passive and active components in diastolic...
TRANSCRIPT

Passive and active components in diastolic function
1
Congenital heart disease and right ventricular failure – The passive vs. the active components in
diastolic dysfunction.
Short title Passive and active components in diastolic function
Version 7
Date 06-04-2014
Coordinating investigators Prof. dr. R.M.F. Berger ([email protected])
Dr. B Bartelds ([email protected])
Principal investigator M.P.H. van Wiechen ([email protected])
Laboratory sites Experimental Cardiology section HPC AB 43
P.O. Box 30.001
9700 RB Groningen
The Netherlands

Passive and active components in diastolic function
2
DUTCH AND ENGLISH SUMMARY
Nederlandse samenvatting
Doel: Rechter ventrikel falen/cor pulmonale (RVF) komt voor in het eind stadium van pulmonale
arteriële hypertensie (PAH) en heeft een slechte prognose. Een langer bestaande drukbelasting van de
rechter kamer leidt tot hypertrofie en uiteindelijk tot falen. Uit eerder onderzoek is gebleken dat falen
wordt veroorzaakt door een verlies van diastolische functie. Het doel van deze studie is om te
onderzoeken of diastolische disfunctie in de rechter kamer voornamelijk wordt veroorzaakt door
verandering in stijfheid of voornamelijk door veranderingen in actieve ontspanning.
Opzet: Het weefsel van 2 bestaande dierproeven is verzameld en hierop is experimenteel onderzoek
gedaan. Het betreft één rattenmodel waarbij dieren een PAB (pulmonary artery banding, het afbinden
van de longslagader) ondergingen. Dit leidt tot een drukbelasting van de rechter ventrikel. Deze ratten
zijn vervolgens ingedeeld op symptomen van RVF. In het tweede rattenmodel is gekeken naar de
invloed van sildenafil behandeling hierop. Met behulp van een RT-PCR, western blot en ELISA zijn
mechanismen achter de diastolische functie onderzocht. Er is gekeken naar de natrium-calcium-
transporter (NCX), phospholamban (PLN) SERCA2, titin-isovormen (N2Ba, N2B) en de activiteit
van protein kinase A en G. (PKA, PKG)
Resultaat: De componenten die stijfheid bepalen in de RV zijn veranderd: er is sprake van minder
fibrose en een toename in de N2Ba/N2B ratio: beide leiden theoretisch tot minder stijfheid. Actieve
componenten zijn verschillend aangedaan: SERCA expressie nam af en PLN expressie nam toe bij
verminderde diastolische functie, NCX bleef onveranderd. PKA activiteit is afgenomen in de ratten
met een PAB. PKG1 activiteit is onveranderd gebleven.
Conclusie: In deze studie is diastolische disfunctie geassocieerd met afgenomen actieve relaxatie en
een veranderde stijfheid door een afname van fibrose en toename van de compliante isovorm van titin.
Summary in English
Objective: Right ventricular failure/cor pulmonale (RVF) is often seen in end-stage pulmonary
arterial hypertension (PAH) and has a bad prognosis. A long-existing pressure load on the right
ventricle (RV) leads to hypertrophy and finally to failure. Previous results suggest that RVF is caused
by diastolic dysfuncton. The goal of this study is to investigate whether diastolic dysfunction is
mainly caused by a change in passive stiffness or by a change in active relaxation.
Design: Tissue of 2 existing animal models was collected and used for experimental research. One of
the two models consisted of rats that underwent pulmonary artery banding (PAB) leading to an
increased pressure load on the RV. Rats were then assessed on clinical symptoms of failure. In the
other model the influence of sildenafil treatment on PAB was investigated. With the use of RT-PCR,
western blot and ELISA; mechanisms of diastolic function were investigated. We looked at the
sodium-calcium transporter (NCX), phospholamban (PLN), SERCA2, titin isoforms (N2Ba, N2B)
and protein kinases A and G activity. (PKA, PKG)
Results: Components determining passive stiffness are changed. Less fibrosis and an increase of the
N2Ba/N2B ratio: both theoretically leading to a less stiff myocardium. The active components are
differentially affected: SERCA2 expression decreased and PLN expression increased with declining
diastolic function, NCX remained unchanged. PKA activity was decreased in the banded animals vs.
control. PKG1 activity was unchanged.
Conclusion: In this study we show that diastolic dysfunction is associated with decreased active
relaxation and a changed stiffness through a decrease in fibrosis and increase in the compliant titin
isoform.

Passive and active components in diastolic function
3
LIST OF ABBREVIATIONS
RA – Right Atrium
RV – Right Ventricle
CHD – Congenital heart disease
CHF – Congestive heart failure
CON – Control Group
PAB – Pulmonary artery binding
PAB-RVF – Right ventricular failure
PAB-RVD – Right ventricular disease
PAB-VEH – Pulmonary artery binding, vehicle treatment
PAB-SIL – Pulmonary artery binding, sildenafil treatment
PAH – Pulmonary arterial hypertension
SR – Sarcoplasmatic Reticulum
Tn - Troponin
SERCA – Sarco/endoplasmic reticulum Ca2+
-ATPase
PLN - Phospholamban
NCX – Na+/Ca
2+ exchanger
Tau – Isovolumic relaxation constant
PCR-RT – Polymerase chain reaction-reverse transcriptase
CAMK-II - Ca2+
/calmodulin-dependent protein kinase
PKA – Protein Kinase A
PKG – Protein Kinase G
IOD – Integrated Optical density

Passive and active components in diastolic function
4
TABLE OF CONTENTS
DUTCH AND ENGLISH SUMMARY .................................................................................... 2
LIST OF ABBREVIATIONS .................................................................................................... 3
1. INTRODUCTION ............................................................................................................. 5
1.1 Active components ...................................................................................................... 6
1.2 Passive Components .................................................................................................... 8
2. MATERIAL AND METHODS ......................................................................................... 9
2.1 Animal models used and definition RVD, RVF ......................................................... 9
2.2 Gene expression N2B/N2Ba, NCX and PLN ........................................................... 10
2.3 Protein expression SERCA, PLN .............................................................................. 10
2.4 Activity Assay PKG .................................................................................................. 10
2.5 Activity Assay PKA .................................................................................................. 11
2.6 Data Analysis ............................................................................................................ 11
3. RESULTS ........................................................................................................................ 12
3.1 Passive Component: Titin Expression....................................................................... 12
3.2 Active components: NCX, SERCA2ATP and PLB .................................................. 12
3.2.1 NCX ................................................................................................................... 12
3.2.2 SERCA2ATPase ................................................................................................ 13
3.2.3 PLN .................................................................................................................... 14
3.2.4 PKA- analysis .................................................................................................... 15
3.2.5 PKG1- analysis .................................................................................................. 15
3.3 Mechanisms involved and the degree of RV relaxation ........................................... 16
3.4 Overview of cellular components ............................................................................. 16
4. DISCUSSION .................................................................................................................. 17
5. CONCLUSION ................................................................................................................ 21
6. REFERENCES ................................................................................................................ 22
7. SUPPLEMENTAL DATA .............................................................................................. 24

Passive and active components in diastolic function
5
1. INTRODUCTION
The estimated prevalence of congenital heart disease (CHD) worldwide is around 9 per 1000
live births. This means that in the Netherlands around 1400 children a year are born with a
congenital heart defect.(1) As the survival of the group of patients with corrected congenital
heart disease improves, new complications come to light. Important complications,
influencing the long term outcome of congenital heart disease (CHD) are right ventricular
failure (RVF) and pulmonary hypertension (PH).(2,3)
RVF is a result of chronic abnormal loading conditions due to the CHD. (4) Under the
influence of an elevated pressure load the RV adapts, but this may progress into a
maladaptation, depending on the clinical presentation. We speak of right ventricular
dysfunction (RVD) or right ventricular failure (RVF). RVF is a maladaptation of the right
ventricle associated with symptoms of fluid retention; (edema and ascites) low cardiac output
(exercise intolerance and fatigue) and atrial or ventricular arrhythmias.(5) RVD describes the
right ventricle which has maladapted as a result of a pathologic condition, but without the
clinical manifestation of RVF. The molecular pathway behind the maladaptation process of
the RV is subject of much research but has not yet been fully explained.
To explain the mechanisms of RVF, our lab has developed an animal model of elevated
pressure load of the RV via a pulmonary artery banding.(6). A PAB leads to stenosis and
hence increased afterload for the RV. In a group of rats with a tight PAB we observed that
after a mean of 52 days half of the animals developed clinical signs of RVF. These rats, PAB-
RVF were compared with a similar group of rats with a PAB but without the clinical signs:
PAB-RVD.
We showed that PAB-RVF is characterized by a higher end-diastolic elestance and end
diastolic pressure, therefore suggesting that RVF is mainly caused by diastolic dysfunction.
Additionally, systolic function was even better in PAB-RVF than PAB-RVD. When further
investigating diastolic function, we can divide the influencing factors into two groups: those
determining resting tension (i.e. Passive relaxation, Fpassive) and those contributing to active
relaxation.(7) Hemodynamic data showed no significant difference in tau (a time constant
and index of ventricular relaxation) between PAB-RVD and PAB-RVF, suggesting that
diastolic dysfunction is mainly caused by an increase in resting tension and not active
relaxation.(6)
The mechanism of the diastolic dysfunction in rats with increased afterload is yet
unknown. To study the contribution of passive- and active relaxation to diastolic dysfunction,
we will seperately analyze most components of the diastolic function of tissues of rats with
PAB-RVD and PAB-RVF.

Passive and active components in diastolic function
6
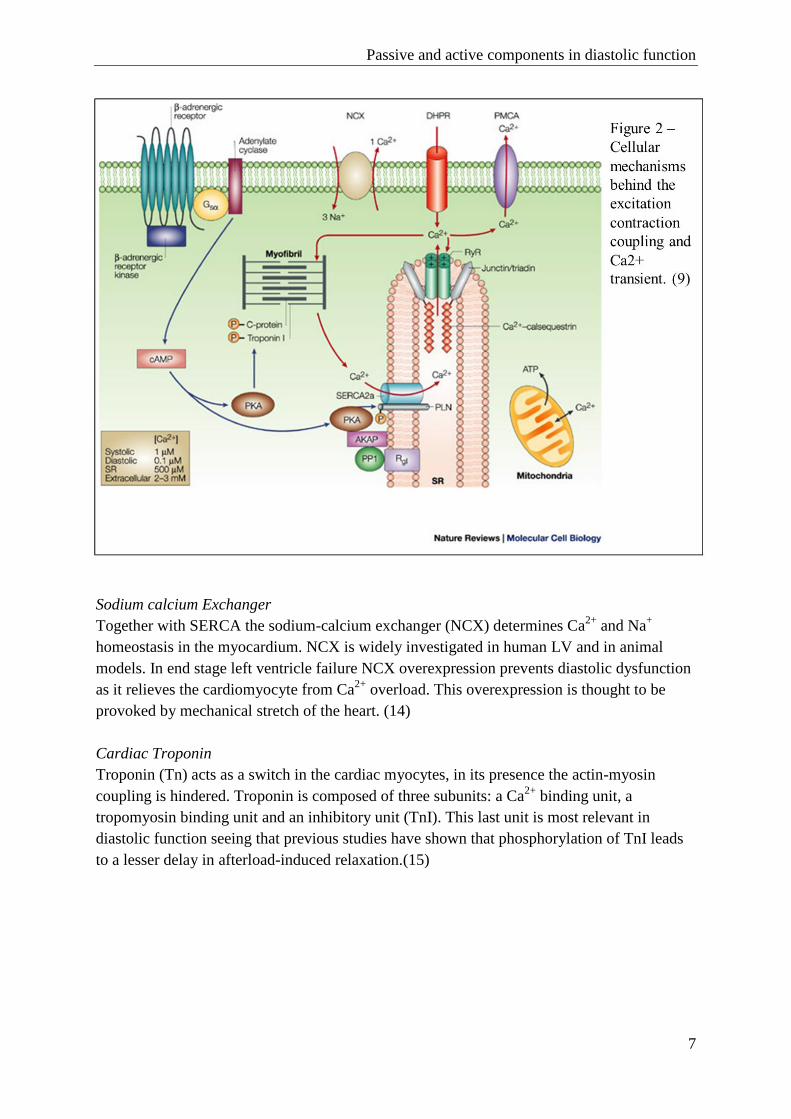
In order for the cardiomyocyte to properly relax actin- and myosinfilaments (the
microfilaments) need to uncouple and Ca2+
needs to be taken up by the Sarcoplasmatic
Reticulum (SR). The cardiomyocyte has several signaling molecules and ion-channels in
order to do so (figure 1). The mechanisms involved and mechanisms determining resting
tension in the myocardium as a whole will be explained from here onwards.(8)
1.1 Active components(8)(8)
SERCA2ATPase/Phospholamban
For microfilament uncoupling, actin and myosin filaments need to relieve their cross-bridges.
This is regulated by the Ca2+
transient in the cardiomyocyte, which in its turn is regulated by
several signaling pathways.
Intracellular, the Ca2+
is taken up by the sarcoplasmatic reticulum through the
Sarco/endoplasmic reticulum Ca2+
-ATPase (SERCA2). In the cardiomyocyte SERCA2
enhances quick Ca2+
uptake by the SR, therefore allowing the cardiomyocyte to relax. When
SERCA2 is not available or when inhibited, the cardiac muscle cells are not able to properly
relax (figure 2).(8)
SERCA2 is modulated by phospholamban (PLN). When dephosphorylated-PLN is bound
to SERCA, SERCA is less active, therefore Ca2+
uptake by the SR is decreased and micro-
filament uncoupling is impaired.(9) PLN can however be phosporylated by protein kinases:
either through PKA (at position serine-16) or through Ca2+
-calmodulin dependent protein
kinase (CAMK-II) (at position threonine-17). In its phosphorylated form PLN relieves its
inhibition and SERCA2 can properly transport Ca2+
through the SR membrane.(10-13) In this
research we compared SERCA2, PLN and PKA-phosphorylated PLN (p-PLN) for the
different groups.
Passive and active components in diastolic function
7
Sodium calcium Exchanger
Together with SERCA the sodium-calcium exchanger (NCX) determines Ca2+
and Na+
homeostasis in the myocardium. NCX is widely investigated in human LV and in animal
models. In end stage left ventricle failure NCX overexpression prevents diastolic dysfunction
as it relieves the cardiomyocyte from Ca2+
overload. This overexpression is thought to be
provoked by mechanical stretch of the heart. (14)
Cardiac Troponin
Troponin (Tn) acts as a switch in the cardiac myocytes, in its presence the actin-myosin
coupling is hindered. Troponin is composed of three subunits: a Ca2+
binding unit, a
tropomyosin binding unit and an inhibitory unit (TnI). This last unit is most relevant in
diastolic function seeing that previous studies have shown that phosphorylation of TnI leads
to a lesser delay in afterload-induced relaxation.(15)

Passive and active components in diastolic function
8
Protein kinase A and G
Many of the intracellular key proteins for contraction and Ca2+
handling are phosphorylated
through protein kinase A or protein kinase G. Both have anti-hypertrophic and inotropic
effects. As described above, PKA can phosphorylate PLN and thereby it enhances Ca2+
uptake through SERCA. This is one of the many proteins in the cardiomyocyte that can be
phosphorylated through PKA. PKG is predominantly antihypertrophic by regulating G-
protein signaling in the cardiomyocyte. Titin for example, is phosphorylated by PKG, where
phosphorylation by PKG leads to posttranslational modification and reduced myocardial
stiffness.(16)
PDE5 inhibitors (such as sildenafil) have recently been approved as treatment for PAH. It
was believed that sildenafil works solely on the pulmonary vasculature, but recent work has
shown that sildenafil improves cardiac output independent from its effect on the pulmonary
vasculature.(17) The pathway by which sildenafil improves cardiac output is thought to
involve both PKG and PKA. It is therefore interesting to see how PKG1- and PKA activity
are changed with failure and after sildenafil treatment.
1.2 Passive Components
Titin
Titin is a very large macromolecule in the cardiomyocyte (~3,8 MDa) preventing it from
overstretching.(18) It is of great importance as it co-determines resting tension in the
myocardium. N2Ba and N2B are the two main titin-isoforms, determined by alternative
splicing. The N2Ba is a compliant form of titin and is co-expressed with N2B, which is a
stiffer isoform. An isoform switch where the compliant N2Ba is replaced by the stiffer N2B
has already been described around birth and in several other animal models with heart
failure.(19-21) A comparable isoform switch in the RV as a result of an increased pressure
load has not yet been described.(22)
Fibrosis
Myocytes are surrounded by a collagen network forming a matrix between cardiomyocytes
and capillaries. In failing myocardium this collagen network is transformed into fibrosis.(7)
Increased collagen in the extracellular matrix of the myocardium results in less space for the
cardiomyocytes to relax and causes a decrease in distensibility. A lot of research on fibrosis
in failing myocardium has been presented, showing a relationship between fibrosis and
cardiac stiffness. Results however remain controversial as some animal models fail to show a
change in myocardial stiffness despite the increase in collagen. The two animal models used
in this study show this discrepancy. The animal model in which sildenafil is used as an
intervention after PAB shows a reduction of fibrosis related to the reduction in end diastolic
pressures. The PAB-RVD/RVF model however fails to show a relationship between RVF and
fibrosis.(6,23)

Passive and active components in diastolic function
9
The above shows that there are many different mechanisms involved in diastolic function of
the heart. The knowledge of the adaptation process in the right ventricle differs per
mechanism. The goal of this study is to further investigate these processes.
To test the hypothesis that changes in passive components are more distinct than changes
in active components we measured titin, SERCA, NCX, PKG and PKA. Our aim is to show
that failing myocardium has a lower N2Ba/N2B ratio and decreased titin phosphorylation
shown by protein kinase A and G (PKA, PKG-1) activity. On the latter we hope to show an
increased activity in sildenafil-treated rats. (PAB-SIL).
2. MATERIAL AND METHODS
2.1 Animal models used and definition RVD, RVF
For this study I used RV tissue that had previously been collected. The first group of tissues
was collected in the study comparing clinical RVD with RVF as described in the
introduction. The second group of rats were rats with PAB-RVD that were treated with a
PDE5e inhibitor (sildenafil) that may interfere with diastolic function via PKG. A short
description of the study protocol is given below.
Chronic Experimental Pressure Load Model(6)
21 Wistar rats (male; 160-180g; Charles River, the Netherlands) were randomized into 2
groups: sham (CON, n=7) or pulmonary artery banding (PAB, n=14). PAB was performed
with a constriction of 1.1mm; sham surgery was equal to the PAB surgery, with the exception
of the actual banding of the pulmonary artery.
From the moment of PAB/sham surgery onward, the animals were daily checked for clinical
signs of RVF according to a predefined ABCDE-checklist, which includes Appearance,
Activity, Bodyweight, Circulation (peripheral), Cyanosis, Dyspnea/tachypnea and Edema,
Effusions. The decision whether RVF was present or not was made by 2 blinded observers.
When a rat developed RVF, echocardiography and pressure-volume analysis were performed
before sacrifice: these rats are the PAB-RVF group (n=5). Four PAB rats that did not show
signs of clinical RVF were analyzed and sacrificed after the same follow-up duration (RV
dysfunction; PAB-RVD group, n=4). At 11 weeks the remaining rats (7 CON and 3 PAB-
RVD) were sacrificed.
After a mean period of 52±5 days, 42% of the animals developed clinical RVF. These
animals are used in our study and compared with the animals developing RVD after the same
follow-up period and with the animals from the control group. (In total n=15, 5 PAB-RVF, 4
PAB-RVD, 6 CON). The hearts were dissected and the RV, interventricular septum, LV and
both atria were separated. The RV-tissue is used for our experiments.

Passive and active components in diastolic function
10
Sildenafil model(23)
For this model 20 Wistar rats (male; 160-180g; Charles River, the Netherlands) were used
and on all PAB was performed. In this study the PAB was less tight so that the rats only
developed RVD and no clinical RVF. After 4 weeks they were analyzed with echo and right
ventricle catheterization to confirm RV dysfunction. They were then randomized into 2
groups: PAB rats who received sildenafil (Pfizer Inc, New York, NY, USA) in their drinking
water (PAB-SIL) and PAB rats who received the drinking water alone (PAB-vehicle
treatment, PAB-VEH). 8 weeks after PAB they were analyzed with echo and RV
catheterization and then sacrificed. Two animals died prematurely of other causes than RVF.
The hearts were dissected and the RV, interventricular septum, LV and both atria were
separated. The RV-tissue is used for our experiments.
2.2 Gene expression N2B/N2Ba, NCX and PLN
The animal tissue of the different groups was used. We extracted RNA using the TRIzol
reagent (Invitrogen Corporation, Carlsbad, CA, USA) and this RNA was converted into
cDNA using QuantiTect Reverse Transcription (Qiagen, Venlo, the Netherlands). Gene
expression was then measured with Absolute QPCR SYBR Green ROX mix (Abgene,
Epsom, UK). The expression of the different genes is quantified using the housekeeping gene
36B4. Primers are listed in supplementary material.
2.3 Protein expression SERCA, PLN
We extracted protein using RIPA buffer and measured protein concentration with the use of
the BioRad DC Protein Assay (Bio-Rad Laboratories B.V., Veenendaal). Protein was put on
a gel and after electrophoresis semi-dry blotting was performed. Protein transfer to the blot
was confirmed with Ponceau S staining. After blotting, the membrane was blocked using 5%
Elk in TBS/0,1% Tween for at least 30 min and incubated with antibody specific to SERCA,
PLN and phosphorylated PLN. Tubulin was used as a loading control. Incubation time was
overnight, after which incubation with the secondary antibodies was performed (antibodies
and dilutions used in supplementary data). We detected and quantified the protein with
ImageQuant LAS 4000 (GE Healthcare Life Sciences).
2.4 Activity Assay PKG
PKG activity was assayed in RV tissue with the cyclex cyclic GMP dependent protein kinase
(cGK) assay kit. (CycLex Co., Nagano, Japan). Samples were prepared in extraction buffer
(50 mM potassium phosphate buffer, 1 mM EDTA, 1 mM EGTA, 5 mM DTT and 4 ul/mL
phosphate inhibitor), potassium phosphate buffer and DE-buffer (20 mM Tris-HCl, 60 mM
NaCl, 0,5 mM EDTA, 1 mM EGTA and 4 uL/mL phosphatase inhibitors) according to the
manufacters protocol.

Passive and active components in diastolic function
11
100 uL of each prepared sample was then added to each well of the plate, precoated with
recombinant G-kinase substrate. After incubation (30 min, 30°C) the wells were washed and
100 uL of HRP conjugated anti-phopho-specific antibody was added, this was incubated for
an hour at room temperature. The plate was then again washed and substrate reagent was
added to incubate for 15 minutes after which stop solution ended the reaction in each well.
The absorbance was read in an ELISA plate reader at dual wavelengths of 450/540 nm. Data
are presented as relative amount of cGK activity per sample. Using the protein concentrations
calculated with the BioRad DC Protein Assay (Bio-Rad Laboratories B.V., Veenendaal) we
can transform the data into quantities of cGK activity expressed in units per ug protein.
2.5 Activity Assay PKA
To measure PKA activity we used the MESACUP Protein Kinase Assay (MBL CO., Ltd,
Nagoya, Japan). This assay is based on a synthetic peptide and a monoclonal antibody
recognizing the phosphorylated form of this peptide.
Sample preparation was done as in PKG sample preparation with extraction buffer (50 mM
potassium phosphate buffer, 1 mM EDTA, 1 mM EGTA, 5 mM DTT and 4 ul/mL phosphate
inhibitor), potassium phosphate buffer and DE-buffer (20 mM Tris-HCl, 60 mM NaCl, 0,5
mM EDTA, 1 mM EGTA and 4 uL/mL phosphatase inhibitors). 100 uL of each prepared
sample was added to each well (incubated for 10 min, 25°C) followed by stop solution. After
washing 100 uL biotinylated antibody 2B9 is added (incubated for 60 min, 25°C) and again
the microplate is washed. The addition of POD-conjugated streptavidin (incubated for 60
minutes at 25°C), substrate solution (incubated for 3 min at 25°C ) and stop solution is
interspersed by washing steps. Finally the microplate is read at a wavelenght of 492 nm. Data
is presented as relative PKA activity.
2.6 Data Analysis
Data are presented as mean and standard deviation (SD). The differences between the
different experimental groups were analyzed using one-way analysis of variance (ANOVA)
with appropriate post-hoc tests or analyzed using t-test. We made two comparisons: Control
rats vs PAB-RVD- and PAB-RVF-rats and rats with PAB-RVD treated with vehicle or
sildenafil. When relevant, data was related to the degree of relaxation with Pearson’s
correlation. P=0,05 was considered significant. All data was analyzed using SPSS (PASW
statistics 20 for Windows, SPSS, Chicago, IL) or graphpad (Prism 5 for windows, GraphPad
Software, Inc. La Jolla, CA).

Passive and active components in diastolic function
12
3. RESULTS
3.1 Passive Component: Titin Expression
The N2Ba/N2B ratio is increased in the PAB-RVF group (figure 9A). This increase in the
direction of the compliant isoform (N2Ba) is due to an elevated level of N2Ba expression as
N2B expression is unchanged in the different experimental groups (data not shown).
Sildenafil treatment does not cause a change in titin-isoform expression after 4 weeks. These
two groups do not differ with regard to N2Ba/N2B changes (figure 9B).
3.2 Active components: NCX, SERCA2ATP and PLB
3.2.1 NCX
NCX expression does not differ amongst the
different experimental groups. (figure 3)

Passive and active components in diastolic function
13
3.2.2 SERCA2ATPase
SERCA2 gene-expression (figure 4A) was obtained from a previously performed gene-array
on the tissue of this study. Gene-expression data shows a significant decrease in SERCA2
expression with failure. When comparing SERCA-expression in sildenafil-treated with
vehicle-treated animals we see no change (Figure 4B).
Western blot results tend to show a decrease in SERCA in PAB-RVF, but this difference did
not reach statistical significance.

Passive and active components in diastolic function
14
3.2.3 PLN
There is a significant increase in PLN gene-expression in the PAB-RVF- compared to the
control group. Also a slight increase in the PAB-RVD group is observed, but this difference
did not reach statistical significance (figure 5A).
There is no significant difference between PAB-
RVD and PAB-RVF. Sildenafil treatment does not
influence PLN expression after banding (figure
5B).
Protein levels of serine 16 phosphorylated PLN
tend to show a decrease in PAB-RVD vs the
control, but we see an increase in the PAB-RVF
group. Due to high diversity in the results the
differences in means lack significance (figure 6A).
Protein levels of threonine 17 phosphorylated
PLN are comparable to the serine 16 results.
Looking at the IOD values there tends to be a
decrease in protein expression in the PAB-RVD
group. But again, due to high variability in results,
the difference is not significant (figure 6B).
To conclude, gene-expression of total
phospholamban is increased in PAB-RVF and
both forms of phosphorylated PLN tend to be
decreased in the PAB-RVD group.
Passive and active components in diastolic function
15
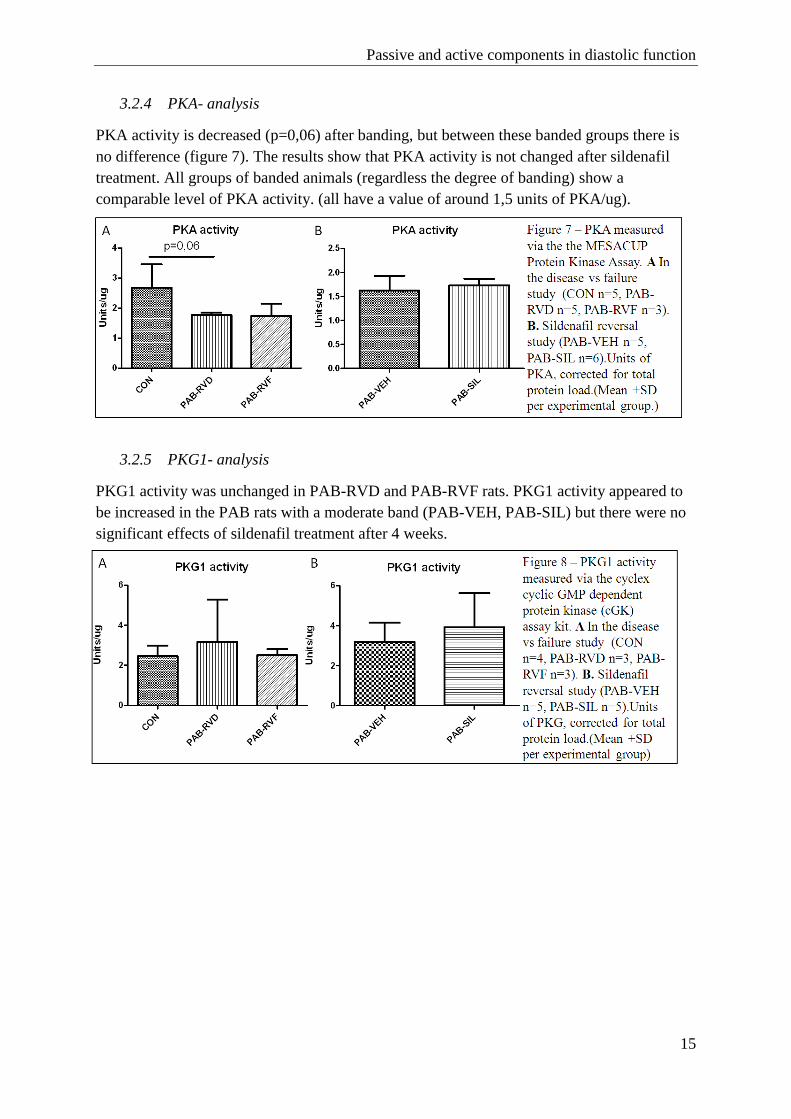
3.2.4 PKA- analysis
PKA activity is decreased (p=0,06) after banding, but between these banded groups there is
no difference (figure 7). The results show that PKA activity is not changed after sildenafil
treatment. All groups of banded animals (regardless the degree of banding) show a
comparable level of PKA activity. (all have a value of around 1,5 units of PKA/ug).
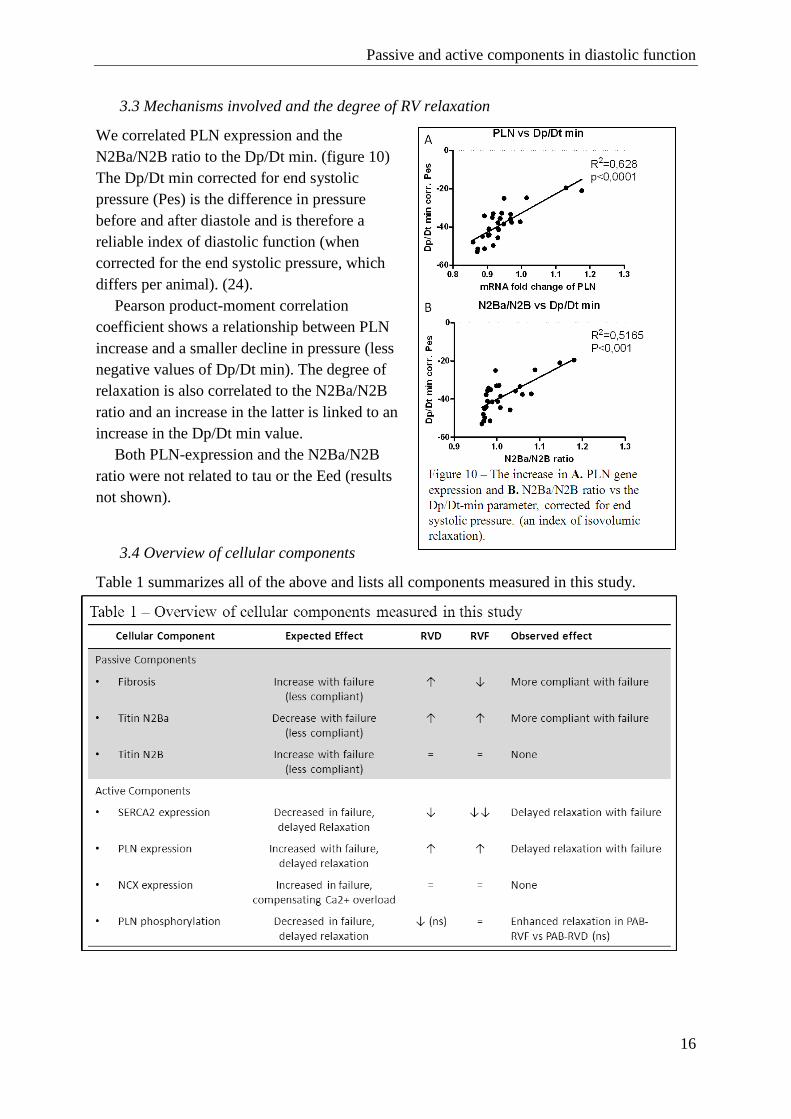
3.2.5 PKG1- analysis
PKG1 activity was unchanged in PAB-RVD and PAB-RVF rats. PKG1 activity appeared to
be increased in the PAB rats with a moderate band (PAB-VEH, PAB-SIL) but there were no
significant effects of sildenafil treatment after 4 weeks.
Passive and active components in diastolic function
16
3.3 Mechanisms involved and the degree of RV relaxation
We correlated PLN expression and the
N2Ba/N2B ratio to the Dp/Dt min. (figure 10)
The Dp/Dt min corrected for end systolic
pressure (Pes) is the difference in pressure
before and after diastole and is therefore a
reliable index of diastolic function (when
corrected for the end systolic pressure, which
differs per animal). (24).
Pearson product-moment correlation
coefficient shows a relationship between PLN
increase and a smaller decline in pressure (less
negative values of Dp/Dt min). The degree of
relaxation is also correlated to the N2Ba/N2B
ratio and an increase in the latter is linked to an
increase in the Dp/Dt min value.
Both PLN-expression and the N2Ba/N2B
ratio were not related to tau or the Eed (results
not shown).
3.4 Overview of cellular components
Table 1 summarizes all of the above and lists all components measured in this study.

Passive and active components in diastolic function
17
4. DISCUSSION
In this study we showed that in rats with a pressure-induced diastolic dysfunction
components of passive relaxation -titin isoforms and RV fibrosis- were changed favoring a
more compliant RV. We also showed that components of active relaxation (SERCA, PLN)
were changed, possibly inducing diastolic dysfunction.
Based on previously obtained tau values of PV-loops after heart catheterization we
hypothesized that diastolic failure is particulary caused by changes in passive components of
the cardiac cycle. Taken all of the above into account we reject this hypothesis. Changes in
passive components are not more evident than those of the active components. In fact, the
increase in N2Ba/N2B and decrease in fibrosis with failure denote a better compliance. This
is in contrast with result of active components, where a decrease of SERCA2 is the case and
where an increase in PLN is directly related to less negative values of Dp/Dt min (i.e.
worsening diastolic function).
Is SERCA-PLN interaction of minor importance, or is tau an insignificant value?
The SERCA-PLN regulatome is very interesting as it not only co-determines diastolic
function in the RV, but also initiates systole by filling the SR with Ca2+
. The results show that
SERCA itself is markedly decreased in gene expression, but only a small (insignificant)
decrease in SERCA follows, when looking at protein expression.
PLN expression is increased in the failing RV. There is not a large difference between
RVD and RVF when looking at means, but more of a lineair increase in PLN with declining
diastolic function. Thus, the existing knowledge of PLN regulation of SERCA in the LV is
also true in the RV: increasing PLN expression is associated with less Ca2+
uptake through
the SERCA2atpase and therefore declining ability to properly relax. The PLN increase is only
correlated to the Dp/Dt min and not correlated to the Dp/Dt max (index of contraction),
confirming that Ca2+
is only taken up through SERCA2 and released from the sarcoplasmatic
reticulum through the RyR-complex.(8).
The question then remains why we did not observe an increase in tau values with failure.
Tau is an exponential time constant of relaxation. With PLN blocking the SR uptake of Ca2+
during diastole, active relaxation is delayed and therefore this tau value should be increased
with higher levels of PLN. This was not observed. The fact that PLN expression correlates to
Dp/Dt min suggests that PLN –being an active component of diastolic function– is in fact
causing a decline in diastolic function. Secondly, fibrosis was less in failure and the
N2Ba/N2B ratio is higher in failure, pointing out that passive stiffness is not increased with
failure. These two findings put tau as a value for assessing the time needed for active
relaxation in a PAB-model under debate.

Passive and active components in diastolic function
18
PLN is a possible target for treatment
Changes in passive tension -such as a titin isoform switch or an increase in fibrosis- are
irreversible and therefore not very suitable as treatment targets. Changes in active
components are reversible and form possible targets for treatment. For that matter our results
are promising.
Reversible SERCA2 inhibition by PLN is an example: it is interesting to see what the
effects of PLN-modulation further down-stream are. To do so we looked at PLN
phosphorylation. PLN can be phosphorylated through PKA and CaMKII at position ser16
and thr17 respectively. The western blot results of both forms of phosphorylated PLN
however lack significance. They do show the same trend: decreasing levels of p-PLN after
PAB and an increase in failing (PAB-RVF) vs. adaption (PAB-RVD). The decrease in
phosphorylated PLN (serine 16 and threonine 17) for PAB-RVD is in line with existing
knowledge. The level of phosphorylated PLN is higher for control, meaning that SERCA
inhibition by PLN is for a large part counterbalanced as a result of PLN-phosphorylation. If a
comparable increase in phosphorylation of PLN could be achieved for RVF through a drug
intervention, this could relieve SERCA of inhibition by PLN and could lead to a better Ca2+
uptake by the SR, thus improving diastolic function.
One way of achieving so could be done through PKA stimulation by sildenafil treatment,
but our PKA result fail to show an increase in PKA after sildenafil treatment. The PKA-
phosphorylated form of phospholamban (Ser16-p-PLN) is not related to PKA activity. This
could be due to the variability in the western blot result: this procedure has a lot of potential
stumbling blocks (sample preparation, gel loading and protein transfer).
The sodium-calcium exchanger is not differentially expressed in failing rat myocardium
Results of previous performed animal models show different result regarding NCX
expression. A study looking at heartfailing rats showed no difference in mRNA- and protein
expression, whereas mice undergoing thoracic aorta banding (TAC model) showed a NCX
upregulation.(25,26) We wondered if an equal NCX upregulation exists in the failing RV.
There is no difference in expression of NCX between the three experimental groups, nor is
there is a decrease or increase when related to Dp/Dt values. NCX seems to be of little
importance in RV pathology, at least according to this model. This data, along with data
presented by Boateng et al. show that an increase in NCX expression to releive the
myocardium from its Ca2+
load is not the case for rat myocardium. (25)
Unchanged NCX expression does not necessarily mean that NCX activity is unchanged.
Perhaps there is not an increase in number of NCX transporters, but an increase in activity.
For that to be proven additional work needs to be done, by for example calculating the
electrochemical potential difference as described by Fujioka et al.(27).
An increase in NCX is described in other animal models, where it prevents further
detoriating diastolic function.(26) The fact that in this model NCX remains unchanged could
perhaps explain the diastolic dysfunctioning of our animals. We would then only expect a
difference in NCX expression between the PAB-RVD and PAB-RVF group. Taken the
unchanged level of NCX per group into account and given the fact that NCX expression is

Passive and active components in diastolic function
19
not related to diastolic function (Dp/Dtmin) we conclude that NCX is of minor importance in
rat RVF.
Titin isoform expression changes the N2Ba/N2B ratio in failure
Passive tension in the sarcomere is for a large part determined by titin filaments. Much
research has been done on
titin expression, signaling
and phosphorylation in
relation to the
cardiomyocyte. (28)For
this research we looked at
titin isoform expression,
which is also widely
investigated: however
mainly in the left ventricle.
The titin isoform switch as
described above has
several times been related
to failure. (19,21,29)
In this research we again show this isoform switch, but now in the right ventricle. When
comparing the groups we see an increase in N2Ba/N2B expression in the PAB-RVF group
versus the control, but no difference between the PAB-RVF and PAB-RVD. Thus the
difference in N2Ba/N2B ratio does not explain why some animals fail and others do not.
However, as for PLN expression, we do show a relationship between Dp/Dt min and the
N2Ba/N2B ratio. An increase in N2Ba/N2B ratio correlated to a worsening diastolic function.
Because of the tau values not increasing with failure we expected the passive tension to be
higher, thus a decrease in N2Ba/N2B ratio was expected. We however show the exact
opposite. This is not as surprisingly, in existing literature on this topic the direction of this
isoform switch with failure is subject of much debate. The direction of the switch described
in our data is in concordance with results published by Neagoe et al. (increase in N2Ba/N2B
ratio in human hearts with dilated cardiomyopathy after coronary artery disease(20)) and
Nagueh et al. (increase in N2Ba/N2B in human heart with nonischemic cardiomyopathy(19)).
This increase in N2Ba/N2B ratio with diastolic failure is shown for the first time in the
RV. When further interpreting N2Ba/N2B increase, we came up with two possible pathways
by which an N2Ba/N2B could change RV properties. One possibility could be that the
increase in N2Ba expression could be a last attempt of the RV to cope with the increased
pressure load. As the system further decompensates and fails to properly relax, N2Ba
expression is increased to cope with the large volume of the RV. This suggests that N2Ba
would act as a last resource to cope with the maladaptation of the RV.
Another possible role of N2Ba could be of pathologic nature, as suggested by Neagoe et
al. They suggest that the increase in N2Ba isoform could explain the failing Frank-Starling
mechanism (an internal mechanism in which stroke volume increases in response to an
increase in the volume of blood filling) (20). Normally an increased stretch of the actin and

Passive and active components in diastolic function
20
myosin chains would result in a better contraction when completely filled, but when titin
would be predominantly available in its compliant form the cardiac myocardium would be
incapable of such a contraction. This however would only explain the rightward shift of the
Frank-Starling curve after PAB (30) and would not explain the inability of the failing RV to
properly relax. (figure 11)
It remains an interesting fact that with failure titin switches to its compliant form and
fibrosis is attenuated. This suggests that in failure the myocardium tries to compensate for its
loss in function by decreasing passive tension.
Study limitations
The PAB model is a good model to study diastolic dysfunction of the RV. This study points
us in the direction of key players in RV pathology, but the number of animals used is too
small to prove and explain the whole moleculair pathway behind diastolic dysfunction.
Additionally this model remains an animal model of disease, it ressembles the human RVF
but all of the observed needs to be varified in human heart.
To relate gene-expression of PLN and titin isoforms to diastolic function we used Dp/Dt
min. The Dp/Dt min is a value inferior to Eed when assessing diastolic function, as Dp/Dt is
load dependant. Eed, which is widely accepted as a load independent index of ventricular
relaxation, was not suitable for this comparison. Eed is a linair estimate of the increase in
pressure for volume during diastole (which is an exponential curve) and these curves shift
greatly when a PAB exists. The Eed was found to be unsuitable to relate mechanisms to Eed,
because of the large diversity between the two animal studies. Dp/Dt was found to be a good
alternative, especially when corrected for end systolic pressure (which makes it an acceptable
load independent index of diastolic function).(31)
Clinical Relevance
Despite the growing interest in RV function and failure, no specific RV treatment is
available. The first step into developing a suitable treatment for RVF is to identify a cellular
mechanism on which a drug can seize. With this study we attempted to find such a cellular
target in the failing RV. In that sense, the fact that our hypothesis got rejected is promising as
active components in diastolic function are easier targets for treatments.(5)
The RV is morphologically and embryological different from the LV and therefore results
obtained from studies on the LV cannot be used to assess RV function.(4) The RV has always
been little investigated and there are currently no treatments for RVF because of lacking
knowledge. This model adds up to the knowledge on the right ventricle that is slowly starting
to grow.

Passive and active components in diastolic function
21
Future prospects
The goal of this study was to identify possible pathologic pathways in the RV. With PLN and
titin-isoforms identified and related to diastolic dysfunction, the next step would be to
individually assess these mechanisms. A gene-knock out model or a model where PLN is
constantly phosphorylated could further explain the meaning of SERCA inhibition by PLN.
The same holds true for titin; individual assessment is needed to describe its function in
the RV. Titin knock-out is however not possible, as this results in a malformation of the thick
filament structure, and therefore a malformation of cardiac muscle. (32) Thus titin isoform
switching needs to be investigated further down-stream: what splicing mechanisms are
involved and how can we prevent alternative splicing in the failing heart? Breakthroughs
have been presented with regard to thyroid hormone and insulin; acting through receptor
kinases and thereby inducing isoform switching (22,28). The problem with all of these
investigated animal model remains that they were all in the LV, which is -as described above-
in many ways different from the RV. A possible way of looking at titin individually could
then be by creating a diabetic- or a hypothyroidic animal model and to look at titin isoform
expression in the RV.
5. CONCLUSION
Many mechanisms are involved in diastolic failure in the RV. Components of passive
stiffness change, underlining their involvement in failure. We hypothesized that PAB-RVF
was associated with increased stiffness but both components in fact cause better compliance
of the RV. This in contrast with active components: the changes in the SERCA-PLN
regulatome cause a delay in relaxation. Based on these findings we rejected our hypothesis.
This has further implications for treatment targets and for the value of tau as a time constant
of relaxation.
The fact that SERCA inhibition by PLN is related to diastolic function and the fact that
failure is for the bigger part caused by diastolic dysfunction makes PLN a promising target
for drugs. Changes in active components are better reversible than changes in passive
components, through phosphorylation of PLN by PKA and CaMKII an improvement of
function could be achieved.
When we related gene-expression of the mechanisms we saw a difference in the PAB-
RVF group vs the control. The increase in expression can however not explain the difference
between PAB-RVD and PAB-RVF (i.e. explain why some animals clinically fail while others
do not). When we looked at diastolic function, there was a linear increase in Dp/Dtmin
corrected for Pes (i.e. declining degree of relaxation) with an increase in PLN and in
N2Ba/N2B ratio. It is therefore advised to relate molecular mechanisms to the degree of
diastolic failure and not to the exerted symptoms. This also shows that there is no cut-off
point for these values that could define RVF.
To conclude, with the improving survival of patients with CHD and PAH the interest in
the RV- and the need for a RV-specific drug is growing. It is a promising fact that in this
study we show that active components are the main cause of diastolic dysfunction. Changes
in active components are reversible; therefore active components are more suitable for

Passive and active components in diastolic function
22
treatment. Individual assessment of RV-diastolic mechanisms is now needed to come to RV-
specific drugs for failure.
6. REFERENCES
(1) van der Linde D, Konings EE, Slager MA, Witsenburg M, Helbing WA, Takkenberg JJ,
et al. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-
analysis. J Am Coll Cardiol 2011 Nov 15;58(21):2241-2247.
(2) van Loon RL, Roofthooft MT, Hillege HL, ten Harkel AD, van Osch-Gevers M, Delhaas
T, et al. Pediatric pulmonary hypertension in the Netherlands: epidemiology and
characterization during the period 1991 to 2005. Circulation 2011 Oct 18;124(16):1755-1764.
(3) D'Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al.
Survival in patients with primary pulmonary hypertension. Results from a national
prospective registry. Ann Intern Med 1991 Sep 1;115(5):343-349.
(4) Fogel MA, Rychik J. Right ventricular function in congenital heart disease: pressure and
volume overload lesions. Prog Cardiovasc Dis 1998 Jan-Feb;40(4):343-356.
(5) Haddad F, Doyle R, Murphy DJ, Hunt SA. Right ventricular function in cardiovascular
disease, part II: pathophysiology, clinical importance, and management of right ventricular
failure. Circulation 2008 Apr 1;117(13):1717-1731.
(6) Borgdorff MA, Bartelds B, Dickinson MG, Steendijk P, deVroomen M, Berger RM.
Right Ventricular failure in chronic experimental pressure load is characterized by diastolic
dysfunction. IN SUBMISSION .
(7) Kass DA, Bronzwaer JG, Paulus WJ. What mechanisms underlie diastolic dysfunction in
heart failure? Circ Res 2004 Jun 25;94(12):1533-1542.
(8) Bers DM. Cardiac excitation-contraction coupling. Nature 2002 Jan 10;415(6868):198-
205.
(9) MacLennan DH, Kranias EG. Phospholamban: a crucial regulator of cardiac contractility.
Nat Rev Mol Cell Biol 2003 Jul;4(7):566-577.
(10) Tada M, Ohmori F, Yamada M, Abe H. Mechanism of the stimulation of Ca2+-
dependent ATPase of cardiac sarcoplasmic reticulum by adenosine 3':5'-monophosphate-
dependent protein kinase. Role of the 22,000-dalton protein. J Biol Chem 1979 Jan
25;254(2):319-326.
(11) Limas CJ, Olivari MT, Goldenberg IF, Levine TB, Benditt DG, Simon A. Calcium
uptake by cardiac sarcoplasmic reticulum in human dilated cardiomyopathy. Cardiovasc Res
1987 Aug;21(8):601-605.
(12) Movsesian MA, Nishikawa M, Adelstein RS. Phosphorylation of phospholamban by
calcium-activated, phospholipid-dependent protein kinase. Stimulation of cardiac
sarcoplasmic reticulum calcium uptake. J Biol Chem 1984 Jul 10;259(13):8029-8032.

Passive and active components in diastolic function
23
(13) Davis BA, Schwartz A, Samaha FJ, Kranias EG. Regulation of cardiac sarcoplasmic
reticulum calcium transport by calcium-calmodulin-dependent phosphorylation. J Biol Chem
1983 Nov 25;258(22):13587-13591.
(14) Schillinger W, Fiolet JW, Schlotthauer K, Hasenfuss G. Relevance of Na+-Ca2+
exchange in heart failure. Cardiovasc Res 2003 Mar 15;57(4):921-933.
(15) Takimoto E, Soergel DG, Janssen PM, Stull LB, Kass DA, Murphy AM. Frequency- and
afterload-dependent cardiac modulation in vivo by troponin I with constitutively active
protein kinase A phosphorylation sites. Circ Res 2004 Mar 5;94(4):496-504.
(16) van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic
growth and remodeling of the heart. J Clin Invest 2013 Jan 2;123(1):37-45.
(17) Nagendran J, Archer SL, Soliman D, Gurtu V, Moudgil R, Haromy A, et al.
Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and
acute inhibition of phosphodiesterase type 5 improves contractility. Circulation 2007 Jul
17;116(3):238-248.
(18) LeWinter MM, Granzier H. Cardiac titin: a multifunctional giant. Circulation 2010 May
18;121(19):2137-2145.
(19) Nagueh SF, Shah G, Wu Y, Torre-Amione G, King NM, Lahmers S, et al. Altered titin
expression, myocardial stiffness, and left ventricular function in patients with dilated
cardiomyopathy. Circulation 2004 Jul 13;110(2):155-162.
(20) Neagoe C, Kulke M, del Monte F, Gwathmey JK, de Tombe PP, Hajjar RJ, et al. Titin
isoform switch in ischemic human heart disease. Circulation 2002 Sep 10;106(11):1333-
1341.
(21) Nelson OL, Robbins CT, Wu Y, Granzier H. Titin isoform switching is a major cardiac
adaptive response in hibernating grizzly bears. Am J Physiol Heart Circ Physiol 2008
Jul;295(1):H366-71.
(22) Kruger M, Linke WA. Titin-based mechanical signalling in normal and failing
myocardium. J Mol Cell Cardiol 2009 Apr;46(4):490-498.
(23) Borgdorff MA, Bartelds B, Dickinson MG, Steendijk P, de Vroomen M, Berger RM.
Sildenafil treatment in established right ventricular dysfunction improves diastolic function
and attenuates interstitial fibrosis independent from afterload. manuscript 2013.
(24) Leeuwenburgh BP, Steendijk P, Helbing WA, Baan J. Indexes of diastolic RV function:
load dependence and changes after chronic RV pressure overload in lambs. Am J Physiol
Heart Circ Physiol 2002 Apr;282(4):H1350-8.
(25) Boateng SY, Naqvi RU, Koban MU, Yacoub MH, MacLeod KT, Boheler KR. Low-dose
ramipril treatment improves relaxation and calcium cycling after established cardiac
hypertrophy. Am J Physiol Heart Circ Physiol 2001 Mar;280(3):H1029-38.

Passive and active components in diastolic function
24
(26) Ito K, Yan X, Tajima M, Su Z, Barry WH, Lorell BH. Contractile reserve and
intracellular calcium regulation in mouse myocytes from normal and hypertrophied failing
hearts. Circ Res 2000 Sep 29;87(7):588-595.
(27) Fujioka Y, Komeda M, Matsuoka S. Stoichiometry of Na+-Ca2+ exchange in inside-out
patches excised from guinea-pig ventricular myocytes. J Physiol 2000 Mar 1;523 Pt 2:339-
351.
(28) Linke WA, Kruger M. The giant protein titin as an integrator of myocyte signaling
pathways. Physiology (Bethesda) 2010 Jun;25(3):186-198.
(29) Wu Y, Bell SP, Trombitas K, Witt CC, Labeit S, LeWinter MM, et al. Changes in titin
isoform expression in pacing-induced cardiac failure give rise to increased passive muscle
stiffness. Circulation 2002 Sep 10;106(11):1384-1389.
(30) Bartelds B, Borgdorff MA, Smit-van Oosten A, Takens J, Boersma B, Nederhoff MG, et
al. Differential responses of the right ventricle to abnormal loading conditions in mice:
pressure vs. volume load. Eur J Heart Fail 2011 Dec;13(12):1275-1282.
(31) Burkhoff D, Mirsky I, Suga H. Assessment of systolic and diastolic ventricular
properties via pressure-volume analysis: a guide for clinical, translational, and basic
researchers. Am J Physiol Heart Circ Physiol 2005 Aug;289(2):H501-12.
(32) van der Ven PF, Bartsch JW, Gautel M, Jockusch H, Furst DO. A functional knock-out
of titin results in defective myofibril assembly. J Cell Sci 2000 Apr;113 ( Pt 8)(Pt 8):1405-
1414.
7. SUPPLEMENTAL DATA
Primers used for qRT-PCR
Forward Primer Reverse Primer
Titin-N2B isoform ccaacgagtatggcagtgtca tgggttcaggcagtaatttgc
Titin-N2Ba isoform cggcagagctcagaatcga gtcaaaggacacttcacactcaaaa
NCX cgtctggtggagatgagtgaga tgttaatgtgaagccaccaagct
PLN ttgtcttcctggcatcatgg cagcttgtcacagaagcatcac
Antibodies used for Western Blot Antibody Cat. number Company Dilution
Monoclonal Anti-a-
Tubulin
T5168 Sigma-Aldrich corp. 1:8000
p-phospholamban (t17) A010-13 Badrilla ltd. 1:5000
p-phospholamban (s16) A010-12 Badrilla ltd. 1:5000
Anti-SERCA2 ATPase
antibody
ab2861 Abcam plc. 1:1000