA
DTNB
DTPA
EPO
GHQ
gm
GSH
HO
H,0,
HQ
hr
I.p.
Kg
LMI
M
mg
min
ml
mM
mmole
MPO
N
ABBREVIATIONS
5 -Aminolevulinic acid
Acute myelogenous leukemia
1,4-Ben2oquinone
Bovine serum albumin
1,2,4-Benzenetriol
Body weight
Chronic myelogenous leukemia
Decilitre
Differential leukocyte count
Deoxyribonucleic acid
5,5'-Dithiobis-(2-nitrobenzoic acid)
Diethylenetriaminepentaacetic acid
Eosinophil peroxidase
Glutathionyl hydroquinone
Gram(s)
Glutathione (reduced)
Hydroxyl radical
Hydrogen peroxide
Hydroquinone
Hour(s)
Intraperitoneal
Kilogram
Low molecular weight iron
Molar
Milligram
Minute(s)
Millilitre
Millimolar
Millimole
Myeloperoxidase
Normal

ii
NADH
nmole
8-OHdG
ppm
RNA
rpm
SD
SE
TBA
TBAR
TCA
WBC
w/v
A°
nm
Mg
Ml
JM
%
x p
6
A
Nicotinamide adenine dinucleotide
Nanomole
8-Hydroxydeoxyguanosine
Part per million
Ribonucleic acid
Rotations per minute
Standard deviation
Standard error
Thiobarbituric acid
Thiobarbituric acid reactive products
Trichloroacetic acid
White blood corpuscle
Weight by volume
Angstrom
Nanometre
Microgram
Microlitre
Micromolar
Percent
Degree centigrade
Beta
Delta
Lembda
Phi

i i i
After the advent of industrial revolution there has tjeen a massive
proliferation of industries to cater to the needs of the modern society But,
unfortunately man has been exposed knowlingly and unknowlingly to many
hazardous chemicals. Benzene is a v /eil known xenobiotic environmental pollutant
and is a natural constituent of crude oil.
Benzene is extensively used as an industrial solvent and a common additive
in gasoline and other solvents. Emissions from burning coal and oil, benzene
waste and storage operations motor vehicle exhaust, evaporation from gasoline
service stations and use of industrial solvents increase benzene level in the air.
Since tobacco contains high level of benzene, tobacco smoke is another source
of benzene in air. People living around hazardous waste sites, petroleum refining
operations, petrochemical manu-facturing sites, or gas station may be exposed to
higher levels of benzene in air.

i v
Benzene causes disorder in the blood and is well known human leukemogen.
People who breath benzene for long periods may experience hannful effects in the
tissues that form blood cells. Understanding and preventing the threat of benzene
to human health is one of the most important environmental issues facing national
and international regulatory authorities. Uncertainties about health effects must
be balanced against the potential for substantial economic and societal costs in
regulating benzene.
Despite the extensive research in the field of benzene induced genotoxicity
and carcinogenesis, the precise mechanism of its toxicity is largely unknown.
Therefore, we studied in this respect to explain the possible mechanisms of
benzene toxicity and role of metal ions (particulariy iron and copper) in benzene
induced genotoxicity and carcinogenesis.

Chapter- I
Benzene an Overvieiv
Introduction
Exposure
Route of Exposure
Biomarkers
Health Hazards
Human Exposure
Metabolism
Toxicity
Mechanism of Toxicity
Current Trend and Future Research

Chapter -1
INTRODUCTION
Benzene (CgHg) is the smallest and most stable aromatic hydrocarbon. It is a
colourless liquid with a sweet odor. Due to high vapour pressure, benzene evaporates
into air very quickly and is highly flammable. Today benzene is commercially recovered
from both coal and petroleum sources. In the past benzene was widely used as a solvent
but this use is now decreasing due to its toxic nature. The major uses of benzene are in
the production of ethylbenzene, cumene, cyclohexane, styrene, caprolactam, linear alkyl
benzene, phenol, maleic anhydride, DDT, BHC and various chemical synthesis. Other
industrial use of the benzene is in the production of rubber plastics, paints, adhesives,
nylon fibers and artificial leather (Eveleth, 1990). Benzene is also a component of
gasoline since it occurs naturally in crude oil and since it is a by product of oil refining
processes. Benzene is especially important for unleaded gasoline because of its
anti-knock characteristics. For this reason, the concentration of aromatics such as
benzene in unleaded fuels has increased (Brief et al., 1980).

Chapter - / 2
EXPOSURE Benzene is a ubiquitous agent. As a component of petroleum it is widely distributed.
Virtually all (99.9%) of the benzene released into the environment is emitted into the air.
Benzene exposure occurs in the workplace, in the general environment, and through the
use of consumer products. Occupational exposures present the highest risks, but most
individuals are exposed through the use of petrochemicals, including gasoline evaporation
and automobile exhaust, and through direct or indirect exposure to cigarette smoke. The
general population may also be exposed to t)enzene in a number of ways.
(a) Cigarette smoking
Cigarette smoking is the most significant source of t>enzene exposure for the general
population from tx)th active and passive smoke. Smoking accounts for about half of the
total population burden of exposure to benzene (Wallace, 1989). Blood benzene levels
have been shown to be higher in smokers than in nonsmokers (Brugnone et al., 1989).
One pack a day contributes about 600 ug benzene to the smoker (WHO, 1987).
(b) Home use of solvents or gasoline
Gasoline is frequently used in daily domestic purpose, particularly those who work
on gasoline-fueled machinery. Gasoline contain variable amount of benzene and can be
a significant source of exposure to the individual, involving dermal exposure as well as
inhalation. Other source of benzene exposure include consumer products such as
paints, adhesives rubber products, tapes and household cleaning agents.
(c) Leaky underground storage tanks
Leakage of underground storage tanks of gasoline is a major source of water
contamination with benzene. Contamination of ground water has occurred, leading to
human exposure through the three routes of ingestion, inhalation and skin absorption.
General population may be exposed with benzene through the supply of contaminated
drinking water supply.

Chapter -1 4
some exposure to benzene, 60-70% indicate a dangerous level of exposure, and less
than 60% indicate an extremely hazardous exposure. Urinary sulphate levels are
however, quite variable, and are not being used to identify exposure levels of benzene
associated with minimal toxic effect. Additionally, the test for urinary sulphates is not
specific for benzene.
Urinary phenol measurements have routinely been used for monitoring occupational
exposure to benzene, and there is some evidence that urinary levels can be correlated
with exposure level (Inoue et al., 1988a; Karacic et at., 1987). However, correlating
urinary phenol with benzene exposure is complicated by potentially high and variable
background levels that result from ingestion of vegetables, exposure to other aromatic
compounds, ingestion of ethanol, and inhalation of cigarette smoke (Nakajima et al.,
1987). In addition, coexposure to toluene, a common solvent, has been shown to inhibit
transformation of benzene to phenol (Inoue et al., 1988a). Analysis of data collected for
49 chemical woricers in the coke oven industry led the examiners to conclude that urinary
phenol could not be used as an indicator of benzene uptake for exposures of about 1
ppm or less (Drummond et al., 1988). The American Conference of Governmental
Industrial Hygienists has established 50 mg phenol/liter urine as the biological exposure
indices for benzene exposure in the workplace (Lowry, 1986),
Other urinary metabolites of benzene have been measured, but it is not clear if any
of these additional metabolites can be used to monitor exposure to benzene. In addition
to phenol, other phenolic metabolites have been investigated as biomarkers of benzene
exposure (Inoue et al., 1988b). Results from data collected on 152 chemical workers
indicated that, for benzene concentrations greater than 10 ppm, there was a linear
relationship between the concentration of benzene in breathing zone air and urinary
concentrations of catechol and quinol. Urinary trans, trans-muconic acid (an open-ring
benzene metabolite) has also been investigated as an indicator of benzene exposure
(Inoue et al., 1989a). In a study of 152 shoe and paint manufacturers exposed to
benzene, it was shown that urinary trans, trans-muconic acid correlated linearty with the
time weighted averages of benzene. Measurements of trans, trans-muconic acid were
able to distinguish the groups of workers exposed to benzene at concentrations of 6-7

Chapter -1 3
(d) Automobile source
Automobile emissions remain a substantial source of community exposure to
benzene. However, release of benzene and ottier gasoline vapors during refueling
remains a significant source of community t>enzene exposure as well as exposure to the
individual doing the refueling. The general population is exposed to t^enzene mainly
through inhalation of contaminated air particularly in areas of heavy motor traffic and
around gas stations. All outdoor source, including automobile exhaust and stationary
source emissions, account for only at>out 20% of the total population exposure to
benzene (Wallace, 1989).
ROUTE OF EXPOSURE
Benzene exposure occurs through inhalation, ingestion and dermal routes.
Inhalation is the dominant pathv*/ay of human exposure, accounting for more than 99%
of the total daily intake of benzene (Hattemer-Frey et al., 1990). Benzene is readily
absorbed in the lung, directly entering the blood stream and taeing distributed to the
tissues. Benzene within the blood is in direct equilibrium with the tsenzene in expired air.
Thus, measurement of end alveolar breath l>enzene concentration is a good indicator
of body benzene concentration. Approximately 50% of benzene taken up into the body
by any route is eventually exhaled, the extent being dependent on benzene dose and the
rate of metabolism and respiratory mechanics. Ingested benzene is also assumed to be
fully absort>ed but the dermal absorption of t>enzene is likely to be minimal (P<1.0%)
(Loden, 1986; Susten et al., 1985).
BIOMARKERS
Biomarkers of exposure are available for tsenzene. One selective test is the urinary
sulphate ratio test which is based on the premise that with increasing exposure there will
be an increase in benzene metabolites conjugated with sulphate moieties (Hammond
and Herman, 1960). Estimates of benzene exposure can be made by comparing the
ratio of inorganic to organic sulphates in the urine. Inorganic sulphate levels amounting
to 80-95% of total urinary sulphates are considered nomnal background, 70-80% indicate

Chapter • I ^
ppm from the group of nonexposed workers. The high sensitivity of trans, trans-muconic
acid is due to the very low urinary background levels of trans, trans-muconic acid among
the general population in contrast to the high background levels found for phenol,
catechol, and quinol. Inoue ef a/., (1989a) determined that, while trans, trans-muconic
acid measurements were useful for determining group exposures to benzene, they could
not t>e used to monitor individual exposure because of the large variations in individuals
urinary trans, trans-muconic acid values. In addition, urinary trans, trans-muconic acid
excretion was suppressed by coexposure \o toluene. However, recent studies (Bartczak
et al., 1994; Weaver et al., 1996) supported that trans, trans-muconic acid may be useful
in the evaluation of environmental benzene exposure. Weaver et al., (1996) assessed
benzene exposure by monitoring urinary trans, trans-muconic add in urban children with
elevated blood lead level. Inoue et al., (1989b) reported that the urinary concentration
of 1,2,4-t)enzenetriol related lineariy to the intensity of exposure to benzene both in men
and women workers exposed to benzene and however was suppressed by toluene
coexposure among male workers exposed to a mixture of benzene and toluene.
Another b>enzene-specific urinary metabolite is S-phenyl-N-acetylcysteine (Jonge-
neelen et al., 1987). No background excretion of S-phenyl-N-acetylcysteine was found
in rats or in humans. However, the authors concluded that biological monitoring of
industrial exposure to benzene by the determination of S-phenyl-N-acetylcysteine in the
urine is not better than the determination of phenol in urine. An improvement in the
biological monitoring of benzene exposure at levels even <1 ppm has been indicated by
determining S-phenylmercapturic acid in the urine (Stommel et al., 1989),
Red and white blood cell counts have been used as an indicator of high occupational
exposures. Monitoring of benzene exposed wori<ers has included monthly blood counts,
with workers being removed from areas of potential exposure when white blood cell
counts fell below 4,000/mm^ or erythrocyte counts fell below 4,000,000/mm' (OSHA,
1987). Additionally, it has been suggested that chromosomal aberrations in the peripheral
lymphocytes and sister chromatid exchanges could be used as monitoring end points for
benzene effects (Van Sitter! and de Jong, 1985). Benzene metabolites have also been
found to form adducts with DNA (Lutz and Schlatter, 1977, Norpoth et al., 1988; Snyder

Chapter • I 6
et ai, 1987). Furthermore, quantitation of benzene derived adducts on haemoglobin
shows potential as a marker for assessing the possible health effects of occupational
exposure to inhaled benzene (Sun et al., 1990). Measurement of protein adducts in
blood also offers the potential for monitoring the bioavailability of reactive, and
presumably toxic metabolites of benzene in workers and other persons. Recently
Yeowell-O'Connell et al., (1996) reported a method for measuring cysteinyl adducts of
the benzene metabolite benzene oxide with haemoglobin in blood from humans or
rodents exposed to benzene. This new procedure is currently being applied to the
analysis of albumin adducts of benzene oxide in rats.
Exposure to benzene causes toxic effects in the bone marrow, either from benzene
or its metabolites (Eastmond et al., 1987). Therefore, it is possible that haematological
tests could be used as markers of haernatotoxicity. To date, surveillance and eariy
diagnosis of benzene haematotoxicity depend primarily on the complete blood count,
including haemoglobin, hematocrit, erythrocyte indices, erythrocyte count, white blood cell
count, and differential and platelet count. In effect, complete blood counts and marrow
examinations should be good for eariy detection of preleukemic lesions. Additionally,
cytogenetic tests of marrow cell are being used more often, but are not yet of diagnostic
significance. Workers exposed to benzene in the air have shown elevated levels of
6-aminolevulinic acid in erythrocytes and elevated levels of coproporphyrin in the urine
(Khan and Muzyka, 1973). These may be biomari<ers for disruption of porphyrin synthesis
and may be eariy indicators of adverse haematological effects.
HEALTH HAZARDS
(I) Acute exposure effects
High concentration of benzene inhaled affect the central nervous system. At high
levels benzene has an anesthetic effect (Walker, 1976). Fatalities due to benzene are
the result of its concentration in the lipid of brain cells, effecting circulating problems,
some instances of convulsions and/or paralysis, the loss of consciousness, coma and
finally death. Damage to brain has been found to be a primary cause of death. At times
death wasprecededby periods of violent convulsion (Feil, 1933). The narcotic threshold

Chapter -1 7
concentration for laboratory animals is approximately 4,000 ppm and concentrations
above 10,000 ppm are usually fatal. Acute LDM value for new bom rats is as low as 1.0
ml/kg, while for adult rats it is in the range of 5.6-6.8 ml/kg (Kimura e a/., 1971). The
experimental results obtained indicate that the putative mechanism of acute benzene
toxicity is the rapid release of the adrenal hormones,epinephrine and nor-epinephrine,
into the blood stream, sensitizing the myocardium to its action and the most prominent
effect is central nervous system stimulation followed by depression and respiratory
failure. Direct exposure to high benzene concentration may lead to tissue destruction
due to its lipid solubility.
(II) Chronic exposure effects
(a) Haematotoxjcity
Benzene is a myelotoxin, chronic exposure of humans and experimental animals
results in a wide vanety of blood dyscrasias including; aplastic anemia, leukopenia,
pancytopenia, thrombocytopenia etc. (Snyder etai, 1977; Aksoy, 1989).
Le Noir (1897) and Selling (1910) were the first to report on haematopoietic disorders
associated with benzene in humans. These disorders were usually manifested by a
decrease in the number of one or more of the three major circulating blood cell types.
Irons et al., (1979) and Snyder et al., (1982) have shown that lymphocytes to be the most
susceptible of all the blood elements and granulocytes to be the most resistant of the
circulating cells to benzene toxicity. Granulocyte levels got depressed only on doses, 880
mg/day for 10 days (Irons et al., 1979). On the other hand exposures to concentration
sufficient to produce lymphocytopenia and anemia, even for life time, failed to effect the
granulocyte levels (Snyder, 1982).
Evidence that benzene acts primarily on committed progenitor cells was indicated
by a decrease in the number of differentiating erythroid and myeloid progenitors in the
bone man-ow (Irons e/a/., 1979). Studies by Bolcsack and Nerland (1983) have shown
that phenol, catechol and hydroquinone reduce the ^'Fe incorporation into developing
erythrocytes. The macrophages, which are a major source of polypeptide growth factors
required for the proliferation, development and survival of progenitor cells may be the
possible target of benzene toxicity (Moore, 1978). Post era/., (1986) demonstrated that

Chapter -1 8
phenol is metabolized in adherent mouse marrow maaophages by a peroxidase activity
to one or more covalently binding species and that micromolar concentrations of
hydroquinone, p-benzoquinone inhibit macrophage RNA synthesis. Later on Lewis et al.,
(1988 a) showed that benzene metabolites, catechol, hydroquinone, p-t)enzoquinone and
1,2,4-benzenetriol have potent and varied toxic effects on macrophage function and
activation. Sabourin et al., (1990) showed that inhalation exposure of mice resulted in
benzene metabolite DNA and haemoglobin adducts formation.
(b) Genotoxicity
Evidence for the genotoxicity of benzene in humans comes from studies of
occupationally exposed populations (Sasiadek et al., 1989; Yardley-Jones et al., 1990).
The convenient parameters that have been monitored include chromosomal abnor
malities, sister chromatid exchanges and micronuclei induction. Benzene induced
cytogenetic effects, including chromosome and chromatid at>errations, sister chromatid
exchanges, and micronuclei, have been consistently found in in vivo animal studies
(Erexson eta!.. 1986, Toftetal., 1982).
Forni et al., (1971) observed chromosomal aberrations in workers occupationally
exposed to benzene. Siou et al., (1981) demonstrated that nicks and breaks were the
most common in the chromosomal aben-ations studied in mice. Tice et al., (1980) have
presented evidence that the sister chromatid exchanges were due to both persistent and
new chromosomal lesions. Morimoto et al., (1983) observed that benzene biotrans
formation is a prerequisite for inducing sister chromatid exchange in human whole blood
cultures. Tunek and coworkers (1982) have related benzene ability to induce micronuclei
in bone marrow cells with benzene biotransformation In vivo. Zhang et al., (1993)
investigated that induction of both kinetochore-positive and kinetochore-negative
micronuclei by 1,2,4-benzenetriol was mediated via active oxygen species formation.
These data indicate the involvement of active oxygen species in 1,2,4-benzenetriol-
induced DNA damage and implicate the potential importance of 1,2,4-benzenetriol-
mediated genetic damage in benzene-induced human leukemia. Toft et al., (1982)
exposed male mice to benzene vapours (1 to 200 ppm) for varying periods of time and
reported that continuous exposure to 14 ppm benzene after one week resulted in a

Chapter -1
significant elevation in micronuclei. Gad-EI-Karim et al., (1984) while studying the
clastogenic effects of benzene, observed two fold increase in miaonuclei in bone man-ow
of CD-1 mice. Luke et al., (1988) evaluated the genotoxic effects of benzene (3,000
ppm) on DBA/2 mice by evaluation of micronuclei frequencies in peripheral blood,
polychromatic erythrocytes and non-chromatic erythrocytes. Delayed cell cycle in mouse
bone marrow and DMA adducts with benzene metabolites in mouse and rat haemoglobin
adducts or rat liver cell DNA adducts were also found after benzene inhalation (Tice et
al.. 1982; Sabourin et al., 1990). Recently Pathak et al., (1995) have noticed the DNA
adduct formation in the bone marrow of Isenzene administered mice. Futher Levay et al.,
(1996) hypothesized that the DNA adduct formation might depend on both the dose and
duration of benzene exposure. DNA adduct formation was also observed in HL-60 cell
culture treated with hydroquinone (Hedii et al., 1996; Pongracz and Bodell, 1996).
Further HedIi e al., (1996) demonstrated that hydroquinone and 1,2,4-t)enzenetriol, each
inhibits retinoic acid-induced granulocytic differentation in HL-60 treated cells but, DNA
adducts formation was only observed in case of hydroquinone. These findings suggest
that adduct formation might play a significant role in hydroquinone but not in 1,2,4-
benzenetriol toxicity, and that different mechanisms might be responsible for the
contributions of each metabolite to benzene toxicity.
(c) Carcinogenicity
Benzene is a well known leukemogenic and carcinogenic agent. Goldstein (1977)
and Aksoy (1985) have demonstrated that acute myelogenous leukemia (AML) and its
variants are commonly associated with t>enzene exposed workers. Paradoxically, the
occun^ence of myeloid leukemia in rats and mice is exceedingly low. But for a few stray
reports of myeloid leukemia in rats, most of them, instead, exhibited tumors. The first
example of malignant tumors experimentally produced by tjenzene was reported by
Maltoni and Scarnato (1979). The predominant tumor was not of haematopoietic origin
but rather a carcinoma of the rat zymbal gland. Further studies by Maltoni et al., (1985),
demonstrated the formation of zymbal gland carcinoma by administering benzene
through different routes. Studies by Cronkite (1987) confirmed that benzene is multipotent
carcinogen in experimental animals. The first experimental demonstration of lympho-

Chapter -1 10
reticular tumors produced by benzene was reported by Snyder et al., (1980). Life time
exposures of C57B1 mice to 300 ppm benzene (6hr/for 5 days/week) produced 6 out of
40 cases of thymic lymphoma. Cronkite et al., (1984) later showed that a shorter
exposure period (16 weeks) to 300 ppm was sufficient to produce thymic lymphoma in
these animals, although the tumor induction time was lengthened
In rodents, benzene induces a variety of tumors in organs including the zymbal
gland, harderian gland, mammary gland, lung, liver, nose and oral cavity (Maltoni e al.,
1983). Despite extensive research the mechanisms of benzene induced myelotoxicity
and leukemogenicity are unclear. Bone marrow cells contain high levels of peroxidase
activity, most of which is attributed to myeloperoxidase (MPO) and eosinophilperoxidase
(EPO) (Kariya et al., 1987). It is interesting that several target organs of benzene induced
carcinogenesis contain appreciable levels of peroxidase activity. Subrahmanyam e al.,
(1991a) have shown that phenolic metabolites of benzene are substrates for human
MPO, and that their interactions result in enhanced production of 1,4-benzoquinone. But
the toxicological significance of such reactions is not clear. They suggested that inter
action of phenolic compounds, presumably by hydrogen-bonding with the activity limiting
distal amino acid residue(s) or with the ferryl oxygen of peroxidase may be an important
contributing factor in the enhanced myeloperoxidase-dependent metabolism of
hydroquinone in the presence of other phenolic compounds. In sum, these provide ample
evidence to demonstrate that benzene is a carcinogen for rats and mice.
(d) Immunotoxicity
Animal studies show that benzene affects humoral and cellular immunity. Benzene
decreases the fonnation of the lymphocytes that produce the seaim immunoglobulins or
antibodies. Exposure to benzene at 10 ppm and above for 6 days decreased the ability
of bone man-ow cells to produce mature B-lymphocytes of C57BL/6 mice (Rozen et al.,
1984). The spleen was also inhibited from forming mature T-lymphocytes at exposure
levels of 31 ppm and above. Mitogen-induced blastogenesis B-and T-lymphocytes was
depressed at 10 ppm and above. Peripheral lymphocyte counts were depressed at all
levels, whereas erythrocyte counts were depressed only at 100-300 ppm (Rozen and

Chapter -I "
Snyder, 1985). A continuation of this line of study for 6 and 23 weeks at 300 ppm
showed continuous decreases in numt)ers of mature B- and T- lymphocytes produced in
the bone marrow, spleen, and thymus. Another series of experiments revealed that
exposures as low as 25 ppm for 2 weeks caused decrease in the numbers of circulating
lymphocytes (Cronkite e a/., 1989). Intemiediate-duration exposure to benzene also
deaeased leukocyte counts in rats and mice exposed to 300 ppm for 2-13 weeks (Ward
ef a/., 1985). Chronic exposure to benzene caused bone marrow hypoplasia,
lymphocytopenia, and anemia in mice exposed to 100 ppm for a lifetime (Snyder e a/.,
1980).
Studies by Irons et al., (1981) and Pfeifer and Irons (1983) have shown that phenol,
hydroquinone and catechol to suppress lymphocyte growth and function in vitro, which
correlates with their capacity to undergo autooxidation and with their concentration in the
bone man ow or lymphoid organs. They also showed that hydroquinone and its oxidation
product, p-benzoquinone inhibit proliferation and differentiation in lectin stimulated
lymphocytes in culture. Further, they showed that these compounds interfere with
microtubule assembly at concentrations that are not cytotoxic, while, phenol and catechol
suppress lymphocyte activation only at cytotoxic concentrations. They postulated that the
suppression of lymphocyte blastogenesis by hydroquinone to be mediated by the
interaction of p-benzoquinone with sulphahydryl groups on tubulin. This binding interferes
with microtubular integrity, which is essential in cell division via spindle formation and in
the regulation of surface receptor movement and signal transduction across the plasma
membrane.
Wierda and Irons (1982) showed that hydroquinone and catechol also to be
immunotoxic in vivo. Benzene, if metabolized in the lymphocyte to a reactive inter
mediate such as p-benzoquinone, could inhibit the production of lymphokines. Post et
al.. (1985) demonstrated that hydroquinone and p-benzoquinone affected the dose-
dependent inhibition of RNA synthesis in mouse spleen lymphocytes in vitro at micromolar
concentrations that were not cytotoxic. They also showed that exposure to p-
benzoquinone completely inhibited the proliferation and production of the T-cell
lymphokine, interieukin-2 by concanavalin A stimulated T-lymphocytes. It has been

Chapter -1 12
observed that benzene can modify both host resistance to a bacterial infectious agent
and T-cell mediated tumor resistance (Rosenthal and Snyder, 1986). Pandya et al.,
(1986) observed that the immunosuppressive effects of benzene have been modulated
by the prior administration of fungal product 6-MFA from Aspergillus ochraceous and
polyinosinic-polycytidilic acid (Pandya et al., 1989) that have interferon inducing
properties.
HUMAN EXPOSURE Inhalation is the dominant pathway of human exposure. Occupational exposure to
benzene has been associated with aplastic anemia, leukemia, and other related blood
disorders (Aksoy, 1988). For certain cancers, increased mortality was noted among
benzene exposed males in comparison with that among unexposed male. Lung cancer,
stomach cancer and primary hepatocarcinoma were reported by numerous investigators
during the benzene exposure, whereas, for female only leukemia occun'ed in excess
among the exposed (Yin et al., 1989). Risk of leukemia rose as duration of benzene
exposure increases. In acute myeloid leukemia (AML) there is diminished production of
normal erythrocytes, granulocytes, and platelets, which leads to death by anemia,
infection, or hemorrhage. Whereas in chronic myeloid leukemia (CML) the leukemic cells
retain the ability to differentiate and perform function; later there is a loss of ability to
differentiate. Case reports and epidemiological studies of workers have established as
a causal relationship between benzene exposure and AML. Study by Yin et al., (1996)
of benzene-exposed workers in China provides further support for the association of
tjenzene exposure with an increased risk for myelogenous leukemia. While some studies
have implicated other types of leukemia or even lymphomas, only the incidence of AML
and its variants has consistently been increased In groups of workers with excess
benzene exposure (Goldstein, 1988). The evidence linking benzene exposure with
leukemia was considered sufficient proof of human carcinogenicity (Dosemeci et al.,
1994). Haematotoxicity among Chinese workers were also observed who were exposed
to high concentration of benzene (Rothman et al., 1996) But precise mechanism for
benzene toxicity is not known.

Chapter -1 13
Benzene and its metabolites do, however, produce chromosomal damage in a
variety of systems. The incidence of chromatid deletions and gaps v\/ere slightly increased
in workers exposed to benzene levels of 10-100 ppm compared with control groups
(Yardley-Jones et ai, 1990). Many investigators (Glatt et at., 1989; Witz et at., 1990;
Yager et ai, 1990) have demonstrated that benzene exposure induce chromosomal
aben'ations, micronuclei and sister chromatid exchanges in human lymphocytes in vitro.
Benzene and some of its metabolites have been tested for genotoxicity in Salmonella
typhimurium and V79 Chinese hamster cells Dihydrodiol, hydroquinone, 1,2,4- benzene-
triol and catechol, the potent benzene metabolites showed genotoxic and mutagenic
effects in V79 cells as well as sister chromatid exchanges (Glatt et al., 1989). It was
further suggested that metabolites of b>enzene can induce heritable functional changes
(gene mutation) in bacterial and mammalian cells. Cytogenetic studies of benzene
exposed wori<ers have reported a similar profile of genotoxicity. Increased frequencies
of structural chromosomal aberration in the lymphocytes of l)enzene-exposed workers
have been reported (Sorsa and Yager, 1987). Fenech and Morley (1988) investigated
the ability of the principal phenolic and quinoid metabolites of benzene to induce staictural
and numerical chromosomal aben-ation by utilizing a cytokinesis-block micronuclei assay
in treated human peripheral lymphocytes. The higher efficacy of hydroquinone in inducing
both total micronuclei and kinetochore-positive miaonucleated cell when compared with
catechol, phenol and 1,4-benzoquinone suggests that hydroquinone is a major contributor
to the clastogenicity and aneuploidy observed in the lymphocytes of benzene-exposed
workers. However, other metabolites of benzene may also contribute to the genetic
effect caused by exposure to benzene.
Since consistent chromosomal aben-ations are often observed in human leukemias,
the ability of phenolic metabolites of benzene to induce chromosomal damage in human
cell also implicates them in benzene induced leukemia (Yager e/a/., 1990). Many studies
have also shown that benzene produces both structural and numerical chromosomal
aberration in the peripheral blood lymphocytes of occupationally exposed individual (Ding
et a/., 1983). Linear regression analysis demonstrated significant decrease in white and
red blood cell counts, as well as haemoglobin content (Kipen et al., 1989).

Chapter • I 14
Because consistent chromosomal aberrations have Ijeen observed in human
leukemias and lymphomas, the induction of chromosomal damage by benzene or its
metabolites may be critical in the development of benzene induced leukemia. However,
the nature of mutations produced at specific gene site in the bone marrow, the target
organ for benzene toxicity, has not been studied to date in humans (Rothman et al.,
1995). Recently, Chen and Eastmond (1995) demonstrated that inhibition of enzymes
involved in DNA replication and repair, such as topoisomerase enzymes, by the
metabolites of benzene represent a potential mechanism for the formation of chro
mosomal al)en-ations. Further in vitro and in vivo studies will be required to demonstrate
the involvement of topoisomerase inhibition in the myelotoxic and carcinogenic effects
of benzene. Earlier, Levay et a!., (1993) demonstrated the DNA adduct fomnation by the
benzene metabolites, hydroquinone and 1,4-benzoquinone in human bone marrow, and
suggested that in human bone marrow also, as in other model system, peroxidase
enzymes play an important role in the activation of hydroquinone to form DNA adducts.
These results support the general hypothesis that benzene metabolites are activated to
form DNA adducts that may contribute to the myelotoxicity and leukemia observed in
workers occupationally exposed to benzene (Cronkite et a!., 1989; Goldstein, 1977).
Besides the genotoxic and carcinogenic effect, adverse immunological effects have
been reported in human with occupational exposure to tjenzene. There are two types of
acquired immunity, humoral and cellular and benzene damages both. First, benzene has
been shown to alter humoral immunity, i.e. to produce changes in blood levels of
antibodies. Painters who were exposed to benzene (3-7 ppm), toluene, and xylene in the
workplace for 1-21 years showed increased serum immunoglobulin values for IgM and
decreased values for IgG and IgA (Lange et a!., 1973a). Other adverse reaction,
characterized by a reaction between leukocyte agglutinins and autoleukocytes, occurred
in 10 out of 35 of these workers (Lange et al., 1973b). One of the problems with these
studies is that the workers were exposed io multiple solvents, so that benzene alone may
not be responsible for the noted effects.
The second type of immunity, cellular immunity, is affected by changes in circulating
leukocytes (White blood cells) and a subcategory of leukocytes, called lymphocytes.

Chapter -I 15
Loss of leukocytes was found in a series of studies of workers exposed to benzene at
levels as low as 30 ppm in various manufacturing processes (Aksoy et ai, 1987). Other
studies in chronically exposed workers also showed loss of lynnphocytes and other blood
elements (Kipen et ai, 1989; Yin et al.. 1987).
METABOLISM
Benzene itself is not the actual toxicant and is relatively inert to confer chemical
reactivity to the aromatic ring. Benzene requires metabolism to induce its toxic effects
and follows similar pathways in humans and animals. The metabolism of benzene
occurs in the liver and, to a lesser extent in the bone marrow by cytochrome P-450
dependent mixed-function oxidase enzymes. Although, the metabolism of benzene is
very complex, but is well understood.
Schrenk et al.. (1941) studied the absorption, distribution and elimination of benzene
after exposure of benzene to the dog. The purpose of the study was to gain better
understanding of the physicochemical phenomena underiying the physiological action of
benzene. They observed that benzene concentration in fat and fatty tissue was many
times than that in blood and vital organs. The fat, acting as a reservoir, apparently is
saturated slowly and loses its benzene slowly. In a way this makes the fat, the master
tissue as regards absorption, distribution, and elimination of benzene by the body tissues
and fluids. This phenomenon may be a partial explanation of the fundamental action of
benzene on the body, namely, its deleterious effect on the haematopoietic system.
Moreover, the high benzene concentration in the urine indicates that benzene is
concentrated by the kidneys. Porteous and Williams (1949) found that phenol, catechol,
quinol and hydroxyquinol are excreted as ethereal sulphates in the unne of rabbits orally
exposed with benzene. However, the first metabolite of benzene is phenol which may
then be oxidized to catechol and quinol. One or both of these dihydric phenols may then
give rise to hydroxyquinol. It is clear that the longer benzene remains in the body, the
greater will be the extent of its oxidation to the toxic polyhydnc phenols, and continuous
feeding to benzene will give rise to a continuous and increased production of catechol
and quinol

Chapter -1 16
According to Fabre (1947a; 1947b) rat liver tissue oxidizes tDenzene to phenol in vitro
more rapidly than any other rat tissue. Under same conditions toluene is not oxidized to
phenolic substances. On the basis of this difference in toxicity between benzene and
toluene and in their metabolic fates, support to the view that benzene is toxic because
of its, phenolic metabolites. Later on urinary benzene metabolites were quantified in a
series of studies by Parke and Williams (1953). Rabbits treated orally with '*C-labelled
benzene, eliminated about 43% of the dose as unchanged benzene in the expired air and
excreted about 35% of the radioactivity in the urine in the form of metabolites. Phenol
accounted for the largest percentage of radioactivity (68%) followed by hydroquinone
(14%), catechol (6%), trans, trans-muconic acid (1%). The similar general profile of
urinary metabolites was subsequently observed in rats (Cornish and Ryan, 1965), mice
(Longacreefa/., 1981), cats and dog (Oehme, 1969), and human (Teisingere/a/., 1952).
Capel et al., (1972) have studied the variation in the metabolic fate of '"C-labelled phenol
in eight different species namely the rat, mouse, jerboes, gerbil, hamster, lemming,
guinea pig and man. In most of the species examined, phenylglucuronide and phenyl
sulphate are indeed the main metabolites except in the case of cat and guinea-pig due
to a defective glucuronic acid and sulphate conjugation mechanism respectively (Dutton,
1966: Capel e/a/., 1972).
Initially, benzene is metabolized to benzene oxide by P-450 IIE1 monooxygenase
enzymes in the liver. Benzene oxide gives rise to phenolic metabolites, such as phenol,
catechol, hydroquinone, 1,2,4-ben2enetriol and open-ring products, such as trans,
trans-muconaldehyde and trans, trans-muconic acid (Subrahmanyam e/a/., 1991b). The
majority of these phenolic metabolites are excreted in unne as glucuronide and sulphate
conjugates . In experiments with rabbits, accumulation of several phenolic metabolites
of benzene have been observed in bone marrow against the concentration gradient
(Greenlee ef a/., 1981a).
The major pathv^ays of benzene metabolism are shown in Figure-1.1. The formation
of benzene oxide is the initial step and it is further metabolized by one of the four
pathways. The first metabolite is formed by rearrangement of the benzene oxide to
phenol spontaneously. Phenol is either conjugated to glucuronide and sulphate directly

-> i
CO CD
TO
D I E o
•D 0) Q. O
• D ra 0) c 0) N c XI »•—
o
03
5 TO Q.
O
m
V

Chapter -1 17
or is oxidized to hydroquinone and followed by conjugation to hydroquinone glucuronide
or sulphate (Huff et al., 1989). The second pathway for benzene oxide metabolism
consisted of reaction with glutathione and subsequent modification of the mercapturic acid
(Henderson et al., 1989). The third pathway for t>enzene metabolism involved in
conversion of benzene oxide into tjenzene dihydrodiol by epoxide hydrase (Tunek et al.,
1981). Benzene dihydrodiol is converted into catechol by oxidation of benzene
dihydrodiol by a soluble NADP*- dependent enzyme, tjenzene dihydrodiol dehydrogenase
(Ayengar et al., 1959) and finally conjugation to glucuronide and sulphate. Benzene
dihydrodiol is also converted into 1,2,4-b)enzenetriol by some unknown mechanism, which
is one of the potent toxic metabolites of t>enzene. However, Inoue et al., (1989b)
observed that phenol and hydroquinone, but not catechol, are precursors of
1,2,4-benzenetriol in urine after the intraperitoneal injection of the three phenolic
compounds to rats followed by urine analysis for 1,2,4-benzenetriol. A similar result was
also observed in case of rabbits (Inoue ef a/., 1989c). The fourth pathway of benzene
oxide metabolism consists of the ring opening reaction resulting in the fonmation of trans,
trans-muconic acid. However, the metabolic pathway of trans, trans-muconic acid in vivo
is yet to be identified (Latriano et al., 1986). The corresponding dialdehyde, trans,
trans-muconaldehyde has been shown to be a precursor of muconic acid in vivo (Witz
etal., 1990).
Although, the pathway of benzene ring opening is not known, but a number of
possible mechanisms for oxidation of benzene ring-opening have been postulated
(Snyder and Chatterjee, 1991). One such postulated pathway involves the formation of
trans, trans-muconaldehyde from benzene dihydrodiol in a Fenton system, presumably
via hydroxyl radical-mediated ring-opening (Latriano et al., 1985; Zhang et al., 1995a).
Zhang et al., (1995b) further demonstrated the role of iron in the mechanism of ring-
opening of benzene in a mouse liver microsomal system.
TOXICITY
Benzene is a well known xenobiotic environmental pollutant. Exposure to benzene
leads to the development of bone marrow depression characterized by progressive

Chapter -1 18
leucopenia, anemia and pancytopenia (Snyder, 1987). The responses to chronic
benzene exposure have been classified as either bone marrow depression and aplastic
anemia or the generation of acute myeloblastic leukemia (AML) and related forms of acute
lymphocytic/non-lymphocytic leukemia. Collectively, these abnonnalities have been
termed as preleukemic syndrome or myelodysplastic syndrome (Tricot, 1991). Irons
(1988) and Tricot (1991) suggested that the spectrum of bone marrow toxicities produced
by exposure to benzene represented a form of myelodysplastic syndrome resulted in
bone marrow depression and that might lead to fatal aplastic anemia. Anon (1982)
reported that benzene is a human leukemogen. Chronic tjenzene poisoning exhibited a
variety of changes in the number of basophils, eosinophils, lymphocytes and other bone
marrow function in workers (Aksoy ef a/., 1971).
Goldstein et al., (1982) reported on the production of a limited number of
myelogenous leukemias in rats and mice exposed to benzene by inhalation. The reports
by Maltoni et al., (1989) and Huff ef al., (1989) of Ijenzene induced carcinogenicity in rats
and mice given benzene orally focussed on solid tumors and multiple carcinogenic foci.
The zymbal gland was a primary carcinogenic site in both sexes of both species. Later
studies using the inhalation route (Maltoni ef al., 1989) also demonstrated the
multipotential carcinogenic activity of benzene. Excluding the studies by Yin ef al., (1987)
there is no strong evidence for benzene-induced solid tumors of these types in human.
Snyder ef al., (1980) also reported the production of thymic lymphoma after inhalation of
benzene in C57B1/6J mice but not in AKR mice. Fanis ef al., (1993) observed the
malignant lymphoma along with preputial gland carcinomas and lung adenomas in
CBA/Ca mice exposed to benzene through inhalation. Neither the Cronkite ef al. (1989)
nor the Fams etal., (1993) studies demonstrated acute granulocytic leukemia in tjenzene
inhaled mice.
Snyder and Chatterjee (1991) and Yardley-Jones ef al., (1991) have studied the
pathway of benzene metabolism and the role of benzene metabolites in the production
of benzene toxicity. Lee ef al., (1981) reported that early erythroblasts are sensitive to
benzene and its metabolites. Benzene has long l5een known to produce chromosomal
abnormalities (Eastmond, 1993) and suggested that benzene may have caused

Chapter • I 19
chromosome breakages and rearrangements in a stem cell that proliferated, leading to
erythroleukemia. Eastmond (1993) also observed that the quinone metabolites of
benzene can cause aneuploidy to chromosome nondisjunction and nonrandom chromo
somal alteration in individuals with advanced forms of Isenzene-induced myelotoxicity.
The association between chromosomal abnormalities and leukemia in tienzene exposed
rats has been reviewed by Le Beau and Larson (1991). Tice et al.. (1980) exposed mice
to benzene by inhalation and demonstrated an increased frequency of sister chromatid
exchanges in bone marrow cells and suggested that benzene metabolites were
responsible for these effects. Muconaldehyde, a hypothesized ring open benzene
metabolite also produced sister chromatid exchanges in B6C3F1 mice (Witz et al.,
1990).
The highly electrophilic metabolites such as 1,4-benzoquinone and others are
generated during Ijenzene exposure in experimental animals and form covalently bound
adducts with both nuclear and mitochondrial DNA (Eastmond, 1993). Kolachana et al.,
(1993) have demonstrated that benzene and its phenolic metabolites; phenol, hydro-
quinone and 1,2,4-benzenetriol induce oxidative damage in the genome in the form of 8-
hydroxydeoxyguanosine in HL-60 promyelocytic leukemic cells and in the bone marrow
of mice. Cheng et al., (1992) demonstrated that benzene induced active oxygen radical
production resulted in 8-hydroxydeoxyguanosine formation in vivo which may play a role
in genotoxicity and leukemogenesis. Formation of 8-hydroxydeoxyguanosine adduct is
known to cause G-T and A-C base substitutions.
MECHANISM OF TOXICITY
Any hypothesis of benzene toxicity must account for the requirement of hepatic
metabolism and selective toxicity of benzene in the bone marrow. As mentioned above,
benzene is metabolized in the liver mainly to one or more phenolic metabolites and
accumulated against the concentration gradient in bone man-ow and exerts its toxic
effects (Smith et al., 1989). It is believed that the effect of benzene is likely to be exerted
through the action of multiple metabolites on multiple targets through multiple biological
pathways (Goldstein, 1989).

Chapter -1 20
Rao and Pandya (1989) observed that polyphenolic metabolites as the toxic inter
mediates due to their capability to undergo autooxidation. One mechanism by which
benzene induces the toxic effects may be mediated by the generation of one more active
oxygen species such as superoxide anion radical (Oj), hydrogen peroxide {Hfii),
hydroxyl radical (HO) and singlet oxygen C^fii) (Subrahmanyam et al., 1991b).
Myeloperoxidase and eosinophil peroxidase are the two principal enzymes in bone
man-ow which are responsible for the formation of quinone and semiquinone free radicals
and activation of oxygen to superoxide radicals. These free radical metabolites of
benzene with dioxygen and cellular targets such as DNA, GSH, protein and lipid are
responsible for the benzene-induced myelotoxicity and leukemia (Subrahmanyam et al.,
1991b). Legathe et al., (1994) have demonstrated pharmacokinetic interaction between
benzene metabolites, phenol and hydroquinone, in B6C3F1 mice. The consequence of
which may be a higher quantity of 1,4-benzoquinone formation from hydroquinone.
Hydroquinone and catechol have been shown to be much more toxic to bone marrow
cell cultures than benzene (Parmentier and Dustin, 1953). Phenol and hydroquinone
have been shown to cause significant decrease in bone man-ow cellularity when
coadminis-tered to mice (Eastmond et al., 1987). Since the bone man'ow is rich in
peroxidative enzymes including myeloperoxidase and potential oxidants such as
hydrogen peroxide derived from leukocytes, it has been suggested (Eastmond et al.,
1987; Smith et al., 1989) that a bone man-ow-localized phenol-dependent stimulation of
hydroquinone metabolism results in the fomnation of 1,4-benzoquinone, the ultimate toxic
metabolite of benzene. 1,4-Benzoquinone has been shown to induce single strand breaks
in DNA of cultured cells (Pellak-Walker and Blumer 1986), genotoxic in V79 cells (Glatt
et al., 1989), inhibit DNA and RNA synthesis (kalf, 1987) and mitogen stimulation of
lymphocyte growth (Wierda and Irons, 1982). Levay et al., (1993) observed the DNA
adduct formation in all cell types tested in presence of 1,4-benzoquinone. Recently Sze
et al., (1996) noticed that 1,4- benzoquinone is more potent to induce DNA strand breaks
in Chinese Hamster Ovary cell. However, hydroquinone also exhibited a significant
effect as well. Although, only less than 1-5% of the benzene inhaled is metabolized to
1,4-benzoquinone in the body (Huff et al., 1989), the amount of 1,4-benzoquinone

Chapter • I 21
produced is perhaps sufficient to cause significant biological effects. On the other hand,
hydroquinone has been known to undergo autooxidation or myeloperoxidase-catalyzed
oxidation to form 1,4-benzoquinone (Subrahmanyam et al., 1991b). As such, the
cytotoxicity of hydroquinone could be due, in part, to the production of 1,4-t)enzoquinone
in the Chinese Hamster Overay cells (Sze et al., 1996).
Quinone and semiquinone metabolites of polyphenols, through their capacity to react
with nucleophilic groups of amino acids are reported to inactivate critical enzymes like
DMA polymerase (Graham et al., 1978), RNA polymerase II (Nagaraja and Shaw, 1982)
and reverse transcriptase (Wick and Fitzgerald, 1981). Quinone or semiquinone
metabolites of hydroquinone and 1,2,4-benzerietriol have been shown to inhibit mRNA
synthesis (Kalf et al., 1982), microtubule polymerization (Irons and Neptun, 1980),
mitogen stimulation of lymphocyte growth (Wierda and Irons, 1982), effect macrophage
function and activation (Lewis et al., 1988a) and form adducts with DlslA (Jowa et al.,
1986). Autooxidation of polyphenolic compounds in presence of low concentrations of
transition metal ion copper resulted in an increased production of active oxygen species
which can release aldehydic products from glutamate or DNA (Rao and Pandya, 1989).
Experimental studies have suggested that involvement of iron or copper in benzene
metabolite induced DNA damage through a mechanism involving the generation of
organic free radicals (Kawanishi et al., 1989; Rao and Pandya, 1989). In vitro studies
by Rahman et al., (1989) and Ahmad et al., (1992) have shown that naturally occurring
flavonoid, quercetin, in the presence of Cu(ll) and molecular oxygen caused breakage
of calf thymus DNA, supercoiled pBR 322 plasmid DNA and single stranded Ml 3 phage
DNA. Copper is distributed throughout the body with the liver and bone marrow being the
two major copper storage organs (Linder, 1991). Copper has been identified as the
essential redox-active centre in a variety of metalloproteins such as ceruloplasmin,
Cu-Zn superoxide dismutase, cytochrome oxidase, dopamine p-hydroxylase, tyrosinase,
lysyloxidase and ascorbate oxidase (Linder, 1991). Since copper also exists in the
nucleus and is closely associated with chromosomes and DNA (particulariy Guanine)
(Prutz et al., 1990) the chemical-metal redox system might be responsible for strand
breaks in vivo during benzene exposure. In vitro, among hydroquinone, 1,2,4-

Chapter - / 22
benzenetriol, catechol and phenol; hydroquinone/Cu^* and 1.2,4-benzenetrioiyCu^* were
the two efficient DNA cleaving systems (Li and Trush, 1993a). Recently Zhang et al.,
(1996) described that the oxidation of 1,2,4-benzenetriol and catalysis by Cu^* may play
an important role in 1,2,4-ben2enetriol-induced genotoxicity.
It was also observed that Cu(ll) strongly induces the oxidation of hydroquinone as
such may be factor involved in the oxidative action and toxic to primary bone marrow
stromal cells of DBA/2 J mice (Li and Trush, 1993b). They further observed that Cu(ll)
is capable of directly reacting with hydroquinone, causing the one electron oxidation of
hydroquinone to semiquinone radical (SQ) and reactive oxygen species generated in
chemical metal redox system were responsible for in vitro plasmid DNA damage (Li et
al., 1995).
Many investigators have shown that t>enzene exposure interferes with iron
metabolism and heme biosynthesis in experimental animals. These studies include
decreased incorporation of ^ Fe into circulating erythrocytes (Lee et al., 1974), inhibition
of heme biosynthesis enzyme at the level of 6-aminolevulinic acid (ALA) dehydratase
resulting in ALA accumulation and its loss in urine (Rao and Pandya, 1980), and
depletion of the regulatory heme pool (Siddiqui et al., 1988). Abraham et al., (1986)
demonstrated that benzene exposure leads to increase in heme oxygenase activity, a
rate limiting enzyme for heme degradation. Existence of an intracellular pool of low
molecular weight iron (LMI) compounds maintaining a dynamic equilibrium between iron
uptake, iron storage and iron incorporation into its final biochemical form have been
described (Jacobs, 1977). Benzene administration led to an accumulation of iron in bone
marrow cells which can catalyze the formation of aldehydic products (base propenal)
from DNA in presence of bleomycin (Rao et al., 1990). In all probability benzene alters
heme metabolism of erythroid cells resulting in the increased internalization of iron
transferrin-receptor complex and uptake of internalized iron is almost entirely restricted
to erythroblasts. In an/n v/fro study, Rao (1991) observed that hemin catalyzed the
autooxidation of hydroquinone or 1,2,4- benzenetriol in the presence of reducing agent
and resulted in the formation of aldehydic products from glutamate, deoxyuridine or DNA
through the formation of reactive oxygen species. Hemin is essential for the erythroid

Chapter -1 23
differentiation process and accumulates in the immature red blood cells in nanomolar
quantities as these cells differentiate (Lo et aJ., 1981). In immature red blood cells in bone
marrow, it has been noted that heme accumulates in the nucleus as these cells
differentiate and nicking of nuclear DMA has been reported as it binds covalently to both
protein and DNA (Mager and Bernstein, 1979; Lo et al., 1981). it has been postulated
that this action of hemin might account for its influence in the differentiation of these and
other cell types (Chen and London, 1981). n view of the accumulation of polyphenolic
metabolites after exposure to benzene and the presence of excessive amounts of heme
in bone marrow cells which may catalyze autooxidation of polyphenols and offer an
alternative mechanism for the bone marrow depressant effect of benzene (Rao, 1991).
CURRENT TREND AND FUTURE RESEARCH
Benzene metabolism in experimental animals are well understood, but the precise
mechanism of benzene toxicity remain largely unknown. The problem of attempting to
extrapolate to the humans is complicated by the lack of metabolism or toxicity data in
humans. In attempt to bridge the data gap, Sabourin et al. (1992) examined benzene
metabolism in cynomolgus monkey and chimpanzee. They concluded that the mouse
forms the highest levels of hydroquinone conjugate and the chimpanzee forms the
lowest. It will be important to determine the sensitivity of the monkeys and the
chimpanzees to benzene. Although, the relative sensitivity of humans to benzene toxicity
is unknown, a study of benzene metabolism in humans exposed in an industrial setting
to evaluate the urinary excretion of phenyl conjugates vs polyhydroxylate and ring open
metabolites might help to estimate how humans relate to test species in this respect
(Sabounnef al., 1992).
Since no single ring hydroxylated metabolite of benzene can produce the whole
spectrum of benzene toxicity, more and more attention has focused on the interactions
of benzene metabolites (Kolachana et al., 1993), reactive ring-opened metabolite i.e.,
trans, trans-muconaldehyde (Goon et al., 1992). Further studies are needed to
demonstrate that trans, trans-muconaldehyde is a precursor of trans, trans-muconic acid
in vivo. Recently Zhang ef al., (1995b) demonstrated the involvement of iron in the

Chapter -1 24
mechanism of ring opening of benzene in mouse liver microsomal system. Although it
is generally believed that there is no "free" iron in vh/o, there is large body of evidence that
supports the existence of a so-called "lov»/-moiecular weight-iron pool" in cells, including
hepatocytes. Earlier Rao et al., (1990) demonstrated the accumulation of bleomycin
detectable low molecular weight iron components in bone marrow in experimental rats
during benzene exposure. The liver is a tissue rich in iron, which is stored in ferritin and
present in many heme containing proteins. Under normal condition, the mobilization of
iron is strictly regulated. However, recently Ahmad et al.. (1995) observed the reductive
release of iron from horse spleen femtin by 1,2,4-benzenetriol. Oxidative stress produces
conditions that interfere with this regulation, and which may result in the release of iron
from femtin. Since there is an animal model for iron accumulation (Nielsen et al., 1993)
and since methods for assessing benzene haematotoxicity and for measuring
metabolism are available, it would be of interest to evaluate the role of iron in benzene
haematotoxicity in vivo.
The molecular mechanism of benzene toxicity is also not very well understood.
Many studies suggest that DNA damage may be playing a role in the myelotoxicity during
benzene exposure. Further studies measuring the formation of both DNA adducts and
oxidative base damage will be required to evaluate the contribution of each of these
forms of DNA damage to the myelotoxic effects of benzene. Accordingly, the interaction
oi phenolic compound with DNA associated copper may result in a spectrum of lesion
in DNA, including site specific oxidative DNA base modifications, DNA strand breaks, and
DNA adducts of phenolic intermediates (Li et al., 1995; Zhang et al., 1996). These
lesions in DNA might contribute to mutation in DNA and to the initiation of carcinogenesis
induced by phenolic compounds. However, benzene induced DNA strand-breaks in vivo
is yet to be identified.
Further studies are needed of large populations of exposed workers to produce
benzene-induced leukemia and to examine the ability of benzene metabolites to induce
mitotic recombination in human cells. Recently Chen and Eastmond (1995) have shown
that a number of polyphenolic metabolites or postulated metabolites (such as 2,2'- and
4,4'-biphenol) of benzene inhibit human topo isomerase II enzyme in vitro when activated

Chapter -1 25
by a peroxidase metabolizing system and hypothesized that the quinone and phenolic
metabolites of benzene might exert their clastogenic effects through inhibition of topo-
isomerase enzymes. Topoisomerases are major chromosomal proteins which modulate
the topological state of DNA by breaking and releasing DNA strands (Pommier and
Bertrand, 1993). These enzymes participate in a range of cellular processes including
DNA replication and transcription (Anderson and Rot)erge, 1992) and DNA repair
(Stevnsner and Bohr, 1993; Thielmann et a/., 1993). Topoisomerase II also play a role
in maintaining genomic stability (Wang et al., 1990). The inhibition or interference with
topoisomerase II activity at critical stages of the cell cycle could lead to chromosome
breakage, aneuploidy or cell death. Several inhibitors of topoisomerase II are quinone
or quinone-forming compounds which exhibit some structural similarity to metabolites of
benzene possessing hydroxyl or keto groups on the aromatic ring (Li et al., 1993;
Stoyanovsky ef a/., 1993). Furthermore, quinone and phenolic compounds have been
identified at relatively high concentrations in the peripheral blood and bone marrow of
rodents following benzene exposure (Rickert et al., 1981). Additional In vitro and In vivo
studies will be required to demonstrate the involvement of topoisomerase inhibition in the
myelotoxic and carcinogenic effects of benzene.
Finally, the possible mechanisms of benzene-induced leukemogenesis are
suggested and areas of further research are indicated that may provide new information
as well as permit the application of existing knowledge to the understanding of the
leukemogenic process.

Chapter - / / Materials & Methods]
L
Chemicals
Instnuncnts
Animals
Experimental Procedures
Statistical Analysis

Chapter - II 26
CHEMICALS
Benzene of high purity was obtained from Glaxo Laboratories India (ANLAR).
Ferritin (horse spleen), ferrozine (3-(2-pyhdyl)-5,6-bis(4-phenyl sulfonic acid)-1,2,4-
tnazine), bathocuproine, dopamine, 6-hydroxydopamine, catalase (bovine liver;
2,000-5,000 units/mg protein), superoxide dismutase (bovine erythrocytes; 2,500-5,000
units/mg protein), bovine serum albumin, calf thymus DNA, 5,5'-dithiobis-(2-nitrobenzoic
acid), Sephadex G-10 beads, agarose gel and L-glutamate monosodium were obtained
from Sigma Chemical Company, St, Louis, MO, USA. Hydroquinone, 1,2,4-benzenethol
and 1,4-benzoquinone were obtained from Aldrich, Milwaukee, USA, Bleomycin
hydrochloride was obtained from Nippon Kayaku Co, Ltd., Japan, 2-Thiobarbituric acid
was obtained from BDH Chemical Limited, England, Supercoiled plasmid pUC 18 DNA
was obtained from Bangalore GENE! Pvt, Ltd., India, whereas tris-hydroxymethyl amino
methane, glutathione reduced, Folin-Ciocalteu reagent and tnchloroacetic acid (TCA)
were obtained from Sisco Laboratohes, India, All other chemicals were either obtained
from CSIR Centre for Biochemicals, New Delhi, India or were of analytical grade.

Chapter - / / 27
INSTRUMENTS Spectronic-2001 spectrophotometer, Bausch and Lomb, Ultra Turrax T-25
homogenizer, UV-transilluminator Poloroid MP4*, gel electrophoretic apparatus, electro
phoresis power supply SPS-3000, mettler balance, digital pH meter, and microcentrifuge
(REMI) were the major instruments used in this study.
ANIMALS Male mice of 25-30 gms bred at the animal house unit of Industrial Toxicology
Research Centre were divided into four groups (10 mice in each group). One group was
kept as control which received only 0.1 ml normal saline. Second and third groups
received hydroquinone or iron (25 mg each/kg body weight) for 20 days (single dose i.p).
The last group received hydroquinone : iron complex (25 mg hydroquinone ; 25 mg
iron/kg body weight) 1:1 ratio intraperitoneally for 20 days (single dose). After the
termination of the treatment blood was collected into clean heparinized tuljes from jugular
vein and kept at 4°C for haematological studies. Liver homogenate was made in 0.15
M KCI for the estimation of sulphahydryl content and lipid peroxidation.
Female albino rats were treated with FeS04.7H20 (100 mg/kg body weight, i.p.) for
three days consecutively. Rats were sacrificed on 4th day and ferritin was isolated from
liver to study the mobilization of iron from ferritin by 1,2,4-benzenetriol. Bone marrow
cells were flushed out by 0.15 M NaCI from four femurs of control female rats to evaluate
the mobilizationof iron by 1,2,4-benzenetriol. The cells of bone marrow pellet were lysed
by resuspension in approximately 1.5 ml distilled water. This suspension was homo
genized in Ultra Turrax T-25 for 30 sec. and the volume was made upto 2.0 ml with
acetate buffer to a final buffer concentration of 0.033 M, Brain homogenate (2%) of
control rat was made in 0.15 M KCI for 1,2,4-t>enzenetriol concentration dependent iron
release and subsequent lipid peroxidation in the presence of ferritin.
Femur bone marrow cells were also isolated from control albino rats and cell lysate
was made to demonstrate the reco\jevj of iron.polyphenol complex from bone marrow
lysate.

Chapter - II 28
Albino rats of either sex (Swiss wistar strain, 100-110 gm) were treated with
benzene (0.5 ml/kg body weight) intraperitoneally daily for 10 days. Control and
experimental rats were sacrificed by cervical dislocation and liver were immediately
excised and liver homogenate (4 gm liver/30 ml of 50 mM Tris -10 mM EDTA buffer, pH
8.0) was prepared to isolate nuclei.
In another set of experiment male mice (25-30 gm) were exposed with glutathionyl
hydroquinone (GHQ) (100 mg/kg body weight) in 0.1 ml normal saline intraperitoneally
daily for one month (single dose) and control group received only 0.1 ml normal saline.
After the termination of the treatment mice were sacrificed and haematological as well
as other parameters were studied.
EXPERIMENTAL PROCEDURES
Release of thiobarbituric acid reactive products (TBAR) from DNA
Iron-catalyzed bleomycin-dependent DNA degradation was performed according to
the method of Gutteridge (1987). Principally, bleomycin in the presence of low molecular
weight iron degrades DNA with the release of base propenals which react with
2-thiobarbituric acid (TBA) to give a chromogen similar to that given by malonaldehyde
on reaction with TBA. Briefly, the assay system in a total volume of 3.0 ml consisted of
0.033 M acetate buffer of pH 5.3, 500 pg calf thymus DNA, 50 pg bleomycin hydro
chloride and the reaction was initiated by the addition of 20 pM iron or a complex of iron
with polyphenol or of endogenous chelators, such as citrate, glucose, fructose, glycine,
glutamic acid, cysteine, aspartic acid, adenosine monophosphate, adenosine diphos
phate, adenosine triphosphate and dithiothreitol(20 pM:40 pM) which were made
immediately before use. The contents were incubated at 37°C for 30 min and the
reaction was terminated by the addition of 1.0 ml of 10% (w/v) ice-cold trichloroacetic acid
(TCA). The contents were centrifuged at 3000 rpm for 15 min. To the clear supematants,
an equal volume of 0.67% (w/v) TBA was added and heated for 15 min in a boiling
watertDath. The resultant TBA chromogen was read at 532 nm using a Spectronic-2001
spectrophotometer. The values are expressed as nmoles of malonaldehyde equivalents
generated by using £532=1.56 x 10^ cm' MV

Chapter - II 29
Formation of thiobarbituric acid reactive products (TBAR) from glutamate
Release of thiobarbituric acid reactive products from glutamate was followed
according to the method described (Gutteridge, 1981). Briefly, the reaction mixture (total
volume 1.5 ml) contained 0.033 M sodium phosphate buffer of pH 7.4 in 0.15 M NaCI,
4 mM glutamate and the reaction was started by the addition of 20 IJM iron or a complex
of iron:polyphenol (20 \iM : 40 \iM). The contents were incubated for 15 min at 37°C and
the reaction was terminated by the addition of 1.0 ml of 10% (w/v) ice-cold TCA. The
contents were centrifuged at 3000 rpm for 15 min and to the clear supematants, an equal
volume of 0,67% (w/v) TBA was added and heated for 15 min in a tx)iling waterbath. The
resultant TBA chromogen formed from glutamate was measured at 532 nm using a
Spectronic-2001 spectrophotometer. The values are expressed as nmoles of
malonaldehyde equivalents generated by using E532 = 1.56 x 10* cm ' MV
Release of Iron from ferritin
Iron release from horse spleen ferritin was investigated by using the assay procedure
previously descrifc)ed by Kadir et al., (1992). Briefly, the assay system (total volume 1.5
ml) contained 100 pg ferritin, 500 pM fen-ozine and the desired concentration of
hydroquinone/1,2,4-benzenetriol/1,2,3-benzenetriol(pyrogallol)/1,3,5-t»enzenetriol(phloro-
glucinol)/1,4-benzoquinone/phenoiyglutathionyl hydroquinone/uric acid (each 330 pM) or
dopamine/6-hydroxydopmaine (125 pM) in 0.033 M acetate buffer of pH 5.6. The reaction
was started by the addition of different reducing agent and the formation of
(Fe(ferrozine)J^* complex was monitored by recording the increase in absorbance at 562
nm with time and the amount of iron released from ferritin is expressed as pM Fe^Vmin.
The effect of oxyradical scavengers was studied by the addition of oxyradical
scavenger before the addition of polyphenol. The complete reaction mixture contained
in a final volume of 1.5 ml, femtin (100 pg), 1,2,4-benzenetriol (330 pM), ferrozine (500
pM), 0.033 M sodium acetate buffer of pH 5.6 and oxygen radical scavengers, such as,
urea and thiourea (5 mM), mannitol (50 mM), albumin, catalase and superoxide dismutase
(150 pg). The time course of formation of [Fe(ferrozine)3]^* release from bone marrow

Chapter - / / 30
cell lysate was assayed in a total volume of 1.5 ml containing 100 pi bone marrow cell
lysate, 500 |JM fen-ozine, 330 pM 1,2,4-benzenetriol in 0.1 M acetate buffer of pH 5.6 (0.5
ml). The amount of iron released is expressed as (JM Fe^Vmg protein.
Lipid peroxidation in brain homogenate
Lipid peroxidation in rat brain homogenate (2% in 0.15 M KCI) was assayed
according to the method of Bemheim et a/., (1948).
(a) In presence of iron or Jron:polyphenol
The assay mixture was incubated either with 20 pM iron, iron-polyphenol (20 pM :
40 pM) or none and 2.0 ml of brain homogenate (2%) in a metabolic shaker at 37°C,
Aliquots (1.0 ml) were taken at intervals and the reaction was terminated by the addition
of 1.0 ml of 10% ice-cold TCA.
(b) In presence of ferritin and 1,2,4-benzenetriol
The assay system (total volume 1.5 ml) contained 100 pg ferritin and 1,2,4-
benzenetriol (0-330 pM) in 0.033 M acetate buffer of pH 5.6. The contents were
incubated for 30 min, after which 2.0 ml of 2% brain homogenate was added and
incubated for 1 hr at 37°C. After 1 hr incubation, aliquots (1.0 ml) were taken out and the
reaction was terminated by the addition of 1.0 ml of 10% ice-cold TCA.
The contents in the above experiments were centrifuged and to the clear
supematants, an equal volume of 0.67% (w/v) TBA was added and heated for 15 min in
a boiling waterbath. The resultant TBA chromogen was read at 532 nm using a
Spectronic-2001 spectrophotometer. The values are expressed as nmoles of malon-
aldehyde equivalents generated by using E^^ = 1.56 x 10^ cm' M'.
Bleomycin-dependent degradation of DNA by 1,2,4-benzenetriol in presence of
ferritin
The effect of increasing concentrations of 1,2,4-benzenetriol on iron catalyzed
bleomycin-dependent degradation of calf thymus DNA in the presence of ferritin was

Chapter -II 31
evaluated. Briefly, the assay system (total volume 1.5 ml) contained 100 Jg ferritin and
1,2,4-benzenetriol (0.165 pM) in 0.033 M acetate buffer of pH 5.6. The contents were
incubated for 30 min, after v\/hich 250 |jg calf thymus DNA and 50 MQ bleomycin-
hydrochloride in 0.3 ml was added and incubated for additional 30 min at 37°C. The
reaction was tenninated by addition of 10% ice-cold TCA and to a dear supernatant add
an equal volume TBA. The resultant TBA chromogen formed from DNA was measured
and the values are expressed as described eariier.
Isolation of ferritin from rat liver and mobilization of Iron by 1,2,4-benzenetriol
Female albino rats (Swiss Wistar strain) were treated with FeSO .THzO (100 mg/kg
body weight, intraperitoneally) for three days consecutively. Rats were sacrificed on 4th
day and ferritin was isolated from liver by the method of Granick (1942) with some minor
modifications.
Liver from two rats was homogenized using Ultra Tun ax T-25 for 30 sec. in distilled
water. Dilute the suspension to 1 gm liver per 4 ml distilled water and heat rapidly with
stirring to 70-80°C for 5 min. The suspension was filtered while hot through a muslin
cloth. Adjust the pH of the filtrate to pH 4.6 with acetate buffer (0.5 M) and centrifuged
at 1500 rpm for 30 min. After discarding the precipitate, ammonium sulphate (30 gm/100
ml) was direclty added to clear red brown supernatant and kept at 4°C for overnight and
the resultant precipitate was centrifuged and dissolved in measured volume of distilled
water. Cadmium acetate (4 gms) was added per 100 ml suspension and centrifuged at
1000 rpm for 10 min to remove any impurity. A red brown supernatant containing
noncrystalline ferritin was obtained and this suspension was used to study the
mobilization of iron from ferritin by 1,2,4-benzenetriol. Release of iron was measured in
a total volume of 1.5 ml contained, 05 ml of sodium acetate buffer (0.1 M, pH 5.6), 330
MM 1.2,4-benzenetriol, 0.5 mM ferrozine and 50 or 100 pi ferritin isolated from rat liver
(0.4 and 0.8 mg equivalent protein). The formation of [Fe(ferrozine)3f* was monitored
(Kadir et al., 1992) by recording the increase in absorbance at 562 nm with time and
amount of iron released from ferritin is expressed as pM Fe^Vmin/mg protein.

Chapter - II 32
Protein estimation
Protein was estimated by the method of Lowry et al., (1951). Protein was
precipitated with 10% ice-cold TCA. The precipitated protein was centrifuged at 3000
rpm for 15 min and the protein pellet was dissolved in 1N NaOH. Finally suitable amount
of dissolved protein was diluted to 1.0 ml with distilled water and 5.0 ml of alkaline
copper reagent was added. After 10 min, 0.5 ml of Folin-Ciocalteu reagent was added.
Absorbance was read at 750 nm after 20 min. Bovine serum albumin (BSA) was used
as standard.
To prepare alkaline copper reagent, solution (A) and (B) were mixed in 1:1 ratio just
prior to use of the reagent. Solution (A) containing 8 gm sodium carbonate anhydrous
dissolved in 100 ml of double distilled water, and (B) containing 120 mg sodium
potassium tartrate and 60 mg of CUSO4.5H2O dissolved in 100 ml of double distilled
water.
Recovery of iron:polyphenol complex
Recovery of iron:polyphenol complex from bone marrow lysate (1 mg protein
equivalent) was monitored by assaying iron: polyphenol complex catalyzed bleomycin-
dependent degradation of DNA as compared to that without bone marrow lysate. Briefly,
the assay system in total volume of 1.0 ml with or without bone marrow suspension (1
mg protein) in phosphate buffer saline (0.1 M, pH 7.4) contained 250 yg calf thymus
DNA, 50 |jg bleomycin and iron: hydroquinone or iron: 1,2,4-benzenetriol (24 |JM:48 pM)
complex. The total reaction mixture was incubated for 30 min at 37°C and the reaction
was terminated by adding 1.0 ml of 10% ice-cold TCA. The contents were centrifuged
at 3000 rpm for 15 min and TBA colour was developed as described eariier. FeS04.7H20
was used to prepare iron and iron:polyphenols by the addition of iron and polyphenol in
1:2 ratio. Iron and iron: polyphenol solutions were prepared just before use.
Optical spectra
The optical spectra of polyphenols with or without the addition of Fe ^ were recorded
on Perkin-Elmer Lambda 15 UVA/IS spectrophotometer in freshly distilled water. The

Chapter - / / 33
incubation mixtures contained 100 pM hydroquinone alone or 1,2,4-benzenetriol alone
or with 200 pM Fe^* and scans were recorded every minute upto 4 min.
Lipid peroxidation in liver homogenate
Lipid peroxidation was estimated in 10% liver homogenate by the method described
by Bemheim e al. (1948). Liver homogenate (10%) was prepared in 0.15 M KCI. Liver
homogenate (10 ml) was taken in 25 ml conical flask and placed in metabolic shaker
waterbath at 37°C. An aliquot of 1.0 ml of homogenate was taken out at different time
interval (i.e. 0 hr and 1 hr) and precipitated by adding 1.0 ml of ice-cold 10% TCA. After
waiting for few min the tubes were centrifuged at 3000 rpm for 15 min. Finally, 1.0 ml
of clear supernatant was taken out and 1.0 ml of 0.67% (w/v) TBA was added. TBA
chromogen colour was developed by boiling the tubes on water bath for 15 min and read
at 532 nm. Results are expressed in terms of malonaldehyde equivalents nmoles/hr/gm
liver using molar extinction coefficient 1.56x10* cm' M' at 532 nm.
Sulphahydryl estimation
Total and free sulphahydryl (glutathione) were estimated using the standard
procedure described by Sedlak and Lindsay (1968).
(a) Total sulphahydryl
Liver homogenate in 0.15 M KCI containing 0.02 M EDTA (0.5 ml of 2%) was mixed
with 0.2M tris buffer containing 0.02 M EDTA (pH 8.2), 0.1 ml of 5,5'-dithiobis-
(2-nitroben2oic acid) (DTNB) (99 mg/25 ml absolute methanol) and kept the reaction
mixture at room temperature for 15 min. After centrifugation at 3000 rpm for 15 min,
absorbance of the supernatant nitromercaptobenzoic acid anion was read at 412 nm.
(b) Free sulphahydryl
To 1.0 ml of 2% liver homogenate (in 0.15 M KCI containing 0.02 M EDTA) was
added 1.0 ml of 10% ice-cold TCA. The test tubes were shaken intermittantly for 15 min
at room temperature and centrifuged at 3000 rpm for 15 min. Supernatant (1.0 ml) was

Chapter - / / 34
mixed with 1.4 ml of 0.4 M tris buffer containing 0.02 M EDTA (pH 8.9). Finally, 0.1 ml
of DTNB was added and mixed well. The absorbance of the nitromercaptobenzoic acid
anion formed was taken at 412 nm.
Sulphahydryl content was calculated from the standard plot of cysteine prepared in
the same manner and expressed in terms oi mmoles/100 gm liver
Haematological parameters
Haemoglobin (Hb) content in male mice was estimated by cyanomethaemoglobin
method (Drabkin and Austin, 1932). The basis of the method is dilution of blood in a
solution containing potassium cyanide and potassium ferricyanide. Haemoglobin is
converted into cyanomethaemoglobin in presence of Drabkin's cyanide - ferricyanide
solution. The absorbance of the solution is then measured in a spectrophotometer at a
wavelength of 540 nm.
Drabkin's reagent was prepared by dissolving 100 mg potassium ferricyanide and
25 mg potassium cyanide in 500 ml of distilled water. The reagent should be clear and
pale yellow in colour and can be stored at room temperature in a brown glass bottle.
When measured against water as blank in a spectrophotometer at wavelength 540 nm,
the absorbance must read zero.
Add 20 pi of blood into 4.0 ml of Drabkin's reagent and mixed well by inverting the
tubes several times. After mixing, the tubes were allowed to stand at room temperature
for atleast 10-20 min to ensure the completion of the reaction. Finally, the tubes were
read at 540 nm against Drabkin's reagent as a blank using Spectronic-2001
spectrophotometer. Cyanomethaemoglobin solution was used as a standard to calculate
the haemoglobin content.
Calculation
A54o0f test sample Cone std. (mg/dl) x dilution factor Hb(g/dl)= X
A540 of standard 1000
Other haematological parameters, such as white blood cell count, differential
leukocyte count and hematocrit value (%) were evaluated using standard procedures.

Chapter - II 35
Packing of Sephadex G-10 column and elution profile of iron alone, 1,2,4-benzenetriol alone and 1,2,4-benzenetriol:iron mixture
Sephadex G-10 (4.0 gm/200 ml) in 0.1 M sodium acetate buffer saline, pH 5.6 was
incubated overnight at room temperature. It was loaded to the column and allowed to
settle down and the gel column volume was 12 ml (12 cm x 1 cm). The column was
washed three times with acetate buffer and iron (as ferrous sulphate) (1 mM in 1.0 ml)
was passed through the column and eluted with 0.1 M acetate buffer of pH 5.6. Flow rate
of 2.5 ml/5 min was maintained and 1 to 30th fractions were collected at every 5 min.
Similarly, ImM in 1.0 ml of 1,2,4-ben2enetriol and 1.0 ml of 1,2,4-benzenetriol : iron
mixture (1 mM : 1 mM) were also passed through the column and 1 to 30th fractions were
collected.
In another set of experiment 1.0 ml of reaction mixture contained 100 pg horse
spleen ferritin, 2mM of 1,2,4-benzenetriol and with or without 1 mM ethylene-
diaminetetraacetic acid (EDTA) were passed through column and fractions were collected
at every 5 min.
Estimation of 1,2,4-benzenetriol and iron
1,2,4-Benzenetriol estimation was followed spectrophotometrically by recording the
optical density at 287 nm using a Spectronic-2001 spectrophotometer and iron estimation
was done according to the method of Peters et al., (1956) by using bathophenanthroline
sulphonic acid. Briefly, 2.0 ml of reaction mixture contained, 0.5 ml of aliquot from each
fraction, 1.5 ml of sodium acetate buffer (0.1 M, pH 5.6), 200 pg ascorbic acid and 50
pg bathophenanthroline sulphonic acid. Reaction mixture was kept at room temperature
for 5 min and absorbance was taken at 535 nm.
Preparation of glutathionyl hydroquinone
Equimolar concentration (200 pM) of 1,4-benzoquinone and glutathione reduced
(GSH) were mixed in distilled water in 1:1 proportion by volume. The nucleophilic
addition of GSH to 1,4-benzoquinone yields glutathionyl hydroquinone and its authenticity
was confirmed by the absorption maximum at 303 nm. No absorption was observed at

Chapter - / / ^
248 nm which is the absorption maximum for 1,4-benzoquinone. Free GSH was also not
demonstrable by Ellman's reagent.
Glutathlonyl hydroquinone and iron dependent TBAR formation from DNA or glutamate or deoxyuridine
of Iron catalyzed bleomycin-dependent release of TBAR from DNA in presence iron
or iron + GSH or iron + GSH + 1,4-ben2oquinone was performed according to the method
of Gutteridge (1987). Briefly, the assay system in a total volume of 1.5 ml consisted of
0.033 M phosphate buffer (pH 7.4), 250 pg calf thymus DNA, 50 jg bleomycin
hydrochloride and with or without oxyradical scavengers, viz. thiourea (5mM), mannitol
(50 mM), dimethylsulfoxide (33 mM), albumin, SOD and catalase (100 pg/ml). The
reaction was initiated by the addition of 15 pM iron (as FeS04.7H20) or 15 pM iron + 15
pM GSH or 15 pM iron + 15 pM GSH + 15 pM 1,4-t>enzoquinone. All these reactants
were dissolved immediately before use. The contents were incubated at 37°C for 30 min
and the reaction was terminated by the addition of 0.75 ml of 10% (w/v) ice-cold TCA.
The contents were centrifuged at 3000 rpm for 15 min and to the clear supernatants an
equal volume of 0.67% (w/v) TBA was added. TBA colour was developed by heating the
tubes for 15 min in a boiling water bath. The resultant TBA chromogen was read at 532
nm using a Spectronic-2001 spectrophotometer. The values are expressed as nmoles
of malonaldehyde equivalents generated by using £532=1.56x10^ cm' MV
Release of TBAR from L-glutamate or deoxyuridine was followed according to the
method of Gutteridge (1981). Briefly, the reaction mixture (total volume 1.5 ml)
contained, 0.033 M sodium phosphate buffer (pH 7.4)in 0.15 M NaCI, 4 mM glutamate
monosodium or 3.2 mM deoxyuridine and the reaction was started by the addition of
glutathlonyl hydroquinone (5.0 pM - 25.0 pM). The contents were incubated for 2 hr at
37°C and the reaction was terminated by the addition of 1.0 ml of 10% (w/v) ice-cold
TCA. TBAR was estimated as described in the case of release of TBAR from DNA.
Recovery of glutathionyl hydroquinone
Recovery of glutathionyl hydroquinone from bone marrow lysate (1 mg protein

Chapter - / / 37
equivalent) was monitored by assaying Iron- catalyzed bleomycin-dependent degradation
of calf thymus DNA as compared to that without bone marrow lysate.
Measurement of Oj'
Release of Oj' from glutathionyl hydroquinone (30 \M) in a total volume of 1.5 ml
was monitored by the superoxide dismutase inhibitable reduction of cytochrome c (80
pM) at 550 nm at 25°C spectrophotometrically using the extinction coefficient value of
reduced cytochrome c as 29,500 cm'M"' (Cumutte et al., 1974). Appropriate blanks were
used to con"ect any cytochrome c reduction in the absence of glutathionyl hydroquinone.
Reaction of glutathionyl hydroquinone with plasmid pUC 18 DNA
Reaction mixture in 50 pi, contained 20 pM phosphate buffered saline (pH 7.4), 300
ng plasmid pUC 18 DNA, 200 pM 1,4-benzoquinone alone or GSH alone or glutathionyl
hydroquinone and with or without oxyradical scavengers, viz. superoxide dismutase,
catalase (54 pg), mannitol, thiourea and sodium benzoate (2 mM). The contents were
mixed well and incubated for 3 hr at 37°C. After 3 hr incubation, 5 pi of solution
containing 40 pM EDTA, 0.05% bromophenol blue tracking dye and 50% (w/v) sucrose
was added and the solution was subjected to electrophoresis on 1% agarose gels in TBE
buffer (89 mM tris base, 89 mM boric acid and 2 mM EDTA, pH 8.0) for 3 hr at 60 mA
current. The gels were stained with ethidium bromide (0.5 pg/ml) viewed and photo
graphed on an UV-transilluminator. One single strand break in supercoiled DNA into
relaxed open circular form or double strand break into linear form, respectively, run
slower than undamaged supercoiled DNA as evidenced by 1% agarose gel
electrophoresis (Toyokuni and Sapripanti, 1993).
Isolation of rat liver nuclei
Approximately 4 gm rat liver was homogenized in 30 ml of Tris-EDTA buffer (50 mM
tris, 10 mM EDTA, pH 8.0) and centrifuged for 10 min at 2000 rpm. Pellet was washed
twice in Tris-EDTA buffer and resuspended to 10 ml with 0.1 M phosphate buffered
saline (pH 7.4) (Mahmutoglu and Kappus, 1987),

Chapter - II 38
Release of TBAR from rat liver nuclei in the presence of different polyphenols and effects of different metal chelator and oxyradical scavengers
The reaction mixture in a total volume of 1.5 ml contained, 0.1 M phosphate buffered
saline (pH 7.4), 0.5 ml isolated rat liver nuclei suspension (12 mg protein), 330 JM
hydroquinone/1,2,4-benzenetriol/6-hydroxydopamine/dopamine and with or without
copper (0.08 and 0.167 mM) or EDTA/DTPA (5 pM) or bathocuproine (5-25 pM) or
different oxyradical scavengers, such as thiourea (2.5 and 5.0 mM)/mannitol (50 mM)/
catalase(150 pg/1.5 ml)/SOD (100 jg/1.5 ml). The contents were incubated for 2 hr
at 37°C with occasional shaking and the reaction was terminated by the addition of 0.5
ml of 10% ice cold TCA. The contents were centrifuged and to 1.0 ml of supematant 0.5
ml of 0.67% thiobarbituhc acid (TBA) was added and heated in a boiling water bath for
15 min and the resultant malonaldehyde: TBA chromogen was read at 532 nm. The
results were expressed as nmoles of TBAR formed per hour per gm protein by using
extinction coefficient 1.56 x 10^ cm' M (Rao and Pandya, 1989; Quinlan and
Gutteridge, 1987).
Metal analysis
Metals were analyzed in liver tissue and isolated rat liver nuclei of rats of benzene
exposed and control group by the method of Berman (1980). Equal weight of liver tissue
or 3.5 ml of isolated nuclei was added into the digestion flask containing 20 ml of
digestion mixture (1:6:: Perchloric acid (60%): nitric acid). The flasks were boiled in the
fuming chamber on hot plate till dry. The dry ash was dissolved in 5.0 ml of 0.1 N nitric
acid and transfered into the test tube. The contents of iron and copper were read using
atomic absorption spectrophotometer. Results were expressed in terms of pg metal/gm
liver tissue or [jg metal/gm protein in liver nuclei.
Nuclear DNA damage by hydroquinone or 1,2,4-benzenetriol
Isolated rat liver nuclei were incubated with hydroquinone or 1,2,4-benzenetriol and
DNA damage was evaluated by agarose gel electrophoresis. Briefly, 50 1 of total
reaction mixture contained, 20 fj| of isolated rat liver nuclei suspension (350 pg protein).

Chapter - II 39
10 tJl of phosphate buffered saline (0.1 M, pH 7.4) and hydroquinone orl ,2,4-benzenetriol
(0.5 mM and 1.0 mM). Reaction mixture was incubated for 4 hr at 37°C and finally mixed
with 5 \i\ of solution containing 40 pM EDTA, 0.05% bromophenol blue tracking dye, 1%
sodium dodecyl sulphate and 50% (w/v) suaose. Total reaction mixture was loaded on
1% agarose gel for electrophoresis in TBE buffer (89 mM tris base, 89 mM boric acid
and 2 mM EDTA, pH 8.0) for 3 hr at 75 mA current. The gels were stained with ethidium
bromide (0.5 pg/ml), viewed and photographed on an UV-transilluminator.
STATISTICAL ANALYSIS
The statistical values and level of significance between the experimental and control
groups of animals were computed by the following formulas :
Standard deviation (SD)
(X, - X)^ SD =
n-1
Where,
X, = individual values X = arithmatic mean of individual values n = sample size
Standard error
SE
t value
t
(SE)
SD
{T
^A • '^B
/ (SEJ^MSEB)^
Where, x^ and Xg are two sample means and SE^ and SEg are the standard
errors.

Chapter • / / 40
Level of significance
After calculating the 't' value the level of significance (p value) was found out from
the table. The p values less than 0.05, 0.02, 0.01 and 0.001 were considered as just
significant, significant, more significant and highly significant respectively.

Chapter - IB Release of Iron from Ferritin
by 1,2,4-benzenetriol Introduction
Results
Discussion

Chapter - III 41
INTRODUCTION
Iron (Fe) is an essential element for living organisms because of its role in major
biological reactions such as the tricarboxylic acid cycle, electron transport, nitrogen
fixation, DMA synthesis and toxication and detoxication reactions (Theil, 1987). Iron in
aqueous solution has ready access to two stable oxidation states, the ferrous, Fe(ll) and
the ferric, Fe(lll). This property underlies the participation of iron in reactions spanning
all of biochemistry including those controlling the flow of electrons through bioenergetic
pathways, the activation of molecular oxygen, the decomposition of noxious derivatives
of oxygen such as peroxide and superoxide and binding of oxygen by haemoglobin,
myoglobin and haemerythrins (Aisen and Listowsky, 1980)
But, the same redox properties that make iron so useful in biocatalysis and oxygen
transport present hazards to the organism. Throughout the biological universe, therefore,
organisms have been compelled to evolve specific iron sequestering molecules to
maintain the element in soluble form available for transport and for biosynthesis to
essential iron proteins and enzymes. By suppressing the catalytic activity of iron in

Chapter - III 42
one-electron redox reactions, such iron-binders may also reduce exposure of cells to
potentially damaging species such as peroxide, superoxide and hydroxyl radicals
(Willson, 1977). In the vertebrate world, these iron-binding functions are managed by
transferrin and ferritin, the transport and storage protein of iron metabolism respectively.
Each of these proteins bind iron sufficiently tightly to resist formation of insoluble
hydroxylitic products. The binding of iron to these proteins is reversible so that the metal
is available upon demand to cells with a specific need so that it is incorporated into its
final biochemical form. The binding of iron by transferrin is to two specific sites with
well-defined stoichiometry, but in fenritin iron exists as polynuclear aggregates of variable
size and compositions (Aisen and Listowsky, 1980).
Storage of iron occurs in two forms; as a soluble protein enveloped core of ferritin,
and as a part of the nondescript insoluble hemosiderin. The latter consists of inorganic
iron associated with protein and other substances in variable proportions, and in fact,
may originate from degraded ferritin (Vidnes and Helgeland, 1973). Mammalian liver,
spleen and bone marrow are especially rich in ferritin but the protein is found in most
other tissues, in serum and in circulating blood cells. Ferritin is synthesized in response
to iron, which it sequesters in accessible form to provide a mobilizable reserve. Thus, the
protein can serve as a source or sink of iron to play a dynamic role in iron metabolism,
with regulation occuning at the level of protein synthesis and turnover, and iron deposition
and release (Aisen and Listowsky, 1980).
Ferritin is a large protein having molecular weight of about 450 kilodatton. Apoferritin
(protein moiety of ferritin only) consists of 24-polypeptide chains, made up of two types
of subunits, H and L, with molecular weight of 22,000 and 19,000 respectively (Biemond
et al.. 1988). The polypeptide chains are arranged in a spherical protein shell, with a
central cavity. X-ray and electron microscopic studies show that the protein shell is
nearly spherical with an external diameter of 120-130 A° and a central cavity of about 75
A° (Hoare et al., 1975). The iron is encapsulated in the central cavity which has eight
shallow pockets (Treffrye^ a/., 1977). The channels facilitate the movement of iron, and
this constitutes an important function of femtin. One molecule of ferritin can store as
many as 4,500 iron atoms (Harrison, 1977). Iron is present in ferritin largely in the form

Chapter - III 43
of ferric oxyhydroxide (FeOOH) together with some phosphate (Theil et al., 1979).
Clinicians monitor the level of ferritin in serum to obtain information about the state of the
reticuloendothelial iron stores, or the presence and progress of some malignant diseases
(Jacobs and Worwood, 1975; Worwood e a/., 1975). Several studies have indicated the
involvement of free radicals in initiation and promotion processess of carcinogenesis
(Wei and Frenkel, 1991). Large human prospective studies suggest that excess body
iron stores estimated by serum ferritin level, serum transferrin level, or transferrin
saturation are associated with carcinogenesis, such as primary hepatocellular carcinoma,
colon cancer, lung cancer, bladder cancer, acute lymphocytic leukemia and neuro
blastoma (Potaznic ef al., 1987; Evans ef a/., 1987). Thus, elucidation of the
mechanisms of iron-mediated oxidative damage may be important for the prevention of
carcinogenesis.
Redox cycling is a characteristic of transition metals such as iron and is closely
associated with the production of reactive oxygen species. In order to link the chemical
reaction with biological system the concept of the "catalytic" or "free" form of iron
(Gutteridge et al., 1982) is important. "Free" iron was originally measured as thiobarbitunc
acid reactive products (TBAR) formation in an in vitro system that contains sample fluid,
calf thymus DNA, bleomycin, ascorbate and HjOj. Bleomycin in the presence of Fe(ll)
degrade DNA to form TBAR. The mechanism of bleomycin-dependent DNA degradation
in presence of iron and reducing agent is shown in Figure-3.1. Although, other ions can
bind to bleomycin, they do not result in DNA degradation (Gutteridge et al., 1982).
Reactive oxygen species in biological system induce oxidative stress. Every cell,
whether using oxygen or not, has elaborate inducible protective mechanisms against
oxidative stress. There are almost always reducing agents, or enzymes associated with
reductants. Reducing molecules or enzymes include ascorbate, glutathione reduced
(GSH) and thiol-specific antioxidant (Chae et al., 1993; Yim et al., 1994). "Free" iron, if
it exists in vivo, can, therefore, t>e quite harmful because there are many reducing agents
mentioned above that can initiate the Fenton reaction via the production of H O as
follows:

O
10
.2 H o ^
5 CO
^ r O X 'J i: o w« QL 03
< + Q 5
< Q 5
<
V I t
< • m
O I
e g o
u > E o
OQ
c (1) O) 03 cn c o 3 •o 0) k. •c c ro c o
0) o c 0)
c o TO
•D TO k_ D) 0)
< Z Q
0) •D C 0) Q. dJ
X)
E o
E w 'c
o
CO
3
iZ
W ^

Chapter - III 44
Fe(lll) + Reductant > Fe(ll) + Oxidized Reductant
2Fe(ll) + O2 + 2H* > 2Fe(lll) + H A
Fe(ll) + H2O2 > Fe(lll) + OH + OH
The mechanism of iron uptake, mobilization, release and utilization are well
understood (Crichton and Ward, 1992). Low molecular weight iron chelates which
maintains a dynamic equilibrium fc>etween iron uptake, storage and utilization have been
descrit>ed (Jacobs, 1977). Iron may be mobilized from ferritin by reduction with agents
such as dithionite, thioglycolate, or other thiols (Crichton, 1973), with removal facilitated
by chelators. Without reductants, chelating agents are either ineffective for releasing iron
from ferritin or remove it only very slowly (Crichton and Roman, 1978). Although
ascorbate, cysteine and reduced glutathione may serve as reductants in vitro, but they
are not efficient enough to be considered possible physiological mediators of iron release
from femtin. However, recently Baaderef a/., (1996) clearty demonstrated that ascorbic
acid is capable of mobilizing iron from ferritin in the cellular system by reduction of the
ferric ion core in neuroblastoma SK-N-SH cells. Superoxide anion radical generated by
xanthine-xanthine oxidase system(Topham e a/., 1982) and many toxicological reductants
which mobilize ferritin iron have been reported. In contrast, ferritin was able to stimulate
free radical damage, possibly by the formation of hydroxyl radicals, in many test systems.
A bulk of evidence obtained in vitro confirms that superoxide radical is also capable of
mobilizing iron from ferritin (Reif, 1992; Monteiro and Winterbourn, 1989a).
Several endogenous and xenobiotic agents have been reported to release Fe(ll)
from ferritin Fe(lll) in vitro (Aust et al., 1985). Thomas et al.. (1985) demonstrated that
xanthine/xanthine oxidase mediated generation of superoxide radical is able to reduce
and release iron from ferritin and concomitantly promote liposome lipid peroxidation.
Redox cycling of paraquat (Thomas and Aust, 1986a), the semiquinone radical of
adriamycin (Thomas and Aust, 1986b), nitric oxide (Reif and Simmons, 1990), 6-
hydroxydopamine (Monteiro and Winterixium, 1989b) and therapeutic and physiological
iron chelators (O'Connell et al., 1989) have been shown to promote mobilization and
reductive release of iron from ferntin. Certain diphenols in the presence of a reductant

Chapter - III 45
have also been shown to release iron from ferritin (Kadir e a/., 1992). It has been
suggested in particular that iron is released from stores under conditions in which
oxidative stress is involved in both systems in vitro (Thomas e a/., 1985) and isolated
perfused organs (Healing et a/., 1990). Ferrali et al., (1992) reported that oxidizing
agents viz. phenyl hydrazine, divicine and isouramil induced iron release and membrane
damage in erythrocytes. The same oxidizing agents were also shown to release iron
from ferritin (Monteiro and Winterboum, 1989a).
Expression of benzene toxicity requires its metabolism to toxic metabolites and
follows similar pathways in humans and animals. It is widely believed that the
polyphenolic metabolites of benzene accumulate against the concentration gradient in
bone marrow (Greenlee e a/., 1981a) and are responsible in the formation of reactive
oxygen species which may play a role in benzene induced toxicity. Hydroquinone and
1,2,4-benzenetriol, the two principal polyphenolic metabolites of benzene have been
shown to be toxic due to their capability to undergo autooxidation (Rao and Pandya,
1989). The suggested mechanism includes free radical formation via superoxide and
covalent binding of semiquinones to DNA, RNA or other cellular macromolecules
(Greenlee et al., 1981b). Although, the mechanism how benzene interferes with iron
metabolism is not well known, but accumulation of bleomycin-detectable low molecular
weight iron (LMI) compound in bone marrow was observed in experimental rats dunng
benzene exposure (Rao et al.. 1990). Earlier studies showed that benzene inhibits heme
biosynthesis at the level of 6-AU\ dehydratase (Freedman et al.. 1977; Rao and Pandya,
1980) and increase in heme oxygenase activity, a rate-limiting enzyme for heme
degradation (Abraham et al., 1986). As a result, depletion in regulatory heme content
(Siddiqui etal., 1988) and accumulation of iron have been reported (Pandya etal., 1990).
Superoxide dependent reductive release of iron from ferritin may increase the LMI pool
capable of undergoing redox reactions (Topham et al.. 1982).
With this available information that accumulation of polyphenolic metabolites of
benzene and formation of reactive oxygen species due to autooxidation of polyphenolic
metabolites, we investigated the efficacy of polyphenols to release iron from horse
spleen ferritin in vitro, in the presence of hydroquinone or 1.2,4-benzenetriol and other

Chapter - III 46
endogenously formed polyhydroxy compounds, and whether the iron released can
catalyze oxidation reactions.
RESULTS Release of iron from ferritin in the presence of polyphenols (hydroquinone or
1,2,4-benzenetriol) and other reducing agents was recorded in Table-3.1. Addition of
hydroquinone (330 pM) did not result in the release of iron from ferritin. However, the
same concentration of 1,2,4-benzenetriol resulted in a significant release of iron (3.2
pM/min) from ferritin. The release of iron was found to be linear upto 15 min and about
20% of total ferritin iron was released. Presence of dopamine failed to release iron from
ferritin, whereas 6-hydroxydopamine released large amounts of iron (4.6 [jM/min) from
ferritin as was reported eariier (Monteiro and Winterbourn, 1989b). Significant amounts
of iron release from femtin was observed in the nitrogen atmosphere, although to a lesser
extent than in aerobic conditions (Table-3.1). The efficacy of other polyphenols such as
1,2,3-t>enzenetriol (Pyrogallol) and 1,3,5-benzenetriol (phloroglucinol) to release iron from
ferritin were studied. Except 1,2,4-benzenetriol, other tnhydroxy benzene derivatives
failed to release iron from ferritin (Table 3.2). The release of iron from ferritin was
concentration dependent on 1,2,4-benzenetriol as is shown in Figure-3.2, although the
increase was not linear with the concentration of 1,2,4-t>enzenetriol under aerobic and
anaerobic conditions.
Table-3.3, represents the effect of oxyradical scavengers on the release of iron from
ferritin in the presence of 1,2,4-benzenetriol The release of iron was substantially
inhibited as was evident from the decreased formation of [Fe(ferrozine)3]^' in the
presence of albumin, catalase and superoxide dismutase (SOD). Thiourea and mannitol
which are hydroxyl radical scavengers were able to inhibit iron release to an extent of
about 15%. This indicates that 1,2,4-benzenetno! mediated release of iron was not
completely due to the mediation of active oxygen species generated during autooxidation
of 1,2,4-benzenetriol.
The effect of increasing concentrations of 1,2,4-benzenetriol on lipid peroxidation of
2% rat brain homogenate in the presence of femtin is shown in Figure-3.3. The increase
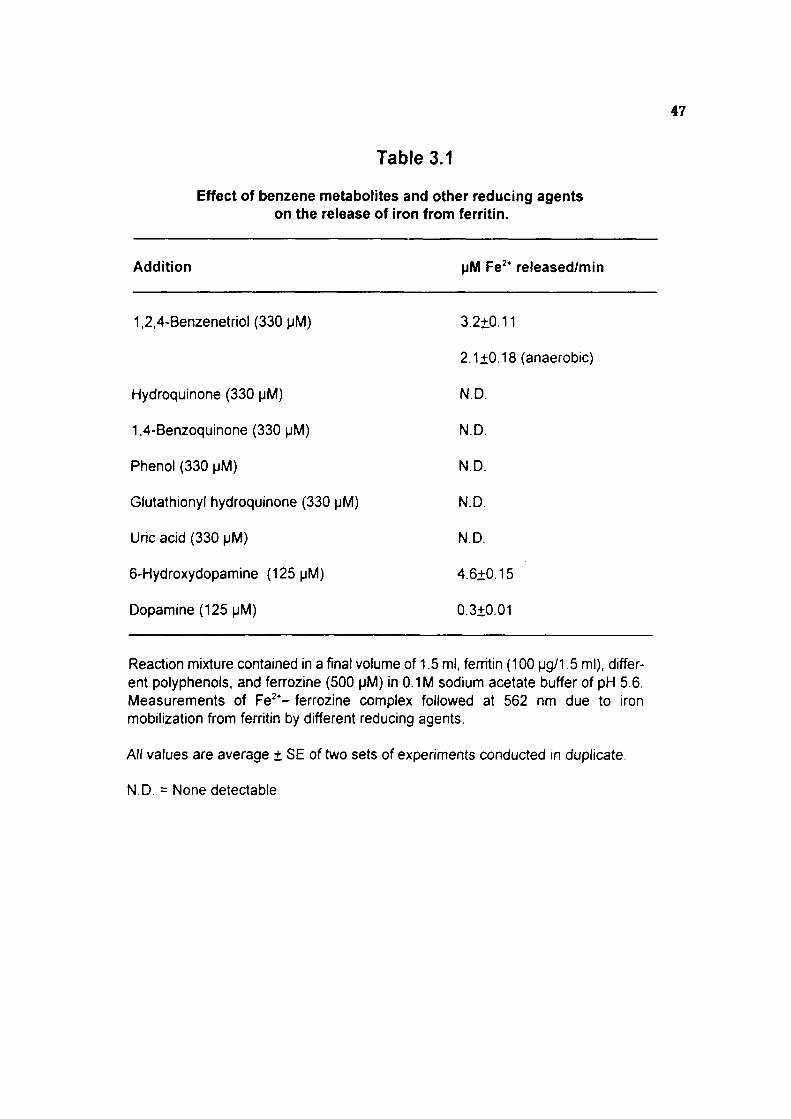
47
Table 3.1
Effect of benzene metabolites and other reducing agents on the release of iron from ferritin.
Addition
1,2,4-Benzenetriol (330 pM)
Hydroquinone (330 (JM)
1,4-Benzoquinone (330 [iM)
Phenol (330 pM)
Glutathionyl hydroquinone (330 pM)
Uric acid (330 pM)
6-Hydroxydopamine (125 pM)
Dopamine (125 pM)
pM Fe * released/min
3.2+0.11
2.1±0.18 (anaerobic)
N.D.
N.D.
ND.
N.D.
N.D.
4.6±0.15
0.3+0.01
Reaction mixture contained in a final volume of 1.5 ml, ferritin (100 pg/1.5 ml), different polyphenols, and ferrozine (500 pM) in 0.1 M sodium acetate buffer of pH 5.6. Measurements of Fe^*- ferrozine complex followed at 562 nm due to iron mobilization from ferritin by different reducing agents.
All values are average + SE of two sets of experiments conducted in duplicate.
N.D. = None detectable

48
Table 3.2
Effect of 1,2,4-benzenetriol and other trihydroxy benzene derivatives on the release of iron from ferritin
Addition \iM Fe^* released/min
1,2,4-Benzenetriol 3.2±0.11
1,2,3-Benzenetriol N.D.
1,3,5-Benzenetriol N.D.
Reaction mixture contained in a final volume of 1.5 ml, fenitin (100 |jg/1.5 ml), polyphenols (330 pM) and ferozine (500 pM) in 0.1 M sodium acetate buffer of pH 5.6. Measurements of Fe^*- ferrozine complex followed at 562 nm. N.D. = None detectable

e ^1 + +
A —Aerobic atmosphere
B —Anaerobic atmosphere
330 B T ( M M )
Figure 3.2 : 1,2,4-Benzenetriol concentration dependent iron release from horse spleen ferritin under (A) Aerobic, and (B) Anaerobic conditions.

49
Table 3.3
Release of iron from ferritin by 1,2,4-benzenetriol and inhibition by oxygen radical scavengers
Reaction mixture pM Fe'* released/min
Complete reaction mixture 3.20±0.11
+ Urea(5mM) 2.88±0.24(10)
+ Thiourea (5 mM) 2.64+0.08(18)
+ Mannitol (50 mM) 2.72±0.10 (15)
+ Albumin (150 pg/l.5 ml) 2.32+0.02(28)
+ Catalase(150[jg/1.5ml) 2.00±0.05(38)
+ SOD (150 ^^g/^ .5 ml) 2.16±0.12(33)
Complete reaction mixture contained in a final volume of 1.5 ml, ferritin (100 |jg/1 5 ml), 1,2,4-benzenetriol (330 pM), fen-ozine (500 pM) and oxygen radical scavengers in 0.1M sodium acetate buffer of pH 5.6. All values are average ± S.E. of t\NO sets of experiments conducted in duplicate. Numbers in paraentheses are percent inhibition.

o en CO
U) •rN' ^ fNI
m CO
•
00
, . ^
2 H,
* - , _ i f r '
H-m
c d) o ^
1^ <U TO N c C m
to fNj x : T—
o ro
If 'c s^ O >»-o o
55 o E ^ m 0) >- i i >»-U X O ^ 2 a> o ^ ^
UJ — Q-
CO
o i . 3
( j q /sd|oaj u ) SHVai

in a?
CM CO
(T> CD
CD iD
ro en
' " • ^
2 d.
*-^ h -m
c s= o ni _ o o ^ c o
1 c N ro Si "D ^ 2
•^ H
o -o
- Q.
8 l o E (-> o
9 -Q ro T^ 2 S< e ro Q o S ^ o c £ 3z 2 ^ U J •
CO
iZ
(jq/Sdiouju)savai

50
Table 3.4
Release of iron from isolated rat liver ferritin
1,2,4-Benzenetriol (MM)
00
330
330
Ferritin (mg equivalent protein)
0.8
0.4
0.8
by 1,2,4-benzenetriol.
pM Fe * released/min/ mg protein
0.00
2.21
3.80
Complete reaction mixture contained in a final volume of 1.5 ml, isolated rat liver ferritin (0.4 and 0.8 mg equivalent protein), 1,2,4-t)enzenetriol and ferrozine (500 (JM) in 0.1 M sodium acetate buffer of pH 5.6. All values are average of two sets of experiments conducted in duplicate with variations of 10% or less.

c
o I—
o.
3
o
o L. U .
o E c o
E o
in O
c o
7.0-
Tfmc ( min)
Figure 3.5 : Time course of formation of [Fe(ferrozine)3p* followed at 562 nm due to iron mobilization from bone marrow cell lysate by 1,2,4-ben2enetriol.

Chapter - III 51
in lipid peroxidation as measured by TBAR formation was almost linear with the increase
in 1,2,4-benzenetriol concentration which shows that there is 1,2,4-benzenetriol
concentration dependent increase in iron release from ferritin. Similarly, the iron release
from ferritin in the presence of increasing concentrations of 1,2,4-ben2enetriol was tested
for the iron catalyzed bleomycin-dependent degradation of calf thymus DNA. There was
a linear increase in TBAR formation from the deoxyribose moiety of DNA with the
increase in concentration of 1,2,4-benzenetriol (0-165 pM) (Figure-3.4).
Ferritin was isolated from female rat liver after inducing the ferritin level by injecting
FeS04.7H20 intraperitoneally. The time course of formation of [Fe(ferrozine)3]^* complex
due to release of iron from isolated rat liver fenitin in presence of 1,2,4-benzenetriol was
recorded. The release of iron from isolated ferritin was found to be linear with increase
in the amount of ferritin (Table-3.4).
Release of iron from bone marrow cell lysate in the presence of hydroquinone and
1,2,4-benzenetriol showed that hydroquinone did not release any iron from bone marrow
cell lysate (results not shown), whereas, 1,2,4-benzenetriol (330 pM) resulted in the
release of iron from bone marrow cell lysate. The increase in the release of iron from
bone marrow lysate in the presence of 1,2,4-benzenetriol as a function of time was also
observed (Figure-3.5).
DISCUSSION
Benzene exposure interferes with iron metabolism which includes, decrease in
incorporation of *®Fe into circulating erythrocytes (Lee ef a/., 1974). Exposure to benzene
leads to decrease in heme biosynthesis enzyme particulariy. 6-AL.A dehydratase
resulting in accumulation of 6-ALA in bone marrow (Rao and Pandya, 1980, Freedman,
et al., 1977), Accumulation of low molecular weight iron (LMI) in bone marrow during
benzene exposure has been demonstrated and it was shown that LMI catalyze
bleomycin-dependent degradation of calf thymus DNA (Pandya ef a/., 1990). However,
the mechanism of how benzene interferes with iron metabolism is not known.
Polyphenolic metabolites of benzene have been shown to accumulate in bone marrow
against the concentration gradient (Greenlee et al., 1981a; Rickert et al., 1979).

Chapter - III 52
Autooxidation of 1,2,4-benzenetriol has been shown to generate superoxide radicals
(Lewis et al., 1988b). Superoxide radical is a potent reductant of ferritin iron and causes
release of iron from ferritin (Topham et al.. 1982).
In the present investigation it was observed that the presence of 1,2,4-benzenetriol
induced iron release from ferritin in a concentration dependent manner. The possible
mechanism of iron release from ferritin by 1,2,4-benzenetriol is shown in Figure-3.6. The
release of iron from ferritin during autooxidation of 1,2,4-benzenetriol in vivo may result
in increased intracellular concentrations of free iron. The inability of 1,2,3-benzenetriol,
1,3,5-benzenetriol, hydroquinone or dopamine to release iron in comparison to 1,2,4-
benzenetriol and 6-hydroxydopamine suggest that hydroxy hydroquinone moiety is
essential. Similariy, dialuric acid, a tetrahydroxypyrimidine and isouramil, a trihydroxy-
pyrimidine have been shown to release iron from ferritin (Monteiro and Winterbourn,
1989a). Although, the physiological mechanism of iron mobilization from ferritin is pooriy
understood, the observed iron release from ferritin by 1,2,4-benzenetriol could occur
through the direct reduction of iron by the hydroxyhydroquinone form or via its
autooxidation intermediates i.e., superoxide radical and semiquinone. The semiquinone
radicals of several antitumorigenic compounds have been shown to reduce and release
ferritin iron (Thomas and Aust, 1986b; Butler et al., 1985).
The presence of oxyradical scavengers i.e., albumin, catalase or superoxide
dismutase caused 28, 38 or 33% inhibition, respectively of 1,2,4-benzenetriol dependent
iron release from ferritin, with the enzymatic activities of catalase and superoxide
dismutase (SOD) being of little importance relative to the protein effect of albumin. The
oxyradical scavenger insensitive rate of iron release from ferritin by 1,2,4-benzenetriol
is probably due to direct reduction of ferritin iron by 1,2,4-benzenetriol, as substantial
amounts of iron were released in a nitrogen atmosphere also. Based on the present
investigation, it is proposed that 1,2,4-benzenetnol induced iron release from ferritin might
be due to direct reduction and the iron released may facilitate the reduction of O2 by
1,2,4-benzenetriol resulting in the generation of superoxide radical. The autooxidation
rate of 1,2,4-benzenetriol has been shown to be 92-fold higher than that of the parent
hydroquinone which lacks an -OH substituent (Brunmark and Cadenas, 1988). The

Ferritin shell
/ \ /
+ ^ 2e" ;+(2H ; OH r
I
I
I
Ferritin ' channel /
Figure 3.6 : The possible mechanism of iron release from ferntin by 1,2.4-benzenetnol

Chapter - HI 53
presence of hydroquinone or dopamine did not result in iron release from ferritin,
probably due to the lack of an -OH substituent and the resulting slow autooxidation.
The results of the present study demonstrate that iron released from ferritin in the
presence of 1,2,4-ben2enetriol is capable of inducing lipid peroxidation and catalyzing
bleomycin-dependent calf thymus DNA degradation. The mechanism by which iron is
released from bone marrow cells and whether it acts as a prooxidant during benzene
toxicity is not known. However, earlier studies had demonstrated that enhanced lipid
peroxidation was observed in experimental animals exposed to benzene (Rao and
Pandya, 1978; Uysal at al., 1986). Polyphenolic metabolites of benzene have been
shown to accumulate in bone marrow in millimolar concentrations during benzene
exposure (Greenlee e/ al., 1981a; Rickert e/a/., 1979). The present study indicated that
a much lower concentration of 1,2,4-benzenetriol (66 pM) is able to release iron from
ferritin. In a recent study (Singh et al., 1994) it was shown that iron in the presence of
1,2,4- benzenetriol was a potent agent to catalyze bleomycin-dependent degradation of
calf thymus DNA. Similariy, the iron released from ferritin by 1,2,4-benzenetriol could
catalyze bleomycin-dependent degradation of calf thymus DNA.
Toyokuni (1996) hypothesized that increase in free iron in the cytoplasm or nucleus
may be associated with an increased risk of cancer and suggested the possible
mechanisms of iron-induced carcinogenesis (Figure-3.7 ). Our present study indicates
that 1,2,4-benzenetriol is a potent reductant of ferric iron of ferritin and also mobilize and
release iron from ferritin core. The release of iron from bone marrow lysate by
1,2,4-benzenetriol may be of toxicological significance as this could lead to disruption of
intracellular iron homeostasis in bone marrow cells. Similariy, iron release was noticed
from ferritin isolated from liver in the presence of 1,2,4-benzenetriol. The release of iron
from ferritin during autooxidation of 1,2,4-benzenetriol In vivo may result in increased
intracellular concentrations of free iron which may attain to genotoxic levels. Involvement
of similar mechanism In vivo as 1,2,4-benzenetriol dependent reduction and release of
iron from storage sites could result in increased generation of active oxygen species which
in turn damage cellular macromolecules and also can interfere in maturation and
differentiation of erythroblasts. In summary, the present observations offer a new

11 K U.
I M O
s l:Al! J O ^ Q
E o
T) B Q. O X3 ro CO (/) <u c QJ O) O c o o
• D (U
o "O
c c o
0) E 0) s: ^^ U CD W CD
ro •,— o
P Q- > . >- O I I-
rs. ro <u 3

Chapter - III ^^
mechanism that the iron released from ferritin by 1,2,4-benzenetriol could contribute to
the better understanding of the toxicity of benzene. In the light of the present results, it
is believed that iron release in bone marrow cells in vivo leads to generation of
superoxide radicals, peroxidation of lipids and probably affects maturation and
differentiation of bone marrow cells.

Chapter - IV Prooxidant and Antioxidant Properties
of Polyphenol - Iron Complex: an Implication for Benzene Toxicity
Introduction
Results
Discussion

Chapter • IV 55
INTRODUCTION
Chronic exposure to benzene is known to induce aplastic anemia, leukemia, and
other related blood disorders (kalf, 1987). However, the precise mechanism by which
benzene induces these effects are unknown. It has been shown that benzene itself is
not actual toxicant. Expression of benzene toxicity requires its metabolism to reactive
intermediate(s). The reactive intermediates responsible for benzene toxicity however,
have not been definitly identified (Snyder et al., 1993). It is believed that the effect of
benzene is likely to be exerted through the action of multiple metabolites on multiple
targets through multiple biological pathways (Goldstein, 1989).
Benzene is metabolized in liver as well as bone marrow by cytochrome P-450
dependent mixed function oxidase enzymes (Huff etal., 1989). Although, the metabolism
of benzene is complex but is well understood. Benzene is primarily metabolized to
phenol, catechol, hydroquinone, 1,2,4-benzenetrio( (Porteous and Williams, 1949) and
open ring products such as trans, trans-muconaldehyde and trans, trans-muconic acid

Chapter - IV 56
(Latriano ef a/., 1986). The pathway of benzene ring opening is not known, but a number
of possible mechanism for oxidative benzene ring opening have t>een postulated (Grotz
etal., 1994),
Earlier reports have shown that hepatic metabolism of benzene results in
accumulation of polyphenolic metabolites against the concentration gradient in bone
marrow and exerts its toxic effects (Smith et al., 1989). Male Fischer rats exposed to
benzene 500 ppm have been shown to accumulate about 1 mM polyphenolic metabolties
in bone man-ow (Rickert et al., 1979). Hydroquinone and 1,2,4-benzenetriol have been
shown to be potent toxic metabolite of t>enzene (Irons, 1985). In an in vitro study, Rao
and Pandya (1989) observed that polyphenolic metakx)lites o1 benzene are autooxidized
faster in the presence of copper ions and resulting in the formation of thiobarbituric acid
reactive products (TBAR) from target molecules such as glutamate and calf thymus
DNA. The suggested mechanism includes free radical formation via superoxide and
covalent binding of the semiquinone metabolites of polyphenol to such molecules.
Greenlee et al., (1981b) demonstrated that the triphenolic metabolite of benzene,
1,2,4-benzenetriol, is the most reactive toward molecular oxygen. Its strong chemical
reactivity toward O2 is consistent with a high biological potency. 1,2,4-Benzenetriol
induces both single and double strand breaks in DNA (Kawanishi et al., 1989), causes
sister chromatid exchange in human lymphocytes (Erexson et al., 1985) and gene
mutations (6-thioguanine resistance) in V79 cells (Glatt, ef al., 1989). Currently in a series
of studies by Zhang et al., (1994) reviewed the roles of quinone, oxygen and transition
metal ions in the genotoxicity of 1,2,4-benzenetriol. Zhang et al., (1996) described the
potential role of metal ions (iron and copper) in the oxidation of 1,2,4-benzenetriol and
may play an important role in benzene-induced genotoxicity. 1,2,4-Benzenetriol has
been shown to induce oxidative DNA damage (formation of 8- hydroxydeoxyguanosine)
both in cultured human cells in vitro (Zhang et al., 1993) and in mouse bone marrow in
vivo (Kolachana et al., 1993). Despite the evidence that 1,2,4-benzenetriol-induced
active oxygen species are cytotoxic and genotoxic, surprisingly little is known regarding
the mechanisms of its reaction with Oj.

Chapter -IV cy
Recent studies revealed that treatment of HL-60 cells with hydroquinone resulted
in the formation of 8-hydroxydeoxyguanosine (8-OHdG) formation, suggesting that,
hydroquinone can induce oxidative DNA base modification in cells (Kolachana et al.,
1993). Although, the mechanism by which hydroquinone causes the above effects,
remains unclear. It is likely that some of these effects, if not all could be mediated by
further oxidative activation of hydroquinone. Similar observation was also obtained in
different cell lines whose peroxidase activation of hydroquinone results in the formation
of DNA adducts (Levay et al,, 1993), Recently Pongracz and Boded (1996) reported the
DNA adduct formation in HL-60 cell culture treated with hydroquinone. The synergistic
interaction of benzene metabolites (i,e, hydroquinone and 1,2,4-ben2enetriol) in the
production of DNA adduct may also play an important role in the genotoxic effects of
benzene in vitro (Levay and Bodell, 1992),
It has been suggested that iron is released from ferritin under conditions in which
oxidative stress is involved in both in vivo and isolated perfused organs (Mazur and
Carleton, 1965; Healing ef a/,, 1990), We observed during an/n wfro study that 1,2,4-
benzenetriol, a trihydroxy metabolite of benzene, was capable of releasing iron from
horse spleen ferritin, which was mediated to a certain extent by superoxide radical
(Ahmad et al., 1995), Similar results were observed by Oteiza (1995) where 8 -amino
levulinic acid (a heme precursor) releases iron from horse spleen and rat liver ferritin in
vitro via free radical formation, and affect iron metabolism in vivo. Available evidence
indicates that benzene induced myelotoxicity and carcinogenicity involve superoxide
radical formation during autooxidation of polyphenolic metabolites of benzene viz
hydroquinone or 1,2,4-benzenetriol (Subrahmanyam et al., 1991b). A fraction of cellular
non haem iron consisting of chelatable low molecular weight iron represent iron species
catalytically active in initiating free radical reactions (Gutteridge, 1987). In various
pathological conditions this decompartmentalized iron is "malplaced" which when bound
to bleomycin and in the presence of oxygen can degrade DNA (Gutteridge et al., 1985).
Conditions inhibiting heme synthesis and uncoupling of oxidative phosphorylation have
been shown to cause a large increase in the low molecular weight iron (LMI) fraction in
the cytosol (Bakkeren e/a/., 1985), Human normal cerebrospinal fluid and synovial fluid

Chapter -IV 58
from rheumatoid patients contain micromolar concentrations of non-protein bound LMI
(<10,000) compounds which are active in catalyzing bleomycin dependent degradation
of DNA and superoxide and hydroxyl radical reactions (Gutteridge et al., 1982). The
presence of LMI pool in various cells and organs including intestinal mucosa, heart, bone
marrow, liver, kidney, spleen, brain and in cultured Chang liver cells have been reported
(Mulligan et al. 1986). LMI were isolated from guinea pig reticulocytes as AMP-Fe and
ATP-Fe complexes which might be involved in cellular iron transport (Weaver and
Pollack, 1989). The interaction between iron-bleomycin and different catecholamines
have been reported (Lovstad, 1990)
We propose in the present studies that the chelatable low molecular weight iron
accumulated during benzene toxicity (Pandya et al., 1990) is the chelate of polyphenolic
metabolites of benzene with iron. In the present study we investigated the efficacy of
hydroquinone or 1,2,4-t)enzenetriol: iron complex to catalyze bloemycin dependent DNA
degradation. We also studied the effect of polyphenol: iron complex to catalyze
glutamate degradation and lipid peroxidation in vitro.
RESULTS
Iron catalyzed and bleomycin dependent degradation of calf thymus DNA was
markedly greater in the presence of iron polyphenol complex than in the presence of iron
alone (Table-4.1). Further, calf thymus DNA degradation was faster with iron: 1,2,4-
benzenetriol complex (2.4 fold) than with iron:hydroquinone complex (1.5 fold). The
degradation of calf thymus DNA showed an increase with increasing concentration of iron
(0-20 JM) or iron:polyphenol complex (1:2 ratio) (Figure-4.1).
The iron-dependent release of TBAR from glutamate exhibited a decrease in the
presence of either hydroquinone or 1,2,4-benzenetriol. The inhibition of iron-dependent
release of TBAR from glutamate was 81% in the presence of hydroquinone and 66% in
the presence of 1,2,4-benzenetriol (Table-4.1). Similariy, the iron-dependent lipid
peroxidation in brain homogenates was substantially inhibited in the presence of
hydroquinone where it was 39% or 1,2,4-benzenetriol where it was 43% (Table-4.1).

59
Table 4.1
Comparison of TBAR formation from DNA by Iron (20 ^M) and Iron: polyphenol (20:40 \ifA) complex in the presence of bleomycin
Parameters Iron lron:HQ lron:BT
Bleomycin dependent degradation 1.52 2.23 3.58 of calf thymus DNA
Iron dependent degradation of 8.25 1.56 2.83 glutamate
Lipid peroxidation 67.14 41.13 38.05
Experimental conditions are as described in the Materials and Methods section. All values are represented as nmoles of TBAR formed and are averages of two sets of experiments conducted in duplicate with variations of 10% or less.

< m »•-O
in CD
"o E c
( f jM)
Figure 4.1 : Linearity of nmoles of TBAR formation from calf thymus DNA with increase in concentration of iron (t •), iron: hydroquinone complex (o—o) or iron; 1,2,4-ben2enetriol complex (A—A). The reaction was initiated by the addition of iron (0-20 pM) or ironpolyphenol (12 ratio)

60
Table 4.2
Recovery of Iron : polyphenol complex (24:48 pM) added to bone marrow lysate by the bleomycin dependent degradation of calf thymus DNA.
Addition lron:HQ lron:BT
-Bone marrow lysate 2.71 5.73
+Bone marrow lysate 3.33 7.67
Bone marrow lysate (1 mg protein) was added to iron.polyphenol cx)mplex (for detail see Materials and Methods section). All values are expressed as nmoles of TBAR formed from DNA and are averages of two sets of expehments conducted in duplicate with variations of 10% or less.

61
Table 4.3
Formation of TBAR from calf thymus ONA by bleomycin in the presence of Iron (20 pM) or iron with the potential iron chelator (1:2 ratio)
Addition
Iron alone
Iron + HQ
+ BT
+ Citrate
+ Glucose
+ Fructose
+ Glycine
+ Glutamic acid
+ Cysteine
+ Aspartic acid
+ AMP
+ ADP
+ ATP
+ Dithiothreitol
Expenmental conditions are as described in the Materials and Methods section. All values are averages of two sets of experiments conducted in duplicate with vanations of 10% or less.
nmoles of TBAR formed/ 30 min
1.52
2.67
3.52
0.19
1.40
1.29
1.03
1.53
1.59
1.09
0.82
0.92
1.49
0.49

o o o o
O O o
E c
o o ^ . g
c 2 8 ^
^ ^
— o o * - <D Si •-00 OJ
0)
5 •D
c
c <u
T O O
'—•" O X o OJ h—
o
fS o
^ 3 .
o o (N x: 5 k -
o V c o nj
O X
"^5
<
u 3

E c
o o
T3 C
TO TJ-
C O
! = M
ll c £:> O 0) •s > n 0)
o
= w .« ro
(U (0 i : • C Q>
" ~ 11
t - O
CO t ;
to o
o a. OT (D
m S
O 1
CD CM "
0)

Chapter - IV 62
The iron:polyphenol complex added to bone marrow lysate was 100% recoverable
when assayed by the bleomycin-dependent degradation of calf thymus DNA (Table-4.2).
Formation of TBAR from calf thymus DNA by bleomycin in the presence of iron,
iron;polyphenol or iron with potential endogenous iron chelators is recorded in Table-4.3.
The endogenous chelators viz. glucose, fructose, glutamic acid, cysteine and adenosine
triphosphate (ATP) did not affect the iron catalyzed bleomycin-dependent TBAR
formation from calf thymus DNA. Whereas, glycine, aspartic acid, adenosine mono-
phospahte (AMP) and adenosine diphosphate (ADP) caused a significant inhibition. The
presence of citrate and dithiothreitol resulted in a drastic inhibition of bleomycin-
dependent iron catalyzed TBAR formation from calf thymus DNA (Table-4.3).
The addition of FeSO^./HjO to hydroquinone or 1,2,4-benzenetriol in freshly distilled
water revealed no change in the optical spectra of hydroquinone. However, that of 1,2,4-
benzenetnol revealed a new absorption peak at 259 nm (Figure-4.2A and 4.2B).
DISCUSSION
Benzene exposure has been shown to generate organic free radicals and superoxide
radicals (Lewis et al., 1988b) and it is generally accepted that benzene requires
metabolism to express its toxicity (Subrahmanyam et al., 1991a). Polyphenolic
metabolites of benzene viz., hydroquinone and 1,2,4-benzenetriol have Ijeen shown to
be toxic and the suggested mechanism includes free radical formation via superoxide
during their autooxidation and the covalent binding of semiquinone to DNA, RNA and
other cellular macromolecules (Wick, 1980; Rushmore et al., 1984). In addition, the
active oxygen radical formed during autooxidation of polyphenolic metabolites has been
shown to be involved in the cytotoxic effects of benzene (Irons, 1985; Karam and Simic,
1989) Exposure to benzene resulted in decrease in antioxidant potential in serum
(Ahmad et al., 1994) and may allow oxygen intermediates such as superoxide and
hydrogen peroxide to survive in plasma long enough. Benzene exposure also leads to
an accumulation of bleomycin sensitive iron in bone marrow (Pandya et al., 1990).
1,2,4-Benzenetriol, a trihydroxy metabolite of benzene was capable of releasing iron from
horse spleen ferritin, which was mediated to a certain extent by superoxide radical

Chapter - IV 63
(Ahmad et al., 1995), probably, this may also be the reason of accumulation of iron in
bone marrow during benzene exposure, as the polyphenolic metalwlites are accumulated
against the concentration gradient in bone marrow.
Certain flavonoids aitin and quercetin as phenolic compounds have affinity for iron
ions and act as iron chelators (Afanas'ev eif al., 1989). For example, plant phenolics
quercetin and myricetin were found to enhance iron ion dependent damage to DNA by
bleomycin (Afanas'ev et al., 1989). This was assumed to be due to reduction of feme
bleomycin to the fen^ous form by the phenols and this ferrous bleomycin complex in the
presence of oxygen, degrade DNA (Laughton et al., 1991). On the other hand,
antioxidant effects of flavonoids rutin and quercetin to scavenge superoxide and form iron
complexes have been reported. The cytoprotective activity of flavonoids catechin,
quercetin and diosmetin have been attributed to their iron-chelating ability (Morel ef a/.,
1993). However,the cytotoxicity of some polyhydroxy pyrimidines viz. divicine, isouramil
and dialuric acid has been attributed to their ability to release iron from fenitin and this
iron was shown to promote lipid peroxidation (Monteiro and Winterbourn, 1989a).
During benzene exposure of experimental animals many oxidation-reduction
reactions have been shown to occur (Irons, 1985; Lewis et al., 1988b) which may release
ferrous iron from ferritin. The ferrous iron may t>e chelated to the polyhydroxy
metabolites of benzene, viz., hydroquinone and 1,2,4-t>en2enetriol. The tentative chemical
structure of polyphenol iron complex for 1,2,4-t)enzenetriol:Fe(lll) is diagrammatically
presented in Figure-4.3.
We propose that the complex of polyphenol with iron (Figure-4.3) may be the low
molecular weight iron in bone marrow which has been reported to accumulate selectively
during the exposure to benzene (Pandya et a!., 1990). In the present study we observed
increase in iron-catalyzed bleomycin-dependent degradation of calf thymus DNA in
presence of hydroquinone or 1,2,4-benzenetriol. The iron.polyphenol complex appears
to be stable and intact as the recovery from bone marrow lysate was complete. It is
also possible that the hydroquinone or 1,2,4-benzenetriol possess combined iron-
chelating and iron ion reducing properties to stimulate calf thymus DNA degradation in
the presence of bleomycin. Further, the iron polyphenol complexes have shown an

-f X
+
X
E O U
c o
m
0) LL
+
X
a E o o c o
CQ
o
0)
o
w "TO o E o
> ro c
CO
CD

Chapter - IV 64
antioxidant activity by inhibiting iron-dependent lipid peroxidation in brain homogenate and
glutamate degradation. This is similar to the antioxidant properties exhibited by
flavonoids due to their ability to chelate iron (Afanas'ev et ai, 1989; Laughton et al..
1991). Thus the flavonoids and polyphenolic chelates limit the availability of iron for lipid
peroxidation and at the same time facilitate bleomycin-dependent degradation of calf
thymus DNA.
It has been shown recently that upon mixing hydroquinone with Cu(ll) the
absorbance of hydroquinone at 289 nm decreased with corresponding increase in
absorbance at 247 nm due to the formation of 1,4-t)enzoquinone (Li and Trush, 1993b).
However, the optical spectra of hydroquinone did not show any change in the presence
of ferrous sulphate. This indicates that hydroquinone is not converted to 1,4-ben2o-
quinone and iron is kept in the ferous state to catalyze oxidation reactions. The addition
of ferrous sulphate to 1,2,4-benzenetriol, on the other hand, revealed a new absorbance
peak at 259 nm with a slight decrease at 286 nm. These observations suggest a
complex formation of polyphenol with iron rather than reduction of iron and oxidation of
polyphenol.
It is concluded from the present study that the phenolic metabolites of benzene
enhance the availability of Fe(ll) to bleomycin. Such Fe(ll) is possibly made available due
to reduction of Fe(lll) by polyphenols, thus protecting the Fe(ll) from hydrolysis to
hydroxides and consequent autooxidation. Recently, a few studies have explained the
mechanisms underlying the involvement of iron in benzene toxicity. Zhang e al., (1995b)
have suggested that benzene ring fission is stimulated by iron. Further, they demonstrated
the oxidant-mediated genotoxicity of 1,2,4-t>enzenetriol in presence of metal ions
(particulariy iron and copper) via production of oxygen-derived active species (Zhang et
al., 1996). Accumulation of LMI and polyphenolic metabolites of benzene in bone man-ow
are well established. Probably iron:polyphenol complex may be the potent toxic
intennediate during the benzene exposure in bone marrow for its genotoxicity. However,
the formation of such complex in vivo is yet to be characterized during benzene
exposure.

Chapter - V Polyphenol'iron Complex: A Probable
Toxic Metabolite of Benzene Introduction
Results
Discussion

Chapter - V 65
INTRODUCTION
The various facts and figures that has accrued so far from survey reports and
experimental evidences has established that exposure of animals and human to benzene
can result in damage to the haematopoietic system leading to leukemia (Snyder and
Kocsis, 1975). Acute exposure to benzene has a strong toxic effect on the
haematopoietic system of both laboratory animals (Tunek et al., 1982), and humans
(Cohen et al., 1978) probably via interaction w/ith undifferen-tiated bone marrow cells.
Chronic exposure is carcinogenic in rats and mice (Maltoni et al., 1989; Huff et al., 1989).
Several studies have shown that benzene must be metabolized to exert its toxic effects
and that the metabolites are responsible for the both its cytotoxic and genotoxic effects
(Kalf, 1987).
Benzene is metabolized in the liver mainly to one or more phenolic metabolites and
accumulates against the concentration gradient in bone marrow and exerts its toxic effects
(Smith ef al., 1989). Bioactivation of benzene in haematopoietic cells may be important
for its leukemogenic effects (Andrews et al., 1979). Also, bioactivation of benzene by

Chapter -V ^
bone marrow and liver microsomal enzyme system appears to be responsible for the
haematopoietic toxicity possibly by covalent binding of a metabolite(s) to DNA (Snyder et
al., 1981). Many studies have also shown that benzene produces both structural and
numerical chromo-somal aberrations in the peripheral blood lymphocytes of occupationally
exposed individuals (Ding et al., 1983). Because consistent chromosomal aberrations
have tjeen observed in human leukemias and lymphomas, the induction of chromo-somal
damage by benzene or its metabolites may be critical in the development of
benzene-induced leukemia (Rothman et al., 1995). Although, haematotoxic and
leukemogenic effects of benzene are generally believed to be caused by its metabolites
as opposed to the parent compound, the ultimate reactive inter-mediate(s) responsible
for benzene toxicity is not known (Goldstein and Witz, 1992).
Hydroquinone and 1,2,4-benzenetriol the two principal polyphenolic metabolites of
benzene have been shown to be toxic and the suggested mechanism include free radical
formation via superoxide and covalent binding of the semiquinone to DNA, RNA and other
cellular macromolecules (Irons, 1985). Eariier it was observed that polyphenolic
metabolites of benzene are autoxidized faster to generate free radicals in the presence
of transition metal ion and can release aldehydic products from target molecules such as
glutamate, calf thymus DNA and damage human DNA (Rao and Pandya, 1989; Kawanishi
etal., 1989; Singh e/a/., 1994).
Many investigators have shown that benzene exposure interferes with iron
metabolism and heme biosynthesis in experimental animals (Lee et al.. 1974; Freedman
et a!., 1977; Rao and Pandya, 1980). Benzene administration also led to an accumulation
of iron in bone marrow cell, which can catalyze the formation of aldehydic products (base
propenal) from DNA in the presence of bleomycin (Rao et al., 1990). In all probability
benzene alters heme metabolism of erythroid cells, resulting in the increased
internalization of iron transferrin receptor complex and uptake of internalized iron is almost
entirely restricted to erythroblasts. In the present studies it was observed (Chapter-I) that
1,2,4-benzenetriol, a trihydroxy metabolite of benzene was capable of releasing iron from
horse spleen ferritin, which was mediated to a certain extent by superoxide radical
(Ahmad et al., 1995). We propose that the low molecular weight iron (LMI) accumulated

Chapter- V 57
during benzene exposure (Pandya et al., 1990) is the chelate of polyphenolic metabolites
of benzene with iron. We investigated the efficacy of hydroquinone and 1,2,4-benzenetriol
in the presence of iron to catalyze bleomycin-dependent calf thymus DNA degradation.
In the presence of hydroquinone or 1.2.4-benzenetriol, iron catalyzed bleomycin
dependent degradation of DNA was markedly enhanced in comparison to alone.
Available evidence indicates that benzene exposure leads to accumulation of LMI
as well as polyphenolic metabolites of benzene in the bone marrow against the
concentration gradient. In the present study we investigated the poly-phenol:iron complex
formation in vitro and methodology standardized to fractionate these complexes by
Sephadex G-10 column chromatography. To demonstrate the polyphenol:iron complex
formation we utilized the system of iron released from ferritin by 1,2,4-benzenetriol. We
further investigated the haematotoxicity of hydroquinone:iron mixture in male mice to
evaluate the possible role of hydroquinone:iron complex in benzene induced
haematotoxicity.
RESULTS
The elution profile of iron, 1,2,4-benzenetriol and 1,2,4-t>enzenetriol:iron mixture were
studied by Sephadex G-10 column chromatography. Fractions were collected at every
5 min. interval (flow rate 0.5 ml/min) and were scanned at 287 nm (the absorption
maximum of 1,2,4-benzenetriol) using Spectronic 2001 spectrophotometer (Figure-5.1).
When iron alone was subjected to Sephadex G-10 column chromatography, the eluted
fractions did not show any peak in any fractions (1-30) at 287 nm. Whereas
1,2,4-benzenetriol alone eluted fractions showed an absorption maximum at 287 nm in
fraction number 21. However, when 1,2,4-t)enzenetriol:iron mixture was subjected to
Sephadex G-10 column chromatography, fraction numt)er 3 showed absorbance
maximum at 287 nm and fraction number 21 did not show any absorbance (Figure-5.1).
Iron content was estimated using colouring reagent bathophenanthroline sulphonic acid
in each fraction and absorbance was followed at 535nm (Figure-5.2). The fractions
collected from 1,2,4-benzenetriol alone subjected to Sephadex G-10 column did not show
any absorbance at 535 nm in any fraction (1-30). But, in case of iron alone and

0.20-1 -O Fc:BT complex ( I ' D - • BT alone •* Fe alone
FRACTION No
Figure 5.1 : Elution profile of iron alone (x x), 1,2,4-benzenetnol alone (• •) and iron:1,2,4-benzenetriol mixture (O—0) subjected to Sephadex G-10 column Fractions were collected at every 5 min interval (flow rate 0.5 ml/mm) and were scanned at 287 nm
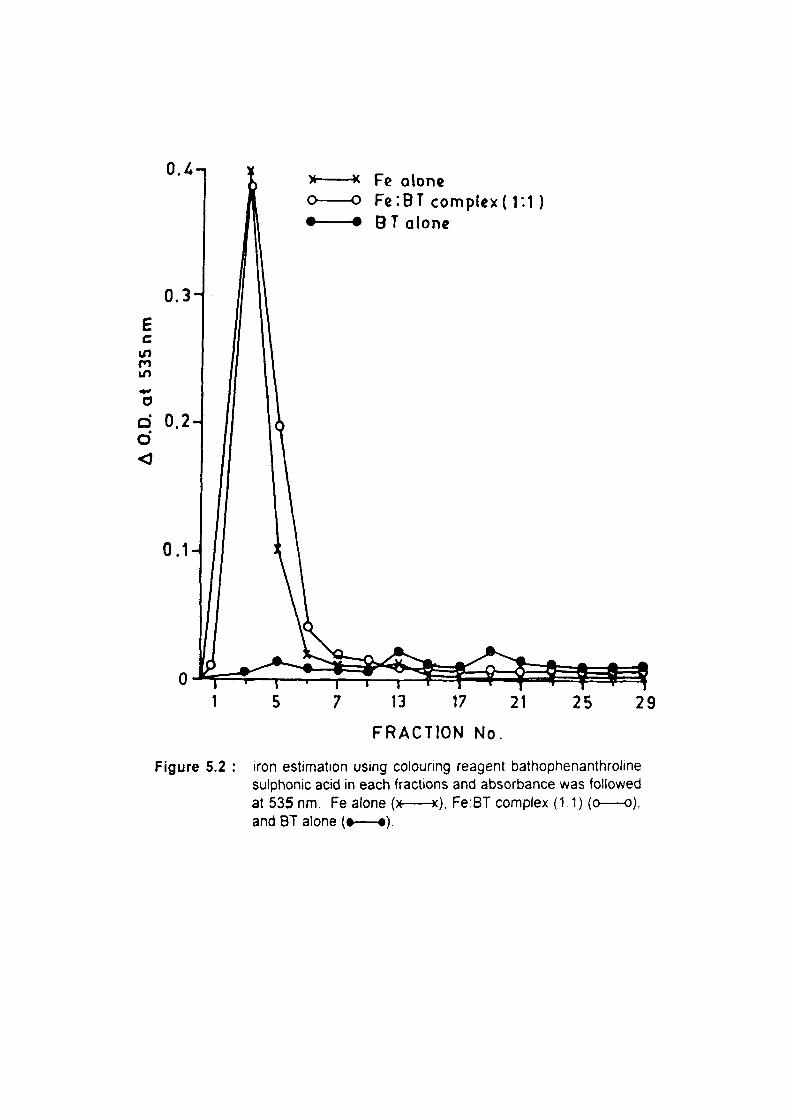
O.^-i -* FG alone -o Fc'.BT complex! l:1) - • BT alone
E c n in *^ o
ft
o o <
13 17 21
FRACTION No.
25 29
Figure 5.2 iron estimation using colounng reagent bathophenanthroline sulphonic acid in each fractions and absorbance was followed at 535 nm. Fe alone (x x), Fe:BT complex (1:1) (o o), and BT alone (• •).

Chapter - V 68
1,2,4-benzenetriol.iron mixture showed an absorbance peak at 535 nm in fraction
number 3.
Further, the elution profile of 1,2,4-t)enzenetriol:iron complex formation was also
studied in presence and absence of ethylenediaminetetraacetic acid (EDTA) during the
release of iron from ferritin by 1,2,4-t3enzenetriol. In the preceding chapter it was reported
that presence of 1,2,4-benzenetriol, a potent toxic metabolite of benzene, resulted in the
release of iron from horse spleen ferritin in 0.1 M acetate buffer at pH 5.6 (Ahmad et al.,
1995). In the absence of EDTA we observed an absorbance peak at 287 nm in fraction
number 3 during the iron release from femtin by 1,2,4-benzenetriol. In addition to
absorbance peak of 1,2,4-benzenetriol.iron complex in fraction number 3, an absorption
peak of low intensity may be due to unchelated 1,2,4-benzenetriol was also observed in
fraction number 21. But in presence of iron chelator (EDTA) the iron released from femtin
by 1,2,4-benzenetriol was chelated with EDTA and eluted in fraction number 3, whereas
an increase in 1,2,4-benzenetriol peak intensity was observed in fraction number 21 at
287 nm (Figure-5.3). This observation indicates that as iron is not available to 1,2,4-
benzenetriol due to the presence of EDTA, all the polyphenol was eluted in fraction
number 21. However, in the absence of EDTA, all the iron released was accessible to
1,2,4-benzenetriol and 1,2,4-benzenetriol eluted in fraction number 3 along with iron.
Studies were conducted to evaluate the haematotoxicity of hydroquinone (25 mg/kg
body weight), iron (25 mg/kg body weight) and hydroquinone:iron complex after exposure
to male mice at 25 mg hydroquinone:25 mg iron complex/kg body weight i.p. in normal
saline for 20 days (single exposure) and also 7 days (twice in a day). Normal saline
treated group served as control.
Enlargement in liver size was observed in hydroquinone:iron complex and iron alone
treated groups compared to the control group. Whereas, increase in spleen weight was
observed only in group treated with hydroquinone.iron complex for 20 days (Table-5.1),
Similariy, significant increase in haemoglobin content was observed in all the three groups
compared to the control group. But hematocrit (%) did not show any change in any group
after 20 days exposure (Table-5.1). The above haematological parameters were also
evaluated after 7 days of i.p. treatment of above mentioned groups, twice a day at the

O O - EDTA
• • +EDTA
E c
00 fM
d o
11 15 19 23
FRACTION No.
Figure 5.3 : Elution profile of iron:BT complex formation during the iron release from ferritin by BT with or without the presence of EDTA Fractions were analyzed at 287 nm

69
Table 5.1
Relative liver/spleen weight, haemoglobin and hematocrit (%) of male mice after 20 days treatment
Group
CONTROL
HQ:lron complex
Iron alone
HQ alone
Relative Liver wt. (per 100 gm)
5.40±0.18
6,84±0.34*
6.15±0.28=
5.02±0,16' s
Relative Spleen wt (per 100 gm)
0.58±0.06
0.81±0.07=
0.87±0.14^^
0.57±0.05'^'
Haemoglobin (gm/dl)
14.3+0.46
17.2±0.45'
16.2±0.25"
16.710.58"
Hematocrit (%)
40.6+1.54
44.2+1.33''5
42.9+1.27^^
40.9+1.56^5
Treatment through single injection daily (i.p. = 25 mg each/kg b.w. in 0.1 ml normal saline and control group received 0.1 ml normal saline only) for 20 days. Values are mean ± S.E. of six mice. Exposed groups were compared with their respective control. P values = a <0.01, b <0.02, c <0.05, NS = Not significant.

70
Table 6.2
Relative liver/spleen weight, haemoglobin and hematocrit (%) of male mice after 7 days exposure, twice in a day
Group Relative Relative Haemoglobin Hematocrit Liver wt Spleen wt (gm/dl) (%) (per 100 gm) (per 100 gm)
CONTROL
HQ.Iron complex
Iron alone
HQ alone
4.83±0.17 0.57±0.06 15.7±0.54
6.35±0.18^ 0.80±0.08^ 15.510.34^^
5.7110.23" 0.57±0.04' s 15.3+0.62^^
5.00±0.23''^ 0.66±0.08''s 14.810.31"
36.5±0.93
35.7±0.54^
35.3±0.83'«
34.7+0.89^
Treatment through double injection daily (i.p. = 25 mg each/kg b.w. in 0.1 ml normal saline and control group received 0.1 ml normal saline only) for 7 days. Values are mean ± S.E. of six mice. Exposed groups were compared with their respective control. P values = a <0.001, b <0.02, c <0.05, NS = Not significant.
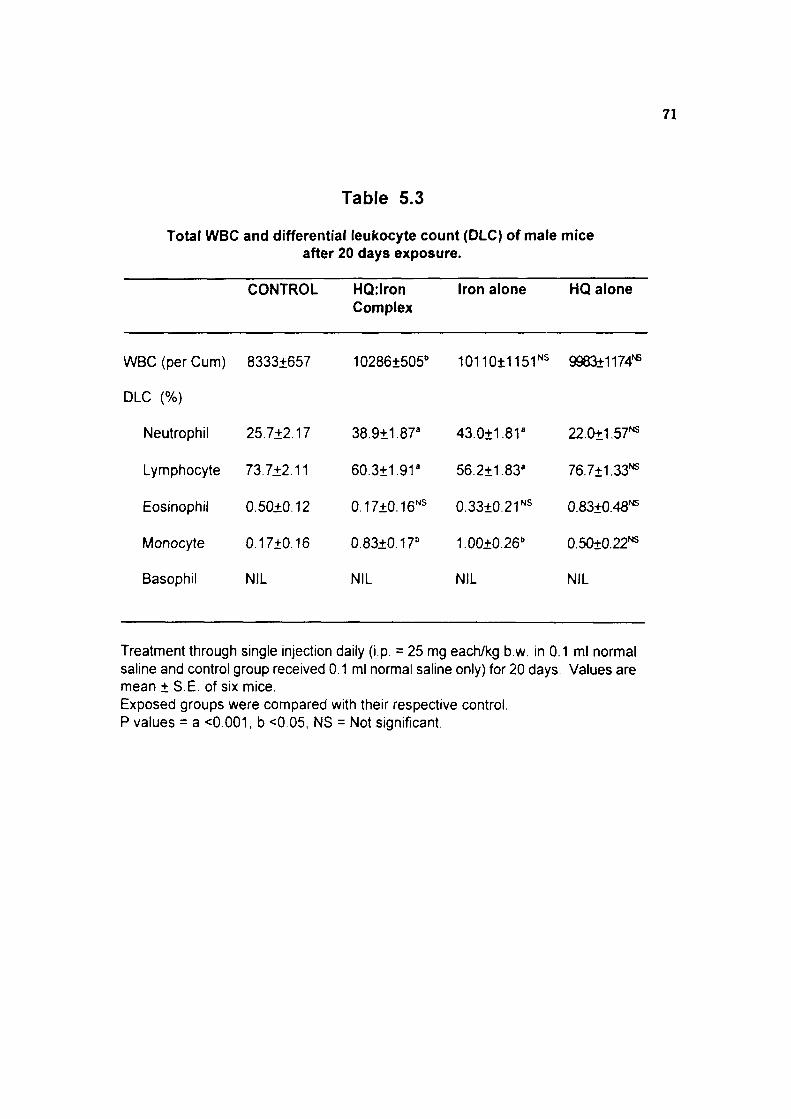
71
Table 5.3
Total WBC and differential leukocyte count (OLC) of male mice after 20 days exposure.
CONTROL HQ:lron Complex
Iron alone HQ alone
WBC (per Cum)
DLC (%)
Neutrophil
Lymphocyte
Eosinophil
Monocyte
Basophil
8333±657
25.7+2.17
73.7+2.11
0.50±0.12
0.17±0.16
NIL
102861505" 10110±115r^ 9983+1174^
38.9+1.87^ 43.0±1.8r
NIL NIL
22.0+1.57'^
60.3±1.9r 56.211.83" 76.7±1.33'«
0.1710.16''^ 0.33±0.2r^ 0.83±0.48'«
0.83+0.17" 1.00+0.26" 0.50±0.22^
NIL
Treatment through single injection daily (i.p. = 25 mg each/kg b.w. in 0.1 ml normal saline and control group received 0.1 ml normal saline only) for 20 days. Values are mean ± S.E. of six mice. Exposed groups were compared with their respective control. P values = a <0.001, b <0.05, NS = Not significant.

72
Table 5.4
Total WBC and differential leukocyte count (DLC) of male mice after 7 days exposure twice in a day
WBC (per Cum)
DLC (%)
Neutrophil
Lymphocyte
Eosinophil
Monocyte
Basophil
CONTROL
7492±320
23.3±2.0
74.7+1.6
1.00±0.4
1.00±0.4
NIL
HQrIron Complex
127061626"
45.912.1"
52.8+1.9"
0.2510.3' ^
1.13±0.4' s
NIL
Iron alone
11717±1066'
43.0±2.5"
54.2±2.0"
0.70±0.4''s
2.20±0.7' s
NIL
HQ alone
6950+539^
25.7±2.3'^
70.7±2.2^
2.20±0.8'^
1.50±0.5^
NIL
Treatment through double injection daily (i.p. = 25 mg each/kg b.w. in 0.1 ml nonnal saline and control group received 0.1 ml normal saline only) for 7 days. Values are mean ± S.E. of six mice. Exposed groups were compared with their respective control. P values = a <0.001, b <0.01, NS = Not significant.

73
Table 5.5
Lipid peroxidation, total and free -SH content in liver of male mice after 20 days exposure
Assay CONTROL HQ:lron Iron Complex alone
HQ alone
Lipid peroxidation 19.3t1.47 46.3+2.44' 44.7±2.80' 19.6+1.72 (nmoles/hr/gm liver)
Total -SH content 2.75±0.07 (mmoles/100 gm liver)
2.2910.06' 2.3710.08" 283±0.W^
Free -SH content 0.74±0.05 (mmoles/100 gm liver)
0.69±0.05''5 0.57±0.07''s 0.9110.04"
Treatment through single injection daily (i.p. = 25 mg each/kg b.w. in 0.1 ml normal saline and control group received 0.1 ml normal saline only) for 20 days. Values are mean iS .E . of six mice. Exposed groups were compared with their respective control. P values = a <0.001, b <0.05, NS = Not significant.

74
Table 5.6
Lipid peroxidation, total and free -SH content in liver of male mice after 7 days exposure twice in a day
Assay CONTROL HQ.Iron Iron HO Complex alone alone
Lipid peroxidation 11.1+2.52 22.3±3.25' 30.013.76" 14,9±1.58'* (nmoles/hr/gm liver)
Total-SH content 3.27±0.07 3.01±0.08^ 2.9310.06" 3.18±0.09^ (mmoles/100 gm liver)
Free-SH content 0.84±0.06 0.75±0.06''^ 0.73±0.05''s 0.74±0.05^ (mmoles/100 gm liver)
Treatment through double injection daily (i.p. = 25 mg each/kg b.w. in 0.1 ml normal saline and control group received 0.1 ml normal saline only) for 7 days. Values are mean ± S.E. of six mice. Exposed groups were compared with their respective control. P values = a <0.001, b <0.02, c <0.05, NS = Not significant.

Chapter - V 75
same dose. Similar pattern of results were observed all the three groups when compared
to control group except the haemoglobin content (Table-5.2). The haemoglobin content
did not show any significant alteration after 7 days exposure twice a day.
Significant increase in WBC was observed in hydroquinone: iron complex treated
male mice after single injection for 20 days exposure (Table-5.3). Similarly, significant
increase in polymorph (%) and monocyte (%) in hydro-quinone:iron complex and iron
alone treated groups with simultaneous decrease in lymphocyte (%) (P<0.001) were
observed when compared to the control group after 20 days of exposure. Whereas,
eosinophil (%) and basophil (%) did not show any alteration after 20 days of exposure.
Further, after 7 days exposure, twice in a day, same pattern of results in WBC and DLC
were observed. Significant increase in WBC was observed in hydroquinone:iron complex
and iron alone treated groups compared to the control group. Similarly, significant
increase in polymorph (%) with simultaneous decrease in lymphocytes were observed in
the groups treated with hydroquinone:iron complex and iron alone. Whereas, eosinophil
(%), monocyte (%) and basophil (%) counts did not show any alteration in any group
(Table-5.4).
We also studied the lipid peroxidation, total and free sulphahydryl content ( -SH
group) in liver of both sets of experiments (Table-5.5 and 5.6). Significant increase in lipid
peroxidation was observed in hydroquinone:iron and iron alone group with simultaneous
decrease in total -SH content in both groups after 20 days (single injection daily) and 7
days (twice in a day). Whereas, no significant alteration in free -SH content was observed
in any group except for the group treated with hydroquinone alone for 20 days (single
injection daily), where a marginal increase in free -SH content was observed (Table-5.5).
DISCUSSION
The haematotoxic effect of benzene in both humans and animals have been well
documented (Snyder, 1987). Chronic exposure of humans to benzene is associated with
vanous types of haematopoietic disorders including several types of leukemia. In
laboratory animals particularly rodents, benzene exposure reduces the number of various
bone marrow cells including the haematopoietic stem cells. The intensity of

Chapter - V 76
haematotoxicity produced by benzene exposure differ between species and even between
strains of the same species. There is abundant evidence that these differences in
response are due to difference in benzene metabolism (Nakajima e a/., 1993). Neun et
al.. (1992) suggested that differences in host target ceil susceptibility may also play a role
in the differential haematotoxic responses to benzene.
It was observed that chronic exposure to several strains of mice to high
concentrations of benzene (100 or 300 ppm) produced a rapid decline in the number of
circulating red cells, whereas the number of circulating granulocytes rose above the
control levels (Snyder et al., 1982). Exposure to t»enzene resulted in a variety of change
in the number of polymorph, lymphocytes, basophil, eosinophil and other bone marrow
cell fractions. The longer exposure to a lower concentration of benzene results in a much
greater haematotoxic effect (Cronkite et al., 1989). Exposure to 300 ppm benzene
caused high mortality, lymphocyto-penia, anemia, neutrophilia, and reticulocytosis in the
mice but only lymphocyto-penia in rats (Snyder et al., 1978). However, the metabolites
of benzene responsible for these effects are not known.
Earlier Greenlee et al., (1981 a) have shown that polyphenolic metabolites of benzene
accumulated in bone marrow against the concentration gradient. We propose that the
complex of polyphenol with iron may be the low molecular weight iron (LMI) in bone
marrow which has been reported to accumulate selectively during the exposure to
benzene (Pandya et al., 1990). In the present studies, we observed that the presence of
polyphenolic metabolites of benzene enhanced iron-catalyzed bleomycin-dependent
degradation of DNA. The polyphenoUron complex appears to be stable and intact as the
recovery from bone marrow lysate was complete (Singh et al., 1994). Although, the
benzene exposure lead to accumulation of iron as well as polyphenol metabolites of
benzene in the bone marrow against the concentration gradient but formation of such
complex in vivo is not known. In the present study, we standardized a methodology to
fractionate these complexes by Sephadex G-10 column chromatography. Results clearly
show the formation of polyphenoMron complex in in vitro condition.
In order to evaluate the haematotoxicity of hydroquinone:iron complex we studied the
effect of 25 mg hydroquinone; 25 mg iron or 25 mg hydroquinone or 25 mg iron/kg body

Chapter - V 77
weight and normal saline in male mice administered i.p. daily for 20 days (single
exposure) and significant alteration in various haematological parameters were observed.
Increase in haemoglobin content, hematocrit and WBC were noticed whereas DLC
showed increase in polymorphs and decrease in lymphocytes in hydroquinone:iron
complex and iron treated groups in comparison to normal saline administered group.
However, the effects were highly significant in hydroquinone:iron treated group and a
marked alterations were also noticed in this group when compared to iron treated group
only. However, no significant changes were observed in hydroquinone alone treated
group m compahson to norma] saline treated group.
The mortality rate in the case of hydroquinone;iron complex treated group was very
high at the dose 50 mg/kg b.w. This shows that the complex formation some how
increase the toxicity. Both absolute and relative organ weights of liver and spleen showed
a significant increase in hydroquinone.iron and iron treated groups, whereas hydroquinone
alone treated group did not show any change. Enlargement in liver and spleen size is one
of the important symptom of blood related disorder. An increase in spleen weight in CD-1
male mice exposed to benzene has also previously been reported (Green e a/., 1981a)
with a concomitant increase in granulocyte count as a consistent finding (Green et al.,
1981b). Similar observations of increased spleen weight and granulocyte number were
noticed in the group of mice treated with hydroquinone:iron complex. Total sulphahydryl
content showed significant decrease, whereas increase in lipid peroxidation were noticed
in both the groups without any change in hydroquinone treated group. This shows the
formation of free radical in vivo during the exposure of such complex and may have
toxicological implication.
Eastmond et al., (1987) have demonstrated that repeated combined exposure of
mice with phenol and hydroquinone led to decrease in bone marrow ceilularity in a
manner similar to the haematotoxic effects expressed by benzene. Following similar
experimental protocol (Guy et al., 1990), it was demonstrated that the radio iron uptake
into erythrocytes was significantly decreased in the group of mice coadministered with
phenol and hydroquinone. The combination of phenol and hydroquinone produced a
decrease in bone marrow ceilularity similar to that produced by benzene. Further Guy et

Chapter - V 78
a/., (1991) extended these observations to other metabolites of benzene, p-l)enzoquinone,
muconaldehyde and hydroqui-none, that may affect haematopoiesis either singly or in
combination. It has been suggested that reduced bone marrow cellulahty during the
coadministration of phenol and hydroquinone might be due to phenol-induced stimulation
of hydroquinone via the peroxidase-dependent metabolic activation in bone marrow and
increased covalent binding of hydroquinone to tissue macromolecules of blood, bone
marrow, liver and kidney (Smith et al., 1989; Subrahmanyam et al., 1990, 1991a).
Muconaldehyde (Witz et al., 1985) and 6-hydroxy-trans, trans- 2,4-hexadienol (Zhang et
al.. 1995b), the putative toxic intermediates of benzene have been shown to cause
decrease in sulphahydryl content, ^'Fe incorporation into circulating erythrocytes. Rao
and Pandya (1978) have shown that benzene increases lipid peroxidation 3 hr after
exposure and it has been reported that lipid peroxidation leads to loss of heme from
microsomes (De Matties and Sparks, 1973). Several fold increase in hepatic lipid
peroxidation was also observed in albino rats of either sex after benzene exposure
intrapehtoneally (0.5 ml/kg body weight) or subcutaneously (1 ml/kg body weight) for 10
days (Ahmad ef a/., 1994).
Similar observations comparable to above observations were noticed in mice treated
with the same dose for 7 days daily i.p. twice in a day. Present studies clearly indicate
that hydroquinone iron chelate showed marginal increase in haematotoxicity in
experimental animals. But the precise mechanism of marginal, yet significant increase
in haematotoxicity is not clear. Atleast some of the effects observed in hydroquinone.iron
treated group showed similarities to some extent with what has been shown to occur
following exposure of mice to benzene. Further studies are required to identify the
complex formation in vivo dunng benzene exposure and to explain the mechanism of
toxicity of such complexes and a possible role of such complexes in benzene toxicity.

Chapter - VI Glutathionyl Hydroquinone: A Possible
Toxic Metabolite of Benzene Introduction
Results\
Discussion

Chapter - VI -o
INTRODUCTION
Benzene is a well known leukemogenic and carcinogenic agent. Benzene induced
cytogenetic effects including chromosome and chromatid aberration, sister chromatid
exchanges and induction in micronuclei have been consistently found in in wVo animal
studies (Erexson et al., 1986). Benzene exposure has also been associated with
myelodysplasia and acute myelogenous leukemia (Snyder, et al.. 1993). Rodents
chronically exposed to benzene display similar effects except that a wider range of
carcinogenic responses have been reported (Maltoni et al.. 1985). Evidences for the
genotoxicity of benzene in human comes from studies of occupationally exposed
populations (Yardley-Jones et al., 1990) Since consistent chromosomal aberrations are
often observed in human leukemias, the ability of phenolic metabolites of benzene to
induce chromosomal damage in human implicates the benzene induced leukemia (Yager
et al., 1990). The evidence linking benzene exposure with leukemia was considered
sufficient proof of human carcinogenicity (Dosemeci ef a/., 1994). Despite extensive
research, the molecular mechanisms responsible for the induction of bone marrow

Chapter - VI gQ
toxicity and leukemia have yet to be fully elucidated. However, there is general
agreement that expression of benzene toxicity requires metabolism and follows similar
pathways in humans and animals exposed to benzene (Snyder e a/., 1993).
Recent report indicates that several metabolites of benzene are capable of reacting
with DNA (Levay and Bodell, 1992). Administration of metabolites of benzene induce
sister chromatid exchanges, micronuclei induction (Zhang et al., 1994), oxidative DNA
damage (Kolachana, 1993), and DNA strand breaks (Li and Trush, 1993a). One
mechanism by which benzene induces these genotoxic effects may be mediated by the
generation of one or more active oxygen species, such as superoxide anion radical
(O2 ) hydrogen peroxide (4 Q). hydroxy! radical ('OH) and singlet oxygen (^t^Q^
(Subrahmanyam et al., 1991b). Damage to DNA is perhaps one of the most crucial
events in cytotoxicity of reactive oxygen species (Halliwell and Aruoma, 1991). DNA
lesions resulting from exposure to reactive oxygen species includes modified bases,
abasic sites, single and double strand breaks and DNA protein crosslinks (Aruoma et al.,
1989a; Aruoma et al., 1989b), although, repair enzymes remove most but not all, of the
lesions formed during endogenous DNA damaged by reactive oxidant (Ames et al..
1993).
The majority of benzene metabolism occurs in liver to its major hydroxylated
metabolites (Subrahmanyam et al., 1991a) and has been reported to accumulate against
concentration gradient in bone marrow (Greenlee e al., 1981a). High levels of
myeloperoxidase in the bone marrow facilitate autooxidation of the polyphenols to quinoid
derivatives (Eastmond et al., 1986). The toxicity of quinoid metabolites of benzene has
been associated with their ability to form adducts with thiols and undergo faster
autooxidation to generate oxygen centered radicals (Subrahmanyam and O'Brien, 1985).
The formation of polyphenolic metabolites of benzene in rabbit exposed to benzene
and their accumulation in bone marrow against the concentration gradient and suggested
mechanism of toxicity has been described earlier (Singh et al., 1994). However, the
reactive intermediates responsible for benzene toxicity have not been definitly identified
(Snyder ef a/., 1993). The toxic effect of benzene is likely to be exerted by multiple of
metabolites through multiple of biological pathways (Goldstein, 1989).

Chapter - VI 81
Li and Trush (1993a) observed that hydroquinone/Cu(ll) and 1,2,4-benzenetriol/
Cu(ll), chemical metal redox were the two most efficient systems which induced strand
breakage in X-174 RF1 plasmid DMA. Recent studies revealed that treatment of HL-60
cells with hydroquinone resulted in the formation of 8-hydroxyguanosine (8-OHdG)
formation, suggesting that hydroquinone can induce oxidative DNA base modification in
cells (Kolachana et al.. 1993), although, the mechanism by which it causes these effects
remain unclear. It is likely that some of these effects, if not all could be mediated by
further oxidative activation of hydroquinone. Similar observation was also obtained in
different cell lines whose peroxidase activation of hydroquinone results in the formation
of DNA adducts (Levay et al., 1993).
The formation of 1,4-benzoquinone has been demonstrated from the oxidation of
hydroquinone by horse radish peroxidase and H2O2 (Yamazaki et al., 1959). Formation
of 1,4-benzoquinone by human myeloperoxidase and HjO; was also observed during the
oxidation of hydroquinone (Smith et al., 1989). 1,4-Benzoquinone has been shown to
induce single strand breaks in DNA of cultured cells (Pellak-Walker and Blumer, 1986),
to decrease the ability of stromal cells to support granulocyte/monocyte colony formation
(Gaido and Wierda, 1984), to form DNA adducts in all cell types tested (Levay et al.,
1993) and to inhibit DNA and RNA synthesis (Kalf, 1987), microtubule polymerization
(Irons and Neptun, 1980) and mitogen stimulation of lymphocyte growth (Wierda and
Irons, 1982).
1,4-Benzoquinone reacts with glutathione (GSH) to form a conjugate, glutathionyl
hydroquinone (Lunte and Kissinger, 1983). Macrophages from bone marrow have been
shown to be capable of peroxidative bioactivation of hydroquinone to 1,4-benzoquinone
which can either form a GSH-conjugate and/or covalently bind to protein or DNA (Thomas
et al., 1990), 1,4-Benzoquinone is a direct-acting alkylating agent. It readily reacts with
sulphahydryls and has been shown to inhibit microtubule assembly by blocking the
thiol-sensitive GTP binding site (Irons et al. 1981). However, the DNA adducts formed
in HL-60 cells treated with 1,4-benzoquinone were shown to be different from those
formed on reaction of purified DNA with 1,4-benzoquinone (Levay e al., 1991),
suggesting that the reactive intermediate in cells is the metabolite of 1,4-benzoquinone

c o
ro
X o o 3 ra
c 00
c g ro E
0) c o c D CT O
c o
u c CD O
O D
£ 0) ro -o Q- > , -D C <u o S£ Q. ro 2 3 Q. en
0)
3 03

Chapter - VI ^^
rather than 1,4-benzoquinone itself. Glutathionyl hydroquinone has been isolated as one
of the urinary metabolites of hydroquinone (Figure-6.1) indicating that glutathionyl
hydroquinone formation occurs in vivo (Nerland and Pierce, 1990). Glutathionyl
hydroquinone has been shown to autooxidize at several fold higher than the parent
hydroquinone to glutathionyl benzoquinone (Brunmark and Cadenas, 1988).
In the present study iron catalyzed bleomycin-dependent degradation of DNA was
investigated in the presence of glutathionyl hydroquinone. We also investigated the
potential of glutathionyl hydroquinone to damage supercoiled DNA in plasmid pUC 18.
Glutathionyl-hydroquinone complex concentration dependent release of aldehydic
products from glutamate or deoxyuridine was also observed. We further investigated the
haematotoxicity of glutathionyl hydroquinone in male mice to evaluate the possible role
of glutathionyl hydroquinone in benzene induced haematotoxicity. The studies revealed
that glutathionyl hydroquinone is a potent prooxidant and this observation may be of
toxicological importance in relation to benzene induced toxicity.
RESULTS
Glutathionyl hydroquinone conjugate was prepared by mixing 1,4-benzoquinone
(BQ) and GSH in distilled water in 1:1 ratio (equimolar concentrations). The nucleophilic
addition of GSH to 1,4-benzoquinone yields glutathionyl hydroquinone and its authenticity
was confirmed by the absorption maximum at 303 nm. No absorption was observed at
248 nm which is the absorption maximum for 1,4-benzoquinone (Figure-6.2).
Iron-catalyzed and bleomycin-dependent degradation of calf thymus DNA was
markedly enhanced (12 fold) in the presence of glutathionyl hydroquinone as compared
to the other experiment in the presence of iron alone (Table-6.1). Presence of 1,4-
benzoquinone did not influence the iron-catalyzed bleomycin- dependent degradation of
calf thymus DNA, however, presence of hydroquinone (HQ) or GSH influenced iron-
catalyzed bleomycin dependent degradation by 1 9 and 3.2 fold respectively (Table- 6.1).
Glutathionyl hydroquinone (GHQ) concentration dependent release of TBAR from target
molecules viz. L-glutamate or deoxyuridine was also observed (Table-6.2). The

o o
o en
o
L2 6 O
o 00 (SI
O
r-•
o
1 CD o
1 rs
• O
1 "? o
• • l
un d
• I •vj
d
1 ro 6
1 • o
I •
o
o fN
•o c m -;
o X O '*--' 0) c o c 'D cr o L_
• D
>< JZ
">. c o sz ra 3 O)
>—V
CQ ^ .^ o CD "—" OJ C
c o O .!= .5 + 3 -, cr J o 5 N O c g 0) D -Q cr ^ O < ^ v _ > >
^ -c CD > ,
i: c o O
W 03
ra ^
a u
•a-o V

83
Table 6.1
Iron-catalyzed bleomycin-dependent release of TBAR from calf thymus DNA in the presence of 1,4-benzoquinone (BQ), hydroquinone (HQ),
glutathione (GSH) or glutathionyl hydroquinone (GHQ).
Addition nmoles of TBAR formed per 30 min
Iron 1.38
Iron + BQ 1.38
Iron + HQ 2.67
Iron + GSH 4.42
GSH 0.00
Iron + GHQ 16.20
GHQ 0.00
Experimental conditions are as described in Materials and Methods section. All values are averages of two sets of experiments conducted in duplicate with variations 10% or less.

84
Table 6.2
Effect of varying concentration of glutathionyl hydroquinone on the release of TBAR from L-glutamate or deoxyuridine
Glutathionyl hydroquinone (pM)
5.0
10.0
15.0
20.0
25.0
nmoles of TBAR formed per 2 hr
L-glutamate
0.27
0.56
0.81
1.15
1.21
Deoxyuridine
0.23
0.46
0.90
1.17
1.40
Experimental conditions are as described in Materials and Methods section.
All values are averages of two sets of experiments conducted in duplicate with variations of 10% or less.

Chapter - VI oc
superoxide anion released during autooxidation of glutathionyl hydroquinone (30 JM)
was found to reduce 2.4 nmoles of cytochrome c per min.
Glutathionyl hydroquinone dependent release of TBAR during iron-bleomycin
degradation of calf thymus DNA did not show any significant increase with the increase
in iron concentration (5-15 [JM) (Table-6.3). Similarly, varying the concentration of 1,4-
benzoquinone did not influence the release of TBAR from calf thymus DNA. However,
a linear increase in the release of TBAR from calf thymus DNA was observed with
increase in GSH concentration (5-15 \M) (Table-6.4). Release of TBAR from DNA in
the presence oi glutathionyl hydroquinone, iron and bleomycin was significantly inhibited
(43-51%) on addtion of oxyradical scavengers, such as thiourea (49%), mannitol (46%),
dimethylsulfoxide (43%), albumin (49%), superoxide dismutase (51%) and catalase
(46%) (Table-6.5). The glutathionyl hydroquinone and iron added to bone marrow cell
lysate was 100% recoverable when assayed by the bleomycin dependent release of
TBAR from calf thymus DNA (Table-6.6).
When the supercoiled plasmid pUC 18 DNA (300 ng) was incubated with 200 (JM
of 1,4-benzoquinone, GSH or glutathionyl hydroquinone for 3 hr at 3 7 ^ , cleavage of
plasmid DNA was observed only with glutathionyl hydroquinone resulting in the formation
of both relaxed open circular and linear form of DNA respectively, which run significantly
slower than supercoiled DNA on 1% agarose gels (Figure-6.3). Presence of DNA alone
(lane a), 1,4-benzoquinone alone (lane b), or GSH alone (lane c) did not result in the
nicking of pUC 18 DNA, Whereas, after 3 hr incubation at 37'C in the presence of
glutathionyl hydroquinone (200 pM), all the supercoiled pUC 18 DNA showed a major
damage in the form of single strand nick to give rise to open circular and a minor damage
in the form of double cut to give rise to linear form of DNA (lane d). The single and
double strand cleavage of pUC 18 DNA by glutathionyl hydroquinone (lane d) is
compared with linear form of pUC 18 DNA obtained by double strand single cut after
EcoRI digestion (lane e). Further, a linear increase in DNA damage was observed in the
presence of increasing concentrations of glutathionyl hydroquinone. After 3 hr incubation
at 37X, the DNA damage was increased with increasing concentrations of glutathionyl
hydroquinone (0-200 \M) (Figure-6.4).

86
Table 6.3
Effect of varying concentration of iron on the iron catalyzed bleomycin dependent release of TBAR from DNA In the
presence of GHQ (15 pM)
Concentration of Iron nmotes of TBAR formed ( pM) per 30 min.
5.0 13.24
7,5 15,27
10.0 16.00
12.5 16.17
15.0 16.20
Experimental conditions are as described in Materials and Methods section. All values are averages of two sets of experiments conducted in duplicate with variations of 10% or less.

87
Table 6.4
Effect of varying concentration of GSH on iron-catalyzed bieomycin-dependent release of TBAR from calf thymus DNA in the
presence of BQ (15 pM) and iron (15 pM).
Concentration of GSH nmoles of TBAR formed (pM) per 30 min.
5.0 4.49
7.5 5.66
10.0 9.66
12.5 12.36
15.0 16.20
Experimental conditions are as described in Materials and Methods section. All values are averages of two sets of experiments conducted in duplicate with variations of 10% or less.

88
Table 6.5
Effect of oxyradical scavengers on iron-catalyzed bleomycin-dependent release of T6AR from calf thymus DNA in the
presence of Iron (5 pM) and GHQ (S pM).
Reaction mixture
Complete reaction mixture
+ Thiourea (5 mM)
+ Mannitol (50 mM)
+ Dimethylsulfoxide (33 mM)
+ Albumin (100 MQ/ml)
+ Catalase(100Mg/ml)
+ SOD (100pg/ml)
Experimental conditions are as described in Materials and Methods section.
All values are averages of two sets of experiments conducted in duplicate with variations of 10% or less. *
Number in parentheses are percent inhibition.
nmoles of TBAR formed per 30 min.
6.73
3.46
3.65
3.85
3.46
3.27
3.65
* (49)
(46)
(43)
(49)
(51)
(46)

89
Table 6.6
Recovery of iron and glutathionyl hydroquinone added to bone marrow cell lysate for the bleomycin-dependent degradation of calf thymus DNA
Addition nmoles of TBAR formed per 30 min.
- Bone marrow lysate 5.38
+ Bone marrow lysate 9.04
Experimental conditions are as described in Materials and Methods section. All values are averages of two sets of expenments conducted in duplicate with variations of 10% or less.

a b
Figure 6.3 : Agarose gel electrophoresis of ethidium bromide stained pUC 18 DNA after treatment with BQ, GSH or GHQ (200 |JM each). Lane (a) DNA alone, lane (b) DNA + BQ, lane (c) DNA+GSH, lane (d) DNA+GHQ, and lane (e) pUC 18 DNA digested with EcoRI. Reaction mixtures were incubated at 37°C for 3 hr. The positions of supercoiled (SC), linear (LIN) and open circular relaxed (OC) DNA are indicated.

Figure 6.4 : Cleavage of pUC 18 DNA by increasing concentrations of GHQ. Lane (a) DNA only, lane (b-f) DNA+GHQ (25, 50, 75, 100 and 200 |iM), and lane (g) EcoRI digested ? DNA molecular marker (3530-21226 bp). Reaction mixtures were incubated at 37°C for 3 hr.

a b
Figure 6.5 : Cleavage of pUC 18 DNA by GHQ in presence of oxyradical scavengers. Lane (a) DNA only, lane (b) DNA+GHQ, and lane (c-g) DNA + GHQ+ SOD/catalase /mannitol/thiourea/sodium benzoate at 10 fold molar excess concentration. Reaction mixtures were incubated at 37°C for 3 hr.

90
Table 6.7
Relative liver/spleen weight, haemoglobin, and hematocrit (%) of male mice after glutathionyl hydroquinone treatment
Parameters Control Treated
Relative liver weight 4.73±0.36 6.00+0.15' (per 100 gm)
Relative spleen weight 0.62+0.08 O.SO+O.IO'' (per 100 gm)
Haemoglobin (gm/dl) 13.9 ±0.65 14.4 ±0.31^^
Hematocrit (%) 41.8 ±1.07 40.5 ±1.10^^
Treatment through single injection daily (i.p. = 100 mg/kg b.w. in 0.1 ml normal saline and control group received 0.1 ml normal saline only) for one month. Values are mean ± S.E. of six mice. Exposed group were compared with respective control. P values; a <0.01, NS = Not significant.

91
Table 6.8
Total WBC and differential leukocyte count (DLC) of male mice after glutathionyl hydroquinone treatment
Parameters Control Treated
WBC (per Cum)
DLC (%)
Neutrophil
Lymphocyte
Eosinophil
Monocyte
Basophil
8000±582
24.0±1.9
71.5±1,9
2.3+0.6
1.3+0.4
NIL
9117±209''^
27.0±3.4^^
71.0±3.3^'
1.8+0.4' ^
0.3+0.2^^
NIL
Treatment through single injection daily (i.p. = 100 mg/kg b.w. in 0.1 ml normal saline and control group received 0.1 ml normal saline only) for one month. Values are mean + S.E. of six mice. Exposed group were compared with respective control. P values; NS = Not significant.

92
Table 6.9
Lipid peroxidation, total and free -SH content in liver of mate mice after glutathlonyl hydroquinone treatment
Parameters Control Treated
Lipid peroxidation 25.0±4.6 33.8±4.5'^^ (nmoles/hr/gm liver)
Total-SH content 3.25±0.13 3.27±0.05''^ (mmoles/100 gm liver)
Free -SH content 0.73±0.07 0.85±0.05' ^ (mmoies/100 gm liver)
Treatment through single injection daily (i.p. = 100 mg/kg b.w. in 0.1 ml normal saline and control group received 0.1 ml normal saline only) for one month. Values are mean ± S.E. of six mice. Exposed group were compared with respective control. P values; NS = Not significant.

Chapter - VI 93
The effect of oxyradical scavengers, such as superoxide dismutase (250 units) and
catalase (1000 units), mannitol, thiourea and sodium t>enzoate (2mM) on pUC 18 DMA
strand cleavage were investigated to evaluate the role of reactive oxygen species. The
results shovi/ed that incubation of pUC 18 DNA with glutathionyl hydroquinone in the
presence of superoxide dismutase (SOD), catalase, mannitol, thiourea and sodium
benzoate showed a differential inhibition of cleavage of pUC 18 DNA (Figure-6.5).
Protection offered by catalase was maximum (more than 80%) (lane, d), whereas
protection by superoxide dismutase was only 30% (lane c) at 10-fold molar higher
concentrations. Mannitol (lane e), thiourea (lane f) and sodium benzoate (lane g) were
able to protect marginal at 10-fold molar excess concentrations to glutathionyl
hydroquinone.
Studies were also conducted to evaluate the haematotoxicity of glutathionyl
hydroquinone after exposure of male mice at 100 mg glutathionyl hydroquinone/kg body
weight i.p. in normal saline for one month.
Significant increase in relative liver weight (P<0.01) was observed in glutathionyl
hydroquinone treated mice compared to control group. Further, increased pattern of
relative spleen weight was observed which was however not significant (Table- 6.7).
Haemoglobin and hematocrit(%) values did not show any significant alteration (Table-6.7).
Slight increase in WBC count was observed in glutathionyl hydroquinone treated mice but
increase was not significant (Table-6.8). Neutrophil, lymphocyte, eosinophil, monocyte
and basophil did not show any significant alteration in treated group (Table-6.9). Further,
lipid peroxidation, total and iree -SH content in rat liver also did not show any significant
alteration in treated group (Table-€.9).
DISCUSSION
Although, the metabolism of benzene is very well understood the metabolite(s)
responsible for myelotoxicity of benzene is not known (Kalf, 1987). Benzene exposure
has been shown to generate organic Uee radicals and superoxide radical and it is
generally accepted that benzene requires metabolism to express its toxicity
(Subrahmanyam ei a/., 1991b). In our earlier studies we proposed that hydroquinone:

Chapter-VI ,4
iron complex may be the toxic intemfiediate of benzene metabolism (Singh et al., 1994).
Recent studies have emphasized on 1,4-benzoquinone as toxic metabolite of benzene
as it has been shown to fomi DMA adduds both in vitro and in vivo (Levay et a!., 1991;
Levay et al., 1993). The additon of GSH to 1,4-ben2oquinone has been shown to catalyze
1,4-reductive addition reaction of the Michael type involving a two-electron transfer to
yield glutathionyl hydroquinone (Brunmark and Cadenas, 1988). It was also observed
that dunng benzene exposure incorporation of iron into circulating erythrocytes was
inhibited (Lee et al., 1974) which might lead to accumulation of LMI components in bone
man-ow (Pandya et al., 1990). Since excess of both glutathionyl hydroquinone and iron
are available in bone marrow cells during benzene exposure, the potential of glutathionyl
hydroquinone to undergo autooxidation and promote iron catalyzed bleomycin dependent
degradation of DNA was studied. Several fold increase in the release of TBAR from DMA
indicates that glutathionyl hydroquinone autooxidation enhances the availability of Fe(ll)
to bleomycin by reducing Fe(lll), thus limiting the Fe(ll) from hydrolysis to hydroxides and
further oxidation. Superoxide release during autooxidation of glutathionyl hydroquinone
can reduce feme iron to ferrous iron (Halliwell et al.. 1992). Earlier we have also shown
that 1,2,4-benzenetriol was capable of releasing iron from horse spleen ferritin, in vitro,
which was mediated to a certain extent by superoxide radical (Ahmad et al., 1995). Iron
mediated generaion of reactive oxygen species have been implicated in the toxicity of
2-bromo-(glutathion-S-yl) hydroquinone to LLC-PK1 cells due to DNA fragmentation and
resultant cytotoxicity (Mertens et al., 1995). Recently, Lee et al., (1995) investigated
that 1,2,4-benzenetriol even in the absence of metal ion resulted in double strand
cleavage of pBR 322 plasmid DNA via active oxygen species formation.
The GSH concentration dependent increase in TBAR release from calf thymus DNA
in the presence of 1,4-benzoquinone and iron shows that the reaction is dependent on
the concentration of reducing equivalents. This particular observation may be of
toxicological importance as GSH is an important intracellular thiol present in millimolar
concentrations (Meister, 1988). GSH is also present in the nucleus and 1,4-benzoquinone
has been shown to form adducts with DNA, it is quite likely that 1,4-benzoquinone may
conjugate with GSH and form adducts with DNA. Probably, for this reason the DNA

Chapter- VI 95
adducts, formed in 1,4— benzoquinone-treated HL-60 cells were different from those
formed with purified DNA on reaction with 1,4-l)enzoquinone. As the autooxidation of
glutathionyl hydroquinone is eight-fold faster than hydroquinone, it is quite likely that
glutathionyl hydroquinone would cause DNA damage including chromosomal breaks
leading to genotoxic and carcinogenic effects which have been reported to occur during
benzene exposure.
The complete recovery of glutathionyl hydroquinone and iron added to bone marrow
lysate further indicates that both iron and glutathionyl hydroquinone are available to
bleomycin to release TBAR from calf thymus DNA. Previous studies have suggested the
involvement of iron or copper in benzene metabolites induced DNA damage through a
mechanism involving the generation of reactive oxygen species (Rao and Pandya, 1989).
Breakage in supercoiled pUC 18 plasmid DNA in the presence of glutathionyl hydro
quinone was concentration dependent. This may have toxicological importance during
benzene exposure where the metabolites of benzene specially 1,4-benzoquinone is
accumulated in bone marrow cells. Since GSH is present at a relatively high
concentrations in mammalian cells nuclei (Ca 19.2 mM) (Bellomo et al., 1992), it might
be expected that the thiol can perform a similar function in v/vo during t)enzene exposure
where 1,4-benzoquinone is converted to glutathionyl hydroquinone after reaction with
GSH and resultant intermediate interact with nuclear DNA. The exact mechanism of
DNA damage by glutathionyl hydroquinone is not known. The DNA damage may be due
to free radicals generation in the reaction mixture or due to covalent modification of DNA
bases by glutathionyl hydroquinone. Quinone-thioethers are known to exhibit redox
cycle with the concomitant release of oxyradicals (Rao et al., 1988; Brown et al.. 1991).
For this we investigated the protection offered by different oxyradical scavengers to DNA
breakage. More than 80% protection was observed in the case of catalase and about
30% by superoxide dismutase which selectively scavenges superoxide radical and
hydrogen peroxide. However, mannitol, thiourea and sodium benzoate which selectively
scavenge the hydroxy! radicals offered marginal protection (10-15%) even at 10-fold
molar excess concentration with respect to glutathionyl hydroquinone (200 \M). This
suggests that glutathionyl hydroquinone mediated strand scission in pUC 18 DNA is

Chapter - VI 96
mediated via superoxide radical and hydrogen peroxide rather than the involvement of
iron-catalyzed Haber-Weiss reaction. These observations indicate that pUC 18 DNA
damage by glutathionyl hydroquinone was not only mediated by free radical but some
other mechanism might also he responsible for plasmid DNA damage in the presence of
glutathionyl hydroquinone.
To establish, glutathionyl hydroquinone as haematotoxin, we exposed male mice at
dose 100 mg/kg body weight, intraperitoneally for one month. No mortality was observed
at this dose in glutathionyl hydroquinone treated group. Inaeased pattern of relative liver
weight (P<0.01) and spleen weight and WBC count were noticed, but these results were
not conclusive to say glutathionyl hydroquinone as potent haematotoxic metabolite of
benzene. The pharmacokinetics and distribution of administered glutathionyl hydro
quinone is not known. Probably, it does not reach the target organ, bone marrow as
glutathionyl hydroquinone form. Other reason may be due to difference in sensitivity of
species towards different benzene metabolites. Chronic toxicity studies by the National
Toxicology Program (Huff, 1983) have shown that B6C3F1 mice are more sensitive to
the haematotoxic and carcinogenic effects to benzene than F344/N rats. The greater
sensitivity of mice compared with rats to benzene toxicity may be related to a greater
metabolism of benzene to toxic intermediates, differences in detoxification of toxic
metabolites or greater inherent susceptibility of target tissues to the action of toxic
metabolites. Earlier,Longacre et al., (1981) showed that there are strain differences in
mice in the metabolism and toxicity of benzene and that saturation of metabolism may
play a role in producing the differences in toxicity.
However, it is concluded from the present in vitro studies that glutathionyl
hydroquinone is a potent prooxidant and due to its faster autooxidation to glutathionyl
t)enzosemiquinone may result in DNA damage. Our experiments showed that under mild
conditions at 37°C with the presence of 200 pM concentration of glutathionyl
hydroquinone, within 3 hr, all the supercoiled pUC 18 DNA has been converted to open
circular relaxed and linear form. Interaction of glutathionyl hydroquinone with DNA may
facilitate the formation of adducts with DNA which is one of the prerequisites of benzene
toxicity and carcinogenicity. Anderson et al., (1995) reported the DNA-damaging ability

Chapter - M 97
of benzene and its metabolites in human lymphocytes using the comet assay. Zhang
et al., (1995) pointed out that interest has focused on the interactions of benzene
metabolites and the toxicological significance of free radicals as ultimate toxic species
mediating benzene toxicity since no single ring-hydroxylated metabolite of benzene can
express all the known haematololgical effects of benzene. Further, experiments are
needed to explain significance and the precise mechanism of DNA damage by
glutathionyl hydroquinone, and the oxyradicals and semiquinone formed during its
autooxidation, in the expression of benzene induced haematotoxicity.

Chapter - VII Role of Transition Metal Ions (Fe, Cu)
During Benzene Toxicity Introduction
Results
Discussion

Chapter - VII 98
INTRODUCTION
Benzene exposure has been associated with many types of genetic damage
including chromosomal aberrations in animals and man, sister chromatid exchanges, and
micronuclei induction both in vitro and in vivo as well as aneuploidy in dividing cells.
However, it has not generally produced point mutations in genotoxicity test system
(Snyder and Kalf, 1994). Benzene is a substrate of cytochrome P-450 IIE1 (CYP2E1)
(Seaton et a/., 1994) and metabolized to phenolic intermediates to exert its toxic effect in
biological system (Kalf, 1987). Hydroquinone and 1,2,4-benzenetriol, the two principal
polyphenolic metabolites of benzene have been shown to be toxic and the suggested
mechanisms include free radical formation via superoxide and covalent binding of the
semiquinone to DNA, RNA and other cellular macromolecules.
Li and Trush (1993a) observed that hydroquinone/Cu(ll) and 1,2,4-benzenethol/
Cu(ll), chemical metal redox were the two most efficient systems which induced strand
breakage in (t)X-174 RF1 plasmid DNA. Studies revealed that treatment of HL-60 cells

Chapter - VII
with hydroquinone resulted in the formation of 8-hydroxydeoxyguanosine (8-OHdG),
suggesting that hydroquinone can induce oxidative DNA base modification in cells
(Kolachana et ai. 1993). The role of DNA adduct formation in the myelotoxic effects of
benzene exposure has not been defined. Levay and Bodell (1992) studies have
suggested that the myeloperoxidase in HL-60 cells can activate hydroquinone to form
DNA adducts. Later on Levay et ai, (1993) demonstrated that peroxidase activation of
hydroquinone results in the formation of DNA adducts in HL-60 cells, mouse bone
marrow, macrophages and human bone marrow. Recently, Pathak et ai, (1995)
demonstrated the formation of DNA adducts in the tx)ne man-ow of B6C3F1 mice treated
with benzene and later on Levay et ai, (1996) hypothesized that the adduct formation
might depend on both the dose and duration of treatment. Low level of benzene
administration failed to adduct fonnation in experimental animals. DNA adduct formation
was also observed in HL-60 cell culture treated with hydroquinone for 16 hr at 250 \M
concentration (Pongracz and Bodell, 1996). Zhang et ai, (1993) demonstrated the
potential role of copper in micronuclei induction and oxidation of DNA damage in human
lymphocyte and HL-60 cell by 1,2,4-benzenetriol.
There are a number of both enzymatic and non-enzymatic mechanisms by which
xenobiotic can be activated to reactive intermediates (Tnjsh and Kensler, 1991). Evidence
is emerging that the generation of active oxygen species in biological systems, either
through normal metabolic pathways or as a consequence of exposure to xenobiotics, such
as chemical carcinogens, is involved in the multistage process of carcinogenesis,
especially the promotion stage (Witz, 1991). Studies have provided evidence that
transition metals including iron and copper are capable of directly mediating the activation
or metabolism of several xenobiotics via a metal redox mechanism, which leads to the
formation of more reactive oxygen and other organic free radicals (Ahmad et ai, 1992;
Yamamoto and Kawanishi, 1992).
Trace amount of the metal ions are present in biological system. An estimated, one
third of all enzymes require a metal ion in one or more phases for their catalytic activity.
Transition metal ions, most notably iron and copper can facilitate the transfer of electrons
to biological macromolecules such as lipid, protein and DNA. In addition to their

Chapter - VII lOo
biological function, metal ions are responsible for tissue damage. In the presence of trace
metal ions the mutagenicity of polyphenolic metabolites of benzene showed an increase
to the tester strain Salmonella typhimurium (Lee and Lin, 1994). Earlier Krsek-Staples
and Webster, (1993) observed the iron-catalyzed oxygen resulting in the formation of
carbonyl derivatives with concomitant loss of enzyme function.
Maintenance of cellular iron homeostasis is a prerequisite for many essentia!
biological processes and for the growth of organisms, and is also a central element in the
regulation of immune function (Weiss et a!., 1995). Over the past decade, free
radical-mediated processes have been established in the pathology of hepatic iron
overload (Bacon and Britton, 1990), and unwinding of hepatic double-strand DNA in rats,
with chronic dietary iron overload was reported (Ediing et al., 1990). Further, Toyokuni
and Sagripanti (1993) demonstrated that Fe-citrate can constitute a powerful DNA
damaging system under physiological conditions.
The biological significance of copper has recently attracted with interest in
connection with carcinogenicity and mutagenicity (Agarwal et a!., 1989). Copper is an
essential component of chromatin (Saucier et al., 1991) and is known to accumulate
preferentially in the heterochromatic regions (Bryan e al., 1976). Copper sulphate
(CUSO4) showed clastogenic effects on the bone marrow cell chromosomes of mice in
vivo (Agarwal et al., 1990). Li et al., (1991) reported that copper accumulated in liver
tissues of LEC (Long-Evans Cinnamon) rats that has spontaneously developed
hepatocellular carcinomas, suggesting that the abnormal copper metabolism is involved
in hepatic carcinogenesis in LEC rats. The exposure of DNA to hydrogen peroxide in the
presence of Cu(l) or Cu(ll) is known to result in the induction of a variety of oxidative
lesions, including DNA strand breaks and base modifications (Kawanishi and Yamamoto,
1991;ltoefa/., 1992).
Previous studies have suggested the involvement of iron or copper in benzene
metabolites induced DNA damage through a mechanism, involving the generation of
reactive oxygen species (Kawanishi etal., 1989; Rao and Pandya, 1989). Although, iron
is known to be capable of catalyzing oxidative damage to DNA, copper has been shown
to be more potent. Copper is distributed throughout the body with liver and bone man-ow

Chapter - VII 101
being the two major storage organs. It is also closely associated with chromosomes and
DNA bases (particularly guanine) and the possible role of copper as a mediator of reactive
oxygen induced cytotoxicity as well as genotoxicity have been proposed (Li and Trush,
1993b). In this respect, the present studies were conducted to quantitate the metal ion
concentrations (iron and coper) in rat liver and isolated liver nuclei in benzene exposed rV
albino rats. Further, we studied the potential role of polyphenolic metabolites of benzene
to release thiobartDituric acid reactive products (TBAR) from isolated liver nuclei with or
without the presence of metal chelators and oxygen radical scavengers to explain the
possible role of metal ions in benzene induced genotoxicity and chromosomal aberration.
RESULTS Total iron and copper contents were estimated in liver tissue of male and female rats
exposed to benzene intraperitoneally (0.5 ml/kg body weight) for 10 days. The iron
content only showed significant increase in male and female rats (P<0.02; P<0.05), which
received benzene and no significant change in copper content was observed in either sex
when compared to respective control groups (Table-7.1). Iron and copper contents were
also analyzed in isolated rat liver nuclei of benzene exposed female rats (i.p). In isolated
rat liver nuclei also only iron content showed significant increase (P<0.05), but no change
was observed in copper content when compared to the respective control groups
(Table-7,2).
Formation of TBAR was observed from isolated rat liver nuclei in presence of
polyphenolic metabolites of benzene, such as hydroquinone and 1,2,4-benzenetriol and
other polyphenols, such as 6-hydroxydopamine and dopamine at 330 pM concentration.
Significant release in TBAR was observed in all cases but the release was maximum in
the presence of hydroquinone (34.6 nmoles TBAR/hr/gm protein) (Table-7.3). Increased
pattern of TBAR formation was observed with increase in hydroquinone concentrations
(0-330 pM), but the release was not linear with increase in hydroquinone concentrations
(Table-7.4).
Further, nuclear DNA damage was evaluated by agarose gel electrophoresis. Rat
liver nuclei were incubated with hydroquinone/1,2,4-benzenetriol (0.5 or 1.0 mM) and

102
Table 7.1
Effect of benzene* administration on total iron and copper metal contents in liver of albino rats
Iron (pg/gm tissue)
Copper (pg/gm tissue)
Male rat
Control
i.p.
Female rat
Control
i.p.
91.50±1.90
118.00+7.85^
115.00±8.58
143.0018.10"
3.24±0.12
3.67+0.19' ^
4.86±0.68
4.86±0.38''5
^Benzene was administered through single injection daily (i.p. = 0.5 ml/kg b.w.) for 10 days. Values are mean ± SE of five rats. Exposed groups were compared with their respective control. P values : a < 0.02; b < 0.05; NS = Not significant.

103
Table 7.2
Effect of benzene* administration on total iron and copper metal content in isolated liver nuclei of albino rats.
Iron (\iglgm protein) Copper (pg/gm protein)
Female rat
Control
l.p.
41.28+4.20
61.10+6.15*
36.70±3.28
42.40±3.9r^
*Benzene was administered through single injection daily (i.p. = 0.5 ml/kg b.w.) for 10 days. Values are mean ± S.E. of five rats. Exposed groups were compared with their respective control P values: a < 0.05; NS = Not significant

104
Table 7.3
Effect of polyphenolic metabolites of benzene and other polyphenols (330 pM) on release of TBAR from isolated rat liver nuclei.
Addition nmoles of TBAR formed/ hr/gm protein
None 0.0
Hydroquinone 34.6
1,2,4-Benzenetriol 19.8
6-Hydroxydopamine 27.2
Dopamine 18,4
Experimental conditions as described in Materials and Methods section. All values are average of two sets of experiments conducted in duplicate with variations 10% or less.

105
Table 7.4
Release of TBAR from isolated rat liver nuclei in the presence of increasing concentration of hydroquinone
Hydroquinone { pM) nmoles of TBAR formed/ hr/gm protein
33 10.9
107 14.1
182 18.9
256 27.0
330 34.6
Experimental conditions as described in Materials and Methods section. All values are average of two sets of experiments conducted in duplicate with variations 10% or less.

Figure 7.1 : Agarose gel electrophoresis of ethidium bromide stained rat liver nuclei treated with HQ/BT (0.5 or 1.0 mM). Lane (a) liver nuclei only, lane (b and c) liver nuclei + HQ (0.5 or LQinM), lane (d-e) liver nuclei + BT (0.5 or 1.0 mM) respectively, and lane (f) EcoRI digested ^DNA molecular marker (3530-21226 bp). Reaction mixtures were incubated at 37°C for 3 hr.

106
Table 7.5
Effect of externally added copper on TBAR release from Isolated rat liver in presence of hydroquinone
Addition nmoles of TBAR formed/hr/gm protein
Hydroquinone alone 34.6
+ Copper (0.08 mM) 37.9
+ Copper (0.167 mM) 36.7
Experimental conditions as described in Materials and Methods section. All values are average of two sets of experiments conducted in duplicate with vanations 10% or less.

107
Table 7.6
Release of TBAR from rat liver nuclei in the presence of hydroquinone and increasing concentration of bathocuproine (0-25 pM)
Components nmoles of TBAR formed/ hr/gm protein
Hydroquinone alone 34.6
+ 5 pM Bathocuproine 16.5 (52%)
+ 10 " " 15.8(54%)
+ 15 " " 14.9(57%)
+ 20 " " 12.9(63%)
+ 25 " " 11.5(67%)
Experimental conditions as described in Materials and Methods section. All values are average of two sets of experiments conducted in duplicate with variations 10% or less. Number in parentheses are percent inhibition.

108
Table 7.7
Release of TBAR from isolated rat liver nuclei in presence of hydroquinone with or without the addition of EDTA and DTPA (5 pM)
Components nmoles of TBAR formed/ hr/gm protein
Hydroquinone alone 34.6
+ EDTA 17.3 (50%)
+ DTPA 15.5 (55%)
Experimental conditions as described in Materials and Methods section. All values are average of two sets of experiments conducted in duplicate with vanations 10% or less. Number in parentheses are percent inhibition.

109
Table 7.8
Effect of oxyradical scavengers on release of TBAR from isolated rat liver nuclei in presence of hydroquinone
Components nmoles of TBAR formed/ hr/gm protein
Hydroquinone alone 34.6
+ Thiourea (2.5 mM) 6.4(81%)'
+ Thiourea (5.0 mM) 3.2(91%)
+ Mannitol (50 mM) 25.1(27%)
+ SOD(100Mg/1.5ml) 28.8(15%)
+ Catalase(150Mg/1.5ml) 34.5(0%)
Expenmental conditions as described in Matenals and Methods section. All values are average of two sets of experiments conducted in duplicate with variations 10% or less. 'Number in parentheses are percent inhibition.

Chapter - VII ^^^
electrophoresis was done on 1% agarose gel (Figure-7.1). When isolated rat liver nuclei
were incubated with hydroquinone for 3 hr at 37°C, nuclear DMA damage was observed
(lane b and c) at 0.5 or 1.0 mM concentration compared to the control nuclei (lane a). Gel
pattern also indicates some damage in nuclear DNA in presence of 1,2,4-benzenetriol
(lane d and e), however hydroquinone is more potent to cause damage to rat liver nuclear
DNA. Lane (f) is EcoRI digested DNA marker (3530-21226 bp) (Figure-7.1).
Externally added copper (0.08 and 0.167 mM) did not show significant increase in
TBAR formation when isolated rat liver nuclei incubated together with hydroquinone (330
pM) at 37°C (Table-7.5). However, inhibition in TBAR fromation was observed in presence
of bathocuproine (5-25 pM) in hydroquinone induced TBAR fonnation from rat liver nuclei.
At 5 pM concentration of bathocuproine, 52% inhibition was observed. Increase in the
concentration of bathocuproine did not show linear decrease in TBAR formation and only
about 67% inhibition was observed at 25 pM concentration of bathocuproine (Table-7.6).
Further, only 50-55% inhibition was observed in TBAR release in the presence of
EDTA/DTPA (5 pM), when isolated rat liver nuclei incubated with hydroquinone (330 pM)
at 37°C (Table-7.7).
Inhibition of TBAR formation from isolated rat liver nuclei was also studied in the
presence of different oxyradical scavengers, such as thiourea (2.5 and 5.0 mM), mannitol
(50 mM) catalase (150 pg/1.5 ml) and SOD (100 pg/1.5 ml). Presence of thiourea 2.5
mM and 5.0 mM resulted in the inhibition of TBAR release from rat liver nuclei to the
extent of 81% and 91% respectively (Table-7.8). Inhibition by mannitol and SOD were
only 27% and 15% respectively. However, presence of catalase failed to exhibit any
inhibition of TBAR formation from rat liver nuclei in the presence of hydroquinone
(Table-7.8).
DISCUSSION
studies related to the effect of hydroquinone or 1,2,4-benzenetriol on isolated rat liver
nuclei were conducted to monitor the damage to DNA, as copper ion is associated with
the heterochromatin. This particular study would help in better understanding of the
chromosomal aberrations/abnormalities associated with benzene exposure. Exposure to

Chapter - VII ^ 111
benzene resulted in significant increase in total iron content in liver tissue, however copper
content did not shov\/ any alteration. Earlier it was observed that benzene exposure
resulted in alteration of heme metabolism (Rao and Pandya, 1980) and accumulation of
low molecular weight iron components (LMI) in bone marrow (Pandya et al., 1990). The
elevated level of LMI in the bone marrow during benzene toxicity may lead to formation
of tissue damaging species like lipid peroxide radical, superoxide anion (Oj), oxyferryl
species and hydroxyl radical which may form reactive metabolite(s) and may react
immediately if they occur in the vicinity of vital molecules like nucleic acids (Lown and Sim,
1977) and polymerase enzymes (Rushmore et al., 1984) which may be responsible for
the ultimate cytotoxicity. During in vitro study Rao and Pandya, (1989) observed that
polyphenolic metabolites of benzene are autooxidized faster in the presence of metal ions
and resulting in the formation of TBAR from target molecules such as glutamate and calf
thymus DNA. The suggested mechanism includes free radical formation via superoxide
and the covalent binding of the semiquinone metabolites of polyphenols to such
molecules. Significant increase in metal ions in rat liver and rat liver nuclei during
benzene exposure may have toxicological implications for cytotoxicity of liver cells and
liver nuclear DNA damage in vivo.
Further in vitro studies revealed that hydroquinone or 1,2,4-benzenetriol at 330 pM
concentrations, released significant amount of TBAR from rat liver nuclei and only 50-55%
inhibition was observed in the presence of iron chelator such as EDTA or DTPA at 5 pM
concentration. Iron overload affects hepatic heme metabolism. However, the biochemical
mechanisms whereby iron overload affects heme metabolism are not completely
understood. Earlier Mahmutoglu and Kappus (1987) demonstrated the DNA damage in
isolated rat liver nuclei in the presence of redox cycling of bleomycin-Fe(lll) complex by
NADH dependent enzyme and suggesed that the enzymatic reduction of a
bleomycin-metal complex in the cell nucleus may be an essential step in the cytotoxic
activity of bleomycin.
Earlier, we demonstrated the prooxidant and antioxidant properties of iron: hydro
quinone and iron: 1,2,4-benzenetriol complex and suggested that iron catalyzed and
bleomycin-dependent degradation of DNA was markedly enhanced in the presence of

Chapter - VII ^ 112
hydroquinone or 1,2,4-benzenetriol than in iron alone (Singh et al.. 1994). We
demonstrated the formation of iron: polyphenol complex in vitro and fractionated on
Sephadex G-10 column (Chapter-Ill) and proposed that iron; hydroquinone complex may
play role in haematotoxicity of benzene. However, formation of such complex in vivo is
not known.
To demonstrate the possible role of copper in TBAR formation in presence of
hydroquinone, the isolated rat liver nuclei were incubated with or without the presence of
bathocuproine, a specific copper ion chelator. In the presence of bathocuproine, about
67% inhibition was observed. Increase in the concentration of bathocuproine (5-25 pM)
resulted in only marginal increase in inhibition of TBAR formation (52-67%). This may be
due to permeability of bathocuproine to cross the nuclear membrane is limiting with
increasing concentrations of bathocuproine as it contains two methyl groups (Mellow-Filho
and Meneghini, 1991). Copper, a natural constituent of cell nuclei has been suggested
to play a key role in structural organization and function of chromosomes (Hu et a/., 1990).
In this context, it appears important to focus attention also on the redox activity of copper
in the vicinity of DNA. Li and Trush, (1993a) demonstrated that Cu(ll) strongly induces
the oxidation of hydroquinone as such may be factor involved in the oxidative action and
toxic to primary bone marrow stromal cells at DBA/2J mice. They further observed that
Cu(ll) is capable of directly reacting with hydroquinone, causing the one electron oxidation
of hydroquinone to semiquinone radical (SQ) and reactive oxygen species generated in
chemical metal redox system were responsible for in vitro plasmid DNA damage (Li ef al..
1995). The oxidative activation of hydroquinone to the electrophilic 1,4-benzoquinone
through a peroxidase/H202 system has been suggested to play an important role in the
hydroquinone-induced bone marrow target cell injury (Smith ef al.. 1989).
Earlier, Pnjtz (1993) has shown that Cu(ll)-induced oxidation of certain compounds
such as ascorbate, catechol and o-phenylenediamine was enhanced by orders of
magnitude in the presence of DNA. DNA in combination with copper could promote the
bioreductive activation of genotoxic compounds and their progeny in the most vulnerable
cell compartment, the nucleus. Recently, Zhang et al., (1996) demonstrated the important
role of Cu^* in 1,2,4-benzenetriol-induced genotoxicity.

Chapter - VII 113
Endogenously generated oxidants that could lead to cellular DMA damage Include
superoxide radicals and H2O2. There have been many reports to indicate that the human
genome is potentially vulnerable to damage (or mutation) by H2O2 (Meneghini and Martin,
1993). Furthermore, with formation of DNA-Cu(l), the DNA becomes predisposed to
oxidative damage by H^Oj via Fenton type reaction that generate strongly oxidizing
radicals such as hydroxyl radical or CuOzH (Masarwa et al., 1988) in situ, i.e. near the site
of Cu(l) fixation within the DNA (Aruoma et al., 1991);
DNA + Cu(l) + H2O2 > DNA:OH + Cu(ll) + OH" I 1
DNA damage
Copper ions are much more efficient at promoting DNA damage by H2O2 than iron ions
(Aruoma et al., 1991), and this is probably due to the high affinity of DNA for Cu (I).
Presence of thiourea, 2.5 mM and 5.0 mM resulted in the inhibition of TBAR release
from rat liver nuclei to the extent of 81% and 91% respectively. Studies by Hanna and
Mason (1992) have shown that thiourea acts as a copper chelator. This further indicates
the possible role of copper in TBAR formation from rat liver nuclei incubated in the
presence of hydroquinone. Further, externally added copper (upto 0.176 mM) to the
reaction mixture did not result in any significant increase in the release of TBAR from rat
liver nuclei by hydroquinone. This particular observation indicates that the copper
associated with nuclear DNA was only responsible to catalyze autooxidation of
hydroquinone or 1,2,4-benzenetriol, during which oxyradicals are released and caused
degradation of the deoxyribose moiety of DNA, resulting in the formation of TBAR
products. Marginal inhibition in TBAR release was observed in the presence of other
oxyradical scavengers, such as mannitol (27%) and SOD (15%) However, catalase did
not show any inhibition in TBAR formation from rat liver nuclei in the presence of
hydroquinone. Probably, nuclear membrane selectively prevent these oxyradical
scavengers to reach the target site. The present in vitro studies, clearly demonstrate that
benzene metabolites (particularly hydroquinone) induced genotoxic effect in presence of
copper which is associated with the nuclear DNA. However, further studies are needed
to explain the precise mechanism of DNA damage and chromosomal aberration in vivo

Chapter - VII ,,.
during benzene exposure and effect of transition metal ions in benzene induced
genotoxicity and human leukemia.

Siimmar)' 115
Small amounts of a large variety of substances normally regarded as foreign to the
body are always existent in the environment. These are often toxic and occur frequently
In the modem world, man is increasingly dependent upon the use of synthetic chemicals
and other domestic products in larger number.
Benzene is frequently used as an industrial solvent of the modem time. Its excellent
solvent properties and its use as a starting material for wide ranging chemicals and
domestic products of commercial importance has made it indispensable. Its value in our
modem society is great, but due to its toxic nature, the use of benzene would undoubtedly
have an impact on our way of life. The use of benzene has been on the rise and
obviously the risk of exposure is also increasing. Though the exposure levels to benzene
can be brought down to acceptable levels but can not be eliminated.
Benzene is a well known genotoxic and carcinogenic agent. But the precise
mechanism of its genotoxicity and carcinogenicity are yet to be known. Therefore, in this
study an attempt has been made to explain the probable metabol(te(s) of benzene to
induce haematotoxicity and role of transition metal ions (iron and copper) to explain the

Summary 116
possible biochemical mechanisms of genotoxicity and carcinogenicity expressed during
benzene exposure.
Release of iron from ferritin by 1,2,4-benzenetriol
Iron is a vital metallic cofactor of several important biomolecules. It is normally
localized in ferritin (main iron storage protein) or transferrin (iron transport protein). But
this iron, when it gets decompartmentalized manifests lethally by triggering off free radical
reactions by means of Fenton and Haber-Weiss type of reactions:
Fe(lll) + Reductant > Fe(ll) + Oxidized Reductant
2Fe(ll) + O2+ 2H* > 2Fe(lll) + H A
Fe(ll) + H2O2 > Fe(lll) +-0H + OH
Large human prospective studies suggest that excess body free iron is associated
with carcinogenesis, such as primary hepatocellular carcinoma, colon cancer, lung cancer,
bladder cancer, acute lymphocytic leukemia and neuroblastoma. Bleomycin detectable
iron has been shown to accumulate in significant concentrations in the bone marrow of
benzene exposed rats. Polyphenolic metabolite of benzene, particulariy 1,2,4-
benzenetriol (a trihydroxy benzene) was found to release significant amount of iron from
horse spleen ferritin under aerobic and anaerobic conditions. The release of iron from
ferritin during autooxidation of 1,2,4-benzenetriol in vivo may result in increased
intracellular concentrations of free iron. Although, the physiological mechanism of iron
mobilization from ferritin is pooriy understood, the observed iron release from ferritin by
1,2,4-benzenetriol could occur through the direct reduction of iron by the hydroxy
hydroquinone form or via its autooxidation intermediates i.e., superoxide radical and
semiquinone. It was observed that the iron released from ferritin in the presence of
1.2,4-benzenetriol was capable of inducing lipid peroxidation and catalyzing
bleomycin-dependent calf thymus DNA degradation. The mechanism by which iron is
released from bone marrow cells and whether it acts as a prooxidant during benzene
toxicity is not known. However, the present observation offer a new mechanism that the
iron released from ferritin by 1,2,4-benzenetnol could contribute to the better

Summary Hy
understanding of the toxicity of benzene. Further, elucidation of the mechanisms of
iron-mediated oxidative damage may be important for the prevention of carcinogenesis.
Prooxidant and antioxidant properties of iron: polyphenol complex
Polyphenolic metabolites of benzene viz. hydroquinone and 1,2,4-benzenetriol have
been shown to be toxic and the suggested mechanism includes free radical formation via
superoxide during their autooxidation and the covalent binding of semiquinone to DNA,
RNA and other cellular macromolecules. Benzene exposure also leads to an
accumulation of bleomycin sensitive iron in bone marrow. 1,2,4-Benzenetriol, a tnhydroxy
metabolite of benzene was capable of releasing iron from horse spleen ferritin, which may
lead to accumulation of iron in bone marrow during benzene exposure, as the
polyphenolic metabolites are accumulated against the concentration gradient in the bone
man^ow. The ferrous iron may be chelated to the polyhydroxy metabolites of benzene viz.
hydroquinone and 1,2,4-benzenetriol.
Studies carried out with hydroquinone/1,2,4-benzenetriol and iron showed several
fold increase in iron-catalyzed bleomycin- dependent degradation of calf thymus DNA as
compared to iron alone. Complex formation some how increase the availability of iron to
degrade DNA. It is also possible that the hydroquinone and 1,2,4-benzenetnol possess
combined iron-chelating and iron ion reducing properties to stimulate DNA degradation
in the presence of bleomycin. Further, iron:polyphenol complex appears to be stable and
intact as the recovery from bone marrow lysate was complete. On the other hand,
iron:polyphenol complexes have shown an antioxidant activity by inhibiting of
iron-dependent lipid peroxidation in rat brain homogenate and glutamate degradation.
This is similar to the antioxidant properties exhibited by flavonoids due to their ability to
chelate iron. Thus the polyphenolic chelates limiting the availability of iron for lipid
peroxidation and at the same time facilitate bleomycin- dependent degradation of DNA.

Summary 118
Polyphenol-iron complex: A probable toxic metabolite of benzene
An attempt was made to examine in vitro formation of polyphenol:iron complex.
Complex formation was fractionated on Sephadex G-10 column chromatography. Results
clearly show the formation of polyphenoUron complex under in vitro condition. Polyphenol
iron complex formation was also observed during the release of iron from ferritin in the
presence of 1,2,4-benzenetriol. Although, benzene exposure leads to accumulation of
iron as well as polyphenoiic metabolites of benzene in the bone marrow against the
concentration gradient, the formation of such complex in vivo is yet to be identified.
We evaluated the haematotoxicity of hydroquinone.iron complex as a possible
haematotoxic intenmediate during benzene exposure. Results indicate that hydroquinone
iron chelate showed marginal increase in haematological parameters in experimental
animals. But the precise mechanism of marginal, yet significant increase in haema
tological indices is not clear. Atleast, some of the effects observed in hydroquinone:iron
treated group showed similarities to some extent with what has been shown to occur
following exposure of benzene to mice.
Glutathionyl hydroquinone: A possible toxic metabolite of benzene
Glutathionyl hydroquinone has been isolated as one of the urinary metabolites of
hydroquinone, indicating that glutathionyl hydroquinone formation occurs in vivo and
autooxidized at several fold higher than the parent hydroquinone to glutathionyl
benzoquinone. We noticed that glutathionyl hydroquinone is a potent prooxidant to
damage both calf thymus as well as plasmid pUC 18 DNA and this observation may be
of toxicological importance in relation to benzene induced genotoxicity.
Haematotoxicity of glutathionyl hydroquinone was studied in male mice at dose 100
mg/kg body weight, intraperitoneally for one month. Increased pattern of relative liver and
spleen weight and WBC were noticed, but these results were not conclusive to explain
GHQ as potent haematotoxic metabolite of benzene. The pharmacokinetics and
distnbution of administered GHQ is not known. Whether GHQ administered i.p. reaches

Sujiimary
the target organ i.e. bone marrow is yet to be evaluated. Other reason may be due to
difference in sensitivity of species towards different t enzene metabolites.
Role of transition metal ions (Fe,Cu) during benzene toxicity
Earlier studies have provided enough evidence that transition metals including iron
and copper are capable of directly mediating the activation or metabolism of several
xenobiotics via a metal redox mechanism, which leads to the formation of more reactive
oxygen and other organic free radicals. We observed that exposure to benzene leads to
increase in total iron content in liver and isolated rat liver nuclei. Benzene metabolites
induced the DNA damage particularly in the presence of iron and copper. This study
would help in better understanding of the chromosomal aberrations/abnormalities
associated with benzene exposure. Copper which is associated with DNA, may play a
potential role in nuclear DNA damage in presence of hydroquinone. However, the
molecular mechanism of its damage is not well understood. Significant increase in iron
content in liver and in isolated liver nuclei during benzene exposure and the copper which
IS associated with DNA may have toxicoiogical implications for cytotoxicity of liver cells
and liver nuclear DNA damage in vivo.
In the light of our investigations following conclusions can be made:
• 1,2,4-Benzenetriol is a potent reductant of feme iron of ferritin and releases iron from
ferritin core. The release of iron from bone marrow lysate by 1,2.4-benzenetriol may
be of toxicoiogical significance during benzene exposure as this could lead to
disruption of intracellular iron homeostasis in bone marrow cells
• The iron:polyphenol chelate, on one hand, is a more potent DNA cleaving agent in
the presence of bleomycin, and on the other hand, it is a less effective free radical
generator as compared to iron alone. However, the formation of such a complex in
vivo is not known. Further studies are needed to characterize such complexes in
vivo and evaluate their toxicoiogical significance during benzene exposure.
• Polyphenol forms complex with iron under in vitro condition and may be a possible
haematotoxic intermediate during benzene exposure

Summary ,_. IZO
The present in vitro studies revealed that glutathionyl hydrcK|uinone is a potent toxic
metabolite of benzene and acts as a prooxidant due to its faster autooxidation to
glutathionyl benzoquinone and oxyradicals which may induce DNA damage.
Exposure to benzene resulted in significant increase in total iron content in liver
tissue and in isolated liver nuclei.
Benzene metabolites (particularly hydroquinone) induced genotoxic effect in
presence of copper which is associated with the nuclear DNA. However, further
studies are needed to explain the precise mechanism of DNA damage and
chromosomal aberration in vivo during benzene exposure and effect of transition
metal ions in benzene induced genotoxicity and human leukemia.

Bibliography ^
Abraham, N.G.; Lutton, J.D.; Freedman, M.L. and Levere, R.D. (1986). Benzene modulation of liver cell structure and heme- cytochrome P-450 metabolism. Am. J. Med. Sci., 292, 81-86.
Afanas'ev, I.B.; Dorozhoko, A.I.; Brodskii, A.V.; Kostyuk, V.A. and Potapovitch, A.I. (1989). Chelating and free radical scavenging mechanisms of inhibitory action of rutin and quercetin and lipid peroxidation. Biochem. Pharmacol., 38, 1763-1769.
Agarwal, K.; Sharma. A. and Talukder, G. (1989). Effects of copper on mammalian cell components. Chem.-Biol. Interact., 69, 1-16.
Agarwal, K.; Sharma. A. and Talukder, G. (1990). Clastogenic effects of copper sulphate on the bone marrov ^ chromosomes of mice in vivo. Mutat. Res., 243, 1-6.
Ahmad, M.S.; Fazal, P.; Rahman, A.; Hadi, S.M. and Parish, J.hi (1992). Activities of flavonoids for the cleavage of DNA in the presence of Cu(ll); Corelation with generation of reactive oxygen species. Carcinogenesis, 13, 605-608.
Ahmad, S.; Singh, V. and Rao, G.S. (1994). Antioxidant potential in serum and liver of albino rats exposed to benzene. Indian J. Exp. Biol., 32, 203-206.

Bibliography 121Q.
Ahmad, S.; Singh, V. and Rao, G.S. (1995). Release of iron from ferritin by 1,2,4-benzenetriol. Chem.-Biol. Interact., 96, 103-111.
Aisen, P. and Listowsky, I. (1980). iron transport and storage proteins. Ann. Rev. Biochem., 49, 357-393
Aksoy, M.; Dincol, K.; Akgun, T.; Erdem, S. and Dincol, G. (1971). Haematological effects of chronic benzene poisoning in 217 workers. Br. J. Ind. Med., 28, 296-302.
Aksoy, M. (1985). Benzene as a leukemogenic and carcinogenic agent. Am. J. Ind. Med.,8, 9-20.
Aksoy, M.; Ozeris, S.; Sabuncu, H.; Inanici, Y. and Yanarday, R, (1987). Exposure to benzene in Turkey between 1983 and 1985: a haematological study on 231 workers. Br. J. Ind Med., 44, 785-787.
Aksoy, M. (1988). In : Benzene carcinogenicity, edited by M. Aksoy, CRC, Boca Raton, FL, 113-148.
Aksoy, M. (1989). Hematotoxicity and carcinogenicity of benzene. Environ. HIth. Perspect., 82, 193-197.
Ames, B.N.; Shigenaga, M.K. and Hagen, T.M. (1993). Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci USA. 90, 7915-7922.
Anderson, D.; Yu, T.W. and Schmezer, P. (1995). An investigation of the DNA-damaging ability of benzene and its metabolites in human lymphocytes, using the comet assay. Environ. Mol. Mutagen., 26, 305-314.
Andrews, L.S.; Sasame, HA. and Gillette, JR. (1979). ^H- Ben::ene metabolism in rabbit bone marrow. Life Sci.. 25, 567- 572
Anderson, H.J. and Roberge. M. (1992). DNA topoisomerase II: A review of its involvement in chromosome structure, DNA replication, transcription and mitosis. Cell Biol. Int. Reports. 16. 717-724
Anon (1982) Benzene, lARC Mongr Eval. Carcinogen. Risks Hum., 29, 93-148.
Arouma, O.I.; Halliwell, B. and Dizdaroglu, M. (1989a). Iron ion-dependent modification of bases in DNA by the superoxide radical-generating system hypoxanthine/xanthine oxidase. J. Biol. Chem., 264, 13024-13028.

Bibliography -,2
Arouma, O.I.; Halliwell, B. and Dizdaroglu, M. (1989b). Damage to bases in DMA induced by hydrogen peroxide and ferric ion chelates. J. Biol. Chem., 264, 20509-20512.
Arouma, 0 1.; Halliwell, B.; Gajewski. E. and Dizdaroglu, M. (1991). Copper-ion-dependent damage to the bases in DNA in the presence of hydrogen peroxide. Biochem. J.. 273, 601-604.
Aust, S.D ; Morehouse. LA. and Thomas, C.E. (1985) Role of metals in oxygen radical reaction. Free Rad. Biol. /Wec/.,1, 3-25.
Ayengar, P.K., Hayaishi, O., Nakajima, M. and Tomida, I. (1959). Enzymic aromatization of 3.5-cyclohexadiene-1,2-diol. Biochim Biophys Acta .ZZ. 111-119.
Badder, S.L.: Bill. E.; Trautwein. AX.; Bruchelt G and Matzanke, B.F. (1996). Mobilization of iron from cellular ferritin by ascorbic acid in neuroblastoma SK-N-SH cells An EPR study FEBS Letters. 381. 131-134
Bacon, BR and Britton, R.S. (1990) The pathology of hepatic iron over load : A free radical mediated process? Hepatology, 11, 127-137
Bakkeren, D.L., de Jeu-Jaspars, C.M.H.; Van der Heul, C and Van Eijk, H.G. (1985) Analysis of iron-binding components in the low molecular weight fraction of rat reticulocyte cytosol. Int J. Biochiem.. 17. 925-930.
Bartczak, A.; Kline, S A.; Yu, R., Weisel, C.P.; Goldstein, B.D ; Witz, G, and Bechtold, WE (1994). Evaluation of assays for the identification and quantitation of muconic acid, a benzene metabolite in human unne. J. Toxicol. Environ. HItti., 42, 245- 258.
Bellomo, G.; Vairetti, M.; Stivala, L.; Mirabelli, F.; Richelmi. P and Orrenius, S. (1992) Demonstration of nuclear compartmentalization of glutathione in hepatocytes. Proc. Natl Acad. Sci. USA. 89, 4412-4416.
Berman. E (1980). In . Toxic metals and their analysis, edited by L.C. Thomas, Hyden. International Topics in Science Senes, London, Hyden,
Bernheim, P.; Bernheim, M.LC, and Willbur. KM. (1948). The reaction between thiobarbitunc acid and the oxidation products of certain lipids. J. Biol. Chem., 174. 257-264.

Bibliography ,23
Biemond, P.; Swaak, A.J.G.; Van Eijk, H.G. and Koster, J.F. (1988). Superoxide dependent iron release from ferritin in inflammatory diseases. Free Rad. Biol. Med., 4, 185-198.
Bolcsack, L. and Nerland, D.E. (1983). Inhibition of erythropoiesis of benzene and benzene metabolites. Toxicol. Appl. Pharmacol., 69, 363-368.
Brief, R.S.; Lynch, J.; Bernath, T. and Scala, R.A. (1980). Benzene in the workplace. Am. Ind. Hyg. Assoc. J , 41, 616-623.
Brown, P.C.; Dulik, D.M. and Jones, T.W. (1991). The toxicity of menadione (2-methyl-1,4-naphthoquinone) and two thioether conjugates studied with isolated renal epithelial cells. Arch. Biochem. Biophys., 285, 187-196.
Bnjgnone, F.; Pertjellini, L.; Faccini, G.B.; Pasini, F.; Dansi, B.; Maranelli, G.; Romeo, L.; Gobbi, M. and Zedde, A. (1989). Benzene in the blood and breath of normal people and occupationally exposed workers. Am. J. Ind. Med., 16, 385-399.
Brunmark, A. and Cadenas, E. (1988). Reductive addition of glutathione to benzoqui-none, 2-hydroxy p-benzoquinone, and p- benzoquinone epoxide. Effects of the hydroxy and glutathionyl substituents on p-benzohydroquinone autooxidation. Chem.-Biol. Interact., 68, 273-298.
Bryan, S.E.; Simon, S.J.; Vizard, D.L. and Hardy, K.J. (1976). Interactions of mercury and copper with constitutive heterochromatin and euchromatin in vivo and in vitro. Biochemistry, 15, 1667-1676.
Butler. J.; Hoey, B.M. and Swallow, A.J (1985) Reactions of the semiquinone free radicals of anti-tumor agents with oxygen and iron complexes. FEBS Lett., 182, 95-98
Capel, i.D ; French, MR.; Millburn, P.; Smith, R.L. and Williams, R.T. (1972). The fate of ['"C] phenol in various species. Xenobiotica, 2, 25-34.
Chae, H.Z.: Kim, I.H.; Kim, K. and Rhee, S.G. (1993). Cloning sequencing, and mutation of thiol-specific antioxidant gene of Saccharomyces cerevisiae, J. Biol. Chem., 268, 16815-16821.
Chen, H. and Eastmond, DA. (1995). Topoisomerase inhibition by phenolic metabolites, a potential mechanism for benzene's clastogenic effects. Carcinogenesis, 16, 2301-2307.

Bibliography I24
Chen, J.J. and London, I.M. (1981). Hemin enhances the differentiation of mouse 3T3 cells to adipocytes. Cell. 26, 117-122.
Cheng, K.C.; Cahill, D.S., Kasai, H.; Nishimura, S. and Loeb, L.A. (1992). 8-Hydroxygua-nine, an abundant form of oxidative DNA damage causes G-T and A-C substitutions. J. Biol. Chem., 267, 166-172.
Cohen, H.S.; Freedman, M.L. and Goldstein, B.D. (1978). The problem of benzene in our environment. Am. J. Med. Sci., 275, 124-136.
Cornish, H. and Ryan, R. (1965). Metabolism of benzene in nonfasted, fasted and aryl hydroxylase inhibited rats. Toxicol. Appl. Pharmacol., 7, 767-771.
Crichton, R.R. (1973). Ferritin. Struc. Bonding Berlin, 17, 67-134.
Crichton, R.R. and Roman, F. (1978). A novel mechanism for ferritin iron oxidation and deposition. J. Mol. Catal., 4, 75- 82.
Crichton, R.R. and Ward, R.J. (1992). Iron metabolism-Nev*/ perspectives in view. Biochemistry, 31, 11255-11264.
Cronkite, E.P.; Bullis, J.; Inoue, T. and Drev\/, R.T. (1984). Benzene inhalation produces leukemia in mice. Toxicol. Appl. Pharmacol., 75, 358-361.
Cronkite, E.P. (1987). Chemical leukemogenesis benzene as a model. Seminar Haematol, 24, 2-1 ^.
Cronkite, E.P.; Drew, R.T.; Inone, T.; Hirayabishi, Y. and Bullis, J.E. (1989). Hemato-toxicity and carcinogenicity of inhaled benzene. Environ. HIth. Perspect., 82, 97-108.
Cumutte, J.D.; Whitten, D.M. and Babior, B.M. (1974). Defective superoxide production by granulocytes from patients with chronic granulomatous disease. New Engl. J. /Wed, 290,593-597.
De Matteis, F. and Sparks, R.G. (1973). Iron-dependent loss of liver cytochrome P-450 heme in vivo and in vitro. FEBS Letters, 29, 141-144.
Ding, X.; Li, Y.; Ding, Y. and Yang, H. (1983). Chromosome changes in patients with chronic benzene poisoning. Chinese Med. J., 96, 681-685.

Bibliography j25
Dosemeci, M.; Li, G,L,; Hayes, R.B.; Yin, S.N.; Linet, M.; Chow, W.H.; Wang, Y.Z.; Jiang, Z.L.; Dai, T.R.; Zhang, W.U.; Chao, X.J.; Ye, P.Z.; Kou, Q.R.; Fan, Y.H.; Zhang, X.C; Lin, X.F,; Meng, JR.; Zho, J.S.; Wacholder, S.; Kneller, R. and Blot, W.J. (1994). Cohort study among workers exposed to benzene in China Exposure assessment. Am. J. Ind. Med., 26, 401-411.
Drabkin, D.L. and Austin, J.H. (1932). Spectrophotometric studies : spectrophotometric constants for common haemoglobin derivatives in human, dog and rabbit blood. J. Biol. Chem., 98, 719-723.
Drummond, L.; Luck, R.; Afacan, A.S. and Wilson, H.K. (1988). Biological monitoring of workers exposed to benzene in the coke oven industry. Br. J. Ind. Med., 45, 256-261.
Dutton, G.J. (1966). In : Glucuronic acid; free and combined, edited by G.J. Dutton, New York and London : Academic Press.
Eastmond, DA.; Smith, M.T.; Ruzo, L.O. and Ross, D. (1986). Metabolic activation of phenol by human myeloperoxidase and horse-radish peroxidase. Mol. Pharmacol., 30, 674-679.
Eastmond, DA.; Smith, M.T. and Irons, R.D. (1987). An interaction of benzene metabolites reproduces the myelotoxicity observed with benzene exposure. Toxicol. Appl. Pharmacol., 91, 85-95.
Eastmond, D.A. (1993). Induction of micronuclei and aneuploidy by the quinone forming agents benzene and 0-phenylphenol. Toxicol. Lett., 67, 105-118.
Edling, J.E.; Britton, R.S.; Gnsham, M.B. and Bacon, BR. (1990). Increased unwinding of hepatic double stranded DNA in rats with chronic dietary iron overload. Gastroenterology, 98, A-585 (Abstract).
Erexson, G.L.; Wilmer, J.L. and Kligerman, AD. (1985). Sister chromatid exchange induction in human lymphocytes exposed to benzene and its metabolites in vitro. Cancer Res., 45, 2471- 2477.
Erexson, G.L.; Wilmer, J.L.; Steinhagen, W.H. and Kligerman, A.D. (1986). Induction of cytogenetic damage in rodents after short term inhalation of benzene. Environ. Mutagen., 8, 29-40.

Bibliography 126
Evans, A.E.; D'angio, G.J.; Propert, K.; Anderson, J. and Hann, H.W.L. (1987). Prognostic factors in neuroblastoma. Cancer, 59, 1853-1859.
Eveleth, W.T. (1990). In : Kline guide to the U.S. Chemical Industry. 5th ed., edited by N.J. Farifield, Kline and Company, Inc., 2-22-2-34.
Fabre, A (1947a). Metabolism of cyclic hydrocarbons. Conditions for oxidation and conjugation in the body as studied with organ homogenates and with hepatectomized rats. Bull. Soc. Chim. Biol.. 29, 318-321.
Fabre, A. (1947b). Changes in the conjugated sulfer-total sulfer ratio (of the unne) dunng chronic posioning with benzene and toluene. Bull. Soc. Chim. Biol., 29, 322-325.
Farris, G.M.; Everitt, J.I.; Irons, R. and Popp, J.A. (1993). Carcinogenicity of inhaled benzene in CBA mice. Fundam. Appl. Toxicol., 20, 503-507.
Feil, A. (1933). Professional benzene poisoning. Presse Med., 41, 129-130.
Fenech, M. and Morley, A.A. (1988). Measurement of micronuclei in lymphocytes. Mutat. Res., 147, 29-36.
Ferrali, M.; Signorini, C; Ciccoli, L. and Comporti, M. (1992). Iron release and membrane damage in erythrocytes exposed to oxidizing agents, phenylhydrazine, divicine and isouramil. Biochem. J., 285, 295-301.
Forni, A.M.; Pacutio, T.O. and Limonta, A. (1971). Chromosome studies in workers exposed to benzene or toluene or both Arch. Environ. HIth., 23, 373-378.
Freedman, M.L.; Wildman, J.M.; Rosman, J.; Eisen, J. and Greenblatt, DR. (1977). Benzene inhibition of in vitro rabbit reticulocyte heme synthesis at 6-ALA synthetase: Reversal of benzene toxicity by pyridoxine. Br J. Haematology, 35, 49-60.
Gad-EI-Karim, M.; Harper, B. and Legator, M. (1984). Modifications in the myelocla-stogenic effect of benzene in mice with toluene, phenobarbital, 3-methyl cholanthrene, Aroclor 1254 and SKF-525A. Mutat. Res.. 135, 225-243.
Gaido, K. and Wierda, D. (1984). In vitro effects of benzene metabolites on mouse bone marrow stromal cells. Toxicol. Appl. Pharmacol., 76, 45-55.

Bibliography j27
Glatt, H : Padykula, R.; Berchtold, G.A„ Ludewig, G.; Piatt, K,L., Klein, J. and Oesch, F. (1989). Multiple activation pathways of benzene leading to products with varying genotoxic charactenstics. Environ. HIth. Perspect., 82, 81-89.
Goldstein, B.D. (1977). Haematotoxicity in humans benzene toxicity a critical evaluation. J. Toxicol. Environ. HIth. Suppl., 2, 69-105.
Goldstein, B.D.; Snyder, C.A.; Laskin, S.; Bromberg, I.; Albert, R.E. and Nelson, N. (1982). Myelogenous leukemia in rodents inhaling benzene. Toxicol. Lett., 13, 169-173.
Goldstein, B.D. (1988). Benzene toxicity : Occupational medicine State of the Art Reviews, 3, 541-554.
Goldstein, B.D. (1989). Introduction : Occam's Razor is Dull. Environ. HIthi. Perspect., 82, 3-6
Goldstein, B.D. and Witz, G. (1992). Benzene. In : Environmental toxicants, human exposure and their health effects, edited by M. Lippman, New York : Van Nostrand Reinhold, 76-97.
Goon, D.; Cheng, X.; Ruth, J.A.; Petersen, D.R. and Ross, D. (1992). Metabolism of trans, trans-muconaldehyde by aldehyde and alcohol dehydrogenases: Identification of a novel metabolite. Toxicol. Appl. Pharmacol., 114, 147-155.
Graham, D.G.; Tiffany, S.M. and Vogel, F.S. (1978). The toxicity of melanin precursors. J. Invest. Dermatol, 70. 113-116.
Granick, S (1942). Physical and chemical properties of horse spleen ferritin. J. Biol. Chem., 146,451-461
Green, J.D.; Snyder, C.A.; LoBue, J.; Goldstein, B.D. and Albert, R.E. (1981a). Acute and chronic dose/response effect of benzene inhalation on the peripheral blood, bone marrow and spleen cells of CD-I male mice. Toxicol. Appl. Phanmacol., 59, 204-214.
Green, J D.: Snyder, C.A , LoBue, J., Goldstein, B.D. and Albert, R.E. (1981b). Acute and chronic dose/response effects of inhaled benzene on multipotential hematopoietic stem (GM-CFU-C) cells in CD-I mice. Toxicol. Appl. Pharmacol., 58, 492-503.

Bibliography 128
Greenlee, W.F.; Gross, E.A. and Irons, R.D. (1981a). Relationship between benzene toxicity and the disposition of ''"C- labelled benzene metabolites in the rats. Chem.-Biol. Interact, 33, 285-299.
Greenlee, W.F.; Sun, J.D. and Bus, J.S. (1981b). A proposed mechanism of benzene toxicity : Formation of reactive intermediates from polyphenol metabolites. Toxicol. Appl. Pharmacol., 59, 187-195.
Grotz, V.L.i Ji, S.; Kline, S.A.; Goldstein, B.D. and W\\z, G. (1994). Metabolism of tjenzene and trans, trans-muconaldehyde in the isolated F)erfused rat liver. Toxicol. Lett. 70, 281-290.
Guttendge, J.M.C. (1981). Thiobarbituric acid-reactivity following iron-dependent free-radical damage to amino acids and carbohydrates. FEBS Lett.. 128, 343-346.
Gutteridge, J.M.C; Rowley, D.A. and Halliwell, B. (1982). Superoxide-dependent formation of hydroxyl radicals and lipid peroxidation in the presence of Iron salts. Detection of 'catalytic' iron and anti-oxidant activity in extracellular fluids. Biochem. J., 206,605-609.
Gutteridge, J.M.C; Rowley, DA.; Griffiths, E. and Halliwell, B. (1985). Low-molecular weight iron complexes and oxygen radical reactions in idiopathic hemochromatosis. Clin. Sci., 68,463-467.
Gutteridge, J.M.C (1987). Bleomycin-detectable iron in knee joint synovial fluid from arthritic patients and its relationship to the extracellular antioxidant activities of ceruloplasmin, transferrin and lactoferrin. Biochem. J., 245, 415-421,
Guy, R.L.; Dimitriadis, E.; Hu, P.; Cooper, K.R. and Snyder, R. (1990). Interactive inhibition of erythroid ^ Fe utilization by benzene metabolites in female mice. Chem.-Biol. Interact., 74, 55-62.
Guy, R.L.; Hu, P.; Witz, G.; Goldstein, B.D. and Snyder, R. (1991). Depression of iron uptake into erythrocytes in mice by treatment with combined benzene metabolites p-benzoquinone, muconaldehyde and hydroquinone. J. Appl. Toxicol., 11, 443-446.
Halliwell, B. and Aruoma. 0.1. (1991). DNA damage by oxygen derived species ; Its mechanism and measurement in mammalian systems. FEBS Letters, 281, 9-19.
Halliwell, B.; Gutteridge, J.M.C and Cross, CE. (1992). Free radicals, antioxidants, and human disease : Where are we now ? J. Lab. Clin. Med., 119, 598-620.

Bibliograplty 129
Hammond, J.W. and Hemian, E.R. (1960). Industrial hygiene features of a petrochemical benzene plant design and operation. Am. Ind. Hyg. Assoc. J., 21, 173-177.
Hanna, P.M. and Mason, R.P. (1992). Direct evidence for inhibition of free radical formation from Cu(l) and hydrogen peroxide by glutathione and other potential ligands using the ERR spin-trapping tecnique. Arch. Biochem. Biophys., 295, 205-213.
Harrison, P.M. (1977). Femtin : an iron storage molecule. Sernin. Haematol., 14, 55-70.
Hattemer-Frey, H.A.; Travis, C.C. and Land. M.L. (1990). Benzene : Environmental partitioning and human exposure. Environ. Res., 53, 221-232.
Healing, G.; Gower, J.; Fuller, B.J. and Green, C.J. (1990). Intracellular iron redistribution; An important determinant of reperfusion damage to rabbit kidneys. Biochem. Pharmacol., 39, 1239-1245.
Hedii, C.C; Rao, N.R.; Reuhl, K.R.; Witmer, CM. and Snyder, R. (1996). Effects of benzene metabolite treatment on granulocytic differentiation and DNA adduct formation in HL-60 cells. Arch. Toxicol., 70, 135-144.
Henderson, R.F.; Sabourin, P.J.; Bechtold. W.E.; Griffith, W.C; Medinsky, M.A.; Birnbaum, L.S. and Lucier, G. (1989). The effect of dose, dose rate, route of administration, and species on tissue and blood levels of benzene metabolites. Environ. HIth. Perspect., 82, 9-17.
Hoare, R.J.; Harrison, P.M. and Hoy, T.G. (1975). Stnjcture of horse-spleen apoferritin at 6A° resolution. Nature, 255, 653- 654.
Hu, S.C, Fuerst, P. and Hamer, D.H. (1990). The DNA and copper binding functions of ACE1 are interdigitated within a single domain. New Biol., 2, 544-555.
Huff, J.E. (1983). National Toxicology Program. Technical Report on the Toxicology and Carcinogenesis Studies of Benzene (CAS No. 71-43-2) in F344 Rats and B6C3F1 Mice (Gavage Studies), Research Triangle Park, NC : National Institute of Environmental Helath Sciences.
Huff, J.E ; Haseman, J.K. and DeMarini, DM. (1989). Multiple- site carcinogenicity of benzene in Fischer 344 rats and B6C3F1 mice. Environ. HIth. Perspect 82 125-163.

Bibliography 130
Inoue, O.; Seiji, K.; Watanabe, T.; Kasahara, M.; Nakatsuka, H.; Yin, S.G.; Li, G.L., Cai, S.X.; Jin, C. and Ikeda, M. (1988a). Mutual metabolic suppression between benzene and toluene in man. Int. Arch. Occup. Environ. HIth., 60, 15-20.
Inoue, O.; Seiji, K.; Kasahara, M.; Nakatsuka, H.; Watanabe, T.; Yin, S.G.; Li, G.L.; Cai, S.X.; Jin, C. and Ikeda, M. (1988b). Detennination of catechol and quinol in the urine of workers exposed to benzene. Br. J. Ind. Med., 45, 487-492.
Inoue, O.; Seiji, K.; Nakatsuka, H.; Watanabe, T.; Yin, S.G.; Li, G.L.; Cai, S.X., Jin, C and Ikeda, M. (1989a). Urinary t,t-muconic add as an indicator of exposure to benzene. Br J. Ind Med., 46, 122-127.
Inoue, O.; Seiji, K.; Nakatsuka, H.; Watanabe, T.; Yin, S.G.; Li, G.L.; Cai, S.X.; Jin, C. and Ikeda, M. (1989b). Excretion of 1,2,4-benzenetriol in the urine of workers exposed to benzene. Br J. Ind Med., 46, 559-565.
Inoue, 0.; Seiji, K. and Ikeda, M. (1989c). Pathway for formation of catechol and 1,2,4-benzenetriol in rabbit. Bull. Environ. Contam. Toxicol., 43, 220-224.
Irons, B.; Heck, H.; Moore, B. and Muirhead, K. (1979). Effects of short-term benzene administration of bone man ow cell cycle kinetics in the rat. Toxicol. Appl.Pharmacol., 51, 399-409.
Irons, R.D. and Neptun, D.A. (1980). Effects of the principal hydroxy-metabolites of benzene on microtubule polymerization. Arch. Toxicol., 45, 297-305.
Irons, R.D.; Neptun, D.A. and Pfeifer, R.W. (1981). Inhibition of lymphocyte transformation and microtubule assembly quinone metabolites of benzene : Evidence for a common mechanism. J. Reticuloendothel. Soc. 30, 359-372.
Irons, R.D. (1985). Quinones as toxic metabolites of benzene. J. Toxicol. Environ. HIth., 16, 673-678.
Irons, R.D.(1988). Studies on the mechanisms of chemically induced leukemia/lymphoma. Chem. Ind. Inst. Activities, 1, 2-8.
Ito, K.; Yamamoto, K. and .Kawanishi, S. (1992). Manganese- mediated oxidative damage of cellular and isolated DNA by isoniazid and related hydrazines: Non-Fenton-Type hydroxyl radical formation. Biochemistry, 31, 11606-11613.

Bibliography 131
Jacobs, A. and Worwood, M. (1975). Feritin : clinical aspects. Excerpta Med. Int. Congr. Ser.. 90-95.
Jacobs, A. (1977). Low molecuair weight intracellular iron transport compounds. Blood, 50, 433-439.
Jongeneelen, F.J.; Dirven, H.A.A.M., Leijdekkers, CM.; Henderson, P.T.; Brouns, R.M.E. and Halm, K, (1987). S-phenyl-N- acetylcysteine in urine of rats and workers after expsoureto benzene. J. Anal. Toxicol., 11, 100-104.
Jowa, L,; Winkle, S.; Kalf, G.; Witz, G. and Snyder, R. (1986). In : Biochemical Reactive Intermediates-Ill. Molecular and Cellular Mechanisms of Action in Animal models and Human Disease, edited by J.J. Kocsis, D.J. Jollow, CM. Witmer, J.O. Nelson, and R. Snyder, Plenum Press, New York, 825-832.
Kadir, F.H.A.; Al-Massad, F.K. and Moore, G.R. (1992). Haem binding to horse spleen ferritin and its effect on the rate of iron release. Biochem. J., 282, 867-870.
Kalf, G.F.; Rushmore, T.R. and Snyder. R. (1982). Benzene inhibits RNA synthesis in mitochondria from liver and bone man-ow. Chem.-Biol. Interact., 42, 353-370.
Kalf, G.F. (1987). Recent advances in the metatx)lism and toxicity of benzene. CRC Cnt. Rev. Toxicol., 18, 141-154.
Karacic, V.; Skender, L. and Prpic-Majic, D. (1987). Occupational exposure to benzene in the shoe industry. Am. J. Ind. Med., 12, 531-536.
Karam, L.R. and Simic, M.G. (1989). Mechanisms of free radical chemistry and biochemistry of benzene. Environ. HIth. Perspect., 82, 37-41.
Kariya, K.; Lee, E,; Hirouchi, M.; Hosokawa, M. and Sayo, H. (1987). Purification and some properties of peroxidases of rat bone man-ow. Biochim. Biophys. Acta. ,911, 95-101.
Kawanishi, S., Inoue, S. and Kawanishi, M, (1989). Human DNA damage induced by 1,2.4-benzenetnol, a benzene metabolite. Cancer Res., 49, 164-168.
Kawanishi, S. and Yamamolo, K. (1991). Mechanism of site- specific DNA damage induced by methylhydrazines in the presence of copper (II) or manganese (III). Biochemistry, 30, 3069-3075.

Bibliography 132
Khan, H. and Muzyka, V. (1973). The chronic effect of benzene on porphyrin metabdism. Work-Environ. Hith., 10, 140-143.
Kimura, E.T.; Elbert, D.M. and Dodge, P.W. (1971). Acute toxicity and limits of solvents residue for sixteen solvents. Toxicol. Appl. Pharmacol., 19, 699-704.
Kipen, H.M,; Cody, R.P. and Goldstein, B.D. (1989). Use of longitudinal analysis of peripheral blood counts to validate historical reconstructions of benzene exposure. Environ. Hith. Perspect., 82, 199-206.
Kolachana, P.; Subrahmanyam, V.V.; Meyer, K.B.; Zhang, L. and Smith, M.T. (1993). Benzene and its phenolic metabolites produce oxidative DNA damage in HL-60 cells in vitro and in the bone marrovj in vivo. Cancer Res., 53, 1023-1026.
Krsek-Staples, J.A. and Webster, R.O. (1993). Cemloplasmin inhibits carbonyl formation in endogenous cell proteins. Free Rad. Biol. Med., 14, 115-125.
Lange, A.; Smolik, R.; Zatonski, W. and Szymanska, J. (1973a). Serum immunoglobulin levels in workers exposed to benzene toluene and xylene. Int. Arch. Arbeitsmed., 31, 37-44.
Lange, A.; Smolik, R.; Zatonski, W. and Glazman, H. (1973b). Leukocyte agglutinins in vi/orkers exposed to benzene, toluene and xylene. Int. Arch. Arteitsmed., 31, 45-50.
Latriano, L., Zaccaria, A., Goldstein, B.D. and Witz, G. (1985) Muconaldehyde formation from '"C-benzene in a hydroxyl radical generating system. Free Rad. Biol. Med., 1, 363-371.
Latnano, L ; Goldstein, B.D. and Witz, G. (1986). Formation of muconaldehyde, an open nng metabolite of benzene, mouse liver microsomes. An additional pathway for toxic metabolites. Proc. Natl Acad. Sci. USA, 83, 8356-8360
Laughton, M.J.; Evans, P.J.; Moroney, M.A.; Hoult, J.R.S. and Halliwell, B. (1991). Inhibition of mammalian 5-lipoxygenase and cyclo-oxygenase by flavonoids and phenolic dietary additives Relationship to antioxidant activity and to iron-reducing ability, Biochem. Pharmacol, 42, 1673-1681
Le Beau, M.M. and Larson, R.A. (1991). In : Hematology Basic Principles and Practice, edited by R. Hoffman, E.J. Benz, S.J. Shattil, B. Furie and H.J. Cohen, Churchil, Levingston, New York, 638.

Bibliography 133
Le Noir, C. (1897). Sur un de purpura attribue a Intoxication par le benzene. Bull. Mem. Soc. Med. Hop. Paris. 14, 1251.
Lee, E.W.; Kocsis, J.J. and Snyder, R. (1974). Acute effect of l enzene on *®Fe incorporation into circulating erythrocytes. Toxicol. Appl. Pharmacol., 27, 431-436.
Lee, E.W.: Kocsis, J.J. and Snyder, R. (1981). The use of ferrokinetics in the study of experimental anemia. Environ. HIth. Perspect., 39, 29-37.
Lee, S.F. and Lin, J.K. (1994). Generation of hydrogen peroxide, superoxide anion and the hydroxy! free radical from polyphenols and active benzene metabolites : their possible role in mutagenesis. J. Biomed. Sci., 1, 125-130.
Lee, S.F.; Liang, Y.C. and Lin, J.K. (1995). Inhibition of 1,2,4-benzenetriol-generated active oxygen species and induction of phase II enzymes by green tea polyphenols. Chem.-Biol. Interact., 98, 283-301.
Legathe, A.; Hoener, B. and Tozer, T.N. (1994). Pharmacokinetic interaction between benzene metabolites, phenol and hydroquinone, in B6C3F1 mice. Toxicol. Appl. Pharmacol., 124, 131-138.
Levay, G.; Pongracz, K. and Bodell, W.J. (1991). Detection of DNA adducts in HL-60 cells treated with hydroquinone and p- benzoquinone by ^^P-postlabelling. Carcinogenesis, 12, 1181- 1186.
Levay, G. and Bodell, W.J. (1992). Potentiation of DNA adduct formation in HL-60 cells by combination of benzene metabolites. Proc. Natl. Acad. Sci. USA, 89, 7105-7109.
Levay, G.; Ross, D. and Bodell, W.J. (1993). Peroxidase activation of hydroquinone results in the formation of DNA adducts in HL-60 cells, mouse bone marrow macrophages and human bone marrow. Carcinogenesis, 14, 2329-2334.
Levay, G.; Pathak, D.N. and Bodell, W.J. (1996). Detection of DNA adducts in the white blood cells of B6C3F1 mice treated with benzene. Carcinogenesis, 17, 151-153.
Lewis, J G.; Odom, B. and Adams, D.O. (1988a). Toxic effects of benzene and benzene metabolites on mononuclear phagocytes. Toxicol. Appl. Pharmacol., 92, 246-254.
Lewis, J.G.; Stewart, W. and Adams, D.O. (1988b). Role of oxygen radicals in induction of DNA damage by metabolites of benzene. Cancer Res., 48, 4762-4765.

BibliograpJiy 134
Li, C.J.; Averboukh, L. and Pardee, A.B. (1993). B-Lapachone, a novel DNA topo-isomerase I inhibitor with a mode of action different from camptothecin. J. Biol. Chem., 268, 22463-22468.
Li, Y.; Togashi, Y.; Sato, S.; Emoto, T.; Kang, J.H.; Takeichi, N.; Kobayashi, H.; Kojima, Y.; Une, Y. and Uchino, J. (1991). Abnormal coper accumulation in non-cancerous tissues of LEC rat developing hereditary hepatitis and spontaneous hepatoma. Jpn. J. Cancer Res., 82, 490-492.
LI, Y. and Trush, MA. (1993a). DNA damage resulting from the oxidation of hydro-quinone by copper: role of a Cu(ll)/Cu(l) redox cycle and reactive oxygen generation. Carcinogenesis, 14, 1303-1311.
Li, Y. and Trush, M.A. (1993b). Oxidation of hydroquinone by coper: Chemical mechanism and biological effects. Arch. Biochem. Biophys., 300, 346-355.
Li, Y.; Kuppusamy, P.; Zweier, J.L. and Trush, MA. (1995). ESR evidence for the generation of reactive oxygen species from the copper-mediated oxidation of the benzene metabolite, hydroquinone : role in DNA damage. Chem. -Biol. Interact., 94, 101-120.
Linder, M.C. (1991). Nutritional Biochemistry and Metabolism. 2nded., Elsevier Science, New York, 231-235.
Lo, S.C; Aft, R. and Mueller, G.C. (1981). Role of non- hemoglobin heme accumulation in the terminal differentiation of Friend erythroleukemia cells. Cancer Res., 41, 864-870.
Loden, M. (1986). The in vitro permeability of human skin to benzene, ethylene glycol, formaldehyde and n-hexane. Acta Pharmacol. Toxicol., 58, 382-389.
Longacre, S.L.; Kocsis, J.J. and Snyder, R. (1981). Infleunce of strain differences in mice on the metabolism and toxicity of benzene Toxicol. Appl. Pharmacol., 60, 398-409
Lovstad, R.A. (1990). Interactionof iron-bleomycin with catecholamines. Int. J. Biochem., 22, 641-644.
Lown, J.W. and Sim, S.K. (1977). The mechanism of the bleomycin- induced cleavage of DNA. Biochem. Biophys. Res. Commun., 77, 1150-1157.

Bibliography , , c
Lowry, L.K. (1986). Biological exposure index as a complement to the TLV. J. Occup. Med., 28, 578-582.
Lowry, OH.; Rosebrough, N.J.; Farr, A.L. and Randall, R.J. (1951). Protein measurement with the Folin Phenol reagent. J. Biol. Chem., 193, 265-275.
Luke, C.A.; Tice, R.R. and Drew, R.T. (1988). The effect of exposure regiment and duration of benzene induced bone marrow damage in mice I sex comparision in DBA/mice. Mutat. Res., 203, 251-272.
Lunte, S.M. and Kissinger, P.T. (1983). Detection and identification of sulphahydrl conjugates of p-benzoquinone in microsomal incubations of benzene and phenol. Chem.-Biol. Interact., 47, 195-212.
Lutz, W.K. and Schlatter, C.H. (1977). Mechanism of the carcinogenic action of tjenzene: Irreversible binding to rat liver DNA. Chem.-Biol. Interact., 18, 241-245.
Mager, D. and Bernstein, A. (1979). The role of heme in the regulation of the late program of friend cell erythroid differentiation. J. Cell Physiol., 100, 467-479.
Mahmutoglu, I. and Kappus, H. (1987). Redox cycling of bleomycin Fe (III) by an NADH-dependent enzyme, and DNA damage in isolated rat liver nuclei. Biochem. Pharmacol, 36, 3677-3681.
Maltoni, C. and Scarnato, C. (1979). First experimental demonstration of the carcinogenic effects of benzene. Long term bioassays on Sprague-Dawley rats by oral administration. Med. Lav., 70, 352-357.
Maltoni, C; Conti, B.; Cotti, G. and Belpoggi, F. (1983). Benzene : A multipotential carcinogen. Results of long term bioassays performed at the Bologna Institute of Oncology. Am. J. Ind. Med,A, 589-630.
Maltoni, C; Conti, B.; Cotti, G. and Belpoggi, F. (1985). Experimental studies on benzene carcinogenicity at the Bologna Institute of Oncology . Current results and ongoing research. Am. J. Ind. Med., 7, 415-446.
Maltoni, C, Ciliberti, A., Cotti, G.; Conti, B. and Belpoggi, F. (1989). Benzene, an experimental multipotential carcinogen . Results of the long-term bioassays performed at the Bologna Institute of Oncology. Environ. HIth. Perspect., 82, 109-124.

Bibliography ^^^
Masarwa, M.; Cohen, H.; Meyerstein, D.; Hickman, D.L.; Bakac, A. and Espenson, J.H. (1988). Reactions of low-valent transition- metal complexes. Are they "Fenton-like" or not? The case of Cu*aq and Cr^^aq. J. Am. Chem. Soc., 110, 4293-4297.
Mazur, A. and Carleton, A. (1965). Hepatic xanthine oxidase and ferritin iron in the developing rat. Blood, 26, 317-322.
Meister, A. (1988). Glutathione metabolism and its selective modification. J. Biol. Chem.,
263, 17205-17208.
Mello-Filho, A.C. and Meneghini, R. (1991). Iron is the intracellular metal involved in the production of DNA damage by oxygen radicals. Mutat Res., 251, 109-113.
Meneghini, R. and Martins, E.L (1993). In . DNA and free radicals, edited by B. Halliwell and 0.1 Aruoma, Ellis Horwood, 83-93.
Mertens J.J.W.M ; Gibson, N.W.; Lau, S.S. and Monks, T.J. (1995). Reactive oxygen species and DNA damage in 2-bromo- (glutathion-S-yl) hydroquinone-mediated cytotoxicity Arch. Biochem. Biophys., 320, 51-58.
Monteiro, H.P. and Winterbourn, C.C. (1989a). Release of iron from ferritin by divicine, isouramil, acid-hydrolyzed vicine, and dialuric acid and initiation of lipid peroxidation. Arch. Biochem. Biophys., 271, 536-545.
Monteiro, H.P. and Winterbourn, C.C. (1989b). 6-hydroxydopamine release iron from ferritin and promotes ferritin-dependent lipid peroxidation. Biochem. Pharmacol., 38, 4177-4182.
Moore, M.A.S. (1978). In : Stem cell and tissue homeostasis, edited by B.I. Lord, C.S. Rotten, and R.J. Cole, Cambridge, Univ. Press, 187-202.
Morel, I.; Lescoat, G.; Cogrel, P.; Sergent, O.; Pasdeloup, N.: Brissot, P.; Cillard, P. and Cillard, J. (1993). Antioxidant and iron-chelating activities of the fiavonoids catechin, quercetin and diosmetin on iron-loaded rat hepatocyte cultures. Biochem. Pharmacol., 45 , 13.
Monmoto, K.; Wolff, S. and Koisumi, A. (1983). Induction of sister-chromatid exchanges in human lymphocytes by microsomal activation of benzene metabolites. Mutat. Res., 119, 355-360.

Bibliography 137
Mulligan, M.; Althaus, B. and Linder, M.C. (1986). Non-ferritin, non-heme iron pools in rat tissues. Int. J. Biochem., 18, 791- 798.
Nagaraja, K.V. and Shaw, P.D. (1982). Inhibition of wheat germ RNIA polymerase II by J2-6 dibromo benzoquinone and related compounds from Aplysina fistularis. Arch. Biochem. Biophys.. 215, 544-550.
Nakajima, T,; Okino, T. and Sato, A. (1987). Kinetic studies on benzene metabolism in rat liver-possible presence of three forms of benzene metabolizing enzymes in the liver. Biochem. Pharmacol., 36, 2799-2804.
Nakajima, T.; Wang, R.; Elovaara, E.; Gelboin, H. and Vainio, H. (1993). Cytochrome P-450 related differences betv\/een rats and mice in the metabolism of benzene, toluene and trichloroethylene in liver microsomes. Biochem. Phamiacol., 45, 1079-1085.
Nerland, D.E. and Pierce, M.M. (1990). Identification of N- acetyl-S- (2,5- dihydroxy-phenyl)-L-cysteine as a urinary metabolite of benzene, phenol and hydroquinone. Drug Metab. Disp., 18, 958-962.
Neun, D.J.; Penn, A. and Snyder, C.A. (1992). Evidence for strain-specific differences in benzene toxicity as a function of host target cell susceptibility. Arch. Toxicol., 66, 11-17.
Nielsen, P.; Dullmann, J.; Wulfhekel, U. and Heinrich, H.C. (1993). Non-transferrin-bound-iron in serum and lov\/-molecular- v\/eight-iron in the liver of dietary iron-loaded rats, Int J. Biochem., 25, 223-232.
Norpoth, K.; Stucker, W.; Krev\/et, E. and Mulier, G. (1988). Biomonitoring of benzene exposure by trace analysis of phenylguanine. Int. Arch. Occup. Environ. HIth., 60, 163-168.
O'Connell, M.J.; Ward, R.J.. Baum, H. and Peters, T.J. (1989). Iron release from haemosiderin and ferritin by therapeutic and physiological chelators. Biochem. J., 260, 903-907.
Oehme, F. (1969). Comparative study of the biotransformation and excreting phenol. Ph.D. Thesis University of Missouri University, Microfilmis, Ann. Arbor, M.I.
OSHA (1987). Occupational exposure to benzene. Final rule U.S. Department of Labor. Occupational Safety and Health Administration Federal Register, 52, 34460-34578.

BibliograpJiy ^^^
Oteiza, P.I.; Kleinman, C.G.; Demasi, M. and Bechara, E.J.H. (1995). 5-Aminolevulinic acid induces iron release from ferritin. Arch. Biochem. Biophys., 316, 607-611.
Pandya, K.P.; Shanker, R.; Gupta, A.; Khan, W.A. and Ray, P.K. (1986). Modulation of benzene toxicity by an interferon inducer (6-MFA). Toxicology, 39, 291-305.
Pandya, K.P.; Khan, S.; Umashankar; Krishnamurthy, R. and Ray, P.K. (1989). Modulation of benzene toxicity by polyinosinic-polycytidilic acid, an interferon inducer. Biochlm. Biophys. Acta., 992, 23-29.
Pandya. K.P.; Rao, G.S.; Khan, S. and Krishnamurthy, R. (1990). Accumulation of low molecular weight (bleomycin detectable)iron in bone marrow cells of rats after benzene exposure. Arch. 7ox/'co/., 64, 339-342.
Parke, D.V. and Williams, R.T. (1953). Studies in detoxication. The metabolism of benzene containing [' C,] benzene. Biochem. J., 54, 231-238.
Pannentier, R. and Dustin, P. Jr. (1953). On the mechanisms of the mitotic abnormalities induced by hydroquinone in animal tissues. Rev Beige. Pathol. Med. Exp., 23, 20-30.
Pathak, D.N.; Levay, G. and Bodell, W.J. (1995). DNA adduct formation in the bone marrow of B6C3F1 mice treated with benzene. Carcinogenesis, 16, 1803-1808.
Pellak-Walker, P. and Blumer, J.L. (1986). DNA damage in L5178YS cells following exposure to benzene metabolites Mol. Pharmacol.. 30, 42-47
Peters, T.; Giovanniello, T J.; Apt, L. and Ross. J.F. (1956). A simple improved method for the determination of serum iron. J. Lab. Clin. Med., 48, 280-288.
Pfeifer, R.W and Irons, R.D. (1983). Alteration of lymphocyte function by quinones through sulphahydryl-dependent disnjption of microtubule assembly. Int. J. Immuno-pharmacol., 5, 463-470.
Pommier, Y and Bertrand, R. (1993) The causes and consequences of chromosomal aberrations, edited by I.R. Kirsch, CRC press, Boca Raton, FL., 277-309.
Pongracz, K. and Bodell, W.J. (1996). Synthesis of N^4- hydroxyphenyl)-2'-deoxygua-nosine 3'-phosphate: Comparison by ^ P- post labeling with the DNA adduct formed in HL-60 cells treated with hydroquinone. Chem. Res. Toxicol.. 9, 593-598.

Bibliography ^^^
Porteous, J.W. and Williams, R.T. (1949). The metabolism of benzene. The isoaltion of phenol, catechol, quinoi and hydroxyquinol from the ethereal sulphate fraction of the urine of rabbit receiving benzene orally. Biochem. J., 44, 56-61.
Post, G.B.; Snyder, R. and Kalf, G.F. (1985). Inhibition of RNA synthesis and interieukin-2 production in lymphocytes in vitro by benzene and its metabolites, hydroquinone and p-benzoquinone. Toxicol. Lett., 29, 161-167.
Post, G.B.; Snyder, R. and Kalf, G.F. (1986). Metatx)lism of benzene in macrophages in vitro and the inhibition of RNA synthesis by benzene metabolites. Cell Biol. Toxicol, 2,231-246.
Potaznic, D.i Groshen, S.; Miller, D.; Bagin, R.; Bhalla, R.; Schwartz, M. and de Sousa, M. (1987). Association of serum iron, serum transferrin saturation and serum ferritin with survival in acute lymphocytic leukemia. Am. J. Pediatr Hematol./Oncol.. 9, 350-355.
Prutz, W.A.; Butler, J. and Land, E.J. (1990). Interaction of copper (I) with nucleic acids. Int. J. Radiat. Biol., 58, 215- 234.
Prutz, W.A. (1993). Cu(ll)-induced oxidation of catechols, ascorbate and o-phenyl-enediamine is promoted by DNA. Z Naturforsch., 48, 872-878.
Quinlan, G.J. and Gutteridge, J.M.C. (1987). Oxygen radical damage to DNA by rifamycin SV and copper ions. Biochem. Pharmacol., 36, 3629-3633.
Rahman, A.; Shahabuddin; Hadi, S.M.; Parish, J.H. and Ainley, K. (1989). Strand scission in DNA induced by quercetin and Cu(ll): role of Cu(l) and oxygen free radicals. Carcinogenesis, 10, 1833-1839.
Rao, D.N.R.; Takahashi, N. and Mason, R. (1988). Characterization of a glutathione conjugate of the 1,4- benzoquinone-free radical formed in rat hepatocytes. J. Biol. Chem., 263, 17981-17986.
Rao, G.S. and Pandya, K.P. (1978). Toxicity of petroleum products ; Effects on alkaline phosphatase and lipid peroxidation. Environ. Res., 16, 174-178.
Rao, G.S. and Pandya, K.P. (1980). Hepatic metabolism of heme in rat after exposure to benzene, gasoline and kerosene Arch. Toxicol.. 46. 313-317

Bibliography 140
Rao, G.S. and Pandya, K.P. (1989). Release of 2-thiobarbituric acid reactive products from glutamate or deoxyribonucleic acid by 1,2,4-benzenetriol or hydroquinone in the presence of copper ions. Toxicology, 59, 59-65.
Rao, G.S.; Goel, S.K. and Pandya, K.P. (1990). Accumulation of bleomycin sensitive iron and spontaneous oxygen radical generation by bone marrow cells of rats exposed to benzene. Clin. Chem. Enzym. Comms.. 3, 313-318.
Rao, G.S. (1991). Hematin catalyzed autooxidation of hydroquinone or 1,2,4-benzenetriol. Chem.-Biol. Interact., 80, 339-347.
Reif, D.W. and Simmons, R.D. (1990). Nitric oxide mediates iron release from ferritin. Arch. Biochem. Biophys.. 283, 537-541.
Reif, D.W. (1992). Ferritin as a source of iron for oxidative damage. Free Rad. Biol. Med., 12, 417-427.
Rickert, D.E.; Baker, T.S.; Bus, J.S.; Barrow, C.S. and Irons, R.D. (1979). Benzene disposition in the rat after exposure by inhalation. Toxicol. Appl. Pharmacol.. 49, 417-423.
Rickert, D E.; Baker, T.S. and Chism, J.P. (1981) Analytical approaches to the study of the disposition of myelotoxic agents Environ HIth Perspect., 39, 5-10
Rosenthal, G.J and Snyder, C.A (1986). Altered T-celi responses in C57BU6J mice following sub-chronic benzene inhalation [Abstract]. Toxicologist, 6. 68.
Rothman, N.; Haas, R.; Hayes, R.B., Li, G.L.; Wiemels, J., Campieman, S.; Quintana, P.J.E., Xi, L.J.; Dosemeci, M ; Titenko- Holland, N.; Meyer, KB.; Lu, W.; Zhang, LP.. Bechtold, W.; Wang, Y.Z.; Kolachana, P.; Yin, S.N.; Blot, W. and Smith M.T. (1995). Benzene induces gene-duplicating but not gene- inactivating mutations at the glycophonn A locus in exposed humans. Proc. Natl. Acad. Sci. USA, 92, 4069-4073.
Rothman, N.; Li, G.L.; Dosemeci, M.; Bechtold, W.E.; Marti, G.E.; Wang, Y.Z.; Linet, M.; Xi, L.Q.; Lu, W.; Smith, M.T.: Tikeno- Holland, N.; Zhang, L.P.; Blot, W.; Yin, S.N. and Hayes, R.B. (1996). Hematotoxicity among Chinese workers heavily exposed to benzene. Am. J. Ind. Med., 29, 236-246.
Rozen, M.G.; Snyder, C.A. and Albert, R.E. (1984). Depression in B and T-lymphocyte mitogen induced blastogenesis in mice exposed to low concentrations of benzene. Toxicol. Lett., 20, 343-349.

Bibliography ^ 1
Rozen, M.G. and Snyder, C.A. (1985). Protracted exposure of C57BLy6 mice to 300 ppm benzene depresses B- and T-lymphocyte numt>ers and mitogen responses; Evidence for thymic and bone marrow proliferation in response to the exposures. 7ox/co/ogy, 37, 13-26.
Rushmore, T.; Snyder, R. and Kalf, G.F. (1984). Covalent binding of benzene and its metabolites to DNA in rabbit bone marrow mitochondria in vitro. Chem.-Biol. Interact, 49, 133-154.
Sabourin, P.J.; Sun, J.D.; Mac Gregor, J.T.; Wehr, CM.; Bimbaum, L.S.; Lucier, G and Henderson, R.F. (1990). Effect of repeated benzene inhalation exposures of benzene metabolism, binding to haemoglobin and induction of micronuclei. Toxicol. Appl. Pharmacol.. 103, 452-462.
Sabourin, P.J.; Muggenburg, B.A.; Couch, R.C.; Letter, D.; Lucier, G.; Bimbaum, L.S. and Henderson, R.F. (1992). Metabolism of ['*C] benzene by cynomolgus monkeys and chimpanzees. Toxicol. Appl. Pharmacol., 114. 277-284.
Sasiadek, M.; Jagielski, J. and Smolik, R. (1989). Localization of breakpoints in the karyotype of workers professionally exposed to benzene. Mutat. Res., 224, 235-240.
Saucier, M.A.; Wang, X.; Re, R.N.; Brown, J. and Bryan, S E. (1991). Effects of ionic strength on endogenous nuclease activity in chelated and non-chelated chromatin. J. Inorg. Biochem., 41, 117-124.
Schrenk, H.H.; Yant, W.P.; Pearce, S.J.; Patty, F.A. and Sayers, R.R. (1941). Absorption, distribution and elimination of benzene by body tissues and fluids of dogs exposed to benzene vapor. J. Ind. Hyg. Toxicol., 23, 20-34.
Seaton, M.J.; Schlosser, P.M.; Bond, J.A. and Medinsky, MA. (1994). benzene metabolism by human liver microsomes in relation to cytochrome P-450 2E1 activity. Carcinogenesis, 15, 1799-1806.
Sedlak, J. and Lindsay, R.H. (1968). Estimation of total, protein bound and non protein -SH groups in tissue with Ellman's reagent. Anal. Biochem., 25, 192-205.
Selling, L. (1910). Preliminary report of some cases of purpura haemorrhagica due to benzol poisoning. Bull. Johs. Hopkins Hosp., 21, 33-37.
Siddiqui, S.M.; Rao, G.S. and Pandya, K.P. (1988). Depletion of liver regulatory heme in benzene exposed rats. Toxicology, 48, 245-251.

Bibliography 1 2
Singh, V.; Ahmad, S. and Rao, G.S. (1994). Prooxidant and antioxidant properties of Iron-hydroquinone and iron-1,2,4- benzenetriol complex. Implications for benzene toxicity. Toxicology, 89, 25-33.
Siou, G.; Canan, L. and El Haitem, M. (1981). Evaluation of the clastogenic action of benzene by oral administration with two cytogenic techniques in mouse Chinese hamster. Mutat. Res., 90, 273-278.
Smith, f^.T.; Yager, J.W.; Steinmetz, K.L. and Eastmond, D.A. (1989). Peroxidase-dependent metabolism of benzene's phenolic metabolites and its potential role in benzene toxicity and carcinogenicity. Environ. Hlth. Perspect., 82, 23-29.
Snyder, C.A.; Goldstein, B.D.; Sellakumar, A.; Wolman, S.R.; Bromberg, I.; Eriichman, M.N. and Laskin, S. (1978). Hematotoxicity of inhaled t»enzene to Sprague-Dawley rats and AKR mice at 300 ppm. J. Toxicol. Environ. Hlth., 4, 605-618.
Snyder, C.A.; Goldstein, B.D.; Sellakumar, A.R.; Bromberg, I.; Laskin, S. and Albert, RE, (1980). The inhalation toxicology of benzene: Incidence of hematopoietic neoplasms and hematotoxicity in AKR/J and C57B1/6J mice. Toxicol. Appl. Ptiarmacol, 54, 323-331.
Snyder, C.A.; Goldstein, B.D.; Sellakumar, A.; Bromberg. I.; Laskin, S. and Albert, RE. (1982). Toxicity of chronic benzene inhalation: GDI mice exposed to 300 ppm. Bull. Environ. Contam. Toxicol., 29, 385-391.
Snyder, C.A. (1987). Benzene. In : Ether Browning's Toxicity and Metabolism of Industrial Solvents, 2nd ed.. Vol. 1, Hydrocartxjns, edited by R. Snyder, Amsterdam. Elsevier, New York, 3-27.
Snyder, R. and Kocsis, J.J. (1975). Cun-ent concepts of chronic benzene toxicity. CRC, Crit. Rev. Toxicol., 3, 265-288.
Snyder, R.; Lee, E.W.; Kocsis, J.J. and Witmer, CM. (1977). Bone marrow depressant and leukemogenic action of benzene. Life Sciences, 21, 1709-1722.
Snyder, R.; Longacre, S.L; Kocsis, J.J.; Witmer, CM.; Lee, E.W. and Sammett, D. (1981). Toxicological and biochemical effects of repeated administration of bezene in mice. J. Toxicol. Environ. Hlth., 7, 223-237.
Snyder, R.; Jowa, L., Witz, G.; Kalf, G.F. and Rushmore, T. (1987). Formation of reactive metabolites from benzene. Arch. Toxicol., 60, 61-64.

Bibliography j ^ j
Snyder, R and Chatterjee, S P. (1991). In: Molecular Aspects of Monooxygenases and Bioactlvation of Toxic Compounds, edited by E. Arinc, J.B. Schenckaman, E. Hodgson, Plenum Press, New York, 375-386.
Snyder, R.; Witz, G. and Goldstein, B.D. (1993). The toxicology of fc)ezene. Environ. HIth. Perspecf, 100, 293-306.
Snyder, R. and kalf, G.F. (1994). A perspective on benzene leukemogenesis. CRC, Crit. Rev. Toxicol., 24, 177-209.
Sorsa, M. and Yager, J.W. (1987). In : Cytogenetic sun/ellance of occupational exposures, edited by G Obe and A. Basler, Berlin Springer-Vertag, Chapt. 17.
Stevnsner, T. and Bohr, V.A. (1993). Studies on the role of topoisomerases in general, gene-and strand-specific DNA repair. Carcinogenesis, 14, 1841-1850.
Stommel, P.; Muller, G.; Stucker, W.; Verkoyen, C; Schobel, S and Norpoth, K. (1989). Determination of S-phenyl mercapturic acid in the unne an improvement in the biological monitonng of benzene exposure. Carcinogenesis, 10, 279-282.
Stoyanovsky, D.; Yalowich, J.; Gantchev, T. and Kagan, V. (1993). Tyrosine-induced phenoxyi radicals of etoposide (VP-16).- interaction with reductants in model systems, K 562 leukemic cell and nuclear homogenates. Free Rad. Res. Commun., 19, 371-386.
Subrahmanyam, V.V. and O'Brien, P.J. (1985). Peroxidase- catalyzed oxygen activation by arylamine carcinogens and phenol Chem.-Biol. Interact.. 56, 185-199.
Subrahmanyam, V.V., Kolachana, P.; Meyer. KB. and Smith, M.T. (1990). Bioactlvation and cytotoxicity of hydroquinone in human promyelocytic leukemia (HL-60) cells. Free Rad. Biol. Med.. 9, abstract No. 3.
Subrahmanyam, V.V.; Kolachana, P. and Smith, M.T. (1991a). Metabolism of hydroquinone by human myeloperoxidase : mechanisms of stimulation by other phenolic compounds. Arch. Biochem. Biophys., 286, 76-84,
Subrahmanyam, V.V.; Ross, D.; Eastmond, D.A. and Smith, M.T (1991b). Potential role of free radicals in benzene-induced myelotoxicity and leukemia. Free Rad. Biol. Med., 11,495-515.

Bibliography j ^
Sun, J.D.; Medinsky, M.A.; Birbaum, L.S.; Lucier, G. and Henderson, R.F. (1990). Benzene hemoglobin adducts in mice and rats: Characterization of formation and physiological modeling. Fundam. Appl. Toxicol., 15, 468-475.
Susten, A.S.; Dames, B.L.; Burg, J.R. and Niemeier, R.W. (1985). Percutaneous penetration of benzene in hairless mice : An estimate of dermal absorption during tire-building operations. Am. J. Ind. Med., 7, 323-335.
Sze, C.C.i Shi, C.Y. and Ong, C.N. (1996). Cytotoxicity and DNA strand breaks induced by benzene and its metabolites in Chinese Hamster Ovary cells. J. Appl. Toxicol., 16,259-264.
Teisinger, J.; Fiserrova-Bergerova, V. and Kudrna, J. (1952). The metabolism of benzene in man. Prac. Lek.,A, 175-188.
Theil, E C ; Sayers, D.E. and Brown, M.A. (1979). Similarity of the stnjcture of ferritin and iron. Dextran (imferon) determined by extended x-ray absorption fine structure analysis. J. Biol. Chem. 254, 8132-8134
Theil, EC (1987). Ferritin; structure, gene regulation and cellular function in animals, plants and microorganisms Ann. Rev. Biochem., 56, 289-315.
Thielmann, H.W.; Popanda, 0.; Gersbach, H. and Gilberg. F. (1993). Various inhibitors of DNA topoisomerases diminish repair-specific DNA incision in UV-irradiated human fibroblasts. Carcinogenesis, 14, 2341-2351.
Thomas. C.E.; Morehouse, LA. and Aust. S.D. (1985). Ferritin and superoxide-dependent lipid peroxidation. J. Biol. Chem.. 260, 3275-3280.
Thomas, C.E. and Aust, S.D. (1986a). Reductive release of iron from ferritin by cation free radicals of paraquat and other bipyndyls. J. Biol. Chem., 261, 13064-13070.
Thomas, C.E. and Aust, S.D. (1986b). Releae of iron from ferritin by cardiotoxic anthra-cycline antibiotics. Arch. Biochem. Biophys., 248, 684-689.
Thomas, D.J.; Sadler, A.; Subrahmanyam, V.V.; Siegel, D.; Reasor, M.J.; Wierda, D. and Ross, D. (1990). Bone marrow stromal cell bioactivation and detoxification of the benzene metabolite hydroquinone : companson of macrophages and fibroblastoid cells. Mol. Pharmacol., 37, 255-262.

Bibliography 145
Tice, R.R.; Costa, D, and Drew, R. (1980). Cytogenic effects of inhaled benzene in murine bone marrow : Induction of sister chromatid exchanges, chromosomal aberrations and cellular proliferation inhibition in DBA/2 mice. Proc. Natl. Acad. Sci. USA, 77, 2148-2155.
Tice, R.R.; Vogt, T.F. and Costa, D.L. (1982). Cytogenetic effects of inhaled benzene in munne bone marrow. In Genotoxic effects of air borne agents. Environ. Sci. Res., 25, 257-275.
Toft, K.; Olofsson, T.; Tunek, A. and Berlin, M. (1982). Toxic effect on mouse bone marrow caused by inhalation of benzene. Arch. Toxicol., 51, 295-302.
Topham, R.W.; Walker, M.C. and Calisch, M.P. (1982). Liver xanthine dehydrogenase and iron mobilization. Biochem. Biophys. Res. Commun., 109, 1240-1246.
Toyokuni, S. and Sagripanti, J.S. (1993). DMA single and double strand breaks produced by ferric nitrilotriacetate in relation to renal tubular carcinogenesis. Carcinogenesis, 14, 223-227.
Toyokuni, S. (1996). Iron-induced carcinogenesis: The role of redox regulation. Free Rad. Biol. Med., 20, 553-566.
Treffry, A.; Banyard, S.H.; Hoare, R.J. and Harnson, P.M., (1977). In Proteins of Iron Metabolism, edited by E.B. Brown,, P. Aisen, J. Fielding, R.R. Crichton, pp. 3-11, New York : Grune & Stratton, 444.
Tricot, G.J.K. (1991) In: Haematology, Basic Principles and Practice: edited by R. Hoffman, E.J. Jr. Benz, S.J. Shattil, B. Furie and H.J. Cohen, Churchill Livingstone, New York, 805.
Trush, MA. and Kensler, T.W. (1991). In: Oxidative Stress: Oxidants and Antioxidants, edited by H. Sies, Academic Press, New York, 277-318.
Tunek, A ; Olofsson, T. and Berlin, M. (1981). Toxic effects of t>enzene and benzene metabolites on granulopoietic stem cells and bone marrow cellularity in mice. Toxicol. Appl. Pharmacol., 59, 149-156.
Tunek, A.; Hogstedt, B. and Olofsson, T. (1982). Mechanism of benzene toxicity. Effects of benzene and benzene metabolites on bone marrow cellularity, number of granulopoietic stem cells and frequency of miaonuclei in mice. Chem.-Biol. Interact., 39, 129-138.

Bibliography ^4^
Uysal, M.: Yalcin, AS.; Kocak-Toker, N.; Keyer-Uysal, M. and Aykae, G.K. (1986). Lipid peroxidation in liver and plasma of rats treated with benzene. IRCS Med. Sci., 14, 772
Van Sittert, N.J. and de Jong, G. (1985). Biomonitoring of exposure to potential mutagens and carcinogens in industrial populations. Fd. Chem. Toxic, 23, 23-31.
Vidnes, A. and Helgeland, L. (1973). Sex and age differences in the hemosiderin content of rat liver. BiochJm. Biophys. Acta., 328, 365-372.
Walker, P. (1976). Health effects of benzene (a review). National Research Council for Environmental Protection Agency, Washington, D.C., PB-254388.
Wallace, LA. (1989). Major sources of tjenzene exposure. Environ. HItfi. Perspect., 82, 165-169.
Wang, J.C, Caron, P.R. and Kim, R.A. (1990). The role of DNA topoisomerases in recombination and genomic stability: a double- edged sword? Cell, 63, 403-406.
Ward, CO.; Kuna, R.A.; Snyder, C.A.; Alsaker, N.K.; Coate, W.B. and Craig, P.H. (1985). Subchronic inhalation toxicity of benzene in rats and mice. Am. J. Ind. Med., 7, 457-473
Weaver. J and Pollack, C. (1989). Low-Mr. iron isolated from guinea pig reticulocytes as AMP-Fe and ATP-Fe complexes. Biochem. J.. 261, 787-792
Weaver. V M.; Davoli, C T ; Heller, P.J.: Fitzwilliam. A.; Peter. H.L.: Sunyer, J.; Murphy, S.E., Goldstein, G.W. and Groopman, J.D. (1996). Benzene exposure, assessed by unnary trans, trans- muconic acid, in urban children with elevated blood lead levels. Environ. HIth. Perspect.. 104, 318-323.
Wei, H and Frenkel, K (1991) In vivo formation of oxidized DNA bases in tumor promoter-treated mouse skin Cancer Res.. 51, 4443-4449
Weiss. G.; Wachter, H. and Fuchs, D (1995). Linkage of cell- mediated immunity to iron metabolism. Immunology Today, 16, 495- 500.
Wick, MM. (1980). Levodopa and dopamine analogs as DNA polymerase inhibitors and antitumor agents in human melanoma. Cancer. Res.. 40, 1414-1418,

Bibliography 147
Wick, MM. and Fitzgerald, G. (1981). Inhibition of reverse transcriptase by tyrosinase generated quinones related to ievodopa and dopamine. Chem.-Biol. Interact., 38, 99-107.
Wierda, D. and Irons, R.D. (1982). Hydroquinone and catechol reduce the frequency of progenitor B lymphocytes in mouse spleen and bone man^ow. Immunophar-macology, 4, 41-54.
Willson, R.L. (1977). Iron, zinc, free radicals and oxygen in tissue disorders and cancer control. Ciba Found. Symp. 51 (NS), 331-354. Amsterdam. Elsevier, 391.
Witz, G.; Rao, G.S. and Goldstein, B.D. (1985). Short-ternn toxicity of trans, trans-muconaldehyde. Toxicol. Appl. Pharmacol., 80, 511-516.
Witz, G.; Gad, S.C; Tice, R.R., Oshiro, Y.; Piper, C.E. and Goldstein, B.D. (1990), Genetic toxicity of the benzene metabolite trans, trans-muconaldehyde in mammalian and bacterial cells. Mutat. Res., 240, 295-306.
Witz, G. (1991). Active oxygen species as factors in multistage carcinogenesis. Proc. Soc. Exp. Biol. Med., 198, 675-682.
World Health Organization (WHO) (1987). Air Quality Guideline for Europe Copenhage: WHO Regional Publications.
Worwood. M.; Aherne, W.: Dawkins, S. and Jacobs, A. (1975). Characteristics of ferritin from human tissues, serum and blood cells. Clin. Sci. Mol. Med., 48, 441-451.
Yager, J.W.; Eastmond, DA.; Robertson, M.L., Paradisin, W.M and Smith, M.T. (1990), Characterization of micronuclei induced in human lymphocytes by benzene metabolites. Cancer Res., 50, 393- 399.
Yamamoto, K. and Kawanishi, S. (1992). Site-specfic DNA damage by phenylhydrazine and phenelzine in the presence of Cu(ll) ion or Fe (III) complexes : roles of active oxygen species and carbon radicals. Chem. Res. Toxicol., 5, 440-446,
Yamazaki, I.; Mason, H.S. and Piette, L. (1959). Identification of intermediate substrate free radicals formed during peroxidate oxidations by electron paramagnetic resonance spectroscopy. Biochem. Biophys. Res. Commun., 1, 336-337.

Bibliography j^g
Yardley-Jones, A., Anderson, D.; Lovell, D.P. and Jenkinson, PC. (1990). Analysis of chromosomal aberrations in workers exposed to low level benzene. Br. J. Ind. Med., 47,48-51.
Yardley-Jones, A.; Anderson, D. and Park, D.V.W. (1991). The toxicity of benzene and its metabolism and molecular pathology in human risk assessment. Br J. Ind. Med., 48, 437-444.
Yeowell-O'Connell, K.; McDonald, T.A. and Rappaport, S.M. (1996). Analysis of hemoglobin adducts of benzene oxide by Gas Chromatography-Mass Spectrometry. Anal. Biochem., 237, 49-55.
Yim, M.B., Chae, H.Z.; Rhee, S.G.; Chock, P.B. and Stadtman, E.R. (1994). On the protective mechanism of the thiol-specific antioxidant enzyme against the oxidative damage of biomacromolecules. J. Biol. Chem., 269, 1621-1626.
Yin, S.N.; Li, Q.; Tian, F.D.; Du, C. and Jin, C. (1987). Occupational Expsoure to benzene in China. Br J. Ind. Med., 44, 192-195.
Yin, S.N., Li, G.L; Tain, F.D.; Fu, Z.I.; Jin, C; Chen, Y.L; Luo, S.J.; Ye, P.Z.; Zhang, J.Z.; Wang, G.C.; Zhang, X.C; Wu, H.N. and Zhong, O.C. (1989). A retrospective cohort study of leukemia and other cancers in benzene workers. Environ HIth. Perspect., 82, 207-213.
Yin, S.N.; Hayes, R.B.; Linet, M.S.; Li, G.L.; Dosemeci, M.: Travis, L.B.; Li, C.Y.; Zhang, Z.N., Li, D.G.; Chow, W.H.; Wacholder, S.; Wang, Y.Z.; Jiang, Z.L.; Dai, T.R.; Zhang, W Y.; Chao, X.J.; Ye, P.Z.; Kou, Q.R.; Zhang, X.C; Lin, X.F.; Meng, J.F.; Ding, C.Y.; Zho, J.S. and Blot, W.J. (1996). A cohort study of cancer among benzene exposed workers in China ; Overall results. Am. J. Ind. Med.. 29, 227-235.
Zhang, L., Robertson, M.: Kolachana, P.; Davison, A.J. and Smith, M.T. (1993). Benzene metabolite, 1.2,4-benzenetriol induces micronuclei and oxidative DNA damage in human lymphocytes and HL- 60 cells Environ. Mol. Mutagen. 21, 339-348
Zhang, L.; Smith, M.T., Bandy, B.; Tamaki, S.J. and Davison, A.J. (1994). Role of quinones, active oxygen species and metals in the genotoxicity of 1,2,4-benzenetriol, a metabolite of benzene In: Free radicals in the environment, medicine and toxicology, edited by H Nohl, H. Esterbauer and C. Rice-Evans, Vol., 8, London Richelieu Press, 521-562.

Bibliography I49
Zhang, L , Bandy, B. and Davison, A.J. (1996). Effects of metals, ligands and antioxidants on the reaction of oxygen with 1,2,4-benzenetriol. Free Rad. Biol. Med., 20, 495-505.
Zhang, Z., Xiang, Q., Goldstein, B.D., Glatt, H., Piatt, K.L. and Witz, G. (1995a). Studies on pathways of ring opening of benzene in a Fenton reaction. Free Rad. Biol. Med.. 18,411-419
Zhang, Z.; Goldstein, B.D. and Witz, G. (1995b). Iron stimulated nng-opening of benzene in a mouse liver microsomal system. Biochem. Pharmacol., 50, 1607-1617.

150
1. Sarfaraz Ahmad, Vinita Singh and G.S. Rao (1994) "Antioxidant Potential in serum & liver of albino rats exposed to benzene". Indian J. of Experimental Biology: Vol. 32, 203-206.
2. Vinita Singh, Sarfaraz Ahmad and G.S. Rao (1994) "Prooxidant and antioxidant properties of iron: hydroquinone and iron: 1,2,4- benzenetnol complex Implication for benzene toxicity". Toxicology, Vol. 89, 25-33.
3. Sarfaraz Ahmad, Vinita Singh and G.S. Rao (1995) "Release of iron from ferritin by 1,2,4-benzenetrior'. Chemico-Biologlcal Interactions, Vol. 96, 103-111.
4. Vinita Singh, Sarfaraz Ahmad and G.S. Rao(1996) "Release of aldehydic product from glutamate or deoxyuridine by S-aminolevu\inic acid in presence of copper ions". Indian J. of Expenmental Biology. Vol. 34, 208-210.
5. Sarfaraz Ahmad and G.S. Rao. "Cleavage of pUC 18 DNA by glutathionyl hydroquinone." Chemico-Biological Interaction, (Communicated in September, 1996).
6. Sarfaraz Ahmad and G.S. Rao. "EDTA: An alternate sp€;ctrophotometric reagent for iron estimation". Current Science (Communicated in September, 1996).
7. Sarfaraz Ahmad and G.S. Rao. "In vitro identification and characterization of polyphenol-iron complex: A possible toxic metatx)lite of benzene", (manuscript under preparation).
8. Sarfaraz Ahmad, R.C. Murty and G.S. Rao. "Role of transition metal ions (iron, Copper) in relation to genetic toxicity dunng benzene exposure." (manuscript under preparation).