rose hoberman roni rosenfeld judith klein-seetharaman
DESCRIPTION
Using physical-chemical properties of amino acids to model site-specific substitution propensities. Rose Hoberman Roni Rosenfeld Judith Klein-Seetharaman. Heterogeneity Across Sites. Substitution rate varies across sites rate parameter assumed to follow a gamma distribution - PowerPoint PPT PresentationTRANSCRIPT

Using physical-chemical properties of amino acids to model site-specific substitution propensities.
Rose HobermanRoni Rosenfeld
Judith Klein-Seetharaman

Substitution rate varies across sites rate parameter assumed to follow a
gamma distribution mathematically convenient little biological justification provides little explanation
HeterogeneityAcross Sites

Rate of substitution varies across sites rate parameter distributed according to a
gamma distribution mathematically convenient little biological justification provides little explanation
Substitution propensities vary across sites leads to an explosion of parameters (400) still no biological explanation
HeterogeneityAcross Sites

Explaining Why Substitution Propensities Vary
Differing substitution propensities are a result of different amino acid preferences (Halpern & Bruno, Koshi & Goldstein) e.g. substitutions to deleterious amino acids
are unlikely Learning amino acid preferences at each
site (~20 vs ~400 parameters) still too many parameters to estimate
accurately still not biologically informative

Our Modeling Assumption
Amino acids preferences are based on which physical and chemical properties are important at each site to the function or structure of the protein
restricts the parameter space (3-5) provides more explanation

A New Statistical Model of Site-Specific Molecular Evolution
1. Learn which properties are important at each site
2. Model amino acid preferences as a function of their properties
3. Determine a mapping from amino acid preferences to substitution propensities
4. Combine property-based substitution propensities with other factors that effect substitutions
nucleotide mutation processes different distances between codons

A New Statistical Model of Site-Specific Molecular Evolution
1. Learn which properties are important at each site don’t rely on structural knowledge about the
protein do not artificially restrict to a few preselected
physical features 2. Model amino acid preferences as a function of
their properties3. Determine a mapping from amino acid
preferences to substitution propensities4. Combine substitution propensities with codon
distance and nucleotide mutation rates

250 Amino Acid Properties
1 Hydrophobicity
2 Volume
3 Net charge
4 Transfer free energy
...
248 Average flexibility
249 Alpha-helix propensity
250 Number of surrounding residues
(Downloaded from http://www.scsb.utmb.edu/comp biol.html/venkat/prop.html)

250 Amino Acid Properties
1 Hydrophobicity
2 Volume
3 Net charge
4 Transfer free energy
...
248 Average flexibility
249 Alpha-helix propensity
250 Number of surrounding residues
A C D E F G H I K L
0.66 2.87 0.10 0.87 3.15 1.64 2.17 1.67 0.09 2.77
M N P Q R S T V W Y
0.67 0.87 1.52 0.00 0.85 0.07 0.07 1.87 3.77 2.67

Visualizing the Amino Acid Distribution
FAMLR...LAMLR...IAMLR...P-EL-...GAELR...PGEIR...L-ELY...L-EVR...I-MLK...WAELR...HAELY...YAILY...WAML-...

Variance
FAMLR...LAMLR...IAMLR...P-EL-...GAELR...PGEIR...L-ELY...L-EVR...I-MLK...WAELR...HAELY...YAILY...WAML-...

Limitations of Variance
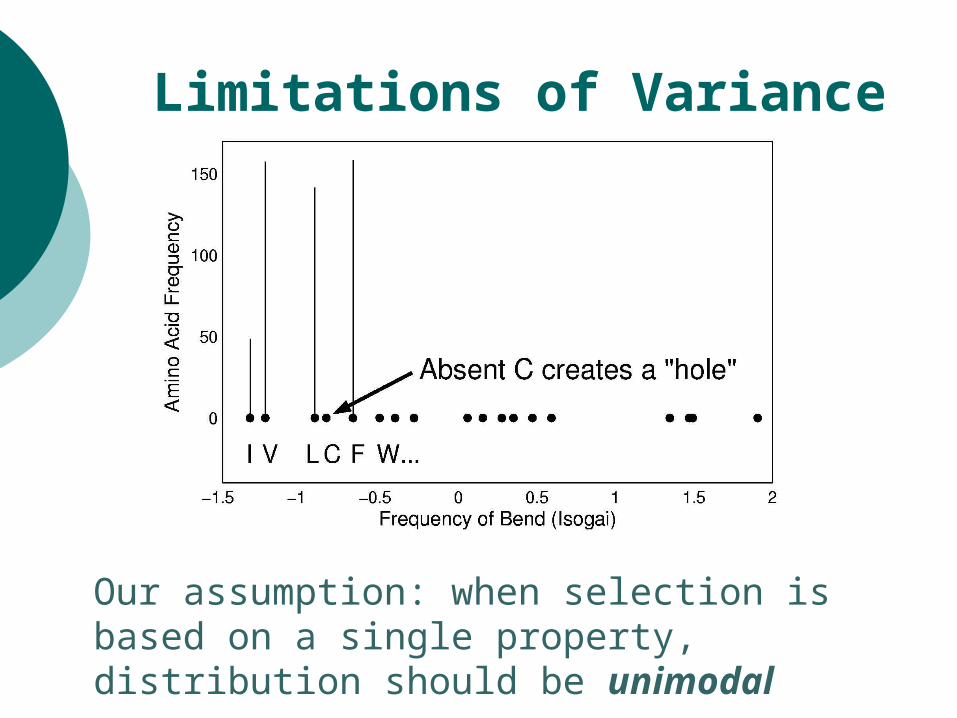
Limitations of Variance
Our assumption: when selection is based on a single property, distribution should be unimodal

Using Gaussian Goodness-of-Fit to Test for Property Conservation
Fit a maximum-likelihood Gaussian to amino acid frequencies in property space
From (discretized) Gaussian calculate expected AA frequencies
Calculate goodness-of-fit to learned Gaussian identifies unimodal distributions penalizes missing amino acids (“holes”)
Use Monte-Carlo method to calculate significance Otherwise will have high false discovery rate when
entropy is low
2

GPCR-A Family
Characterized by 7 TM segments
Responds to a large variety of ligands
Ligand binding allows binding and activation of a G protein
Diversity in sequences Believed to share similar
structure Only known structure is for
Rhodopsin



Results for GPCR
0
2
4

Estimating the False Discovery Rate (FDR)
Properties
Tested
Significance
Threshold
Significant Positions FDR
Number Expected
Number Detected
5 0.0005 0.63 76 0.8%
5 0.001 1.26 85 1.5%
5 0.005 6.26 136 4.6%
50 0.0005 6.25 103 6.1%
50 0.001 12.34 130 9.5%
240 0.0005 28.61 154 18.6%
FDR = # false positives / # predicted positives

Initial Validation
Charge conserved at 134 part of D/E R Y motif of importance to binding and
activation of G-protein Size conserved at 54, 80, 87, 123, 132, 153, 299
helix faces one or two other helices Cluster of dynamics properties conserved in third
cytoplasmic loop in Rhodopsin this is the most flexible interhelical loop

Continuing Work
Use multivariate Gaussian to model selection pressure from multiple properties
Derive substitution propensities from amino acid preferences and combine these with codon distance effects and nucleotide mutation rates

Thank You
Roni RosenfeldJudith Klein-SeetharamanNSF

Summary
Proposed a new approach for modeling heterogeneity of the evolutionary process across sites
Designed a test that is able to identify which properties are conserved at different sites
Promising approach for modeling site-specific substitution propensities in a biologically-realistic and computationally tractable way

Significance
Problem: for positions with low entropy, every property will have low variance very high false positive rate: any combination of 1
more more properties can explain this! actual explanation may involve several properties
In this case, multiple property constraints Cannot determine which one property is
conserved Need to condition on entropy

Significance Testing
What is the probability of a property having low variance in this position purely by chance?
Generate a large set of “random” (shuffled) property scales
show examples of shuffling Calculate variance for each random property The distribution of this statistic can be used to calculate a
threshold for acceptability of false-positives Show picture here? add error bars?

Gaussian Significance I


Halpern & Bruno 1997 Koshi & Goldstein 1998
Related Work

New Model Model of One Fitness Class
Model of Multiple Sequences from one Protein Family

Abstract
Existing models of molecular evolution capture much of the variability in mutation rates across sites. More biologically realistic models also seek to explain site-specific differences in substitution propensities between residue pairs, leading to more accurate and informative models of evolutionary dynamics. Toward this end, we describe a procedure for systematically characterizing the conservation of each position in a multiple sequence alignment in terms of specific physical and chemical properties. We use a Monte-Carlo method to ascertain the statistical significance of the findings and to control the False Discovery Rate. We use our method to annotate the diverse GPCRA family with a selection pressure profile. We demonstrate the computational and statistical significance of the properties we have identified, and discuss the biological significance of our findings. The latter include confirmation of experimentally determined properties as well as novel testable hypotheses.
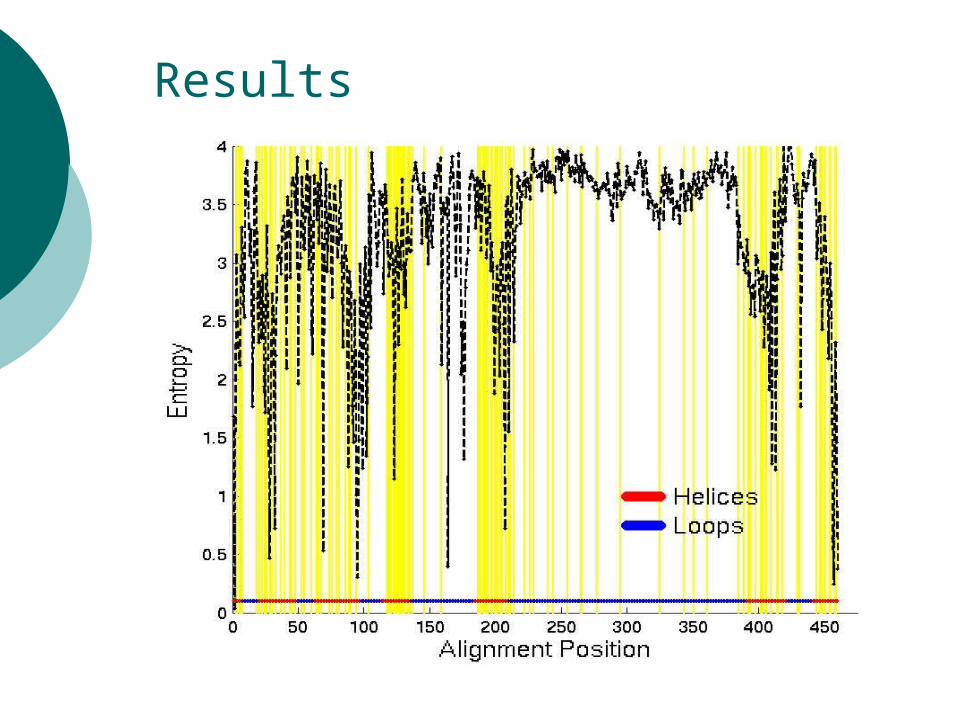
Results

Novel Hypothesis
175 and 265 highly similar conservation patterns
Both tryptophans in rhodopsin Trp265 in direct contact with retinal ligand, but
when exposed to light, crosslinks to Ala169 instead.
Trp161 has been proposed to contribute to this process
The property conservation patterns suggest Trp175 has a more significant role
This hypothesis can be tested experimentally