chromogranin a is preferentially cleaved into pro...
TRANSCRIPT

1
Chromogranin A is preferentially cleaved into pro-angiogenic peptides in the bone marrow of multiple myeloma patients
Mimma Bianco1, Anna Maria Gasparri1, Barbara Colombo1, Flavio Curnis1, Stefania
Girlanda1, Maurilio Ponzoni1, Maria Teresa Sabrina Bertilaccio1, Arianna Calcinotto3,
Angelina Sacchi1, Elisabetta Ferrero1, Marina Ferrarini1, Marta Chesi2, P. Leif Bergsagel2,
Matteo Bellone3, Giovanni Tonon1, Fabio Ciceri1, Magda Marcatti1, Federico Caligaris-
Cappio1, 4, Angelo Corti1
Division of 1Experimental Oncology and 3Immunology, San Raffaele Scientific Institute, Milan,
Italy. 2Mayo Clinic, Scottsdale, USA. 4Università Vita-Salute San Raffaele, Milan, Italy.
Running title: Chromogranin A in multiple myeloma
Key words: Multiple myeloma, chromogranin A, angiogenesis, proteolytic processing,
plasmin, vasostatin-1, PAI-1
Corresponding author: Angelo Corti, Department of Molecular Oncology, San Raffaele
Scientific Institute, via Olgettina 58, 20132 Milan, Italy (Tel. +39 02 26434802; Fax +39 02
26434786; E-mail: [email protected])
Word counts for text: 5152 Word counts for abstract: 191
Figure counts: 7
Reference count: 39 Conflict-of-interest: No competing financial interest.
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

2
Abstract 1 Angiogenesis has been postulated to be critical for the pathogenesis of multiple myeloma 2 (MM), a neoplastic disease characterized by abnormal proliferation of malignant plasma cells 3 in the bone marrow (BM). Cleavage of the N- and C-terminal regions of circulating 4 chromogranin A (CgA, CHGA), classically an anti-angiogenic protein, can activate latent anti- 5 and pro-angiogenic sites, respectively. In this study, we investigated the distribution of CgA-6 derived polypeptides in MM patients and the subsequent implications for disease progression. 7 We show that the ratio of pro-/anti-angiogenic forms of CgA is altered in MM patients 8 compared with healthy subjects, and that this ratio is higher in BM plasma compared with 9 peripheral plasma, suggesting enhanced local cleavage of the CgA C-terminal region. 10 Enhanced cleavage correlated with increased VEGF and FGF2 BM plasma levels and BM 11 microvascular density. Using the Vk*MYC mouse model of MM, we further demonstrate that 12 exogenously administered CgA was cleaved in favor of the pro-angiogenic form and was 13 associated with increased microvessel density. Mechanistic studies revealed that MM and 14 proliferating endothelial cells can promote CgA C-terminal cleavage by activating the 15 plasminogen activator/plasmin system. Moreover, cleaved and full-length forms could also 16 counter-balance the pro-/anti-angiogenic activity of each other in in vitro angiogenesis assays. 17 These findings suggest that the CgA-angiogenic switch is activated in the BM of MM patients 18 and prompt further investigation of this CgA imbalance as a prognostic or therapeutic target.19
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

3
INTRODUCTION 1 Multiple myeloma (MM) is a plasma cell malignancy characterized by abnormal clonal 2 proliferation and accumulation of plasma cells in the bone marrow (BM). This results in a 3 variety of clinical manifestations including anemia, osteolytic bone lesions, hypercalcemia 4 and renal failure. Most cases of myeloma also feature the production of a paraprotein (also 5 called "M protein"), an abnormal immunoglobulin that can cause kidney problems (1). The 6 cross-talk between myeloma and endothelial cells in the BM and the consequent activation of 7 the angiogenesis process (i.e. the formation of new blood vessels from pre-existing vessels) is 8 critical for the pathogenesis of MM (2-4). Accordingly, BM microvessel density increases 9 parallel to disease progression, is an independent prognostic factor for survival, and correlates 10 with established parameters of disease activity in patients (3-11). 11 Physiological and pathological angiogenesis are tightly regulated by the coordinated action 12 of anti- and pro-angiogenic factors (12-14). Among the wide range of angiogenesis regulators 13 so far discovered, recent studies have shown that chromogranin A (CgA) may have an 14 important role in the regulation of angiogenesis (15, 16). CgA is a glycosylated, sulfated and 15 phosphorylated protein, 439 residue-long, stored in the secretory vesicles of many 16 neuroendocrine cells and neurons (17) and exocytotically released in circulation together with 17 the co-stored hormones, to reach 0.5-1 nM levels in normal conditions (18). Tissue-specific 18 intra-granular and extra-cellular proteolytic processing of CgA leads to production of various 19 bioactive peptides involved in the regulation of angiogenesis, metabolism and cardiovascular 20 system (18). Regarding angiogenesis, it has been recently shown that CgA contains: a) a 21 functional anti-angiogenic site in the C-terminal region 410-439; b) a latent anti-angiogenic 22 site in the N-terminal region 1-76 and c) a latent pro-angiogenic site in the region 352-372 23 (15, 19, 20). These sites are activated by proteolytic cleavage of Q76-K77 and R373-R374 24 bonds, respectively. Accordingly, full-length CgA1-439 and the N-terminal fragment CgA1-76 25 (called vasostatin-1) inhibit angiogenesis in various angiogenesis assays, whereas the 26 fragment CgA1-373 can stimulate angiogenesis (15). Mechanistic studies have shown that full-27 length CgA and the N-terminal fragment vasostatin-1 can inhibit endothelial cell migration, 28 motility, sprouting, invasion and capillary-like structure formation induced by vascular 29 endothelial growth factor (VEGF), a potent pro-angiogenic factor, as well as the pro-30 angiogenic activity of basic fibroblast growth factor (FGF2), another important factor 31 involved in the regulation of angiogenesis (15)(21). This fragment can also inhibit the nuclear 32 translocation of hypoxia inducible factor (HIF)-1α, a master regulator of angiogenesis, in 33 endothelial cells (22). On the other hand, the fragments CgA1-373 and CgA352-372 can induce the 34
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

4
release of FGF2 from endothelial cells (15, 19). Hematological studies have shown that 1 biologically relevant levels of full-length CgA1-439 and fragment CgA1-76 plus lower levels of 2 other fragments lacking the C-terminal region are present in circulation in normal subjects. 3 Blood coagulation causes, in a thrombin-dependent manner, conversion of CgA1-439 into the 4 pro-angiogenic fragment CgA1-373 (15). Thus, CgA-related circulating polypeptides form a 5 balance of anti- and pro-angiogenic factors tightly regulated by N- and C-terminal proteolysis. 6 Although abnormal levels of immunoreactive CgA have been detected in the blood of 7 patients with neuroendocrine tumors and in several other pathological conditions, including 8 renal failure, heart failure, rheumatoid arthritis, atrophic gastritis, inflammatory bowel disease, 9 sepsis, or in subjects treated with proton pump inhibitors (a class of drugs commonly used to 10 treat acid peptic disorders) (18, 23, 24), little information is available regarding the levels of 11 pro- and anti-angiogenic CgA-derived polypeptides in the blood of cancer patients and no 12 information at all regarding hematological malignancies, including MM. 13 To fill this gap, we investigated whether the balance of CgA-derived polypeptides (which 14 may favor angiostasis in normal condition) is altered in MM patients. To this aim we analyzed 15 the extent of N- and C-terminal proteolytic processing of CgA in MM patients, using plasma 16 samples obtained from peripheral blood and BM, and in immunocompetent Vk*MYC 17 transgenic mice, a model that has demonstrated high biologic fidelity to the human disease 18 (25). We show that the balance of anti-/pro-angiogenic CgA polypeptides is tipped toward a 19 pro-angiogenic state in the BM, with potentially important pathophysiological implications. 20 21 22 PATIENTS, MATERIALS AND METHODS 23 24 Patients and plasma samples 25 Peripheral blood plasma samples and BM aspirates were obtained from 31 patients with 26 MM at diagnosis (active, n=25; smoldering, n=5; MGUS=1; age 64.06 + 12.7 years (mean + 27 SD)), after informed written consent and with ethical approval from the institutional review 28 board. Patient's characteristics, disease staging, treatment with proton pump inhibitors (PPIs) 29 and other therapies are reported in Supplementary Table 1. BM plasma samples were 30 obtained by centrifugation of BM aspirates and cryopreserved in the gas-phase of liquid 31 nitrogen. Normal plasma samples were obtained from 25 normal donors (males/females, 32 13/12; age, 57.8 ± 13.9 years (mean ± SD)) that were not taking proton pump inhibitors or 33 other drugs. 34
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

5
1 Myeloma and endothelial cells 2 Human myeloma cell lines (KMS12-PE, KMS28-BM, U266), genotyped and tested for 3 Mycoplasma infection, were cultured in RPMI 1640 medium (Lonza) containing 10% fetal 4 bovine serum. Human umbilical vein endothelial cells (HUVECs) were cultured in endothelial 5 cell growth medium (EGM)-2 (Lonza, Walkersville, MD). 6 7 CgA fragments and antibodies 8 Full-length human CgA1-439, three fragments lacking the C-terminal region (CgA1-409, 9 CgA1-400, CgA1-373) and one fragment corresponding to the N-terminal region (CgA1-76) were 10 prepared by recombinant DNA technology as described (15) (see Fig. 1 A for a schematic 11 representation of CgA). Monoclonal antibodies (mAb) B4E11 and 5A8, and polyclonal 12 antisera α-439, α-410-439, α-373, and α-76 were described previously (15)(26, 27). The 13 polyclonal antiserum α-373 was prepared by immunizing a rabbit with the CgA368-373 peptide 14 coupled to keyhole-limpet hemocyanin. The antiserum, after pre-adsorption on a 15 chromatographic column bearing full-length CgA1-439, recognizes the fragments CgA1-373, but 16 not full-length CgA (Supplementary Fig. 1 A). Full details on antibody preparation and 17 epitopes can be found in Supplemental Methods and Supplementary Table 2. 18 19 Mass spectrometry analysis of CgA fragments 20 Samples were desalted by reverse-phase chromatography using C18 ZipTip resin 21 (Millipore). Mass Spectrometry analysis was performed using an LTQ-Orbitrap XL mass 22 spectrometer (Thermo Scientific, Bremen, Germany) equipped with a nanoelectrospray ion 23 source (Proxeon Biosystems, Odense, Denmark). Full scan mass spectra were acquired in the 24 LTQ Orbitrap mass spectrometer in the mass range m/z 700 to 1500 Da. 25 26 Immunoassays 27 Human full-length CgA and CgA fragments in plasma samples were detected using various 28 sandwich ELISAs, called *439−, *439+436−, *76−, and *439+436+FRs−ELISA, described 29 previously (15). These assays, based on the use of antibodies against different epitopes of CgA 30 (see Supplementary Table 2), were calibrated using CgA1-439 (*439−, *439+436−, and 31 *439+436+FRs−ELISA) or with CgA1-76 (*76−ELISA). These ELISAs can selectively detect: 32 a) N-terminal fragments cleaved after residue Q76 (*76−ELISA); b) full-length CgA 33
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

6
(*439−ELISA); c) full-length CgA with or without the C-terminal tripeptide sequence 437-439 1 (*439+436−ELISA) and d) full-length CgA plus fragments containing the N-terminal and all 2 or part of the central and C-terminal regions (*439+436+FRs−ELISA) (15). 3 The fragment CgA1-373 was analyzed by a new sandwich-ELISA (called *373−ELISA) 4 based on the use of mAb-B4E11 in the capture step, the antibody α-373 in the detection step, 5 and CgA1-373 as a standard. Assay validation experiments showed that this ELISA selectively 6 detects CgA1-373, but not CgA1-439, CgA1-409, CgA1-400, and CgA1-76 (Supplementary Fig. 1 B). 7 All these assays can not detect murine CgA and CgA fragments. A schematic representation of 8 the five ELISAs used in this study is shown in Fig. 1 B. 9 VEGF and FGF2 were analyzed using the ELISA Quantikine kits (R&D Systems). 10 11 Rat aortic ring (RAR) angiogenesis assay 12 The rat aortic ring assay, based on measurement of the number of capillary-like structures 13 spontaneously sprouting from rat aorta rings after 6 days in cell culture, was performed as 14 described in Supplementary Methods. Basal angiogenesis, obtained without addition of FGF2 15 or VEGF, was examined in the absence or presence of various doses of CgA polypeptides. 16 17 In vivo studies in the Vk*MYC model 18 Studies on animal models (Vk*MYC mice) were approved by the Ethical Committee of the 19 San Raffaele Scientific Institute and done according to the prescribed guidelines. Vk*MYC 20 transgenic mice spontaneously develop clinically relevant MM as a result of activation of the 21 c-myc oncogene in maturing B cells under control of the kappa light chain (Vk) promoter (25). 22 Disease development was monitored by measuring the levels of paraprotein (M-spike) in the 23 serum, by serum protein electrophoresis, as previously described (28). Vk*MYC mice develop a 24 monoclonal gammopathy starting at 30 weeks of age that progress slowly over time. Mice 55-25 80 weeks old were injected with 3 µg of recombinant human CgA1-439. Blood was collected 26 after 1 and 24 h in heparinized tubes and immediately centrifuged (2000xg, 15 min) using a 27 refrigerated centrifuge. Plasma samples were analyzed by *439− and 28 *439+436+FRsELISAs to assess human CgA fragmentation. Serum samples were also 29 prepared in parallel for M-spike quantification. 30 31 Immunohistochemical analysis of BM microvascular density 32
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

7
BM microvessel density in Vk*MYC mice was analyzed by immunofluorescence 1 microscopy as follows: bones were fixed with formalin, decalcified with Fix Decal (Pro-Eco), 2 embedded in paraffin and sectioned (4 μm). Vessels were stained by incubation with anti-3 CD31 antibody (1:200, overnight) (Neomarkers, Fremont, CA) followed by biotinylated 4 secondary antibody and avidin-horseradish peroxidase (BioCare, UK). Slides were then 5 incubated for 5 min with the 3,3’–diaminobenzidine (DAB) and, after washing, with Mayer-6 Hematoxylin (BioOptica, Italy). All sections were blindly evaluated by an expert 7 hematopathologist. 8 Analysis of BM microvessel density in MM patients and, as a control, in uninvolved BM 9 biopsies from patients with nodal diffuse large B-cell lymphoma, was performed using 10 formalin-fixed, paraffin embedded and decalcified sections (3 µm thick) of BM biopsies 11 stained with an anti-CD34 antibody (Novocastra, UK, diluted 1:200), after antigen retrieval 12 with a TRIS-EDTA buffer at pH 9. 13 14
15 RESULTS 16
17 Full-length CgA and various fragments are present in the peripheral blood of normal 18 subjects and MM patients (at diagnosis) in variable amounts 19 To assess the extent of proteolytic processing of circulating CgA in normal subjects and 20 MM patients we have analyzed samples of peripheral blood plasma with five different 21 sandwich ELISAs specific for various CgA-derived polypeptides (see Supplementary Table 22 2 for antibody and assay description, analyte sequences and codes, and Fig. 1 A and B for a 23 schematic representation of CgA, antibody epitopes and assays). 24 The results showed that various CgA-derived polypeptides are present in the peripheral 25 blood (PB) of normal subjects, including: a) full-length CgA1-439 (called *439) b) fragments 26 lacking residues 437-439 (CgA1-436); c) extremely low amounts of CgA1-373 (called *373); d) 27 CgA1-76 (called *76) and e) fragments lacking the C-terminal region 410-439 but containing 28 central region epitopes (FRs) (Fig. 1 C, first bars). 29 Analysis of PB plasma from MM patients at diagnosis revealed that the pro-angiogenic 30 fragment *373, but not the antiangiogenic *439 and *76 polypeptides, were increased (Fig. 1 31 C, second bars). As a consequence, the *373/*439 and *373/*76 ratios (i.e. the ratio of pro-32 /anti-angiogenic forms) were significantly increased in the peripheral blood of patients 33 compared to normal subjects (Fig. 1 E, first and second bars). These data point to increased 34
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

8
cleavage of CgA C-terminal region in MM patients, particularly at residue R373. 1 2 Full-length CgA is cleaved at R373 in the BM of MM patients 3 To assess whether cleavage of CgA at R373 occurred within secretory cells, during its 4 transport in the blood, or within the bone marrow (BM), we then analyzed plasma samples 5 obtained from the BM of MM patients by ELISA. Interestingly, we observed that BM plasma 6 contained even lower levels of *439 and higher levels of *373, compared to PB plasma (Fig. 1 7 C, third bars, and Fig. 1 D). These data suggest that cleavage at R373 occurred in the BM. In 8 contrast, no changes of *76 was observed. Thus, the *373/*439 and *373/*76 ratios were 9 further increased in the BM of patients (Fig. 1 E, third bars), suggesting that the balance of 10 pro-/anti-angiogenic CgA polypeptides was tipped toward a pro-angiogenic state in the BM. 11 12 Treatment with proton pump inhibitors and renal failure are major causes of increased 13 secretion of CgA in MM patients 14 The cause and the source of the increased CgA levels, observed in certain MM patients, 15 were then investigated. No CgA was detected in the supernatant of various cultured myeloma 16 cell lines (data not shown), arguing against a role of myeloma cells as a source of CgA. More 17 likely, CgA was released in circulation by the neuroendocrine system. Considering that a large 18 fraction of our study population were taking proton pump inhibitors (PPIs) or had renal failure 19 (RF), two conditions known to enhance the circulating levels of CgA, we evaluated each 20 analyte in the BM of patients who were not taking PPIs and had no RF. Although these 21 patients had normal levels of total CgA (*439+436+FRs) (Fig. 1 C bottom, compare the last 22 bar with the first bar) they had low levels of *439 and high levels of *373 (Fig. 1 C) and, 23 consequently, markedly elevated *373/*439 and *373/*76 ratios (Fig. 1 D, last bars). These 24 data strongly suggest that the CgA normally released in circulation by the neuroendocrine 25 system was proteolytically processed at residue R373 in the BM of MM patients. 26 To assess the specific effects of PPI and RF on CgA levels in PB and BM plasma we then 27 stratified the patients according to these variables. As expected, RF tended to increase all 28 variables in PB and BM (Supplementary Fig. 2 A). PPI-treatment was associated with 29 increased levels of *439 and other fragments, but not of *76 (Supplementary Fig. 2 B). These 30 data and the results of previous studies showing that RF and PPI treatment are associated with 31 increased CgA levels (29-32) suggest that both these factors were important causes of the 32 increased circulating levels of CgA in MM patients. However, considering that RF can be 33 associated with more advanced disease (as also the need of PPI-treatment) we cannot exclude 34
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

9
a direct role of the disease itself. Furthermore, as PPI-treated patients were also taking steroids 1 and/or other drugs (Supplementary Table 1), we cannot exclude that also other 2 pharmacological treatments could have contributed to the increased CgA in those patients. 3 The associations of CgA and fragments with disease parameters and stage were also 4 analyzed. *76 or *439+436+FRs negatively correlated with M-spike, whereas *439 positively 5 correlated with creatinine (Supplementary Table 3). No significant association was observed 6 between each analyte and the International or Durie and Salmon Staging Systems (ISS and 7 DSS) for all patients (Supplementary Fig. 3 A). However, we observed a higher *373/*439 8 ratio in 3 out of 6 PPI-untreated patients at DSS III (Supplementary Fig. 3 B, right). 9
10 The cleavage of CgA C-terminal region is associated with FGF2 and VEGF levels and 11 increased microvessel density in the BM of MM patients 12 We then investigated whether CgA cleavage in the BM of MM patients for which BM 13 biopsis were available was associated with established parameters of angiogenesis activation. 14 Interestingly, the enhanced ratio of pro-/anti-angiogenic forms (e.g. *373/*439) in BM plasma 15 was associated with enhanced production of VEGF and FGF2 in the BM, two potent pro-16 angiogenic cytokines (Fig. 2 A and B, and Supplementary Table 4). Of note, *373/*439, 17 VEGF and FGF2 tended to be higher in patients with advanced disease stage (Fig. 2 A and B). 18 No significant correlation was observed between VEGF and FGF2 (r=0.412, p=0.057). 19 Furthermore, the sum of standardized values of the ratios between FRs (which includes *373 20 and other fragments lacking the C-terminal region, like CgA1-372, potentially pro-angiogenic) 21 and *439 or *76 (anti-angiogenic) significantly correlated with the microvessel density (MVD) 22 in patients (Fig. 3 A and B). No statistically significant values were obtained with single ratios 23 or with single variables, suggesting that the overall balance of pro- and anti-angiogenic CgA 24 polypeptides is more important than single factors. These data indicate that proteolytic 25 cleavage of CgA C-terminal region and the consequent change in the relative levels of pro-26 /anti-angiogenic forms of CgA was associated with angiogenesis in the BM of patients. 27 In contrast, no significant correlation was observed between MVD and VEGF or FGF2 28 (r=0.176, p=0.484; r=0.135, p=0.595, respectively). 29 30 The anti-angiogenic and pro-angiogenic forms of CgA can regulate each other in the 31 RAR angiogenesis assay 32 To assess whether CgA cleavage and changes in the relative levels of pro-/anti-angiogenic 33 forms of CgA may regulate angiogenesis we analyzed the effect of full-length CgA and CgA1-34
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

10
373, alone and in combination, on the spontaneous capillary sprouting from rat aorta rings 1 (RAR) cultured in collagen gels, a widely used angiogenesis assay. In a previous study we 2 showed that 0.2-1 nM CgA1-439 can inhibit the pro-angiogenic effects of FGF2 and VEGF, as 3 well as basal angiogenesis, whereas CgA1-373 can promote spontaneous angiogenesis at 1-9 4 nM concentrations (15). As expected, CgA1-373, alone (3 nM), significantly enhanced the 5 number of capillary-like structures spontaneously outgrowing from rat aorta rings, whereas 1 6 nM CgA1-439 decreased the number of capillary-like structures (Fig. 4 A). When we added 7 both components to the cultures we observed neutralization of each other. Similar 8 experiments were performed with CgA1-76, a fragment previously shown to inhibit the 9 angiogenic effects of FGF2 and VEGF (15). Although this fragment could not inhibit the 10 spontaneous sprouting of capillaries from aortic rings, 1 nM CgA1-76 could neutralize CgA1-373 11 (Fig. 4 B). These data suggest that changes in the relative levels of anti-/pro-angiogenic CgA 12 fragments, as observed in the BM of patients, can regulate the anti-/pro-angiogenic activity of 13 each other and, consequently, can contribute to promote angiogenesis in the BM of patients. 14 In particular the increase of CgA1-373 and the concomitant decrease of CgA1-439 in the BM of 15 patients may tip the balance toward a pro-angiogenis state in the BM microenvironment. On 16 the other hand the inhibitory activity of CgA1-76 against CgA1-373 , FGF2 and VEGF suggests 17 that this fragment, which is produced also in physiological conditions, may serve to buffer the 18 pro-angiogenic effects of these agents at a systemic level. 19 20 Human CgA systemically administered to transgenic Vk*MYC mice is proteolytically 21 processed at the C-terminal region 22 To provide further experimental evidence that MM is associated with enhanced cleavage of 23 CgA and angiogenesis we performed a study using Vk*MYC transgenic mice, a clinically 24 relevant model of MM. Notably, in this model, mice having an M-spike >12% of total serum 25 proteins, i.e. mice with more advanced disease (25), had a BM microvascular density 2-fold 26 greater than those with M-spike <12% (Fig. 5 A), suggesting that angiogenesis was active in 27 these mice. 28 When we injected recombinant human CgA1-439 (3 µg, i.v) to 55-80 week-old mice with M-29 spike >12% we observed that the *373/*439 ratio in plasma samples taken 1 h later was 30 increased, compared to mice with M-spike <12% (Fig. 5 B), pointing to proteolytic cleavage 31 of CgA in the C-terminal region. Accordingly, a similar experiment performed with normal 32 and Vk*MYC mice showed that the *439/*439+436+FRs ratio (which is equivalent to 1 when 33
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

11
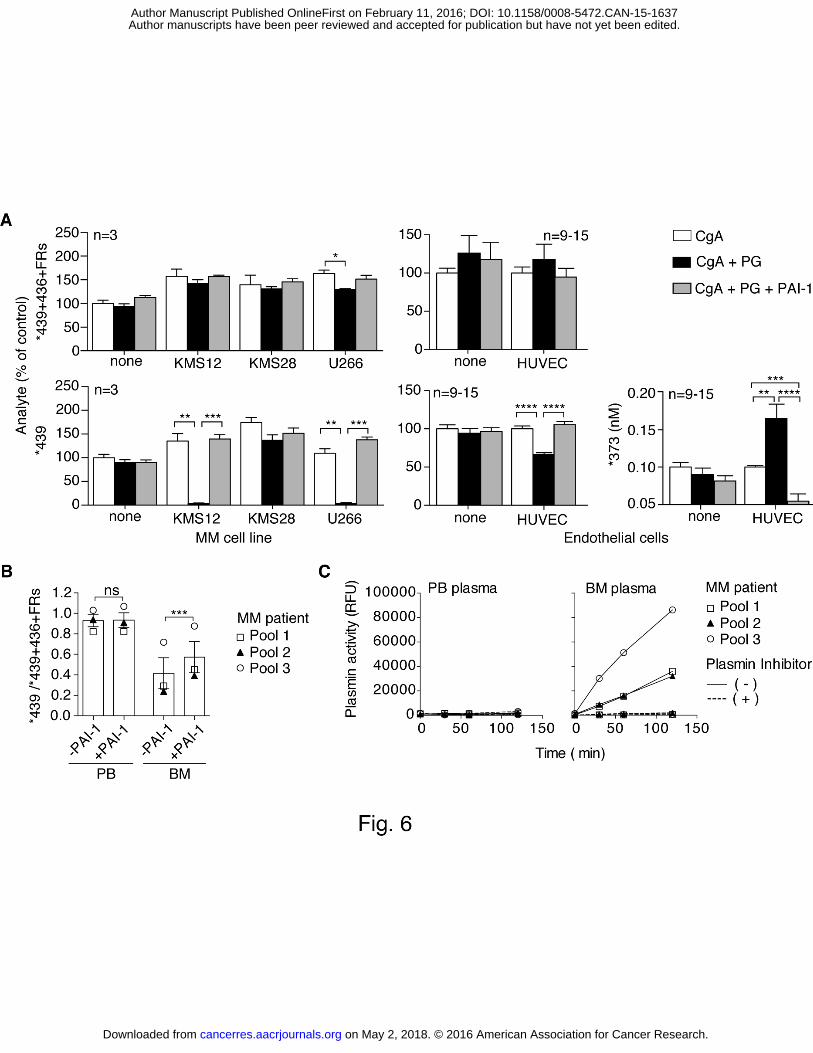
CgA is intact and to 0 when it is totally cleaved in the C-terminal region) significantly and 1 inversely correlated with the M-spike (Fig. 5 C). These results support the hypothesis that 2 disease progression is associated with cleavage of CgA. Considering that CgA was injected 3 into mice intravenously, these data also support the hypothesis that the cleavage of CgA 4 observed in patients occurred in the extracellular environment, likely the BM 5 microenvironment, and not in neuroendocrine secretory cells. 6 7 Myeloma and endothelial cells can promote CgA C-terminal cleavage by activating 8 plasminogen to plasmin 9 We then tested the hypothesis that MM cells and/or proliferating endothelial cells, both 10 present in the BM of patients with enhanced angiogenesis, could cleave CgA. To test these 11 hypotheses we incubated full-length CgA with three human myeloma cell lines (KMS12, 12 KMS28, U266) or with endothelial cells (HUVECs) and monitored CgA fragmentation by 13 ELISA (6 h later). No cleavage was observed, arguing against a direct role of these cells in 14 CgA fragmentation. However, when we added plasminogen to the cultures we observed that 15 two myeloma cell lines (KMS12, U266) as well as endothelial cells caused significant loss of 16 immunoreactivity in the *439-ELISA, but not in the *439+436+FRs-ELISA, pointing to 17 cleavage of CgA C-terminal region (Fig. 6 A). It appears therefore that both myeloma and 18 endothelial cells can promote CgA cleavage, although to a different extent. No significant 19 degradation of CgA was observed with cultured HS-5 bone marrow stromal cells or with 20 human peripheral blood mononuclear cells (data not shown). Interestingly, the fragment *373 21 was detected in the supernatant of endothelial cells, but not of myeloma cells. Furthermore, 22 cleavage was completely prevented by the addition of plasminogen-activator inhibitor-1 (PAI-23 1) to the cultures. Based on these data we hypothesized that myeloma and endothelial cells 24 produced a plasminogen activator and that plasmin, thereof, was the proteolytic enzyme 25 responsible for CgA cleavage. 26 In agreement with this hypothesis, western blot analysis of myeloma and endothelial cells 27 showed that both cell types could produce the urinary-type plasminogen activator, an enzyme 28 that can efficiently convert plasminogen to plasmin (Supplementary Fig. 4). 29 To assess whether the plasminogen activator/plasmin system was activated in the BM of 30 MM patients to an extent sufficient to cleave CgA we spiked PB or BM plasma samples with 31 human full-length CgA and analyzed each sample by *439-ELISA and *439+436+FRs-32 ELISA after incubation for 2 h in the absence or presence of PAI-1. Little or no C-terminal 33 degradation occurred in PB plasma samples, as suggested by the observation that the 34
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

12
*439/*439+436+FRs ratio remained close to 1 in these conditions (Fig. 6 B). In contrast, this 1 ratio dropped to 0.4 upon incubation in BM plasma, pointing to C-terminal degradation. 2 Notably, degradation was significantly inhibited by PAI-1 (Fig. 6 B) supporting the 3 hypothesis that plasminogen activators were present in the BM in an amount sufficient to 4 cause plasmin formation and CgA cleavage. Accordingly, when we measured the plasmin 5 activity with the Sensolyte Rh110 fluorimetric assay, a very sensitive commercial kit, we 6 observed that a) BM plasma, but not PB plasma, could cleave the plasmin fluorogenic 7 substrate and b) cleavage was completely inhibited by the selective plasmin inhibitor D-Val-8 Phe-Lys chloromethyl ketone (Fig. 6 C). These findings further support the hypothesis that 9 the plasminogen activator/plasmin system was activated in the BM of patients. 10 11 The R373R374 dibasic site of CgA C-terminal region is efficiently cleaved by plasmin 12 To verify that plasmin can indeed cleave the C-terminal region of CgA and to identify 13 cleavage sites we incubated full-length CgA with low amounts of plasmin-Sepharose and 14 monitored CgA fragmentation by SDS-PAGE, western blotting, ELISAs, and mass 15 spectrometry. SDS-PAGE and western blot analysis with various antibodies against the N-16 terminal (mAb B4E11), central (α-FRs, α-373) and C-terminal region (α-410-439, α-439) 17 showed that plasmin can indeed cleave CgA to generate large fragments lacking the C-18 terminal region, including, albeit not limited to, CgA1-373 (Fig. 7 A). This view was supported 19 by the results of ELISA and mass spectrometry analysis (Fig. 7 B and C). Interestingly, 20 although various cleavage sites were identified in the C-terminal region of CgA by mass 21 spectrometry (Fig. 7 C and D), cleavage at R373 occurred faster than other sites (Fig. 7 B 22 and C). Of note, prolonged incubation with plasmin led to loss of immunoreactivity in the 23 *373-ELISA, likely because of excessive cleavage at multiple sites. This may explain the lack 24 of *373 in the supernatant of MM cells that efficiently cleaved CgA. 25 These findings, overall, suggest that both MM and endothelial cells can contribute to CgA 26 cleavage and that the plasminogen activator/plasmin system is an important mechanism for 27 CgA fragmentation. 28
29 30
DISCUSSION 31 We have previously shown that circulating CgA1-439 (an anti-angiogenic protein) can work 32 as an angiogenic switch positively activated by cleavage at R373 and negatively regulated by 33 cleavage at Q76, with CgA1-373 and CgA1-76 being capable of exerting pro- and anti-angiogenic 34
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

13
activities, respectively (15). The present work provides experimental evidence to suggest that 1 the CgA-angiogenic switch is positively activated in MM. 2 In particular, the results of hematological studies performed with a series of ELISAs capable 3 of discriminating between intact and cleaved molecules show that various CgA-derived 4 polypeptides are present in the peripheral blood of MM patients at diagnosis, including CgA1-5 439 (called *439), CgA1-373 (*373), CgA1-76 (*76) and other fragments. Remarkably, the relative 6 levels of circulating pro- and anti-angiogenic molecules (e.g. *373/*439 and *373/*76) were 7 higher in patients than in normal subjects, suggesting that a proteolytic mechanism capable of 8 "turning-on" the CgA-angiogenic switch was active in MM patients. Furthermore, the results 9 of hematological studies show that the BM plasma of patients contained higher levels of *373 10 and lower levels of *439 than peripheral-blood plasma, suggesting that cleavage at R373 11 occurred in the BM. At variance, cleavage at Q76 likely occurred in secretory neuroendocrine 12 cells (and/or in circulation) and only in minor part in the BM, as suggested by the modest 13 increase of *76 observed in the BM plasma compared to peripheral-blood plasma. 14 The results also show that certain MM patients had higher circulating levels of total CgA 15 compared to normal subjects. Which are the sources and the mechanisms underlying such 16 increase? As cultured myeloma cells do not release CgA in the supernatant (not shown) it is 17 likely that abnormal CgA was produced by the (neuro)endocrine system. Notably, in our study 18 population, patients having abnormal CgA were taking PPIs and/or had renal failure, i.e. two 19 conditions known to be associated with increased circulating CgA. Indeed, PPIs, which are 20 drugs commonly used in the treatment of acid peptic disorders, can enhance 2 to 3 times, and 21 in certain subjects even up to 10 times, the circulating levels of CgA (depending on 22 administration schedule), by inducing CgA-positive enterochromaffin-like cell hyperplasia 23 (30-32). Thus, enterochromaffin-like cells were likely the major source of increased CgA 24 levels in PPI-treated patients. The mechanism of CgA increase consequent to renal failure is 25 different, being likely related to a decline of glomerular filtration rate and accumulation of the 26 CgA pool released by the diffuse neuroendocrine system, as previously reported (29). Thus, 27 the cellular sources of aberrant CgA in PPI-treated patients and in patients with RF were likely 28 different. We cannot exclude, however, that in some patients also the disease, by itself, may 29 cause CgA elevation, considering that disease progression may be associated with occurrence 30 of RF and increased need for PPI treatment. 31 Remarkably, patients who were not taking PPIs and had no RF had normal levels of total CgA, 32 still had abnormally low levels of *439 and high levels of *373 in the BM (hence high 33 *373/*439 ratio). This observation lends support to the concept that even the normal pool of 34
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

14
circulating CgA was cleaved in the BM of patients, changing the local balance of pro-/anti-1 angiogenic forms. 2 These findings raise the question as to whether changes in the balance of pro-/anti-3 angiogenic CgA molecules in the BM might contribute to promote local angiogenesis. The 4 following considerations suggest that CgA and its fragments may indeed have a role in the 5 regulation of angiogenesis in patients. First, we have previously shown that CgA1-439 and 6 CgA1-76 can inhibit angiogenesis in various in vivo and in vitro angiogenesis assays, whereas 7 the fragment CgA1-373 promotes angiogenesis, at pathophysiologically relevant concentrations 8 (15). Second, the results of the angiogenesis assays of the present study show that CgA1-439 9 and CgA1-373 can counterbalance the activity of each other. Thus, the increased cleavage of 10 CgA at residue R373 and other sites in the C-terminal region may tip the balance toward pro-11 angiogenic effects. Third, the present results of hematological studies in MM patients show 12 that the *373/*439 ratio correlates with the BM plasma levels of VEGF and FGF2, two potent 13 pro-angiogenic factors known to play key roles in the cross-talk between myeloma cells, 14 stromal cells and endothelial cells (4, 33). Notably, in previous studies we have shown that 15 CgA1-439 can inhibit the anti-angiogenic activity of FGF2 and VEGF, whereas CgA1-373 can 16 promote the release of FGF2 from cultured endothelial cells (15). Finally, the results of 17 microvascular density (MVD) analysis in BM tissue sections obtained from patients show 18 positive correlation between MVD and the extent of proteolytic cleavage of CgA C-terminal 19 region. These findings suggest that CgA and its fragments may indeed represent new players 20 in the regulation of angiogenesis in the BM microenvironment of MM patients. 21 Regarding the proteolytic mechanism responsible for the cleavage of the CgA C-terminal 22 region in MM patients, the results of in vitro experiments performed with cell cultures suggest 23 that MM cells and proliferating endothelial cells can induce CgA cleavage in a plasminogen-24 dependent manner and that this effect can be inhibited by PAI-1, a potent inhibitor of 25 plasminogen activators. These findings point to the plasminogen activator/plasmin system as 26 an important component of the CgA cleavage mechanism. Accordingly, the results of 27 biochemical and immunological studies of CgA after treatment with low doses of plasmin 28 show that indeed this enzyme can cleave CgA and generate large fragments lacking the C-29 terminal region, including CgA1-373. Interestingly, previous studies showed that myeloma cells 30 express the urinary-type plasminogen activator (uPA) and the uPA receptor, a cell surface 31 protein (34). This system is present also in the angiogenic endothelium and its activation is 32 known to be important for angiogenesis (35, 36). It is therefore possible that plasmin 33 activation, which may likely occur in the BM microenvironment of MM patients (because of 34
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

15
the presence of both myeloma cells and angiogenic endothelial cells) can start a vicious cycle 1 involving local cleavage of circulating CgA, activation of angiogenesis, plasma cell 2 proliferation, CgA cleavage and so on. According to this view, we observed that Vk*MYC 3 transgenic mice with advanced disease (having levels of M-spike >12%) and high MVD could 4 cleave exogenous CgA more efficiently than mice whose M-spike was <12%. Furthermore, 5 we detected plasmin activity in the BM plasma of patients, but not in their PB plasma. We 6 cannot exclude, however, that other proteases in addition to plasmin (e.g. thrombin, known to 7 cleave the R373-R374 bond (15) or other proteases produced by osteoclasts or other cells of the 8 immune system present in the BM), are brought into play in the regulation of CgA activity. 9 Notably, the high conservation of R373-R374 dibasic cleavage site, flanking residues, and 10 adjacent CgA352-372 proangiogenic site, previously identified (15, 19), in human, mouse, rat, 11 bovine, and horse CgA (Supplementary Fig. 5) further suggests that this proteolytic 12 mechanism is biologically relevant. 13 We observed previously that the anti-angiogenic activity of CgA occurs with U-shaped 14 dose-response curve, the effect being lost at high CgA concentrations (> 5 nM) (15). However, 15 a dose-dependent response is likely to occur in MM, as lower concentrations were measured 16 (range 0.01-1.3 nM). Furthermore, no U-shaped observed with CgA1-373 up to 9 nM, or with 17 CgA1-76 up to 5 nM (15), i.e. with concentrations within the range of circulating CgA in MM 18 patients. The U-shaped curve and loss of activity could be relevant for neuroendocrine tumors 19 in which CgA is released locally by neuroendocrine cancer cells in very high amounts, which is 20 not the case of MM. 21 In conclusion, the results of the present study indicate that CgA, after being released in 22 circulation by the neuroendocrine system, is proteolytically cleaved in the BM of MM patients, 23 tipping the local balance of anti-/pro-angiogenic CgA polypeptides toward a pro-angiogenic 24 state. This finding may have important pathological implications and may stimulate clinical 25 studies aimed at assessing whether detection of CgA levels and its fragmentation have 26 prognostic values in terms of response to therapy and patient survival, and/or to assess whether 27 pharmacological alteration of the CgA balance, e.g. by administration of anti-angiogenic 28 fragments, might have therapeutic effects. 29 30 Acknowledgements 31 This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC, Special 32 Program Molecular Clinical Oncology 5x1000-9965 and IG-14338). We thank Annapaola 33 Andolfo for performing mass spectrometry analysis of CgA fragments. 34
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

16
1 Authorship 2 Contribution: Mi.B., B.C., A.G., F.C., M.P., A.C., M.T.S.B., A.S. designed and performed 3 experiments; Mi.B. analyzed data and wrote figures; S.G., F.C., and M.M. provided plasma 4 samples and clinical data; M.C. and P.L.B. provided the Vk*MYC experimental model; M.C. 5 and P.L.B., Ma.B., M.P., E.F., M.F., F.C.C., G.T., provided intellectual input; A.C. conceived 6 and directed the project and wrote the paper. 7
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

17
References 1. Dimopoulos MA, Kastritis E, Anagnostopoulos A. Hematological malignancies: myeloma. Ann Oncol. 2006;17 Suppl 10:x137-43. 2. Vacca A, Ribatti D. Bone marrow angiogenesis in multiple myeloma. Leukemia. 2006;20:193-9. 3. Ria R, Reale A, De Luisi A, Ferrucci A, Moschetta M, Vacca A. Bone marrow angiogenesis and progression in multiple myeloma. Am J Blood Res. 2011;1:76-89. 4. Jakob C, Sterz J, Zavrski I, Heider U, Kleeberg L, Fleissner C, Kaiser M, Sezer O. Angiogenesis in multiple myeloma. Eur J Cancer. 2006;42:1581-90. 5. Vacca A, Ribatti D, Roncali L, Ranieri G, Serio G, Silvestris F, Dammacco F. Bone marrow angiogenesis and progression in multiple myeloma. Br J Haematol. 1994;87:503-8. 6. Sezer O, Niemoller K, Jakob C, Zavrski I, Heider U, Eucker J, Kaufmann O, Possinger K. Relationship between bone marrow angiogenesis and plasma cell infiltration and serum beta2-microglobulin levels in patients with multiple myeloma. Ann Hematol. 2001;80:598-601. 7. Rajkumar SV, Mesa RA, Fonseca R, Schroeder G, Plevak MF, Dispenzieri A, Lacy MQ, Lust JA, Witzig TE, Gertz MA, Kyle RA, Russell SJ, Greipp PR. Bone marrow angiogenesis in 400 patients with monoclonal gammopathy of undetermined significance, multiple myeloma, and primary amyloidosis. Clin Can Res. 2002;8:2210-6. 8. Xu JL, Lai R, Kinoshita T, Nakashima N, Nagasaka T. Proliferation, apoptosis, and intratumoral vascularity in multiple myeloma: correlation with the clinical stage and cytological grade. J Clin Pathol. 2002;55:530-4. 9. Pruneri G, Ponzoni M, Ferreri AJ, Decarli N, Tresoldi M, Raggi F, Baldessari C, Freschi M, Baldini L, Goldaniga M, Neri A, Carboni N, Bertolini F, Viale G. Microvessel density, a surrogate marker of angiogenesis, is significantly related to survival in multiple myeloma patients. Br J Haematol. 2002;118:817-20. 10. Kumar S, Witzig TE, Greipp PR, Rajkumar SV. Bone marrow angiogenesis and circulating plasma cells in multiple myeloma. Br J Haematol. 2003;122:272-4. 11. Munshi NC, Wilson C. Increased bone marrow microvessel density in newly diagnosed multiple myeloma carries a poor prognosis. Semin Oncol. 2001;28:565-9. 12. Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6:273-86. 13. Italiano JE, Jr., Richardson JL, Patel-Hett S, Battinelli E, Zaslavsky A, Short S, Ryeom S, Folkman J, Klement GL. Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood. 2008;111:1227-33. 14. Ribatti D. Endogenous inhibitors of angiogenesis: a historical review. Leuk Res. 2009;33:638-44. 15. Crippa L, Bianco M, Colombo B, Gasparri AM, Ferrero E, Loh YP, Curnis F, Corti A. A new chromogranin A-dependent angiogenic switch activated by thrombin. Blood. 2013;121:392-402. 16. Helle KB, Corti A. Chromogranin A: a paradoxical player in angiogenesis and vascular biology. Cell Mol Life Sci. 2014. 17. Corti A, Ferrero E. Chromogranin A and the endothelial barrier function. Curr Med Chem. 2012;19:4051-8. 18. Helle KB, Corti A, Metz-Boutigue MH, Tota B. The endocrine role for chromogranin A: a prohormone for peptides with regulatory properties. Cell Mol Life Sci. 2007;64:2863-86. 19. Theurl M, Schgoer W, Albrecht K, Jeschke J, Egger M, Beer AG, Vasiljevic D, Rong S, Wolf AM, Bahlmann FH, Patsch JR, Wolf D, Schratzberger P, Mahata SK, Kirchmair R. The neuropeptide catestatin acts as a novel angiogenic cytokine via a basic fibroblast growth factor-dependent mechanism. Circ Res. 2010;107:1326-35.
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

18
20. Maestroni S, Maestroni A, Ceglia S, Tremolada G, Mancino M, Sacchi A, Lattanzio R, Zucchiatti I, Corti A, Bandello F, Zerbini G. Effect of chromogranin A-derived vasostatin-1 on laser-induced choroidal neovascularization in the mouse. Acta Ophthalmol (Copenh). 2014. 21. Belloni D, Scabini S, Foglieni C, Veschini L, Giazzon A, Colombo B, Fulgenzi A, Helle KB, Ferrero ME, Corti A, Ferrero E. The vasostatin-I fragment of chromogranin A inhibits VEGF-induced endothelial cell proliferation and migration. The FASEB J. 2007;21:3052-62. 22. Veschini L, Crippa L, Dondossola E, Doglioni C, Corti A, Ferrero E. The vasostatin-1 fragment of chromogranin A preserves a quiescent phenotype in hypoxia-driven endothelial cells and regulates tumor neovascularization. FASEB J. 2011;25:3906-14. 23. Ceconi C, Ferrari R, Bachetti T, Opasich C, Volterrani M, Colombo B, Parrinello G, Corti A. Chromogranin A in heart failure; a novel neurohumoral factor and a predictor for mortality. Eur Heart J. 2002;23:967-74. 24. Corti A. Chromogranin A and the tumor microenvironment. Cell Mol Neurobiol. 2010;30:1163-70. 25. Chesi M, Robbiani DF, Sebag M, Chng WJ, Affer M, Tiedemann R, Valdez R, Palmer SE, Haas SS, Stewart AK, Fonseca R, Kremer R, Cattoretti G, Bergsagel PL. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell. 2008;13:167-80. 26. Corti A, Longhi R, Gasparri A, Chen F, Pelagi M, Siccardi AG. Antigenic regions of human chromogranin A and their topographic relationships with structural/functional domains. Eur J Biochem. 1996;235:275-80. 27. Ratti S, Curnis F, Longhi R, Colombo B, Gasparri A, Magni F, Manera E, Metz-Boutigue MH, Corti A. Structure-activity relationships of chromogranin A in cell adhesion. Identification of an adhesion site for fibroblasts and smooth muscle cells. J Biol Chem. 2000;275:29257-63. 28. Chesi M, Matthews GM, Garbitt VM, Palmer SE, Shortt J, Lefebure M, Stewart AK, Johnstone RW, Bergsagel PL. Drug response in a genetically engineered mouse model of multiple myeloma is predictive of clinical efficacy. Blood. 2012;120:376-85. 29. O'Connor DT, Mahata SK, Taupenot L, Mahata M, Livsey Taylor CV, Kailasam MT, Ziegler MG, Parmer RJ. Chromogranin A in human disease. Adv Exp Med Biol. 2000;482:377-88. 30. Vlasveld LT, van 't Wout J, Castel A. False elevation of chromogranin A due to proton pump inhibitors. Netherl J Med. 2011;69:207. 31. Giusti M, Sidoti M, Augeri C, Rabitti C, Minuto F. Effect of short-term treatment with low dosages of the proton-pump inhibitor omeprazole on serum chromogranin A levels in man. Eur J Endocrinol. 2004;150:299-303. 32. Sanduleanu S, Stridsberg M, Jonkers D, Hameeteman W, Biemond I, Lundqvist G, Lamers C, Stockbrugger RW. Serum gastrin and chromogranin A during medium- and long-term acid suppressive therapy: a case-control study. Aliment Pharmacol Ther. 1999;13:145-53. 33. Ribatti D, Nico B, Vacca A. Importance of the bone marrow microenvironment in inducing the angiogenic response in multiple myeloma. Oncogene. 2006;25:4257-66. 34. Hjertner O, Qvigstad G, Hjorth-Hansen H, Seidel C, Woodliff J, Epstein J, Waage A, Sundan A, Borset M. Expression of urokinase plasminogen activator and the urokinase plasminogen activator receptor in myeloma cells. Br J Haematol. 2000;109:815-22. 35. Pepper MS, Montesano R, Mandriota SJ, Orci L, Vassalli JD. Angiogenesis: a paradigm for balanced extracellular proteolysis during cell migration and morphogenesis. Enzyme Protein. 1996;49:138-62. 36. Breuss JM, Uhrin P. VEGF-initiated angiogenesis and the uPA/uPAR system. Cell Adh Migr. 2012;6:535-615. 37. Corti A, Sanchez LP, Gasparri A, Curnis F, Longhi R, Brandazza A, Siccardi AG, Sidoli A. Production and structure characterisation of recombinant chromogranin A N-terminal fragments (vasostatins) -- evidence of dimer-monomer equilibria. Eur J Biochem / FEBS. 1997;248:692-9.
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

19
38. Colombo B, Curnis F, Foglieni C, Monno A, Arrigoni G, Corti A. Chromogranin a expression in neoplastic cells affects tumor growth and morphogenesis in mouse models. Cancer Res. 2002;62:941-6. 39. Ratti S, Curnis F, Longhi R, Colombo B, Gasparri A, Magni F, Manera E, Metz-Boutigue MH, Corti A. Structure-activity relationships of chromogranin A in cell adhesion. Identification and characterization of an adhesion site for fibroblasts and smooth muscle cells. J Biol Chem. 2000;275:29257-63.
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

20
Figure legends 1 Fig. 1. Sandwich ELISAs and levels of different CgA-related analytes in peripheral-2 blood (PB) and bone marrow (BM) plasma samples obtained from normal subjects and 3 MM patients 4 (A) Schematic representation of CgA and of its N-terminal, central and C-terminal regions. 5 (B) Schematic representation of the sandwich-ELISAs used in hematological studies: mAb 6 B4E11 or 5A8 (capture antibodies); α-439, α-410-439, α-373, α-76, and α-FRs (detection 7 antibodies). 8 (C) Levels of different CgA-related analytes (*439, *439+436, *373, *76, and 9 *439+436+FRs) as measured by ELISA in PB plasma obtained from normal subjects (n=25) 10 or paired PB and BM plasma from multiple myeloma (MM) patients (n=31). CgA levels in 11 BM plasma samples from patients untreated with proton pump inhibitors and without renal 12 failure (-PPI, -RF) (n= 11) are also shown. The absolute values (without normalization) are 13 shown. 14 (D) Levels of *373 in PB and BM plasma of patients with levels higher than those found in 15 normal subjects (n=14). Lines show paired values. 16 (E) Levels of analyte *373 relative to *439 or *76 (analyte ratios). 17 Dots represent the analyte values for each patients; bars (mean ± SEM). Statistical analysis: 18 Mann-Whitney test, except for the comparison of paired samples (paired PB and BM plasma 19 values of MM patients) (Wilcoxon test). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 20 0.0001. 21 22 Fig. 2. The proteolytic processing of CgA is associated with increased levels of VEGF 23 and FGF2 in the BM of patients with MM 24 Levels of VEGF (A) and FGF2 (B) in the BM of MM patients at diagnosis with low (<2, n=8) 25 or high (>2, n=9) *373/*439 ratio (an index of CgA fragmentation). VEGF, FGF2, *373 and 26 *439 analytes were measured by ELISA (box-plots with median, interquartile and 5-95 27 percentile values). 28 ***, P < 0.001 (Mann-Whitney test). 29 30 Fig. 3. The cleavage of CgA C-terminal region is associated with increased microvessel 31 density in the BM of MM patients 32
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

21
(A) BM microvessel density in MM (black bars) and controls (i.e., uninvolved BM biopsies 1 taken at staging in patients with nodal diffuse large B-cell lymphoma) (white bars). BM 2 microvessel density was analyzed by immunohistochemistry after staining with an anti-CD34 3 antibody (Bars: mean ± SD). (B) Linear regression analysis and correlation (r= 0.478; p= 4 0.045, Pearson test) between microvessel density and the sum of the standardized values (Z) 5 of the ratios between FRs and *76 or *439. Z-score of FRs/76 and FRs/439 was calculated as 6 Z=(X-m)/s where X= value, m= mean and s= standard deviation of population (n=18 MM 7 patients). 8 9 Fig. 4. Effect of CgA1-439, CgA1-373 and CgA1-76 (alone or in combination) on angiogenesis 10 in the rat aortic ring (RAR) assay 11 (A and B) Rat aortic rings were incubated with the indicated CgA fragments. Open circles 12 correspond to number of capillaries sprouting from each aortic ring at day 6. Box-plots with 13 median, interquartile and 5-95 percentile values are also shown. The number of aorta rings 14 tested is also indicated (n). 15 *, P < 0.05; **, P < 0.01; ****, P < 0.0001 (Mann-Whitney test). 16 17 Fig. 5. BM microvessel density, M-spike and proteolytic processing of exogenous CgA in 18 transgenic Vk*myc mice 19 (A) BM microvessel density in 55-80 week-old Vk*myc mice (n=10) with M-spike < or 20 >12% of total serum proteins. BM microvessel density was analyzed by immunofluorescence 21 microscopy after staining with anti-CD31 antibodies. 22 (B) Proteolytic cleavage of exogenous CgA in Vk*myc mice (n=39) with M-spike < or >12% 23 of total serum proteins, as evaluated by measuring *373 and *439 by ELISA 1 h after 24 administration (i.v) of 3 µg of human CgA1-439. The *373/*439 ratio is shown. 25 (C) Proteolytic cleavage of exogenous CgA in normal mice (n=5) and Vk*myc mice (n=29) 26 with different levels of M-spike. Plasma samples were collected from each mouse, 24 h after 27 administration of 3 µg of human CgA1-439, and analyzed by *439- and *439+436+FRs-28 ELISA. The correlation between *439/*439+436+FRs ratio (an index of C-terminal 29 fragmentation) and M-spike is shown. 30 Bars (mean ± SEM); *, P < 0.05; **, P < 0.01; ***P<0.001 (t-test, two tails). 31 32 Fig. 6. Myeloma and endothelial cells induce CgA C-terminal cleavage by activating 33 plasminogen to plasmin 34
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

22
(A) Effect of myeloma cells, HUVECs, plasminogen and PAI-1 on CgA fragmentation. 1 HUVECs and human myeloma cell lines KMS12, KMS28, U266 (5x105 cells/well in 100 µl 2 of PBS containing 0.5% of bovine serum albumin) were cultured for 24 h at 37 °C in the 3 presence or absence of plasminogen (PG, 8.8 µg/ml) and PAI-1 (1 µg/ml). Then, CgA1-439 4 (300 ng/ml) was added to the cell supernatants and further incubated for 6 h. The supernatants 5 were collected and analysed by *439- and *439+436+FRs-ELISAs. The levels of each analyte 6 are shown as % of control. Bars (mean ± SEM); *, P < 0.05; **, P < 0.01; ***P<0.001; 7 ****P<0.0001 (t-test, two tails). 8 (B) Degradation of exogenous CgA1-439 upon incubation in PB and BM plasma samples. Full-9 length CgA1-439 (300 ng/ml) was added to three different pools of PB and BM plasma samples 10 from 6 MM patients (2 patients/pool) in the presence or absence of PAI-1, left to incubate for 11 2 h at r.t., and analyzed by *439- and *439+436+FRs-ELISAs. The analyte 12 *439/*439+436+FRs ratio (an index of C-terminal degradation) is shown (mean + SEM). BM, 13 but not PB plasma, caused CgA degradation. Degradation was significantly inhibited by PAI-14 1. 15 Bars (mean ± SEM); ***P<0.001 (Wilcoxon test, paired +/- PAI-1 values) 16 (C) Measurement of plasmin activity in PB and BM plasma of MM patients (Pool 1, 2 and 3, 17 as indicated). Plasmin assay was performed using the Sensolyte Rh110 fluorimetric assay 18 (AnaSpec), based on cleavage of a synthetic fluorogenic substrate in the absence (solid line) 19 and the presence (dashed line) of the selective plasmin inhibitor D-Val-Phe-Lys chloromethyl 20 ketone. Product formation was monitored using a spectro-fluorimeter (RFU, relative 21 fluorescence units). 22 23 Fig. 7. Cleavage of CgA C-terminal region by plasmin 24 (A-C) SDS-PAGE, western blotting, ELISA and mass spectrometry analysis of CgA before 25 and after incubation with plasmin-sepharose. CgA (150 µg/ml, in 680 µl of PBS) was 26 incubated, for the indicated times, with 300 µl of a plasmin-sepharose suspension (1:3 in 27 PBS), centrifuged, and analyzed by SDS-PAGE and western blotting (A) with the following 28 antibodies: mAb B4E11, α-FRs, α-373, α-410-439, and α-439 (see Materials and Methods and 29 Fig. 1 B for epitope location). Samples were also analyzed by ELISA as indicated (B) and, in 30 the case of 2 and 24 h digestions, by electrospray mass spectrometry (C). The sequence of 31 CgA and the identified plasmin cleavage sites at 24 h (arrows) are also shown (D). 32
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637

Published OnlineFirst February 11, 2016.Cancer Res Mimma Bianco, Anna Maria Gasparri, Barbara Colombo, et al. peptides in the bone marrow of multiple myeloma patientsChromogranin A is preferentially cleaved into pro-angiogenic
Updated version
10.1158/0008-5472.CAN-15-1637doi:
Access the most recent version of this article at:
Manuscript
Authoredited. Author manuscripts have been peer reviewed and accepted for publication but have not yet been
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/early/2016/02/10/0008-5472.CAN-15-1637To request permission to re-use all or part of this article, use this link
on May 2, 2018. © 2016 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on February 11, 2016; DOI: 10.1158/0008-5472.CAN-15-1637