modern tools of drug discovery combinatorial synthesis efficient way to make structurally diverse...
TRANSCRIPT
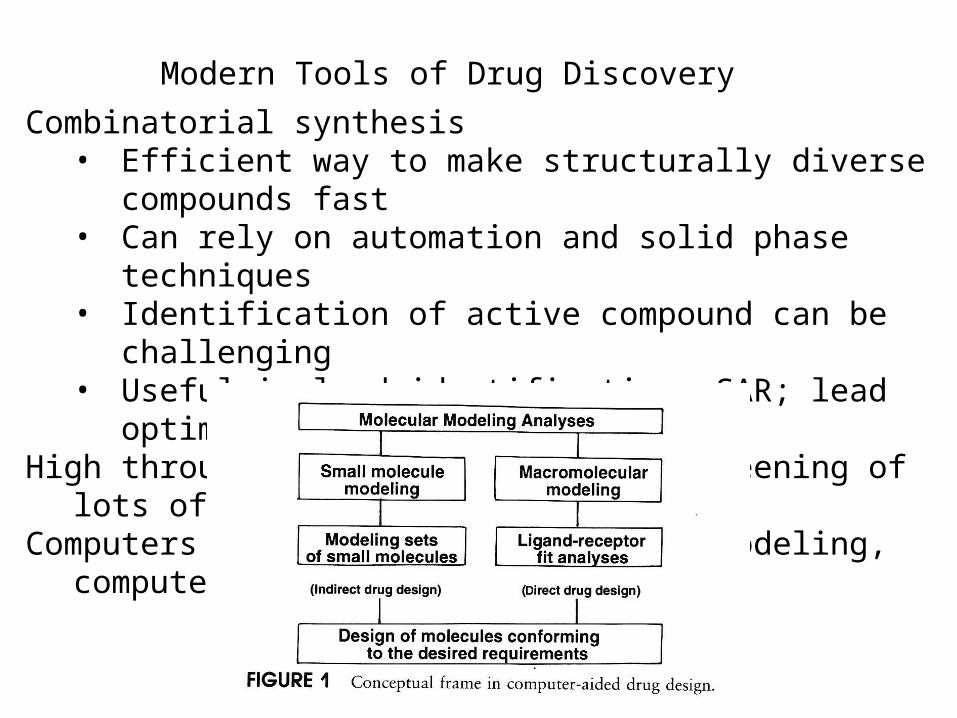
Modern Tools of Drug Discovery
Combinatorial synthesis• Efficient way to make structurally diverse compounds fast• Can rely on automation and solid phase techniques• Identification of active compound can be challenging• Useful in lead identification; SAR; lead optimization
High throughput screening - rapid screening of lots of compoundsComputers in drug design: Molecular modeling, computer algorithms

Computers in drug design: Where do they play a role?
A. Molecular visualizationB. Energy of different conformations (Conformational
analysis). Bioactive conformation determinationC. 3D Pharmacophore generation, molecular mimicry
• alignment of molecules• Calculation of/comparison of physiochemical features
such as electrostatic potential or logPD. Receptor-based drug design (docking; de novo construction)E. Receptor MappingF. Quantitative Structure-Activity Relationships (QSAR)

Two major Computational methods for the Calculation of Structure and Property Data
1. Molecular mechanics: • atoms are spheres, bonds are springs
• Giant classical mechanics (classical physics) problem• Calculated Energies/”force fields” are relative - good for
comparing two conformations of the same molecule• Fast• Cannot be used to calculate electronic properties (no
electrons in the model!)• Useful for: Energy minimization, Identification of stable
conformations, Energy calculations for specific conformations, Molecular motion

Two major computational methods for the Calculation of Structure and Property Data
2. Quantum mechanics• Nuclei and electrons of the molecules are considered• Giant quantum physics problem
H = E• Requires many approximations to make these problems
tractable. (Example: nuclei are motionless, electrons move; electrons move independently of one another)
• Two quantum mechanical approachesAb initio - more rigorous, from first principles (no stored
parameters or data), takes a long time, restricted to small molecules
Semi-empirical - faster, but less accurate, can be used on larger molecules (MNDO, AM1, PM3)
• Useful for MO energies, partial charges, electrostatic potentials, dipole moments

Computers in drug design: Where do they play a role?A. Molecular visualization - 3D experimental data from
crystal structures or NMR (x, y, z coordinates)
Acetylcholinesterase + Aricept(anti-Alzheimer drug)


Computers in drug design: Where do they play a role?A. Molecular visualization - 3D structure from 2D drawing.
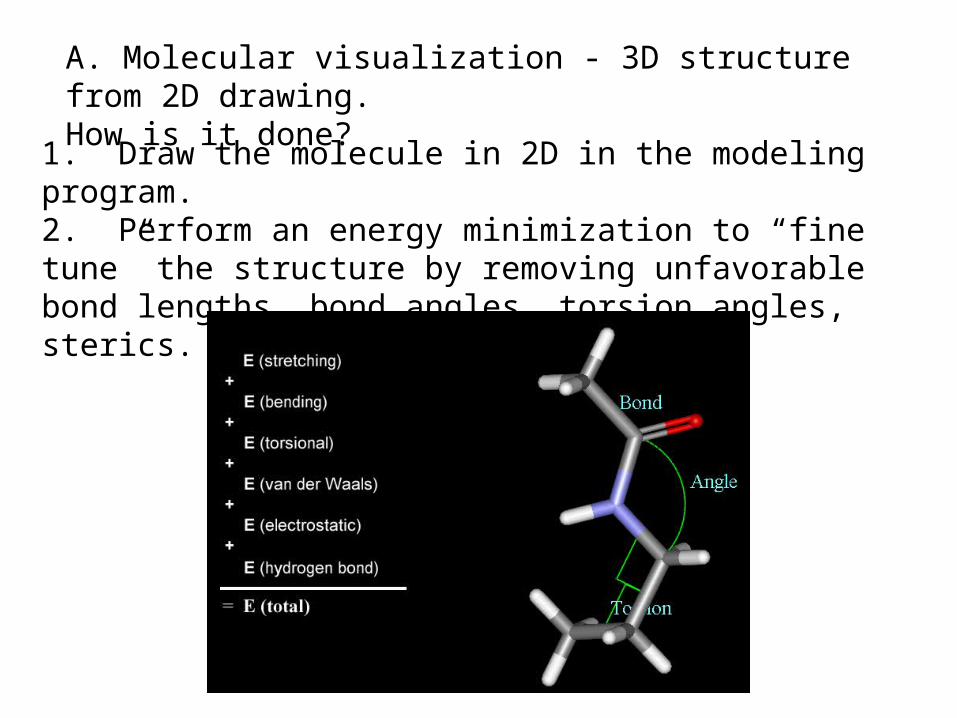
A. Molecular visualization - 3D structure from 2D drawing. How is it done?
1. Draw the molecule in 2D in the modeling program.2. Perform an energy minimization to “fine tune” the structure by removing unfavorable bond lengths, bond angles, torsion angles, sterics. See movie on MC A5.5.1-8

Bond Stretching: Bond angle bending: Torsion angles:
Minimize energy of molecule by optimizing the following:
A. Molecular visualization - 3D structure from 2D drawing. Energy minimization (continued)

Van der Waals Interactions: Dipole-dipole Interactions
Hydrogen Bonding:
Minimize energy of molecule by optimizing the following:
A. Molecular visualization - 3D structure from 2D drawing. Energy minimization (continued)

After performing this energy minimization, you have a reasonable structure for the molecule in 3D.
Note: Energy minimization stops at the first stable conformation it finds - closest in structure to the starting structure. This may be a local minimum, and not the global minimum. It may or may not be the bioactive conformation.
A. Molecular visualization - 3D structure from 2D drawing. Energy minimization (continued)

Spartan: Example of molecular modeling software
1. Build structure in 2D 2. Minimize (molecular mechanics)
3. Set up and perform quantumcalculations
4. Examine results: here, an electrostatic potential surface

Computers in drug design: Where do they play a role?
B. Energy of different conformations (Conformational analysis). Bioactive conformation determination. In general, one wants to know the lowest energy conformation plus all other conformations within 12kJ/mole of the global minimum.
Recall a flexible molecule may have more than one stable conformation.
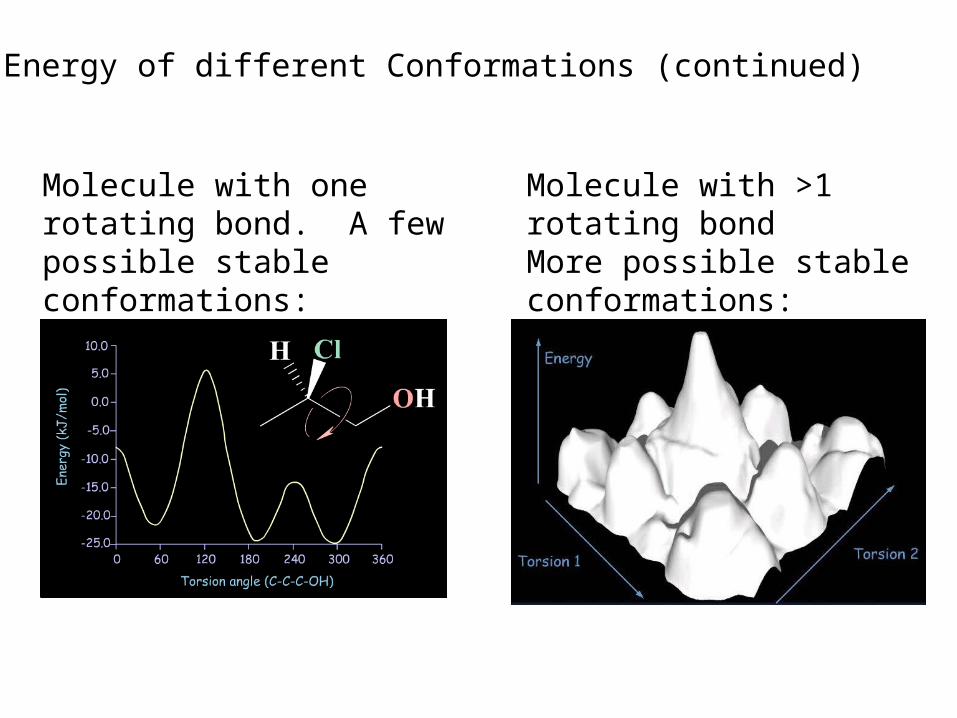
Molecule with one rotating bond. A few possible stable conformations:
Molecule with >1 rotating bondMore possible stable conformations:
B. Energy of different Conformations (continued)

Stable conformations occur at local and global minima:
B. Energy of different conformations (continued)
Many, MANY possibleStable conformations

Goal: To generate by computer all possible different stable conformations of the molecule (to help identify possible bioactive conformations, for example).
Method 1: molecular dynamics = simulated heating and cooling of the molecule to accumulate all possible conformations.
Method 2: stepwise rotation of bonds (rotate each bond of the molecule by a set number of degree increments).
B. Energy of different conformations (continued)

The resulting representative conformations for each local minimum should be minimized to obtain the lowest energy conformation.
Energy
Minimization
B. Energy of different Conformations (continued)
•Now a group of conformations with structures and energies at local and global minima has been generated. •They can be analyzed to determine the bioactive conformation (systematically compare reasonable minimized conformations of all active compounds; look at X-ray data; compare activity of rigid and nonrigid analogs).•Better molecules can be synthesized based on this information.

B. Energy of different Conformations (continued)
A group of stable conformations (structures local and global minima) were generated by computation for all active species and compared. The overlap of all active compounds reveals a bioactive conformation.
Example: Captopril
Rigid analogs were designed to keep amide and CO2 in place, but providing a range of locations for SH; tested for activity...

Computers in drug design: Where do they play a role?
C. 3D Pharmacophore generation, molecular mimicryRecall alignment of molecules to generate a pharmacophore:
Example: Overlay of procaine and cocaine: 2D overlap is misleading
A computer algorithm can then use the 3D pharmacophore to perform a 3D database search to find new leads!

Calculation of/comparison of physiochemical features such as electrostatic potential or logP
C. 3D Pharmacophore generation, molecular mimicry (continued)
The electrostatic potential surface above explains why binding a cationic (positively charged) compound through the cation-pi interaction decreases from left to right. The cation-pi interaction is electrostatic, so the additional fluorines substituted on the aromatic ring decrease the negative charge that stabilizes the cationic compound.
Example 1:

Calculation of/comparison of physiochemical features such as electrostatic potential or logP
C. 3D Pharmacophore generation, molecular mimicry (continued)
Example 2: Prediction of pharmacokinetic properties (ADMET)
Prescreening based on “Rule of Five”: MW<500; logP<5; no more than 5 H-bond donors; no more than 10 H-bond acceptors

D. Receptor-based drug design (docking; de novo construction)• 3D structure of the receptor is known:
Computers in drug design: Where do they play a role?

D. Receptor-based drug design (continued)
De novo design by 3D database searching computer algorithmsComputer-based structure building or linking (atoms, fragments)
building
linking
Example: Thymilidate synthase inhibitors bind at coenzyme binding site of the target enzyme - antitumor agents that block DNA synthesis.
de novo structure

D. Receptor-based drug design (continued)
Docking provides information about:Role of each functional group in the ligand and/or binding siteActive conformation of ligandModifications of known ligands that will enhance bindingPrediction of activities of unknown compounds - virtual screening
of libraries of compounds. Challenge: comparison of binding energies of structurally different ligands (called “scoring”).
Docking. If the binding groups on the ligands and the target are known (or automatically selected by the program), a program can move a rigid ligand around a rigid binding site to optimize binding interactions. Once the docking is finished, the energy of the system is minimized (ligand and binding site).
Docking

E. Receptor Mapping
Computers in drug design: Where do they play a role?
If primary amino acid sequence of the receptor is known and the X-ray structure of a related protein has been determined: Construct a model receptor:
1. The known structure is used as a template. The backbone of the new receptor is constructed on a computer to match that of the known protein. 2. Side chains are added in favorable conformations, and the energy is minimized by computer. 3. Key residues in the new receptor are identified and tested experimentally (site-directed mutagenesis).

E. Receptor Mapping (continued)
If no homologous proteins are known: Construct a model binding site: 1. Use experimental activity data for a diverse range of small molecules to construct a 3D pharmacophore. 2. Compute interaction energies for hydrophobic, ionic, polar interactions all around the molecule. Significant interactions are highlighted by “isoenergy contours.” SAR confirms these. 3. Suitable amino acids can be positioned around the molecule. 4. This model can be tested by comparing experimental binding data to calculated binding energies of docked ligands to this model binding site.
After constructing and testing the validity of a model receptor or model binding site, new molecules can be designed for improved binding, selectivity, or other properties.

Case study: Development of a drug using multiple techniques
Target: serotonin receptor. An antagonist will help treat anxiety, depression, migraines. This antagonist should be selective for a certain type and subtype of receptor (for 5HT2C over 5HT2A) to avoid side effects.
Lead: drug already developed by Lilly; it was insoluble. Modified structure to improve solubility.
What is the chemical modification here? How does it increase solubility?

N
N
(CH2)n
OHN
NCH3
Case study (continued)
This structure was also modified: What is the modification and why?
If n=5, affinity increased; if n = 6, affinity decreased. Molecular modeling was used to understand why. Minimized structures:
n = 5 n = 6Better affinity, better selectivity; but metabolic instability...
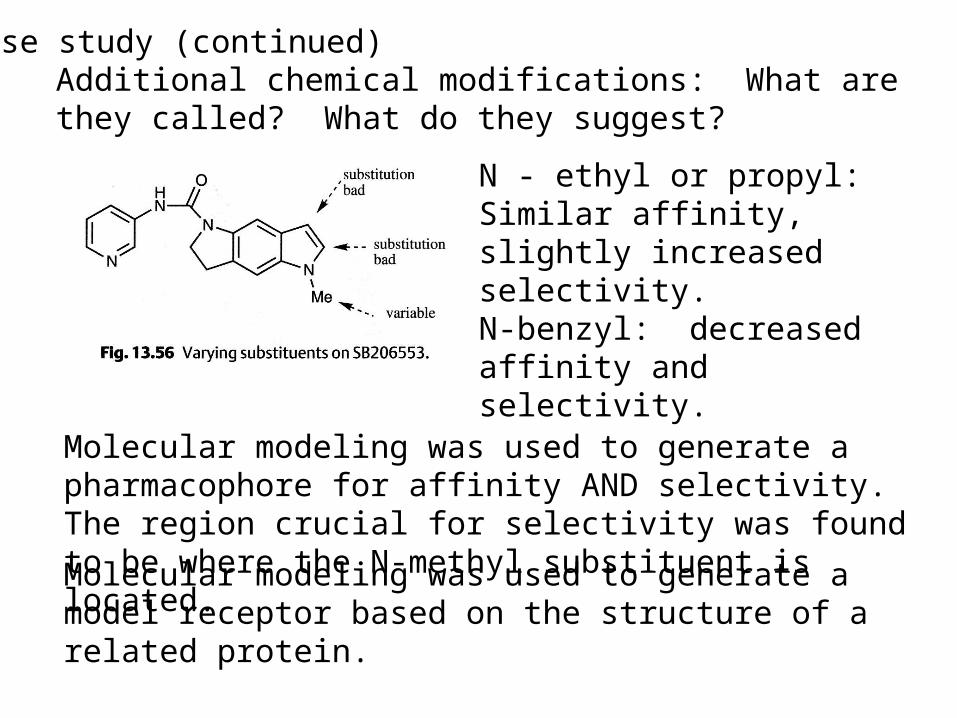
Case study (continued)Additional chemical modifications: What are they called? What do they suggest?
N - ethyl or propyl:Similar affinity, slightly increased selectivity.N-benzyl: decreased affinity and selectivity.
Molecular modeling was used to generate a pharmacophore for affinity AND selectivity. The region crucial for selectivity was found to be where the N-methyl substituent is located.
Molecular modeling was used to generate a model receptor based on the structure of a related protein.

Case study (continued)
Molecular modeling was used to generate a model receptor based on the structure of a related protein.
It was thought that the serine residues H-bond to the carbonyl group, while the hydrophobic pockets fit the rings. Docking calculations were performed to determine the most stable binding mode.

Case study (continued)
Note: N-methyl group is in a pocket with two valine residues. The other receptor subtype has leucine residues there. How might that explain the increased selectivity with N-ethyl vs. N-methyl?
Best binding mode:
Next, combinatorial synthesis was used to generate multiple structures for testing to improve selectivity

Case study (continued)Quantitative structure-activity relationships show these substituents lead to the best compromise of binding affinity and selectivity
Docking was performed again:Note: -CF3 acts as a conformational blocker (forcing SCH3 into hydrophobic pocket for selectivity enhancement)
What further changes in structure could improve binding affinity?
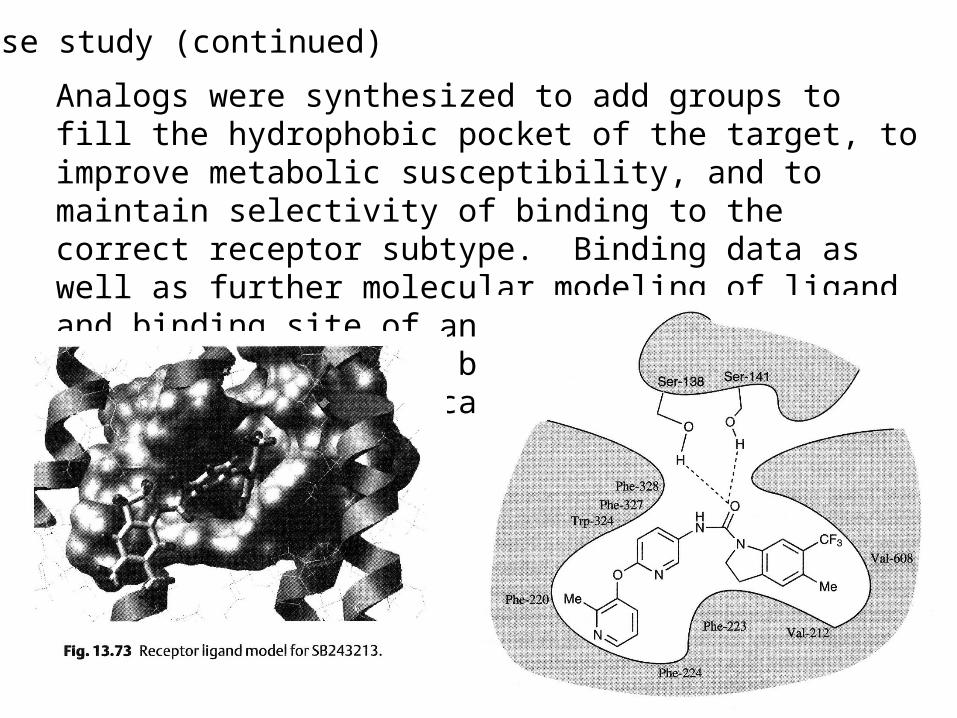
Case study (continued)
Analogs were synthesized to add groups to fill the hydrophobic pocket of the target, to improve metabolic susceptibility, and to maintain selectivity of binding to the correct receptor subtype. Binding data as well as further molecular modeling of ligand and binding site of analogs were used to develop the compound below. That compound was sent on to clinical trials.

Case study 2: J. Med. Chem. 2001, 44, 4615-4627Cyclin-dependent kinases (Cdk): regulate the cell cycle. Cancer is associated with abnormal cell cycle regulation. Inhibitors for G1 phase of cell cycle (over other phases) are desirable: must inhibit Cdk4.
Possible structure-based approaches:3D database search: commercially available, so synthetically accessiblePerhaps not correct ionization (databases are neutral)Not all conformations are searchedNo novel structures
De novo:Computers generate structures that are difficult to synthesize
So, these investigators chose de novo plus a program to find synthetically accessible related compounds.

Inhibitors of Cdk2 (A - C) and predicted binding region for Cdk4 (D)
Step 1: Use homologous protein structure of Cdk2 to create a receptor model on computer. (DON’T FORGET: Cdk4 is the TARGET!)

De novo design program LEGEND is used to generate scaffolds: builds atom-by-atom.
Below, I is one output of LEGEND. II is a simpler structure to use in 3D computer database searches.

382 compounds were purchased and screened for activity
Automated de novo structure building program
Special program to find easier compounds to synthesize

Hits from the computer database search were divided into four structural classes

One of the classes with multiple hits that also will lend itself to parallel synthesis was picked: Diaryl ureas
Structure-activity relationships (SAR)studies were performed. One compound was chosen as the new lead…

This lead was docked to the model site of Cdk4 on computer. Multiple conformations and binding modes were examined and minimized.

Comparison of the four modeled binding modes (A-D) to experimental data (SAR) ruled out two of the modes. The remaining binding modes were compared, and mode A was determined to be a stronger binding mode.
Examining this mode revealed a steric repulsion in the ligand as well as two areas to “grow” the ligand to better fit the site. New analogs were synthesized and tested.

One ligand was chosen as a new lead IC50 for Cdk4 = 0.042 MHowever, there was no selectivity for Cdk4 over Cdk1/2.
Future work involved comparing receptor structures to design an inhibitor with increased selectivity for Cdk4 over other subtypes.
Xray structure of the new lead compound with Cdk2.

Patrick, G. L. An Introduction to Medicinal Chemistry; Oxford University Press: New York, NY, 2001.
Guidebook on Molecular Modeling in Drug Design; Claude Cohen, N. Ed.; Academic Press: San Diego, CA, 1996.
Chemoinformatics; Gasteiger, J.; Engel, T., Eds.; Wiley-VCH: Weinheim 2003.
Molecular Conceptor Volume A
References