interaction of apicoplast-encoded elongation factor (ef) ef-tu with nuclear-encoded ef-ts mediates...
TRANSCRIPT

International Journal for Parasitology 41 (2011) 417–427
Contents lists available at ScienceDirect
International Journal for Parasitology
journal homepage: www.elsevier .com/locate / i jpara
Interaction of apicoplast-encoded elongation factor (EF) EF-Tuwith nuclear-encoded EF-Ts mediates translation in the Plasmodium falciparumplastid
Subir Biswas a, Erin E. Lim b,1, Ankit Gupta a,1, Uzma Saqib a, Snober S. Mir a, Mohammad Imran Siddiqi a,Stuart A. Ralph b, Saman Habib a,⇑a Division of Molecular and Structural Biology, Central Drug Research Institute, P.O. Box 173, Chattar Manzil, Mahatma Gandhi Marg, Lucknow 226001, Indiab Department of Biochemistry and Molecular Biology, Bio21 Molecular Science and Biotechnology Institute, The University of Melbourne, Victoria 3010, Australia
a r t i c l e i n f o a b s t r a c t
Article history:Received 28 September 2010Received in revised form 8 November 2010Accepted 8 November 2010Available online 14 December 2010
Keywords:Plasmodium falciparumApicoplastTranslationEF-TuEF-TsKirromycin
0020-7519/$36.00 � 2010 Australian Society for Paradoi:10.1016/j.ijpara.2010.11.003
⇑ Corresponding author. Tel.: +91 522 2612411/ 4182623938/2629504.
E-mail address: [email protected] (S. Habib1 These authors made equal contribution to the man
Protein translation in the plastid (apicoplast) of Plasmodium spp. is of immense interest as a target forpotential anti-malarial drugs. However, the molecular data on apicoplast translation needed for optimi-sation and development of novel inhibitors is lacking. We report characterisation of two key translationelongation factors in Plasmodium falciparum, apicoplast-encoded elongation factor PfEF-Tu and nuclear-encoded PfEF-Ts. Recombinant PfEF-Tu hydrolysed GTP and interacted with its presumed nuclear-encoded partner PfEF-Ts. The EF-Tu inhibitor kirromycin affected PfEF-Tu activity in vitro, indicating thatapicoplast EF-Tu is indeed the target of this drug. The predicted PfEF-Ts leader sequence targeted GFP tothe apicoplast, confirming that PfEF-Ts functions in this organelle. Recombinant PfEF-Ts mediated nucle-otide exchange on PfEF-Tu and homology modeling of the PfEF-Tu:PfEF-Ts complex revealed PfEF-Ts-induced structural alterations that would expedite GDP release from PfEF-Tu. Our results establishfunctional interaction between two apicoplast translation factors encoded by genes residing in differentcellular compartments and highlight the significance of their sequence/structural differences from bacte-rial elongation factors in relation to inhibitor activity. These data provide an experimental system tostudy the effects of novel inhibitors targeting PfEF-Tu and PfEF-Tu.PfEF-Ts interaction. Our finding thatapicoplast EF-Tu possesses chaperone-related disulphide reductase activity also provides a rationalefor retention of the tufA gene on the plastid genome.
� 2010 Australian Society for Parasitology Inc. Published by Elsevier Ltd. All rights reserved.
1. Introduction
The malaria parasite Plasmodium falciparum and relatedapicomplexans contain a relict plastid called the apicoplast. Theapicoplast is essential for parasite survival (Fichera and Roos,1997; McFadden and Roos, 1999) and several biochemical path-ways appear to function within the organelle (Ralph et al., 2004).In addition to these pathways, the housekeeping processes ofapicoplast DNA replication, transcription and translation havebeen recognised as sites for drug intervention (Ralph et al., 2001;Foth and McFadden, 2003). Although some such inhibitors causea delayed-death effect, the irreversible lethal effects of loss ofplastid function nevertheless makes some of them plausiblechemotherapeutic agents. In particular, there is interest in theuse of known and novel prokaryotic translation inhibitors that
sitology Inc. Published by Elsevier
x4282; fax: +91 522 2623405/
).uscript.
specifically target the apicoplast in anti-malarial therapy (Goodmanet al., 2007; Dahl and Rosenthal, 2008). Indeed, two such inhibitors,doxycycline and clindamycin, are already in use; the former isrecommended as a malaria prophylactic for travellers to endemicareas while the latter is in advanced human clinical trials incombination with another drug with apicoplast-specific action(Borrmann et al., 2006; Ruangweerayut et al., 2008).
The evidence for active translation in the P. falciparum apicoplastis compelling. The 35 kb circular DNA genome (plDNA) of the orga-nelle carries genes encoding 16S and 23S rRNAs, 25 tRNAs, transla-tion elongation factor Tu (tufA) and 29 other proteins, including 18ribosomal proteins (Wilson et al., 1996). As the complement ofapicoplast-encoded ribosomal proteins is insufficient for 70S ribo-somal assembly, it is believed that a large component of ribosomalproteins is nuclear-encoded and imported by the apicoplast (Wilsonet al., 1996). A number of these have been identified to contain thebipartite import signal characteristic of apicoplast-targeted proteins(Foth et al., 2003) and apicoplast localisation of the nuclear-encodedribosomal proteins S9 and L28 has been demonstrated in the relatedapicomplexan Toxoplasma gondii (Waller et al., 1998). 70S
Ltd. All rights reserved.

418 S. Biswas et al. / International Journal for Parasitology 41 (2011) 417–427
ribosome-like particles in the apicoplast are apparent by electronmicroscopy (McFadden et al., 1996). Ribosome-like particles thatcarry plastid-specific mRNA and rRNAs have also been detected(Roy et al., 1999). Conclusive evidence for active translation in theorganelle has come from the detection of the tufA gene product inthe apicoplast and the inhibitory effect of the prokaryotic translationinhibitor thiostrepton on elongation factor Tu (EF-Tu) levels(Chaubey et al., 2005). Clindamycin, an inhibitor that binds bacteriallsrRNA, inhibits Plasmodium growth, and a single point mutation inthe lsrRNA gene of the T. gondii apicoplast confers clindamycin resis-tance in vitro (Camps et al., 2002). Azithromycin resistance gener-ated in parasite lines has been attributed to a point mutation inthe P. falciparum apicoplast lsrRNA gene as well as the gene encodingthe apicoplast-encoded ribosomal protein subunit Rpl4 (Sidhu et al.,2007). In addition, apicoplast-specific effects such as disruption ofprotein import into the organelle by clindamycin and tetracycline(Goodman et al., 2007) have been observed. Doxycycline apparentlyblocks expression of the apicoplast genome resulting in the distribu-tion of non-functional apicoplasts during erythrocytic schizogony(Dahl et al., 2006a). A recent study (Stanway et al., 2009) using livemicroscopy on Plasmodium berghei has shown that microbial trans-lation inhibitors also block development of the apicoplast duringexo-erythrocytic schizogony in liver stages, leading to impaired par-asite maturation.
Despite strong interest in the process of apicoplast translation asa target for anti-malarial drugs, there is minimal information onfunctional and structural aspects of initiation, and elongation factorsparticipating in the process. Although they remain to be functionallycharacterised, translation factors that may participate in proteinsynthesis in the apicoplast are listed in Supplementary Table S1.During protein synthesis in prokaryotes, amino acyl-tRNA (aa-tRNA)is delivered to the ribosomal A-site by EF-Tu which hydrolyses GTPand releases the tRNA after codon recognition (Schmeing et al.,2009). The second elongation factor, EF-Ts, binds to the structurallydistinct EF-Tu.GDP form that is released from the ribosome andmediates GDP release, thus regenerating EF-Tu for another roundof GTP binding and aa-tRNA delivery. For catalysis of the nucleotideexchange reaction, EF-Ts binds to EF-Tu.GDP and GDP is rapidly re-leased from the unstable EF-Tu.GDP.EF-Ts complex. Subsequentbinding of GTP forms the EF-Tu.GTP.EF-Ts complex which then dis-sociates into EF-Ts and the active EF-Tu.GTP form (Dahl et al.,2006b). EF-G binds the ribosome sequentially after EF-Tu and acti-vates translocation, the movement of deacylated-tRNA from theribosome P-site to the E-site and concurrent transfer of the pepti-dyl-tRNA from the A-site to the P-site, thus advancing the mRNAone codon in the 30 direction. The A-site is thus freed for entry of an-other EFTu.GTP.aa-tRNA ternary complex (Yu et al., 2009). Of thethree elongation factors required for apicoplast translation onlyone, EF-Tu, is encoded by the apicoplast genome. It is highly similarto other EF-Tu molecules, particularly to the orthologous functionaldomains (Wilson et al., 1996; Sato et al., 2000). No EF-Ts or EF-G areencoded by the apicoplast genome but the current PlasmoDB anno-tation identifies genes encoding EF-Ts (PFC0225c) and EF-G(PFF0115c) homologs on chromosomes 3 and 6 of P. falciparum,respectively, with an apparent bipartite apicoplast-targeting leaderencoded at the 50 end of each (Foth et al., 2003) (Supplementary Ta-ble S1).
Among the drugs that target EF-Tu is kirromycin, a complex lin-ear polyketide. The antibiotic blocks protein synthesis by inhibitingthe release of bacterial EF-Tu.GDP from the ribosome. The EF-Tu.GDP.kirromycin complex locks bacterial EF-Tu in an EFTu.GTP-like conformation that remains on the ribosome, thus preventingthe peptidyl transferase reaction and subsequent translocation(Parmeggiani and Swart, 1985; Vogeley et al., 2001). Kirromycinalso alters the behaviour of Escherichia coli EF-Tu such that theactivities elicited by cellular effectors are mimicked (Mesters
et al., 1994). Kirromycin is capable of enhancing GDP release frombacterial EF-Tu in the absence of EF-Ts and stimulates GTP hydro-lysis in the absence of ribosome and aa-tRNA (Wolf et al., 1974;Mesters et al., 1994). The drug has been shown to exhibit anti-malarial activity in P. falciparum blood culture with an IC50 of�50 lM and can bind to the P. falciparum apicoplast EF-Tu.GDP(Clough et al., 1999).
Attempts at functional characterisation of the P. falciparum api-coplast EF-Tu (PfEF-Tu) and its interaction with potential inhibitorshave been made in the past but have met with limited success dueto difficulties associated with recombinant expression of the pro-tein as a functionally active molecule (Clough et al., 1999; Chaubeyet al., 2005). We describe the use of re-folded functional apicoplastEF-Tu, expressed in E. coli as a fusion protein with maltose bindingprotein (MBP), to study P. falciparum EF-Tu (PfEF-Tu) function, itsinteraction with PfEF-Ts as well as the effect of kirromycin on itsactivity. Our results establish apicoplast localisation of nuclear-encoded PfEF-Ts and its ability to mediate nucleotide exchangeon PfEF-Tu. We believe this provides the first functional character-isation of two critical components of the translation machinery ofthe Plasmodium apicoplast.
2. Materials and methods
2.1. Parasite culture and cDNA preparation
Plasmodium falciparum (strain 3D7) was cultured as described byJensen and Trager (1978). Parasite genomic DNA was isolated byphenol/chloroform extraction. Total parasite RNA was isolated withTrizol Reagent (Invitrogen) and reverse transcribed (RT) using theSuperScript First Strand Synthesis System for RT-PCR (Invitrogen).
2.2. Recombinant protein expression and purification
Primers were designed to PCR-amplify the nuclear DNA se-quence encompassing the two exons encoding PfEF-Ts (PlasmoDBID PFC0225c) as well as only the first exon of the gene. The entireexon 1 was PCR-amplified using upstream (50-CGCGGATCCAAATTGTTTTACTTTTTCTTGT TAAGC-30) and downstream (50-CGCGTCGACCTGCTCTAGGAATACTATG-30) primers carrying BamHI and SalItags (underlined) with genomic DNA as template. The amplifiedDNA was cloned into pET23a vector with a polyhistidine (His)-tag at the N-terminus generating the construct pET-Tsexon1. E. coliBL-21 DE3 cells were co-transformed with pET-Tsexon1 and theRIG plasmid (gift from Prof. W.G.J. Hol, Howard Hughes MedicalInstitute, USA). The protein was expressed as inclusion bodiesand PAGE-separated, electroeluted protein (Ram et al., 2008) wasused to generate antibodies against PfEF-Ts in rabbit. Approvalfor animal use was given by the Institutional Animal Ethics Com-mittee of the Central Drug Research Institute, India. Maintenanceand care of animals was in accordance with Government of Indiaguidelines.
The DNA sequence encoding processed EF-Ts (protein lackingthe predicted signal-transit sequence for apicoplast targeting)was amplified using upstream (50-CGCGGATCCGATCATCTAAAACTATTAAAATATG-30) and downstream (50-CGCGTCGACTTCCATAAGAA CGTTTTTTTCCCC-30) primers carrying BamHI and SalI tags.cDNA prepared from total parasite RNA was used as template.The amplified sequence was cloned into the pGEX-KG vector andthe resultant pGEX-Tsf vector and the RIG plasmid were co-trans-formed into E. coli XL-1 blue cells. The GST-PfEFTs fusion protein(�65 kDa) with N-terminal GST was expressed in the soluble frac-tion and was purified on a glutathione Sepharose affinity column(GE Healthcare). For removal of the GST-tag, fusion protein boundto the affinity column was cleaved with thrombin. The eluted

S. Biswas et al. / International Journal for Parasitology 41 (2011) 417–427 419
fractions containing cleaved PfEF-Ts were dialysed in 50 mM Tris–HCl pH 8.0, 50 mM KCl, 5 mM EDTA, 1 mM phenylmethanesulfonylfluoride (PMSF), 2 mM DTT and 5% glycerol. The cleaved proteinwas further purified on a S200 column to obtain the monomericPfEF-Ts fraction. Although processed PfEF-Ts has a predicted sizeof �39 kDa, the purified recombinant EF-Ts protein ran at�35 kDa on SDS–PAGE.
EF-Tu encoded by the apicoplast genome had been previouslycloned and expressed as a fusion protein with MBP (Chaubeyet al., 2005). Our attempts at recombinant expression of soluble,stable apicoplast-encoded PfEF-Tu (�47 kDa) with His and GSTaffinity tags in the E. coli expression system were not successful.Very low levels of intact protein were expressed with the His tag(in the pQE30 vector). Although PfEF-Tu was expressed as a solubleprotein with the GST tag and the tag was cleaved spontaneously,the untagged protein degraded immediately upon purification.We also attempted stabilizing PfEF-Tu by co-expression of theEF-Tu interacting factor, EF-Ts, in a dual expression vector (modi-fied version of pETDuet-1). However, S-tagged EFTu was not stabi-lized by co-expression of EF-Ts. Hence, the MBP-EFTu fusionprotein (Chaubey et al., 2005) was expressed as inclusion bodiesin E. coli. In order to obtain functional MBP-EF-Tu, inclusion bodiesof the �89 kDa fusion protein were solubilised and re-foldedfollowed by affinity purification on amylose resin. Briefly, E. coliTG-1 cells were co-transformed with the pMAL-tufA construct(Chaubey et al., 2005) and the RIG plasmid. After induction with0.5 mM isopropyl b-D-thiogalactopyranoside (IPTG), the inclusionbody fraction was washed twice with a buffer containing 20 mMTris–HCl, pH 7.5, 10 mM EDTA and 1% Triton X-100 followed bysolubilisation with a buffer containing 0.3% N-lauroylsarcosineand 2 mM DTT in 50 mM N-cyclohexyl-3-aminopropane-sulphonicacid (CAPS), pH 11.0, according to the manufacturer’s instructions(Protein Re-folding Kit, Novagen). After centrifugation at 10,000gfor 10 min at room temperature, the solubilised protein was addedto re-folding buffer (50 mM Tris, pH 7.6, 200 mM NaCl, 1 mMEDTA, 1 mM DTT and 0.5 M non-detergent sulfobetaines-256(NDSB-256) at the ratio of 1:10 (v:v) with fast mixing and incu-bated at 4 �C for 1 h. This was followed by dialysis in re-foldingbuffer lacking NDSB-256, followed by two changes of re-foldingbuffer lacking NDSB-256 and DTT. The re-folded protein was puri-fied on amylose resin (New England Biolabs). Purified PfEF-Tu wasdialysed in 50 mM Tris pH 7.5, 200 mM NaCl, 1 mM EDTA, 1 mMPMSF and 1% glycerol, and concentrated using Centricon units(Millipore). Re-folding of the protein was confirmed by circulardichroism (CD) spectroscopy (Supplementary Fig. S1A).
2.3. EF-Tu mutagenesis
The EF-Tu W196G and T62A mutants were generated using thepMAL-tufA as template. The QuickChange XL Site-Directed Muta-genesis Kit (Stratagene) was used to mutate the tufA gene. Themutations were confirmed by DNA sequencing. The PfEFTuT62A
protein also expressed as inclusion bodies in E. coli and was re-folded and purified as described for wild-type PfEFTu. The purifica-tion profile of the mutants was similar to the wild-type protein(data not shown).
2.4. Western blotting
For preparation of total parasite lysate, parasite cultures at 6–8%parasitaemia were harvested when cells were predominantly atthe trophozoite stage. Parasites were released by 0.05% saponin ly-sis, washed with PBS, and suspended in 1� SDS loading buffer con-taining protease inhibitors (Protease Arrest, GBiosciences, USA).After 30 min incubation on ice followed by brief vortexing, the celllysate was separated on a 10% SDS–PAGE. Western blotting was
carried out (Chaubey et al., 2005) and the blot was developed usinga chemiluminescent system (GE Healthcare).
2.5. GTPase assay
GTP hydrolysis by wild-type and mutant PfEF-Tu was measuredusing the EnzCheck Phosphate Assay Kit (Molecular Probes). Onemillimolar GTP and 1 lM PfEF-Tu or PfEF-TuT62A were added tothe reaction mixture (60 mM Tris, pH 7.5, 30 mM KCl, 30 mMNH4Cl, 10 mM MgCl2 and 2 mM DTT) and assayed for Pi releaseas per the manufacturer’s instructions. Plasticware and cuvettesused in the assay were pre-treated with ‘Pi-mop’ (1 U/ml nucleo-tide phosphorylase, 750 lM 7-methyl guanosine, Sigma–Aldrich).
The effect of kirromycin on the intrinsic GTPase activity ofPfEF-Tu was assayed using c32P-GTP (Jonaki, India) as described byMesters et al. (1994). Briefly, 2 lM PfEF-Tu was incubated with0.2 lM c32P-GTP, 2 lM GDP, 64 mM Tris–HCl pH 7.6, 10 mM MgCl2,80 mM NH4Cl, 10 mM b-mercaptoethanol, 83 lM phosphoenolpyruvate, 40 lg/ml pyruvate kinase/lactate dehydrogenase at37 �C for 15 min followed by addition of kirromycin to the final30 ll reaction volume and further incubation at 37 �C for 15 min.The reaction was stopped by adding 15 ll of 25% (v/v) formic acid.Aliquots were spotted on 3MM Whatman paper and chromato-graphed (Schwemmle and Staeheli, 1994). Phosphorimager signalswere quantitated by densitometric analysis.
2.6. Nucleotide release assay
The interaction of GDP with PfEFTu as well as GDP release medi-ated by EF-Ts were assayed using 20-/30-O-(N0-Methylanthraniloyl)-GDP (mant-GDP) (Molecular Probes). For the nucleotide binding as-say, PfEF-Tu was incubated with 1.6 lM mant-GDP at 37 �C for15 min in nucleotide binding (NB) buffer containing 60 mM Tris–HCl pH 7.6, 30 mM KCl, 30 mM NH4Cl, 10 mM MgCl2 and 2 mMDTT. Tryptophan was excited at 280 nm. Mant-GDP bound to PfEF-Tu was excited by fluorescence resonance energy transfer (FRET)from tryptophan (mant-GDP excitation at 366 nm) and the emissionof mant-GDP (emission wavelength of 450 nm) was recorded in scanspectra using a LS50B spectrofluorimeter (Perkin Elmer).
The effect of kirromycin on PfEF-Tu.GDP dissociation was esti-mated by measuring the dissociation of the PfEF-Tu.mant-GDPcomplex with increasing kirromycin concentrations. The PfEF-Tu.-mant-GDP complex was prepared by incubation of PfEF-Tu (1 lM)and 15 lM mant-GDP for 15 min at 37 �C. GDP (25 lM) andincreasing concentrations of kirromycin were added, followed byincubation at 0 �C for 40 min. Mant-GDP emission as a result ofFRET was recorded at each kirromycin concentration. Backgroundcorrection was made by subtraction of emission spectra of 15 lMmant-GDP in NB buffer.
In order to assay EF-Ts mediated GDP release from EF-Tu, thePfEF-Tu.mant-GDP complex was prepared by adding threefold mo-lar excess (1.6 lM) of mant-GDP to PfEF-Tu in NB buffer as above.Increasing molar ratios of EF-Ts were added to the complex to-gether with 50 lM GDP to prevent re-binding of mant-GDP toPfEF-Tu. The change in mant-GDP emission was recorded.
The kinetics of GDP release from EF-Tu mediated by EF-Ts wasstudied by fluorescence stopped-flow measurements on a JASCOJ-810 spectro-polarimeter with a stopped-flow attachment (SFM-300/S, BioLogic Science Instruments, France) in a buffer containing50 mM Tris–HCl pH 7.5, 70 mM NH4Cl, 30 mM KCl, 7 mM MgCl2
and 1 mM DTT at 20 �C. The change in fluorescence of the mant-GDP.PfEF-Tu complex in the presence of PfEF-Ts was recorded.The fluorescence of mant-GDP bound to PfEF-Tu was excited viaFRET from tryptophan excited at 280 nm and measured afterpassing through a 400 nm filter (BioLogic). Experiments were per-formed by rapid mixing of the mant-GDP.EF-Tu complex (0.5 lM

420 S. Biswas et al. / International Journal for Parasitology 41 (2011) 417–427
PfEF-Tu) and 4 lM of PfEF-Ts (in buffer containing 25 lM GDP) andfluorescence was monitored over time. Time course profiles de-picted in Fig. 1B were obtained by averaging three individual tran-sients. The rate constant (ki) was determined by fitting the data toan exponential function of the form y(t) = at + b + RAi e�k
it where y
is the fluorescence at time t and the slope (a) and offset (b) corre-spond to the linear drift after the reaction; the best-fitting ampli-tude (Ai) and apparent rate constant (Ki) were determined withBio-Kine software (BioLogic) (Huecas et al., 2007).
2.7. Assay of insulin disulphide reduction
The reduction of insulin by a disulphide oxidoreductase was as-sessed spectrophotometrically by recording absorbance at 650 nm(Bardwell et al., 1991). Insulin contains two polypeptide chains Aand B that are linked by two interchain disulphide bonds. Whenthese bonds are broken upon reduction, the insoluble free B chainprecipitates, leading to an increase in absorbance at 650 nm. Forthe assay reaction, the incubation mixture contained the followingin a final volume of 100 ll: 0.1 M N2-equilibrated potassium phos-phate pH 6.6, 0.3 mM EDTA, 0.13 mM porcine pancreas insulin and0.3 mM DTT in the presence or absence of 2 lM PfEF-Tu. Insulinstock solutions of 10 mg/ml (1.67 mM) were prepared by
0 2 4 6
0.90
0.92
0.94
0.96
0.98
1.00
1.02
2
1
1 0.5µM PfEF-Tu.mantGDP2 0.5µM PfEF-Tu.mantGDP
Rel
ativ
e fl
uo
resc
ence
(F
RE
T)
Time (sec)
A
B
300 350 4000
200
400
600
800
1000
Flu
ore
scen
ce in
ten
sity
Wavelength
Buffer+ PfEF-T PfEF-T PfEF-T PfEF-T
Fig. 1. Plasmodium falciparum nuclear-encoded translation elongation factor (PfEF)-Telongation factor (PfEF)-Tu. (A) 20-/30-O-(N0-Methylanthraniloyl)-GDP (mant-GDP) relearesonance energy transfer (FRET) from Trp-196 of PfEFTu in the presence of PfEF-Ts. PfEF-Tand excess (50 lM) GDP in EF-Tu:EF-Ts molar ratios of 1:1, 1:2 and 1:5. (B) Fluorescencaddition of PfEF-Ts. Residuals of the fit are shown in the small panel. Trace 1, PfEF-Tu.matime course profile obtained by averaging three individual transients is shown.
suspending 50 mg of insulin in 4 ml of 0.05 M Tris–Cl, pH 8.0 andthe pH was adjusted to pH 2–3 by addition of 1.0 M HCl. This wasfollowed by rapid titration of the solution back to pH 8.0 with1.0 M NaOH. Finally, the volume was adjusted to 5 ml with water.The solution of insulin was perfectly clear and was stored at�20 �C.
2.8. Structural modeling of the PfEF-Tu.PfEF-Ts complex
The three-dimensional model of the PfEF-Tu.PfEF-Ts complexwas constructed using E. coli EF-Tu.EF-Ts as template (PDB: 1EFUchains A and B) (Kawashima et al., 1996). The alignment of PfEF-Tsand PfEF-Tu was performed using ClustalW, as implemented at theEBI server (http://www.ebi.ac.uk/Tools/clustalw2/index.htmlhtml). The resulting model was subsequently energetically mini-mised with 500 steps of steepest descent minimisation, followedby 2000 steps of conjugate gradient minimisation to remove thegeometrical strain. GDP and Mg2+ were manually docked to thePfEF-Tu binding site based on previous structural information ofthe template (Song et al., 1999). The model was constructed usingthe program MODELLER (Sali and Blundell, 1993) interfaced withInsightII. MODELLER is an implementation of an automatedapproach to comparative modeling by satisfaction of spatial re-straints (Sali and Overington, 1994; Sali, 1995). In order to assess
8 10
+ 4µM PfEFTs
450 500
(nm)
mantGDPu.mantGDPu.mantGDP+PfEF-Ts (1:1)u.mantGDP+PfEF-Ts (1:2)u.mantGDP+PfEF-Ts (1:5)
s mediates nucleotide release from P. falciparum apicoplast-encoded translationse from PfEF-Tu was monitored by measuring excitation of mant by fluorescenceu (0.5 lM) was complexed with mant-GDP (1.6 lM) followed by addition of PfEF-Ts
e stopped flow rapid kinetic measurements of PfEF-Tu.mant-GDP dissociation uponnt-GDP alone; trace 2, PfEF-Tu.mant-GDP with PfEF-Ts in the molar ratio of 1:8. The

S. Biswas et al. / International Journal for Parasitology 41 (2011) 417–427 421
the overall stereochemical quality of the modeled proteinRamachandran plot analysis was performed using the programPROCHECKv3.4.4 (Morris et al., 1992; Laskowski et al., 1993).
2.9. Molecular dynamics
The PfEF-Tu.Mg.GDP.PfEF-Ts ternary complex was further sol-vated by using Explicit Spherical Boundary with harmonic restraints(sphere of 50 radius). This solvated complex was subjected to energyminimisation first by 500 steps of steepest descent followed by 2000steps of conjugate gradient method. The system was heated from 50to 300 K over a period of 50 psec with a time step of 1 fsec and thevelocities being reassigned in the system every 0.05 psec. The sys-tem was further equilibrated with a 1 fsec time step, for 100 psecso that the energy of the system achieves complete stability. Produc-tion runs were performed at 300 K and carried out under a constantnumber of particles, volume and temperature conditions for 1 nsecwith a 1 fsec time step. All of the bonds involving hydrogen atomswere constrained using the SHAKE algorithm in all simulations(Ryckaert et al., 1977). The molecular trajectory for the systems gen-erated by the molecular dynamic simulations were analysed usingVMD (Humphrey et al., 1996) software and the CHARMM program(Brooks et al., 1983). All molecular dynamic simulations were car-ried out with CHARMM program using the CHARMM force field.
2.10. Parasite transfection and microscopy
The sequence encoding the predicted signal-transit peptide ofPfEF-Ts (amino acids 1–54) was PCR-amplified using the followingforward and reverse primers: 50-GATCTCGAGATGAAGTTGTTTTATTTTTTCTTGTTAAG-30 and 50-GATCCCGGGATTGTTTGTAGAATATAATCTATTTTT-30. The sequence was cloned as an XhoI–XmaI insertinto the pGlux.1 vector (Boddey et al., 2009) (gift from Prof. AlanCowman, Walter and Eliza Hall Institute, Australia) such that thesignal and transit peptides were fused in frame with GFP. One hun-dred microgram of the EF-Tsleader-GFP construct was transfectedinto ring stages of 3D7 strain parasites by electroporation as previ-ously described (Crabb and Cowman, 1996) and selected using theWR99210 antifolate (Waller et al., 1998). For colocalisation withthe apicoplast marker acyl carrier protein (ACP), parasites werefixed as previously described (Tonkin et al., 2004) (without the so-dium borohydride treatment), and labeled using a 1:500 dilution ofanti-ACP anti-sera (Waller et al., 1998), (kindly provided by Prof.G.I. McFadden, University of Melbourne, Australia) and an Alexa-fluor goat-anti-rabbit IgG (Invitrogen, A-11012). Unfixed parasiteswere also stained with 20 nM Mitotracker Red (Invitrogen) tocompare distribution of the EF-Tsleader-GFP with the mitochondria.Parasites were imaged using an Zeiss AxioPlan 2 fluorescencemicroscope and individual images were merged using ImageJ(National Institutes of Health, USA) and Adobe Photoshop v10(Adobe Systems). For detection of the fusion protein by Westernblotting, parasite cultures were lysed with saponin and parasiteprotein extracted in SDS loading buffer containing DTT. Proteinfrom approximately 10 million parasites was loaded per lane anddetected using anti-GFP antibody (Roche).
3. Results
3.1. PfEF-Tu exhibits GTPase activity
Apicoplast-encoded PfEF-Tu was expressed as a fusion proteinwith MBP with most of the protein partitioning in the insoluble frac-tion as described by Chaubey et al. (2005). Later attempts to expressstable PfEF-Tu with other fusion tags were unsuccessful (describedin methods Section 2.2). Hence, inclusion bodies containing
89 kDa MBP-PfEFTu were solubilised, re-folded and purified on anamylose affinity resin (Supplementary Fig. S2A). Storage of the puri-fied fusion protein at�80 �C generated auto-cleaved EF-Tu (47 kDa)with the average MBP-EFTu: cleaved EF-Tu ratio of 2:1 to 1:1 (Sup-plementary Fig. S2B). The identity of cleaved PfEF-Tu was confirmedby mass spectrometry (Supplementary Fig. S2C). As the cleavedPfEF-Tu was unstable upon further purification, the re-folded pro-tein containing both tagged and cleaved EF-Tu was used for experi-ments and is referred to as PfEF-Tu.
The ability of apicoplast PfEF-Tu to interact with GDP wasexplored by FRET analysis using mant-GDP [20/30-O-(N-methyl-anthraniloyl-GDP)]. As in E. coli, Euglena gracilis and Thermus ther-mophilus EF-Tu, PfEF-Tu contains a single tryptophan residue inhelix F of domain I (Trp-196 in PfEF-Tu) (Sato et al., 2000) andproximity of this residue to the GDP-binding site enables excita-tion of mant by FRET (Rodnina et al., 1995; Dahl et al., 2006b).Incubation of mant-GDP with PfEF-Tu resulted in enhancedmant-fluorescence (maximum emission at 440 nm) (Fig. 2A).E. coli (Ec) EF-Tu was used as a positive control. Control experi-ments with the PfEF-Tu W196G mutant and purified MBP (Supple-mentary Fig. S1B and C) did not result in enhanced emission frommant-GDP confirming that the observed enhancement of mantfluorescence by PfEF-Tu was a result of FRET from Trp-196 and thatMBP in the PfEF-Tu fusion protein did not interact with the nucle-otide. These results indicate specific binding of PfEF-Tu to GDP.
The GTPase activity of PfEF-Tu was assayed spectrophotometri-cally. PfEF-Tu hydrolysed GTP in a protein concentration-depen-dent manner (Fig. 2B). The observed GTPase activity was notattributable to any contaminating E. coli protein as the identi-cally-purified PfEF-TuT62A mutant had negligible GTPase activity(Fig. 2C). The conserved Thr-62 residue is involved in the coordina-tion of the c-phosphate of GTP, a Mg2+ ion and a water molecule inE. coli and T. thermophilus, and has been shown to be critical for theintrinsic and ribosome-induced GTP hydrolysis of EF-Tu (Berchtoldet al., 1993; Ahmadian et al., 1995). Our results also indicate a crit-ical role of this residue in the intrinsic GTPase activity of PfEF-Tu.
3.2. Kirromycin alters PfEF-Tu activity in vitro
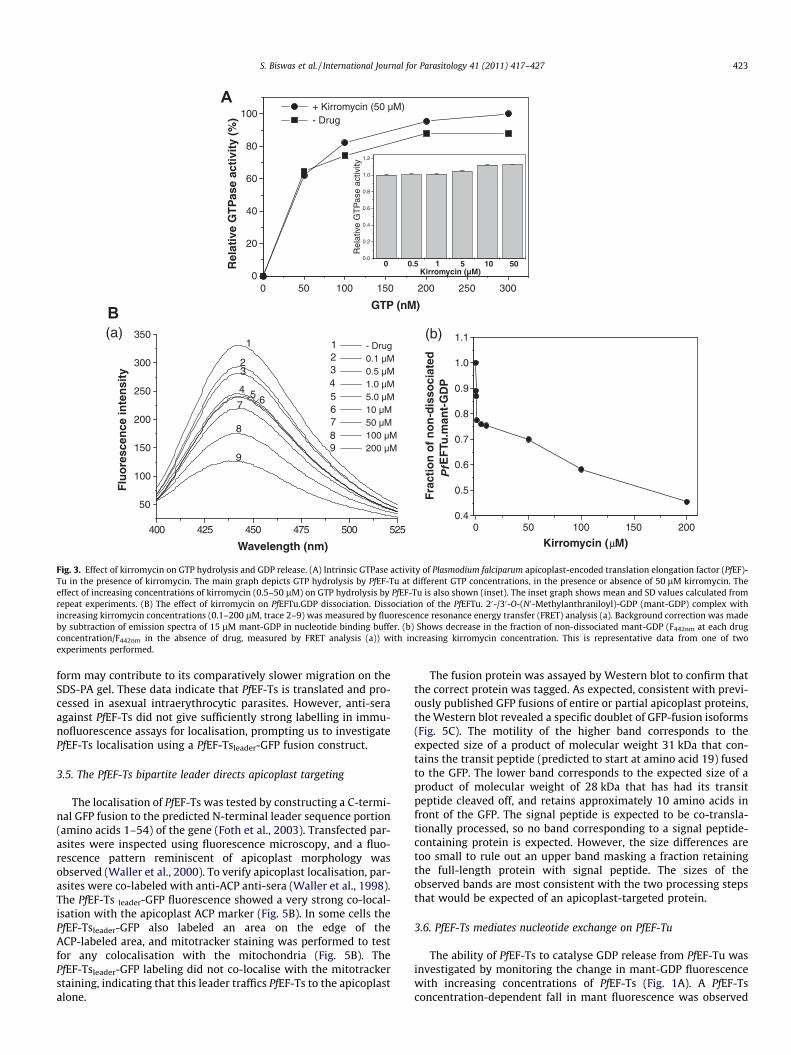
We investigated the effect of kirromycin on the intrinsic GTPaseactivity of PfEF-Tu as well as GDP release from PfEF-Tu in the absenceof EF-Ts. GTP hydrolysis by PfEF-Tu was not significantly affected bykirromycin (Fig. 3A). There was little enhancement of GTPase activ-ity with increasing concentrations of kirromycin (0.1 to 50 lM). Onthe other hand, kirromycin clearly stimulated the release of GDPfrom the EF-Tu.GDP complex (Fig. 3B) as measured by FRET usingmant-GDP. The fraction of non-dissociated PfEF-Tu.mant-GDP re-duced with increasing concentrations of kirromycin, indicatingenhancement of mant-GDP release from PfEF-Tu by the drug.
3.3. PfEF-Tu exhibits chaperone activity
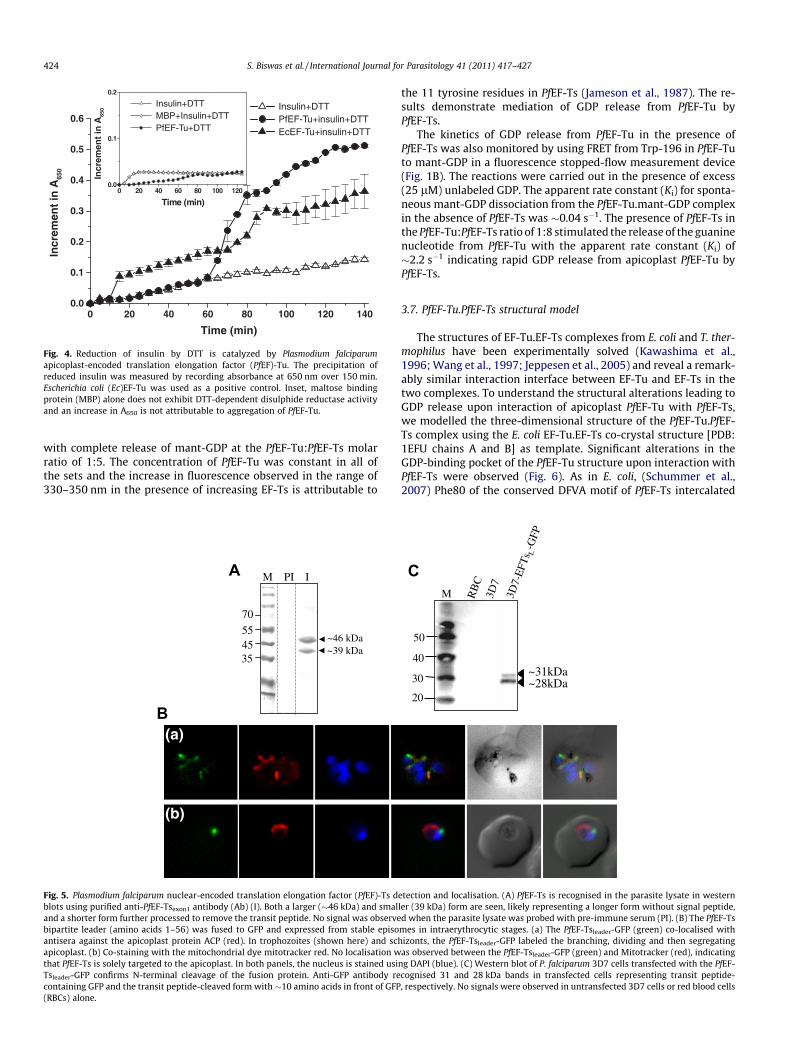
Bacterial EF-Tu has been shown to possess chaperone-like prop-erties in protein folding and renaturation after stress, and itsin vitro protein disulphide isomerase activity has been demon-strated (Kudlicki et al., 1997; Caldas et al., 1998, 2000; Richarme,1998). We thus investigated the chaperone function of PfEF-Tuby assaying its DTT-dependent disulphide reductase activity(Fig. 4). Insulin, which contains two interchain disulphide bondsand precipitates upon disulphide reduction, was used as substrate.Enhanced DTT-dependent reduction of insulin resulting in in-creased insulin precipitation was observed in the presence ofPfEF-Tu. Thus PfEF-Tu may function as a protein disulphide isomer-ase in the apicoplast, thereby also assisting protein folding in addi-tion to its role in aa-tRNA entry and proof-reading activity duringtranslation.

0 20 40 60 80 100 120 140
0.0
0.1
0.2
0.3
0.4
1µM PfEF-Tuwt+ 1mM GTP
1µM PfEF-TuT62A+ 1mM GTPA36
0
Time (sec)
C
0 100 200 300 400 5000.0
0.1
0.2
0.3
0.4
Pi r
elea
sed
(n
mo
les)
PfEF-Tu (nmoles)
350 400 450 5000
50
100
150
200
250
300
350
400
450 Buffer+mant-GDP EcEF-Tu+mant-GDP(1:3) PfEF-Tu+mant-GDP (1:3) PfEF-Tu+mant-GDP (2:3) PfEF-Tu+mant-GDP (1:1)
Flu
ore
scen
ce in
ten
sity
Wavelength (nm)
A
B
Fig. 2. Plasmodium falciparum apicoplast-encoded translation elongation factor (PfEF)-Tu binds guanine nucleotides and hydrolyses GTP. (A) Excitation of 20-/30-O-(N0-Methylanthraniloyl)-GDP (mant-GDP) by the intrinsic tryptophan of PfEF-Tu by fluorescence resonance energy transfer (FRET). Emission from mant is seen at �450 nm.Different concentrations of PfEF-Tu were incubated with 1.6 lM mant-GDP followed by excitation at 280 nm. Background emission of 1.6 lM mant-GDP in binding buffer isalso shown. Representative data from one of two repeat experiments is presented. (B) GTPase activity of PfEF-Tu as a function of increasing protein concentration. Meanand ±SD calculated from three experiments is plotted. (C) GTP hydrolysis by PfEF-Tu and PfEF-TuT62A. One mM GTP and 1 lM PfEFTu or PfEFTuT62A were added to the reactionmixture and assayed for Pi release using the EnzCheck Phosphate Assay Kit (Molecular Probes). Absorbance at 360 nm was recorded over 150 s.
422 S. Biswas et al. / International Journal for Parasitology 41 (2011) 417–427
3.4. Nuclear-encoded EF-Ts is targeted to the apicoplast
The spontaneous release of GDP from bacterial EF-Tu is too slow(0.002 s�1) to be physiologically relevant (Wagner et al., 1995) andthe action of EF-Ts is required for rapid nucleotide exchange onEF-Tu. The P. falciparum apicoplast genome does not encode EF-Ts.However, an EF-Ts homolog (PFC0225c) (hereafter named PfEF-Ts)that displays 31% identity to the Fulvimarina pelagi (a pigmentedmarine bacterium) EF-Ts and 23% identity to EcEF-Ts is encoded bychromosome 3 of the P. falciparum nuclear genome. PfEF-Ts has anN-terminal extension containing an apparent bipartite apicoplasttargeting sequence (Foth et al., 2003) of approximately 50 aminoacids (Supplementary Fig. S3A). The PfEF-Ts sequence is longer thanE. coli and F. pelagi EF-Ts and carries a 33–42 amino acid insertionafter the conserved DFVA domain that is involved in interaction withEF-Tu (Schummer et al., 2007). Homology modeling of the predictedprocessed PfEF-Ts on the crystal structure of EcEF-Ts revealed con-servation of major protein folds with differences observed in the ori-entation of the helices of the N-terminal domain of PfEF-Ts(Supplementary Fig. S3B). One major (42 residues) and three minor(five, seven and 11 residues) loops representing amino acid stretchesabsent in EcEF-Ts extruded from the PfEF-Ts structure with the
major loop positioned on the face opposite the conserved DFVAsequence. The major loop lacked homology with any other sequenceof known structure and could not be modeled.
The sequence encoding the predicted processed form (aftercleavage of the signal and transit peptides) of PfEF-Ts was ex-pressed as a GST-fusion protein in E. coli. The GST tag of the solublepurified protein (�64 kDa) was cleaved and the resultant PfEF-Tsmigrated at the approximate size of 35 kDa (SupplementaryFig. S4). Exon-1 of the EF-Ts coding sequence was also expressedas a His-tag fusion in E. coli. However, PfEF-Tsexon1 was expressedas inclusion bodies. Antibodies against GST-PfEF-Ts and His-PfEF-Tsexon1 were raised in mice and a rabbit, respectively. The rabbitanti-EF-Ts antibody was used to detect PfEF-Ts in parasite lysates.A specific signal of two bands (�46 and �39 kDa) was seen in Wes-tern blots, (Fig. 5A) which presumably corresponds to unprocessedand processed forms of the protein, respectively. The expected sizeof the signal peptide-cleaved form containing the transit peptide(TP) and mature protein (MP) is 42.7 kDa. The slightly bigger sizeshift between this MP + TP form (apparent molecular weight of�46 kDa instead of 42.7 kDa) and processed form (MP only) maybe explained by the presence of a very basic transit peptide inthe former. The reduced overall negative charge in the MP + TP
400 425 450 475 500 525
50
100
150
200
250
300
350
99
88
7
7 66
5 54
43 3
22
1 1 - Drug 0.1 µM 0.5 µM 1.0 µM 5.0 µM 10 µM 50 µM 100 µM 200 µM
Flu
ore
scen
ce in
ten
sity
Wavelength (nm)
A
0 50 100 150 200 250 3000
20
40
60
80
100 + Kirromycin (50 µM) - Drug
Rel
ativ
e G
TP
ase
acti
vity
(%
)
GTP (nM)
0.0
0.2
0.4
0.6
0.8
1.0
1.2
5010510.50Kirromycin (µM)
Rel
ativ
e G
TP
ase
activ
ity
B(a) (b)
0 50 100 150 2000.4
0.5
0.6
0.7
0.8
0.9
1.0
1.1
Fra
ctio
n o
f n
on
-dis
soci
ated
PfE
FTu
.man
t-G
DP
Kirromycin (µM)
Fig. 3. Effect of kirromycin on GTP hydrolysis and GDP release. (A) Intrinsic GTPase activity of Plasmodium falciparum apicoplast-encoded translation elongation factor (PfEF)-Tu in the presence of kirromycin. The main graph depicts GTP hydrolysis by PfEF-Tu at different GTP concentrations, in the presence or absence of 50 lM kirromycin. Theeffect of increasing concentrations of kirromycin (0.5–50 lM) on GTP hydrolysis by PfEF-Tu is also shown (inset). The inset graph shows mean and SD values calculated fromrepeat experiments. (B) The effect of kirromycin on PfEFTu.GDP dissociation. Dissociation of the PfEFTu. 20-/30-O-(N0-Methylanthraniloyl)-GDP (mant-GDP) complex withincreasing kirromycin concentrations (0.1–200 lM, trace 2–9) was measured by fluorescence resonance energy transfer (FRET) analysis (a). Background correction was madeby subtraction of emission spectra of 15 lM mant-GDP in nucleotide binding buffer. (b) Shows decrease in the fraction of non-dissociated mant-GDP (F442nm at each drugconcentration/F442nm in the absence of drug, measured by FRET analysis (a)) with increasing kirromycin concentration. This is representative data from one of twoexperiments performed.
S. Biswas et al. / International Journal for Parasitology 41 (2011) 417–427 423
form may contribute to its comparatively slower migration on theSDS-PA gel. These data indicate that PfEF-Ts is translated and pro-cessed in asexual intraerythrocytic parasites. However, anti-seraagainst PfEF-Ts did not give sufficiently strong labelling in immu-nofluorescence assays for localisation, prompting us to investigatePfEF-Ts localisation using a PfEF-Tsleader-GFP fusion construct.
3.5. The PfEF-Ts bipartite leader directs apicoplast targeting
The localisation of PfEF-Ts was tested by constructing a C-termi-nal GFP fusion to the predicted N-terminal leader sequence portion(amino acids 1–54) of the gene (Foth et al., 2003). Transfected par-asites were inspected using fluorescence microscopy, and a fluo-rescence pattern reminiscent of apicoplast morphology wasobserved (Waller et al., 2000). To verify apicoplast localisation, par-asites were co-labeled with anti-ACP anti-sera (Waller et al., 1998).The PfEF-Ts leader-GFP fluorescence showed a very strong co-local-isation with the apicoplast ACP marker (Fig. 5B). In some cells thePfEF-Tsleader-GFP also labeled an area on the edge of theACP-labeled area, and mitotracker staining was performed to testfor any colocalisation with the mitochondria (Fig. 5B). ThePfEF-Tsleader-GFP labeling did not co-localise with the mitotrackerstaining, indicating that this leader traffics PfEF-Ts to the apicoplastalone.
The fusion protein was assayed by Western blot to confirm thatthe correct protein was tagged. As expected, consistent with previ-ously published GFP fusions of entire or partial apicoplast proteins,the Western blot revealed a specific doublet of GFP-fusion isoforms(Fig. 5C). The motility of the higher band corresponds to theexpected size of a product of molecular weight 31 kDa that con-tains the transit peptide (predicted to start at amino acid 19) fusedto the GFP. The lower band corresponds to the expected size of aproduct of molecular weight of 28 kDa that has had its transitpeptide cleaved off, and retains approximately 10 amino acids infront of the GFP. The signal peptide is expected to be co-transla-tionally processed, so no band corresponding to a signal peptide-containing protein is expected. However, the size differences aretoo small to rule out an upper band masking a fraction retainingthe full-length protein with signal peptide. The sizes of theobserved bands are most consistent with the two processing stepsthat would be expected of an apicoplast-targeted protein.
3.6. PfEF-Ts mediates nucleotide exchange on PfEF-Tu
The ability of PfEF-Ts to catalyse GDP release from PfEF-Tu wasinvestigated by monitoring the change in mant-GDP fluorescencewith increasing concentrations of PfEF-Ts (Fig. 1A). A PfEF-Tsconcentration-dependent fall in mant fluorescence was observed
0 20 40 60 80 100 120 1400.0
0.1
0.2
0.3
0.4
0.5
0.6 Insulin+DTT PfEF-Tu+insulin+DTT EcEF-Tu+insulin+DTT
Incr
emen
t in
A65
0
Time (min)
0 20 40 60 80 100 1200.0
0.1
0.2
Insulin+DTT MBP+Insulin+DTT PfEF-Tu+DTT
Incr
emen
t in
A65
0
Time (min)
Fig. 4. Reduction of insulin by DTT is catalyzed by Plasmodium falciparumapicoplast-encoded translation elongation factor (PfEF)-Tu. The precipitation ofreduced insulin was measured by recording absorbance at 650 nm over 150 min.Escherichia coli (Ec)EF-Tu was used as a positive control. Inset, maltose bindingprotein (MBP) alone does not exhibit DTT-dependent disulphide reductase activityand an increase in A650 is not attributable to aggregation of PfEF-Tu.
424 S. Biswas et al. / International Journal for Parasitology 41 (2011) 417–427
with complete release of mant-GDP at the PfEF-Tu:PfEF-Ts molarratio of 1:5. The concentration of PfEF-Tu was constant in all ofthe sets and the increase in fluorescence observed in the range of330–350 nm in the presence of increasing EF-Ts is attributable to
~46 kDa~39 kDa
B
M PI I
70
554535
A
B(a)
(b)
Fig. 5. Plasmodium falciparum nuclear-encoded translation elongation factor (PfEF)-Ts dblots using purified anti-PfEF-Tsexon1 antibody (Ab) (I). Both a larger (�46 kDa) and smaland a shorter form further processed to remove the transit peptide. No signal was observebipartite leader (amino acids 1–56) was fused to GFP and expressed from stable episoantisera against the apicoplast protein ACP (red). In trophozoites (shown here) and schapicoplast. (b) Co-staining with the mitochondrial dye mitotracker red. No localisation wthat PfEF-Ts is solely targeted to the apicoplast. In both panels, the nucleus is stained usiTsleader-GFP confirms N-terminal cleavage of the fusion protein. Anti-GFP antibody recontaining GFP and the transit peptide-cleaved form with �10 amino acids in front of GFP(RBCs) alone.
the 11 tyrosine residues in PfEF-Ts (Jameson et al., 1987). The re-sults demonstrate mediation of GDP release from PfEF-Tu byPfEF-Ts.
The kinetics of GDP release from PfEF-Tu in the presence ofPfEF-Ts was also monitored by using FRET from Trp-196 in PfEF-Tuto mant-GDP in a fluorescence stopped-flow measurement device(Fig. 1B). The reactions were carried out in the presence of excess(25 lM) unlabeled GDP. The apparent rate constant (Ki) for sponta-neous mant-GDP dissociation from the PfEF-Tu.mant-GDP complexin the absence of PfEF-Ts was �0.04 s�1. The presence of PfEF-Ts inthe PfEF-Tu:PfEF-Ts ratio of 1:8 stimulated the release of the guaninenucleotide from PfEF-Tu with the apparent rate constant (Ki) of�2.2 s�1 indicating rapid GDP release from apicoplast PfEF-Tu byPfEF-Ts.
3.7. PfEF-Tu.PfEF-Ts structural model
The structures of EF-Tu.EF-Ts complexes from E. coli and T. ther-mophilus have been experimentally solved (Kawashima et al.,1996; Wang et al., 1997; Jeppesen et al., 2005) and reveal a remark-ably similar interaction interface between EF-Tu and EF-Ts in thetwo complexes. To understand the structural alterations leading toGDP release upon interaction of apicoplast PfEF-Tu with PfEF-Ts,we modelled the three-dimensional structure of the PfEF-Tu.PfEF-Ts complex using the E. coli EF-Tu.EF-Ts co-crystal structure [PDB:1EFU chains A and B] as template. Significant alterations in theGDP-binding pocket of the PfEF-Tu structure upon interaction withPfEF-Ts were observed (Fig. 6). As in E. coli, (Schummer et al.,2007) Phe80 of the conserved DFVA motif of PfEF-Ts intercalated
RB
C3D
73D
7-E
FTs L
-GFP
~31kDa~28kDa
20
M
30
40
50
C
etection and localisation. (A) PfEF-Ts is recognised in the parasite lysate in westernler (39 kDa) form are seen, likely representing a longer form without signal peptide,d when the parasite lysate was probed with pre-immune serum (PI). (B) The PfEF-Tsmes in intraerythrocytic stages. (a) The PfEF-Tsleader-GFP (green) co-localised withizonts, the PfEF-Tsleader-GFP labeled the branching, dividing and then segregatingas observed between the PfEF-Tsleader-GFP (green) and Mitotracker (red), indicating
ng DAPI (blue). (C) Western blot of P. falciparum 3D7 cells transfected with the PfEF-cognised 31 and 28 kDa bands in transfected cells representing transit peptide-, respectively. No signals were observed in untransfected 3D7 cells or red blood cells

S. Biswas et al. / International Journal for Parasitology 41 (2011) 417–427 425
between the His85 and His119 residues of helix B and C of PfEF-Tu,respectively, and caused movement of the two helices. Movementof the P-loop away from GDP was also seen, although the flip inthe backbone of the P-loop that breaks the H-bond to the b-phos-phate as a result of movement of Val20 in E. coli (Schummer et al.,2007) was not observed. In addition, PfEF-Ts induced a prominentshift of the Asn136 and Asp139 residues, which form H-bonds andstabilise the guanine base, thereby relaxing the interactions of theseresidues with the nucleotide. Hence, a combination of changes thatcause distancing of residues critical for GDP binding in PfEF-Tu isindicated in the structural model and provides a mechanistic expla-nation for mediation of GDP release by PfEF-Ts.
4. Discussion
Translation of proteins encoded by the apicoplast genomewould require coordinated activity of translation initiation, elonga-tion and release factors to catalyze the multiple steps of peptidesynthesis on organellar ribosomes. Apart from the apicoplast-en-coded PfEF-Tu, all other factors must be encoded by the parasitenuclear DNA. The P. falciparum nuclear genome contains predictedopen reading frames (ORFs) for many of these proteins, althoughtheir organellar targeting and functions remain to be confirmed(Supplementary Table S1). Our results establish the guanine nucle-otide binding and hydrolysis activity of apicoplast EF-Tu, targetingof the nuclear-encoded putative guanine nucleotide exchange
Fig. 6. Predicted structural alterations in the GDP-binding site of Plasmodiumfalciparum apicoplast-encoded translation elongation factor (PfEF)-Tu upon P.falciparum nuclear-encoded translation elongation factor (PfEF)-Ts binding. Arrowsindicate change in orientation of PfEF-Tu in EF-Tu.GDP (cyan) or PfEF-Tu.PfEF-Ts(dark blue) complexes. His85 His119, Asp139 and Asn136 are shown in cyan stick inEF-Tu-GDP and dark blue stick in EF-Tu-EF-Ts. Phe80 (maroon) of PfEF-Tsintercalates between His85 and His119 of PfEF-Tu. GDP and coordinated Mg2+ ionare depicted in atom colour and magenta, respectively.
factor EF-Ts to the apicoplast, and involvement of EF-Ts in organel-lar translation via interaction with apicoplast EF-Tu.
Our data indicate that translation elongation in the apicoplast isfacilitated by the plastid-encoded EF-Tu in combination with anuclear-encoded but apicoplast-targeted EF-Ts, and presumablyby an apicoplast-targeted EF-G (PFF0115c). The picture in the mito-chondrion is less clear. There are only three proteins encoded by themitochondrial genome – none are involved in translation. A nuclearencoded EF-Tu (MAL13P1.164) bears an N-terminal extension that ispredicted to be a mitochondrial transit peptide, as does an EF-G(PFL1590c), however an obvious mitochondrial EF-Ts is lacking.The related apicomplexans Toxoplasma and Babesia each possesstwo bacterial EF-Ts genes that may service both the apicoplast andmitochondrion, but only a single EF-Ts is apparent in each of themultiple Theileria and Plasmodium spp. The extremely reduced mito-chondrial translation may persist without an EF-Ts, relying insteadon the slow recycling of EF-Tu.GDP to EF-Tu.GTP for elongation.Alternatively, a small fraction of EF-Ts may reach the mitochondriaby an unknown route.
As a critical factor for translation in the apicoplast, PfEF-Tu is apotential target for development of anti-malarial drugs. TheEF-Tu-binding antibiotic, kirromycin, exhibits anti-malarial activityin culture with an IC50 value of 50 lM (Clough et al., 1999). Due tovariable permeability of the drug in various bacterial systems, it isdifficult to compare the inhibitory effect of kirromycin in P. falcipa-rum and bacteria. However, the IC50 of kirromycin in P. falciparum ishigh compared with other antibiotics that potentially target EF-Tu(Clough et al., 1999). Our results on the effect of kirromycin onGTP hydrolysis and GDP release from PfEF-Tu reveal differences withits observed effects on EcEF-Tu. As with bacterial EF-Tu, kirromycinhas an action analogous to EF-Ts and increases GDP release from thePfEF-Tu.GDP complex, possibly by weakening of the Asp51/water/Mg2+ interaction (Vogeley et al., 2001). Conversely, kirromycin doesnot significantly enhance PfEF-Tu GTPase activity. Analysis of kirro-mycin resistance mutants has revealed the involvement of sevenamino acid residues along the opposing interfaces of domains Iand III in the EF-Tu.GTP form (Abdulkarim et al., 1994; Mesterset al., 1994). One of these, Ala375 (numbering according to theE. coli sequence), has been replaced by serine (Ser392) in PfEF-Tu.An alteration at this position in E. coli has been shown to lower thebinding affinity of kirromycin to EF-Tu.GTP as well as reduce kirro-mycin-induced enhancement of GTPase activity of EF-Tu (Mesterset al., 1994). Ala375S has also been suggested to sterically interferewith binding of the aliphatic extension of the goldinoic acid moietyof kirromycin in its hydrophobic pocket in EFTu.GDP (Abdulkarimet al., 1994; Vogeley et al., 2001). However, previously reportedbinding of PfEF-Tu.GDP to kirromycin (Clough et al., 1999) as wellas kirromycin-induced stimulation of GDP release from PfEF-Tu (thiswork) indicates that the A375S replacement in PfEF-Tu does notsignificantly alter binding of kirromycin to the PfEF-Tu.GDP form.This may be due to structural differences attributable to the lack ofconservation in the amino acid stretch immediately precedingSer392 in PfEF-Tu. Plasmodium falciparum mitochondrial EF-Tu, theother possible target for kirromycin, also carries the A375S muta-tion. Overall, our analysis indicates that sequence/structural differ-ences between apicoplast and bacterial EF-Tu may cause differentialinhibitory effects of kirromycin-type antibiotics, and may explainthe high IC50 for kirromycin in Plasmodium. Evaluation of the effectsof other EF-Tu inhibitors such as GE2270A and pulvomycin onPfEF-Tu function may reveal additional differences with the bacte-rial elongation factor.
The slow rate (�0.002 s�1) of spontaneous dissociation of GDPfrom bacterial EF-Tu.GDP is physiologically irrelevant (Gromadskiet al., 2002) and requires the catalytic action of EF-Ts for rapidGDP dissociation. In E. coli, rapid protein synthesis is supportedby the EF-Ts-mediated enhancement of GDP dissociation from

426 S. Biswas et al. / International Journal for Parasitology 41 (2011) 417–427
EF-Tu by a factor of 60,000 (Gromadski et al., 2002). The rate ofspontaneous dissociation of GDP from PfEF-Tu was �0.04 s�1, a va-lue 20-fold higher than for EcEF-Tu. This may be explained by thelower affinity of PfEF-Tu for GDP that would result from fewerhydrogen bonds with GDP predicted in the PfEF-Tu.GDP structuremodeled on EcEF-Tu.GDP (Supplementary Fig. S5). Of the Lys136,Asn135, Asp138 and Ser173 residues (E. coli numbers) that formstrong hydrogen bonds with the ribose and guanine ring of GDPin EcEF-Tu (Supplementary Fig. S5A) only the correspondingAsn136 and Asp139 residues (PfEF-Tu numbers) are predicted toform hydrogen bonds in the PfEF-Tu structural model (Supplemen-tary Fig. S5B).
The apparent rate constant for PfEF-Ts mediated GDP releasefrom PfEFTu at the PfEFTu: PfEF-Ts molar ratio of 1:8 was enhanced�55-fold (0.04 s�1 compared with 2.2 s�1) compared with�30,000-fold at the same molar ratio of the E. coli factors (0.002 s�1 comparedwith 60 s�1) (Dahl et al., 2006b). Interestingly, the rate constants ofspontaneous GDP dissociation and PfEF-Ts stimulated release of GDPin PfEF-Tu are comparable with those reported for the helix C H118Amutant of EcEF-Tu at the EcEF-Tu: EcEF-Ts ratio of 1:8 (Dahl et al.,2006b). The H118A mutant has also been shown to increase therelease rate of both GDP and GTP, an effect that has been attributedto the loss of His118 interactions with Gly18 in the EcEF-Tu P-loop(Schummer et al., 2007). Although PfEF-Tu has conserved His andGly residues at the corresponding positions, variations in otherresidues of helix C and surrounding structural elements may havea general effect on the subunit interface, resulting in weaker nucle-otide binding.
The contact between the N-terminal domain of EcEF-Ts and he-lixD of EcEF-Tu has been suggested to play an important role in theformation of the EF-Tu.EF-Ts complex (Zhang et al., 1998; Wiedenet al., 2002). The formation of contacts between the two domains isthe rate limiting step for complex formation (Schummer et al.,2007) and disruption of this interaction decreases the rate of nucle-otide release �2- to 13-fold (Wieden et al., 2002). The structuralmodel of PfEF-Ts (Supplementary Fig. S3B) suggests that there isa change in orientation of the N-terminal domain of the proteinthat would compromise its interaction with helix D of PfEF-Tu. Inaddition, of the six interacting residues each on EF-Tu helixD andEF-Ts N-terminal domain (Wieden et al., 2002), only three residueson the EF-Tu side and two residues on the EF-Ts side are conservedin the P. falciparum sequences. The relatively slow rate of nucleo-tide release by PfEF-Ts (2.2 s�1 in P. falciparum versus 60 s�1 inE. coli) could be attributed to these differences in the Plasmodiumtranslation factors compared with their E. coli counterparts.
In addition to exploring structure–function relationships of twocritical apicoplast translation factors, this study also demonstratesfunctional interaction between two components of the apicoplasttranslation machinery that are encoded by genes located in differ-ent cellular compartments. Genes for apicoplast EF-Ts and otherputative translation initiation, elongation and release factors ofthe organelle transferred to the parasite nucleus during evolution.However, the tufA gene is retained in all plastid-carrying apicom-plexans and in many other plastid genomes. EF-Tu does not fulfilthe requirement of the ‘hydrophobicity hypothesis’ for retentionof organellar genes (Daley and Whelan, 2005). However, the disul-phide reductase activity exhibited by the Plasmodium apicoplastEF-Tu presents a case for its role in response to the organellar re-dox system and thus fulfilment of the requirements of the ‘CORR(co-location of redox regulation) hypothesis’ (Allen, 2003). Proteinswith chaperone activities such as EF-Tu could destabilise disul-phide-forming folding intermediates that may appear as a resultof the changing redox potential of the organelle. Thus, EF-Tu chap-erone function may have a role in rapid organellar response to pro-tein misfolding or related stress in the apicoplast where it may aidin maintaining protein solubility, folding and assembly. Such a
rapid and specific response may require the location of the encod-ing gene within the organelle as suggested by Allen (2003). Anadditional apicoplast-encoded chaperone, ClpC, has been impli-cated in protein import into the organelle (Wilson, 2005; Tonkinet al., 2008).
In conclusion, our results establish the interaction between twoapicoplast translation factors, EF-Tu and EF-Ts, encoded by differentcellular compartments in Plasmodium. Although EF-Tu is a target forseveral known antibiotics that inhibit Plasmodium apicoplast trans-lation (Fleige and Soldati-Favre, 2008), the EF-Tu.EF-Ts interactioninterface that is outside the context of the ribosome has not beenexplored for rational design of translation inhibitors. Sequence andstructural differences between plasmodial and the well-studiedbacterial factors would require careful biochemical analyses of theinhibitory effects of candidate drugs on activities of the targetproteins. In addition, molecular characterisation of other apicoplasttranslation elongation, initiation and release factors is likely to offernewer insights into the process and help identify additional sites fordrug intervention against malaria.
Acknowledgements
We are grateful to Dr. Charlotte Knudsen (Aarhus University,Denmark) for the E. coli EF-Tu expression clone, Prof. Alan Cowman(Walter and Eliza Hall Institute, Australia) for the pGlux.1 vector,Prof. G.I. McFadden (University of Melbourne, Australia) for anti-ACP antibody, Prof. W.G.J. Hol (Howard Hughes Medical Institute,USA) for the RIG plasmid and Dr. P. Guptasarma (Institute of Micro-bial Technology, India) for help with stopped-flow fluorimetry. SB,AG and US received scholarships from the University GrantsCommission and the Council for Scientific and Industrial Research,Government of India. SAR was funded by a CR Roper Fellowshipfrom the University of Melbourne, Australia and National Healthand Medical Research Council, Australia project grant 628704. Thiswork has been conducted as part of the MEPHITIS project andpartially funded by the European Community’s Seventh Frame-work Programme (FP 7/2007-2013) under the grant agreementnumber HEALTH-F3-2009-223024. This is Central Drug ResearchInstitute, India communication number 7897.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.ijpara.2010.11.003.
References
Abdulkarim, F., Liljas, L., Hughes, D., 1994. Mutations to kirromycin resistance occurin the interface of domains I and III of EF-Tu.GTP. FEBS Lett. 352, 118–122.
Ahmadian, M.R., Kreutzer, R., Blechschmidt, B., Sprinzl, M., 1995. Site-directedmutagenesis of Thermus thermophilus EF-Tu: the substitution of threonine-62by serine or alanine. FEBS Lett. 377, 253–257.
Allen, J.F., 2003. The function of genomes in bioenergetic organelles. Philos. Trans. R.Soc. Lond. B. Biol. Sci. 358, 19–37.
Bardwell, J.C., McGovern, K., Beckwith, J., 1991. Identification of a protein requiredfor disulfide bond formation in vivo. Cell 67, 581–589.
Berchtold, H., Reshetnikova, L., Reiser, C.O., Schirmer, N.K., Sprinzl, M., Hilgenfeld, R.,1993. Crystal structure of active elongation factor Tu reveals major domainrearrangements. Nature 365, 126–132.
Boddey, J.A., Moritz, R.L., Simpson, R.J., Cowman, A.F., 2009. Role of the Plasmodiumexport element in trafficking parasite proteins to the infected erythrocyte.Traffic 10, 285–299.
Borrmann, S., Lundgren, I., Oyakhirome, S., Impouma, B., Matsiegui, P.B., Adegnika, A.A.,Issifou, S., Kun, J.F., Hutchinson, D., Wiesner, J., Jomaa, H., Kremsner, P.G., 2006.Fosmidomycin plus clindamycin for treatment of pediatric patients aged 1 to 14years with Plasmodium falciparum malaria. Antimicrob. Agents Chemother. 50,2713–2718.
Brooks, B.R., Bruccoleri, R.E., Olafson, B.D., States, D.J., Swaminathan, S., Karplus, M.,1983. CHARMM: a program for macromolecular energy, minimization, anddynamics calculations. J. Comput. Chem. 4, 187–217.

S. Biswas et al. / International Journal for Parasitology 41 (2011) 417–427 427
Caldas, T., Laalami, S., Richarme, G., 2000. Chaperone properties of bacterialelongation factor EF-G and initiation factor IF2. J. Biol. Chem. 275, 855–860.
Caldas, T.D., El Yaagoubi, A., Richarme, G., 1998. Chaperone properties of bacterialelongation factor EF-Tu. J. Biol. Chem. 273, 11478–11482.
Camps, M., Arrizabalaga, G., Boothroyd, J., 2002. An rRNA mutation identifies theapicoplast as the target for clindamycin in Toxoplasma gondii. Mol. Microbiol.43, 1309–1318.
Chaubey, S., Kumar, A., Singh, D., Habib, S., 2005. The apicoplast of Plasmodiumfalciparum is translationally active. Mol. Microbiol. 56, 81–89.
Clough, B., Rangachari, K., Strath, M., Preiser, P.R., Wilson, R.J., 1999. Antibioticinhibitors of organellar protein synthesis in Plasmodium falciparum. Protist 150,189–195.
Crabb, B.S., Cowman, A.F., 1996. Characterization of promoters and stabletransfection by homologous and nonhomologous recombination inPlasmodium falciparum. Proc. Natl. Acad. Sci. USA 93, 7289–7294.
Dahl, E.L., Shock, J.L., Shenai, B.R., Gut, J., DeRisi, J.L., Rosenthal, P.J., 2006a.Tetracyclines specifically target the apicoplast of the malaria parasitePlasmodium falciparum. Antimicrob. Agents Chemother. 50, 3124–3131.
Dahl, E.L., Rosenthal, P.J., 2008. Apicoplast translation, transcription andgenome replication: targets for antimalarial antibiotics. Trends Parasitol.24, 279–284.
Dahl, L.D., Wieden, H.J., Rodnina, M.V., Knudsen, C.R., 2006b. The importance ofP-loop and domain movements in EF-Tu for guanine nucleotide exchange. J.Biol. Chem. 281, 21139–21146.
Daley, D.O., Whelan, J., 2005. Why genes persist in organelle genomes. Genome Biol.6, 110.
Fichera, M.E., Roos, D.S., 1997. A plastid organelle as a drug target in apicomplexanparasites. Nature 390, 407–409.
Fleige, T., Soldati-Favre, D., 2008. Targeting the transcriptional and translationalmachinery of the endosymbiotic organelle in apicomplexans. Curr. Drug Targets9, 948–956.
Foth, B.J., McFadden, G.I., 2003. The apicoplast: a plastid in Plasmodium falciparumand other apicomplexan parasites. Int. Rev. Cytol. 224, 57–110.
Foth, B.J., Ralph, S.A., Tonkin, C.J., Struck, N.S., Fraunholz, M., Roos, D.S., Cowman,A.F., McFadden, G.I., 2003. Dissecting apicoplast targeting in the malariaparasite Plasmodium falciparum. Science 299, 705–708.
Goodman, C.D., Su, V., McFadden, G.I., 2007. The effects of anti-bacterials on themalaria parasite Plasmodium falciparum. Mol. Biochem. Parasitol. 152, 181–191.
Gromadski, K.B., Wieden, H.J., Rodnina, M.V., 2002. Kinetic mechanism of elongationfactor Ts-catalyzed nucleotide exchange in elongation factor Tu. Biochemistry41, 162–169.
Huecas, S., Schaffner-Barbero, C., Garcia, W., Yebenes, H., Palacios, J.M., Diaz, J.F.,Menendez, M., Andreu, J.M., 2007. The interactions of cell division protein FtsZwith guanine nucleotides. J. Biol. Chem. 282, 37515–37528.
Humphrey, W., Dalke, A., Schulten, K., 1996. VMD: visual molecular dynamics. J.Mol. Graph. 14 (33–38), 27–38.
Jameson, D.M., Gratton, E., Eccleston, J.F., 1987. Intrinsic fluorescence of elongationfactor Tu in its complexes with GDP and elongation factor Ts. Biochemistry 26,3894–3901.
Jensen, J.B., Trager, W., 1978. Plasmodium falciparum in culture: establishment ofadditional strains. Am. J. Trop. Med. Hyg. 27, 743–746.
Jeppesen, M.G., Navratil, T., Spremulli, L.L., Nyborg, J., 2005. Crystal structure of thebovine mitochondrial elongation factor Tu.Ts complex. J. Biol. Chem 280,5071–5081.
Kawashima, T., Berthet-Colominas, C., Wulff, M., Cusack, S., Leberman, R., 1996. Thestructure of the Escherichia coli EF-Tu.EF-Ts complex at 2.5 A resolution. Nature379, 511–518.
Kudlicki, W., Coffman, A., Kramer, G., Hardesty, B., 1997. Renaturation of rhodaneseby translational elongation factor (EF) Tu. Protein refolding by EF-Tu flexing. J.Biol. Chem 272, 32206–32210.
Laskowski, R.A., Moss, D.S., Thornton, J.M., 1993. Main-chain bond lengths and bondangles in protein structures. J. Mol. Biol. 231, 1049–1067.
McFadden, G.I., Reith, M.E., Munholland, J., Lang-Unnasch, N., 1996. Plastid inhuman parasites. Nature 381, 482.
McFadden, G.I., Roos, D.S., 1999. Apicomplexan plastids as drug targets. TrendsMicrobiol. 7, 328–333.
Mesters, J.R., Zeef, L.A., Hilgenfeld, R., de Graaf, J.M., Kraal, B., Bosch, L., 1994. Thestructural and functional basis for the kirromycin resistance of mutant EF-Tuspecies in Escherichia coli. Embo J. 13, 4877–4885.
Morris, A.L., MacArthur, M.W., Hutchinson, E.G., Thornton, J.M., 1992.Stereochemical quality of protein structure coordinates. Proteins 12, 345–364.
Parmeggiani, A., Swart, G.W., 1985. Mechanism of action of kirromycin-likeantibiotics. Annu. Rev. Microbiol. 39, 557–577.
Ralph, S.A., D’Ombrain, M.C., McFadden, G.I., 2001. The apicoplast as an antimalarialdrug target. Drug Resist. Updat. 4, 145–151.
Ralph, S.A., van Dooren, G.G., Waller, R.F., Crawford, M.J., Fraunholz, M.J., Foth, B.J.,Tonkin, C.J., Roos, D.S., McFadden, G.I., 2004. Tropical infectious diseases:metabolic maps and functions of the Plasmodium falciparum apicoplast. Nat.Rev. Microbiol. 2, 203–216.
Ram, E.V., Naik, R., Ganguli, M., Habib, S., 2008. DNA organization by the apicoplast-targeted bacterial histone-like protein of Plasmodium falciparum. Nucleic AcidsRes. 36, 5061–5073.
Richarme, G., 1998. Protein-disulfide isomerase activity of elongation factor EF-Tu.Biochem. Biophys. Res. Commun. 252, 156–161.
Rodnina, M.V., Fricke, R., Kuhn, L., Wintermeyer, W., 1995. Codon-dependentconformational change of elongation factor Tu preceding GTP hydrolysis on theribosome. Embo J. 14, 2613–2619.
Roy, A., Cox, R.A., Williamson, D.H., Wilson, R.J., 1999. Protein synthesis in theplastid of Plasmodium falciparum. Protist 150, 183–188.
Ruangweerayut, R., Looareesuwan, S., Hutchinson, D., Chauemung, A., Banmairuroi,V., Na-Bangchang, K., 2008. Assessment of the pharmacokinetics and dynamicsof two combination regimens of fosmidomycin–clindamycin in patients withacute uncomplicated falciparum malaria. Malar. J. 7, 225.
Ryckaert, J.P., Ciccitti, G., Berendsen, H.J.C., 1977. Numerical integration of thecartesian equations of motion of a system with constrains: molecular dynamicsof n-alkanes. J. Comp. Phys. 23, 327–341.
Sali, A., Blundell, T.L., 1993. Comparative protein modelling by satisfaction of spatialrestraints. J. Mol. Biol. 234, 779–815.
Sali, A., Overington, J.P., 1994. Derivation of rules for comparative protein modelingfrom a database of protein structure alignments. Protein Sci. 3, 1582–1596.
Sali, A., 1995. Comparative protein modeling by satisfaction of spatial restraints.Mol. Med. Today 1, 270–277.
Sato, S., Tews, I., Wilson, R.J., 2000. Impact of a plastid-bearing endocytobiont onapicomplexan genomes. Int. J. Parasitol. 30, 427–439.
Schmeing, T.M., Voorhees, R.M., Kelley, A.C., Gao, Y.G., Murphy, F.V.T., Weir, J.R.,Ramakrishnan, V., 2009. The crystal structure of the ribosome bound to EF-Tuand aminoacyl-tRNA. Science 326, 688–694.
Schummer, T., Gromadski, K.B., Rodnina, M.V., 2007. Mechanism of EF-Ts-catalyzedguanine nucleotide exchange in EF-Tu: contribution of interactions mediated byhelix B of EF-Tu. Biochemistry 46, 4977–4984.
Schwemmle, M., Staeheli, P., 1994. The interferon-induced 67-kDa guanylate-binding protein (hGBP1) is a GTPase that converts GTP to GMP. J. Biol. Chem.269, 11299–11305.
Sidhu, A.B., Sun, Q., Nkrumah, L.J., Dunne, M.W., Sacchettini, J.C., Fidock, D.A., 2007.In vitro efficacy, resistance selection, and structural modeling studies implicatethe malarial parasite apicoplast as the target of azithromycin. J. Biol. Chem. 282,2494–2504.
Song, H., Parsons, M.R., Rowsell, S., Leonard, G., Phillips, S.E., 1999. Crystal structureof intact elongation factor EF-Tu from Escherichia coli in GDP conformation at2.05 A resolution. J. Mol. Biol. 285, 1245–1256.
Stanway, R.R., Witt, T., Zobiak, B., Aepfelbacher, M., Heussler, V.T., 2009.GFP-targeting allows visualization of the apicoplast throughout the life cycleof live malaria parasites. Biol. Cell 101, 415–430.
Tonkin, C.J., van Dooren, G.G., Spurck, T.P., Struck, N.S., Good, R.T., Handman, E.,Cowman, A.F., McFadden, G.I., 2004. Localization of organellar proteins inPlasmodium falciparum using a novel set of transfection vectors and a newimmunofluorescence fixation method. Mol. Biochem. Parasitol. 137, 13–21.
Tonkin, C.J., Kalanon, M., McFadden, G.I., 2008. Protein targeting to the malariaparasite plastid. Traffic 9, 166–175.
Vogeley, L., Palm, G.J., Mesters, J.R., Hilgenfeld, R., 2001. Conformational change ofelongation factor Tu (EF-Tu) induced by antibiotic binding. Crystal structure ofthe complex between EF-Tu.GDP and aurodox. J. Biol. Chem. 276, 17149–17155.
Wagner, A., Simon, I., Sprinzl, M., Goody, R.S., 1995. Interaction of guanosinenucleotides and their analogs with elongation factor Tu from Thermusthermophilus. Biochemistry 34, 12535–12542.
Waller, R.F., Keeling, P.J., Donald, R.G., Striepen, B., Handman, E., Lang-Unnasch, N.,Cowman, A.F., Besra, G.S., Roos, D.S., McFadden, G.I., 1998. Nuclear-encodedproteins target to the plastid in Toxoplasma gondii and Plasmodium falciparum.Proc. Natl. Acad. Sci. USA 95, 12352–12357.
Waller, R.F., Reed, M.B., Cowman, A.F., McFadden, G.I., 2000. Protein trafficking tothe plastid of Plasmodium falciparum is via the secretory pathway. Embo J. 19,1794–1802.
Wang, Y., Jiang, Y., Meyering-Voss, M., Sprinzl, M., Sigler, P.B., 1997. Crystalstructure of the EF-Tu.EF-Ts complex from Thermus thermophilus. Nat. Struct.Biol. 4, 650–656.
Wieden, H.J., Gromadski, K., Rodnin, D., Rodnina, M.V., 2002. Mechanism ofelongation factor (EF)-Ts-catalyzed nucleotide exchange in EF-Tu.Contribution of contacts at the guanine base. J. Biol. Chem. 277, 6032–6036.
Wilson, R.J., Denny, P.W., Preiser, P.R., Rangachari, K., Roberts, K., Roy, A., Whyte, A.,Strath, M., Moore, D.J., Moore, P.W., Williamson, D.H., 1996. Complete gene mapof the plastid-like DNA of the malaria parasite Plasmodium falciparum. J. Mol.Biol. 261, 155–172.
Wilson, R.J., 2005. Parasite plastids: approaching the endgame. Biol. Rev. Camb.Philos. Soc. 80, 129–153.
Wolf, H., Chinali, G., Parmeggiani, A., 1974. Kirromycin, an inhibitor of proteinbiosynthesis that acts on elongation factor Tu. Proc. Natl. Acad. Sci. USA 71,4910–4914.
Yu, H., Chan, Y.L., Wool, I.G., 2009. The identification of the determinants of thecyclic, sequential binding of elongation factors Tu and G to the ribosome. J. Mol.Biol. 386, 802–813.
Zhang, Y., Yu, N.J., Spremulli, L.L., 1998. Mutational analysis of the roles of residuesin Escherichia coli elongation factor Ts in the interaction with elongation factorTu. J. Biol. Chem. 273, 4556–4562.