prion infection dynamics- an analysis of conversion mechanisms to characterize propagation of prion...
TRANSCRIPT

H. Williams Page 1 4/28/14
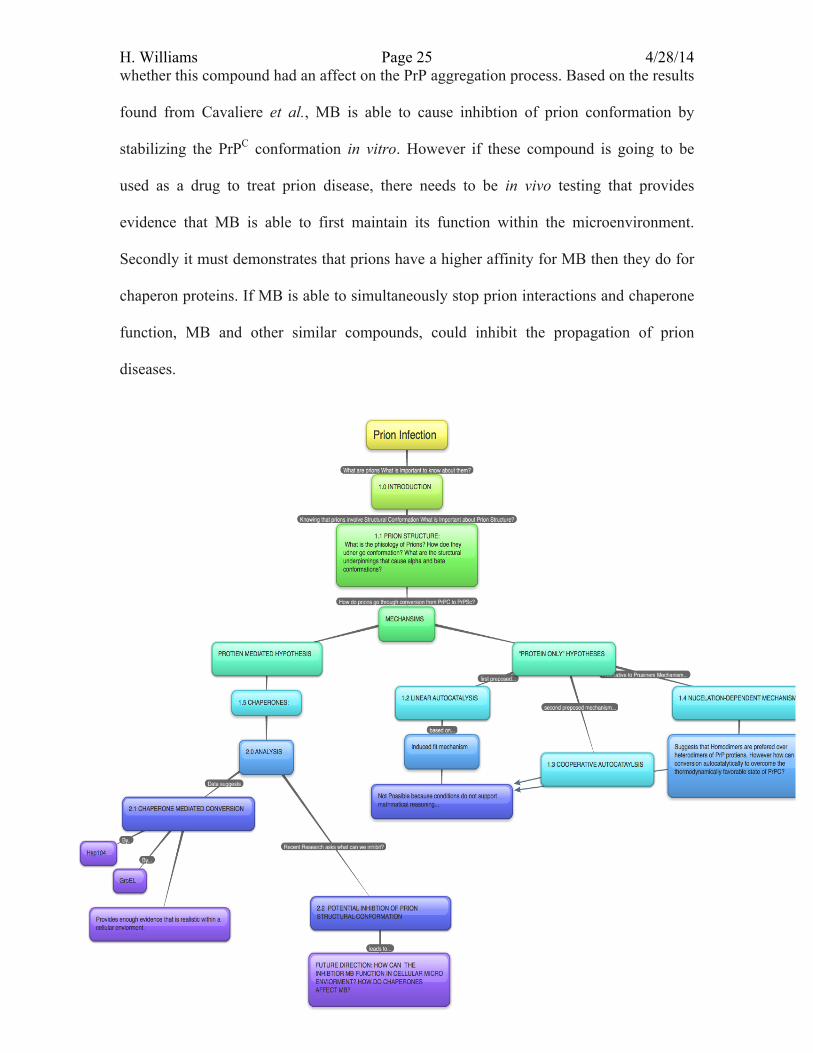
Prion Infection Dynamics: An Analysis of Prion Conversion Mechanisms to Characterize the Propagation of Prion Disease
Henry W. Williams
Department of Biochemistry, Hobart and William Smith, Geneva, NY 14456 Submitted by: April 27, 2014
1.0 Introduction
Common cognitive diseases such as transmissible Spongiform Encephalopathy
(TSE), Scrapie, Creutzfeldt-Jacob disease, Gertmann-Straussler-Scheinker disease,
Familia Insomnia, Kuru, and Alzheimer’s Disease, have all been linked to fibrilization
caused by prion structural conversion. Proteinaceous infectious particles or prions (PrP),
are a distinguished family of pathogens that, unlike viruses or bacteria, do not use nucleic
acids for replication. Historically it was believed that infectious pathogenic agents require
nucleic acids to replicate and cause disease. Toxic prions however, are able to induce
conformational changes among other healthy prion proteins to adopt an infectious
structure causing the proliferation of amyloid fibers and progressive loss of structure and
function in nerve cells, resulting in neurodegeneration. The degeneration of neurons in
the brain, particularly in elderly populations, is a serious health concern resulting in wide
spread neurodegenerative diseases, severe brain damage, and even death.
Over the two past decades there has been substantial research on the
conformational change of healthy cellular prion (PrPC) to the toxic prion isoform (PrPSc).
Neurodegenerative diseases have been found to share a common prion conformational
transition that contributes to neural toxicity. This has stimulated further research to better
understand prion structural dynamics. A mechanistic understanding of prion conversion
could provide essential new information for future medical treatment for prion related
diseases. To fully appreciate how infectious prions aggregate, one needs to understand

H. Williams Page 2 4/28/14 prion interaction at the structural level and the evolution of prion conversion
mechanisms.
1.1 Prion Structure and Toxic Characteristics:
Prions are a gene family that consists of three members that comprise of Prnd
which encodes Doppel, a testis-specific protein involved in male reproduction, Sprn, a
prion protein expressed in the central nervous system, and Prnp which encodes for PrPC
the precursor to prion disease (Watts et al., 2007). Human Cellular Prion Protiens (PrPC)
are 253 amino acids peptides possessing one disulfide bond and three to four alpha
helical structures. PrPC is homologous protein, with similar prions found in bovine,
sheep, mice, and other mammals. Though highly expressed within the central nervous
system (CNS) of mammals, their function still remains unclear. The ability of PrPC to
bind to copper (II) ions with relatively high affinity is thought to play a role in
intracellular signaling within neurons (Steele et al., 2006). PrPC proteins are glycosylated
and integrated to the plasma membrane of neurons and can be readily digested by
proteinase K (Eigen, 1996). In fact, proteases function to regulate the concentration of
PrPC in the CNS via the selective cleavage of the glycophosphatidylinositol (GPI)
glycolipid anchor which attaches PrPC to the cellular surface of neurons (Eigen, 1996).
Cleavage results in the digestion of PrPC, allowing for the body to control the
concentration of prions from reaching toxic levels. However prions that have adopted the
toxic PrPSc conformation become particularly resistant to proteases digestion, disrupting
the normal concentration of prions. For a prion infection to occur, there needs to be
enough accumulation of the protease resistant PrPSc proteins to allow for development of
plaque in the brain.

H. Williams Page 3 4/28/14 While PrPSc and PrPC have very different effects on cell viability, structurally
prions only differ in secondary and tertiary structure. Edman sequencing and mass
spectrometry showed very similar amino acid sequences for both PrPC or PrPSc (Harrison
et al., 1997). In contrast fourier transform infrared (FTIR) and circular dichroism (CD)
spectroscopy has revealed that PrPC is comprised of primarily alpha helices, while PrPSc
has a characteristically high beta sheet content (Harrison et al., 1997). When the healthy
PrPC is converted to its toxic PrPSc form, the N terminus end of PrP proteins contains
approximately 90 residues that are resistant to proteinase-K activity. This inhibition of
proteinase activity is thought to be caused
by the conversion of four different alpha
helical regions of the prion protein to beta
sheets.
Spectroscopic studies have further
demonstrated that there are three predicted
alpha helical regions in healthy PrPC (figure
1) (Harrison et al., 1997). Each of these
regions can undergo conformational
transitions from alpha helices to beta sheet
structures. However, data has shown
that the PrP(109-122) region is the
essential for the conversion of PrPC to
PrPSc. The adoption of the beta sheet
Figure 1: Ribbon Representation of Healthy PrP. 3D diagram of structural composition of PrPC. The Binding surface is the shadowed region. The cavity is comprised of three alpha helices H1 in blue (F144, N146, E149), H2 in green (H180, V183, N184, V187, K188, T191, V192, T194, T195), and helix H3 in red (E203, I206, K207, E210, R211, E214, Q215). (B) Is the same structure except it is rotated by about 90°.

H. Williams Page 4 4/28/14 structure in this region would therefore be toxic. This conformation change to beta-
sheets is initiated by the interactions of PrP(109-122) with fragments PrP(104-122) and
PrP(129-141) (Harrison et. al., 1997).
This change in the structural conformation alters the way in which prion proteins
cooperate. These abnormal interactions result in the change in conformation of healthy
PrPC to the toxic isoform of PrPSc. Since toxic PrPSc is resistant to proteinase K, the build
up of PrPSc causes aggregation, forming amyloid fibers that build up and cause plaque in
brain tissue. The specific mechanism for this process has been highly controversial. A
progression of hypotheses has contributed to the evolutionary understanding of how
prions interact and whether prions undergo autocatalysis or enzymatic conversion.
1.2 “Prusiner Mechanism”-Linear Auto Catalysis In the 1990’s Stanley Benjamin
Prusiner proposed one of the first
mechanisms for prion conformation through
an autocatalytic approach (Eigen, 1996).
Prusiner hypothesis suggested that the toxic
PrPSc product of the catalytic conversion of
PrPC facilitates the conversion of more PrPC
to its infectious conformation. The
secondary, tertiary, and even quaternary
conformations are the determinants for a
prions’ pathological structure. Prusiner first
proposed a model that resembled an “induced fit process”, which later became known as
the Prusiner mechanism for linear autocatalysis. This process involves a PrPSc monomer
or dimer acting as a template to mediate the conformational change of PrPC to PrPSc.
Figure 2: Prusiner’s Linear Autocatalysis. Induced fit mechanism in causing prion conversion to toxic structure. (A) represents PrPC conformation, while (B) is indicative of the toxic PrPSc structure.

H. Williams Page 5 4/28/14 According to Prusiner, the binding of these two monomers lowers the free energy barrier
between PrPC and PrPSc structures (Harrison, 1997). Upon formation of the new PrPSc,
more PrPC monomers are recruited for further conversion, resulting from PrPC having a
high binding affinity for PrPSc.
Since beta-sheets are more stable than alpha helices the conversion of PrPC to
PrPSc is thermodynamically favored; however it is usually prevented due to a high kinetic
barrier. When PrPSc is present the activation energy for the conversion of healthy PrPC to
the toxic PrPSc is then lowered. Prusiner suggests that these conditions, provide enough
driving force to cause the conversion of PrPC to PrPSc (figure 2) (Eigen, 1996).
To better understand conformational kinetics of this mechanism, in regards to
prion infection, the propagation of prions at low concentrations must be examined over
time. At the initial stage of infection, the concentration of PrPSc is assumed to be zero.
Under the assumption that the original population of PrPC is on the order of ~1024, and
the PrPSc population was close to zero, then the non-catalytic rate kAB would have to be
between 10-22 or 10-23 s-1. This rate has to be small because if it was any higher,
spontaneous conversions could cause prion disease when an infection is not present.
However In order for an infection to occur the rate of turn over (kT = [A]/KM) (figure 2)
of the PrPSc-PrPSc dimer would have to be large enough to create an accumulation of
monomeric PrPSc. To cause disease, the linear autocatalysis mechanism would require kT
to be on the order of 10-8 to 10-7 s-1 to accumulate enough of the PrPSc. An examination of
the ratio of KT/KAB provides valuable information regarding the rate of enhancement of
the prion conversion. Here, Eigen et al. suggests that the ratio of KT/KAB is
approximately 1015, which they express to be unrealistic for a non-cooperative
autocatalytic mechanism. Eigen et al. states that there is no non-cooperative enzymatic

H. Williams Page 6 4/28/14 reactions that could have a turnover that high. Though the linear autocatalysis mechanism
was one of the first accepted models for prion conversion, it was unable accurately
express the true rate of toxic prion conversion. To correct this, Prusiner also suggest a
second hypothesis to provide a better representation of how toxic prions replicate. In this
second mechanism, prions undergo structural transition via cooperative autocatalysis.
1.3 “Prusiner Mechanism”-Cooperative Autocatalysis A second mechanism proposed by Prusiner is
the cooperative autocatalysis mechanism. The
cooperative autocatalysis model, suggests that PrPSc
is still thermodynamic favorable and causes the
decrease in the activation energy barrier in the
conversion of PrPC. However the two PrPSc
monomers remain bound after conversion is
complete. This accumulation of bound PrPSc results
in further recruitment of PrPC, lowering the activation
energy of conversion for each new PrPSc
that is converted (figure 3).
Each time a new PrPC binds to the
existing oligomer, the amount of energy
needed to convert to the toxic form decreases (Eigen et al., 1996). This method of
conversion is very similar to the process found in subunit cooperativitiy in hemoglobin
(Egen et al., 1996). As a result of this exponential decrease in activation energy, the rate
of conversion increases as PrPSc accumulates. However the cooperativity in binding
depends on the stability of the subunits.
Figure 3: Prusiner’s Cooperative Autocatalysis. Analyzing the catalyst affects of oligomeric Prion proteins. Cooperative Autocatalysis suggests PrPSc conformer is thermodynamically favored. Reaction is driven concentration of previous reaction step.

H. Williams Page 7 4/28/14 Figure 3 fails to account for how PrPC can affect the stability of other nearby
prions in the PrPSc and PrPC conformation (Laurent, 1997). According to Laurent et. al,
there are two hypothesis that have been formulated as a means of possibly determining
whether there are two different binding domains that bind PrPSc and PrPC or if PrPSc and
PrPC are competing for the same binding domain. In linear autocatalysis, there would
only be one binding domain. In this case the opposite isoform should have a stronger
binding affinity for that site. Meaning that PrPSc should have a higher binding affinity for
PrPC, while PrPC has a higher affinity for
PrPSc. While for cooperative autocatalysis
there could be heterogeneous and
homogenous binding domains.
Heterogenous binding domains would
allow for specific binding between PrPSc
and PrPC. While homogenous binding
domains would be a set of identical
domains binding, which could bind
without specificity. To understand the
difference between linear autocatalysis,
PrPSc-PrPC, and PrPSc-PrPSc coopertivity
autocatalysis, one has to compare between
the catalytic efficiency between each type
of aggregation.
Figure 4: Comparing Linear Autocatalysis to Cooperative Autocatalysis. Comparing the catalytic efficiency between three forms of autocatalysis to determine, catalytic activator causing increased PrPSc turnover. (A)Linear Autocatalysis is compared to autocatlysis with the formation of a PrP heterodimer (B) or PrP homodimer (C) .

H. Williams Page 8 4/28/14 In Prusiner’s cooperative autocatalysis hypothesis, either a heterodimer can form
from PrPSc and PrPC, or a homodimer can arise from two PrPSc with PrPSc. Each of these
dimers influences the catalytic efficiency of prion propagation differently. Luranet et al.
describes that understanding the differential activity of heterodimers and homodimers
reveals the key influences of prion propagation based on the relative binding affinities
that PrPSc has for either itself or PrPC. In figure 4, Luranet et al. compares the linear
autocatalytic efficiency (Figure 4.A) with the cooperative catalytic efficiency of when
PrPSc has a higher binding efficiency for PrPC ( Figure 4.B) or PrPSc ( Figure 4.C).
The formation of either a homo or a hetero-catalytic oligomer can have different
kinetic effects on prion propagation. Figure 4.B demonstrates that a heterodimer would
provide an excellent catalyst to initiate the infection. However this would mean that PrPSc
has a weak affinity for itself, which would, inhibits PrPSc monomers from assembling
into amyloid fibers. Assuming that monomeric PrPSc is incapable of catalyzing
aggregation, the cooperative autocatalysis of heterodimers would increase affinity for
binding of PrPC. Each PrPC conversion to PrPSc causes an increase in the affinity of PrPC
binding to PrPSc. In contrast, the homodimer illustrates that the PrPSc has a higher affinity
to PrPSc. Based on the information provided by Luranet et al., this hypothesis is less
likely to occur due to the fact that interactions between PrPSc would be too great,
decreasing PrPSc binding affinity for PrPC. Homodimers of PrPSc would dramatically
decreases the number of healthy PrPC converted to toxic PrPSc. Thus the heterodimer
hypothesis illustrates the most likely means by which PrPSc binds to PrPC, causing
elevated propagation of PrPSc in a cooperative way. However there is more scientific
evidence that illustrates that prions could propagate based on this homodimer hypothesis
through a Nucleation-dependent Mechanism.

H. Williams Page 9 4/28/14 1.4 Nucleation-Dependent Mechanism One of the most widely excepted hypotheses within “protein-only” aggregation
mechanism is the nucleation-dependent model. According to this model, PrPSc and PrPC
differ in their quaternary structures. Lansbury suggests that nucleation of four PrPSc
monomers must occur to accelerate the fibrilization and progression of disease.. Prusiner
and Luranet originally suggested that accumulation of PrPSc was based on the formation
of PrPC-PrPSc heterodimers subunits. According to Lansbury, PrPSc has a higher affinity
for PrPSc than PrPC, as a result of its monomeric instability. Lansbury assumes that PrPSc
is stabilized by the intermolecular (allosteric) interactions between other PrPSc proteins.
Unlike Prusiner, where the concentration of monomeric PrPSc concentration determines
the speed of the conversion and development of disease, Lansbury suggests that the
formation of PrPSc oliogmers determines rate of fibrilization. Lansbury suggest that the
accelerated conformational change and fibrilization are due to the development of nuclei,
which then polymerize together to form long fibril like structures. In order for there to be
rapid aggregation, a nucleation step is needed to stabilize PrPSc and facilitate the
polymerization of oligomers into fibril structures. This oligomerization step has been
seen numerous recent studies involving how prevent the conversion of prions (Cavaliere
et al., 2013).
In short, there are three fundamental modifications from cooperative autocatlysis
that make up the nucleation-dependent mechanism (NDM) (Eigen et al., 1997). The first
requires NDM to produce quaternary structures to catalyze the formation of fibrils.
Second, in the NDM, unlike Prusiner model, PrPC is the thermodynamically preferred
state relative to PrPSc.. Finally, according to NDM PrPSc monomers are less stable than
PrPC, however in their dimeric state, bound PrPSc is more stable than PrPC. Eigen et al.,

H. Williams Page 10 4/28/14 explains that free PrPSc particles can be stable, however their stability is increased upon
binding to other PrPSc molecules (forming a nucleus) (Eigen et al, 1996).
Eventually, according to NDM, these PrPSc homodimers would form a sextet
(seed), which causes a shift in the equilibrium to form fibrils (figure 5). The nucleation
step of these seeds limits the development of disease. PrPC is more stable than
monomeric PrPSc, preventing PrPC from spontaneously converting to PrPSc. However
PrPSc gains greater stability when oligomerized. The nucleation of PrPSc into seeds then
causes a shift in the activation energy barrier, resulting in rapid polymerization into
fibrils. This rapid fibirlization of seeds accounts for the rapid growth in the disease phase.
While the nucleation-dependent mechanism provides a possible alternative to
accumulation of PrPSc in an autocatalytic fashion, this hypothesis still does not take into
account whether there are other proteins involved that could stimulate the conformation
of these prion proteins. It is known that there are chaperon proteins within the cell that
facilitate the aggregation of proteins when under various stresses.
Figure 5: The Nucleation-dependent mechanism as a means to convert PrPC to PrPSc. This mechanism results from the idea that prion oliogmers must form first before the accelerated aggregation. According to Lansbury, prion aggregation is dependent on two steps. A oliogmerization step and secondly a accelerated fibrilization step.

H. Williams Page 11 4/28/14 1.5 Which mechanism is the correct one? Eigen et al. demonstrates that there is a logical progression from prusiner’s
original linear autocatalysis to Lansbury’s nucleation mediated mechanism. Prusiner’s
autocatalysis mechanisms are capable of causing exponential growth of toxic prions.
However mathematical analysis of each mechanism revealed that the method of
conversion is unrealistic to conditions found in a cell. The issues with Prusiner’s
autocatalysis method is that the rate of production of PrPSc is either slower than the rate
of degradation, meaning disease can never occur, or spontaneous conversion of PrPC to
PrPSc occurs far too readily resulting in the development of disease every time (Eigen et
al., 1997). Both of these inaccuracies illustrate that the prusiner models fail to accurate
represent the true method of prion conversion. Both the cooperative autocatalysis and
linear autocatalysis method do not have the ability to control spontaneous growth of PrPSc
and don’t provide realistic conditions for PrPSc to accumulate non-spontaneously.
Adapting from Prusiner’s cooperative autocatalysis mechanism, the NDM asserts
that there needs to be a nucleation step that develops seeds which can then activate the
accelerated formation of fibrils. Based on the fact that seed formation decreases the
activation energy barrier in order to cause development of the disease, the nucleation step
acts as a threshold preventing or allowing accelerated fibrilization. Where either
accumulation of PrPSc is to low to form seeds, preventing disease from spontaneously
occurring, or the concentration is high enough allowing forward progression of
fibrilization due to the presence of PrPSc oliogmers. Though research demonstrates that
the NDM can occur under cellular conditions, Eigen et al. questions whether the
mechanism can occur autocatalylitcally. Though nucleation provides a barrier to prevent
the onset of spontaneous disease, Egien et al. questions how the cellular environment is

H. Williams Page 12 4/28/14 able to provide the conditions needed to cause the formation of seeds when PrPC is the
thermodynamically favored state. According to Eigen et al., there has be skepticism on
whether the accumulation of PrPSc is enough to overcome the kinetic barrier needed to
convert PrPC to PrPSc or if there is a separate agent that could be catalyzing prion
aggregation (Eigen et al., 1996).
1.6 Chaperons Proteins: In more recent studies, researchers have adopted an alternative theory that
diverges from “protein-only” hypotheses, previously described by Prusiner and Lansbury.
It has been proposed that there are molecular chaperones that can regulate the
conformational transitions between the two PrP structural states. This is a radically
different proposal compared to the previous propagation models, which demonstrate that
prions facilitate their own conformational change through autocataylsis.
Molecular chaperons are proteins that assist the non-covalent folding or unfolding
of other proteins. Though their primary function is to prevent polypeptide chain or
protein subunits from aggregating, chaperones have been found to mediate the misfolding
of secondary and tertiary structures. Many of these chaperons are heat-shock proteins
(Hsp) because proteins have a tendency to aggregate more readily when under heat-
related stress. Heat-shock proteins attempt to prevent aggregation by facilitating protein
interactions. Chaperone proteins are typically activated due to infections, inflammation,
hypoxia, or other related stresses to a cell.
There is evidence to suggest that heat shock proteins facilitate the conversion of
PrPC to PrPSc conformation, specifically the chaperons, GroEL and Hsp104 (DeBurman
et al., 1997). This chaperone mediated conversion hypothesis of prions provides a new
understanding of the nature of PrP conversion. If chaperones help to elevate
conformational change of PrPC to PrPSc then this would change our understanding of the

H. Williams Page 13 4/28/14 nature of PrP intermediates. In doing so, researchers have shifted how prions propagation
is viewed as a disease, suggesting that the conversion of prions is more complex than we
originally thought.
Hsp104 is a protein-remolding factor that belongs to the AAA+ (Adenosine
triphosphatases Associative with diverse Activities) family. This family of proteins is
able to hydrolyze ATP to cause conformational changes. AAA+ proteins are found in all
organisms. These proteins are essential for many cellular functions such as DNA
replication, protein degradation, membrane fusion, signal transduction, and the regulation
of gene expression (Grimminger-Marquardt et al., 2010). As a molecular chaperone
HsP104 has a central role in the digestion of aggregates after heat shock (Grimminger-
Marquardt et al., 2010). However Hsp 104 has the capacity to catalyze protein
aggregation as disaggregase causing the misfolding of proteins. This disaggregation
activity has been linked to prion propagation. In yeast Hsp104 is able to cutting prionic
fibrils creating prion seeds (propagons). These seeds are then inherited to the next
generation of cells, when the parent cell divides its cytoplasm into two daughter cells.
This phenomenon is known as inherited prionic disease.
GroEL belongs to a family of chaperons that is found in a number of bacteria
including Escherichia coli. To function properly GroEL requires GroES as a cochaperone
protein to prevent the undesired aggregation of proteins (Martin et al., 1993). GroEL is
both structurally functionally similar to Hso60 and Hsp10 found in eukaryotes. Within
the cell the GroEl/ES complex accelerates the binding, conformation, and release of
various protein reactions. In order for cause a change in the conformation, GroEL needs
to bind to the substrate protein and then bind to ATP. The assembly of this complex then
recruits GroES, which causes the substrate protein to be fold. Hydrolysis of ATP and the
H. Williams Page 14 4/28/14 binding of a new substrate cause the protein to be released into the cytosol. However the
ATP-dependent release of GroEL in the absence of GroES results in aggregation rather
than favorable folding (Martin et al., 1993). Both GroEL and Hsp104 have significantly
influenced prion propagation and assembly of fibril aggregates in Prion disease.
2.0 Analysis
2.1 The Involvement of Chaperon Proteins in Prion Conformation. To asses the role chaperone proteins play in prion conversion, DebBurman et al.
examined seven major cellular chaperones GroES (Hsp10), Hsp26, Hsp40,
GroEL(Hsp60), Hsp70, Hsp90, and Hsp104 for their ability to activate the change in
conformation and physical states of other proteins (DebBurman et al,. 1997). First, they
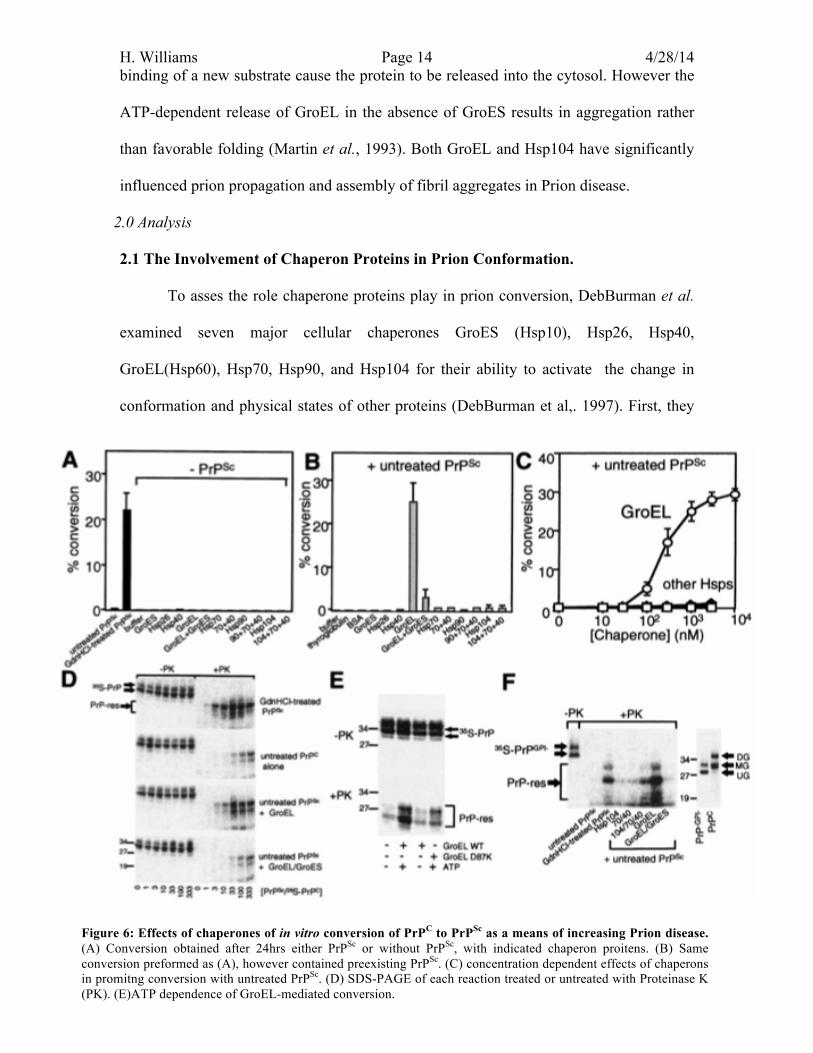
Figure 6: Effects of chaperones of in vitro conversion of PrPC to PrPSc as a means of increasing Prion disease. (A) Conversion obtained after 24hrs either PrPSc or without PrPSc, with indicated chaperon proitens. (B) Same conversion preformed as (A), however contained preexisting PrPSc. (C) concentration dependent effects of chaperons in promitng conversion with untreated PrPSc. (D) SDS-PAGE of each reaction treated or untreated with Proteinase K (PK). (E)ATP dependence of GroEL-mediated conversion.

H. Williams Page 15 4/28/14 confirmed that none of the chaperon proteins could induce the conformational change of
PrPC into PrPSc in the absence of PrPSc (figure 6.A). In contrast chaperones can induce the
conversion of PrP proteins, in the presence of PrPSc (figure 6.B). This finding illustrates
that preexisting PrPSc is needed within the system to causes an infection (DebBurman
eal., 1997).
Currently it has been found that in order for PrPC to convert PrPSc, preexisting
PrPSc need to be slightly denatured in order to facilitate the transition between the two
conformations. Looking at the SDS-Page gel in figure 6.D, the denaturation of PrPSc (6M
Gdn-HCl pretreatment) caused a large accumulation of PrPSc. When this occurs,
proteinase K cannot degrade toxic prions. This is unlike PrPC, which can easily be
degraded by proteinase K. Knowing this DebBurman et al., wanted to test whether
chaperones were capable of converting undenatured PrPSc. It was found that many of the
chaperons were incapable of reproducing PrPSc (figure 6.B) in the presence of preexisting
untreated PrPSc monomers except for GroEL. GroEL was able to convert around 25-30%
and occasionally 50-100% of PrPC to PrPSc(Figure 6.B). Looking back at figure 6.D, they
found that GroEL was able to yield the similar PrPSc accumulations as the completely
denatured PrPSc (6M Gdn-HCl pretreatment). This demonstrated that GroEL was capable
of reducing the quantity of preexisting PrPSc required for detectable conversion and it
raised maximal number of conversions by ten fold (DebBurman et al., 1997). By
comparing different doses of GroEL and other Hsps, researchers found similar results
(figure 6.C), illustrating that GroEL has the ability to promote the conversion of untreated
PrPSc to a statistically significant higher rate of conversion. Interestingly, the activity of
GroEL was stunted by either the presence of GroES or lack of ATP.

H. Williams Page 16 4/28/14 PrP conversion was not observed in the absence of ATP or in the presence of
GroES. Looking at figure 6.E the SDS PAGE gel shows that in the presence of ATP and
the wild type (WT) GroEL, PrPSc was able to accumulate in the presence of Proteinase K.
However when GroEL contains a mutation that blocks the release of the substrate protein
(D87K) or when ATP was not present, a reduction was seen in ability of GroEL to
convert PrPC to PrPSc (figure 6.E). It is known that GroEL hydrolyzes ATP to release the
old substrate protein to bind to a new substrate. Thus if ATP or the binding domain is
unable to release the substrate protein, there is an inhibition in the function of GroEL.
The same issue occurs in the presence of GroES. The SDS-PAGE gel in Figure 6.D,
illustrates that when proteinase K is active and both GroES and GroEL are present, there
is a decrease in the accumulation of PrPSc. Comparing this result to GroEL is alone, there
is greater aggregation when GroES is not present. This is because in the absence of
GroES, GroEL is able to make errors, resulting in aggregation rather than favorable
folding (Martin et al., 1993).
To gain insight about the chaperone-mediated processes of GroEL, researchers
analyzed kinetics of the conversion of PrPC to PrPSc by evaluating protease resistance and
insolubility of PrPSc. It was found that in the presence of GroEL and monomeric PrPSc,
the rate of nucleation didn’t change from reactions where GroEL was absent(DebBurman
et al., 1997). However when PrPSc has been nucleated into oligomers and GroEL is
present, GroEL is able to increase the rate of the PrPSc forming a polymer. GroEL is able
to facilitate the conversion of PrPC by bringing PrPSc oligomers and PrPC in close
proximity. DebBurman et al.’s protease resistance analysis provided evidence that the
conversion of PrPC is a two-step process. the initial formation of nucleus which then
leads to a chaperone driven reaction which promotes PrPSc propagation. This data that

H. Williams Page 17 4/28/14 DebBurman et al. presents demonstrates chaperones have a significant effect on prion
propagation. Based on SDS-PAGE and percent conversion analysis, DebBurman et al.
had shown that GroEL has the ability to faciltate rate of the conversion of PrPC to PrPSc.
Although there were no other proteins that facilitated the conversion in the presence of
untreated PrPSc, Hsp104 demonstrated that it could convert PrPC when preexisting PrPSc
was slightly denatured.
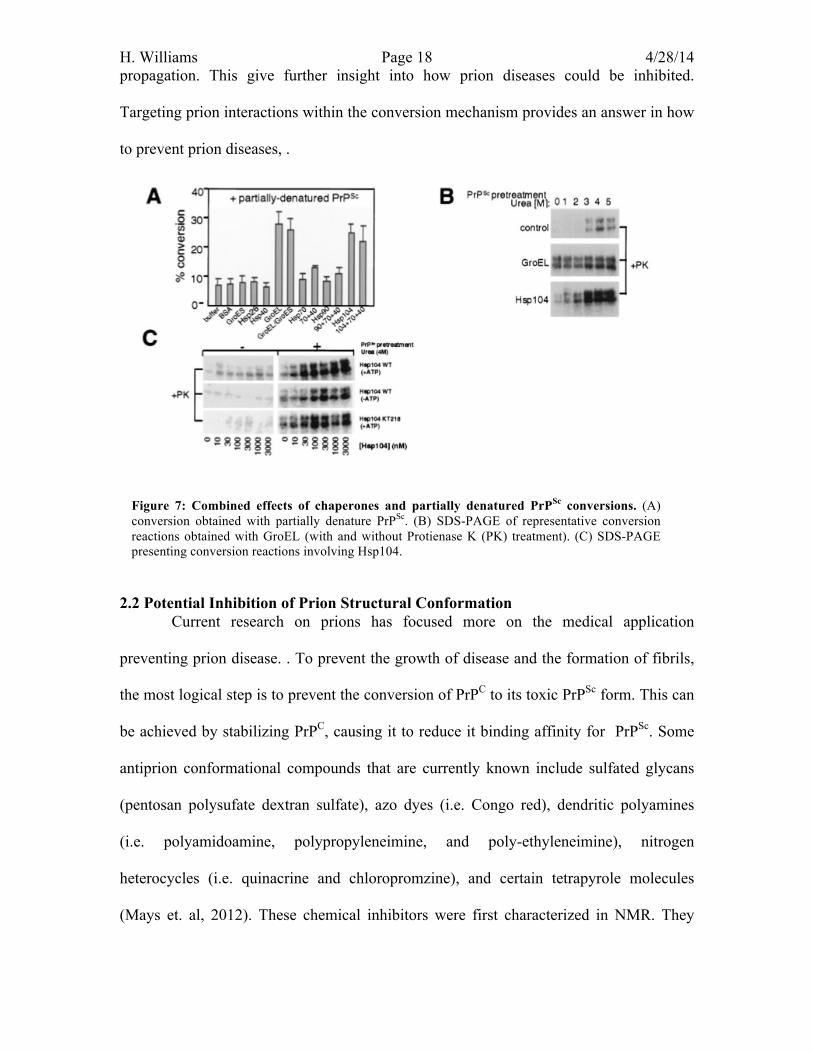
For this reaction PrPSc was exposed to urea which is a milder denaturant because
Gdn-HCl could inhibit the function of certain chaperones. When PrPSc is partially
denatured chaperones only stimulate conversion to a small degree. However Hsp104 was
found to strongly stimulated conversion (Figure 7.A), contributing to 20-30% (and
occasionally more than 50%) of the total PrPC converted (DebBurman et al., 1997).
DebBurman et. al, then compared the activity of GroEL and Hsp104 to one another in the
presence of protienase K and a pretreatment of Urea. It was found from the SDS-PAGE
gel in figure 7.B that GroEL and Hsp104 could accumulate PrPSc under the same
conditions to a significantly higher level then the control (which did not contain any
chaperones). DebBurman et al. then tested the ATP dependence of Hsp104. Figure 7. C,
demonstrates Hsp104 does not depend on ATP. Even with ATPase-deficient Hsp104
mutants (KT218 and KT620), the conversion of PrPSc was the same as the wild type
(DebBurman et al., 1997).
The data that DebBurman presents not only provides significant evidence that
GroEL and Hsp104 affect prion conversion but they also illustrate that the state of which
prions are denatured can improve the ability of chaperones to facilitate conversion. This
data provides noteworthy insight about how the state of prions affects how chaperone
function. This demonstrates that many chaperones can have a primary role in prion
H. Williams Page 18 4/28/14 propagation. This give further insight into how prion diseases could be inhibited.
Targeting prion interactions within the conversion mechanism provides an answer in how
to prevent prion diseases, .
2.2 Potential Inhibition of Prion Structural Conformation Current research on prions has focused more on the medical application
preventing prion disease. . To prevent the growth of disease and the formation of fibrils,
the most logical step is to prevent the conversion of PrPC to its toxic PrPSc form. This can
be achieved by stabilizing PrPC, causing it to reduce it binding affinity for PrPSc. Some
antiprion conformational compounds that are currently known include sulfated glycans
(pentosan polysufate dextran sulfate), azo dyes (i.e. Congo red), dendritic polyamines
(i.e. polyamidoamine, polypropyleneimine, and poly-ethyleneimine), nitrogen
heterocycles (i.e. quinacrine and chloropromzine), and certain tetrapyrole molecules
(Mays et. al, 2012). These chemical inhibitors were first characterized in NMR. They
Figure 7: Combined effects of chaperones and partially denatured PrPSc conversions. (A) conversion obtained with partially denature PrPSc. (B) SDS-PAGE of representative conversion reactions obtained with GroEL (with and without Protienase K (PK) treatment). (C) SDS-PAGE presenting conversion reactions involving Hsp104.

H. Williams Page 19 4/28/14 were found to stabilize the PrPC conformation, and prevent the structural change of PrPC
to PrPSc (Hosokawa-Muto et. al, 2009). In order to determine a viable drug for prion
disease, researchers need to look at current compounds that have similar characteristics to
antiprion compounds.
In a recent study Methylene blue (MB), which is commonly used for treatment of
several medical conditions concerning memory consolidation and neuro-protective
effects, has been under investigation for its potential anti-prion characteristics (Cavaliere
et al., 2013). This FDA approved oral and I.V. administered compound is a heterocylic
aromatic phenothianzine compound that is water-soluble. It is able to pass the blood-
brain barrier, making it suitable to reach neuronal targets such as prions (Cavaliere et al.,
2013). This particular compounds has been shown to have anti-aggregating properties in
relation to Alzehimers Disease (AD), and has been shown to restrict protein chaperons
such as hsp70/hsp90 (Cavaliere et al., 2013). Knowing that this compound has potential
to provide treatment of neurodegenerative disease and prevent protein aggregation,
Cavaliere et al. tested whether this compound had an affect on the PrP aggregation
process.
In order to explore whether MB has the capability of interacting with the PrPC
ground state, interactions with monomeric PrPC were analyzed through NMR
experiments at ph 4.6 and 7.0. This allowed for researchers to map out the region of
where MB could interact with PrPC. From this NMR study they found that there were 20
residues located around the helix H1, H2, and the N-terminal region helix H3 that have
the potential to interact with MB. Out of the nine residues that showed chemical shifts
N246, K188, 1T191, T194, T195, and Q215, all fell in the active binding domain of PrPC
(figure 1). This high number of ionizable residues (1 Arg, 2 Lys, 1 His, and 5 Glu), could
H. Williams Page 20 4/28/14 potentially affect the attraction of the positively charged MB molecule within a pH of
4.6-7.0 (Cavaliere et al., 2013).
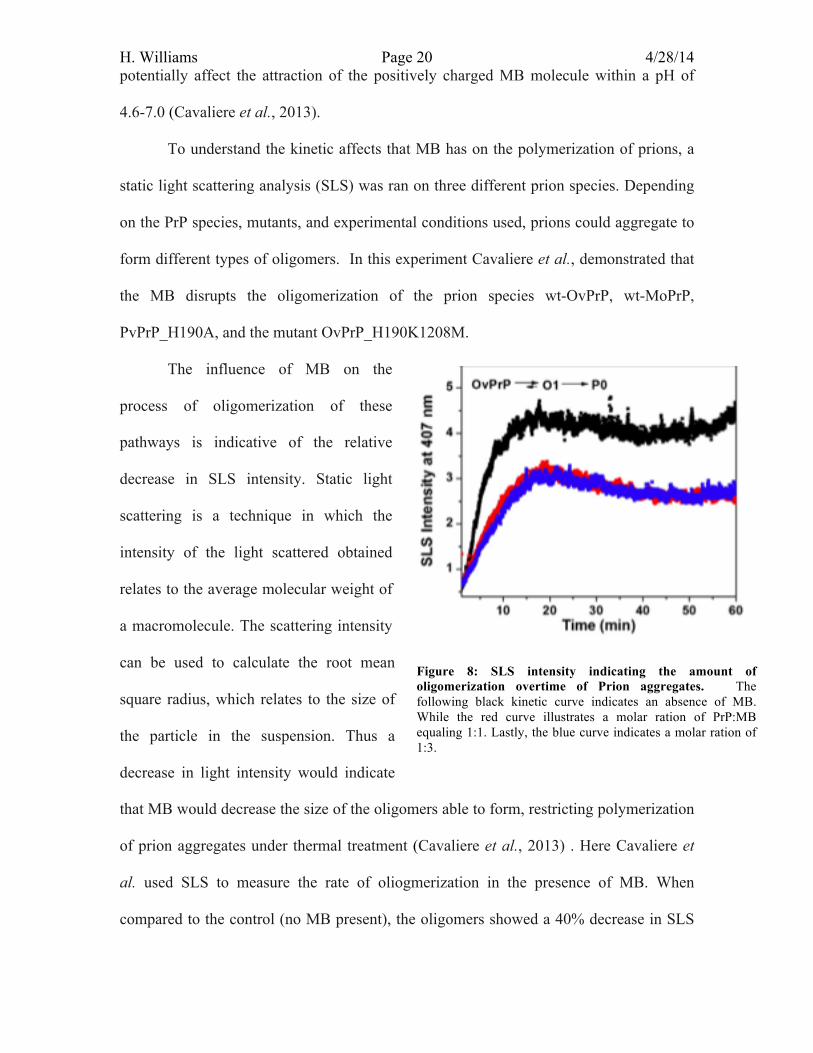
To understand the kinetic affects that MB has on the polymerization of prions, a
static light scattering analysis (SLS) was ran on three different prion species. Depending
on the PrP species, mutants, and experimental conditions used, prions could aggregate to
form different types of oligomers. In this experiment Cavaliere et al., demonstrated that
the MB disrupts the oligomerization of the prion species wt-OvPrP, wt-MoPrP,
PvPrP_H190A, and the mutant OvPrP_H190K1208M.
The influence of MB on the
process of oligomerization of these
pathways is indicative of the relative
decrease in SLS intensity. Static light
scattering is a technique in which the
intensity of the light scattered obtained
relates to the average molecular weight of
a macromolecule. The scattering intensity
can be used to calculate the root mean
square radius, which relates to the size of
the particle in the suspension. Thus a
decrease in light intensity would indicate
that MB would decrease the size of the oligomers able to form, restricting polymerization
of prion aggregates under thermal treatment (Cavaliere et al., 2013) . Here Cavaliere et
al. used SLS to measure the rate of oliogmerization in the presence of MB. When
compared to the control (no MB present), the oligomers showed a 40% decrease in SLS
Figure 8: SLS intensity indicating the amount of oligomerization overtime of Prion aggregates. The following black kinetic curve indicates an absence of MB. While the red curve illustrates a molar ration of PrP:MB equaling 1:1. Lastly, the blue curve indicates a molar ration of 1:3.

H. Williams Page 21 4/28/14 intensity when the ratio of PrP to MB is 1:1 and 1:3 (figure 8). Figure 8 demonstrates that
the ability of PrP to oligomerize in the presence or absence of MB. By comparing the
SLS intensities of PrP oligomerization without MB (black line) with the intensities of
different ratios (red and blue lines) of PrP to MB, it can be seen that MB is able to reduce
the amount of oligomeization. By restricting the number of oligomers forming prevents
or slows the process of fibrillation.
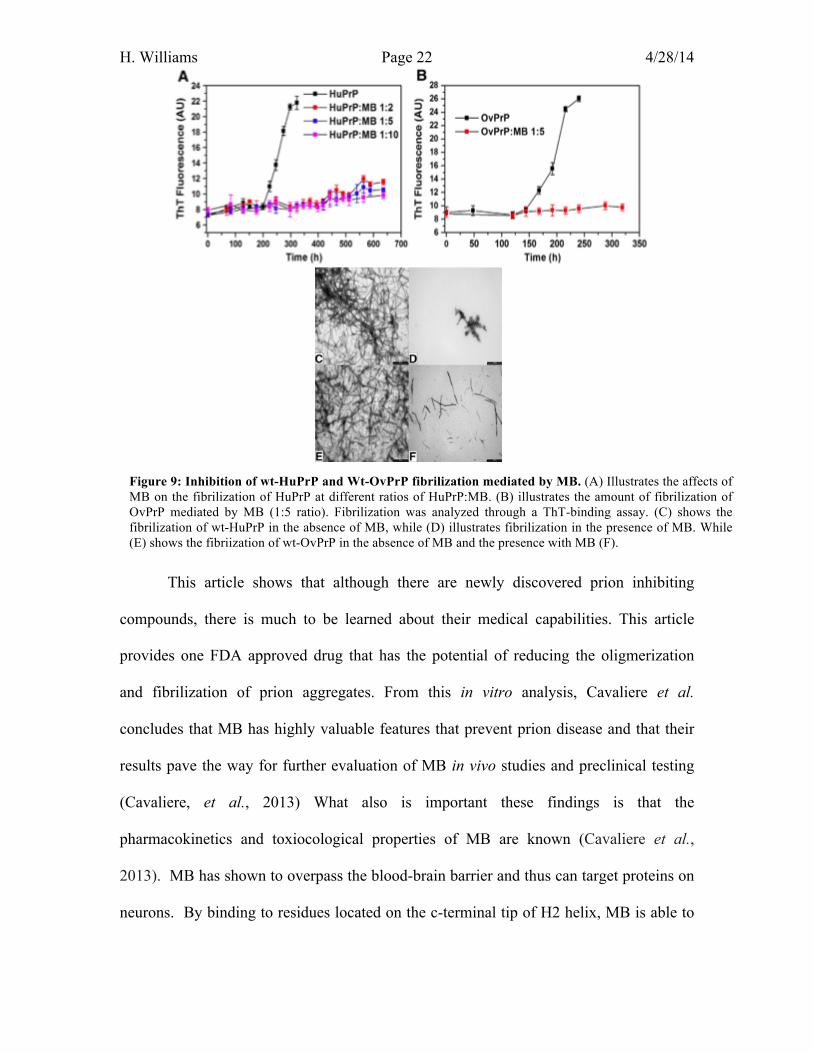
The last experiment preformed by Cavaliere et al. studied the affects of MB on
fibril formation. As a control, two prion fibril species, wt-HuPrp and wt-OvPrP, were
generated at pH 6.0 in the absence and presence of MB. Prion fibril formation was
monitored using Thiolavin T (ThT) spectroscopic assay. Figure 9.A and 9.B illustrates
the differences in binding curves of wt-HuPrP and wt-OvPrP at different molar ratios of
PrP:MB. In both species, in the absence of MB, the ThT signal indicated that there was a
sigmodial in protein aggregation and fibrilization (black line). In both cases, there was a
significant decreases in fibrilization when MB was present in any concentration (colored
lines) (Figure 9 A and B). This suggests that MB is able to suppress fibril formation. This
conclusion can be supported by TEM images, showing that both wtHPrP and wt-OPrP in
the presence and absence of MB (figures 9.C,D,E, and F). Figure 9.C illustrates the
fibrilization of wt-HuPRp in the absence of MB, while figure D illustrates the fibrilzation
in the presence of MB. The same was done for wt-OvPrP, where figure 9.E illustrated
fibrilization with MB and Figure 9.F illustrated with fibrillation. In both Cases (figures
9.D and 9.E), MB was able to inhibit the formation of fibrils. This alone demonstrates
that MB could prevent the onset of prion disease.
H. Williams Page 22 4/28/14
This article shows that although there are newly discovered prion inhibiting
compounds, there is much to be learned about their medical capabilities. This article
provides one FDA approved drug that has the potential of reducing the oligmerization
and fibrilization of prion aggregates. From this in vitro analysis, Cavaliere et al.
concludes that MB has highly valuable features that prevent prion disease and that their
results pave the way for further evaluation of MB in vivo studies and preclinical testing
(Cavaliere, et al., 2013) What also is important these findings is that the
pharmacokinetics and toxiocological properties of MB are known (Cavaliere et al.,
2013). MB has shown to overpass the blood-brain barrier and thus can target proteins on
neurons. By binding to residues located on the c-terminal tip of H2 helix, MB is able to
Figure 9: Inhibition of wt-HuPrP and Wt-OvPrP fibrilization mediated by MB. (A) Illustrates the affects of MB on the fibrilization of HuPrP at different ratios of HuPrP:MB. (B) illustrates the amount of fibrilization of OvPrP mediated by MB (1:5 ratio). Fibrilization was analyzed through a ThT-binding assay. (C) shows the fibrilization of wt-HuPrP in the absence of MB, while (D) illustrates fibrilization in the presence of MB. While (E) shows the fibriization of wt-OvPrP in the absence of MB and the presence with MB (F).

H. Williams Page 23 4/28/14 stabilize this structurally unstable region, leading to the prevention of unfolding and
aggregation of health PrPC proteins. MB was also found to significantly affects the rate of
oligomerization for each of the prion forms tested. This limits the amount of oligomers
formed during heat-induced unfolding processes, and completely suppress fibril
formation (Cavaliere et al., 2013). The following experiment provides strong evidence
for future studies in vivo to advance research in furthering clinical trials to develop a drug
or treatment to combat prion diseases.
3.0 Summary Prions are an infectious pathogen that is responsible for numerous fatal
neurodegenerative diseases. These PrP proteins are transmissible particles that lack
nucleic acids and seem to be composed of exclusively modified proteins. PrPC and PrPSc
are identical with respect to all chemical features however compared to the PrPC form,
PrPSc has a higher proportion of beta sheets rather than alpha helices. Each of theses
structural conformations have an effect on the way in which prion proteins cooperate.
These abnormal interactions result in the change in conformation of healthy PrPC to the
toxic isoform of PrPSc. Since toxic PrPSc isoforms are resistant to proteinase K, the build
up of PrPSc causes aggregation, forming amyloid fibers accumulate and create plaque in
brain tissue. The cause of the initial infection is still undetermined. It is believed to occur
through a spontaneous conformation or through the interaction of prions from different
species.
Though characterization of prion structure is important for the initial infection.
Even more important for disease prevention is the further characterization of the
conformational transition from PrPC to PrPSc toxic aggregates essential for the prevention
of the propagation of disease. The mechanism by which the PrPC protein converts to its
toxic conformation has evolved from many suggested hypotheses. The linear

H. Williams Page 24 4/28/14 autocatalysis model (heterodimer mechanism) stated that a single PrPSc protein is able to
change the conformation of another PrPC molecule (Eigen et al., 1996). The cooperative
autocatalysis, implied conformational change through symmetry conversion (Eigen et
al., 1996). Here, conformation of PrPC to PrPSc is compared to the cooperitivity of
hemoglobin through allosteric interactions, causing each subunit to aggregate to the
stable PrPSc form. Leading to the nucleation-dependent aggregation mechanism (NDM
mechanism), which illustrates how monomeric PrPSc aggregates into oliogmeric PrPSc
and then is polymerized to cause the formation of fibril structures(Eigen et al., 1996). By
comparing the nucleation model of growth to elements of the cell, current research has
suggested a the chaperone-assisted model, which illustrates that chaperone proteins
facilitate the nucleation or the polymerization of fibril formation. Unlike the previous
models the chaperone-mediated aggregation provides a more realistic method for how
prions can aggregate. Each of the autocatalysis methods seams to be lacking the
understanding that in cellular environment, where there are high incidences for
interaction and protein interaction. Knowing that chaperone proteins are able to facilitate
the prion interactions, questions arise concerning how can one inhibit prion conformation
and prevent prion disease.
Methlyene blue is a FDA approved drug that has been shown to support cognitive
function. Where it is able to pass the blood-brain barrier, making it suitable to reach
neuronal targets such as prions (Cavaliere et al., 2013). This particular compounds has
been shown to have anti-aggregating properties in relation to Alzehimers Disease (AD)
and restricts protein chaperons such as GroEL and Hsp104 (Cavaliere et al., 2013).
Knowing that this compound has significant nuerological applications in terms of treating
neurodegenerative disease and protein aggregation, Cavaliere et al. wanted to test
H. Williams Page 25 4/28/14 whether this compound had an affect on the PrP aggregation process. Based on the results
found from Cavaliere et al., MB is able to cause inhibtion of prion conformation by
stabilizing the PrPC conformation in vitro. However if these compound is going to be
used as a drug to treat prion disease, there needs to be in vivo testing that provides
evidence that MB is able to first maintain its function within the microenvironment.
Secondly it must demonstrates that prions have a higher affinity for MB then they do for
chaperon proteins. If MB is able to simultaneously stop prion interactions and chaperone
function, MB and other similar compounds, could inhibit the propagation of prion
diseases.

H. Williams Page 26 4/28/14 4.0 The Next Experiment: Analyzing the effect of Chaperon Proteins on Antiprion Inhibition Compounds PrPC is involved in the neuroprotective response of the brain. In cases such as the
cellular oxidative stress response, prions protect against cerebral ischaemia and traumatic
brain injury. Prions can’t be knocked out as a therapeutic treatment due to their role in
regulating presynaptic copper concentrations, calcium homeostasis, as well as activation
and proliferation of lymphocytes, astroytes, and signal transduction (Roettger et al.,
2013). PrPC is essential in interacting with synaptic proteins, cell adhesion molecules, and
apoptosis regulator Bcl-2 proteins. PrPC is able to activate these signally pathways by
activating protein kinases such as cAMP-protein kinase A to ensure neural survival
(Roettger et al, 2013). In order to prevent disease while maintaining PrPC function,
conversion of PrPC to PrPSc must be inhibited.
Current research has been explored concerning prion inhibition by targeting the
prion binding domains that facilitates conversion of PrPC to PrPSc. From what was
previously discussed, Methelyene Blue (MB) has shown promising results that suggest
MB has the potential to inhibit both prion oliogmerization and fibrilization. By inhibiting
the ability of prions to form oligomers and prevent the formation of fibril structures, MB
allows for the suppression of prion related diseases. As previously discussed, prion
disease is related to the formation of fibril structures that are incapable of being broken
down by protienase K. This resistance to proteases causes an accumulation of plaque in
the brain causes the death of neurons. By preventing this process from occurring,
compounds able to occupy the prion binding domains of PrPC could provide therapeutic
value to prevent the growth of protease-resistant aggregates. Though MB has been shown
to have antiprion capabilities, tests have only been done in vitro. There has yet to be an in
vivo study that measures the affects of how the microenvironment affects MB’s ability to

H. Williams Page 27 4/28/14 prevent prion aggregation. Prions may have a higher binding affinity for chaperones
rather than MB. It has been found that a majority of chaperone proteins bind to the same
binding site as MB (Moran et al., 2013). The relative binding affinity of prions to
chaperones, in contrast to other proteins within the cellular environment, could result in
the repression of MB as an inhibitor. In vitro studies have demonstrated that MB has the
capabilities to prevent accumulation of prion aggregates. In vivo experimentation could
further validate that MB has the characteristic need to become a preventative treatment
for prion diseases.
4.1 Hypothesis: It is hypothesized that MB would be able to inhibit prion aggregation even in the
presence of chaperones in vivo. If MB was analyzed through an in vivo Thiofalvin T
assay (ThT-binding assay) then the results should indicate no fibrilization within the cell
because MB is able to target prion proteins directly by blocking the PrPC binding site for
PrPSc. This would prevent the nucleation step from occurring, which would restrict the
rate at which chaperones could facilitate oliogmerization and fibrilization.
4.2 Methods: This experiment would be carried out in a similar manor as the in vitro study
conducted by Cavaliere et al. In order for the results to be comparable with Cavaliere et
al., the same prion species will be tested from the Cavaliere et al experiment. Full length
PrP from sheep OvPrP (A136 R154 Q171 variant) , mouse PrP (MoPrP), and human PrP
(HuPrP) would be amplified through PCR using cDNA or genomic DNA. The PCR
product will be placed into a pET22bC vector and the plasmids would then be transfected
into mice using an i.v. or direct injection into the brian. To test for successful
transfection, an immunofluorescence assay will use fluorescently tagged antibodies with
specific binding to each prion species. A mouse from each prion plasmid transfection will

H. Williams Page 28 4/28/14 be sacrificed and brain tissue will be harvested. Subjecting brain tissue to these specific
antibodies will mark the eah type of PrPC, because healthy PrPC is attached on the surface
of neurons thorugh the GPI anchor. If the microphotography indicates fluorescence on
the surface of cells this would indicate that successful transfection of prions had occurred.
DAPI dye will indicate the location of the nucleus to reference where the prions are
located in relation to the cell.
Since there are four different types of prions being tested, 4 different groups of
mice would be used, each injected with a different plasmid. Each of these groups would
then be split into two different groups, a control group where no MB was administered,
and an experimental group where MB was injected into body. Once MB and the plasmids
were injected, mice will killed and their brain tissue will be subjected to an in vivo ThT-
assay to measure the amount of prion protein aggregation occurring in the presence and
in the absence of MB over a 30 day period. Mice from each group of transfections would
be sacrificed every 100hrs to perform a ThT-assay to determine the amount of
fibrilization present. Typically a ThT-assay is meant to visualize and quantify the
presence of fibrilization of misfolded protein aggregates both in vitro and in vivo. A
comparison between the ThT-assays of the control and experimental groups, would allow
one to compare how fibrilization was occurring between each strain of prion with or
without MB present. The results of this experiment could lead to confirmation that MB
has the ability inhibit prion propagation in the cellular environment, or it would illustrate
that something in the cellular environment restricts MB’s ability to inhibit prion
conversion.
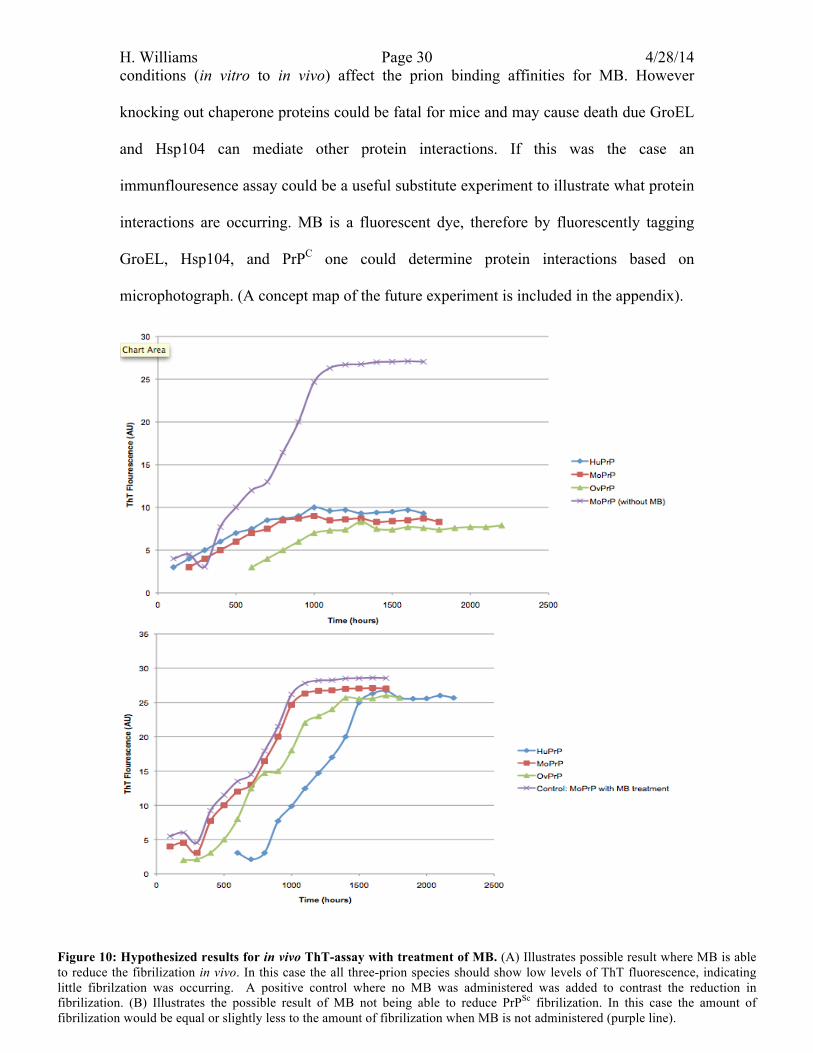
4.3 Outcomes and Interpretations: If MB was able to prevent fibrilization the results of the ThT-assay could
resemble what is found in figure 10.A. If MB was able to prevent fibrilization in vivo this

H. Williams Page 29 4/28/14 result could provide grounds for future clinical and human testing. However MB were
unable to restrict the conversion of prions, a second experiment would be conducted
using knockouts of either GroEL or Hsp104 chaperones to determine whether chaperones
were causing MB to become inhibited. Figure 10.B illustrates the possible results that
would be characteristic of this conclusion. A large sigmodal increase in ThT fluorescence
illustrates fibrilization is present (Cavaliere et al., 2013). As previously described,
chaperone proteins have been found to facilitate oligomerization and fibrilization of
prions and bind to the same active sites as MB (DebBurman et al., 1997). Chaperones
could compete with MB for prion binding sites. Prions may have a stronger binding
affinity for chaperones than MB. By knocking out potential prion chaperones, one could
learn whether prions have a higher binding affinity to chaperones or another cellular
component.
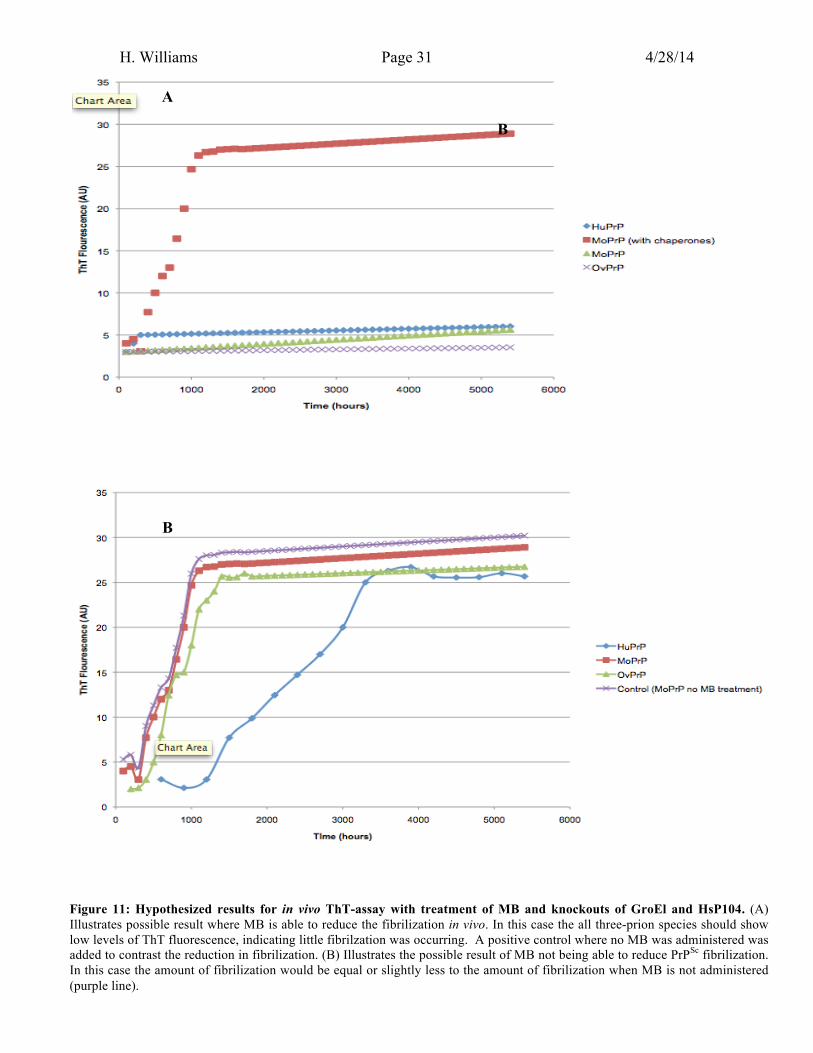
A second ThT-assay would have to be done using chaperone deficient mice, to
see if prion fibrilization is affected. If no fibrilization is present then this would suggest
MB was able to inhibit prion-prion interactions in the absence of chaperones. Figure 11.A
illustrates evidence that would characteristic of this result. When MB treatment in the
absence of chaperones, the amount of fibrilization should be significantly lower than
when mice are treated with MB. Figure 11.A indicates that each prion species shows
lower ThT-flouresence when MB is present compared to the control (when MB is absent
(purple line)). This would demonstrate that the decreased fibrilization is due to prions
having a higher binding affinity for MB. If fibrilization does occur then there is likely
something in the cellular environment that MB has a higher affinity for. Thus inhibiting
MB’s ability to interact with PrPC. This would indicate that further research is needed to
understand how MB is interacting with the cellular environment and whether changes in
H. Williams Page 30 4/28/14 conditions (in vitro to in vivo) affect the prion binding affinities for MB. However
knocking out chaperone proteins could be fatal for mice and may cause death due GroEL
and Hsp104 can mediate other protein interactions. If this was the case an
immunflouresence assay could be a useful substitute experiment to illustrate what protein
interactions are occurring. MB is a fluorescent dye, therefore by fluorescently tagging
GroEL, Hsp104, and PrPC one could determine protein interactions based on
microphotograph. (A concept map of the future experiment is included in the appendix).
Figure 10: Hypothesized results for in vivo ThT-assay with treatment of MB. (A) Illustrates possible result where MB is able to reduce the fibrilization in vivo. In this case the all three-prion species should show low levels of ThT fluorescence, indicating little fibrilzation was occurring. A positive control where no MB was administered was added to contrast the reduction in fibrilization. (B) Illustrates the possible result of MB not being able to reduce PrPSc fibrilization. In this case the amount of fibrilization would be equal or slightly less to the amount of fibrilization when MB is not administered (purple line).
H. Williams Page 31 4/28/14
B
Figure 11: Hypothesized results for in vivo ThT-assay with treatment of MB and knockouts of GroEl and HsP104. (A) Illustrates possible result where MB is able to reduce the fibrilization in vivo. In this case the all three-prion species should show low levels of ThT fluorescence, indicating little fibrilzation was occurring. A positive control where no MB was administered was added to contrast the reduction in fibrilization. (B) Illustrates the possible result of MB not being able to reduce PrPSc fibrilization. In this case the amount of fibrilization would be equal or slightly less to the amount of fibrilization when MB is not administered (purple line).
A
B

H. Williams Page 32 4/28/14 Bibliography: Cavaliere, Paola, et al. "Binding of methylene blue to a surface cleft inhibits the oligomerization and fibrillization of prion protein." Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 1832.1 (2013): 20-28. Eigen, Manfred. "Prionics or the kinetic basis of prion diseases." Biophysical chemistry 63.1 (1996): A1-A18. Harrison, Paul M., et al. "The prion folding problem." Current opinion in structural biology 7.1 (1997): 53-59. Grimminger‐Marquardt, Valerie, and Hilal A. Lashuel. "Structure and function of the molecular chaperone Hsp104 from yeast." Biopolymers 93.3 (2010): 252-276. Hosokawa-Muto, Junji, et al. "Variety of antiprion compounds discovered through an in silico screen based on cellular-form prion protein structure: Correlation between antiprion activity and binding affinity." Antimicrobial agents and chemotherapy 53.2 (2009): 765-771. Laurent, Michel. "Autocatalytic processes in cooperative mechanisms of prion diseases." FEBS letters 407.1 (1997): 1-6. Martin, Jörg, et al. "The reaction cycle of GroEL and GroES in chaperonin-assisted protein folding." Nature 366.6452 (1993): 228-233. Mays, Charles E., et al. "Prion inhibition with multivalent PrP< sup> Sc</sup> binding compounds." Biomaterials 33.28 (2012): 6808-6822. Roettger, Yvonne, et al. "Immunotherapy in prion disease." Nature Reviews Neurology 9.2 (2013): 98-105. Taguchi, Yuzuru, and Hermann M. Schätzl. "Identifying critical sites of PrPc-PrPSc interaction in prion-infected cells by dominant-negative inhibition." Prion 7.6 (2013): e1003466-1. Steele, Andrew D., et al. "Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis." Proceedings of the National Academy of Sciences of the United States of America 103.9 (2006): 3416-3421. Watts, Joel C., and David Westaway. "The prion protein family: diversity, rivalry, and dysfunction." Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 1772.6 (2007): 654-672. Wills, Peter R. "Autocatalysis, information and coding." Biosystems 60.1 (2001): 49-57.
H. Williams Page 33 4/28/14 5.0 Appendix:

H. Williams Page 34 4/28/14