importance of the amino-acid composition of the shutter region of plasminogen activator inhibitor-1...
TRANSCRIPT

Importance of the amino-acid composition of the shutter region ofplasminogen activator inhibitor-1 for its transitions to latent andsubstrate forms
Martin Hansen, Marta N. Busse and Peter A. Andreasen
Laboratory of Cellular Protein Science, Department of Molecular and Structural Biology, University of Aarhus, Denmark
The serpins are of general protein chemical interest due to
their ability to undergo a large conformational change
consisting of the insertion of the reactive centre loop (RCL)
as strand 4 of the central b sheet A. To make space for the
incoming RCL, the ‘shutter region’ opens by the b strands
3A and 5A sliding apart over the underlying a helix B. Loop
insertion occurs during the formation of complexes of
serpins with their target serine proteinases and during
latency transition. This type of loop insertion is unique to
plasminogen activator inhibitor-1 (PAI-1). We report here
that amino-acid substitutions in a buried cluster of three
residues forming a hydrogen bonding network in the shutter
region drastically accelerate a PAI-1 latency transition; that
the rate was in all cases normalized by the PAI-1 binding
protein vitronectin; and that substitution of an adjacent b
strand 5A Lys residue, believed to anchor b strand 5A to
other secondary structural elements, had differential effects
on the rates of latency transition in the absence and the
presence of vitronectin, respectively. An overlapping, but
not identical set of substitutions resulted in an increased
tendency to substrate behaviour of PAI-1 at reaction with its
target proteinases. These findings show that vitronectin
regulates the movements of the RCL through conformation-
al changes of the shutter region and b strand 5A, are in
agreement with RCL insertion proceeding by different
routes during latency transition and complex formation, and
contribute to the biochemical basis for the potential use of
PAI-1 as a therapeutic target in cancer and cardiovascular
diseases.
Keywords: cancer; extracellular proteolysis; fibrinolysis;
proteinase inhibitors; serine proteinases.
The serpins constitute a protein family of which the bestcharacterized members are serine proteinase inhibitors,including antithrombin III, a1-antitrypsin, and plasminogenactivator inhibitor-1 (PAI-1). The serpins are globularproteins consisting of nine a helices and three b sheets(reviewed in [1–3]). Serpins are of general protein chemicalinterest due to their ability to undergo a large confor-mational change with the insertion of the surface-exposedreactive centre loop (RCL) as strand 4 of the large central bsheet A as the main event (Fig. 1). The RCL insertionresults in a considerable stabilization compared to the nativeserpin structure, and is often referred to as the stressed-to-relaxed transition (for a review, see [2]). This stabilization
forms the basis for the mechanism behind the inhibitoryfunction of serpins. After cleavage of the P1–P1
0 peptidebond in the RCL, the active site serine of the proteinaseremains attached to the carboxyl group of the P1 residue byan ester bond [4–6]. The subsequent RCL insertion intob sheet A therefore results in an < 7-nm translocation ofthe proteinase from the position of its initial encounter withthe RCL to the other pole of the serpin [7–10]. Thetranslocation results in distortion of the proteinase [11] andinactivation of the enzymatic machinery [10]. Delayed RCLinsertion results in hydrolysis of the ester bond, the serpinthus behaving as an ordinary substrate [12]. The stabil-ization caused by RCL insertion also underlies the uniqueconversion of active PAI-1 to the latent state, in which theN-terminal part of the intact RCL is inserted as b strand 4Awithout cleavage of any peptide bonds, and the C-terminalpart is stretched along the surface of the molecule [13](Fig. 1).
In order to make space for the incoming new strandduring RCL insertion, a fragment of the structure consistingof b strands 1A, 2A, 3A, and a helix F (the small serpinfragment) must slide away from the rest of the structure (thelarge serpin fragment). During the b sheet opening, theregion around a helices D and E forms a flexible joint, andb strands 3A and 5A slide apart in a shutter-like manner overthe underlying a helix B [14]. The central part of b strands3A and 5A and the N-terminal part of a helix B is thereforereferred to as the shutter region [2]. By high resolution X-raycrystal structure analysis of the native form of the serpinplasminogen activator inhibitor-2 (PAI-2) and the P1–P1
0
cleaved form of horse leukocyte elastase inhibitor, a buried
Enzymes: Urokinase-type plasminogen activator (EC 3.4.21.73).
Note: plasminogen activator inhibitor-1 and vitronectin have the NCBI
accession numbers P05121 and P04004, respectively.
Note: a website is available at http://www.mbio.aau.dk
Correspondence to M. Hansen, Laboratory of Cellular Protein Science,
Department of Molecular and Structural Biology, University of Aarhus,
10C Gustav Wieds Vej, 8000 Aarhus C, Denmark.
Fax: þ 45 86123178, Tel.: þ 45 89425079,
E-mail: [email protected]
(Received 16 July 2001, revised 5 October 2001, accepted
8 October 2001)
Abbreviations: HEK293T, the human embryonic kidney cell line 293T;
LMW-uPA, low Mr uPA; PAI-1, plasminogen activator inhibitor-1;
PAI-2, plasminogen activator inhibitor-2; RCL, reactive centre loop;
S-2444, L-5-pyroglutamyl-glycyl-L-arginine-p-nitroaniline; uPA,
urokinase-type plasminogen activator.
Eur. J. Biochem. 268, 6274–6283 (2001) q FEBS 2001

cluster with a complicated hydrogen bonding network wasseen to be present in the shutter region, although differentlyorganized, in both the stressed and the relaxed confor-mations [15]. The network involves the side chains of theamino acids in positions 53 and 56 in a helix B, 186 in b
strand 3A, and position 334 in b strand 5A (Fig. 1; thenumbering of amino acids in PAI-1 is according to thea1-antitrypsin template numbering scheme [1,3]). Sequencealignments of 219 serpins showed that residue 53 is a Ser in92% of the cases; residue 56 is a Ser in 74% of the cases;residue 186 an Asn in 87% of the cases; and residue 334 aHis in 80% of the cases [3]. In addition, residue 54 is a Pro in89% of the cases. The importance of the identity of theresidues present in these and adjacent positions aresupported by the clustering of disease-causing mutationsin the shutter region [16,17].
PAI-1 differs from most other serpins with respect to theidentity of the residues in the buried cluster in the shutterregion, having a Gly in position 56 and a Gln in position334 (Fig. 1). This composition of amino acids inpositions 53/56/334 is present in only 5% of the serpins,for example PN-1, RASP-1, TSA2004, and the viral serpinsSPI-1, M2L, and H14-B [3]. A few previous studies haveaddressed the importance of the shutter region for themovements of the RCL in PAI-1. Berkenpas et al. [18]demonstrated that Ser and Thr substitutions of Pro54delayed latency transition. We showed that a Q334Hsubstitution accelerated latency transition [19]. We also
implicated the region of b strand 5A overlying the buriedcluster in RCL movements by demonstrating that increasedproteolytic susceptibility of the peptide bonds Gln331–Ala332, Ala332–Leu333, and Lys335–Val336 accom-panied a transition to substrate behaviour in detergent-containing buffers at low temperatures [20,21]; and that aK335A substitution potentiated activity-neutralization ofPAI-1 by some monoclonal antibodies [22]. Substitutions ofLys335 in a1-antitrypsin, a1-antichymotrypsin, and anti-thrombin III resulted in an increased conformationalstability and a decreased specific inhibitory activity[23,24]. Lys335, localized in b strand 5A, points outwardfrom the hydrophobic core and is conserved in 66% ofserpins, the remaining serpins having Gln (10%), Ala (5%),and Arg (5%) in this position [3].
In order to investigate the importance of the shutter regionfor the unique types of RCL insertion in PAI-1, we have nowundertaken a number of substitutions in the shutter regionand b strand 5A of PAI-1 and studied their effect on thetransition to latent and substrate forms and on the stabilizingeffect of vitronectin, a flexible joint region-binding acofactor known to delay PAI-1 latency transition (reviewedin [25,26]). Both transitions to latent and substrate formswere strongly but differently influenced by the amino-acidcomposition of the shutter region. Surprisingly, we foundthat substitution of Lys335 to Ala affected the rate of latencytransition differently in the absence and presence ofvitronectin.
Fig. 1. The buried cluster and Lys335 in the
shutter region of PAI-1. The top panel shows
ribbon diagrams of active (left) and latent
PAI-1 (right). Secondary structure elements are
indicated as follows: blue, b sheet A; red,
a helix B; green, gate region; yellow, RCL and
b strand 1C in active PAI-1 and RCL inserted
as b strand 4A in latent PAI-1. The P1 Arg is
displayed as a stick. The lower panel shows the
three-dimensional structure of the shutter
region of active PAI-1 (left) and latent PAI-1
(right). The molecules were rotated < 908
around a horizontal axis compared to the top
panel. The colour code for secondary structure
elements are as in the top panel. Presented
amino-acid residues are: green, shutter region
residues Ser53, Gly56, and Gln334; grey,
Asn186 in b strand 3A; yellow, Lys335; purple,
potential interaction partners for Lys335, i.e.
Glu294 in b strand 6A and the backbone of
Asn171 in the a helix F/b strand 3A loop.
Note: SWISSPDB VIEWER uses the same
signature for a helices and the short 310-helix
found in the a helix F/b strand 3A loop of
active PAI-1.
q FEBS 2001 PAI-1 shutter region mutations (Eur. J. Biochem. 268) 6275

M A T E R I A L S A N D M E T H O D S
PAI-1
In order to generate recombinant wild-type and mutatedPAI-1, PAI-1 cDNA [27] was cloned into the expressionvector pcDNA3.1(–) (Invitrogen) by use of standardtechniques. The generated expression plasmid was denotedpcDNA3.1(–)PAI-1. Relevant fragments of the PAI-1sequence were transferred to the mutagenesis vectorLITMUS 28 (New England Biolabs). Point mutationswere introduced into the PAI-1 cDNA fragment inserted intoLITMUS 28 by use of the PCR-based QuickChangee Site-Directed Mutagenesis kit (Stratagene). The mutagenesisprimers were from DNA Technology (Aarhus, Denmark),had a melting point above 60 8C, and were designed with thedesired mutation(s) in the middle of their sequence. Aftermutagenesis, the fragments were moved back intopcDNA3.1(–)PAI-1 by the use of unique restriction sites.All mutations were verified by DNA sequencing of bothstrands of the PCR produced fragment after transfer backto pcDNA3.1(–)PAI-1, by use of either the ThermoSequenasee II dye terminator cycle sequencing kit(Amersham Pharmacia Biotech AB) or the ABI PRISMe
dye terminator cycle sequencing ready reaction kit(PerkinElmer).
Recombinant PAI-1 variants were expressed in humanembryonic kidney 293T cells (phenotype 293tsA1609neo)[28], grown in Dulbecco’s modified Eagle’s medium, bytransient transfection using the calcium/phosphate precipi-tation technique [28]. Briefly, 1 h prior to transfection, newmedium with 10% fetal bovine serum and 25 mM
chloroquine was added to cells grown to 90% confluencein a 15-cm culture dish. Transfection was carried out bymixing 30 mg DNA (H2O added to a total of 1752 mL),248 mL 2 M CaCl2, and 2 mL 42 mM Hepes, pH 7.05,274 mM NaCl, 10 mM KCl, 1.5 mM Na2HPO4, 11 mM
D-(þ)-glucose. After 1–2 min, this mixture was addeddropwise to the cell medium and carefully distributed. Freshmedium without fetal bovine serum and chloroquine wasadded after 9–11 h of incubation. The conditioned mediumwas harvested after 48 and 96 h. Nontransfected or mocktransfected HEK293T cells were shown not to express eitherPAI-1 or uPA by standard ELISA with monoclonal andpolyclonal antibodies as capture and detection antibodies,respectively. Recombinant PAI-1 variants were purifiedfrom serum-free conditioned medium of the transfectedcells by immunoaffinity chromatography in one step[29,30]. After purification, the variants were dialysedagainst NaCl/Pi (0.01 M NaH2PO4, pH 7.4, 0.14 M NaCl)and concentrated to < 1 mg·mL21.
Other proteins and miscellaneous materials
The following materials were purchased from the indicatedsources: BSA (Sigma); media components for HEK293Tculturing (Life Technology); Qiaquick gel extraction kit(Qiagen); Rapid DNA ligation kit (Boehringer Mannheim);restriction enzymes (New England Biolabs Inc.; orAmersham Pharmacia Biotech AB; or BoehringerMannheim); L-5-oxopropyl-glycyl-L-arginine-p-nitro-anilide (S-2444, Chromogenix AB); SDS (Serva); humanurokinase-type plasminogen activator (uPA; Wakamoto
Pharmaceutical Co.); vitronectin (Becton Dickinson; orHaemochrom AB). All other chemicals and reagents were ofthe highest quality commercially available.
Activation of latent PAI-1
Unless otherwise indicated, latent PAI-1 was converted tothe active conformation by denaturation with 0.1% SDS for1 h at room temperature and refolding by a . 50-folddilution in 0.1 M Tris, pH 8.1 (37 8C), containing either 1%BSA or 0.2% Triton X-100. Alternatively, latent PAI-1 wasreactivated by denaturation with guanidinium chloride andrefolded by dialysis against NaCl/Pi.
Assays for measuring specific inhibitory activity of PAI-1
The specific inhibitory activity of the reactivated PAI-1variants was measured by titration against uPA in a directpeptidyl anilide assay at 37 8C, in the presence or absence ofa slight excess of vitronectin over PAI-1 [30]. A twofolddilution series of PAI-1, with or without vitronectin, wasmade immediately after refolding, to avoid loss of activitydue to fast latency transition. The dilution series ofdenatured and refolded PAI-1 (0–20 mg·mL21, 0–370 nM)were quickly (in less than 1 min) mixed with an equalvolume (100 mL) of 0.25 mg·mL21 (4.3 nM) uPA, 0.1 M
Tris, pH 8.1, 1% BSA or 0.2% Triton X-100. The finalconcentrations of uPA was 0.125 mg·mL21 (2.15 nM), ofPAI-1 in the range 0–10 mg·mL21 (0–185 nM), and ofvitronectin in the range 0–15 mg·mL21 (0–200 nM). Uponcompletion of the uPA inhibition reaction (. 5 min), theremaining uPA activity in the reaction mixture wasdetermined by use of L-5-oxopropyl-glycyl-L-arginine-p-nitroanilide (S-2444), a chromogenic peptidyl anilidesubstrate for uPA. The amount of active PAI-1, and thus thespecific inhibitory activity, was calculated from the totalamount of PAI-1 that had to be present to inhibit half of theuPA activity in the assays.
PAI-1 latency transition assay
Denatured and refolded PAI-1 wild-type and variants, in aconcentration of 20 mg·mL21 (370 nM), were incubated at37 8C in 0.1 M Tris, pH 8.1, 1% BSA in the presence orabsence of 30 mg·mL21 (400 nM) vitronectin. Followingincubation for different time periods, the specific inhibitoryactivity of PAI-1 wild-type and variants were determined asdescribed above, and the functional half-lives of the variantswere calculated.
Analysis of functional behaviour of PAI-1 by reaction withlow Mr uPA (LMW-uPA) and SDS/PAGE
PAI-1 portions (30 mg each) were denatured with 3 mL 1%SDS, refolded by dilution to 1200 mL with 0.1 M Tris,pH 8.1, 1% BSA and incubated at 37 8C. At various timepoints, samples of 5 mg PAI-1 were mixed with 7.5 mgLMW-uPA and incubated for at least 2 min at 37 8C. BSAwas then removed by the following procedure, performed atroom temperature unless otherwise indicated: One-hundredmicrograms of monoclonal murine anti-(PAI-1) IgG fromhybridoma clone 2 [31], coupled to Sepharose-4B, wastransferred to Ultrafreew-MC 0.22-mm filter units for
6276 M. Hansen et al. (Eur. J. Biochem. 268) q FEBS 2001
centrifugal filtration (Millipore, USA) and washed twicewith 0.1 M Tris, pH 8.1. The sample (PAI-1, LMW-uPA,BSA) was then added and incubated for at least 30 minfollowed by four washes with 0.1 M Tris, 1 M NaCl, pH 8.1,and one wash with 0.1 M Tris, pH 8.1. PAI-1 was eluted byincubation with 400 mL 3 M ammoniumthiocyanat(NH4SCN) for at least 30 min at 37 8C before centrifugationat 16 500 g for 10 min. The samples were precipitatedwith trichloroacetic acid, and subjected to 6–16% gradientSDS/PAGE.
Determination of second order rate constants for thereaction between PAI-1 and uPA
The second order rate constants were determined asdescribed previously [32]. The calculation of the secondorder rate constants is based on the assumption that theconcentration of active PAI-1 is unchanged during the assay.As most of these variants have significantly shorterfunctional half-lives (see below) than wild-type, thecalculated second order rate constants for the variantswere expected to be somewhat lower than their real values.
Gel filtration
Thirty-microgram portions of PAI-1 wild-type and variantswere analysed by FPLC gel filtration on a Superdex 200 HR10/30 column (Pharmacia) in 0.1 M Tris, pH 8.1, 0.5 M
NaCl at 4 8C, using a flow rate of 0.3 or 0.4 mL·min21. Thefollowing marker proteins were used: BSA (Mr 67 000),murine IgG (Mr 150 000), and b-galactosidase (Mr
540 000).
Molecular graphics
SWISSPDB VIEWER [33] was used to display the three-dimensional X-ray structure of active [34] and latent [13]PAI-1.
Statistical analysis
Data were evaluated by Student’s t-test.
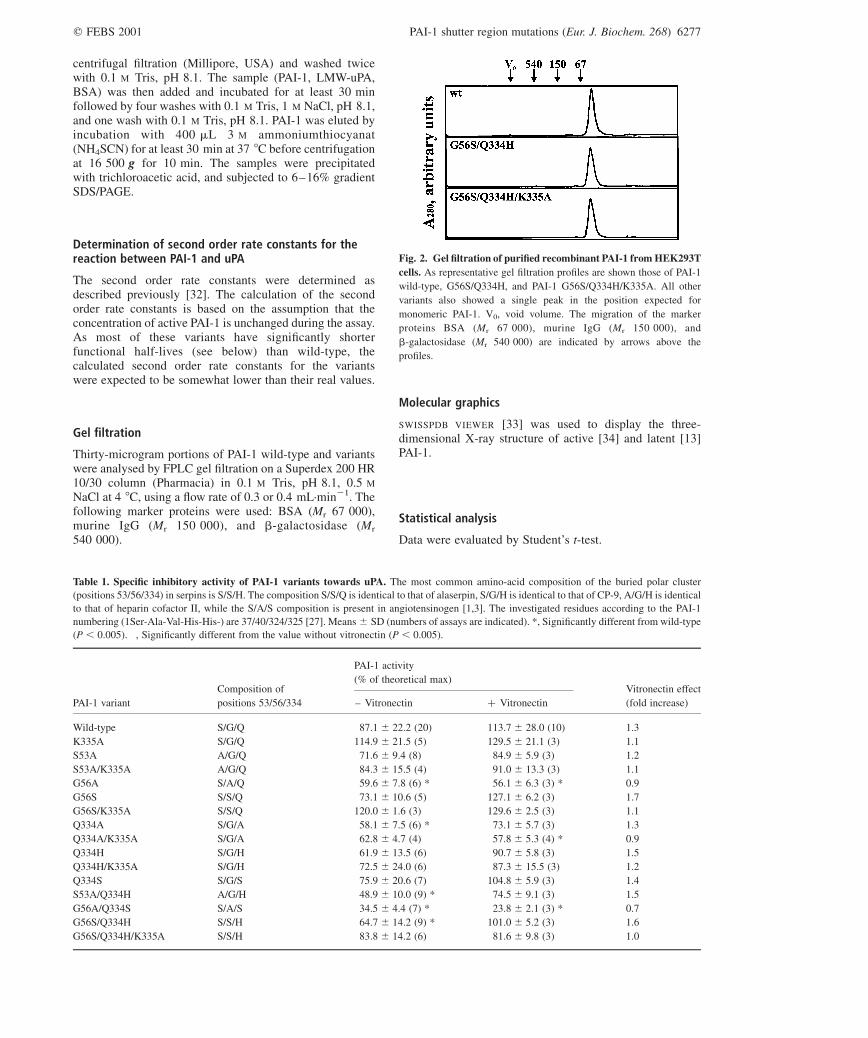
Fig. 2. Gel filtration of purified recombinant PAI-1 from HEK293T
cells. As representative gel filtration profiles are shown those of PAI-1
wild-type, G56S/Q334H, and PAI-1 G56S/Q334H/K335A. All other
variants also showed a single peak in the position expected for
monomeric PAI-1. V0, void volume. The migration of the marker
proteins BSA (Mr 67 000), murine IgG (Mr 150 000), and
b-galactosidase (Mr 540 000) are indicated by arrows above the
profiles.
Table 1. Specific inhibitory activity of PAI-1 variants towards uPA. The most common amino-acid composition of the buried polar cluster
(positions 53/56/334) in serpins is S/S/H. The composition S/S/Q is identical to that of alaserpin, S/G/H is identical to that of CP-9, A/G/H is identical
to that of heparin cofactor II, while the S/A/S composition is present in angiotensinogen [1,3]. The investigated residues according to the PAI-1
numbering (1Ser-Ala-Val-His-His-) are 37/40/324/325 [27]. Means ^ SD (numbers of assays are indicated). *, Significantly different from wild-type
(P , 0.005). †, Significantly different from the value without vitronectin (P , 0.005).
PAI-1 variant
Composition of
positions 53/56/334
PAI-1 activity
(% of theoretical max)Vitronectin effect
(fold increase)– Vitronectin þ Vitronectin
Wild-type S/G/Q 87.1 ^ 22.2 (20) 113.7 ^ 28.0 (10) † 1.3
K335A S/G/Q 114.9 ^ 21.5 (5) 129.5 ^ 21.1 (3) 1.1
S53A A/G/Q 71.6 ^ 9.4 (8) 84.9 ^ 5.9 (3) 1.2
S53A/K335A A/G/Q 84.3 ^ 15.5 (4) 91.0 ^ 13.3 (3) 1.1
G56A S/A/Q 59.6 ^ 7.8 (6) * 56.1 ^ 6.3 (3) * 0.9
G56S S/S/Q 73.1 ^ 10.6 (5) 127.1 ^ 6.2 (3) † 1.7
G56S/K335A S/S/Q 120.0 ^ 1.6 (3) 129.6 ^ 2.5 (3) † 1.1
Q334A S/G/A 58.1 ^ 7.5 (6) * 73.1 ^ 5.7 (3) 1.3
Q334A/K335A S/G/A 62.8 ^ 4.7 (4) 57.8 ^ 5.3 (4) * 0.9
Q334H S/G/H 61.9 ^ 13.5 (6) 90.7 ^ 5.8 (3) 1.5
Q334H/K335A S/G/H 72.5 ^ 24.0 (6) 87.3 ^ 15.5 (3) 1.2
Q334S S/G/S 75.9 ^ 20.6 (7) 104.8 ^ 5.9 (3) 1.4
S53A/Q334H A/G/H 48.9 ^ 10.0 (9) * 74.5 ^ 9.1 (3) † 1.5
G56A/Q334S S/A/S 34.5 ^ 4.4 (7) * 23.8 ^ 2.1 (3) *† 0.7
G56S/Q334H S/S/H 64.7 ^ 14.2 (9) * 101.0 ^ 5.2 (3) † 1.6
G56S/Q334H/K335A S/S/H 83.8 ^ 14.2 (6) 81.6 ^ 9.8 (3) 1.0
q FEBS 2001 PAI-1 shutter region mutations (Eur. J. Biochem. 268) 6277
R E S U L T S
Expression and purification of recombinant PAI-1 inHEK293T cells
PAI-1 wild-type and the substitution variants were expressedin HEK293T cells and purified from their conditionedmedium by immunoaffinity chromatography. The yields ofpurified protein were 1.4–11.4 mg protein per L ofconditioned medium. PAI-1 wild-type, K335A, S53A/K335A, G56S/K335A, and Q334H/K335A were obtainedin the greatest yields. All variant preparations were . 95%pure, as evaluated by SDS/PAGE, and migrated as a singlesharp peak in the position expected for monomeric PAI-1 ingel filtration (Fig. 2). N-Terminal sequencing of theproduced PAI-1 showed two distinct sequences in almostequal amounts, SAVHHPPS and VHHPPSYV, in agreementwith the previously reported N-terminal heterogeneity ofnatural PAI-1 [27]. The purified recombinant PAI-1 was inthe latent form, but could be reactivated by denaturation andrefolding, either by SDS and refolding by dilution into abuffer with 1% BSA, or by guanidinium chloride andrefolding by dialysis against NaCl/Pi. In this study, PAI-1was routinely reactivated using SDS, as some of the variantshad a very fast latency transition (see below) and wouldtherefore lose all activity during the dialysis used forrefolding after guanidinium chloride denaturation. Thespecific inhibitory activities of most PAI-1 variants, whendenatured with SDS and refolded in BSA-containing buffer,were 60–80% of the theoretical maximum, and thusindistinguishable from recombinant PAI-1 wild-type(Table 1). However, the recombinant variants G56A,Q334A, S53A/Q334H, G56A/Q334S, and G56S/Q334Hshowed a small, but statistically significant (P , 0.005)reduction in specific inhibitory activity as compared to wild-type. All variants except the variant G56A/Q334S had asecond order rate constant differing less than 2.5-fold fromthat of wild-type (data not shown). The second order rateconstant for G56A/Q334S was 3.8-fold lower than that ofthe wild-type, but this can be ascribed to a fast decrease ininhibitory activity of this variant during the experiment (seebelow). Vitronectin caused a small, but statisticallysignificant increase of the specific inhibitory activity ofthe PAI-1 wild-type and the variants G56S, S53A/Q334H,G56A/Q334S, G56S/Q334H, and G56S/K335A. Interest-ingly, the specific inhibitory activity of G56A/Q334S wasslightly decreased by vitronectin.
Latency transition of PAI-1 wild-type and PAI-1 variants inthe absence and the presence of vitronectin
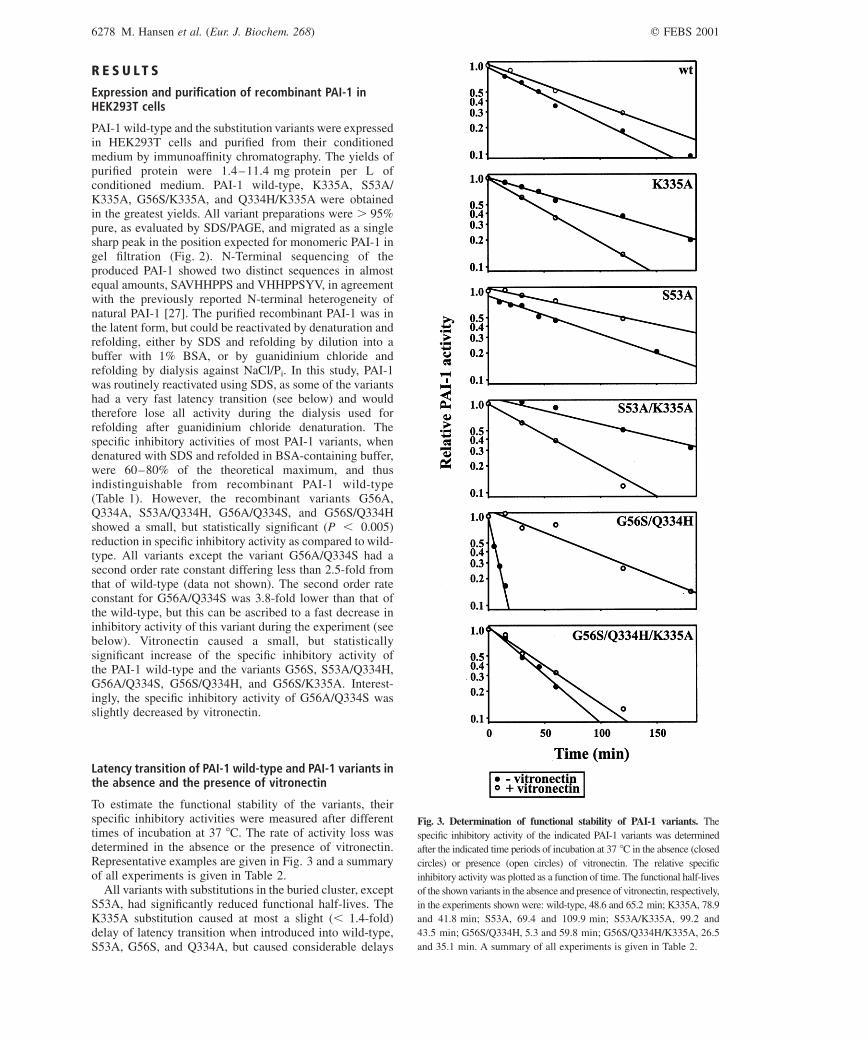
To estimate the functional stability of the variants, theirspecific inhibitory activities were measured after differenttimes of incubation at 37 8C. The rate of activity loss wasdetermined in the absence or the presence of vitronectin.Representative examples are given in Fig. 3 and a summaryof all experiments is given in Table 2.
All variants with substitutions in the buried cluster, exceptS53A, had significantly reduced functional half-lives. TheK335A substitution caused at most a slight (, 1.4-fold)delay of latency transition when introduced into wild-type,S53A, G56S, and Q334A, but caused considerable delays
Fig. 3. Determination of functional stability of PAI-1 variants. The
specific inhibitory activity of the indicated PAI-1 variants was determined
after the indicated time periods of incubation at 37 8C in the absence (closed
circles) or presence (open circles) of vitronectin. The relative specific
inhibitory activity was plotted as a function of time. The functional half-lives
of the shown variants in the absence and presence of vitronectin, respectively,
in the experiments shown were: wild-type, 48.6 and 65.2 min; K335A, 78.9
and 41.8 min; S53A, 69.4 and 109.9 min; S53A/K335A, 99.2 and
43.5 min; G56S/Q334H, 5.3 and 59.8 min; G56S/Q334H/K335A, 26.5
and 35.1 min. A summary of all experiments is given in Table 2.
6278 M. Hansen et al. (Eur. J. Biochem. 268) q FEBS 2001

(2.9-fold and 4.6-fold, respectively) of the very unstablevariants Q334H and G56S/Q334H.
At the routinely used pH of 8.1, there is only a slighteffect of vitronectin on the rate of latency transition of PAI-1wild-type, and the use of this pH therefore allowedoptimization of the difference in the effect of vitronectinon wild-type and the unstable variants. The presence ofvitronectin during the incubations had a stabilizing effect onall PAI-1 variants with substitutions in the buried cluster ofthe shutter region. The most pronounced effect wasobserved on the most unstable variants, so that theirfunctional half-lives in the presence of vitronectin increasedtowards that of PAI-1 wild-type (Table 2). Surprisingly, thestabilizing effect of vitronectin was abolished by the K335Asubstitution. None of the variants with this substitution hadlonger half-lives in the presence of vitronectin, and withmost of them, vitronectin even accelerated the activity loss.Hence, while the K335A substitution had a stabilizingeffect in the absence of vitronectin, it had a destabilizingeffect in the presence of vitronectin. Importantly, theK335A substitution did not result in altered affinity of PAI-1to vitronectin (T. Wind, & P.A. Andreasen, Department ofMolecular and Structural Biology, Aarhus University,Denmark, personal communication).
Although the assays were routinely performed in a bufferof 0.1 M Tris, pH 8.1, similar results were obtained with abuffer of 0.1 M Tris, pH 7.4. In addition, when examiningthe results obtained with SDS-activated PAI-1 vs.guanidinium chloride-activated PAI-1 with wild-type andtwo of the most stable variants (S53A and K335A), nodistinguishable difference was observed.
In order to ensure that the loss of activity during theincubations at 37 8C was due to latency transition, PAI-1,that had been incubated for various time periods at 37 8C,was reacted with an excess of LMW-uPA, and the reactionproducts were analysed by SDS/PAGE. Representative
experiments are shown in Fig. 4. Nonincubated wild-typereacted to form the expected < 80 000-Da LMW-uPA–PAI-1 complex. A fraction of wild-type reacted in asubstrate-manner, giving rise to the < 50 000-DaN-terminal fragment resulting from P1–P1
0 cleavage, the< 4000-Da C-terminal fragment not being recovered bythe gel system used here. During incubation at 37 8C, theamount of a PAI-1 form inert to reaction with LMW-uPAincreased with time, and the complex formation andsubstrate reaction decreased, in agreement with anincreasing fraction of PAI-1 being in the latent state. Thesame was true for the PAI-1 variants, except that theaccumulation of inert PAI-1 occurred faster, in agreementwith the activity measurements. It should also be noted thata fraction of all tested variants seemed to be present in astable substrate form [30,35,36], the amount of which didnot decrease during the incubations at 37 8C. On this basis,we concluded that the loss of activity during incubations at37 8C was caused by latency transition, and that the effectsof the substitutions and of vitronectin on the functionalhalf-lives was caused by changes in the rate of latencytransition.
Effect of shutter region substitutions on PAI-1 active tosubstrate transition
Nonionic detergents induce substrate behaviour in glycosy-lated PAI-1 at 0 8C, but much less so at 37 8C [20,21], andinduce substrate behaviour in nonglycosylated PAI-1 at37 8C as well as at 0 8C [37]. Therefore, to study the effectof shutter region substitutions on the transition of PAI-1 to asubstrate form, we replaced the BSA in the assay buffer with0.2% Triton X-100. The variants with a S53A, Q334A, orQ334S substitution all had significantly reduced specificinhibitory activity in Triton X-100 containing buffer at37 8C as compared to PAI-1 wild-type (Table 3). Analysis of
Table 2. Stability of specific inhibitory activity of PAI-1 variants at 37 8C. Means, SDs, and numbers of experiments are indicated. *, Significantly
different from wild-type (P , 0.005). †, Significantly different from the value without vitronectin (P , 0.005). ‡, Significantly different from the
corresponding variants without the K335A substitution (P , 0.02).
PAI-1 variant
Functional half-lives
(min)Vitronectin effect
(fold increase)– Vitronectin þ Vitronectin
Wild-type 54.7 ^ 13.5 (16) 63.4 ^ 11.6 (10) 1.2
K335A 76.9 ^ 11.6 (4) *‡ 35.3 ^ 6.7 (3) *†‡ 0.5
S53A 64.0 ^ 8.6 (5) 100.7 ^ 9.4 (3) *† 1.6
S53A/K335A 85.1 ^ 20.5 (4) * 32.0 ^ 10.0 (3) *†‡ 0.4
G56A 26.1 ^ 4.4 (4) * 36.5 ^ 6.8 (3) * 1.4
G56S 19.7 ^ 1.7 (4) * 54.9 ^ 4.1 (3) † 2.8
G56S/K335A 25.5 ^ 6.3 (3) * 12.3 ^ 2.1 (3) *‡ 0.5
Q334A 10.9 ^ 1.5 (4) * 39.9 ^ 10.2 (3) *† 3.7
Q334A/K335A 12.9 ^ 2.9 (3) * 14.1 ^ 0.9 (3) *‡ 1.1
Q334H 10.9 ^ 1.4 (4) * 52.3 ^ 6.0 (3) † 4.8
Q334H/K335A 32.1 ^ 2.7 (5) *‡ 29.4 ^ 1.2 (3) *‡ 0.9
Q334S 23.4 ^ 2.5 (5) * 75.2 ^ 15.8 (3) † 3.2
S53A/Q334H 18.5 ^ 3.2 (7) * 78.3 ^ 12.5 (3) † 4.2
G56A/Q334S 9.7 ^ 1.6 (4) * 72.9 ^ 17.7 (3) † 7.5
G56S/Q334H 6.1 ^ 1.5 (7) * 62.4 ^ 15.6 (4) † 10.2
G56S/Q334H/K335A 27.8 ^ 2.6 (5) *‡ 34.3 ^ 2.3 (3) *‡ 1.2
q FEBS 2001 PAI-1 shutter region mutations (Eur. J. Biochem. 268) 6279

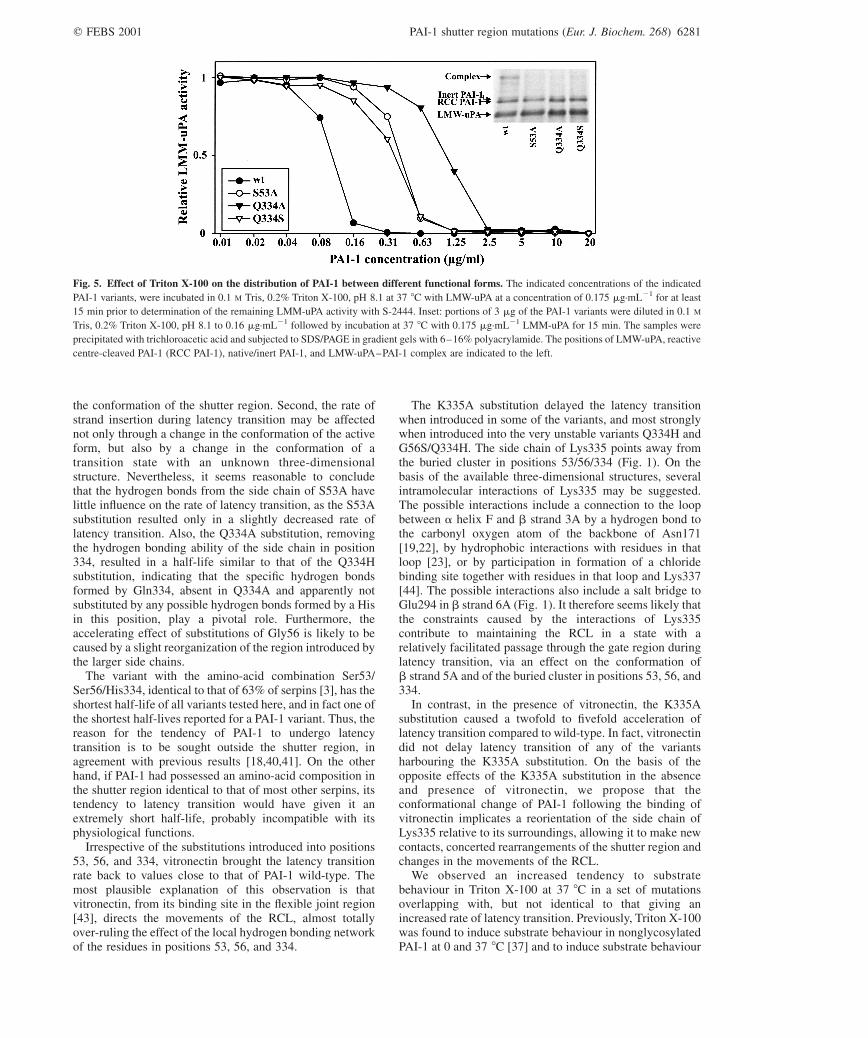
the distribution of PAI-1 between different functional formsby the use of reaction with LMW-uPA and SDS/PAGEconfirmed that the decreased specific inhibitory activity wascaused by an increased tendency to substrate behaviourand not the generation of an inert form, as shown by therepresentative experiments shown in Fig. 5. The K335Asubstitution did not counteract the increased tendency tosubstrate behaviour (Table 3).
D I S C U S S I O N
In this report, we show that the combination of amino acidsin positions 53, 56, and 334 in the shutter region of PAI-1 isan important determinant of the latency transition rate.Except one, all tested deviations from the wild-typecombination of amino acids in these positions resulted inan accelerated latency transition. Substitution of the Lysresidue in position 335 counteracted the accelerating effectof some of the substitutions in positions 53, 56, and 334. Thesubstitutions also had specific effects on the vitronectin andTriton X-100 induced changes in PAI-1 latency transitionand specific inhibitory activity, respectively.
Based on our observations we propose that thesubstitutions in positions 53, 56, and 334 affect the latency
transition by changing the local conformation of theseresidues, including their hydrogen bonds. As there is noP1–P1
0 cleavage during latency transition, strand insertionmust imply the passage of the intact RCL through the ‘gateregion’, which is situated between (a) the turn between b
strands 3C and 4C (residues 204–219) and (b) the turnbetween b strands 3B and a helix G (residues 257–259)[13,38,39] (Fig. 1). Because of steric reasons, it is not verylikely that the RCL can surround the turn between b strands3C and 4C without having a completely stretched-outconformation. Only after the RCL has passed this turn canthe final insertion into b sheet A proceed [39]. Consideringthat RCL insertion into b sheet A is several orders ofmagnitude faster during complex formation than duringlatency transition, it seems reasonable to presume that thepassage of the RCL through the gate region is rate limitingfor latency transition. This presumption is supported by theobservation that substitutions of basic residues in the turnbetween b strands 3C and 4C with acidic residues acceleratelatency transition [40,41]. On this basis, we reach theconclusion that the substitutions in the shutter region affectthe rate of latency transition by affecting the rate of passageof the RCL through the gate region. Based on the amino-acid sequence of the RCL and b strand 5A being directlycontinuous, it may be proposed that movements of the RCLduring passage through the gate region are coupled tomovements of b strand 5A and therefore sensitive to theinteractions of b strand 5A with the underlying structure. Analternative, but with the presently available information, aless likely explanation is that passage of the RCL throughthe gate region is rapid and reversible, and that it is the b
sheet A opening and the final insertion of RCL as b strand4A that is rate limiting for latency transition.
Two facts complicate the interpretation of our resultson the basis of detailed structural considerations. First,the three-dimensional structure available for active PAI-1[34,42] is that of a mutant with a strongly delayed latencytransition. The stabilizing mutations may well have affected
Fig. 4. Analysis of the functional behavior of PAI-1 by SDS/PAGE.
PAI-1 wild-type or Q334H (25 mg·mL21) were SDS-denatured and
incubated in a buffer of 0.1 M Tris, 1% BSA, pH 8.1 at 37 8C. After the
indicated time periods, samples corresponding to 5 mg PAI-1 were
incubated with 7.5 mg LMW-uPA for at least 2 min at 37 8C at a PAI-1
concentration of 20 mg·mL21 and an LMW-uPA concentration of
30 mg·mL21 before inert PAI-1, reactive center-cleaved PAI-1, and
LMW-uPA–PAI-1 complex were isolated from the reaction mixture by
immunoaffinity chromatography, removing most of the BSA and most
of the excess of LMW-uPA. The samples were then precipitated with
trichloroacetic acid and subjected to SDS/PAGE in gradient gels with
6–16% polyacrylamide. The positions of LMW-uPA, reactive centre-
cleaved PAI-1 (RCC PAI-1), native/inert PAI-1, BSA, and LMW-uPA-
PAI-1 complex (complex) are indicated to the right. N, PAI-1 incubated
for at least 3 h at 37 8C without LMW-uPA added. The apparently low
fraction of nonincubated PAI-1 forming a complex is related to a
somewhat higher tendency to substrate behavior at the high PAI-1 and
LMW-uPA concentrations used in this assay [22].
Table 3. Effect of 0.2% Triton X-100 on specific inhibitory activity
of PAI-1 at 37 8C. The specific inhibitory activity of each variant is
given as a fraction of the specific inhibitory activity of the same variant
in 1% BSA. Means, SDs, and numbers of experiments are indicated. *,
Significantly different from wild-type (P , 0.005).
PAI-1 variant Specific inhibitory activity
Wild-type 0.87 ^ 0.12 (7)
K335A 0.95 ^ 0.09 (3)
S53A 0.20 ^ 0.02 (3)*
S53A/K335A 0.17 ^ 0.02 (3)*
G56A 0.65 ^ 0.11 (3)
G56S 1.17 ^ 0.16 (3)
G56S/K335A 1.06 ^ 0.03 (3)
Q334A 0.12 ^ 0.01 (3)*
Q334A/K335A 0.18 ^ 0.03 (3)*
Q334H 0.61 ^ 0.08 (3)*
Q334H/K335A 0.77 ^ 0.01 (3)
Q334S 0.21 ^ 0.05 (3)*
S53A/Q334H 0.13 ^ 0.01 (3)*
G56A/Q334S 0.10 ^ 0.02 (3)*
G56S/Q334H 0.45 ^ 0.03 (3)*
G56S/Q334H/K335A 0.74 ^ 0.09 (3)
6280 M. Hansen et al. (Eur. J. Biochem. 268) q FEBS 2001
the conformation of the shutter region. Second, the rate ofstrand insertion during latency transition may be affectednot only through a change in the conformation of the activeform, but also by a change in the conformation of atransition state with an unknown three-dimensionalstructure. Nevertheless, it seems reasonable to concludethat the hydrogen bonds from the side chain of S53A havelittle influence on the rate of latency transition, as the S53Asubstitution resulted only in a slightly decreased rate oflatency transition. Also, the Q334A substitution, removingthe hydrogen bonding ability of the side chain in position334, resulted in a half-life similar to that of the Q334Hsubstitution, indicating that the specific hydrogen bondsformed by Gln334, absent in Q334A and apparently notsubstituted by any possible hydrogen bonds formed by a Hisin this position, play a pivotal role. Furthermore, theaccelerating effect of substitutions of Gly56 is likely to becaused by a slight reorganization of the region introduced bythe larger side chains.
The variant with the amino-acid combination Ser53/Ser56/His334, identical to that of 63% of serpins [3], has theshortest half-life of all variants tested here, and in fact one ofthe shortest half-lives reported for a PAI-1 variant. Thus, thereason for the tendency of PAI-1 to undergo latencytransition is to be sought outside the shutter region, inagreement with previous results [18,40,41]. On the otherhand, if PAI-1 had possessed an amino-acid composition inthe shutter region identical to that of most other serpins, itstendency to latency transition would have given it anextremely short half-life, probably incompatible with itsphysiological functions.
Irrespective of the substitutions introduced into positions53, 56, and 334, vitronectin brought the latency transitionrate back to values close to that of PAI-1 wild-type. Themost plausible explanation of this observation is thatvitronectin, from its binding site in the flexible joint region[43], directs the movements of the RCL, almost totallyover-ruling the effect of the local hydrogen bonding networkof the residues in positions 53, 56, and 334.
The K335A substitution delayed the latency transitionwhen introduced in some of the variants, and most stronglywhen introduced into the very unstable variants Q334H andG56S/Q334H. The side chain of Lys335 points away fromthe buried cluster in positions 53/56/334 (Fig. 1). On thebasis of the available three-dimensional structures, severalintramolecular interactions of Lys335 may be suggested.The possible interactions include a connection to the loopbetween a helix F and b strand 3A by a hydrogen bond tothe carbonyl oxygen atom of the backbone of Asn171[19,22], by hydrophobic interactions with residues in thatloop [23], or by participation in formation of a chloridebinding site together with residues in that loop and Lys337[44]. The possible interactions also include a salt bridge toGlu294 in b strand 6A (Fig. 1). It therefore seems likely thatthe constraints caused by the interactions of Lys335contribute to maintaining the RCL in a state with arelatively facilitated passage through the gate region duringlatency transition, via an effect on the conformation ofb strand 5A and of the buried cluster in positions 53, 56, and334.
In contrast, in the presence of vitronectin, the K335Asubstitution caused a twofold to fivefold acceleration oflatency transition compared to wild-type. In fact, vitronectindid not delay latency transition of any of the variantsharbouring the K335A substitution. On the basis of theopposite effects of the K335A substitution in the absenceand presence of vitronectin, we propose that theconformational change of PAI-1 following the binding ofvitronectin implicates a reorientation of the side chain ofLys335 relative to its surroundings, allowing it to make newcontacts, concerted rearrangements of the shutter region andchanges in the movements of the RCL.
We observed an increased tendency to substratebehaviour in Triton X-100 at 37 8C in a set of mutationsoverlapping with, but not identical to that giving anincreased rate of latency transition. Previously, Triton X-100was found to induce substrate behaviour in nonglycosylatedPAI-1 at 0 and 37 8C [37] and to induce substrate behaviour
Fig. 5. Effect of Triton X-100 on the distribution of PAI-1 between different functional forms. The indicated concentrations of the indicated
PAI-1 variants, were incubated in 0.1 M Tris, 0.2% Triton X-100, pH 8.1 at 37 8C with LMW-uPA at a concentration of 0.175 mg·mL21 for at least
15 min prior to determination of the remaining LMM-uPA activity with S-2444. Inset: portions of 3 mg of the PAI-1 variants were diluted in 0.1 M
Tris, 0.2% Triton X-100, pH 8.1 to 0.16 mg·mL21 followed by incubation at 37 8C with 0.175 mg·mL21 LMM-uPA for 15 min. The samples were
precipitated with trichloroacetic acid and subjected to SDS/PAGE in gradient gels with 6–16% polyacrylamide. The positions of LMW-uPA, reactive
centre-cleaved PAI-1 (RCC PAI-1), native/inert PAI-1, and LMW-uPA–PAI-1 complex are indicated to the left.
q FEBS 2001 PAI-1 shutter region mutations (Eur. J. Biochem. 268) 6281

in glycosylated PAI-1 at 0 8C, but not at 37 8C [20,21]. Onthe basis of our present findings, we propose that TritonX-100 acts by destabilizing the shutter region, thishappening more readily with less perfect interactionsbetween the side chains, resulting in a delay in strandinsertion during reaction with the target proteinase. On theother hand, the Triton X-100-induced substrate behaviourdid not seem to implicate the interactions of the Lys335 sidechain, in contrast to antibody-induced substrate behaviourthat was potentiated by the K335A substitution [19,22]. Theobservation of latency transition and complex formationbeing affected differently by mutations in the shutter regionand b strand 5A is in agreement with RCL insertionfollowing different routes in the two cases.
PAI-1 is a potential target for antithrombotic [45] andanticancer therapy [46,47]. The biochemical mechanism ofaction of a few PAI-1 neutralisers has been characterized,including monoclonal antibodies and organochemicalcompounds. These compounds neutralize PAI-1 either bysteric hindrance, by inducing conversion to the latent state,by inducing substrate behaviour, and/or by inducingconversion to inert polymers [48–52]. The present resultsprompt further studies into the role of the shutter region andb strand 5A in PAI-1 in conformational changes leading toneutralization.
A C K N O W L E D G E M E N T S
Dr Kees Rodenburg is thanked for fruitful discussions in the early phase
of this work. Dr Claus Oxvig is acknowledged for providing the
HEK293T cell line. This work was supported financially by the Danish
Cancer Society, the Danish Research Agency, the Danish Heart
Foundation, the NOVO-Nordisk Foundation, and the Danish Cancer
Foundation.
R E F E R E N C E S
1. Huber, R. & Carrell, R.W. (1989) Implications of the three-
dimensional structure of a1-antitrypsin for structure and function of
serpins. Biochemistry 28, 8951–8966.
2. Carrell, R.W. & Stein, P.E. (1996) The biostructural pathology of
the serpins: critical function of sheet opening mechanism. Biol.
Chem. Hoppe Seyler 377, 1–17.
3. Irving, J.A., Pike, R.N., Lesk, A.M. & Whisstock, J.C. (2000)
Phylogeny of the serpin superfamily: implications of patterns of
amino acid conservation for structure and function. Genome Res.
10, 1845–1864.
4. Lawrence, D.A., Ginsburg, D., Day, D.E., Berkenpas, M.B.,
Verhamme, I.M., Kvassman, J.O. & Shore, J.D. (1995) Serpin-
protease complexes are trapped as stable acyl-enzyme inter-
mediates. J. Biol. Chem. 270, 25309–25312.
5. Wilczynska, M., Fa, M., Ohlsson, P.I. & Ny, T. (1995) The
inhibition mechanism of serpins. J. Biol. Chem. 270,
29652–29655.
6. Egelund, R., Rodenburg, K.W., Andreasen, P.A., Rasmussen, M.S.,
Guldberg, R.E. & Petersen, T.E. (1998) An ester bond linking a
fragment of a serine proteinase to its serpin inhibitor. Biochemistry
37, 6375–6379.
7. Shore, J.D., Day, D.E., Francis-Chmura, A.M., Verhamme, I.,
Kvassman, J., Lawrence, D.A. & Ginsburg, D. (1995) A fluorescent
probe study of plasminogen activator inhibitor-1. Evidence for
reactive center loop insertion and its role in the inhibitory
mechanism. J. Biol. Chem. 270, 5395–5398.
8. Stratikos, E. & Gettins, P.G. (1999) Formation of the covalent
serpin-proteinase complex involves translocation of the proteinase
by more than 70A and full insertion of the reactive center loop into
b-sheet A. Proc. Natl Acad. Sci. USA 96, 4808–4813.
9. Fa, M., Bergstrom, F., Hagglof, P., Wilczynska, M., Johansson,
L.B. & Ny, T. (2000) The structure of a serpin–protease complex
revealed by intramolecular distance measurements using donor–
donor energy migration and mapping of interaction sites. Struct.
Fold Des. 8, 397–405.
10. Huntington, J.A., Read, R.J. & Carrell, R.W. (2000) Structure of a
serpin–protease complex shows inhibition by deformation. Nature
407, 923–926.
11. Egelund, R., Petersen, T.E. & Andreasen, P.A. (2001) A serpin-
induced extensive proteolytic susceptibility of urokinase-type
plasminogen activator implicates distortion of the proteinase
substrate-binding pocket and oxyanion hole in the serpin inhibitory
mechanism. Eur. J. Biochem. 268, 673–685.
12. Lawrence, D.A., Olson, S.T., Muhammad, S., Day, D.E., Kvass-
man, J.O., Ginsburg, D. & Shore, J.D. (2000) Partitioning of serpin-
proteinase reactions between stable inhibition and substrate
cleavage is regulated by the rate of serpin reactive center loop
insertion into b-sheet A. J. Biol. Chem. 275, 5839–5844.
13. Mottonen, J., Strand, A., Symersky, J., Sweet, R.M., Danley, D.E.,
Geoghegan, K.F., Gerard, R.D. & Goldsmith, E.J. (1992) Structural
basis of latency in plasminogen activator inhibitor-1. Nature 355,
270–273.
14. Stein, P. & Chothia, C. (1991) Serpin tertiary structure
transformation. J. Mol. Biol. 221, 615–621.
15. Harrop, S.J., Jankova, L., Coles, M., Jardine, D., Whittaker, J.S.,
Gould, A.R., Meister, A., King, G.C., Mabbutt, B.C. & Curmi, P.M.
(1999) The crystal structure of plasminogen activator inhibitor 2 at
2.0A resolution: implications for serpin function. Structure 7,
43–54.
16. Stein, P.E. & Carrell, R.W. (1995) What do dysfunctional serpins
tell us about molecular mobility and disease? Nat. Struct. Biol. 2,
96–113.
17. Davis, R.L., Shrimpton, A.E., Holohan, P.D., Bradshaw, C., Feiglin,
D., Collins, G.H., Sonderegger, P., Kinter, J., Becker, L.M.,
Lacbawan, F., Krasnewich, D., Muenke, M., Lawrence, D.A.,
Yerby, M.S., Shaw, C.M., Gooptu, B., Elliott, P.R., Finch, J.T.,
Carrell, R.W. & Lomas, D.A. (1999) Familial dementia caused by
polymerization of mutant neuroserpin. Nature 401, 376–379.
18. Berkenpas, M.B., Lawrence, D.A. & Ginsburg, D. (1995)
Molecular evolution of plasminogen activator inhibitor-1 func-
tional stability. EMBO J. 14, 2969–2977.
19. Kirkegaard, T., Jensen, S., Schousboe, S.L., Petersen, H.H.,
Egelund, R., Andreasen, P.A. & Rodenburg, K.W. (1999)
Engineering of conformations of plasminogen activator inhibitor-
1. A crucial role of b-strand 5A residues in the transition of active
form to latent and substrate forms. Eur. J. Biochem. 263, 577–586.
20. Kjøller, L., Martensen, P.M., Sottrup-Jensen, L., Justesen, J.,
Rodenburg, K.W. & Andreasen, P.A. (1996) Conformational
changes of the reactive-centre loop and b-strand 5A accompany
temperature-dependent inhibitor-substrate transition of plasmino-
gen-activator inhibitor 1. Eur. J. Biochem. 241, 38–46.
21. Andreasen, P.A., Egelund, R., Jensen, S. & Rodenburg, K.W.
(1999) Solvent effects on activity and conformation of plasminogen
activator inhibitor-1. Thromb. Haemost. 81, 407–414.
22. Schousboe, S.L., Egelund, R., Kirkegaard, T., Preissner, K.T.,
Rodenburg, K.W. & Andreasen, P.A. (2000) Vitronectin and
substitution of a b-strand 5A lysine residue potentiate activity-
neutralization of PA inhibitor-1 by monoclonal antibodies against
a-helix F. Thromb. Haemost. 83, 742–751.
23. Im, H. & Yu, M.H. (2000) Role of Lys335 in the metastability and
function of inhibitory serpins. Protein Sci. 9, 934–941.
24. Im, H., Seo, E.J. & Yu, M.H. (1999) Metastability in the inhibitory
mechanism of human a1-antitrypsin. J. Biol. Chem. 274,
11072–11077.
6282 M. Hansen et al. (Eur. J. Biochem. 268) q FEBS 2001

25. Preissner, K.T. & Jenne, D. (1991) Structure of vitronectin and its
biological role in haemostasis. Thromb. Haemost. 66, 123–132.
26. Deng, G., Royle, G., Seiffert, D. & Loskutoff, D.J. (1995) The PAI-
1/vitronectin interaction: two cats in a bag? Thromb. Haemost. 74,
66–70.
27. Andreasen, P.A., Riccio, A., Welinder, K.G., Douglas, R., Sartorio,
R., Nielsen, L.S., Oppenheimer, C., Blasi, F. & Danø, K. (1986)
Plasminogen activator inhibitor type-1: reactive center and amino-
terminal heterogeneity determined by protein and cDNA sequen-
cing. FEBS Lett. 209, 213–218.
28. DuBridge, R.B., Tang, P., Hsia, H.C., Leong, P.M., Miller, J.H. &
Calos, M.P. (1987) Analysis of mutation in human cells by using an
Epstein–Barr virus shuttle system. Mol. Cell. Biol. 7, 379–387.
29. Munch, M., Heegaard, C., Jensen, P.H. & Andreasen, P.A. (1991)
Type-1 inhibitor of plasminogen activators. Distinction between
latent, activated and reactive centre-cleaved forms with thermal
stability and monoclonal antibodies. FEBS Lett. 295, 102–106.
30. Munch, M., Heegaard, C.W. & Andreasen, P.A. (1993)
Interconversions between active, inert and substrate forms of
denatured/refolded type-1 plasminogen activator inhibitor. Bio-
chim. Biophys. Acta 1202, 29–37.
31. Nielsen, L.S., Hansen, J.G., Andreasen, P.A., Skriver, L., Danø, K.
& Zeuthen, J. (1983) Monoclonal antibody to human 66,000
molecular wieght plasminogen activator from melanoma cells.
Specific enzyme inhibition and one-step affinity purification.
EMBO J. 2, 115–119.
32. Petersen, H.H., Hansen, M., Schousboe, S.L. & P.A.A. (2001)
Localisation of epitopes of monoclonal anti-urokinase-type
plasminogen activator antibodies. Relationship to antibody effects
on molecular interactions of the enzyme. Eur. J. Biochem. 268,
4430–4439.
33. Guex, N. & Peitsch, M.C. (1997) SWISS-MODEL and the Swiss-
PdbViewer: an environment for comparative protein modeling.
Electrophoresis 18, 2714–2723.
34. Nar, H., Bauer, M., Stassen, J.M., Lang, D., Gils, A. & Declerck,
P.J. (2000) Plasminogen activator inhibitor 1. Structure of the
native serpin, comparison to its other conformers and implications
for serpin inactivation. J. Mol. Biol. 297, 683–695.
35. Declerck, P.J., De Mol, M., Vaughan, D.E. & Collen, D. (1992)
Identification of a conformationally distinct form of plasminogen
activator inhibitor-1, acting as a noninhibitory substrate for tissue-
type plasminogen activator. J. Biol. Chem. 267, 11693–11696.
36. Urano, T., Strandberg, L., Johansson, L.B. & Ny, T. (1992) A
substrate-like form of plasminogen-activator-inhibitor type 1.
Conversions between different forms by sodium dodecyl sulphate.
Eur. J. Biochem. 209, 985–992.
37. Gils, A. & Declerck, P.J. (1998) Modulation of plasminogen
activator inhibitor 1 by Triton X-100-identification of two
consecutive conformational transitions. Thromb. Haemost. 80,
286–291.
38. Aertgeerts, K., De Bondt, H.L., De Ranter, C.J. & Declerck, P.J.
(1995) Mechanisms contributing to the conformational and
functional flexibility of plasminogen activator inhibitor-1. Nat.
Struct. Biol. 2, 891–897.
39. Kruger, P., Verheyden, S., Declerck, P.J. & Engelborghs, Y. (2001)
Extending the capabilities of targeted molecular dynamics:
simulation of a large conformational transition in plasminogen
activator inhibitor 1. Protein Sci. 10, 798–808.
40. Gils, A., Lu, J., Aertgeerts, K., Knockaert, I. & Declerck, P.J.
(1997) Identification of positively charged residues contributing to
the stability of plasminogen activator inhibitor 1. FEBS Lett. 415,
192–195.
41. Vleugels, N., Leys, J., Knockaert, I. & Declerck, P.J. (2000) Effect
of stabilizing versus destabilizing interactions on plasminogen
activator inhibitor-1. Thromb. Haemost. 84, 871–875.
42. Sharp, A.M., Stein, P.E., Pannu, N.S., Carrell, R.W., Berkenpas,
M.B., Ginsburg, D., Lawrence, D.A. & Read, R.J. (1999) The
active conformation of plasminogen activator inhibitor 1, a target
for drugs to control fibrinolysis and cell adhesion. Structure 7,
111–118.
43. Lawrence, D.A., Berkenpas, M.B., Palaniappan, S. & Ginsburg, D.
(1994) Localization of vitronectin binding domain in plasminogen
activator inhibitor-1. J. Biol. Chem. 269, 15223–15228.
44. Stout, T.J., Graham, H., Buckley, D.I. & Matthews, D.J. (2000)
Structures of active and latent PAI-1: a possible stabilizing role for
chloride ions. Biochemistry 39, 8460–8469.
45. Juhan-Vague, I. & Alessi, M.C. (1996) Fibrinolysis and risk of
coronary artery disease. Fibrinolysis 10, 127–136.
46. Andreasen, P.A., Kjøller, L., Christensen, L. & Duffy, M.J. (1997)
The urokinase-type plasminogen activator system in cancer
metastasis: a review. Int. J. Cancer 72, 1–22.
47. Andreasen, P.A., Egelund, R. & Petersen, H.H. (2000) The
plasminogen activation system in tumor growth, invasion, and
metastasis. Cell. Mol. Life Sci. 57, 25–40.
48. Debrock, S. & Declerck, P.J. (1997) Neutralization of plasminogen
activator inhibitor-1 inhibitory properties: identification of two
different mechanisms. Biochim. Biophys. Acta 1337, 257–266.
49. Bjorquist, P., Ehnebom, J., Inghardt, T., Hansson, L., Lindberg, M.,
Linschoten, M., Stromqvist, M. & Deinum, J. (1998) Identification
of the binding site for a low-molecular-weight inhibitor of
plasminogen activator inhibitor type 1 by site-directed mutagen-
esis. Biochemistry 37, 1227–1234.
50. Friederich, P.W., Levi, M., Biemond, B.J., Charlton, P., Templeton,
D., van Zonneveld, A.J., Bevan, P., Pannekoek, H. & ten Cate, J.W.
(1997) Novel low-molecular-weight inhibitor of PAI-1 (XR5118)
promotes endogenous fibrinolysis and reduces postthrombolysis
thrombus growth in rabbits. Circulation 96, 916–921.
51. Wind, T., Jensen, M.A. & Andreasen, P.A. (2001) Epitope mapping
for four monoclonal antibodies against human plasminogen
activator inhibitor type-1: implications for antibody-mediated
PAI-1-neutralization and vitronectin-binding. Eur. J. Biochem. 268,
1095–1106.
52. Egelund, R., Einholm, A.P., Pedersen, K.E., Nielsen, R.W.,
Christensen, A., Deinum, J. & Andreasen, P.A. (2001) A regulatory
hydrophobic area in the flexible joint region of plasminogen
activator inhibitor-1, defined with fluorescent activity-neutralizing
ligands. Ligand-induced serpin polymerization. J. Biol. Chem. 276,
13077–13086.
q FEBS 2001 PAI-1 shutter region mutations (Eur. J. Biochem. 268) 6283