last lecture summary. new generation sequencing (ngs) the completion of human genome was just a...
TRANSCRIPT

Last lecture summary

New generation sequencing (NGS)• The completion of human genome was just a start of
modern DNA sequencing era – “high-throughput next generation sequencing” (NGS).
• New approaches, reduce time and cost.• Holly Grail of sequencing – complete human genome
below $ 1000.• 1st generation – Sanger dideoxy method• 2nd generation – sequencing by synthesis
(pyrosequencing)• 3rd generation – single molecule sequencing

cDNA, EST libraries• cDNA – reverse transcriptase, containsonly expressed genes (no introns)cDNA library – a collection of different DNA sequences that have been incorporated into a vector
• EST – Expressed Sequence Tag• short, unedited (single-pass read),
randomly selected subsequence (200-800 bps) of cDNA sequence generated either from 5’ or from 3’
• higher quality in the middle
• cDNA/EST – direct evidence of transcriptome

What is sequence alignment ?
CTTTTCAAGGCTTA GGCTTATTATTGC
CTTTTCAAGGCTTA GGCTATTATTGC
CTTTTCAAGGCTTA GGCT-ATTATTGC
Fragments overlaps

What is sequence alignment ?
CCCCATGGTGGCGGCAGGTGACAG CATGGGGGAGGATGGGGACAGTCCGG TTACCCCATGGTGGCGGCTTGGGAAACTT TGGCGGCTCGGGACAGTCGCGCATAAT CCATGGTGGTGGCTGGGGATAGTA TGAGGCAGTCGCGCATAATTCCG
TTACCCCATGGTGGCGGCTGGGGACAGTCGCGCATAATTCCG
“EST clustering”
CCCCATGGTGGCGGCAGGTGACAGCATGGGGGAGGATGGGGACAGTCCGG TTACCCCATGGTGGCGGCTTGGGAAACTTTGGCGGCTCGGGACAGTCGCGCATAATCCATGGTGGTGGCTGGGGATAGTATGAGGCAGTCGCGCATAATTCCG
consensus
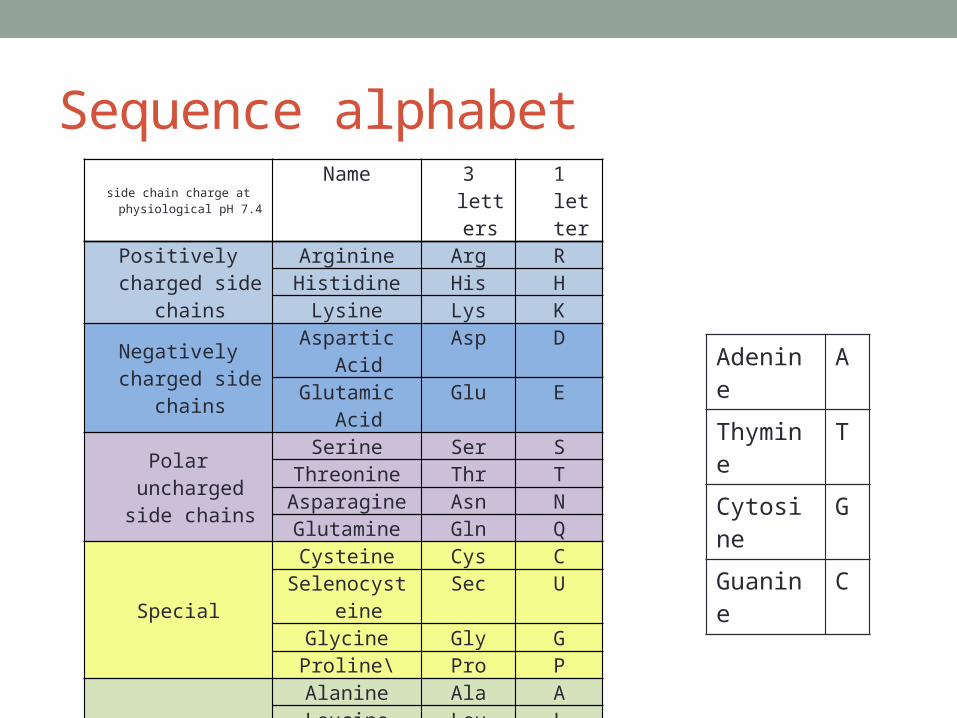
Sequence alphabetside chain charge at physiological
pH 7.4 Name 3 letters 1 letter
Positively charged side chains
Arginine Arg RHistidine His H
Lysine Lys KNegatively charged
side chainsAspartic Acid Asp DGlutamic Acid Glu E
Polar uncharged side chains
Serine Ser SThreonine Thr TAsparagine Asn NGlutamine Gln Q
Special
Cysteine Cys CSelenocysteine Sec U
Glycine Gly GProline\ Pro P
Hydrophobic side chains
Alanine Ala ALeucine Leu L
Isoleucine Ile IMethionine Met M
Phenylalanine Phe FTryptophan Trp W
Tyrosine Tyr YValine Val V
Adenine A
Thymine T
Cytosine G
Guanine C

Sequence alignment• Procedure of comparing sequences• Point mutations – easy
• More difficult example
• However, gaps can be inserted to get something like this
ACGTCTGATACGCCGTATAGTCTATCTACGTCTGATTCGCCCTATCGTCTATCT
ACGTCTGATACGCCGTATAGTCTATCTCTGATTCGCATCGTCTATCT
ACGTCTGATACGCCGTATAGTCTATCT----CTGATTCGC---ATCGTCTATCT
gapless alignment
gapped alignmentinsertion × deletionindel

Why align sequences – continuation• The draft human genome is available• Automated gene finding is possible
• Gene: AGTACGTATCGTATAGCGTAA• What does it do?
• One approach: Is there a similar gene in another species?• Align sequences with known genes• Find the gene with the “best” match

Flavors of sequence alignment
pair-wise alignment × multiple sequence alignment

Flavors of sequence alignment
global alignment × local alignment
global
local
align entire sequence
stretches of sequence with the highest density of matches are aligned, generating islands of matches or subalignments in the aligned sequences

New stuff

Evolution
wikipedia.org
common ancestors

Evolution of sequences• The sequences are the products of molecular evolution.• When sequences share a common ancestor, they tend to
exhibit similarity in their sequences, structures and biological functions.
Similar functionSequence similarity Similar 3D structure
Protein1 Protein2
DNA1 DNA2
However, this statement is not a rule. See Gerlt JA, Babbitt PC. Can sequence determine function? Genome Biol. 2000;1(5) PMID: 11178260
Similar sequences produce similar proteins

Homology• During the time period, the molecular sequences undergo
random changes, some of which are selected during the process of evolution.
• Selected sequences accumulate mutations, they diverge over time.
• Two sequences are homologous when they are descended from a common ancestor sequence.
• Traces of evolution may still remain in certain portions of the sequences to allow identification of the common ancestry.
• Residues performing key roles are preserved by natural selection, less crucial residues mutate more frequently.

Orhology, paralogy I• Orthologs – homologous proteins from different species
that possess the same function (e.g. corresponding kinases in signal transduction pathway in humans and mice)
• Paralogs – homologous proteins that have different function in the same species (e.g. two kinases in different signal transduction pathways of humans)
However, these terms are controversially discussed:Jensen RA. Orthologs and paralogs - we need to get it right. Genome Biol. 2001;2(8), PMID: 11532207 and references therein

Orthology, paralogy II• Orthologs – genes separated by the
event of speciation• Sequences are direct descendants of a
common ancestor.• Most likely have similar domain structure, 3D structure and
biological function.
• Paralogs – genes separated by the event of genetic duplication• Gene duplication: An extra copy of a gene. Gene duplication is a
key mechanism in evolution. Once a gene is duplicated, the identical genes can undergo changes and diverge to create two different genes.
http://www.globalchange.umich.edu/globalchange1/current/lectures/speciation/speciation.html

Gene duplication
1. Unequal cross-over
2. Entire chromosome is replicated twice• This error will result in one of the daughter cells having an extra
copy of the chromosome. If this cell fuses with another cell during reproduction, it may or may not result in a viable zygote.
3. Retrotransposition• Sequences of DNA are copied to RNA and then back to DNA
instead of being translated into proteins resulting in extra copies of DNA being present within cell.
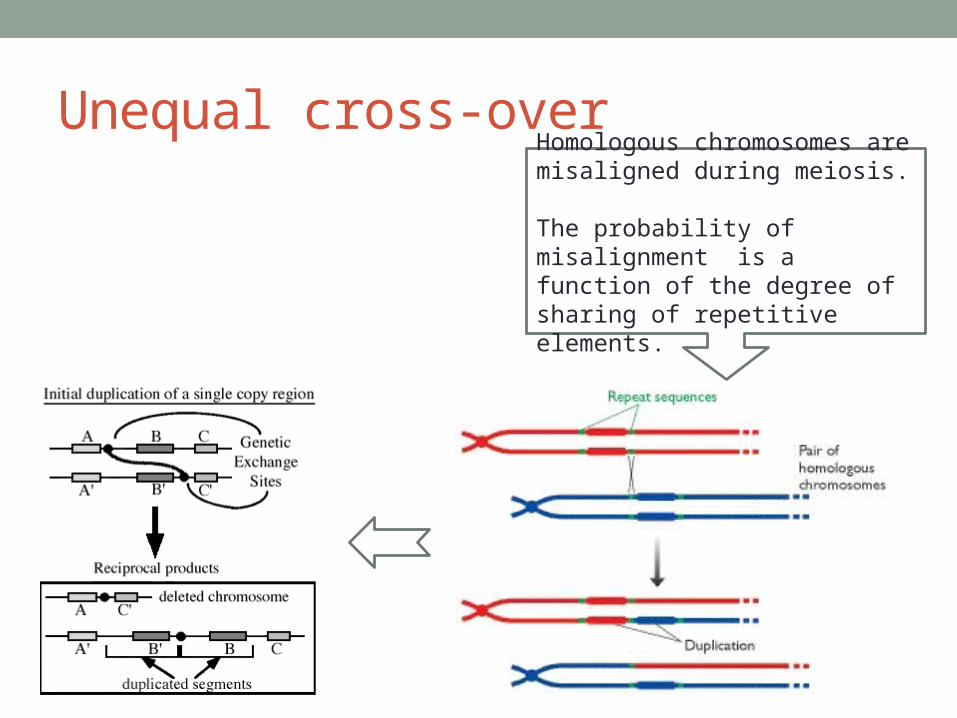
Unequal cross-overHomologous chromosomes are misaligned during meiosis.
The probability of misalignment is a function of the degree of sharing of repetitive elements.

• Comparing sequences through alignment – patterns of conservation and variation can be identified.
• The degree of sequence conservation in the alignment reveals evolutionary relatedness of different sequences
• The variation between sequences reflects the changes that have occurred during evolution in the form of substitutions and/or indels.
• Identifying the evolutionary relationships between sequences helps to characterize the function of unknown sequences.
• Protein sequence comparison can identify homologous sequences from common ancestor 1 billions year ago (BYA). DNA sequences typically only 600 MYA.

Identity matrix
Scoring systems I• DNA and protein sequences can be aligned so that the
number of identically matching pairs is maximized.
• Counting the number of matches gives us a score (3 in this case). Higher score means better alignment.
• This procedure can be formalized using substitution matrix.
A T T G - - - TA – - G A C A T
A T C G
A 1
T 0 1
C 0 0 1
G 0 0 0 1
How looks such a substitution matrix for proteins?
20x20 unity matrix.

Scoring systems II• For nucleotide sequences identity matrix is usually good
enough.• For protein sequences identity matrix is not sufficient to
describe biological and evolutionary proceses.• It’s because amino acids are not exchanged with the same
probability as can be conceived theoretically.• For example substitution of aspartic acids D by glutamic acid E
is frequently observed. And change from aspartic acid to tryptophan W is very rare.
• Why is that?1. Triplet-based genetic code
GAT (D) → GAA (E), GAT (D) → TGG (W)
2. Both D and E have similar properties, but D and W differ considerably. D is hydrophylic, W is hydrophobic, D → W mutation can greatly alter 3D structure and consequently function.

Genetic code
http://www.doctortee.com/dsu/tiftickjian/bio100/gene-expression.html

Gaps or no gaps

Scoring DNA sequence alignment (1)• Match score: +1• Mismatch score: +0
• Gap penalty: –1
•
ACGTCTGATACGCCGTATAGTCTATCT ||||| ||| || ||||||||----CTGATTCGC---ATCGTCTATCT
• Matches: 18 × (+1)• Mismatches: 2 × 0• Gaps: 7 × (– 1)
Score = +11

Length penalties• We want to find alignments that are evolutionarily likely.• Which of the following alignments seems more likely to
you?
ACGTCTGATACGCCGTATAGTCTATCTACGTCTGAT-------ATAGTCTATCT
ACGTCTGATACGCCGTATAGTCTATCTAC-T-TGA--CG-CGT-TA-TCTATCT
• We can achieve this by penalizing more for a new gap, than for extending an existing gap

Scoring DNA sequence alignment (2)• Match/mismatch score: +1/+0
• Origination/length penalty: –2/–1
•
ACGTCTGATACGCCGTATAGTCTATCT ||||| ||| || ||||||||----CTGATTCGC---ATCGTCTATCT
• Matches: 18 × (+1)• Mismatches: 2 × 0• Origination: 2 × (–2)• Length: 7 × (–1)
Score = +7

Substitution matrices• Substitution (score) matrices show scores for amino acids
substitution. Higher score means higher probability of mutation.
• Conservative substitutions – conserve the physical and chemical properties of the amino acids, limit structural/functional disruption
• Substitution matrices should reflect:• Physicochemical properties of amino acids.• Different frequencies of individual amino acids occuring in proteins.• Interchangeability of the genetic code.

PAM matrices I• How to assign scores? Let’s get nature – evolution –
involved!• If you choose set of proteins with very similar sequences,
you can do alignment manually.• Also, if sequences in your set are similar, then there is high
probability that amino acid difference are due to single mutation.
• From the frequencies of mutations in the set of similar protein sequences probabilities of substitutions can be derived.
• This is exactly the approach take by Margaret Dayhoff in 1978 to construct PAM (Accepted Point Mutation) matrices.
Dayhoff, M.O., Schwartz, R. and Orcutt, B.C. (1978). "A model of Evolutionary Change in Proteins". Atlas of protein sequence and structure (volume 5, supplement 3 ed.). Nat. Biomed. Res. Found.. pp. 345–358.

PAM matrices II• Alignments of 71 groups of very similar (at least 85%
identity) protein sequences. 1572 substitutions were found.• These mutations do not significantly alter the protein
function. Hence they are called accepted mutations (accepted by natural selection).
• Probabilities that any one amino acid would mutate into any other were calculated.
• If I know probabilities of individual amino acids, what is the probability for the given sequence?• Product
• Thus probabilities are converted to logarithms, and an alignment score can be calculated by summation.
Excellent discussion of the derivation and use of PAM matrices: George DG, Barker WC, Hunt LT. Mutation data matrix and its uses. Methods Enzymol. 1990,183:333-51. PMID: 2314281.

PAM matrices III• Dayhoff’s definition of accepted mutation was thus based
on empirically observed amino acids substitutions.• The used unit is a PAM. Two sequences are 1 PAM apart
if they have 99% identical residues.• PAM1 matrix is the result of computing the probability of
one substitution per 100 amino acids.• PAM1 matrix represents probabilities of point mutations
over certain evolutionary time.• in Drosophila 1 PAM corresponds to ~2.62 MYA• in Human 1 PAM corresponds to ~4.58 MYA

PAM1 matrix
numbers are multiplied by 10 000

Higher PAM matrices• What to do if I want get probabilities over much longer
evolutionary time? • i.e. I want to align sequences with far less than 85%
identity.• Dayhoff proposed a model of evolution that is a Markov
process.• We already met (in Lin Alg lecture) linear dynamical
system, which is a case of Markov process.

Linear dynamical system IA new species of frog has been introduced into an area where it has too few natural predators. In an attempt to restore the ecological balance, a team of scientists is considering introducing a species of bird which feeds on this frog. Experimental data suggests that the population of frogs and birds from one year to the next can be modeled by linear relationships. Specifically, it has been found that if the quantities Fk and Bk represent the populations of the frogs and birds in the kth year, then
The question is this: in the long run, will the introduction of the birds reduce or eliminate the frog population growth?

Linear dynamical system II
• So this system evolves in time according to x(k+1) = Ax(k). Such a system is called discrete linear dynamical system, matrix A is called transition matrix.
• If we need to know the state of the system in time k = 50, we have to compute x(50) = A50 x(0).
• And the same is true for Dayhoff’s model of evolution.• If we need to obtain probability matrices for higher
percentage of accepted mutations (i.e. covering longer evolutionary time), we do matrix powers.
• Let’s say we want PAM120 – 120 mutations fixed on average per 100 residues. We do PAM1120.

Linear dynamical system III• PAM1120.• How to avoid multiplications?• Diagonalization: A = SΛS-1
• Which property of PAM1 matrix helps us in its diagonalization?
• Its symmetry. And why does it help?• It means that eigenvectors are orthonormal. S is
orthogonal matrix Q.• And what is Q-1?• Q-1 = QT !• PAM1120 = (QΛQT)120 = QΛ120QT

Higher PAM matrices• Biologically, the PAM120 matrix means that in 100 amino
acids there have been 50 substitutions, while in PAM250 there have been 2.5 amino acid mutation at each side.
• This may sound unusual, but remember, that over evolutionary time, it is possible that an alanine was changed to glycine, then to valine, and then back to alanine.
• These are called silent substituions.

PAM 120
small, polar
small, nonpolar
polar or acidic
basic
large, hydrophobic
aromatic
Zvelebil, Baum, Understanding bioinformatics.
Positive score – frequency of substitutions is greater than would have occurred by random chance.
Zero score – frequency is equal to that expected by chance.
Negative score – frequency is less than would have occurred by random chance.

PAM matrices assumptions• Mutation of amino acid is independent of previous
mutations on the same position (Markov process requirement).
• Only PAM1 was “measured”, all other are extrapolations (i.e. predictions based on some model).
• Each amino acid position is equally mutable.• Mutations are assumed to be independent of surrounding
residues.• Forces responsible for sequence evolution over short time
are the same as these over longer times.• PAM matrices are based on protein sequences available
in 1978 (bias towards small, globular proteins)• New generation of Dayhoff-type – e.g. PET91

How to calculate score?Selzer, Applied bioinformatics.
substitution matrix
2