penicillium chrysogenum - journal of biological chemistry
TRANSCRIPT

THE JOURNAL OF BIOLOGICAL CHEMISTRY Q 1992 by The American Society for Biochemistry and Molecular Biology, Inc.
Vol. 267, No. 8, Issue of March 15, pp. 5474-5481,1992 Printed in U.S.A.
Isolation and Characterization of the Acetyl-coA Synthetase from Penicillium chrysogenum INVOLVEMENT OF THIS ENZYME IN THE BIOSYNTHESIS OF PENICILLINS*
(Received for publication, July 29, 1991)
Honorina Martinez-Blanco, Angel Reglero, Martiniano Fernhdez-Valverde, Miguel A. Ferrero, Miguel A. Moreno$, Miguel A. Peiialvatll, and J o i M. LuengoJI From the Departamento de Bwquimica y Biologiu Molecular, Facultad de Veterinuriu, Uniuersidad de Ledn, 24007 Ledn, the $Departamento de Bioquimica, Antibidticos S. A. Ledn, and the SCentro de Investigaciones Biolhgicas, Consejo Superior de Inuestigacwnes Cientificas, Veldzquez 144,28006 Madrid, Spain
Acetyl-coA synthetase (ACS) of Penicillium chrys- ogenum was purified to homogeneity (746-fold) from fungal cultures grown in a chemically defined medium containing acetate as the main carbon source. The en- zyme showed maximal rate of catalysis when incubated in 50 mM HC1-Tris buffer, pH 8.0, at 37 “C. Under these conditions, ACS showed hyperbolic behavior against acetate, CoA, and ATP; the K,,, values calcu- lated for these substrates were 6.8, 0.18, and 17 mM, respectively. ACS recognized as substrates not only acetate but also several fatty acids ranging between Cz and CS and some aromatic molecules (phenylacetic, 2- thiopheneacetic, and 3-thiopheneacetic acids). ATP can be replaced by ADP although, in this case, a lower activity was observed (37%). ACS is inhibited by some thiol reagents (5,6’-dithiobis(nitrobenzoic acid), N- ethylmaleimide, p-chloromercuribenzoate) and diva- lent cations (Zn2+, CuZ+, and Hg2+), whereas it was stimulated when the reaction mixtures contained 1 mM dithiothreitol, reduced glutathione, or 2-mercaptoeth- anol. The calculated molecular mass of ACS was 139 f 1 kDa, and the native enzyme is composed of two apparent identical subunits (70 kDa) in an a2 oligo- meric structure. ACS activity was regulated “in vivo” by carbon catabolite inactivation when glucose was taken up by cells in which the enzyme had been pre- viously induced. This enzyme can be coupled “in vitro” to acyl-CoA:6-aminopenicillanic acid acyltransferase from P. chrysogenum, thus allowing the reconstitution of the functional enzymatic system which catalyzes the two latter reactions responsible for the biosynthesis of different penicillins. The ACS from Aspergillus nidu- l a m can also be coupled to 6-aminopenicillanic acid acyltransferase to synthesize penicillins. These results strongly indicate that this enzyme can catalyze the activation (to their CoA thioesters) of some of the side- chain precursors required in these two fungi for the production of several penicillins. All these data are reported here for the first time.
* Work in J. M. Luengo’s laboratory was supported in part by Grant PB-89-0387 from the Direcci6n General de Investigaci6n Cien- tifica y TCnica (Madrid, Spain) and Grant FISss (Fondo de Inves- tigaciones Sanitaria8 de la Seguridad Social, Madrid, Spain). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “aduertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
(I Supported by Grant BIO-244/88 (CICYT, Espaiia). 11 To whom correspondence should be addressed. Fax: 987-291194.
The biosynthetic pathway of L-lysine and penicillins in Penicillium chrysogenum and in Aspergillus nidulans is a branched route which starts with the condensation of an acetyl-coA molecule with a-ketoglutaric acid, leading to the formation of homocitric acid which is the first biosynthetic intermediate (1-4). Later, through a series of reactions similar to those reported for the tricarboxylic acid cycle, L-a-ami- noadipic acid is produced. In the specific branch of penicillins L-a-aminoadipic acid is linked with another two amino acids (L-cysteine and L-valine) generating a tripeptide molecule [6-(L-a-aminoadipyl)-L-cysteinyl-D-valine], without antibac- terial activity and commonly named ACV’ (5). In a second step, this compound is cyclized to isopenicillin N (IPN) by the enzyme IPN synthase (6-9). The low antibacterial activity of IPN is greatly increased by replacing the L-a-aminoadipic acid moiety by another acyl-chain (A3-hexenoic, hexanoic, octanoic phenylacetic, and phenoxyacetic acids), thus gener- ating different penicillins (10, 11). These transference reac- tions are catalyzed by a single enzyme, acyl-CoA:6-APA acyl- transferase (AT), which requires the previous activation of the acyl-chains to their CoA derivatives (12-14). It has been reported (15-16) that P. chrysogenum catalyzes the activation of phenylacetic acid (PAA) (the side-chain precursor of ben- zylpenicillin G ) , to phenylacetyl-coA (PA-CoA) by a phen- acyl-CoA ligase (PCL) in the presence of M P , ATP, CoA, and PAA according to the following reaction:
PAA + CoA + ATP pcL > PA-CoA + AMP + PPi (1)
However, this protein was never purified, nor has the activ- ity been characterized in detail. We have attempted to assay PCL in different low producing or industrial strains of P. chrysogenum but we failed to find it-by the reported proce- dures (15,16). However, we found a similar enzyme in a strain of Pseudomonas putidu (U) able to grow in a chemically defined medium containing PAA as the sole carbon source (17). Recently, Smith et al. (18) have shown that the PCL gene is not linked to the other genes which code for ACV synthase, IPN synthase, and AT. They suggested that PCL could be a nonspecific penicillin biosynthetic enzyme, i.e. a general one involved in the primary metabolism of the fungus. To test this hypothesis we have purified and studied, as an obvious candidate, the acetyl-coA synthetase (ACS) of P.
M%+
The abbreviations used are: ACV, 6-(L-or-aminoadipy1)-L-cystei- nyl-D-valine; IPN, isopenicillin N; AT, acyl-CoA6-aminopenicillanic acid acyltransferase; HPLC, high performance liquid chromatogra- phy; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electro- phoresis.
5474

P. chrysogenum Acetyl-coA Synthetase 5475
chrysogenum; its optimal physicochemical assay conditions, catalytic parameters, substrate specificity, and some aspects of its regulation were established. As a result of our investi- gations, the participation of ACS in the biosynthetic pathway of benzylpenicillin ( G ) and natural penicillins (DF, F, and K) is reported and discussed.
EXPERIMENTAL PROCEDURES
Materials-Nucleotides, sodium acetate, CoA, phenylmethylsulfo- nyl fluoride, dithiothreitol (DTT), reduced glutathione, 2-mercapto- ethanol, and cycloheximide were supplied by Sigma. [U-14C]Leucine (351 mCi/mmol) was purchased from the Radiochemical Centre (Amersham, England). [ l-14C]Phenylacetic acid (24 mCi/mmol) was from CEA (France). @-Lactamase from Bacillus cereus was acquired from Difco (Detroit, MI). 6-Aminopenicillanic acid (6-APA), A'- pentenylpenicillin (F, 1510 units/mg), n-amylpenicillin (DF, 1625 units/mg), n-hexylpenicillin (1540 units/mg), n-heptylpenicillin (K, 2400 units/mg), A'-heptenylpenicillin (1760 units/mg), benzylpeni- cillin (G, 1590 units/mg), 2-thiopheneacetylpenicillin (1540 units/ mg), and 3-thiopheneacetylpenicillin (1510 units/mg) were either gifts from Antibioticos S. A. (Lebn, Spain) or synthesized by us. All other products were of analytical quality or HPLC grade.
Microorganism-P. chrysogenum Wis 54-1255 (ATCC 28089) was obtained from the American Type Culture Collection, Aspergillus nidulans biA1, veAl was from our collection, and Micrococus luteus ATCC 9341 was used for the determination of the different penicillins by bioassay (19). The strains were maintained lyophilized or impreg- nated in Silica Gel.
Culture Media and Growth Conditions-P. chrysogenum Wis 54- 1255 was maintained in sporulation medium with the following com- position (grams/liter): Malt extract, 20; bactopeptone, 1; glucose, 20; bactoagar, 20. Incubations were carried out on slants of 3 X 20 cm containing 25 ml of medium at 25 "C for 7 days. Spores from the slants were collected by adding 10 ml of sterile saline solution and filtering the suspension through a glass fiber filter. The final spore suspension, containing lo6 $pores/ml, was used to inoculate the liquid medium. Each 2-liter Erlenmeyer flask containing 250 ml of a chem- ically defined medium (see below) was seeded with 2.5 ml of the spore suspension and incubated in a rotary shaker at 250 rpm and 25 "C for 54 h or the time specified in each set of experiments. The liquid medium contained (grams/liter): lactose, 15; sucrose, 5; sodium ace- tate, 23; phenylacetic acid, 2; ethylamine, 3; citrate, 10; (NH&SO,, 5; KHzP04, 1; Na2HP04, 1; FeS04.7Hz0, 0.05; MgSO,. 7Hz0, 0.5; ZnSO.. 7H20, 0.01; CuSO,. 5H20, 0.01; MnClz .4H20, 0.01; CoS04. 7H20, 0.005 and NaCl, 0.001. The medium (basal medium) was adjusted to pH 6.5 before autoclaving and lactose and sucrose were added separately. When required glucose (1.5%, w/v) was added to the cultures.
Aspergillus nidulans biA1, veAl was grown in the same basal medium containing 50 pg/ml of biotin and in similar conditions. However, in this case the growth temperature was 37 "C.
Determinution of the "in Vivo Half-life of Acetyl-coA Synthetase- Cells were grown as above and at different times protein synthesis was stopped by adding cycloheximide (125 pg/ml). From these times up to 60-h ACS activity was evaluated at intervals.
Acetyl-coA Synthetase Assay-ACS activity was evaluated by measuring the rate of acetylhydroxamate formation in the presence of ATP, CoA, acetate, M e , and neutral hydroxylamine as has been described for other acyl-CoA activating enzymes (15-17).
The reaction mixture contained MgClz (0.2 M, 12.5 pl); ATP (0.1 M, 50 pl); CoA (20 mM, 30 pl) ; sodium acetate (0.2 M, 30 pl) and hydroxylamine solution (prepared as described below) 50 pl. All substrates except M&lZ (water) were dissolved in 50 mM Tris and adjusted to pH 8.0. When required acetate or ATP were replaced by other molecules (fatty acids, aromatic compounds, or nucleotides). After 5 min of temperature equilibration in a water bath at 37 "C (or the required temperature) 100 pl of enzyme solution was added to the tubes and incubated for different times (usually 30 min). Reactions were stopped by adding 450 pl of the ferric chloride reagent (see below) and kept on ice for 10 min. At this time the tubes were centrifuged in an Eppendorf 5414 microcentrifuge for 2 min and the red-purple color generated was measured at 540 nm with a Shimadzu UV-120-02 spectrophotometer. The extinction coefficient of acetyl- hydroxamate under these conditions was 0.96 mM" cm". In the standards test, acetate, ATP, CoA, or MgCl, was omitted. In these cases formation of acetylhydroxamate was not detected. Only in the
absence of CoA, and after longer incubation times (3 h), was some acetylhydroxamate (about 10%) formed.
One unit of enzyme activity is defined as the catalytic activity leading to the formation of 1 nmol of acetylhydroxamate in 1 min (unit). Specific activity is given as units/mg of protein.
Protein was measured by the method of Bradford (20) using bovine serum albumin as standard. Buffers identical to those containing the protein samples were used as blanks.
Neutral hydroxylamine solution, pH 8.0, was prepared by mixing 1 ml of 4 M hydroxylamine hydrochloride and 1 ml of 4 M KOH (17).
The ferric chloride reagent was prepared by mixing 0.37 M ferric chloride, 20 mM trichloroacetic acid and 0.66 M hydrochloric acid, as previously described (21).
Acetyl-coA Synthetase (ACS) and Acyl-CoA: 6-APA Acyl Transfer- ase (AT) Coupled Assay-To establish whether some of the substrates activated by ACS to CoA thioesters can be transformed into penicil- lins, ACS and AT were incubated "in vitro." The assay mixture contained, in a total volume of 126.5 pl: 50 mM HC1-Tris buffer, pH 8.0; 10 mM MgCl2; 20 mM A T P 2.4 mM CoA; 12 mM sodium acetate (or the corresponding penicillin side-chain precursor); 2 mM D T T 30 p~ 6-APA (free acid); pure ACS (3 pg); and pure AT (10 pg). The reactions were incubated at 30 "C for 60 min and stopped by the addition of a similar volume of methanol (126.5 pl). The antibiotics generated were measured by bioassay against M. luteus (19). Control reactions were carried out under the same conditions without ATP, CoA, M e , 6-APA, or the side-chain precursor.
Determination of Protein Synthesis-Protein synthesis was fol- lowed in P. chrysogenum by measuring the incorporation of [U-"C] leucine into trichloroacetic acid-insoluble material, as previously re- ported (4). The effect of cycloheximide on protein synthesis was studied by adding this antibiotic (125 pg/ml) to the flasks at different times (39 and 43 h).
Carbon Compound Determinations-Residual D-glucose was meas- ured by the glucose oxidase enzymatic test (22). The quantity of acetate remaining in the broths was measured by the method of Holz (23).
Gel Electrofocusing-Isoelectric focusing was carried out as re- ported by Wrigley (24).
Electrophoresis-Polyacrylamide gel electrophoresis (PAGE) un- der denaturing conditions (SDS-PAGE) was performed in 7% slab gels (25) using phosphorylase b (M, 94,000), bovine serum albumin (M, 67,000), ovalbumin (M, 43,000), and carbonic anhydrase (Mr 29,000) as molecular weight standards.
Starvation Medium-In some experiments (regulation by glucose, see "Results and Discussion") cultures of P. chrysogenum grown in a medium containing acetate and glucose were harvested at different times, washed with sterile saline solution, and resuspended in the same chemically defined medium without glucose and deprived of the nitrogen sources. Under these conditions cellular growth was halted.
Determination of Oil in the Broth-Residual oil was evaluated by extracting 5 ml of culture (without cells) with 5 ml of n-hexane (twice). The aqueous and the two organic phases were separated and these latter (containing oil) were mixed and dried in an oven for 24 h at 40 "C.
PAA Transport System-Mycelia of P. chrysogenum grown in the above medium and conditions were harvested at different times and washed five times with sterile distilled water. Aliquots of 35 mg (wet weight; about 10 mg of dry weight) were suspended in 25-ml Erlen- meyer flasks containing 1.4 ml of 0.06 M phosphate buffer, pH 6.5, and preincubated at 25 'C for 5 min in a thermostatically controlled bath at 160 strokes/min before adding PA (6.4 p~ containing 1.4 p~ of labeled PA). Incubations were carried out for 30 s, or the required time, halted by addition of 10 volumes of water, rapidly filtered through Millipore filters (0.45-pm pore size), and washed with 3 X 10 ml of sterile distilled water. The filters were dissolved in 10 ml of scintillation fluid and counted as reported. [I4C]PA uptake is given as picomoles/min.
Determination of Penicillins by HPLC-The different penicillins produced by P. chrysogenurn grown in the minimal medium reported above for 84 h were analyzed by HPLC. Samples of culture broth (10 ml) were either filtered and lyophilized (for the determination of the total @-lactam antibiotics, including 6-APA) or adjusted to pH 2.0, extracted with isobutylacetate and the antibiotic-rich fraction (or- ganic phase) transferred to 50 mM phosphate buffer (l/lO, v/v) pH 7.0 (11). The organic and the aqueous phases were separated and the latter was lyophilized. The powder obtained in both cases was resus- pended in 1 ml of distilled water, and 20 p1 were injected in a high performance liquid chromatograph (Spectra-Physic SP 8450)

5476 P. chrysogenum Acetyl-coA Synthetase
equipped with a variable-wavelength UV-visible detector (SP 8450), a computing integrator (SP 4290), and a Nucleosil Cl8 column (250- X 4.6-mm inner diameter). The mobile phase was: A, sodium acetate 0.01 M, pH 7.0, and B, sodium acetate 0.01 M, pH 7.0, and acetonitrile (6040, v/v). The gradient system was: from 0 to 10 min, 100% A; from 20 to 40 min, 60% A and 40% B; from 40 to 60 min, 100% B; and from 60 to 70 min, 100% A. The wavelength was 219 nm. In these conditions the retention times were: 6-APA, 8 min; PAA, 11 min; peniciloic acid, 20 min; and penicillin, G, F, DF, and K, 28, 31, 34, and 57 min, respectively.
Purification of ACS of P. chrysogenum-Mycelia from this fungus, cultured under the above conditions, were harvested at 54 h of growth, filtered, washed with 3 1 of sterile saline solution and resuspended in the following buffer: 0.5 M KH2P04/Na2HP04, pH 8.5 containing phenylmethylsulfonyl fluoride (1 mM), EDTA (4 mM), and 2-mercap- toethanol (5 mM). Cells (1.5 g of wet mycelium/lO ml of buffer) were disrupted with glass beads using a Braun MSK mechanical disinte- grator (10). Cell debris was eliminated by centrifugation at 17,0000 X g, 10 min at 2 "C. The pellet was discarded and the supernatant fluid, containing the ACS activity, was precipitated with ammonium sul- fate. The fraction precipitating between 35 and 65% was collected, dissolved in the same buffer containing ammonium sulfate (20% saturation) adjusted to pH 9.2, and ultracentrifuged at 250,000 X g for 45 min at 2 "C. The precipitate was eliminated and the superna- tant was used for the following steps. Stability of ACS was good in this buffer. Ultracentrifuged extracts were applied to a phenyl-Seph- arose CL-4B column (Pharmacia LKB Biotechnology Inc.) (2.0 X 20 cm) equilibrated with the above buffer slightly modified (containing 30% saturation of ammonium sulfate). The column was washed with 250 ml of buffer and eluted with a 10 mM KH2P04/Na2HP04, pH 8.5, buffer containing the following: ascorbate (1 mM), DTT (4 mM), EDTA (4 mM), MgC12 (5 mM), and glycerol (20%, v/v) (PG buffer). The lack of glycerol in the buffer led to rapid inactivation of this enzyme (more than 70% over 12 h a t 2 "C and almost 100% at 20 "C). Aliquots of 2.26 ml were collected at a flow rate of 20 ml/h. ACS eluted between the fractions 7-18, showing a peak of activity in tube 12. Fractions 9-16 were mixed and applied to a concanavalin A- Sepharose (Pharmacia) column (1.8 X 8 cm) equilibrated with PG buffer. ACS was not retained in the column suggesting that it is not a glycoprotein. However, this step did eliminate some proteins that handicapped the purification to homogeneity of this enzyme. Aliquots of 2.25 ml of the nonretained sample were collected (flow rate, 10 ml/ h) and analyzed. Those fractions containing activity (fractions 3-13, with a peak in tube 5) were mixed and precipitated with ammonium sulfate (65% saturation). The precipitate was dissolved in 1 ml of PG buffer and applied to a Sephacryl S-300 (Pharmacia) column (2.5 X 33.5 cm) equilibrated with the same buffer containing 20% saturation of ammonium sulfate. The absence of this salt causes a quick deac- tivation of the enzyme in this step. Aliquots of 2.15 ml were collected (flow rate, 15 ml/h) and assayed. ACS eluted between tubes 44 and 59 with a peak of activity in tube 50. Fractions 46-56 were mixed and precipitated again with ammonium sulfate (65% saturation). The precipitate was dissolved in 1.5 ml of PG buffer and applied to a Sephadex G-25 PD-10 column (Pharmacia), equilibrated with PG buffer to eliminate the excess of ammonium sulfate. Fractions of 1 ml were collected, and those containing ACS activity (3-6) were mixed and applied to a Blue-Sepharose CL-GB (Pharmacia) column (1.8 X 8 cm). The column was washed with 60 ml of PG buffer and eluted with a KC1 gradient (0.18-0.35 M) (aliquots of 2.2 ml). ACS eluted between tubes 4 and 12 with a peak of activity in fraction 6 (0.23 M KCl). Tubes 5-10 were mixed, precipitated again with am- monium sulfate (65% saturation), and treated as above (filtration through Sephadex G-25 PD-10 column) to eliminate the excess of salt. The fractions containing activity (fractions 3-6) were mixed and applied to a DEAE-5PW FPLC (Waters) column (0.8 X 7.5 cm) equilibrated with PG buffer. The column was washed with 30 ml of buffer (flow rate, 10 ml/h) and eluted with a KC1 gradient (0.20-0.26 M). Aliquots of 0.6 ml were collected and assayed. ACS activity eluted between fractions I7 and 24 with an activity peak in fraction 20 (0.22 M KCl) which contains 50 pg of homogeneity pure protein. Using this procedure (summarized in Table I) we purified this enzyme 745-fold. The excess of KC1, which could have interfered in some experiments, was eliminated by filtration through an Amicon membrane (exclusion size, <30,000) and after diluting the sample twice to 20 ml with PG buffer, it was filtered and concentrated to the original or to the required volume.
RESULTS AND DISCUSSION
Time Course of the Appearance of Acetyl-coA Synthetase in P. chrysogenum-Acetyl-coA synthetase from P. chryso- genum (acetate:CoA ligase (AMP forming), EC 6.2.1.1) is an enzyme that catalyzes the activation of acetic acid to acetyl- CoA in the presence of M%+, CoA, ATP, and acetate, accord- ing to the following equation
M%+ Acetate + CoA + ATP > Acetyl-coA + AMP + PPi (2)
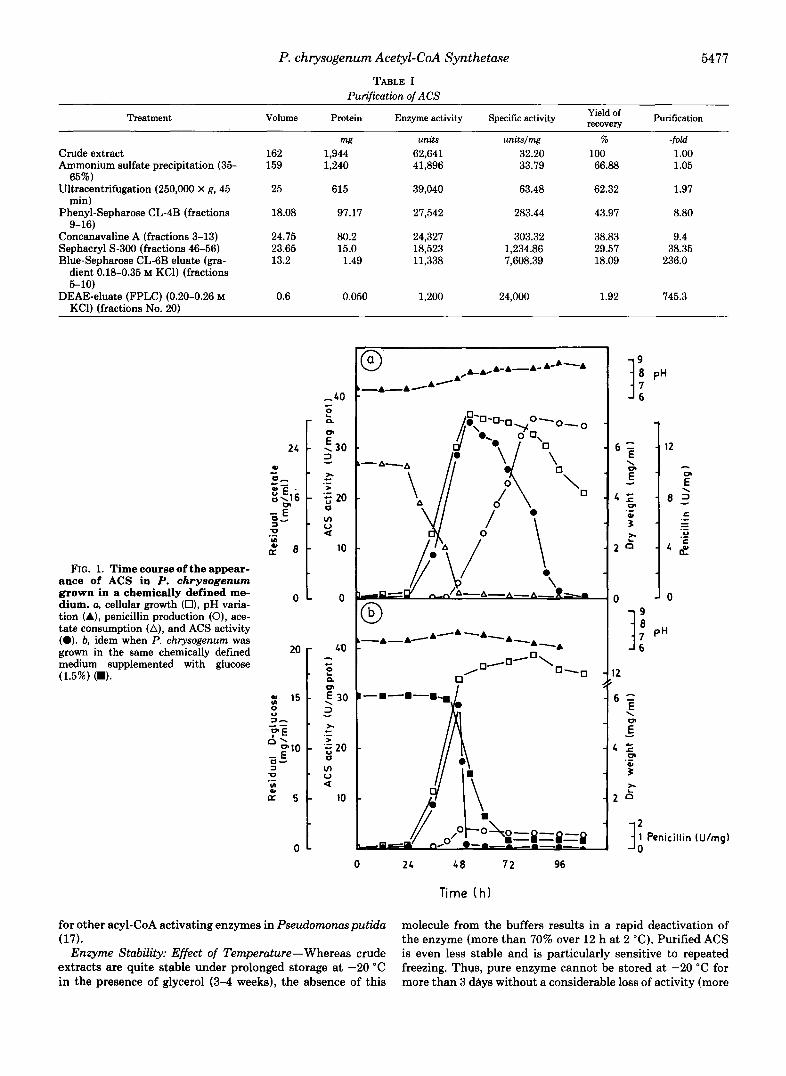
We have studied this enzyme in mycelia of P. chrysogenum grown in a chemically defined medium containing acetate as the main carbon source. ACS began to be synthesized at the early logarithmic phase of growth (30 h) and its activity increased linearly during the exponential phase, reaching a maximal level a t 54 h. Between 54 and 96 h ACS activity decreased rapidly, probably because acetate exhaustion oc- curred (Fig. la). Extracellular penicillin accumulation started to be detectable at 48 h, being maximal a t 84 h. From this time to 108 h penicillin titers remained constant (Fig. la). A similar curve was found when the phenylacetic acid transport system was measured (Fig. 2). The fact that PAA transport system induction and penicillin production started when the acetate was almost exhausted (Figs. la and 2) suggests that they are two directly related processes both regulated by acetate. The analysis of the penicillins accumulated in the broths by HPLC showed that about 90% of the total 8-lactams produced was penicillin G, the rest being 6-APA. It is worth noting that omission of PAA from the broths drastically changes the nature of the penicillin accumulated since, under these conditions, A2-pentenyl penicillin (F) is the only peni- cillin produced (data not shown).
In view of the above data we decided to take fungal cells grown for 54 h as the source of ACS.
Physicochemical Properties of ACS-Purified ACS (see "Materials and Methods") runs in 7% SDS-PAGE slab gels as a single band with an R F of 0.358. The calculated molecular mass for the denatured protein was 70 kDa (Fig. 3). However, by filtration through a Sephadex G-200 (Pharmacia) column (2.8 X 34.5 cm) the molecular mass of the enzyme was 139 k 1 kDa suggesting that ACS, in the native form, is a dimer composed of two apparently identical subunits in an a2 oli- gomeric structure. Similar molecular weights for both the native enzyme and subunits have been reported in Methan- othrix soehngenii (26) and in Saccharomyces cerevisiae (27). Contrariwise, the ACS purified from rat liver seems to be formed of a single monomer of 62 kDa (28). Additionally, it has been shown that in A. nidulum and Neurospora crassa the ACS gene codes for a polypeptide chain of about 670 and 606-626 residues of amino acids, respectively (29). Similar results have been reported for the long-chain acyl-CoA syn- thetase from rat liver (30).
ACS activity was maximal at 37 "C and at a pH value of 8.0. Under these conditions the reaction was linear over 30 min (Fig. 4). The enzyme showed hyperbolic behavior for ATP, CoA, and acetate, the K, calculated for each being 17, 0.18, and 6.8 mM, respectively. All these experiments were carried out at saturating concentrations of M$+. This ion can be replaced by Mn2+, but in this case a lower rate of catalysis is observed (about 64%).
ACS shows low activity in the presence of the ampholites contained in the buffers required for chromatofocusing. The enzyme migrates in isoelectric focusing gels as a diffuse band between pH 5.6 and 6.0. Similar results have been reported
P. chrysogenum Acetyl-CoA Synthetase 5477
TABLE I Purification of ACS
Treatment Volume Protein Enzyme activity Specific activity 2:;: Purification
w units unitslmg % -fold Crude extract 162 1,944 62,641 32.20 100 1 .oo Ammonium sulfate precipitation (35- 159 1,240 41,896 33.79 66.88 1.05
Ultracentrifugation (250,000 X g, 45 25 615 39,040 63.48 62.32 1.97
Phenyl-Sepharose CL-4B (fractions 18.08 97.17 27,542 283.44 43.97 8.80
Concanavaline A (fractions 3-13) 24.75 80.2 24,327 303.32 38.83 9.4 Sephacryl S-300 (fractions 46-56) 23.65 15.0 18,523 1,234.86 29.57 38.35 Blue-Sepharose CL-GB eluate (gra- 13.2 1.49 11,338 7,608.39 18.09 236.0
65% )
min)
9-16)
dient 0.18-0.35 M KCl) (fractions 5-10)
DEAE-eluate (FPLC) (0.20-0.26 M 0.6 0.050 1,200 24,000 1.92 745.3 KCl) (fractions No. 20)
FIG. 1. Time course of the appear- ance of ACS in P. chrysogenum grown in a chemically defined me- dium. a, cellular growth (O), pH varia- tion (A), penicillin production (O), ace- tate consumption (A), and ACS activity (0). b, idem when P. chrysogenum was grown in the same chemically defined medium supplemented with glucose (1.5%) (W).
0 t o
20
0 0
f IS
" a- "E
- E d 2,o 0 -
-0 3
cn a#
.- a s
0
\ 0 \
0
".".".~ %L
0 2s 4 8 7 2 96
Time (h)
for other acyl-CoA activating enzymes in Pseudomonasputida molecule from the buffers results in a rapid deactivation of (17). the enzyme (more than 70% over 12 h at 2 "C). Purified ACS
Enzyme Stability: Effect of Temperature-Whereas crude is even less stable and is particularly sensitive to repeated extracts are quite stable under prolonged storage at -20 "C freezing. Thus, pure enzyme cannot be stored at -20 "C for in the presence of glycerol (3-4 weeks), the absence of this more than 3 days without a considerable loss of activity (more

5478 P. chrysogenum Acetyl-coA Synthetase
than 50%). ACS is also very sensitive to temperatures above 0 degrees, showing very rapid deactivation kinetics when incubated for 30 min at temperatures higher than 30 “C (Fig. 5). Similar results have been reported for other related en- zymes (17).
Effect of Cations and Other Molecules-The effect of several cations on ACS activity was studied by adding them to the assays at a final concentration of 1 mM. Whereas monovalent cations (K+, Na+, Li+) did not cause any significant effect, certain divalent cations (Zn2+, Cu2+, and H e ) strongly inhib- ited the enzyme (97,98, and loo%, respectively).
ACS was also inhibited by several agents that react with thiol groups [5,6’-dithiobis(2-nitrobenzoic acid), p-chloro- mercuribenzoate, and N-ethylmaleimide] when added to the reaction mixtures at a concentration of 1 mM (93, 97, and loo%, respectively). However, DTT, reduced glutathione, and
- C . _
300 E - 0
E a
- 1; 200 a VI C 0 L - a 100 a
0 26 Cb 7 2
Time (h)
FIG. 2. Uptake of phenylacetic acid when P. chrysogenum Wis 54-1255 was grown in minimal medium. PAA uptake (0); acetate consumption (A).
kDa kDa 941 - - 94
61 - ACS - 67 -
431 - - 43
I d
29’ -- k. ,29 ”_
FIG. 3. Electrophoretic mobility of purified ACS in SDS- PAGE, ACS, acetyl-coA synthetase (5 pg). a and b are molecular mass standards proteins (phosphorylase b, 94 kDa; bovine serum albumin, 67 kDa; ovalbumin, 43 kDa; and carbonic anhydrase, 30 kDa). Proteins were stained with Coomassie Brilliant Blue R-250.
2-mercaptoethanol (1 mM) increased ACS activity (114, 122, and 140%) respectively). These results suggest that, as re- ported for phenylacetyl-coA ligase from P. putida (17)) some SH groups of ACS are essential for catalysis to occur.
Detergents (0.5% w/v) also affected the activity of this enzyme, SDS being the compound that elicited a strongest degree of inhibition (100%). Other molecules such as Tween 20, Tween 80, Tween 100, and Nonidet P40 inhibited activity to a lower extent (24, 30, 36, and 14%) respectively); this effect was similar to that reported for other enzymes or enzymatic systems (17, 31-32).
Substrate Specificity-In an attempt to characterize the specificity of this enzyme, different molecules were tested as substrates of ACS. Using the hydroxylamine procedure as the assay method (see “Materials and Methods”), we found that ACS recognizes as substrates: acetic, propionic, butyric, and valeric acids but with different efficiencies (100, 48, 20, and 6.7%) respectively). Taking into account the substantial levels of catalysis with substrates other than acetate, we have de- signed a highly sensitive method to evaluate low levels of conversion of some rare substrates related to penicillin bio- synthesis. By coupling in vitro ACS and AT, these substrates can be converted into penicillins and evaluated by bioassay (see “Materials and Methods”). The results presented in Table I1 and Fig. 6 indicate that the substrate specificity of this enzyme is quite broad and that ACS can activate many other molecules to acyl-CoA derivatives: hexanoic, A3-hex- enoic, heptanoic, octanoic, A3-octenoic, phenylacetic, 2-thio- phene acetic, and 3-thiopheneacetic acids. These acyl-CoA derivatives can be used by P. chrysogenum to produce in vivo the corresponding penicillins, implicating that ACS could play an important role in the biosynthetic pathway of such anti- biotics. Moreover, the fact that the acetyl-coA synthetase obtained from the p-lactam producer A. niduluns also acti- vates these kind of compounds (Table 11)) strongly reinforces the above conclusion.
It can be observed that ACS recognizes better as substrates A3-hexenoic acid and A3-octenoic acids than their saturated molecules (hexanoic and octanoic acids) (see Table 11). These results contrast with the higher titers of saturated side chain- containing penicillins (DF and K) reported by us (11) when P. chrysogenum was cultured in industrial broths in the ab- sence of PAA. We suggest three alternative explanations to justify this observation: (i) the inhibitory effect (owing to their detergent properties) on the enzymatic system (ACS- AT) caused by hexanoic and octanoic acids would lead to a lower rate of catalysis when these acids are used as substrates; (ii) the synthesis of hexanoyl- and octanoyl-CoA (required by AT for the biosynthesis of penicillins DF and K) might be catalyzed in vivo by a different enzymatic system (fatty acid &oxidation or directly through a reaction catalyzed by a different acyl-CoA synthetase) and (iii) the structure of fatty acids containing double bonds at the C3 position (A3-hexenoic
FIG. 4. Effect of time (a), temper- ature (b) , and pH (c), 50 mM citric- citrate buffer (0); 50 mM phosphate buffer (A); 50 mM HC1-Tris buffer (0); and glycine-NaOH buffer (0); on the activity of ACS. v) u
U
1050
900
750
450
300
150
0 20 LO 60 0 10 20 30 LO 50 0 4 5 6 7 8 9
l i m e ( m i n ) l e P C ) pH

P. chrysogenum Acetyl-coA Synthetase 5479
Time lm in )
FIG. 5. Effect of temperature on ACS activity. Enzyme in- activation rate when incubated at different temperatures. After in- cubation at these temperatures, ACS was assayed for 30 min at 37 “C under the conditions described under “Materials and Methods.”
TABLE I1 In uitro biosynthesis of penicillins
ACS from P. chrysogenum (I) or from A. nidulans (11) was coupled in uitro with AT from P. chrysogenum in the presence of the different substrates. Each penicillin was evaluated against its standard (see “Materials and Methods”).
Side-chain urecursor Penicillin uroduction mM
Hexanoic acid 12 Hexanoic acid 3 Hexanoic acid 0.6 A’-Hexenoic acid 12 A3-Hexenoic acid plus acetate 12 + 12 Heptanoic acid 12 Octanoic acid 12 Octanoic acid 3 Octanoic acid 0.6 A3-Octenoic acid 12 PAA 12 PAA plus acetate 12 + 12 PAA plus acetate 12 + 1 PAA plus acetate 12 + 0.05 2-Thiopheneacetic acid 12 3”Thio~heneacetic acid 12
I I1 unitslml
0.62 0.51 0.80 0.63 0.70 0.40 5.0 4.0 0 0 0.75 0.8 0.12 0.10 0.25 0.23 0.36 0.30 0.45 0.5 1.5 0
1.2 0
0.2 0.15 0.8 0.60 1:o 0.82 0.7 0.65
and A3-octenoic) would permit the acquisition of an appro- priate configuration (more rigid) which would facilitate the binding of the substrates at the active site. A similar structural requirement was reported when the substrate specificity of a different penicillin biosynthetic enzyme (AT) from P. chrys- ogenum was studied (33).
In order to shed light on these points, several experiments were performed. We have tested different concentrations of hexanoic and octanoic acids in the ACS-AT reaction mixture to establish whether they are able to act as inhibitors of the enzyme (see Table 11). We observed that when the final concentration of octanoic acid was reduced (0.6 and 3 mM) it was added more efficiently to 6-APA (as measured by the generation of the corresponding penicillins). This effect was less pronounced when lower concentrations of hexanoic acid were used (see Table 11). These results suggest that, in uiuo, P. chrysogenum can utilize ACS to obtain all the acyl-CoA
Sequence of reactlons
R-COOH + COA +ATP R-C-CoA + AMP+ PPI’ Mg2+
D l 1 CoA
Penicil l ins
FIG. 6. Bioassay against M. Zuteus ATCC 9341 of the prod- ucts generated after incubating ACS and AT in the presence of different substrates. A: 1, hexanoic; 2, A’-hexenoic; 3, heptanoic; and 4 , octanoic acids. B: I , A’-octenoic; 2, phenylacetic; 3, P-thiophe- neacetic acid; and 4, 3-thiopheneacetic acids. When the reactions were carried out in the absence of penicillin side-chain precursor, CoA, ATP, M P , or 6-APA no bioactivity was detected. When A3- hexenoic acid was tested as a substrate ( A , 2), the reaction product was diluted 2.5-fold before analyzing the penicillin formed by bioas- say. All the antibiotics produced were sensitive to 8-lactamase attack.
thioesters required to synthesize penicillins G, F, DF, and K. However, we cannot rule out that other acyl-CoA activating enzyme(s), which also could serve to synthesize hexanoyl- and octanoyl-CoA, might be induced in P. chrysogenum when this fungus was cultured in complex industrial broths.
In summary, we demonstrate that the lack of catalysis observed (according to the hydroxylamine assay method) when ACS was incubated with acyl-acids whose carbon length ranges between Cs and Cs, does not actually mean that this enzyme was fully unable to activate these substrates; it is possible that the sensitivity of the method was too low. The quantities of the acyl-CoA products generated in the reactions were quite sufficient to be detected by bioassay against M. luteus after being transformed into penicillins (see Table 11).
The high quantity of penicillin F obtained when A3-hex- enoic acid was tested as a substrate (Table 11), indicates that this enzymatic system (ACS-AT) could probably produce in uiuo higher amounts of this penicillin if the appropriate side chain precursors (or unsaturated fatty acids) were present in the raw materials routinely used in industrial fermentations. This high capacity to synthesize penicillin F could be the reason why this antibiotic is the only penicillin produced in the absence of PAA (not shown), and was the first penicillin isolated from the cultures of P. chrysogenum (34). Therefore,

5480 P. chrysogenum Acetyl-coA Synthetase
the fact that penicillin F was not accumulated when PAA is added to the chemically defined medium, suggests that A3- hexenoyl-CoA is not synthesized in the presence of phenyl- acetic acid.
The comparative study of the substrate specificity of ACS from P. chrysogenum and A. niduluns (Table 11) suggests that it is quite different from that reported for phenylacetyl-coA ligase (PCL) from P. putida (17). Although these three en- zymes recognize similar substrates, the enzyme of P. putida preferentially activated phenylacetic acid and also, but with a lower efficiency, different acyl-acids, indicating that they are two gifferent enzymes. The relevance of these results will be discussed below.
When ATP was replaced by other nucleotides (ADP, UTP, UDP, CTP, CDP, GTP, or GDP) ACS activity was only detected when ADP, UTP, CTP, and GTP were used as substrates. The structural similarity between ATP and ADP, to a certain extent (37%) permitted the synthesis of acetyl- CoA, whereas in the other cases activity was almost negligible (10, 7, and 3%, respectively). Similar results have been re- ported for phenylacetyl-coA ligase from P. putida (17) and benzoyl-CoA synthetase from Rhodopseudomonas pulustris (35).
Carbon Catabolite Inactivation of ACS-Fig. l b shows that ACS was synthesized at a similar rate when P. chrysogenum was grown in the basal medium containing acetate or acetate and glucose (1.5% w/v) for 48 h, whereas later, when (due to the exhaustion of acetate) glucose started to be catabolized, ACS activity decreased rapidly. The lack of consumption of glucose seems to be due to the preferential utilization of acetate by this fungus when a mixture of these two carbon sources (acetate and glucose) was present in the medium. The preferential utilization of acetate has been reported both in P. chrysogenum and A. niduluns (36,37). Similar results were obtained when soy bean oil (1% w/v) was supplied together with acetate in the chemically defined medium. Under these conditions, P. chrysogenum did not start to consume the oil until the acetate had been exhausted (at 24 h 100% of oil remains; at 48 h 80%; whereas at 60 h, due to the consumption of acetate, only 20% of the initial oil was detected). These experiments indicate that in the presence of acetate, fatty acids (oils), like glucose, are not (or are very poorly) taken up by the cells. When glucose consumption started (at about 42 h of growth) a rapid loss on the measurable activity of ACS was observed (Fig. lb). Although the ACS of A. niduluns has been shown to be controlled at the level of mRNA accumu- lation by carbon catabolite repression (38), the rapid loss of activity of the enzyme from P. chrysogenum cannot be ex- plained by this mechanism alone. The in vivo half-life of ACS, evaluated by halting protein synthesis in 39- and 43-h-old cultures with cycloheximide (125 pglml), was estimated to be 10 h (Fig. 7a), i.e. higher than the time required to inactivate ACS when glucose is catabolized (about 2 h, Fig. lb). This glucose effect cannot be explained by transcriptional repres- sion (38,39), and we conclude that a faster posttranscriptional mechanism of control of the enzymatic activity does exist: carbon catabolic inactivation (40-46). To study further this effect, 125 pg/ml cycloheximide was added to cultures grown for 42 h in minimal medium containing acetate and glucose, and the rate of the disappearance of ACS was studied. Fig. 7b shows that in both cases (with and without CH) the rate of disappearance was quite similar, suggesting that protein syn- thesis is not required for ACS inactivation. Similar results have been reported for a different enzyme (fructose-1,6-di- phosphatase) in yeasts (47, 48). To establish whether the effect caused by glucose on ACS activity was or was not
b
lome l h l Timr t h l
FIG. 7. Inactivation of ACS by glucose. a, half-life of ACS (0) when P. chrysogenum was grown in the basic chemically defined medium for 39 and 43 h (V). Arrows indicate the time at which cycloheximide (CH) (125 pg/ml) was added. After antibiotic addition, protein synthesis is completely and immediately halted (as we assured by [“Clleucine incorporation). b, rate of ACS deactivation when P. chrysogenum was grown for 42 h in a chemically defined medium containing acetate (0) or acetate and glucose (0). Idem when at 42 h. CH (125 pg/ml) was added (A, m). Some cultures grown in the glucose containing medium were harvested at different times (42.5 and 43 h), washed to eliminate the glucose, and resuspended in starvation medium, and ACS was measured at short intervals (V). The arrow indicates the time at which CH was added.
reversible, cultures incubated for 42 h in the presence of acetate and glucose were harvested at different times (42.5 h and 43 h), washed, resuspended in starvation medium (see “Materials and Methods”) and incubated in it for different times. ACS activity was measured in vitro at short intervals (Fig. 7b). In no case was the restoration of the lost activity observed nor after Sephadex G-25 filtration, dialysis, heat treatment, or by incubating the extracts with different phos- phodiesterases. All these results strongly suggest that an irreversible change in the structure of the enzyme had been produced that handicapped the condensation of acetate to acetyl-coA.
In summary, the absence or low titers of natural penicillins and penicillin G found in the culture broths of P. chrysogenum when glucose was added at the very beginning of the fermen- tation (see Fig. l b ) could be explained by the following over- lapping mechanisms: (i) by carbon catabolite inactivation of ACS when this enzyme has been synthesized during growth on acetate (this report), and also by carbon catabolite repres- sion of the gene encoding this enzyme, as has been reported in A. nidulans (38); (ii) by glucose repression of IPN synthase (49); and (iii) by repression of the phenylacetic acid transport system (50-52).
ACS and PTS regulation could be explained by a common mode of control. Thus, repression of PTS could be an indirect effect of the inactivation of ACS, since if PAA is not activated, phenylacetyl-coA (which seems to be the true inducer of PTS) (52), is not produced and therefore phenylacetic acid cannot be taken up by the cells (inducer exclusion). The fact that PTS was not induced until acetate was exhausted (Fig. 2) fits well with this interpretation.
Role of ACS in the Biosynthesis of Penicillins-The synthe- sis of different penicillins when ACS and AT were coupled in vitro (Table I1 and Fig. 6), indicates that ACS could be the enzyme actually responsible for the activation, to their CoA derivatives, of the different side-chain precursors required by P. chrysogenum and A. nidulans for the production of such antibiotics in vivo. This result is consistent with the absence of penicillin when acetate is being catabolized as carbon source (see Fig. 1). The direct involvement of ACS in the biosynthesis of penicillin G is supported by the fact that

P. chrysogenum Ace
formation of this antibiotic, when the ACS-AT coupled sys- tem was incubated in vitro with CoA, ATP, Mg+, 6-APA, DTT, phenylacetate, and acetate, was not observed (see Table 11). In this reaction, mainly acetyl-coA (but no or very little phenylacetyl-CoA) is formed and taking into account that acetyl-coA is not a substrate of AT (11,12), no penicillin can be produced. On the contrary, we detected synthesis of peni- cillin G when the concentration of acetate in the reaction was reduced (0.05 and 1 mM, see Table 11). Similar results have been obtained when this enzymatic system was incubated with A3-hexenoic acid and acetate (Table 11), strongly sup- porting the hypothesis that ACS also catalyzes in vivo the activation (to their CoA derivatives) of other molecules used as penicillin-side chain precursors. However, this situation (an excess of acetate or other acyl-acids with respect to PAA) would be a rare event in industrial fermentations of penicillin G , since the continuous addition of PAA to the fermenters ensures the presence of high quantities of this compound compensating for the lower affinity of ACS for phenylacetic acid.
Acknowledgments-We are gratefully indebted to Dr. M. J. Alonso and R. Sanchez Barber0 for their participation on the elaboration of this manuscript and to N. S. D. Skinner for revising the English version.
REFERENCES 1. Masurekar, P. S., and Demain, A. L. (1972) Can. J. Microbiol.
2. Demain, A. L., and Masurekar, P. S. (1974) J. Gen. Microbiol.
3. Friedrich, C. G., and Demain, A. L. (1977) J. Antibiot. 3 0 , 760-
4. Luengo, J. M., Revilla, G., L6pez-Nietdyl. J., Villanueva, J. R.,
5. Fawcett, P., and Abraham, E. P. (1975) Methods Enzymol. 43,
6. O’Sullivan, J., Bleaney, R. C., Huddleston, J., and Abraham, E. P. (1979) Biochem. J. 184 , 421-426
7. Konomi, T., Herchen, S., Baldwin, J. E., Yoshida, M., Hunt, N. A., and Demain, A. L. (1979) Biochem. J. 184,427-430
8. Rambn, D., Carramolino, L., Patiiio, C., Sinchez, F., and Peiialva, M. A. (1987) Gene 6 7 , 171-181
9. Ramos, F. R., L6pez-Nieto, M. J., and Martin, J. F. (1985) Antimicrob. Agents Chemother. 27,380-387
10. Luengo, J. M., Iriso, J. L., and L6pez-Nieto, M. J. (1986) J. Antibiot. 3 9 , 1565-1573
11. Luengo, J. M., Iriso, J. L., and Mpez-Nieto, M. J. (1986) J. Antibiot. 39,1754-1759
12. Alonso, M. J., Bermejo, F., Reglero, A., Fernindez-Caii6n, J. M., Gonztilez de Buitrago, G., and Luengo, J. M. (1988) J. Antibiot.
13. Martin-Villacorta, J., Reglero, A., and Luengo, J. M. (1989) J. Antibiot. 42 , 1502-1505
14. Whiteman, P. A., Abraham, E. P., Baldwin, J. E., Fleming, M. D., Schofield, C. J., Sutherland, J. D., and Willis, A. C. (1990)
15. Brunner, R., and Rohr, M. (1975) Methods Enzymol. 4 3 , 476-
16. Kogekar, R., and Deshpande, V. N. (1983) Indian J. Biochern.
17. Martinez-Blanco, H., Reglero, A., Rodriguez-Aparicio, L. B., and
18,1045-1048
82,143-151
761
and Martin, J. F. (1980) J. Bacteriol. 144,869-876
471-473
41,1074-1084
FEBS Lett. 262,342-344
481
Biophys. 20,208-212
Luengo, J. M. (1990) J. Biol. Chem. 2 6 5 , 7084-7090
tyl-CoA Synthetase 5481
18. Smith, D. J., Burnham, M. K. R., Edwards, J., Early, A. J., and Turner, G. (1990) BiolTechnology 8,39-41
19. Luengo, J. M., Alemany, M. T., Salto, F., Ramos, F., Mpez-Nieto, M. J., and Martin, J. F. (1986) BiolTechnology 4,44-47
20. Bradford, M. M. (1976) Anal. Biochem. 7 2 , 248-254 21. Lipmann, F., and Tuttle, L. C. (1945) J. Biol. Chem. 169 , 21-28 22. Keston, A. S. (1956) 5th Meeting of the American Chemical
23. Holz, G., and Bergmeyer, H. U. (1974) Methods Enzym. Anal. 3,
24. Wrigley, C. W. (1971) Methods Enzymol. 22,559-564 25. Weber, K. J., and Osborn, M. (1969) J. Biol. Chem. 2 4 4 , 4406-
26. Jetten, M. M., Stams, A. J. M., and Zehnder, A. J. B. (1989) J.
27. Frenkel, E. P., and Kitchens, R. L. (1977) J. Biol. Chem. 252 ,
28. Imesch, E., and Rons, S. (1984) Znt. J. Biochem. 16,875-881 29. Connerton, I. F., Fincham, J. R. S., Sandeman, R. A., and Hynes,
M. J. (1990) Mol. Microbiol. 4 , 451-460 30. Suzuki, H., Kawarabayasi, Y., Kondo, J., Abe, T., Nishkawa, K.,
Kimura, S., Hashimoto, T., and Yamamoto, T. (1990) J. Biol.
31. Wallach, D., and Pastan, I. (1976) J. Biol. Chem. 261,5802-5809 32. Bador, H., Morelis, R., and Louisot, P. (1984) Biochimie 66,223-
233 33. Martin-Villacorta, J., Reglero, A., Ferrero, M. A., and Luengo, J.
M. (1990) J. Antibiot. 43,1559-1563 34. Florey, H. W., Chain, E. B., Heatley, N. G., Jennings, M. A.,
Sanders, A. G., Abraham, E. P., and Florey, M. E. (eds) (1949) in Antibiotics, Vol. 2, Oxford University Press, London
35. Geisller, J. F., Harwood, C. S., and Gibson, J. (1988) J. Bacteriol.
36. Burnett, J. H. (1976) Fundamentals of Mycology, pp. 238-248, E.
37. Romano, A. H., and Kornberg, H. L. (1969) Proc. Roy. SOC. B.
38. Sandeman, R. A., and Hynes, M. J. (1989) Mol. Gen. Genet. 218 ,
39. Magasanik, B. (1961) Cold Spring Harbor Symp. Quunt. Biol. 26 ,
40. Duntze, W., Neumann, D., and Holzer, H. (1966) Eur. J . Biochern.
41. Gorts, C. P. M. (1969) Biochim. Biophys. Acta 184 , 299-305 42. Gancedo, C. (1971) J. Bacteriol. 107,401-405 43. Brown, H. D., Satynarayama, T., and Umbarger, H. E. (1975) J.
44. Gancedo, C., and Schwerzmann, K. (1976) Arch. Microbiol. 104 ,
45. Gancedo, C., and Serrano, R. (1989) in The Yeasts (Rose, A. H., and Harrison, J. S., eds) Vol. 3, pp. 205-259, Academic Press, New York
46. Mattern, H., and Holzer, H. (1977) J. Biol. Chem. 262 , 6399- 6402
47. Lenz, A. G., and Holzer, H. (1980) FEBS Lett. 109 , 271-274 48. Mazon, M. J., Gancedo, J. M., and Gancedo, C. (1982) Eur. J.
Biochem. 127,605-608 49. Revilla, G., Ramos, F. R., L6pez-Nieto, M. J., Alvarez, E., and
Martin, J. F. (1986) J. Bacteriol. 168,947-952 50. Fern&ndez-Caii6n, J. M., Reglero, A., Martinez-Blanco, H., and
Luengo, J. M. (1989) J. Antibiot. 42,1398-1409 51. Fernandez-Caiih, J. M., Reglero, A., Martinez-Blanco, H.,
Ferrero, M. A., and Luengo, J. M. (1989) J. Antibiot. 42,1410- 1415
52. Martinez-Blanco, H., Reglero, A., Ferrero, M. A., Fernindez- Caiibn, J. M., and Luengo, J. M. (1989) J. Antibiot. 42 , 1416- 1423
Society, Dallas, TX, pp. 310-314 (Abstr. 129)
1528-1532
4412
Bacteriol. 171,5430-5435
504-507
Chem. 266, 8681-8685
170,1709-1714
Arnold, London
173,475-490
87-92
249-256
3,326-331
Bacteriol. 121 , 954-969
221-225