roles of hho1p, esc2p, and the post -translational
TRANSCRIPT

Roles of Hho1p, Esc2p, and the Post-Translational Modification of Sir2p in Transcriptional Silencing in Saccharomyces cerevisiae
by
Holly Kuzmiak-Ngiam
Submitted in Partial Fulfillment of the
Requirements for the Degree
Doctor of Philosophy
Supervised by
Professor Xin Bi
Department of Biochemistry
School of Medicine and Dentistry
University of Rochester
Rochester, NY
2011

ii
CURRICULUM VITAE
The author was born in Newton, New Jersey on April 27, 1982. She attended
Drew University from 2000 to 2004 and graduated with a Bachelor of Arts degree in
Biochemistry. She came to the University of Rochester in the Fall of 2004 and
began graduate studies in Biochemistry. She pursued her research under the
direction of Professor Dr. Xin Bi in the Department of Biology and received a Master
of Science degree from the University of Rochester in 2007. During her tenure as a
graduate student, she was appointed to the Cellular, Biochemical, and Molecular
Science Training Program, and received the Graduate Alumni Fellowship Award in
2004 and the Stotz Graduate Fellowship Award in Biochemistry in 2006.

iii
PUBLICATIONS
Stockwell SB, Kuzmiak-Ngiam H, Beach NM, Miyamoto D, Fernandez R, Temple L. (2011) The autotransporter protein from Bordetella avium, Baa1, is involved in host cell attachment. Microbiology Research. [In Press.]
Yu Q, Kuzmiak H, Olsen L, Kulkarni A, Fink E, Zou Y, Bi X. (2010) Saccharomyces cerevisiae Esc2p interacts with Sir2p through a small ubiquitin-like modifier (SUMO)-binding motif and regulates transcriptionally silent chromatin in a locus-dependent manner. Journal of Biological Chemistry. 285(10):7525-36.
Yu Q, Kuzmiak H, Zou Y, Olsen L, Defossez PA, Bi X. (2009) Saccharomyces cerevisiae linker histone Hho1p functionally interacts with core histone H4 and negatively regulates the establishment of transcriptionally silent chromatin. Journal of Biological Chemistry. 284(2):740-50.
Sabaihia M, Preston A, Maskell DJ, Kuzmiak HA, Connell TD, King ND, Orndorff PE, Miyamoto DM, Thomson NR, Harris D, Goble A, Lord A, Murphy L, Quail MA, Rutter S, Squares R, Squares S, Woodward J, Parkhill J, and Temple LM. (2006) Comparison of the genome sequence of the poultry pathogen Bordetella avium with those of B. bronchiseptica, B. pertussis, and B. parapertussis reveals extensive diversity in surface structures associated with host interaction. Journal of Bacteriology. 133 (15): 6002-15.
Kuzmiak HA, Maquat LE. (2006) Applying nonsense-mediated mRNA decay to the clinic: progress and challenges. Trends in Molecular Medicine. 12 (7): 306-316.

iv
ACKNOWLEDGMENTS
None of this would have been possible if Dr. Xin Bi had not welcomed me into
his lab four and a half years ago. His support, expertise, and tireless editing have
guided me to where I am today. Behind both of us were my committee members, Dr.
Cheeptip Benyajati, Dr. Jeffrey Hayes, and Dr. Eric Phizicky. Their questions,
critiques, and suggestions helped shape the work that I have presented here.
Members of the Bi lab, past and present, have been both colleagues and
friends. I am indebted to Yanfei Zou and Qun Yu, who taught me absolutely
everything I needed to know when I first joined the lab, and to Lars Olsen, who kept
the lab running smoothly, freeing us to focus on research. I owe special thanks to
undergraduate summer student Emma Fink, who generated plasmids and helped
with the subsequent Esc2-Sir2p yeast two-hybrid screen, and to Genetics graduate
student Lindy McClelland, who made a crucial breakthrough on the ubiquitination
project during her rotation in the Bi lab.
I have been lucky enough to be a part of both the Biology and Biochemistry
Departments here – thanks to the faculty, post-docs, technicians, fellow students,
and administrative staff of both, and particularly the Yeast and Chromatin Groups, for
discussions, suggestions, and reagents.
But graduate school isn’t always thrilling discoveries and beautiful data, and
when things were the hardest, I was saved by dinner dates with Melanie Baker,
Jennifer Newell, and Melissa Yu; I was saved by phone dates and message
exchanges with Drewids and Sussex County folks; and I was saved by running dates
with my Fleet Feet family.
Last, but not least – words cannot express the gratitude and love I have for
the Kuzmiak-Ngiam family: For my Singaporean family, who were my long-distance
cheerleaders through every Ph.D. milestone. For my parents, who didn’t really
understand what I was studying, but were unwavering in their certainty that I could
study it. For my “little” sister Sarah, a brilliant Ph.D. student herself, for our lengthy
science debrief sessions. And finally – for my husband Kee-Min, who listened a lot,
then laughed, cried, ranted, and danced with me, as necessary, every step of the
way.

v
ABSTRACT
In Saccharomyces cerevisiae, silencing of the cryptic mating type loci HML
and HMR and regions near telomeres is mediated by the formation of
heterochromatin-like transcriptionally silent chromatin, which serves as a tractable
model for the study of eukaryotic chromatin domains. Silencing is established
through the combined actions of cis-acting silencers, which flank the silent loci and
recruit the SIR silencing complex composed of Sir2p, Sir3p and Sir4p that
propagates along the nucleosomes to maintain the silent state. While the essential
players in silencing are known, many other chromatin-associated proteins are also
involved in this process, but the mechanisms of their functions remain poorly
understood. This thesis examines two such proteins, Hho1p and Esc2p, as well as
the post-translational modification of Sir2p, and provides new insights into how they
may finely tune transcriptional silencing.
In Chapter 1, I describe the negative regulation of silencing by the linker
histone Hho1p. I show that Hho1p functionally interacts with Sir1p, a protein
involved in the initiation of silencing, and negatively regulates the de novo
establishment, but not the maintenance, of silent chromatin.
In Chapter 2, I dissect the interaction of Sir2p with Esc2p, a protein that
differentially regulates silencing at telomeric and HM loci. I identify a putative SUMO
(Small Ubiquitin-like Modifier)-binding motif in Esc2p that is necessary and sufficient
for interaction with Sir2p and for the function of Esc2p in silencing.
In Chapter 3, I find that Sir2p is both sumoylated and ubiquitinated. I
investigate the potential of these post-translational modifications to affect the role of
Sir2p in transcriptional silencing. I use mutational and deletion analyses in an effort
to identify the sites of modification in Sir2p. I also develop and optimize an
alternative approach for the identification of modified residue(s) that involves
purification of modified Sir2p followed by mass spectrometry.

vi
Table of Contents
Curriculum Vitae ........................................................................................................ ii
Publications............................................................................................................... iii
Acknowledgments ..................................................................................................... iv
Abstract ..................................................................................................................... v
Table of Contents ..................................................................................................... vi
List of Tables........................................................................................................... viii
List of Figures ........................................................................................................... ix
Foreword ................................................................................................................... 1
Introduction ............................................................................................................... 2
Chapter 1: The role of Saccharomyces cerevisiae linker histone Hho1p in transcriptionally silent chromatin
1.1 Abstract ............................................................................................................. 7
1.2 Introduction ....................................................................................................... 8
1.3 Results ............................................................................................................ 11
1.4 Discussion ...................................................................................................... 24
1.5 Materials and Methods ................................................................................... 26
Chapter 2: The role of Saccharomyces cerevisiae Esc2p protein in transcriptional silencing
2.1 Abstract ........................................................................................................... 28
2.2 Introduction ..................................................................................................... 29

vii
2.3 Results ............................................................................................................ 31
2.4 Discussion ...................................................................................................... 45
2.5 Materials and Methods ................................................................................... 49
Chapter 3: Post-translational modification of Saccharomyces cerevisiae histone deacetylase Sir2p
3.1 Abstract ........................................................................................................... 52
3.2 Introduction ..................................................................................................... 53
3.3 Results ............................................................................................................ 59
3.4 Discussion ...................................................................................................... 84
3.5 Materials and Methods ................................................................................... 87
References.............................................................................................................. 93
Appendix 1: Catalog of strains .............................................................................. 103
Appendix 2: Results of screen for synthetic interactors with hho1∆ .................... 105

viii
List of Tables
Table 1-1 Deletions screened for synthetic effect on silencing with hho1∆ ........... 23
Table 3-1 Sir2p point mutations screened for sumoylation and ubiquitination ...... 66
Table A-1 Catalog of Strains ................................................................................ 103

ix
List of Figures
Fig. I-1 Schematic representation of the MAT and HM loci on Chromosome III in S. cerevisiae .................................................................................................................. 3
Fig. I-2 Model for the establishment of silent chromatin at HM loci in yeast ........... 5
Fig. 1-1 HHO1 deletion suppresses the silencing defect caused by sir1∆ ............ 13
Fig. 1-2 HHO1 negatively regulates the de novo establishment of transcriptionally silent chromatin ....................................................................................................... 15
Fig. 1-3 HHO1 does not affect the stability of preexisting silent chromatin ........... 17
Fig. 1-4 Hho1p physically interacts with Orc6p ..................................................... 19
Fig. 1-5 Using the TelVII-L URA3 reporter construct to screen for synthetic interactions with hho1∆ ........................................................................................... 21
Fig. 2-1 Schematic of Esc2p .................................................................................. 30
Fig. 2-2 Esc2p interacts with Sir2p in vivo ............................................................. 31
Fig. 2-3 Esc2p(aa 116-135) are necessary and sufficient for binding to Sir2p ....... 34
Fig. 2-4 Esc2p(aa116-135) are necessary and sufficient for interaction with SUMO ................................................................................................................................ 36
Fig. 2-5 Esc2p-Sir2p interaction is independent of SIR3 and SIR4 ...................... 37
Fig. 2-6 Recombinant Esc2p and Sir2p interact directly and in an SBM1-dependent manner .................................................................................................................... 38
Fig. 2-7 Recombinant Sir2p missing any of its three domains does not bind Esc2p in vitro ......................................................................................................................... 40
Fig. 2-8 Targeted silencing by Esc2p is mediated by SBM1 ................................. 43
Fig. 2-9 SBM1 of Esc2p is required for telomeric silencing but not cellular tolerance of genotoxic stress .................................................................................................. 44

x
Fig. 3-1 Schematic of the conjugation pathway for post-translational modification by ubiquitin and Ubls ................................................................................................... 54
Fig. 3-2 Constructs and protocol for denaturing Ni2+-NTA purification experiment to confirm Ubl modification of protein of interest ........................................................ 58
Fig. 3-3 Sir2p and Sir3p are sumoylated in vivo .................................................... 60
Fig. 3-4 Sir2p and Sir3p are ubiquitinated in vivo .................................................. 63
Fig. 3-5 Predicted sites of ubiquitination and sumoylation in Sir2p ....................... 65
Fig. 3-6 Search for Sir2p sumoylation site(s) by KR mutagenesis..................... 67
Fig. 3-7 Search for Sir2p sumoylation site(s) with C-terminal truncations ............. 69
Fig. 3-8 Search for Sir2p sumoylation site(s) with N-terminal truncations ............. 70
Fig. 3-9 Search for Sir2p ubiquitination site(s) by KR mutagenesis................... 72
Fig. 3-10 Search for Sir2p ubiquitination site(s) with N-terminal truncations ........ 74
Fig. 3-11 Sumoylation and ubiquitination of Sir2p fused to a biotinylatable peptide (BIOpep) ................................................................................................................. 76
Fig. 3-12 High levels of biotinylation requires growth in rich media or minimal media supplemented with biotin ........................................................................................ 78
Fig. 3-13 Optimizing purification of HA-Sir2p-BIOpep with Streptavidin-Agarose . 80
Fig. 3-14 Comparison of HA-His6-Sir2p-BIOpep binding to Ni2+-NTA resin and Streptavidin-Agarose in the presence of different concentrations of imidazole ...... 83

1
Foreword
In Chapter 1, Y. Zou performed the mating experiment shown in Fig. 1-1B.
Q. Yu performed the experiments shown in Fig. 1-3 and Fig. 1-5A.
In Chapter 2, plasmids in Fig. 2-3, 2-4, 2-5, 2-8, and 2-9 were generated as a
collaborative effort among myself, E. Fink (under my supervision), A. Kulkarni, and L.
Olsen. L. Olsen performed the spotting for these figures.
In Chapter 3, the experiment and analysis depicted in Fig. 3-4B was
performed by L. McClelland, under my supervision. L. Olsen assisted with Site-
Directed Mutagenesis to generate KR mutations in pHK84 (plasmids pLO225 –
pLO229) and with the cloning of the plasmid-based N-terminal truncations of SIR2
(plasmids pLO242 – pLO245). Dr. E. Phizicky provided advice and guidance during
development and optimization of purification protocols for the BIOpep constructs.

2
Introduction
I.1 Chromatin domains in eukaryotes
Chromatin is a general term used to describe the complexes of protein and
DNA found in the nucleus of a eukaryotic cell. Nucleosomes are the fundamental
unit of chromatin, and consist of ~147 base pairs of DNA wrapped around a histone
octamer. A histone octamer is composed of two copies each of histones H2A, H2B,
H3, and H4 (Luger et al. 1997). Nucleosomes are separated by stretches of DNA
called linker DNA, whose length varies from organism to organism (Woodcock et al.
2006). Linker histones, also referred to as histone(s)H1, bind linker DNA where it
enters and exits the nucleosome and facilitate chromatin condensation in vitro
(Robinson et al. 2006). Oligonucleosome arrays can be folded into higher order
chromatin structures, which include the 30 nm fiber and thicker fibers with complex
tertiary structure (Woodcock et al. 2001). Chromatin structure is dynamic, and
constitutes a major mechanism for DNA regulation in eukaryotic cells, influencing not
only gene transcription, but also DNA replication, recombination, and repair (Grewal
et al. 2003).
Chromatin structure is not uniform across the genome, and can be divided
into two major classes based on its cytological and molecular properties.
Euchromatin is generally more loosely packed, accessible to DNA binding factors,
and is associated with gene expression. Heterochromatin tends to be more tightly
packed, inaccessible, and transcriptionally silent. Heterochromatic domains are
found at centromeres and telomeres, and also interspersed throughout the genome
(Grewal et al. 2003). However, heterochromatin is also both stable and heritable,
making it ideal for the maintenance of repression over a long period of time and/or
many cell divisions. For example, heterochromatin is responsible for dosage

3
compensation in female mammals, the process by which one of the two X
chromosomes is inactivated in somatic cells. This inactive state is maintained and
inherited through many cell divisions (Avner et al. 2001).
I.2 Transcriptionally silent chromatin in yeast
Transcriptionally silent chromatin in the budding yeast Saccharomyces
cerevisiae is analogous to the heterochromatin found in multi-cellular eukaryotes.
The DNA in heterochromatin has been shown to replicate late in S phase (Reynolds
et al. 1989). The nucleosomes in this condensed chromatin have reduced
acetylation compared to nucleosomes in active regions (Braunstein et al. 1993), and
the DNA is highly compact and inaccessible to many DNA modifying agents (Bi et al.
1997; Singh et al. 1992; Gottschling 1992). Because of its structural and functional
similarities with heterochromatin in higher organisms, transcriptionally silent
chromatin in yeast has long served as a useful model for the study of
heterochromatin .
Yeast silent chromatin is found at telomeres and the HM loci (Fig. I-1), where
it functions in telomere maintenance and mating-type maintenance and switching
(Rusche et al. 2003). Silent chromatin is found at three distinct loci in S. cerevisiae:
rDNA, telomeres, and the HM loci (Fig. I-1). For the purposes of this work, the focus
will remain on transcriptionally silent chromatin at telomeres and HM loci.
Fig. I-1. Schematic representation of the MAT and HM loci on Chromosome III in S. cerevisiae. HML, HMR, and MAT loci are shown in grey, with the two genes in each (α1/α2 or a1/a2) depicted as black arrows. Silencers are indicated by open boxes, and each contains some combination of binding sites for Rap1p, Abf1p, and ORC.

4
Transcriptional silencing is generally location-dependent, not sequence-
dependent, and genes inserted at the HM loci are also subject to silencing.
Transcriptional silencing at both telomeres and HM loci is established and
maintained by a combination of cis-acting DNA elements and trans-acting proteins
(Rusche et al. 2003). At HML and HMR, the cis-acting sequences are the E and I
silencers that flank the HM loci (Fig. I-1). The silencers consist of combinations of
binding sites for proteins Rap1p (Repressor/Activator Protein 1) and Abf1p (ARS
Binding Factor 1), and ORC (Origin Recognition Complex for DNA replication)
(Rusche et al. 2003). The trans-acting factors include the proteins that bind to these
silencers: Rap1p, Abf1p, and the ORC complex (Fig. I-2A). ORC is composed of six
subunits, Orc1p – Orc6p, all of which are essential proteins (Giaever et al. 2002).
Additional trans-acting factors include the four SIR (Silent Information
Regulator) proteins, Sir1p – Sir4p. Three of these - Sir2p, Sir3p, and Sir4p - are
essential for silencing and interact with one another to form the SIR complex, which
constitutes the basic structural component of silent chromatin (Rusche et al. 2003).
Sir4p interacts strongly with both Sir2p and Sir3p, and serves as the scaffold of the
complex (Moazed 2001; Strahl-Bolsinger et al. 1997; Hoppe et al. 2002). The
enzymatically active member of this complex is Sir2p, an NAD+-dependent histone
deacetylase (Moazed 2001).
To initiate de novo silencing, the silencer-binding proteins at the E and I
silencers recruit the SIR complex. Rap1p interacts with Sir3p and Sir4p, and ORC
recruits the SIR complex indirectly, via interaction with Sir1p, which binds Sir4p
(Rusche et al. 2003). As SIR complexes are recruited to the silencer, Sir2p
deacetylates the histone tails of the adjacent nucleosome. Sir3p and Sir4p, which
interact with the N-terminal tails of histones, have a higher affinity for hyperacetylated
histones. This fact, coupled with high local concentration of the SIR complex, results
in propagation of the SIR complex along the chromatin in cycles of deacetylation and
SIR protein binding (Moazed 2001) (Fig. I-2).
Transcriptionally silent chromatin at the telomeres is structurally and
functionally similar to that at the HM loci. The primary difference is in how the SIR
complex is recruited during the initiation of silencing. The SIR complex is recruited
by the combined action of multiple Rap1p binding sites and the yeast DNA end-

5
binding complex Ku70/Ku80 (Gottschling et al. 1990; Luo et al. 2002). Once the SIR
complex is brought to the locus, it can deacetylate and propagate as it does at the
HM loci.
Sir2p, but not the other SIR proteins, is also involved in a unique form of DNA
silencing found only at rDNA arrays. At this locus, Sir2p acts with Net1p and Cdc14p
as part of the RENT complex (Straight et al. 1999).
Fig. I-2. Model for the establishment of silent chromatin in HM loc in yeast. (A) A silencer (HMR-E is shown here) includes DNA sequences that bind the silencer binding proteins Rap1p, Afb1p, and ORC. ORC binds Sir1p. Together, these proteins recruit the SIR complex (Sir2p, Sir3p, and Sir4p) to the silencer. Sir2p deacetylates the adjacent nucleosome (curved arrow). (B) This deacetylated nucleosome (yellow) recruits another SIR complex, which binds and deacetylates the adjacent nucleosome. (C) In this way, the SIR complex spreads, silencing the HMR locus. Blue circles represent nucleosomes in active chromatin; yellow circles represent nucleosomes in silent chromatin.

6
The goal of this work is to use S. cerevisiae as a model organism to examine
the roles of several chromatin-associated proteins and post-translational
modifications in the regulation of silent chromatin. In Chapter 1, I examine the effect
that the yeast linker histone has on the establishment of transcriptional silencing.
Chapter 2 describes my investigation into how Sir2p interacts with the protein Esc2p,
a regulator of transcriptional silencing. Finally, in Chapter 3, I provide evidence that
Sir2p is subject to covalent modification by the peptide modifiers ubiquitin and
SUMO, and attempt to delineate the mechanisms of Sir2p modification.

7
Chapter 1
The role of Saccharomyces cerevisiae linker histone Hho1p in transcriptionally silent chromatin
1.1 Abstract Saccharomyces cerevisiae linker histone Hho1p is a non-essential protein whose
deletion results in no obvious phenotypes in cell growth or transcriptional silencing.
Little is known about how Hho1p functions in vivo. In this work, I demonstrate that
hho1∆ suppresses defects in HML silencing and changes in chromatin structure
caused by deletion of SIR1, which encodes a protein involved in the initiation of the
formation of silent chromatin. I also show that HHO1 negatively regulates the de
novo establishment of silent chromatin. Finally, I conduct a targeted screen for other
genes whose mutations exhibit a synthetic effect on transcriptional silencing with
hho1∆. I propose several models for how Hho1p could function in transcriptionally
silent chromatin.

8
1.2 Introduction Nucleosomes form the most basic unit of chromatin and are composed of 147
base pairs of DNA wrapped around a complex of core histones. The DNA between
nucleosomes is called linker DNA, and its length varies from organism to organism
(Woodcock et al. 2001). Linker histones (also called histone H1) bind linker DNA
where it enters and exits the nucleosome, and facilitate chromatin condensation and
regulate the 30 nm chromatin fiber in vitro (Robinson et al. 2006). Work done in the
late 1980’s and early 1990’s suggested that linker histones repress chromatin
transcription in vitro (Hannon et al. 1984; Shimamura et al. 1989; Laybourn et al.
1991), leading to the hypothesis that linker histones act as global transcription
repressors in vivo. This hypothesis has come into question recently, as there is
evidence indicating that although H1 is essential in multicellular eukaryotes, it does
not play a significant role in regulating transcription. In mice, a 50% reduction in the
level of histone H1 results in embryonic lethality and significant changes in chromatin
structure, including less chromatin compaction, decreased distance between
nucleosomes, and reduction of certain core histone modifications (Fan et al. 2003).
However, microarray experiments showed that the expression of very few genes was
affected by the reduction of histone H1 (Fan et al. 2005). Histone H1 is not essential
in Tetrahymena thermophila, but as in mouse cells, its deletion reduces overall
chromatin compaction and affects transcription of a limited number of genes, but
without any major effect on global transcription (Shen et al. 1996; Shen et al. 1995).
Structurally, linker histones can be classified into two families: tripartite and
single domain (Kasinsky et al. 2001). Members of the tripartite H1 family are usually
found in multicellular eukaryotes, and contain a conserved globular domain flanked
by unstructured lysine-rich N- and C-terminal tails. The single domain family is found
in certain protists (e.g. Tetrahymena) and lacks the N-terminal tail and globular
domain – these linker histones are composed of only the C-terminal tail (Kasinsky et
al. 2001).
A query of the Saccharomyces cerevisiae genome with known linker histone
sequences identified one gene (HHO1) with significant homology to the conserved
globular domain of histone H1 (Landsman 1996; Ushinsky et al. 1997). In fact,
closer examination of the sequence of HHO1 suggests that it actually contains two

9
globular domains that are predicted to form similar secondary structures (Kasinsky et
al. 2001). Several lines of evidence suggest that Hho1p is a bona fide linker histone.
Fluorescent imaging performed on cells expressing Hho1p fused to green
fluorescent protein shows that Hho1p localizes to the nucleus (Ushinsky et al. 1997).
Furthermore, recombinant Hho1p has similar biochemical properties to tripartite
linker histones. Addition of recombinant Hho1p to purified, H1-stripped nucleosomes
increases the length of DNA protected during treatment with micrococcal nuclease,
from ~146 base pairs to ~168 base pairs, an addition of 10-12 base pairs on each
side of the nucleosome (Patterton et al. 1998). Recombinant Hho1p also forms a
stable complex with reconstituted dinucleosomes in a 1:1 ratio in vitro (Patterton et
al. 1998). However, it should be noted that this ratio would not be maintained in vivo,
as the molar ratio of Hho1p:nucleosome cores is much lower than 1:1 (Freidkin et al.
2001).
To date, little is known about the in vivo function of Hho1p. Chromatin
immunoprecipitation (ChIP) experiments have shown that Hho1p is bound broadly to
the yeast genome (Freidkin et al. 2001; Downs et al. 2003), and a study performed in
our lab indicates that Hho1p is associated without apparent preference to both active
and silent chromatin (Yu et al. 2009). Deletion of HHO1 results in no obvious defects
in growth, mating, or sporulation, and HHO1 is not required for telomeric silencing or
basal transcriptional repression (Ushinsky et al. 1997; Patterton et al. 1998; Escher
et al. 1997). Microarray analyses comparing gene expression in wild type and hho1∆
strains has shown that there is no upregulation of genes in response to HHO1
deletion. Rather, there is a general decrease in transcription, although only 27 out of
~6,200 genes are downregulated by more than 2-fold in hho1∆ cells, and these
genes do not belong to any common pathway or process (Hellauer et al. 2001). In
two independent studies, Hho1p has been shown to inhibit homologous
recombination – the first demonstrated that Hho1p is inhibitory to both DNA repair by
homologous recombination and to the recombination-dependent mechanism of
telomere maintenance (Downs et al. 2003). The second showed that Hho1p
represses recombination at the rDNA locus in a Sir2-independent manner (Li et al.
2008).

10
Despite being dispensable for cell growth, Hho1p, as a linker histone with a
unique structure, remained of interest in the chromatin field. Hho1p is a
nucleosome-interacting protein and thus has the potential to influence the formation
and/or maintenance of silent chromatin. It had been previously shown that
overexpression of HHO1 has an inhibitory effect on silencing (Veron et al. 2006). It
is formally possible that the role of Hho1p is redundant with or antagonistic to the
function(s) of other factors, and is therefore not readily apparent. To test whether
this was true with respect to transcriptional silencing, our lab screened a library of
histone mutations for any that had a synthetic effect with ∆hho1. My labmate Qun
Yu identified a histone H4 mutation, Y88G, whose phenotypes are partially
suppressed by deletion of HHO1 (Yu et al. 2009). Tyr-88 is found in a region of the
globular domain of histone H4 that is important for the interactions between the
H3/H4 tetramer and H2A/H2B dimer (Luger et al. 1997). Mutation of this residue to
glycine (H4-Y88G) results in temperature sensitivity, MMS sensitivity, and decreased
transcriptional silencing. Partial rescue of the H4-Y88G mutation by ∆hho1 suggests
that Hho1p negatively regulates these functions but is normally counteracted by
intact histone H4 (Yu et al. 2009).
Consistent with our finding that ∆hho1 suppresses the silencing defect
induced by the H4-Y88G mutation, we also showed that ∆hho1 suppresses changes
in silent chromatin structure caused by H4-Y88G (Yu et al. 2009). Taken together,
these data suggest a negative role for Hho1p in transcriptional silencing – but one
that only manifests when silent chromatin is already compromised. We next asked
whether the negative role of Hho1p in silencing could also be revealed in other
situations where silencing is weakened or eliminated. Ultimately, we found deletion
of HHO1 to partially rescue the defect in silencing caused by SIR1 deletion.
In this Chapter, I provide evidence that Hho1p negatively regulates
transcriptional silencing by interfering with the de novo establishment of silent
chromatin, rather than altering the stability of existing silent chromatin structure.
Additionally, I discuss several hypotheses that could explain how Hho1p functions.
Finally, I describe a targeted screen I performed to identify other genes whose
mutations exhibit a synthetic effect on transcriptional silencing with hho1∆.

11
1.3 Results 1.3.1 hho1∆ suppresses the silencing defect caused by sir1∆ To better understand and characterize the role(s) of linker histone H1 in S.
cerevisiae, our lab identified a synthetic interaction between hho1∆ and the histone
H4 mutation Y88G. The H4-Y88G mutation results in a silencing defect that is
partially rescued by hho1∆, revealing a negative role for HHO1 in silencing [Yu
2009]. To better understand how Hho1p functions, we looked for other conditions in
which hho1∆ could rescue a silencing defect.
We identified sir1∆ as such a mutation (Fig. 1-1B). In this experiment, we
measured HML silencing by evaluating the mating efficiency of a MATa strain. In
wild type and hho1∆ cells (Strains 1-1s and 1-2s), the HM loci are silent and genes
for just one mating type – in this case, MATa – are expressed. When these cells are
plated on a lawn of a MATα tester strain, they mate and can grow on minimal
medium, which specifically selects for diploids (‘Mating’ in Fig. 1-1B). Deletion of
SIR1 (Strain 1-3s) reduces HM silencing, leading to partial expression of HMLα
genes, which decreases the mating efficiency of the host. The mating efficiency of
the hho1∆ sir1∆ double mutant is higher than that of the hho1∆ or sir1∆ single
mutant, indicating that hho1∆ partially reverses the decrease in HML silencing
caused by sir1∆ (compare Strain 1-3s with 1-4s).
It is important to note several significant differences between the subunits of
the SIR complex (Sir2p, Sir3p, and Sir4p) and Sir1p. The SIR complex propagates
along chromatin to deacetylate the nucleosomes, and thereby becomes an integral
part of the resultant silent chromatin. Furthermore, the Sir2p, Sir3p, and Sir4p
proteins are absolutely required for HM and telomeric silencing, as deletion of any
one of the three results in complete derepression at these loci (Rine et al. 1987). On
the other hand, Sir1p is only involved in the initiation stage of the formation of silent
chromatin, when it is recruited to silencers via the silencer binding complex ORC
(Triolo et al. 1996). Sir1p then recruits the SIR complex via interaction with Sir4p,
but itself remains associated with the silencers, not throughout the silenced region.
A sir1∆ culture consists of two populations of mitotically stable cells, one in which
HML is silenced, the other in which HML is derepressed (Pillus et al. 1989; Xu et al.
2006) (Fig. 1-1A).

12
There are two primary ways that we study silent chromatin in yeast: by
measuring silencing at a particular locus via expression of a reporter at that locus,
and by using a topology-based assay to examine the chromatin structure. The
topology of DNA is determined primarily by the density and configuration of
nucleosomes, which determines the degree of negative supercoiling of the DNA. For
example, DNA at HML and HMR becomes more negatively supercoiled when the
regions are silenced than when they are derepressed, and so we can use
supercoiling as an indicator of the state of silent chromatin (Bi et al. 1997; Cheng et
al. 1998). In practice, we examine the topology of HML DNA using a strain whose
HML locus (including or excluding silencers) is flanked by copies of the Flp1p
recombination target (FRT) for the site-specific recombinase Flp1p. Flp1p is under
the control of a galactose-inducible promoter, so we grow cells, and then induce
Flp1p with galactose, which results in the excision of HML on a minichromosome
circle. We then isolate these circles and separate them by gel electrophoresis in the
presence of the DNA intercalator chloroquine (Bi et al. 1997).
When we perform this assay on the hho1∆ and sir1∆ strains, we see that the
topoisomers of the HML circle in a sir1∆ strain contain a mixture of two populations,
one resembling those from SIR+ cells where HML was silenced and the other
resembling those from sir- cells where HML was derepressed (Fig. 1-1C, compare
Strain 1-7s with 1-5s and 1-9s). Deletion of HHO1 in SIR1 cells had no effect on the
distribution of HML topoisomers (Fig. 1-1C, compare Strain 1-5s with 1-6s), but
deletion of HHO1 in sir1∆ cells significantly increases the proportion of cells with
silenced HML circles (Fig. 1-1C, compare Strain 1-7s with 1-8s). These results
support the hypothesis that hho1∆ partially suppresses the defect in the formation of
silent chromatin caused by sir1∆.

13
1.3.2 hho1∆ negatively regulates the de novo establishment of silent chromatin Given the role of SIR1 in the initiation of silencing and the functional
interaction between SIR1 and HHO1 described above, I hypothesized that HHO1
plays a role in the establishment of silencing. I tested this model by monitoring the
chromatin structure of a silencer-bearing HML circle in a strain containing sir3-8, a
temperature sensitive allele of SIR3 that is nonfunctional at 30°C but functional at
23°C (Miller et al. 1984). HML circles excised from sir3-8 cells growing at 30°C
Fig. 1-1. HHO1 deletion suppresses the silencing defect caused by sir1∆. (A) Summary of HM silencing phenotypes in cells deleted for different SIR proteins. In a culture of wild type cells, all exhibit fully functional silencing (closed circles), and in a culture of sir3∆ cells, none exhibit silencing (open circles). However, in a culture of sir1∆ cells, some exhibit wild type silencing and others have no silencing. (B) Growth of MATa Strains 1s-4s on SC medium (‘Growth’) and synthetic minimum medium lacking amino acids, but coated with cells of the MATα tester strain DC17 (MATα his1) (‘Mating’). Only MATa strains with silent HM loci are able to mate with the tester strain, and only these diploid cells can grow on the ‘Mating’ plate. Strain genotypes appear on the left. (C) Effects of hho1∆ and sir1∆ single and double deletions on the supercoiling of HML DNA. The schematic at the top depicts the modified HML locus that is found in Strains 1-5s through 1-9s. FRT recombination sites flank the entire HML locus, including the –E and –I silencers. Induction of Flp1p recombinase results in excision of the HML circles, and resultant DNA samples are analyzed by agarose gel electrophoresis in the presence of 13 µg/ml chloroquine. N, nicked form of HML circle. SIR+ and sir-, topoisomers of HML from wild type and sir3∆ strains, respectively.

14
contain active chromatin and have the same reduced negative supercoiling found in
a sir3- strain. After temperature shift to 23°C, Sir3p becomes active, and the
negative supercoiling of the HML circle gradually increases to a level similar to that
of HML circles observed in SIR+ cells (Xu et al. 2006). Using this system, the
change in chromatin structure that occurs as silencing is established can be
observed. Fig. 1-2A describes how I performed this experiment. I used a sir3-8 strain
with the HML locus, including its silencers, flanked by FRTs. I grew cells to late log
phase in YPR medium at 30°C, then induced expression of PGAL10-FLP1 with the
addition of galactose for 2.5 hrs. Because the sir3-8 allele is non-functional at 30°C,
the Flp1p recombinase excises active HML loci during this induction. I then switched
the cells to YPD media to turn off FLP1 expression and moved the cultures to 23°C,
the temperature at which the sir3-8 mutant becomes functional. I removed aliquots
at various time points, isolated DNA, and fractionated the topoisomers of HML circle
on a chloroquine gel. In the sir3-8 HHO1 strain (Strain 1-10s) at time 0, Sir3p had
not yet become functional, so the circles assume the topology seen in a sir- strain
(Fig. 1-2B, compare Lane 1 with 14). As the cells grew for longer at 23°C, Sir3p
became functional, silencing was established on the circles, and the HML circles in
an increasing number of cells assumed the topology seen in a SIR+ strain (Fig. 1-2B,
Lanes 2-6). In the hho1∆ strain (Strain 1-11s), SIR+ topoisomers accumulated more
quickly than in the HHO1 strain (Fig. 1-2B, compare Lanes 8-12 with Lanes 2-6). For
example, after 7.5 hrs at 23°C, nearly all of the circles in the hho1∆ strain are of the
SIR+ variety, while in the HHO1 strain, the ratio of the two topoisomers is about equal
(Fig. 1-2B, compare Lane 11 with 5). These results provide evidence suggesting
that HHO1 negatively regulates the de novo establishment of silencing.

15
(10s) (11s)
Fig 1-2. HHO1 negatively regulates the de novo establishment of transcriptionally silent chromatin. (A) Schematic of an experiment to follow the kinetics of the de novo establishment of silent chromatin on HML circles. Experimental details appear in Materials and Methods, Section 1.5.2. Shaded circles, nucleosomes in active chromatin. Black circles, nucleosomes in silent chromatin. (B) Effect of hho1∆ on the kinetics of establishment of silent chromatin on HML circles in HHO1 (Strain 1-10s) and hho1∆ (Strain 1-11s) backgrounds. After growth and induction of HML circle excision at 30ºC, cells were moved to YPD media and grown at 23ºC for up to 20 hrs. Aliquots of the cultures were harvested at 0, 1, 2.5, 4.5, 7.5, and 20 hrs. DNA was isolated from all samples and fractionated by agarose gel electrophoresis in the presence of 13 µg/ml chloroquine. N and L, nicked and linear forms of the HML circle, respectively. (C) Profiles of topoisomers in Lanes 1, 4-6, 7, and 10-14 as determined by NIH Image. Shaded lines mark the center of distribution of topoisomers in Lanes 13 (SIR+) and 14 (sir-) and the position of the linear form of the circle (L).
9s 5s

16
1.3.3 hho1∆ does not affect the stability of pre-existing silent chromatin We also tested whether HHO1 could regulate the stability of silent chromatin.
Previous work has shown that an HML circle excised without silencers will gradually
lose its silent state and SIR+ topology as the host cells grow (Bi et al. 1997), and the
kinetics of the loss of the silent state of HML circle is a measure of the stability of
HML silent chromatin structure. We wanted to determine whether HHO1 had any
effect on the stability of silent chromatin. The construct used in this experiment
contains HML, excluding its silencers, flanked by FRT sites (Fig. 1-3A). We grew
cells in YPR medium, induced PGAL10-FLP1 with galactose, then shifted cells to YPD,
removed aliquots at various time points, isolated DNA from these aliquots, and
visualized the topoisomers of the HML circles on a chloroquine gel. In the 9 hrs
following excision, the distribution of topoisomers gradually shifts from mostly SIR+ to
mostly sir- (Fig. 1-3B, compare Lanes 1, 3, 5, 7, and 9). This shift is unaffected by
hho1∆ (Fig. 1-3B, compare Lanes 2, 4, 6, 8 with Lanes 1, 3, 5, 7). Therefore, hho1∆
does not affect the stability of HML silent chromatin structure.

17
Fig. 1-3. HHO1 does not affect the stability of preexisting silent chromatin. (A) Schematic of an experiment designed to examine the stability of silent chromatin on silencer-free HML circles. Experimental details appear in Materials and Methods, Section 1.5.2. Shaded circles, nucleosomes in active chromatin. Black circles, nucleosomes in silent chromatin. (B) Effect of hho1∆ on the kinetics of conversion of HML silent chromatin to active chromatin after its excision from the genome in HHO1 (Strain 12s) and hho1∆ (Strain 13s) backgrounds. After growth and induction of HML circle excision, cells were moved to YPD media and grown at for 9 hrs. Aliquots of the cultures were harvested at 0, 2, 3, 5, and 9 hrs. DNA was isolated from all samples and fractionated by agarose gel electrophoresis in the presence of 13 µg/ml chloroquine. N and L, nicked and linear forms of the HML circle, respectively. Asterisk, cross-hybridizing DNA. Topoisomers from sir3∆ (Strain 9s) in Lane 11 are marked sir-.

18
1.3.3 Hho1p and Orc6p physically interact in vitro All of the data presented so far suggest that Hho1p is inhibitory to the
establishment of transcriptionally silent chromatin. We can envision several ways
that Hho1p could be acting to have such an effect. (1) Hho1p, as a chromatin binding
protein, could compete with the SIR complex for binding to nucleosomes in silent
chromatin. (2) Hho1p could interfere with the association of silencer binding proteins
with the silencer, or during the initial recruitment of the SIR complex.
To test hypothesis 1, we attempted to use a gel shift assay to examine the
effect of Hho1 on the kinetics of Sir3p binding to nucleosomes with the assistance of
Tamara Caterino in Dr. Jeff Hayes’ lab in the Biochemistry Department at the
University of Rochester Medical Center. We purified Sir3p-His6-6xHA-Protein A from
yeast and reconstituted mononucleosomes (generated by the Hayes lab). However,
we were not able to observe robust, convincing nucleosome-Sir3p interaction via gel
shift assay.
During the establishment of silent chromatin, silencers adjacent to the HM
loci and telomeres recruit silencer binding proteins including Rap1p, Abf1p and the
ORC complex (Rusche et al. 2003). These proteins then recruit the SIR complex,
which deacetylates the first nucleosomes, and thus begins the process of
deacetylation and SIR complex spreading across the entire region. Sir1p functions
in the establishment of silencing by bridging the SIR complex and the silencer-
binding ORC complex (Triolo et al. 1996), which consists of the six essential proteins
Orc1p – Orc6p (Giaever et al. 2002). Interestingly, a large-scale screen that utilized
TAP purification and subsequent mass spectrometry to identify protein-protein
interactions in S. cerevisiae purified Orc6p in a pull-down of Hho1p (Krogan et al.
2006). It is possible that Hho1p inhibits ORC-Sir1 interaction through binding to
Orc6, which is in line with hypothesis 2.

19
I verified the interaction between Orc6p and Hho1p using a GST pull-down
assay (Fig. 1-4). In this experiment, I bound bacterially expressed GST-Hho1p or,
as a negative control, GST, to glutathione sepharose resin and washed away
unbound material. To this resin, I then added lysate from either wild type yeast or
yeast expressing Orc6p-HA. Fig. 1-4 clearly demonstrates that both GST (Lanes 5
and 6, Coomassie) and GST-Hho1p (Lanes 7 and 8, Coomassie) can be purified on
glutathione resin. However, only GST-Hho1p can co-purify Orc6-HA (compare Lane
6 with 8, western blot). This result is consistent with the result of the large-scale
screen, and suggests that the Hho1p-Orc6p interaction may be physiologically
Fig. 1-4. Hho1p physically interacts with Orc6p. Lysates from bacteria expressing GST or GST-Hho1p (Input, Lanes 1 and 2, respectively) and from yeast expressing either untagged Orc6p or Orc6p-HA (Input, Lanes 3 and 4, respectively) are shown on the left. Results from the pull-down experiment appear on the right (Lanes 5 – 8). GST (Lanes 5 and 6) or GST-Hho1p (Lanes 7 and 8) was bound to glutathione-sepharose beads, which were washed, then mixed with lysate from yeast expressing either untagged Orc6p (Lanes 5 and 7) or Orc6p-HA (Lanes 6 and 8). Resin was washed again, and proteins were eluted by boiling in Laemmli and separated by SDS-PAGE. GST and GST-Hho1p were detected by Coomassie staining and Orc6p-HA was detected by western blotting with anti-HA antibody.

20
relevant. While this result alone does not offer conclusive proof of how Hho1p acts
in silencing, it certainly raises the interesting possibility that Hho1p could affect the
establishment of silencing by interfering with the recruitment of the SIR complex by
ORC and Sir1p.
1.3.4 Screen for synthetic interactors with hho1∆
The data above provide clear evidence that HHO1 negatively regulates the
establishment of silencing, but fail to provide a mechanistic explanation for how
Hho1p opposes silencing. In an effort to better understand how Hho1p acts, I
employed a targeted synthetic interaction screen. Synthetic interaction screens have
many variations, but all are built on the same principle – Deletion phenotypes of
antagonistic or redundant proteins may manifest only when combined with other
mutations or deletions. The work I presented earlier in this chapter is an ideal
example: The antagonistic effect of Hho1p on silencing was only uncovered when
silencing was already weakened. An even simpler example is redundancy: If two
proteins have redundant or overlapping functions, single deletion of either may have
no effect, but a combination of both deletions may yield a clear phenotype.
I designed a synthetic interaction screen to test whether hho1∆ combined
with a second gene deletion had any effect on transcriptional silencing at the
telomeres. The screen was built from the MATa version of the Open Biosystems
Yeast Knockout Collection, a collection of strains in which each non-essential yeast
gene is replaced by the kanMX cassette (Giaever et al. 2002). I selected
approximately 100 genes from this collection that may work in the same process(es)
as Hho1p, including those involved in chromatin remodeling, histone modification,
and transcription, as well as genes with published genetic or physical interactions
with HHO1 (Krogan et al. 2006; Tarassov et al. 2008; Lim et al. 2007; Collins et al.
2007)(Table 1-1). Starting with the single deletion of each candidate gene from the
Open Biosystems collection, I replaced the endogenous Tel VII-L (left arm of
chromosome VII) with a synthetic telomere construct containing a URA3 reporter
(Fig. 1-5A). With these strains, I could easily assay telomeric silencing in each of the
single deletions with a simple spot test. Then, I replaced HHO1 in each strain with
LEU2 (hho1∆::LEU2) and assayed silencing on -Ura and FOA plates.

21
This system works robustly, as demonstrated by the results from the strain
containing the H4-Y88G mutation. Introducing this mutated H4 in place of wild type
H4 results in a significant decrease in telomeric silencing, as seen by loss of growth
on the FOA plate (Fig. 1-5A, compare Strain 1-14s with 1-16s). This silencing defect
is partially rescued by hho1∆ (Fig. 1-5A, compare Strain 1-16s with 1-17s).
Furthermore, additional experiments supported this data and indicated that hho1∆
does, in fact, partially rescue structural changes in chromatin that are caused by the
H4-Y88G mutation. Thus, this synthetic system is capable of identifying conditions in
which weakened silencing can be rescued by hho1∆.
Fig. 1-5. Using the TelVII-L URA3 reporter construct to screen for synthetic interactions with hho1∆. (A) Top, diagram of the reporter construct that replaces the left arm of Telomere VII with a URA3 reporter. Bottom, silencing phenotype of the Y88G mutant and its suppression by hho1∆ (Strains 1-16s and 1-17s, respectively). Cells of each strain were grown to late log phase, 10-fold serial diluted, and spotted on plates. SC, Synthetic complete plate. FOA, SC supplemented with 1 mg/ml 5-fluoroorotic acid. (B) Silencing phenotype in strains with different nucleosome remodeling complex components deleted alone (Strains 1-20s, 1-22s, and 1-24s) and in combination with hho1∆ (Strains 1-21s, 1-23s, and 1-25s). Experiment was performed as described in (A).

22
Of the approximately 100 deletions I tested for synthetic interaction with
hho1∆, I was unable to identify any combinations with synthetic silencing defects.
Representative results are shown in Fig. 1-5B. Snf2p, Ubp8p, and Rsc1p are each
part of a different nucleosome remodeling complex. Single deletion of each gene
does not affect silencing (compare Strain 1-18s with Strains 1-20s, 1-22s, and 1-24s,
FOA plates), although Strain 1-24s does exhibit a slight growth defect (compare
Strains 1-18s and 1-24s, SC plates). Deletion of HHO1 has no effect on growth or
silencing (compare Strains 1-20s with 1-21s, 1-22s with 1-23s, and 1-24s with 1-
25s). A complete set of images appears in Appendix 2.

23
Table 1-1. Deletions screened for synthetic effect on silencing with hho1∆ Name Classification
Name Classification
ARP5 Chromatin Remodeling
BDF1 Transcription ARP8 Chromatin Remodeling
CCR4 Transcription
ASF1 Chromatin Remodeling
DPB3 Transcription CHZ1 Chromatin Remodeling
SIN4 Transcription
HIR1 Chromatin Remodeling
BUR2/SVG2 Transcription Regulation HTZ1 Chromatin Remodeling
CHD1 Transcription Regulation
IES1 Chromatin Remodeling
MOT2 Transcription Regulation IES2 Chromatin Remodeling
NGG1/SWI7 Transcription Regulation
IES3 Chromatin Remodeling
ROX1 Transcription Regulation IES4 Chromatin Remodeling
RPN4 Transcription Regulation
IES5 Chromatin Remodeling
SGF11 Transcription Regulation IES6 Chromatin Remodeling
SPT3 Transcription Regulation
IOC2 Chromatin Remodeling
SPT7 Transcription Regulation RSC1 Chromatin Remodeling
SSN6/CYC8 Transcription Regulation
RSC2 Chromatin Remodeling
TUP1 Transcription Regulation RTT102 Chromatin Remodeling
UBP8 Transcription Regulation
SNF2 Chromatin Remodeling
CIN4 Chromosome Stability SNF6 Chromatin Remodeling
APN1 DNA Damage Repair
SWC2/VPS72 Chromatin Remodeling
DDR48 DNA Damage Repair SWC3 Chromatin Remodeling
MAG1 DNA Damage Repair
SWC5 Chromatin Remodeling
RAD9 DNA Damage Repair SWC7 Chromatin Remodeling
MPH1 DNA Damage Repair/Rec.
SWR1 Chromatin Remodeling
MUS81 DNA Damage Repair/Rec. TAF14 Chromatin Remodeling
POL32 DNA Replication
VPS71/SWC6 Chromatin Remodeling
FOB1 Recombination YAF9 Chromatin Remodeling
ARO1 Genetic Interaction w/ HHO1
RTT106 Chromatin Remodeling
BUB3 Genetic Interaction w/ HHO1 ACS1 Histone Modification (Ac)
CSM3 Genetic Interaction w/ HHO1
EAF3 Histone Modification (Ac)
RAS2 Genetic Interaction w/ HHO1 EAF6 Histone Modification (Ac)
UBA3 Genetic Interaction w/ HHO1
ELP3 Histone Modification (Ac)
VPS21 Genetic Interaction w/ HHO1 NAT4 Histone Modification (Ac)
GAL4 Physical Interaction w/ HHO1
RTT109 Histone Modification (Ac)
HRB1 Physical Interaction w/ HHO1 ELP4 Histone Modification (Ac)?
MOT3 Physical Interaction w/ HHO1
CTI6 Histone Modification (HDAC)
NHP6A Physical Interaction w/ HHO1 HOS3 Histone Modification (HDAC)
DSK2 MISC
HPA2 Histone Modification (HDAC)
ELC1 MISC HST2 Histone Modification (HDAC)
GAS1 MISC
RPD3 Histone Modification (HDAC)
HSC82 MISC RXT2 Histone Modification (HDAC)
HSP82 MISC
SDS3 Histone Modification (HDAC)
PAC2 MISC SIN3 Histone Modification (HDAC)
POP2 MISC
UME6 Histone Modification (HDAC)
ARD1 Protein Modification (Ac) DOT1 Histone Modification (Me)
NAT1 Protein Modification (Ac)
FPR4 Histone Modification (Me)
ESC8 Silencing HMT1 Histone Modification (Me)
SLX5 SUMO-related
SET2 Histone Modification (Me)
UBC9 SUMO-related SWD1 Histone Modification (Me)
TEL1 Telomere Length
JHD1 Histone Modifcation (deMet)
LGE1 Unknown JHD2 Histone Modification (deMet)
SET6 Unknown
BRE1 Histone Modification (Ub)

24
1.4 Discussion Linker histones are abundant chromatin binding proteins that have long been
thought to stabilize higher order chromatin structure. However, reduction (in mouse
cells) or deletion (in Tetrahymena and S. cerevisiae) of H1 has little effect on global
transcription (Fan et al. 2005; Shen et al. 1996; Ushinsky et al. 1997). In fact,
deletion of HHO1 in S. cerevisiae results in no obvious phenotype. However,
increasing HHO1 dosage inhibits silencing (Veron et al. 2006), which suggests that
Hho1p might play a negative role in regulating silencing. In fact, when silencing is
weakened by introduction of an H4-Y88G mutation, deletion of HHO1 partially
rescues both the transcriptional silencing defects and the changes in silent chromatin
structure caused by H4-Y88G (Yu et al. 2009).
In this work, I have shown a similar effect in sir1∆ hho1∆ cells. A sir1∆
culture contains a mixture of two populations of cells – one with silenced HML, one
with derepressed HML (Pillus et al. 1989; Xu et al. 2006). I have shown that deletion
of HHO1 results in an increase in the proportion of cells with silenced HML (Fig. 1-1),
and furthermore, that hho1∆ accelerates the establishment of silent chromatin on
HML circles (Fig. 1-2). Thus, Hho1p appears to inhibit the de novo establishment of
silent chromatin. We envision several possible modes of action for Hho1p that are
consistent with these data. It is possible that Hho1p binding to nucleosomes
competes with or blocks the spreading of the SIR complex across a region that
should be silenced. Alternatively, Hho1p could interfere with the initial recruitment of
silencer binding proteins or the SIR complex. I have presented evidence that Hho1p
physically interacts with Orc6p, one member of the silencer-binding ORC complex
(Fig. 1-4). The ORC complex binds Sir1p, which then recruits the SIR complex to
initiate silencing. It is possible that Hho1p acts by interfering with ORC recruitment
or the ORC-Sir1p interaction. However, the design and implementation of further
genetic experiments to investigate the functional relevance of this interaction proved
difficult, as all six proteins that constitute the ORC complex are essential (Giaever et
al. 2002).
Any of these scenarios place Hho1p acting during the de novo establishment
of silent chromatin. Evidence suggests that the structure of silent chromatin, once
achieved, is quite robust (Rusche et al. 2003). It is easy to believe that the loss of

25
the SIR complex from one or a few nucleosomes in a wild type cell could easily be
remedied by recruitment of more SIR proteins by the adjacent, intact silencing
machinery. Thus, in wild type cells, intact silencing machinery offsets any inhibitory
effects of Hho1p. Why, then, do we see the effects of hho1∆ in cells containing the
H4-Y88G or sir1∆ mutations? In these cells, the silencing machinery is
compromised and may be more susceptible to small amounts of damage. Thus,
when a SIR complex is lost, it cannot be replaced as quickly, and this small
perturbation may result in large scale disruptions of silent chromatin. Repair of such
disruptions would require the de novo establishment of silent chromatin, which I have
clearly shown is negatively regulated by HHO1 (Fig. 1-2).
Finally, in an effort to better understand the mechanism of action of Hho1p, I
performed a targeted screen for genes that exhibited a synthetic effect on telomeric
silencing when combined with hho1∆ (Fig. 1-5). For this targeted screen, I selected
single deletions of genes involved in defining and maintaining chromatin structure
and gene regulation, for example genes involved in chromatin remodeling, histone
modification, and transcription (Table 1-1). I was expecting to find either single
deletions whose silencing defects were partially rescued by hho1∆ (like sir1∆), or
possibly single deletions whose silencing defect was exacerbated by hho1∆.
However, I was unable to identify any genes that exhibited a synthetic silencing
effect with hho1∆. Of course, it is formally possible that I simply did not select the
correct gene/set of genes for my screen, or that other genes that would have shown
a synthetic effect are essential and thus, not available in the knockout collection. It is
also possible that there are subtle changes in silencing occurring that my reporter
construct is not sensitive enough to detect. A larger-scale SGA (synthetic genetic
analysis) screen employing a more sensitive reporter for silencing may yield genes
that play redundant or antagonistic roles with Hho1p. Such an experiment would
allow for a “blind” screen for HHO1 interactors, and therefore would not be skewed
by the investigator’s hypotheses of how Hho1p acts.

26
1.5 Materials and Methods 1.5.1 Plasmids and Strains Plasmid pHK34 for bacterial expression of GST-Hho1p was made by
inserting an EcoRI-HHO1-XhoI fragment into pGEX6p-1 (Amersham Biosciences).
Plasmids pMS337 and pMS364 are CEN-LEU2 plasmids encoding HHT1-HHF1 and
HHT1-hhf1-Y88G, respectively.
Strains 1-2s and 1-3s were constructed by replacing HHO1 and SIR1,
respectively, with kanMX in Strain 1-1s. Strain 1-4s was constructed by replacing
HHO1 with LEU2 in Strain 1-3s. Construction of Strains 1-5s, 1-7s, 1-9s, 1-10s, and
1-12s has been described elsewhere (Xu et al. 2005; Bi et al. 2004). Strains 1-6s, 1-
8s, and 1-11s were constructed by replacing HHO1 with NatMX in Strains 1-5s, 1-7s,
and 1-10s, respectively. Strain 1-13s was constructed by replacing HHO1 with
URA3 in Strain 1-12s. Strains 1-14s and 1-16s were made by transforming Y1838
(MATα ura3-52 leu2-3,112 his3∆ trp1-289 (hht1-hhf1)∆ (hht2-hhf2)∆ pMS329, from
J. Broach) with plasmid pMS337 and pMS364, respectively. Strains 1-15s and 1-17s
were constructed by replacing HHO1 with KanMX in Strains 1-14s and 1-16s,
respectively. Strains 1-18s, 1-20s, 1-22s, and 1-24s were made by transforming
BY4741 or the appropriate knockout strain from the Open Biosystems Collection
(Giaever et al. 2002) to Ura+ with EcoRI-SalI-digested pT1 (Bi et al. 2004). Strains 1-
19s, 1-21s, 1-23s, and 1-25s were generated by replacing HHO1 with LEU2 in
Strains 1-18s, 1-20s, 1-22s, and 1-24s, respectively. Strain 1-26s was generated by
transforming BY4741 with a PCR-produced fragment encoding 6xHA linked to
NatMX, amplified from pYM17 (Janke et al. 2004) using primers encoding 40 base
pairs of homology to the 3’ end of HHO1 (5’ primer) and to the sequence immediately
after the HHO1 STOP codon (3’ primer).
1.5.2 Analysis of DNA Topology Cells were grown in YPR medium (1% yeast extract, 2% bacto-peptone, and
2% raffinose) to mid-log phase, then treated with galactose (2%) for 2.5 hrs to induce
the expression of PGAL10-FLP1. For a steady state topology experiment (Fig. 1-1C),
cells were harvested directly after induction. For kinetic experiments, cells were
washed with water and YPD, then resuspended in YPD (2% glucose) and grown at

27
either 23°C (for establishment experiment, Fig. 1-2B) or 30°C (for stability
experiment, Fig. 1-3B). In all experiments, nucleic acids were isolated using the
glass bead method and fractionated on agarose gels supplemented with 13 µg/ml
chloroquine. DNA was visualized by Southern blotting. The profiles of topoisomers
were determined using NIH Image software.
1.5.3 GST Pull-Down Escherichia coli BL21 cells bearing pGEX6p-1 or pHK34 were treated with
isopropyl 1-thio-β-D-galactopyranoside (1 mM) for 4 hrs to induce the expression of
GST or GST-Hho1p, respectively. Cells were washed and resuspended in E. coli
B150 buffer (50 mM Tris pH 7.4, 150 mM NaCl, 0.2% Triton X-100) plus protease
inhibitors and lysed by treatment with lysozyme (0.5 mg/ml) for 30 min on ice,
followed by sonication. Extracts were clarified by centrifugation at 13,000 rpm for 15
min, and the protein concentration of the supernatant was determined by Bradford
assay. Whole cell extract (1 mg) was bound to equilibrated glutathione conjugated
beads (50 µl packed, Amersham Biosciences) for 2 hrs at 4°C. Beads were washed
five times with B150 and mixed with yeast lysate prepared from BY4741 or Strain 1-
26s in Yeast Lysis Buffer (1% Triton X-100, 50 mM Tris pH 7.4, 300 mM NaCl, 5 mM
EDTA, plus protease inhibitors) by the glass bead method. Proteins were bound for
2 hrs at 4°C, washed five times with B150, and eluted by boiling for 5 min in Laemmli
buffer. Proteins were separated by SDS-PAGE and detected by Coomassie staining
(GST) or western blotting with anti-HA antibody (Sigma).

28
Chapter 2:
The role of Saccharomyces cerevisiae Esc2p protein in transcriptional silencing
2.1 Abstract The S. cerevisiae histone deacetylase Sir2p forms complexes with Sir3p,
Sir4p, and Net1p to effect transcriptional silencing at different loci; it also physically
interacts with Esc2p, Esc8p, Slx5p, and Mcm10p, but the physiological relevance of
these interactions is less clear. In this Chapter, I examine the mechanism of the
interaction of Sir2p with Esc2p. I first confirm this interaction with a co-purification
experiment, and then use a yeast two-hybrid system to identify a 20 amino acid
sequence in Esc2p that is necessary and sufficient to support interaction with Sir2p.
This sequence contains the consensus sequence for a SUMO-binding motif (SBM),
and is necessary and sufficient for interaction of Esc2p with the yeast SUMO protein
Smt3p. I also demonstrate that this SBM is required for the function of Esc2p in
transcriptional silencing. Finally, I show that, despite the SBM-dependence of the
Esc2p-Sir2p interaction, the two proteins can interact directly and without any
SUMO-modification on Sir2p.

29
2.2 Introduction Transcriptionally silent chromatin in S. cerevisiae is found at three distinct
loci: telomeres, rDNA, and the HM loci. The chromatin structure in these regions is
akin to the heterochromatin of higher eukaryotes and serves as an experimentally
tractable model for the study of silent chromatin across species (Rusche et al. 2003).
Silencing at all three loci requires Sir2p, an NAD+-dependent histone deacetylase
(Braunstein et al. 1993; Moazed 2001), and Sir2p functions in at least two different
silencing complexes. The SIR complex contains Sir2p, Sir3p, and Sir4p, and
mediates silencing at telomeres and the HM loci (Liou et al. 2005). The RENT
complex contains Sir2p, Net1p, and Cdc14p, and mediates silencing at rDNA
(Straight et al. 1999).
In addition to Sir3p, Sir4p, and Net1p, Sir2p also interacts with Esc2p, Esc8p,
Slx5p, and Mcm10p (Cuperus et al. 2002; Liachko et al. 2009; Darst et al. 2008).
Our interest was piqued by the Sir2p-Esc2p interaction. Initially, Esc2p was
identified as a factor required for robust silencing at telomeres and HMR (Dhillon et
al. 2000). Other work showed that Esc2p plays a role in homologous recombination
repair during DNA replication and in sister chromatid cohesion (Sollier et al. 2009;
Mankouri et al. 2009; Ohya et al. 2008). This is consistent with results showing that
esc2∆ strains are more sensitive to the genotoxic agent methyl methanesulfonate
(MMS) (Yu et al. 2010). We discovered that Esc2p actually affects transcriptional
silencing in a locus-dependent manner. Deletion of ESC2 decreases silencing at
telomeres and rDNA, but increases silencing at the HM loci. ChIP experiments to
examine the abundance of Sir2p at silent loci in ESC2 and esc2∆ cells demonstrated
that there is less Sir2p at the telomeres in esc2∆ cells (Yu et al. 2010).
Esc2p is a member of an evolutionarily conserved family of proteins that
contain two small ubiquitin-like modifier (SUMO)-like domains (Novatchkova et al.
2005). SUMO is a small (~11 kDa) polypeptide that is post-translationally
conjugated to lysine residues in target proteins (Verger et al. 2003). SUMO and
ubiquitin have only 18% sequence identity, but their folded structures are almost
identical, and they share the same general chemistry for lysine conjugation (Bayer et
al. 1998; Schwartz et al. 2003). While ubiquitination is most commonly associated
with the targeting of a protein for degradation, there is no similarly unifying activity

30
triggered by sumoylation, which is associated with a collection of seemingly
unrelated cellular processes. However, in many situations, addition of SUMO to a
substrate affects its interactions with other proteins, usually by creating or blocking
binding sites or interacting domains. Sumoylation has been associated with diverse
cellular processes, including transcription, DNA repair, nuclear transport, signal
transduction, and cell cycle regulation (Verger et al. 2003). Vertebrates contain three
SUMO genes, while S. cerevisiae contains only one, SMT3 (Johnson et al. 1997).
Throughout Chapters 2 and 3, “SUMO” and “Smt3p” are used interchangeably when
referring to the S. cerevisiae SUMO protein.
What is unusual about the Esc2p-containing family of proteins is that the
proteins themselves contain two domains with sequence homology to the SUMO
polypeptide. Whether these domains can behave as SUMO and associate with a
SUMO-binding motif is unknown. Interestingly, Esc2p also contains four putative
SUMO-binding motifs (SBMs) (Sollier et al. 2009; Raffa et al. 2006) (Fig. 2-1). There
is currently no data available on whether Esc2p associates with any sumoylated
proteins. In this Chapter, I show that one particular SUMO-binding motif in Esc2p is
necessary and sufficient for interaction with Sir2p and SUMO. I also provide
evidence that this motif is required for the function of Esc2p in transcriptional
silencing. Finally, I demonstrate that, although the region of Esc2p essential for
interaction with Sir2p contains a SUMO-binding motif, sumoylation of Sir2p is not
required for interaction with Esc2p.
Fig. 2-1. Schematic of Esc2p. SUMO-like domains (open boxes) and SUMO-binding motifs (diamonds) are indicated. The zigzag line indicates a long helical region conserved among Esc2p and its close relatives (Novatchkova et al. 2005).

31
2.3 Results 2.3.1 Esc2p and Sir2p can be co-purified from yeast.
Esc2p was previously shown to interact with Sir2p by yeast two-hybrid
analysis (Cuperus et al. 2002). I confirmed this interaction with a pull-down
experiment in which I purified Esc2p-His6-6xHA-Protein A (abbreviated Esc2p-His-
HA) or, as a negative control, Cyt2p-His-HA (similarly abbreviated), in a yeast strain
expressing untagged Sir2p or Sir2p-9xMyc (abbreviated Sir2p-Myc). Cyt2p is
cytochrome c1 heme lyase, a protein that has no known association with Sir2p, silent
chromatin, or any of the silencing machinery. All proteins were well expressed and
relatively stable in yeast lysate (Lanes 1-3, anti-HA and anti-Myc). Purification on
Ni2+-NTA resin efficiently purified both Esc2p-HIS-HA and Cyt2p-HIS-HA (Lanes 1’-
3’, anti-HA), and Sir2p-Myc only co-purified with Esc2p-HIS-HA (Lanes 1’-3’, anti-
Myc), indicating a specific interaction between Esc2p and Sir2p.
Fig. 2-2. Esc2p interacts with Sir2p in vivo. Proteins isolated from Strain 2-2s expressing either Esc2p-HIS-HA (from pESC2-MORF) or Cyt2p-HIS-HA (from pCYT2-MORF) were bound to Ni2+-NTA resin. Bound proteins were washed, eluted by boiling, separated on SDS-PAGE, and detected by western blotting with anti-Myc and anti-HA antibodies.

32
2.3.2 A putative SUMO-binding motif of Esc2p is necessary and sufficient for interaction with Sir2p and SUMO.
Previous work showed that the N-terminal 195 amino acids of Esc2p are
necessary and sufficient for two-hybrid interaction with Sir2p (Ohya et al. 2008). We
used the yeast two-hybrid system to define the minimum domain of Esc2p required
for interaction with Sir2p. We constructed plasmids encoding a series of Esc2p
fragments fused to the Gal4p DNA binding domain (GBD) (Fig. 2-3, left) and assayed
their ability to interact with Sir2p fused to the Gal4p activation domain (GAD-Sir2p)
(Fig. 2-3, right). GBD and GAD plasmids were co-transformed into the two-hybrid
reporter Strain 2-3s, which contains two genomic reporters for interaction of the
GBD- and GAD-fusion proteins: GAL1-HIS3 and GAL7-ADE2. GAL1-HIS is
generally easier to activate than GAL7-ADE2, therefore we interpret HIS3 expression
as an indicator of moderate interaction, and ADE2 expression as an indicator of
strong interaction (James et al. 1996). In this assay, -Ura-Leu plates serve as a
control for cell growth, and I monitor protein-protein interaction by observing growth
on two reporter plates: -Ura-Leu-His+3-aminotriazol (3-AT, added to increase the
stringency of the HIS3 selection) (abbreviated -His), and -Ura-Leu-Ade (abbreviated -
Ade).
We utilized this system to identify the smallest fragment of Esc2p that still
retained the ability to bind Sir2p. First, we confirmed published data showing that
full-length Esc2p and Esc2p(aa1-195) both strongly interact with Sir2p (Fig. 2-3,
Rows 2 and 3, growth on -His and -Ade) (Ohya et al. 2008). We further narrowed
the binding site by assaying additional Esc2p fragments: a larger C-terminal
truncation, Esc2p(aa1-115) did not interact with Sir2p (Fig. 2-3, Row 4, no growth on
reporter plates). An N-terminal truncation, Esc2p(aa116-456) still interacted, but
Esc2p(aa195-456) failed to interact with Sir2p (Fig. 2-3, compare Row 5 with 6, -His).
This indicated that Esc2p amino acids 116-195 were important for the interaction,
and in fact, GBD fused to just these 80 amino acids interacted with Sir2p (Fig. 2-3,
Row 8, growth on -His), while Esc2p lacking these 80 amino acids did not (Fig. 2-3,
Row 13, no growth on reporter plates). I then divided these 80 amino acids into two
40 amino acid GBD-fusions and found that only Esc2p(aa116-155) interacted with
Sir2p. I further halved that region into two 20 amino acid GBD-fusions, and showed

33
that only Esc2p(aa116-135) interacted with Sir2p (Fig. 2-3, compare Row 9 with 10
and Row 11 with 12, growth on -His). Deletion of these 20 amino acids from Esc2p
rendered Esc2p unable to bind Sir2p (Fig. 2-3, Row 16, no growth on reporter
plates). While it is formally possible that this loss of binding results from misfolding
or other disruption of Esc2p structure caused by the missing amino acids, we believe
this is unlikely. Esc2p missing aa156-195 or aa136-155 is still able to interact with
Sir2p (Fig. 2-3, Rows 15 and 17, respectively, growth on -His and -Ade), suggesting
that such a small deletion does not result in gross misfolding of Esc2p. Furthermore,
an anti-GBD western blot performed on lysates of all strains showed that each GBD-
Esc2p fusion was well expressed (data not shown). Thus, we conclude that amino
acids 116-135 of Esc2p constitute a specific binding site for Sir2p.

34
Fig. 2-3. Esc2p(aa 116-135) are necessary and sufficient for binding to Sir2p. Two-hybrid analysis of interactions between GAD-Sir2p and GBD-Esc2p fragments. The Esc2p constructs used are shown on the left. Two-hybrid reporter Strain 2-3s (James et al. 1997) was transformed with GAD-Sir2p and one of the Esc2p plasmids indicated. Two independent clones from each transformation were grown to late log phase in -Ura-Leu media, and 10-fold dilutions were spotted on -Ura-Leu (growth control), -Ura-Leu-His+3-AT (growth indicates interaction), and -Ura-Leu-Ade (growth indicates strong interaction). SD, SUMO-like domain. SBM, SUMO-binding motif (only SBM1 is shown). Asterisk, denotes Esc2p(V120,121A) mutant.

35
We were surprised to have identified such a short sequence of Esc2p
responsible for Sir2p binding, and we next asked whether there was anything special
about these 20 amino acids. Interestingly, this sequence
(116MKESVVEINSSESDLDEDKN135) contains one of Esc2p’s four putative SUMO-
binding motifs, SBM1. Amino acids 119-133 (in bold, above) encode a perfect
SUMO-binding consensus site, which consists of a hydrophobic core (V/I)(V/I)X(V/I),
where X represents any residue (120VVEI123 in Esc2p), followed by an acidic patch
(127ESDLDED133 in Esc2p) (Kerscher et al. 2006; Hecker et al. 2006). To test
whether SBM1 itself is required for Sir2p binding, we made two point mutations in the
hydrophobic core of full-length Esc2p, changing valines 120 and 121 to alanine
(V120,121A), altering the hydrophobic core from 120VVEI123 to 120AAEI123. This SBM1
mutant peptide failed to interact with Sir2p (Figure 2-3, compare Row 2 with 18,
growth on -His and -Ade).
The importance of Esc2p’s SBM1 in Sir2p binding raised the question of
whether SBM1 exhibited SUMO binding activity. Although Sollier et al. had
previously shown that Esc2p interacts with Smt3p (the SUMO protein in yeast), they
did not dissect Esc2p to determine the relative contributions of each SBM to SUMO-
binding (Sollier et al. 2009). To address this question, we tested our GBD-fused
Esc2p constructs in a yeast two-hybrid assay, using pGAD-SUMO in place of pGAD-
Sir2 (Fig. 2-4).

36
As expected, full-length Esc2p interacted with SUMO (Fig. 2-4, Row 2,
growth on -His). Esc2p(aa1-195) also interacted with SUMO, seemingly even more
strongly than the full-length Esc2p construct (Fig. 2-4, compare Row 3 with 2, growth
on -His and -Ade). However, Esc2p(aa196-456) did not interact with SUMO (Fig. 2-
4, Row 6, no growth on reporter plates), indicating that SBMs 2-4 and the SUMO-like
domains are not sufficient for interaction with SUMO. Furthermore, the small SBM1-
containing fragment of Esc2p(aa116-135) is both necessary and sufficient for
interaction with SUMO (Fig. 2-4, Rows 11 and 16, growth and no growth on reporter
plates, respectively). Finally, disruption of SBM1 in Esc2p(V120,121A) eliminated
interaction between Esc2p and SUMO (Fig. 2-4, Row 18, no growth on reporter
plates).
These results raise the interesting possibility that sumoylation, either of Sir2p
or of a bridging protein, mediates the SBM1-dependent interaction of Sir2p and
Esc2p. Several large-scale screens for sumoylated proteins in yeast have detected
sumoylated Sir3p and Sir4p, but not Sir2p (Wohlschlegel et al. 2004; Denison et al.
2005). One possible explanation for the SBM-dependence of Esc2p binding is that
Esc2p binds sumoylated Sir3p and/or Sir4p, which then recruit Sir2p. However, Fig.
Fig. 2-4. Esc2p(aa116-135) are necessary and sufficient for interaction with SUMO. Two-hybrid analysis of interactions between GAD-SUMO (Smt3p) and GBD-Esc2p fragments. The Esc2p constructs used are shown on the left. This experiment was performed as described in the legend to Figure 2-3. SD, SUMO-like domain. SBM, SUMO-binding motif. Asterisk, denotes Esc2p(V120,121A) mutant.

37
2-5 shows that the Esc2p-Sir2p interaction is SIR3 and SIR4 independent. In a
sir3∆sir4∆ background, Esc2p(aa116-135) is still both necessary and sufficient for
Sir2p binding (Fig. 2-5, Rows 11 and 17, respectively). In fact, it appears that the
Esc2p-Sir2p interaction is stronger in the sir3∆sir4∆ background (Fig. 2-3 and Fig. 2-
5, Rows 5 and 8, compare -Ade plates between Figures). This raises the possibility
that Sir3p or Sir4p could actually compete with Esc2p for Sir2p binding.
2.3.3 Recombinant Esc2p and full-length Sir2p interact directly in an SBM1-dependent manner.
To better understand the mechanism of Esc2p-Sir2p interaction, we turned to
an in vitro interaction system. We imagined three possible explanations for how Sir2p
interacts with SBM1 of Esc2p: (1) the two proteins interact via a third, sumoylated
protein; (2) they interact directly via a SUMO modification on Sir2p; or (3) they
interact directly without sumoylation. To distinguish among these possibilities, we
asked whether sumoylation of Sir2p was required for its interaction with Esc2p. To
Fig. 2-5. Esc2p-Sir2p interaction is independent of SIR3 and SIR4. Two-hybrid analysis of interactions between GAD-Sir2p and GBD-Esc2p fragments in Strain 2-4s (two-hybrid reporter strain, with SIR3 and SIR4 deleted). The Esc2p constructs used are shown on the left. This experiment was performed as described in the legend to Figure 2-3. SD, SUMO-like domain. Asterisk, denotes Esc2p(V120,121A) mutant.

38
this end, I performed an in vitro binding assay with recombinant Esc2p and Sir2p. E.
coli lacks the machinery required for sumoylation; thus, bacterially-expressed
recombinant proteins are guaranteed to be free of SUMO. In this experiment, I show
that E. coli expressed GST-Sir2p, but not GST, can interact directly with recombinant
His6-HA-Esc2p (Fig. 2-6, compare Lane 1 with 3, anti-HA). However, in the same
assay, recombinant His6-HA-Esc2p(V120,121A) is not pulled down with Sir2p (Fig. 2-
6, compare Lane 3 with 4, anti-HA). Thus, it seems that sumoylation of Sir2p is not
required for the Esc2p-Sir2p interaction in vitro, even though the integrity of SBM1 is
essential for interaction. Furthermore, the fact that this in vitro interaction maintains
its SBM1-dependence suggests that it genuinely mimics the in vivo interaction, and
is not simply an artifact of mixing high concentrations of recombinant proteins in vitro.
Sir2p is the founding member of a family of enzymes that catalyze NAD+-
dependent lysine deacetylation. This family includes the four S. cerevisiae Hst
proteins (Hst1p – Hst4p) and seven human SirT proteins (SirT1 – SirT7) (Frye 2000).
These protein deacetylases share a conserved catalytic core domain of ~270 amino
acids that is flanked by highly variable N- and C- terminal extensions (Fig. 2-7A),
which can provide functional specificity and sites for unique protein-protein
interactions (Frye 2000). Sir2p has many known binding partners: Sir3p, Sir4p,
Fig. 2-6. Recombinant Esc2p and Sir2p interact directly and in an SBM1-dependent manner. Lysates from bacteria expressing GST (Lanes 1 and 2) or GST-Sir2p (Lanes 3 and 4) were incubated with glutathione-sepharose resin. Resin was washed, then mixed with lysate from bacteria expressing either wild type HA-Esc2p (Lanes 1 and 3) or HA-Esc2p(V120,121A). The resin was washed again, and proteins were eluted by boiling in Laemmli. Eluates were separated by SDS-PAGE and subject to Western Blotting with anti-GST and anti-HA antibodies.

39
Esc2p, Net1p, Slx5p, and Mcm10p (Cuperus et al. 2002; Liachko et al. 2009; Darst
et al. 2008), and identifying which domains of Sir2p are important for each of its
binding interactions would improve our understanding of how Sir2p is regulated.
Technical difficulties prevented us from using the yeast two-hybrid system to address
this question, but the in vitro GST-binding assay provided a viable alternative.
Bacterially expressed Esc2p and Sir2p were abundant, soluble, and relatively stable.
Furthermore, the Esc2p-Sir2p interaction in vitro mimics the in vivo interaction with
respect to its dependence on SBM1.
To map the domain(s) of Sir2p involved in Esc2p binding, I first generated
plasmids for bacterial expression N-terminal GST fusions of each combination of
contiguous Sir2p domains (N-Cat-C, N-, N-Cat, Cat, Cat-C, -C; where N is the N-
terminal extension, Cat is the Conserved Catalytic domain, and C is the C-terminal
extension) (Fig. 2-7B, top). These Sir2p fragments were expressed and subject to
the same in vitro GST pull-down assay described above, with recombinant Esc2p.
All GST-Sir2p fragments were well-expressed and purified on glutathione resin (Fig.
2-7B, see asterisks on anti-GST blot), but only full-length GST-Sir2p was able to pull-
down HA-Esc2p (Fig. 2-7B, compare Lane 2 with all other Lanes).

40
2.3.3 Targeted silencing promoted by Esc2p is mediated by SBM1.
We had so far characterized Esc2p as a protein that was important for
transcriptional silencing at certain loci and that interacted directly, via its SBM1, with
Sir2p, a member of the SIR silencing complex. The next natural step was to
determine whether this SBM1-Sir2p interaction is important for silencing.
Fig. 2-7. Recombinant Sir2p missing any of its three domains does not bind Esc2p in vitro. (A) Schematic of Sir2p domains. Sir2p can be divided into three domains: the N-terminal domain (N, open box) includes aa 1-255, the conserved Catalytic domain (Cat, light grey box) includes aa 256-497, and the C-terminal domain (C, dark grey box) includes aa 498-562. (B) GST pull-down of recombinant Esc2p with GST-Sir2p fragments. Lysates from bacteria expressing GST or the indicated GST-Sir2p fragment were incubated with glutathione-sepharose resin. Resin was washed, and all samples were mixed with lysate from bacteria expressing wild type HA-Esc2p. The resin was washed again, and proteins were eluted by boiling in Laemmli. Eluates were separated by SDS-PAGE and subject to western blotting with anti-GST and anti-HA antibodies. Asterisks highlight the location of each Sir2p construct on the anti-GST blot.

41
Transcriptional silencing at HMR and HML is mediated by the E and I
silencers, which are composed of combinations of binding sites for Rap1p, Abf1p,
and the ORC complex, all of which function to recruit the SIR complex (Rusche et al.
2003; Brand et al. 1985; Mahoney et al. 1989). The HMR-E silencer includes each
of these binding sites (Fig. I-2), and deletion of two or more sites abolishes HMR
silencing. Silencing can be restored by directly targeting the SIR complex to the
mutated HMR-E silencer (Chien et al. 1993). By artificially tethering a protein of
interest to a defective silencer and monitoring expression of a reporter gene placed
at HMR, we can test the ability of that protein to recruit the SIR complex. In the
strain that we use for this assay (Strain 2-5s), the ORC site remains intact, but the
Rap1p and Abf1p sites at HMR-E are replaced with three copies of UASg, the Gal4p-
binding site (Fig. 2-8, top). To monitor silencing at HMR, the TRP1 reporter gene
has been inserted at HMR.
When the empty pGBDU-C1 vector is transformed into the host, only GBD is
expressed and recruited to HMR-E, the SIR complex is not recruited to HMR, the
HMR locus is derepressed, and cells grow well on -Ura-Trp (abbreviated -Trp) (Fig.
2-8, Row 1). However, expression of GBD-Sir4p(aa732-1358) or GBD-Sir1p results
in HMR silencing and no cell growth on -Trp (Fig. 2-8, Rows 19 and 20, lack of
growth on -Trp). Therefore, targeting Sir4p(aa732-1358) or Sir1p to the defective
HMR-E silencer is sufficient to restore HMR silencing.
Previous work showed that GBD-Esc2p can mediate targeted silencing in the
above assay (Andrulis et al. 2004), and we confirmed this result (Fig. 2-8, Row 2, no
growth on -Trp). We further determined that amino acids 116-195 of Esc2p are
necessary and sufficient for targeted silencing: Esc2p(aa1-195), Esc2p(aa116-456),
and Esc2p(aa116-195) all support targeted silencing, whereas Esc2p(aa1-115) and
Esc2p(∆116-195) do not (Fig. 2-8, compare Rows 3 and 5 with Row 4, and compare
Row 8 with 13, growth on -Trp). Next, we mapped targeted silencing activity to
Esc2p(aa 116-155), then down to the 20-amino acid fragment Esc2p(aa116-135).
The 40 and 20 amino acid fragments both retain significant silencing activity (Fig. 2-
8, Rows 9 and 11, lack of growth on -Trp). Furthermore, Esc2p deleted for amino
acids 116-135 and Esc2p(V120,121A) fail to support targeted silencing (Fig. 2-8,
Rows 16 and 18, growth on -Trp). Comparing results from Figs. 2-3 and 2-8, we

42
note an absolute correlation between an Esc2p fragment’s ability to bind Sir2p and
its ability to direct targeted silencing. This evidence strongly supports the hypothesis
that Esc2p induces targeted silencing by recruiting Sir2p through its SBM1 motif.

43
Fig. 2-8. Targeted silencing by Esc2p is mediated by SBM1. Top, HMR locus in the targeted silencing reporter construct, Strain 2-5s (Chien et al. 1993). Bottom, Strain 2-5s was transformed with each of the indicated GBD-Esc2p plasmids, GBD-Sir4p(aa732-1358), or GBD-Sir1p. Two independent clones from each transformation were grown to late log phase in -Ura media, and 10-fold dilutions were spotted on -Ura (growth control) and -Ura-Trp (growth indicates loss of silencing at HMR).

44
2.3.4 The function of Esc2p in transcriptional silencing is dependent on SBM1. Finally, we tested whether SBM1 is necessary for the function of Esc2p in
transcriptional silencing. Early experiments in our lab indicated that deletion of ESC2
reduced telomeric silencing and rendered strains sensitive to the genotoxic agent
methanesulfonate (MMS) (Yu et al. 2010), so we tested the ability of GBD-Esc2p and
GBD-Esc2p(V120,121A) to restore telomeric silencing in an esc2∆ strain. To do this
experiment, an esc2∆ strain that also encodes the Tel VIIL-URA reporter construct
was transformed with plasmids 1’, 2’ and 18’ encoding GBD, GBD-Esc2p and GBD-
Esc2p-V120,121A, respectively (Fig. 2-9). These plasmids are identical to 1, 2, and
18, except the URA3 marker has been replaced by LEU2. The strain expressing
GBD alone had weak telomeric silencing and was sensitive to MMS (Fig. 2-9, Row
1’, lack of growth on FOA and MMS). Expression of GBD-Esc2p restored URA3
silencing and MMS resistance (Fig. 2-9, compare Row 1’ with 2’, FOA and MMS
plates), proving that the plasmid-expressed GBD-Esc2p construct suppresses esc2∆
phenotypes. However, GBD-Esc2p(V120,121A) expressed in esc2∆ cells rescued
growth on MMS, but was unable to restore telomeric silencing (Fig. 2-9, compare
Row 1’ with 18’, FOA and MMS plates). This indicates that SBM1 is necessary for
the role of Esc2p in telomeric silencing, most likely for the recruitment of Sirp2p, but
is not required for the role of Esc2p in tolerating genotoxic stress.
Fig. 2-9. SBM1 of Esc2p is required for telomeric silencing but not cellular tolerance of genotoxic stress. Schematic at the top shows the TelVIIL-URA3 construct used to monitor silencing in this experiment. Strain 2-6s, bearing this telomeric silencing reporter and esc2∆ were transformed with Plasmid 1’, 2’, or 18’ (identical to Plasmids 1, 2, and 18 except with a LEU2 marker). Two independent clones from each transformation were grown to late log phase in -Leu media, and 10-fold dilutions were spotted on -Leu (growth control), -Leu+FOA (growth indicates silencing at telomere), and -Leu+MMS (growth indicates resistance to genotoxic stress).

45
2.4 Discussion
S. cerevisiae Sir2p is a histone deacetylase that interacts with Sir4p in the
SIR complex to effect silencing at the telomeres and HM loci. In addition, Sir2p is
known to physically interact with other proteins, including Esc2p, Esc8p, Slx5p, and
Mcm10p (Liachko et al. 2009; Darst et al. 2008; Cuperus et al. 2000). How Sir2p
interacts with any of these proteins has not been resolved.
Esc2p was identified as a factor required for robust silencing at telomeres
and HMR (Dhillon et al. 2000). It is a member of a conserved family of proteins that
encode two SUMO-like domains in the ORF of the protein. Esc2p also encodes four
putative SUMO-binding motifs (Fig. 2-1). The role(s) of these regions in Esc2p
function has not been characterized.
In this Chapter, I first demonstrated that Sir2p and Esc2p physically interact
in yeast cells (Fig. 2-2). Using a yeast two-hybrid assay, I then showed that the 20
amino acids of Esc2p that contain SBM1 (Esc2p aa116-135) are necessary and
sufficient for interaction with both Sir2p and SUMO (Figs. 2-3 and 2-4). SUMO-
binding consensus motifs are composed of a hydrophobic core flanked by an acidic patch, and SBM1 is a perfect fit for this consensus sequence
(119VVEINSSESDLDED132). Mutating the hydrophobic core of SBM1 (V120,121A)
abolished these interactions and rendered Esc2p non-functional in silencing (Figs. 2-
3, 2-4, 2-8, and 2-9).
We considered several possible explanations for the SBM1-dependence of
the Esc2p-Sir2p interaction. First, it is possible that the interaction is mediated by a
third, sumoylated protein. Likely candidates included Sir3p and Sir4p, which have
been identified in several large-scale screens for sumoylated proteins in yeast
(Wohlschlegel et al. 2004; Denison et al. 2005). However, the Esc2p-Sir2p
interaction was maintained, and even enhanced, in a sir3∆sir4∆ strain, leading us to
conclude that neither of these proteins is required for the interaction (Fig. 2-5). In
fact, quite the opposite was true – it seems that Sir3p/Sir4p may actually compete
with Esc2p for binding to Sir2p. Furthermore, the fact the Esc2p is not enriched at
transcriptionally silent loci (Yu et al. 2010), indicates that it most likely interacts with
Sir2p that is not bound to chromatin. Taken together, these data suggest that Esc2p
could affect silencing by modulating the availability of “free” Sir2p.

46
A second possible explanation for the SBM1 dependence of the Esc2p-Sir2p
interaction is that Esc2p might interact with sumoylated Sir2p. Sir2p was not
identified in any large-scale screens for sumoylated proteins, but such experiments
must balance sensitivity with specificity, and it is not uncommon for a genuine target
to be missed (Wohlschlegel et al. 2004; Denison et al. 2005). To determine whether
Esc2p and Sir2p could interact directly in a sumoylation independent fashion, I
performed an in vitro co-purification experiment with bacterially expressed Sir2p and
Esc2p. I found that the two recombinant proteins did interact, and did so in an
SBM1-dependent manner, as bacterially expressed Esc2p bound Sir2p, but
Esc2p(V120,121A) did not (Fig. 2-6). E. coli lack sumoylation machinery, therefore
these results indicate that Esc2p and Sir2p interact directly, in a sumoylation-
independent manner. This result is actually supported by the co-purification
experiment in yeast, where there is only one band observed for Sir2p-Myc pulled
down by His6-HA-Esc2p (Fig. 2-2). One SUMO peptide is approximately ~10 kDa,
but addition of the SUMO moiety to a protein often retards the protein considerably
more than 10 kDa on an SDS-PAGE gel, resulting in an obvious shift on the gel
(Ulrich et al. 2009). However, just one Sir2p-Myc band appears in both the Input and
Pull-down lanes. Furthermore, quantitative comparison of the percentage of co-
purification in the in vivo and in vitro experiments is difficult, and so we cannot
absolutely exclude the possibility that sumoylated Sir2p contributes to the binding of
Esc2p. (For further discussion and data regarding the sumoylation of Sir2p, see
Chapter 3.)
The fact that recombinant Sir2p and Esc2p can associate with each other led
us to the third hypothesis that Sir2p interacts directly with SBM1 of Esc2p
independent of Sir2 sumoylation. It is possible that the hydrophobic and acidic
residues that form SBM1 interact with certain hydrophobic and basic residues in
Sir2p. To test this hypothesis, I attempted to isolate a domain in Sir2p that bound to
Esc2p, using the in vitro GST pull-down protocol. Although the fragments that I
chose were well expressed and relatively stable, none showed significant Esc2p
binding activity (Fig. 2-7). It is possible that the truncated Sir2p constructs expressed
in bacteria are incorrectly or incompletely folded. Another possibility is that a non-
continuous combination of Sir2p sequences is required for binding to Esc2p.

47
According to data from S. Gasser’s lab, both the N- and C-terminal domains of Sir2p
(aa 94-198 and 422-562) are required for its interaction with Sir4p (Cockell et al.
2000). This could be explained if there are multiple points of interaction between the
two proteins, or these separated sequences are brought in close proximity in the final
folded protein. If non-continuous sequences were also required for Sir2p interaction
with Esc2p, it is possible that the constructs designed (Fig. 2-7) would not contain
the necessary pieces together
Although there is much information on interaction domains of Sir3p and Sir4p
in the literature [summarized in (Gasser et al. 2001)], little is known about the
domain(s) of Sir2p responsible for interacting with any of its partners (Cockell et al.
2000). Thus, it is tempting to speculate that something inherent to Sir2p – its
dynamics of folding, its higher order structure, or another unknown factor –
complicates attempts to generate stable, functional fragments and/or truncations for
analysis in binding studies.
What are the implications of the Esc2p-Sir2p interaction on silencing? Esc2p
plays a role in DNA repair during replication and in transcriptional silencing (Dhillon
et al. 2000; Sollier et al. 2009; Mankouri et al. 2009; Ohya et al. 2008). We have
determined that ESC2 is inhibitory to silencing at HM loci, but is required for efficient
telomeric silencing. Furthermore, ESC2 is involved in maintaining Sir2p association
with chromatin near the telomeres, but not at the HM silent loci (Yu et al. 2010).
These data are complex and seemingly contradictory; however, we propose
related, but locus-specific explanations for the role of Esc2p at the silent loci: (i) At
the HM loci, Esc2p negatively regulates silencing, but has no effect on the level of
Sir2p. One of my labmates has shown that ESC2 regulates the stability of HML
silent chromatin, making it more sensitive to disruption (Yu et al. 2010). Perhaps
Esc2p can bind to Sir2p in silent chromatin and “pull” it away reducing the stability of
silent chromatin. It is possible that the Sir2p level at the HM loci (as measured by
ChIP) could still appear unchanged in these cells, particularly if such a disruption
only occurs at only a portion of each locus. In line with this, the Sir2p-Esc2p
interaction appears to be increased in a sir3∆ sir4∆ strain, suggesting that Sir3p/4p
and Esc2 could compete for Sir2p binding. (ii) At telomeres, Esc2p positively
regulates silencing and the level of Sir2p. Although many of the same mechanisms

48
act to establish and maintain silencing at the HM loci and at telomeres, we speculate
that Esc2p may have a telomere-specific positive effect on silencing that outweighs
the negative effect described in (i). We suggest that Esc2p binds Sir2p and presents
it for binding to telomere-specific silencing factors, like the Ku70/Ku80 complex. In
∆esc2 cells, Sir2p is no longer being recruited to telomeres by Esc2p, so the level of
Sir2p there drops, and silencing is decreased.
Clearly, we have just begun to appreciate how the Esc2p-Sir2p interaction
affects transcriptional silencing and the overall regulation of Sir2p. As we begin to
understand how this protein pair acts at different silent loci, we hope to contribute to
a greater understanding of how, when, and why Sir2p interacts with its various
binding partners, and how these interactions regulate Sir2p activity.

49
2.5 Materials and Methods 2.5.1 Plasmids and Strains Plasmids pESC2-MORF and pCYT2-MORF are the Esc2p- and Cyt2p-
encoding members of a yeast plasmid library that expresses each yeast open
reading frame under the control of a GAL1 promoter, and C-terminally fused to a
tandem affinity tag encoding His6-6xHA-Protein A•ZZ Domain (Gelperin et al. 2005).
Plasmids encoding Gal4p DNA binding domain (GBD)-fused Esc2p are numbered 2-
18 (Fig. 2-3). Plasmid 1 is pGBDU-C1 (James et al. 1996). Plasmids 2-10 were
made by inserting PCR-generated, BamHI-SalI flanked full-length or truncated ESC2
fragments into BamHI-SalI digested pGBDU-C1. Plasmids 11 and 12 were
constructed similarly, but the short inserts were ordered as two complimentary
oligonucleotides that generated BamHI-SalI sticky ends after annealing. The
annealed oligonucleotides treated with T4 Polynucleotide Kinase (NEB), and ligated
to BamHI-SalI digested pGBDU-C1. Plasmid 13 was made using Quik Change
mutagenesis (Stratagene) to delete the sequence encoding amino acids 115-195 of
Esc2p from Plasmid 2. Plasmids 14 and 16 were made by inserting a fragment
encoding Esc2p amino acids 156-456 (Plasmid 14) or 136-456 (Plasmid 16) as a
SalI-PstI fragment into Plasmid 4. Plasmids 15 and 17 were constructed similarly,
using the appropriate two fragments joined at a SalI site. Plasmid 18 was made
using Quik Change mutagenesis to generate two single-nucleotide substitutions in
the ESC2 coding sequence, T359A and T362A. Plasmid 19 was made by inserting
the sequence encoding the C-terminal half of Sir4p(aa732-1358) into pGBDU-C1 as
an EcoRI-SalI fragment. Plasmids 1’, 2’, and 18’ were made by replacing the URA3
gene in Plasmids 1, 2, and 18 with a LEU2 cassette.
Plasmid pGAD-SMT3 was generated by inserting PCR-amplified SMT3 as a
BamHI-SalI fragment into pGAD-C1 (James et al. 1996). pGAD-SIR2 was a gift from
B. Tye and has been previously described (Liachko et al. 2009). Plasmid 20,
encoding pGBD-Sir1p, is pJR1815 from J. Rine.
Plasmid pHK49 encoding GST-Sir2p was made by inserting an EcoRI-SIR2-
XhoI fragment into pGEX6p-1 (Amersham Biosciences). Plasmids encoding GST-
Sir2p fragments are as follows: pHK58 (Sir2p aa1-255), pHK59 (Sir2p aa1-497),
pHK60 (Sir2p aa256-497), pHK61 (Sir2p aa256-562), and pHK62 (Sir2p aa498-562).

50
These plasmids were made by inserting PCR-generated truncated SIR2 fragments
flanked by EcoRI-XhoI sites into EcoRI-XhoI digested pGEX6p-1. Plasmid pHK52
encoding His6-HA-Esc2p was made by inserting an NdeI-HA-ESC2-XhoI fragment
into pET16b (Novagen). Plasmid pHK65 encoding His6-HA-Esc2p(V120,121A) was
made from pHK52 using Quik Change mutagenesis to generate two single-
nucleotide substitutions in the ESC2 coding sequence, T359A and T362A.
Strain 2-2s was generated by transforming Strain 2-1s with a PCR-produced
fragment encoding 9xMyc linked to KanMX, embedded in a sequence spanning the
3’ region of the SIR2 coding sequence. Strain 2-4s was generated from Strain 2-3s
by replacing SIR3 with NatMX and SIR4 with KanMX. Strain 2-6s was made from
YHK53 (Strain 1-18s) by replacing ESC2 with KanMX.
2.5.2 Pull-down Analyses 2.5.2.1 Ni2+-NTA Pull-Down: Yeast cells were grown and induced as
described (Gelperin et al. 2005). Briefly, cells were grown in -Ura liquid containing
2% raffinose to an OD600 ~ 0.8. Expression from pESC2-MORF and pCYT2-MORF
was induced by the addition of 3x YPGal (3% yeast extract, 6% bacto-peptone, and
6% galactose), to bring the culture to a final concentration of 1x YPGal (1% yeast
extract, 2% bacto-peptone, and 2% galactose). After 24 hrs induction, cells were
harvested, washed, and lysed by the glass bead method in TALON Extraction buffer
(5% Glycerol, 150 mM NaCl, 20 mM HEPES, pH7.5) plus protease inhibitors.
Protein concentration was determined by Bradford assay, and 7 mg of total protein
lysate was added to 90 µl equilibrated Ni2+-NTA resin (Qiagen) and incubated for 2
hrs at 4°C. Resin was washed with Wash Buffer (Extraction buffer without protease
inhibitors), and proteins were eluted by boiling for 5 min in Laemmli buffer. Proteins
were separated by SDS-PAGE and detected by western blotting with anti-HA
(Sigma) or anti-Myc (Roche) antibody.
2.5.2.2. GST Pull-Down: Escherichia coli BL21 cells bearing pGEX6p-1,
pHK49, pHK52, or pHK65 were treated with isopropyl 1-thio-β-D-galactopyranoside
(1 mM) for 4 hrs at room temperature to induce the expression of GST, GST-Sir2p,
His6-HA-Esc2p, or His6-HA-Esc2p(V120,121A), respectively. Cells were washed and
resuspended in E. coli Lysis Buffer (20 mM Tris pH 8.0, 350 mM NaCl, 1% Nonidet

51
P-40, 10 mM dithiothreitol) plus protease inhibitors and lysed by treatment with
lysozyme (0.5 mg/ml) for 30 min on ice, followed by sonication. Extracts were
clarified by centrifugation, and GST-tagged protein abudance was determined by
Coomassie staining. Lysate containing an equivalent amount of GST or GST-Sir2p
was bound to 100 µl of glutathione-sepharose slurry (Amersham Biosciences) for 2
hrs at 4°C. Resin was washed four times with 1 ml E. coli Lysis Buffer. Lysate
containing equivalent amounts of His6-HA-Esc2p and His6-HA-Esc2p(V120,121A)
was added to the resin and incubated for 2 hrs at 4°C. Resin was washed five times
with 1 ml E. coli Lysis Buffer and eluted by boiling in Laemmli. Proteins were
separated by SDS-PAGE. GST tagged proteins were detected by Coomassie
staining or immunoblotting with anti-GST antibody (Sigma). HA-tagged proteins
were detected by immunoblotting with anti-HA antibody (Sigma).
2.5.3 Two-hybrid Analysis Two-hybrid analysis was performed as described (James et al. 1996), using
their reporter strain PJ69-4α (Strain 2-3s, for the purposes of this thesis). This strain
was transformed with a plasmid encoding a GBD-fused protein (URA3 marked) and
a plasmid encoding a GAD-fused protein (LEU2 marked). Two independent
transformants were selected and grown overnight in -Ura-Leu liquid to late log
phase. Cultures were 10-fold serial diluted and spotted on -Ura-Leu, -Ura-Leu-His+3
aminotriazol (AT), and -Ura-Leu-Ade plates. 3-AT is a competitive inhibitor of His3p
activity and thus increases the stringency of the HIS3 selection. 3-AT was used at a
concentration of 2mM.

52
Chapter 3:
Post-translational modification of Saccharomyces cerevisiae histone deacetylase Sir2p
3.1 Abstract Post-translational modification of proteins, including the covalent attachment
of small peptides like ubiquitin and SUMO, is used by cells to extend and expand the
functionality of a limited proteome. In this Chapter, I demonstrate that the histone
deacetylase Sir2p is subject to both ubiquitination and sumoylation in vivo. In order
to examine the mechanism and function of the ubiquitination and sumoylation of Sir2,
I attempt to map the sites of Sir2 ubiquitination and sumoylation with mutational and
deletion analyses, as well as mass spectrometry analysis of purified Sir2p protein.

53
3.2 Introduction Protein post-translational modifications come in many varieties, and include
structural changes, chemical alterations of constituent amino acids, addition of small
chemical groups, and covalent modification by small polypeptides. All of these
extend the range of function of an organism’s proteome, and modifications affect
virtually all aspects of protein control, including localization, binding interactions,
stability, and activation. To say that every cellular process in eukaryotes is affected
by post-translational modification of one or more proteins is hardly an overstatement.
One class of protein modifications that has recently gained attention is
covalent modification by small peptides including ubiquitin and small ubiquitin-like
modifier (SUMO). Ubiquitin is a highly conserved, 76-residue protein that is the best-
characterized member of this group. There are at least ten other ubiquitin-like
proteins (Ubls) that can also act as post-translational protein modifiers, including
NEDD8, ISG15, and SUMO, among others (Xirodimas 2008; Ritchie et al. 2004).
These small, modifying polypeptides vary in sequence – for example, SUMO and
ubiquitin have only 18% sequence identity – although many of their folded structures
are similar (Bayer et al. 1998; Schwartz et al. 2003). What ubiquitin and the Ubls do
share is a common chemistry for conjugation to lysine residues in their substrates
(Fig 3-1).
This conjugation reaction requires that the Ub or Ubl be passed through
several intermediate enzymes before being conjugated onto the target substrate. Ub
and most Ubls are synthesized as inactive precursors that are C-terminally
processed by a protease to reveal a glycine carboxylate. This exposed site is then
adenylated in an ATP-dependent manner by a specific activating enzyme (E1) and
attached to a cysteine on the E1 via a high-energy E1-Ubl thioester linkage. The Ubl
is passed to a cysteine residue on a Ub-conjugating, or E2, enzyme. Finally, an E3
protein ligase transfers the Ubl from E2 onto the ε-amino group of a lysine in the
substrate (Kerscher et al. 2006) (Fig 3-1). Cells balance economy with specificity by
using one or two E1s and one-to-several E2s for economy, and several-to-many E3s
to confer substrate specificity. In S. cerevisiae, there are currently 1 E1, 11 E2s, and
50 E3s identified for ubiquitination and 1 E1, 1 E2, and 4 E3s identified for

54
sumoylation (Bergink et al. 2009). Additional modifying enzymes may exist, but have
not yet been identified as such.
The work in this chapter focuses on Sir2p modification by ubiquitin and
SUMO. Ubiquitination is most popularly known as the modification that targets
proteins for proteasomal degradation. This is certainly one of its crucial roles, but
ubiquitination has also been shown to affect a number of cellular processes in a
degradation-independent manner. These processes include protein internalization at
the plasma membrane, vesicle trafficking within the cell, nuclear transport, DNA
damage repair, histone modification and gene silencing (Hicke 2001; Katzmann et al.
Fig. 3-1. Schematic of the conjugation pathway for post-translational modification by ubiquitin and Ubls. First, a protease processes the C-terminus of the Ubl to reveal a glycine carboxylate (-GG). Next, the Ubl-Activating Enzyme (E1) catalyzes the conjugation of the Ubl onto itself through a high-energy thioester bond (represented by an ‘S’) in an ATP-dependent manner. This high energy bond is transferred to a cysteine residue in the Ubl Conjugating Enzyme (E2). Finally, the Ubl Ligase (E3) acts as a binding platform to bring together the E2-Ubl complex and the Substrate. The Ubl is transferred from its high-energy bond to a stable linkage on the ε-amino group of one of the Substrate’s lysine residues.

55
2002; Sobko et al. 2002; Hoege et al. 2002; Sun et al. 2002). S. cerevisiae encodes
four different ubiquitin genes (UBI1-UBI4); three are translated as fusions to
unrelated cellular proteins, and one is translated as five head-to-tail polyubiquitin
precursors (Ozkaynak et al. 1987). As the first step of the conjugation cycle,
individual ubiquitin molecules are cleaved from the rest of the fusion protein to their
mature form with an exposed C-terminal glycine carboxylate. Note that the same
protease also acts to remove ubiquitin from ubiquitinated proteins (Kerscher et al.
2006). The individual ubiquitin molecules are then cycled through the E1, E2, and E3
enzymes for conjugation to a substrate (Fig. 3-1).
Modification by sumoylation does not directly target proteins for degradation.
Addition of SUMO to a substrate may instead affect its interactions with other
proteins, usually by creating or blocking binding sites or interacting domains. In this
way, SUMO affects a seemingly unrelated collection of cellular processes, including
transcription, DNA repair, nuclear transport, signal transduction, and cell cycle
regulation (Verger et al. 2003). In S. cerevisiae, SUMO is expressed from a single
gene, SMT3, and must be pre-processed by a protease to be competent for
conjugation (Johnson et al. 1997).
Post-translational modification by Ub and Ubls is far more complex than a
simple binary (modified-or-not-modified) choice. For example, substrate proteins can
have a single ubiquitin added (monoubiquitination), ubiquitin added to more than one
lysine (multi-ubiquitination), or chains of ubiquitin added to one or more lysine
(polyubiquitination). Ubiquitin has 7 lysine residues, and the fate of a
polyubiquitinated protein often depends on which of these lysines a chain is
generated. Linkage through Lys11 or Lys 48 signals for targeting to the proteasome,
while linkage through Lys63 appears to mediate interaction with binding partners.
The effect of linkage through other lysine residues is not yet known (Ye et al. 2009).
In addition, although one lysine residue can only be conjugated to one Ubl at a time,
there is precedent for a single lysine to be the target of more than one modifier with
different physiological effects (Hoege et al. 2002). Finally, modification by one Ubl
may affect recognition by or availability for modification by another Ubl. For
example, a recently identified class of modifying enzymes known as SUMO-Targeted
Ubiquitin Ligases (STUbLs) recognize a sumoylated protein and target it for

56
ubiquitination and proteasomal degradation (Uzunova et al. 2007). Clearly,
conjugation to multiple and varied Ubls presents almost limitless possible
combinations and a highly tunable control system.
The existence of so many combinatorial possibilities begs the question:
Which lysine residues are targets of modification by a particular Ubl? Previous
examinations of known ubiquitination sites revealed general trends in amino acid
composition surrounding a ubiquitinated lysine. These trends include: an abundance
of charged and polar amino acids (especially aspartate and glutamate), a depletion
of hydrophobic residues (leucine, isoleucine, proline, phenylalanine), and an
absence of other lysine residues nearby (Radivojac et al. 2010). Based on these
trends, computer programs such as UbPred have been designed to predict potential
ubiquitination sites in proteins (Radivojac et al. 2010). In the case of sumoylation, a
consensus site has been empirically determined and is found in many, although not
all, physiological SUMO targets, particularly when the sequence resides in an
unstructured loop. The consensus sequence motif is ΨKX(E/D), where Ψ represents
a bulky aliphatic residue (isoleucine, leucine, or valine) and X represents any residue
(Johnson et al. 1999; Bernier-Villamor et al. 2002). The SUMOplot program
analyzes an amino acid sequence for cognate and near-cognate matches to the
consensus sumoylation site. Since not all predicted sites are actual modification
sites, and there are precedents of lysines residing in non-consensus sites being
subject to modification, any predictions must be validated experimentally (Johnson
2004).
In general, there are two types of experiments that can be done to confirm
the in vivo modification of a protein. One is to affinity purify the protein of interest and
assay for the modifier by western blotting with an antibody for the Ubl. The other is to
affinity purify all the proteins modified by the Ubl, and then examine the presence of
the protein of interest in the precipitate by western blotting. Both of these
approaches present two notable technical challenges. The first is proving that the
modification occurs directly on the protein of interest, and not on a co-purifying
binding partner. The second is preserving the modification during cell lysis and
subsequent purification procedures, because de-modifying enzymes are very active
in cell lysates (Wohlschlegel 2009). To make matters worse, the level of Ubl

57
modification of proteins are often intrinsically low under normal conditions. Both of
these problems can be solved by working under denaturing conditions that disrupt
protein-protein interactions and inactivate de-modifying enzymes. A standard
protocol is described in detail in the Materials and Methods, Section 3.5.2 and is
summarized in Fig. 3-2.
We became interested in the potential modification of Sir2p by SUMO after
determining that Sir2p interacts with Esc2p through a stretch of 20 amino acids in
Esc2p that contain a SUMO-binding motif (Chapter 2). However, previous large-
scale screens for sumoylated proteins in yeast identified Sir3p and Sir4p, but not
Sir2p, as sumoylation targets (Wohlschlegel et al. 2004; Denison et al. 2005). Given
that such screens often yield false negative results, it is formally possible that Sir2p
sumoylation was missed in these screens. Interestingly, Sir2p physically interacts
with the STUbL Slx5p (Prudden et al. 2007). It is possible that Sir2p is sumoylated,
and is subject to regulation by Slx5p.
In this Chapter, I show for the first time that Sir2p is both ubiquitinated and
sumoylated in vivo. I take several approaches to map the site(s) of each
modification on Sir2p, including analysis of Sir2p point mutants and truncations. I
adapt existing purification schemes into a denaturing double purification system that
should allow for efficient purification of modified Sir2p for analysis by mass
spectrometry.

58
Fig. 3-2. Constructs and protocol for denaturing Ni2+-NTA purification experiment to confirm Ubl modification of protein of interest (YFP). (A) Description of constructs. Plasmid expressing the His6, Epitope-Tagged Ubl under the control of an inducible promoter (pINDUCIBLE-His6-Tag1-UBL) or, as a negative control, a Vector Control (either untagged UBL or empty vector), is transformed into either wild-type yeast or yeast expressing YFP with a second Epitope Tag (YFP-Tag2). Three resulting constructs are used in subsequent experiments: YFP + pINDUCIBLE-His6-Tag1-UBL (negative control), YFP-Tag2 + Vector Control (negative control), and YFP-Tag2 + pINDUCIBLE-His6-Tag1-UBL (experimental sample). (B) Outline of steps in protocol. BME, β-mercaptoethanol; NaOH, sodium hydroxide; TCA, trichloroacetic acid; GuHCl, guanidine hydrochloride buffer; TI, Tris-Imidazole buffer.

59
3.3 Results 3.3.1 Sir2p and Sir3p are sumoylated in vivo.
Chapter 2 of this thesis describes the identification of a SUMO-binding motif
(SBM1) as the region of Esc2p that interacts with Sir2p, and so we next asked
whether Sir2p could be post-translationally modified by SUMO in vivo. To answer
this question, I examined if Sir2p was present in a pool of sumoylated proteins that
were affinity-purified from cell extracts. To improve the efficiency of this purification, I
expressed a His6-tagged version of SUMO in the host cells. Specifically, a plasmid
encoding pGAL-His6-FLAG-SMT3 (pHF-SMT3) or, as a negative control, pRS415,
was transformed into yeast cells expressing Sir2p-6xHA, Sir3p-6xHA, or, as a
negative control, no HA-tagged protein (Strains 3-1s, 3-2s, and 1-1s, respectively).
After addition of galactose to induce the expression of His6-FLAG-SMT3 (HF-
SUMO), cells were lysed under denaturing conditions, and proteins conjugated to
HF-SUMO were affinity purified on nickel resin (see Fig. 3-2 and Materials and Methods Section 3.5.2 for protocol details). An anti-HA western blot of the Input
samples shows that Sir2-HA was well-expressed and runs at ~72 kDa (Fig. 3-3A,
Lanes 2-4). An anti-FLAG blot of the pull-down samples revealed global sumoylation
by HF-SUMO, and confirms the successful purification of HF-SUMO conjugated
proteins (Fig. 3-3A, Lanes 1’-4’, anti-FLAG). Western blot of these same samples
with anti-HA antibody revealed a single band at ~95 kDa in cells expressing both
Sir2p-HA and HF-SUMO, but not in cells expressing either one alone (Fig. 3-3A,
compare Lane 3’ with Lanes 1’ and 2’, anti-HA). The band is strong, specific, and
highly reproducible and we believe it represents mono-sumoylated Sir2p.

60
Fig. 3-3. Sir2p and Sir3p are sumoylated in vivo. (A) Standard purification of His6-FLAG-SUMO in HA-tagged strains. Cell lysate prepared in GuHCl was made from Strains 1-1s, 3-1s, and 3-2s (None, Sir2p-HA, Sir3p-HA, respectively) expressing either His6-FLAG-SUMO (+) or empty vector control (-). His6-FLAG-SUMO and any proteins to which it was covalently bound were purified on Ni2+-NTA resin under denaturing conditions. Bound proteins were washed, eluted by boiling, separated by SDS-PAGE, and detected by western blotting with anti-FLAG and anti-HA antibodies. Asterisk marks a sumoylated product (B) Same as (A) except with TAP-tagged strains Strains BY4741, 3-3s, and 3-4s (None, Sir2p-TAP, Cdc11p-TAP, respectively) and detected by western blotting with anti-Protein A antibody.

61
Sir2p-HA and HF-SUMO have predicted molecular weights (MWs) of 72 and
11 kDa, respectively. The Sir2p-HA band in the pull-down runs at approximately 95-
100 kDa, larger than the expected MW of 83 (72 + 11) kDa for Sir2p-HA plus a single
molecule of HF-SUMO. However, molecular mass determination via SDS-PAGE is
often inaccurate, and high levels of imidazole (200 mM) in the elution may affect
protein migration. Furthermore, there is evidence suggesting that the addition of Ubl
moieties can differentially affect protein migration. For example, SUMO conjugates
tend to run more slowly than would be expected based on their molecular weights,
and conjugation of SUMO to different lysines in the same protein may slow its
migration on SDS-PAGE to different extents (Ulrich et al. 2009). As such, we think
that the HA-Sir2 band of 95-100 kDa represents the monosumoylated from.
However, it is also formally possible that this band is a di-sumoylated form.
We also detected Sir3p-HA in the pool of HF-SUMO modified proteins pulled
down by Ni2+-NTA column (Fig. 3-3A, compare Lanes 4 and 4’, anti-HA). This result
is consistent with previously work showing that Sir3p is sumoylated (Denison et al.
2005; Johnson et al. 1999).
To test if the sumoylation of Sir2p-HA observed above was an artifact that
resulted from tagging Sir2p with HA, I performed essentially the same experiment,
but with cells expressing Sir2p C-terminally tagged with TAP. This TAP tag consists
of a calmodulin binding peptide, a TEV cleavage site, and two IgG binding domains
of Staphylococcus auerus Protein A (Ghaemmaghami et al. 2003). Strains
expressing TAP-tagged Cdc11 and untagged proteins were used as positive and
negative controls in this experiment, respectively. Cdc11p is a component of the
septin ring that is required for cytokinesis that has been shown to be sumoylated to a
significant level during mitosis (Johnson et al. 1999). In fact, the level of Cdc11p-
TAP sumoylation was so high that sumoylated Cdc11p-TAP was readily detectable
even in the Input sample (Fig. 3-3B, Lane 7). After purification, I detected many
strong higher molecular weight bands in the Cdc11p-TAP sample and a single higher
molecular weight band in the Sir2p-TAP sample (Fig. 3-3B, Lanes 10, 11, and 13).
No bands were detected in pull-down from cells lacking TAP-tagged protein or pHF-
SMT3 (Fig. 3-3B, Lanes 8 and 9). These results demonstrate that, similar to Sir2p-
HA, Sir2p-TAP is also subject to sumoylation in vivo.

62
3.3.2 Sir2p and Sir3p are ubiquitinated in vivo.
Sir2p interacts directly with Slx5p, a protein with characterized activity as a
SUMO-targeted ubiquitin ligase (Darst et al. 2008), so we next tested whether Sir2p
and Sir3p were ubiquitinated. This experiment was performed identically to the
sumoylation test, except with plasmids bearing His6-Myc-tagged ubiquitin (HM-Ub),
rather than HF-SUMO. Strains 1-1s, 3-1s, and 3-2s (untagged, Sir2p-HA, and Sir3p-
HA, respectively) were transformed with a plasmid encoding pCUP-His6-Myc-UB
(pHM-UB) or pCUP-UB (untagged UB). Pull-down of HM-Ub ubiquitin was efficient
(Fig. 3-4A, compare Lanes 1’, 2’ and 4’ with Lanes 3’ and 5’, anti-Myc), and western
blot analysis of the pull-down samples with anti-HA antibody revealed multiple higher
molecular weight bands for both Sir2p-HA and Sir3p-HA (Fig. 3-4A, compare Lanes
1’ and 4’ with 2’, 3’, and 5’, anti-HA). In fact, there was a ladder of bands, presumably
representing mono-, di-, and tri- ubiquitinated Sir2p-HA in the pull-down sample (Fig.
3-4A, Lane 1’, anti-HA). Sir2p-HA•mono-Ub is actually visible in the Input sample, as
a light band running just above the strong unmodified band (Fig. 3-4A, Lane 1). This
band is most likely ubiquitinated Sir2p, as it runs in approximately the same place as
the first band in the pull-down sample (Fig. 3-4A, compare Lane 1 with 1’), and
importantly, it is slightly larger than the faint, higher molecular weight band seen in
Lane 3 (Fig. 3-4A, compare Lanes 1 with 3). This sample is from Sir2p-HA
transformed with the vector control, which overexpresses untagged ubiquitin, a
slightly smaller moiety than the tagged version. Corresponding modified bands are
not present in the pull-down because there is no His6 tag on the Ub construct,
therefore no ubiquitinated bands are purified by Ni2+-NTA. These results indicate that
both Sir2p and Sir3p are subject to ubiquitination in vivo, and at least Sir2p can be
modified by multiple ubiquitin peptides.
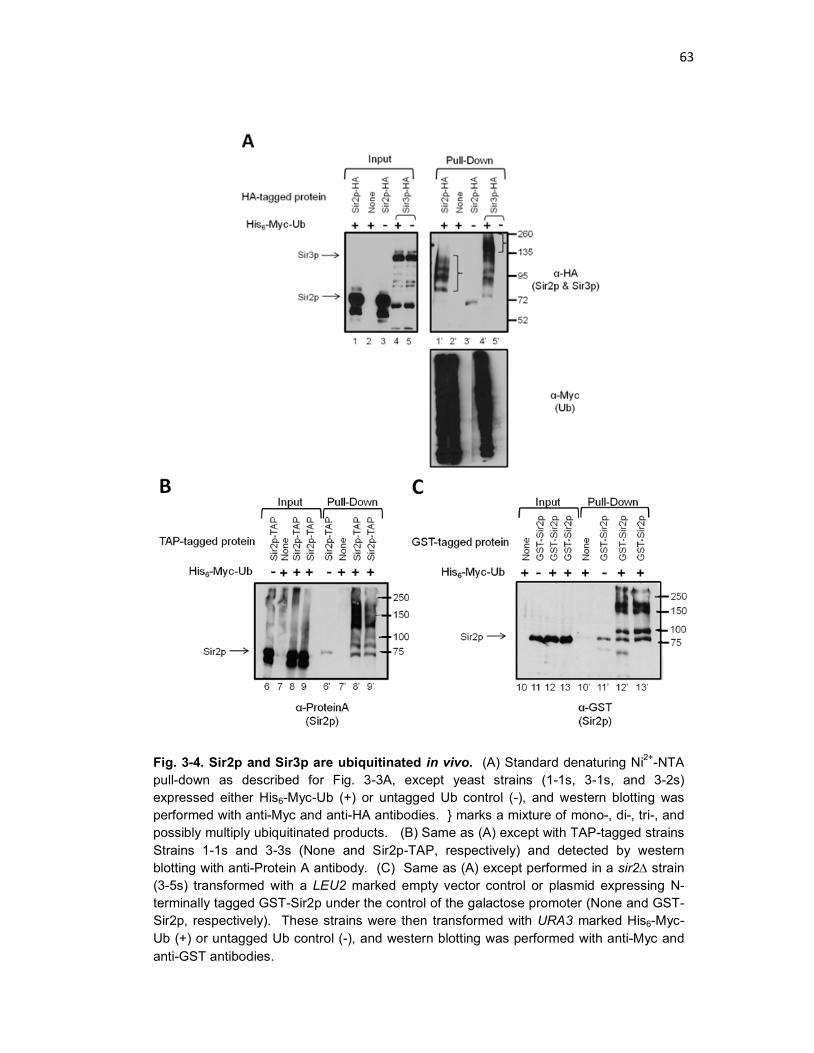
63
Fig. 3-4. Sir2p and Sir3p are ubiquitinated in vivo. (A) Standard denaturing Ni2+-NTA pull-down as described for Fig. 3-3A, except yeast strains (1-1s, 3-1s, and 3-2s) expressed either His6-Myc-Ub (+) or untagged Ub control (-), and western blotting was performed with anti-Myc and anti-HA antibodies. } marks a mixture of mono-, di-, tri-, and possibly multiply ubiquitinated products. (B) Same as (A) except with TAP-tagged strains Strains 1-1s and 3-3s (None and Sir2p-TAP, respectively) and detected by western blotting with anti-Protein A antibody. (C) Same as (A) except performed in a sir2∆ strain (3-5s) transformed with a LEU2 marked empty vector control or plasmid expressing N-terminally tagged GST-Sir2p under the control of the galactose promoter (None and GST-Sir2p, respectively). These strains were then transformed with URA3 marked His6-Myc-Ub (+) or untagged Ub control (-), and western blotting was performed with anti-Myc and anti-GST antibodies.

64
To test if the ubiquitination observed on Sir2p-HA was an artifact that resulted
from the HA tag, I repeated the above experiment in two other systems expressing
Sir2p-TAP and GST-Sir2p. I obtained evidence indicating that both Sir2p-TAP and
GST-Sir2p can be modified by multiple ubiquitin molecules (Fig. 3-4B, Lanes 8’ and
9’ and Fig. 3-4C, Lanes 12’ and 13’).
3.3.3 The effect of point and truncation mutations on Sir2p sumoylation.
Having demonstrated the ubiquitination and sumoylation of Sir2p, my next
goal was to understand the functional relevance of these modifications. This can be
achieved by assaying for the phenotype of a non-modifiable version of Sir2p. In
general, a non-modifiable protein can be generated by identifying the modified lysine
residue and changing it to an arginine (KR). Lysine and arginine share similar
properties, but arginine cannot be modified by Ubls. However, identifying the
modified residue(s) in a protein is often a challenging task. Sir2p, for example,
contains 49 lysines, each of which is a candidate for modification.
I started by using two programs to computationally predict the probability of
modification on each lysine residue by Ub or SUMO. The UbPred program predicts
five ubiquitination sites with high confidence. These are, in order of descending
probability, K28, K17, K33, K82, and K70. It also predicts additional ubiquitination
sites with medium (K106, K111, K548, K560) or low (K47, K50, and K414)
confidence. The SUMOplot program predicts, in order of descending probability,
K106, K439, K111, K33, and K523 as sumoylation sites in Sir2p (Fig. 3-5). While the
most probable sites for ubiquitination are clustered within the first 100 amino acids of
Sir2p, the sumoylation sites are spread throughout. Note that K33 appears on both
“high” probability lists. It is important to note that, although one lysine cannot be
simultaneously conjugated to both Ub and SUMO, there is precedent for a single
lysine to be targeted for multiple modifications, at different times, and with different
physiological effects (Hoege et al. 2002).

65
I set out to mutate the predicted lysine targets for ubiquitination and
sumoylation to arginines individually or in combinations, then examine their
modification phenotypes. To this end, I first generated plasmid pHK84, a HIS3
marked plasmid encoding SIR2 under its endogenous promoter and C-terminally
tagged with 6xHA. In all situations, I transformed pHK84 into a sir2∆ strain (Strain 3-
5s). The levels of Sir2p-HA expressed from the plasmid and genomic copies were
comparable, and pHK84-encoded Sir2p-HA is ubiquitinated and sumoylated (data
not shown). With the SIR2 gene on a plasmid, it was feasible to generate a
Fig. 3-5. Predicted sites of ubiquitination and sumoylation in Sir2p. The SIR2 sequence was analyzed by UbPred and SUMOplot to determine which lysines most closely fit the consensus sequence for ubiquitination and sumoylation, respectively. The five lysines with the greatest probability of ubiquitination appear as red asterisks on the schematic of Sir2p and in red text in the Sir2p sequence at the bottom. These are, in descending order of probability, K28, K17, K33, K82, and K70. The five lysines with the greatest probability of sumoylation are similarly marked, but in blue. These are, in descending order of probability, K106, K439, K111, K33, and K523. Note that K33 is a predicted target for both modifications. The schematic of Sir2p at the top denotes its three domains: the N-terminal domain (N, open box, aa 1-255), the conserved Catalytic domain (Cat, light grey box, aa 256-497), and the C-terminal domain (C, dark grey box, aa 498-562).

66
collection of individual and multiple KR point mutants (Table 3-1). I assayed these
constructs for sumoylation in Strain 3-5s bearing pHK84 and its derivatives.
Table 3-1. Sir2p point mutations screened for sumoylation and ubiquitination. Point mutation is noted in the left hand column, and color coded as a predicted sumoylation site (green) or a predicted ubiquitination site (purple). Note that K33 is predicted for both modifications. Y, indicates Sir2p with the point mutation is modified; N/D, Not Done.
Is the SIR2 mutant:
K --> R Mutation
Sumoylated? Ubiquitinated? Plasmid Name
K12,17R Y Y pLO227 K28,33R Y Y pLO225
K33R Y N/D pHK99 K47,48,50R Y Y pLO229
K70R Y Y pLO226 K82R Y Y pHK102 K106R Y Y pHK85 K111R Y Y pLO228 K414R Y N/D pLO232 K439R Y Y pHK86 K523R Y Y pLO230 K548 Y Y pLO231
Fig. 3-6B shows representative results of a sumoylation pull-down for 7
different KR mutation(s). From the Input panel on the left, it is clear that Sir2p-HA
is detectable in all samples except the “No Sir2p-HA” control (Lane 1). The Pull-
Down panel on the right shows the higher molecular weight Sir2p-HA•SUMO band at
~95 kDa in all samples expressing tagged Sir2p and HF-SUMO, but not in the empty
vector control (Fig. 3-6B, Lane 2’). There is a small amount of unmodified Sir2p-HA
in many of the pull-down samples; I observed this frequently in such experiments
(also see Fig. 3-3A and Fig. 3-4A, B, C). This is probably due to background pull
down of unmodified Sir2p-HA by Ni2+-NTA column. Regardless, because of the
significant shift in the modified version of Sir2p-HA, a small amount of background
binding at the “unmodified” size does not interfere with the detection of sumoylated
Sir2p-HA. .

67
All of the Sir2p mutant constructs shown in Fig. 3-6 appear to be sumoylated.
Although there are differences in Sir2p expression level (Fig. 3-6B, compare Lanes 5
and 6, for example), up-shifted sumoylated bands were visible in all pull-down
sample (Fig. 3-6B, Lanes 3’-10’). Thus, none of these mutations eliminates Sir2p
sumoylation, suggesting that the corresponding lysine is not the primary target of, or
not essential for, sumoylation. Moreover, a rough estimation of the levels of Input vs.
sumoylated Sir2p for each point mutant suggests that these mutations do not
significantly affect the level of sumoylation. Similar results were obtained for
additional Sir2p point mutations, as summarized in Table 3-1.
Fig. 3-6. Search for Sir2p sumoylation site(s) by KR mutagenesis. (A) The five highest probability sumoylation sites are noted in blue on the schematic of Sir2p. (B) Standard denaturing Ni2+-NTA purification to test sumoylation of SIR2 KR point mutants. Strain 3-5s (∆sir2) was transformed with one of the following HIS3 marked plasmids: pRS413, as a negative control (Lane 1); pHK84, which expresses Sir2p-HA under the control of its endogenous promoter (Lanes 2 and 3); or a pHK84 derivative bearing one or more KR point mutations (Lanes 4-10). Cells also expressed His6-FLAG-SUMO (+) or empty vector (-). Triangle at the upper right of each panel indicates the loading of 2-fold dilutions of the sample in Lane 10.

68
To complement the mutational analyses of the sumoylation site(s) of Sir2p, I
also examined the effects of N- and C-terminal truncations of Sir2p on its
sumoylation. Sir2p contains a particularly high concentration of lysine residues
(22%) in its C-terminal 40 amino acids. If the entire Sir2p sequence is considered,
lysine constitutes only 8.7% of all the amino acids (7.4% is average for yeast
proteins) (Akashi 2003). To determine whether one or more of these C-terminal
lysines was the target of modification by SUMO, I generated three small C-terminal
truncations on the genomic copy of Sir2p by inserting the C-terminal HA-tag 41, 26,
or 14 amino acids from the end of wild-type Sir2p, generating constructs ∆C-41, ∆C-
26, or ∆C-14 (Fig. 3-7A). These deletions removed 9, 6, and 1 of the C-terminal
lysines, respectively. However, of the three truncated versions of Sir2p-HA, only ∆C-
14 remained stably expressed (Fig. 3-7B, compare Lanes 2-4 with Lanes 5 and 6).
Analysis of the pull-down samples indicates that the ∆C-14 construct is still
sumoylated, suggesting that K560 is not required for Sir2p sumoylation (Fig. 3-7B,
Lane 4’). However, the lack of expression of the ∆C-26 and ∆C-41 truncations of
Sir2p-HA prevented me from examining the roles of the remaining C-terminal lysines
in Sir2p sumoylation.

69
Fig. 3-7. Search for Sir2p sumoylation site(s) with C-terminal truncations. (A) Schematic of the early termination sites for the three N-terminal Sir2p truncations assayed in this experiment. (B) Standard denaturing Ni2+-NTA purification to test sumoylation of Sir2p C-terminal truncations. Strains expressed Sir2p from its genomic locus with: no HA-tag (Strain 1-1s, Lane 1); HA-tagged, Full Length Sir2p (Strain 3-1s, Lanes 2 and 3); or one of three HA-tagged C-terminal SIR2 truncations (Strains 3-6s, 3-7s, and 3-8s encode ∆C-14, ∆C-26, ∆C-41, respectively) (Lanes 4, 5, and 6). Cells also expressed either His6-FLAG-SUMO (+) or empty vector (-).

70
I also tested the effects of N-terminal deletions of Sir2p on its sumoylation.
Three progressive deletions, ∆N-33, ∆N-84, and ∆N-121, were constructed from the
SIR2-6xHA on plasmid pHK84. The ∆N-33, ∆N-84, and ∆N-121 truncations
removed 5, 10, and 13 lysine residues, respectively (Materials and Methods, Section 3.5.1) (Fig. 3-8A). Note the ∆N-121 construct removed 3 of the 5 highly
probable sumoylation sites predicted by SUMOplot.
Fig. 3-8. Search for Sir2p sumoylation site(s) with N-terminal truncations. (A) Schematic of the start sites of the three N-terminal Sir2p truncations assayed in this experiment. (B) Standard denaturing Ni2+-NTA purification to test sumoylation of Sir2p N-terminal truncations. Strain 3-5s (sir2∆) was transformed with one of the following HIS3 marked plasmids: pRS413, as a negative control (Lane 3); pHK84, which expresses Sir2p-HA under the control of its endogenous promoter (Lanes 1, 2, and 4); or a pHK84 derivative with an N-terminal truncation (plasmids pLO243, pLO244, and pLO245 encode ∆N-30, ∆N-84, ∆N-121, respectively) (Lanes 5-7, 8-10, 11 and 12). Cells also expressed either His6-FLAG-SUMO (+) or empty vector (-). Triangle at the upper left of the Input panel indicates the loading of a 2-fold dilution of the sample in Lane 1.

71
The full length, ∆N-33, and ∆N-84 versions of Sir2p were well expressed and
ran according to their predicted molecular weight (Fig. 3-8B, Lanes 1 – 10). On the
other hand, the ∆N-121 construct was barely detectable (Fig. 3-8B, compare Lanes
4-10 with Lanes 11 and 12). Analysis of the Pull-Down samples reveals that the ∆N-
33 Sir2p construct is sumoylated (Fig. 3-8B compare Lanes 1’ and 2’ with Lanes 5’-
7’). The ∆N-84 construct is also sumoylated, and in contrast to full length Sir2p, it
appears to have two sumoylated variants, likely representing mono- and di-
sumoylated Sir2p-HA based on their apparent molecular weights (Fig. 3-8B,
compare Lanes 1’ and 2’ with Lanes 8’-10’). There are no bands visible in the Pull-
Down of the ∆N-121 construct, which is not unexpected, given that this truncation is
barely detectable in the Input. It is, therefore, impossible to draw any conclusion
about the modification state of the ∆N-121 Sir2p-HA construct.
Taken together, the above analyses of sumoylation of both N- and C-terminal
truncations of Sir2p demonstrate that lysines within the N-terminal 84 amino acids as
well as K560 are not essential for Sir2p sumoylation. The fact that the ∆C-28, ∆C-
41, and ∆N-121 truncations of Sir2p-HA are barely detectable makes it impossible to
examine the effects of larger N- or C-terminal deletions of Sir2p on its sumoylation.
3.3.4 The effect of point and truncation mutations on Sir2p sumoylation I assayed the Sir2p KR mutants listed in Table 3-1 for ubiquitination.
These included the lysines predicted to have a high probability of ubiquitination: K17,
K28, K33, K70, and K82 (Fig. 3-5). Representative results for 5 of these mutant
proteins are shown in Fig. 3-9B. Bands on the Input and Pull-Down gels are specific
to samples expressing Sir2p-HA, and there is no band detectable in the No Sir2p-HA
sample (Lanes 1 and 1’). However, just as in the sumoylation experiment described
in Section 3.3.3 above, all of the point mutants are ubiquitinated to detectable levels
(Fig. 3-9B, Pull-Down, lower panel). Furthermore, similar to wild type Sir2p-HA, all
Sir2p mutant proteins can be modified by multiple ubiquitin molecules (Fig. 3-9B,
compare Lane 2’ to Lanes 3’-7’). Similar results were obtained for the other point
mutants tested (results summarized in Table 3-1). Thus, just as in the sumoylation
assay, none of the KR point mutations of Sir2p eliminates its modification by
ubiquitin.

72
The five lysines of Sir2p with the highest probability of ubiquitination (K17,
K28, K33, K70 and K82) and two low confidence lysines (K47 and K50) all fall within
the first 82 amino acids of Sir2p (Fig. 3-9A) . However, changing any one of them to
arginine does not abolish Sir2p ubiquitination (Fig. 3-9B). It is possible that two or
more of these lysines can be ubiquitinated. To test this possibility, I examined the
effect of deleting the N-terminal 84 residues containing all 7 of these potential
ubiquitination targets on Sir2p ubiquitination. As shown in Fig. 3-10B, the ∆N-84
Fig. 3-9. Search for Sir2p ubiquitination site(s) by KR mutagenesis. (A) The five highest probability ubiquitination sites are noted in red on the schematic of Sir2p. (B) Standard denaturing Ni2+-NTA purification to test ubiquitination of SIR2 KR point mutants. Strain 3-5s (∆sir2) was transformed with one of the following HIS3 marked plasmids: pRS413, as a negative control (Lane 1); pHK84, which expresses Sir2p-HA under the control of its endogenous promoter (Lane 2); or a pHK84 derivative with a KR point mutation (Lanes 3-7). Cells also expressed His6-Myc-Ub (+).

73
allele of Sir2p is still ubiquitinated, suggesting that the predicted ubiquitination sites
K17, K28, K33, K47, K50, K70 and K82 are not the targets, or at least not the sole
targets, for ubiquitination (Lanes 8’-11’). As expected, Sir2p-∆N-33 also remains
ubiquitinated (Fig. 3-10B, Lanes 5’-7’).
In addition to the aforementioned five high confidence and two low
confidence ubiquitination sites in Sir2p, K106 and K111 are predicted to be medium
confidence sites. I tested whether Sir2∆N-121 lacking a total of 13 lysines, including
all the predicted ubiquitination sites, is still ubiquitinated. As shown in Fig. 3-10B,
although the abundance of Sir2∆N-121 is lower than that of intact Sir2p, it is still
ubiquitinated (Lanes 12’ and 13’). There are two possible explanations for this result.
First, it is possible that the physiological ubiquitination site(s) are not within the first
121 amino acids of Sir2p. Second, perhaps deletion of the regular ubiquitination
sites in the N-terminus exposes cryptic target(s) in the rest of the protein, possibly as
a result of structural changes in Sir2p caused by the truncation.

74
3.3.5 Generation and purification of Sir2p fused to a biotinylatable peptide.
Given that my effort to use mutational and deletion analyses to map Sir2p
ubiquitination and sumoylation sites has not been successful, I also took a mass-
spectrometry based alternative approach. This method is similar to the technique
used for large-scale identification of sumoylated or ubiquitinated proteins, except that
a single protein is purified, then subject to trypsin digest and analyzed by mass
spectrometry. The resultant peptides are analyzed for any with a higher-than-
expected mass, indicating conjugation to a trypsin-digested Ubl fragment
(Wohlschlegel 2009).
The greatest challenge with such an experiment is obtaining a sufficient
amount of modified protein that is pure enough for mass spectrometry. I modeled
Fig. 3-10. Search for Sir2p ubiquitination site(s) with N-terminal truncations. (A) Schematic of the start sites of the three N-terminal Sir2p truncations assayed in this experiment. (B) Standard denaturing Ni2+-NTA purification to test ubiquitination of Sir2p N-terminal truncations. Strain 3-5s (sir2∆) was transformed with one of the following HIS3 marked plasmids: pRS413, as a negative control (Lane 3); pHK84, which expresses Sir2p-HA under the control of its endogenous promoter (Lanes 1, 2, and 4); or a pHK84 derivative with an N-terminal truncation (plasmids pLO243, pLO244, and pLO245 encode ∆N-30, ∆N-84, ∆N-121, respectively) (Lanes 5-7, 8-10, 11 and 12). Cells also expressed either His6-Myc-Ub (+) or untagged-Ub (-).

75
my approach from a published protocol (Wohlschlegel 2009), which combines Ni2+-
NTA purification of the Ubl with a second purification step on Streptavidin-Agarose.
In this experiment, the protein of interest must be fused to a biotinylatable peptide
(abbreviated BIOpep) (Tagwerker et al. 2006). The biotin-streptavidin interaction is
both incredibly strong and resistant to denaturation, even in the presence of 8 M urea
or 6 M GuHCl (Tagwerker et al. 2006). Thus, this system provides the advantages of
purity conferred by a tandem purification scheme with the ability to perform two
purification steps under denaturing conditions in order to preserve as much modified
protein as possible.
To use this purification scheme, I made two constructs with the SIR2 gene
fused to the biotinylatable peptide. Strains 3-10s and 3-12s encode Sir2p-HA-
BIOpep and HA-Sir2p-BIOpep, respectively (Fig. 3-11A). HA-Sir2p-BIOpep was put
under the control of the pTEF promoter (see Materials and Methods, Section 3.5.1). Experiments to assess the relative levels of Sir2p in these two constructs
indicated that they are expressed within ~ 2-3 fold of each other.
I first confirmed that the two BIOpep-containing Sir2p constructs were still
sumoylated and ubiquitinated (Fig. 3-11B, compare Lanes 4’ and 5’ with Lane 3’).
Note that the C-terminal HA tag is slightly larger than the N-terminal HA tag, thus
explaining the slight differences in mobility between the two constructs (Fig. 3-11B,
compare Lanes 3 and 4 with Lane 5, and Lanes 3’ and 4’ with Lane 5’). Both
constructs are also ubiquitinated (Fig. 3-11C, Compare Lanes 9’ and 10’ with Lane
8’). Furthermore, the BIOpep-tagged constructs maintain the same ubiquitination
profile observed for Sir2p-HA. Finally, the BIOpep constructs are purified specifically
by Streptavidin-Agarose (Fig. 3-11C, compare Lanes 8”-10” with Lanes 6” and 7”),
indicating that BIOpep is biotinylated and that the purification works in GuHCl Buffer.
Therefore, tagging Sir2p with BIOpep does not affect its sumoylation and
ubiquitination, fulfilling a prerequisite for using the aforementioned tandem
purification scheme for modified Sir2p.

76
Fig. 3-11. Sumoylation and ubiquitination of Sir2p fused to a biotinylatable peptide (BIOpep). (A) Schematic of the constructs used in this experiment, representing Strains 1-1s, 3-1s, 3-10s, and 3-12s (top to bottom). All are expressed from the Sir2p chromosomal locus. (B) Standard denaturing Ni2+-NTA purification to test sumoylation of Sir2p constructs. Strains shown in (A) expressed either His6-FLAG-SUMO (+) or empty vector (-). Strains were prepared in -Ura media. (C) Standard denaturing Ni2+-NTA purification to test ubiquitination of Sir2p constructs (Lanes 1’-5’). Strains shown in (A) expressed either His6-Myc-Ub (+) or untagged-Ub (-). Lysates were also subject to purification on Streptavidin-Agarose (Lanes 1”-5”). Strains were prepared in -Ura media.

77
To ensure maximum purification of the BIOpep-fused constructs, I tested the
efficiency of Sir2p purification from both strains 3-10s and 3-12s with Streptavidin-
Agarose. Although I did obtain a small amount of purified Sir2p (as in Fig. 3-11C,
Lanes 8”-10”), I found that a significant amount remained in the unbound fraction
(data not shown). There were two possible explanations for this result. One is that
the conditions were not optimum for Streptavidin-Agarose to bind the biotinylated
peptide. The other is that the BIOpep construct itself was not fully biotinylated. It
has been shown that synthetic media (which I had been using to grow cells) may not
have sufficient biotin to support complete biotinylation of a BIOpep construct
(Tagwerker et al. 2006).
I tested the effect of growth media on the level of biotinylation of the BIOpep-
fused constructs. I used detection by Streptavidin conjugated to Horseradish
Peroxidase (Strep-HRP) to measure the relative levels of biotinylation of the two
Sir2p-BIOpep fusion constructs grown in synthetic media, rich media (YPD),
synthetic media supplemented with rich media for the final 4 hours of growth, and
synthetic media supplemented with increasing levels of Biotin (Fig. 3-12B). It was
obvious that minimal media did not support high levels of biotinylation of BIOpep
(Fig. 3-12B, compare Lanes 3 and 9 with all other Lanes). It also appeared that
increasing the level of Biotin increased the level of BIOpep biotinylation (Fig. 3-12B,
Lanes 10-12). Finally, growth in YPD and in -Ura supplemented with YPD for the
final 4 hours of growth resulted in the highest levels of biotinylation (Fig. 3-12B,
compare Lanes 1, 2, 7, and 8 with all other Lanes). In order to maintain plasmid
selection for as long as possible, I chose to grow cells in -Ura supplemented with 3x
YP (for a final concentration of 1x YPD) at the time of plasmid induction.

78
I next examined the efficiency of BIOpep biotinylation. While the
Streptavidin-HRP detection was ideal for the comparison of biotinylation levels, it did
not reveal the amount of un-biotinylated BIOpep in the sample. I performed a
Streptavidin-Agarose binding titration, using Strain 3-12s. The lysate sample was
divided into binding reactions with different amounts of Streptavidin-Agarose (25, 50,
75, and 100 µl slurry). Following binding, a fraction of the unbound sample was
reserved, and the resin was washed and the bound proteins eluted. I analyzed the
Fig. 3-12. High levels of biotinylation requires growth in rich media or minimal media supplemented with biotin. (A) Schematic of the constructs used in this experiment. All are expressed from the Sir2p chromosomal locus. (B) Overnight cultures of Strains 3-10s and 3-12s transformed with His6-Myc-Ub were used to inoculate fresh media: YPD (Lanes 1 and 7), -Ura (Lanes 2, 3, 8, and 9), or -Ura supplemented with biotin (Lanes 4-6, 10-12) to an OD600 ~ 0.1. Cells were grown to OD600 ~ 0.8 and Myc-Ub expression was induced with 0.5 mM CuSO4. Samples in Lanes 2 and 8 were also supplemented with 3x YP to a final overall concentration of 1x YPD. After 4 hrs, cells were harvested and lysates prepared. Proteins were separated by SDS-PAGE and analyzed by Coomassie stain or by transfer and detection with Streptavidin-HRP. Triangle at the upper left of the panel indicates the loading of a 2-fold dilution of the sample in Lane 1.

79
unbound fraction (Flow Through) and the bound fraction (Pull-Down) by western blot
(Fig. 3-13A). The blot is detected within the linear range, judging from the signals of
a two-fold serial dilution of the Pull-Down sample (Fig. 3-13A). Clearly, increasing
the amount of Streptavidin-Agarose slurry increased the amount of HA-Sir2p-BIOpep
purified (Fig. 3-12A, decreasing band intensity in Lanes 2 through 5, increasing band
intensity in Lanes 2’ through 5’), and binding appears near saturation with 100 µl of
resin slurry (Fig. 3-12A, compare Lane 4’ with 5’). Note that, although there are
twice the cell equivalents loaded in the Flow Through sample, binding is still efficient.
The completeness of the HA-Sir2p-BIOpep binding to Streptavidin-Agarose indicates
that most of the BIOpep is biotinylated, and furthermore, that the purification
efficiency is excellent.

80
Fig. 3-13. Optimizing purification of HA-Sir2p-BIOpep with Streptavidin-Agarose. (A) Titration of HA-Sir2p-BIOpep binding to Streptavidin-Agarose. ~1000 ODs of Strain 3-12s were grown in YPD and subject to denaturing lysis and resuspension in GuHCl. Lysate was divided into 4 aliquots and bound overnight to 25, 50, 75, or 100 µl of pre-washed Streptavidin-Agarose slurry. After binding, a sample of unbound protein for each binding reaction was precipitated and washed in ice-cold ethanol and resuspended by boiling in Laemmli. Bound proteins were subject to standard wash protocol, eluted by boiling, separated by SDS-PAGE, and detected by western blotting with anti-HA antibodies. Lanes 2-5 represent approximately 2 x more cell equivalents than Lanes 2’-5’. Triangle at the upper right of the panel indicates the loading of 2-fold dilutions of the sample in Lane 5’. (B) Effect of resin washing on Streptavidin-Agarose pull-down of HA-Sir2p-BIOpep. ~750 ODs of Strains 3-11s (-BIOpep) and 3-12s (+BIOpep) were grown in YPD and subject to denaturing lysis and resuspension in GuHCl. Lysates were divided into 2 aliquots and bound overnight to 100 µl Streptavidin-Agarose slurry that had been pre-washed either 3 x quick in 15 mL 1x PBS (1x PBS, standard washing condition in protocol) or 3 x quick in 15 mL 1x PBS followed by 2 x 30 min in 15 mL GuHCl Buffer (GuHCl Buffer). After binding, each sample was separated into 2 tubes. One set was subject to standard wash conditions (7 washes: 3 x GuHCl Buffer, 3 x GuHCl/TI Buffer, 1 x TI Buffer) and the other to increased wash conditions (11 washes: 5 x GuHCl Buffer, 5 x GuHCl/TI Buffer, 1 x TI buffer). All samples were eluted by boiling in 50 µl Laemmli buffer, and 16 µl (representing ~50 ODs) was run on SDS-PAGE gel and analyzed by silver staining.

81
Next I designed a set of experiments to optimize the number and stringency
of washing steps, in order to generate as clean a purification as possible. The goal
was to remove as many contaminants from the resin as possible before applying the
protein sample. This is necessary because the biotin-streptavidin interaction is so
strong that boiling in Laemmli buffer is needed to elute bound proteins from
Streptavidin-Agarose, and any contaminants on the resin would also be eluted under
this condition. I also tested different post-binding wash conditions.
As shown in Fig. 3-13B, the resin itself is very clean (Lanes 1, 4, 7, and 9).
There is a single band at ~95 kDa that appears in each +BIOpep (but not –BIOpep)
lane (Fig. 3-13B, compare Lane 2 with 3, 5 with 6, etc.). This is the HA-Sir2p-
BIOpep protein. There are two strong bands, one ~135 kDa, and one between 42
and 52 kDa, that appear in both the + and -BIOpep lanes. It is likely that these
bands correspond to endogenous biotinylated yeast proteins. Increased washing of
the bound resin reduced the contaminating bands in the eluted sample (Fig. 3-13B,
compare Lanes 2, 3, 5, and 6 with Lanes 8, 9, 10, and 11) without decreasing the
level of purified Sir2p (Fig. 3-13, compare Lane 3 with 9 and Lane 6 with 12). Pre-
washing the resin in GuHCl deos not seem to improve the purity of the eluted sample
(Fig. 3-13B, compare Lanes 2 and 3 with 5 and 6, and Lanes 8 and 9 with 11 and
12).
Having optimized the conditions for cell growth and purification by
Streptavidin, I was nearly ready to perform a double purification of modified HA-His6-
Sir2p-BIOpep. In this experiment, all Ubl-modified proteins are first pulled down by
Ni2+-NTA resin, and then subjected to Streptavidin-Agarose chromatography to purify
Ubl-modified Sir2p. The tandem purification method requires the elution of Ubl-
modified proteins bound to Ni2+-NTA resin with imidazole.
I tested whether imidazole at the concentrations required for elution from Ni2+-
NTA resin affected binding to Streptavidin-Agarose. I prepared cell lysate from
Strains 3-11s (HA-Sir2p) and 3-14s (HA-His6-Sir2p-BIOpep) in GuHCl buffer
(containing 10 mM imidazole), divided the lysate into aliquots, and supplemented
some samples with of 80 or 240 mM imidazole. I then applied the lysates to
Streptavidin-Agarose and eluted the samples. I analyzed these samples by silver
stain after SDS-PAGE and determined that additional imidazole did not affect the

82
binding of Sir2p-BIOpep to Streptavidin-Agarose (Fig. 3-14, Lanes 8-11). I also
tested the purification of the His6-tagged construct on two different metal-chelate
resins: the Ni2+-NTA (nickel) resin I had been using (Qiagen), and a cobalt resin
(TALON, Clontech) in the presence of 10, 25, and 50 mM imidazole. The same
amount of lysate was bound to the resin in these samples as was used in the Strep-
Agarose binding experiments. These His6-binding resins do not provide a very clean
purification – at 10 mM imidazole, there is a significant amount of background
binding, independent of Sir2p expression (Fig. 3-14, Lanes 3 and 5). Increasing the
concentration of imidazole decreases the background, but also causes a significant
loss in HA-His6-Sir2p-BIOpep binding (Fig. 3-14, Lanes 3-5, see Sir2p at arrow).
The TALON resin purification did not contain any Sir2p (Fig. 3-14, Lane 2). In
conclusion, this experiment confirmed that Streptavidin-Agarose will still bind the
BIOpep construct in the presence of high concentrations of imidazole, and that His6
tagged Sir2p can be purified with nickel resin, although only at a maximum imidazole
concentration of ~10 mM.

83
I transformed Strains 3-12s and 3-14s (HA-Sir2p-BIOpep and HA-His6-Sir2p-
BIOpep, respectively) with pHM-UB and prepared cells for double-purification using
the growth conditions described above (3x YP added at induction). After induction, I
was unable to detect the expression of Myc-Ub in these samples. A comparison of
induction in the presence and absence of YPD indicated that Myc-Ub expression
was not induced in the presence of YPD, but was induced in cultures grown without
the addition of YPD. Thus, YPD has an inhibitory effect on the induction of HM-Ub
from the pCUP-HM-UB plasmid, and future experiments will need to be performed
with biotin-supplemented media in place of YPD.
Fig. 3-14. Comparison of HA-His6-Sir2p-BIOpep binding to Ni2+-NTA resin and Streptavidin-Agarose in the presence of different concentrations of imidazole. Strains 3-11s (-BIOpep) and 3-14s (+BIOpep) were grown in YPD and subject to denaturing lysis and resuspension in GuHCl Buffer (with 10 mM Imidazole). Lysates were divided into aliquots representing 100 ODs each and imidazole was added to reach the noted concentration. Lysates were bound overnight to 30 µl of the resin indicated, pre-washed according to standard protocol. After binding, each sample was subject to standard wash conditions, eluted by boiling in 30 µl Laemmli buffer, and 15 µl (representing ~50 ODs) was run on SDS-PAGE gel and analyzed by silver staining. Asterisk marks HA-HIS6-Sir2p-BIOpep.

84
3.4 Discussion The projects described in this Chapter began while we were examining the
interaction between Esc2p and Sir2p. Deletion of ESC2 results in silencing defects
in yeast, and we hypothesized that Esc2p might play a role in the regulation of Sir2p
(Yu et al. 2009). As part of the studies described in Chapter 2, I identified a 20
amino acid sequence in Esc2p that is necessary and sufficient for interaction with
Sir2p. These 20 amino acids constitute a perfect consensus sequence for a SUMO-
binding motif (SBM). As this SBM in Esc2p binds both SUMO and Sir2p, we
considered the possibility that Sir2p was post-translationally modified by SUMO. I
demonstrated in this chapter that Sir2p is indeed subject to both sumoylation and
ubiquitination in vivo.
I demonstrated the modification of Sir2p by SUMO or ubiquitin by identifying
Sir2p in pool of affinity-purified sumoylated or ubiquitinated proteins. The abundance
of sumoylated or ubiquitinated Sir2p is low relative to unmodified Sir2p. It is possible
that sumoylation or ubiquitination only occurs on a small pool of special (e.g.,
misfiled or damaged) Sir2p molecules. Sir2p is most likely mono-sumoylated, and
multiply-ubiquitinated. Note that none of the previous global screens for sumoylated
or ubiquitinated proteins has identified Sir2p, probably due to a lack of sufficient
sensitivity of the approaches used (Wohlschlegel et al. 2004; Denison et al. 2005;
Zhou et al. 2004). To understand the physiological functions of Sir2p sumoylation and
ubiquitination, it is first necessary to identify the residues in Sir2p to which SUMO or
ubiquitin is attached. I undertook several approaches to map the sites of sumoylation
and ubiquitination within Sir2p.
One approach was to test whether KR mutations of candidate modification
sites affect Sir2p sumoylation or ubiquitination. The UbPred program predicts K28,
K17, K33, K82, and K70 as high probability ubiquitination sites in Sir2p. However, I
found changing these lysines to arginines, individually or in combinations of 2 or 3,
does not abolish Sir2p ubiquitination. Similar results were obtained for some of the
medium or low confidence sites of ubiquitination predicted by UbPred. The
SUMOplot program predicts K106, K439, K111, K33, and K523 as sumoylation sites
in Sir2p, but mutating these sites to arginines does not eliminate Sir2p sumoylation.

85
There are many published examples in which KR mutagenesis of a single
lysine within a modification consensus site results in a clear decrease or elimination
of modification (Ulrich et al. 2009; Zhu et al. 2011; Hang et al. 2011). Clearly,
identifying the modification sites on Sir2p has not proven to be so straightforward. It
is possible that the proper combination of KR mutations at sites with a high
probability of modification, or in close proximity to each other, would yield an
unmodified Sir2p construct. There have been examples where non-modifiable
proteins have been made by mutating all of the lysines in a particular region or
domain to arginine (Sekiguchi et al. 2011).
Given that many of the high probability ubiquitination sites are clustered at
the N-terminus of the Sir2p, we instead opted to test for modification of Sir2p
constructs with small N- or C- terminal truncations (Figs. 3-7, 3-8, and 3-10). This
approach would test the effect of removing many high probability modification sites at
one time, and without leaving adjacent lysines that could be used as secondary
modification sites. Through analysis of these constructs, I eliminated K560 and the
first 33 amino acids of Sir2p as sumoylation sites. For ubiquitination, I found that
Sir2p lacking amino acid residues 1-121 is still ubiquitinated at a comparable level
and with a similar profile as wild type Sir2p. Deletion of 121 amino acids from the N-
terminus of Sir2p removes 9 out of the 13 predicted ubiquitination sites (including the
5 high probability sites). Thus, either Sir2p is ubiquitinated at a low or non-
consensus site(s), or these truncations trigger aberrant ubiquitination, resulting from
either exposure of a modification site normally sequestered in the full-length protein,
or the targeting of a misfolded or unstable protein for proteasomal degradation by
ubiquitination.
Unfortunately, the steady state levels of the Sir2p C∆-28 and C∆-41
truncations were too low for analysis of sumoylation or ubiquitination, and the
steadys-state level of the N∆-121 truncation was too low for analysis of sumoylation.
It is likely that most of the Sir2p sequence is required for its stability so that the
steady state level of Sir2p drops dramatically when even modestly-sized pieces are
removed from either end. This may be the reason why there is a lack of structural
and/or functional analyses of Sir2p fragments in the literature, despite the
tremendous interest in Sir2p.

86
As identifying the modification site(s) for SUMO and ubiquitin on Sir2p by
mutational and deletion analyses proved challenging, I took an alternative approach
aimed at purifying modified Sir2p for analysis by mass spectrometry. I have
designed a tandem affinity purification approach for purifying Sir2p modified by
SUMO or ubiquitin, and have optimized the procedures involved (Section 3.3.4). In a
pilot experiment, I was able to purify a very clean fraction of ubiquitinated Sir2p from
120 ml of cell culture using this tandem purification approach. Since only a small
amount of cells were used in this experiment, the amount of modified Sir2p obtained
was insufficient for mass spec analysis. . However, the scalability of the protocol will
easily allow for purification from 10-100 times more cells, which would likely yield
enough modified Sir2p for mass spec analysis.
Modification of Sir2p by SUMO or ubiquitin may regulate its stability and/or
functions. Given that Sir2p interacts with a SUMO-binding motif of Esc2p, it is
tempting to speculate that sumoylation of Sir2p modulates this interaction. Although
I found that Sir2p sumoylation is not essential for Esc2p-Sir2p interaction in vitro, it is
still possible that sumoylation of Sir2p increases its avidity to Esc2p. This could be
particularly important in vivo, when Esc2p must compete with other proteins for
binding to Sir2p. It is also possible that Sir2 sumoylation affects its interaction with
Sir4p, thereby regulating the stability and/or function of the SIR complex. Sir2p
ubiquitination may serve to target damaged or misfolded forms to degradation by the
proteasome, or regulate its function independent of the degradation pathway.
Interestingly, Sir2p also interacts with Slx5p, a SUMO-targeted ubiquitin ligase that
mediate the crossover of the sumoylation and ubiquitination pathways (Darst et al.
2008; Prudden et al. 2007). Given the fact Sir2p can be both sumoylated and
ubiquitinated, it is possible that sumoylation of Sir2p marks it as a target for
ubiquitination by the STUbL Slx5p. This hypothesis predicts that Sir2p ubiquitination
is dependent on its ability to be sumoylated, which can be easily tested once we
identify the sumoylation site(s) of Sir2p.

87
3.5 Materials and Methods 3.5.1 Plasmids and Strains
The plasmid encoding pGAL-His6-FLAG-SMT3 (pHF-SMT3) with a LEU2
marker was a gift from E. Johnson. Plasmids encoding pCUP-His6-Myc-UB
(pUb221), pCUP-Myc-UB, (pUB210), and pCUP-UB (pUb175) were a gift from D.
Finley. Plasmid pFA6a-BIO-kan was a gift from P. Kaiser, and is also available from
Addgene (Tagwerker et al. 2006).
Plasmid pGLC16 (Cuperus et al. 2000) encodes SIR2 flanked by
endogenous sequences on a LEU2-marked plasmid. Plasmid pHK82 was made by
transforming Strain 3-5s carrying pGC16 with a PCR-produced fragment encoding
6xHA linked to NatMX, amplified from pYM17 (Janke et al. 2004) using primers
encoding 40 base pairs of homology to the 3’ end (5’ primer) and to the sequence
immediately after the STOP codon (3’ primer) of SIR2. Plasmid pHK84 was
generated by inserting the ApaI-5’flank-SIR2-6xHA-XcmI fragment from pHK82 into
ApaI/XmaI digested pRS413. HA-tagged SIR2 point mutants (Table 3-1) were made
using Quik Change mutagenesis to change a lysine codon (AAA or AAG) to the AGA
arginine codon.
To generate plasmid pLO242, pHK84 was digested with XhoI, which cuts
upstream of the SIR2 insertion, and BsrGI, which cuts after nucleotide 693 of SIR2.
Then, a XhoI/BsrGI flanked PCR fragment encoding all of the DNA upstream of the
SIR2 start in the vector was re-inserted. The resulting pLO242 plasmid contains the
same sequences upstream of SIR2 as in pHK84, followed directly by the BsrGI site
at SIR2 nucleotide 694. To generate the N-terminal truncations of Sir2p-HA, PCR
products encoding SIR2 nts 100-692, nts 253-692, or nts 364-692 were amplified by
primers encoding a 5’ ATG and flanked by BsrGI sites. These fragments were each
inserted into BsrGI-digested pLO242 to generate pLO243 (∆N-33), pLO244 (∆N-84),
and pLO245(∆N-120).
Plasmid pHK97 was generated by inserting the SalI-GST-SIR2-XhoI fragment
from pHK49 (for description, see Chapter 2, Materials and Methods, Section 2.5.1)
into SalI/XhoI digested pYEp55-A (Rose et al. 1990).The resulting plasmid
expresses GST-Sir2p under the control of a galactose promoter from a plasmid with
a LEU2 marker.

88
Strains 3-1s and 3-2s were generated by transforming BY4741 with a PCR-
produced fragment encoding 6xHA linked to KanMX, amplified from pYM14 using
primers encoding 40 base pairs of homology to the 3’ end (5’ primer) and to the
sequence immediately after the STOP codon (3’ primer) of SIR2 (Strain 3-1s) and
SIR3 (Strain 3-2s). Strains 3-6s, 3-7s, and 3-8s contain HA-tagged C-terminal Sir2p
truncations and were generated by transforming in a fragment encoding the C-
terminal HA tag at the desired site of truncation in Strain 1-1s. Specifically, BY4741
(Strain 1-1s) was transformed with a PCR-produced fragment encoding 6xHA linked
to NatMX, amplified from pYM17 using primers encoding 40 base pairs of homology
upstream of the truncation (5’ primer) and to the sequence immediately after the
STOP codon (3’ primer) of SIR2. Strain 3-9s was generated from the same plasmid
template, but with the primers used for Strain 3-1s to yield 6xHA tagged SIR2,
marked with that NatMX cassette. Strain 3-11s was made by transforming BY4741
with a PCR-produced fragment encoding the NatMX cassette upstream of the TEF
promoter (pTEF), a start codon, and a 6xHA tag, amplified from pYM-N20 using
primers encoding 40 base pairs of homology to the sequence immediately upstream
of the SIR2 START (5’ primer) and to the first 40 nucleotides of SIR2 (3’ primer).
Strains 3-10s, 3-12s, and 3-13s were made by transformation of Strains 3-9s, 3-11s,
and 1-1s, respectively, with a PCR-produced fragment encoding a biotinylatable
peptide (BIOpep) linked to KanMX, amplified from pFA6a-BIO-kan using primers
encoding 40 base pairs of homology to the 3’ end of the HA-tag (Strain 3-10s) or to
the 3’ end of SIR2 (Strains 3-12s and 3-13s) (5’ primer) and to the sequence
immediately after the STOP codon of SIR2 (3’ primer). Strain 3-14s was made by
transforming Strain 3-13s with a PCR-produced fragment encoding the NatMX
cassette in front of the TEF promoter (pTEF), a start codon, a 3xHA tag, and a His6
tag amplified from pYM-N20 using primers encoding 25 base pairs of homology to
the sequence immediately upstream of the SIR2 START and the His6 tag (5’ primer)
and to the first 40 nucleotides of SIR2 (3’ primer)
All pYM plasmids used as PCR templates are part of the Euroscarf PCR
Toolbox collection (Janke et al. 2004).

89
3.5.2 Cell growth and denaturing Ni2+-NTA purification of sumoylated and ubiquitinated proteins in vivo.
For ubiquitin pull-down experiments, 150 ml of yeast cells were grown
overnight in -Ura liquid containing 2% glucose to an OD600 of ~0.8. Expression of
pCUP-His6-Myc-UB or pCUP-UB was induced by the addition of CuSO4 to a final
concentration of 500 µM. For SUMO pull-down experiments, 150 ml of yeast
cells were grown overnight in -Leu liquid containing 2% raffinose (from a starter
grown in -Leu with 2% raffinose) to an OD600 of ~0.8. Expression of pHis6-FLAG-
SMT3 was induced by the addition of 1.5 g galactose powder. In both experiments,
plasmid expression was induced for 4 hrs, then cells were harvested (reserve one
1.5 ml fraction separately for Cell Pellet Input), washed in 50 mL cold water, and
spun down. Pellet was stored at -80°C.
To prepare lysates for pull-down, frozen pellets were thawed on ice and
resuspended in 2 ml cold water. 4 ml of cold, freshly prepared BME/NaOH solution
(7.4% β-mercaptoethanol, 1.85 M NaOH) was added to each sample. Samples were
vortexed well and incubated on ice for 30 min. 4 mL cold 50% TCA solution was
added, samples were vortexed, and incubated on ice for 30 min. Lysates were spun
for 8 min at 12,000 rpm to pellet precipitated protein. The protein pellet was washed
twice in 10 ml cold acetone. In order to remove as much TCA as possible, pellets
should be crushed, shaken, and vortexed until resuspended as very small pieces
after each addition of acetone washes after addition of acetone both times. After
removal of the final acetone wash, protein pellets were allowed to dry for 2-3 min at
room temperature, then resuspended in GuHCl Buffer (6 M GuHCl, 100 mM Sodium
Phosphate, 10 mM imidazole; pH = 8.0) by crushing, vortexing, and finally shaking
for 1 hr at 30°C. Undissolved material was spun out by centrifuging for 8 min at
12,000 rpm. The resulting supernatant was adjusted to pH 8.0 (with the addition of
~15 µl of 10 M NaOH) and used in subsequent experiments. When necessary,
protein concentration was determined by Bradford Assay. To reserve a Cell Lysate
Input sample, 20 µl lysate was mixed with 60 µl water and 20 µl Laemmli buffer and
boiled for 5 min. This sample was reboiled prior to each loading on SDS-PAGE.
Each pull-down required 75 µl of Ni2+-NTA resin slurry (Qiagen). Up to 3 ml
of resin was prepared together by pre-washing three times in 15 ml 1x PBS: 1x PBS

90
was added, sample was inverted 2-3 times to mix, resin was spun down for 1 min at
1,000 rpm at room temperature, and washing buffer removed, x3. Resin was
resuspended in GuHCl Buffer and added to cell lysates. Binding was performed at
room temperature for at least 4 hrs, but often as long as overnight (~16 hrs). To
save a post-binding Flow Through sample, resin was spun down, and 20 µl lysate
was removed and prepared as described above for Cell Lysate Input. Standard
Conditions for resin washing were: 3 times with GuHCl Buffer, 3 times with GuHCl/TI
Buffer (1 volume GuHCl Buffer:3 volumes TI Buffer), and 1 time with TI Buffer (25
mM Tris, pH 6.8, 20 mM imidazole). Washes were performed as follows: resin was
resuspended in 1 mL buffer, rotated 4 min at room temperature, centrifuged for 1 min
at 1,000 rpm (preferably in swinging bucket rotor), and wash solution was removed,
x7. After the final wash, as much liquid as possible was removed using a syringe
needle, and resin was resuspended in 75 µl Laemmli buffer (with 200 mM imidazole),
and boiled for 5 min. To separate eluate from resin, samples were spun through
glass wool for 10 seconds at low speed. For detection on the same western blot,
approximately 40x more cell equivalents are were loaded for Pull-Down samples, as
compared with Input samples. For example, Input samples were either Cell Lysate
Input (described above) or lysate prepared directly from cells via the method
described in Materials and Methods, Section 3.5.3. In either case, SDS-PAGE
gels were loaded with ~5 µl (out of 100 µl total) Input sample and ~15 µl (out of 75
µl) Pull-Down sample. Proteins were separated by SDS-PAGE and detected by
immunoblotting with anti-HA (Sigma), anti-Myc (Roche), anti-Protein A (Sigma), anti-
GST (Sigma) or anti-FLAG (Sigma) antibody. Detection of biotinylated proteins was
performed using HRP-conjugated Streptavidin (Thermo Scientific). Note that
membranes probed with Streptavidin-HRP were not exposed to milk; instead 5%
BSA in 1x PBS or TBS + 0.05% Tween-20 was used for blocking and antibody
inucbations.
3.5.3 Preparation of protein lysates
Small amounts of cell lysate were prepared for western blot using the fast,
denaturing protocol described elsewhere (von der Haar 2007). Briefly, ~4-5 ODs of
cells were spun down, washed with cold water, resuspended in 100 µl VDH Lysis

91
Buffer (100 mM NaOH, 50 mM EDTA, 2% SDS, 2% BME) and heated at 95°C for 10
min. Samples were neutralized by addition of 2.5 µl of 4 M acetic acid, vortexed for
30 sec, and heated at 95°C for 10 min. 25 µl VDH Loading Dye (250 mM Tris-HCl
pH 6.8, 50% glycerol, 0.05% bromphenol blue) was added to each sample. These
lysates are suitable for analysis by Coomassie stain, western blot, or silver staining.
3.5.4 Denaturing purification of biotinylated Sir2p-BIOpep
Cell growth, lysis, and Streptavidin-agarose purification of Sir2p fused to a
biotinylatable peptide (BIOpep) was performed as described in Section 3.5.2 with
the following modifications: Cells were grown in selective media to an OD600 of ~0.8,
then induced with CuSO4 (if necessary) and supplemented with 3x YP for a final
concentration of 1x YPD for 4 hrs. Cells were harvested, lysed with BME/NaOH, and
proteins were TCA-precipitated and resuspended in GuHCl Buffer (no imidazole,
unless otherwise noted). For 50 mL cell growth, 50 µl of Streptavidin-Agarose resin
(Thermo Scientific) was pre-washed, mixed with soluble lysate fraction, washed, and
eluted in 50 µl Laemmli buffer as described above, unless otherwise noted. Use
more or less culture volume as necessary, but maintain ratio of culture volume:resin
slurry. For analysis, load ~2 µl for detection by anti-HA western blot, or ~10-15 µl for
detection by silver stain.
3.5.5 Silver Staining
Silver staining was performed on standard SDS-PAGE gels cast on
scrupulously clean glass plates (detergent wash, thorough rinse, 8M nitric acid soak,
thorough rinse) and with ultra-pure reagents. Gels were minimally manipulated,
using only glass rods or gel releasers. This protocol was obtained from the
University of Rochester Proteomics Center, and it produces silver-stained bands
compatible with subsequent analysis by mass spectrometry. All solutions were
prepared fresh, on the day of use, in MilliQ water. Briefly, after electrophoresis, gels
were incubated (always with shaking) for 1 hr in Fixing Solution (40% ethanol/10%
acetic acid), followed by incubation for 30 min in Sensitizing Solution (30%
ethanol/0.2% sodium thiosulphate/6.8% sodium acetate). Gels were washed 3
times, 5 min each with enough ddH2O to be covered. Gels were shaken for 20 min

92
in Silver Nitrate Solution (0.25% silver nitrate in water) and washed 2 times, 1 min
each with ddH2O. Developer Solution (2.5% sodium carbonate, 0.015%
formaldehyde) was added and gels were shaken until bands reached the desired
intensity. Developer Solution can be changed if further development is necessary.

93
References
Akashi, H. 2003. Translational selection and yeast proteome evolution. Genetics 164 (4) (Aug): 1291-303.
Andrulis, E. D., D. C. Zappulla, K. Alexieva-Botcheva, C. Evangelista, and R. Sternglanz. 2004. One-hybrid screens at the saccharomyces cerevisiae HMR locus identify novel transcriptional silencing factors. Genetics 166 (1) (Jan): 631-5.
Avner, P., and E. Heard. 2001. X-chromosome inactivation: Counting, choice and initiation. Nature Reviews.Genetics 2 (1) (Jan): 59-67.
Bayer, P., A. Arndt, S. Metzger, R. Mahajan, F. Melchior, R. Jaenicke, and J. Becker. 1998. Structure determination of the small ubiquitin-related modifier SUMO-1. Journal of Molecular Biology 280 (2) (Jul 10): 275-86.
Bergink, S., and S. Jentsch. 2009. Principles of ubiquitin and SUMO modifications in DNA repair. Nature 458 (7237) (Mar 26): 461-7.
Bernier-Villamor, V., D. A. Sampson, M. J. Matunis, and C. D. Lima. 2002. Structural basis for E2-mediated SUMO conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP1. Cell 108 (3) (Feb 8): 345-56.
Bi, X., and J. R. Broach. 1997. DNA in transcriptionally silent chromatin assumes a distinct topology that is sensitive to cell cycle progression. Molecular and Cellular Biology 17 (12) (Dec): 7077-87.
Bi, X., Q. Yu, J. J. Sandmeier, and Y. Zou. 2004. Formation of boundaries of transcriptionally silent chromatin by nucleosome-excluding structures. Molecular and Cellular Biology 24 (5) (Mar): 2118-31.
Brand, A. H., L. Breeden, J. Abraham, R. Sternglanz, and K. Nasmyth. 1985. Characterization of a "silencer" in yeast: A DNA sequence with properties opposite to those of a transcriptional enhancer. Cell 41 (1) (May): 41-8.
Braunstein, M., A. B. Rose, S. G. Holmes, C. D. Allis, and J. R. Broach. 1993. Transcriptional silencing in yeast is associated with reduced nucleosome acetylation. Genes & Development 7 (4) (Apr): 592-604.
Cheng, T. H., Y. C. Li, and M. R. Gartenberg. 1998. Persistence of an alternate chromatin structure at silenced loci in the absence of silencers. Proceedings of the National Academy of Sciences of the United States of America 95 (10) (May 12): 5521-6.

94
Chien, C. T., S. Buck, R. Sternglanz, and D. Shore. 1993. Targeting of SIR1 protein establishes transcriptional silencing at HM loci and telomeres in yeast. Cell 75 (3) (Nov 5): 531-41.
Cockell, M. M., S. Perrod, and S. M. Gasser. 2000. Analysis of Sir2p domains required for rDNA and telomeric silencing in saccharomyces cerevisiae. Genetics 154 (3) (Mar): 1069-83.
Collins, S. R., K. M. Miller, N. L. Maas, A. Roguev, J. Fillingham, C. S. Chu, M. Schuldiner, et al. 2007. Functional dissection of protein complexes involved in yeast chromosome biology using a genetic interaction map. Nature 446 (7137) (Apr 12): 806-10.
Cuperus, G., R. Shafaatian, and D. Shore. 2000. Locus specificity determinants in the multifunctional yeast silencing protein Sir2. The EMBO Journal 19 (11) (Jun 1): 2641-51.
Cuperus, G., and D. Shore. 2002. Restoration of silencing in saccharomyces cerevisiae by tethering of a novel Sir2-interacting protein, Esc8. Genetics 162 (2) (Oct): 633-45.
Darst, R. P., S. N. Garcia, M. R. Koch, and L. Pillus. 2008. Slx5 promotes transcriptional silencing and is required for robust growth in the absence of Sir2. Molecular and Cellular Biology 28 (4) (Feb): 1361-72.
Denison, C., A. D. Rudner, S. A. Gerber, C. E. Bakalarski, D. Moazed, and S. P. Gygi. 2005. A proteomic strategy for gaining insights into protein sumoylation in yeast. Molecular & Cellular Proteomics : MCP 4 (3) (Mar): 246-54.
Dhillon, N., and R. T. Kamakaka. 2000. A histone variant, Htz1p, and a Sir1p-like protein, Esc2p, mediate silencing at HMR. Molecular Cell 6 (4) (Oct): 769-80.
Downs, J. A., E. Kosmidou, A. Morgan, and S. P. Jackson. 2003. Suppression of homologous recombination by the saccharomyces cerevisiae linker histone. Molecular Cell 11 (6) (Jun): 1685-92.
Escher, D., and W. Schaffner. 1997. Gene activation at a distance and telomeric silencing are not affected by yeast histone H1. Molecular & General Genetics : MGG 256 (4) (Oct): 456-61.
Fan, Y., T. Nikitina, E. M. Morin-Kensicki, J. Zhao, T. R. Magnuson, C. L. Woodcock, and A. I. Skoultchi. 2003. H1 linker histones are essential for mouse development and affect nucleosome spacing in vivo. Molecular and Cellular Biology 23 (13) (Jul): 4559-72.
Fan, Y., T. Nikitina, J. Zhao, T. J. Fleury, R. Bhattacharyya, E. E. Bouhassira, A. Stein, C. L. Woodcock, and A. I. Skoultchi. 2005. Histone H1 depletion in

95
mammals alters global chromatin structure but causes specific changes in gene regulation. Cell 123 (7) (Dec 29): 1199-212.
Freidkin, I., and D. J. Katcoff. 2001. Specific distribution of the saccharomyces cerevisiae linker histone homolog HHO1p in the chromatin. Nucleic Acids Research 29 (19) (Oct 1): 4043-51.
Frye, R. A. 2000. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochemical and Biophysical Research Communications 273 (2) (Jul 5): 793-8.
Gasser, S. M., and M. M. Cockell. 2001. The molecular biology of the SIR proteins. Gene 279 (1) (Nov 14): 1-16.
Gelperin, D. M., M. A. White, M. L. Wilkinson, Y. Kon, L. A. Kung, K. J. Wise, N. Lopez-Hoyo, et al. 2005. Biochemical and genetic analysis of the yeast proteome with a movable ORF collection. Genes & Development 19 (23) (Dec 1): 2816-26.
Ghaemmaghami, S., W. K. Huh, K. Bower, R. W. Howson, A. Belle, N. Dephoure, E. K. O'Shea, and J. S. Weissman. 2003. Global analysis of protein expression in yeast. Nature 425 (6959) (Oct 16): 737-41.
Giaever, G., A. M. Chu, L. Ni, C. Connelly, L. Riles, S. Veronneau, S. Dow, et al. 2002. Functional profiling of the saccharomyces cerevisiae genome. Nature 418 (6896) (Jul 25): 387-91.
Gottschling, D. E. 1992. Telomere-proximal DNA in saccharomyces cerevisiae is refractory to methyltransferase activity in vivo. Proceedings of the National Academy of Sciences of the United States of America 89 (9) (May 1): 4062-5.
Gottschling, D. E., O. M. Aparicio, B. L. Billington, and V. A. Zakian. 1990. Position effect at S. cerevisiae telomeres: Reversible repression of pol II transcription. Cell 63 (4) (Nov 16): 751-62.
Grewal, S. I., and D. Moazed. 2003. Heterochromatin and epigenetic control of gene expression. Science (New York, N.Y.) 301 (5634) (Aug 8): 798-802.
Hang, L. E., X. Liu, I. Cheung, Y. Yang, and X. Zhao. 2011. SUMOylation regulates telomere length homeostasis by targeting Cdc13. Nature Structural & Molecular Biology 18 (8) (Jul 10): 920-6.
Hannon, R., E. Bateman, J. Allan, N. Harborne, and H. Gould. 1984. Control of RNA polymerase binding to chromatin by variations in linker histone composition. Journal of Molecular Biology 180 (1) (Nov 25): 131-49.

96
Hecker, C. M., M. Rabiller, K. Haglund, P. Bayer, and I. Dikic. 2006. Specification of SUMO1- and SUMO2-interacting motifs. The Journal of Biological Chemistry 281 (23) (Jun 9): 16117-27.
Hellauer, K., E. Sirard, and B. Turcotte. 2001. Decreased expression of specific genes in yeast cells lacking histone H1. The Journal of Biological Chemistry 276 (17) (Apr 27): 13587-92.
Hicke, L. 2001. Protein regulation by monoubiquitin. Nature Reviews.Molecular Cell Biology 2 (3) (Mar): 195-201.
Hoege, C., B. Pfander, G. L. Moldovan, G. Pyrowolakis, and S. Jentsch. 2002. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419 (6903) (Sep 12): 135-41.
Hoppe, G. J., J. C. Tanny, A. D. Rudner, S. A. Gerber, S. Danaie, S. P. Gygi, and D. Moazed. 2002. Steps in assembly of silent chromatin in yeast: Sir3-independent binding of a Sir2/Sir4 complex to silencers and role for Sir2-dependent deacetylation. Molecular and Cellular Biology 22 (12) (Jun): 4167-80.
James, P., J. Halladay, and E. A. Craig. 1996. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 144 (4) (Dec): 1425-36.
Janke, C., M. M. Magiera, N. Rathfelder, C. Taxis, S. Reber, H. Maekawa, A. Moreno-Borchart, et al. 2004. A versatile toolbox for PCR-based tagging of yeast genes: New fluorescent proteins, more markers and promoter substitution cassettes. Yeast (Chichester, England) 21 (11) (Aug): 947-62.
Johnson, E. S. 2004. Protein modification by SUMO. Annual Review of Biochemistry 73 : 355-82.
Johnson, E. S., and G. Blobel. 1999. Cell cycle-regulated attachment of the ubiquitin-related protein SUMO to the yeast septins. The Journal of Cell Biology 147 (5) (Nov 29): 981-94.
Johnson, E. S., I. Schwienhorst, R. J. Dohmen, and G. Blobel. 1997. The ubiquitin-like protein Smt3p is activated for conjugation to other proteins by an Aos1p/Uba2p heterodimer. The EMBO Journal 16 (18) (Sep 15): 5509-19.
Kasinsky, H. E., J. D. Lewis, J. B. Dacks, and J. Ausio. 2001. Origin of H1 linker histones. The FASEB Journal : Official Publication of the Federation of American Societies for Experimental Biology 15 (1) (Jan): 34-42.
Katzmann, D. J., G. Odorizzi, and S. D. Emr. 2002. Receptor downregulation and multivesicular-body sorting. Nature Reviews.Molecular Cell Biology 3 (12) (Dec): 893-905.

97
Kerscher, O., R. Felberbaum, and M. Hochstrasser. 2006. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annual Review of Cell and Developmental Biology 22 : 159-80.
Krogan, N. J., G. Cagney, H. Yu, G. Zhong, X. Guo, A. Ignatchenko, J. Li, et al. 2006. Global landscape of protein complexes in the yeast saccharomyces cerevisiae. Nature 440 (7084) (Mar 30): 637-43.
Landsman, D. 1996. Histone H1 in saccharomyces cerevisiae: A double mystery solved? Trends in Biochemical Sciences 21 (8) (Aug): 287-8.
Laybourn, P. J., and J. T. Kadonaga. 1991. Role of nucleosomal cores and histone H1 in regulation of transcription by RNA polymerase II. Science (New York, N.Y.) 254 (5029) (Oct 11): 238-45.
Li, C., J. E. Mueller, M. Elfline, and M. Bryk. 2008. Linker histone H1 represses recombination at the ribosomal DNA locus in the budding yeast saccharomyces cerevisiae. Molecular Microbiology 67 (4) (Feb): 906-19.
Liachko, I., and B. K. Tye. 2009. Mcm10 mediates the interaction between DNA replication and silencing machineries. Genetics 181 (2) (Feb): 379-91.
Lim, M. K., V. Tang, A. Le Saux, J. Schuller, C. Bongards, and N. Lehming. 2007. Gal11p dosage-compensates transcriptional activator deletions via Taf14p. Journal of Molecular Biology 374 (1) (Nov 16): 9-23.
Liou, G. G., J. C. Tanny, R. G. Kruger, T. Walz, and D. Moazed. 2005. Assembly of the SIR complex and its regulation by O-acetyl-ADP-ribose, a product of NAD-dependent histone deacetylation. Cell 121 (4) (May 20): 515-27.
Luger, K., A. W. Mader, R. K. Richmond, D. F. Sargent, and T. J. Richmond. 1997. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389 (6648) (Sep 18): 251-60.
Luo, K., M. A. Vega-Palas, and M. Grunstein. 2002. Rap1-Sir4 binding independent of other sir, yKu, or histone interactions initiates the assembly of telomeric heterochromatin in yeast. Genes & Development 16 (12) (Jun 15): 1528-39.
Mahoney, D. J., and J. R. Broach. 1989. The HML mating-type cassette of saccharomyces cerevisiae is regulated by two separate but functionally equivalent silencers. Molecular and Cellular Biology 9 (11) (Nov): 4621-30.
Mankouri, H. W., H. P. Ngo, and I. D. Hickson. 2009. Esc2 and Sgs1 act in functionally distinct branches of the homologous recombination repair pathway in saccharomyces cerevisiae. Molecular Biology of the Cell 20 (6) (Mar): 1683-94.

98
Miller, A. M., and K. A. Nasmyth. 1984. Role of DNA replication in the repression of silent mating type loci in yeast. Nature 312 (5991) (Nov 15-21): 247-51.
Moazed, D. 2001. Common themes in mechanisms of gene silencing. Molecular Cell 8 (3) (Sep): 489-98.
Novatchkova, M., A. Bachmair, B. Eisenhaber, and F. Eisenhaber. 2005. Proteins with two SUMO-like domains in chromatin-associated complexes: The RENi (Rad60-Esc2-NIP45) family. BMC Bioinformatics 6 (Feb 7): 22.
Ohya, T., H. Arai, Y. Kubota, H. Shinagawa, and T. Hishida. 2008. A SUMO-like domain protein, Esc2, is required for genome integrity and sister chromatid cohesion in saccharomyces cerevisiae. Genetics 180 (1) (Sep): 41-50.
Ozkaynak, E., D. Finley, M. J. Solomon, and A. Varshavsky. 1987. The yeast ubiquitin genes: A family of natural gene fusions. The EMBO Journal 6 (5) (May): 1429-39.
Patterton, H. G., C. C. Landel, D. Landsman, C. L. Peterson, and R. T. Simpson. 1998. The biochemical and phenotypic characterization of Hho1p, the putative linker histone H1 of saccharomyces cerevisiae. The Journal of Biological Chemistry 273 (13) (Mar 27): 7268-76.
Pillus, L., and J. Rine. 1989. Epigenetic inheritance of transcriptional states in S. cerevisiae. Cell 59 (4) (Nov 17): 637-47.
Prudden, J., S. Pebernard, G. Raffa, D. A. Slavin, J. J. Perry, J. A. Tainer, C. H. McGowan, and M. N. Boddy. 2007. SUMO-targeted ubiquitin ligases in genome stability. The EMBO Journal 26 (18) (Sep 19): 4089-101.
Radivojac, P., V. Vacic, C. Haynes, R. R. Cocklin, A. Mohan, J. W. Heyen, M. G. Goebl, and L. M. Iakoucheva. 2010. Identification, analysis, and prediction of protein ubiquitination sites. Proteins 78 (2) (Feb 1): 365-80.
Raffa, G. D., J. Wohlschlegel, J. R. Yates 3rd, and M. N. Boddy. 2006. SUMO-binding motifs mediate the Rad60-dependent response to replicative stress and self-association. The Journal of Biological Chemistry 281 (38) (Sep 22): 27973-81.
Reynolds, A. E., R. M. McCarroll, C. S. Newlon, and W. L. Fangman. 1989. Time of replication of ARS elements along yeast chromosome III. Molecular and Cellular Biology 9 (10) (Oct): 4488-94.
Rine, J., and I. Herskowitz. 1987. Four genes responsible for a position effect on expression from HML and HMR in saccharomyces cerevisiae. Genetics 116 (1) (May): 9-22.

99
Ritchie, K. J., and D. E. Zhang. 2004. ISG15: The immunological kin of ubiquitin. Seminars in Cell & Developmental Biology 15 (2) (Apr): 237-46.
Robinson, P. J., and D. Rhodes. 2006. Structure of the '30 nm' chromatin fibre: A key role for the linker histone. Current Opinion in Structural Biology 16 (3) (Jun): 336-43.
Rose, A. B., and J. R. Broach. 1990. Propagation and expression of cloned genes in yeast: 2-microns circle-based vectors. Methods in Enzymology 185 : 234-79.
Rusche, L. N., A. L. Kirchmaier, and J. Rine. 2003. The establishment, inheritance, and function of silenced chromatin in saccharomyces cerevisiae. Annual Review of Biochemistry 72 : 481-516.
Schwartz, D. C., and M. Hochstrasser. 2003. A superfamily of protein tags: Ubiquitin, SUMO and related modifiers. Trends in Biochemical Sciences 28 (6) (Jun): 321-8.
Sekiguchi, T., T. Sasaki, M. Funakoshi, T. Ishii, Y. H. Saitoh, S. Kaneko, and H. Kobayashi. 2011. Ubiquitin chains in the Dsk2 UBL domain mediate Dsk2 stability and protein degradation in yeast. Biochemical and Biophysical Research Communications 411 (3) (Aug 5): 555-61.
Shen, X., and M. A. Gorovsky. 1996. Linker histone H1 regulates specific gene expression but not global transcription in vivo. Cell 86 (3) (Aug 9): 475-83.
Shen, X., L. Yu, J. W. Weir, and M. A. Gorovsky. 1995. Linker histones are not essential and affect chromatin condensation in vivo. Cell 82 (1) (Jul 14): 47-56.
Shimamura, A., M. Sapp, A. Rodriguez-Campos, and A. Worcel. 1989. Histone H1 represses transcription from minichromosomes assembled in vitro. Molecular and Cellular Biology 9 (12) (Dec): 5573-84.
Singh, J., and A. J. Klar. 1992. Active genes in budding yeast display enhanced in vivo accessibility to foreign DNA methylases: A novel in vivo probe for chromatin structure of yeast. Genes & Development 6 (2) (Feb): 186-96.
Sobko, A., H. Ma, and R. A. Firtel. 2002. Regulated SUMOylation and ubiquitination of DdMEK1 is required for proper chemotaxis. Developmental Cell 2 (6) (Jun): 745-56.
Sollier, J., R. Driscoll, F. Castellucci, M. Foiani, S. P. Jackson, and D. Branzei. 2009. The saccharomyces cerevisiae Esc2 and Smc5-6 proteins promote sister chromatid junction-mediated intra-S repair. Molecular Biology of the Cell 20 (6) (Mar): 1671-82.

100
Strahl-Bolsinger, S., A. Hecht, K. Luo, and M. Grunstein. 1997. SIR2 and SIR4 interactions differ in core and extended telomeric heterochromatin in yeast. Genes & Development 11 (1) (Jan 1): 83-93.
Straight, A. F., W. Shou, G. J. Dowd, C. W. Turck, R. J. Deshaies, A. D. Johnson, and D. Moazed. 1999. Net1, a Sir2-associated nucleolar protein required for rDNA silencing and nucleolar integrity. Cell 97 (2) (Apr 16): 245-56.
Sun, Z. W., and C. D. Allis. 2002. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature 418 (6893) (Jul 4): 104-8.
Tagwerker, C., H. Zhang, X. Wang, L. S. Larsen, R. H. Lathrop, G. W. Hatfield, B. Auer, L. Huang, and P. Kaiser. 2006. HB tag modules for PCR-based gene tagging and tandem affinity purification in saccharomyces cerevisiae. Yeast (Chichester, England) 23 (8) (Jun): 623-32.
Tarassov, K., V. Messier, C. R. Landry, S. Radinovic, M. M. Serna Molina, I. Shames, Y. Malitskaya, J. Vogel, H. Bussey, and S. W. Michnick. 2008. An in vivo map of the yeast protein interactome. Science (New York, N.Y.) 320 (5882) (Jun 13): 1465-70.
Triolo, T., and R. Sternglanz. 1996. Role of interactions between the origin recognition complex and SIR1 in transcriptional silencing. Nature 381 (6579) (May 16): 251-3.
Ulrich, H. D., and A. A. Davies. 2009. In vivo detection and characterization of sumoylation targets in saccharomyces cerevisiae. Methods in Molecular Biology (Clifton, N.J.) 497 : 81-103.
Ushinsky, S. C., H. Bussey, A. A. Ahmed, Y. Wang, J. Friesen, B. A. Williams, and R. K. Storms. 1997. Histone H1 in saccharomyces cerevisiae. Yeast (Chichester, England) 13 (2) (Feb): 151-61.
Uzunova, K., K. Gottsche, M. Miteva, S. R. Weisshaar, C. Glanemann, M. Schnellhardt, M. Niessen, et al. 2007. Ubiquitin-dependent proteolytic control of SUMO conjugates. The Journal of Biological Chemistry 282 (47) (Nov 23): 34167-75.
Verger, A., J. Perdomo, and M. Crossley. 2003. Modification with SUMO. A role in transcriptional regulation. EMBO Reports 4 (2) (Feb): 137-42.
Veron, M., Y. Zou, Q. Yu, X. Bi, A. Selmi, E. Gilson, and P. A. Defossez. 2006. Histone H1 of saccharomyces cerevisiae inhibits transcriptional silencing. Genetics 173 (2) (Jun): 579-87.
von der Haar, T. 2007. Optimized protein extraction for quantitative proteomics of yeasts. PloS One 2 (10) (Oct 24): e1078.

101
Wohlschlegel, J. A. 2009. Identification of SUMO-conjugated proteins and their SUMO attachment sites using proteomic mass spectrometry. Methods in Molecular Biology (Clifton, N.J.) 497 : 33-49.
Wohlschlegel, J. A., E. S. Johnson, S. I. Reed, and J. R. Yates 3rd. 2004. Global analysis of protein sumoylation in saccharomyces cerevisiae. The Journal of Biological Chemistry 279 (44) (Oct 29): 45662-8.
Woodcock, C. L., and S. Dimitrov. 2001. Higher-order structure of chromatin and chromosomes. Current Opinion in Genetics & Development 11 (2) (Apr): 130-5.
Woodcock, C. L., A. I. Skoultchi, and Y. Fan. 2006. Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosome Research : An International Journal on the Molecular, Supramolecular and Evolutionary Aspects of Chromosome Biology 14 (1): 17-25.
Xirodimas, D. P. 2008. Novel substrates and functions for the ubiquitin-like molecule NEDD8. Biochemical Society Transactions 36 (Pt 5) (Oct): 802-6.
Xu, E. Y., X. Bi, M. J. Holland, D. E. Gottschling, and J. R. Broach. 2005. Mutations in the nucleosome core enhance transcriptional silencing. Molecular and Cellular Biology 25 (5) (Mar): 1846-59.
Xu, E. Y., K. A. Zawadzki, and J. R. Broach. 2006. Single-cell observations reveal intermediate transcriptional silencing states. Molecular Cell 23 (2) (Jul 21): 219-29.
Ye, Y., and M. Rape. 2009. Building ubiquitin chains: E2 enzymes at work. Nature Reviews.Molecular Cell Biology 10 (11) (Nov): 755-64.
Yu, Q., H. Kuzmiak, L. Olsen, A. Kulkarni, E. Fink, Y. Zou, and X. Bi. 2010. Saccharomyces cerevisiae Esc2p interacts with Sir2p through a small ubiquitin-like modifier (SUMO)-binding motif and regulates transcriptionally silent chromatin in a locus-dependent manner. The Journal of Biological Chemistry 285 (10) (Mar 5): 7525-36.
Yu, Q., H. Kuzmiak, Y. Zou, L. Olsen, P. A. Defossez, and X. Bi. 2009. Saccharomyces cerevisiae linker histone Hho1p functionally interacts with core histone H4 and negatively regulates the establishment of transcriptionally silent chromatin. The Journal of Biological Chemistry 284 (2) (Jan 9): 740-50.
Zhou, W., J. J. Ryan, and H. Zhou. 2004. Global analyses of sumoylated proteins in saccharomyces cerevisiae. induction of protein sumoylation by cellular stresses. The Journal of Biological Chemistry 279 (31) (Jul 30): 32262-8.

102
Zhu, M., M. P. Torres, J. B. Kelley, H. G. Dohlman, and Y. Wang. 2011. Pheromone- and RSP5-dependent ubiquitination of the G protein beta subunit Ste4 in yeast. The Journal of Biological Chemistry 286 (31) (Aug 5): 27147-55.

103
Table A-1. Catalog of strains Number Name Description Reference
1-1s BY4741 MATa his3∆1 leu2∆0 met15∆0 ura3∆0
1-2s BY4741, hho1∆::KanMX Yu et al. 2009 1-3s BY4741, sir1∆::KanMX Yu et al. 2009 1-4s YYZ333 BY4741, sir1∆::KanMX hho1∆::LEU2 Yu et al. 2009 1-5s YXB10 MATa ura3-52 ade2-1 lys1-1 his5-1
can1-100 LEU2-GAL10-FLP1 FRT-E-hml::β1-I-FRT [cir°]
Bi et al. 2004
1-6s YHK13 YXB10, hho1∆::NatMX Yu et al. 2009 1-7s YXB810 YXB10, sir1∆::URA3 Yu et al. 2009 1-8s YHK14 YXB810, hho1∆::NatMX Yu et al. 2009 1-9s YXB10s YXB10, sir3∆::URA3 Bi et al. 2004 1-10s YXB814 YXB10s, sir3-8-KanMX Xu et al. 2005 1-11s YHK16 YXB814, hho1∆::NatMX Yu et al. 2009 1-12s YXB803 MATa ura3-52 ade2-101 lys1-1 his5-
1 can1-100 trp1∆901 his3∆200 his5-1LEU2-GAL10-FLP1 hhf1∆::HIS3 hhf2∆::LEU2 E-FRT-hml::β1-FRT-I pRS414-HHF1 (CEN-TRP1-HHF1) [cir°]
1-13s YQY566 YXB803, hho1∆::URA3 Yu et al. 2009 1-14s YQY385 MATα ura3-52 leu2-3,112 his3∆
trp1-289 (hht1hhf1)∆ (hht2hhf2)∆ pMS337 (CEN-LEU2-HHT1-HHF1), Tel VII-L-URA3
Yu et al. 2009
1-15s YQY386 yQY385, hho1∆::KanMX Yu et al. 2009 1-16s YQY355 MATα ura3-52 leu2-3,112 his3∆
trp1-289 (hht1hhf1)∆ (hht2hhf2)∆ pMS364 (CEN-LEU2-HHT1-hhf1-Y88G), Tel VII-L-URA3
Yu et al. 2009
1-17s YQY356 yQY355, hho1∆::KanMX Yu et al. 2009 1-18s YHK53 BY4741, Tel VII-L-URA3 Yu et al. 2010 1-19s YHK140 YHK53, hho1∆::LEU2 This work 1-20s YHK-SS-68T Open Biosystems Yeast Knockout
Collection (snf2∆), TEL VII-L-URA3 This work
1-21s YHK-SS-68Td YHK-SS-68T, hho1∆::LEU2 This work 1-22s YHK-SS-66T Open Biosystems Yeast Knockout
Collection (ubp8∆), TEL VII-L-URA3 This work
1-23s YHK-SS-66Td YHK-SS-66T, hho1∆::LEU2 This work 1-24s YHK-SS-71T Open Biosystems Yeast Knockout
Collection (rsc1∆), TEL VII-L-URA3 This work
1-25s YHK-SS-71Td YHK-SS-71T, hho1∆::LEU2 This work 1-26s YHK17 BY4741, ORC6-6xHA-NatMX This work

104
Number Name Description Reference 2-1s YXB76 MATa ura3-52 leu2-3,112 ade2-1
lys1-1 his5-2 can1-100 E-HML-Iinverted
Chiu et al. 2003
2-2s yJS537 YXB76, sir2-6xMyc-kanMX Yu et al. 2010 2-3s PJ69-4α MATα trp1-901 leu2-3,112 ura3-52
his3-200 gal4gal80 LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ
James et al. 1996
2-4s YLO55 PJ69-4α, sir3∆::NatMX sir4∆::KanMX Yu et al. 2010 2-5s YSB35 MATα ade2-1 can 1-100 his3-11,15
leu2-3,112 trp1-289 ura3-1 gal4::LEU2 Aeb::UASg
Chien et al. 1993
2-6s YHK56 YHK53, esc2::KanMX Yu et al. 2010
3-1s YHK174 BY4741, SIR2-6xHA-KanMX This work 3-2s YHK175 BY4741, SIR3-6xHA-KanMX This work 3-3s Open Biosystems Yeast TAP-Fusion
Collection, CDC11-TAP Ghaemmaghami et al. 2003
3-4s Open Biosystems Yeast TAP-Fusion Collection, SIR2-TAP
Ghaemmaghami et al. 2003
3-5s Open Biosystems Yeast Knockout Collection (sir2∆)
Giaever et al. 2002
3-6s YHK271 BY4741, SIR2(∆C-14)-6xHA-NatMX, HA tag inserted after nt 1644
This work
3-7s YHK306 BY4741, SIR2(∆C-26)-6xHA-NatMX, tag inserted after nt 1608
This work
3-8s YHK307 BY4741, SIR2(∆C-41)-6xHA-NatMX, tag inserted after nt 1563
This work
3-9s YHK430 BY4741, SIR2-6xHA-NatMX This work 3-10s YHK474 BY4741, SIR2-6xHA-BIOpep-
KanMX This work
3-11s YHK472 BY4741, NatMX-pTEF-3xHA-SIR2 This work 3-12s YHK475 BY4741, NatMX-pTEF-3xHA-SIR2-
BIOpep-KanMX This work
3-13s YHK478 BY4741, SIR2-BIOpep-KanMX This work 3-14s YHK509 BY4741, NatMX-pTEF-3xHA-HIS6-
SIR2-BIOpep-KanMX This work

105
Appendix 2:
Results for Screen for Synthetic Interactors with hho1∆

106

107

108

109

110