na /vacancy disordering promises high-rate na-ion...
TRANSCRIPT

SC I ENCE ADVANCES | R E S EARCH ART I C L E
ELECTROCHEM ISTRY
1Chinese Academy of Sciences (CAS) Key Laboratory of Molecular Nanostructure andNanotechnology, CAS Research/Education Center for Excellence in MolecularSciences, Institute of Chemistry, CAS, Beijing 100190, P. R. China. 2University ofCAS, Beijing 100049, P. R. China. 3Beijing National Laboratory for Condensed MatterPhysics, Institute of Physics, CAS, Beijing 100190, P. R. China.*Corresponding author. Email: [email protected] (Y.-G.G.); [email protected] (Y.-X.Y.);[email protected] (L.G.)
Wang et al., Sci. Adv. 2018;4 : eaar6018 9 March 2018
Copyright © 2018
The Authors, some
rights reserved;
exclusive licensee
American Association
for the Advancement
of Science. No claim to
originalU.S. Government
Works. Distributed
under a Creative
Commons Attribution
NonCommercial
License 4.0 (CC BY-NC).
Dow
nloaded from
Na+/vacancy disordering promises high-rateNa-ion batteriesPeng-Fei Wang,1,2 Hu-Rong Yao,1,2 Xin-Yu Liu,3 Ya-Xia Yin,1,2* Jie-Nan Zhang,3 Yuren Wen,3
Xiqian Yu,3 Lin Gu,3* Yu-Guo Guo1,2*
As one of the most fascinating cathode candidates for Na-ion batteries (NIBs), P2-type Na layered oxides usuallyexhibit various single-phase domains accompanied by different Na+/vacancy-ordered superstructures, depend-ing on the Na concentration when explored in a limited electrochemical window. Therefore, their Na+ kineticsand cycling stability at high rates are subjected to these superstructures, incurring obvious voltage plateaus inthe electrochemical profiles and insufficient battery performance as cathode materials for NIBs. We show thatthis problem can be effectively diminished by reasonable structure modulation to construct a completelydisordered arrangement of Na-vacancy within Na layers. The combined analysis of scanning transmission elec-tron microscopy, ex situ x-ray absorption spectroscopy, and operando x-ray diffraction experiments, coupledwith density functional theory calculations, reveals that Na+/vacancy disordering between the transitionmetal oxide slabs ensures both fast Na mobility (10−10 to 10−9 cm2 s−1) and a low Na diffusion barrier (170 meV)in P2-type compounds. As a consequence, the designed P2-Na2/3Ni1/3Mn1/3Ti1/3O2 displays extra-long cycle life(83.9% capacity retention after 500 cycles at 1 C) and unprecedented rate capability (77.5% of the initial capacityat a high rate of 20 C). These findings open up a new route to precisely design high-rate cathode materials forrechargeable NIBs.
http
on Septem
ber 16, 2018://advances.sciencem
ag.org/
INTRODUCTIONNa-ion batteries (NIBs) are enjoying renewed interest as a viablealternative to Li-ion batteries especially for future large-scale energystorage in light of the low cost, nontoxicity, and large abundance ofNa resources (1–3). Among the cathode candidates, NaxTMO2 layeredoxides (where TM refers to transition metal) have attracted intensiveattention due to their high capacities and feasible synthesis (4, 5).Depending on the atomic environment of Na (prismatic or octahedral)and stacking sequence of O layers (ABBA or ABCABC), they can becommonly categorized into P2 and O3 phases (6). In contrast withO3 counterparts, P2-type layered frameworks own better structure sta-bility during the sodium extraction process owing to direct Na-ion dif-fusion and more open prismatic paths within TMO2 slabs. However,they usually undergo undesired P2-O2 phase transition in high-voltageregions, which leads to large volume change and rapid capacity decay(7, 8). Recently, this problem has been significantly mitigated by dop-ing inactivemetal (Li, Mg, Cu, or Zn) in the TM layers, with the aim ofeliminating phase transformations during cycling and extending the solid-solution zone over a wider Na intercalation range, although at the ex-pense of some capacity (9–13). Apart from the issue of phase transition,there are three kinds of ordering in P2-type oxides containing more thanone TM species: Na+/vacancy ordering in the Na layer, charge ordering,and TM1/TM2 cationic ordering on the TM lattice (14–16). It is wellacknowledged that Na+/vacancy ordering, which is closely related tothe Na+ ion mobility in the interlayer, occurs frequently when the Fermilevels (ca. redox potentials) between the TM1 and TM2 species aresimilar (17–21). In this manner, most P2-type oxides differ from oneanother in commensurable or incommensurable Na+/vacancy-ordered
superstructures between the TMO2 slabs within single-phase do-mains upon Na+ extraction/insertion (22, 23). Because the presenceof ordered intermediate phases caused by Na ordering rearrangementin these compounds affects both voltage plateaus in electrochemicalcurves and Na diffusion path, understanding intercalation orderingis indispensable for tailoring the material to achievable electrochemicalapplications.
In this context, our interest focused on the electrochemical (de)in-tercalation of sodium in P2-Na2/3Ni1/3Mn2/3O2 (NaNM), which has arelatively high operating voltage based on Ni2+/3+/4+ redox reactionsand a highly symmetric crystal structure free from the Jahn-Teller activecenter (24). This material exhibits a complete reversibility and single-phase region between the Na2/3Ni1/3Mn2/3O2 and Na1/3Ni1/3Mn2/3O2
compositions within a lower voltage than 4.2 V (25). There are twoobvious voltage plateaus at 3.3 and 3.7 V, which might imply the re-arrangement of differentNa+/vacancy ordering across theNa extraction/insertion range, thus making it possible to embark on the effects of TMmixing on the Na ordering mechanism using it as a model system.Considering the same valence, smaller ionic radii ratio, and substantialdifference in the Fermi level between Ti4+ (60.5 pm) andMn4+ (53 pm),substituting a number of Mn4+ with Ti4+ in TMO2 slabs might be ef-fective for suppressing cationic ordering, charge ordering withinTMO2 slabs, and Na-vacancy ordering in Na layers.
Hence, Ti-free P2-NaNM and Ni/Mn/Ti-mixed P2-Na2/3Ni1/3Mn1/3Ti1/3O2 (NaNMT) compounds were synthesized to present a com-prehensive study on the influence of Na+/vacancy ordering on theirelectrochemical properties, structural evolution, and Na+ kineticstowardNa(de)intercalation.The substitutionofTi forMnin theP2-NaNMcrystal framework enlarges the distance of Na layers with the contrac-tion of the TMO6 octahedron; thus, electron delocalization is limited,and charge coupled to Na+/vacancy ordering is thoroughly broken.Consequently, the two voltage plateaus in the charge/discharge pro-files of P2-NaNM are fully eliminated without affecting the capacity,manifesting characteristic sloping curves for complete solid-solution re-action. The Na-vacancy–disordered P2-NaNMT material exhibits a
1 of 9
SC I ENCE ADVANCES | R E S EARCH ART I C L E
specific capacity of 88 mA·hour g−1 with a high average voltage of ca.3.5 V versus Na+/Na, excellent cycle performance (ca. 83.9% capacityretention at 1 C after 500 cycles), a very small decrease in the volume(0.64%), and outstanding rate capability (ca. 87.9 and 77.5% capacityretention at high rates of 10 and 20 C, respectively). Via Na+ kineticsanalysis using an electrochemical characterization technique [galvano-static intermittent titration technique (GITT)], the nature of rateperformance, together with the use of density functional theory (DFT)calculations, both experimental and theoretical information are providedto reveal the relationship between Na-vacancy ordering/disorderingmechanisms and Na diffusion properties. These results indicate thatNa+/vacancy disordering increases the degree of freedom ofNa+ uponcycling, thus ensuring fastNa-ionmobility (average, 10−10 cm2 s−1) and alow diffusion barrier (170 meV) in the interlayer, which successfully es-tablish the linkage betweenNa-vacancy disordering and high-rate cath-ode materials for NIBs.
http:D
ownloaded from
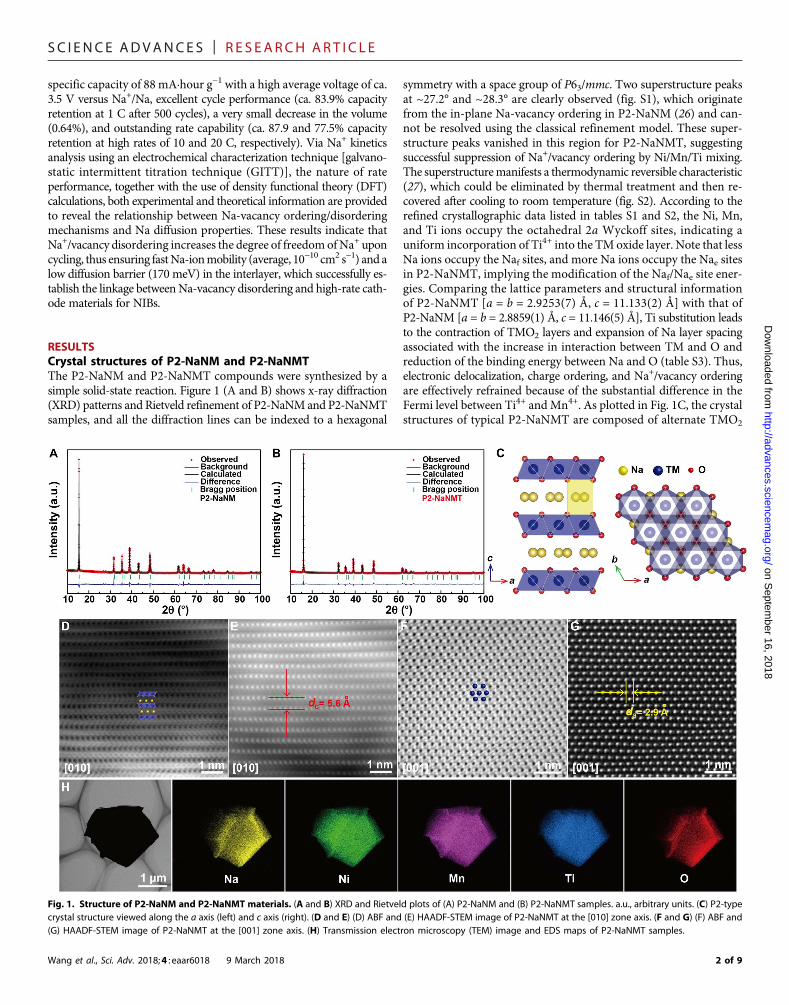
RESULTSCrystal structures of P2-NaNM and P2-NaNMTThe P2-NaNM and P2-NaNMT compounds were synthesized by asimple solid-state reaction. Figure 1 (A and B) shows x-ray diffraction(XRD) patterns and Rietveld refinement of P2-NaNMand P2-NaNMTsamples, and all the diffraction lines can be indexed to a hexagonal
Wang et al., Sci. Adv. 2018;4 : eaar6018 9 March 2018
symmetry with a space group of P63/mmc. Two superstructure peaksat ~27.2° and ~28.3° are clearly observed (fig. S1), which originatefrom the in-plane Na-vacancy ordering in P2-NaNM (26) and can-not be resolved using the classical refinement model. These super-structure peaks vanished in this region for P2-NaNMT, suggestingsuccessful suppression of Na+/vacancy ordering by Ni/Mn/Ti mixing.The superstructuremanifests a thermodynamic reversible characteristic(27), which could be eliminated by thermal treatment and then re-covered after cooling to room temperature (fig. S2). According to therefined crystallographic data listed in tables S1 and S2, the Ni, Mn,and Ti ions occupy the octahedral 2a Wyckoff sites, indicating auniform incorporation of Ti4+ into the TM oxide layer. Note that lessNa ions occupy the Naf sites, and more Na ions occupy the Nae sitesin P2-NaNMT, implying the modification of the Naf/Nae site ener-gies. Comparing the lattice parameters and structural informationof P2-NaNMT [a = b = 2.9253(7) Å, c = 11.133(2) Å] with that ofP2-NaNM [a = b = 2.8859(1) Å, c = 11.146(5) Å], Ti substitution leadsto the contraction of TMO2 layers and expansion of Na layer spacingassociated with the increase in interaction between TM and O andreduction of the binding energy between Na and O (table S3). Thus,electronic delocalization, charge ordering, and Na+/vacancy orderingare effectively refrained because of the substantial difference in theFermi level between Ti4+ and Mn4+. As plotted in Fig. 1C, the crystalstructures of typical P2-NaNMT are composed of alternate TMO2
on Septem
ber 16, 2018//advances.sciencem
ag.org/
Fig. 1. Structure of P2-NaNM and P2-NaNMT materials. (A and B) XRD and Rietveld plots of (A) P2-NaNM and (B) P2-NaNMT samples. a.u., arbitrary units. (C) P2-typecrystal structure viewed along the a axis (left) and c axis (right). (D and E) (D) ABF and (E) HAADF-STEM image of P2-NaNMT at the [010] zone axis. (F and G) (F) ABF and(G) HAADF-STEM image of P2-NaNMT at the [001] zone axis. (H) Transmission electron microscopy (TEM) image and EDS maps of P2-NaNMT samples.
2 of 9

SC I ENCE ADVANCES | R E S EARCH ART I C L E
on Septem
ber 16, 2018http://advances.sciencem
ag.org/D
ownloaded from
(TM=Ni,Mn, andTi) layers andNa layerswithABBAoxygen stackingpatterns. Na ions occupy two kinds of trigonal prismatic sites, eithersharing edges with the six TMO6 octahedrons (Nae, 2d Wyckoff sites)or sharing two faces with the lower and upper TMO6 octahedrons(Naf, 2b Wyckoff sites).
Detailed atomic-scale structural information on P2-NaNMT com-pounds was confirmed by aberration-corrected scanning transmissionelectronmicroscopy (STEM)with annular bright-field (ABF) and high-angle annular dark-field (HAADF) techniques. The positions of the TM(Ni, Mn, and Ti) columns are revealed by the dark-dot contrast in theABF-STEM images and bright-dot contrast in the HAADF-STEMimages. The faint dark-dot contrast with the interlayer positions inthe ABF-STEM images corresponds to the light element Na and Ocolumns in the layered structure (28, 29). The ABF-STEM observationsof octahedral TMO2 at the [010] zone axis are highly consistent with theP2 atomic model, as inserted in Fig. 1D for convenient visualization.The Na atoms are clamped by layered TMO2, the O columns followa distinct ABBA-stacking mode, and every two layers of TMO2 displayamirroring symmetry. The distance of the adjacent layer dc inHAADF-STEM images (Fig. 1E) is measured to be ca. 0.56 nm, which isconsistent with the interslab distance by refined crystallographic data.TheABF-STEM images (Fig. 1F) at the [001] zone axis demonstrate thearrangement of the TM atoms with a hexagonal symmetry. In addition,the distance of the adjacent TM atoms in corresponding HAADF-STEM images is ca. 0.29 nm (Fig. 1G), which matches well with thevalue of cell parameter a. The particle size of the samples is about1 to 5 mmwith plate-like morphology (fig. S3). The width of 2.09 Å be-tween neighboring lattice fringes revealed by high-resolution transmis-sion electron microscopy (HRTEM) image corresponds to the (103)planes of P2-NaNMT sample (fig. S4). Energy-dispersive spectroscopy(EDS) mapping, as shown in Fig. 1H, demonstrates uniform Na, Ni,Mn, Ti, and O element distribution in the P2-NaNMT particle.
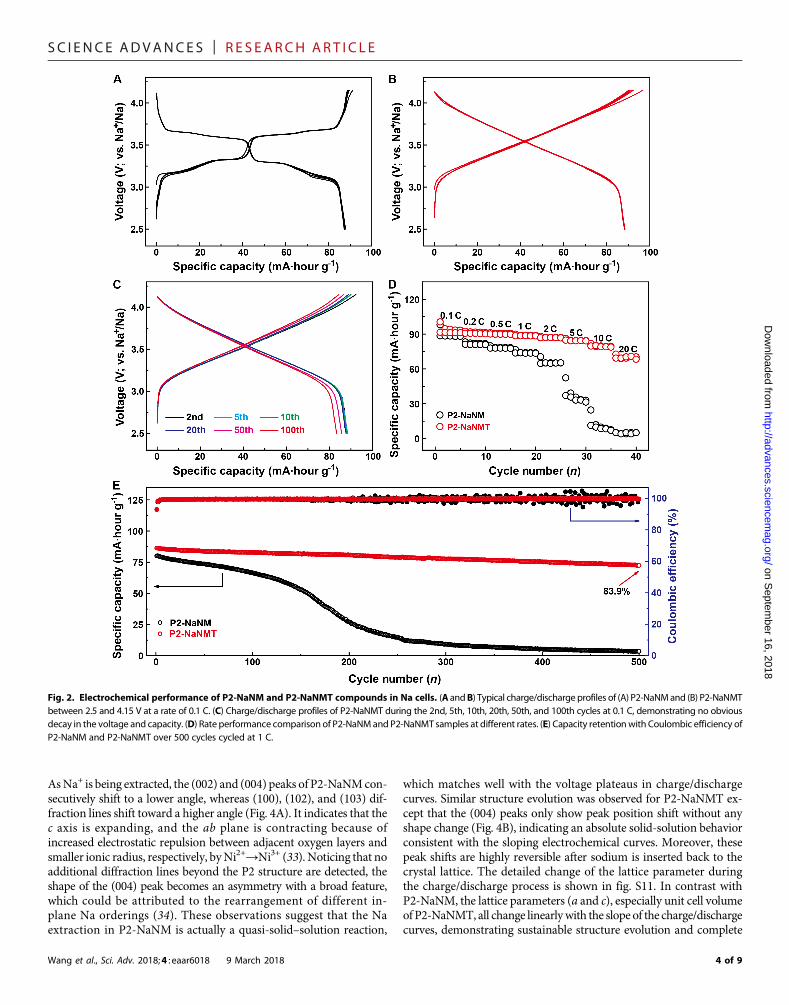
Electrochemical performanceThe electrochemical properties of P2-NaNM and P2-NaNMT wereexamined in aNa half-cell between 2.5 and 4.15V at the current densityof 0.1 C (1 C = 173mA g−1). As shown in Fig. 2 (A and B), the galvano-static charge/discharge profiles of P2-NaNM obviously involve twovoltage plateaus at 3.3 and 3.7 V, respectively, suggesting differentNa-vacancy ordering arrangements between neighboring TMO2 slabs.In contrast, the typical charge/discharge curves of P2-NaNMT becomecompletely sloping lines, and two voltage plateaus totally disappearedwithout affecting the capacity, which results from a solid-solution reac-tion without a Na+/vacancy-ordered superstructure, as discussed later.Disordered P2-NaNMT displays a reversible capacity of 88 mA·hour g−1
with a high average voltage of 3.5 V, smooth charge/discharge profiles,and little polarization (Fig. 2B). There is no obvious capacity decayand voltage drop over extended cycles (Fig. 2C), showing its greatpotential for future practical application. P2-NaNMT shows the samecycling performance as P2-NaNM (94.6% versus 95.2%) at a low rate of0.1 C after 100 cycles (fig. S5). However, as compared in the rateperformance with a loading mass of 3 to 4 mg cm−2 in Fig. 2D, theP2-NaNMT electrode can still deliver ca. 87.9 and 77.5% of its initialcapacity even cycled at 10 and 20C, respectively, whereas the P2-NaNMelectrode is approaching 0mA·hour g−1. It can be seen that Na-vacancydisordering favors charge/discharge capability at high current density.Therefore, when cycled at a high rate of 1 C, the P2-NaNMT cathodedemonstrates remarkable improvement in its cycle stability, as shown inFig. 2E, delivering an outstanding capacity retention of 83.9% even after
Wang et al., Sci. Adv. 2018;4 : eaar6018 9 March 2018
500 cycles with a high Coulombic efficiency of >99% due to the slightside reactions occurring between electrode materials and electrolyteduring cycling. By comparison, the P2-NaNM electrode exhibitsvery fast capacity decay from 80 mA·hour g−1 in the first cycle to3.5 mA·hour g−1 after 500 cycles, corresponding to only 4.4% capacityretention. In addition, the morphology of the P2-NaNMT cathode iswell maintained, whereas P2-NaNM shows pulverization after longcycling (fig. S6). Moreover, the P2-NaNMT cathode also exhibits obvi-ous improvement in its cycling performance in awider potential range of2.5 to 4.3V due to the suppressed P2-O2 phase transition at high voltageregions, delivering a capacity retention of 83.4% at 0.1 C after 100 cycles(figs. S7 and S8). In total, Ti incorporation into P2-NaNM couldsuppress Na+/vacancy ordering, smooth the charge/discharge curves,and extend solid-solution zone over a wider compositional range, sofaster ionic transport between the interlayers is achieved under Na+
(de)intercalation, resulting in better battery performance at high rates.
Charge compensation mechanismThe cyclic voltammograms (CVs) in Na half-cells were tested at a scanrate of 0.1 mV s−1, as shown in fig. S9. Consistent with the voltageplateaus in charge/discharge curves, the P2-NaNM electrode showstwo pairs of reversible redox peaks located at 3.39/3.23 and 3.72/3.52 V,respectively, which arise from multiple Na-vacancy ordering in sodiumlayers. In marked contrast to the P2-NaNM electrode, the observed CVbehavior for P2-NaNMT electrode resembles that of capacitors with onlyone large andbroad oxidation/reductionpeak, further demonstrating thatthe Na+/vacancy ordering is successfully prevented in the Na intercala-tion process after Ni/Mn/Ti mixing into the crystal lattice. Ex situ x-rayabsorption spectroscopy (XAS) (Fig. 3), combined with x-ray photo-electron spectroscopy (XPS) (fig. S10), was performed to elucidate thecharge compensation mechanism of Na1−xNMT upon Na+ extraction/insertion. As displayed in the x-ray absorption near-edge structure(XANES) spectra at Ni and Mn K-edges, the Ni K-edge absorptionspectrum (Fig. 3A) shows apparent shifts toward the higher-energyregion (~2 eV), demonstrating the oxidation of Ni2+ to Ni3+ as chargeincreases from 2.5 to 4.15 V (30, 31). In contrast, the Mn K-edges (Fig.3B) show no apparent changes, indicating that the tetravalent manga-nese ions are electrochemically inactive. So, it indicates that theNi2+/Ni3+
redox couple is responsible for the charge compensation mechanism(1 e− transfer) within the explored 2.5 to 4.15 V voltage window,which is highly consistent with CV results. The interatomic distanceinformation of Na1−xNMT was also revealed by extended x-ray ab-sorption fine structure (EXAFS) spectra at the Ni and Mn K-edges(Fig. 3, C and D). In the first TM-O coordination shell of Na1−xNMT,lowered Fourier transform magnitude and shortened interatomic dis-tance are observed at the Ni K-edge, whereas the Mn K-edge showsalmost no change upon Na removal from the crystal lattice. In thisregion, the electrochemically active nickel ions compensate for thecharge balance, and the manganese ions always maintain the +4 state(32), which are consistentwith the ex situ XANES results. In the secondTM-TM coordination shells, both Ni andMn K-edges confirm a simi-lar shift of the decreased in-plane interatomic distances after electro-chemical oxidation, which is in good agreementwith the change of ahexlattice parameters upon Na ion extraction by in situ XRD observationin the latter parts.
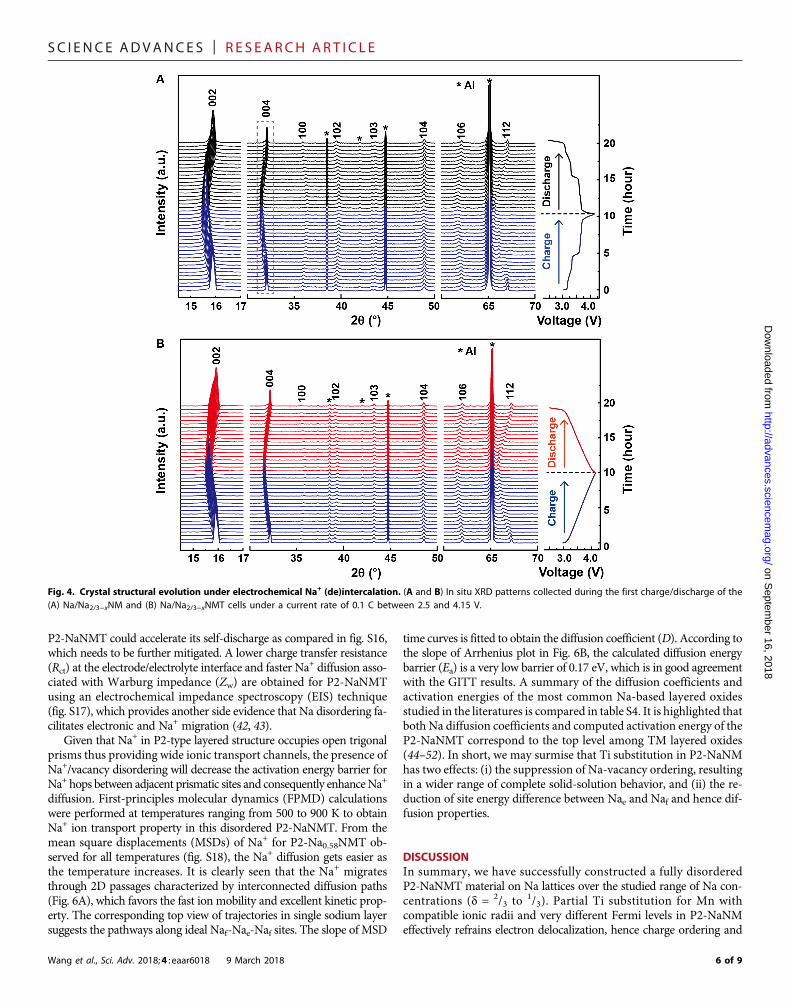
Structural evolution and reaction mechanismTo understand the structural evolution during the charge/dischargeprocess, in situ XRD experiments were carried out, as shown in Fig. 4.
3 of 9
SC I ENCE ADVANCES | R E S EARCH ART I C L E
on Septem
ber 16, 2018http://advances.sciencem
ag.org/D
ownloaded from
AsNa+ is being extracted, the (002) and (004) peaks of P2-NaNMcon-secutively shift to a lower angle, whereas (100), (102), and (103) dif-fraction lines shift toward a higher angle (Fig. 4A). It indicates that thec axis is expanding, and the ab plane is contracting because ofincreased electrostatic repulsion between adjacent oxygen layers andsmaller ionic radius, respectively, byNi2+→Ni3+ (33). Noticing that noadditional diffraction lines beyond the P2 structure are detected, theshape of the (004) peak becomes an asymmetry with a broad feature,which could be attributed to the rearrangement of different in-plane Na orderings (34). These observations suggest that the Naextraction in P2-NaNM is actually a quasi-solid–solution reaction,
Wang et al., Sci. Adv. 2018;4 : eaar6018 9 March 2018
which matches well with the voltage plateaus in charge/dischargecurves. Similar structure evolution was observed for P2-NaNMT ex-cept that the (004) peaks only show peak position shift without anyshape change (Fig. 4B), indicating an absolute solid-solution behaviorconsistent with the sloping electrochemical curves. Moreover, thesepeak shifts are highly reversible after sodium is inserted back to thecrystal lattice. The detailed change of the lattice parameter duringthe charge/discharge process is shown in fig. S11. In contrast withP2-NaNM, the lattice parameters (a and c), especially unit cell volumeof P2-NaNMT, all change linearlywith the slope of the charge/dischargecurves, demonstrating sustainable structure evolution and complete
Fig. 2. Electrochemical performance of P2-NaNM and P2-NaNMT compounds in Na cells. (A andB) Typical charge/discharge profiles of (A) P2-NaNMand (B) P2-NaNMTbetween 2.5 and 4.15 V at a rate of 0.1 C. (C) Charge/discharge profiles of P2-NaNMT during the 2nd, 5th, 10th, 20th, 50th, and 100th cycles at 0.1 C, demonstrating no obviousdecay in the voltage and capacity. (D) Rate performance comparison of P2-NaNMand P2-NaNMT samples at different rates. (E) Capacity retentionwith Coulombic efficiency ofP2-NaNM and P2-NaNMT over 500 cycles cycled at 1 C.
4 of 9

SC I ENCE ADVANCES | R E S EARCH ART I C L E
on Septem
ber 16, 2018http://advances.sciencem
ag.org/D
ownloaded from
single-phase reaction upon Na+ extraction and insertion. Furthermore,the unit cell volume change of P2-NaNMT before and after Na extrac-tion is merely −0.64%, which is close to zero-strain characteristics andresponsible for the excellent cycling stability cycled at 1 C over 500 cycles.Not only could a half substation of Ti forMn in P2-NaNMeliminate in-plane orderings within sodium layers but it also could suppress the fre-quent P2-O2phase transition at 4.2Vbypreventing theTMO2 slabs fromgliding to form the O2 phase, as illustrated in fig. S12. It is evidenced thatthe O2 characteristic (002′) and (112′) peaks that are located at about 21°and 70° (24), respectively, disappeared in P2-Na1−xNMT electrodes evencharged to 4.3 V (fig. S13), resulting in an improved cycling performancein a wider potential range of 2.5 to 4.3 V.
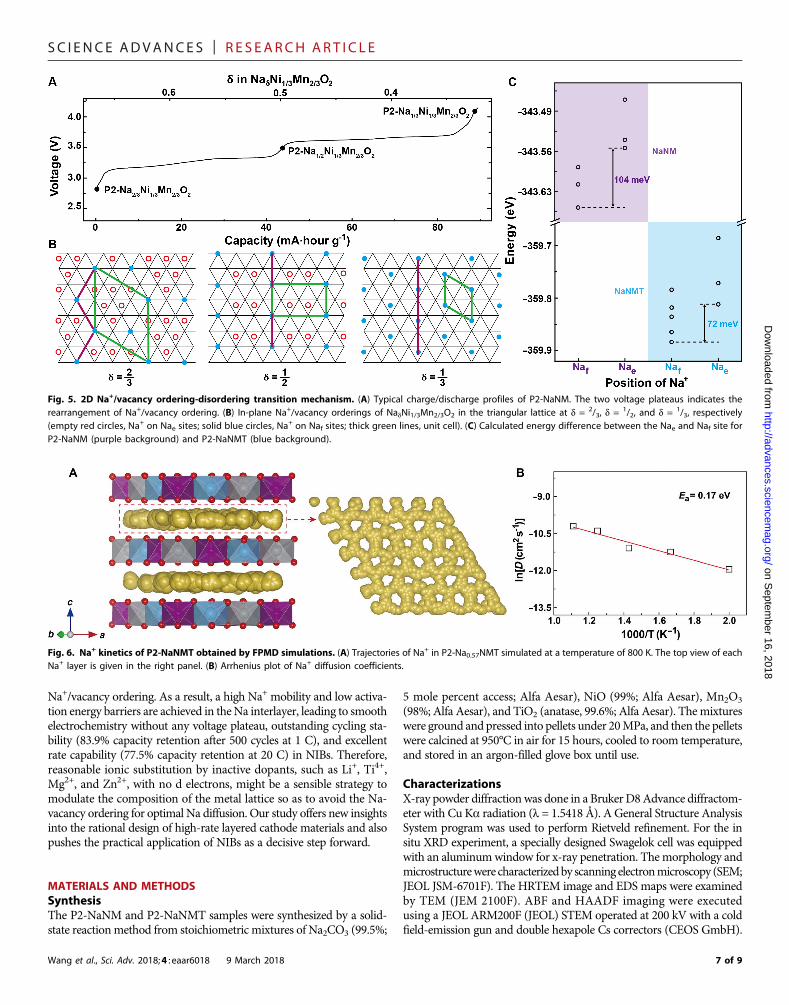
Combining in situ XRD results and electrochemical curves, there aretwo major intermediate phases in P2-NadNM at 3.5 and 4.0 V, whichcorresponds to the Na concentration of 1/2 and
1/3 with different in-plane ordering, respectively. Figure 5 (A and B) illustrates the two-dimensional (2D) in-plane Na ordering patterns with respect to thetypical charge/discharge curves of P2-NadNM, which consist of Nafconnecting in a very intriguing pattern (25, 35–37). In “large zigzag”(LZZ) pattern for d = 2/3, Naf sites form a zigzag pattern with distance2|ahex| between the nearest Naf sites. When the concentration is re-duced to 1/2, the ordering is converted from LZZ to rows in which Naorders alternate in one row of Naf and two rows of Nae in the plane.At d = 1/3, either Naf or Nae arranges in rows within a single layer. Itcan be concluded that two voltage plateaus located at 3.3 and 3.7 Vin P2-NadNM arise from the in-plane ordering rearrangement of LZZ-row-row (38, 39). As discussed above, there are two types of Na sites that
Wang et al., Sci. Adv. 2018;4 : eaar6018 9 March 2018
areNaf andNae in theP2 structure. The calculated totalDFTenergies (Fig.5C) demonstrate that Ni/Mn/Ti mixing effectively decreases the siteenergy difference between Nae and Naf, thus suppressing the Na-vacancyordering, which could refrain the formation of strongly orderedintermediate phases, and promote complete solid-solution behavior overa wider range ofNa content. So, the Na+/vacancy ordering-disorderingtransition phenomenon in this systemmainly originates from the differ-ent Nae-Naf site energy after Ti substitution into the lattice.
Na+ kinetics studyOwing to different Na-vacancy in-plane orderings at differentNa contents in P2-NaNM, it can be assumed that such a fast self-arrangement upon charge/discharge might sacrifice Na-ion mobility.To verify this hypothesis, the apparent diffusion coefficients of Na+ ofthe two compounds were investigated by GITT (40, 41). The equilib-rium voltage of P2-NaNMT shows a continuous change, confirmingthe single-phase reaction process during Na+ (de)intercalation between2.5 and 4.15 V (fig. S14). Figure S15 compares the calculated Na diffu-sion coefficient (DNa
+) in P2-NaNMand P2-NaNMT samples based onthe GITT curves. The Na-ion mobility in P2-NaNMT is increased bytwo orders of magnitude with diffusion coefficients ranging from10−10 to 10−9 cm2 s−1, whereas P2-NaNM showed diffusion coeffi-cients of 10−12 to 10−11 cm2 s−1 in the Na range of 2/3 ≤ d ≤ 1/3. More-over, no jumps in the Na diffusion coefficient values are observed forP2-NaNMT, which could be attributed to Na-vacancy disordering inthe compound and consistent with the sloping characteristic of theelectrochemical curves. Noticeably, this fast diffusivity of Na ions in
Fig. 3. Charge compensation mechanism of P2-Na2/3−xNMT upon Na+ extraction/insertion. (A and B) Ex situ XANES spectra at the (A) Ni K-edge and (B) Mn K-edgeof Na2/3−xNMT electrodes collected at different charge/discharge states. (C and D) Corresponding ex situ EXAFS spectra at the (C) Ni K-edge and (D) Mn K-edge of Na2/3−xNMTelectrodes collected at different charge/discharge states. FT, Fourier transform.
5 of 9
SC I ENCE ADVANCES | R E S EARCH ART I C L E
on Septem
ber 16, 2018http://advances.sciencem
ag.org/D
ownloaded from
P2-NaNMT could accelerate its self-discharge as compared in fig. S16,which needs to be further mitigated. A lower charge transfer resistance(Rct) at the electrode/electrolyte interface and faster Na
+ diffusion asso-ciated with Warburg impedance (Zw) are obtained for P2-NaNMTusing an electrochemical impedance spectroscopy (EIS) technique(fig. S17), which provides another side evidence that Na disordering fa-cilitates electronic and Na+ migration (42, 43).
Given that Na+ in P2-type layered structure occupies open trigonalprisms thus providing wide ionic transport channels, the presence ofNa+/vacancy disordering will decrease the activation energy barrier forNa+ hops between adjacent prismatic sites and consequently enhanceNa+
diffusion. First-principles molecular dynamics (FPMD) calculationswere performed at temperatures ranging from 500 to 900 K to obtainNa+ ion transport property in this disordered P2-NaNMT. From themean square displacements (MSDs) of Na+ for P2-Na0.58NMT ob-served for all temperatures (fig. S18), the Na+ diffusion gets easier asthe temperature increases. It is clearly seen that the Na+ migratesthrough 2D passages characterized by interconnected diffusion paths(Fig. 6A), which favors the fast ion mobility and excellent kinetic prop-erty. The corresponding top view of trajectories in single sodium layersuggests the pathways along ideal Naf-Nae-Naf sites. The slope of MSD
Wang et al., Sci. Adv. 2018;4 : eaar6018 9 March 2018
time curves is fitted to obtain the diffusion coefficient (D). According tothe slope of Arrhenius plot in Fig. 6B, the calculated diffusion energybarrier (Ea) is a very low barrier of 0.17 eV, which is in good agreementwith the GITT results. A summary of the diffusion coefficients andactivation energies of the most common Na-based layered oxidesstudied in the literatures is compared in table S4. It is highlighted thatboth Na diffusion coefficients and computed activation energy of theP2-NaNMT correspond to the top level among TM layered oxides(44–52). In short, we may surmise that Ti substitution in P2-NaNMhas two effects: (i) the suppression of Na-vacancy ordering, resultingin a wider range of complete solid-solution behavior, and (ii) the re-duction of site energy difference between Nae and Naf and hence dif-fusion properties.
DISCUSSIONIn summary, we have successfully constructed a fully disorderedP2-NaNMT material on Na lattices over the studied range of Na con-centrations (d = 2/3 to 1/3). Partial Ti substitution for Mn withcompatible ionic radii and very different Fermi levels in P2-NaNMeffectively refrains electron delocalization, hence charge ordering and
Fig. 4. Crystal structural evolution under electrochemical Na+ (de)intercalation. (A and B) In situ XRD patterns collected during the first charge/discharge of the(A) Na/Na2/3−xNM and (B) Na/Na2/3−xNMT cells under a current rate of 0.1 C between 2.5 and 4.15 V.
6 of 9
SC I ENCE ADVANCES | R E S EARCH ART I C L E
on Septem
ber 16, 2018http://advances.sciencem
ag.org/D
ownloaded from
Na+/vacancy ordering. As a result, a high Na+ mobility and low activa-tion energy barriers are achieved in theNa interlayer, leading to smoothelectrochemistry without any voltage plateau, outstanding cycling sta-bility (83.9% capacity retention after 500 cycles at 1 C), and excellentrate capability (77.5% capacity retention at 20 C) in NIBs. Therefore,reasonable ionic substitution by inactive dopants, such as Li+, Ti4+,Mg2+, and Zn2+, with no d electrons, might be a sensible strategy tomodulate the composition of the metal lattice so as to avoid the Na-vacancy ordering for optimal Na diffusion. Our study offers new insightsinto the rational design of high-rate layered cathode materials and alsopushes the practical application of NIBs as a decisive step forward.
MATERIALS AND METHODSSynthesisThe P2-NaNM and P2-NaNMT samples were synthesized by a solid-state reactionmethod from stoichiometric mixtures of Na2CO3 (99.5%;
Wang et al., Sci. Adv. 2018;4 : eaar6018 9 March 2018
5 mole percent access; Alfa Aesar), NiO (99%; Alfa Aesar), Mn2O3
(98%; Alfa Aesar), and TiO2 (anatase, 99.6%; Alfa Aesar). Themixtureswere ground and pressed into pellets under 20MPa, and then the pelletswere calcined at 950°C in air for 15 hours, cooled to room temperature,and stored in an argon-filled glove box until use.
CharacterizationsX-ray powder diffractionwas done in a BrukerD8Advance diffractom-eter with Cu Ka radiation (l = 1.5418 Å). A General Structure AnalysisSystem program was used to perform Rietveld refinement. For the insitu XRD experiment, a specially designed Swagelok cell was equippedwith an aluminumwindow for x-ray penetration. Themorphology andmicrostructurewere characterizedby scanning electronmicroscopy (SEM;JEOL JSM-6701F). The HRTEM image and EDS maps were examinedby TEM (JEM 2100F). ABF and HAADF imaging were executedusing a JEOL ARM200F (JEOL) STEM operated at 200 kV with a coldfield-emission gun and double hexapole Cs correctors (CEOS GmbH).
Fig. 6. Na+ kinetics of P2-NaNMT obtained by FPMD simulations. (A) Trajectories of Na+ in P2-Na0.57NMT simulated at a temperature of 800 K. The top view of eachNa+ layer is given in the right panel. (B) Arrhenius plot of Na+ diffusion coefficients.
Fig. 5. 2D Na+/vacancy ordering-disordering transition mechanism. (A) Typical charge/discharge profiles of P2-NaNM. The two voltage plateaus indicates therearrangement of Na+/vacancy ordering. (B) In-plane Na+/vacancy orderings of NadNi1/3Mn2/3O2 in the triangular lattice at d = 2/3, d = 1/2, and d = 1/3, respectively(empty red circles, Na+ on Nae sites; solid blue circles, Na+ on Naf sites; thick green lines, unit cell). (C) Calculated energy difference between the Nae and Naf site forP2-NaNM (purple background) and P2-NaNMT (blue background).
7 of 9

SC I ENCE ADVANCES | R E S EARCH ART I C L E
on Septem
ber 16, 2018http://advances.sciencem
ag.org/D
ownloaded from
XPSmeasurements were carried out with a ESCALAB 250Xi spectrom-eter (Thermo Fisher Scientific) using anAl Ka achromatic x-ray source.XAS measurements were done at beamline BL14W1 of the ShanghaiSynchrotron Radiation Facility (SSRF), China, operating with a Si(111)double-crystal monochromator.
ElectrochemistryThe working electrode was prepared by mixing 75% active material,15%SuperP carbon, and10%polyvinylidenedifluoride (Sigma-Aldrich)binder onto aluminum foil, followed by drying at 80°C in vacuumovernight. The loading mass of active material was 3 to 4 mg cm−2.The electrolyte was 1 M NaPF6 in ethylene carbonate and diethylcarbonate (1:1 in volume; 5% fluoroethylene carbonate). Sodium foiland porous glass fiber (GF/D) were used as the counter electrode andseparator, respectively. Coin cells (CR2032)were assembled in an argon-filled glove box (H2O and O2 < 0.1 parts per million). An Arbin batterytest system was used to perform discharge and charge measurementsover a voltage range of 2.5 to 4.15 V and 2.5 to 4.3 V at a rate of 0.1 Cat 25°C. CV measurements were performed on an Autolab PG302Nelectrochemical workstation at a scan rate of 0.1 mV s−1. For the GITTtests, the batteries were charged at a current of 17 mA g −1 for 30 min,followed by open-circuit relaxation for 10 hours.
D ¼ 4p
iVm
zAFS
� �2 dE=dd
dE=dffiffit
p� �2
where i is the pulse current (A),Vm is the molar volume (cm3mol−1),zA is the charge number (1 for Na/Na+), F is the Faraday’s constant(Cmol−1; 96485 Cmol−1), S is the surface area of electrode (cm2), dE/ddis the change of voltage with respect to composition during currentpulse, and dE/dt1/2 is the change of voltage with respect to the squareroot of time during the period in between current pulse.
For the self-discharge tests, the batteries were charged to 4.15 V at0.1 C, followed by open-circuit relaxation at 45°C for 24 hours, andthen the open-circuit voltage (OCV) was recorded at room tempera-ture every 15 min.
DFT calculationsTo calculate the energy difference between the Nae and Naf sites, the3a × 3b × 1c supercell with one Na ion was constructed to avoid theCoulombic repulsion between different Na ions. The difference of siteenergy is given by the following equation
DE ¼ EðNaeÞ � EðNaf Þ
where E(Nae) and E(Naf) are the total DFT energies of bulk modelswith one ion occupying Nae and Naf site, respectively.
To simulate the Na ion transport properties in P2-Na0.58NMT, theDFT calculations were performed within the 3a × 3b × 1c supercellas implemented in Viena ab initio simulation package. The projectoraugmented-wave method and generalized gradient approximation(GGA) in the Perdew-Burke-Ernzerhof parameterization were used.GGA + U was applied in this work, in which the applied effectiveU value given to Ni ions was 6.1 eV, to Mn was 4.0 eV, and to Ti was2.3 eV. For ensuring the configuration, an energy cutoff of 520 eV and ak-mesh of 0.03 Å−1 were used. The force convergence criterion for therelaxationwas 0.01 eVÅ−1. For the FPMD calculations of each tempera-ture, a time step of 1 fs was used to simulate a total time of 10 ps by a
Wang et al., Sci. Adv. 2018;4 : eaar6018 9 March 2018
Nose-Hoover thermostat. An energy cutoff of 450 eV was used with ak-point sampling at the G point. The ionic diffusion behavior can becalculated by a time-dependent MSD
MSDðtÞ ¼ riðtÞ � rið0Þj j2� �
in which ri(t) is the site of the i-th Na+ at the time t. According to theEinstein equation, the diffusion coefficientD is givenby the slope ofMSD
D ¼ 16limt→∞
ddt
r2ðtÞ� �
Thus, the activation energy barrier is correlated with the diffusioncoefficients at various temperatures based on Arrhenius equation.
SUPPLEMENTARY MATERIALSSupplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/3/eaar6018/DC1fig. S1. Powder XRD patterns of as-synthesized P2-NaNM and P2-NaNMT samples.fig. S2. In situ XRD patterns of P2-NaNM and P2-NaNMT powders at different temperatures.fig. S3. SEM images of P2-NaNM and P2-NaNMT sample.fig. S4. TEM and HRTEM images of P2-NaNMT sample.fig. S5. Cycling performance comparison of P2-NaNM and P2-NaNMT electrodes at 0.1 C in thevoltage range of 2.5 to 4.15 V during 100 cycles.fig. S6. SEM images of P2-NaNM and P2-NaNMT electrodes after long cycles.fig. S7. Charge/discharge profiles of P2-NaNM and P2-NaNMT electrodes between 2.5 and 4.3 V.fig. S8. Cycling performance comparison of P2-NaNM and P2-NaNMT electrodes at 0.1 C in thevoltage range of 2.5 to 4.3 V during 100 cycles.fig. S9. CVs of P2-NaNM and P2-NaNMT electrodes.fig. S10. XPS spectra of P2-NaNM and P2-NaNMT powders.fig. S11. Variation of lattice constants and unit cell volume during cycling.fig. S12. Schematic illustration for P2-O2 phase transition.fig. S13. In situ XRD patterns of P2-NaNM and P2-NaNMT electrodes between 2.5 and 4.3 V.fig. S14. GITT curves and the quasi-equilibrium potential as a function of the stoichiometry.fig. S15. Calculated Na chemical diffusion coefficients from GITT.fig. S16. OCV decay of P2-NaNM and P2-NaNMT electrodes.fig. S17. Nyquist plots of EIS and the fit for P2-NaNM and P2-NaNMT electrodes.fig. S18. MSD curves for each kind of ions in P2-Na0.58NMT.table S1. Crystallographic parameters of P2-NaNM refined by the Rietveld method.table S2. Crystallographic parameters of P2-NaNMT refined by the Rietveld method.table S3. Atomic distances, slab thickness, and d-spacing of the Na layer and interslab distancefor as-prepared materials.table S4. Summary of Na+ diffusion coefficients and activation energies in layered oxides.
REFERENCES AND NOTES1. M. Armand, J.-M. Tarascon, Building better batteries. Nature 451, 652–657 (2008).
2. N. Yabuuchi, K. Kubota, M. Dahbi, S. Komaba, Research development on sodium-ionbatteries. Chem. Rev. 114, 11636–11682 (2014).
3. X. Dong, L. Chen, J. Liu, S. Haller, Y. Wang, Y. Xia, Environmentally-friendly aqueous Li (or Na)-ion battery with fast electrode kinetics and super-long life. Sci. Adv. 2, e1501038 (2016).
4. H. Park, J. Kwon, H. Choi, T. Song, U. Paik, Microstructural control of new intercalationlayered titanoniobates with large and reversible d-spacing for easy Na+ ion uptake.Sci. Adv. 3, e1700509 (2017).
5. H. Yoshida, N. Yabuuchi, K. Kubota, I. Ikeuchi, A. Garsuch, M. Schulz-Dobrick, S. Komaba,P2-type Na2/3Ni1/3Mn2/3−xTixO2 as a new positive electrode for higher energy Na-ionbatteries. Chem. Commun. 50, 3677–3680 (2014).
6. C. Delmas, C. Fouassier, P. Hagenmuller, Structural classification and properties of thelayered oxides. Physica 99B, 81–85 (1980).
7. R. J. Clément, P. G. Bruce, C. P. Grey, Review—Manganese-based P2-type transition metaloxides as sodium-ion battery cathode materials. J. Electrochem. Soc. 162, A2589–A2604(2015).
8. K. Kubota, S. Komaba, Review—Practical issues and future perspective for Na-ionbatteries. J. Electrochem. Soc. 162, A2538–A2550 (2015).
8 of 9

SC I ENCE ADVANCES | R E S EARCH ART I C L E
on Septem
ber 16, 2018http://advances.sciencem
ag.org/D
ownloaded from
9. J. Xu, D. H. Lee, R. J. Clément, X. Yu, M. Leskes, A. J. Pell, G. Pintacuda,X.-Q. Yang, C. P. Grey, Y. S. Meng, Identifying the critical role of Li substitution inP2-Nax[LiyNizMn1−y−z]O2 (0 < x, y, z < 1) intercalation cathode materials for high-energyNa-ion batteries. Chem. Mater. 26, 1260–1269 (2014).
10. G. Singh, N. Tapia-Ruiz, J. M. Lopez del Amo, U. Maitra, J. W. Somerville,A. R. Armstrong, J. Martinez de Ilarduya, T. Rojo, P. G. Bruce, High voltage Mg-dopedNa0.67Ni0.3−xMgxMn0.7O2 (x = 0.05, 0.1) Na-ion cathodes with enhanced stability and ratecapability. Chem. Mater. 28, 5087–5094 (2016).
11. P.-F. Wang, Y. You, Y.-X. Yin, Y.-S. Wang, L.-J. Wan, L. Gu, Y.-G. Guo, Suppressing theP2–O2 phase transition of Na0.67Mn0.67Ni0.33O2 by magnesium substitution for improvedsodium-ion batteries. Angew. Chem. Int. Ed. 55, 7445–7449 (2016).
12. X. Wu, J. Guo, D. Wang, G. Zhong, M. J. McDonald, Y. Yang, P2-type Na0.66Ni0.33−xZnxMn0.67O2
as new high-voltage cathode materials for sodium-ion batteries. J. Power Sources 281,18–26 (2015).
13. L. Wang, Y.-G. Sun, L.-L. Hu, J.-Y. Piao, J. Guo, A. Manthiram, J. Ma, A.-M. Cao,Copper-substituted Na0.67Ni0.3−xCuxMn0.7O2 cathode materials for sodium-ion batterieswith suppressed P2–O2 phase transition. J. Mater. Chem. A 5, 8752–8761 (2017).
14. C. Zheng, B. Radhakrishnan, I.-H. Chu, Z. B. Wang, S. P. Ong, Effects of transition-metal mixingon Na ordering and kinetics in layered P2 oxides. Phys. Rev. Appl. 7, 064003 (2017).
15. B. Mortemard de Boisse, G. Liu, J. Ma, S.-i. Nishimura, S.-C. Chung, H. Kiuchi, Y. Harada,J. Kikkawa, Y. Kobayashi, M. Okubo, A. Yamada, Intermediate honeycomb ordering to triggeroxygen redox chemistry in layered battery electrode. Nat. Commun. 7, 11397 (2016).
16. X. Li, X. Ma, D. Su, L. Liu, R. Chisnell, S. P. Ong, H. Chen, A. Toumar, J.-C. Idrobo, Y. Lei, J. Bai,F. Wang, J. W. Lynn, Y. S. Lee, G. Ceder, Direct visualization of the Jahn–Teller effectcoupled to Na ordering in Na5/8MnO2. Nat. Mater. 13, 586–592 (2014).
17. Y. Wang, R. Xiao, Y.-S. Hu, M. Avdeev, L. Chen, P2-Na0.6[Cr0.6Ti0.4]O2 cation-disorderedelectrode for high-rate symmetric rechargeable sodium-ion batteries. Nat. Commun. 6,6954 (2015).
18. J. Vinckevičiu̅tė, M. D. Radin, A. Van der Ven, Stacking-sequence changes and Na orderingin layered intercalation materials. Chem. Mater. 28, 8640–8650 (2016).
19. G. J. Shu, A. Prodi, S. Y. Chu, Y. S. Lee, H. S. Sheu, F. C. Chou, Searching for stableNa-ordered phases in single-crystal samples of g−NaxCoO2. Phys. Rev. B 76, 184115 (2007).
20. P. Zhang, R. B. Capaz, M. L. Cohen, S. G. Louie, Theory of sodium ordering in NaxCoO2.Phys. Rev. B 71, 153102 (2005).
21. G. J. Shu, F. C. Chou, Sodium-ion diffusion and ordering in single-crystal P2-NaxCoO2.Phys. Rev. B 78, 052101 (2008).
22. R. Berthelot, D. Carlier, C. Delmas, Electrochemical investigation of the P2–NaxCoO2 phasediagram. Nat. Mater. 10, 74–80 (2011).
23. M. Guignard, C. Didier, J. Darriet, P. Bordet, E. Elkaïm, C. Delmas, P2-NaxVO2 system aselectrodes for batteries and electron-correlated materials. Nat. Mater. 12, 74–80 (2013).
24. Z. Lu, J. R. Dahn, In situ x-ray diffraction study of P2-Na2/3[Ni1/3Mn2/3]O2. J. Electrochem.Soc. 148, A1225–A1229 (2001).
25. D. H. Lee, J. Xu, Y. S. Meng, An advanced cathode for Na-ion batteries with high rate andexcellent structural stability. Phys. Chem. Chem. Phys. 15, 3304–3312 (2013).
26. L. Zheng, J. Li, M. N. Obrovac, Crystal structures and electrochemical performance ofair-stable Na2/3Ni1/3−xCuxMn2/3O2 in sodium cells. Chem. Mater. 29, 1623–1631 (2017).
27. X. N. Ying, Z. C. Xu, High temperature sodium ordering transition in NaxCoO2 studied bymechanical spectrum. J. Appl. Phys. 105, 063504 (2009).
28. X. Lu, Y. Wang, P. Liu, L. Gu, Y.-S. Hu, H. Li, G. P. Demopoulos, L. Chen, Direct imaging oflayered O3- and P2-NaxFe1/2Mn1/2O2 structures at the atomic scale. Phys. Chem. Chem.Phys. 16, 21946–21952 (2014).
29. S. Guo, P. Liu, Y. Sun, K. Zhu, J. Yi, M. Chen, M. Ishida, H. Zhou, A high-voltage andultralong-life sodium full cell for stationary energy storage. Angew. Chem. Int. Ed. 54,11701–11705 (2015).
30. X. Yu, Y. Lyu, L. Gu, H. Wu, S.-M. Bak, Y. Zhou, K. Amine, S. N. Ehrlich, H. Li, K.-W. Nam,X.-Q. Yang, Understanding the rate capability of high-energy-density Li-rich layeredLi1.2Ni0.15Co0.1Mn0.55O2 cathode materials. Adv. Energy Mater. 4, 1300950 (2014).
31. P.-F. Wang, H.-R. Yao, X.-Y. Liu, J.-N. Zhang, L. Gu, X.-Q. Yu, Y.-X. Yin, Y.-G. Guo,Ti-substituted NaNi0.5Mn0.5-xTixO2 cathodes with reversible O3–P3 phase transition forhigh-performance sodium-ion batteries. Adv. Mater. 29, 1700210 (2017).
32. S. Komaba, N. Yabuuchi, T. Nakayama, A. Ogata, T. Ishikawa, I. Nakai, Study on thereversible electrode reaction of Na1–xNi0.5Mn0.5O2 for a rechargeable sodium-ion battery.Inorg. Chem. 51, 6211–6220 (2012).
33. Y. H. Jung, A. S. Christiansen, R. E. Johnsen, P. Norby, D. K. Kim, In situ x-ray diffractionstudies on structural changes of a P2 layered material during electrochemicaldesodiation/sodiation. Adv. Funct. Mater. 25, 3227–3237 (2015).
34. K. Kubota, Y. Yoda, S. Komaba, Origin of enhanced capacity retention of P2-typeNa2/3Ni1/3-xMn2/3CuxO2 for Na-ion batteries. J. Electrochem. Soc. 164, A2368–A2373 (2017).
35. Y. S. Meng, Y. Hinuma, G. Ceder, An investigation of the sodium patterning in NaxCoO2
(0.5 ≤ x ≤ 1) by density functional theory methods. J. Chem. Phys. 128, 104708 (2008).36. M. Roger, D. J. P. Morris, D. A. Tennant, M. J. Gutmann, J. P. Goff, J.-U. Hoffmann,
R. Feyerherm, E. Dudzik, D. Prabhakaran, A. T. Boothroyd, N. Shannon, B. Lake, P. P. Deen,
Wang et al., Sci. Adv. 2018;4 : eaar6018 9 March 2018
Patterning of sodium ions and the control of electrons in sodium cobaltate. Nature 445,631–634 (2007).
37. T. Wu, K. Liu, H. Chen, G. Wu, Q. L. Luo, J. J. Ying, X. H. Chen, Rearrangement of sodiumordering and its effect on physical properties in the NaxCoO2 system. Phys. Rev. B 78,115122 (2008).
38. I. R. Mukhamedshin, H. Alloul, Na order and Co charge disproportionation in NaxCoO2.Physica B 460, 58–63 (2015).
39. Y. Hinuma, Y. S. Meng, G. Ceder, Temperature-concentration phase diagram ofP2-NaxCoO2 from first-principles calculations. Phys. Rev. B 77, 224111 (2008).
40. N. Bucher, S. Hartung, J. B. Franklin, A. M. Wise, L. Y. Lim, H.-Y. Chen, J. N. Weker,M. F. Toney, M. Srinivasan, P2–NaxCoyMn1–yO2 (y = 0, 0.1) as cathode materials insodium-ion batteries—Effects of doping and morphology to enhance cycling stability.Chem. Mater. 28, 2041–2051 (2016).
41. S. Guo, H. Yu, Z. Jian, P. Liu, Y. Zhu, X. Guo, M. Chen, M. Ishida, H. Zhou, A high-capacity,low-cost layered sodium manganese oxide material as cathode for sodium-ion batteries.ChemSusChem 7, 2115–2119 (2014).
42. J.-j. Zhang, P. He, Y.-y. Xia, Electrochemical kinetics study of Li-ion in Cu6Sn5 electrode oflithium batteries by PITT and EIS. J. Electroanal. Chem. 624, 161–166 (2008).
43. K. Tang, X. Yu, J. Sun, H. Li, X. Huang, Kinetic analysis on LiFePO4 thin films by CV, GITT,and EIS. Electrochim. Acta 56, 4869–4875 (2011).
44. N. A. Katcho, J. Carrasco, D. Saurel, E. Gonzalo, M. Han, F. Aguesse, T. Rojo, Origins ofbistability and Na ion mobility difference in P2-and O3-Na2/3Fe2/3Mn1/3O2 cathodepolymorphs. Adv. Energy Mater. 7, 1601477 (2017).
45. P.-F. Wang, Y. You, Y.-X. Yin, Y.-G. Guo, An O3-type NaNi0.5Mn0.5O2 cathode forsodium-ion batteries with improved rate performance and cycling stability. J. Mater. Chem. A4, 17660–17664 (2016).
46. H.-R. Yao, P.-F. Wang, Y. Wang, X. Yu, Y.-X. Yin, Y.-G. Guo, Excellent comprehensiveperformance of Na-based layered oxide benefiting from the synergetic contributions ofmultimetal ions. Adv. Energy Mater. 7, 1700189 (2017).
47. J.-L. Yue, Y.-N. Zhou, X. Yu, S.-M. Bak, X.-Q. Yang, Z.-W. Fu, O3-type layered transition metaloxide Na(NiCoFeTi)1/4O2 as a high rate and long cycle life cathode material for sodiumion batteries. J. Mater. Chem. A 3, 23261–23267 (2015).
48. Y. Wang, X. Yu, S. Xu, J. Bai, R. Xiao, Y.-S. Hu, H. Li, X.-Q. Yang, L. Chen, X. Huang,A zero-strain layered metal oxide as the negative electrode for long-life sodium-ion batteries.Nat. Commun. 4, 2365 (2013).
49. S. Guo, Y. Sun, J. Yi, K. Zhu, P. Liu, Y. Zhu, G.-z. Zhu, M. Chen, M. Ishida, H. Zhou,Understanding sodium-ion diffusion in layered P2 and P3 oxides via experiments andfirst-principles calculations: A bridge between crystal structure and electrochemicalperformance. NPG Asia Mater. 8, e266 (2016).
50. Z. Y. Li, J. C. Zhang, R. Gao, H. Zhang, L. R. Zheng, Z. B. Hu, X. F. Liu, Li-substituted Co-freelayered P2/O3 biphasic Na0.67Mn0.55Ni0.25Ti0.2−xLixO2 as high-rate-capability cathodematerials for sodium ion batteries. J. Phys. Chem. C 120, 9007–9016 (2016).
51. Z.-Y. Li, R. Gao, L. Sun, Z. Hu, X. Liu, Designing an advanced P2-Na0.67Mn0.65Ni0.2Co0.15O2
layered cathode material for Na-ion batteries. J. Mater. Chem. A 3, 16272–16278 (2015).52. X.-H. Zhang, W.-L. Pang, F. Wan, J.-Z. Guo, H.-Y. Lu, J.-Y. Li, Y.-M. Xing, J.-P. Zhang, X.-L. Wu,
P2–Na2/3Ni1/3Mn5/9Al1/9O2 microparticles as superior cathode material for sodium-ionbatteries: Enhanced properties and mechanisam via graphene connection. ACS Appl.Mater. Interfaces 8, 20650–20659 (2016).
AcknowledgmentsFunding: Thisworkwas supportedby the Basic Science Center Project ofNational Natural ScienceFoundation of China (grant no. 51788104), the National Natural Science Foundation ofChina (grant nos. 51772301 and 21773264), the National Key R&D Program of China(grant no. 2016YFA0202500), and the “Strategic Priority Research Program” of the ChineseAcademy of Sciences (grant no. XDA09010100). The authors acknowledge the support by thebeamline BL14W1 of the SSRF, China. Author contributions: Y.-G.G. and Y.-X.Y. proposedand supervised the project. Y.-G.G. and P.-F.W. conceived and designed the experiments withthe help fromY.-X.Y. P.-F.W. carried out the synthesis, electrochemical experiments, and in situ XRDmeasurements. L.G. performed STEM observation. X.-Y.L. and Y.W. analyzed the images withL.G. J.-N.Z. performed ex situ XAS measurements, and X.Y. analyzed the XAS data. H.-R.Y.performed the first-principles calculations. All authors participated in analyzing the experimentalresults and preparing the manuscript. Competing interests: The authors declare that theyhave no competing interests. Data and materials availability: All data needed to evaluate theconclusions in the paper are present in the paper and/or the Supplementary Materials. Additionaldata related to this paper may be requested from the authors.
Submitted 28 November 2017Accepted 1 February 2018Published 9 March 201810.1126/sciadv.aar6018
Citation: P.-F. Wang, H.-R. Yao, X.-Y. Liu, Y.-X. Yin, J.-N. Zhang, Y. Wen, X. Yu, L. Gu, Y.-G. Guo,Na+/vacancy disordering promises high-rate Na-ion batteries. Sci. Adv. 4, eaar6018 (2018).
9 of 9

/vacancy disordering promises high-rate Na-ion batteries+NaPeng-Fei Wang, Hu-Rong Yao, Xin-Yu Liu, Ya-Xia Yin, Jie-Nan Zhang, Yuren Wen, Xiqian Yu, Lin Gu and Yu-Guo Guo
DOI: 10.1126/sciadv.aar6018 (3), eaar6018.4Sci Adv
ARTICLE TOOLS http://advances.sciencemag.org/content/4/3/eaar6018
MATERIALSSUPPLEMENTARY http://advances.sciencemag.org/content/suppl/2018/03/05/4.3.eaar6018.DC1
REFERENCES
http://advances.sciencemag.org/content/4/3/eaar6018#BIBLThis article cites 52 articles, 6 of which you can access for free
PERMISSIONS http://www.sciencemag.org/help/reprints-and-permissions
Terms of ServiceUse of this article is subject to the
registered trademark of AAAS.is aScience Advances Association for the Advancement of Science. No claim to original U.S. Government Works. The title
York Avenue NW, Washington, DC 20005. 2017 © The Authors, some rights reserved; exclusive licensee American (ISSN 2375-2548) is published by the American Association for the Advancement of Science, 1200 NewScience Advances
on Septem
ber 16, 2018http://advances.sciencem
ag.org/D
ownloaded from