master medicina cosmetica y del …semcc.com/master/files/dieta aporte proteico - dres. delgado...
TRANSCRIPT

MASTER EN
MEDICINA COSMÉTICA Y DEL ANTIENVEJECIMIENTO
Estudio de la Masa Grasa Visceral en el tratamiento del Síndrome Metabólico, mediante Dieta de Aporte Proteico. Autores: Dr. Juan Manuel Delgado Ribas
Dr. Manuel Nieto Fernández – Arroyo
Tutores: Dr. Victor García Jiménez
Dr. Jaume Alijotas Reig
Vilanova i la Geltrú, 17 Abril 2015

RESUMEN
Introducción:
El Síndrome Metabólico (SM), como pluripatología coincidente en la Obesidad, se ha
mostrado como un proceso de categoría pandémica en el siglo XX, aunque
infradiagnosticado. Ya en el siglo XXI la connivencia científica en considerar a la masa grasa
visceral como un factor de riesgo de salud “per se”, ha llevado a considerar dicho síndrome,
más como patología orgánica general que como elementos aislados de patología unitaria a
tratar. Las acciones encaminadas a la resolución de cada uno de sus cuadros metabólicos
concomitantes por separado, no han logrado mejorar el pronóstico cardiovascular y
degenerativo a largo plazo, ya consumidos 15 años del nuevo siglo. La Dieta de Aporte
Proteico (DAP), está siendo un puntal de mejora general de todos y cada uno de dichos
factores de riesgo total y en el general del SM.
Objetivos:
Estudiaremos cómo mediante una dieta específica proteica, la “Dieta de aporte
proteico” (DAP), podemos influir determinantemente en la disminución de la Masa Grasa
Visceral (MGV), y por ende en la mejora de los factores de riesgo cardiovascular y
degenerativos, así como demostrar la buena tolerancia y seguimiento de dicha dieta por los
pacientes, logrando un balance clínico, biológico y ponderal muy correcto en el tiempo,
utilizando herramientas de la vanguardia nutricional actual como la Crononutrición, donde la
importancia de cuándo administramos un alimento, el número de veces diarias y en qué
momento del día, nos ayuda según los micronutrientes que incluyan los alimentos, incluso a
producir neuro modulación, para así mantener un bienestar general sin ansiedad pre
pandrial y a la vez obtener de ellos la vitalidad necesaria para desarrollar nuestras acciones
diarias óptimamente.

Material y Métodos:
Se han estudiado un total de 32 pacientes con IMC superior a 29.9, mediante la
utilización de los parámetros antropométricos y de Impedianciometria con el aparato Body
Composition Analyzer In Body 720 Bioespace. Los datos analizados, incluyen el seguimiento
de los pacientes finales desde enero de 2012 hasta junio de 2014. Los parámetros biológicos
fueron obtenidos mediante analítica sanguínea en ayunas.
Resultados:
Hemos encontrado una significación estadística de una magnitud elevada en la
disminución del parámetro en estudio, el Area de Grasa Visceral (AGV), así como del resto de
parámetros paralelos estudiados, como por ejemplo el diámetro de cintura e IMC, en todos
los pacientes tratados mediante la Dieta de Aporte Proteico.
Conclusiones:
El parámetro en estudio más importante, el AGV, mejoró en todos los pacientes con
Síndrome Metabólico tratados. Hemos observado que mediante la Dieta de Aporte Proteico,
se han obtenido una disminución importante y estadísticamente significativa de los
parámetros biológicos analizados, a la par observamos una mejora clínica general en cuanto
a estado osteomuscular, agilidad mental y estado físico, además de que durante el
tratamiento nutricional a que se sometieron los pacientes, tuvieron una observancia y
adherencia cómoda al tratamiento. El parámetro que nos ocupa preferentemente en el
estudio, el área de Grasa Visceral mejoró de forma sustancial, probablemente en nuestra
experiencia mucho más que con cualquier otro tipo de dieta hipocalórica, si bien no
estudiamos paralelamente ésta variable de manera estadística. Dicha variable comparativa

sería de sumo interés en ser estudiada en adelante, dado que existen pocos estudios al
respecto.
Palabras Clave:
Area Grasa Visceral (AGV); Síndrome Metabólico (SM); Dieta de Aporte Proteico (DAP)

ÍNDICE DE CAPÍTULOS
CAPÍTULO 1. INTRODUCCIÓN……………………………………………………………………………..6 1.1 Síndrome Metabólico
1.1.1 Definición evolutiva y Concepto ………………………………………………………………………9
1.1.2 Patogenia de la Masa Grasa Visceral ………………………………………………………………16
1.1.3 Obesidad y Resistencia Insulínica ……………………………………………………………………18
1.1.4 Alteraciones del Metabolismo Lipídico ………………………………………………………….23
1.1.5 Hipertensión Arterial y Riesgo Cardiovascular ……………………………………………….26
1.1.6 Otras alteraciones clínicas asociadas al Síndrome Metabólico…………………….…28
1.1.7 Alteración Membranaria celular …………………………………………………………………….29
1.1.8 Alteración de los Genes Reloj ………………………………………………………………….…….30
1.2 Dieta de Aporte Proteico
1.2.1 Orígenes……………………………………………………………………………………………………….….36
1.2.2 Concepto…………………………………………………………………………………………………….……38
1.2.3 Fisiología…………………………………………………………………………………………………..………41
1.2.4 Beneficios Biológicos……………………………………………………………………………..…………43
1.2.5 Implementación en el Síndrome Metabólico……………………………………………..……45
1.2.6 Equilibrio Alimentario…………………………………………………………………………….…………48
1.2.7 Interés de la Crononutrición en el Síndrome Metabólico……………………………..…51
CAPÍTULO 2. MATERIAL Y MÉTODOS…………………………………………………………………....…59
CAPÍTULO 3. RESULTADOS…………………………………………………………………………………….…68
CAPÍTULO 4. DISCUSIÓN………………………………………………………………………………………....78
CAPÍTULO 5. CONCLUSIONES………………………………………………………………………..…………80
Conflicto de Intereses…………………………………………………………………………………………………83
BIBLIOGRAFÍA……………………………………………………………………………………………………..………84

CAPÍTULO 1. INTRODUCCIÓN
1.1 SÍNDROME METABÓLICO
La obesidad, epidemia mundial del siglo XXI y asociado a ella también el SM(1,2) tienen
una prevalencia e intensidad en continuo aumento, incluso en los países emergentes. Por
primera vez en la historia, el número de individuos con sobrepeso en el mundo, supera al
número de personas que pasan hambre. En 2005 la OMS constataba en más de 1600
millones el número de adultos con sobrepeso y en 400 millones el número de sujetos
obesos(3,4).
La transición nutricional que ocurre en algunos países que viven de modo acelerado,
supone cambios muy rápidos al respecto: en 1975, Brasil registraba 2 casos de malnutrición
por 1 caso de obesidad y en 1997 más de 2 obesos por persona desnutrida (6-8). En Europa y
según las zonas, entre el 30 y el 80% de los adultos tienen sobrepeso. Alrededor del 20% de
los niños padecen sobrepeso y un tercio de éstos es obeso. (9-11)
Numerosos estudios han demostrado la relación directa del sobrepeso, aumentando
los riesgos de morbilidad y mortalidad. La obesidad, está vinculada a una amplia lista de
enfermedades: patologías cardiovasculares, dislipemias, diabetes, patologías articulares y
respiratorias, cánceres y de manera muy directa, con el Síndrome Metabólico. Dicho
síndrome, afecta a un 20-25% de la población mundial, multiplicando por tres la probabilidad
de sufrir un evento cardiovascular a quien lo padece y por dos la probabilidad de morir por
dicho evento. Los niños y jóvenes, se ven igualmente afectados: en EEUU, el 30% de los
adolescentes con sobrepeso, sufre de SM. (13-17)

En cuanto a la diabetes, contamos actualmente con un número cercano a los 200
millones de personas en el mundo, pero lo más preocupante es que se espera que dicha cifra
llegue a los 333 millones para el año 2025. Sabemos ya, que la obesidad constituye la
primera causa de muerte por cáncer en los países desarrollados.
Toda ésta situación, también comporta consecuencias económicas, estimándose que el
coste del sobrepeso, representa entre el 2 y el 8% del gasto sanitario en nuestro país. Un
estudio prospectivo en EEUU, estima que en el 2030, el 86% de los adultos padecerá
sobrepeso u obesidad e implicará así un costo del 16% del gasto sanitario.(18-21)
Evidentemente, esta epidemia no es transmisible en el sentido puramente genético o
patológico del término. Sin embargo es obvio que, si en una familia alguno o los dos padres
son obesos, sus hábitos alimentarios favorecerán de forma determinante la obesidad o el
sobrepeso de sus hijos e hijas, perpetuando así esta epidemia.
Se cree que el principal factor higiénico dietético, condicionante para la aparición del
SM, es el cambio en el estilo de vida actual, con incremento en la ingesta de grasas
saturadas, la vida sedentaria y las situaciones de estrés crónico que nos acosan
diariamente(22).
La prevalencia del SM es muy alta y va en aumento en todas las sociedades
occidentales, como consecuencia de los cambios en el estilo de vida que llevan al sobrepeso
y la obesidad.
En Estados Unidos en el periodo 2003-2006, según la Encuesta Nacional de Examen de
Salud, teniendo en cuenta los criterios de la NCEP: ATPIII, sobre el 34% de las personas
estudiadas, presentaban un SM. Y si tenemos en cuentas los criterios de la IDF, la cifra llega
hasta el 39,1%(23).

En España, la prevalencia de SM es muy elevada. En uno de los estudios
epidemiológicos más importantes realizados hasta la actualidad, el estudio DARIOS(24),
analizando 24.640 individuos, mediante 11 estudios poblacionales en 10 Comunidades
Autónomas, sobre personas comprendidas entre los 35 y 74 años, se halló una prevalencia
de SM del 32% en varones y del 29% en mujeres. En este estudio se aplicó la definición de
consenso (IDF/AHA), considerando los perímetros de cintura de corte en 102 cm para los
varones y 88 cm para las mujeres. También se estudió la prevalencia de SM premórbido,
excluyendo del diagnóstico de SM a los pacientes con diabetes mellitus y/o enfermedad
cardiovascular sintomática, dando unas prevalencias del 26% en los varones y del 24% en las
mujeres.
Si tenemos en cuenta los criterios diagnósticos de SM por separado, en el citado
estudio DARIOS, podemos observar que:
• Tensión arterial ≥ 130/85 mm Hg, estuvo presente en el 89% de los
varones y en el 87% de las mujeres
• Aumento del perímetro de la cintura (≥102 en varón, ≥88 en mujer)
estuvo presente en el 95% de las mujeres y el 77% de los varones
• Triglicéridos ≥ 150 mg/dl apareció en 62% de los varones y 44% en las
mujeres.
• Colesterol HDL disminuido (≤40mg/dl en varones y ≤50mg/dl en
mujeres) se halló en el 41% de los varones y en el 58% de las mujeres.
• Glucemia en ayunas ≥ 100 mg/dl o en tratamiento por ello, se
encontró en el 80% de los varones y en el 71% de las mujeres.
En éste y otros estudios también se ha podido comprobar que la prevalencia del SM
aumenta con la edad(25).

La prevalencia de SM también aumenta cuando se incrementa el índice de masa
corporal (IMC). Tanto en hombres como mujeres con sobrepeso, el riesgo de padecer SM es
6 veces superior en los varones y 5,5 veces en las mujeres, que el de la población con
normopeso. Cuando se llega al grado de obesidad, el riesgo se multiplica, llegando a 32 veces
en el varón y 17 veces en la mujer, si los comparamos con individuos en normopeso(26).
1.1.1 Definición evolutiva y Concepto de Síndrome Metabólico
Se denomina Sindrome Metabólico (SM) (también conocido como síndrome X,
síndrome plurimetabólico, síndrome de insulinorresistencia, síndrome de Reaven o CHAOS
en Australia) al conjunto de enfermedades o factores de riesgo en un mismo individuo que
aumentan su probabilidad de padecer una enfermedad cardiovascular o diabetes
mellitus(27,28).
Se caracteriza fundamentalmente por la presencia de una resistencia a la insulina con
un hiperinsulinismo compensador, asociado a trastornos como la alteración en el
metabolismo de los carbohidratos, dislipemia, hipertensión arterial y obesidad.
Suele existir una relativa confusión, entre la definición conceptual del síndrome
metabólico y los parámetros clínicos y valores de corte propuestos por diferentes
organizaciones (NCEPT-ATP III, IDF, OMS, etc...) para identificar pacientes con Síndrome
Metabólico. He aquí las definiciones evolutivas y la definición de consenso utilizada para este
estudio finalmente.

I. Criterios diagnósticos de Síndrome Metabólico según la Organización Mundial de la
Salud (OMS). (Año 1998)(29 )
A- Criterios mayores
Diabetes mellitus tipo 2 o Intolerancia oral a la glucosa (glicemia en ayunas ≥ a
110mg/dl y/o 2 hs postcarga ≥ a 140 mg/dl) y/o resistencia a la insulina(captación de
glucosa por debajo del P25 en clamp .).
B- Criterios menores
Hipertensión arterial (Presión arterial ≥ a 140-90 mmHg)
Dislipemia (TG> a 150 mg/dl y/o colesterol HDL <35 -39 mg/dl en hombres y mujeres).
Obesidad (índice cintura/cadera >0.90(varón)-0.85(mujer) y/o índice de masa
corporal > 30 kg/m2)
Microalbuminuria (excreción urinaria de albúmina ≥ 20 mg/min).
La OMS señala que es indispensable para el diagnóstico de Síndrome Metabólico (SM)
la presencia de resistencia a la insulina y/o alteración en la tolerancia a la glucosa. A esto debe
sumarse al menos dos de los criterios menores siguientes: hipertensión arterial, dislipemia,
obesidad, microalbuminuria. Es importante destacar que la microalbuminuria es, para la OMS
un importante predictor de riesgo cardiovascular.
Debido a que es necesaria la utilización de técnicas de alto costo, complejas y de no
tan sencilla aplicación, esta definición es una herramienta poco aplicable en la práctica
médica diaria, resultando útil en investigación.

II. Criterios diagnósticos de Síndrome Metabólico según el National Cholesterol Education
Program (NCEP) Adult Treatment Panel III (ATP III.)Año 2001(30)
Obesidad abdominal (perímetro abdominal > 102 cm en hombres y >88 cm
en mujeres)
Dislipemia: Hipertrigliceridemia (TG ≥150 mg/dl)
HDL-colesterol < 40 mg/dl en hombres y < 50 mg/dl en mujeres.
Presión arterial ≥ 130-85 mmHg
Glicemia basal en ayunas ≥ 110 mg/dl
La definición de la National Cholesterol Education Program (NCEP) se basa en la
coexistencia de cualquier combinación de tres alteraciones de las cinco descritas: en
la distribución de grasa corporal, presión arterial, triglicéridos, HDL y glicemia en ayunas.
En 2004 con la actualización de la American Diabetes Association se modificó la
glucemia basal de corte a 100mg/dl (5,6 mmol/l)(31)
A diferencia de lo establecido por la OMS, la NCEP no recomienda
una medición rutinaria de la insulinemia por no considerarla esencial para el diagnóstico de
SM. Se tienen en cuenta parámetros clínicos mucho más accesibles y asequibles. Así, se
puede llegar a un diagnóstico con tan sólo una cinta métrica y un tensiómetro.
III. Criterios diagnósticos de Síndrome Metabólico según la American Association of Clinical
Endocrinologists (AAEC). (Año 2003)(32)
A- Criterios mayores.-
Resistencia a la Insulina (medida por hiperinsulinemia)
Acantosis nigricans

Obesidad abdominal (circunferencia abdominal ≥ 102 cm en hombres y ≥ de 88 cm en mujeres). Dislipemia (colesterol HDL ≤ 45 mg/dl en mujeres y ≤ 35 mg/dl en hombres o TG ≥ 150 mg/dl)
Hipertensión arterial
Intolerancia a la glucosa o diabetes mellitus tipo II
Hiperuricemia
B- Criterios menores.-
Hipercoagulabilidad
Síndrome del ovario poliquístico
Disfunción endotelial
Microalbuminuria
Enfermedad cardíaca coronaria
La American Association of Clinical Endocrinologists (AACE) volvió a rescatar el papel
central de la resistencia a la insulina y de nuevo lo denominó como síndrome de resistencia a
la insulina.
La AACE consideraba como criterio imprescindible el de la resistencia a la insulina, a la
que se añadían, pero sin dar un número mínimo, varios de los otros criterios mayores.
Según la AACE, también se considera que cuando se ha realizado un diagnóstico de
diabetes mellitus tipo 2, no se puede aplicar el término de síndrome de resistencia a la
insulina.

IV. Criterios diagnósticos de Síndrome Metabólico según el European Group for Study of
Insulin Resistance (EGIR)(33)
A- Criterios mayores
Existencia de Resistencia a la insulina o hiperinsulinemia en ayunas superior al
percentil 75
B- Criterios menores
Glucemia plasmática basal ≥110 mg/dl (sin alcanzar rango diabético)
Hipertensión arterial (≥ 140/90) o seguir tratamiento para HTA)
Dislipemia: Hipertrigliceridemia (TG≥ 180 mg/dl) o
Colesterol HDL ≤ 40 mg/dl
Obesidad central: IMC ≥ 30kg/m2 o Cociente cintura/cadera ≥ 94 (varones), ≥
80(mujeres)
El diagnóstico se realiza mediante la suma de 1 criterio mayor y dos o más criterios
menores.
V. Criterios diagnósticos de Síndrome Metabólico según la International Diabetes
Federation (IDF) (Berlin, abril 2005)(34,35)
Se da como criterio necesario la existencia de Obesidad Centroabdominal
Perímetro de cintura ≥ 94 cm en Varones
Perímetro de cintura ≥ 80 cm en mujeres
Acompañado de 2 o más de los siguientes criterios:

Hipertrigliceridemia (≥ 150 mg/dl) o en tratamiento por ello
HDL-colesterol bajo (≤ 40mg/dl en varones, ≤ 50mg/dl en mujeres) o estar
siendo tratado por ello
Hipertensión arterial (P.Sist. ≥ 130 mmHg o P.Diast. ≥ 85 mmHg) o en
tratamiento por ello
Diabetes Mellitus Tipo 2 o hiperglucemia en ayunas (glucosa ≥ 100mg/dl)
Se da como criterio necesario la obesidad centroabominal y señala la importante
correlación entre el perímetro abdominal y resistencia a la insulina(36).
Tenemos que destacar la variación interracial de la valoración de la circunferencia
abdominal, para un correcto diagnóstico del Síndrome Metabólico según la étnia del
paciente.(35)(Tabla 1)
Para una correcta medición del perímetro de la pared abdominal tenemos que
estandarizar el método:
El individuo debe estar de pie y el examinador se coloca a su derecha, localiza
palpando la cresta ilíaca y justo sobre lo más alto del borde lateral de la cresta ilíaca derecha
se traza una línea horizontal y se cruza con una línea vertical trazada a través de la línea
media axilar. La cinta de medir se coloca en un plano horizontal alrededor del abdomen a la
altura de este punto por el lado derecho del tronco. El plano de la cinta es paralelo al suelo y
la cinta debe estar ajustada pero no debe comprimir la piel. La medición se hace en
inspiración normal.

ORIGEN ÉTNICO CRITERIO ADOPTADO POR VARONES MUJERES EUROPEOS IDF ≥94 ≥80 CAUCÁSICOS OMS ≥94 ≥80 ESTADOUNIDENSES AHA/NHLBI ≥102 ≥88 EUROPEOS EAS/ESC ≥102 ≥88 ASIÁTICOS IDF ≥90 ≥80
JAPONESES Sociedad Japonesa de Obesidad ≥85 ≥90
ORIENTE MEDIO IDF ≥94 ≥80 SUBSAHARIANOS IDF ≥94 ≥80
Tabla 1. Perímetro de cintura según étnias
VI. Criterios diagnósticos de consenso entre la IDF, la American Heart Association (AHA) y el
Nacional Heart, Lung, and Blood Institute (NHLBI) en 2009 (37)
Es la definición más utilizada en la actualidad
Para definir el SM se deben cumplir 3 o más de los siguientes criterios:
Perímetro de la cintura elevado
Triglicéridos ≥150 mg/dl
Colesterol HDL ≤ 40 mg/dl en varones o ≤ 50 en mujeres
Tensión Arterial ≥ 130/85 mm Hg
Glucosa en ayunas ≥ 100 mg/dl

1.1.2 Patogenia de la Masa Grasa Visceral en el Síndrome Metabólico
Hay una estrecha correlación de la obesidad abdominal y los factores de riesgo que
definen el SM, especialmente la hipertrigliceridemia(38,39), así como entre la obesidad
abdominal y la RI(40).
Según el estudio “International Day for the Evaluation of Abdominal Obesity” (IDEA),
sobre la Prevalencia de Obesidad Abdominal, aleatorio y transversal a nivel internacional, con
un reclutamiento de muestra final de 182.970 pacientes, de los cuales un 11% eran pacientes
españoles (19.912) y estando incluida España en la región Europa Sur, junto a Portugal, Italia,
Grecia y Turquía, se encontró una prevalencia general del 50,7% de obesidad abdominal
siguiendo los criterios de medida de la circunferencia abdominal de la ATPIII.(41) (Figura 1).
Figura 1. Estudio IDEA. Prevalencia de la Obesidad Abdominal en el mundo

Algunos autores consideran que el almacenamiento disfuncional de energía del obeso
es el punto clave para el desarrollo del SM. Según esta teoría, la RI es consecuencia de
alteraciones en el procesado y almacenamiento de ácidos grasos y triglicéridos (TG)
(moléculas básicas de reserva energética).
La tendencia fisiológica es el almacén de TG en adipocitos pequeños periféricos, pero
cuando la capacidad de estas células se sobrepasa, se acumulan en el músculo y causan RI a
la insulina de dichos tejidos (42,43).
El aumento del tejido adiposo intraabdominal o visceral provoca un aumento del flujo
de AGL hacia la circulación esplácnica, mientras que los derivados del tejido subcutáneo
evitan el paso hepático y sus consecuencias (aumento de la producción de glucosa, síntesis
de lípidos y secreción de proteínas protrombóticas).
También se ha comprobado que el depósito patológico puede realizarse en adipocitos
periféricos anormalmente grandes, como se demuestra en un estudio realizado en indios
pima. El efecto del tamaño del adipocito en el riesgo del desarrollo de DM parece ser
independiente y aditivo al efecto de la insulinorresistencia(44).
Los síndromes lipodistróficos constituyen un buen ejemplo de las consecuencias de la
incapacidad de almacén del exceso de TG en los depósitos fisiológicos. Como consecuencia,
en estos individuos se producen hipertrigliceridemias severas, hígado graso y DM. Del
mismo modo ocurre en los pacientes infectados por el VIH en tratamiento con inhibidores de
la proteasa, que muestran algunas características del SM.
El tejido graso, se considera un órgano endocrino dinámico que secreta multitud de
mediadores de la inflamación y la inmunidad conocidos como “adipoquinas”. La
disrregulación de esta secreción de adipoquinas, producida sobre todo en el tejido graso

visceral, más que en el subcutáneo, es un factor determinante para desencadenar la
resistencia a la insulina, incrementando así el riesgo de diabetes y enfermedades
cardiovasculares en el Sd. Metabólico (45).
1.1.3 Obesidad y Resistencia insulínica en el Síndrome Metabólico
Tradicionalmente, se ha considerado como hipótesis fisiopatológica subyacente al
Síndrome Metabólico(SM) la Resistencia a la insulina (RI), que se define como un defecto en
la acción de la insulina, en los tejidos periféricos, con descenso en la captación y utilización
de glucosa(46), que provoca aumento de la insulina basal para mantener la glucemia en un
rango normal. (Figura 2)
Adaptado de Deprès J .P., Ann.End. 2000
1. ↑lipólis is →↑secreción AGL →↑flujo de AGL hacia la vena porta →↓degradación hepá tica de la insulina →↑INS ULINE MIA
2. ↑oxidación de lípidos →↑neog lucogénes is →↑g lucosa hepá tica →INTOLE R ANCIA A LA GLUCOS A
3. ↑INS ULINE MIA compensa toria pa ra mantener la g lucemia norma l
5.1. TAV e híg ado: ins tauración de la insulinorres is tencia
↑ Glucos a hep(Intolerancia a la g lucosa)
↑Insulina(Hiperinsulinemiacompens atoria)
↑AGL
Acumulaciónimportantede tejido
adipos o vis cera l
↑LH
Vena porta
Ins tauración de la insulinorres is tencia
Figura 2. Adaptado de Deprès J.P., Ann. End. 2000
La RI , considerada el trastorno base común de todos los componentes del SM(47),no es
tan sólo una alteración del metabolismo glucídico sino que está relacionada con un trastorno
en la función del tejido adiposo, asociado en la mayoría de los casos con un acúmulo excesivo

del mismo. Los principales reguladores del metabolismo de los ácidos grasos son: el tejido
adiposo, el hígado y el músculo esquelético(48) .
La Resistencia a la insulina, tiene especial relevancia en estos tres órganos(49):
1.-En el tejido adiposo. El tejido adiposo, más allá de un mero depósito de reserva de
combustible, es un verdadero órgano endocrino, que produce una serie de sustancias
como las adipoquinas, como son: Factor de necrosis tumoral alfa (FNT-α), Leptina,
resistina, visfatina, Interleukina-6 (IL-6), Factor inhibidor de la activación del
plasminógeno (PAI-1), etc… Todos ellos promueven la instauración de un estado
proinflamatorio crónico, protrombótico y de resistencia a la insulina en el SM(50).
Asimismo produce adiponectina(51,52), que es una citoquina antiinflamatoria exclusiva
de los adipocitos. La disrregulación de esta secreción de adipoquinas, producida sobre
todo en el tejido graso visceral, más que en el subcutáneo, es un factor determinante
para desencadenar la resistencia a la insulina, incrementando así el riesgo de diabetes
y enfermedades cardiovasculares en el Sd Metabólico (53). (Figura 3)
Figura 3. Gelfand EV et al, 2006; Vasudevan AR et al, 2005

El tamaño de los adipocitos en el riesgo del desarrollo de DM, también parece
jugar un papel relevante, independiente y aditivo al efecto de la
insulinorresistencia(54).
En estado normal la insulina tiene efecto lipogénico, favoreciendo el acúmulo
de ácidos grasos en los adipocitos, pero en presencia de resistencia a la insulina hay
más efecto lipolítico y se liberan más ac. grasos vía portal que van en mayor cantidad
al hígado. Al desarrollarse la RI, aumenta la liberación de AGL en el tejido adiposo que,
a su vez, inhiben los efectos antilipolíticos en la insulina.
2.-En el hígado. Se produce un aumento en la liberación de glucosa
(gluconeogénesis), un aumento en la síntesis de triglicéridos y lipoproteínas de baja
densidad (VLDL y LDL), favorecida a su vez por la llegada incrementada de ac.grasos
procedentes del tejido adiposo, así como una disminución en la producción de HDL.
3.-En el músculo esquelético. La tendencia fisiológica es el almacén de TG en
adipocitos pequeños periféricos, pero cuando la capacidad de estas células se
sobrepasa, se acumulan en el músculo y causan RI en dichos tejidos (55). Se produce un
acúmulo de grasa en el músculo, que será utilizada como fuente de energía,
disminuyendo así el consumo de glucosa y contribuyendo a un incremento de la
glicemia, que sumada a la sobreproducción hepática conduce a un hiperinsulinismo. En
el músculo, en pacientes resistentes a la insulina, obesos y con diabetes mellitus tipo 2
se han encontrado defectos intracelulares en la fosforilación oxidativa de las
mitocondrias que se relacionan con la ocupación de las vías metabólicas por los lípidos,
llegando incluso a su acumulación en forma de TG.
El mecanismo concreto por el cual la obesidad central provoca resistencia hepática a la
insulina no está del todo definido pero existen varias hipótesis que lo podrían explicar:

• Hipótesis portal, apunta a que un incremento en la entrega de ácidos grasos
libres directamente desde el tejido adiposo visceral a través de la vena porta al
hígado provocaría la resistencia hepática a la insulina(56). El aumento del tejido
adiposo intraabdominal o visceral provoca un aumento del flujo de AGL hacia
la circulación esplácnica, mientras que los derivados del tejido subcutáneo
evitan el paso hepático y sus consecuencias (aumento de la producción de
glucosa, síntesis de lípidos y secreción de proteínas protrombóticas).
• Hipótesis endocrina, consistiría en considerar la secreción de adipoquinas
como las responsables de la inducción de la resistencia hepática a la insulina.
• Hipótesis inflamatoria. Actualmente se tiende a considerar el SM como un
“proceso inflamatorio de bajo grado” debido a la alteración analítica que
presentan estos pacientes con elevación de citoquinas proinflamatorias:
Interleukina-6 (IL-6), factor de necrosis tumoral alfa (TNF-α) (57) y factor
inhibidor de la activación del plasminógeno (PAI-1):
*La interleukina-6 (IL-6), es un importante marcador de infección y de
estados inflamatorios. Se ha encontrado una considerable secreción de
IL-6, a nivel de vena porta, en pacientes obesos, proveniente de la
grasa visceral y en estrecha correlación entre IL-6 y los niveles de PCR
sistémica(58)
*El factor de necrosis tumoral alfa (FNT-α) es sintetizado por el
músculo esquelético y cardiaco, así como por los adipocitos. Tiene dos
funciones primordiales, por un lado en el adipocito inhibe la actividad
de la lipoproteinlipasa y por otro, activa la insulina mediante
fosforilación del receptor de ésta. El FNT-α induce resistencia a la
insulina de las células endoteliales(59)

Se ha comprobado en varios estudios que la sensibilidad a la insulina
se mejora con la neutralización, mediante tratamiento con anticuerpo
FNT-α monoclonal Infliximab, en pacientes con artritis reumatoide o
espondilitis anquilosante.(60,61) Esto nos indicaría, aunque fuese sólo en
parte, la responsabilidad de esta adipoquina en el estado de
resistencia a la insulina. Aunque otros estudios no reflejan los mismos
resultados(62)
*El factor inhibidor de la activación del plasminógeno (PAI-1), es una
proteína reguladora de la fibrinólisis, la migración celular, la
angiogénesis y la remodelación de los tejidos(63). El tejido adiposo,
concretamente la parte del estroma vascular de la grasa visceral(64,65),
es una fuente importante de producción de PAI-1. Se ha podido
demostrar en ratones que la deficiencia de PAI-1, inducida por la dieta,
mejora el perfil metabólico, atenúa la obesidad y la resistencia a la
insulina(66)
La elevación de los marcadores proinflamatorios, no se da en todos los casos; ya que
hay obesos con marcadores normales y delgados con los marcadores elevados. En estos
casos se ha sugerido la existencia de cierta predisposición genética que favorecería una
optimización energética con una ingesta mínima, denominada genotipo ahorrador(67). Esta
predisposición genética, muy útil en tiempos de escasez de alimentos, en épocas de gran
abundancia y poco ejercicio como la actual, favorecería el desarrollo de la obesidad, la
diabetes mellitus tipo 2 y el SM.
La proteína C reactiva (PCR), es un marcador de inflamación que aumenta en el
plasma en estados de inflamación crónica subclínica. El incremento de PCR produce una

disminución en la angiogénesis, favorece la apoptosis de las células endoteliales con
disminución en la supervivencia y diferenciación de las células endoteliales progenitoras e
incrementan la expresión endotelial de moléculas de adhesión. Para su medición se analizan
los niveles de proteína C reactiva ultrasensible (PCR-us). Estos niveles elevados en la PCR-us
se asocian fuertemente con enfermedades relacionadas con el estilo de vida y el SM (68)
Aunque parece evidente que la resistencia a la insulina es la alteración clave asociada
con el perfil de aterogénesis, protrombosis e inflamación, cada día hay más observaciones
que señalan, como el factor común más habitual entre todos los pacientes con síndrome
metabólico, es la obesidad abdominal, especialmente la adiposidad visceral abdominal.
Se ha visto que tras la eliminación de cantidades importantes de grasa subcutánea
mediante liposucción, no se han logrado mejorar los parámetros de SM(69); en cambio se ha
podido advertir en un ensayo en pacientes a los que se realizó una exéresis de grasa
intraabdominal por laparoscopia, el efecto positivo, a largo plazo, de esta extirpación de grasa
visceral, sobre el SM(70)
1.1.4 Alteraciones del Metabolismo Lipídico en el Síndrome Metabólico
La dislipemia en el SM se caracteriza por elevación de los triglicéridos (TG) y
lipoproteínas de muy baja densidad (VLDL), descenso de lipoproteínas de alta (HDL) y
aumento de las lipoproteínas de baja densidad (LDL) pequeñas y densas, lo que se ha
denominado fenotipo lipoproteínico aterogénico(71) . En cambio, las cifras de colesterol total
y colesterol LDL no suelen estar aumentados, pero este último está constituido por partículas
más pequeñas y densas llamado “patrón de LDL tipo B”. De hecho, la mayoría de pacientes

con SM y TG por encima de 150 mg/dl, las LDL predominantes corresponden al fenotipo
“B”(72)
Entre las alteraciones en el perfil lipídico también podemos encontrar las siguientes:
• Aumento en la concentración de Apolipoproteína B (Apo-B) (proteína del col-
LDL) por encima de los 120 mg/dl
• Aumento del colesterol transportado en partículas remanentes.
• Aumento en la actividad de las enzimas CETP (proteína transferidora de
esteres de colesterol).
• Aumento de la Lipoproteinlipasa
En la práctica clínica habitual sólo se recomienda para el seguimiento y control del SM,
la medición del colesterol-HDL y los TG.(73)
En el metabolismo lipídico normal se produce una liberación de ácidos grasos libres
(AGL) desde los adipocitos a la sangre circulante, hacia el hígado y el músculo. En el hígado,
una parte es oxidada y la mayoría reesterificada a TG. Hay un transporte continuo de AGL
entre tejido adiposo e hígado; sin embargo, si el proceso de reesterificación se satura, la
acumulación de TG puede conducir al hígado graso.
En los pacientes con obesidad central presentan un acúmulo de grasa visceral, mayor
que subcutánea. Esta grasa visceral está relacionada con la resistencia a la insulina y el SM. El
mecanismo que induce a la resistencia la insulina en los pacientes con acúmulo de grasa
visceral parece estar mediado por la liberación, por parte de los adipocitos, de adipoquinas
proinflamatorias, como el FNT-α, IL-6, etc…(74)(Figura 4)

Figura 4. Trayhurn P. Endocrine and signalling role of adipose tissue: new perspectives on fat. Acta Physiol Scand.
2005 Aug;184(4):285-93.
En presencia de insulinorresistencia, el exceso de grasa visceral produce un hiperaflujo
de AGL al hígado y como consecuencia un aumento de la síntesis de TG y de VLDL ricas en TG y
apo B. Este hecho explicaría la hipertrigliceridemia(75) que presentan estos individuos. Sin
embargo en condiciones normales, la insulina inhibe la secreción de VLDL a la circulación. En el
tejido adiposo y en el músculo se produce un descenso de la actividad lipoproteinlipasa (LPL),
por lo que no se aclaran los TG de las VLDL y favorece la acumulación de lipoproteínas de
densidad intermedia (IDL) y LDL. La vida media de dichas partículas se alarga, favoreciendo su
exposición a la CETP (cholesteryl ester transfer protein) .
Los TG de las VLDL se intercambian con ésteres de colesterol en las HDL por acción de
la CETP(76)y la mayoría de dichos ésteres vuelven al hígado en forma de remanentes, una vez

que se hidrolizan las VLDL por la LPL. Diversos estudios han encontrado una relación
inversamente proporcional entre la disminución de CETP en el plasma y el incremento de
eventos cardiovasculares(77-79)
Las HDL pequeñas son aclaradas de la circulación con mayor facilidad que sus
homólogas, de lo que resulta una disminución del HDL y de la apo A1 (ambas
antiaterogénicas).
Las LDL pequeñas y densas también son más aterogénicas por ser más tóxicas, debido
a su mayor capacidad de penetración en la íntima y buena adherencia a los
glucosaminoglicanos y por su mayor susceptibilidad a la oxidación y su unión selectiva a los
receptores basureros de los macrófagos.
1.1.5 Hipertensión Arterial y Riesgo Cardiovascular en el Síndrome Metabólico
La extraordinaria importancia clínica del SM y de su detección precoz radica, no sólo
en su prevalencia cada vez mayor, sino en el elevado riesgo cardiovascular que conlleva.
Es conocida desde años la relación existente entre hiperinsulinismo e hipertensión
arterial (HTA)(80-84). Pero aunque no siempre los pacientes hipertensos desarrollan resistencia
a la insulina (RI), ni todos los pacientes con RI son hipertensos(85), se ha podido conocer que
la insulinorresistencia y el hiperinsulinismo son factores predictivos para el desarrollo de
HTA, sobre todo cuando forman parte del SM(86-90) .
La insulina posee acciones vasomotoras(91):
• Por un lado, efectos vasopresores:

Estimula el sistema nervioso simpático por hiperactividad del eje hipotálamo-
hipofisario-adrenal con aumento de intercambio Na+/H+ y un incremento de la
reabsorción tubular de Na+(92)
Aumenta la respuesta a la angiotensina II
La insulina estimula la bomba Na+/K+-ATPasa (encargada de mantener el
balance de K+ intra y extracelular) y también regula la bomba Ca++-ATPasa
(que mantiene el Ca++ intracelular). Cuando esta bomba de calcio falla debido
a la resistencia a la insulina facilita la acumulación de calcio intracelular,(93) lo
cual facilita una hiperrrectividad vascular, contractura de la musculatura lisa de
la pared vascular, aumento de resistencias periféricas, con la consecuente
hipertensión y calcificaciones arteriales.
• Por otro lado produce vasodilatación, mediante la estimulación de la
producción endotelial de óxido nítrico(94).
En el sujeto sano los efectos vasopresores y dilatadores se compensan, pero en
estados patológicos como la obesidad y el SM, el equilibrio puede romperse y lo suele hacer
hacia el lado presor, por el incremento de la actividad simpática debido a la
hiperinsulinemia(95).
En un meta-análisis reciente en el que se incluían la mayoría de estudios
epidemiológicos prospectivos sobre pacientes diagnosticados de SM, se constató que el SM
multiplica por dos el riesgo de padecer una enfermedad cardiovascular, aumentando hasta el
50% la mortalidad por todas las causas, incluso excluyendo a los casos de diabetes o a los
sujetos con enfermedad cardiovascular previa(96).

1.1.6 Otras alteraciones clínicas asociadas al Síndrome Metabólico
• Síndrome del ovario poliquístico (SOP). El SOP es la patología endocrina más
frecuente entre las pacientes jóvenes. Afecta a un 6-10% de las mujeres en
edad fértil y cursa con obesidad, anovulación crónica e hiperandrogenismo. El
SOP comparte con el SM el mismo mecanismo fisiopatológico de resistencia a
la insulina(97), por lo cual presentan sintomatologías superponibles. La mayoría
de las pacientes con SOP además de sus alteraciones en el área de la fertilidad,
presentan alteraciones a nivel metabólico y cardiovascular: obesidad,
resistencia a la insulina, intolerancia a la glucosa, dislipemia e hipertensión. Se
ha observado que la pérdida de peso en estas pacientes mejora sus ciclos
menstruales, su fertilidad y también los parámetros diagnósticos del SM(98).
• Infertilidad masculina.
• Hiperuricemia debido a la disminución en la eliminanción de ácido úrico.
• Microalbuminuria.
• Esteatosis hepática no alcohólica. El hígado se ve afectado en el SM por un
exceso de aporte de ácidos grasos, debido al aumento de actividad de los
adipocitos de la grasa visceral(47), que saturan la capacidad del hígado para
poder procesarlos. La prevalencia de la esteatosis hepática en el SM se sitúa
alrededor del 55-70% de los casos.
• Síndrome de Apnea del Sueño, se asocia habitualmente con el sobrepeso, la
resistencia a la insulina y las dislipemias. Se ha podido comprobar que el
tratamiento de la apnea con CPAP (presión positiva continua en la vía aérea)
mejora los componentes metabólicos del SM(99).

1.1.7 Alteraciones de la Señalización Membranaria en el Síndrome Metabólico
La Leptina, producida por los adipocitos diferenciados, controla la ingesta alimentaria
mediada por el SNC, de forma que a más cantidad de Leptina, más saciedad y así menor
ingesta alimentaria. Pero a su vez, a más masa grasa visceral, más cantidad de Leptina y dado
que pertenece a la misma familia de los receptores de las citokinas, provoca mayor estado
inflamatorio. Es por ello, que el acúmulo de Tejido Adiposo Visceral en exceso provoca una
disfunción celular, mediada por dos mecanismos: alteración de la señalización membranaria
celular y fenómenos de glicación de proteínas. La inflamación debida al aumento de la Masa
Grasa Visceral, genera una rigidez de la membrana adipocitaria que provoca a nivel de sus
receptores una comunicación celular inadaptada con respuestas exageradas sobre los
receptores membranarios (Figura 5):
§ Receptores de Insulina → Insulinorresistencia (100,101 )
§ Receptores de Leptina → Lep�norresistencia (102,103)
§ Receptores adrenérgicos → ↓ lipólisis y ↑ lipogénesis (104,105)
Figura 5. Ahima RS, Saper CB, Flier JS, Elmquist JK. Leptin regulation of neuroendocrine systems. Front
Neuroendocrinol. 2000 Jul;21(3):263-307
Los receptores de insulina y de las vías de señalización de otros receptores están muy
próximos en la superficie de la membrana, así que las prostaglandinas inflamatorias afectan

de manera aleatoria a ambos. Las quinasas activan entonces los procesos de inflamación a
nivel de membrana.
Existe también una glicación no enzimática de las proteínas ó Reacción de Maillard(106),
donde son generados los Productos Terminales de Glicación (PTG), en los que ocurre la
interacción en presencia de calor, entre azúcares (fructosa, glucosa…) y grupos de
aminoácidos, generando éste proceso macromoléculas por reacción de enlaces cruzados.
Esta reacción la encontramos siempre en presencia de hiperglicemias, donde éstos PTG y en
presencia de éstos glúcidos se transforman en los Advance Glycation End products (AGE)(107)
siendo muy reactivos e impidiendo los intercambios entre vasos sanguíneos y células,
provocando finalmente vasoconstricción e impermeabilidad vascular.
1.1.8 Alteraciones de los Genes Reloj en el Síndrome Metabólico
La ingesta de alimentos, constituye un fenómeno cíclico en la mayoría de los animales,
habiéndose descrito ritmos de diferentes frecuencias. En los humanos, los ritmos de ingesta
de alimentos están condicionados por factores de índole cultural, sin embargo la relativa
estabilidad de esos ritmos entre culturas, refuerza la existencia de fuertes condicionantes
biológicos. Así, que se han descrito ritmos circadianos, en la mayoría de los factores
neuroendocrinos implicados en la regulación de la ingesta(108).
El que un determinado nutriente no sea igualmente aprovechado metabólicamente
según las horas del día en que es ingerido, es un hecho ampliamente demostrado. Los ritmos
de apetito, digestión, absorción y actividad enzimática son responsables de ello. Los horarios
de comida, influyen en factores tan variados como el incremento del peso, glucemia,
intolerancia a la glucosa, trigliceridemia, riesgo cardiovascular(109), etc. Los núcleos

supraquiasmáticos del hipotálamo son los encargados del mantenimiento del orden
temporal interno relacionado con los ritmos de ingesta y metabolismo de los nutrientes(110),
mediante la activación de las vías simpáticas y parasimpáticos (Figura 6) que actúan
selectivamente sobre los diferentes compartimentos del organismo(111,112) y mediante la
producción rítmica de señales humorales como la melatonina, hormona pineal cuya
concentración plasmática aumenta durante la noche en todas las especies, induciendo los
cambios metabólicos propios del periodo nocturno.
Figura 6. Buijs RM, Kreier F. The metabolic syndrome: a brain disease? J Neuroendocrinol. 2006
Sep;18(9):715-6
La ritmicidad circadiana no sólo es el resultado de la actuación del marcapasos central
hipotalámico, sino que también es el resultado de la actividad oscilatoria de varios tejidos
periféricos como consecuencia de la actividad de sus propios genes reloj(113). Así, hoy sabemos
que el mismo tejido adiposo posee genes reloj generadores de ritmos circadianos en su
funcionamiento. Esto no es raro, dado que el mismo tejido adiposo, genera una serie de

secreciones endocrinas que realizan funciones importantes y no es sólo un almacén de
energía. Las más importantes de estas secreciones en nuestra causa a estudio, son la leptina,
adiponectina y resistina, también llamadas adipoquinas, que no sólo muestran amplios ritmos
circadianos en sus concentraciones plasmáticas, sino que su ritmicidad se ve fuertemente
atenuada ó abolida en individuos obesos. Estas adipoquinas constituyen un punto de unión
molecular entre el aumento de adiposidad y el desarrollo de diabetes tipo II y/o el SM(114)
Hasta el momento los genes implicados en el tejido adiposo estudiados por Ando y
col. en 2005 (Dpb, Per1, Per2, Bmal1, Cry1 y Cry2) muestran ritmos circadianos significativos en
su expresión en el tejido adiposo del ratón. Las concentraciones de Per 1 y Per 2 muestran un
pico en la última parte del día y en Bmal1 una disminución. Sin embargo la expresión de estos
genes por el tejido adiposo pueden verse alteradas en ciertas situaciones patológicas como
ocurre en ratones diabéticos y obesos.
Recientemente se ha encontrado que el ratón con el gen Clock mutante, es hiperfágico
y obeso, desarrollando además un SM consistente en hiperlipidemia, hiperglicemia e
hiperleptinemia. La ritmicidad de la expresión de los genes reloj, puede ser muy importante
para la preservación de la salud, al igual que la expresión atenuada de los mismos puede
causar también Diabetes Tipo II.
Así, este modelo de ritmicidad circadiana, se basa en bucles autorreguladores (115,116) y
los gestionan por ello dos estructuras proteicas:
• Clock y Bmal1, que genera un bucle de retroacción positiva, activando la transcripción
de otros genes reloj.
• Cry y Per, que forman parte del bucle de retroacción negativa, inhibiendo la
transcripción.

Es por ello, que éstas señales rítmicas de salida, van a ser utilizadas por el reloj
supraquiasmático para distribuir mensajes temporales al cerebro y la periferia (Figura 7).
Figura 7. Green CN, Takahashi JS, Bass J, Cell. 2008, 134(5):728-42. Genes implicados en las señales circadianas de
salida celular y sus efectos periféricos.
Con la ayuda de éstas señales, los tejidos son preparados de manera orquestada para
los periodos de actividad ó de reposo y por ende, las alteraciones de la fisiología normal de
dichas señales, van a ser capaces de producir alteraciones metabólicas y enfermedad en el
tiempo. Así por ejemplo, el Infarto de miocardio, la muerte súbita, la insuficiencia cardiaca,
debido al aumento del factor protrombótico PAI-1, suelen sobrevenir a primera hora de la
mañana (papel heterodímero Clock-Bmal1). (Figura 8)

Figura 8. Organización molecular de los relojes central y periféricos.
Así mismo, según la señal de salida existente a nivel celular, resultante de la
interacción de los genes reloj y las intromisiones existentes en los sincronizadores orgánicos
y celulares según la patología previa que esté presentando el individuo, van a poder provocar
enfermedad por desincronización de su fisiología básica compensada. De esta manera,
podremos ver por ej. que si hay una alteración de desincronización a nivel del reloj
hipotalámico, puede desencadenar alteración de la percepción de la saciedad(117); si existe
una desincronización en los relojes linfocitarios, puede provocar inflamación(118); si la
desincronización es del reloj específico cardíaco, puede presentar el individuo enfermedad
cardiovascular(119,120); si el órgano el cual tiene alterada la sincronización de su reloj es el
hígado, entonces podremos encontrar insulinoresistencia y/o esteatosis hepática(121); si la
desincronización el del reloj pancreático, podremos ver así un déficit insulínico y diabetes
posteriormente; si es en el reloj muscular la desincronización, veremos también
insulinoresistencia; y si finalmente la desincronización ocurre en el reloj del tejido adiposo,
veremos obesidad y alteraciones endocrinas varias(122).

Podemos ver así en resumen las anormalidades funcionales del sistema circadiano y
los desórdenes metabólicos que irán apareciendo en el individuo en el tiempo al tener
señales circadianas anómalas y alteradas(123). (Figura 9)
Figura 9. Challet E. Clock genes, circadian rhythms and food intake. Pathol Biol (Paris). 2007 Apr-May;55(3-
4):176-7.

1.2 DIETA DE APORTE PROTEICO
1.2.1 Orígenes
Los antecedentes iniciales de dietas proteicas prácticamente se remontan a los albores
del siglo XX. Fue el Dr. Wilder, quien observó en niños afectos de epilepsia, que no
respondían correctamente a los fármacos existentes en aquellos tiempos , si eran sometidos
a dieta cetogénica(124), o bien disminuían sus crisis comiciales o prácticamente desaparecían
éstas, durante el tiempo en que estaban sometidos a las condiciones cetogénicas de la dieta.
Esto ocurría hacia el año 1921. Posteriormente, durante los años 30´, 40´ y 50´ con el
advenimiento de nuevos fármacos antiepilépticos más eficaces, la dieta proteica empezó a
ser olvidada. Hacia los años 70´, cuando se empezaron a purificar las proteínas comenzó
nuevamente a resurgir. Fué cuando se acuñó el término de Protein Sparing Modified Fast
(PSMF) (125,126), observándose entonces que había una protección de la pérdida de masa
muscular, existente con dietas hipocalóricas al uso y que añadido a la generación de los
cuerpos cetónicos producidos por la dieta proteica, existía un estado de bienestar que hacía
soportar la ingesta diaria de cómo mucho 50 gr. de hidratos de carbono al día, sin padecer
hambre. Fué a partir de aquí y mediante un seguimiento médico estricto, que la generación
de las proteínas cada vez más purificadas y sometidas al proceso de cracking ó rotura
proteica, mejoraron enormemente los aminogramas de los productos al uso.
La Dieta Proteica no es un invento actual ni una última moda. Hacia finales de los 70´
empezó a ser utilizada médicamente en los deportistas de élite, para optimizar su
musculatura y perder la grasa acompañante que pudieran tener, para así mejorar el
rendimiento en competición.
Está basada firmemente en la tesis doctoral del Dr. Blackburn(127,128), endocrino de la
Harvard Medical School, quien en 1973 determinó la cantidad exacta de proteínas que un

paciente debía ingerir para mantener la masa muscular y a su vez perder única y
exclusivamente grasa.
Posteriormente fue generalizándose su uso, debido a las características antes
descritas, en la medicina deportiva, empleándose inicialmente en los púgiles de boxeo.
Rápidamente se fue extendiendo a otros ámbitos del deporte y al observarse no sólo la
correcta implementación proteica muscular que realizaba, sino también la pérdida de masa
grasa que conllevaba, se trasladó a los tratamientos por sobrepeso y obesidad.
Fue comprobada su eficacia gracias a los buenos resultados obtenidos y actualmente
es el método de adelgazamiento más eficaz, rápido y con óptimos beneficios para la salud
global, física, biológica y psíquica del paciente, siendo en la actualidad cuando su utilidad en
el SM es más que evidente (129).
Este cálculo exacto de la cantidad de proteínas necesarias para mantener la masa
muscular y perder peso sólo a expensas de las reservas grasas, se diseñó para evitar los
problemas de salud por todos conocidos, surgidos con las antiguas y conocidas dietas hiper-
proteicas (Dieta Atkins), en las que el aporte de proteínas se limitaba a ingerir cantidades
ingentes de proteínas de origen animal con su grasa incluida.
Por ello, el Dr. Blackburn en su tesis doctoral estableció las bases de la dieta proteica
tal como hoy día se preconizan, es decir: demostró la necesidad de mantener un balance
calórico negativo, manteniendo un balance nitrogenado equilibrado; que privándonos
limitadamente de los hidratos de carbono, neutralizábamos el efecto anabólico de la insulina
sobre el metabolismo lipídico; que la insulinemia se mantenía baja, pero a niveles
fisiológicos; que este acontecimiento ponía en marcha el catabolismo de los triglicéridos; que
los cuerpos cetónicos generados aportaban al organismo las necesidades energéticas y que la
glicemia se mantenía baja, pero en niveles fisiológicos.

A partir de entonces se han ido mejorando los aminogramas de los productos
utilizados, al igual que se han logrado producir alimentos proteicos diversos para poder llevar
a cabo la dieta sin encontrar a faltar ningún tipo de gusto ni textura.
1.2.2 Concepto
Dentro de las dietas hipocalóricas, merecen nuestra especial atención desde el punto
de vista del síndrome metabólico (SM), las denominadas dietas proteicas, dietas proteinadas
o dietas de aporte proteico (DAP), englobadas dentro del tipo de dietas VLDC (de muy bajo
contenido calórico)(130) donde el componente proteico nutricional es una proteína
manufacturada de características químicas excelentes, por la composición de su
aminograma, compuesto de todos los aminoácidos esenciales y por otro lado el aporte
disminuido de los otros macronutrientes, los lípidos y los glúcidos.
La DAP, provoca una disminución de la masa grasa sin menoscabar la masa magra, con
lo que nos proporciona un mantenimiento del metabolismo basal y por ende una capacidad
de mantenimiento del peso en el tiempo mucho más acorde que otras dietas hipocalóricas
tradicionales, donde existe una disminución de la masa muscular y a partir de ahí el rebote
calórico en muchas de las ocasiones.
De hecho, el estudio que hemos llevado a cabo sometiendo a una cantidad sesgada de
pacientes afectos de SM al tratamiento con DAP, es para evaluar dicha disminución de grasa
intrabdominal, como génesis primaria y acompañante de los factores de peor pronóstico
cardiovascular y a su vez, proferirle con dicho método una capacidad de mantenimiento
correcto de dicha grasa intraabdominal, con la base de un mantenimiento en su metabolismo
basal, mediante la preservación de su masa muscular junto a la incentivación de ejercicio
moderado y de estrategias ritmonutricionales basadas en la cronobiología alimentaria.

Refiriéndonos a la DAP, el procedimiento se basa en la ingesta de una serie de
preparados proteicos de diferentes gustos, texturas y densidades, que se ingieren desde el
inicio del programa de disminución ponderal, asociados a verduras y en sucesivas fases a
proteínas animales, siendo en ésta fase inicial del proceso donde se pierde la mayor parte del
peso corporal deseado. Posteriormente a la consecución de dicho peso es necesario
reintroducir los alimentos de forma programada y escalonada en cuanto a la progresión del
índice glicémico de los alimentos, para evitar la hiperinsulinización y posteriormente
proceder a la estrategia de mantenimiento que comentaremos más adelante.
Presentan en general una composición de 15 a 18 gr. de proteína por producto,
destinados a la pérdida de peso bajo control médico, imprimiéndoles desde el primer
momento un enfoque cronobiológico de estabilización del peso. Son proteínas de alto valor
biológico que contienen todos los aminoácidos esenciales por producto y en las proporciones
adecuadas al ser humano, con un índice químico de 100 ó superior lo que comporta su fácil
absorción, metabolización y biodisponibilidad orgánica.
La procedencia de éstas proteínas es múltiple, proviniendo del mundo animal y del
vegetal. Comúnmente son caseinatos y lactoserum de leche, proteínas de la clara del huevo,
del guisante, de la soja e incluso de la alfalfa.
Las recomendaciones de la OMS en cuanto a las necesidades proteicas del ser humano,
hacen sostenible definir a la DAP como dieta no hiperproteica sino euproteica, ya que dichas
cantidades están en línea de la alimentación proteica actual de las sociedades
industrializadas.

§ Necesidad de Aporte de proteico diario *
♀: P (peso en g) = 1.2 * 25 * T² (en metros)
♂: P (peso en g) = 1.5 * 25 * T² (en metros)
* según las recomendaciones de la OMS y la AFSSA-2008
En cuanto a las cantidades de lípidos que contiene, es entre unos 10 a 20 gr. de aceite
de oliva diario, necesario para poder evitar una eventual colelitiasis. Esto implica como
hándicap inicial y debido a que las grasas forman parte de la lubricación fisiológica del tubo
digestivo, una tendencia a la constipación intestinal. Por ello la recomendación insistente al
paciente de promover un consumo de verduras importante junto con una ingesta hídrica de
no menos de 2 l de agua diarios, que junto a la fibra vegetal minimizan dicho problema.
Los aportes de glúcidos deben estar muy controlados y por ello, no deberemos de
sobrepasar la ingesta de 50 gr. al día (entre los productos proteicos y los vegetales). Por
encima de estos aportes, revertiría la B Oxidación de los ácidos grasos libres (AGL) y el
paciente saldría de la cetosis autoinducida, como veremos en el apartado de fisiología.
Además, nos asegura una glicemia e insulinemia correctas, que aunque en límites bajos son
fisiológicamente muy bien toleradas.
Así mismo y una vez con la proporción de macronutrientes necesarios para inducir la
cetosis, es preciso hacer una complementación exacta de los déficits inherentes generados a
la composición del producto proteico preformado y al estado cetósico inducido durante el
tratamiento, es decir que deberemos suplementar con Cloruro Sódico, Potasio, Vitaminas y
Minerales, Magnesio, Calcio y Acidos Grasos Omega3/6.(131,132)

1.2.3 Fisología
Al iniciar una dieta hipocalórica como la DAP, que está dentro del grupo de las VLDC
(very low diet calory) y representa en su fase más estricta un aporte de unas 700kcal.
ocurren tres fenómenos progresivamente: las reservas glucídicas del glucógeno intrahepático
principalmente, se agotan en las primeras 24 a 48 h. Nunca hay utilización de la proteína
orgánica ni muscular debido a la utilización por el organismo de las proteínas purificadas,
hecho importantísimo para poder mantener adecuadamente el metabolismo basal (133)y no
inducir como la gran parte de dietas hipocalóricas al efecto rebote de las mismas por
disminución inherente del metabolismo.
Al tener una restricción glucídica controlada, permite al organismo iniciar el
catabolismo de los triglicéridos: la lipasa, activada por la APO CII en los capilares hidroliza los
triglicéridos en Glicerol y Acidos Grasos Libres (AGL), el 40% de los cuales son directamente
consumidos por el músculo y el 60% restante son B Oxidados en Hígado y transformados en
Acetil CoA. Por cada dos moléculas de Acetil CoA que se condensan, generan el Acido Acetil
Acético que será transformado en los Cuerpos Cetónicos, Acetona y Acidos Alfa y Beta
Hidroxibutírico, siendo éste último el que provoca la activación del centro hipotalámico de la
saciedad y a su vez el efecto psicotónico de vitalización durante el tratamiento.
Las moléculas de Acetil CoA, penetran en el Ciclo de Krebs donde son oxidadas y
generan el 75% de la energía utilizada por los órganos vitales. El 25% restante de la energía
necesaria por el organismo, proviene de la Neoglucogénesis hepática y renal básicamente.
(Figura 10)

HÍGADO
RIÑÓN
VÍAS METABÓLICAS DE LA DAP↓ APORTE GLUCÍDICO TOTAL
< 50 g/día
Hipoglucemia leve
PÁNCREAS S.N.C.
↓Insulina y ↑Glucagón Catecolaminas+Ahorro de glucosa
GLUCOGENOLISIS(unas horas)
Glucosa1º
Tejidos glucodependientesCEREBROERITROCITOSMÉDULA SUPRARRENAL
GlicerolADIPOCITOS
Lipasas
Ác. Grasos Libres
β-Oxidación
Ác. Grasos Libres
β-Oxidación
Ác. Grasos Libres
β-Oxidación
Ác. Grasos Libres
β-Oxidación
Ác. Grasos Libres
β-Oxidación
Ác. Grasos Libres
β-Oxidación
2º NEOGLUCOGÉNESIS(permanente pero insuficiente)
MÚSCULOProtección masa magra
3º CETOGÉNESIS(es constante y suficiente mientras se hace DAP cetogénica)
Acetil Co AAcetona + Ac Hidroxibutírico
ENERGÍA
75%
Glutamina liberada por el músculo
4º NEOGLUCOGÉNESIS
25%
Tejidos no glucodependientes
SaciedadEf. psicotónico
Autorregulación funcional
Pérdida de peso continuada
↑ APORTE PROTEICO
Aminoácidos
Figura 10. Vías Metabólicas de la DAP
Por encima de éstos aportes glucídicos antes señalados, existe una oxidación reversible
del proceso. Así mismo, al estar hablando de la existencia de pacientes con una masa
pancreática de células B correcta, sólo con la existencia de un 20% de ésa mínima necesidad
insulínica, nos va a tamponar el proceso para evitar que nunca podamos entrar en un estado
de cetoacidosis por acumulación de los cuerpos cetónicos.
Las ventajas que nos proporciona la utilización de la DAP, son múltiples: permitir al
hígado la oxidación de gran cantidad de AGL; la producción de un 75% de energía de los
metabolitos generados, los cuerpos cetónicos, que además nos provocan un estado de

saciedad; a su vez, los cuerpos cetónicos pueden ser utilizados por los tejidos que no pueden
satisfacer directamente sus requerimientos energéticos a partir de los AGL.
En consecuencia, el organismo sólo utiliza las reservas grasas para producir la energía
necesaria y provocar una pérdida de peso importante y de características muy volumétricas
por las características de la densidad grasa.
La cetogénesis generada es baja, del orden como máximo de 5 mmol/dl(134),
comparativa a la generada por un deportista después de un esfuerzo físico intenso.
Como todo procedimiento médico, tiene sus contraindicaciones formales, que hay que
testar siempre con anterioridad a su puesta en marcha: embarazo; lactancia e infancia;
estados patológicos graves evolutivos como en el cáncer, insuficiencia renal, cardiaca ó
hepática; infecciones graves; IAM ó AVC de menos de 6 meses del evento cardiovascular;
alteraciones psiquiátricas severas; diabetes mellitus insulino dependiente tipo 1; pacientes
con potasio lábil o no controlado por el motivo fisiopatológico o farmacológico previo
existente.
1.2.4 Beneficios biológicos
Las ventajas funcionales de practicar la DAP, son múltiples: pérdida de peso rápida y de
grasa principalmente; ausencia de hambre; sensación de vitalidad; rápida normalización de
las alteraciones clínicas y biológicas, incidiendo en éste aspecto en las alteraciones
metabólicas del SM; la preservación de la relación entre el gasto energético basal y la masa
magra, en que al preservar dicha masa magra, proporciona el factor vital de estabilización del
peso. Así y siguiendo la ecuación de Boulier para el cálculo del metabolismo basal *(kcal/dia):

Mtb. Basal (Kcal/día) = 21 x Masa Muscular + 500*
De aquí la importancia de proporcionar al organismo un aporte proteico adecuado, así
como la necesidad de realizar ejercicio físico para favorecer el mantenimiento de la masa
muscular, durante la dieta y en el equilibrio alimentario.
Los Beneficios clínicos de practicar la DAP, también son muy importantes para la
mejora de todos y cada uno de los factores pronósticos del compendio sindrómico del SM:
disminuye obviamente por utilización grasa, el volumen de la circunferencia de la cintura
corporal; mejora y/o normaliza en todos los casos las cifras de sufrimiento del hepatocito
debido a la ocupación grasa de él como ocurre en la esteatosis hepática(135-137), demostrables
por la cifra de transaminasas; disminuye las cifras de glicemia y la hemoglobina glicosilada,
procediendo a la normalización de la insulinoresistencia y por ende a la disminución o
abolición farmacológica de los hipoglucemiantes y/o insulina; disminuye las cifras de
colesterol total a expensas casi exclusivamente del LDL y de los TG; provoca la disminución
de las cifras tensionales en los pacientes hipertensos tratados y con ello la disminución de
dosis total de fármacos hipotensores.
Hay otra serie de mejoras biológicas que no van a ser las que tratemos actualmente,
como por ejemplo la abolición de los ronquidos y del temido síndrome de apnea del sueño;
mejoras en el dolor crónico osteoartrósico; y otros muchas comunes a otras formas de
disminución de la masa corporal con otras dietas hipocalóricas, principalmente de
características funcionales.
A destacar también las mejoras estéticas que ocurren con la DAP, como la
esculturización de la silueta corporal, más que con ningún otro sistema de pérdida de peso,
dado que al preservar la musculatura y perder sólo la grasa, provoca una definición excelsa
de los contornos corporales.

1.2.5 Implementación en el Síndrome Metabólico
Siempre y como en todo acto médico, procedemos a realizar una historia clínica
exahustiva que va incluir también una exploración física completa y antropométrica, un
análisis sanguíneo con todos los perfiles biológicos, así como un estudio de la composición
corporal mediante bioimpedianciometría, que en su conjunto nos va a indicar la
conveniencia o no de la práctica de la DAP en el paciente afecto de SM. Ante alguna duda de
la indicación o no, siempre podemos recurrir a la práctica de una exploración
complementaria (Eco Abdominal, ECG…) ó una interconsulta específica.
Tal como sabemos, todos estos pacientes estudiados sufren de SM, es decir además de
tener la circunferencia abdominal medida a la altura de la cintura por encima de los criterios
definidos de 94 cm en el hombre y 80 cm en la mujer, presentan cifras de TG por encima de
150 mg/dl y/o cifras de glicemia en ayunas mayores a 100 mg/dl ó ya es conocido como
diabético tipo II y/o cifras de HDL menores de 40 en el hombre ó menores de 50 en la mujer
y/o cifras de tensión arterial por encima de 130 mm sistólica y/o 85 mm diastólica. En cada
uno de estos casos han de darse 2 ó más criterios de los señalados, siempre asociados al
diámetro alterado de cintura. También a tener presente que muchos de éstos pacientes
tenían cifras normales de TG, HDL, Glicemia y T. Arterial al estar tratados
farmacológicamente para ello. Aún así, son considerados criterios positivos para
diagnosticarlos de SM.

FASE ACTIVA(Estricta F1 + Mixtas F2, F3)
FASE DE REINTRODUCCÍÓN.
ALIMENTARIA(Fases de Transición
F4, F5)
FASE DE EQUILIBRIO ALIMENTARIO(Mantenimiento F6)
L L L L
3. FASES en laDAP-Ritmonutrición®
cetosis no cetosis
stop cetosis
Figura 11. Fases de la DAP
A partir de aquí, definimos una serie de fases cetósicas por donde podemos empezar el
tratamiento, que serían la 1, 2 y 3. (Figura 11) En estas fases hemos de aplicar un criterio de
cronobiología alimentaria con 5 ingestas diarias. Iniciamos el tratamiento habitualmente por
la Fase 1, que consta de 5 ingestas al día de proteínas purificadas e ingestas de vegetales en
comida y cena. Dichos vegetales pueden ser sin límite de un grupo donde su carga glicémica
es de unos 3 gr de hidratos de carbono por cada 100 gr. de vegetales y otro grupo, con una
carga glicémica de unos 6 gr de hidratos de carbono, siendo éste último limitada su ingesta a
200 gr/día pesadas en crudo, pudiendo ser mezclados vegetales de ambos grupos. Esta fase
se ve continuada por las Fases 2 y 3, a demanda de la rapidez del proceso, necesidades
sociales, familiares, laborales o sencillamente de tolerancia psicológica a dicho tratamiento.
Es decir que podían pasar a realizar la Fase 2 ó 3, donde existían entonces 4 y 3 productos de
proteína purificada diaria respectivamente, al substituir uno ó dos de ellos en comida y/o
cena por proteína animal. Fueron unos 150 gr. de carnes y/o 200 gr de pescados, mariscos,
crustáceos ó 2 huevos. Existía siempre una limitación de lípidos en base a 1 a 2 cucharadas
FASES EN LA DIETA DE APORTE PROTEICO
PROTEICO

soperas de aceite de oliva, siempre para aderezar. En éstas fases cetósicas es cuando la
pérdida grasa subcutánea y visceral fue más rápida, así mismo y directamente relacionado
con las mejoras biológicas y clínicas presentadas.
Las Fases 4 y 5 son no cetósicas y se comportan como una dieta hipocalórica normal,
donde el aporte de proteína purificada está ubicada sólo en media mañana y media tarde y
responden a fases denominadas de reintroducción alimentaria donde el aporte de la carga
glucídica introducida va de menos a más, evitando siempre los hidratos de carbono
refinados, ayudándonos a evitar el incremento insulínico rápido y por ello una menor
sensación de hambre posterior. En la fase 4 introducimos un desayuno con pan integral con
queso, jamón o huevos y además una pieza de fruta asociada a la proteína purificada de la
tarde; en la fase 5 además añadimos legumbres y/o cereales integrales a la hora de la
comida, incorporando ya cualquier tipo de verdura u hortaliza, más una pieza de fruta de
postre en la comida.
Durante las fases cetósicas del proceso es donde el paciente siente saciedad y
vitalización, mientras practica una pauta alimentaria de bajas calorías pero muy fácilmente
sostenible.
En las fases no cetósicas aún persiste la sensación de vitalización y saciedad, aunque
menor, pero permite continuar fácilmente el proceso hasta la fase final de equilibrio
alimentario ó de mantenimiento.
Tanto en las fases cetósicas como en las no cetósicas fue necesario suplementar los
requerimientos básicos para el funcionamiento celular y la homeostasis interna(138,139), dado
que la individualización del proceso así lo requiere según los parámetros biológicos
detectados, las patologías previas conocidas en la anamnesis y las variaciones clínicas que
pudiera presentar durante el proceso. Por ello, debimos prescribir de forma sistemática la

ingesta de suplementos de Cloruro sódico, Bicarbonato Potásico, Vitaminas y Minerales,
Bicarbonato cálcico, Magnesio y Acidos Omega 3/6.
1.2.6 Equilibrio Alimentario
En todos los casos, al acabar la reducción ponderal se procedió a una estrategia de
consolidación alimentaria ritmonutricional y de características cronobiológicas, tal y como en
nuestra práctica clínica habitual hacemos, tanto en procesos de disminución del peso
corporal, como en casos de nutrición deportiva y como no en el caso que nos ocupa, del
tratamiento del SM.
La base de dicha estrategia o procedimiento, constaba en continuar la ritmonutrición
alimentaria manteniendo para siempre las 5 ingestas diarias, disminuyendo al máximo los
hidratos de carbono refinados, aumentando las grasas saludables ricas en Omega3 y
observando siempre la ingesta abundante de frutas y verduras. Siempre en dicho proceso
existía la recomendación de caminar diariamente al menos 1h/día o bien contabilizar los
pasos realizados diarios, teniendo en cuenta que por debajo de 10.000 pasos/día se
considera a una persona sedentaria. Todos ellos fueron conminados a ejecutar mínimamente
dichos pasos. Para hacer fácil dicho proceso, se les recomendó la utilización en su
Smartphone de una App llamada “10.000 pasos al día” o cualquier otro sistema
contabilizador podométrico en su móvil que le proporcionara la facilidad de ejecutarlo.
Dentro de éste esquema, fue muy importante dotar a los pacientes de una
neuromodulación de las sensaciones de bienestar y control del hambre, siendo por ello muy
importante incluir alimentos que contengan más cantidad de tirosina por la mañana y los que
contengan más cantidad de triptófano por la tarde(140-142). Por ello, se insistió en la ingesta en

el desayuno sobretodo de huevos, jamón o queso y por las tardes fruta más frutos secos o
chocolate negro. (Figura 12)
Melatonina
Noradrenalina
12
18
159 24 hTriptófano
PROTEÍNAS
Tirosina
Dopamina
Serotonina
Figura 12. Neuromodulación alimentaria básica.
Esto facultaba el hecho de que al incluir alimentos ricos en el aminoácido tirosina a
primera hora de la mañana, dicha tirosina al transformarse primero en dopamina y
posteriormente en noradrenalina, implicaba un estímulo importante de bienestar la primera
parte del día y a su vez una disminución de la tendencia al picoteo antes de la comida.
Además, nos prepara mejor a la primera parte de la jornada diaria con el aporte de energía
vital. Con la inclusión de más alimentos ricos en triptófano en la merienda, al transformarse
dicho aminoácido en serotonina y posteriormente con la marcha de la luz solar en
melatonina, por un lado disminuyó la predisposición a la bulimia por dulces en ésa segunda
parte del día y por otra, mejoraron también con el incremento de la melatonina una
sensación de mayor bienestar y mejor conciliación del sueño nocturno, disminuyendo a su
vez en varias de las personas que se sometieron al tratamiento las crisis de “eating night

syndrome”(143)que también padecían con anterioridad e incluso disminuyendo también la
ingesta de hipnóticos en quienes los tomaban.
El proceso se completó con el seguimiento mensual de dichas estrategias, donde el
paciente nos aportaba los pesos tomados en su domicilio de los lunes y viernes nada más
levantarse, dado que lo importante para él considerábamos, era conocer la variabilidad
fisiológica del peso que ocurre los fines de semana, donde se permitieron diversas licencias
de alimentos más ricos en otro tipo de grasas animales, al igual que otros cereales y glúcidos.
El objetivo constaba en hacer un equilibrio ponderal en cuanto que el peso que se
incrementaba el fin de semana, fuera capaz de ser disminuido con las estrategias de
alimentación equilibrada de lunes a viernes y con parámetros de crononutrición y
cronobiología alimentaria como hemos visto. Dicho equilibrio fue conseguido en un plazo
más que razonable entre unos 3 y 6 meses, donde el metabolismo basal fué mantenido en la
gran mayoría de los casos. Con la práctica de la bioimpedianciometría durante las visitas de
control, nos aseguramos no iniciaran vicios dietéticos diarios que irían aumentando el peso
de manera imperceptible (Figura 13) (144) y aunque intentaran hacer compensación de ellos el
resto de semana, incluso constatando un mantenimiento en su peso, en realidad ocurriera
un intercambio de patrones, es decir que aumentaran grasa y disminuyeran musculatura,
provocando entonces un cambio en la composición corporal a favor del aumento de grasa.

Figura 13. Rosenbaum M, Leibel RL, Hirsch J. Efecto acumulativo de errores diarios menores en al aumento de la grasa corporal. Obesity. N Engl J Med 1997; 337: 396-408
1.2.7 Interés de la Crononutrición en el Síndrome Metabólico
Las primeras observaciones cronobiológicas relacionadas con la nutrición, se remontan
al siglo XVII, donde Sanctorius hizo construir una gran balanza donde vivió durante meses y
fue registrando su peso y el alimento ingerido, recolectando a su vez las heces y orina. Entre
otras obsevaciones, detectó un ritmo circamensual en la turbidez de la orina y un ritmo
diario y mensual en el peso corporal. La crononutrición, debemos entenderla como el
estudio de la organización temporal de los procesos implicados en la nutrición de los
organismos y de la influencia que la alimentación tiene sobre los ritmos biológicos(145), sin
embargo aún hoy en día constituye uno de los campos de la cronobiología más complejos
por acabar de desvelar.

La Cronobiología es la ciencia que estudia los ritmos biológicos. En efecto, todo el
organismo desde la célula hasta el órgano, obedece a un funcionamiento rítmico. Estos
ritmos biológicos son de origen endógeno y genético, estando controlados por relojes
biológicos internos, el principal de los cuales se halla en los núcleos supraquiasmáticos
cerebrales y que son reajustados cada día por:
• Sincronizadores externos (Figura 14):
Medioambientales: el principal es el sincronizador fótico (alternancia día/noche)
Sociales: el horario de trabajo, el horario de las comidas,…
Figura 14. Sincronizadores externos: Medioambientales y Sociales
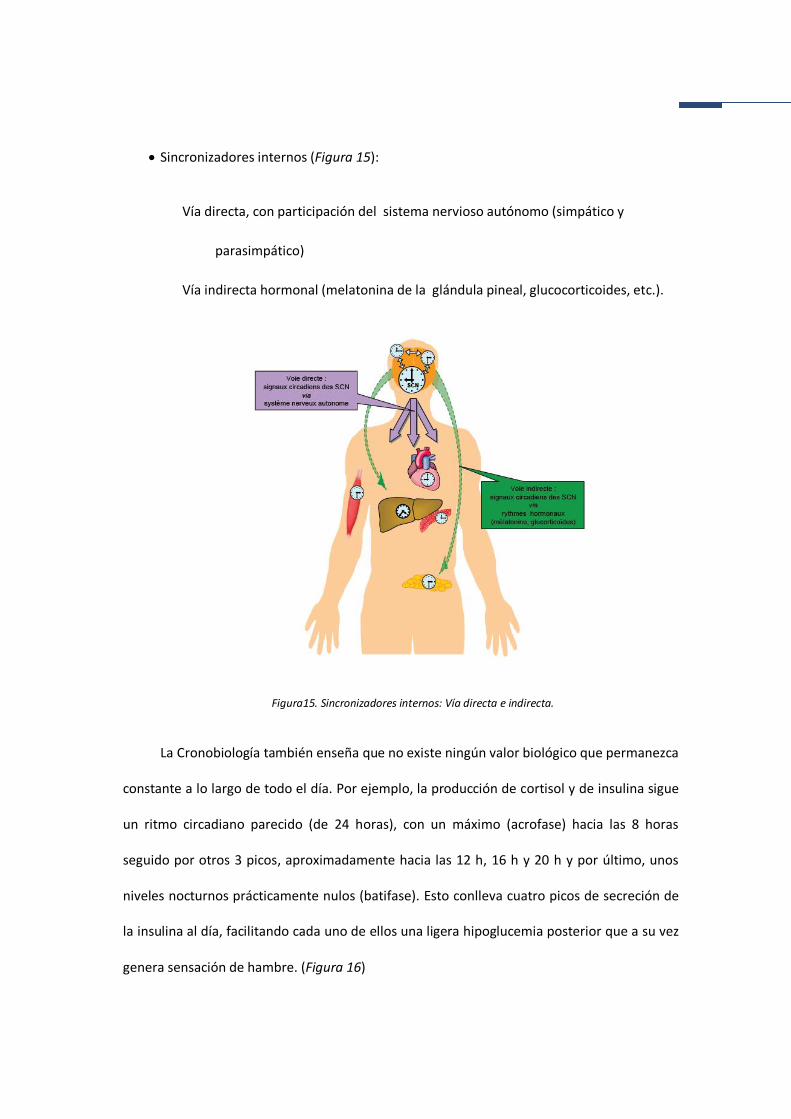
• Sincronizadores internos (Figura 15):
Vía directa, con participación del sistema nervioso autónomo (simpático y
parasimpático)
Vía indirecta hormonal (melatonina de la glándula pineal, glucocorticoides, etc.).
Figura15. Sincronizadores internos: Vía directa e indirecta.
La Cronobiología también enseña que no existe ningún valor biológico que permanezca
constante a lo largo de todo el día. Por ejemplo, la producción de cortisol y de insulina sigue
un ritmo circadiano parecido (de 24 horas), con un máximo (acrofase) hacia las 8 horas
seguido por otros 3 picos, aproximadamente hacia las 12 h, 16 h y 20 h y por último, unos
niveles nocturnos prácticamente nulos (batifase). Esto conlleva cuatro picos de secreción de
la insulina al día, facilitando cada uno de ellos una ligera hipoglucemia posterior que a su vez
genera sensación de hambre. (Figura 16)

Figura 16. Relojes biológicos: central y periféricos
Sólo implementando estos conocimientos a la nutrición de los pacientes afectados de
SM, mejoraremos su pronóstico de forma importante(146).
Tomando en cuenta estas realidades fisiológicas para prevenir o tratar la obesidad,
parece insoslayable una intervención nutricional con las siguientes características y objetivos:
• reducir al máximo los aportes de lípidos y particularmente de glúcidos,
• aumentar la ración de proteínas,
• tener en cuenta la Cronobiología de los nutrientes, de la insulina y de sus receptores para
establecer los horarios favorables a la ingesta de los glúcidos,

• favorecer, en el momento adecuado, la síntesis de los neuromediadores, reguladores del
humor y de la saciedad
• utilizar, de forma óptima, suplementos alimenticios específicos que aporten cofactores
necesarios en todos los mecanismos participantes en la regulación del peso: precursores de
los neuromediadores
• micronutrientes específicos para luchar contra el estrés
• vitaminas o minerales, cofactores del metabolismo de la insulina,…
La Cronobiología Nutricional consiste en aplicar todos estos datos científicos a la
Nutrición, cuyo funcionamiento responde a una serie de parámetros organizados de forma
rítmica:
• La secreción de neuromediadores, determinantes para la regulación del hambre y la
saciedad (catecolaminas y serotonina),
• La biodisponibilidad y la utilización metabólica de los nutrientes,
• Las secreciones hormonales (por ejemplo, la leptina, que regula también la saciedad),
• Los diferentes modos de funcionamiento de los receptores hormonales,
• Las interacciones alimentarias y la competición entre nutrientes en el proceso de
absorción, etc.
En base a lo expuesto, podríamos decir, de manera general, que:
En la primera mitad del nictámero (aproximadamente el periodo que comprende
desde las 5 hasta las 17 horas), los nutrientes aportados se utilizan preferentemente para
cubrir el gasto energético del organismo y de la célula, así como el de las diversas rutas
metabólicas.

En la segunda mitad (aproximadamente desde las 17 hasta las 5 horas), los nutrientes
se almacenan y se utilizan preferentemente para la reparación, la regeneración celular, la
multiplicación tisular o el crecimiento. Es decir, podría decirse que nos reparamos durante la
noche. Es en ese periodo cuando tenemos picos de secreción de algunas hormonas
específicas como la GH.
Todo ello nos deja entrever la conveniencia de organizar la ingesta de los nutrientes de
manera que nos permita optimizar su utilización siguiendo estos principios.
Basada en estos conocimientos científicos, así consiste en sincronizar los aportes
nutricionales con los biorritmos para optimizar la utilización de los nutrientes, mejorando los
componentes patológicos del SM(147).
Para tratar el sobrepeso, habrá que aportar en el momento propicio:
• Los aminoácidos específicos que favorecen la síntesis proteica y la síntesis de
neuromediadores del humor, del hambre y de la saciedad,
• Los glúcidos adecuados para regular el apetito y el almacenamiento de grasas,
siempre respetando los momentos de secreción de la insulina,
• Los ácidos grasos necesarios para controlar la lipogénesis de novo y favorecer su
integración en las membranas celulares,
• Aporte de los micronutrientes, cofactores indispensables en todas las reacciones
metabólicas, caso que pudiéramos apreciar déficit,
• Las tomas diarias de alimentos, que se distribuirán en cuatro o cinco comidas
(según los ritmos de cortisol e insulina) (Figura 17).

Figura 17. Rapin JR. Chronobiologie et nutrition. AIM, 2002 80 : 21-23
Numerosos estudios han demostrado que este ritmo de 4 ó 5 comidas favorece la
estabilidad del peso(148)(Figura 18).
Figura 18. Chapelot D. Consequence of omitting or adding a meal in man on body composition, food intake and metabolism. Obesity 2006;14:215-227.
Optimizada gracias a una buena gestión de los neuromediadores, a una adecuada
suplementación micronutricional y al respeto constante de las pautas establecidas por la

Cronobiología Nutricional, la Dieta de Aporte Proteico ofrece una solución plausible a la
solución del SM en su fase de tratamiento integral inicial.
La hora de alimentación posee efectos sincronizadores(149) y por otro la restricción
alimentaria también. No obstante es el Núcleo Supraquiasmático del hipotálamo quien
regula la sincronización de los ritmos alimentarios totales. La frecuencia de comidas y los
intervalos entre ellas, son modulados a lo largo del ciclo luz-oscuridad(150,151).
El que un determinado nutriente no sea igualmente aprovechado según las horas del
día en que se ingiere está ampliamente demostrado(152). Los ritmos de apetito, digestión,
absorción y actividad de las enzimas clave de su metabolismo son en gran medida
responsables de ello. Estos efectos son especialmente manifiestos en trabajadores en turno
nocturno, en los que no se llega a producir una total sincronización de los ritmos biológicos al
horario nocturno de alimentación (153) y por ello presentan una mayor tendencia a padecer
SM.
Los núcleos supraquiasmáticos del hipotálamo son los principales encargados del
mantenimiento del orden temporal interno, relacionado con los ritmos de ingesta y
metabolismo de los nutrientes. Esto lo consigue de dos maneras, mediante la activación de
vías simpática y parasimpáticas que actúan selectivamente sobre los diferentes
compartimentos del organismo y mediante la producción rítmica de señales humorales como
la melatonina, hormona pineal cuyas concentraciones plasmáticas aumentan por la noche,
induciendo cambios metabólicos propios del periodo nocturno(154,155).

CAPITULO 2. MATERIAL Y METODOS
Se ha realizado un estudio prospectivo sobre 32 pacientes que acuden
espontáneamente a la consulta para bajar peso y son diagnosticados de Sd. Metabólico
según la Definición de Consenso de la IDF de 2005 y que estaban dentro de los criterios de
inclusión señalados posteriormente. Inicialmente la reclutación fue de 51 pacientes afectos
de SM, pero que no cumplían con los criterios de inclusión señalados posteriormente ó bien
que estaban dentro de alguno de los criterios de exclusión del mismo. Del total de estos 51
pacientes, fueron reclutados finalmente 32, de los cuales 24 acabaron el estudio que se les
propuso durante los 30 meses de duración. No concluyeron el estudio 8 de ellos por los
siguientes motivos: 1 de ellos argumentó causa de fuerza mayor por marchar fuera de la
ciudad, debido a motivos laborales; 5 más por considerar que la pérdida ponderal ya había
cumplido todas sus expectativas antes de acabar el estudio y decidieron no proseguirlo; los 2
últimos no manifestaron ninguna causa.
Las visitas fueron semanales en Fase 1, cada 10 días en Fase 2 y quincenales de la Fase
3 en adelante. Las determinaciones analíticas fueron antes del inicio del estudio, cada 2
meses y al final del mismo. Se han tenido en cuenta los parámetros biológicos estudiados de
inicio y final del tratamiento propuesto en cada caso para elaborar los resultados, siendo los
parámetros intermedios los utilizados para el seguimiento clínico durante el tratamiento.
Se determinaron una serie de características de inclusión y de exclusión de los
pacientes a estudiar, teniendo en cuenta que todos los testados y analizados eran
etiquetados de inicio como obesos y con patología asociada acompañante, ya que
consideramos era el perfil de paciente que más beneficios podría obtener de dicho
procedimiento en estudio.

Los criterios de inclusión utilizados fueron:
• Pacientes de edad entre 18 y 70 años
• Pacientes con IMC ≥ 29,9
• Pacientes que quedaban estabilizados en su peso con otras dietas
• Pacientes bajo medicación correctora de la multifactorialidad patológica en el SM
• Pacientes que habían fracasado con otros tipos de dietas
• Pacientes con patología asociada ó agravada por exceso de peso, como: DM, HTA,
Hipotiroidismo, Dislipemias, Artrosis, Sdr. Apnea del Sueño, Ovarios Poliquísticos.
Los criterios de exclusión utilizados fueron:
• Patología cancerosa activa
• Patología cardiovascular tipo ECV reciente, de menos de 3 meses de evolución
• Embarazo, Lactancia
• Pacientes menores de 18 años y mayores de 70
• Insuficiencia Renal, cardiaca (no por postcarga) y hepática (no por Hígado graso)
• Pacientes con alteraciones psiquiátricas severas
• Pacientes con Diabetes Mellitus tipo 1
Parámetros bioquímicos que se determinaron en el estudio comparativo:
• Colesterol Total; HDL; LDL; Triglicéridos; Glucemia; HbA1c; PCR(us); Homocisteína.
Parámetros clínicos y antropométricos determinados en el estudio comparativo:
• Tensión Arterial: Se utilizó para su medición el aparato OMRON Health Care M6.

• Peso Seco: Fue utilizada la báscula In Body 720®Bioespace.
• Indice Masa Corporal (IMC): Fue determinado por la misma báscula In Body 720®
• Perímetro Cintura: Para una correcta medición del perímetro de la pared abdominal se
estandarizó el método con cinta métrica de la siguiente manera:
El individuo debe estar de pie y el examinador se coloca a su derecha, localiza
palpando la cresta ilíaca y justo sobre lo más alto del borde lateral de la cresta ilíaca
derecha se traza una línea horizontal y se cruza con una línea vertical trazada a través
de la línea media axilar. La cinta de medir se coloca en un plano horizontal alrededor
del abdomen a la altura de este punto por el lado derecho del tronco. El plano de la
cinta es paralelo al suelo y la cinta debe estar ajustada pero no debe comprimir la
piel. La medición se hace en inspiración normal.
• Area de Grasa Visceral (AGV): Se determinó mediante impedianciometría, utilizando el
analizador de composición corporal Biospace InBody 720® de la casa Microcaya.
(Figura 19)
Figura 19. Biospace InBody 720®

El cuerpo humano está compuesto por agua corporal, proteínas, grasa corporal y
minerales básicamente. Usando diferentes intervalos de frecuencia desde 1kHz hasta
1MHz, Inbody 720 mide la cantidad de agua corporal con exactitud. Particularmente el
Inbody 720 es la primera versión que usa el método de análisis de reactancia, ésta es una
tecnología punta para el análisis de la composición corporal.
El Metabolismo Basal indica la cantidad mínima de energía requerida para realizar las
funciones vitales esenciales en reposo. El Inbody720 hace posible el cálculo del Metabolismo
Basal usando una conocida ecuación de regresión basada en la Masa Libre de Grasa. La Masa
Libre de Grasa está íntimamente relacionada con el Metabolismo Basal.
El Metabolismo Basal se calcula normalmente usando la Calorimetría Indirecta, lo que
es decir, la demanda de oxígeno empleado. Sin embargo, el InBody 720 calcula el
Metabolismo Basal teniendo en cuenta la Masa Libre de Grasa de la siguiente manera:
Metabolismo Basal = 21.6 FFM(kg) + 370 FFM(kg) ⅹ 10,11
FFM (Free Fat Mass) = Masa Libre de Grasa (kg)
Por ejemplo, si la persona examinada ha ganado masa libre de grasa durante el
programa de control de peso, su Metabolismo Basal también aumenta. Este es un resultado
deseable en cualquier programa de disminución ponderal, ya que nos indica que la masa
grasa almacenada en el cuerpo ha disminuido como resultado del Metabolismo Basal.
Para detectar con exactitud los cambios en la composición corporal es importante
mantener el entorno y las condiciones de metodología constantes. Minimizando las variables
ambientales como la temperatura de la habitación del test, o la hora del test para que los

resultados sean fiables. Así por ello, siguiendo las recomendaciones específicas del
fabricante, procedimos a:
*Asegurarnos que el paciente no hubiera practicado deporte las horas anteriores
previas a la determinación. La práctica de ejercicio riguroso o de actividades físicas
cambia la composición corporal temporalmente.
*Se intentó que la mayoría de los pacientes se realizaran el test por la mañana, para
que estuvieran en ayunas y a su vez, el proceso fisiológico de fluencia de agua hacia
la parte inferior del cuerpo no fuera muy importante.
*También se recomendó al paciente que no tomara un baño ó hiciera una sauna
previamente al test, ya que el sudar afecta temporalmente la composición corporal.
*La temperatura de la habitación siempre estuvo entre 20°C y 25°C. El cuerpo
humano alcanza su equilibrio químico en este intervalo de temperaturas. Las
temperaturas por encima y por debajo de éstas provocan cambios en la composición
corporal.
*Se conminaba al paciente a ir al baño antes de practicarle el test. Los
excrementos en el organismo reducen la exactitud del test.
El Inbody 720 nos determinó la siguiente información de resultados (Figura 20).
1 Diagnóstico de Obesidad (Obesity Diagnosis)
2 Grado de Edema (Degree of Edema)
3 Evaluación Nutricional (Nutritional Evaluation)
4 Evaluación del Peso (Weight Management)
5 Balance Corporal (Body Balance)
6 Area de Grasa Visceral (Visceral Fat)

7 Fuerza Corporal (Body Strength)
8 Control de Peso (Weight Control)
9 Grado Atlético (Fitness)
Figura 20. Informe de resultados obtenidos con In Body 720
¿Con qué precisión se miden los distintos segmentos?
El estudio comparativo entre resultados obtenidos con DEXA y con InBody 720(156,157)
demostró una alta correlación, siendo así un sistema de detección muy fiable (Figura 21).

Figura 21. S Kriemler, J Puder, L Zahner, R Roth, C Braun-Fahrländer and G Bedogni. Cross-validation of bioelectrical impedance analysis for the assessment of body composition in a representative sample of 6- to 13-year-old
children. European Journal of Clinical Nutrition (2009) 63, 619–626; doi:10.1038/ejcn.2008.19; published online 20 February 2008
En el análisis de la composición corporal de Inbody 720 utilizamos la determinación de
la Masa Grasa Corporal (kg), Porcentaje de Grasa Corporal y el del Area de Grasa Visceral
(AGV). El 100% de la masa grasa corporal significa que el sujeto está en el peso y el
porcentaje ideal de masa grasa. Comparada con la masa muscular, la grasa corporal varía
mucho entre los individuos. El Porcentaje de Grasa Corporal , es considerado como normal el
intervalo de grasa corporal en los varones es 15 ±5%, y 23 ±5% para las mujeres. El intervalo
normal de porcentaje de grasa corporal en la mujer es siempre el mismo para todos los
grupos de edades, mientras que para los varones no es constante. Comenzando desde la
edad de 7 años, el porcentaje de grasa decaerá en un 0.5% anualmente hasta la edad de 17.
Esto resulta en un porcentaje promedio de grasa corporal de 15% para los varones adultos.
EL Área de Grasa Visceral, es una de las medidas que más ponderamos en éste estudio,
donde dicha área va a ser determinante en el desarrollo de la patología concomitante en el
SM(158,159).

Figura 22. Area de Grasa Visceral (AGV), superficie al corte en cm².
AGV (Visceral Fat Area= Área de Grasa Visceral) es la sección cruzada del área de grasa
visceral obtenida de la TC (Tomografía Computerizada) en un corte de la región abdominal
(Figura 22).
Normal: <100 cm²
Alto: 100~150 cm²
Extremadamente alto: >150 cm²
Figura 23. Area de Grasa Visceral
Área de grasa visceral se define aquí como el área de la sección cruzada de grasa
visceral que se encuentra en el abdomen (Figura 23). Cuando el área de grasa visceral se
extiende a más de 100 cm2, esto se conoce como obesidad abdominal. La grasa, dependiendo
de su localización, se puede dividir en grasa visceral, subcutánea e intramuscular. En esta

sección lo que se calcula es el área de grasa visceral. La parte sombreada del gráfico indica el
área de la sección cruzada por grupos de edad, lo que revela que el valor de la sección cruzada
del área de grasa visceral es proporcional a la edad.
Habitualmente, los niños tienden a tener un área de grasa visceral menor que los
adultos, pese a que tienen una mayor relación cintura-cadera. Esto es debido a que la grasa
subcutánea de la mayoría de los niños está bastante desarrollada. Por el contrario, según la
gente se hace mayor tiende a desarrollar un área de grasa visceral mayor.
Exactitud de la sección cruzada del área de grasa visceral
Estudios comparativos de la exactitud de la sección cruzada del área de grasa visceral
presentado por el InBody 720 cuyo CT es r=0.922 (n=332, SEE=17.3cm2) con otros sistemas
de medición, indican una alta exactitud en su medición comparativa.
Así que en las condiciones clínicas actuales, el uso del InBody 720 en vez del sistema de
tomografía CT notablemente más caro, permite obtener datos sobre la grasa visceral de una
manera más simple y económica.

CAPITULO 3. RESULTADOS
La muestra final del estudio, está constituida así por 24 participantes que acabaron
definitivamente todo el estudio propuesto, de los cuales un 37.5% son hombres y un 62.5%
mujeres (edades comprendidas entre los 19 años hasta los 70 años) con una edad media de
46.1 años (DT=16.2).
Dentro del estudio que hemos realizado, el interés máximo en cuanto a significación
estadística a comprobar, está basado sobretodo en los parámetros analizados de IMC,
Cintura y AGV, siendo éste último sobre el que se basa el grueso del análisis.
En las medidas pre test (Tabla 2), el IMC medio fue de 37.3 (DT=5.6), con un mínimo de
30 y un máximo de 51. Las dimensiones en el pre test estaban distribuidas de forma normal
(p>0.05). En cuanto a la Cintura media fue de 115.9cm. (DT=12.8), con un mínimo de 95 y un
máximo de 138. Relativo al AGV, el nivel medio fué de 191.1cm2 (DT=39.8), con un mínimo
de 148 y un máximo de 283.
Tabla 2: Descriptivo dimensiones PRE
N Mínimo Máximo Media DT Normalidad Shapiro-Wilk
IMC 24 30 51 37.3 5.6 0.610
Cintura 24 95 138 115.9 12.8 0.980
AVG 24 148 283 191.1 39.8 0.328
Colest 24 162 303 206.1 41.5 0.175
HDL 24 32 53 42.4 5.9 0.789
LDL 24 101 222 153.7 33.4 0.567
TG 24 101 254 181 36 0.861
Glicemia 24 77 162 107.5 21.8 0.580
HbA1c 24 5 7.3 6.0 0.7 0.549
PCRus 24 0.6 13 4.7 3.2 0.620
Homocisteina 24 0.8 25 12.2 6.8 0.985

En las medidas post test (Tabla 3), el IMC medio fue de 25.7 (DT=1.8) con un mínimo
de 23 y un máximo de 29. Siendo las dimensiones en el post test distribuidas de forma
normal (p>0.05). En cuanto a la Cintura media fué 91.3cm. (DT=7.9), con un mínimo de 79 y
un máximo de 91.3. El AGV medio fue de 112.9cm2. (DT=17.1) con un mínimo de 89 y un
máximo de 149.
Tabla 3: Descriptivo dimensiones POST
N Mínimo Máximo Media DT Normalidad Shapiro-Wilk
IMC 24 23.0 29.0 25.7 1.8 0.703
Cintura 24 79.0 110.0 91.3 7.9 0.878
AVG 24 89.0 149.0 112.8 17.1 0.457
Colest 24 127.0 210.0 169.3 18.3 0.739
HDL 24 32.0 56.0 41.8 6.8 0.984
LDL 24 88.0 174.0 121.4 22.6 0.441
TG 24 73.0 185.0 120.8 29.2 0.967
Glicemia 24 69.0 102.0 85.5 9.7 0.394
HbA1c 24 5.0 6.1 5.5 0.4 0.859
PCRus 24 0.6 10.0 3.4 2.3 0.537
Homocisteina 24 0.8 17.0 9.0 5.3 0.336
En cuanto a la comparación de las medias pre y post y el tamaño de su efecto (Tabla 4;
Figura 24) el resultado de la prueba t [t23=12.1; p=<0.001] nos indica que la diferencia entre
los niveles medios de IMC es estadísticamente significativa, (p<0.001). Ambas medias
difieren en el sentido de una reducción significativa del IMC (11.6), en el pretest (37.3) a la
del postest (25.7). Finalmente, para evaluar la significación práctica del resultado hemos
obtenido el valor de la diferencia media tipificada, donde el valor resultante d=2.5, nos
informa de que la disminución en los niveles promedio de IMC es de una magnitud elevada

y por tanto clínicamente relevante. Para el AVG, el nivel medio en el pretest fue de 191.1
(DT=39.8) frente a un 112.8 (DT=17.1) en el postest. La diferencia entre el pretest y el
postest es de 78.3, siendo esta diferencia estadísticamente significativa en el sentido de
una reducción del AVG del pretest al postest [t23=13.216; p=<0.001]. La magnitud del efecto
(d=2.7) nos indica que la disminución en los niveles promedio de AVG es de una magnitud
elevada y por tanto clínicamente muy relevante.
Tabla 4: Comparación medias entre PRE y POST. Tamaño del efecto
Prueba t muestras relacionadas
Media Media Dif. ET IC95% diferencia t23 p d†
IMC_pre 37.3 11.6
1.
0 (9.6 - 13.6)
12.10
0
<0.001**
* 2.5
IMC_post 25.7
Cintura_pre 115.9 24.7
1.
9 (20.7 - 28.7)
12.70
9
<0.001**
* 2.6
Cintura_post 91.3
AVG_pre 191.1 78.3
5.
9 (66.1 - 90.6)
13.21
6
<0.001**
* 2.7
AVG_post 112.8
Colest_pre 206.1 36.9
6.
8 (22.8 - 50.9) 5.436
<0.001**
* 1.1
Colest_post 169.3
HDL_pre 42.4 0.5
0.
7 (-0.9 - 2) 0.775 0.446 0.2
HDL_post 41.8
LDL_pre 153.7 32.3
7.
3 (17.2 - 47.5) 4.415
<0.001**
* 0.9
LDL_post 121.4
TG_pre 181.0 60.2
7.
4 (44.9 - 75.5) 8.157
<0.001**
* 1.7
TG_post 120.8
Glicemia_pre 107.5 22.0
3.
4 (15 - 28.9) 6.527
<0.001**
* 1.3
Glicemia_post 85.5
HbA1c_pre 6.0 0.5
0.
1 (0.3 - 0.7) 5.736
<0.001**
* 1.2
HbA1c_post 5.5
PCRus_pre 4.7 1.2
0.
3 (0.7 - 1.8) 4.729
<0.001**
* 1.0
PCRus_post 3.4
Homocisteina_pre 12.2 3.3
0.
7 (1.8 - 4.7) 4.652
<0.001**
* 0.9
Homocisteina_pos 9.0
***p=0,001 †Tamaño del efecto de Cohen

El resto de parámetros analizados, vierten también resultados positivos en su
conjunto. Así, el Colesterol Total, el nivel medio en el pre test fue de 206,1 (DT = 41,5),
frente a un nivel medio del post test de 169,3 (DT = 18,3). La diferencia entre el pre test y el
post test es de 36,9 (t ₂₃= 5,436; p< 0,01) y la magnitud del efecto d=1,1 nos indica que
también es una disminución relevante del Colesterol Total. (Figura 24)
Así mismo, en cuanto a los TG, su nivel medio en el pre test fue de 181 (DT = 36 ),
frente a un nivel medio del post test de 120,8 (DT = 29,2 ). La diferencia entre el pre test y
el post test es de 60,2 (t₂₃= 8,157 ; p< 0,01) y su magnitud del efecto es incluso mayor que
la del Colesterol total, de un d= 1,7 que nos indica también una muy buena magnitud del
efecto observado.
Figura 24: Comparación medias pre-post. Las barras representan la media±ET
IMC
Pre Post20
25
30
35
40
37.3
25.7
p<0.001
CINTURA
Pre Post80
90
100
110
120
115.9
91.25
p<0.001
AVG
Pre Post
120
140
160
180
200
191.1
112.8 p<0.001
COLESTEROL
Pre Post160
180
200
220
206.1
169.3
p<0.001

LDL
Pre Post
120
140
160
153.7
121.4
p<0.001
TG
Pre Post
120
140
160
180 181.0
120.8
p<0.001
GLICEMIA
Pre Post80
90
100
110
120
107.5
85.54
p<0.001
HbA1c
Pre Post5.0
5.5
6.0
6.5
6.0
5.5
p<0.001
PCRus
Pre Post
3
4
5
4.7
3.4
p<0.001
HOMOCISTEINA
Pre Post6
8
10
12
14
12.2
8.97
p<0.001
Valoración del efecto de la intervención.
En la Tabla 5 y Figura 25, se presentan los descriptivos y contrastes estadísticos para
las variables de resultado considerando dicho análisis para la variable AVG.

Para el grupo de hombres la DT disminuyó progresivamente del pre al post, lo mismo
sucedió para el grupo de mujeres aunque las diferencias entre sexos no fueron
estadísticamente significativas: F(1,22)=2.35; p>0.05.
La AVG de los pacientes en el estudio varió significativamente a través del tiempo,
independientemente del sexo F(1,22)=179.22; p<0.05. El eta2 parcial(160) fue en todas las
variables mayor al 0.89, que significa que al menos el 89% de la varianza en la variable AVG
está asociada con el tiempo transcurrido de una medida para la siguiente y por tanto,
clínicamente muy relevante. En otras palabras la AVG de los pacientes del estudio varió
significativamente a través del tiempo, independientemente del sexo.
Respecto a la interacción observamos que el paso del tiempo influyó de igual manera
en el paciente, independientemente del sexo F(1,22)=1.89; p>0.05, de modo que los efectos
principales se interpretan separadamente.
Tabla 5: MLG: Medias (DT) y contrastes estadísticos entre grupos en la variable AVG.
AVG MLG: Efectos
Variable Pre Post SC Tipo III F1,22 p (eta2) Media (DT) Media (DT)
Sexo Sexo 3234.27 2.35 0.140 (0.096)
Hombre† 206.9 (44.0)
118.2 (20.3) Tiempo 72721.80 179.22 <0.001 (0.891)
Mujer‡ 181.7 (35.2)
109.5 (14.7) Sexo*Tiempo 768.80 1.89 0.183 (0.079)
†Homogeneidad asumida: Prueba Levene F(1,22)=1.441;p=0.243
‡Homogeneidad asumida: Prueba Levene F(1,22)=1.251;p=0.275

Figura 25: Efecto de la intervención antes y después en la variable AVG.
En cuanto a la valoración del IMC, la Tabla 6 y Figura 26, nos muestran los datos obtenidos. El
IMC varió significativamente también a través del tiempo, independientemente del sexo.
F(1,22)=1.46;p<0.05.
Tabla 6: MLG: Medias (DT) y contrastes estadísticos entre grupos en la variable IMC.
IMC MLG: Efectos
Variable Pre Post SC Tipo III F1,22 p (eta2)
Media (DT) Media (DT) Sexo Sexo 22.76 0.99 0.330 (0.043)
Hombre† 38.9 (7.2) 25.9 (2.3) Tiempo 1584.20 146.15 <0.001 (0.869)
Mujer‡ 36.3 (4.3) 25.6 (1.5) Sexo*Tiempo 14.45 1.33 0.261 (0.057)
†Homogeneidad asumida: Prueba Levene F(1,22)=4.061;p=0.056
‡Homogeneidad asumida: Prueba Levene F(1,22)=3.660;p=0.069

Figura 26: Efecto de la intervención antes y después en la variable IMC.
En cuanto a la variable, perímetro de cintura, según los datos obtenidos, Tabla 7 y Figura
27, la disminución de dicho perímetro, varió también significativamente
independientemente del sexo, F(1,22)=3.95;p<0.05.
Tabla 7: MLG: Medias (DT) y contrastes estadísticos entre grupos en la variable Cintura.
Cintura MLG: Efectos
Variable Pre Post SC Tipo III F1,22 p (eta2)
Media (DT) Media (DT) Sexo Sexo 634.69 3.95 0.059 (0.152)
Hombre† 121.6 (14.3) 95.0 (5.7) Tiempo 7056.27 153.10 <0.001 (0.874)
Mujer‡ 112.5 (11.0) 89.0 (8.3) Sexo*Tiempo 25.69 0.56 0.463 (0.025)
†Homogeneidad asumida: Prueba Levene F(1,22)=0.753;p=0.395
‡Homogeneidad asumida: Prueba Levene F(1,22)=0.739;p=0.399

Figura 27: Efecto de la intervención antes y después en la variable Cintura.
ANÁLISIS DE RESULTADOS
Para el análisis general de las variables se han empleado los métodos descriptivos
básicos para las variables cualitativas, obteniendo el número de casos presentes en cada
categoría y el porcentaje correspondiente y para las variables cuantitativas hemos obtenido
el máximo, mínimo, media y desviación típica.
El análisis estadístico se realizó con el programa SPSS 21.0 para Windows. Las
diferencias consideradas estadísticamente significativas son aquellas cuya p<0.05.
Para la comparación de medias entre el pretest y el postest se ha empleado el test de
t-Student para muestras relacionadas bajo el supuesto de normalidad comprobada con el
test de Shapiro-Wilk (n<30)(161). La evaluación de la significación práctica del resultado, se ha
llevado a cabo calculando el tamaño del efecto (d) propuesto por Cohen(162)según el cual los

valores de 0.2, 0.5 y 0.8 corresponden a un efecto típicamente bajo, medio y alto,
respectivamente.
Para contrastar si el cambio entre el pre y el post depende del sexo, se ha realizado el
análisis Modelo Lineal General (MLG): ANOVA(163) de un factor con medidas repetidas.
La significación estadística en estos modelos, se ha complementado con la
interpretación del índice eta2 para obtener una valoración de la significación clínica de los
efectos y que permite evaluar qué porcentaje de la varianza de la variable dependiente está
asociada con el tiempo transcurrido de una medida para la siguiente. La interpretación de
dicho índice la realizamos con el criterio propuesto por Cohen, según el cual en modelos
ANOVA sería preciso que el facto sea capaz de explicar al menos el 10% de la varianza de la
variable dependiente, para poder considerar que el factor tiene relevancia práctica o clínica,
mientras que un valor en torno al 25% ya sería indicativo de una magnitud alta o
clínicamente muy relevante.

CAPITULO 4. DISCUSIÓN
El objetivo del estudio que hemos realizado, tiene su génesis a razón de que durante
nuestra práctica médica de muchos años, tratando personas obesas gran parte de ellas con
SM y mediante la Dieta de Aporte Proteico, hemos ido observando los beneficios clínicos y
biológicos que se producían de una manera no sólo global sino que concretamente podíamos
ir objetivando una disminución del AGV, que no lográbamos con otros tipos de abordajes del
SM, tanto higiénico-dietéticos, farmacológicos ó conductuales.
Así, observamos que con el paso del tiempo mejoran enormemente, no sólo su
aspecto físico por pérdida de masa grasa general, sino que los parámetros biológicos
inherentes a dicho síndrome, en cuanto a la importante pérdida de grasa intravisceral, van
acercándose cada vez más a la normalidad, surgiendo entonces la necesidad de bajar é
incluso eliminar la ingente farmacopea a que se ven sometidos éstos pacientes, comúnmente
multidisciplinar en base a hipotensores, hipoglucemiantes y lípido reguladores
fundamentalmente, ya que hay una rápida mejora metabólica sólo con la disminución
ponderal generada con la DAP. Obviamente esto no ocurría en todos los casos en cuanto a la
eliminación de dichos fármacos, pero sí en cambio en cuanto a la disminución de dosis
siempre. Aún así, la proporción existente observada en cuanto a retirada total de fármacos
en general por líneas de patología/tratamiento, es muy importante.
Nuestro propósito posteriormente a la finalización del estudio realizado y que aquí
presentamos, es iniciar otro estudio observacional de dicho fenómeno y acotar la
significancia estadística de dicha variable en cuanto a la impronta de la mejora en los
parámetros biológicos y disminución ó eliminación de los fármacos que anteriormente
precisaban, con la ventaja añadida de disminuir ó incluso eliminar gran parte de los efectos
secundarios que comportan dichos fármacos, como: mareos, disfunción eréctil, pérdida de

masa muscular, pérdida de memoria, depresión, inflamación de bajo grado, etc., todos ellos
relacionados con una morbi-mortalidad aumentada y una pérdida de bienestar general, en
una población comúnmente añosa.
La muestra estudiada es estadísticamente significativa en cuanto al AGV,
independientemente del sexo, siendo así que podemos considerar que el carácter del objeto
de estudio ha sido enormemente eficaz. Esto nos hace plantear, la necesidad de empezar a
considerar otros sistemas analíticos en cuanto a la definición del verdadero actor en
mayúsculas, el AGV, directamente implicado en la enfermedad cardiovascular, cáncer,
enfermedad de bajo grado y por ello empezar a considerar al IMC y RCC (relación
cintura/cadera) como actores secundarios y no directamente implicados en la patogenia
antes mencionada, puesto que sus mediciones nos aportan sólo caracteres secundarios en
todas las funciones.

CAPITULO 5. CONCLUSIONES
*La Dieta de Aporte Proteico, mejora todos y cada uno de los parámetros estudiados
de manera significativa, sobretodo el AGV objeto del estudio principal con elevada magnitud
en cuanto a significación, así como IMC y Diámetro de Cintura, directamente relacionados
con la primera, siendo dicha actuación y objetivo demostrado en gran parte de todo el
trabajo aquí desarrollado.
*La medida del diámetro de circunferencia abdominal, es un parámetro fiable que nos
ayuda enormemente a identificar con más celeridad los obesos tributarios de padecer el SM,
sobretodo en las Áreas de Asistencia Primaria, donde el momento interventivo es crucial
para ser eficaces en su diagnóstico y tratamiento. Aunque su observación no está
directamente relacionada con la enfermedad cardiovascular, sí que es el primer paso para no
considerar al paciente únicamente obeso, sino que nos obliga a incidir mucho más en el
diagnóstico de SM, con la implicación pronóstica que ello comporta. Obviamente cuando
pudiera generalizarse el estudio del área de grasa intraabdominal y proferirle unos grados de
normalidad/severidad baremizados, al igual que ocurre con el IMC y sus grados, podremos
mediante una sencilla máquina de impedianciometría, que nos mida dicha grasa
intraabdominal, tener un parámetro definitivo para iniciar un tratamiento del paciente como
verdadero SM ó no y con ello poder mejorar el pronóstico final del SM, sobretodo a nivel de
los centros de asistencia primaria.
*El resto de parámetros biológicos estudiados, tanto metabólicos (HbA1c, Glicemia,
Colesterol T, HDL, LDL, TG y Tensión Arterial) como de inflamación (PCRus, Homocisteína),
también mejoraron de manera significativa mediante la DAP, presumiendo así que la
capacidad proinflamatoria del tejido graso visceral había decrecido y con ello los problemas
orgánicos derivados de dicha acción sobre ellos.

*La disminución ponderal de predominio abdominal, mejora el pronóstico
cardiometabólico, cerebrodegenerativo, vascular y neoformativo en todos los casos. No
obstante, pese a considerar la DAP como la dieta más eficaz para provocar dicha mejora de
grasa intravisceral, cualquier paciente diagnosticado de SM, se vería con cierta seguridad
beneficiado clínicamente de una disminución ponderal con cualquier otra medida higiénico
dietética que provocara disminución grasa general y como proceso previo siempre a la
práctica habitual de la excesiva medicación que procuramos a los pacientes insistentemente.
*Al mejorar de una forma muy eficaz y rápida la grasa intraabdominal, obtenemos
también una mejora rápida y eficaz de la insulino-resistencia y la leptino-resistencia, factores
directamente implicados en toda la patología metabólica que venimos tratando.
*Desde un punto de vista de praxis, llamar la atención a todo el colectivo médico,
independientemente de la especialidad que practique, para considerar cualquier paciente
con obesidad abdominal como un paciente metabólico y con serios números de padecer un
SM en toda regla, pudiendo ser más interventivos desde el diagnóstico que hayamos
realizado, o bien derivándolo rápidamente a otro facultativo que en su especialidad troncal
posea la nutrición, ya sea endocrinología, medicina interna, medicina general ó medicina
estética y nutrición.
*La DAP, dada su fácil implementación en un equipo médico bien entrenado,
proporciona al paciente un seguimiento muy satisfactorio por su rápida pérdida de grasa,
falta de hambre y ansiedad por la comida, bienestar general con vitalización y
consecuentemente con ello y en definitiva una observancia al tratamiento muy elevada,
siendo así muy exitosa en la mayoría de pacientes tratados con SM.
*La Crononutrición y la aplicación de la Cronobiología en general, son una ayuda
inestimable a la consecución de la neuromodulación necesaria, en un primer momento para

ayudar al paciente a soportar la restricción calórica de manera óptima en el inicio del
tratamiento dietético, como en la transición lógica hacia el mantenimiento del peso,
pudiéndole generar unas expectativas de pronóstico exitoso y razonable en el tiempo.
*Siendo conscientes de la multifactorialidad existente en el paciente obeso, que se
acaba transformando en SM, no podemos banalizar en pensar que exista un único
tratamiento que pueda tratar dicha pluripatología de manera exitosa al 100%. Obviamente
no debemos olvidar el ejercicio físico moderado promoviendo un balance energético
equilibrado, nutrición no inflamatoria, entendiendo con ello la disminución de la ingesta de
alimentos procesados tan ricos en grasas trans que nos generan inflamación celular y sobre
todo las políticas nutricionales que deberían existir al respecto, promocionando los cultivos
orgánicos y ecológicos, entre muchas otras.

CONFLICTO DE INTERESES
Declaramos que no hemos tenido ningún conflicto de intereses en la realización del
presente trabajo.
No hemos recibido subvenciones ni favores de ningún tipo, ni desde ninguna
institución diferente a nuestro centro de trabajo.
Así pues, nada ha podido desde nuestra conciencia y honor, afectar ni positiva ni
negativamente en la consecución de los objetivos que nos planteamos desde el inicio de la
propuesta del trabajo inicial.

BIBLIOGRAFIA
1. Rössner, S (2002) Obesity: the disease of the twenty-first century. Int J Obes Relat Metab Disord;26 Suppl 4:S2-4.
2. World Health Organisation (2000). Obesity: preventing and managing the global epidemic. WHO Technical report Series 894. Geneva.
3. World Health Organisation (2006). Obesity and overweight. Fact sheet N° 311, September 2006.
4. Prevalence of overweight and obesity. The International Obesity Task Force. www.iotf.org.
5. Omran AR (1971) The epidemiologic transition. A theory of the epidemiology of population change. Milbank Mem Fund Q 1;49:509-38.
6. Moreno LA et al. (2002) The nutrition transition in Spain: a European Mediterranean country. European Journal of Clinical Nutrition, 56:992-1003.
7. Popkin BN (1994) The nutrition transition in low-income countries: an emerging crisis. Nutr Rev 52:185-98.
8. Monteiro CA (2004) The burden of disease from undernutrition and overnutrition in countries undergoing rapid nutrition transition: a view from Brasil. Am J Public Health; 94:433-4.
9. World Health Organisation (2007). The challenge of obesity in the WHO European Region and the strategies for response.
10. Charles MA (2008) Monitoring the Obesity Epidemic in France: The Obepi Surveys 1997-2006 Obesity, doi:10.1038/oby.2008.285.
11. Aranceta-Batrina J et al. (2005) Prevalencia de obesidad en España. Medicina Clinica, 125:32-38.
12. Serra Majem L et al. (2003) Obesidad infantil y juvenil en España: resultados del estudio enKid (1998-2000) Medicina Clinica, 121:725-732.
13. Visscher TL, Seidell JC. (2001) The public health impact of obesity. Annual Review of Public Health, 2001, 22:355-375
14. Flegal KM (2002) Prevalence and trends in obesity among US adults, 1999-2000. JAMA, 288:1723-27.
15. Peeters A et al. (2003) Obesity in adulthood and its consequences for life expectancy: a life-table analysis. Annals of Internal Medicine, 138(1):24-32.
16. Overweight, obesity, and health risk. National Task Force on the Prevention and Treatment of Obesity, Arch Intern Med 2000;160:898-904.

17. Cook S (2003). Prevalence of a metabolic syndrome phenotype in adolescents: findings from the third National Health and Nutrition Examination Survey, 1988-1994, Archives of Pediatrics & Adolescent Medicine, 157(8):821-827.
18. Avenell A (2004) Systematic review of the long-term effects and economic consequences of treatments for obesity and implications for health improvement. Health Technol Assess. 8(21):iii-iv, 1-182.
19. Wolf AM (1998) Current estimates of the economic cost of obesity in the United States. Obes Res.6(2):97-106.
20. Detournay B (2000) Obesity morbidity and health care costs in France: an analysis of the 1991-1992 Medical Care Household Survey. Int J Obes Relat Metab Disord, 24:151-155.
21. Wang Y (2008) Will All Americans Become Overweight or Obese? Estimating the Progression and Cost of the US Obesity Epidemic. Obesity (Silver Spring) Jul 24.
22. Costa B, Cabré JJ, Martin F. Síndrome metabólico, resistencia a la insulina y diabetes.
¿Qué se oculta bajo la punta del iceberg? Aten Primaria 2003;31(7):436-45.
23. Ervin RB. Prevalence of metabolic síndrome among adults 20 years of age and over, by
sex, age, race and ethnicity, and body mass index: United Status, 2003-2006. Natl Healt Stat
report. 2009;13:1-7.
24. Fernández-Bergés D, Cabrera de León A, Sanz H, Elosua R, Guembe MJ, Alzamora M, et al.
Metabolic syndrome in Spain: prevalence and coronary risck associated with harmonized
definition and WHO proposal. DARIOS study. Rev Esp Cardiol. 2012;65:241-8.
25. Gutierrez-Fisac JL, Rodruiguez-Artalejo F, Regidor E. Increasing prevalence of over-
weightand obesity among spanish adults, 1987-1997. Int J Obes 2000; 24:1677-82.
26. Kassi E, Pervanidou P, Kaltsas G, Chrousos G. Metabolic syndrome: definitions and
controversies. BMC Med. 2011;9:48.
27. Reaven GM. Banting lecture 1988: role of insulin resistance in human disease. Diabetes.
1988;37:1595-1607.
28. Liese AD, Mayer-Davis EJ, Haffner SM. Development of the multiple metabolic syndrome:
an epidemiologic perspective. Epidemiol Rev. 1998;20:157-172.
29. Alberti KG, Zimmet PZ, for the WHO Consultation. Definition, diagnosis and classification
of diabetes mellitus, personal report of a WHO consultation. Diabetes Med. 1998;15:539-53.

30. Executive Summary of The Third Report of the National Cholesterol Education Program
(NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in
Adults (Adult Treatment Panel III). JAMA. 2001;285:2486-97.
31. Genuth S, Alberti KG, Bennet P, Buse J, Defronzo R, Hahn R, et al. Expert Committee on
the Diagnosis and Classification of Diabetes Mellitus-American Diabetes Association. Follow-
up report on the diagnosis of diabetes mellitus. Diabets Care 2003; 26:3160-7.
32. Einhom D, Reaven GM, Cobin RH, Ford E, Ganda OP, Handelsman Y, et al. American
College of Endocrinology position statement on the insulin resistance syndrome. Endocr
Pract. 2003; 9: 237-52.
33. Balkau B, Charles MA, Drivsholm T, et al. The European Group for the Study of Insulin
Resistance (EGIR). Frequency of the WHO metabolic syndrome in European cohorts, and an
alternative definition of an insulin resistance syndrome. Diabetes Med 2002;28:364-76.
34. International Diabetes Federation. The IDF consensus worldwide definition of metabolic
syndrome. www.idf.org/webdata/docs/IDF Metasyndrome definition. pdf.
35. Zimmet et al. Una nueva definición mundial del Síndrome Metabólico propuesta por la
Federación Internacional de Diabetes: fundamento y resultados. Rev. Esp. Cardiología 2005;
58:1371 – 1376.
36. Pouliot MC, Després JP, Lemieux S, Moorjani S, Bouchard C, Trembloy A, et al. Waist
circumference and abdominal sagittal diameter, best simple anthropometric indexes of
abdominal visceral adipose tissue accumulation and related cardiovascular risk in men and
women. Am J Cardiol. 1994; 73: 460-8.
37. Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, et al. International
Diabetes Federation Task Force on Epidemiology and Prevention, National Heart, Lung, and
Blood Institute, American Heart Association, World Heart Federation, International
Atherosclerosis Society, International Association for the Study of Obesity. Harmonizing the
metabolic syndrome: a joint interim statement of the International Diabetes Federation Task
Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American
Heart Association; World Heart Federation; International Atherosclerosis Society; and
International Association for the Study of Obesity. Circulation. 2009; 120:1640-5.
38. Onat A, Can G, Örnek E, Sansoy V, Aydın M, Yüksel H. Abdominal obesity with hypertriglyceridaemia, lipoprotein(a) and apolipoprotein A-I determine marked cardiometabolic risk.Eur J Clin Invest. 2013 Nov;43(11):1129-39. doi: 10.1111/eci.12150. Epub 2013 Sep 11.

39. Okosun IS, Liao Y, Rotimi CN, Prewitt TE, Cooper RS. Abdominal adiposity and clustering of multiple metabolic syndrome in white, black and hispanic americans. Ann Epidemiol. 2000;10:263-70.
40.Premanath M, Basavanagowdappa H, Mahesh M, Suresh M.
Correlation of abdominal adiposity with components of metabolic syndrome, anthropometric
parameters and Insulin resistance, in obese and non obese, diabetics and non diabetics: A
cross sectional observational study. (Mysore Visceral Adiposity in Diabetes Study). Indian J
Endocrinol Metab. 2014 Sep;18(5):676-82. doi: 10.4103/2230-8210.139231.
41. Balkau et al. International Day for the Evaluation of Abdominal Obesity (IDEA): A Study of
Waist Circumference, Cardiovascular Disease, and Diabetes Mellitus in 168000 primary care
patients in 63 countries. Circulation 2007; 116:1942-1951.
42. Patsouris D, Cao JJ, Vial G, Bravard A, Lefai E, Durand A, Durand C, Chauvin MA, Laugerette F, Debard C, Michalski MC, Laville M, Vidal H, Rieusset J. Insulin Resistance is Associated with MCP1-Mediated Macrophage Accumulation in Skeletal Muscle in Mice and Humans. PLoS One. 2014 Oct 22;9(10):e110653. doi: 10.1371/journal.pone.0110653. eCollection 2014.
43. Miranda JP, De Fronzo RA, Califf RM, Guyton JR. Metabolic syndrome: Definition,
pathophysiology and mechanisms. Am Heart J. 2005;149:33-45.
44.Weyer C, Foley JE, Bogardus C, Tataranni PA, Pratley RE. Enlarged subcutaneous abdominal
adipocyte size, but not obesity itself, predicts type II diabetes independent of insulin
resistance. Diabetologia. 2000;43:1498-506.
45. Klöting N, Blüher M.Rev Endocr Metab Disord. 2014 Dec;15(4):277-87. doi: 10.1007/s11154-014-9301-0. 46. American Diabetes Association. Consensus Development Conference on Insulin Resistance. Diabetes Care 1998; 21:310-4.
47. Martinez de Moratín BE, Rodriguez MC, Martínez JA. Síndrome metabólico, resistencia a la
insulina y metabolismo tisular. Endocrinol Nutr 2003;50(8):324-33.

48. Fernández-Majías C. Nuevo panorama para el entendimiento de los vínculos moleculares
entre la obesidad y la diabetes tipo 2. Rev Invest Clin 2001;53:209-11.
49. Bremer AA, Mietus-Snyder M, Lustig RH. Toward a unifying hipótesis of metabolic
síndrome. Pediatrics. 2012;129:557-70.
50. Moller DE, Kaufman KD. : Metabolic syndrome: a clinical and molecular perspective. Annu
Rev Med 2005; 56:45-62.
51. Hutley L, Prins JB.: Fat as an endocrine organ: relationship to the metabolic syndrome.
Am J Med Sci 2005; 330:280-289.
52. Matsuzawa Y.: adipocytokines and metabolic syndrome. Semin Vasc Med 2005;5:34-39.
53. Phillips LK, Prins JB. The link between abdominal obesity and the Metabolic syndrome. Curr Hypertens Rep. 2008 Apr;10(2):156-64. Review. PubMed PMID:18474184.
54. Weyer C, Foley JE, Bogardus C, Tataranni PA, Pratley RE. Enlarged subcutaneous
abdominal adipocyte size, but not obesity itself, predicts type II diabetes independent of
insulin resistance. Diabetologia. 2000;43:1498-506.
55. Miranda JP, De Fronzo RA, Califf RM, Guyton JR. Metabolic syndrome: Definition,
pathophysiology and mechanisms. Am Heart J. 2005;149:33-45.
56. Kabir M, Catalano KJ, Ananthnarayan S, Kim SP, Van Citters GW, Dea MK, Bergman RN.:
Molecular evidence supporting the portal theory: a causative link between visceral adiposity
and hepatic insulin resistance. Am J Physiol Endocrinol Metab 2005; 288: E454-E461.
57. Das UN. Metabolic síndrome X: an inflamatory condition? Curr Hypertens Rep
2004;123(10):381-2.
58. Fontana L, Eagon JC, Trujillo ME, Scherer PE, Klein S. Visceral fat adipokine secretion is
associated with systemic infflammation in obese humans. Diabetes 2007;56: 1010-1013.
59. Li G, Barrett EJ, Barrett MO, Cao W, Liu Z. Tumor necrosis factor-alpha induces insulin
resistance in endothelial cells via a p38 mitogen-activated protein kinase-dependent
pathway. Endocrinology 2007;148:3356-3363.

60. Gonzalez-Gay MA, De Matias JM, Gonzalez-Juanatey C, Garcia-Porrua C, Sanchez-Andrade
A, Martín J, Llorca J. Anti-tumor necrosis factor-alpha blockade improves insulin resistance in
patients with rheumatoid arthritis. Clin Exp Rheumatol 2006;24:83-86.
61. Kiortsis DN, Mavridis AK, Vasakos S, Nikas SN, Drosos AA. Effects of infliximab treatment
on insulin resistance in patients with rheumatoid arthritis and ankylosing spondylitis. Am
Rheum Dis 2005;107:765-766.
62. Paquot N, Castillo MJ, Lefebvre PJ, Scheen AJ. No increased insulin sensitivity after a
single intravenous administration of a recombinant human tumor necrosis factor receptor: Fc
fusion protein in obese insulin-resistant patients. J Clin Endocrinol Metab 2000;85:1316-
1319.
63. Lijnen HR. Pleiotropic functions of plasminogen activator inhibitor-1. J Thromb Haemost
2005;3:35-45.
64. He G, Pedersen SB, Bruun JM, Lihn AS, Jensen PF, Richelsen B. Differences in plasminogen
activator inhibitor 1 in subcutaneous versus omental adipose tissue in non-obese and obese
subjects. Hor Metab Res 2003;35:178-182.
65. Mavri A, Alessi MC, Bastelica D, Geel-Georgelin O, Fina F, Sentocnik JT, Stegnar M, Juhan-
Vague I. Subcutaneous abdominal, but not femoral fat expression of plasminogen activator
inhibitor-1 (PAI-1) is related to plasma PAI-1 levels and insulin resistance and decreases after
weight loss. Diabetologia 2001;44:2025-2031.
66. Ma LJ, Mao SL, Taylor KL, Kanjanabuch T, Guan Y, Zhang Y, Brown NJ, Swift LL,
McGuinness OP, Wasserman DH, et al. Prevention of obesity and insulin resistance in mice
lacking plasminogen activator inhibitor 1. Diabetes 2004;53:336-346.
67. Oya M. Inflamación y síndrome metabólico. Med Clin 2004;123(10):381-2.
68. Yoshikane H, Yamamoto T, Ozaki M, Matsuzaki M. Clinical significance of high-sensitivity
C-reactive protein in lifestyle-related disease and metabolic syndrome. J Cardiol 2007;50(3):
175-82.
69. Klein S, Fontanta L, Young VL, Coggan AR, Kilo C, Patterson BW, Mohammed BS.:
Absence of an effect of liposuction on insulin action and risk factors for coronary heart
disease. N Engl J Med 2004; 350(25):2549-2557.

70. Thörne A, Lönnqvist F, Apelman J, Hellers G, Arner P.: A pilot study of long-term effects of
a novel obesity treatment: omentectomy in connection with adjustable gastrc banding. Int J
Obes 2002;26: 193-199.
71. Ginsberg HN. Insulin resistance and cardiovascular disease. J Clin Invest. 2000;106:453-8.
72. Adiels M, Olofsson SO, Taskinen MR, Borén J. Overproduction of very low-density
lipoproteins is the hallmark of the dyslipemia in the metabolic syndrome. Arterioscler
Thromb Vasc Biol. 2008;28:1225-36.
73. Ayyobi AF, Brunzell JD. Lipoprotein distribution in the metabolic syndrome, type 2
diabetes mellitus, and familial combined hyperlipemia. Am J Cardiol 2003;92:27-33.
74. Oda E. Metabolic síndrome: its history, mechanisms, and limitations. Acta Diabetol.
2012;49:89-95.
75. Okosun IS, Liao Y, Rotimi CN, Prewitt TE, Cooper RS. Abdominal adiposity and clustering
of multiple metabolic syndrome in white, black and hispanic americans. Ann Epidemiol.
2000;10:263-70.
76. Fielding CJ, Fielding PE: Molecular physiology of reverse cholesterol transport. J Lipid Res
1995;36(2):211-28.
77. Vasan RS, Pencina MJ, Robins SJ, et al.: Association of circulating cholesteryl ester
transfer protein activity with incidence of cardiovascular disease in the community.
Circulation 2009;120(24):2414-20.
78. Duwensee K, Breitling LP, Tancepski I, et al.: Cholesteryl ester transfer protein in patients
with coronary heart disease. Eur J Clin Invest. 2010;40(7):616-22.

79. Robins SJ, Lyass A, Brocia RW, et al.: Plasma lipid transfer proteins and cardiovascular
disease. The Framingham Heart Study. Atherosclerosis. 2013;228(1):230-6.
80. Welborn TA, Breckenride A, Rubinstein AH, Dollery CT, Fraser TR. Serum insulin in
essential hypertension and in peripheral vascular disease. Lancet 1966;1:1336-7.
81. Modan M, Halkin H, Almog S, et al. Hyperinsulinemia. A link between obesity and glucose
intolerance. J Clin Invest 1985;75:809-17.
82. Lucas CP, Estigarribia JA, Darga LL, Reaven GM. Insulin and blood pressure in obesity.
Hypertension 1985;7:702-6.
83. Minicardi V, Camellini L, Belloidi G, Ferrannini E. Evidence for an association of high blood
pressure and hyperinsulinemia in obese man. J Clin Endocrinol Metab 1986;62:1302-4.
84. Ferrannini E, Buzzigoli G, Bonnadonna R, et al. Insulin resistence in essential
hypertension. N Engl J Med 1987 ;317 :350-7.
85. Reaven GM. The metabolic syndrome or the insulin resistence syndrome ? Different
names, different concepts, and different goals. Endocrinol Metab Clin N Am. 2004;(33):283-
303.
86. Skarfors ET, Lithell HO, Selinus I. Risk factors for the development of hypertension: a 10-
year longitudinal study in middle-aged men. J Hypertens 1991;9:217-23.
87. Lissner L, Bengtsson C, Lapidus L, Kristiansson K, Nedel M. Fasting insulin in relation to
subsequent blood pressure changes and hypertension in women. Hypertension 1992;20:797-
801.

88. Rantala AO, Kauma H, Lilja M, Savolainen MJ, Reunanen A, Kesaniemi YA. Prevalence of
the metabolic syndrome in drug-treated hypertensive patients and control subjects. J Intern
Med 1999;245(2):163-74.
89. Liese AD, Mayer-Davis EJ, Chambless LE, et al. Elevated fasting insulin predicts incident
hypertension : the ARIC study. Atherosclerosis Risk in Communities Study Investigators. J
Hypertens 1999;17:1169-77.
90. McLaughlin T Reaven G. Insulin resistence and hypertension. Patients in double jeopardy
for cardiovascular disease. Geriatrics 2000;55:28-32,35.
91. Anderson EA, Hoffman RP, Balon TW, Sinkey CA, Mark AL. Hyperinsulinemia produces
both sympathetic neural activation and vasodilation in normal humans. J Clin Invest.
1991;87:2246-52.
92. Fruehwald E, Schultes B. Hiperinsulimemia causes activation of the hypothalamus-
pituitary-adrenal axis in human. Int J Obes Relat Metab Disord. 2001; Suppl 1:538-40.
93. DeFronzo RA, Ferrannini E. Insulin resistence. A multifaceted syndrome responsible for
NIDDM, obesity, hypertension, dyslipemia, and atherosclerotic cardiovascular disease.
Diabetes Care. 1991;14:173-94.
94. Petrie JR, Ueda S, Webb DJ, Elliott HL, Connell JM. Endothelial nitric oxide production and
insulin sensitivity. A physiological link with implications for pathogenesis of cardiovascular
disease. Circulation. 1996;93:1331-3.
95. Laine H, Knuuti MJ, Ruotsalainen U, Raitakari M, Lida H, Kapanen J, et al. Insulin
resistence in essential hypertension is characterized by impaired insulin stimulation of blood
flow in skeletal muscle. J Hypertens. 1998;16:211-9.

96. Mottillo S, Filion KB, Genest J, Joseph L, Pilote L, Poirier P, et al. The metabolic syndrome
and cardiovascular risk: a systematic review and meta-analysis. J Am Coll Cardiol.
2010;56:1113-32.
97. García Romero de Tejada G, Escobar Monrrealle HF. Hiperandrogenismo en la mujer
diabética: rol de la resistencia insulínica y de la hiperinsulinemia. Endocrinol Nutr.
2003;50:363-8.
98. Lim SS, Norman RJ, Davis MJ, Moran LJ. The effect of obesity on polycystic ovary
syndrome: a systematic review and meta-analysis. Obes Rev. 2013;14:95-109.
99. Mannarino MR, Di Filippo F, Pirro M. Obstructive sleep apnea syndrome. Eur J Intern
Med. 2012;23:586-93.
100. Jager J. Grémeaux T, Cormont M, Le Marchand-Brustel Y, Tanti JF. Interleukin-1beta-
induced insulin resistance in adipocytes through down-regulation of insulin receptor
substrate-1 expression. Endocrinology.2007 Jan;148(1):241-51, Epub 2006 Oct 12.
101. Vázquez Carballo A, Ceperuelo-Mallafré V, Chacón MR, Maymó-Massip E, Lorenzo M.
Porras A. Vendrell J, Fernández-Veledo S. Tweak prevents TNF-α-induced insulin resistance
through PP2A activation in human adipocytes. Am J Physiol Endocrinol Metab. 2013 Jul 1;
305(1):E101-12. Doi:10.1152/ajpendo.00589,2012. Epub 20134 May 7.
102. Chedraui P, Pérez-López FR, Escobar GS, Palla G, Montt-Guevara M, Genazzani AR,
Simoncini T, Research Group for the Omega Women´s Health Project. Circulating leptin,
resistin, adiponectin, visfatin, adipsin, and ghrelin levels and insulin resistance in
postmenopausal women with and without the metabolic syndrome. Maturitas, 2014
Sep;79(1):86-90. doi;10.1016/j. maturitas.2014.06.008. Epub 2014 Jun18.

103. Ahima RS, Saper CB, Flier JS, Elmquist JK. Leptin regulation of neuroendocrine systems.
Front Neuroendocrinol. 200 Jul;21(3):263-307.
104. Madec S, Rossi C, Chiarugi M, Santini E, Salvati A, Ferrannini E, Solini A. Adipocyte P2X7
receptors expression: a role in modulating inflamatory response in subjects with metabolic
síndrome? Atherosclerosis.2011 Dec;219(2):552-8.doi
10.101016/j.atherosclerosis.2011.09.012. Epub 2011 Sep 16.
105. Rega G, Kaun C, Weiss TW, Demyanets S, Zorn G, Kasti SP, Steiner S, Seidinger D, Kopp
CW, Frey M, Roehle R, Maurer G, Huber K, Wojta J. Inflamatory cytokines interleukin-6 and
oncostatin m induce plasminogen activator inhibitor-1 in human adipose tissue. Circulation.
2005 Apr 19; 111(15): 1938-45.
106. Robert L, Labat-Robert J. The metabolic síndrome and the Maillard reaction- An
introcuction. Pathol BIOL (Paris). 2006 Sep;54(7):371-4. Epub 2006 Sep15.
107. Hudson BI, Dong C, Gardener H, Elkind MS, Wright CB, Glodberg R, Saco RL, Rindek T.
Serum levels of soluble receptor for advance glycation end-products and metabolic
syndrome: the Northern Manhattan Study. Metabolism. 2014 Sep:63(9):1125-30.
Doi:10.1016/j.metabol.2014.05.011. Epub 2014 Jun14.
108. Reinberg A. 1983. Chronobiology and nutrition. (Ed. Reinberg A, Smolensky MH).
Springer Verlag, New York. pp.265-300.
109. Kreier F, Kalsbeek A, Ruiter M, Yilmaz A, Romijn JA, Sauerwein HP, Fliers E, Buijs RM.
2003. Central nervous determination of food storage a daily switch from conservation to
expenditure: implications for the metabolic syndrome. European J. Pharmacol. 480:51-65.

110. Strubbe JH, van Dijk G, 2002. The temporal organization of ingestive behaviour and its
interaction with regulation of energy balance. Neuros. Biobehav. Rev. 26: 485-498.
111. Buijs RM, Kreier F. The metabolic síndrome: a brain disease?. Neuroendicronol. 2006
Seo:18(9):715-16.
112. Vieira E, G Ruano, C Figueroa AL, Aranda G, Momblan D, Carmona F, Gomis R, Vidal J,
Hanzu FA. Altered clock gene expression in obese visceral adipose tissue is associated with
metabolic síndrome. PLoS One.2014 Nov 3; 9(11):e111678. doi:
10.1371/journal.pone.0111678.eCollection 2014.
113. Shostak A, Husse J, Oster H. Circadian regulation of adipose function. Adipocite.2013 Oct
1;2(4):201-6. doi: 10.4161/adip.26007. Epub 2013 Aug 13.
114. Garaulet M, Madrid JA,. Chronobiology, genetics and metabolic syndrome. Curr Opin
Lipidol, 2009. Apr; 20(2):127-34.
115. Kume K, Zylka MJ, Sriram S, Shearman LP, Weaver DR, Jin X, Maywood ES, Hastings MH,
Rep pert SM. 1999. mCRY1 and mCRY2 are essential components of the negative limb of the
circadian clock feedback loop. Cell 98:193-205.
116.Albrecht U, Sun ZS, Eichele G, Lee CC. 1997. A differential response of two putative
mammalian circadian regulators, mPer1 and mPer2, to light. Cell 91:1055-1064.
117. Dardente H, Cermakian N. Molecular circadian rhythms in central and peripheral clocks
in mammals. Chronobiol. Int. 2007; 24: 195-213.
118. Haus E, Smolensky M. 1999. Biologic rhythms in hematology. Path. Biol. 44: 618-630.

119. Young ME, Bray MS. Potential role for peripheral circadian clock dyssynchrony in
thepathogenesis of cardiovascular dysfunction. Sleep Med. 2007. Sep;8(6):656-67.
120. Scott EM, Carter AM, Grant PJ. Diabetes and cardiovascular deisease:related disorders
created by disturbances in the endogenous clock. J Indian Med Assoc. 2008 Nov;
106(11)736-8,740.
121. Xiazo F, Yu J, Liu B, Guo Y, Li K, Deng J, Zhang J, Wang C, Chen S, Du Y, Lu Y, Xiao Y, Zhang
Z, Guo F. A novel function of microRNA 130a-3p in hepatic insulin sensivity and liver
steatosis. Diabetes. 2014 Aug;63(8):2631-42. doi: 10.2337/db13-1689. Epub 2014 Mar 27.
122. Ando H. Circadian clocks and lifestyle-related diseases. Rinsho Byori. 2013 Nov;
61(11)1044-50. Review. Japanese.
123. Challet E. Clock genes, circadian rhythms and food intake. Pathol Biol (Paris). 2007 Apr-
May;55(3-4):176-7.
124. Freeman JM, Millicient K, Freeman JB. The epilepsy diet treatment: an introduction to
the ketogenic diet. New York: Demos Vermande, 1996.
125. Walters JK, et al. The Protein Sparing Modified Fast for obesity-related medical
problems. Cleveland Clinical J Med (1997) 64:242-243.
126. Yudkin J, Carey M. The treatment of obesity by a high fat diet- the inevitavility of
calories. Lancet (1960) 939-941.
127. Blackburn GL. Role of a protein-sparing modified fast in a comprehensive weight
reduction program Howard, AN eds. Recent Advances in Obesity Research; (1975)279-281
Newman Publishing London, UK.

128. Blackburn GL. Protein sparing therapy during periods of starvation with sepsis and
trauma Ann Surg; (1973)177:588-594.
129. Su HY, Lee HC, Cheng WY, Huang SY. A calorie-restriction diet supplemented with fish oil
and high-protein powder is associated with reduced severity of metabolic syndrome in obese
women. Eur J Clin Nutr. 2014. Sep 24. Doi:10.1038/ejcn. 2014.196.
130. Mustajoki P. Very low energy diets in the treatment of obesity. Obes Rev, (2001) 2: 61-
72.
131.Hercberg S. The SU.VI.MAX Study. A Randomized, Placebo-Controlled Trial of the Health
Effects of Antioxidant Vitamins and Minerals. Arch Intern Med;(2004) 164:2335-2342.
132. Parra D. A diet rich in long chain omega-3 fatty acids modulates satiety in overweight
and obese volunteers during weight loss. Appetite. (2008) Nov;51(3):676-80.
133. Carbone JW, McClung JP, Pasiakos SM. Skeletal muscle responses to negative energy
balance: effects of dietary protein. Am J Pathol. (2012) Mar 1;3(2):119-26. Doi: 10.3945/an.
111.001792.
134. Mitchell GA et al. Medical aspects of ketone body metabolism. Clinical & Investigative
Medicine (1995) 18:193-216.
135. Robinson AM, Williamson DH. Physiological roles of ketone bodies as substrates and
signals in mammalian tissues. Physiol Rev. (1980)60:143-187.
136. Porta N, Vallée L, Boutry E, Auvin S. The ketogenic diet and its variants: state of the art.
Rev Neurol (París) 2009 May; 165(5):430-9. Doi: 10.1016j.neurol.2008.10.007. Epub 2008
Nov 21.

137. Garcia Caraballo SC, Comhair TM, Houten SM, Dejong CH, Lamers WH, Koehler SE. High
protein diets prevent steatosis and induce accumulation of monomethyl branched chain fatty
acids. J Nutr Biochem.2014 Sep 16.pii:S0955-2863(14)00169/jnutbio.2014.07.005
138. Stock A, Yudkin J. Nutrient intake of subjects of low carbohydrate diet used in treatment
of obesity. Am J Clin Nutr (1970)23:948-952.
139. Hercberg S. The SU.VI.MAX Study. A Randomized, Placebo-Controlled Trial of the Health
Effects of Antioxidant Vitamins and Minerals. Arch Intern Med(2004);164:2335-2342.
140. Wurtman RJ. Effects of their nutrient precursors on the synthesis and release of
serotonin, the catecholamines, and acetylcholine: implications for behavioral disorders. Clin
Neuropharmacol (1988);11 Suppl 1:S187-93.
141. Wurtman RJ. Effects of normal meals rich in carbohydrates or proteins on plasma
tryptophan and tyrosine ratios. Am J Clin Nutr (2003);77:128-32.
142. Asarian L, Bächler T, Neuroendocrine control of satiation. Horm Mol Biol Clin Investig.
2014 Sep 1;19(3):163-92. Doi: 10.1515/hmbci-2014-0010.
143. Lundgren JD, Amsterdam J, Newberg A, Allison KC, Wintering N, Stunkard AJ. Differences
in serotonin transporter binding affinity in patients with major depressive disorder and night
eating syndrome. Eat Weight Disord. 2009 Mar; 14(1):45-50.
144. Rosenbaum M, Leibel RL, Hirsch J. Obesity..Cumulative effect on BMI of daily minor
errors in the energy balance. N Engl J Med 1997; 337: 396-408.
145. Oike H, Oishi K, Kobori M. Nutrients, Clock Genes, and Chrononutrition. Curr Nutr Rep.
2014 Apr 27;3:204-212.

146. Zulet MA, Bondia-Pons I, Abete I, de la Iglesia R, López-Legarrea P, Forga L, Navas-
Carretero S, Martinez JA. The reduction of the metabolic síndrome in Navarra-Spain-
(RESMENA-S) study: a multidisciplinary strategy base don chrononutrition and nutritional
education, together with dietetic and psychological control.Nutr Hosp. 2011 Jan-
Feb;26(1):16-26.
147. Andersen CJ, Blesso CN, Lee J, Barona J, Shah D, Thomas MJ, Fernandez ML. Egg
consumption modulates HDL lipid composition and increases the cholesterol accepting
capacity of serum in metabolic syndrome. Lipids. 2013 Jun;48(6):557-67.
148. Smith KL, Straker LM, Kerr DA, Smith AJ. Overweight adolescents eat what? And when?
Analysis of consumption patterns to guide dietary message development dor intervention. J
Hum Nutri Diet.2014 Aug 26. Doi:10. 1111/jhn.12263.
149. Mistlberger RE. Circadian food anticipatory activity: formal models and physiological
mechanisms. Neurosci. Biobehav. 1994. Rev. 18:171-195.
150. Rajaratnan SMW, Arendt J. Health in a 24 h society. The Lancet 2001. 358:999-1005.
151. Mrosovsky N. Beyond the suprachiasmatic nucleus. Chronobiol. Internat. 2003. 20:1-8.
152. Reinberg A. Chronobiology and nutrition.1983. Biological Rhythms and Medicine.
(Ed.Reinberg A, Smolensky MH). Springer Verlag, New York. Pp.265-300.
153. Aschoff J, von Goetz C, Wildgruber C, Weber RA. Meal timing in humans during isolation
without time cues. 1986. J. Biol. Rhythms 1:151-162.
154. Leibowitz SF. Neurochemical-neuroendocrine systems in the brain controlling
macronutrient intake and metabolism. 1992. TINS 15:491-497.

155. Russel J. Reiter. Melatonin: clinical relevance.2003. Best Practice & Research Clinical
Endocrinology and Metabolism. Vol.17, No.2,pp. 273-285.
156. G. Bedogni et al, Cross-calibration of eight-polar bioelectrical impedance analysis versus
dual-energy X-ray absorptiometry for the assessment of total and appendicular body
composition in healthy subjects aged 21-82 years, Annals of Human Biology. 2003. Vol. 30,
No. 4, pp. 380-391.
157. S Kriemler, J Puder, L Zahner, R Roth, C Braun-Fahrländer and G Bedogni. Cross-
validation of bioelectrical impedance analysis for the assessment of body composition in a
representative sample of 6- to 13-year-old children. European Journal of Clinical Nutrition
(2009) 63, 619–626; doi:10.1038/ejcn.2008.19; published online 20 February 2008.
158. Pouliot MC, Despres JP, Lemieux S et al. Waist circumference and abdominal sagittal
diameter: best simple anthropometric indexes of abdominal visceral adipose tissue
accumulation and related cardiovascular risk in men and women. Am J. Cardiol 1994; 73:460-
8.
159. Klein S, Allison DB, Heymsfield SB, et al.Waist circumference and cardiometabolic risk: a
consensus statement from shaping america´s health: Association for weight management
and obesity prevention; NASSO, The Obesity Society; the American Society for Nutrition and
American Diabetes Association. Obesity Vol. 15 No. 5 May 2007. 1061.
160. Thompson B. Foundations of behavioral statistics: An insight based approach.2006. New
York: Guilford.
161. Shapiro S Wilk MB. An analysis of variance test for normality complete samples).1965.
Biometrika 52 (3-4):pp591-611.

162. Cohen J. Statistical Power Analysis for the Behavioral Sciences. 1988. 2nd. edit.Hillsdale.
Erlbaum NJ.(1th edition, 1977 New York: Academic Press).
163. Spiegel MR, Schiller J, Srinivasan RA. Analisis de la varianza. Probabilidad y estadística
(Schaum´s Outline of Theory and Problems of Probability and Statistics) Schaum(2ª,edición).
México DF:McGraw-Hill. Pp.335-371.
