presented by dr. shazzad hosain asst. prof. eecs, nsu
DESCRIPTION
Multiple Sequence Alignment Motif Finding and Gene Prediction. Presented By Dr. Shazzad Hosain Asst. Prof. EECS, NSU. What is a Multiple Sequence Alignment?. characterize protein families by identify shared regions of homology molecular evolution analysis using Phylogenetic methods - PowerPoint PPT PresentationTRANSCRIPT

Presented ByDr. Shazzad Hosain
Asst. Prof. EECS, NSU
Multiple Sequence AlignmentMotif Finding and Gene Prediction

What is a Multiple Sequence Alignment?
• characterize protein families by identify shared regions of homology
• molecular evolution analysis using Phylogenetic methods• tell us something about the evolution of organisms• Homologous genes (genes with share evolutionary origin) have
similar sequences• Uncover changes in gene structure• Look for evidence of selection

Motivation
• Let n number of sequences• A new sequence i.e. gene/protein comes up• Wants to find its family

Methods of MSA
• Exact method• Heuristic methods

Exact method
• Sequence Alignment (two sequences)
0 -2 -4 -6 -8 -10
-2
-4
-6
A C G T A
A
G
T
F(i, j) = F(i-1, j-1) + s(xi ,yj)
F(i, j) = max F(i, j) = F(i-1, j) - d F(i, j) = F(i, j-1) - d
2
00

Exact method (Dynamic Programming)
V S N
—
S
S
—
N A —
A S— — —
V S N S
S
N
A
AS
Start

Dynamic Programming for Three Sequences
• There are 7 ways to get to C[i,j,k]
C[i,j,k]
C[i-1,j-1,k-1]
C[i-1,j,k-1]
C[i-1,j,k-1]
For 3 seqs. of length n, time is proportional to n3
Enumerate all possibilities and choose the best one

Dynamic programming cont.• More then three sequences• Four dimension
No deterministic polynomial time algorithm to find optimal solutionMSA complexity is NPSo, Heuristics algorithms for near optimal solution

Heuristics for MSA
• Iterative pair-wise alignment• Motif / Anchor – based alignment
• Divide and conquer Algorithm• Statistical methods like Hidden Markov Model

Divide and Conquer Algorithm

Iterative Pairwise Alignment
• Let four strings to align• MASH, MESH, SQUASH, SQUAMISH
MASHMESH
M_ _ASHM_ _ESHSQUASH
M_ _A_ _SHM_ _E_ _SHSQUA_ _SHSQUAMISH

Iterative Pairwise Alignment cont.
• In other way
MASHMESH
SQUAMISHSQUA_ _SH
SQUAMISHSQUA_ _SH_M_A _ _SH_M_E _ _SH

Regulatory Motifs in DNA Sequences

The Immune system
• Immunity genes are usually dormant• When infected, somehow get switched on• When these genes are turned on, they
produce proteins that destroy the pathogen, usually curing the infection

Immune System in Fruit Flies
• Fruit flies do not have sophisticated immune system as humans
• Have small set of immunity genes, usually dormant
• But when infected, somehow get switched on• For fruit flies, let we like to know which genes
are switched on as an immune response

Regulatory Motif
• Regulatory motif is a short sequence of string, where the transcription factors, a protein that encourages RNA polymerase to transcribe the downstream genes, bind
• Regulatory motif triggers gene activation• Also known as NF-κB binding sites• Immunity genes in fruit fly genome have strings that are
reminiscent of TCGGGGATTTCC
ACGTCGCGTACGTAAACGCTCGCTAAACGCTCGCTAAACGCTCGCT
Regulatory Motif
Upstream downstream

The Fruit Fly Experiment
• Which genes are switched on as an immune response?– Infect the fly, grind it up, collect a set of upstream regions
form the genes in the genome– Each region contains at least one NF-κB binding sites
• NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) is a protein complex that controls the transcription of DNA
– Suppose we do not know what the NF-κB pattern looks like, nor do the position
– So, given a set of sequences from a genome, can we find short substrings that seem to occur surprisingly often.

Profiles

Profiles

Profiles

Profile Matrix

Motif Finding Problem

Gene Prediction Problem

Genome Complexities
• Human genome is larger than bacterial genomes, seems logical
• But Salamander genome is ten times larger than the human genome– Junk DNA or introns are more in Salamander

cDNA Problem
cDNA

Similar genesAcross species

Genome Complexities
• Jumps are inconsistent across species• A gene in an insect edition is differently organized than a related
gene in a worm genome• The number of parts (exons) may be different• Information that appears in one part of human edition may be
broken up into two in the mouse version or vice versa• So, quite different in terms of part structure.
Does it mean intron exon lengths are same across species?

Genome Complexities
• Human genes constitute only 3% of the human genome
• No existing in silico gene recognition algorithm provides completely reliable gene recognition.
• Roughly two approaches of gene prediction– Statistical methods– Similarity based approach

Similarity Based Approach The Exon Chaining Problem
• This approach uses previously sequenced genes and their protein products as a template
• Find a set of potential exons, putative exons, by local alignment
• The exon set may be overlapping• The problem is to choose the best subset of non-
overlapping substrings as a putative exon structure
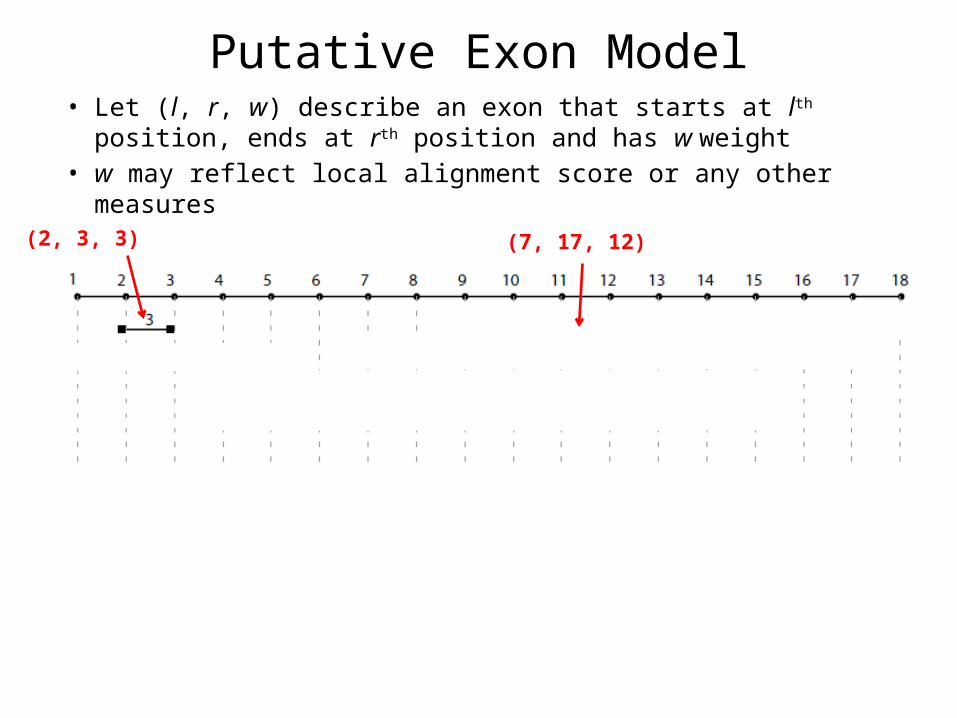
Putative Exon Model• Let (l, r, w) describe an exon that starts at lth position, ends at
rth position and has w weight• w may reflect local alignment score or any other measures
(2, 3, 3) (7, 17, 12)

Putative Exon Model• Let (l, r, w) describe an exon that starts at lth position, ends at
rth position and has w weight• w may reflect local alignment score or any other measures
0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
or
i is the current locationj is the left end of the current location
3
5
6 110
7
12
4
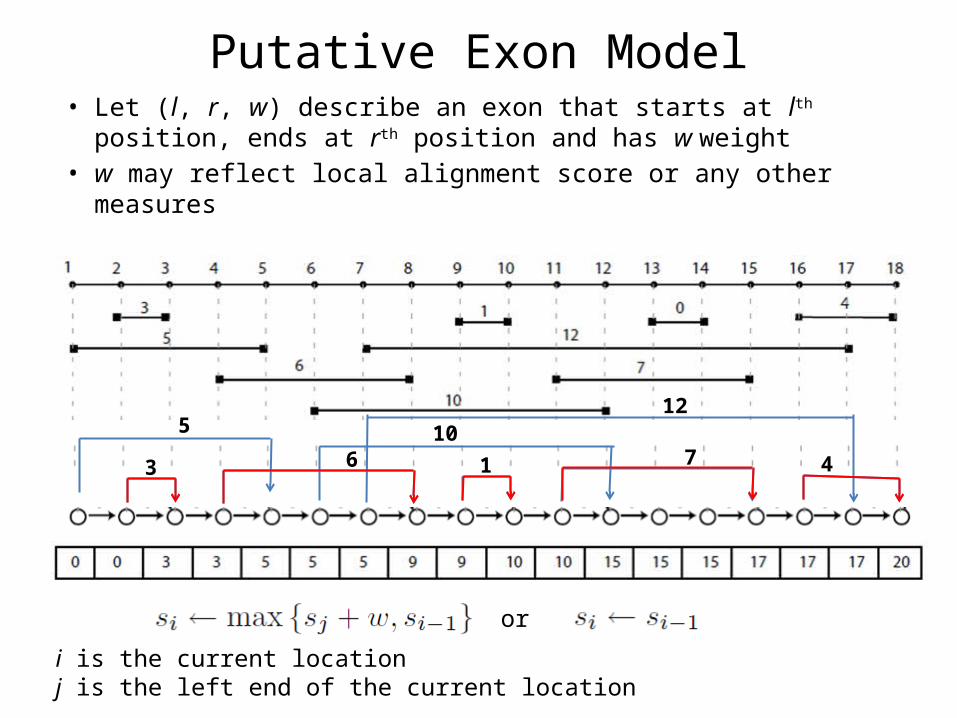
Putative Exon Model• Let (l, r, w) describe an exon that starts at lth position, ends at
rth position and has w weight• w may reflect local alignment score or any other measures
or
i is the current locationj is the left end of the current location
3
5
6 110
7
12
4

Exon Chaining Algorithm

Reference
• Multiple Sequence Alignment: No specific Reference, Use Web Resources
• Motif Finding Problem: Chapter 4.4, Introduction to Bionformatics – by Pavel Pevzner
• Gene Prediction Problem: Chapter 6.11, Introduction to Bionformatics – by Pavel Pevzner