molecular dynamics tutorial with applications to … kemia... · molecular dynamics tutorial ......
TRANSCRIPT

Molecular dynamics tutorial with applications to aqueous systems
Garold Murdachaew
1400-1600, 13-14 October 2015
Chemicum A122
1

Outline
Why should I learn about molecular simulations?
Why should I learn about aqueous systems?
CP2K package for molecular simulations
VMD package for molecular visualization and analysis
gnuplot and bash scripts and fortran codes for analysis of MD trajectories
Hands-on MD exercises at CSC (taito) and on your local linux machine using
CP2K and VMD
2

Why should I learn about molecular simulations?
Another tool in your toolbox to study systems more complex than clusters
Simulations are computer experiments
Simulations allow one to see atomic detail and discover reaction mechanisms
Simulations allow one to model difficult conditions or processes:
not possible in lab (e.g., high P, high T, etc.)
too dangerous (e.g., deactivation/breakdown of nerve agents)
too expensive
Always keep in mind:
“The purpose of computing is insight, not numbers.”
– Richard Hamming, Numerical Methods for Scientists and Engineers
3
Why should I learn about aqueous systems?
Water is ubiquitous
Atmospheric and environmental
chemistries (one example: molecular
adsorption and chemical reactions on wet
and icy surfaces can lead to ozone holes)
Catalysis
Astrochemistry
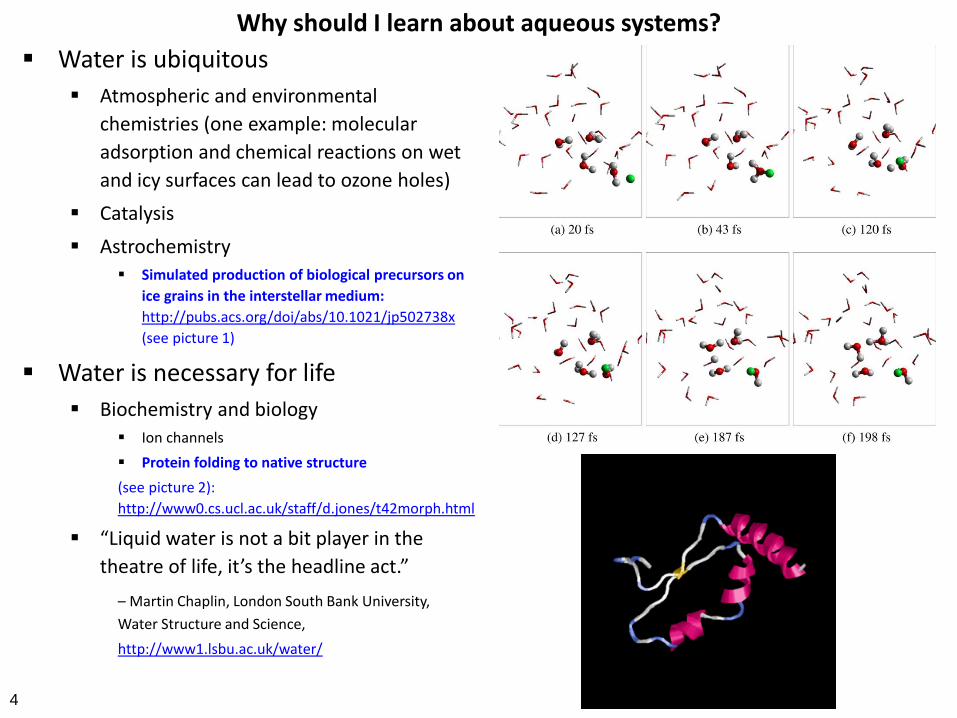
Simulated production of biological precursors on
ice grains in the interstellar medium:
http://pubs.acs.org/doi/abs/10.1021/jp502738x
(see picture 1)
Water is necessary for life
Biochemistry and biology
Ion channels
Protein folding to native structure
(see picture 2):
http://www0.cs.ucl.ac.uk/staff/d.jones/t42morph.html
“Liquid water is not a bit player in the
theatre of life, it’s the headline act.”
– Martin Chaplin, London South Bank University,
Water Structure and Science,
http://www1.lsbu.ac.uk/water/
4
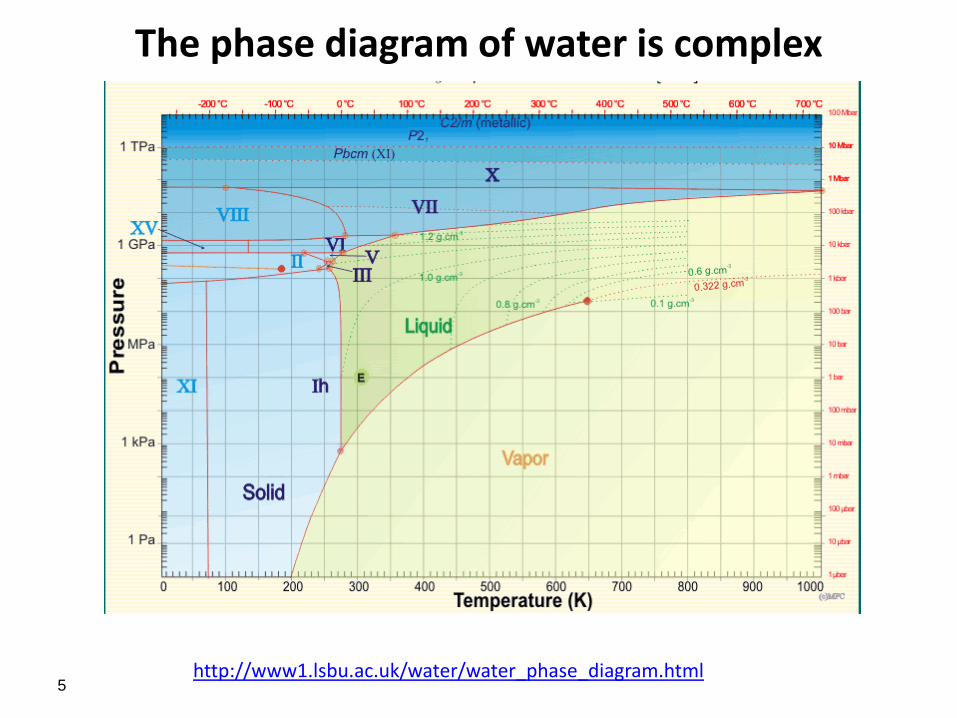
The phase diagram of water is complex
http://www1.lsbu.ac.uk/water/water_phase_diagram.html
5

CP2K package for molecular simulations
CP2K is free, open source (Fortran 2003), capable, and versatile package with a
large, active user and developer base
Some key parts of CP2K (we will use the bolded capabilities in the exercises)
FIST: classical molecular mechanics
Quickstep: density functional calculations
QM/MM: quantum mechanics and classical mechanics
Molecular dynamics, Monte Carlo, and much more
See: http://www.cp2k.org/
Science with CP2K: http://www.cp2k.org/science
Upcoming CECAM workshop: http://www.cecam.org/workshop-1122.html
Previous CECAM workshop: http://www.cecam.org/workshop-273.html
Tutorials: http://www.cp2k.org/tutorials
Exercises: http://www.cp2k.org/exercises
Input manual: http://manual.cp2k.org/trunk/CP2K_INPUT.html
Google Groups: https://groups.google.com/forum/#!forum/cp2k
On taito:
module load cp2k-env/2.5
sbatch cp2k_script.bash
6

VMD package for molecular visualization & analysis VMD is free to download
Can be used for visualization and also analysis (gpu acceleration possible)
Can handle large systems and long trajectories in many formats (xyz, etc.)
Can produce publication quality snapshots and movies in many popular formats
Can be run interactively or using a script
See: http://www.ks.uiuc.edu/Research/vmd/
Tutorials: http://www.ks.uiuc.edu/Research/vmd/current/docs.html#tutorials
Documentation: http://www.ks.uiuc.edu/Research/vmd/current/docs.html
Mailing list for questions: http://www.ks.uiuc.edu/Research/vmd/mailing_list/vmd-l/
On taito:
module load vmd
vmd system.xyz
or
vmd -e vmd_script.vmd
7

Exercises Hands-on exercises at CSC (taito) and on your local linux machine (ask if you
wish to run locally) using CP2K and VMD. (Note that all examples are already
equilibrated but you should confirm this.)
Structure and dynamics of ambient bulk liquid water using—
Example 1: Classical potential (exercise4)
Example 2: Density functional theory (exercise5)
Calculate: Internal energy (enthalpy); Structure (RDFs); Diffusion coefficient (Einstein
relation); IR spectrum. Compare to experiment.
Example 3: Rare instance of formic acid dissociation at the air-water
interface studied with DFT (exercise6)
Timescale of deprotonation; Grotthus migration of the proton defect; Mechanisms; RDFs.
Extra examples (ask if interested): Minimum energy structures of water
clusters (H2O)n=1-21 from density functional theory; Sulfuric acid deprotonation
on wet quartz surface using DFT; etc.
8

Important CP2K and theory references
Quickstep: http://www.sciencedirect.com/science/article/pii/S0010465505000615 (paper1)
Performance of BLYP-D2 for water and effectiveness in reproducing the hydrogen bond:
http://pubs.acs.org/doi/abstract/10.1021/jp901990u (paper2); see also:
https://en.wikipedia.org/wiki/Hydrogen_bond ; https://en.wikipedia.org/wiki/Water_model
Grotthuss mechanism: http://www.sciencedirect.com/science/article/pii/000926149500905J (paper3); see also:
https://en.wikipedia.org/wiki/Grotthuss_mechanism
Grimme’s DFT-D2: http://onlinelibrary.wiley.com/doi/10.1002/jcc.20495/abstract (paper4) or see:
https://en.wikipedia.org/wiki/London_dispersion_force
Books: M. P. Allen, D. J. Tildesley, Computer Simulation of Liquids (1989)
Donald McQuarrie, Statistical Mechanics (1976, 2000) Dominik Marx, Jürg Hutter, Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods (2009)
Mark Tuckerman, Statistical Mechanics: Theory and Molecular Simulation (2010)
View the wiki links; then download and start reading these papers, starting with the Quickstep paper (paper1), while
you are waiting for calculations to finish. Finish the reading at home. papers5,6,7 (see next page) may also be helpful.
Some of you may already have backgrounds in these areas, some do not. Thus I included the wiki links to give a quick
flavor.
9

Recent publications from Halonen group using CP2K Relevant papers:
Relevant to Examples 1 and 2: Simulated with semiempirical method (NDDO): “Semiempirical
Self-Consistent Polarization Description of Bulk Water, the Liquid-Vapor Interface, and Cubic Ice”
http://pubs.acs.org/doi/abs/10.1021/jp110481m (paper5)
Relevant to Example 3: Simulated with DFT and shows acid deprotonation and Grotthus
mechanism: “Dissociation of HCl into Ions on Wet Hydroxylated (0001) α-Quartz”
http://pubs.acs.org/doi/abs/10.1021/jz4017969 (paper6)
Relevant to Example 3: Simulated with classical potentials and shows molecular scattering :
“Nitrogen dioxide at the air–water interface: trapping, absorption, and solvation in the bulk and at
the surface” http://pubs.rsc.org/en/content/articlehtml/2012/cp/c2cp42810e (paper7)
Other papers:
Ice slab and proton hopping example using DFT from Sampsa Riikonen: “Ionization of Acids on the
Quasi-Liquid Layer of Ice” http://pubs.acs.org/doi/abs/10.1021/jp505627n
Simulated with DFT and shows acid deprotonation and Grotthus mechanism: “First and second
deprotonation of H2SO4 on wet hydroxylated (0001) α-quartz”
http://pubs.rsc.org/en/content/articlehtml/2014/cp/c4cp02752c
10

CP2K example 1: Water with classical potential @SET BASE_NAME run
@SET ID 01
&GLOBAL
PROJECT liq
PREFERRED_FFT_LIBRARY FFTW
PRINT_LEVEL LOW
RUN_TYPE GEOMETRY_OPTIMIZATION
&END GLOBAL
&MOTION
&GEO_OPT
TYPE minimization
OPTIMIZER BFGS
MAX_ITER 400 ! 200 is default
&END GEO_OPT
&END MOTION
&FORCE_EVAL
METHOD FIST
&MM
&POISSON
&EWALD
EWALD_TYPE spme
ALPHA .44
GMAX 25 25 25
O_SPLINE 6
&END EWALD
&END POISSON
&FORCEFIELD
&SPLINE
EMAX_ACCURACY 500.0
EMAX_SPLINE 1.0E15 ! 10000000000.0
EPS_SPLINE 1.0E-9
&END SPLINE
&BEND
ATOMS H O H
K 0.
THETA0 1.8
&END BEND
&BEND
ATOMS O H H
K 0.
THETA0 1.8
&END BEND
&BOND
ATOMS O H
K 0.
R0 1.8
&END BOND
&BOND
ATOMS H H
K 0.
R0 1.8
&END BOND
&CHARGE
ATOM O
CHARGE -0.8476
&END CHARGE
&CHARGE
ATOM H
CHARGE 0.4238
&END CHARGE
11
&NONBONDED
&LENNARD-JONES
ATOMS O O
EPSILON 78.198 ! this is K, = 0.155 kcal/mol = 0.650 kJ/mol
SIGMA 3.166
RCUT 11.4
&END LENNARD-JONES
&LENNARD-JONES
ATOMS O H
EPSILON 0.0
SIGMA 3.6705
RCUT 11.4
&END LENNARD-JONES
&LENNARD-JONES
ATOMS H H
EPSILON 0.0
SIGMA 3.30523
RCUT 11.4
&END LENNARD-JONES
&END NONBONDED
&END FORCEFIELD
&END MM
&SUBSYS
&SUBSYS
&CELL
ABC 12.4138 12.4138 12.4138
&END CELL
&COORD
O 12.25967785390 1.34872474190 12.42975017890 H2O
H 12.28658481340 1.45497852510 11.43794042330 H2O
H 12.12685964540 2.28501721350 12.78165108500 H2O
...
H 10.52064998830 9.65806143920 9.70630308870 H2O
&END COORD
&TOPOLOGY
&GENERATE
! BONDLENGTH_MAX 2.0
BONDPARM_FACTOR 0.9
&END GENERATE
&END TOPOLOGY
&KIND O
ELEMENT O
&END KIND
&KIND H
ELEMENT H
&END KIND
&CELL
&END CELL
&END PRINT
&END SUBSYS
&GRID_INFORMATION
&END GRID_INFORMATION
&END PRINT
&END FORCE_EVAL
!&EXT_RESTART
! RESTART_FILE_NAME ./run-01.restart
!&END EXT_RESTART

CP2K example 1: Water with classical potential
As you can see, the cp2k input file can have four
major sections (order of the sections is not
important). Note that ”!” or ”#” comments out the
line.
&GLOBAL
PROJECT liq
PREFERRED_FFT_LIBRARY FFTW
PRINT_LEVEL LOW
RUN_TYPE GEOMETRY_OPTIMIZATION
&END GLOBAL
&MOTION
&GEO_OPT
TYPE minimization
OPTIMIZER BFGS
MAX_ITER 400 ! 200 is default
&END GEO_OPT
&END MOTION
12
&FORCE_EVAL
METHOD FIST
&MM
&POISSON
&EWALD
…
&END MM
&SUBSYS
&CELL
ABC 12.4138 12.4138 12.4138
&END CELL
&COORD
O 12.25967785390 1.34872474190 12.42975017890 H2O
H 12.28658481340 1.45497852510 11.43794042330 H2O
H 12.12685964540 2.28501721350 12.78165108500 H2O
…
&END COORD
….
&END FORCE_EVAL
!&EXT_RESTART
! RESTART_FILE_NAME ./run-01.restart
!&END EXT_RESTART
Running example 1
1. login to taito (you are going to be doing calculations in the queue, thus have open in a web
browser for reference: https://research.csc.fi/taito-user-guide)
2. cd $WRKDIR
3. cp –pr /wrk/murdacha/md_class . (copy directories with fortran analysis codes and examples to
your WRKDIR)
4. cd md_class/ANALYZE_PROGRAMS (compile two simple fortran-2003 analysis programs; later try
to understand these programs since you may run them)
5. module load gcc
6. cd src-analyze-water
7. make analyze.x
8. cd ../src-rdf-water
9. make rdf.x
10. cd $WRKDIR/liq_spce (this is the input we just went over = Exercise4 for the class)
11. sbatch runit.bash
1. But first: edit if needed the input and script; module load vmd; vmd geometry.xyz or vmd –e liq.vmd to see the starting
geometry
12. Examine the output:
1. Use gnuplot on the *.ener file to check energy conservation (plot column 2 versus 4, then column 2 versus 5 and 6)
2. Use vmd to view the trajectory: module load vmd; vmd run-01.xyz or use the vmd script (may need to edit)
13

Running example 1
13. Now do the short MD NVE run but first clean
the directory (rm some files), and edit liq.inp
replacing:
1. RUN_TYPE GEOMETRY_OPTIMIZATION by RUN_TYPE
MD (this means GEO_OPT stuff will be ignored)
2. Add these lines (see file md_lines) after the line
&END GEO_OPT : &MD
ENSEMBLE NVT ! NVE
STEPS 1000
TIMESTEP 1.0
TEMPERATURE 300.0
&THERMOSTAT
TYPE NOSE
REGION MOLECULE
&NOSE
LENGTH 3
YOSHIDA 3
TIMECON 100
MTS 2
&END NOSE
&END THERMOSTAT
&PRINT ON
&ENERGY
&EACH
MD 1
&END EACH
FILENAME =${BASE_NAME}-${ID}.ener
&END ENERGY
&END PRINT
&END MD
14
3. Do the run: sbatch runit.bash
4. Examine the output:
1. Use gnuplot on the *.ener file to check energy
conservation (plot column 2 versus 4, then column 2
versus 5 and 6)
2. Use vmd to view the trajectory: module load vmd;
vmd run-01.xyz or use the vmd script (may need to
edit)
3. How does an MD run at 300 K differ from a GEO_OPT
run (at 0K)?
&TRAJECTORY ON
&EACH
MD 10
&END EACH
FILENAME =${BASE_NAME}-${ID}.xyz
FORMAT XYZ
&END TRAJECTORY
&VELOCITIES ON
&EACH
MD 10
&END EACH
FILENAME =${BASE_NAME}-${ID}_vel.xyz
FORMAT XYZ
&END VELOCITIES
&FORCES ON
&EACH
MD 10
&END EACH
FILENAME =${BASE_NAME}-${ID}_force.xyz
FORMAT XYZ
&END FORCES
&RESTART_HISTORY
&EACH
MD 1000
&END EACH
&END RESTART_HISTORY
&RESTART ON
BACKUP_COPIES 1
&EACH
MD 1
&END EACH
FILENAME =${BASE_NAME}-${ID}.restart
&END RESTART
&END PRINT

Running example 1
14. Now do the MD NVT production run, first clean the directory (rm some files), and edit liq.inp
replacing:
1. ENSEMBLE NVE by ENSEMBLE NVT
2. STEPS 1000 by STEPS 100000 (100 ps run)
3. VELOCITIES ON by VELOCITIES OFF
4. FORCES ON by FORCES OFF
15. Do the run and then examine the output:
1. Use gnuplot on the *.ener file to check energy conservation (plot column 2 versus 4, then column 2 versus 5 and 6)
2. Use vmd to view the trajectory: module load vmd; vmd run-01.xyz or use the vmd script (may need to edit)
3. Is the energy conserved? This the canonical ensemble (NVT). Should energy be conserved? Do you see oscillations?
4. Is your water liquid? How can you tell? Is it equilibrated? Hwne does equlibration occur?
5. Obtain RDFs using vmd
6. cd to the ANALYZE subdir, edit the *.in files, and do the analysis (use the bash script)
7. How do your results (structures in the form of the RDFs—plot against Soper experimental RDFs; internal
energy/enthalpy) compare to the literature, see for example: http://pubs.acs.org/doi/abs/10.1021/jp110481m
8. The SPC/E potential you have used is from Berendsen et al., see: https://en.wikipedia.org/wiki/Water_model
and https://dx.doi.org/10.1021%2Fj100308a038
Do you expect the results you obtained?
If you have time, you can use the end point of your (hopefully fully equilibrated) NVT trajectory to do an NVE run. That can
be analyzed in a similar way but also to obtain dynamical quantities like diffusion coefficient, IR spectra, etc. Speak with me
and I will help you out. Note that the SPC/E water molecule is rigid. We can do a run using TIP3P-F flexible water to get a
view of the internal IR vibrations.
15

CP2K example 2: Water with DFT @SET BASE_NAME run
@SET ID 01
&GLOBAL
PROJECT ${BASE_NAME}-${ID}
RUN_TYPE MD
&END GLOBAL
&MOTION
&MD
ENSEMBLE NVT
STEPS 20 ! Now you are calculating dft on the fly, it will be much slower
TIMESTEP 0.5
TEMPERATURE 300.0
&THERMOSTAT
TYPE NOSE
REGION MASSIVE
&NOSE
LENGTH 3
YOSHIDA 3
TIMECON [wavenumber_t] 2300
MTS 2
&END NOSE
&END THERMOSTAT
&PRINT ON
&ENERGY
&EACH
MD 1
&END EACH
FILENAME =${BASE_NAME}-${ID}.ener
&END ENERGY
&END PRINT
&END MD
16
&TRAJECTORY ON
&EACH
MD 1
&END EACH
FILENAME =${BASE_NAME}-${ID}.xyz
FORMAT XYZ
&END TRAJECTORY
&VELOCITIES ON
&EACH
MD 1
&END EACH
FILENAME =${BASE_NAME}-${ID}_vel.xyz
FORMAT XYZ
&END VELOCITIES
&FORCES ON
&EACH
MD 1
&END EACH
FILENAME =${BASE_NAME}-${ID}_force.xyz
FORMAT XYZ
&END FORCES
&RESTART ON
&EACH
MD 1
&END EACH
FILENAME =${BASE_NAME}-${ID}.restart
&END RESTART
&END PRINT
&END MOTION

CP2K example 2: Water with DFT (note how sections in blue differ from classical potential example) &FORCE_EVAL
METHOD QS
&DFT
POTENTIAL_FILE_NAME ./GTH_POTENTIALS
BASIS_SET_FILE_NAME ./GTH_BASIS_SETS
! WFN_RESTART_FILE_NAME ./run-01-RESTART.wfn
&MGRID
CUTOFF 280
&END MGRID
&SCF
MAX_SCF 20
EPS_SCF 1.0E-7
SCF_GUESS RESTART
&OUTER_SCF
EPS_SCF 1.0E-7
MAX_SCF 20
&END
&OT T
MINIMIZER DIIS
N_DIIS 7
&END OT
&RESTART ON
&END RESTART
&RESTART_HISTORY OFF
&END RESTART_HISTORY
&END PRINT
&END SCF
&QS
EPS_DEFAULT 1.0E-12
MAP_CONSISTENT
EXTRAPOLATION ASPC
EXTRAPOLATION_ORDER 3
&END QS
17
&XC
&XC_GRID
XC_SMOOTH_RHO NN10
XC_DERIV SPLINE2_SMOOTH
&END XC_GRID
&XC_FUNCTIONAL BLYP
&END XC_FUNCTIONAL
&vdW_POTENTIAL
DISPERSION_FUNCTIONAL PAIR_POTENTIAL
&PAIR_POTENTIAL
TYPE DFTD2
REFERENCE_FUNCTIONAL BLYP
R_CUTOFF 40.0
&END PAIR_POTENTIAL
&END vdW_POTENTIAL
&END XC
&END DFT
&SUBSYS
&CELL
ABC 12.4138 12.4138 12.4138
&END CELL
&COORD
O 1.2025696987709971E+01 1.2412376840360351E+00 1.1100847567157336E+01
H 1.1959096889663195E+01 1.3409373770618183E+00 1.0106406672798471E+01
H 1.1593234139420252E+01 2.0327876480659519E+00 1.1421274324532323E+01
…
O 1.2024298671712041E+01 9.9218625553065536E+00 9.2400384614568534E+00
H 1.2053386790559529E+01 9.6994663967598260E+00 1.0223617621157310E+01
H 1.1277449073604592E+01 9.4150658994176109E+00 8.9496605424081750E+00
&END COORD
&KIND O
BASIS_SET TZV2P-GTH
POTENTIAL GTH-BLYP-q6
&END KIND
&KIND H
BASIS_SET TZV2P-GTH
POTENTIAL GTH-BLYP-q1
&END KIND
&END SUBSYS
&END FORCE_EVAL
!&EXT_RESTART
! RESTART_FILE_NAME ./run-01.restart
!&END EXT_RESTART

Running and analyzing example 2
1. cd $WRKDIR/liq_blypd2_tzv2p_short (this is the input we just went over = Exercise5 for the class)
2. sbatch runit.bash
1. But first: edit if needed the input and script; module load vmd; vmd geometry.xyz or vmd –e liq.vmd to see the starting
geometry
3. While the run is happening, continue the readings or ask questions
4. Examine the output:
1. Use gnuplot on the *.ener file to check energy conservation (plot column 2 versus 4, then column 2 versus 5 and 6)
2. Use vmd to view the trajectory: module load vmd; vmd run-01.xyz or use the vmd script (may need to edit)
3. We only did an extremely short run. Why? Compare timings in the *.ener file to the classical case. How many processor
cores are we using now? How much more costly is Born-Oppenheimer MD with DFT compared to that with a classical
potential 2-body Lennard-Jones plus charges potential?
5. Since this is so costly, you only ran 20 steps to get a feel for DFT-MD. Now you will analyze a pre-
computed long trajectory:
6. cd $WRKDIR/liq_blypd2_tzv2p (this is the identical input but this run went longer)
7. Examine the files as before. Use gnuplot, vmd, etc. You can cd to ANALYZE sub-dir and do analysis.
8. Finally, compare the results of the classical simulation with the DFT one and also with experiment.
You can use gnuplot to plot RDFs obtained from SPC/E and BLYP-D2 and the experimental ones
(Soper files). How do the plots look? What about enthalpy? Put some results together to show
the whole class.
18

Running and analyzing example 3 (formic acid at air-water interface)
1. cd $WRKDIR/water_slab_with_formic_acid_blypd2_dzvp_nve300_short . How does the input file compare to
the one for DFT liquid water? (Hint: use the linux sdiff command: ’sdiff –aw 192 file file2 |less’). What does the
system look like (use: ’vmd geometry.xyz’)? What is the purpose of the vacuum? The constraints?
2. Run it: sbatch runit.bash
3. While the run is happening, continue the readings or ask questions
4. Examine the output
1. Use gnuplot on the *.ener file to check energy conservation (plot column 2 versus 4, then column 2 versus 5 and 6)
2. Use vmd to view the trajectory
3. The formic acid starts to fall. How can we monitor its height above the water surface? (hint ’use grep C position_file > C’,
then use gnuplot) . (Ask me for a gnuplot file to make a good plot.)
4. We only did an extremely short run. Why?
5. Since this is so costly, you only ran 50 steps to get a feel for this problem. Now you will analyze a pre-computed
longer trajectory:
6. cd $WRKDIR/water_slab_with_formic_acid_blypd2_dzvp_nve300 (this is the identical input but this run went
longer, to 10 ps)
7. Examine the files as before. Use gnuplot, vmd (use the scripts and try to understand them), etc. You can cd to
ANALYZE sub-dir and do analysis (first do: ’ssh taito-gpu’, vmd will run faster on gpus). Note that the analyze.x
code called now is slightly different. (You may need to compile it.) Also, vmd is used for calculating RDFs.
8. Is there any chemistry happening? If yes, what are the mechanisms and time scales? (Formic acid is a weak acid
so the deprotonation was not expected. Out of 50 trajectories, I only saw two deprotonate.) Make some nice
vmd snaphots of the Grotthus steps and present to the class. Compare to this Lee et al. paper.
19