structure of the catalytic and ubiquitin-associated ... · include par-1 in c. elegans and...
TRANSCRIPT

1
Structure 14, 173-183 (2006) DOI 10.1016/j.str.2005.09.022
Structure of the catalytic and ubiquitin-associated domains of the protein kinase MARK / Par-1
Saravanan Panneerselvam*, Alexander Marx*, Eva-Maria Mandelkow, Eckhard Mandelkow§
Max-Planck-Unit for Structural Molecular Biology
c/o DESY, Notkestrasse 85, 22607 Hamburg, Germany
Keywords: Tau, Ubiquitin, UBA domain, Cell polarity, Kinase Abbreviations: MARK = MAP-microtubule affinity regulating kinase; MR = molecular replacement; SeMet = selenomethionine; UBA = ubiquitin-associated domain; Running title: Structure of MARK/Par-1 * SP and AM contributed equally to this work § corresponding author: [email protected]

2
ABSTRACT The Ser/Thr kinase MARK2 phosphorylates tau protein at sites that cause the detachment from microtubules in Alzheimer neurofibrillary degeneration. Homologues of MARK2 include Par-1 in C. elegans and Drosophila which generates embryonic polarity. We report the X-ray structure of the catalytic and ubiquitin-associated domains of human MARK2. The activity was altered by mutations in the ATP binding site and/or activation loop. The catalytic domain shows the small and large lobes typical of kinases. The substrate cleft is in an inactive, open conformation in the inactivated and the wild type structure. The UBA domain is attached via a taut linker to the large lobe of the kinase domain and leans against a hydrophobic patch on the small lobe. The UBA structure is unusual because the orientation of its third helix is inverted, relative to previous structures. Possible implications of the structure for the regulation of the kinase activity are discussed.

3
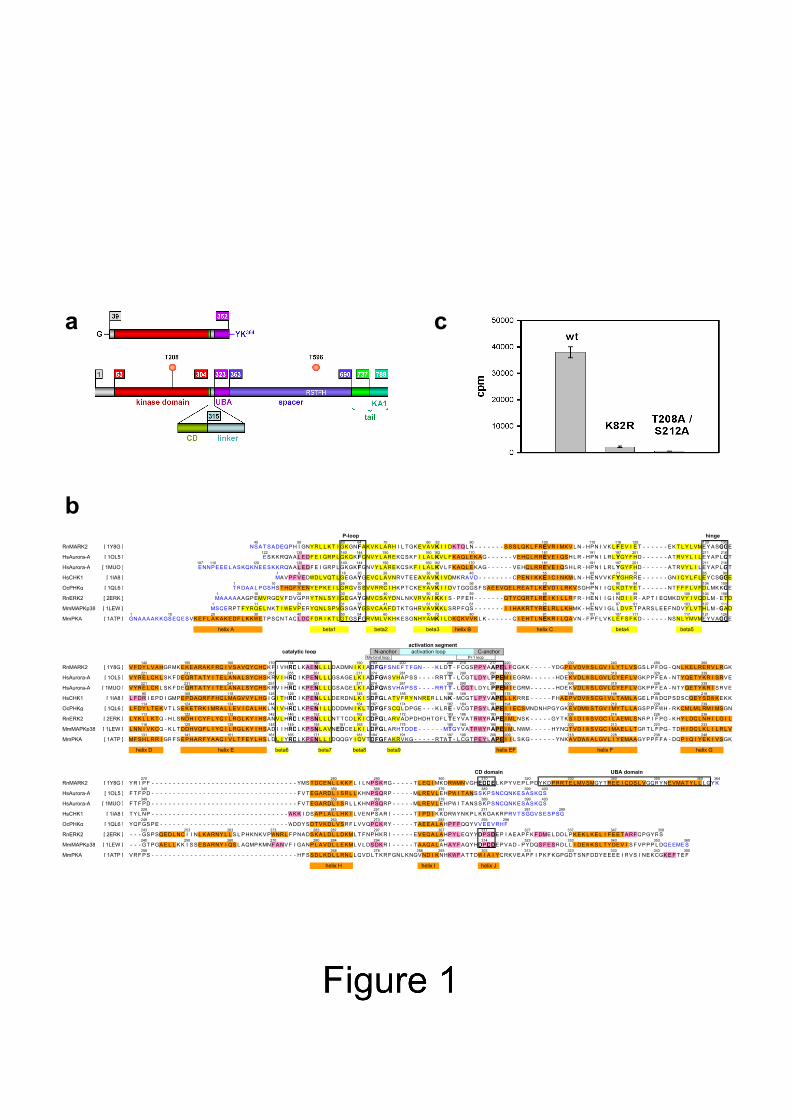
INTRODUCTION The activity of the protein kinase MARK was initially noticed in the search for pathological phosphorylation sites of tau protein (Biernat et al., 1993). Tau is a microtubule-associated protein prominent in the brain, particularly in the axonal compartment of neurons, where it helps to stabilize microtubules. The tau-microtubule interaction is regulated by phosphorylation, especially at the KXGS motifs in the repeat domain of tau which represents the core of the microtubule-binding domain. The same domain also forms the core of the abnormal tau aggregates ("paired helical filaments") in Alzheimer's disease. Microtubule binding and PHF assembly are efficiently suppressed when tau is phosphorylated by MARK. Thus, excess activation of MARK in cells leads to microtubule breakdown because they are not properly stabilized. A second function of tau is its interference with motor proteins moving along microtubules; this function is also fine-tuned in axons by MARK (Mandelkow et al., 2004). Both MARK and tau are important for the establishment of neuronal polarity (Biernat et al., 2002). Compared to other kinases, MARK is a large protein (~720-790 aa) which contains an N-terminal leader sequence, a kinase catalytic domain, an ubiquitin-associated domain (UBA), a spacer, and a tail domain containing the KA1 (kinase-associated) motif characteristic for the family of kinases ending with the ELKL motif (Fig. 1a). Four isoforms of MARK (1-4) were found in rats, encoded by different genes, with additional splicing variants (Drewes, 2004; Drewes et al., 1997). MARK family members show a striking homology to kinases of the Par-1 family which are best known for their role in defining embryonic polarity in Drosophila and C. elegans (reviews: Kemphues, 2000; Tomancak et al., 2000). Members of the MARK/Par-1 family occur in most organisms examined so far. Analysis of the human genome showed that there are four members of the human MARK family which belong to the class of CaMKII kinases (Manning et al., 2002). Known targets of MARK family members include tau, MAP2, MAP4, doublecortin, PTPH1, Cdc25C, KSR1, plakophilin, dishevelled, oskar, Raf1; these proteins are typically involved in the regulation of the cytoskeleton and in signaling (review: Drewes, 2004). Like other kinases, MARK family members can be activated by phosphorylation in the "activation loop" which controls the access of the substrate to the catalytic center (reviews, e.g. Huse and Kuriyan, 2002). This can be achieved by the protein kinase MARKK which phosphorylates T208 in MARK2 and the corresponding residues in MARK1, 3, and 4 (Timm et al., 2003). The same kinase was found in the context of activation of MEKs and named TAO-1 (Hutchison et al., 1998). MARK can also be activated by LKB1 which plays a role in tumor suppression (Lizcano et al., 2004). A notable feature of MARK isolated from brain tissue is its double phosphorylation in the activation loop (at T208 and S212 in MARK2; Drewes et al., 1997). While phosphorylation of T208, the target of MARKK or LKB1, activates the kinase, phosphorylation of S212 is inhibitory (Timm et al., 2003). A further level of regulation lies in the association with other proteins and domains. In the related kinase KIN1 in yeast, the tail domain motif is thought to bind and inactivate the catalytic domain (Elbert et al., 2005). MARK in turn phosphorylates other partners which then bind to 14-3-3, such as Cdc25C, KSR1, plakophilin, or Raf-1 (Benton et al., 2002; Muller et al., 2003). Finally, the presence of an UBA domain adjacent to the catalytic domain suggests potential interactions with proteins involved in the ubiquitin proteosome pathway, DNA repair, or cell signaling (Brajenovic et al., 2004; Hofmann and Bucher, 1996; Mueller and Feigon, 2002). Because of the potential importance of MARK in basic physiological processes and human pathology we embarked on a structural analysis of MARK family members as a step towards

4
identifying interaction sites or inhibitors. Here we report the X-ray structure of the catalytic domain of MARK2, combined with the UBA domain. The catalytic domain shows the typical features expected for an inactive kinase with a disordered activation loop. The UBA domain is tethered to the large lobe via an extended linker comprising a structural motif similar to the common docking domain of the MAPK family that provides an interaction site for upstream or downstream signalling molecules. RESULTS The fragment of human MARK2 described here (residues N39-K364) comprises part of the N-terminal header (N39-N52), the catalytic domain (Y53-M304), CD motif and linker N305-D322, and the UBA domain (Y323-K362) (Fig. 1a, b). Crystals of the wild type construct and of two inactive mutants were analyzed, K82R and T208A/S212A. K82 is essential for catalysis, T208 is the primary phosphorylation site in the activation loop, and S212 was also found to be phosphorylated in MARK2 from brain (Drewes et al., 1997). Phosphorylation of T208 is required for full activation, but phosphorylation of S212 or mutation to alanine completely abolish the kinase activity of MARK2 (Timm et al., 2003). Many Ser/Thr kinases have a conserved threonine at the position S212 in MARK2 which contributes to substrate specificity and assists in catalysis (Chen et al., 2000; Min et al., 2004; Taylor et al., 1995). Figure 1c shows that the wild type construct has a basal kinase activity even without phosphorylation of T208, while the activities of the mutants are largely diminished. The three variants and a SeMet derivative of the T208A/S212A double mutant crystallized in the hexagonal space group P61. Two similar crystal forms were observed, which differ only by the length of the c-axis: c = 106.0 Å for the double mutant, c = 99.7 Å for the wild type and the K82R mutant; the wild type was found in both forms. All crystal structures are similar in crystal packing and folding of the molecules. The general structure will be described by reference to the T208A/S212A mutant which has the highest resolution. Catalytic domain The crystals contain two molecules per asymmetric unit (Cα-traces, Fig. 2). The kinase domain has a bilobed structure like many other kinases. The smaller, N-terminal lobe (residues ~53-130; N-lobe) consists of 5 β-strands and an α-helix (helix C according to PKA), whereas the C-terminal lobe (residues ~135 to 304; C-lobe) is composed mainly of α-helices. A structural sequence alignment with other kinases is presented in Figure 1b, including the notation used for the structural elements. The 11 initial residues (G38-P48) are invisible due to disorder. Residues D193-K205 including the DFG motif and most of the activation loop are also disordered. Figures 3a shows details of the electron density map around L206, the first residue following the invisible part of the activation segment. The two lobes are linked by a flexible segment of 6 amino acids (130-135) including two glycines, G134-G135. Further, H-bonds between the lobes connect residues in the loop between helix C and strand β4 (C-lobe) with residues in helix E and the loop between β7 and β8 (N-lobe). Both the covalent link and the H-bond interactions are restricted to a narrow region at the back of the catalytic cleft and serve as a hinge that allows the small lobe to librate, thereby opening and closing the catalytic cleft.

5
Conformation of the activation loop While most of the activation loop is disordered in the wild type and the K82R structures (D193-C210), the T208A/S212A mutant reveals five more residues at the end of the activation loop (L206-C210). This includes the P+1 loop (F209-A217) which is thought to recognize the substrate by specific interaction with the residue following the phosphorylation site. The end of the activation segment (P213-E219 including APE motif) is well defined in all MARK2 structures. The conformation of the structured parts of the activation segment indicates that it folds away from the C helix, in the opposite direction to most active kinases (Nolen et al., 2004), and occupies the area below the P-loop (Fig. 4, and see Discussion). Intermolecular disulfide bridge In the double mutant, the end of the activation loop (206-212) is stabilized by an intermolecular disulfide bridge between cysteines C210 of two adjacent molecules that are related by noncrystallographic symmetry (Fig. 3a). C210 is located in the midst of T208A and S212A, the two phosphorylatable sites that distinguish the double mutant from the wild type. Formation of the disulfide bridge is probably a crystallisation artifact. It appears that the S-S bridge is essential for crystallization, as crystals do not form in the presence of DTT. This holds for all variants of the protein, although the disulfide bridge is visible only in the T208A/S212A structure. Dimerization The MARK2 crystals contain two molecules per asymmetric unit (A and B) that interact via multiple contacts and form a dimer with a proper two-fold non-crystallographic symmetry (Fig. 5). The catalytic domains in a dimer face each other with their active sites. The most variable and disordered portion of the activation segment is close to center of the dimer, encircled by the four lobes of the catalytic domains. Interactions between the monomers are concentrated in three zones (Fig. 5). Zone 1 in the C-lobe and zone 3 in the N-lobe of one molecule form a wide-open entrance to the catalytic cleft. Helix G of the other molecule (zone 2) inserts into this space, making contacts to both rims. In the T208A/S212A mutant, 30 residues of each molecule are involved in intra-dimer contacts, while the wildtype and K82R mutant show only 18 contact residues, and the S-S bond at C210 is not visible. Thus, the dimer-forming interaction in these constructs appear to be weakened compared to the double mutant. This could explain the difference in crystal packing between wild type and double mutants (see Supplement). UBA domain The UBA domain (Y323-K362) is a small, globular domain that consists of three short helices (α1-α3). Helices α1 and α3 are roughly antiparallel (folding reminiscent of a "U", Fig. 6). This conformation is unexpected since the helices of other UBA domains solved so far (Fig. 6a, structural sequence alingment) alternate so that α1 and α3 are almost parallel to each other (as in an "N"). Figure 6b compares stereo views of the MARK2 UBA domain (yellow) with the UBA domain of HHR23A (green, PDB-ID: 1IFY; Mueller and Feigon, 2002) which is representative for the other UBA domains listed in Figure 6a. In MARK2, the UBA domain binds to the N-lobe of the catalytic domain close to the hinge, opposite to the cleft (i.e. at the "back side"). The interaction is predominantly hydrophobic and mainly due to helix α3 (see Fig. 6a, c). At the catalytic domain, the interaction involves

6
residues L115, F116 at the beginning of β4. Other hydrophobic interactions involve residues at the N-terminus of the N-lobe (Y53) and at the β2-β3 turn, e.g. L74 interacts with M335 and Y337 between helices α1 and α2 of the UBA domain which belong to the MGF/Y motif characteristic for UBA domains. In addition to the hydrophobic interactions, residue K105 at the end of helix C and the side chain amino group of K114 form hydrogen bonds to Y351 in the UBA domain. Most UBA domains contain one or two leucines near the end of α3 (three in the case of MARK2, L359-L361). The first one (corresponding to L359) is highly conserved. It is important for the internal cohesion of the UBA domain by fitting into a hydrophobic pocket formed by residues of α1-α2 and the MGF/Y motif. In MARK2, the conserved L359 lies on the outside of the reversed helix α3 and makes hydrophobic contacts with the N-lobe of the catalytic domain. Instead of L359, L361 forms hydrophobic intra-UBA interactions with side chains of helix α1 and the MGF/Y motif. The hydrophobic pocket normally occupied by the conserved leucine is narrowed by a ~30° inward tilt of Y337. The remaining space is filled with the side chain of V354 at the start of α3 which – because of the inversion of the helix – ends up roughly at the same place as the conserved leucine in normal UBA structures. In the position observed here, the UBA domain would not be able to interact with ubiquitin, judging by the published structures of mono- or poly-ubiquitin docked onto UBA domains of other proteins (Ohno et al., 2005; Varadan et al., 2005). There would be a steric clash with the N-lobe (Fig. 6d). Consistent with this, attempts to identify a MARK2-ubiquitin complex biochemically have failed so far (data not shown). UBA linker and common docking domain for kinase activators The UBA domain is linked to the catalytic domain by ~20 residues (~305 to 322, Fig. 2, 7). The first half contains a motif similar to the "common docking" motif (CD) of MAP kinases, characterized by a cluster of negative surface charges (DxxD/E; Tanoue et al., 2000). In MARK2 the motif E309DDE312 and surrounding residues fold into a loop similar to the CD domain of MAP kinases (Fig. 7). Together with the "ED site" (corresponding to residues A185-D186 in MARK2) at the tip of the β7-β8 turn, the CD domain is thought to form a docking groove for upstream and downstream signalling molecules on the back surface of the catalytic domain opposite to the active site (Tanoue and Nishida, 2003), although the exact location of the docking groove is still a matter of debate (Chang et al., 2002). The presence of these features in MARK2 suggests a similar function, but the putative docking partners are not known so far. The second half of the stretch tethering the UBA and catalytic domain ("linker", ~315-322) assumes an extended conformation. Remarkably, the linker has little contact to the lobes of the catalytic domain, in fact L320 and adjacent residues are surrounded by interstitial water (Fig. 3b). Accordingly, the B-factors are high in this region (main chain B-factor about 60), but the electron density was sufficiently well defined (Fig. 3b) to trace the linker unambiguously all the way from the end of the catalytic domain up to the UBA domain. However, the loose attachment suggests the possibility that the linker and UBA domain could swing away from the catalytic domain and thus alter the regulatory state of the domains.

7
DISCUSSION MARK kinases constitute a subfamily of the AMPK/Snf1 family of kinases within the CAMK group of Ser/Thr kinases (Manning et al., 2002). Apart from a highly conserved catalytic domain, the MARK kinases stand out in that they contain a C-terminal KA1 domain and a UBA domain adjacent to the catalytic domain (Fig. 1). MARK occurs in 4 isoforms and several splicing variants. Here we present the structure of the MARK2 catalytic and UBA domain, including the connecting sequence of amino acids with a motif that may be involved in protein-protein recognition and regulation. By structural comparison of MARK2 with other kinases using the program CE (Shindyalov and Bourne, 1998), Aurora-A and Aurora-B were consistently found at or close to the top of the ranking, even above CHK1, which is more closely related to MARK by sequence and which has been used for molecular replacement. The kinase domain of Aurora-A has been solved in an inactive form (in complex with adenosine; PDB-ID: 1MUO; Cheetham et al., 2002), as well as in active and "half-activated" forms (ATPγS complexes with/without a fragment of the activating protein TPX2; PDB-ID: 1OL5 and 1OL7, respectively; Bayliss et al., 2003) and, thus, lends itself as a paradigm for the discussion of the MARK2 structure. Activation segment The inactive and the fully activated form of Aurora-A mainly differ by the conformation of the activation segment and by a 6.7° tilt of the minor lobe (Fig. 4b, c). In the inactive form, the activation loop is partially disordered; the N- and C-termini of the activation segment indicate that the activation loop points in a direction opposite to the active conformation (to the left side in Fig. 4b), passing close below the P-loop. In the MARK2 structures, only the C-terminal residues of the activation segment are visible. They adopt a conformation similar to the inactive form of Aurora-A, suggesting that the activation loop of inactive MARK2 also folds to the left side (Fig. 4a). By folding in this way, cysteine residues C210 of the two monomers can meet and form a disulfide bridge. Superposition of MARK2 with the structure of the phosphorylated c-AMP dependent protein kinase (PKA) in complex with peptides derived from protein kinase inhibitor PKI (Knighton et al., 1991; Madhusudan et al., 2002) reveals that the C-terminal part of the MARK2 activation loop occludes the space required for substrate binding. Thus, in the inactive state observed in the MARK2 structure, the substrate cannot bind in a productive way because of steric interference with the activation loop. Notably, threonine T208 (the primary phosphorylation site of the activation loop) is close to the position of the phosphorylation site (serine S21) of the peptide substrate in the PKA complex (Cα distance 3.5 Å) indicating the possibility of autophosphorylation. In phosphorylase kinase (PHK), another example of a kinase domain in complex with a substrate peptide (Lowe et al., 1997), the substrate binds in an extended conformation analogous to the PKA-PKI complex. As in the case of PKA, binding of the substrate to MARK2 in the same position as in PHK would result in a steric clash with residues 208 to 210 – at least according to the double mutant, where these residues are stabilized by the disulfide bridge between cysteines C210. Catalytic cleft and nucleotide binding site The catalytic cleft in the MARK2 structure is extremely wide open, compared to other active or inactive kinases. It is 1-2 Å wider than the cleft of Aurora-A in the inactive form, judged by the distance between β1 (N-lobe) and β6 (C-lobe). In the activated form of Aurora-A, as in

8
other active kinases, helix C of the N-lobe contributes to nucleotide binding by a conserved glutamic acid (E100 in MARK2) that forms a salt bridge with a strictly conserved lysine in strand β3 (K82 in MARK2, Fig. 4f) and positions this lysine for proper coordination of the nucleotide's α- and β-phosphates. In the crystal structure of the MARK2 double mutant, the side chains of K82 and E100 are not aligned correctly for interaction with the nucleotide (Fig. 4d). In the wild type structure and especially in that of the K82R mutant, the side chains of K82 and E100 are less well ordered (by B-factor) or even invisible. It is not surprising that long side chains facing the activation loop are affected by disorder. This applies also to methionine M104 which reveals a double conformation that could be identified due to the high electron density of the selenium atom in the SeMet structure. There is no hint at a specific interaction between K82 and E100 in any of the four MARK2 structures. Conformation of the catalytic loop Activation of Aurora-A involves phosphorylation of threonines T287 and T288 in the activation loop. In the fully activated state, the phospho group of pT288 (primary phosphorylation site, corresponding to T208 in MARK2) is engaged in ion pair interactions with R255 in the catalytic loop (R174 in MARK2), adjacent to the catalytically active aspartate D256 (D175 in MARK2, Fig. 4f). This interaction stabilizes the catalytic loop and positions the aspartate towards the attacking OH-group of the substrate. In MARK2, the overall fold and conformation of the catalytic loop is the same; there are, however, significant differences that culminate at the RD motif (R174, D175; Fig. 4d-f): the side chain of D175 is too far from N180 further down the catalytic loop to form a hydrogen bond which is important for coordination of a divalent cation. The absence of the hydrogen bond between D175 and N180 is probably a consequence of the unusual main chain conformation of the RD motif. Possibly the catalytic loop adopts several, slightly different conformations which cannot be described adequately by a single conformation. This would agree with the assumption that the catalytic loop needs stabilization by interaction with the primary phospho-site. The unusual conformation of the RD motif in MARK2 could also be induced by interaction of the two monomers of the dimer: helix G of one molecule protrudes toward the catalytic loop of the other molecule, with the side chain of N254 at the tip of helix G approaching the catalytic aspartate (minimum distance 3.3 Å; Fig. 5). C-terminal extension, UBA domain, and regulation of MARK2 Many kinases comprise C-terminal extensions of the catalytic core that wrap around the core domain and terminate in a subdomain that binds to the N-lobe. They are probably involved in regulation of the kinase activity, comparable to regulatory proteins of other kinases, like cyclins for CDKs or TPX2 and INCENP in the case of Aurora-A and B, respectively. In PKA, for instance, the C-terminal extension spirals up in a right-handed rotation, in the MAP kinases ERK2 and p38 it winds in the opposite direction around the core domain; in either case, the terminal subdomain ends up at a similar location, close to helix C of the N-lobe. In MARK2, a corresponding extension consists of the UBA domain which is linked to the catalytic domain by a sequence of about 20 amino acids, comprising a bulge with a cluster of negatively charged residues (~N305-P315) and a straight section heading for the UBA domain (linker, ~P315-D322). This is roughly similar to ERK2 except that the UBA domain binds at some distance to the C helix (Fig. 7). In MAP kinases, the bulge residues (~L311-P321 in ERK2 and ~F308-P318 in p38) have been proposed as a common docking domain ("CD domain") for many upstream and downstream interaction partners (Tanoue et al., 2000;

9
Tanoue and Nishida, 2003). The similarity in position, conformation and amino acid composition suggests that the bulge in the structure of MARK2 may also play a role in protein-protein recognition. The UBA domain is linked to the potential CD domain by a stretch of about seven residues in extended conformation. As the UBA domain binds opposite to the catalytic cleft, at the back of the hinge region, closure of the catalytic cleft by rotation of the N-lobe (with the UBA domain attached on it) around the hinge would require further elongation of the linker (e.g. by unfolding of the CD domain) or detachment of the UBA domain from the N-lobe. Thus it seems, that the crystal structure of MARK2 represents a state with the kinase domain locked in an open (inactive) conformation. A similar mechanism for regulation of the kinase activity has been proposed for Aurora-B (Sessa et al., 2005). In the crystal structure of Aurora-B with part of the activating protein INCENP, the C-terminal tail of the kinase domain assumes an extended conformation and connects back to the N-lobe, similar to MARK2. In the case of Aurora-B, the contact to the N-lobe is mediated by INCENP, in the case of MARK2 by the UBA domain. The INCENP peptide consists of three helices that wind around the N-lobe. Interestingly, the second helix (B) binds to the same groove at the surface of the N-lobe as the UBA domain. Thus, in a superposition of the structures, helix B of INCENP and helix α3 of the UBA domain would overlap to a large extent. The role of the UBA domain in MARKs is unknown, but its presence is suggestive of a ubiquitin-related function, such as protein degradation or others (Buchberger, 2002). Other possibilities include an autoregulatory role, reminiscent of Ca/calmodulin regulated kinases whose C-terminal tail binds into the catalytic cleft, or members of the PAK or MAP kinase familes which have extra helices that bind to the N-lobe. Superposition of the MARK2-UBA structure with published UBA-ubiquitin complexes (Fig. 6d; Ohno et al., 2005; Varadan et al., 2005) shows an overlap between the catalytic domain and ubiquitin, indicating that binding of the two would be mutually exclusive. While the MARK sequence complies with the Prosite profile of the UBA domain (Fig. 6a; Hofmann and Bucher, 1996), the crystal structure reveals an unexpected conformation as the third helix is inverted compared to the known structures. It is conceivable that the inversion of helix α3 is evoked by interaction with the N-lobe, while the free UBA domain (after detachment from the catalytic domain) could adopt the normal conformation. Alternatively, the unusual conformation of the UBA domain could be a specific feature of MARK2 and related kinases. We note that the structures of UBA domains vary considerably, complementary to the variable interactions with mono- or polyubiquitin, and the different linkage modes of polyubiquitin (Chim et al., 2004). This opens a range of potential regulatory interactions which awaits further analysis. EXPERIMENTAL PROCEDURES Protein preparation, labeling, and crystallization Fragments of MARK2 from rat (GenBank No. CAB06295; Drewes et al., 1997) were cloned and expressed in E. coli strain BL21 AI (Invitrogen) by using the manufacturer's protocol. The MARK2 fragment N39-K364 described here was identified by limited proteolysis, and it is identical in sequence to human MARK2. The construct includes an N-terminal glycine (G38) left over from TEV protease cleavage of a His6 tag. All point mutations

10
(T208A/S212A, K82R) were subcloned from existing plasmids and the proteins were purified as described (Timm et al., 2003). Selenomethionine labeling was done by expression in methionine auxotrophic E.coli strain B834 (Invitrogen) using M9 minimal medium supplemented with all amino acids except methionine and 40 mg SeMet (Arcos Organics) per liter. Crystals were grown by vapor diffusion by mixing 2 µl of protein (20 mg/ml) with 2 µl of a reservoir solution containing 7-10 % PEG 3350, 0.1 M Bis-Tris pH 6.5, 0.2 M ammonium sulfate or 7-10 % PEGMME 5000, 0.1 M Bis-Tris pH 6.5, 7.5 % tacsimate at room temperature. Heavy atom derivatives were obtained by soaking in 1mM KAuCl4 or Yb(NO3)3. The kinase activities of the MARK2 constructs were assayed as described (Drewes et al., 1997) using a substrate peptide from the first repeat of tau containing S262 in the KXGS motif (TR1 peptide NVKSKIGSTENLK). Data collection, phasing, and model building X-ray data were obtained at the synchrotron beamline of the X13 Consortium at DESY, Hamburg. Crystals were flash frozen and kept at 100 K in cold nitrogen. Data reduction, statistical analysis, phasing and refinement was performed using programs of the HKL data processing system V1.97.2 (Otwinowski and Minor, 1997) and the CCP4 program package (Collaborative Computer Project Number 4, 1994). All crystals had a similar shape (short hexagonal rods) and belonged to space group P61, they fall, however, into two distinct classes differing by the length of the c-axis (see Table I). The double mutant consistently crystallized in the form with the longer c-axis, while the wild type crystallized in both forms, with a preference for the shorter c-axis. A potential MR solution was obtained with the checkpoint kinase CHK1 as a search model (PDB-ID: 1IA8; Chen et al., 2000), using the program PHASER Version 1.2 (Storoni et al., 2004) which found two molecules in the asymmetric unit. The solution was confirmed by its ability to identify heavy atom sites in weak Au- and Yb-derivatives using phases calculated from the MR solution. Heavy atom parameters were refined and experimental phases were calculated with MLPHARE, including the anomalous signals of both derivatives. Occupancies of the heavy atoms ranged between 0.27 and 0.40. Experimental phases from MLPHARE were improved with DM by solvent flattening, histogram mapping, and NCS averaging. The overall mean figure of merit after phase extension to 2.7 Å with DM was 0.79. Several rounds of manual model building using O (Jones et al., 1991), local real space refinement with RSRef2000 (Korostelev et al., 2002), and phase combination of model and experimental phases were followed by automatic refinement with CNS 1.1 (Brunger et al., 1998) for simulated annealing, switching to REFMAC5 (Murshudov et al., 1997) at later stages of refinement. At that time, higher quality crystals of the SeMet derivative became available. This SeMet derivative was the first of four MARK2 structures to be refined to the end, using the partially refined model of the double mutant as start model. The final R-factor was 0.198 (Rfree = 0.268) using all reflections up to 2.5 Å (mean I/σ(I) > 2). Structures of the double mutant with native methionine, wild type, and the K82R mutant were modelled after the SeMet derivative using the same subset of reflections for crossvalidation. The quality of the models was checked with the programs PROCHECK (Laskowski et al., 1993) and WHAT_CHECK (Hooft et al., 1996) using DSSP secondary structure assignments (Kabsch and Sander, 1983).

11
Acknowledgements: We thank the X13 consortium (Univ. of Hamburg and EMBL Outstation Hamburg) for synchrotron beam time, in particular Drs. C. Betzel, M. Perbandt, and W. Rypniewski as well as Drs. J. Müller and Th. Timm (Max-Planck Unit, Hamburg) for helpful discussions. We thank Dr. Gunter Stier (EMBL Heidelberg) for the gift of TEV protease expression plasmid. Accession Numbers: The atomic coordinates of the MARK2 structures have been deposited in the Protein Data Bank with the accession codes 1Y8G, 1ZMU, 1ZMV, and 1ZMW.

12
a, b, c: Cell constants of the individual crystals used for data collection. Average cell constants (sd: standard deviation, n: number of observations): Class 1 crystals: a = b = 119.58 Å (sd = 0.43 Å), c = 105.97 Å (sd = 0.37 Å), n = 15 Class 2 crystals: a = b = 120.93 Å (sd = 0.51 Å), c = 99.69 Å (sd = 0.23 Å), n = 8 Values in parentheses refer to the high resolution shells.
Table I Data collection and refinement statistics Construct wild type T208A, S212A double mutant Space group P61 P61 Cell constants [Å]: a = b 120.3 119.3
c 99.5 105.7 Resolution range [Å] 46.14 - 2.90 73.88 - 2.80 Data collection High resolution shell [Å] 2.95 - 2.90 2.85 - 2.80 No of observations 122003 132608 No of unique reflections 18222 21073 Completeness [%] 99.9 (100) 99.9 (100) Redundancy 6.6 (6.6) 6.2 (4.8) Rsym 0.067 (0.626) 0.102 (0.576) <I>/<sigI> 27.8 ( 3.3) 17.1 ( 2.8) Refinement High resolution shell [Å] 2.976 - 2.90 2.875 - 2.80 No of reflections working set 17281 19981 test set 942 1092 R 0.195 (0.335) 0.203 (0.313) Rfree 0.270 (0.429) 0.262 (0.353) No of residues (total 654) 589 604 No of atoms (total) 4664 4885

13
FIGURE LEGENDS Fig. 1: Domain organization of MARK2. (a) Bar diagram of MARK2 domain structure. Residue numbers refer to the longest isoform of human MARK2 (Swiss-Prot entry Q7KZI7). Phosphorylation of T596 by atypical PKC downregulates membrane localization and kinase activity (Hurov et al., 2004). The short bar diagram above that of the full length protein represents the construct used for x-ray structure analysis. The fragment used is identical in rat and human MARK2. (b) Structural sequence comparison of MARK2 and related kinases. Secondary structure is color coded (orange = α-helices, pink = 310 helices, yellow = β strands) and numbered in the usual kinase convention. Special elements are boxed (P-loop in active site, hinge between lobes, catalytic loop containing RD motif, activation segment with N-anchor, P+1 loop, and C-anchor, CD domain, UBA domain). Blue residues are not visible due to disorder. (c) Kinase activities of the MARK2 constructs. Relative activities were assayed as described (Drewes et al., 1997) using a substrate peptide from the first repeat of tau containing S262 in the KXGS motif (TR1 peptide NVKSKIGSTENLK); data show averages of 4 experiments (error bars = s.e.m.). Fig. 2: Folding of the catalytic and UBA domain of MARK2. Stereo view of an overlay of Cα-traces of molecules A and B in the asymmetric unit, based on the SeMet double mutant T280A/S212A (A blue, B with different colors depending on distance to A, blue to red). The superposition was calculated using residues 135-309 (C-lobe). Not shown are residues 38-47, 193-205 (activation loop, indicated by dashed line), and 363-364. Besides the initial residues (48-51), the largest shift in Cα positions occurs in the UBA domain (maximum 1.87 Å at residue M335). Figures 2, 4, 5, 6, and 7 were prepared with Deep View Swiss-PDB Viewer (Guex and Peitsch, 1997) and POVray for Windows (Persistence of Vision Pty. Ltd. 2004, Persistence of Vision Raytracer Version 3.5, retrieved from http://www.povray.org/). Fig. 3: Electron density maps of the SeMet T208A/S212A double mutant (stereo view). Wire-frame representation of weighted 2Fo-Fc maps calculated with REFMAC5 (Murshudov et al., 1997) and contoured at 1 σ level. Superimposed to the electron density is the final model. (a) View on the C-terminal end of the activation segment. Residues L206-S212A were omitted before calculation of the map. The view is perpendicular to the two-fold NCS axis relating molecules A and B. Close to the center are cysteines C210 of both molecules, which form a disulfide bridge (orange) in the structures of the MARK2 double mutant. (b) View on the C-terminal part of the peptide stretch that links the UBA domain to the catalytic domain. Residues Y316 to K324 were omitted before calculation of the map. Although there is virtually no contact to the catalytic domain or to another molecule, the electron density is well defined. The figure was generated with PyMOL (DeLano, 2002). Fig. 4: Comparison of MARK2 and Aurora-A kinase domains. a, d: MARK2 double mutant, b, e: inactive Aurora-A (PDB-ID: 1MUO), c, f: Aurora-A in the fully activated state (PDB-ID: 1OL5) with a framgent of the activating protein TPX2 (gray). In all panels, the catalytic loop is red and the activation segment purple. Catalytically important residues are shown in stick model representation. All three structures are in the same orientation by least-squares superposition of their catalytic loops (residues 171-183 in MARK2, 252-264 in Aurora-A). a-c: Front view, showing the open (a, b; inactive state) or closed (c, active state) cleft between the lobes. In the active state (c) the activation loop is ordered and points to the right side, in the inactive state, it is disordered and presumably leans to the left side. In a and b, plausible conformations of the invisible part of the activation loop are indicated by dotted lines. d-f: Side view, showing a close-up of the nucleotide binding site with catalytic loop and helix C; the P-loop has been omitted to allow an unobstructed view on the invariant ion pair

14
(K82 and E100 in MARK2, K162 and E181 in Aurora-A) that coordinates α- and β-phosphates of the bound nucleotide in the active state (f). In the inactive structures (d, e) the ion pair interaction is disrupted. Part of the catalytic loop around the enzmatically active aspartate (D175 in MARK2, D256 in Aurora-A) and the preceding arginine (R174 in MARK2, R255 in Aurora-A) is represented by its Cα-trace to show the subtle differences in the conformations of MARK2 and Aurora-A. In the active state (c, f) the arginine of the conserved RD motif is hydrogen bonded to phosphothreonine pT288. Fig. 5: Intermolecular contacts in MARK2 dimers (stereo view). Ribbon diagram of the wild type dimer viewed along the non-crystallographic two-fold symmetry axis (molecule A blue, B purple). Residues involved in intermolecular contacts (at least one atom closer than 4 Å to an atom of the other molecule) are shown in stick model representation. Contact residues are color-coded after their distance from cyan (4 Å) to red (~2 Å). The shortest contact (2.07 Å distance) is between cysteines C210 in the double mutant (see Supplement, Fig. S9) which form an interchain disulfide bridge. No disulfide bridge is observed in the wild type structure due to disorder. Most of the contact residues are in two zones: C-terminal anchor of the activation loop and the following loop preceding helix F (zone 1, residues 206-227) and helix G and part of its N-terminal loop (zone 2, residues 251-261). In the double mutant (not shown, see Supplement Fig. S9), zone 1 comprises 15 contact residues in the range from D207 to D227, zone 2 all but one residue in the range D251-R261. For the wild type the corresponding numbers are lower (9 residues in zone 1, 5 residues in zone 2), due to disordered residues L206 to C210. Residues S92, S93, and K96 at the N-terminus of helix C form another cluster of contact residues (zone 3). Helix G and the preceding loop (zone 2) in one molecule inserts into the space between zones 1 and 3 of the other molecule, making extensive contacts with both of these zones and approaching the RD motif (R174, D175) in the catalytic loop. Fig. 6: UBA domain. a: Structural sequence alignment of UBA domains. Parts of the sequences determined by NMR or X-ray analysis are highlighted by grey and colored background (orange for helices α1, α2, and α3, pink for other helices; boundaries determined with Promotif (Hutchinson and Thornton, 1996)). Leading or trailing residues on white background were not part of the constructs. The parts enclosed by the black line correspond to the UBA domain identified by Prosite scans (Release 19.2, Bucher and Bairoch, 1994). Residues at the C-terminus of MARK2 are printed in purple to indicate that the structural alignment breaks down for this part of the sequence: starting with R350, the polypeptide chain diverges from the common trace of the other UBA structures resulting in an orientation of the final helix that is reversed compared to normal UBA domains. Residues that have been shown to interact with ubiquitin are marked by blue underlines (for HHR23A, PDB-ID 1DVO, only residues of the primary interaction site are highlighted). In the case of MARK2, residues in contact with the N-lobe of the kinase domain are marked. Columns: source, protein name, domain, Prosite score, PDB-ID. A plus sign following the Prosite score indicates that binding to ubiquitin has been reported. The list is sorted according to the Prosite score, sequences at the end of the list are not recognized as UBA domains (no score), although they are structurally similar. b: (stereo view) Overlay of the MARK2 UBA domain (yellow) with that of HHR23A (green; PDB-ID: 1IFY; Mueller and Feigon, 2002) after least-squares superposition of 9 residues in helix α1. Residues M335 and Y337 of the MGY motif are shown in stick model representation. Helix α2 of MARK2 is tilted outwards by about 20° compared to HHR23A. This is accompanied by a change in the main chain conformation of the MGY loop that translates into a ~30° inward rotation of the aromatic ring of tyrosine Y337. At the end of

15
helix α2, the peptide chains bend in different directions, in such a way that helix α3 ends up at almost the same position but with reversed orientation. c: (stereo view) Details of the binding interactions between the UBA domain and the N-lobe of the catalytic domain of MARK2. All three leucines at the end of helix α3 are involved in hydrophobic interactions with the N-lobe. The final leucine (L361) is also engaged in hydrophobic interactions with helix α1, and plays an important role for the cohesion of the UBA domain. In normal UBA structures it is the almost invariant leucine (L359 in MARK) that is most important for the internal interactions. d: (stereo view) UBA domain of MARK2 overlaid with that of Dsk2p in complex with ubiquitin. The UBA domains are green (Dsk2p, PDB-ID: 1WR1; Ohno et al., 2005) and purple (MARK2), ubiquitin red, and MARK2 kinase domain yellow. In the overlay by least-squares fit of 10 Cα atoms of helices α1, the ubiquitin locates above the MARK2 kinase domain, with a small overlap in the β2-β3 region of the N-lobe. Fig. 7: Common docking domain and ED site of MAP kinases compared to MARK2. The structures of (a) MARK2 and (b) ERK2 (PDB-ID: 2ERK; Canagarajah et al., 1997) are shown in the same orientations after least-squares superposition of 35 residues from helix E to the catalytic loop. The common docking domain (CD, in red) according to (Tanoue and Nishida, 2003) is C-terminal to the kinase domain and corresponds in MARK to the first half of the tether connecting the kinase domain to the UBA domain (residues ~305-315). The C-terminal extensions following the CD domain (linker and UBA domain in MARK2) are shown in purple. Characteristic for the CD domain is a cluster of negatively charged residues exposed to the surface, located in a bulge at the end of the catalytic domain (stick model representation).

16
REFERENCES Bayliss, R., Sardon, T., Vernos, I. and Conti, E. (2003) Structural basis of Aurora-A
activation by TPX2 at the mitotic spindle. Mol Cell, 12, 851-862. Benton, R., Palacios, I.M. and St Johnston, D. (2002) Drosophila 14-3-3/PAR-5 is an
essential mediator of PAR-1 function in axis formation. Dev Cell, 3, 659-671. Biernat, J., Gustke, N., Drewes, G., Mandelkow, E.M. and Mandelkow, E. (1993)
Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron, 11, 153-163.
Biernat, J., Wu, Y.Z., Timm, T., Zheng-Fischhofer, Q., Mandelkow, E., Meijer, L. and Mandelkow, E.M. (2002) Protein kinase MARK/PAR-1 is required for neurite outgrowth and establishment of neuronal polarity. Mol Biol Cell, 13, 4013-4028.
Brajenovic, M., Joberty, G., Kuster, B., Bouwmeester, T. and Drewes, G. (2004) Comprehensive proteomic analysis of human Par protein complexes reveals an interconnected protein network. J Biol Chem, 279, 12804-12811.
Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., Read, R.J., Rice, L.M., Simonson, T. and Warren, G.L. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr, 54, 905-921.
Buchberger, A. (2002) From UBA to UBX: new words in the ubiquitin vocabulary. Trends Cell Biol, 12, 216-221.
Bucher, P. and Bairoch, A. (1994) A generalized profile syntax for biomolecular sequence motifs and its function in automatic sequence interpretation. Proc Int Conf Intell Syst Mol Biol, 2, 53-61.
Canagarajah, B.J., Khokhlatchev, A., Cobb, M.H. and Goldsmith, E.J. (1997) Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell, 90, 859-869.
Chang, C.I., Xu, B.E., Akella, R., Cobb, M.H. and Goldsmith, E.J. (2002) Crystal structures of MAP kinase p38 complexed to the docking sites on its nuclear substrate MEF2A and activator MKK3b. Mol Cell, 9, 1241-1249.
Cheetham, G.M., Knegtel, R.M., Coll, J.T., Renwick, S.B., Swenson, L., Weber, P., Lippke, J.A. and Austen, D.A. (2002) Crystal structure of aurora-2, an oncogenic serine/threonine kinase. J Biol Chem, 277, 42419-42422.
Chen, P., Luo, C., Deng, Y., Ryan, K., Register, J., Margosiak, S., Tempczyk-Russell, A., Nguyen, B., Myers, P., Lundgren, K., Kan, C.C. and O'Connor, P.M. (2000) The 1.7 A crystal structure of human cell cycle checkpoint kinase Chk1: implications for Chk1 regulation. Cell, 100, 681-692.
Chim, N., Gall, W.E., Xiao, J., Harris, M.P., Graham, T.R. and Krezel, A.M. (2004) Solution structure of the ubiquitin-binding domain in Swa2p from Saccharomyces cerevisiae. Proteins, 54, 784-793.
Collaborative Computer Project Number 4. (1994) The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. D, 50, 760-763.
DeLano, W.L. (2002) PyMOL. DeLano Scientific, San Carlos, CA, USA. Drewes, G. (2004) MARKing tau for tangles and toxicity. Trends Biochem Sci, 29, 548-555. Drewes, G., Ebneth, A., Preuss, U., Mandelkow, E.M. and Mandelkow, E. (1997) MARK, a
novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell, 89, 297-308.
Elbert, M., Rossi, G. and Brennwald, P. (2005) The yeast par-1 homologs kin1 and kin2 show genetic and physical interactions with components of the exocytic machinery. Mol Biol Cell, 16, 532-549.

17
Guex, N. and Peitsch, M.C. (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis, 18, 2714-2723.
Hofmann, K. and Bucher, P. (1996) The UBA domain: a sequence motif present in multiple enzyme classes of the ubiquitination pathway. Trends Biochem Sci, 21, 172-173.
Hooft, R.W., Vriend, G., Sander, C. and Abola, E.E. (1996) Errors in protein structures. Nature, 381, 272.
Hurov, J.B., Watkins, J.L. and Piwnica-Worms, H. (2004) Atypical PKC phosphorylates PAR-1 kinases to regulate localization and activity. Curr Biol, 14, 736-741.
Huse, M. and Kuriyan, J. (2002) The conformational plasticity of protein kinases. Cell, 109, 275-282.
Hutchinson, E.G. and Thornton, J.M. (1996) PROMOTIF - a program to identify and analyze structural motifs in proteins. Protein Sci, 5, 212-220.
Hutchison, M., Berman, K.S. and Cobb, M.H. (1998) Isolation of TAO1, a protein kinase that activates MEKs in stress-activated protein kinase cascades. J Biol Chem, 273, 28625-28632.
Jones, T.A., Zou, J.Y., Cowan, S.W. and Kjeldgaard. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A, 47, 110-119.
Kabsch, W. and Sander, C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers, 22, 2577-2637.
Kemphues, K. (2000) PARsing embryonic polarity. Cell, 101, 345-348. Knighton, D.R., Zheng, J.H., Ten Eyck, L.F., Xuong, N.H., Taylor, S.S. and Sowadski, J.M.
(1991) Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science, 253, 414-420.
Korostelev, A., Bertram, R. and Chapman, M.S. (2002) Simulated-annealing real-space refinement as a tool in model building. Acta Crystallogr D Biol Crystallogr, 58, 761-767.
Laskowski, R.A., MacArthur, M.W., Moss, D.S. and Thornton, J.M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr., 26, 283-291.
Lizcano, J.M., Goransson, O., Toth, R., Deak, M., Morrice, N.A., Boudeau, J., Hawley, S.A., Udd, L., Makela, T.P., Hardie, D.G. and Alessi, D.R. (2004) LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. Embo J, 23, 833-843.
Lowe, E.D., Noble, M.E., Skamnaki, V.T., Oikonomakos, N.G., Owen, D.J. and Johnson, L.N. (1997) The crystal structure of a phosphorylase kinase peptide substrate complex: kinase substrate recognition. Embo J, 16, 6646-6658.
Madhusudan, Akamine, P., Xuong, N.H. and Taylor, S.S. (2002) Crystal structure of a transition state mimic of the catalytic subunit of cAMP-dependent protein kinase. Nat Struct Biol, 9, 273-277.
Mandelkow, E.M., Thies, E., Trinczek, B., Biernat, J. and Mandelkow, E. (2004) MARK/PAR1 kinase is a regulator of microtubule-dependent transport in axons. J Cell Biol, 167, 99-110.
Manning, G., Whyte, D.B., Martinez, R., Hunter, T. and Sudarsanam, S. (2002) The protein kinase complement of the human genome. Science, 298, 1912-1934.
Min, X., Lee, B.H., Cobb, M.H. and Goldsmith, E.J. (2004) Crystal structure of the kinase domain of WNK1, a kinase that causes a hereditary form of hypertension. Structure (Camb), 12, 1303-1311.
Mueller, T.D. and Feigon, J. (2002) Solution structures of UBA domains reveal a conserved hydrophobic surface for protein-protein interactions. J Mol Biol, 319, 1243-1255.

18
Muller, J., Ritt, D.A., Copeland, T.D. and Morrison, D.K. (2003) Functional analysis of C-TAK1 substrate binding and identification of PKP2 as a new C-TAK1 substrate. Embo J, 22, 4431-4442.
Murshudov, G.N., Vagin, A.A. and Dodson, E.J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr, 53, 240-255.
Nolen, B., Taylor, S. and Ghosh, G. (2004) Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell, 15, 661-675.
Ohno, A., Jee, J., Fujiwara, K., Tenno, T., Goda, N., Tochio, H., Kobayashi, H., Hiroaki, H. and Shirakawa, M. (2005) Structure of the UBA domain of Dsk2p in complex with ubiquitin molecular determinants for ubiquitin recognition. Structure (Camb), 13, 521-532.
Otwinowski, Z. and Minor, W. (1997) Processing of X-ray Diffraction Data Collected in Oscillation Mode. In Carter, C.W. and Sweet, R.M. (eds.), Methods in Enzymology, Vol. 276: Macromolecular Crystallography, part A. Academic Press, pp. 307-326.
Sessa, F., Mapelli, M., Ciferri, C., Tarricone, C., Areces, L.B., Schneider, T.R., Stukenberg, P.T. and Musacchio, A. (2005) Mechanism of Aurora B activation by INCENP and inhibition by hesperadin. Mol Cell, 18, 379-391.
Shindyalov, I.N. and Bourne, P.E. (1998) Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng, 11, 739-747.
Storoni, L.C., McCoy, A.J. and Read, R.J. (2004) Likelihood-enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr, 60, 432-438.
Tanoue, T., Adachi, M., Moriguchi, T. and Nishida, E. (2000) A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat Cell Biol, 2, 110-116.
Tanoue, T. and Nishida, E. (2003) Molecular recognitions in the MAP kinase cascades. Cell Signal, 15, 455-462.
Taylor, S.S., Radzio-Andzelm, E. and Hunter, T. (1995) How do protein kinases discriminate between serine/threonine and tyrosine? Structural insights from the insulin receptor protein-tyrosine kinase. Faseb J, 9, 1255-1266.
Timm, T., Li, X.Y., Biernat, J., Jiao, J., Mandelkow, E., Vandekerckhove, J. and Mandelkow, E.M. (2003) MARKK, a Ste20-like kinase, activates the polarity-inducing kinase MARK/PAR-1. Embo J, 22, 5090-5101.
Tomancak, P., Piano, F., Riechmann, V., Gunsalus, K.C., Kemphues, K.J. and Ephrussi, A. (2000) A Drosophila melanogaster homologue of Caenorhabditis elegans par-1 acts at an early step in embryonic-axis formation. Nat Cell Biol, 2, 458-460.
Varadan, R., Assfalg, M., Raasi, S., Pickart, C. and Fushman, D. (2005) Structural determinants for selective recognition of a Lys48-linked polyubiquitin chain by a UBA domain. Molecular Cell, 18, 687-698.
a c
bP-loop hinge
40 50 60 64 70 80 82 90 100 110 116 120 130 135RnMARK2 [ 1Y8G ] NSA T SADEQPH I GNYR L L K T I GKGNFAKVK L ARH I L TGKEVAVK I I DK TQL N - - - - - - - SSS L QK L F REVR I MKV L N - HPN I VK L FEV I E T - - - - - - EK T L Y L VMEYASGGE
122 130 140 144 150 160 162 170 181 191 197 201 211 216HsAurora-A [ 1OL5 ] E SKKRQWA L EDF E I GRP L GKGKFGNVY L AREKQSK F I L A LKV L F KAQL EKAG - - - - - - VEHQL RREVE I QSH L R - HPN I L R L YGY F HD - - - - - - A T RVY L I L EYAP LGT
107 110 120 130 140 144 150 160 162 170 181 191 197 201 211 216HsAurora-A [ 1MUO ] ENNPEEE L ASKQKNE ESKKRQWA L EDF E I GRP L GKGKFGNVY L AREKQSK F I L A LKV L F KAQL EKAG - - - - - - VEHQL RREVE I QSH L R - HPN I L R L YGY F HD - - - - - - A T RVY L I L EYAP LGT
1 6 16 20 26 36 38 46 55 65 71 75 85 90HsCHK1 [ 1IA8 ] MAVP F V EDWD L VQT L GEGAYGEVQL A VNRV T EEAVAVK I VDMKRAVD - - - - - - - - CPEN I KKE I C I NKML N - HENV V K FYGHRRE - - - - - - GN I QY L F L EYCSGGE
1 10 16 26 30 36 46 48 56 73 84 90 94 104 109OcPHKg [ 1QL6 ] T RDAA L PGSHS T HGF Y ENY EPKE I L GRGVSSVVRRC I HKP T CKEYAVK I I DV TGGGS F SAEEVQE L REA T L KEVD I L RKVSGHPN I I QL KDT YE T - - - - - - NT F F F L V F D LMK KGE
1 10 20 30 34 40 50 52 59 69 79 85 89 100 104 108RnERK2 [ 2ERK ] MA AAAA AGP EMVRGQV F DVGPRY T N L SY I GEGAYGMVCSAYDN L NKVRVA I KK I S - P F EH - - - - - - - QT YCQRT L RE I K I L L RF R - HEN I I G I ND I I R - AP T I EQMKDVY I VQD LM - E T D
1 11 21 31 35 41 51 53 61 71 81 87 91 103 107 111MmMAPKp38 [ 1LEW ] MSQERP T F YRQE L NK T I WE VPERYQN L SPVGSGAYGSVCAA F DT K TGHRVAVKK L SRP FQS - - - - - - - I I HAKRT YRE L R L L KHMK - HENV I GL L DV F T P ARS L EE F NDVY L V T H LM - GAD
1 10 20 30 40 50 54 60 70 72 80 91 101 107 111 117 121 126MmPKA [ 1ATP ] GNAAAAKKGSEQE SVKE F L A KAKEDF L K KWE T P SQNT AQL DQF DR I K T L GTGSFGRVML VKHKESGNHYAMK I L DKQKVVK L K - - - - - - Q I EHT L NEKR I L QAVN - F P F L VK L E F S F KD - - - - - - NSN L YMVMEYVAGGE
helix A beta1 beta2 beta3 helix B helix C beta4 beta5
activation segmentcatalytic loop N-anchor activation loop C-anchor
Mg-bind loop P+1 loop140 150 160 170 174 180 190 193 200 208 210 217 220 230 240 250 260
RnMARK2 [ 1Y8G ] V F DY L VAHGRMK EKEARAK F RQ I VSAVQYCHQK F I VHRDL KAENL L L DADMN I K I ADFGF SNE F T FGN - - - K L DT - F CGSPPYAAPE L FQGKK - - - - - YDGPEVDVWS L GV I L Y T L VSGS L P F DG - QN L K E L RERV L RGK221 231 241 251 255 261 271 274 281 288 290 297 300 309 319 329 339
HsAurora-A [ 1OL5 ] VYRE L QK L SK F DEQRT A T Y I T E L ANA L S YCHS KRV I HRD I K PENL L L GSAGE L K I ADFGWSVHAPSS - - - - RRT T - L CGT L DY L PPEM I EGRM - - - - - - HDEKVD L WS L GV L CYE F L VGKPP F EA - NT YQE T Y KR I SRVE221 231 241 251 255 261 271 274 281 288 290 297 300 309 319 329 339
HsAurora-A [ 1MUO ] VYRE L QK L SK F DEQRT A T Y I T E L ANA L S YCHS KRV I HRD I K PENL L L GSAGE L K I ADFGWSVHAPSS - - - - RRT T - L CGT L DY L PPEM I EGRM - - - - - - HDEKVD L WS L GV L CYE F L VGKPP F EA - NT YQE T Y KR I SRVE95 105 115 125 129 135 145 148 155 166 168 175 178 188 198 208 219
HsCHK1 [ 1IA8 ] L F DR I EPD I GMP EPDAQRF F HQLMAGVVY L HG I G I T HRD I K PENL L L DERDN L K I SDFGL A T V F RYNNRER L L NK - MCGT L PYVAPE L L KRRE - - - - - F HAEPVDVWSCG I V L T AML AGE L PWDQPSDSCQEYSDWKEKK114 124 134 144 148 154 164 167 174 182 184 191 194 209 219 229 239
OcPHKg [ 1QL6 ] L F DY L T EKV T L S EKE T RK I MRA L L EV I CA L HK L N I VHRDL KPEN I L L DDDMN I K L TDFGF SCQL DPGE - - - K L RE - VCGT PSY LAPE I I ECSMNDNHPGYGKEVDMWS TGV I MY T L L AGSPP FWH - RKQMLML RM I MSGN113 122 132 142 146 152 162 165 172 183 186 193 196 206 216 226 236
RnERK2 [ 2ERK ] L Y K L L K TQ - H L SNDH I CY F L YQ I L RGL K Y I HSANV L HRDL KPSNL L L NT T CD L K I CDFGL ARVADPDHDHTGF L TEYVA T RWYRAPE I ML NSK - - - - - GY T KS I D I WSVGC I L AEML SNRP I F PG - KHY L DQL NH I L G I L116 125 135 145 149 155 161 165 168 175 180 183 190 193 203 213 223 233
MmMAPKp38 [ 1LEW ] L NN I V KCQ - K L T DDHVQF L I YQ I L RGL K Y I HSAD I I HRDL KPSNL A VNEDCE L K I LDFGL ARHT DDE - - - - - - MTGYVA T RWYRAPE I ML NWM - - - - - HYNQT VD I WSVGC I MAE L L TGRT L F PG - T DH I DQL K L I L R L V131 141 151 161 165 171 181 184 191 197 199 206 209 218 228 238 248
MmPKA [ 1ATP ] MF SH L RR I GRF S EPHARF Y AAQ I V L T F E Y L HS L D L I YRDL KPENL L I DQQGY I QV TDFGF A KRVKG - - - - - RTWT - L CGT PEY LAPE I I L SKG - - - - - - YNKAVDWWA L GV L I YEMAAGYPP F F A - DQP I Q I YEK I VSGK
helix D helix E beta6 beta7 beta8 beta9 helix EF helix F helix G
CD domain UBA domain270 280 290 300 310 320 330 340 350 360 364
RnMARK2 [ 1Y8G ] YR I P F - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - YMS T DCEN L L KK F L I L NPSKRG - - - - - T L EQ I MKDRWMNVGHEDDE L KPYVEP L PDYKDPRRT E LMV SMGY T REE I QDS L VGQRYNEVMA T Y L L L GY K349 359 369 379 389 399 403
HsAurora-A [ 1OL5 ] F T F PD - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - F V T EGARD L I SR L L KHNPSQRP - - - - - ML REV L EHPW I T ANSSKPSNCQNKESASKQS349 359 369 379 389 399 403
HsAurora-A [ 1MUO ] F T F PD - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - F V T EGARD L I SR L L KHNPSQRP - - - - - ML REV L EHPW I T ANSSKPSNCQNKESASKQS229 241 251 261 271 281 289
HsCHK1 [ 1IA8 ] T Y L NP - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - WKK I DSAP L A L L HK I L VENPSAR I - - - - - T I PD I KKDRWYNKP L KKGAKRPRV T SGGVSESPSG249 263 273 283 293 298
OcPHKg [ 1QL6 ] YQFGSPE - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - WDDY SDT V KD L VSRF L VVQPQKRY - - - - - T AEEA L AHP F FQQYVVEEVRHF243 253 263 273 283 287 297 307 317 327 337 347 358
RnERK2 [ 2ERK ] - - - GSPSQED L NC I I N L K ARNY L L S L PHKNK V PWNR L F PNADSKA L D L L DKML T F NPHKR I - - - - - EVEQA L AHPY L EQYYDPSDEP I AEAP F K F DME L DD L PKEK L KE L I F EE T ARFQPGYRS240 250 260 270 280 284 294 304 314 323 333 343 353 360
MmMAPKp38 [ 1LEW ] - - - GT PGAE L L KK I S SESARNY I QS L AQMP KMNF ANV F I GANP L A VD L L E KML V L DSDKR I - - - - - T AAQA L AHAY F AQYHDPDDEPVAD - PYDQS F E SRD L L I DEWKS L T YDEV I S F VPPP L DQEEMES258 268 278 288 293 303 313 323 333 343 350
MmPKA [ 1ATP ] VRF PS - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - HF SSD L KD L L RN L L QVD L T KRFGN L KNGVND I KNHKWF A T T DW I A I YQRKVEAP F I PK F KGPGDT SNF DDYEEEE I RVS I NEKCGKE F T E F
helix H helix I helix J

Figure 2

Figure 3

Figure 4

Figure 5

194 206 230 243
Hs TDRD3 UBA 14.79 [ 1WJI ] G V Y R E L V D E K A L K H I T E M - G F - - S K E A S R Q A L M D N G N N - L E A A L N V L L T S N K Q K P V M G P P156 174 198 204
Hs HHR23A UBA1 14.72 + [ 1IFY ] T L V T G S E Y E T M L T E I M S M - GY - - E R E R V V A A L R A S Y N N - P H R A V E Y L L T G I P G651 683 707 710
At UBP14 UBA2 14.23 [ 1WIV ] L L S H M D D P D I D A P I S H Q T S D I D Q S S V D T L L S F - G F - - A E D V A R K A L K A S G G D - I E K A T DW V F N N P N342 374 398 401
At RSGI RUH-014 UBA 13.75 [ 1VG5 ] S R Q A P I A N A A V L P Q S Q G R V A A S E E Q I Q K L V A M - G F - - D R T Q V E V A L A A A D D D - L T V A V E I L M S Q Q A2 14 39 52
Mm U33K UBA 13.20 [ 1WHC ] M A E L T A L E S L I E M - G F - - P R G R A E K A L A L T G N Q G I E A A M DW L M E H E D D P D V D E P L319 331 355 363
Hs HHR23A UBA2 12.88 + [ 1DVO ] Y I Q V T P Q E K E A I E R L K A L - G F - - P E S L V I Q A Y F A C E K N - E N L A A N F L L S Q N F D D E318 331 355 363
Hs HHR23A (P333E) UBA2 12.88 + [ 1F4I ] Y I Q V T P Q E K E A I E R L K A L - G F - - E E S L V I Q A Y F A C E K N - E N L A A N F L L S Q N F D D E381 403 427 430
Hs UBAP1 UBA1 12.19 [ 1WGN ] A Y S E L Q M L S P S E R Q C V E T V V N MG - Y - - S Y E C V L R A M K K K G E N - I E Q I L D Y L F A H G Q328 343 368 373
Sc Dsk2p UBA 12.14 + [ 1WR1 ] T R P P E E R Y E H Q L R Q L N D M - G F - F D F D R N V A A L R R S G G - S V Q G A L D S L L N G D V
469 501 525 538
Mm NUB1 UBA3 12.09 [ 1VEG ] N P H MWW L Q D A D P E N N S R Q A S P S Q E S I N Q L V Y M - G F - - D T V V A E A A L R V F G G N - V Q L A A Q T L A H H G G S L P P D L Q F 594 626 651 664
At UBP14 UBA1 11.41 [ 1VEK ] G E E L L P D G V P E E V M E S A Q P V A N E E I V A Q L V S M - G F - - S Q L H C Q K A A I N T S N A G V E E A M NW L L S H M D D P D I D A P I323 336 350 359 364
Rn MARK2 UBA 9.94 [ 1Y8G ] . . . P Y V E P L P D Y K D P R R T E L M V S M - GY - - T R E E I Q D S L V G Q R Y N - - E VMA T Y L L L GY K387 405 431 436
Hs p62 UBA 9.61 + [ 1Q02 ] P P E A D P R L I E S L S Q M L S M - G F S D E G GW L T R L L Q T K N Y - D I G A A L D T I Q Y S K H1 29 54
Mm RSGI RUH-013 UBA 7.58 [ 1VDL ] M T V E Q N V L Q Q S A A Q K H Q Q T F L N Q L R E I T G I N - D A Q I L Q Q A L K D S N G N - L E L A V A F L T A K N A K T P P Q E E T77 92 116
Archaea AENAC * * [ 1TR8 ] M E I P E D D I E L V M N Q T G A - - S R E D A T R A L Q E T G G D - L A E A I M R L S137 154 177
Sc SWA2P * * + [ 1PGY ] A L V D E V K D M E I A R L M S L - G L - - S I E E A T E F Y E N D V T - - Y E R Y L E I L K S K Q K E1 17 42
Rn p47 * * + [ 1V92 ] M A E E R Q D A L R E F V A V T G A - - E E D R A R F F L E S A GW D L Q I A L A S F Y E D G G561 579 603
Hs TAP * * [ 1OAI ] P T L S P E Q Q E M L Q A F S T Q S GM N - - L EW S Q K C L Q D N NW D - Y T R S A Q A F T H L K A K G E I P E V A F M K
alpha 1 alpha 2 alpha3
Figure 6 a

Figure 6 b,c,d

Figure 7

1
Structure 14, 173-183 (2006) DOI 10.1016/j.str.2005.09.022
Structure of the catalytic and ubiquitin-associated domains of the protein kinase MARK / Par-1
Saravanan Panneerselvam*, Alexander Marx*, Eva-Maria Mandelkow, Eckhard Mandelkow§
Max-Planck-Unit for Structural Molecular Biology
c/o DESY, Notkestrasse 85, 22607 Hamburg, Germany
SUPPLEMENT

2
EXPERIMENTAL PROCEDURES Cloning, protein expression and purification Crystallizable fragments of MARK2 (GeneBank No. CAB06295; Drewes et al., 1997) were identified by expressing different constructs of the protein and subjecting them to limited proteolysis using various proteases (e.g. trypsin, chymotrypsin, GluC, AspN, thermolysin). Stable fragments were identified by N-terminal sequencing and mass spectrometry. The fragment N39-K364 was cloned into the GATEWAY pDEST17 expression vector (Invitrogen). The protein has an N-terminal tag consisting of six histidines which was cleaved off with TEV protease after purification. All point mutations (T208A/S212A, K82R) were subcloned from existing plasmids (Timm et al., 2003). Proteins (wild type and mutant) were expressed in E.coli strain BL21 AI (Invitrogen). Cells were induced overnight at 24°C by adding 0.2% arabinose at OD600 ≈ 0.6 . Cells harvested from a 1.5 L culture were resuspended in 40 ml lysis buffer (50mM Hepes pH 7.2, 300 mM NaCl, 5% glycerol supplemented with one tablet EDTA-free complete protease inhibitor cocktail (Roche)) and lysates were prepared by passing twice through a French press cell. The expressed proteins were purified in four steps: Ni-NTA affinity chromatography, TEV protease cleavage to remove the His-tag, ion exchange chromatography and gel filtration. Clarified lysates were applied to a Ni-NTA affinity column of 5ml Ni-NTA beads (Qiagen) and purified according to the manufacturer's protocol. The proteins were eluted with an imidazole gradient in buffer A (50 mM Hepes pH 7.2, 300 mM NaCl and 5% glycerol). Pure protein fractions were pooled and mixed with purified recombinant tobacco etch virus (TEV) protease in a ratio of 1:15-1:20 to cleave off the His-tag. The protein mixtures were dialyzed overnight against buffer B (50 mM Hepes pH 7.2, 100 mM NaCl, 5% glycerol, 1 mM EGTA, 1 mM DTT). Around 1 ml Ni-NTA beads were added to the dialyzed proteins to bind the His-tagged TEV protease and the uncleaved MARK2 protein. After one hour, the Ni-NTA beads were removed and the salt concentration was reduced to 100 mM by dilution with salt free buffer (50 mM Hepes pH 7.2 and 5% glycerol). The proteins were further purified by cation exchange chromatography (MonoS HR 10/10 column, Amersham Biosciences) using a 100 to 1000 mM NaCl gradient. Pooled fractions were concentrated using Ultrafree-30 concentrators and applied to a gel filtration column (Hiload 16/60, Amersham Biosciences) equilibrated with 50 mM Bis-Tris sulphate pH 6.5, 250 mM NaCl, 5% glycerol. Peak fractions were pooled and concentrated to ~20 mg/ml. Selenomethionine labelling SeMet labelled protein of the double mutant MARK2 was prepared by expression in methionine auxotrophic E.coli strain B834 (Invitrogen) using M9 minimal medium supplemented with all amino acids except methionine and 40 mg SeMet (Arcos Organics) per liter. The purification procedure was essentially the same as for the unlabelled proteins. The incorporation of SeMet was estimated (nearly 100%, i.e. 10 SeMet residues per molecule) by mass spectrometry. Kinase activity Kinase activities were assayed in 50 mM Tris-HCl pH 7.5, 5 mM MgCl2, 2 mM EGTA, 0.5 mM PMSF, 0.5 mM DTT, 0.5 mM benzamidine for 30 minutes at 30°C. Final concentration of [32P] ATP (3.7*107 MBq/mol) and substrate peptide were 100 µM. A peptide derived

3
from the first repeat of tau protein containing S262 in the KXGS motif (TR1-peptide NVKSKIGSTENLK, Drewes et al. 1997) was used as substrate. Reactions were stopped by addition of half the volume of 30 %(w/v) TCA. After centrifugation the supernatant was applied to phosphocellulose-paper discs, washed with phosphoric acid (0.1 M), dried by air and radioactivity was measured in a scintillation counter (Tricarb 1900 CA, Packard). Data show averages of 4 experiments (error bars = s.e.m.). Crystallization Initial crystallization trials were performed by the vapour diffusion technique using commercially available screening kits (Hampton Research). After optimization, suitable crystals were grown by mixing 2 µl of protein (20 mg/ml) with 2 µl of a reservoir solution containing 7-10 % PEG 3350, 0.1 M Bis-Tris pH 6.5, 0.2 M ammonium sulfate or 7-10 % PEGMME 5000, 0.1 M Bis-Tris pH 6.5, 7.5 % tacsimate at room temperature. SeMet protein crystals were grown under similar conditions. Data collection and phasing X-ray diffraction data were collected with synchrotron radiation at the Consortium beamline X13, EMBL Outstation, HASYLAB (DESY, Hamburg). Before data collection, crystals were soaked in cryoprotectant solution (15 % PEG 3350, 0.1 M Bis-Tris 0.2 M ammonium sulphate and 15% glycerol or 7- 10 % PEGMME 5000, 0.1 M Bis-Tris 7.5 % Tacsimate and 15% glycerol) for a few minutes and rapidly cooled to 100 K in a stream of cold nitrogen (Oxford Cryosystems). Data reduction and statistical analysis (Table SII) was performed with programs DENZO, XDISPLAYF, and SCALEPACK of the HKL data processing system V1.97.2 (Otwinowski and Minor, 1997). Programs of the CCP4 package and its graphical user interface CCP4i (Potterton et al., 2003) were used for further analysis, phasing, and model refinement. All crystals had a similar shape (short hexagonal rods) and belonged to space group P61. The crystals fall into two distinct classes differing by the length of the c-axis (hexagonal axis). The double mutant consistently crystallized in the form with the long c-axis (c = 106.0 Å, sd = 0.37 Å, based on 15 crystals including crystals grown from SeMet protein and crystals soaked with heavy atom salts), while the wild type crystallized in both forms, with a preference for the short c-axis (c = 99.7 Å, sd = 0.23 Å, 8 out of 10 crystals). The crystal of the K82R mutant had also a short c-axis (c = 99.66 Å). An MR solution was obtained with the human checkpoint kinase CHK1 as a search model (PDB-ID: 1IA8; Chen et al., 2000), using the program PHASER Version 1.2 (Storoni et al., 2004) which found two molecules in the asymmetric unit (corresponding to a Matthews parameter of 2.9 Å3/Da). Both of the enantiomorphic space groups P61 and P65 were allowed for MR solutions. Using phases from the MR solution, crystals of the double mutant soaked in 1mM of KAuCl4 or Yb(NO3)3 were found to have heavy atoms incorporated at distinct sites, one site per molecule. Using these sites and the MR phases, heavy atom parameters were refined and experimental phases were calculated with MLPHARE (Otwinowski, 1991), including the anomalous signals of both derivatives. Occupancies of the heavy atoms ranged between 0.27 and 0.40. The overall figure of merit of the phases calculated with MLPHARE was 0.26 (using reflections up to 3 Å resolution). Experimental phases from MLPHARE were improved with DM (Cowtan, 1994) by solvent flattening, histogram mapping, and NCS averaging using automatically generated NCS masks and NCS operator refining, starting with the NCS operator derived from the MR solution. The overall mean figure of merit after phase extension to 2.7 Å with DM was 0.79.

4
Model building and refinement Manual model building was performed with the graphics software TURBO-FRODO (Roussel and Cambillau, 1989) and O (Jones et al., 1991). The electron density map calculated with phases obtained with DM revealed some secondary structure elements, mostly helices. A model was built to ~80% by several rounds of manual model building, local real space refinement with RSRef2000 (Korostelev et al., 2002), and phase combination of the model with experimental phases by using SIGMAA (Read, 1986), followed by density modification with DM. The two molecules were treated independently. After that cycles of automatic refinement and manual rebuilding were performed, first using CNS 1.1 (Brunger et al., 1998) for simulated annealing, switching to REFMAC5 (Murshudov et al., 1997) at later stages of refinement. During this stage, higher quality crystals of the SeMet derivative became available (resolution of 2.5 Å, mean I/σI > 2). This SeMet derivative was the first of four MARK2 structures to be refined to the end, using the partially refined model of the double mutant as start model. The final model contains 604 of 654 residues in two independend molecules, 20 Se atoms in SeMet residues, and 158 water molecules. Eight residues were modelled with dual conformation, amongst them four of the SeMet side chains (MSE 104A/B and MSE 187A/B). In each of the molecules, ten or eleven residues at the N-termini and 13 residues close to the catalytic cleft were not modelled due to weak or uninterpretable electron density, three residues have missing side chains (Glu260A, Arg264A, Arg268A). The final R-factor was 0.198 (Rfree = 0.268) using all reflections up to 2.5 Å. Structures of the double mutant with native methionine, wild type, and the K82R mutant were modelled after the SeMet derivative. To reduce model bias, composite omit maps were calculated with CNS prior to refinement. In all cases, the same subset of reflections was used for crossvalidation. To avoid overfitting, NCS restraints were applied for all structures except the SeMet derivative. All structures were refined with restrained individual isotropic B-factors, except for the K82R mutant which was refined with overall B-factor and TLS parameters for six groups (N-terminal and C-terminal lobes of the catalytic domain and UBA domains, three groups per molecule); see Table SII for refinement statistics. The quality of the models was checked with the programs PROCHECK (Laskowski et al., 1993) and WHAT_CHECK (Hooft et al., 1996) using DSSP secondary structure assignments (Kabsch and Sander, 1983). Interatomic contacts and solvent accessible surface areas were calculated with the software modules CONTACT and AREAIMOL of the CCP4 package. CRYSTAL PACKING AND INTERMOLECULAR CONTACTS The crystals of the MARK2 constructs contain twelve molecules per unit cell, two per asymmetric unit (A and B). They form hollow cylindrical, helical tubes in a close-packed hexagonal arrangement (Fig. S8). The molecules of a tube are related by one of the six-fold screw-axes of the crystal symmetry and by two-fold, noncrystallographic symmetry (NCS) axes almost perpendicular to the main symmetry axis. Because of the close contact between NCS related molecules A and B (Fig. 5 and Fig. S9), they may be considered as a dimer. In the MARK2 double mutant, the two monomers of an NCS dimer are covalently linked by a disulfide bridge between cystein residues C210 (Fig. S9). Interactions of one single NCS dimer with surrounding dimers within the same tube are much more numerous than its interactions with other tubes. Although the numbers of contacts provide only rough estimates of the strength of interactions between the dimers, they indicate that the dominant interactions are those within the tube wall. An isolated tube would be a helix with approximately six dimers per turn and with dyad axes perpendicular to the helix

5
axis. The packing of tubes into a hexagonal array affects the symmetry of the tubes in two ways: first, it locks the helical symmetry of the tubes to become an exact 61 screw rotation compatible with the six-fold crystallographic symmetry; and second, the two-fold symmetries perpendicular to the tube axis are broken and degraded to local NCS symmetries due to perturbations of the internal interactions by intertubular crystal contacts. The dyad axes are rotated by about 7° from the orientation (Fig. S8a) of true (crystal) symmetries in P6122. Thus, the crystal packing is close to that of space group P6122, which is one of the factors that complicated the solution of the crystal structure by molecular replacement. If the crystal packing is mainly determined by the propensity of the molecules to form dimers with two-fold symmetry, then the expected space group is P61, as observed, rather than P6122. The higher symmetry would appear accidental. By contrast, if we assume that the special arrangement of the molecules with the observed proper two-fold NCS is just the consequence of crystal packing, this would be a remarkable coincidence unless the space group was P6122 (which is definitely not the case). In conclusion, the crystal packing suggests that the molecules have a high propensity to form dimers, either in solution or during the process of crystallization. DIFFERENCES IN CRYSTAL PACKING The structures of the MARK2 wild type and mutant constructs are very similar to each other in terms of crystal symmetry and overall packing of the molecules. However, the unit cell dimensions differ systematically (Table SII and Fig. S8). While the unit cell of the double mutant is about 105 Å in the direction of the c-axis, the wild type and the K82R mutant prefer another crystal form with a unit cell that is 5-6% shorter in this direction. Dimers are formed by contacts between the catalytic domains of the two molecules that are involved. The UBA domains do not participate in intra-dimer interactions because they are located on opposite sides of a dimer. With the UBA domains at the extreme poles, the dimers adopt an elongated shape and are aligned with their long axis roughly parallel to the axes of the helical tubes. Thus, two dimers related by one complete helix turn (corresponding to a translation along the c axis by one unit cell) are facing each other with their UBA domains, in particular with helices α1 and α2 of the UBA domains (Fig. S8b-e and Fig. S10). In the double mutant, the interactions between adjacent UBA domains are rather weak, involving only three residues (P326, E330, and R339) on both sides. The conformation of these residues is symmetric in both UBA domains, with E330 and R339 forming two intra-molecular salt bridges (Fig. S10a). The symmetry is broken in the wild type structure (Fig. S10b). There is a small shift in the position of the UBA domains in opposite directions and perpendicular to the helix axis, allowing the side chains at the front sides to interdigitate. This is accompanied by an approach in axial direction by 4-5 Å, which accounts for most of the change in the cell dimension in this direction. As a consequence, the number of contacts between the UBA domains increases. Furthermore, an inter-dimer salt bridge is formed between R339(A) and D322(B) at the end of the linker. Thus, the interaction between UBA domains is stronger in the wild type structure with the short cell constant c compared to the double mutant with the large c constant. The analysis of more than 20 crystals of the MARK2 wild type and mutant constructs shows that there is a strong correlation between cell dimensions (i.e. the overall packing of molecules) and the nature of the side chains of residues 208 and 210 which are buried in the

6
center of the dimer (whether these residues are changed to alanine or not). How can mutations of these residues affect the crystal packing? The crystal structure is a compromise between the tendency of the dimers to form helical tubes of high symmetry on the one hand and crystal contacts which force the tubes to adopt hexagonal symmetry and degrade the dyad axes perpendicular to the hexagonal axis on the other hand. In the double mutant, the dimer forming interaction is strong and the intrinsic symmetry of the helical tubes is not much affected by crystal packing effects; in the wild type structure, the intra-dimer interactions are relatively weak and the symmetry-breaking effect of the crystal contacts is rather strong.

7
Table S II Data collection and refinement statistics Construct wild type K82R mutant T208A, S212A T208A, S212A double mutant double mutant Se-methionine Space group P61 P61 P61 P61 Cell constants [Å]: a = b 120.3 121.2 118.8 119.3
c 99.5 99.7 105.4 105.7 Resolution range [Å] 46.14 - 2.90 51.78 - 3.10 46.30 - 2.50 73.88 - 2.80 Data collection High resolution shell [Å] 2.95 - 2.90 3.15 - 3.10 2.54 - 2.50 2.85 - 2.80 No of observations 122003 88139 214839 132608 No of unique reflections 18222 14814 29259 21073 Completeness [%] 99.9 (100) 98.2 (99.3) 99.9 (100) 99.9 (100) Redundancy 6.6 (6.6) 5.7 (4.0) 7.1 (7.1) 6.2 (4.8) Rsym 0.067 (0.626) 0.109 (0.639) 0.099 (1.066) 0.102 (0.576) <I>/<sigI> 27.8 ( 3.3) 14.0 ( 2.0) 17.9 ( 2.0) 17.1 ( 2.8) Refinement High resolution shell [Å] 2.976 - 2.90 3.185 - 3.10 2.57 - 2.50 2.875 - 2.80 No of reflections working set 17281 14072 27745 19981 test set 942 740 1514 1092 R 0.195 (0.335) 0.200 (0.287) 0.198 (0.295) 0.203 (0.313) Rfree 0.270 (0.429) 0.251 (0.385) 0.268 (0.359) 0.262 (0.353) No of residues (total 654) 589 589 604 604 No of atoms (total) 4664 4667 5071 4885 No of water molecules - - 158 - B-factors individ., isotrop. overall, TLS individ., isotrop. individ., isotrop. NCS restraints 3 groups 3 groups - 3 groups MC tight, SC loose MC, SC tight MC tight, SC loose TLS: TLS refinement assuming 6 rigid groups (minor and larger lobes, and UBA domains for two molecules). MC: main chain atoms, SC side chain atoms. Values in parentheses are for the high resolution shells.

8
FIGURE LEGENDS Fig. S8: Packing of molecules in MARK2 double mutant and wild type crystals. (a) View along the hexagonal axis (c-axis). Twelve of the molecules representing the total contents of one unit cell are drawn with various colors. The molecules form helical tubes in close-packed hexagonal arrangement. Arrows radiating from the hexagonal axis in the centre of the colored tube indicate proper, two-fold noncrystallographic symmetry (NCS) operators. There are two classes of NCS operators represented by long, blue (NCS-1) and short, black (NCS-2) arrows, six of each kind. Molecules related by long, blue NCS-1 axes form dimers and are shown in the same color. The space group of the MARK2 crystals is P61, however, due to the presence of NCS operators the packing of the molecules is very similar to P6122. Rotation of the tubes by 6-7° around their axes would transform the space group symmetry into P6122. (b, c, d, e) Crystal packing of the double mutant (b and d) and the wild type (c and e) in orthographic projections perpendicular to the hexagonal c-axis. The projections correspond to views along the short arrows in the lower left corner of panel a (black arrow for b and c, blue arrow for d and e). Only molecules of the colored tube in panel a are shown. Molecules are shown in light gray except for the molecules in the front which are shown in the same colors as in panel a. Also shown in these projections are arrows indicating NCS operators as in panel a. The NCS operators are almost, but not exactly perpendicular to the hexagonal axis, the deviations being more substantial in the wild type crystal than in the crystal of the double mutant. The length of the c-axis, i.e. the pitch of the helical tubes, corresponds roughly to the extent of the dimer in axial direction. Nevertheless, the difference in the length of the c-axes between wild type and double mutant is largely due to a change in the distance between dimers along the c-axis, as shown by the dashed lines (for details see Fig. S10). This change is accompanied by a slight movement of the UBA domains and a rearrangement of their side chains. Figures S8, S9, and S10 were prepared with Deep View Swiss-PDB Viewer (Guex and Peitsch, 1997) and POVray for Windows (Persistence of Vision Pty. Ltd. (2004), Persistence of Vision Raytracer Version 3.5, retrieved from http://www.povray.org/). Fig. S9: Intermolecular contacts in dimers of the MARK2 double mutant (stereo view). Ribbon diagram showing a close-up of the central part of the dimer. The view is along the non-crystallographic two-fold symmetry axis (molecule A blue, B purple). Residues involved in intermolecular contacts (at least one atom closer than 4 Å to an atom of the other molecule) are in stick model representation. The shortest contact (2.07 Å distance) is between cysteines C210 which form an interchain disulfide bridge. The contact residues concentrate in three zones: zone 1 comprises 15 contact residues in the range from D207 to D227 at the C-terminal anchor of the activation loop and the adjacent loop preceding helix F, zone 2 comprises all but one residue in the range D251-R261 in helix G and part of its N-terminal loop, and zone 3 consists of residues S92, S93, and K96 clustered at the N-terminus of helix C. Fig. S10: Interaction of UBA domains (stereo view). Ribbon diagrams of MARK2 double mutant (a) and wild type (b) superimposed on stick model representations of the UBA domains and adjacent residues. The view corresponds to that of Figure S8d and e. Arrows in blue and black represent local NCS operators as in Figure S8. The molecules interacting via UBA-UBA contacts are drawn in red and pink. In the case of the double mutant (a) the conformation of the two UBA domains is characterized by symmetric interaction involving residues P326, E330, and R339 in both molecules, with intramolecular salt bridges between E330 and R339. The symmetry is broken in the wild type structure (b). This is most obvious in the formation of an intermolecular salt bridge between R339 of one molecule (A) and D322 at the end of the linker in the other molecule (B). This salt bridge is highlighted with a yellow

9
bond between the side chains. The Cα-distance between R339(A) and D322(B) is 11.9 Å. The corresponding distance between R339(B) and D322(A) is more than 2 Å larger, and a salt bridge between these residues is not formed. Instead of that, R339(B) is engaged in an intramolecular salt bridge with E330(B), similarly to the double mutant. As a result of the broken symmetry between the UBA domains combined with a small sideward shift relativ to each other, the side chains interdigitate and make more intermolecular contacts in the wild type. This results in a shortening by 4 - 5 Å of the center-to-center distance between adjacent molecules.

10
REFERENCES Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W.,
Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., Read, R.J., Rice, L.M., Simonson, T. and Warren, G.L. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr, 54, 905-921.
Chen, P., Luo, C., Deng, Y., Ryan, K., Register, J., Margosiak, S., Tempczyk-Russell, A., Nguyen, B., Myers, P., Lundgren, K., Kan, C.C. and O'Connor, P.M. (2000) The 1.7 A crystal structure of human cell cycle checkpoint kinase Chk1: implications for Chk1 regulation. Cell, 100, 681-692.
Cowtan, K. (1994) 'dm': An automated procedure for phase improvement by density modification. Joint CCP4 and ESF-EACBM Newsletter on Protein Crystallography, 31, p34-38.
Drewes, G., Ebneth, A., Preuss, U., Mandelkow, E.M. and Mandelkow, E. (1997) MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell, 89, 297-308.
Hooft, R.W., Vriend, G., Sander, C. and Abola, E.E. (1996) Errors in protein structures. Nature, 381, 272.
Jones, T.A., Zou, J.Y., Cowan, S.W. and Kjeldgaard. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A, 47, 110-119.
Kabsch, W. and Sander, C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers, 22, 2577-2637.
Korostelev, A., Bertram, R. and Chapman, M.S. (2002) Simulated-annealing real-space refinement as a tool in model building. Acta Crystallogr D Biol Crystallogr, 58, 761-767.
Laskowski, R.A., MacArthur, M.W., Moss, D.S. and Thornton, J.M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr., 26, 283-291.
Murshudov, G.N., Vagin, A.A. and Dodson, E.J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr, 53, 240-255.
Otwinowski, Z. (1991) Maximum likelihood refinement of heavy atom parameters. Daresbury Study Weekend Proceedings, 80-87.
Otwinowski, Z. and Minor, W. (1997) Processing of X-ray Diffraction Data Collected in Oscillation Mode. In Carter, C.W. and Sweet, R.M. (eds.), Methods in Enzymology, Vol. 276: Macromolecular Crystallography, part A. Academic Press, pp. 307-326.
Potterton, E., Briggs, P., Turkenburg, M. and Dodson, E. (2003) A graphical user interface to the CCP4 program suite. Acta Crystallogr D Biol Crystallogr, 59, 1131-1137.
Read, R.J. (1986) Improved fourier coefficients for maps using phases from partial structures with errors. Acta Cryst. A, 42, 140-149.
Roussel, A. and Cambillau, C. (1989) TURBO-FRODO. In Silicon Graphics Geometry Partners Directory. Silicon Graphics, Mountain View, CA, USA.

11
Storoni, L.C., McCoy, A.J. and Read, R.J. (2004) Likelihood-enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr, 60, 432-438.
Timm, T., Li, X.Y., Biernat, J., Jiao, J., Mandelkow, E., Vandekerckhove, J. and Mandelkow, E.M. (2003) MARKK, a Ste20-like kinase, activates the polarity-inducing kinase MARK/PAR-1. Embo J, 22, 5090-5101.

Figure S8

Figure S9

Figure S10