sequential headful packaging and fate of the cleaved dna ends in bacteriophage spp1
TRANSCRIPT

J. Mol. Biol. (1996) 264, 954–967
Sequential Headful Packaging and Fate of theCleaved DNA Ends in Bacteriophage SPP1
Paulo Tavares*, Rudi Lurz, Asita Stiege, Beate Ru ¨ ckert andThomas A. Trautner
Max-Planck-Institut fur The virulent Bacillus subtilis bacteriophage SPP1 packages its DNA fromMolekulare Genetik a precursor concatemer by a headful mechanism. Following disruption ofIhnestrabe 73, D-14195 mature virions with chelating agents the chromosome end produced by the
headful cut remains stably bound to the phage tail. Cleavage of thisBerlin, Germanytail-chromosome complex with restriction endonucleases that recognizesingle asymmetric positions within the SPP1 genome yields several distinctclasses of DNA molecules whose size reflects the packaging cycle theywere generated from. A continuous decrease in the number of moleculeswithin each class derived from successive encapsidation rounds indicatesthat there are several packaging series which end after each headfulpackaging cycle. The frequency of molecules in each packaging classfollows the distribution expected for a sequential mechanism initiatedunidirectionally at a defined position in the genome (pac). Theheterogeneity of the DNA fragment sizes within each class reveals animprecision in headful cleavage of 02.5 kb (5.6% of the genome size).The number of encapsidation events in a packaging series (processivity)was observed to increase with time during the infection process.
DNA ejection through the tail can be induced in vitro by a variety of milddenaturing conditions. The first DNA extremity to exit the virion isinvariably the same that was observed to be bound to the tail, implyingthat the viral chromosome is ejected with a specific polarity to penetratethe host. In mature virions a short segment of this chromosome end(55 to 67 bp equivalent to 187 to 288 Å) is fixed to the tail area proximalto the head (connector). Upon ejection this extremity is the first to movealong the tail tube to exit from the virion through the region where the tailspike was attached.
7 1996 Academic Press Limited
Keywords: bacteriophage SPP1; DNA packaging; virus structure;*Corresponding author connector; DNA ejection
Introduction
The majority of tailed icosahedral bacteriophagespackage their chromosome from a concatemerprecursor into a pre-assembled pro-capsid struc-ture. Encapsidation is normally initiated at aspecific nucleotide sequence (cos or pac) andproceeds unidirectionally. After DNA is packagedinside the viral pro-capsid either a site specific (e.g.in l, f105, T3, T7) or sequence independent(headful packaging; e.g. in T4, P22, T1, P1, SPP1)
terminal endonucleolytic cleavage occurs definingthe mature chromosome size (reviewed by Black,1989). Subsequent packaging events occur in aprocessive fashion starting from the DNA endgenerated by such a cut.
After DNA encapsidation the filled phage head isstabilized by binding of additional proteins. One ormore of these bind to the portal protein, aturbine-like oligomer with a central pore throughwhich DNA is believed to move into and out of thecapsid (Bazinet & King, 1985; Dube et al., 1993;Valpuesta & Carrascosa, 1994). This interactionleads to closure of the portal pore and creates thecorrect structural arrangement for tail attachmentoccurring at the unique vertex of the head wherethe portal protein is located (Casjens & Hendrix,
Abbreviations used: EM, electron microscopy;dsDNA, double-stranded DNA; EDAC,1-ethyl-3-(3-dimethylaminopropyl) carbodiimide; pfu,plaque-forming units.
0022–2836/96/500954–14 $25.00/0 7 1996 Academic Press Limited

Headful Packaging and Fate of the Cleaved DNA Ends 955
1988). The multiprotein complex assembled at theportal vertex is named connector (Hendrix, 1978).Binding of the tail finishes morphogenesis yieldingthe infective virion, a metastable structure highlyresistant to environmental challenges but alsodesigned to deliver the viral chromosome in aregulated and efficient way upon interaction withthe host.
The virulent Bacillus subtilis bacteriophage SPP1packages its DNA by a headful mechanism. Thephenomenology of DNA encapsidation (Morelliet al., 1979; Humphreys & Trautner, 1981),characterization of the target sequence (pac) forinitiation of the process (Deichelbohrer et al., 1982;Bravo et al., 1990), recognition and cleavage at pacby the terminase which is a multimer of gp1 andgp2 (Chai et al., 1992, 1994, 1995), and participationof the SPP1 portal protein (gp6) in determination ofchromosome size (Tavares et al., 1992, 1995) werepreviously investigated. Here we describe studieson the processivity of the general packagingreaction, investigate the topology of the DNAmolecule ends in the phage particle, and discuss itspotential implications for chromosome ejection.
Results
The DNA pac distal end is attached to thephage tail
During early studies on bacteriophage SPP1 itwas observed that exposure of viral particles tochelating agents leads to disassembly of the virioninto partially disrupted heads and tails (Esche et al.,1975). When SPP1 particles are disrupted, one endof the viral chromosome is found tightly bound tothe tail region where the connector is located(Figure 1). In order to determine whether aparticular extremity of the DNA molecule isattached and, if this is the case, which one of the
Figure 1. SPP1 particles disrupted after treatment withEDTA. Two empty head shells (arrowheads) and onetail-chromosome complex can be observed. The DNA isbound to the head proximal end of the tail (connector,arrow) while the tail spike is present at the otherextremity. The bar represents 0.5 mm. The material for EMwas prepared by adsorption to mica.
two ends, we employed the strategy outlined inFigure 2A. The phage chromosome-tail complexwas digested with BglII which cuts at a singleposition located asymmetrically within the SPP1genome using our previous convention for itspresentation (Humphreys & Trautner, 1981), ap-proximately 13 kb from the packaging initiationsequence (pac; Bravo et al., 1990; Chai et al., 1992).This procedure allowed us to distinguish the pacproximal and distal DNA ends. To facilitate theanalysis we eliminated the effect of circularpermutation in the chromosome population by
Figure 2. Identification of theDNA end bound to the tail. A,Experimental strategy. The heavybar represents a precursor concate-mer from which encapsidation oc-curs. The origin of packaging (pac),the BglII site (B), and the directionof packaging are indicated. The thinlines below represent series of mole-cules generated after sequentialpackaging initiated at the right mostpac site, and cleavage with BglII ofSPP1wt and SPP1sizS DNAs. Up-ward thick lines symbolize tailsattached to the DNA moleculesextremity generated by the headfulcut. B, EM measurement of individ-ual DNA molecules associated withthe phage tail after disruption ofSPP1sizS with EDTA and sub-sequent BglII cleavage. Molecules
are aligned by the end bound to the tail and ordered by increasing size. The upper thick bar represents the chromosomesize (44.1 kb; Tavares et al., 1992). Complexes were prepared for EM measurements by the droplet technique.

Headful Packaging and Fate of the Cleaved DNA Ends956
using SPP1sizS, a mutant characterized by a smallerchromosome size which is virtually identical to thatof the genome (44.6 kb; Tavares et al., 1992),implying that the DNA molecules packaged have avery short terminal redundancy. Thus, in contrastto the wild-type situation, a relatively constanttopology was expected (Figure 2A). Electronmicroscopy (EM) measurements of the chromo-some-tail complexes cleaved with BglII showed thata relatively homogeneous population of longfragments (31 to 35 kb) remained attached to the tail(Figure 2B) demonstrating that the pac distal end isbound to the tail.
Processivity of headful packaging
The tail attached to a specific end of SPP1 DNAprovides a fixed reference to align the population ofphage chromosomes. This feature enabled us tocharacterize several properties of the packagingreaction which preceded the interactions betweenSPP1 DNA and the tail. Since mature DNA mole-cules are generated by sequential headful encapsida-tion initiated at pac (Tye et al., 1974; Gill & MacHattie,1976; Morelli et al., 1979), we expected that severalclasses of DNA molecules shall be present when thetail-chromosome complex is digested with arestriction enzyme cutting at a single asymmetricposition within the SPP1 genome. Each class wouldinclude molecules with similar size and this sizewould depend on the packaging cycle from whichthey were generated. If the endonucleolytic cleavageoccurs very distal to the pac extremity, the smallestmolecules bound to the phage tail would be thoseoriginating from the first event of the encapsidationseries and the next classes would be derived fromsuccessive packaging cycles (Figure 3A). Further-more, the size step between each class would reflectthe extent of terminal redundancy. Our experimen-tal approach required only the measurement ofdouble-stranded DNA (dsDNA). It was thereforeconsiderably more accurate than previous sizedeterminations which involved measurements ofmolecules with both single and double-strandedregions (Tye et al., 1974; Morelli et al., 1979).
The tail-chromosome complexes obtained byEDTA treatment of SPP1wt virions were cleavedwith SnaBI that recognizes a single sequence withinfragment EcoRI-1 of the SPP1 wild-type genome(040 kb from the pac extremity, which correspondsto 4893 bp before the following pac sequence in theconcatemer used as substrate for packaging (Chaiet al., 1993; Figure 3A). EM measurements of DNAmolecules from SPP1wt yielded a distribution offragments attached to the tails where different sizeclasses were discernable. The distinction betweenthese classes, however, was hampered by the smallsteps between successive classes (not shown).Therefore, we repeated the experiment with amutant carrying deletion X (3427 bp; Chai et al.,1993) located between the SnaBI site and pac of theSPP1 genome. Since reduction in SPP1delX110chromosome size is compensated by an increase in
terminal redundancy (Tavares et al., 1992), thespacing between the genomic location of successiveheadful cleavages is increased accordingly (fromabout 1.6 kb, which is the average terminalrepetition for SPP1wt chromosomes, to approxi-mately 5.0 kb; Figure 3A). This strategy, also usedin the seminal work from Tye et al. (1974), alloweda clear distinction of five discrete classes of DNAmolecules and a few extra long fragments (Figure3B). In addition to the visual inspection procedurewe also used an analytical criterion to defineclasses. The terminal redundancy was calculated bysubtracting the distance between the SnaBI site andpac (1459 bp in SPP1delX110) from the average sizeof molecules in the first class of Figure 3B to D andthis value (5.05 kb) was used as the step betweenclasses. The mean value between the size expectedfor two successive classes (vertical arrows in thegraphics of Figure 3B to D) was used as a cut-offlength to assign molecules to a specific class.
The distribution in Figure 3B shows that 48% ofthe SPP1delX110 molecules measured were derivedfrom the first encapsidation cycle while this per-centage was only 032% for SPP1wt (not shown).Classes derived from subsequent packaging cyclesof SPP1delX110 are characterized by a continuousreduction in the number of elements (27, 9, 8 and6% for classes 2, 3, 4 and 5, respectively; Figure 3B).The extent of circular permutation in the completepopulation of mature chromosomes can be calcu-lated by subtracting the size of the smallest DNAfragment in the first class from the length of thelargest molecule measured (29 kb (70% of thegenome size) for SPP1delX110 and 16 kb (36%) forSPP1wt). This value divided by the terminalredundancy size gives the packaging series length(i.e. the number of sequential headfuls). Based onthe data obtained with our phage preparations wecalculated a maximum number of 6 and 12 headfulsfrom a single packaging series for the deletionmutant and wild-type, respectively.
Comparison between SPP1wt and SPP1delX110,as well as other quantifications not shown, revealedthat the frequency of SPP1 chromosomes derivedfrom the first encapsidation cycle varied amongdifferent lysates. Therefore, we determined if thisfrequency changes during SPP1 infection as foundin the case of bacteriophage P22 (Adams et al.,1983). Host cells infected with SPP1delX110 werelysed at 20 and 120 minutes after infection byaddition of lysozyme and chloroform to culturesamples. Assembled phages were purified and thetopology of their chromosomes was investigated asdescribed above. The distribution of size classeswas considerably different at the two stages ofinfection (Figure 3C, D): while within 20 minutesthe vast majority of packaged DNA molecules isderived from the first encapsidation cycle (82%)and the few remaining from the second (16%) andthird (1%) cycles, at least eight classes can bedistinguished and full circular permutation of thechromosome population is observed after 120minutes. As would be expected considering the

Figure 3. Processive headful packaging in bacteriophage SPP1. A, Sequential encapsidation initiated at pac. The heavylines in the centre represent the concatemeric substrate DNA for encapsidation from SPP1wt (top) and SPP1delX110(bottom). Deletion end points marked in the SPP1wt sequence are connected to the site of deletion in SPP1delX110 andthe deleted region is also highlighted in the SPP1wt concatemer (black boxes). SnaBI (S) and pac cleavage sequencesare indicated. Vertical arrows show the position of SmaI cleavage (Santos et al., 1986) in the first genomic unit of theSPP1wt concatemer. The thicker arrow indicates three close sites. All restriction sites shown for the SPP1wt concatemerare found at identical positions in SPP1delX110 (not represented for simplification of the scheme). The pac terminalfragment generated by SmaI cleavage (SmaI-4) is represented by a bar. Ensembles of mature molecules cleaved withSnaBI are depicted above (SPP1wt) or below (SPP1delX110) the concatemeric structures using the convention of Figure2A. B, Electron microscopic measurement of individual DNA molecules bound to the phage tail after disruption ofSPP1delX110 (standard lysate) with EDTA and cleavage with SnaBI. All molecules present in a pre-defined area of thecarbon grid were measured to provide an unbiased statistical distribution but only those bound to tail structures aredepicted. Vertical arrows indicate the size limits used to assign molecules to a specific packaging cycle (class) usingthe criterion described in the text. Data presentation and methods are as in Figure 2B. C, D, Results from experimentssimilar to B except that the SPP1delX110 phage population was analysed 20 and 120 minutes after the initiation ofinfection, respectively. E, Increase in the number of packaging events per encapsidation series (average series length)during infection with SPP1wt (input multiplicity, 5 phage/bacterium). Beginning of infection is time 0. The averageseries length is calculated from the molar ratio between the SmaI-3 restriction fragment and the pac terminated fragment(SmaI-4; arrow on the insert) in restriction patterns (insert) as described in the text (experimental time points arerepresented by diamonds and the full line is a visual aid fitted by hand). The yield in viable phage progeny determinedby titration with B. subtilis YB886 is also shown (triangles).

Headful Packaging and Fate of the Cleaved DNA Ends958
sequential character of the process, the number ofmolecules derived from successive packagingcycles decreased continuously in all the popu-lations analysed. This reduction, however, is muchless pronounced for the chromosomes of phagesassembled late in infection suggesting that theencapsidation series length increases during infec-tion due to a higher packaging processivity. Thedistribution observed for the SPP1 lysate describedabove (Figure 3B) is intermediate between theextreme cases shown in Figure 3C and D and mightrepresent the most common situation for lysatesproduced routinely under laboratory conditions.
One feature of the set of individual molecules ofthe same class is the considerable variability in size,normally within a range of 2 to 3 kb (Figure 3B toD). This variation is not only due to experimentalerror in the measurements but, more importantly,to inaccuracy of the headful cleavage in SPP1(Humphreys & Trautner, 1981). To assess thecontribution of the two factors we performedparallel measurements of replicative forms of phageM13mp18 chromosomes linearized with EcoRI orHindIII. Since these dsDNA molecules have aconstant length (7250 bp; Yanisch-Perron et al.,1985), the variation observed gives an estimate ofthe accuracy of our measurements. An average sizeof 7.25(20.12) kb (01.7% variation) was found fora dataset of 65 M13 molecules (data not shown). Incomparison the length of fragments of the first classin Figure 3C is 6.66(20.69) kb (010.3% variation).The experimental data follow a normal (gaussian)distribution. Comparable results were obtainedwith measurements of DNA molecules adsorbed tomica. We could thus conclude that the variation indimensions observed within the first class of SPP1fragments (02.5 kb; 5.6% of the genome size) wasdue essentially to imprecision in headful cleavage.Heterogeneity in size of molecules produced fromsubsequent encapsidation cycles is normally alsowithin a range of 2 to 3 kb but increases in the caseof the last classes, an effect attributable to additiveimprecision from several packaging rounds. Thisvariability in chromosome size is larger than thestep between classes (terminal redundancy) in thecase of SPP1wt preventing the discrimination ofclear size classes. Furthermore, it raises also theprobability of error to define groups of moleculesgenerated from late packaging cycles in longencapsidation series from SPP1delX110.
The imprecision of headful cleavage in SPP1 iscomparable to the reports for other phagespackaging their DNA by a headful mechanism (Mu:02.7 kb, Chow & Bukhari, 1977; P22: 01.5 kb,Casjens & Hayden, 1988) and yields a randomdistribution of molecular ends at the level ofresolution of the EM measurements.
The processivity of DNA packaging increasesduring infection
Complementary information on the processivityof packaging was obtained from quantification of
the pac terminated restriction fragment in encapsi-dated DNA (Figure 3A, E). This segment of DNAhas one end derived from cleavage at pac and theother from digestion with the restriction enzymeused. It is produced, therefore, only once perpackaging series and its molar ratio to otherfragments reveals the average frequency of packag-ing cycles initiated at pac relative to the totalnumber of encapsidation events (Bachi & Arber,1977; Jackson et al., 1978; Ratcliff et al., 1979). Thisratio is inversely proportional to the averagenumber of encapsidation events per packagingseries (average series length = 1/pac fragmentratio).
Densitometric analysis of restriction patternsobtained by SmaI cleavage of packaged viral DNAshowed that the ratio between the pac terminalfragment (SmaI-4; nomenclature according toSantos et al., 1986; Figure 3A) and equimolarfragments derived from the SmaI digest (SmaI-3, 5or 6) varied among different phage lysatesindependently of the SPP1 strain. Since the averageseries length was observed to increase with timeafter P22 infection (Adams et al., 1983) and our EMdata also demonstrated a temporal variation in thecase of SPP1 we followed the rise of this parameterafter infection with SPP1wt (Figure 3E). A valueof 1.8 (equivalent to a pac ratio of 0.56) wasdetermined at 20 minutes post-infection, indicatingthat most packaged chromosomes were derivedfrom the first encapsidation cycle. The series lengththen increased continuously reaching 5.4 (pac ratio0.19) for the last time point tested in the experimentof Figure 3E (120 minutes). Early after infection thenumber of progeny virions increased rapidly untilstarting to stabilize around 40 minutes post-infec-tion (Figure 3E), as would be expected for anintracellular growth curve (Klotz & Spatz, 1971).Interestingly, between 40 and 120 minutes post-in-fection, an interval characterized by a lower rise invirion number, there is a significant increase inseries length. This observation suggests that amajority of phages assembled during this periodcarry chromosomes derived from long packagingseries. The effect of the increase in packaging serieslength with time on the full population of progenychromosomes can best be observed by comparingthe class distributions of SPP1delX110 moleculesshown in Figure 3B to D. Determination of theaverage series length based on the pac ratio for thismutant, however, is biased by the large circularpermutation which implies that headful cleavagesoccur spread within most of the genome reducingalso the representativity of ‘‘true’’ restrictionfragments.
The monotonic increase in the average packagingseries length is very similar to the one reported byAdams et al. (1983) for P22, although, in contrast tothe experimental design of these authors, we couldnot prevent normal lysis of the host cell (startingafter 30 minutes under the infection conditionsused) and consequent stabilization of the progenyparticle titre (Figure 3E). Thus, for interpretation of

Headful Packaging and Fate of the Cleaved DNA Ends 959
the data we have to take into consideration that thenumber of host bacteria is reduced during theexperiment and that, at late time points, the rise inaverage series length in the entire population ofviral chromosomes is due to phage particlesproduced by only a fraction of the initial infectedcells. Consequently, the values presented are, infact, an underestimation of the series length for latepackaging events. We did not observe considerablere-infection, another phenomenon that couldcomplicate data analysis, as confirmed by themodest rise in progeny after the first cycle of phagegrowth (Figure 3E).
The DNA end to be packaged last isejected first
In order to distinguish whether the DNA endfixed to the tail structure is the first or last to exitthe virion upon infection we took advantage of theobservation that DNA ejection through the phagetail can be triggered in vitro by several milddenaturing conditions (e.g. 2.5 M sodium iodide;see below) leaving, in a large number of cases, oneend of the chromosome still bound to the tail regiondistal to the head (Figure 4A). This end wasidentified by restriction with endonucleases (BglIIor SnaBI) and measurement of the DNA moleculesremaining attached to the phage tail, a strategysimilar to the one employed in the experiment ofFigure 2. SPP1sizS was used due to the virtualabsence of circular permutation, a feature whichsimplifies the analysis (see Figure 2A).
DNA ejection from SPP1sizS particles wastriggered by NaI treatment. After cross-linking withglutaraldehyde and digestion with the endonucle-ase, the material was prepared for EM. DNA boundto the tails of empty phages was then measured.The size distributions obtained show that thechromosome extremity associated with the tail ismore distal to the SnaBI site and closer to the BglIIcleavage site (Figure 4B). Thus, the first end to exitthe tail is the one generated by headful cleavage.The finding of a minor population of moleculesconsiderably smaller than expected is most prob-ably due to partial ejection and not to heterogeneityin chromosome size as it was not detected when thesame phage preparation was used for EDTAtreatment (Figure 2; data not shown). Absence ofany molecules significantly larger than expected inthe case of the BglII experiment (13 kb) demon-strates that the chromosome extremity attached tothe connector (generated by headful cleavage;Figures 1, 2) is invariably the first to exit the virionupon ejection.
Size of the DNA segment protected by the tail
To measure the size of the chromosome fragmentassociated to the tail of virions disrupted withEDTA, we performed a DNAase protection assay.Following incubation with the chelating agent, thetail-DNA complex generated was exhaustively
Figure 4. Chromosome ejection from SPP1sizS par-ticles. A, Ejection triggered in vitro by NaI (2.5 M). Twoghosts can be observed with DNA leaving the tail enddistal from the head (arrowheads). The bar represents0.5 mm. The material for EM was prepared by adsorptionto mica. B, Identification of the first chromosome end thatis ejected. The upper thick line represents the SPP1chromosome. Cleavage positions for the endonucleasesBglII (B) and SnaBI (S), and the initial cut at pac performedby the terminase are represented by vertical arrows.Packaging is from right to left. The lower part showsresults from EM measurements of individual DNAmolecules associated with the phage tail after ejectionfrom SPP1sizS triggered by NaI. Measurements of intactand restricted molecules (SnaBI and BglII) are depicted.Data presentation and methods are as in Figure 2B.

Headful Packaging and Fate of the Cleaved DNA Ends960
Figure 5. Size of the DNA fragment protected by thetail after disruption with EDTA. Nuclease treatedcomplexes, not deproteinized or extracted with phenol,were radioactively labelled and separated in a 8% nativegel PAGE together with appropriate molecular weightmarkers (Mr; pBR322 digested with HaeIII).
that it is buried by protein (Figure 5). Evidence forthe association between this fragment and the tailwas obtained from sedimentation of the nucleasetreated material in a sucrose gradient. Tail and thesmall DNA segment co-sedimented while the bulkof the disrupted heads moved considerably fasterand disaggregated material remained at the top ofthe gradient as monitored by several techniques(extraction with phenol and DNA end-labelling,SDS-PAGE, Western blot, electron microscopy; datanot shown). The size of the protected fragment (55to 67 bp, equivalent to 187 to 228 A for DNAconformation B) is considerably shorter than thetail length. Thus we hypothesize that it is locatedonly in the connector region, most probablyextending from the portal protein pore (0105 Aheight; Dube et al., 1993; Tavares et al., 1995) to thelower connector part bound to the helical tail(unpublished results). In the case of bacteriophagesT4 and T7, Zachary & Black (1992) observed alsothe protection of a short segment of DNA (040 bpfor T4) in portal protein-DNA complexes isolatedfrom acid treated virions.
Disruption of the mature phage particle andDNA ejection
The experiments described above were based onthe availability of reproducible methods to disas-semble virions. We found two distinct types ofpartial disruption. First, treatment with chelatingagents (e.g. EDTA, citrate) caused separation ofheads from tails or head disruption (Figure 1; datanot shown), an effect that was proportional to themass of DNA present inside the virion head(Table 1). No further disaggregation of thestructures was observed when the concentration ofthe chemical was increased. Second, incubationwith a variety of denaturing agents (KSCN [e2 M],NaI [e2.5 M], guanidinium hydrochloride [e3 M],formamide [e35%]) led to the appearance of emptyphage particles (ghosts) the majority of whichejected their DNA through the tail (Figure 4A,
treated with nucleases after which part of it wasextracted with phenol. Both samples, with orwithout deproteinization, were then radioactivelylabelled using T4 polynucleotide kinase to identifyfree 5' DNA ends. A small segment of labelledDNA, ranging in size from 55 to 67 bp (minorvariations within these values were observed inindependent experiments), was found exclusivelyin samples extracted with phenol, demonstrating
Table 1. Characterization of virion disruption triggered by EDTA or NaIElectron microscopy
Tails with DNA boundto the region proximal to
Disruption Phage Intact Empty headsconditions Phage viability phages or ghosts Tails Head Tail spike Both sides +Glu
100 mM EDTA X110 − + + + + + + − − NS + + + + + − − + − − NDX + + + + + + + + + − + − − ND
2.5 M NaI X110 + − + + − − + + + PS + − + + − − + + + + NDX + − + + − − + + − ND
Phages with different amounts of packaged DNA (SPP1delX110 [45.6 kb], SPP1sizS [44.1 kb] and SPP1delX [43.1 kb] Tavares et al.,1992) were treated with 100 mM EDTA for 30 minutes at 37°C or with 2.5 M NaI for ten minutes at 30°C. The effect of cross-linkingthe virions with glutaraldehyde ( + Glu) before chemical treatment is also shown. Phage viability, expressed as percentage of the titrefrom the initial lysate, was evaluated by titration with YB886. EM quantifications were based on samples prepared by mica adsorption(100 to 200 structures from each preparation were classified): + + + +, more than 75%; + + +, more than 50%; + +, more than 20%;
+, more than 5%; −, below 5%. P, indicates that pre-treatment with glutaraldehyde prevents disruption, N, reveals no effect, andND was not determined. The cross-linking reaction causes a severe reduction in phage viability (<10−5 %).

Headful Packaging and Fate of the Cleaved DNA Ends 961
Figure 6. DNA ejection triggered by NaI (2.5 M). Kinetics of DNA ejection from SPP1 mutants with variouschromosome sizes (SPP1wt [45.9 kb], SPP1sizS [44.1 kb], SPP1delX [43.1 kb] and SPP1delX110 [45.6 kb]; Tavares et al.,1992). Ejection was stopped by cross-linking with EDAC at the time points shown above each gel lane and releasedDNA was probed by restriction with EcoRI. Digested material (equivalent to 02 × 108 pfu/well) was resolved in a 1%agarose gel and stained with ethidium bromide. The material retained in the slot and the smear in the upper part ofthe gel observed for short time points likely reveals DNA present in intact phages. S, SPP1wt purified DNA digestedwith EcoRI; P, phage sample not exposed to NaI but processed like the other samples; 0', sample taken and cross-linkedimmediatelly after manual mixing with NaI (<15 seconds).
Table 1 and data not shown). In this case, raisingthe concentration of denaturant caused disassem-bly of the head and ultimately of the tail. Chemicalcross-linking of the phage particle with glutaralde-hyde or 1-ethyl-3-(3-dimethylaminopropyl)carbo-diimide (EDAC) prior to exposure to denaturingagents (e.g. 2.5 M NaI) prevented DNA fromexiting while it did not avoid disruption by EDTA(Table 1). Increase in temperature from 30 to 37°Cto 45 to 50°C, which by itself does not affect SPP1viability, was observed to have a synergistic effectin both disruption methods (not shown).
When chromosome ejection from phage particleswas stopped by cross-linking at various times afteraddition of NaI and released DNA was probed withendonuclease EcoRI, we observed an increase in theamount of digested DNA after long periods ofexposure to the chemical (Figure 6). However, thestoichiometry between fragments in the restrictionpattern remained constant, being similar to ratiosfound for purified DNA. Thus, ejection seems to bean all or nothing process which, after beinginitiated, leads to release of virtually the fullchromosome (Figures 4, 6). Cross-linking can stopthe process by preventing the trigger and/orinitiation but is most probably not able to freeze themovement of DNA through the tail tube once it hasstarted. The level of headfilling apparently does nothave a major effect on the kinetics of chromosomerelease (at least in the minute time range) as judgedby the near identical amounts of DNA ejected fromvarious SPP1 mutants with different chromosomesize (Figure 6).
The majority of particles treated with NaI ejectedmost of the chromosome except for the endpackaged last (Figure 4). This association betweenthe tail extremity distal to the head and SPP1 DNAis relatively strong as a significant population of theprotein-DNA complexes are maintained duringequilibrium isopycnic centrifugation in CsCl gradi-ents. It is also further stabilized by glutaraldehydecross-linking (not shown). However, the interactiondoes not promote any detectable protection of theattached DNA against nuclease treatment (data notshown). Binding of DNA to the tail tip after ejectionthus seems to not be specific, either because it isweaker or because the stretch of DNA associated tothe tail has a heterogeneous size. The nature of thisinteraction and its potential physiological role, ifany, remain to be determined.
Discussion
Organization of the packaged DNA in thevirion and functional implications
SPP1 virions are highly resistant structures. Theirdisruption by artificial treatments involves eitherseparation of head and tail (Figure 1) or ejection ofDNA through the tail tube (Figure 4). Each effect istriggered by distinct chemicals which targetdifferent components of the phage particle.Incubation with chelating agents leads to the firsttype of disassembly. Its severity, as observed alsofor other bacteriophages, is proportional to theamount of DNA inside the phage head (l:

Headful Packaging and Fate of the Cleaved DNA Ends962
Parkinson & Huskey, 1971; P22: Casjens et al., 1992;SPP1: Tavares et al., 1992). Such correlation betweenthe level of headfilling and the requirement fordivalent cations can be interpreted as if these smallmolecules could act to shield the repulsive forcesbetween the phosphate backbones closely packedin the confines of the phage capsid. A tenfoldincrease in the local Mg2+ concentration in the DNApackaged state was indeed reported for bacterio-phage P22 (Aubrey et al., 1992).
SPP1 DNA ejection through the tail can betriggered by a variety of chemicals whose uniquecommon feature appears to be to act as denaturingagents. We believe that, instead of a particulareffect, their destabilizing action mimics the naturalsignal initiated by adsorption of SPP1 to the B.subtilis cell and induces the conformational changesleading to DNA exit by the ‘‘legitimate way’’. It isinteresting to note that, at least in vivo, the signalwhich is initiated at the phage adsorptionapparatus (tail spike?) has to be communicated tothe opposite end of the tail (connector) where thefirst DNA end to exit is bound (Figures 1, 5).Chromosome movement out of the virion isbelieved to be entropically driven by the highconcentration of DNA inside the phage capsid (cf.Earnshaw & Casjens, 1980) even though we couldnot detect an evident correlation between the levelof headfilling and the kinetics of ejection in vitro(Figure 6). DNA ejection leads to loss of the tailspike, the tail tube becomes empty if all DNA isreleased (stain penetrates in negative stainingpreparations), and the phage head appears partlycollapsed indicating that presence of nucleic acid onits interior is an important requirement forstructural stability (Figure 4A and EM observationsnot shown).
The DNA end created by headful cleavage (lastto be packaged) during SPP1 morphogenesis isinvariably associated to the phage tail region whichbinds the icosahedral head (connector; Figure 1)showing that the two DNA extremities of the viralchromosome have a distinct topology in the virion.This feature was also reported for other phagesystems using chemical cross-linking (Thomas,1974; Chattoraj & Inman, 1974) suggesting that itmay play an important role in viral physiology, themost obvious being to facilitate the polar exit ofDNA through the tail during viral infection. Thefindings that the DNA end attached to the tail is thefirst to exit when ejection is triggered in vitro(Thomas, 1974; Figure 4) and that l mutantsdefective in this interaction are not able to eject theirDNA (Thomas et al., 1978) favour such interpret-ation. Since the extremities of the SPP1 chromo-some are permuted, the relevant structural featuremight be that one of these ends is positioned forejection and the choice between the two is dictatedby the way DNA is packaged (cf. Chattoraj &Inman, 1974; Earnshaw & Casjens, 1980). Theinteraction between tail and DNA is apparentlystronger in SPP1 than in other phages as it does notrequire any cross-linking to be stably conserved
upon virion disruption. The absence of anyobservable structure bound to the first DNA endencapsidated (Figure 1) suggests that it mightremain free inside the phage head. We presently donot know whether this is the case or if some looseassociation fixes this extremity during DNAencapsidation. The distinction between both possi-bilities is a relevant issue to understand how SPP1DNA is being packed while it is translocated intothe pro-capsid.
Sequential headful packaging
Mature SPP1 phages have the DNA endgenerated by headful cleavage bound to the tail. Intail-DNA complexes produced by disruption ofvirions with chelating agents (Figure 1) thisassociation provides a useful physical marker tocharacterize the topology of individual maturechromosomes by EM, particularly when it is usedin combination with cleavage at unique positionsof the genome by restriction endonucleases (Fig-ures 2, 3). The alignment of restricted tail-DNAcomplexes allows one to distinguish size classeswhose order reflects the sequential packagingcycles from which the population of partiallycircularly permuted chromosomes was generated(Figure 3B). Thus, these studies provide quantitat-ive information on the mechanism of sequentialheadful packaging by SPP1.
The distribution of the viral chromosomepopulation according to their original encapsida-tion cycle (packaging classes) shown in Figure 3supports the concept that DNA packaging initiatesat a unique position within the SPP1 genome (pac)and proceeds unidirectionally in a sequentialfashion (Morelli et al., 1979; Bravo et al., 1990). Thisfact implies that the relative frequency of moleculesgenerated after the first packaging cycle (classesC2, C3, . . . , Cn in Figure 7A) depends on thefrequencies of molecules derived from encapsida-tion cycles occurring before in the packaging seriesand is, ultimately, a function of the percentage ofmature chromosomes derived from the firstencapsidation event (C1). It is thus possible topredict the full distribution of chromosomes if it isdictated by a strictly sequential process and nomajor biases are introduced by factors inherent tothe encapsidation mechanism or infection con-ditions that would favour, a priori, the occurrenceof packaging series with a defined size. In this casethe probability (P) that after each encapsidationround another packaging cycle will follow isidentical for any cycle in the packaging series (i.e.P = P1 = P2 = P3 = . . . = Pn ; see also Casjens &Hayden, 1988). P can be calculated from thepercentage of molecules (C1) generated in the firstpackaging cycle (P = 1 − C1; see formulation inFigure 7), a value easily derived from the frequencyof pac terminal fragments in endonuclease restric-tion profiles of phage DNA (Results and Figure 3E).The distribution for the full population ofchromosomes can then be determined based on P.
Headful Packaging and Fate of the Cleaved DNA Ends 963
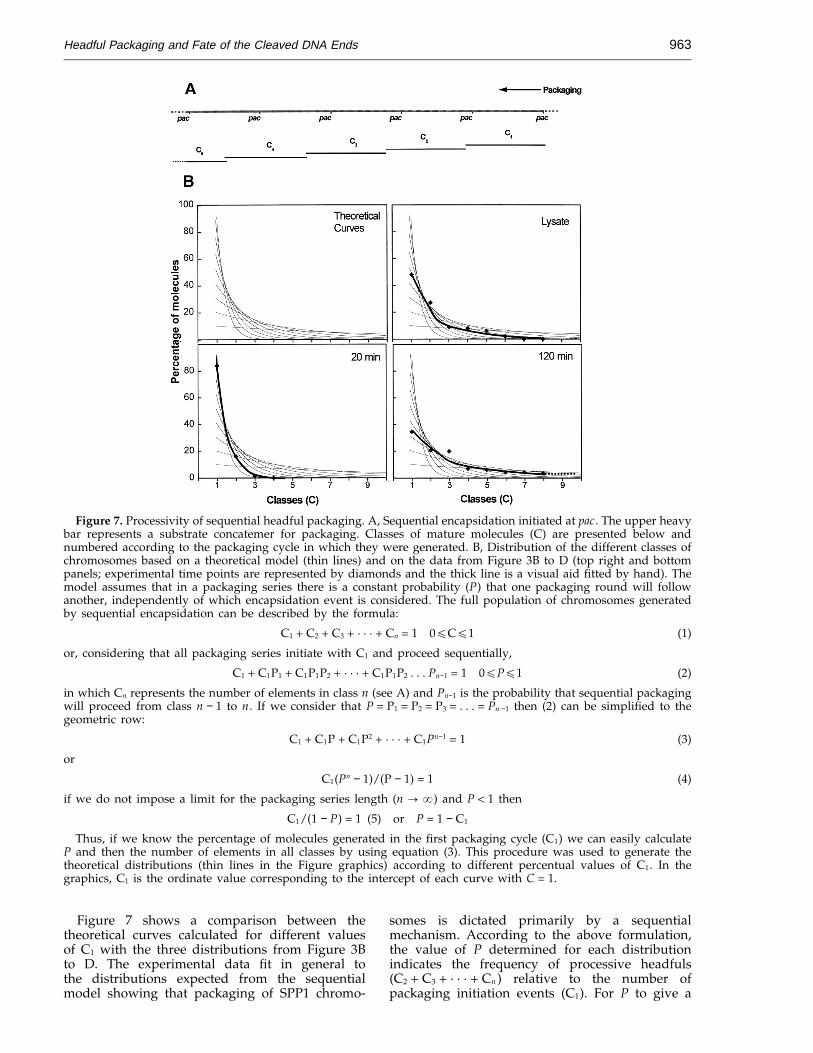
Figure 7. Processivity of sequential headful packaging. A, Sequential encapsidation initiated at pac. The upper heavybar represents a substrate concatemer for packaging. Classes of mature molecules (C) are presented below andnumbered according to the packaging cycle in which they were generated. B, Distribution of the different classes ofchromosomes based on a theoretical model (thin lines) and on the data from Figure 3B to D (top right and bottompanels; experimental time points are represented by diamonds and the thick line is a visual aid fitted by hand). Themodel assumes that in a packaging series there is a constant probability (P) that one packaging round will followanother, independently of which encapsidation event is considered. The full population of chromosomes generatedby sequential encapsidation can be described by the formula:
C1 + C2 + C3 + · · · + Cn = 1 0ECE1 (1)
or, considering that all packaging series initiate with C1 and proceed sequentially,
C1 + C1P1 + C1P1P2 + · · · + C1P1P2 . . . Pn−1 = 1 0EPE1 (2)
in which Cn represents the number of elements in class n (see A) and Pn−1 is the probability that sequential packagingwill proceed from class n − 1 to n. If we consider that P = P1 = P2 = P3 = . . . = Pn −1 then (2) can be simplified to thegeometric row:
C1 + C1P + C1P2 + · · · + C1Pn−1 = 1 (3)
or
C1(Pn − 1)/(P − 1) = 1 (4)
if we do not impose a limit for the packaging series length (n : a) and P < 1 then
C1/(1 − P) = 1 (5) or P = 1 − C1
Thus, if we know the percentage of molecules generated in the first packaging cycle (C1) we can easily calculateP and then the number of elements in all classes by using equation (3). This procedure was used to generate thetheoretical distributions (thin lines in the Figure graphics) according to different percentual values of C1. In thegraphics, C1 is the ordinate value corresponding to the intercept of each curve with C = 1.
Figure 7 shows a comparison between thetheoretical curves calculated for different valuesof C1 with the three distributions from Figure 3Bto D. The experimental data fit in general tothe distributions expected from the sequentialmodel showing that packaging of SPP1 chromo-
somes is dictated primarily by a sequentialmechanism. According to the above formulation,the value of P determined for each distributionindicates the frequency of processive headfuls(C2 + C3 + · · · + Cn ) relative to the number ofpackaging initiation events (C1). For P to give a

Headful Packaging and Fate of the Cleaved DNA Ends964
measurement of the absolute processivity of thepackaging machinery, however, the duration of acomplete encapsidation cycle would have to benegligible relative to the duration of the infectiouscycle. This condition is not fulfilled during normalinfections. A main limiting factor is cell lysis thatcauses premature termination of the packagingseries. Long infection periods are indeed associatedto an increase in the frequency of processive versusinitiation packaging events that implies the rise ofP from 0.16 (20 minutes) to 0.65 (120 minutes) forthe distributions shown in Figure 7B. Thus ifthe distribution of viral chromosomes would bedictated uniquely by the processivity of thepackaging apparatus higher P values should beobserved.
Some deviations of the experimental distri-butions relative to the theoretical curves fromFigure 7B are observed in the case of the twodatasets representative of late stages in infection.Since P increases as the infectious cycle proceeds,we attribute this effect to asynchronous lysis of partof the infected host cells. A sub-population ofprogeny phages would thus have chromosomesgenerated from packaging series with different Pvalues. Their contribution, however, is apparentlynot very significant as it does not bias much thetotal distribution of mature DNA molecules,particularly in the case of virion populationsderived from cultures lysed artificially (20 and 120minutes in Figure 7B).
Sequential headful packaging requires seriesinitiation (pac cleavage and first encapsidationcycle) and extension events (processive headfuls).As proposed by Adams et al. (1983) based onstudies with bacteriophage P22, the size of theencapsidation series is dictated by the relativefrequency of each of the two processes. Thefrequency of initiation events is high at early stagesduring SPP1 infection while later extension eventspredominate as revealed by the rise from 1.8 tomore than five average sequential headfuls perpackaging series (Figure 3E). Our data do not allowus to discriminate whether this increase is due tovariation on the physical length of the substrateconcatemer, to alterations in the properties of thepackaging machinery or to the kinetics of packag-ing (i.e. the time required to complete a fullencapsidation round). It is also possible that allthese factors contribute to some extent to thebalance between initiation at pac and processivity.
It is presently not known how DNA concatemersare generated (rolling circle replication and/orrecombination) during SPP1 infection. Indepen-dently of the mechanism, their formation andpackaging shall occur, at least partly, in parallel.The concatemer sizes would thus be defined bycompetition between both processes. Terminasecleavage at pac, the first event in DNA encapsida-tion, is one of the factors that reduces the size of theconcatemer. Chai et al. (1992) demonstrated that theterminase endonucleolytic activity is regulatedsince the frequency of all pac sequences cut
(packaged or free in the host cytoplasm) is keptrelatively constant between 12 and 30 minutes afterinitiation of SPP1 infection even when no DNApackaging occurs. Maintenance of a low steady-state level of cleaved pac sites is essential to ensuresequential packaging. However, even when only 22to 27% of all the pac sequences are cut (Chai et al.,1992), the size of the substrate DNA will beconsiderably limited if cleavage occurs at pac sitesdistributed randomly within the SPP1 DNAconcatemers. Random cuts would especially pre-vent the occurrence of long packaging series asthose observed in Figure 3B, D as well astransduction of plasmid concatemers carrying thepac sequence (Bravo & Alonso, 1990). It is thereforepossible that the frequency of pac cleavage dropslate during infection and a mechanism could beoperative which prevents further pac cleavage onconcatemers where sequential packaging wasinitiated. In the case of coliphage P1, Sternberg &Coulby (1987) demonstrated that the substratedimension is indeed one main factor to limit thenumber of sequential packaging events. Theseauthors inserted pac in a molecule of ‘‘infinite size’’,the host chromosome, and observed that such aconstruct leads to the unidirectional encapsidationof five to ten consecutive headfuls of host DNAinstead of the three to four headfuls normallyobserved for P1 DNA.
Burger & Trautner (1978) observed that afterSPP1 encapsidation was initiated (from 16 minutespost-infection on) the majority of phage DNAmolecules present in the infected cell have a sizesimilar to mature chromosomes. To conciliate thisresult with the occurrence of long packaging seriesrequiring substrates larger than eight to tenfold thesize of the SPP1 chromosome, we propose that afterthe first cut at pac the packaging apparatus followsclosely the replication machinery and most of thenew pac sites being generated would be encapsi-dated before attack by the terminase. A relatedstrategy would also be possible if formation of SPP1concatemers involves recombination events, es-pecially if cross-talk occurs between the recombina-tion machinery and the packaging apparatus (seeWu et al., 1995 for a terminase-dependentrecombination event). Long encapsidation series atlate stages of infection would be a naturalconsequence of the mechanism proposed.
Differences in packaging processivity duringinfection can also be due to variations in the relativeintracellular pools of terminase, substrate DNA andpro-capsids. These could affect the length of theencapsidation series in different ways. If cleavage atpac is a rate limiting step, initiation would requirehigher concentrations of terminase than processivepackaging. Accordingly, Adams et al. (1983)showed that a reduction in terminase levels,particularly of its major subunit, affects thefrequency of packaging initiation rather thanprocessivity during bacteriophage P22 encapsida-tion. An opposite effect would be expected ifpro-capsids are present in limiting amounts

Headful Packaging and Fate of the Cleaved DNA Ends 965
implying that molecules cleaved at pac areencapsidated but subsequent packaging cycleswould be rare due to the lack of pro-capsids.Furthermore, the rate of sequential packagingevents is limited by the speed of DNA translocationinto the pro-capsid and the time of formation of anew complex between the substrate DNA andanother pro-capsid.
In summary, DNA packaging is a dynamicprocess occurring pari passu with the generation ofsubstrate concatemers. Various factors contribute todefine the distribution of progeny chromosomes.Under conditions favouring SPP1 fast multipli-cation the encapsidation series are short due to apredominance of packaging events initiated at pacand to early host lysis. If infection lasts a long timea large number of sequential headfuls is ensured bythe high processivity of the packaging machinery.
Material and Methods
Bacterial and phage strains
B. subtilis and SPP1 strains were as described (Tavareset al., 1992).
Materials and standard methods
Endonucleases SphI, SmaI and SnaBI were purchasedfrom New England Biolabs and BglII and EcoRI werefrom Boehringer-Mannheim. T4 polynucleotide kinasewas obtained from New England Biolabs. Micrococcalnuclease was obtained from Pharmacia Biotech andBenzonase from Eurogentec. Glutaraldehyde and EDACwere purchased from Fluka and Sigma, respectively.EDTA and NaI were obtained from Merck. All chemicalswere of analytical grade.
Titration and amplification of bacteriophage SPP1wild-type and mutants, viral particle purification andphage DNA extraction were as described (Chai et al.,1992; Tavares et al., 1992). All phage preparations usedwere purified by centrifugation through a CsCl stepgradient with the exception of the lysates used for theexperiment of Figure 3E.
Anti-gp6 and anti-SPP1 polyclonal sera were obtainedby immunization of rabbits with purified gp6 (Tavares,1992; Dube et al., 1993) and caesium chloride purifiedSPP1 particles, respectively.
Determination of the pac fragment molar ratio inDNA packaged during SPP1 infection
Exponentially growing YB886 were infected withSPP1wt (input multiplicity = 5) following our standardprocedure (Tavares et al., 1992). Samples were taken atdefined times and lysis was induced by addition of 2%(v/v) chloroform and 0.5 mg/ml lysozyme, shaken forfive additional minutes at 37°C, and kept on ice until theend of the kinetics. Lysate processing and DNAextraction were as described by Tavares et al. (1992).DNA digested with SmaI was resolved in agarose gels(0.8%, w/v) stained with ethidium bromide or SyberGreenTM (Molecular Probes), scanned in a FluorImager(Molecular Dynamics), and quantified using the Image-Quant software (Molecular Dynamics). Representativityof the pac fragment (SmaI-4) was determined by com-parison of the corresponding band signal with the
intensities of SmaI-3, 5 and 6, taking into account the ratiobetween the Mrs of the species being analysed.
Phage particles disruption and cross-linking
The chemicals to be tested were diluted from a stocksolution in TBT buffer (Biswal et al., 1967) to theconcentration required (8 ml total volume), equilibratedbriefly at 30°C and the reaction was initiated by additionof 2 ml from SPP1wt phages (01012 pfu/ml). Incubationwas for 30 minutes at 30°C. The reaction was stopped by50-fold dilution with TBT or phosphate buffer (forcross-linking, see below) followed by characterization:DNA loss from the viral capsid was tested by spotting10 ml of the suspension in a 2 ml drop of ethidium bromide(10 mg/ml) and visualization under ultraviolet light, andphage viability was evaluated by spotting serial dilutionsof the suspension (done in microtitre plates) on a lawn ofB. subtilis YB886. After this preliminary screening,selected samples were observed in the electron micro-scope using negative staining, direct adsorption to micaor cytochrome c techniques (see EM methods).
For cross-linking, phage particles or disrupted material(01012 pfu/ml) in phosphate buffer (10 mM sodiumphosphate (pH 7.5), 10 mM MgCl2) were mixed, with anidentical volume of a 1% (w/v) solution of glutaralde-hyde or 50 mM freshly prepared EDAC. After incubationfor five minutes at 30°C the reaction was quenched with100 mM glycine or 50-fold dilution in TBT. Aliquots werethen further treated with NaI or EDTA, or characterizedas described above. Buffer changes were done by dialysisin 0.025 mm VS filters (Millipore) when required. Resultsobtained with either glutaraldehyde or EDAC cross-link-ing were essentially identical.
Complexes of phage DNA bound to the connectorregion of the tail for electron microscopy were obtainedby treatment of virions (1010 to 1011 pfu) with 100 mMEDTA for 45 minutes at 45°C. The chelating agent waseliminated by dialysis against bi-distilled water for 90minutes in 0.025 mm pore size VS filters (Millipore) andthe sample was then used for EM studies or digestedwith an endonuclease (BglII, SphI or SnaBI), normallyovernight, before preparation for EM measurements todetermine the sizes of DNA molecules. Complexes formeasurement of the size of the DNA fragment protectedby the tail structure were prepared identically except thathigher amounts of phages were used (01011 pfu), and thesample was then treated overnight with Benzonase (150units/ml) and micrococcal nuclease (75 units/ml). AfterDNA digestion, half of the material was extracted twicewith phenol and once with chloroform. Residual organicsolvents were eliminated by dialysis against TE (10 mMTris-HCl (pH 7.5), 1 mM EDTA) in 0.025 mm filters. DNApresent in both deproteinized and non-treated sampleswas end-labelled with [g-32P]ATP using T4 polynucle-otide nuclease (Sambrook et al., 1989). Non-incorporatedlabel was removed by filter dialysis. The size of theradioactive species was estimated by running thesamples and appropriate Mr standards in native ordenaturing PAGE (Sambrook et al., 1989) followed byanalysis of the dried gels using a PhosphorImager(Molecular Dynamics). When required, the nucleases-treated disrupted phages (1012 pfu) were concentrated ina Centricon-50 (Amicon) and applied to 5% to 20%sucrose (w/v) gradients in 0.5 × TBT buffer preparedwith a Biocomp apparatus. Centrifugation was at35,000 rpm for 90 minutes in a SW50.1Ti rotor (4°C).Individual fractions were dialysed against 0.1 × TBT,concentrated tenfold under vacuum and an aliquot was

Headful Packaging and Fate of the Cleaved DNA Ends966
processed as above. Additionally, all fractions werecharacterized by SDS-PAGE (Laemmli, 1970), Westernblot with anti-gp6 and anti-SPP1 sera (Tavares et al.,1995), and electron microscopy.
Complexes of phage DNA ejected through the tailstructure were prepared from SPP1sizS. Ejection wastriggered by incubation with 2.5 M NaI for ten minutesat 30°C and the complexes were normally cross-linkedwith glutaraldehyde or EDAC. The ghost-DNA com-plexes were then used for EM measurements (see below)or analysed by equilibrium isopycnic centrifugation. Forthe latter experiment the particle suspension was mixedwith a CsCl solution (1.533 g/cm3) and centrifuged for40 hours at 35,000 rpm in a SW50.1Ti rotor (20°C).Individual fractions were dialysed against 10 mMTris-HCl (pH 7.6), 10 mM MgCl2 and characterized byEM observation and/or restriction analysis.
Kinetics of DNA ejection from various SPP1 strains(01010 pfu/reaction) triggered by NaI were performed asdescribed above except that the reaction was stopped bycross-linking at the desired time points (see Figure 6).After quenching with glycine and dialysis against 10 mMTris-HCl (pH 7.6), 10 mM MgCl2, the DNA released wasprobed by restriction with EcoRI. Digestion productswere resolved in 1% (w/v) agarose gels. Quantificationsof bands in the restriction patterns were done from directscans of the ethidium bromide stained gels (Fluorimager)or from negatives of photographed gels (densitometer[Molecular Dynamics]) using the ImageQuant software.
Electron microscopy
Negative staining with 1% (w/v) uranyl acetate wasperformed as described by Valentine et al. (1968).Preparation of the SPP1 phages for EM after treatmentwith different chemicals to release the DNA was done bydirect adsorption to mica (Portmann et al., 1974) and bycytochrome c spreading with the droplet technique (Lang& Mitani, 1970) essentially as described by Spiess & Lurz(1988). For length measurements we preferred samplesprepared by the droplet technique because most of themolecules could be traced easily due to unfolding of theDNA in the cytochrome c surface film. Except for somecontrol experiments done to measure all DNA of a givenarea, we selected only those DNA molecules which hada tail or an empty phage attached at one end.
Data analysis and graphics
Statistical analysis was done using the programStatistica (StatSoft). The graphics derived from thepolynomial formulation presented in Figure 7 and alldata graphics were constructed using MicrografxCharisma or Microsoft Excel.
AcknowledgementsWe are thankful to Gerhild Luder for performing part
of the electron microscopy experiments. We thank ElmarDroge (Institut fur Festkorperphysik, TU Berlin) for helpwith the mathematical formulation and generation of thetheoretical curves from Figure 7, Anja Droge fordiscussions, and Angie Hofmann for critical reading ofthe manuscript. We also acknowledge Mark Achtman forstatistical analysis. P.T. was partly supported by an ECfellowship (Contract ERBBIOTCL1923104).
ReferencesAdams, M. B., Hayden, M. & Casjens, S. (1983). On the
sequential packaging of bacteriophage P22 DNA.J. Virol. 46, 673–677.
Aubrey, K. L., Casjens, S. R. & Thomas, G. J., Jr (1992).Secondary structure and interactions of the pack-aged dsDNA genome of bacteriophage P22 investi-gated by Raman difference spectroscopy.Biochemistry, 31, 11835–11842.
Bachi, B. & Arber, W. (1977). Physical mapping of BglII,BamHI, EcoRI, HindIII, and PstI restriction fragmentsof bacteriophage P1 DNA. Mol. Gen. Genet. 153,311–324.
Bazinet, C. & King, J. (1985). The DNA translocatingvertex of dsDNA bacteriophage. Annu. Rev. Micro-biol. 39, 109–129.
Biswal, N., Kleinschmidt, A. K., Spatz, H. C. & Trautner,T. A. (1967). Physical properties of the DNA ofbacteriophage SP50. Mol. Gen. Genet. 100, 39–55.
Black, L. W. (1989). DNA packaging in dsDNAbacteriophages. Annu. Rev. Microbiol. 43, 267–292.
Bravo, A. & Alonso, J. C. (1990). The generation ofconcatemeric plasmid DNA in Bacillus subtilis as aconsequence of bacteriophage SPP1 infection. Nucl.Acids Res. 18, 4651–4657.
Bravo, A., Alonso, J. C. & Trautner, T. A. (1990).Functional analysis of the Bacillus subtilis bacterio-phage SPP1 pac site. Nucl. Acids Res. 18, 2881–2886.
Burger, K. J. & Trautner, T. A. (1978). Specific labelling ofreplicating SPP1 DNA. Analysis of viral DNAsynthesis and identification of phage dna-genes. Mol.Gen. Genet. 166, 277–285.
Casjens, S. & Hayden, M. (1988). Analysis in vivo of thebacteriophage P22 headful nuclease. J. Mol. Biol. 199,467–474.
Casjens, S. & Hendrix, R. (1988). Control mechanisms indsDNA bacteriophage assembly. In The Bacterio-phages, (Calendar, R., ed.), vol. 1, pp. 15–91, PlenumPress, New York.
Casjens S., Wyckoff, E., Hayden, M., Sampson, L., Eppler,K., Randall, S., Moreno, E. & Serwer, P. (1992).Bacteriophage P22 portal protein is part of the gaugethat regulates packing density of intravirion DNA.J. Mol. Biol. 224, 1055–1074.
Chai, S., Bravo, A., Luder, G., Trautner, T. A. & Alonso,J. C. (1992). Molecular analysis of the B. subtilisbacteriophage SPP1 region encompassing genes 1 to6. The products of gene 1 and gene 2 are required forpac cleavage. J. Mol. Biol. 224, 87–102.
Chai, S., Szepan, U., Luder, G., Trautner, T. A. & Alonso,J. C. (1993). Sequence analysis of the left end of theBacillus subtilis bacteriophage SPP1 genome. Gene,129, 41–49.
Chai, S., Kruft, V. & Alonso, J. C. (1994). Analysis of theBacillus subtilis bacteriophages SPP1 and SF6 gene 1product: a protein involved in the initiation ofheadful packaging. Virology, 202, 930–939.
Chai, S., Lurz, R. & Alonso, J. C. (1995). The small subunitof the terminase enzyme of Bacillus subtilis bacterio-phage SPP1 forms a specialized nucleoproteincomplex with the packaging initiation region. J. Mol.Biol. 252, 386–398.
Chattoraj, D. K. & Inman, R. B. (1974). Location of DNAends in P2, 186, P4 and lambda bacteriophage heads.J. Mol. Biol. 87, 11–22.
Chow, L. T. & Bukhari, A. I. (1977). Bacteriophage Mugenome: structural studies on Mu DNA and Mumutants carrying insertions. In DNA Insertion

Headful Packaging and Fate of the Cleaved DNA Ends 967
Elements, Plasmids, and Episomes (Bukhari, A. I.,Shapiro, J. A. & Adhya, S. L., eds), pp. 295–306, ColdSpring Harbor Laboratory Press, Cold SpringHarbor, NY.
Deichelbohrer, I., Messer, W. & Trautner, T. A. (1982).Genome of Bacillus subtilis bacteriophage SPP1:structure and nucleotide sequence of pac, the originof DNA packaging. J. Virol. 42, 83–90.
Dube, P., Tavares., P., Lurz, R. & van Heel, M. (1993).Bacteriophage SPP1 portal protein: a DNA pumpwith 13-fold symmetry. EMBO J. 12, 1303–1309.
Earnshaw, W. C. & Casjens, S. (1980). DNA packaging bythe double-stranded DNA bacteriophages. Cell, 21,319–331.
Esche, H., Schweiger, M. & Trautner, T. A. (1975). Geneexpression of bacteriophage SPP1. I. Phage directedprotein synthesis. Mol. Gen. Genet. 142, 45–55.
Gill, G. S. & MacHattie, L. A. (1976). Limitedpermutations of the nucleotide sequence in bacterio-phage T1 DNA. J. Mol. Biol. 104, 505–515.
Hendrix, R. W. (1978). Symmetry mismatch and DNApackaging in large bacteriophages. Proc. Natl Acad.Sci. USA, 75, 4779–4783.
Humphreys, G. O. & Trautner, T. A. (1981). Maturationof bacteriophage SPP1 DNA: limited precision in thesizing of mature bacteriophage genomes. J. Virol. 37,832–835.
Jackson, E. N., Jackson, D. A. & Deans, R. J. (1978). EcoRIanalysis of bacteriophage P22 DNA packaging.J. Mol. Biol. 118, 365–388.
Klotz, G. & Spatz, H. Ch. (1971). A biological assay forintracellular SPP1 DNA. Mol. Gen. Genet. 110,367–373.
Laemmli, U. K. (1970). Cleavage of structural proteinsduring the assembly of the head of bacteriophage T4.Nature, 227, 680–685.
Lang, D. & Mitani, M. (1970). Simplified quantitativeelectron microscopy of biopolymers. Biopolymers, 9,373–379.
Morelli, G., Fisseau, C., Behrens, B., Trautner, T. A., Luh,J., Ratcliff, S. W., Allison, D. P. & Ganesan, A. T.(1979). The genome of B. subtilis phage SPP1: thetopology of DNA molecules. Mol. Gen. Genet. 168,153–164.
Parkinson, J. S. & Huskey, R. J. (1971). Deletion mutantsof bacteriophage lambda. I. Isolation and initialcharacterization. J. Mol. Biol. 56, 369–384.
Portmann, R., Sogo, J. M., Koller, T. & Zillig, W. (1974).Binding sites of E. coli RNA polymerase on T7 DNAas determined by electron microscopy. FEBS Letters,45, 64–67.
Ratcliff, S. W., Luh, J., Ganesan, A. T., Behrens, B.,Thompson, R., Montenegro, M. A., Morelli, G. &Trautner, T. A. (1979). The genome of Bacillus subtilisphage SPP1: the arrangement of restriction endonu-
clease generated fragments. Mol. Gen. Genet. 168,165–172.
Santos, M. A., Almeida, J., Lencastre, H., Morelli, G.,Kamke, M. & Trautner, T. A. (1986). Genomicorganization of the related Bacillus subtilis bacter-iophages SPP1, 41c, r15 and SF6. J. Virol. 60, 702–707.
Sambrook, J., Fritsch, E. F. & Maniatis, T. (1989). MolecularCloning. A Laboratory Manual, 2nd edit., Cold SpringHarbor Laboratory Press, Cold Spring Harbor, NY.
Spiess, E. & Lurz, R. (1988). Electron microscopic analysisof nucleic acids and nucleic acid-protein complexes.Methods Microbiol. 20, 293–323.
Sternberg, N. & Coulby, J. (1987). Recognition andcleavage of the bacteriophage P1 packaging site(pac). I. Differential processing of the cleaved endsin vivo. J. Mol. Biol. 194, 453–468.
Tavares, P. (1992). Func� ao da proteına portal naencapsidac� ao do DNA pelo bacteriofago SPP1. PhDthesis, Universidade de Coimbra, Portugal.
Tavares, P., Santos, M. A., Lurz, R., Morelli, G., Lencastre,H. L. & Trautner, T. A. (1992). Identification of a genein Bacillus subtilis bacteriophage SPP1 determiningthe amount of packaged DNA. J. Mol. Biol. 225,81–92.
Tavares, P. Droge, A., Lurz, R., Graeber, I., Orlova, E.,Dube, P. & van Heel, M. (1995). The SPP1 connection.FEMS. Microbiol. Rev. 17, 47–56.
Thomas, J. O. (1974). Chemical linkage of the tail to theright-hand end of bacteriophage lambda DNA.J. Mol. Biol. 87, 1–9.
Thomas, J. O., Sternberg, N. & Weisberg, R. (1978).Altered arrangement of the DNA in injection-defec-tive lambda bacteriophage. J. Mol. Biol. 123, 149–161.
Tye, B. K., Huberman, J. A. & Botstein, D. (1974).Non-random circular permutation of phage P22DNA. J. Mol. Biol. 85, 501–532.
Valentine, R. C., Shapiro, B. M. & Stadtman, E. R. (1968).Regulation of glutamine synthetase. XII. Electronmicroscopy of the enzyme from Escherichia coli.Biochemistry, 7, 2143–2152.
Valpuesta, J. M. & Carrascosa, J. L. (1994). Structure ofviral connectors and their function in bacteriophageassembly and DNA packaging. Quart. Rev. Biophys.27, 107–155.
Wu, C. H. H., Lin, H. & Black, L. W. (1995). BacteriophageT4 gene 17 amplification mutants: evidence forinitiation by the T4 terminase subunit gp16. J. Mol.Biol. 247, 523–528.
Yanisch-Perron, C., Vieira, J. & Messing, J. (1985).Improved M13 phage cloning vectors and hoststrains: nucleotide sequences of the M13mp18 andpUC19 vectors. Gene, 33, 103–119.
Zachary, A. & Black, L. W. (1992). Isolation andcharacterization of a portal protein-DNA complexfrom dsDNA bacteriophage. Intervirology, 33, 6–16.
Edited by J. Karn
(Received 7 August 1996; accepted 4 October 1996)