inborn errors and cmo
TRANSCRIPT

Diagnostic Approaches to Pediatric Cardiomyopathy of Metabolic Genetic Etiologies and Their Relation to Therapy
Ped. Cardiology 2007
Gerald F. Cox, MD, PhD
Dr. Lalit G. L.

Introduction
Pediatric cardiomyopathy has a number of different causes which poses diagnostic challenge.
This review will focus on specific group of genetic disorders, (IEM), that cause cardiomyopathy.
Most IEM are caused by defects in enzymes involved in intermediary metabolism or energy production.
Many of IEM are currently treatable and in many cases cardiomyopathy may be reversed.

Epidemiology and Genetics of IEM:
IEM occur in approximately 1 in 4000 newborns.
Approximately 5% of IEM associated with cardiomyopathy.
In some cardiomyopathy dominates clinical picture & is major cause of death, whereas in others it may be an incidental finding.
Rarely heart is the only affected organ, as in cardiac phosphorylase kinase deficiency.

Most IEM are inherited in autosomal recessive manner.
Recurrence risk is 25% with each future pregnancy.
Only few IEM causing cardiomyopathy are X-linked. Affecting males & h/o affected uncles, grandfathers from maternal side.
Specific diagnosis is important to optimally treat child, accurate genetic counseling, family planning, prenatal diagnosis & screening of younger siblings.

IEM Causes of Cardiomyopathy
Pediatric Cardiomyopathy Registry (PCMR) data.Two-thirds children had an unknown cause &
classified as idiopathic disease. One-third of cases was diagnosed with
myocarditis or a variety of genetic causes (29.1%),
familial isolated cardiomyopathy (24.2%), neuromuscular disease (22.2%), IEM (15.4%),genetic syndrome (8.8%).
Thus, approximately 5% of all cases of cardiomyopathy and 15% with known causes were due to an IEM.

Frequencies of Known Causes of Cardiomyopathy:
The most common adaptive response of heart in IEM is hypertrophy, with or without dilatation.
Categories of known causes presented by functional type of cardiomyopathy. IEM accounted for 26.8% of known causes of
hypertrophic cardiomyopathy, 6.8% of cases of dilated cardiomyopathy, 0% of restrictive cardiomyopathy, and 26.9% of mixed/other type of
cardiomyopathy.

Inborn Error of Metabolism Disease Groups:
With exception of mitochondrial disorders, each IEM is associated with specific functional type of cardiomyopathy.
Nearly half cases of hypertrophic cardiomyopathy caused by IEM were due to glycogen storage diseases, most commonly Pompe disease.

Pompe disease:
Lysosomal storage disorder-deficiency of the α-glucosidase.
Classical infantile form: Presents in first few months of life with hypotonia, muscle weakness, enlarged tongue, & CHF.
ECG: short PR interval with tall QRS waves.
Cardiac silhouette is massively enlarged and the heart is extremely thickened (left ventricular mass Z-score > 6).
LVOT obstruction may be present.
Untreated, children typically die by 1yr of age.

Non-classical infantile Pompe disease:
Approximately 10% of infants with Pompe disease.
Mild cardiomyopathy and severe skeletal myopathy.
May survive for several years.

In dilated cardiomyopathy group, oxidative phosphorylation defects & systemic carnitine deficiency are the most common causes aprox. 40% of cases each.
Mitochondrial syndromes MERFF, MELAS & Kearns-Sayre syndrome affect synthesis of multiple mitochondrial proteins & respiratory chain complexes will cause cardiomyopathy.

Within mixed/other group, oxidative phosphorylation defects accounts for 70% of cases, several of them were boys with Barth syndrome.
Defect in remodeling of cardiolipin, lipid that is localized on inner membrane of mitochondria, is important for oxidative phosphorylation.
This condition is likely under-diagnosed.

Barth syndrome:
Classic features: endocardial fibroelastosis, neutropenia with frequent infections, skeletal myopathy, growth retardation, abnormal mitochondria in biopsies.
Some may have only dilated cardiomyopathy without other systemic findings, cardiomyopathy may be very severe during infancy, often responds well to standard cardiac medications.

Pathophysiology:
Three main pathophysiological mechanisms.
1. Bulk storage & infiltration of substrate causing mechanical effect on cardiomyocyte disrupting alignment of myofibrils required for efficient contraction.
Involve large macromolecules, i.e., triglycerides, glycogen, and lysosomal substrates.

2. Impaired energy production, which results in decreased ATP to meet needs of the cell.
Main sources of energy in heart fatty acids & secondarily glycogen.
Patients who have defects in oxidative phosphorylation are unable to efficiently make ATP from any energy source.
Adaptive response of heart to inefficient contraction is hypertrophy- hypertrophic cardiomyopathy.

3. Production of toxic metabolites by organic acidemias, amino acidurias, Refsum disease, and disorders of oxidative phophorylation.
Accumulated metabolites exert their deleterious effect by lowering the cellular pH, inhibiting intermediary metabolism or oxidizing mitochondrial components such as DNA, lipids, and proteins structures (free radicals).

Clinical Presentation and Approach to Diagnosis:
Cardiomyopathy: presenting or dominant clinical feature.
Clinical features include a distinctive physical appearance: coarse facial features, cloudy corneas, slow growth and hepatosplenomegaly.
Neurologic findings: acute or chronic encephalopathy, developmental delay & seizures.
Myopathy: hypotonia & weakness.
Skeletal findings or liver dysfunction.

IEM impairing energy production or produce toxic metabolites: CHF often occurs in setting of acute metabolic decompensation triggered by energy stressors, like illness, surgery, infection, fasting, or physical exertion.
These events confound initial presentation suggest an alternative diagnosis for cardiomyopathy, e.g., viral myocarditis.
Basic laboratory abnormalities: metabolic acidosis with increased anion gap, hypoglycemia, hyperammonemia, ketosis, elevated liver function tests.

Acute encephalopathy is typically accompanied by metabolic abnormalities e.g., metabolic acidosis, hyperammonemia & hypoglycemia- characteristic of small molecule diseases.
Chronic encephalopathy is often associated with mitochondrial or lysosomal storage disorders.


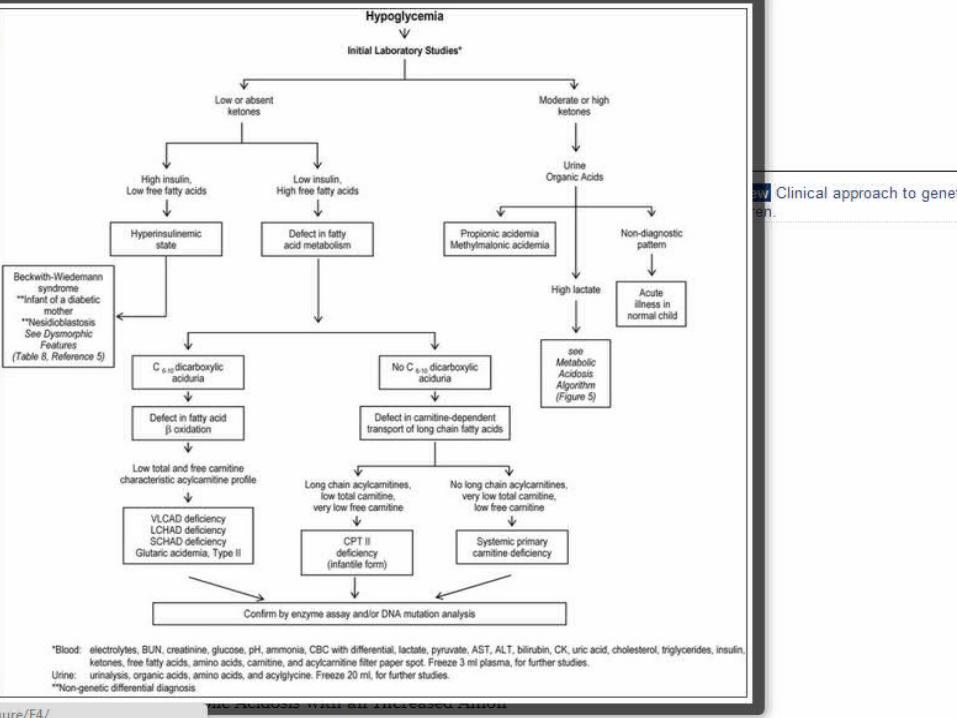
Evaluation of hypoglycemia:
Measurement of ketones, insulin, free fatty acids, urine organic acids (including dicarboxylic acids), and carnitine (total and free).
Assist in determination of whether the hypoglycemia is associated with a defect in mobilizing triglycerides (overgrowth syndromes and infant of a diabetic mother).
Inability to convert fatty acids into ketones (fatty acid oxidation defects or carnitine-transport defects), organic acidemias, or GSD III.

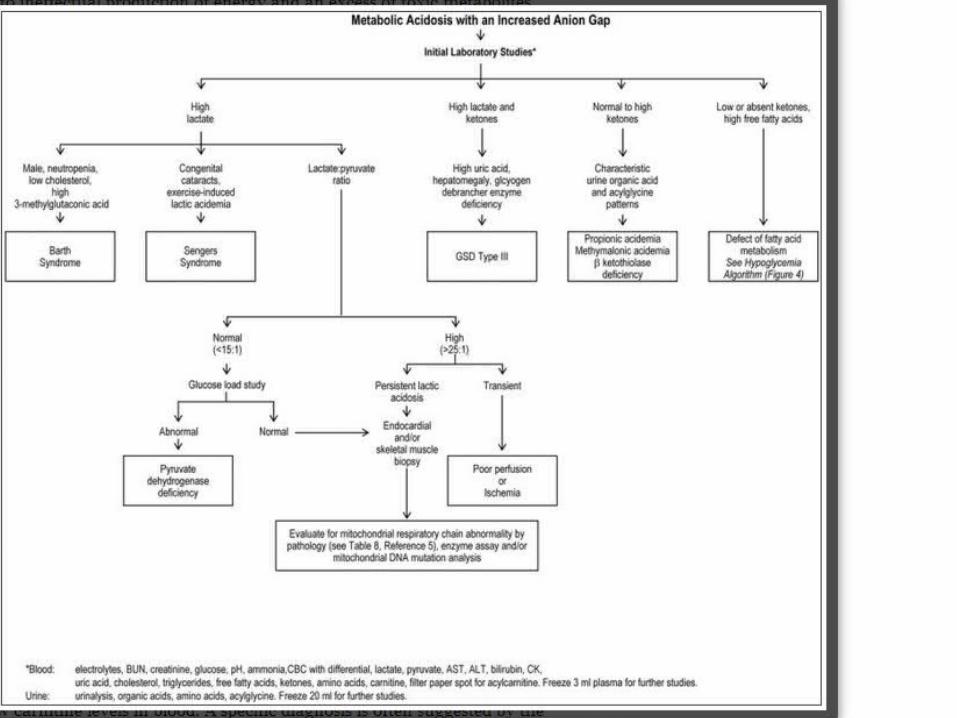
Metabolic acidosis with an increased anion gap (> 15 mEq/L) focus on determining the identity of the offending anion, which can be lactic acid, ketoacids, more complex organic acids (propionic acid), or fatty acids.
The different acids are distinguished by direct measurement in blood by routine clinical testing (lactic, ketoacids, free fatty acids) or by tandem-mass spectrometry as acylcarnitine derivatives.
When the lactate level is increased, the lactate:pyruvate ratio (L:P) can help to distinguish pyruvate dehydrogenase deficiency (L:P < 15) and oxidative phosphorylation defects (L:P > 25).

Neuromuscular symptoms that point towards an IEM:
Weakness/hypotonia beginning in infancy & ataxia.
Pompe disease a major cause of “floppy baby syndrome”, hypotonia is severe that can mimic spinal muscular atrophy.
Diagnosis of Barth syndrome is suggested in a male, neutropenia, low cholesterol level & 3-methylglutaconic aciduria.
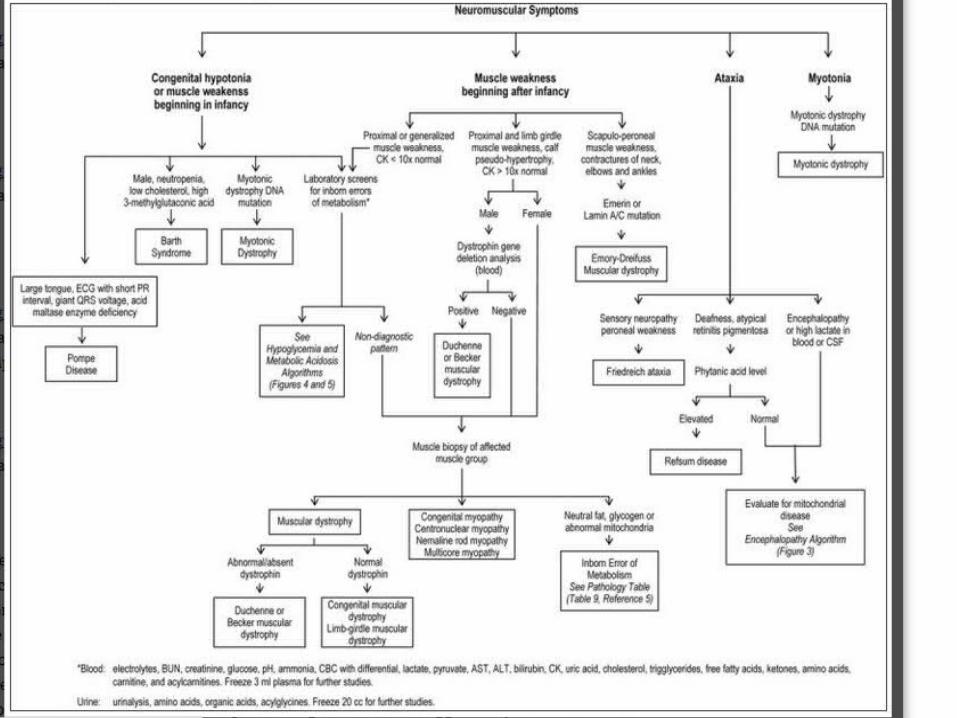

Mitochondrial disorders: Ataxia, chronic encephalopathy, cardiac conduction defects, and arrhythmias.
Refsum disease: Ataxia, deafness, atypical retinitis pigmentosa, and elevated plasma phytanic acid levels.

Diagnosis:
Ideal time- first few days of life prior to symptom onset, newborn screening followed by confirmatory enzyme assay.
Tandem mass spectroscopy can diagnose more than 40 different IEM on basis of their characteristic acylcarnitine & amino acid profiles.

Newborn screen negative- certain abnormal metabolites may not be elevated in first two days of life- fatty acid oxidation defects.
IEM unable to diagnose by current tandem spectrometry assays-glycogen storage diseases, oxidative phosphorylation defects & lysosomal storage disorders.

Child presenting with cardiomyopathy requires evaluation of diagnostic metabolites, demonstration of deficient enzyme activity in appropriate cell type or bodily fluid & DNA mutation analysis.
IEM with excessive acid production, best time for testing is when metabolism is stressed & underlying metabolic defects are drawn out.CHF, infection, illness, post-surgery, fasting,
during metabolic decompensation (hypoglycemia, metabolic acidosis, acute encephalopathy).

Detection of metabolites can be suggestive-increased urinary glycosaminoglycans in MPS & elevated C14:1 acylcarnitine by tandem mass spectrometry in VLCAD deficiency.
Gold standard for diagnosis-enzyme assay. Level of enzymatic activity in wbc, serum, or
plasma.Cultured fibroblasts from a skin biopsy-
requires several weeks.
Enzyme assay for Pompe disease can reliably performed using a dried blood spot on a newborn screening card, shortens time for diagnosis.

Despite newborn screening, IEM are under diagnosed
In hypertrophic cardiomyopathy, metabolic blood & urine testing and skeletal muscle biopsy useful in establishing cause of cardiomyopathy.
Mitochondrial disorder, skeletal muscle biopsy is gold standard for diagnosis provides pathologic findings. e.g., ragged red fibers by Gomori trichrome staining & abnormal-appearing mitochondria by electron microscopy.

Treatment:
Dextrose 10% with IV fluids: flush out accumulated metabolites- corrects hypoglycemia and metabolic acidosis.
If metabolic acidosis is present, carnitine will help to replenish low free reserves, bind acidic compounds, liberate CoA from acyl-CoA & help to restore intermediary metabolism.

Chronic treatment:
Reducing intake of non-metabolizable substrates from diet -branch chain amino acids in propionic acidemia & long-chain fatty acids in VLCAD and multiple acyl-CoA dehydrogenase deficiencies.
Avoidance of fasting, In infancy fasting beyond 6 hours should be
avoided. After age 1, a cornstarch-supplemented drink
at bedtime provides a slow release of glucose throughout night.

Alternative, metabolizable energy source- medium chain triglycerides for long-chain fatty acid oxidation defects.
Antioxidants to neutralize free radicals in oxidative phosphorylation defects & carnitine to liberate CoA from acylCoA compounds.
Residual enzyme activity can be enhanced by using pharmacologic doses of vitamin cofactors, biotin for propionic acidemia & riboflavin for multiple acyl-CoA dehydrogenase deficiency.

Oxidative phosphorylation defects treated empirically with combinations of antioxidants water soluble [Vitamin C] and lipid-soluble [CoEnzyme Q10] to prevent free radical damage.
Carnitine supplements are very effective for treating systemic carnitine deficiency & dilated cardiomyopathy.
Enzyme replacement therapy- Gaucher disease, Fabry disease, Pompe disease, and MPSI, II, VI & Pompe disease.

In clinical trials, reduction of myocardial hypertrophy has been demonstrated in Pompe disease and MPS I with enzyme replacement therapy.
Beneficial effects of hematopoietic stem cell transplantation seen in MPS I and VI.

Future Directions
Improving the outcomes of children with IEM- timely diagnosis and the use of disease-specific therapies.
Use of dried blood spots for metabolite screening (amino acid and acylcarnitine profiles) and enzyme assays (Pompe disease) to facilitate the early screening.
Replacing the missing enzymes- recombinant enzymes, stem cells, organ transplantation or gene therapy.

Thank you…