learning goals for this file - the medical university of...
TRANSCRIPT

Pharmacology Unit 1 Page 1 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
Learning goals for this file:
1) Definition of terms 2) Group pharmacodynamic concepts in one part of your brain, for instance:
a. Efficacy b. Potency c. Dose-response curves d. Toxicity e. Safety f. Adverse effects
3) Group pharmacokinetic concepts in another part of your brain a. Absorption b. Distribution c. Biotransformation (phase I, phase II)
4) Drug-drug interactions a. CYP interaction (inducer, inhibitor, substrate) b. Other reasons for drug-drug interactions
5) Drug dosing 6) Monitoring 7) Bioavailability 8) Cultural awareness in pharmacotherapy 9) Pharmacogenomics

Pharmacology Unit 1 Page 2 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
First PHARMACODYNAMICS, an excursion to ADRs, then PHARMACOKINETICS… PHARMACODYNAMICS: effects of drugs on the body 1. EFFICACY:
• the degree to which a drug (or class of drug) is able to induce maximal effects of a defined clinical outcome (e.g. pain relief, lowering of BP)
• allows us to compare different drugs and drug classes (e.g. different pain relievers or classes of pain relievers).
• this has NOTHING to do with the amount (dose) of drug used for this maximal effect. • Example: morphine may have the ability to completely relieve post-op pain (moderate
to severe pain), while ibuprofen cannot achieve this maximal effect regardless of the dose of ibuprofen used (less efficacious than morphine and cannot manage severe pain).
2. POPULATION STUDIES FOR DRUG EFFICACY AND TOXICITY:
• Biologic variables usually graph out as a normal or “bell-shaped” curve • The mean = the median • Data clusters around the mean/median • Could be descriptive variables (e.g. physical linear height), or dose at which
persons exhibit toxicity to drugs. • Note that “normal” is usually defined as within 2 SD (standard deviations) of the
mean and this includes 95% of the population studied • Those falling outside the 2 SD range are often called “outliers” and include the
other 5% of the population • Thus, response to a drug that is experienced by the “normal” 95% of the population
may NOT be what your particular patient experiences – they may be in the upper or lower outlying area of this normal distribution curve – and there is no way to predict this
We are usually measuring what are called continous biologic variables that occur within a particular range. These usually will plot out as a “normal” bell-shaped curve. (Example: serum cholesterol, blood pressure, temperature)

Pharmacology Unit 1 Page 3 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
3. DOSE-RESPONSE CURVES: plots dose of a drug vs. response • In general, as dose increases, concentration of drug at receptor sites increases and the
maximal response to a drug occurs when all receptors are occupied by the drug • in general, as the dose increases, the response increases
o for most drugs, at some point the response “plateaus” as all available receptor sites are occupied and increases in dose won’t give any greater response
o for some drugs, increasing doses continue to provide increasing response o for some drugs, a “flat” (plateau) response occurs with the dose that initially
gives the desired clinical response – there is only one dose of drug (e.g. Lescol-XL 80 mg gives the desired clinical response for LDL-C lowering, but raising the dose beyond this does not provide additional response)
See more on ED50 below…
Here, the dose-response curve is looking for the shock strength to convert (terminate) ventricular fibrillation. The shock strength in this case is like our drug dose BUT in this case the response curve is more linear.
Note that the response eventually reaches a plateau (“flattens out”) because all the receptors are occupied by the drug and no further increase in drug action is possible beyond a certain dose)

Pharmacology Unit 1 Page 4 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
4. POTENCY: ED50 & EC50
• DEFINITION: the dose (ED50) or concentration (EC50) of a drug required to produce 50% of that drug's maximal effect (there is another ED50 with a different meaning, see below).
• Allows you to compare drug doses (compare mg to mg effect) when both drugs are able to achieve the same target outcome (same endpoint efficacy) – this ED50 is usually what prescribers mean when they use this term – potency.
5. EFFECTIVENESS (the other ED50):
• ED50 means the dose at which 50% of the population exhibits the desired effect. • This distributes into the usual "bell shaped curve" for the population & will yield a value
called the ED50 = the median effective dose. • This is NOT the same ED50 as when used for measurement of potency! • THUS, THERE ARE TWO ED50 VALUES, MEANING TWO DIFFERENT THINGS !
In this picture, compare morphine (Drug A) & codeine (Drug B) – both can completely relieve severe pain (same EFFICACY), but compare their POTENCY by dose (compare mg for mg).

Pharmacology Unit 1 Page 5 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
6. DRUG DISCONTINUATION: • Some drugs cannot be abruptly discontinued • a withdrawal or discontinuance syndrome develops • the symptoms of the discontinuance syndrome vary according to the type of drug • as we study drugs, many will have this warning and the patient must be educated as to
this risk • the drug will need to be weaned over time to be discontinued
o often, another drug will be started to treat the condition as the initial drug is weaned
o titration is adjusting the dose up or down in incremental (small) amounts o the initial drug would be titrated down and the new drug would be titrated up o an example is seizure drugs – as the initial drug is titrated down, another
seizure drug is started at low dose and titrated up to therapeutic dose • Examples:
o suddenly stopping many anti-hypertensives (especially peripheral vasodilators) can cause acute hypertensive crisis
o suddenly stopping SSRI/SNRI antidepressants often causes withdrawal symptoms
o suddenly stopping epilepsy drugs may induce seizures 7. CLINICAL PARAMETERS AFFECTING DRUG RESPONSE:
• Clinical history & exam provides this information: o Age, sex, reproductive status (pregnant, lactating, birth control) o Medical comorbidities (the medical history) o Lifestyle choices (smoking, alcohol – the social history) o Organ function (usually the most important are liver & kidney) o Medication history – prescribed & OTC drugs/herbals o Genetic factors (if available)(pharmacogenomics)
• This determines a tailored therapeutic strategy for each patient o this is the prescriber’s responsibility o this is the opposite of “cookbook” medicine where every patient with the same
diagnosis gets the same treatment

Pharmacology Unit 1 Page 6 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
8. CULTURAL PARAMETERS AFFECT PHARMACOTHERAPEUTICS:
• Sometimes called ethnopharmacology • Considered part of health literacy • Essential to reduce health disparities • May be included as a social determinant of health
o What are determinants of health? o Biology & genetics, individual behavior, social environment, physical
environment, health services • Cultural competence vs. cultural knowledge vs. cultural awareness?
o From CDC: “Culture is the blended patterns of human behavior that include "language, thoughts, communications, actions, customs, beliefs, values, and institutions of racial, ethnic, religious, or social groups." Cultural competence is "a set of congruent behaviors, attitudes, and policies that come together in a system, agency, or among professionals that enables effective work in cross-cultural situations." "Competence" in the term cultural competence implies that an individual or organization has the capacity to function effectively "within the context of the cultural beliefs, behaviors, and needs presented by consumers and their communities."
o https://www.cdc.gov/nchhstp/socialdeterminants/definitions.html • Cultural competence vs cultural knowledge vs cultural awareness?
o From CDC: “Cultural competence emphasizes the idea of effectively operating in different cultural contexts, and altering practices to reach different cultural groups. Cultural knowledge, sensitivity, and awareness do not include this concept. Although they imply understanding of cultural similarities and differences, they do not include action or structural change”
o https://npin.cdc.gov/pages/cultural-competence#difference • Knowledge of cultural background may help:
o predict adherence to recommended treatment regimens o provide clues to treatment preferences o prompt diagnosis
• example: infant lead poisoning from use of “tiro” (eye cosmetic containing lead from the archives of the MMWR: http://www.cdc.gov/mmwr/preview/mmwrhtml/mm6130a3.htm
• Culturally competent care in pharmacotherapy: o Overcome language barriers (e.g., translators) and remember privacy issues in
using family translators o Communicate effectively in the context of cultural norms
• Hispanic cultures value eye contact as a measure of respect • Some Native American cultures feel that eye contact is disrespectful • Some Asian cultures nod for politeness, not to indicate agreement or
understanding • Be prepared to discuss culture-specific CAM
• Resources: o AHRQ: https://www.ahrq.gov/topics/topic-cultural-competence.html o AHRQ health literacy in pharmacy: https://www.ahrq.gov/professionals/quality-
patient-safety/pharmhealthlit/index.html o CDC: https://npin.cdc.gov/pages/cultural-competence

Pharmacology Unit 1 Page 7 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
ADVERSE DRUG REACTION (ADR): LOTS OF SYNONYMS – CAN BE CONFUSING • ALSO called Adverse Drug Event (ADE) • ALSO called Adverse Event (AE) or Adverse Effect (AE) • A big reason for emergency department visits:
http://jamanetwork.com/journals/jama/article-abstract/2585977 1. ALL ABOUT ADRs (ADEs)(AEs): • Definition:
o a reaction (clinical event) occurs that is not part of the expected therapeutic response to a drug (NOT predicted or expected)
o there are different TYPES of ADR (ADE)(AE) • Medical Error (Mistake):
o according to the Institute of Medicine (IOM), a medical error is any mistake made in diagnosis or treatment
o a type of medical error is a medication error, meaning mistakes made in prescribing transcribing, dispensing or administering mediation near miss: a mistake that has not caused harm preventable adverse event: harm is caused by a mistake
• ADR (ADE)(AE): o Definition: harm has been caused but NO mistake or error has been made
and the event could NOT have been prevented o Consider this as having occurred: any new clinical event involving a patient
on drug therapy o Clinician Response:
o stop one drug at a time to “de-challenge” o if this is NOT reported on the drug’s prescribing information, report
confirmed ADEs to the FDA; publish the ADE if possible Types of ADR (ADE)(AE): toxic (dose related), idiosyncratic, allergic, side effect • Dose-related:
o these are also called “TOXIC” o as the dose increases, so does the potential for adverse drug reaction
• Unpredictable: the typical ADR o either idiosyncratic (non-immunologic) o OR allergic (immunologic)
• Predictable drug reaction: o sometimes also called a side effect of the drug o an example is the dry mouth from anticholinergic effects of antihistamine drug o this is directly related to the drug’s mechanism of action (MOA) o this really isn’t an ADR

Pharmacology Unit 1 Page 8 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
More on predictable ADR (ADE)(AE) – also called SIDE EFFECTS: • can be predicted by the actual known pharmacologic MOA of the drug • Examples:
o thrush while taking antibiotics o hepatotoxicity from methotrexate (which is a cytotoxic drug) o skin photo-reactions to certain drugs
More on upredictable non-immunologic (idiosyncratic) ADR (ADE)(AE): • idiosyncratic means “unrelated to pharmacologic action” • Examples:
o hemolytic anemia from alpha-methyldopa (Aldomet) due to G-6-PD deficiency o drug-induced thrombocytopenia (DIT) from any drug (especially heparin,
quinidine, H2 blockers especially cimetidine) o non-immune anemia from antimalarial drugs
2014 Clinical Practice Guideline on Drug Allergy: https://www.guideline.gov/summaries/summary/48564/ Some good articles on this topic: • A good overview article (2003) from AAFP:
http://www.aafp.org/afp/2003/1101/p1781.html • An article on ethnicity & ADR from AAFP (2006):
http://www.aafp.org/afp/2006/1015/p1410a.html • An article on minimizing ADRs in older patients from AAFP (2007):
http://www.aafp.org/afp/2007/1215/p1837.html • Inappropriate prescribing in older patients and minimizing ADEs with information on
European STOPP/START (2009) http://www.biomedcentral.com/content/pdf/1471-2318-9-5.pdf
• Exanthematous drug eruptions (N Engl J Med, 28 June 2012): http://www.nejm.org/doi/full/10.1056/NEJMcp1104080
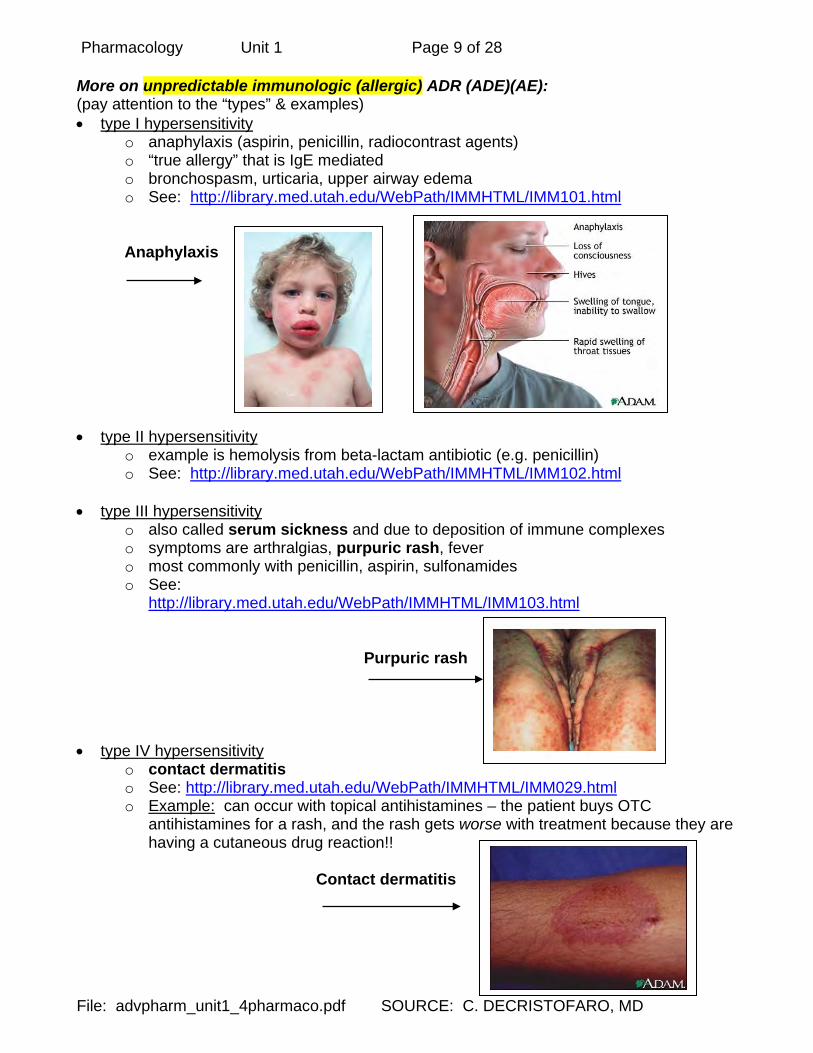
Pharmacology Unit 1 Page 9 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
More on unpredictable immunologic (allergic) ADR (ADE)(AE): (pay attention to the “types” & examples) • type I hypersensitivity
o anaphylaxis (aspirin, penicillin, radiocontrast agents) o “true allergy” that is IgE mediated o bronchospasm, urticaria, upper airway edema o See: http://library.med.utah.edu/WebPath/IMMHTML/IMM101.html
Anaphylaxis • type II hypersensitivity
o example is hemolysis from beta-lactam antibiotic (e.g. penicillin) o See: http://library.med.utah.edu/WebPath/IMMHTML/IMM102.html
• type III hypersensitivity
o also called serum sickness and due to deposition of immune complexes o symptoms are arthralgias, purpuric rash, fever o most commonly with penicillin, aspirin, sulfonamides o See:
http://library.med.utah.edu/WebPath/IMMHTML/IMM103.html
Purpuric rash • type IV hypersensitivity
o contact dermatitis o See: http://library.med.utah.edu/WebPath/IMMHTML/IMM029.html o Example: can occur with topical antihistamines – the patient buys OTC
antihistamines for a rash, and the rash gets worse with treatment because they are having a cutaneous drug reaction!!
Contact dermatitis

Pharmacology Unit 1 Page 10 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
• T-cell activation (a separate type) fixed drug reactions: o cutaneous drug reaction independent of IgE effects – these are exanthema-like
(macular & papular) eruptions – also called a fixed drug reaction o typical is the morbilliform drug rash of ampicillin & sulfa drugs
Fixed drug eruption (morbilliform rash) (morbilliform means resembling measles, since morbilli is Latin for measles)

Pharmacology Unit 1 Page 11 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
• Fas/ligand-induced apoptosis (a separate type) – erythema multiforme & SJS:
o erythema multiforme which can progress to Stevens-Johnson syndrome (SJS) with skin ulcerations and blisters (severe, can cause blindness)
o often after sulfa, allopurinol & anticonvulsants o Article: https://www.uspharmacist.com/article/cutaneous-adverse-reactions-
stevens-johnson-syndrome o extraordinarily important & the only time your instructor was ever sued for
malpractice (dismissed from the case as non-culpable – ask me if interested) SJS Corneal involvement Erythema multiforme SJS rash • Other poorly-defined types:
o lupus-like syndrome & anemia (procainamide, quinidine, phenytoin, hydralazine) o anticonvulsant hypersensitivity syndrome (fever, rash & hepatitis) o drug reaction with eosinophilia and systemic symptoms (DRESS) – may occur
several weeks after starting drug, relapses even with discontinuation, and may reactivate herpes virus infection: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3718748/
• Drug-induced photosensitivity (photoallergy): o Drug has a photosensitizer that when combined with UV radiation causes a
phototoxic (photoallergic) reaction – can be topical OR systemic drugs o See: http://www.dermnetnz.org/reactions/drug-photosensitivity.html and
https://www.ismp.org/newsletters/ambulatory/archives/200704_2.asp

Pharmacology Unit 1 Page 12 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
2. MORE ON DOSE-RELATED “TOXIC” ADR (ADE)(AE): Steep dose-response curves: • What is wrong with a “steep” dose-response curve?
o doesn't allow incremental dosage variations to achieve a graded response o a small increase in dose causes a large increase in response o the toxic side effects ALSO go up rapidly with even small increases in dose
• For most drugs, toxicity starts to show up as the dose goes higher o many adverse effects are linked to the therapeutic effect of the drug o lowering the dose of the drug usually reduces adverse effects but may also
reduce the therapeutic benefit o the way to lower the dose of a drug and still maintain its therapeutic
effectiveness is to add an adjunctive drug – this allows the use of the PRIMARY drug at a LOWER dose with LOWER toxicity
o typical examples are chemotherapy combinations, where the primary drug is very toxic and would not be tolerated without addition of adjunctive drugs that allow use of the primary drug at a lower dose
• Examples of dose-related “toxic” predictable ADR: o seizure from excessive lidocaine dosage o drug-drug interactions that cause toxicity due to higher serum level of a drug
such rhabdomyolysis from statin drug when taking simvastatin (Zocor) with other drugs using the CYP3A4 biotransformation system (e.g. verapamil)
Pharmacologic effects and toxic AEs: • Often, toxic (side) effects are a direct pharmacologic extension of the therapeutic
(desired) actions of the drug. • Primary pharmacologic effect:
o caused by same mechanism responsible for drug's primary effect. o Example: for Aspirin, pain reduction is from prostaglandin inhibition, and the GI
distress or bleeding is from the same cause. • Secondary pharmacologic effects:
o other responses that are not related to the drugs mechanism of action. o Example: Heparin causing hair loss and drug-induced thrombocytopenia (DIT)
Nutritional correlate: even nutrients can be thought of as drugs, with responses ranging from deficiency states, to optimal health at correct intake doses, to toxicity from excess.

Pharmacology Unit 1 Page 13 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
TD50 = Median Toxic Dose: • dose at which 50% of the population studied will show the toxic effect. LD50 = Median Lethal Dose: • dose at which 50% of the population will die • this is a value obtained through animal studies (& extrapolated to humans) or from
accidental overdoses in humans Therapeutic Index (TI) = Margin of Safety: • this compares the TD50 (or LD50) with the ED50, to determine the MARGIN OF SAFETY
for the drug. • this is distance away from the therapeutic dose and the toxic (or lethal) dose. • Narrow margin of safety may be tolerated when treating fatal illnesses, or illnesses for
which there is so far no cure (e.g. AIDS drugs, anti-cancer drugs) Adjunctive drugs: • Add an adjunctive drug to a primary drug which is toxic at therapeutic doses • Allows the use of the PRIMARY drug at a LOWER DOSE WITH LOWER TOXICITY • Enables you to utilize the primary drug • Often seen in chemotherapy combinations

Pharmacology Unit 1 Page 14 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
3. THERAPEUTIC WINDOW (THERAPEUTIC RANGE): efficacy vs. safety • because we know that there is a dose-response curve for the drug, and also possible
toxicity from the drug, we find out what dose range gives us benefit without toxicity o Too much drug toxicity o Too little drug subtherapeutic o Just the right amount therapeutic o Like Goldilocks & the Three Bears
• Example: o theophylline has a therapeutic window of a serum level from 5-15 mcg/mL (the
older range was 10-20 mcg/mL, but using this range caused a lot of toxicity from increased serum levels)

Pharmacology Unit 1 Page 15 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
PHARMACOKINETICS: The effects of the body on drugs (what the body does to the drug) • knowledge of pharmacokinetics of drugs allows you to better predict
o drug effects o drug-drug interactions o need for specific monitoring o drug dosing
1. OVERVIEW OF SOME PHARMACOKINETIC TERMS: • Addition:
o response of combined drugs is additive (1 + 1 = 2) o Example: each BP drug drops the BP 5 points, together BP drops 10 points
• Synergism: o effect by using combined drugs is greater than would be predicted than by
simple additive effects (greater than the combined responses of the individual drugs).
o This means that it isn’t 1 + 1 = 2…with synergism it is 1 + 1 > 2 o Example: each BP drug drops the BP 5 points, together BP drops 15 points
• Potentiation: o a drug with no effect will enhance the effect of another drug o Example: adjuvant drugs used in combination with the primary therapeutic drug
• Antagonism: o one drug inhibits the effect of another drug
• Disease effects: o disease condition may affect metabolism of drugs. o Example: reduced biotransformation of drugs in liver disease (e.g. cirrhosis) as
well as in cardiac disease (reduced liver blood flow) – higher drug levels result • Bioavailability:
o amount of unchanged drug that reaches the systemic circulation. o Factors include absorption from gut, plasma protein binding (albumin inert
binding sites), first pass (hepatic) biotransformation, presence of other drugs, lifespan issues, dosing regimen, clinical morbidities, intestinal and cellular P-glycoprotein (P-gp) transporters – affects onset AND duration of drug action
o Example: P-gp transporters (P-glycoprotein transporters) Cell membrane transporters that carry toxins & drugs out of the cell and
also found on the intestinal cells, pumps the drug BACK into the intestine and reduces systemic absorption
drug-drug and herbal-drug interactions may result from using/inhibiting the transporter

Pharmacology Unit 1 Page 16 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
• Drug elimination (excretion)(clearance):
o elimination of the drug from the body using various routes – renal (urine), hepatic, pulmonary, perspiration total clearance is the sum of all the clearance processes in the body impairment of any of these systems results in reduced drug clearance
o Typical clearance is by first order kinetics: A constant fraction of the drug in the body is eliminated per unit time The rate of elimination is proportional to the amount of drug in the body the MORE drug in the plasma, the longer time it takes to eliminate the LESS drug in the plasma, the shorter time it takes to eliminate The majority of drugs are eliminated in this way
o Another type of clearance is by zero order kinetics:
some special situation exists that causes the rate of elimination to be “constant” and not be related to the drug concentration in plasma.
This may occur when elimination processes are “saturated” (reached their functional limit) such as saturation of degrading enzymes in the liver, or saturation of transporters in the kidney.
First-order kinetics for drug clearance. • The fraction of the drug in the body
eliminated per unit time is determined by the elimination constant (kel).
• This is represented by the slope of the line of the log plasma concentration versus time.
An example of zero order kinetics is alcohol (ethanol) elimination. • The metabolic pathways
responsible for alcohol metabolism are rapidly saturated and that clearance is determined by how fast these pathways can work.
• The metabolic pathways work to their limit.
• A constant amount of drug is eliminated per unit time
Pharmacology Unit 1 Page 17 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
• Drug Dosing:
o Remember the elimination half-life: amount of time needed for the plasma concentration to drop by 50%
after the drug is discontinued determines how long the drug remains in the system often written as T1/2 or t1/2
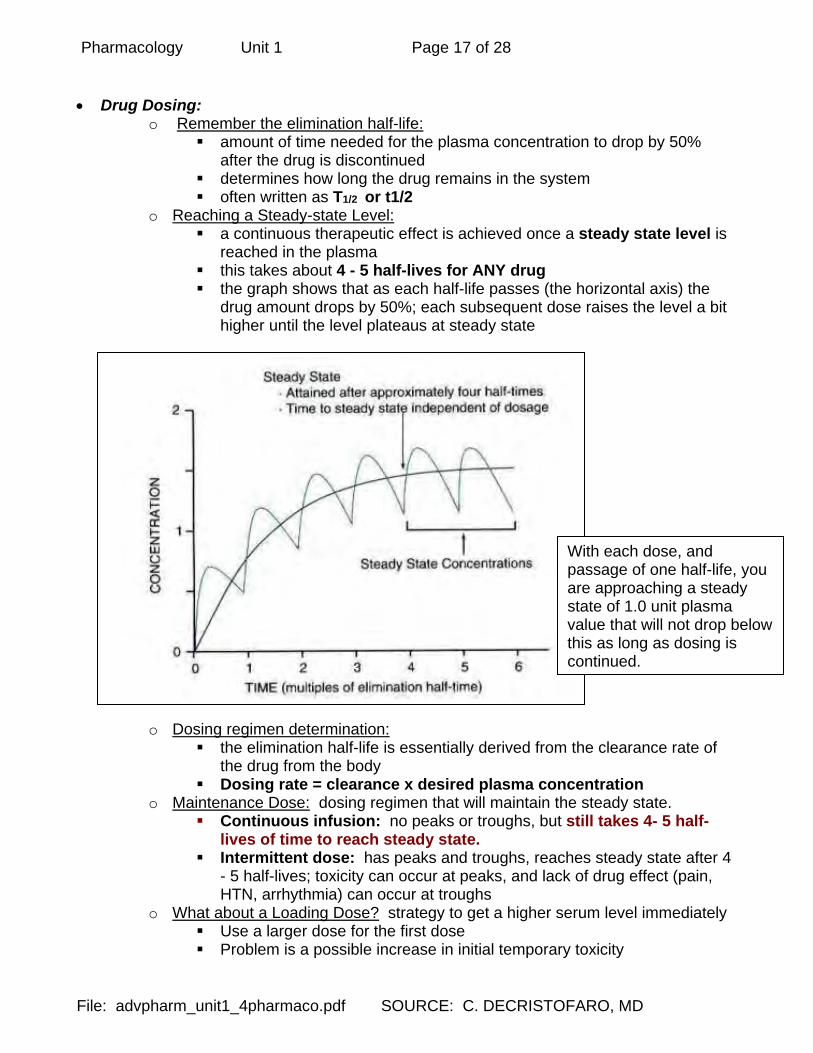
o Reaching a Steady-state Level: a continuous therapeutic effect is achieved once a steady state level is
reached in the plasma this takes about 4 - 5 half-lives for ANY drug the graph shows that as each half-life passes (the horizontal axis) the
drug amount drops by 50%; each subsequent dose raises the level a bit higher until the level plateaus at steady state
o Dosing regimen determination:
the elimination half-life is essentially derived from the clearance rate of the drug from the body
Dosing rate = clearance x desired plasma concentration o Maintenance Dose: dosing regimen that will maintain the steady state.
Continuous infusion: no peaks or troughs, but still takes 4- 5 half-lives of time to reach steady state.
Intermittent dose: has peaks and troughs, reaches steady state after 4 - 5 half-lives; toxicity can occur at peaks, and lack of drug effect (pain, HTN, arrhythmia) can occur at troughs
o What about a Loading Dose? strategy to get a higher serum level immediately Use a larger dose for the first dose Problem is a possible increase in initial temporary toxicity
With each dose, and passage of one half-life, you are approaching a steady state of 1.0 unit plasma value that will not drop below this as long as dosing is continued.

Pharmacology Unit 1 Page 18 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
2. OVERVIEW ON PHARMACOKINETICS: What is “ADME”? • Sometimes taught as ADME • Absorption • Distribution • Metabolization • Excretion ABSORPTION: affects the rate and amount of drug transfer to the blood • Barriers to absorption of oral drugs:
o GI dysfunction o P-gp transporters prevent absorption by intestinal cells (more on these later) o Even patient noncompliance can be thought of as a type of absorption problem
DISTRIBUTION TO TISSUES: • can the drug reach all the body compartments ?? • Factors in distribuation:
o blood flow – tissues with the highest blood flow receive the drug first o protein binding – drugs stuck to plasma proteins can only go where the proteins go
(stay in the vascular compartment) – mainly due to inert binding sites on albumin • if more than one drug is used, and both are protein bound, they will
compete for the inert binding sites • this will DISPLACE drug from the sites and RAISE the amount of free
(active) drug in the serum – may result in toxic effects o lipid solubility – ionized or not ionized at body temperature & pH?
• if the drug is not ionized (neutral electrical charge) it will be more lipid soluble and can pass through cell membrane phospholipid
• if the drug is ionized, then cell membranes (including vascular membranes) will stop its distribution
• see more below

Pharmacology Unit 1 Page 19 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
BIOTRANSFORMATION = METABOLIZATION: • What is biotransformation?
o The body CHANGES the drug chemically o Either inactivates the drug or makes it more water-soluble for easier renal
excretion o Multiple pathways used by the body for this biotransformation (more below)
• A lot has to do with how the drug is administered o Oral administration: absorbed from the GI tract and go to the liver via the hepatic
portal vein – the liver sees everything first (factors include liver blood flow, type of drug, other drugs being taken, genetic & disease states) – a first pass effect or first pass elimination occurs in the liver
o Non-oral administration (e.g. IV, IM, transdermal) does NOT have the first-pass effect, thus drug dosing is usually LOWER
o Rectal administration: • Absorption through the lower half of the rectum drains directly to the IVC &
therefore has its absorption directly into systemic blood (no first pass effects)
• BUT usually the suppository slips upward and is absorbed into portal circulation via superior hemorrhoidal vein (& then to liver -- with first pass effects) – due to anastomoses of veins there is often up to a 50% first pass effect
• Knowledge of biotransformation processes can be EXPLOITED by using pro-drugs o Pro-drugs: if the parent drug is a pro-drug, biotransformation actually converts it
into active drug o Example: amitriptyline (Elavil) is converted to nortriptyline (Pamelor) o Example: sulfasalazine is broken down by bacteria in the colon into two active
products, 5-aminosalicylic acid (5ASA) and sulfapyridine o Example: carisoprodol (Soma) is a non-CS muscle relaxant that is converted to
meprobamate, which is a CS sedative/hypnotic • The metabolite (the chemical that results from biotransformation) may be toxic
o Example: Aspartame (artificial sweetener) has as one of its metabolites the poison, formaldehyde.
EXCRETION: usually renal, but also includes perspiration, GI routes.

Pharmacology Unit 1 Page 20 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
MORE DETAILS ON DRUG IONIZATION AND LIPID SOLUBILITY: Ionization and lipid solubility: • Electrical charge and lipid solubility:
o IONIZED (electrically charged, polar) molecules are more hydrophilic o Non-ionized (electrically neutral, nonpolar) molecules are more lipophilic
• Lipophilic distribution: o a drug that is lipophilic will be better absorbed from the gut o also, a lipophilic drug is better able to move across compartments because the
lipophilic drug can dissolve into cell membrane phospholipid o even special barriers in the body such as the blood brain barrier (BBB) cannot
keep this type of drug from entering that body compartment o hydrophilic drugs dissolve well into the vascular compartment, and are
distributed well via the bloodstream, but may not be able to get out into the tissues to exert drug action on cells
o Clinical example: at body pH, morphine is ionized, fentanyl is not – thus, fentanyl narcotic has a faster onset of action (distributed faster to the tissues)
• What makes a molecule ionized or not? o this is due to the relationship between a drugs pKa and the pH of the
surroundings – the pKa is the the pH at which the drug is 50% dissociated (acids and bases “dissociate” or break apart based on the local pH & the drug pKa)
o most drugs are either weak acids or weak bases & they act opposite of each other – RULE OF THUMB: Acids are most highly ionized (dissociated, polar, hydrophilic) at a high
pH (alkaline environment) and are neutral (undissociated, nonpolar, lipophilic) at low pH (acid environment) – remember acids/lipophilic/acid environment
Bases are most highly ionized (hydrophilic) at a low pH (acidic environment) and are neutral (lipophilic) at high pH (alkaline environment) – remember bases/lipophilic/basic environment
Ionization and drug absorption & drug action: • Example of ionization and lipophilicity on local anesthetic action:
o local anesthetics are weak bases and the closer the pKa of the local anesthetic to the local tissue pH, the MORE un-ionized it is (more lipophilic)
o lidocaine with pKa 7.7 has a faster onset than bupivicaine with pKa 8.3 o if the local tissue is alkalinized (adding bicarbonate to the local anesthetic
infiltration) then tissue pH is closer to bupivicaine pKa & faster onset results • Example of ionization and drug absorption:
o Weakly acid drug such as aspirin in the stomach (an acidic environment) will be more unionized (undisocciated, nonpolar, lipophilic) & better absorbed
o However, a weak base such as quinidine in the acid stomach environment is mostly ionized (dissociated, polar, hydrophilic) & thus less stomach absorption
o However, a weak acid in the duodenum (a more alkaline pH) is more ionized (dissociated, polar, hydrophilic) and this would inhibit absorption – this would be offset by the large duodenal surface area that will enhance absorption

Pharmacology Unit 1 Page 21 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
DETAILS ON BIOTRANSFORMATION (METABOLIZATION): pharmacokinetics • First we talk about Phase I, then about Phase II. • BE CAREFUL
o This doesn’t mean that Phase I always comes before Phase II o Sometimes there is Phase I followed by Phase II; sometimes there is only
Phase I or only Phase II and Phase II can come before Phase I A. “PHASE I” “NON-SYNTHETIC” BIOTRANSFORMATION REACTIONS:
• Chemical reactions that occur due to activity of the microsomal P450 enzymes in the liver and intestine
• Other non-microsomal enzymes elsewhere in the body also do Phase I non-synthetic biotransformation reactions
• Non-synthetic means that the enzyme action is NOT “making” (synthesizing) a new organic molecule.
• The phrase “Phase I” doesn’t mean that it always occurs as the first chemical reaction affecting drugs – don’t be confused by the term.
1, What are P450 CYP microsomal enzymes? And how do we get the name?
• drugs are attached to a hemoprotein called cytochrome P450 or CYP450 o these proteins are enzymes that have “heme” o the same as is found in hemoglobin, myoglobin and other enzymes o Why “450” ? The "450" is because these chromic (colored) molecules
absorb light at this wavelength. o Why “CYP”? The CY comes from the word “cytochrome” and the P
comes from “protein” o Put it together: Cytochrome P-450 (their light spectrum absorption)
• These hemoprotein enzymes are capable of microsomal Phase I non-synthetic reactions:
o oxidation (adds oxygen) o hydroxylation (adds hydroxyl group, -OH) o reduction (adds Hydrogen, removes oxygen)
• These enzymes are located mostly in the LIVER and also in the INTESTINES 2. Lifespan issues:
• elderly & preadolescents have less ability to perform biotransformation • thus at risk of dose-related toxicity since dugs are not metabolized as well
3. What are the common CYP enzymes involved in drug biotransformation: • most commonly CYP3A4, CYP2D6, CYP2C19 (and others) • a drug that is biotransformed by a specific CYP enzyme is called a SUBSTRATE of
the enzyme • this is part of the pharmacokinetic information that is important to know for drugs • Importance in pharmacology:
o CYP enzyme issues create many drug-drug interaction problems o be on the lookout for these CYP enzymes to be mentioned as we study
specific drugs – this alerts you to the possibility of drug interaction

Pharmacology Unit 1 Page 22 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
Please SEE the SEPARATE notes file about CYP enzymes and drug-drug interactions 5. Drug-drug interactions due to CYP-450 enzyme issues:
• Biotransformation reaction pathways are one of the most common causes of drug-drug interactions
• related to enzyme induction, enzyme inhibition, and shared pathways Enzyme induction:
• Some drugs induce (increase the amount) the amount of biotransformation enzymes in the liver.
• AS A RESULT, ANY DRUG USING THIS BIOTRANSFORMATION PATHWAY (substrate of the CYP enzyme) WILL BE DEGRADED AT A MORE RAPID RATE AND ITS SERUM LEVEL WILL BE LOWERED
• Inducing substance: o pollutants (charbroiled meats become carcinogenic nitrosamines by this
process) o ethanol (drinking alcohol) o cigarette smoking
• Drug interaction issue: o taking a primary drug along with an inducer drug will increase the
biotransformation of the primary drug and DECREASE the amount of primary drug released to the body (serum), possibly resulting in a lower therapeutic effect.
o this results in the need for an INCREASE in the dose of the primary drug o BUT what if the inducing substance is stopped – such as the patient
discontinues smoking – then the prescribed drug will be at a dose that is TOO HIGH and dose-related toxicity can occur
Enzyme inhibition:
• Some drugs inhibit (reduce the activity) biotransformation enzymes in the liver • AS A RESULT, ANY DRUG USING THIS BIOTRANSFORMATION PATHWAY
(substrate of the CYP enzyme) WILL BE DEGRADED AT A LESS RAPID RATE AND ITS SERUM LEVEL WILL BE INCREASED
• Inhibiting substance: o Grapefruit juice causes a 24-hour inhibiting effect o Many SSRI drugs are inhibitors o Example of a drug deliberately designed to create this effect in HIV treatment
(combine cobicistat with other HIV drugs to increase their benefit): http://content.govdelivery.com/accounts/USFDA/bulletins/d16150
• Drug interaction issue: o Taking a primary drug along with an inhibitor drug will reduce the
biotransformation of the primary drug and INCREASE the amount of primary drug, possibly resulting in a dose-related toxicity
Acronym mnemonic: A-SLAVED-LIVER (amiodarone, simvastatin, lovastatin, atorvastatin, verapamil, erythromycin, diltiazem, cLarithromycin, itraconazole, voriconazole, colchicinE, ritonavir)

Pharmacology Unit 1 Page 23 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
Drug interaction checkers:
• CYP information for drug-drug interactions: http://medicine.iupui.edu/clinpharm/ddis/ (chart has substrates, inhibitors, inducers)
• FDA drug interaction checker: http://www.drugs.com/drug_interactions.html • Other web drug interaction checkers:
o http://www.rxlist.com/drug-interaction-checker.htm • Use smartphone/tablet hand-held point of care pharmacology apps check for
these interactions • Printable pocket card:
https://static.medicine.iupui.edu/divisions/clinpharm/content/p450_Table_Oct_11_2009.pdf
Example of CYP 3A4 enzyme drug-drug interaction: • Erythromycin is a CYP34 INHIBITOR
o THUS, statins may be more toxic when administered with erythromycin (statin levels rise since the degrading CYP enzyme is inhibited, thus potentially increasing statin toxicity)
• Grapefruit juice (and pomegranate juice) is a known CYP3A4 INHIBITOR o can cause an increase in the level of some statins (e.g. simvastatin) possibly
increasing statin toxicity o this fruit juice can inhibit BOTH intestinal CYP enzymes as well as affecting liver
CYP enzymes (the intestinal enzymes are inhibited by as little as 4 ounces of juice)
o the effect of the grapefruit juice lasts 24 hours
AND LASTLY – another way to get a drug-drug interaction from CYP enzyme issues Shared Biotransformation Pathways and drug-drug interactions: • The situation: two or more drugs share the same CYP pathway for biotransformation
o if two people are in a room and they want to leave (get biotransformed and eliminated) they have to leave by the SAME shared doorway
o the people have to WAIT to pass through the doorway and their elimination will take LONGER
o the same thing occurs with drugs – if drugs are SHARING a biotransformation pathway, they will be both be biotransformed by the same pathway and thus eliminated at a SLOWER RATE and the level of all the affected drugs sharing the pathway will be RAISED
• Example: Cimetidine (Tagamet) inhibits the metabolism of anticoagulants & sedatives, increasing their blood levels to potentially toxic levels; strategy choose another H2B (H2-blocker) such as Axid or Pepcid which don’t have this issue.

Pharmacology Unit 1 Page 24 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
NOTE: (continue to read more) • not ALL drugs use the CYP system, this is just ONE type of Phase I biotransformation • and also there are Phase II biotransformation systems • ALSO, some drugs do not use EITHER of these systems
o Example: thyroxine (Synthroid) undergoes metabolism by de-iodination & renal excretion
• AND drug interactions may occur due to other issues (e.g., P-glycoprotein transport system, see more below)
6. Non-microsomal Phase I non-synthetic biotransformation systems: • these are not usually inducible • they CAN be blocked (inhibited) by other drugs or substances • Examples of non-microsomal biotransformation reactions:
o hyodrolysis in plasma by esterases (suxamethonium by cholinesterase) o dehydrogenase enzymes in liver such as alcohol and aldehyde
dehydrogenase (works on ethanol) o mitochondrial monoamine oxidase enzyme (biotransforms tyramine,
noradrenalin, dopamine) o xanthene oxidase that degrades 6-mercaptopurine (6-MP), and also produces
uric acid o other specific enzymes for specific drugs (e.g. tyrosine hydroxylase, dopa-
decarboxylase) • Examples of substances that can BLOCK (inhibit) these non-microsomal enzymes:
o monoamine oxidase inhibitor (MAOI) which is the first (oldest) antidepressant drug
o anticholinesterase drugs that are used to treat dementia
WHAT HAPPENS AFTER “PHASE I”? • It can proceed to Phase II • OR It may be the only biotransformation that occurs • OR It may even be PRECEDED by Phase II. • Thus, these different biotransformation reactions can occur in any order and do not
need to all be performed for every drug. SEE BELOW…

Pharmacology Unit 1 Page 25 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
B. “PHASE II” “SYNTHETIC” BIOTRANSFORMATION REACTIONS: • usually occurs in sequence after the Phase I microsomal (or non-microsomal)
reactions, but not always — may occur first. • The term synthetic means that a brand-new organic chemical compound is made due
to the action of enzymes on the drug. 1. Types of “synthetic” chemical reactions:
• Conjugation reactions: o synthesis of a new compound by enzymatically adding organic
compounds o examples of these organic compounds that are chemically added are
glucuronide, sulfates, glutathion o the hepatic enzymes that do this are called the UGT enzymes
• Acetylation reactions: o acetyl-Co-A donates an acetyl group to the drug.
2. Clinical correlate:
• Drug overdose: o if too much drug has been taken in all at once, there aren't enough
organic substances in the liver to metabolize them. o Example: in acetaminophen overdose, intermediate "reactive"
metabolites accumulate that may even cause cell death (liver failure). • Genetics:
o inherited differences in amounts of available synthetic Phase II biotransformation enzymes
o this is a part of the study of pharmacogenomics o Example:
in inherited pseudocholinesterase deficiency, the post-op patient can't metabolize succinyl choline
remains paralyzed for hours due to persistence of this drug in their body
don’t pull out the ET tube until you are sure the patient is breathing spontaneously
followup care includes genetic counseling of the patient’s family members

Pharmacology Unit 1 Page 26 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
C. P-GLYCOPROTEIN (P-GP) TRANSPORTERS: • a cell membrane transporter that exists to transport toxins out of our cells • often called an “efflux pump” since it pumps drugs OUT of the cell or body compartment • Location in the body:
o on specialized cells serving as barriers to body compartments o occurs on gut intestinal cells and pumps drugs BACK into the intestine (reducing
absorption of drugs) o occurs in the brain, pumping drug OUT of the brain (part of the BBB)
• Overall effect: o to REDUCE the bioavailability of the drug to the body or to the cell
Genetic variability in P-gp transporters: • Depending on our genetics, this transporter exists in larger or smaller amounts on our
cells, and also exists in different amounts on different cell tissue types (e.g. may be very large expression in the brain and less so in other body regions)
P-gp action and drug bioavailabilty: • If two substances uses this transporter, there will be competition for the transporter
o will result in higher level of both substances reaching the body or body compartment & potentially toxic levels
• If a substance is an inhibitor of P-gp o higher levels of any substance using the P-gp can enter the body or body
compartment, resulting in potentially toxic levels • Example:
o protease inhibitor drugs (PIs) for HIV infection are both substrates (“use” the P-gp transporter) and inhibitors of the transporter! This can help explain the very large differences in penetration of the tissues with these drugs among patients. A specific issue is lack of penetration into the CSF in the brain.
o Even garlic supplements can interact with PIs, due to the intestinal P-gp transporter interaction
The blood brain barrier (BBB), also called the hematoencephalic barrier, prevents substances from entering the brain AND pumps substances out of the brain if they’ve managed to get in: 1) tight junctions in epithelial cells prevent
polar (ionized) substances from getting into the brain
2) nonpolar (unionized) substances are lipophilic and are able to enter, as are gases
3) P-gp transporters pump substances OUT of the brain if they’ve managed to get in

Pharmacology Unit 1 Page 27 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
MINIMIZING DRUG-DRUG INTERACTIONS AND THEIR EFFECTS: • continued awareness of potential for drug interactions increased monitoring • possibly choose a different drug if interactions are a concern due to CYP or other issues • educate patients regarding dietary influences on drugs
o everything from grapefruit and pomegranate juice to garlic • use new technology that analyzes DNA genetic variations in expression of CYP enzymes
o example is the SensitivityDetect test that measures genetic variations in o measures variations in the CYP2C9 and VKORC1 genes which determine
sensitivity to warfarin • Warn patients about potential drug-drug interactions and educate regarding symptoms of
toxicity or symptoms of drug ineffectiveness • Avoid polypharmacy by carefully evaluating need for each drug and reducing amounts of
different drugs being used at the same time in the same individual ANOTHER ISSUE WITH DRUGS – LQTS (LONG QT INTERVAL SYNDROME): • Causes sudden loss of consciousness (LOC) and death • EKG findings are QT interval prolongation and tachycardia • The fatal arrhythmia is “torsades de pointes” • This syndrome can be inherited (more predisposed, even without drugs) (congenital
LQTS) • This syndrome may occur in ANYONE with certain drugs or drug combinations • More common in antibiotic drugs (discussed further in infectious disease Module) TIMING OF CLINICAL BLOOD SAMPLING FOR DRUG MONITORING: Peak measurements: • absorption usually occurs within 2 hours of taking a drug orally • peak is after at least 2 hours has passed. Steady state measurements: • after about 3 - 5 half-lives take serum sample in the middle of the dosing interval • this gives you the steady state value. Unusual behaving drugs: • must memorize the appropriate timing • examples:
o Lithium level taken right before next dose is due o Digoxin level taken at least 6 hrs after last dose
Special Toxicity monitoring: • peak & trough levels are taken • Example:
o aminoglycoside level taken 1 hour after administration for peak, then just before next dose for trough

Pharmacology Unit 1 Page 28 of 28
File: advpharm_unit1_4pharmaco.pdf SOURCE: C. DECRISTOFARO, MD
PHARMACOGENOMICS – PERSONALIZED MEDICINE – A BRAND-NEW AGE!! • Understanding the human genome to BOTH explain and PREDICT drug reactions:
o adverse drug reactions o differences in efficacy between individuals and groups o tailoring drug therapies based on genetic information
• Many genes have been identified as markers for certain conditions (e.g. multiple myeloma, multiple sclerosis, restless legs syndrome, stroke):
o NIH clinical genome primer : https://www.nih.gov/news-events/news-releases/new-report-offers-primer-doctors-use-clinical-genome-exome-sequencing
o In Iceland, the “deCODE” project tied genes to stroke risk: http://www.gene.ch/genet/2003/Sep/msg00101.html
o In the USA, the NIH has dbGaP for “genome-wide” association risk analysis: http://www.nlm.nih.gov/archive//20120510/news/press_releases/dbgap_launchPR06.html
• What genetic information is important? most of the variability between people and groups resides in “single nucleotide polymorphisms” (SNPs)(pronounced “snips”)
o Read about SNPs: http://ghr.nlm.nih.gov/handbook/genomicresearch/snp o All the publicly available genome sequences at the NIH GenBank:
http://www.ncbi.nlm.nih.gov/Genbank/GenbankOverview.html • Pharmacogenomics Overview ::
o Good overview FAQs : https://www.genome.gov/27530645/ o Precision medicine : http://learn.genetics.utah.edu/content/precision/ o Pharmacogenomics database from Stanford University:
http://www.pharmgkb.org/ (check out some examples at https://www.pharmgkb.org/search/knownPairs.action )
o Beta-blocker drug response & genetics : http://www.ncbi.nlm.nih.gov/pubmed/21965354
o Children’s Hospital of Philadelphia CNV project : http://cnv.chop.edu/ (copy number variants are associated with some childhood conditions, such as autism)
• What are some examples of pharmacogenomics in current practice? o Those with pseudocholinesterase deficiency should not receive the muscle
relaxant succinylcholine (Anectine) – can receive genetic test for the disorder o Some Asians may need reduced dose of rosuvastatin (Crestor) o Bidil (isosorbide + hydralazine) approved for HF in African-Americans as an
identified racial subgroup o AmpliChip pharmacogenomic test DNA to genotype for the CYP2D6 &
CYP2C19 (involved in many psychiatric drug biotransformations): http://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/DeviceApprovalsandClearances/Recently-ApprovedDevices/ucm078879.htm
o Certain HLA alleles (HLA-B*1502), usualy in Asians & South Asian Indians, have been found to be more common in patients who get Stevens-Johnson-Syndrome (severe drug reaction) with carbamazepine (test available)
o Extreme weight gain with antipsychotics: http://www.nimh.nih.gov/science-news/2012/gene-variants-implicated-in-extreme-weight-gain-associated-with-antipsychotics.shtml