qualitative proteomics (obtaining high-confidence high ... proteomic… · (how to obtain...
TRANSCRIPT

Qualitative Proteomics Qualitative Proteomics (how to obtain high(how to obtain high--confidence confidence
highhigh--throughput protein identification!)throughput protein identification!)
James A. Mobley, Ph.D.James A. Mobley, Ph.D.Director of Research in UrologyDirector of Research in UrologyAssociate Director of Mass SpectrometryAssociate Director of Mass Spectrometry(contact: (contact: [email protected]@uab.edu))

The The ““BirthBirth”” of Proteomicsof Proteomics
•• ProteomicsProteomics was not was not ““bornborn”” as many will say, but was indeed as many will say, but was indeed ““ignitedignited””with the with the ““discoverydiscovery”” of of ““veryvery soft ionizationsoft ionization”” techniques.techniques.
•• For those in the field at the time, For those in the field at the time, this was this was ““hugehuge””, but still didn, but still didn’’t really t really take of until the mid to late 90take of until the mid to late 90’’s!!s!!
•• However, prior to this However, prior to this ……. ..both . ..both ““hard and soft ionizationhard and soft ionization”” processes processes were well established! These include, were well established! These include, electron ionizationelectron ionization (EI), (EI), chemical ionizationchemical ionization (CI), (CI), fast atom bombardmentfast atom bombardment (FAB), (FAB), liquid liquid secondary ion MSsecondary ion MS (LSIMS), and (LSIMS), and plasma plasma desorptiondesorption MSMS (PDMS). (PDMS).
•• The newer techniques were initiated byThe newer techniques were initiated by…………and and ““still includestill include”” matrix matrix associated associated desorptiondesorption ionization (MALDI)ionization (MALDI)-- [[HillenkampHillenkamp, et. al. 1988], et. al. 1988]and and electrosprayelectrospray ionization (ESI)ionization (ESI)-- [[FennFenn et. al. 1989] based sources.et. al. 1989] based sources.
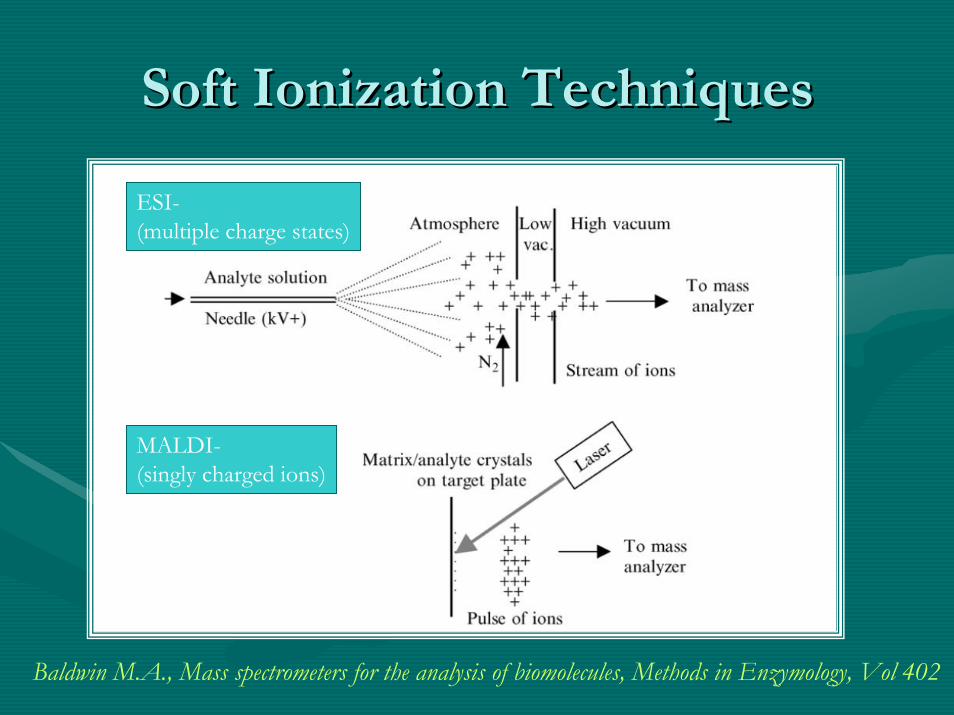
Soft Ionization TechniquesSoft Ionization Techniques
MALDI-(singly charged ions)
ESI-(multiple charge states)
Baldwin M.A., Mass spectrometers for the analysis of biomolecules, Methods in Enzymology, Vol 402

Protein CharacterizationProtein Characterization
•• SoSo…….There are .There are just three simple pointsjust three simple points to remember in to remember in the business of protein identification/ characterization.the business of protein identification/ characterization.–– 1) 1) Generation of a peptideGeneration of a peptide--based mass spectra.based mass spectra.
•• [[MSMS11 onlyonly]; peptide mass fingerprinting [PMF] (protein must be ]; peptide mass fingerprinting [PMF] (protein must be nearly pure)nearly pure)
•• [[MSMS22]; sequence data (high purity is not necessarily required)]; sequence data (high purity is not necessarily required)–– 2) 2) Analysis.Analysis.
•• Matching algorithmsMatching algorithms based on based on inin--silicosilico digestion (MSdigestion (MS11) & ) & fragmentation (MSfragmentation (MS22) of known genes ) of known genes
•• DenovoDenovo Sequencing (MSSequencing (MS22))–– 3) 3) Validation.Validation.
•• ImmunoImmuno--affinity affinity techniques (Western analysis, techniques (Western analysis, ImmunoImmuno--histochemistryhistochemistry, ELISA, , ELISA, LuminexLuminex, etc)., etc).
•• Mass SpectrometryMass Spectrometry (standard heavy isotope tags)(standard heavy isotope tags)

Proteins to PeptidesProteins to Peptides……..
•• Even today, we are Even today, we are highly limitedhighly limited by decreased detection, by decreased detection, resolving power, and poor fragmentation of resolving power, and poor fragmentation of ““wholewhole”” proteinsproteins!!
•• Therefore, we Therefore, we ““digestdigest”” proteins to peptidesproteins to peptides prior to MS analysis.prior to MS analysis.•• Many chemical and enzymatic techniques have been published; Many chemical and enzymatic techniques have been published;
however, however, trypsintrypsin remains the most commonly utilized enzyme remains the most commonly utilized enzyme for use in proteomics!for use in proteomics!
•• This enzyme This enzyme cleaves at cleaves at argininearginine and lysineand lysine, yielding peptides that , yielding peptides that are easily detected and fragmented in the most common mass are easily detected and fragmented in the most common mass analyzers today.analyzers today.
•• Keeping in mind that utilizing Keeping in mind that utilizing multiple digestionmultiple digestion procedures procedures carried out on the same sample can be carried out on the same sample can be very complementing!very complementing!
http://donatello.ucsf.edu/http://donatello.ucsf.edu/A lot of information hereA lot of information here…………Take a look at Protein ProspectorTake a look at Protein Prospector…………..
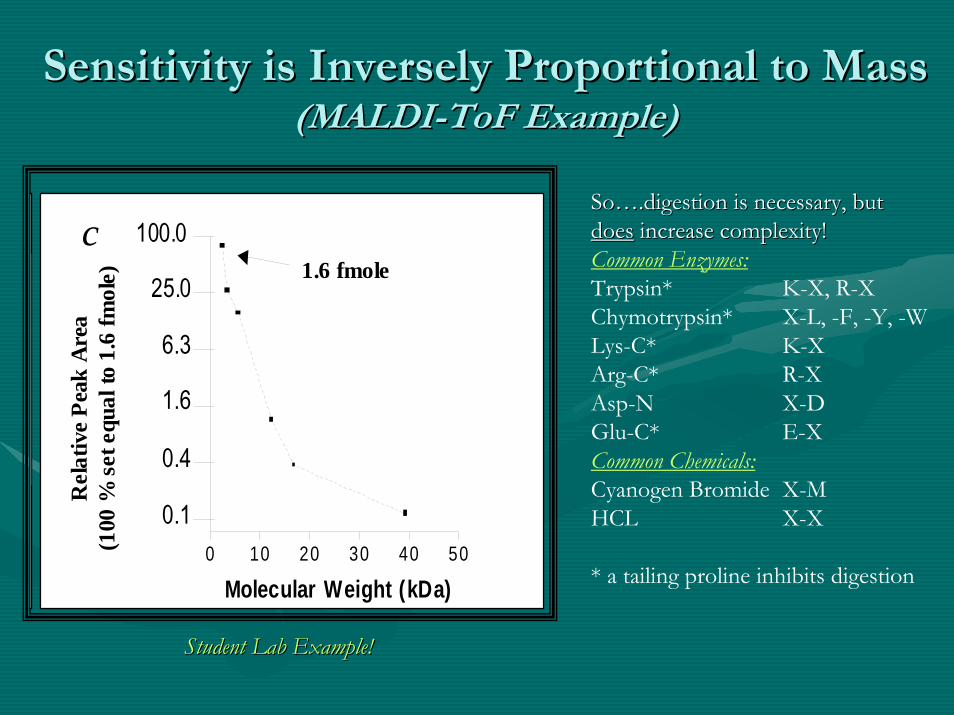
Sensitivity is Inversely Proportional to MassSensitivity is Inversely Proportional to Mass(MALDI(MALDI--ToFToF Example)Example)
0 10 20 30 40 50
Molecular Weight (kDa)
100.0
25.0
6.3
1.6
0.4
0.1
Rel
ativ
e Pe
ak A
rea
(100
% =
1.6
fmol
e)
1.6 femptomole3.9 pg/ μL
c
Rel
ativ
e Pe
ak A
rea
(100
% se
t equ
al to
1.6
fmol
e) 1.6 fmole
0 10 20 30 40 50
Molecular Weight (kDa)
100.0
25.0
6.3
1.6
0.4
0.1
Rel
ativ
e Pe
ak A
rea
(100
% =
1.6
fmol
e)
1.6 femptomole3.9 pg/ μL
c
Rel
ativ
e Pe
ak A
rea
(100
% se
t equ
al to
1.6
fmol
e) 1.6 fmole
Student Lab Example!Student Lab Example!
SoSo…….digestion is necessary, but .digestion is necessary, but doesdoes increase complexity!increase complexity!Common Enzymes:Trypsin* K-X, R-XChymotrypsin* X-L, -F, -Y, -WLys-C* K-XArg-C* R-XAsp-N X-DGlu-C* E-XCommon Chemicals:Cyanogen Bromide X-MHCL X-X
* a tailing proline inhibits digestion

Concept of PMF [MSConcept of PMF [MS11]]
•• MSMS11 approachesapproaches –– spectra containing peptide parent molecules spectra containing peptide parent molecules only!only!
•• This type of unambiguous protein ID is referred to as This type of unambiguous protein ID is referred to as ““peptide peptide mass fingerprintingmass fingerprinting”” (PMF) introduced ~1990.(PMF) introduced ~1990.
•• In this case, In this case, no sequence information is generatedno sequence information is generated, but it is very , but it is very sensitive when very little sample is sensitive when very little sample is availibleavailible!!
•• The downfall is that the sample The downfall is that the sample must be very puremust be very pure! Highly ! Highly complementing for 2D PAGE work.complementing for 2D PAGE work.
•• However, However, high mass accuracyhigh mass accuracy is a must as well!is a must as well!•• Overall, these daysOverall, these days………………unless absolutely necessaryunless absolutely necessary……………………. .
PMF should not be used!PMF should not be used!•• There are simply There are simply too many matches possibletoo many matches possible with this technique with this technique
even with access to high resolution instruments.even with access to high resolution instruments.
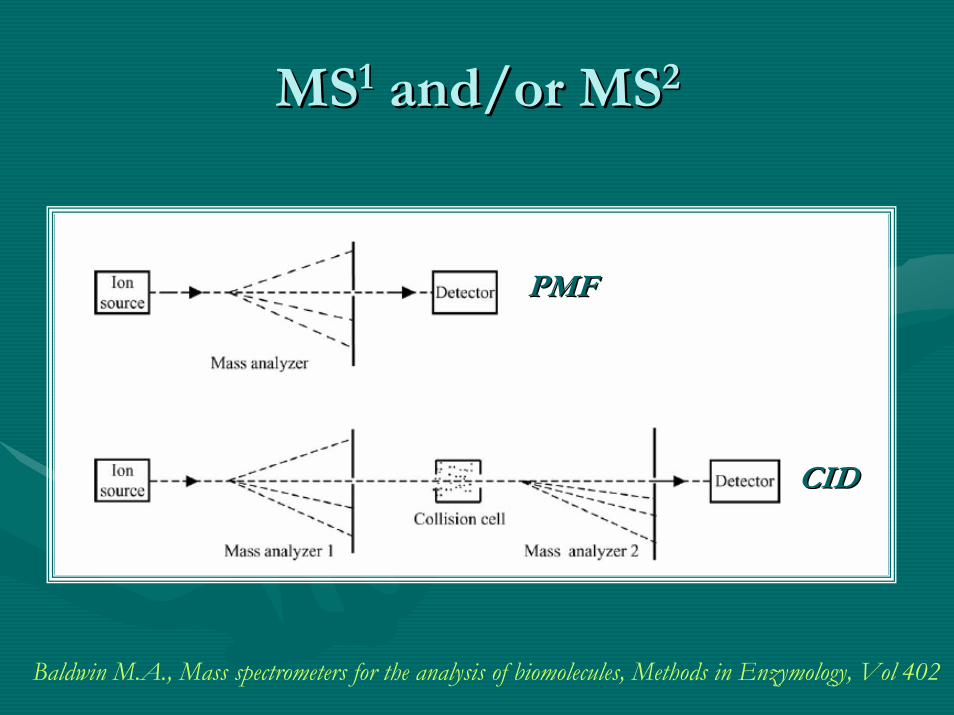
MSMS11 and/or MSand/or MS22
Baldwin M.A., Mass spectrometers for the analysis of biomolecules, Methods in Enzymology, Vol 402
PMFPMF
CIDCID
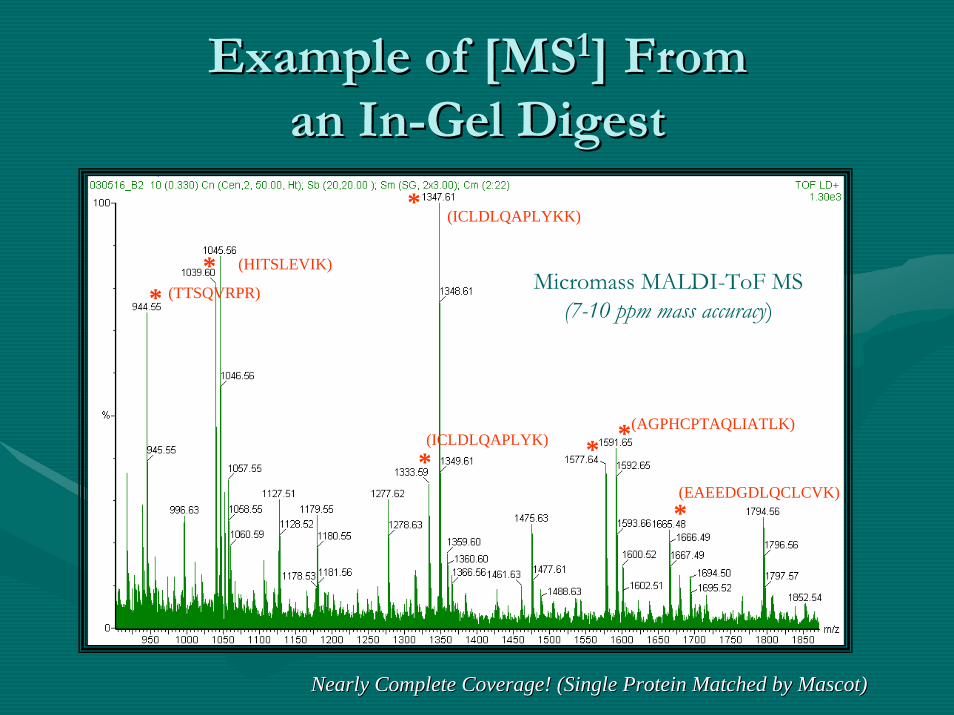
Example of [MSExample of [MS11] From] Froman Inan In--Gel DigestGel Digest
Micromass MALDI-ToF MS(7-10 ppm mass accuracy)
Nearly Complete Coverage! (Single Protein Matched by Mascot)Nearly Complete Coverage! (Single Protein Matched by Mascot)
*
**
*
* *
*
(TTSQVRPR)
(HITSLEVIK)
(ICLDLQAPLYK)
(ICLDLQAPLYKK)
(AGPHCPTAQLIATLK)
(EAEEDGDLQCLCVK)

PMF Match!PMF Match!
Predicted and determined mass for CXC4 (~ 8 Predicted and determined mass for CXC4 (~ 8 kDakDa Protein)Protein)
4632-0.1980AcryAGPHCPTAQLIATLK1591.84741591.6497
6150-0.1321AcryKICLDLQAPLYK1347.73461347.60286151-0.1960CAM-CICLDLQAPLYK1461.81391461.61804632-0.2070CAM-CAGPHCPTAQLIATLK1577.84741577.6407
1665.710
1333.71901039.6152944.5278
Theoretical Mass (mi)
EAEEDGDLQCLCVK
ICLDLQAPLYKHITSLEVIKTTSQVRPR
Peptide Sequence
CAM-C
Modi.
0
000
MC
-0.227
-0.131-0.016+ 0.021
Δm (Da)
1
512315
Start
22944.5486311039.5997611333.5878
141665.4830
EndMeasured m/z
4632-0.1980AcryAGPHCPTAQLIATLK1591.84741591.6497
6150-0.1321AcryKICLDLQAPLYK1347.73461347.60286151-0.1960CAM-CICLDLQAPLYK1461.81391461.61804632-0.2070CAM-CAGPHCPTAQLIATLK1577.84741577.6407
1665.710
1333.71901039.6152944.5278
Theoretical Mass (mi)
EAEEDGDLQCLCVK
ICLDLQAPLYKHITSLEVIKTTSQVRPR
Peptide Sequence
CAM-C
Modi.
0
000
MC
-0.227
-0.131-0.016+ 0.021
Δm (Da)
1
512315
Start
22944.5486311039.5997611333.5878
141665.4830
EndMeasured m/z
(1)EAEEDGDLQC LCVKTTSQVR PRHITSLEVI KAGPHCPTAQ LIATLKNGRK ICLDLQAPLY KKIIKKLLES (70)

Protein Fragmentation [MSProtein Fragmentation [MS22]]
•• Both ESI and MALDI based Both ESI and MALDI based tandem instrumentstandem instruments are are common in most core settings, each with a common in most core settings, each with a combination of mass analyzers.combination of mass analyzers.
•• Each source and combination of Each source and combination of mass analyzersmass analyzers have have their selective advantages worthy of a second talk!their selective advantages worthy of a second talk!
•• Fragmentation is generally similarFragmentation is generally similar, primarily with the , primarily with the generation of eithergeneration of either………………..–– b and y ions;b and y ions; collision induced decay (CID) & infrared collision induced decay (CID) & infrared
multiphotonmultiphoton dissociation (IRMPD)dissociation (IRMPD)–– z and c ions;z and c ions; electron transfer dissociation (ETD) & electron electron transfer dissociation (ETD) & electron
capture dissociation (ECD)capture dissociation (ECD)
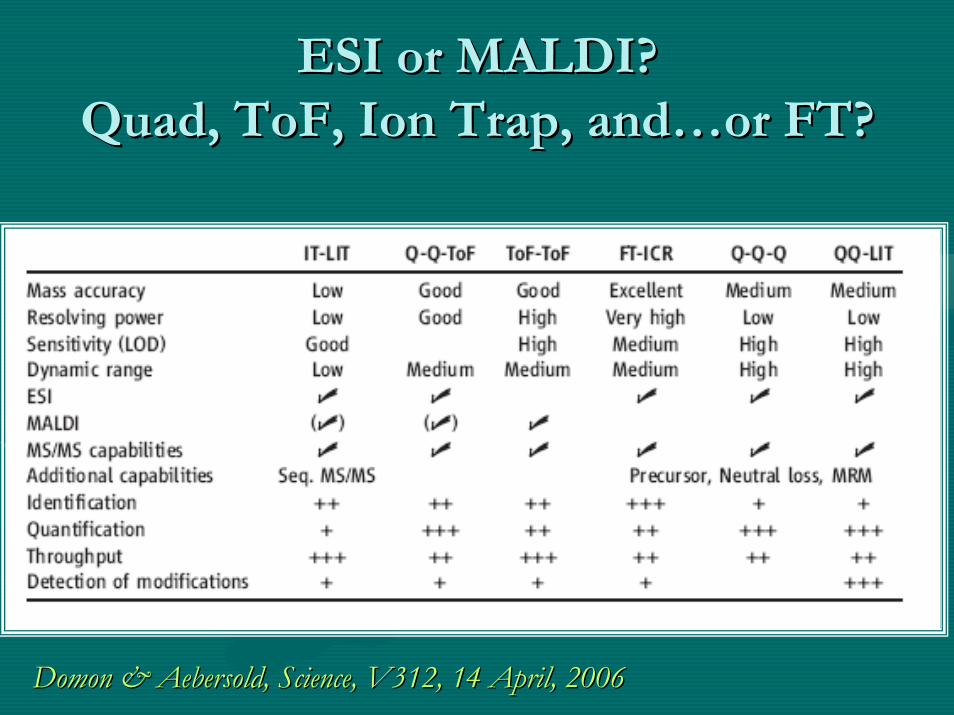
ESI or MALDI?ESI or MALDI?Quad, Quad, ToFToF, Ion Trap, and, Ion Trap, and……or FT?or FT?
DomonDomon & & AebersoldAebersold, Science, V312, 14 April, 2006 , Science, V312, 14 April, 2006

Peptide FragmentationPeptide Fragmentation
CID/ IRMPDCID/ IRMPD ETD/ECDETD/ECD
0 1 0 2 0 3 0 4 0 5 0T i m e ( m i n )
0
0
0
0
0
0
0
0
0
0
03 2 . 9 0
1 9 . 3 0
2 0 . 4 1
2 1 . 0 6
2 3 . 0 4
2 3 . 6 12 . 6 1 3 0 . 6 22 . 8 5 4 2 . 6 93 3 . 6 81 8 . 3 46 . 8 0 4 3 . 3 1 5 1 . 4 8
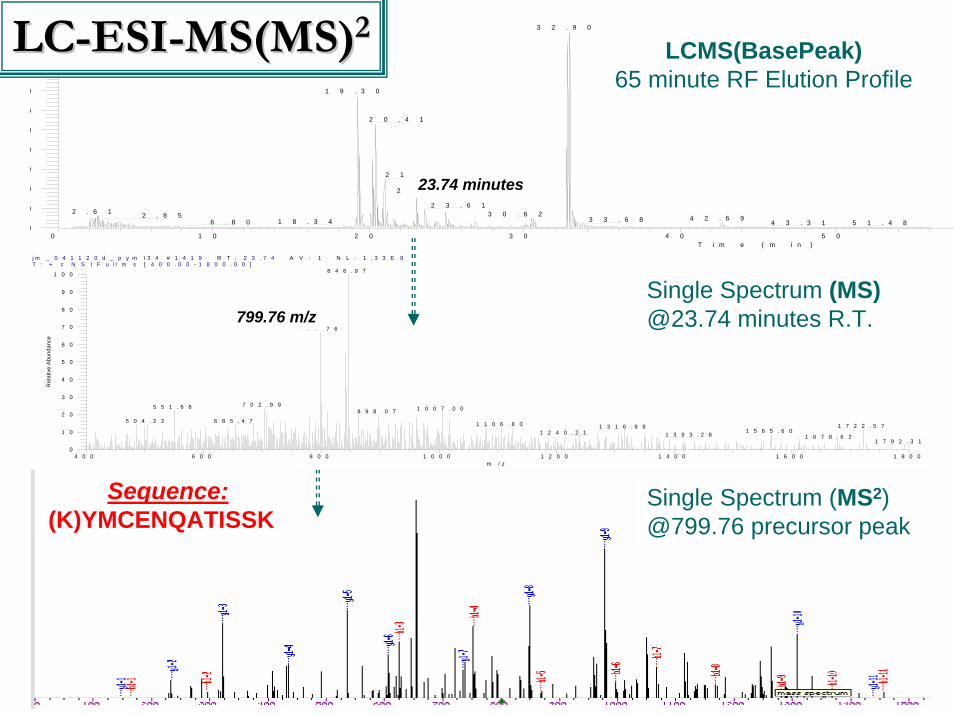
Single Spectrum (MS2)@799.76 precursor peak
LCMS(BasePeak)65 minute RF Elution Profile
23.74 minutes
j m _ 0 4 1 1 2 0 d _ p y m t 3 4 # 1 4 1 9 R T : 2 3 . 7 4 A V : 1 N L : 1 . 3 3 E 8T : + c N S I F u l l m s [ 4 0 0 . 0 0 - 1 8 0 0 . 0 0 ]
4 0 0 6 0 0 8 0 0 1 0 0 0 1 2 0 0 1 4 0 0 1 6 0 0 1 8 0 0m / z
0
1 0
2 0
3 0
4 0
5 0
6 0
7 0
8 0
9 0
1 0 0
Rel
ativ
e A
bund
ance
8 4 6 . 9 7
7 9 9 . 7 6
7 0 2 . 9 95 5 1 . 6 8 1 0 0 7 . 0 08 9 8 . 0 7
6 8 5 . 4 75 0 4 . 2 2 1 1 0 6 . 8 0 1 7 2 2 . 5 71 3 1 6 . 9 91 5 6 5 . 6 01 2 4 0 . 2 1 1 3 9 3 . 2 8 1 6 7 8 . 6 2
1 7 9 2 . 3 1
799.76 m/z
Sequence:(K)YMCENQATISSK
LCLC--ESIESI--MS(MS)MS(MS)22
Single Spectrum (MS)@23.74 minutes R.T.
800 1000 1200 1400 1600 1800 2000 2200Mass
0
20000
40000
60000
80000
100000
Clus
ter A
rea
800 1000 1200 1400 1600 1800 2000 2200Mass
0
20000
40000
60000
80000
100000
Clus
ter A
rea
2383.96
2281.08
2254.06
2224.09
2194.332125.88
2088.00
2056.08
2052.921994.02
1953.00
1883.89
1872.871838.89
1831.931794.78
1791.77
1765.73
1716.85
1707.77
1664.78
1641.77
1630.83
1584.67
1529.74
1493.711487.74
1472.781451.74
1410.731383.68
1365.64
1336.64
1320.60
1310.68
1274.711265.58
1233.60
1232.60
1198.63
1182.59
1172.56
1136.56
1118.58
1104.57
1086.55
1019.53
999.47987.51
959.53
947.52917.28
900.09
882.89
870.26
832.31
817.49
789.04
759.42746.09
696.09
679.52
669.00
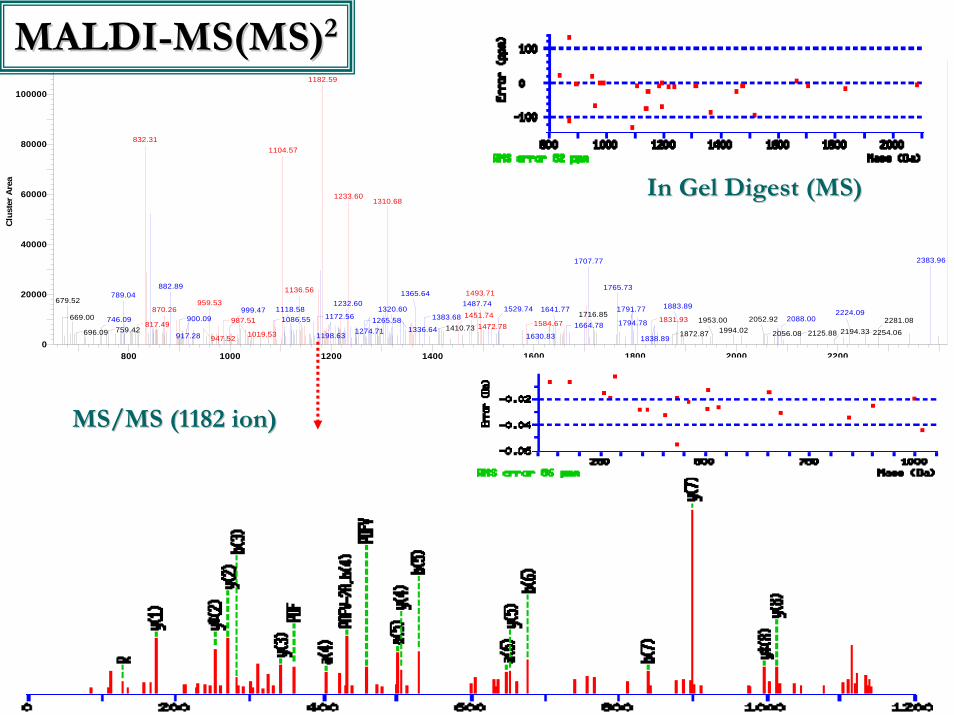
MS/MS (1182 ion)MS/MS (1182 ion)
In Gel Digest (MS)In Gel Digest (MS)
MALDIMALDI--MS(MS)MS(MS)22
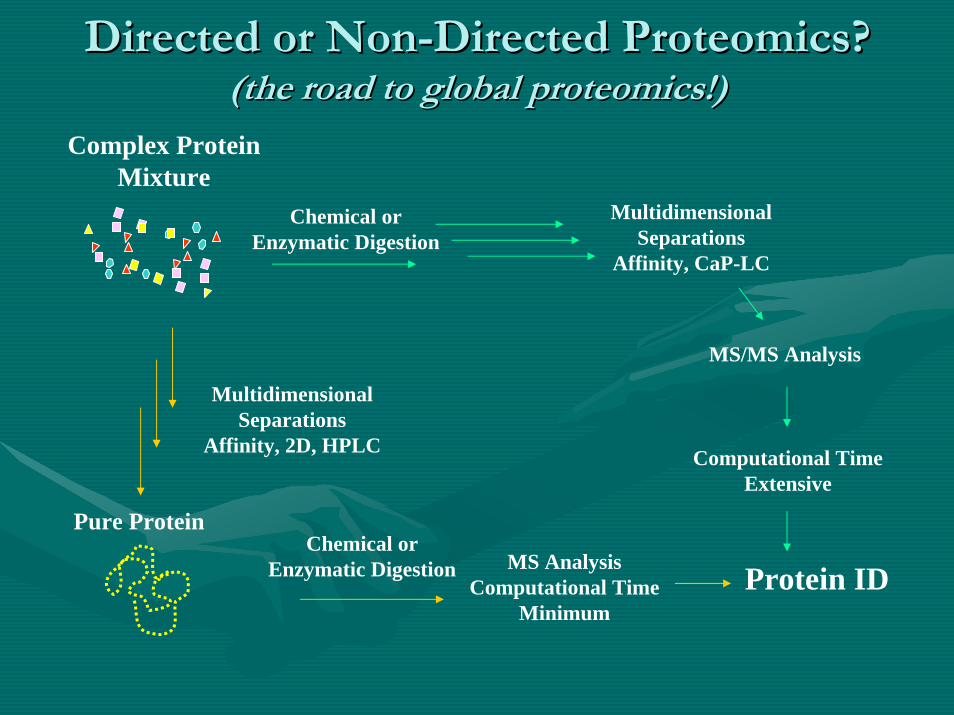
Directed or NonDirected or Non--Directed Proteomics?Directed Proteomics?(the road to global proteomics!)(the road to global proteomics!)
Pure Protein
MultidimensionalSeparations
Affinity, 2D, HPLC
MultidimensionalSeparations
Affinity, CaP-LC
MS/MS Analysis
Computational TimeExtensive
MS AnalysisComputational Time
MinimumProtein ID
Complex ProteinMixture
Chemical or Enzymatic Digestion
Chemical or Enzymatic Digestion
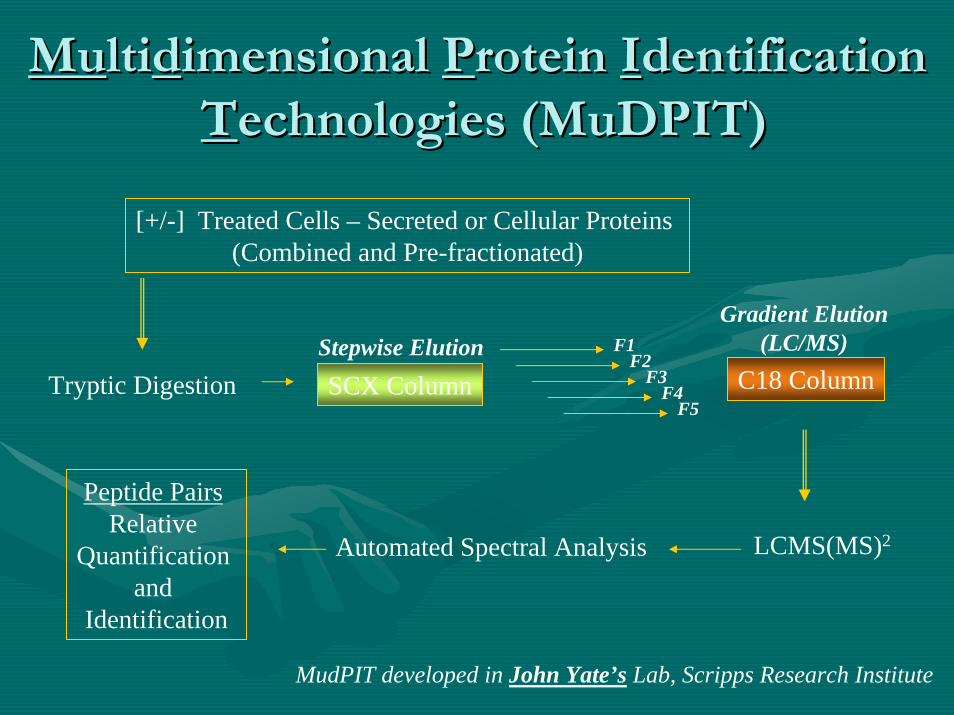
MuMultiltiddimensional imensional PProtein rotein IIdentification dentification TTechnologies (echnologies (MuDPITMuDPIT))
MudPIT developed in John Yate’s Lab, Scripps Research Institute
SCX Column C18 ColumnTryptic Digestion
[+/-] Treated Cells – Secreted or Cellular Proteins (Combined and Pre-fractionated)
LCMS(MS)2
Stepwise Elution F1F2
F3F4
F5
Automated Spectral Analysis
Peptide Pairs Relative
Quantification and
Identification
Gradient Elution(LC/MS)

Data AnalysisData Analysis
•• A standard 1D LCA standard 1D LC--ESI run may have as many as ESI run may have as many as 4,0004,000--6,000 MS files!!6,000 MS files!!
•• A A MuDPITMuDPIT run may contain run may contain 25,00025,000--60,000 60,000 files!!files!!
•• While LCWhile LC--MALDI runs generate far fewer data MALDI runs generate far fewer data files, they still contain files, they still contain tootoo--much data to analyze much data to analyze by hand!by hand!
•• Therefore, Therefore, automated data analysis is requiredautomated data analysis is required!!!!

Data AnalysisData Analysis
•• Common Matching Algorithms; Common Matching Algorithms; –– SequestSequest, MASCOT, , MASCOT, XTandemXTandem
•• Automated Automated DenovoDenovo Sequence Tools;Sequence Tools;–– Peaks, Rapid Peaks, Rapid DenovoDenovo, , DenovoXDenovoX, Mascot Distiller, , Mascot Distiller,
PepNovoPepNovo, others, others……....•• Statistical SoftwareStatistical Software
–– Scaffold, Protein Profit, Scaffold, Protein Profit, FinniganFinnigan, others, others……•• Standardizing the Field!Standardizing the Field!
–– Trans Proteomic Pipeline (Sashimi Project; Trans Proteomic Pipeline (Sashimi Project; mzXMLmzXML based based universal software package)universal software package)

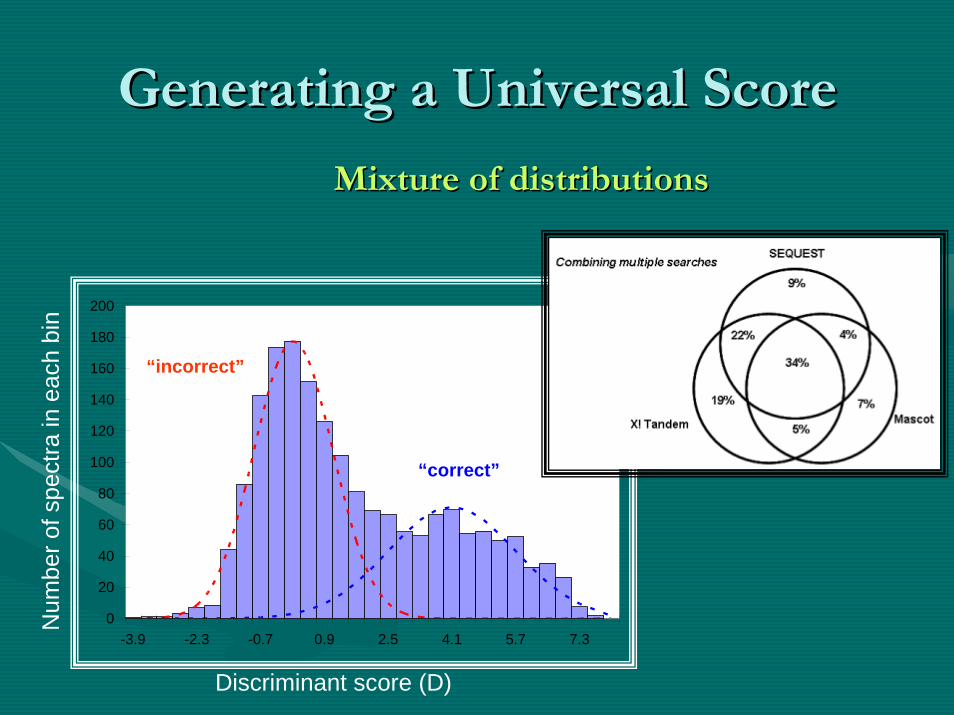
Matching AlgorithmsMatching Algorithms
•• All matching algorithms are based on generating All matching algorithms are based on generating a score based on a score based on ““closeness of fitcloseness of fit”” between the between the peptides measuredpeptides measured in the mass spectrometer and in the mass spectrometer and thethe inin--silicosilico digestion of known genesdigestion of known genes or proteins or proteins in a database.in a database.–– The two most commonly used databases include: The two most commonly used databases include:
NCBINCBI--NR and NR and UniprotUniprot
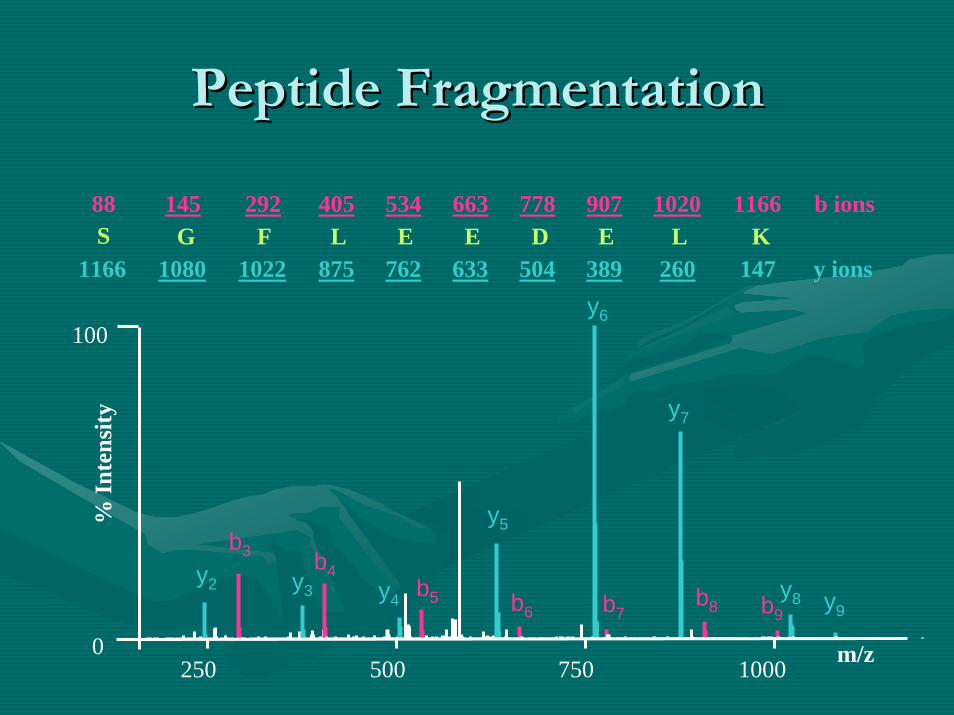
Peptide FragmentationPeptide Fragmentation
K1166
L1020
E907
D778
E663
E534
L405
F292
G145
S88 b ions
100
0250 500 750 1000 m/z
% In
tens
ity
147260389504633762875102210801166 y ionsy6
y7
y2 y3 y8b9
b4y4
y5
y9
b3
b5 b6 b7b8
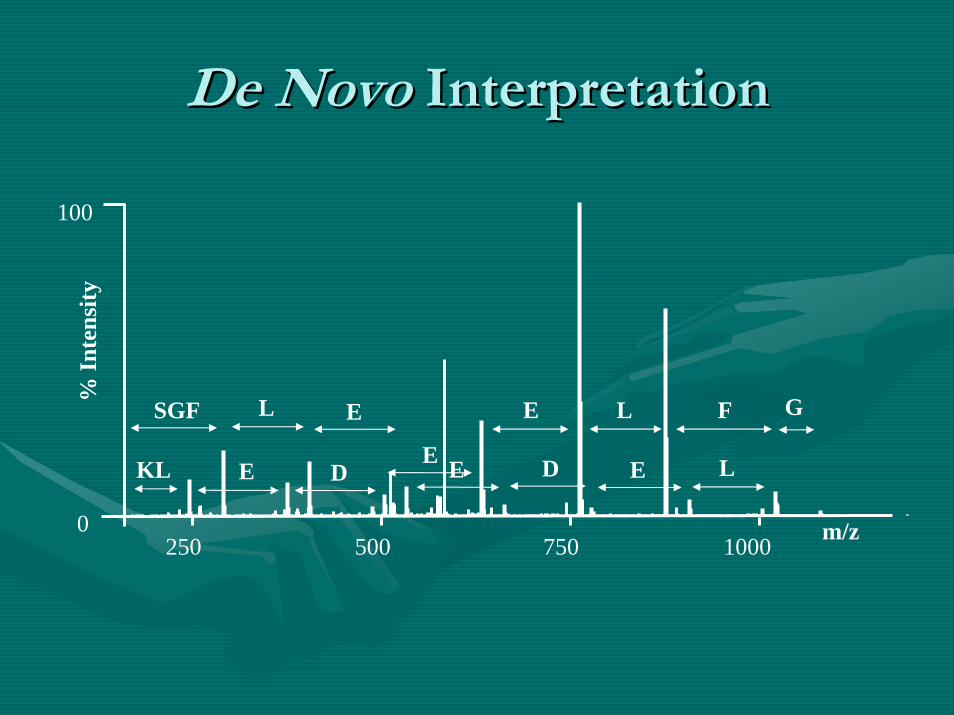
De Novo De Novo InterpretationInterpretation
100
0250 500 750 1000 m/z
% In
tens
ity
E L F
KL
SGF G
E DE
L E
E D E L
0
20
40
60
80
100
120
140
160
180
200
-3.9 -2.3 -0.7 0.9 2.5 4.1 5.7 7.3
“correct”
“incorrect”
Discriminant score (D)
Num
ber o
f spe
ctra
in e
ach
bin
Mixture of distributionsMixture of distributions
Generating a Universal ScoreGenerating a Universal Score

SoSo…….We Have .We Have IDID’’ss, Now What?, Now What?
•• ValidationValidation…………..•• ValidationValidation……………………....•• ValidationValidation………………………………..
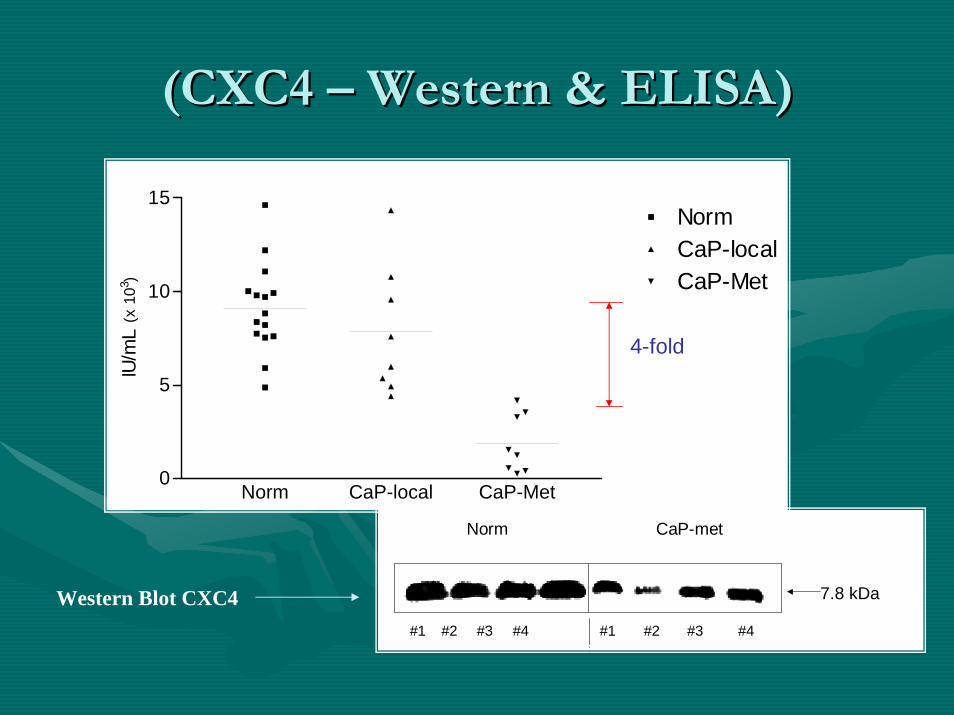
(CXC4 (CXC4 –– Western & ELISA)Western & ELISA)
Norm CaP-local CaP-Met0
5
10
15NormCaP-localCaP-Met
IU/m
L(x
103 )
Norm CaP-met
7.8 kDa
#1 #2 #3 #4 #1 #2 #3 #4
Western Blot CXC4
4-fold
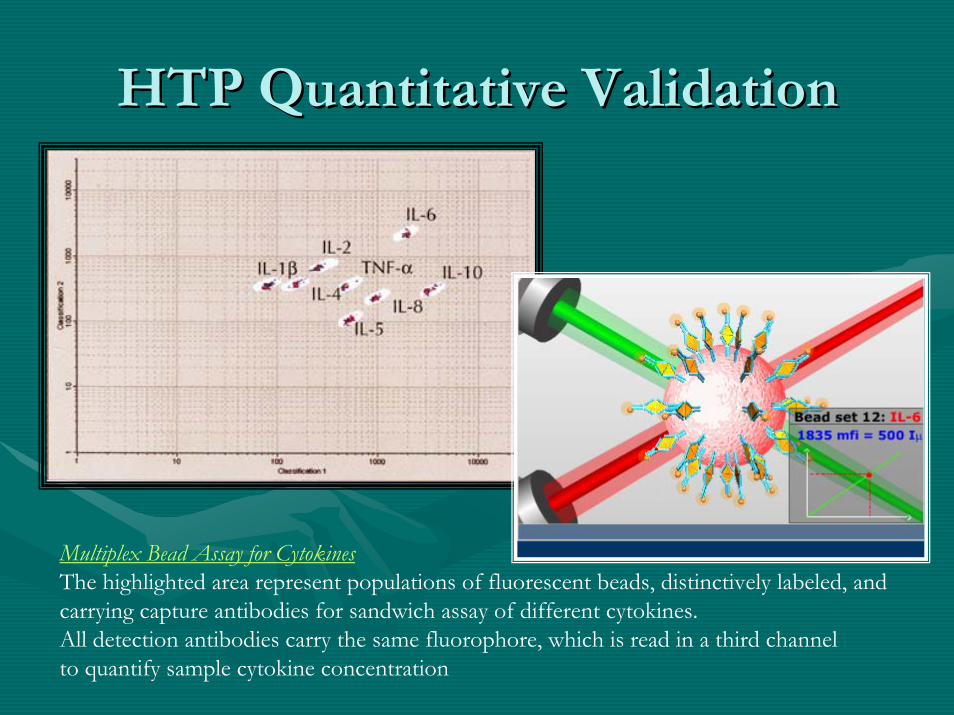
HTP Quantitative ValidationHTP Quantitative Validation
Multiplex Bead Assay for Cytokines The highlighted area represent populations of fluorescent beads, distinctively labeled, and carrying capture antibodies for sandwich assay of different cytokines. All detection antibodies carry the same fluorophore, which is read in a third channel to quantify sample cytokine concentration

SummarySummary
•• Whether or not you do the MS work yourselfWhether or not you do the MS work yourself………….. .. –– Know the specificsKnow the specifics…………!!–– Know the limitationsKnow the limitations……..!..!
•• Sample Prep is always importantSample Prep is always important……..but ..but which instrument you which instrument you have access to is also important!have access to is also important!
•• Similarly importantSimilarly important…….how is your data analyzed?? .how is your data analyzed?? –– DenovoDenovo??–– Matching Algorithm??Matching Algorithm??–– Mix of techniques??Mix of techniques??–– What are the cut off points and does it make sense?What are the cut off points and does it make sense?
•• Keep in mind that Keep in mind that validationvalidation must be part of your workflow!!must be part of your workflow!!•• So, make sure you have So, make sure you have confidenceconfidence in your choices before going in your choices before going
forward!forward!

Useful Links!Useful Links!
•• ii--mass.commass.com ionsource.comionsource.com•• spectroscopynow.comspectroscopynow.com bruker.combruker.com•• expasy.chexpasy.ch/tools/tools thermo.comthermo.com•• cprmap.comcprmap.com appliedbiosystems.comappliedbiosystems.com•• psidev.sourceforge.netpsidev.sourceforge.net shimadzu.comshimadzu.com•• prospector.ucsf.eduprospector.ucsf.edu luminexcorp.comluminexcorp.com•• jeolusa.com/ms/docs/ionize.htmljeolusa.com/ms/docs/ionize.html•• asms.orgasms.org (become a member!)(become a member!)•• hupo.orghupo.org•• matrixscience.commatrixscience.com•• proteomecenter.org/software.phpproteomecenter.org/software.php

Questions??Questions??