haematologica. volume 101, issue 5
DESCRIPTION
ÂTRANSCRIPT


Editor-in-ChiefJan Cools (Leuven)
Deputy EditorLuca Malcovati (Pavia)
Managing DirectorAntonio Majocchi (Pavia)
Associate EditorsHélène Cavé (Paris), Ross Levine (New York), Claire Harrison (London), Pavan Reddy (Ann Arbor), AndreasRosenwald (Wuerzburg), Juerg Schwaller (Basel), Monika Engelhardt (Freiburg), Wyndham Wilson (Bethesda), PaulKyrle (Vienna), Paolo Ghia (Milan), Swee Lay Thein (Bethesda), Pieter Sonneveld (Rotterdam)
Assistant EditorsAnne Freckleton (English Editor), Cristiana Pascutto (Statistical Consultant), Rachel Stenner (English Editor), Kate O’Donohoe (English Editor)
Editorial BoardOmar I. Abdel-Wahab (New York); Jeremy Abramson (Boston); Paolo Arosio (Brescia); Raphael Bejar (San Diego); ErikBerntorp (Malmö); Dominique Bonnet (London); Jean-Pierre Bourquin (Zurich); Suzanne Cannegieter (Leiden);Francisco Cervantes (Barcelona); Nicholas Chiorazzi (Manhasset); Oliver Cornely (Köln); Michel Delforge (Leuven);Ruud Delwel (Rotterdam); Meletios A. Dimopoulos (Athens); Inderjeet Dokal (London); Hervé Dombret (Paris); PeterDreger (Hamburg); Martin Dreyling (München); Kieron Dunleavy (Bethesda); Dimitar Efremov (Rome); SabineEichinger (Vienna); Jean Feuillard (Limoges); Carlo Gambacorti-Passerini (Monza); Guillermo Garcia Manero(Houston); Christian Geisler (Copenhagen); Piero Giordano (Leiden); Christian Gisselbrecht (Paris); AndreasGreinacher (Greifswals); Hildegard Greinix (Vienna); Paolo Gresele (Perugia); Thomas M. Habermann (Rochester);Claudia Haferlach (München); Oliver Hantschel (Lausanne); Christine Harrison (Southampton); Brian Huntly(Cambridge); Ulrich Jaeger (Vienna); Elaine Jaffe (Bethesda); Arnon Kater (Amsterdam); Gregory Kato (Pittsburg);Christoph Klein (Munich); Steven Knapper (Cardiff); Seiji Kojima (Nagoya); John Koreth (Boston); Robert Kralovics(Vienna); Ralf Küppers (Essen); Ola Landgren (New York); Peter Lenting (Le Kremlin-Bicetre); Per Ljungman(Stockholm); Francesco Lo Coco (Rome); Henk M. Lokhorst (Utrecht); John Mascarenhas (New York); Maria-VictoriaMateos (Salamanca); Simon Mendez-Ferrer (Madrid); Giampaolo Merlini (Pavia); Anna Rita Migliaccio (New York);Mohamad Mohty (Nantes); Martina Muckenthaler (Heidelberg); Ann Mullally (Boston); Stephen Mulligan (Sydney);German Ott (Stuttgart); Jakob Passweg (Basel); Melanie Percy (Ireland); Rob Pieters (Rotterdam); Stefano Pileri (Milan);Miguel Piris (Madrid); Andreas Reiter (Mannheim); Jose-Maria Ribera (Barcelona); Stefano Rivella (New York);Francesco Rodeghiero (Vicenza); Richard Rosenquist (Uppsala); Simon Rule (Plymouth); Claudia Scholl (Heidelberg);Martin Schrappe (Kiel); Radek C. Skoda (Basel); Gérard Socié (Paris); Kostas Stamatopoulos (Thessaloniki); David P.Steensma (Rochester); Martin H. Steinberg (Boston); Ali Taher (Beirut); Evangelos Terpos (Athens); Takanori Teshima(Sapporo); Pieter Van Vlierberghe (Gent); Alessandro M. Vannucchi (Firenze); George Vassiliou (Cambridge); EdoVellenga (Groningen); Umberto Vitolo (Torino); Guenter Weiss (Innsbruck).
Editorial OfficeSimona Giri (Production & Marketing Manager), Lorella Ripari (Peer Review Manager), Paola Cariati (Senior GraphicDesigner), Igor Ebuli Poletti (Senior Graphic Designer), Marta Fossati (Peer Review), Diana Serena Ravera (Peer Review)
Affiliated Scientific SocietiesSIE (Italian Society of Hematology, www.siematologia.it)SIES (Italian Society of Experimental Hematology, www.siesonline.it)
haematologicaJournal of the European Hematology Association
Published by the Ferrata Storti Foundation


Information for readers, authors and subscribers
Haematologica (print edition, pISSN 0390-6078, eISSN 1592-8721) publishes peer-reviewed papers on all areas of experi-mental and clinical hematology. The journal is owned by a non-profit organization, the Ferrata Storti Foundation, andserves the scientific community following the recommendations of the World Association of Medical Editors(www.wame.org) and the International Committee of Medical Journal Editors (www.icmje.org).
Haematologica publishes editorials, research articles, review articles, guideline articles and letters. Manuscripts should beprepared according to our guidelines (www.haematologica.org/information-for-authors), and the Uniform Requirementsfor Manuscripts Submitted to Biomedical Journals, prepared by the International Committee of Medical Journal Editors(www.icmje.org).
Manuscripts should be submitted online at http://www.haematologica.org/.
Conflict of interests. According to the International Committee of Medical Journal Editors (http://www.icmje.org/#conflicts),“Public trust in the peer review process and the credibility of published articles depend in part on how well conflict ofinterest is handled during writing, peer review, and editorial decision making”. The ad hoc journal’s policy is reported indetail online (www.haematologica.org/content/policies).
Transfer of Copyright and Permission to Reproduce Parts of Published Papers. Authors will grant copyright of their articles to theFerrata Storti Foundation. No formal permission will be required to reproduce parts (tables or illustrations) of publishedpapers, provided the source is quoted appropriately and reproduction has no commercial intent. Reproductions with com-mercial intent will require written permission and payment of royalties.
Detailed information about subscriptions is available online at www.haematologica.org. Haematologica is an open accessjournal. Access to the online journal is free. Use of the Haematologica App (available on the App Store and on GooglePlay) is free.For subscriptions to the printed issue of the journal, please contact: Haematologica Office, via Giuseppe Belli 4, 27100Pavia, Italy (phone +39.0382.27129, fax +39.0382.394705, E-mail: [email protected]).
Rates of the International edition for the year 2016 are as following:Institutional Personal
Print edition Euro 500 Euro 150
Advertisements. Contact the Advertising Manager, Haematologica Office, via Giuseppe Belli 4, 27100 Pavia, Italy (phone+39.0382.27129, fax +39.0382.394705, e-mail: [email protected]).
Disclaimer. Whilst every effort is made by the publishers and the editorial board to see that no inaccurate or misleadingdata, opinion or statement appears in this journal, they wish to make it clear that the data and opinions appearing in thearticles or advertisements herein are the responsibility of the contributor or advisor concerned. Accordingly, the publish-er, the editorial board and their respective employees, officers and agents accept no liability whatsoever for the conse-quences of any inaccurate or misleading data, opinion or statement. Whilst all due care is taken to ensure that drug dosesand other quantities are presented accurately, readers are advised that new methods and techniques involving drug usage,and described within this journal, should only be followed in conjunction with the drug manufacturer’s own publishedliterature.
Direttore responsabile: Prof. Edoardo Ascari; Autorizzazione del Tribunale di Pavia n. 63 del 5 marzo 1955.Printing: Tipografia PI-ME, via Vigentina 136, Pavia, Italy. Printed in April 2016.
haematologicaJournal of the European Hematology Association
Published by the Ferrata Storti Foundation


New Drugs In HematologySocietà Italiana di Ematologia (SIE)Chair: PL ZinzaniMay 9-11, 2016Bologna, Italy
ESH-EBMT 20th Training Course on Haemopoietic Stem CellTransplantationESH EBMTChairs: J Apperley, E Carreras, E Gluckman, T MassziMay 11-14, 2016Budapest, Hungary
21st Congress of the European Hematology AssociationEuropean Hematology AssociationJune 9-12, 2016Copenhagen, Denmark
Hematology Tutorial on managing complications in patients withhematologic malignancies in the era of new drugsEHA-ROHS-RSHChairs: E Parovichnikova, I Poddubnaya, R FoàJuly 1-3, 2016Moscow, Russian Federation
Summer School of Personalised Medicine for Health CareProfessionalsEuropean Alliance for Personalised Medicine (EAPM)July 4-7, 2016Cascais, Portugal
EHA Scientific Conference on Bleeding DisordersScientific Program Committee: C Balduini (Chair), A Falanga (Chair),F Rodeghiero,I Pabinger, M MakrisSeptember 14-17, 2016Barcelona, Spain
2nd International Conference on New Concepts in B-CellMalignanciesEuropean School of Haematology (ESH)Chairs: M Hallek, L Staudt, S Stilgenbauer, A Thomas-TikhonenkoSeptember 9-11, 2016Estoril, Portugal
10th Hodgkin SymposiumUniversity hospital of CologneChairs: A Engert, B von Treskow, B BöllOctober 22-25, 2016Cologne, Germany
Calendar of Events updated on April 1, 2016
calendar of events
haematologicaJournal of the European Hematology Association
Published by the Ferrata Storti Foundation




haematologicaJournal of the European Hematology Association
Published by the Ferrata Storti Foundation
Cover FigureStem cell transplantation (Image created by www.somersault1824.com)
Editorials515 Next generation research and therapy in red blood cell diseases
Roberta Russo, et al.
518 Innovations in treatment and response evaluation in multiple myelomaRuth Wester and Pieter Sonneveld
Review Articles521 Nonmyeloablative allogeneic hematopoietic cell transplantation - Leaders in Hematology review series
Rainer Storb and Brenda M. Sandmaier
531 Role of the tumor microenvironment in mature B-cell lymphoid malignanciesNathan H. Fowler, et al.
541 Chronic myeloid leukemia: reminiscences and dreamsTariq I. Mughal, et al.
ArticlesRed Cell Biology & Its Disorders559 ATP11C is a major flippase in human erythrocytes and its defect causes congenital hemolytic anemia
Nobuto Arashiki, et al.
566 Cannabinoid receptor-specific mechanisms to alleviate pain in sickle cell anemia via inhibition of mast cell activation and neurogenic inflammation Lucile Vincent, et al.
Blood Transfusion578 Metabolic pathways that correlate with post-transfusion circulation of stored murine red blood cells
Karen de Wolski, et al.
587 Impaired killing of Candida albicans by granulocytes mobilized for transfusion purposes: a role for granule components Roel P. Gazendam, et al.
Myeloproliferative Disorders597 Long-term serial xenotransplantation of juvenile myelomonocytic leukemia recapitulates human disease in Rag2–/–γc–/– mice
Christopher Felix Krombholz, et al.
Volume 101, Issue 5: May 2016Table of Contents
Haematologica 2016; vol. 101 no. 5 - May 2016http://www.haematologica.org/

Haematologica 2016; vol. 101 no. 5 - May 2016http://www.haematologica.org/
Acute Myeloid Leukemia607 Association of acute myeloid leukemia’s most immature phenotype with risk groups and outcomes
Jonathan M. Gerber, et al.
Plasma Cell Disorders616 Pre-clinical evaluation of CD38 chimeric antigen receptor engineered T cells for the treatment of multiple myeloma
Esther Drent, et al.
Cell Therapy & Immunotherapy626 Effects of anti-NKG2A antibody administration on leukemia and normal hematopoietic cells
Loredana Ruggeri, et al.
Stem Cell Transplantation634 Reduced intensity haplo plus single cord transplant compared to double cord transplant: improved engraftment
and graft-versus-host disease-free, relapse-free survivalKoen van Besien, et al.
644 Allogeneic unrelated bone marrow transplantation from older donors results in worse prognosis in recipients with aplastic anemiaYasuyuki Arai, et al.
Letters to the EditorLetters are available online only at www.haematologica.org/content/101/5.toc
e164 Using zebrafish to model erythroid lineage toxicity and regeneration Anna Lenard, et al.http://www.haematologica.org/content/101/5/e164
e168 ε-globin expression is regulated by SUV4-20h1 Yadong Wang, et al.http://www.haematologica.org/content/101/5/e168
e173 Anti-hemojuvelin antibody corrects anemia caused by inappropriately high hepcidin levelsSuzana Kovac, et al.http://www.haematologica.org/content/101/5/e173
e177 Impaired formation of erythroblastic islands is associated with erythroid failure and poor prognosis in a significant proportion of patientswith myelodysplastic syndromesGuntram Buesche, et al.http://www.haematologica.org/content/101/5/e177
e182 Pegylated interferon alpha-2a for essential thrombocythemia during pregnancy: outcome and safety. A case seriesYan Beauverd, et alhttp://www.haematologica.org/content/101/5/e182
e185 Acute myeloid leukemia patients’ clinical response to idasanutlin (RG7388) is associated with pre-treatment MDM2 protein expression in leukemic blastsBernhard Reis, et al.http://www.haematologica.org/content/101/5/e185
haematologicaJournal of the European Hematology Association
Published by the Ferrata Storti Foundation


Haematologica 2016; vol. 101 no. 5 - May 2016http://www.haematologica.org/
e189 Structural modeling of JAK1 mutations in T-cell acute lymphoblastic leukemia reveals a second contact site between pseudokinase andkinase domainsKirsten Canté-Barrett, et al.http://www.haematologica.org/content/101/5/e189
e192 Activity of the Janus kinase inhibitor ruxolitinib in chronic lymphocytic leukemia: results of a phase II trial David E. Spaner, et al.http://www.haematologica.org/content/101/5/e192
e196 Safety and efficacy of lenalidomide in combination with rituximab in recurrent indolent non-follicular lymphoma: final results of a phaseII study conducted by the Fondazione Italiana LinfomiStefano Sacchi, et al.http://www.haematologica.org/content/101/5/e196
e200 Identification of a novel stereotypic IGHV4-59/IGHJ5-encoded B-cell receptor subset expressed by various B-cell lymphomas with highaffinity rheumatoid factor activityRichard J. Bende, et al.http://www.haematologica.org/content/101/5/e200
e204 Early Th1 immunity promotes immune tolerance and may impair graft-versus-leukemia effect after allogeneic hematopoietic cell transplantationBrian G. Engelhardt, et al.http://www.haematologica.org/content/101/5/e204
e209 Natural killer cell licensing after double cord blood transplantation is driven by the self-HLA class I molecules from the dominant cordbloodNicolas Guillaume, et al.http://www.haematologica.org/content/101/5/e209
CommentsComments are available online only at www.haematologica.org/content/101/5.toc
e213 Why should hemophilia B be milder than hemophilia A?Shrimati Shetty, et al.http://www.haematologica.org/content/101/5/e213
e214 Failure to effectively treat chronic graft-versus-host disease: a strong call for prevention Andrea Bacigalupo, et al.http://www.haematologica.org/content/101/5/e214
haematologicaJournal of the European Hematology Association
Published by the Ferrata Storti Foundation


haematologica | 2016; 101(5)
EDITORIALS
515
Next generation research and therapy in red blood cell diseasesRoberta Russo,1,2 Immacolata Andolfo,1,2 and Achille Iolascon1,2
1Dipartimento di Medicina Molecolare e Biotecnologie Mediche, Università degli Studi di Napoli Federico II; and 2CEINGE Biotecnologie Avanzate, Napoli, Italy
E-mail: [email protected] doi:10.3324/haematol.2015.139238
Pathogenetic studies on red blood cell (RBC) diseases havealways represented a powerful model for the study ofmedical genetics and for technology innovation in both
diagnostics and research. This has mainly been due to theavailability of these cells compared to others, such as neurons,myocytes, and so on, that are not so easily available. Indeed,the first and the best molecular characterization of genetic dis-eases was carried out in RBC disorders.It is now over 50 years since the first pioneer studies on
abnormal globin and glucose 6-phosphate dehydrogenase(G6PD) genes, the forerunners of the current research andmolecular diagnosis of Mendelian disorders, and completionof the Human Genome Project was a crucial milestone in thediagnosis and research of genetic disorders. The assembly andrefinement of the reference genome provide the mainstay forcurrent knowledge in the field of human genetics. In recentyears, scientists have spent much time and effort in identifyinggenes and mutations that are causative of several diseases,with great success. Although the identification of these geneticvariants has improved our knowledge of disease etiology,there is still a considerable gap in our understanding of thegenetic factors that modify disease severity. In this context, itis important to consider that there has been a substantial evo-lution in diagnostic and research technologies. The implemen-tation of the new technologies is changing the approach todiagnosis and research. We started out using Sanger sequenc-ing, and we are now embracing next generation sequencing(NGS), moving from a monogenic approach to an oligo/multi-genic one. The application of next generation approaches willincrease our knowledge of genetic and genomic differencesamong individuals, gradually leading to a shift in the clinicalmanagement and the therapeutic plan from a population-based approach to personalized therapy for the individualpatient. The ‘next generation’ era in the field of RBC physiopatholo-
gy provided important insights into the molecular mecha-nisms of normal and diseased RBC homeostasis. These find-ings generated several novel therapeutic approaches that arenow being examined in clinical trials.In the last few years, several studies have supplied new con-
cepts about the regulation of erythrocyte volume. In particular,PIEZO1 has been discovered to be the causative gene of hered-itary xerocytosis, also known as dehydrated hereditary stom-atocytosis (DHS, OMIM 194380), an autosomal dominanthemolytic anemia characterized by primary erythrocyte dehy-dration.1,2 Piezo proteins have recently been identified as ionchannels mediating mechanosensory transduction in mam-malian cells.3 Mutations in PIEZO1 show a partial gain-of-function phenotype with delayed inactivation of the channelsuggesting increased cation permeability that leads to erythro-cyte dehydration.1,2 In 2015, a second causative gene of DHSwas identified, KCNN4, encoding a Gardos channel (a Ca2+sensitive, intermediate conductance, potassium selective chan-
nel).4-6 Similarly to gain-of-function genetic variants in PIEZO1,heterozygous dominantly inherited mutations in the KCNN4gene lead to greater activity of the channel when compared tothe wild type.4 The identification of PIEZO1 and KCNN4 vari-ants in DHS patients strongly indicates that both genes play acritical role in normal erythrocyte deformation and in mainte-nance of erythrocyte volume homeostasis. Moreover, theidentification of variants in these genes will open up new stud-ies on their role in the improvement or worsening of RBChydration in patients with primary (DHS) and secondary ery-throcyte hydration disorders such as sickle cell disease (SCD).Thus, the routine introduction of NGS targeted panels wouldmost likely facilitate, not only the diagnosis, but also the prog-nostic evaluation of these patients.Among the disorders of secondary erythrocyte hydration,
recent advances in the pathophysiology of SCD and β-tha-lassemia have elucidated new possible therapeutic approach-es. A clinical trial on senicapoc (ICA-17043), a potent blockerof the Gardos channel, demonstrated that treatment of SCDpatients resulted in increased hemoglobin and reduced mark-ers of hemolysis, strongly suggesting that the survival of sicklered blood cells was improved.7 Despite the lack of any reduc-tion in the frequency of pain episodes, the increasing recogni-tion that hemolysis contributes to the development of severalSCD-related complications suggests that senicapoc may bebeneficial in this disease by decreasing hemolysis.7 Thus,blockers of Gardos and PIEZO1 channels could be used infuture clinical practice for the treatment of primary and sec-ondary disorders of erythrocyte hydration.Likewise, another promising approach for the treatment of
both β-thalassemia and SCD is gene replacement therapy. Inthis approach, samples of multipotent hematopoietic stemprogenitor cells (HSPCs) are collected from the patient andsubsequently modified to express a β-like globin gene in ery-throid precursors; these cells are then re-infused.8 The modi-fied HSPCs will reconstitute the hematopoietic system, thusproducing normal, gene-corrected RBCs. This approach stillpresents many challenges: i) to reduce the tendency of inte-grated viral vectors; ii) to activate nearby genes; and also iii) tofurther increase β-like globin expression. Early results of a clin-ical trial in β-thalassemia major patients treated with improvedvectors are promising, and it is hoped that they will lead toadvances in the treatment of thalassemic patients.9
The application of gene therapy to treat erythroid disordersregards not only β-thalassemia and hemoglobinopathies. Forexample, gene therapy has been investigated for DiamondBlackfan anemia (DBA) and other erythroid diseases, such asred cell enzyme disorders, including severe forms of G6PD andpyruvate kinase deficiency.10,11
For most of the anemias due to RBCs defects, blood transfu-sion therapy or treatment by erythropoiesis stimulating agents(ESAs), such as recombinant EPO, are the front-line therapies.However, neither of these treatment approaches is without

Editorials
516 haematologica | 2016; 101(5)
risks and they are not effective in all cases. For example,patients with ineffective erythropoiesis do not respond toEPO. Thus, there is a clinical need for novel agents with adifferent mechanism of action from current ESAs. Members of the transforming growth factor beta (TGF-β)
superfamily, which include activins (A-B), growth differen-tiation factors (GDFs), and bone morphogenetic proteins(BMPs), have been studied as potential regulators of ery-thropoiesis, iron regulation and globin expression. Somerecent studies have investigated the role played by twodrugs, an activin receptor IIA (ActRIIA) ligand trap (ACE-011 or sotatercept) and a modified ActR type IIB (ActRIIB)ligand trap (ACE-536) in the regulation of late-stage ery-thropoiesis. It has been recently demonstrated that a mouseversion of both drugs, termed RAP-011 and RAP-536, isable to induce differentiation of erythroid cells, improveineffective erythropoiesis, correct anemia, and limit ironoverload in a mouse model of β-thalassemia intermedia.12,13
Both drugs act through inhibition of GDF11, a newly iden-tified regulator of erythropoiesis that will contribute signif-icantly to the understanding of the fine regulation of ery-thropoiesis and iron metabolism, and to the developmentof new drugs. So far, two phase II clinical trials have provid-ed proof of the importance of ActR ligand trap molecules inthe use of sotatercept in adults with β-thalassemia (clinical-trials.gov identifier: 01571635) and in transfusion-dependentDBA patients (clinicaltrials.gov identifier: 01464164).Finally, another no less interesting approach for future
therapy in RBC diseases is represented by genome editingtechnologies. These have mainly been used to study genefunction, in the discovery of therapeutic targets, and todevelop disease models in several disorders. Considerable
progress has been made in genome editing in the pastdecade via the use of either engineered nucleases systems,such as zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), or of the RNA-guidedengineered nucleases based on CRISPR-Cas9 (clustered reg-ularly inter-spaced short palindromic repeats/CRISPR-asso-ciated nuclease 9).14-16 The most promising progress hasbeen seen in the use of CRISPR/Cas9 technology forgenome correction of specific DNA sequences includingchanges in either coding or non-coding regions of autolo-gous cell genome.17 This system has become a simple-to-design and cost-effective tool for various genome editingpurposes, including gene therapy studies; indeed, it offersseveral advantages, the main one being its ability to editmultiple genes simultaneously.18 Current challenges forgenome editing of HSPCs include optimizing the deliveryof gene-editing tools, improving the efficiency of introduc-ing targeted modifications, and avoiding the creation ofpotentially harmful off-target mutations. β-thalassemiamutations have been corrected by gene editing in inducedpluripotent stem cells (iPSCs) by converting β-thalassemicmutations from homozygous to heterozygous state, thusrestoring HBB gene expression in erythrocytes differentiat-ed from the corrected iPSCs.19 This gene editing strategywill provide a crucial step to cure monogenic disease bygenetic repair of patient-specific iPSCs. We can envision afuture in which the functional integration between nextgeneration technologies for genomic screening and genom-ic editing will allow us to achieve our goal of targeted diag-nosis and therapy (Figure 1).The importance and the advantages of next generation
technologies are obvious. However, despite the widespread
Figure 1. Integration between technological updates and clinical applications in diagnosis and therapy of red blood cell (RBC) diseases. Adult hematopoietic stemprogenitor cells (HSPCs) or induced pluripotent stem cells (iPSCs) can be used for gene-therapy approaches. DNA extracted from a peripheral blood sample can beused to identify genetic variations by next generation sequencing (NGS). The causative role of these variations can be validated by in vitro/in vivo functional studiesand then the commonly used CD34+ HSPCs may be corrected directly by gene therapy or genome editing by CRISPR/CAS9 technology. Alternatively, somatic cellscan be isolated by fibroblasts of the patient and reprogrammed to pluripotency, with the resulting iPSCs then being corrected by gene therapy or genome editingand differentiated through erythroid lineage.

use of these tools in clinical practice, some considerationson their limitations and/or disadvantages should be made.On the one hand, will the different stages of data process-ing represent a major limitation of NGS genome screening,or will the need to accurately profile and control the off-tar-get effects of genome editing compromise its use in genetherapy? Unlike ex vivo cell therapies, genome-editing tech-nologies can potentially affect the human germline, andinternational committees to study the ethical, legal, andsocial implications of human gene editing have alreadybeen appointed. For example, the Hinxton Group is work-ing to guide decision-makers on the use of these technolo-gies in humans (http://www.hinxtongroup.org/). Thus, theeducation and training of all professional figures involved inthe clinical practice of molecular medicine still remains oneof the main aims of the scientific community.
References
1. Zarychanski R, Schulz VP, Houston BL, et al. Mutations in the mechan-otransduction protein PIEZO1 are associated with hereditary xerocyto-sis. Blood. 2012;120(9):1908-1915.
2 Andolfo I, Alper SL, De Franceschi L, et al. Multiple clinical forms ofdehydrated hereditary stomatocytosis arise from mutations in PIEZO1.Blood. 2013;121(19):3925-3935.
3. Ge J, Li W, Zhao Q, et al. Architecture of the mammalian mechanosen-sitive Piezo1 channel. Nature. 2015;527(7576):64-69.
4. Andolfo I, Russo R, Manna F, et al. Novel Gardos channel mutationslinked to dehydrated hereditary stomatocytosis (xerocytosis). Am JHematol. 2015;90(10):921-926.
5. Rapetti-Mauss R, Lacoste C, Picard V, et al. A mutation in the Gardoschannel is associated with hereditary xerocytosis. Blood.2015;126(11):1273-1280.
6. Glogowska E, Lezon-Geyda K, Maksimova Y, Schulz VP, Gallagher PG.Mutations in the Gardos channel (KCNN4) are associated with hered-itary xerocytosis. Blood. 2015;126(11):1281-1284.
7. Ataga KI, Reid M, Ballas SK, et al. Improvements in haemolysis andindicators of erythrocyte survival do not correlate with acute vaso-occlusive crises in patients with sickle cell disease: a phase III random-ized, placebo-controlled, double-blind study of the Gardos channelblocker senicapoc (ICA-17043). Br J Haematol. 2011;153(1):92-104.
8. Kohn DB, Pai SY, Sadelain M. Gene therapy through autologous trans-plantation of gene-modified hematopoietic stem cells. Biol BloodMarrow Transplant. 2013;19:S64-S69.
9. Thompson AA, Rasko EJ, Hongeng S, et al. Initial Results from theNorthstar Study (HGB-204): A Phase 1/2 Study of Gene Therapy for β-Thalassemia Major Via Transplantation of Autologous HematopoieticStem Cells Transduced Ex Vivo with a Lentiviral βA-T87Q-GlobinVector (LentiGlobin BB305 Drug Product). Blood. 2014;124(21):549.
10. Rovira A, De Angioletti M, Camacho-Vanegas O, et al. Stable in vivoexpression of glucose-6-phosphate dehydrogenase (G6PD) and rescueof G6PD deficiency in stem cells by gene transfer. Blood.2000;96(13):4111-4117.
11. Meza NW, Alonso-Ferrero ME, Navarro S, et al. Rescue of pyruvatekinase deficiency in mice by gene therapy using the human isoenzyme.Mol Ther. 2009;17(12):2000-2009.
12. Dussiot M, Maciel TT, Fricot A, et al. An activin receptor IIA ligand trapcorrects ineffective erythropoiesis in β-thalassemia. Nat Med.2014;20(4):398-407.
13. Suragani RN, Cawley SM, Li R, et al. Modified activin receptor IIB lig-and trap mitigates ineffective erythropoiesis and disease complicationsin murine β-thalassemia. Blood. 2014;123(25):3864-3872.
14. Cong L, Ran FA, Cox D, et al. Multiplex genome engineering usingCRISPR/Cas systems. Science. 2013;339(6121):819-823.
15. Doulatov S, Vo LT, Chou SS, et al. Induction of multipotentialhematopoietic progenitors from human pluripotent stem cells viarespecification of lineage-restricted precursors. Cell Stem Cell.2013;13(4):459-470.
16. Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineer-ing via Cas9. Science. 2013;339(6121):823-826.
17. Lupiáñez DG, Kraft K, Heinrich V, et al. Disruptions of topologicalchromatin domains cause pathogenic rewiring of gene-enhancer inter-actions. Cell. 2015;161(5):1012-1025.
18. Xiao-Jie L, Hui-Ying X, Zun-Ping K, Jin-Lian C, Li-Juan J. CRISPR-Cas9:a new and promising player in gene therapy. Med Genet.2015;52(5):289-296.
19. Xie F, Ye L, Chang JC, et al. Seamless gene correction of β-thalassemiamutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac.Genome Res. 2014;24(9):1526-1533.
Editorials
haematologica | 2016; 101(5) 517

Editorials
518 haematologica | 2016; 101(5)
Innovations in treatment and response evaluation in multiple myelomaRuth Wester and Pieter SonneveldErasmus MC Cancer Institute (EMC), Rotterdam, The Netherlands.
E-mail: [email protected] doi:10.3324/haematol.2016.142737
Multiple myeloma (MM) is still an incurable dis-ease. Recently, overall survival (OS) and progres-sion-free survival (PFS) have improved with the
introduction of immunomodulatory agents (IMIDs) andproteasome inhibitors (PI). Overall, an increase in 5-yearrelative survival from 28.8% to 34.7% was reportedbetween 1990-1992 and 2002-2004 by Brenner et al.1
Palumbo et al. reported a 10-year OS of 30% in transplanteligible patients.2 Innovative agents (i.e. monoclonal anti-bodies) may further increase response rates and the qualityof responses. Consequently, there will be a need for a moresensitive response assessment and risk-adapted treatmentschedules.In this editorial we will discuss the role of two innovative
approaches to evaluate response in MM, minimal residualdisease (MRD) and response evaluation with positronemission tomography-computed tomography (PET-CT), inthe context of recent treatment innovations.
Prognostic factorsThe International Staging System (ISS) has recently been
revised (R-ISS)3 to facilitate stratification of patients withdifferent clinical outcome. The R-ISS is a combination ofISS with chromosomal abnormalities (CA) and serum lac-tate dehydrogenase (LDH). CA t(4;14), t(14;16), del(17p),and potentially del(1p) and gain(1q), are associated with anadverse outcome.4
At present, a dichotomy arises between patients withpoor CA and patients with potential long PFS and OS.Reliable, sensitive techniques for response assessment are
needed to identify patients who require additional therapy. The International Myeloma Working Group (IMWG)
defined uniform response criteria for MM in 2006. In 2011,two new categories, stringent complete response (sCR) andvery good partial response (VGPR) were added.5 However,the current definition of complete response (CR) fails topredict a distinct overall outcome. Using MRD for responseevaluation may give a better prediction of OS.6,7 With mul-tiparameter flow cytometry (FCM) or next generationsequencing (NGS) it is possible to detect a tumor load of 10-
5 (Figure 1).5,6,8-10 This is clinically relevant since time to pro-gression (TTP) in patients with MRD below 10-5 is signifi-cantly better than in patients with MRD between 10-5 to 10-3 or above 10-3 (80 vs. 48 vs. 27 months).11 MRD combinedwith cytogenetics gives a better prediction of outcome thanstandard CR.7 Therefore, MRD has now been incorporatedinto several clinical trials.
Evaluation by PET-CT Bone marrow infiltration in patients with MM can be
patchy. This implies that because of sampling error, MRDmay be negative even in the presence of extramedullarydisease (EMD). Therefore imaging techniques are increas-ingly applied to assess EMD.12 Magnetic resonance imaging(MRI) seems the most sensitive imaging technique fordetection of bone involvement in the spine;6 however,EMD may not be visualized with this technique. PET-CTcan detect bone involvement as well as EMD. Patients withpersistence of abnormal 18F-fluorodeoxyglucose (FDG)uptake following high-dose therapy and stem cell trans-
Figure 1. In the last two decades,response criteria have changedbecause novel treatments haveimproved the quality of response.

Editorials
haematologica | 2016; 101(5) 519
plantation (SCT) have a poor prognosis.13 While smalldefects may be missed because of low spatial resolution,the use of PET-CT in detection of MRD seems promisingenough to warrant further evaluation in clinical trials.
Novel agents and treatment strategiesTreatment modalities have greatly expanded in the last
two decades and we will discuss some of the novel agentsin the context of new treatment strategies. IMIDs such aslenalidomide and thalidomide have increased OS and PFSin newly diagnosed multiple myeloma (NDMM).14,15
Pomalidomide is a next generation IMID. It has directantiproliferative, pro-apoptotic, and antiangiogenic effects,as well as modulatory effects on bone resorption, theimmune system and the bone marrow microenviron-ment.16-18 The pivotal phase III trial assessed the efficacy andsafety of pomalidomide with/without low-dose dexam-ethasone in patients with relapsed/refractory multiplemyeloma (RRMM). At a follow up of 14.2 months, medianPFS was 4.2 versus 2.7 months (HR=0.68; P=0.003), overallresponse rates (ORRs) were 33% and 18% (P=0.013),median response duration was 8.3 and 10.7 months, andOS was 16.5 and 13.6 months, respectively.19,20
The other class of novel agents is made up of proteasomeinhibitors (PI). Bortezomib has improved CR rate, PFS andOS in elderly patients (VMP, VD) and in transplant eligibleMM (PAD, VCD, VTD); as an example, in theHOVON65/GMMG-HD4 trial, addition of bortezomibincreased CR from 25% in controls to 36% (P<0.001) andPFS was also superior (28 vs. 35 months; P=0.002).21
Novel PIs have emerged: carfilzomib, oprozomib, mari-zomib and ixazomib. Carfilzomib is an epoxyketone pro-teasome inhibitor that binds selectively and irreversibly tothe constitutive proteasome and immunoproteasome. TheASPIRE trial evaluated safety and efficacy of adding carfil-zomib to lenalidomide/dexamethasone (RD) versus RDalone in patients with relapsed MM. PFS was significantlybetter with carfilzomib versus control group (26.3 vs. 17.6months, respectively).22 The ENDEAVOR trial comparedcarfilzomib with bortezomib in patients with RRMM; PFSwas 18.7 months with carfilzomib versus 9.4 months withbortezomib (P<0.0001).23
Ixazomib is a reversible boronic ester prodrug PI. Pre-
clinical studies have shown activity in myeloma cellsresistent to bortezomib. Combination of ixazomib withRD gave good responses also in unfavorable CA.24,25
Monoclonal antibodies [daratumumab, SAR650984(SAR) and elotuzumab] have set the stage for a new treat-ment modality in MM. Elotuzumab is a monoclonal anti-body targeting signaling lymphocytic activation moleculeF7 (SLAMF7). This is a cell surface glycoprotein highlyexpressed on MM cells and normal plasma cells. A phase IIItrial was recently performed in patients with RRMM.Patients were randomized between treatment with RDwith/without elotuzumab. Median PFS was 19.4 months inthe elotuzumab group versus 14.9 months in the controlgroup (P<0.001). OS in the elotuzumab group was 79% ver-sus 66% in the control group (P<0.001).26
Daratumumab is an anti-CD 38 monoclonal antibody. Itinduces cell killing by multiple mechanisms: complement-dependent cytotoxicity, antibody-dependent cellular cyto-toxicity and antibody-dependent cellular phagocytosisthrough activation of complement proteins, natural killercells, and macrophages, respectively.27,28 A phase I/II studyin heavily pre-treated patients with RRMM inducedresponse in 42% of patients.29 Daratumumab is currentlyunder investigation in several phase III trials, including theIFM2015/HOVON131 randomized phase III trial inNDMM who are transplant eligible. This study investigatesthe efficacy of the combination of daratumumab with VTDfor induction and consolidation followed by daratumumabmaintenance treatment. During this trial, assessment ofMRD will be performed using NGS on bone marrow andperipheral blood samples collected from subjects whoachieve at least VGPR (Figure 2). Histone deacetylase inhibitors (panobinostat, vorinostat
and ricolinostat) inhibit cell growth and induce apoptosis.In the PANORAMA-1 trial, treatment with bortezomib,dexamethasone plus panobinostat resulted in significantlylonger PFS (12 months vs. 8 months; P<0.0001).30
ConclusionsDuring the last two decades, diagnostic methods and treat-
ment modalities in MM have greatly improved. In decidinghow to treat a particular patient, prognostic factors such ascytogenetic abnormalities are becoming more important.
Figure 2. IFM2015/HOVON 131.Patients are randomized betweentreatment with VTD with/withoutdaratumumab followed by high-dose melphalan (HDM) and autol-ogous stem cell transplantation(ASCT). After ASCT, patientsreceive two consolidation cycles.Patients with at least a partialresponse (PR) will be randomizedafter determination of response atapproximately day 100 after ASCT,and will enter the MaintenancePhase. Minimal residual disease(MRD) assessment will be per-formed before the first inductioncycle, before ASCT, at day 100after ASCT, and during mainte-nance in patients who achieve atleast a very good partial response(VGPR).

Treatment schedules should be adapted to these prognosticfactors. This requires further evaluation in clinical trials. Novel agents induce deeper responses. This implies the
need for a more sensitive response assessment such asdetermination of MRD by FCM or NGS. Therefore, clinicaltrials with novel agents should include standard panels forcytogenetics, MRD, and optimal imaging.
References
1. Brenner H, Gondos A, Pulte D. Recent major improvement in long-term survival of younger patients with multiple myeloma. Blood.2008;111(5):2521-2526.
2. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med.2011;364(11):1046-1060.
3. Palumbo A, Avet-Loiseau H, Oliva S, et al. Revised InternationalStaging System for Multiple Myeloma: A Report From InternationalMyeloma Working Group. J Clin Oncol. 2015;33(26):2863-2869.
4. Hebraud B, Magrangeas F, Cleynen A, et al. Role of additional chromo-somal changes in the prognostic value of t(4;14) and del(17p) in multi-ple myeloma: the IFM experience. Blood. 2015;125(13):2095-2100.
5. Durie BG, Harousseau JL, Miguel JS, et al. International uniformresponse criteria for multiple myeloma. Leukemia. 2006;20(9):1467-1473.
6. Paiva B, van Dongen JJ, Orfao A. New criteria for response assessment:role of minimal residual disease in multiple myeloma. Blood.2015;125(20):3059-3068.
7. Rawstron AC, Gregory WM, de Tute RM, et al. Minimal residual dis-ease in myeloma by flow cytometry: independent prediction of sur-vival benefit per log reduction. Blood. 2015;125(12):1932-1935.
8. Blade J, Samson D, Reece D, et al. Criteria for evaluating diseaseresponse and progression in patients with multiple myeloma treated byhigh-dose therapy and haemopoietic stem cell transplantation.Myeloma Subcommittee of the EBMT. European Group for Blood andMarrow Transplant. Br J Haematol. 1998;102(5):1115-1123.
9. Cavo M, Rajkumar SV, Palumbo A, et al. International MyelomaWorking Group consensus approach to the treatment of multiplemyeloma patients who are candidates for autologous stem cell trans-plantation. Blood. 2011;117(23):6063-6073.
10. Durie BG, Miguel JF, Blade J, Rajkumar SV. Clarification of the defini-tion of complete response in multiple myeloma. Leukemia.2015;29(12):2416-2417.
11. Martinez-Lopez J, Lahuerta JJ, Pepin F, et al. Prognostic value of deepsequencing method for minimal residual disease detection in multiplemyeloma. Blood. 2014;123(20):3073-3079.
12. Dimopoulos MA, Hillengass J, Usmani S, et al. Role of magnetic reso-nance imaging in the management of patients with multiple myeloma:a consensus statement. J Clin Oncol. 2015;33(6):657-664.
13. Caers J, Withofs N, Hillengass J, et al. The role of positron emissiontomography-computed tomography and magnetic resonance imagingin diagnosis and follow up of multiple myeloma. Haematologica.2014;99(4):629-637.
14. Palumbo A, Hajek R, Delforge M, et al. Continuous lenalidomide treat-ment for newly diagnosed multiple myeloma. N Engl J Med.2012;366(19):1759-1769.
15. Benboubker L, Dimopoulos MA, Dispenzieri A, et al. Lenalidomideand dexamethasone in transplant-ineligible patients with myeloma. NEngl J Med. 2014;371(10):906-917.
16. Ruchelman AL, Man H-W, Zhang W, et al. Isosteric analogs of lenalido-mide and pomalidomide: Synthesis and biological activity. Bioorg MedChem Lett. 2013;23(1):360-365.
17. Richardson PG, Mark TM, Lacy MQ. Pomalidomide: newimmunomodulatory agent with potent antiproliferative effects. CritRev Oncol Hematol. 2013;88 Suppl 1:S36-44.
18. Shortt J, Hsu AK, Johnstone RW. Thalidomide-analogue biology:immunological, molecular and epigenetic targets in cancer therapy.Oncogene. 2013;32(36):4191-4202.
19. Richardson PG, Siegel DS, Vij R, et al. Pomalidomide alone or in com-bination with low-dose dexamethasone in relapsed and refractory mul-tiple myeloma: a randomized phase 2 study. Blood. 2014;123(12):1826-1832.
20. San Miguel J, Weisel K, Moreau P, et al. Pomalidomide plus low-dosedexamethasone versus high-dose dexamethasone alone for patientswith relapsed and refractory multiple myeloma (MM-003): a ran-domised, open-label, phase 3 trial. Lancet Oncol. 2013;14(11):1055-1066.
21. Sonneveld P, Schmidt-Wolf IG, van der Holt B, et al. Bortezomib induc-tion and maintenance treatment in patients with newly diagnosed mul-tiple myeloma: results of the randomized phase III HOVON-65/GMMG-HD4 trial. J Clin Oncol. 2012;30(24):2946-2955.
22. Stewart AK, Rajkumar SV, Dimopoulos MA, et al. Carfilzomib,lenalidomide, and dexamethasone for relapsed multiple myeloma. NEngl J Med. 2015;372(2):142-152.
23. Dimopoulos MA, Moreau P, Palumbo A, et al. Carfilzomib and dexam-ethasone versus bortezomib and dexamethasone for patients withrelapsed or refractory multiple myeloma (ENDEAVOR): a randomised,phase 3, open-label, multicentre study. Lancet Oncol. 2016;17(1):27-38.
24. Moreau P, Masszi T, Grzasko N, et al. Ixazomib, an Investigational OralProteasome Inhibitor (PI), in Combination with Lenalidomide andDexamethasone (IRd), Significantly Extends Progression-Free Survival(PFS) for Patients (Pts) with Relapsed and/or Refractory MultipleMyeloma (RRMM): The Phase 3 Tourmaline-MM1 Study(NCT01564537). Blood. 2015;126(23)(Abstract 727).
25. Kumar SK, Berdeja JG, Niesvizky R, et al. Safety and tolerability of ixa-zomib, an oral proteasome inhibitor, in combination with lenalidomideand dexamethasone in patients with previously untreated multiplemyeloma: an open-label phase 1/2 study. Lancet Oncol.2014;15(13):1503-1512.
26. Lonial S, Dimopoulos M, Palumbo A, et al. Elotuzumab Therapy forRelapsed or Refractory Multiple Myeloma. N Engl J Med. 2015;373(7):621-631.
27. Overdijk MB, Verploegen S, Bogels M, et al. Antibody-mediatedphagocytosis contributes to the anti-tumor activity of the therapeuticantibody daratumumab in lymphoma and multiple myeloma. MAbs.2015;7(2):311-321.
28. de Weers M, Tai YT, van der Veer MS, et al. Daratumumab, a noveltherapeutic human CD38 monoclonal antibody, induces killing of mul-tiple myeloma and other hematological tumors. J Immunol.2011;186(3):1840-1848.
29. Lokhorst HM, Laubach J, Nahi H, et al. Dose-dependent efficacy ofdaratumumab (DARA) as monotherapy in patients with relapsed orrefractory multiple myeloma (RR MM). ASCO Annual MeetingProceedings. 2014;2014:8513.
30. San-Miguel JF, Hungria VT, Yoon SS, et al. Panobinostat plus borte-zomib and dexamethasone versus placebo plus bortezomib and dex-amethasone in patients with relapsed or relapsed and refractory multi-ple myeloma: a multicentre, randomised, double-blind phase 3 trial.Lancet Oncol. 2014;15(11):1195-1206.
Editorials
520 haematologica | 2016; 101(5)

haematologica | 2016; 101(5) 521
Received: December 29, 2015.
Accepted: February 5, 2016.
Pre-published: no prepublication.
©2016 Ferrata Storti Foundation
Check the online version for the most updatedinformation on this article, online supplements,and information on authorship & disclosures:www.haematologica.org/content/101/5/521
Material published in Haematologica is cov-ered by copyright. All rights reserved to FerrataStorti Foundation. Copies of articles areallowed for personal or internal use. A permis-sion in writing by the publisher is required forany other use.
Correspondence: [email protected]
Nonmyeloablative allogeneic hematopoieticcell transplantationRainer Storb and Brenda M. SandmaierFred Hutchinson Cancer Research Center and the University of Washington, Seattle, WA, USA
Ferrata StortiFoundation
EUROPEANHEMATOLOGYASSOCIATION
Haematologica 2016Volume 101(5):521-530
REVIEW ARTICLELeaders in Hematology review series
doi:10.3324/haematol.2015.132860
Most hematological malignancies occur in older patients. Untilrecently these patients and those with comorbidities werenot candidates for treatment with allogeneic hematopoietic
transplantation because they were unable to tolerate the heretoforeused high-dose conditioning regimens. The finding that many of thecures achieved with allogeneic hematopoietic transplantation weredue to graft-versus-tumor effects led to the development of less toxicand well-tolerated reduced intensity and nonmyeloablative regimens.These regimens enabled allogeneic engraftment, thereby setting thestage for graft-versus-tumor effects. This review summarizes theencouraging early results seen with the new regimens and discussesthe two hurdles that need to be overcome for achieving even greatersuccess, disease relapse and graft-versus-host disease.
ABSTRACT
Introduction
Conditioning for allogeneic hematopoietic cell transplantation (HCT) in the treat-ment of hematologic malignancies has traditionally involved high doses of totalbody irradiation (TBI) and/or chemotherapy. The dual purpose of conditioning hasbeen to reduce the patients’ burden of malignant cells before HCT and suppresstheir immune system so that the allogeneic grafts are not rejected. The high inten-sity of the traditional regimens has precluded using allogeneic HCT in olderpatients or those with comorbidities because of unacceptable toxicities. This hasbeen unfortunate, given that the median ages of patients at the time of diagnosis ofmost candidate malignancies, e.g. acute myelocytic leukemia (AML) or non-Hodgkin lymphoma (NHL), range from 65 to 75 years. The finding that the cure ofhematologic malignancies not only results from intense conditioning but also inlarge part from the killing of tumor cells by transplanted donor immune cells,termed “graft-vs.-tumor” (GVT) effect, set the stage for the development ofreduced-intensity conditioning (RIC) regimens. Such regimens need to be immuno-suppressive enough to allow sustained engraftment, thereby enabling GVT effects.The markedly reduced toxicities associated with these novel regimens haveallowed for the extension of allogeneic HCT to include older and medically infirmpatients. The relative intensities of individual conditioning regimens vary consider-ably as far as their immunosuppressive and myelosuppressive properties are con-cerned (Figure 1). The choice of a given regimen may, in part, be dictated by thenature of the underlying malignancy and, in part, by comorbidities. The results oftrials using RIC or nonmyeloablative (NMA) regimens have been surprisinglyencouraging. However, all the trials share two major problems that have limitedtrial outcomes. These are non-relapse mortality (NRM), mainly related to concur-rent or preceding graft-vs.-host disease (GVHD) and its treatment, and relapse mor-tality.This review will describe the preclinical basis for some of the RIC and NMA reg-
imens, address GVT effects, summarize trial results with HLA-matched and mis-matched grafts, address the use of older sibling donors, and explore ways to reducethe risks of GVHD and relapse.

Pre-clinical studiesWe used a canine model of major histocompatibility
complex (MHC=DLA)-matched marrow grafts to developa minimal-intensity or NMA conditioning regimen. Wefound that 2 Gy TBI either without postgrafting immuno-suppression or with monotherapy using cyclosporine(CSP) did not enable consistently sustained engraftment.1However, when a short course of mycophenolate mofetil(MMF) was combined with CSP following 2 Gy TBI, syn-ergism between the two drugs was noted, host T-cellswere prevented from rejecting the donor marrow, and sus-tained engraftment was seen.2 Similar synergism wasobserved with rapamycin used in lieu of MMF.3 In otherstudies, which substituted 4.5 Gy irradiation targeted tothe cervical, thoracic, and upper abdominal lymph nodechain for 2 Gy TBI, we saw sustained engraftment in non-irradiated marrow and lymph node spaces, suggesting thatthe donor T-lymphocytes created space for grafts tohome.4 The results of the canine studies were the basis forthe successful clinical introduction of an NMA regimen of2 Gy TBI combined with fludarabine (FLU) before andMMF/calcineurin inhibitor after HLA-matched related andunrelated HCT.Further canine work focused on replacing or augment-
ing 2 Gy TBI with radiolabeled monoclonal antibodies(mAbs).5 Current clinical studies have already employedmAb to CD45 or CD20 coupled to beta-emitting radionu-clides such as iodine-131 (131I)6or yttrium-90 (90Y);7 howev-er, the disadvantages of the beta-emitters became appar-ent, and included relatively long path lengths, long half-lives, and low energy. Therefore, we turned to alpha-emit-ting radionuclides, including bismuth-213 (213Bi)8 and asta-tine-211 (211At).9 211At coupled to an anti-CD45 mAb turnedout to be more effective than 213Bi.10 Other advantages of211At include that it is produced at the University ofWashington Cyclotron Facility, has a short half-life of 7.2hours, has high energy, and, importantly, a very short pathlength of approximately 0.04-0.06 mm, thereby reducingthe risk of off-target effects. Dose-finding toxicity studiesin dogs have been completed, and DLA-identical marrowgrafts successfully established using a 211At-labeled anti-CD45 mAb.9 Clinical studies are in preparation that areaimed at increasing tumor cell kill in patients with hema-tologic malignancies and replacing systemic chemo/radia-tion therapy in those with nonmalignant diseases.In 1991 Japanese investigators showed that treating
MHC-mismatched murine recipients with high-dosecyclophosphamide (CY) after HCT induced tolerance ofthe grafted lymphocytes to host tissues, while not impair-ing hematopoietic engraftment.11 This has been possiblesince hematopoietic stem cells are protected against thetoxic effect of CY metabolites by the presence withinthese cells of aldehyde dehydrogenase. These observa-tions and those by investigators from Johns HopkinsMedical School12,13 set the stage for the development of aneffective HLA-haploidentical transplant protocol. The pro-tocol utilized the basic FLU/2 Gy TBI NMA regimen withtwo additional small doses of CY for conditioning.14Patients were then given one or two high doses of CY ondays 3 and/or 4 post-grafting, followed by MMF/cal-cineurin inhibitor.
Clinical resultsHLA-matched related and unrelated HCT. The choice of
conditioning regimen intensity depends in part on the
underlying malignancy, disease burden, and comorbidi-ties. The effects these variables can have on transplanta-tion outcome are illustrated by results in 1,092 patientswith advanced hematologic malignancies given a uniformNMA regimen of FLU/2 Gy TBI, which allowed for thepurest assessment of GVT effects apart from conditioningand the best determination of GVHD not augmented bytoxicities related to the regimen.15 Patients were eitherolder or had serious comorbidities. Their median age was56 (range 7 to 75) years. Thirty-five percent of patientswere older than 60 years. Six hundred and eleven patientshad HLA-matched related donors and 481 had unrelateddonors (one HLA allele-level mismatch was permitted).Diseases and disease stages are shown in Table 1. Twentypercent of patients had failed high-dose autologous orallogeneic HCT or had developed a secondary, usuallymyeloid malignancy after autologous HCT for anothermalignancy. Forty-five percent of patients had HCT-Comorbidity Index (CI) scores of 3 or greater. Cumulativeincidence rates of acute GVHD were 37% for grade 2, 9 %for grade 3, and 4% for grade 4, respectively; the rateswere lower for related than for unrelated recipients. Table1 divides patients based on low, standard, or high-risk ofrelapse as assessed by relapse rate per patient year. It isevident that disease and disease burden were major deter-minants for relapse risk. For example, patients with high-grade NHL in remission had a relapse rate of 0.16 perpatient year in years 1-2, while those not in remission hada rate of 0.48. Similar findings were made for other dis-eases. These data suggested that reducing the tumor bur-den in certain diseases and disease stages before HCTmight reduce the risk of relapse after HCT. Most relapsesoccurred in the first 2 years, and relapse rates in subse-quent years were generally low. Five-year relapse mortali-ty rates ranged from 18% to 50% depending on relapserisk (Figure 2). Of note, 5-year overall relapse mortalitywas the same among related and unrelated recipients, at34.5% for both. Figure 2 also shows 5-year overall sur-vivals which ranged from as low as 25% in patients withhigh relapse risk and high comorbidity scores to 60% inpatients with low relapse risk and low comorbidity scores.Unrelated recipients had a significantly increased risk ofGVHD-associated NRM compared to related recipients.Of note, a single HLA allele-level mismatch at class I didnot adversely affect HCT outcome. Five-year overall NRMwas 24% (20% related to preceding or concurrentGVHD), ranging from 14.7% (12% related to GVHD)among related recipients with low comorbidity scores to36% (31.8% related to GVHD) among unrelated recipi-ents with comorbidity scores of 3 and higher. A phase II randomized clinical trial was carried out as
part of an ongoing effort to optimize control of acuteGVHD without reducing the GVT effect after unrelatedHCT.16 Patients were randomized between three differentpost-HCT immunosuppressive regimens. In arm 1,tacrolimus was administered for 180 days and MMF for 95days (n=69). In arms 2 (n=71) and 3 (n=68), tacrolimus andMMF were administered for 150 and 180 days, respective-ly, with the addition of 80 days of sirolimus in arm 3.Grade II-IV acute GVHD rates in the 3 arms were 64%,48% and 47% at day 150. Steroid use was significantlylower at day 150 in arm 3 (32% vs. 55% in arm 1 and 49%in arm 2; and the day 150 incidence of cytomegalovirusreactivation was significantly lower in arm 3 (arm 1, 54%;arm 2, 47%; arm 3, 22%) (Figure 3). Currently a 2-arm
R. Storb and B.M. Sandmaier
522 haematologica | 2016; 101(5)

phase III trial is ongoing using cyclosporine and MMFwith and without sirolimus, in order to further evaluatethe role of sirolimus.Table 2 shows results with RIC or NMA regimens
reported by registries or individual transplant centers.Most regimens used were more intense and relied less onGVT effects than the NMA regimen used in studiesshown in Table 1 and Figures 2 and 3. Information oncomorbidity scores were generally not provided. The twoNMDP studies focused on results with unrelated donors.Five-year outcomes in the former of the two included38% NRM, 42% relapse, and 23% overall survival.17 Thesecond study had a median follow-up of 3 years andshowed that outcomes after RIC were comparable tothose after NMA regimens, with approximately 34%NRM, 37% relapse, and 32% overall survival in bothgroups.18 A large French registry study included slightlyyounger patients receiving grafts from related or unrelateddonors.19 Median follow-up was short at 1.75 years. Eventhough NRM was low at 15 %, overall survival was only42%. A Dana-Farber report included 433 related and unre-lated recipients given RIC.20 The median follow-up was 2 years. NRM rates were 6% for related and 8% for unre-lated recipients, relapse rates were 65% and 52%, andoverall survival rates were 50% and 56%, respectively. Alarge CIBMTR study of RIC and either T-replete or in vivoT-depleted (ATG or Campath) grafts from related or unre-lated donors reported results with a median follow-up of3 years.21 NRM ranged from 21% to 26%, relapse from38% to 51%, and survival from 38% to 50%, respectively,with slightly better outcomes seen with T-replete grafts. A
smaller single-center study from Marseille had a medianfollow-up of 5 years with grafts from related donors afterRIC. NRM was 25%, relapse 22%, and survival 60%.22Among other comparisons, a second large CIBMTR studycompared results with marrow and PBSC grafts after RICto grafts after NMA conditioning.23 Donors were eitherrelated or unrelated. With a median follow-up of 3 years,NRM ranged from 33.5% to 38%, relapse from 35% to40%, and survival from 35% to 40%. An EBMT registrystudy in younger patients given either related or unrelatedgrafts after RIC, showed a 2-year NRM rate of 35%,relapse of 34% and event-free survival of 29%. In summa-ry, the median follow-up in these studies was 3 (range,1.75 to 5) years.24 Across the studies the median eventrates were 43% (range, 22–65%) for relapse, 34% (range,6–38%) for NRM and 38% (range, 22–65%) for overallsurvival.A phase III trial investigating conditioning intensity by
the Blood and Marrow Transplant Clinical Trials Network(BMT CTN)25 randomized patients with MDS or AML toeither a RIC regimen (FLU/BU2 or FLU/Mel) or a myeloab-lative conditioning (MAC) regimen (FLU/BU4, BU4Cy, orCyTBI). Inclusion criteria included <5% blasts, beingbetween 18-65 years of age, an HCT-CI of < 4, both relat-ed and unrelated donors with 7/8 or 8/8 HLA loci match-ing, and either marrow or PBSC. The primary diagnosiswas AML (80 %) and 92 % of patients received PBSC. Thestudy was stratified by center. The primary endpoint was18 months overall survival. The DSMB closed the studyearly at the second interim analysis after 272 patients wereenrolled (MAC n=135; RIC n=137). Overall survival and
Nonmyeloablative allogeneic hematopoietic cell transplantation
haematologica | 2016; 101(5) 523
Table 1. Relapse rates per patient year among 1,092 patients.15
Diagnosis* Stage No. of Patients Relapse RateYears 1 and 2 Years 3-5
Low-risk MPN Any 18 0.10 0.00CLL CR 9 0.11 0.14Waldenström’s syndrome Any 10 0.13 0.06NHL Any stage of mantle cell and low-grade; aggressive CR 140 0.16 0.02ALL CR1† 28 0.17 0.04MM CR 38 0.19 0.06Standard-risk CLL No CR 113 0.24 0.05CML CP1 24 0.24 0.00MM No CR 179 0.32 0.17AML CR‡ 191 0.33 0.02MDS RA / RARS 30 0.35 0.00High-risk NHL Aggressive; no CR 50 0.48 0.00AML No CR; evolved from MDS 98 0.65 0.04HL After failed autologous HCT 61 0.61 0.14MDS RAEB; CMML; second 62 0.65 0.04CML CP2; AP; BC 23 0.71 0.07ALL ≥ CR2; no CR 18 1.03 -
ALL: acute lymphoblastic leukemia; AML: acute myeloid leukemia; AP: accelerated phase; BC: blast crisis; CLL: chronic lymphocytic leukemia; CML: chronic myelocytic leukemia;CMML: chronic myelomonocytic leukemia; CP: chronic phase; HCT: hematopoietic cell transplantation; HL: Hodgkin lymphoma; MDS: myelodysplastic syndrome; MPN: myelopro-liferative neoplasms; MM: multiple myeloma; NHL: non-Hodgkin lymphoma; RAEB: refractory anemia with excess blasts; RARS: refractory anemia with ring sideroblasts. *Therewere 243 patients in the low-risk group (53% related and 47% unrelated donors); 537 patients in the standard-risk group (58% related and 42% unrelated donors), and 312 patientsin the high-risk group (54% related and 46% unrelated donors). †Before HCT, 14% of patients had minimal residual disease. ‡Before HCT, 13% of patients had minimal residual dis-ease. Reprinted with permission. From: Storb R, et al. Graft-versus-host disease and graft-versus-tumor effects after allogeneic hematopoietic cell transplantation. J Clin Oncol 31(12),2013; 1530-1538. ©2013 American Society of Clinical Oncology. All rights reserved.
progression-free survival at 18 months were 77.4% and68.8% (MAC) and 67.7% and 47.3% (RIC), respectively(P=0.07; P<0.01). The incidences of both acute and chronicGVHD were significantly higher in MAC patients(P=0.024 and P=0.019, respectively). The primary causesof death were GVHD in the MAC arm (52%) and relapsein the RIC arm (82%). The conclusion was that MACremains the treatment of choice for younger patients withMDS or AML. HLA-mismatched unrelated HCT. While many patients
who would benefit from HCT have a HLA-matcheddonor, a substantial number will not, particularly thosewho do not have white European ancestry. An analysiswas performed on data from the NMDP unrelated donorand cord blood registries to predict the likelihood of iden-tifying suitable donors for U.S. patients.26 The likelihood offinding an 8/8 HLA loci match ranged from 75% in whiteEuropeans to 16-19% for Black/African racial groups. Ifone accepts a 7/8 HLA loci matched donor, the numbersincrease to 97% and 66-76%, respectively. The CIBMTRcompared outcomes in 563 recipients of a single HLAlocus mismatch with 2,025 recipients of 8/8 HLA locihigh-resolution matched unrelated RIC HCT.27 Therewere more grades II-IV acute GVHD, higher NRM andlower disease free survival and overall survival in recipi-ents of 7/8 HLA loci matched URD. Interestingly, therewas no difference in chronic GVHD or relapse. Thedecreases in overall and disease free survival using a 7/8HLA loci matched donor were slightly less than those inthe myeloablative setting, suggesting a role of tissue dam-age in mortality following higher dose regimens. The find-ings in this large registry study are consistent with anothersmaller prospective study.28Taken together these studies show that relapse and
NRM, mostly related to GVHD, represent the two majorobstacles for patients given RIC or NMA regimens thatneed addressing in future trials.HLA-haploidentical HCT. Many patients, particularly
members of ethnic minorities, lack HLA-matched unrelat-ed donors; however, most patients have a relative who isHLA-haploidentical. The development of low-toxicityregimens sufficient to overcome the immunologic barriersto engraftment is equally important for such patients.Johns Hopkins University and the Fred HutchinsonCancer Research Center investigated a novel HLA-hap-loidentical marrow transplant trial using the fludarabineand 2 Gy TBI regimen and additional immunosuppressionwith CY both before and after HCT for the treatment ofhematologic malignancies.14 This regimen was well toler-ated and, considering the strong immunological barriersthat needed to be overcome, the rejection incidence waslow. In addition, the incidences of severe acute and chron-ic GVHD were encouragingly low. These results wereconfirmed in a multi-site trial conducted by the BMTCTN29 which also showed a relatively high relapse rate. Acurrently ongoing randomized study, BMT CTN Protocol1101, compares HLA-haploidentical marrow vs. cordblood as a stem cell source.A recent European publication noted a pronounced
increase in the use of HLA-haploidentical family donorsand a concurrent decrease in the use of cord blooddonors.30 More than twice the number of HLA-haploiden-tical grafts have been reported since 2010 compared tocord blood transplants. CIBMTR is reporting similartrends in North America. A recent CIBMTR study compared outcomes in 2,174
patients with AML given grafts from HLA-matched unre-lated (n=1,982) or HLA-haploidentical related donors
R. Storb and B.M. Sandmaier
524 haematologica | 2016; 101(5)
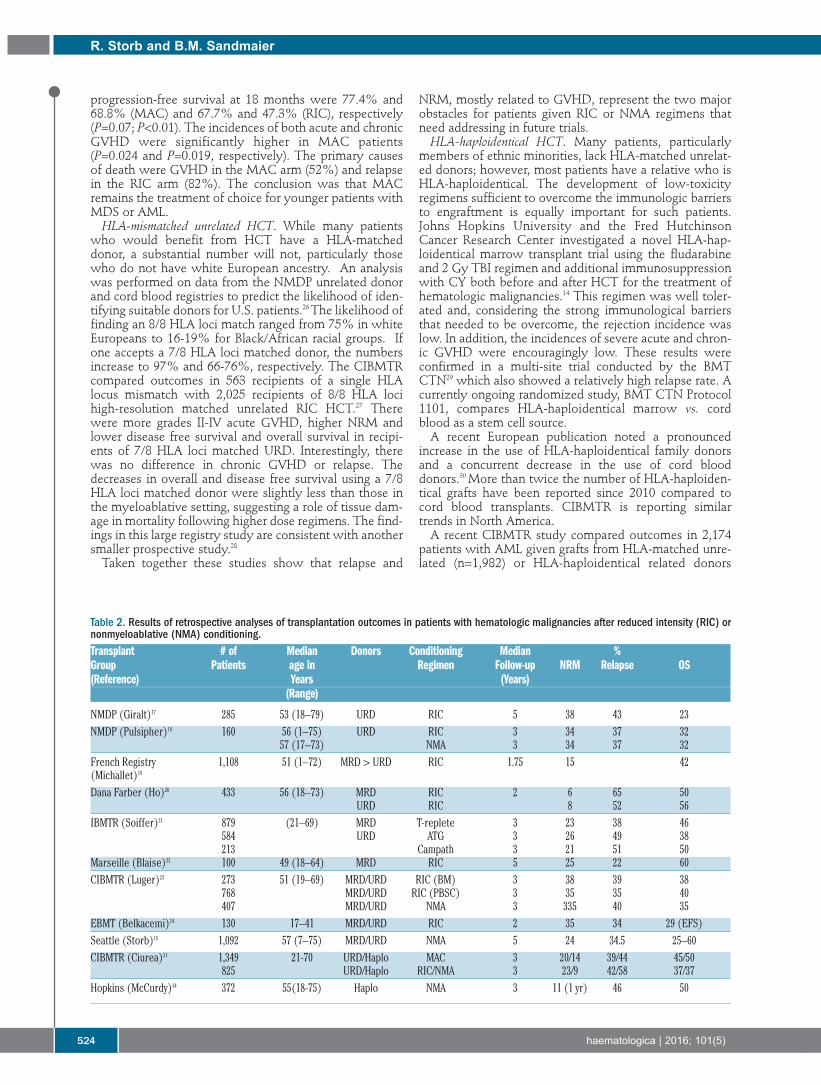
Table 2. Results of retrospective analyses of transplantation outcomes in patients with hematologic malignancies after reduced intensity (RIC) ornonmyeloablative (NMA) conditioning.Transplant # of Median Donors Conditioning Median %Group Patients age in Regimen Follow-up NRM Relapse OS(Reference) Years (Years)
(Range)
NMDP (Giralt)17 285 53 (18–79) URD RIC 5 38 43 23NMDP (Pulsipher)18 160 56 (1–75) URD RIC 3 34 37 32
57 (17–73) NMA 3 34 37 32French Registry 1,108 51 (1–72) MRD > URD RIC 1.75 15 42(Michallet)19
Dana Farber (Ho)20 433 56 (18–73) MRD RIC 2 6 65 50URD RIC 8 52 56
IBMTR (Soiffer)21 879 (21–69) MRD T-replete 3 23 38 46584 URD ATG 3 26 49 38213 Campath 3 21 51 50
Marseille (Blaise)22 100 49 (18–64) MRD RIC 5 25 22 60CIBMTR (Luger)23 273 51 (19–69) MRD/URD RIC (BM) 3 38 39 38
768 MRD/URD RIC (PBSC) 3 35 35 40407 MRD/URD NMA 3 335 40 35
EBMT (Belkacemi)24 130 17–41 MRD/URD RIC 2 35 34 29 (EFS)Seattle (Storb)15 1,092 57 (7–75) MRD/URD NMA 5 24 34.5 25–60CIBMTR (Ciurea)31 1,349 21-70 URD/Haplo MAC 3 20/14 39/44 45/50
825 URD/Haplo RIC/NMA 3 23/9 42/58 37/37Hopkins (McCurdy)34 372 55(18-75) Haplo NMA 3 11 (1 yr) 46 50

given regimens using post-HCT Cy (n=192).31 The studyincluded patients with myeloablative (unrelated n=1,245;HLA-haploidentical n=104) and RIC/NMA conditioning(unrelated n=737; HLA-haploidentical n=88). There wasno difference in overall and disease free survival betweenthe different donor types in either the myeloablative orRIC/NMA recipients (Table 2 and Figure 4). There was sig-nificantly more acute and chronic GVHD in recipients ofunrelated grafts but a lower risk of NRM (P=0.01), and aborderline increase risk of relapse (P=0.05) in RIC/NMA-conditioned recipients of HLA-haploidentical relatedgrafts. A similar CIBMTR study compared outcomes in917 patients with NHL receiving HLA-haploidentical relat-ed versus HLA-matched unrelated HCT, the latter eitherwith or without ATG.31 There was no significant differ-ence in overall survival between the 3 groups but therewas inferior survival in those unrelated patients whoreceived ATG. In a single center series of 372 patients,patients were stratified by the refined Disease Risk Index(DRI)32,33 and evaluated for outcomes. By refined DRI, 3-year progression-free survival in low, intermediate andhigh/very high-risk groups were 65%, 37% and 22%,respectively (Table 2).34 These results are similar to thosehistorically seen with HLA-matched HCT, suggesting thatprospective randomized trials are warranted to evaluatethe use of alternative donors given the lower incidence ofchronic GVHD seen after HLA-haploidentical HCT.It has been suggested that the use of PBSC may reduce
the risk of relapse among HLA-haploidentical recipientswithout increasing the risk of GVHD. Concurrent studiesusing PBSC were carried out at 4 centers and analyzedtogether.35 Grades 2 and 3 acute GVHD developed in 53%and 8% of patients, respectively, and the 2 year incidenceof chronic GVHD was 18%. The 2 year rates of NRM and
relapse were 23% and 28%, respectively, suggesting thatPBSC can be substituted for marrow in HLA-haploidenti-cal HCT. Other strategies to prevent, preempt or treatrelapse include planned donor lymphocyte infusions.36 Amore novel approach includes preemptive infusions ofdonor NK cells. Thirty-six heavily pre-treated patientswith hematologic malignancies, median age of 46 (range8-75) years, were given donor NK cells on day 7 afterHLA-haploidentical HCT.37 Patients had a median timefrom cancer diagnosis to transplant of 2.1 (0.3 – 9.9) years,including 7 patients with prior autologous HCT and 6patients with 1 or more prior allogeneic HCT. Overall andrelapse-free survivals at 1 year of 74% and 69%, and at 2years of 63% and 51% were observed, respectively.
Engraftment kinetics and donor chimerismThe overall goal in malignant disorders is to achieve
high levels of or even complete donor T-cell chimerismearly after HCT, as this has been associated with lowerrisks of graft rejection and relapse.38-40 While completedonor chimerism develops rapidly following myeloabla-tive allogeneic HCT, varying degrees of mixed donor hostchimerism are seen initially following NMA conditioning,though the majority of patients will have full donorchimerism by day 100 after HCT. Many of the RIC regi-mens that are more myelosuppressive have kinetics ofdonor engraftment similar to those of myeloablative regi-mens. In addition to regimen intensity, other factors influ-ence the kinetics of engraftment including the use of PBSCand in vivo T-cell depleting agents (such as ATG or alem-tuzumab) and HLA disparity between donor and recipi-ent. Patients who received myelosuppressive chemothera-py or a preceding autologous HCT had a more rapidengraftment of donor T-cells. An association between high
Nonmyeloablative allogeneic hematopoietic cell transplantation
haematologica | 2016; 101(5) 525
Figure 1. Reproducedfrom: Sandmaier BM,Storb R. Reduced-intensityallogeneic transplantationregimens, Chapter 21, In:Thomas’ HematopoieticCell Transplantation, 5th
Edition. Forman SJ, NegrinRS, Antin JH, andAppelbaum FR, Eds.,©John Wiley & Sons, Ltd.,in press.

levels of donor T-cell chimerism and GVHD has beenobserved using different conditioning regimens.39,40 Whenboth NK and T-cell chimerism were modeled as continu-ous variables, only early donor T-cell chimerism was asso-ciated with acute GVHD, whereas high levels of NKchimerism were significantly associated with lowerrelapse rates but not with increased GVHD.41 A phase IIItrial among patients treated with 2 Gy TBI alone vs. TBIwith fludarabine 90mg/m2 showed that adding fludara-bine contributed to a more rapid T and NK cell chimerismand significantly less relapse (40 % vs. 55%), resulting insuperior survival (60 % vs. 54% at 3 years).42 This support-ed the previous observations of higher donor chimerismbeing protective for relapse.
Toxicities and infectionsHigh-dose conditioning is associated with higher NRM
from organ toxicities and infectious complications. Theformer include hepatic sinusoidal obstruction syndrome/veno-occlusive disease (SOS / VOD) and idio-pathic pneumonia syndrome (IPS). No cases of SOS wereobserved among 193 patients given NMA conditioning.43Acute renal failure (ARF) (defined as a >50% decrease inglomerular filtration rate) occurred less often in patientsgiven NMA HCT compared to myeloablative condition-ing (43% vs. 73%), despite greater age and comorbidities
among NMA recipients.44 A separate multivariate analysisrevealed that ARF during the first 100 days was associatedwith the development of chronic kidney disease (CKD).CKD was defined as at least a 25% reduction in GFR frombaseline. Previous autologous HCT, long-term calcineurininhibitor use and extensive chronic GVHD were inde-pendently associated with CKD. CKD following NMAHCT appears to be a distinct clinical entity and likely notrelated to radiation nephritis.45 Pulmonary function wasevaluated in patients before, at day 100, and 1 year afterHCT.46 Results suggested that, despite having worse pre-transplant lung function, NMA patients experienced lesspulmonary toxicity than myeloablative patients. The inci-dences and outcomes of IPS among NMA (n=183) versusmyeloablative (n=917) patients were compared. Thecumulative incidence of IPS was significantly lower at 120days after NMA conditioning (2.2% vs. 8.4%). IPSoccurred early after transplant, progressed rapidly, and hada high mortality rate (75%) despite aggressive support.These findings support the concept that lung damage fromconditioning regimen plays a crucial role in IPS after HCT.Following NMA conditioning, patients have less cytope-
nias including less neutropenia. Significantly fewer NMArecipients (n=503) required platelet transfusions (25% vs.99%) and red blood cell transfusions (64% vs. 96%) thanmyeloablative (n=1,353) recipients.47 Among the NMA
R. Storb and B.M. Sandmaier
526 haematologica | 2016; 101(5)
Figure 2. Five-year relapse mortality and overall survival of patients with advanced hematologic malignancies who were conditioned with FLU/2 Gy TBI before HLA-matched related or unrelated HCT and post-grafting immunosuppression with MMF/calcineurin inhibitor. Survival is shown depending on relapse risk andhematopoietic comorbidity scores (HCT-CI).
A
C
B
D

patients, platelet and RBC transfusions were less frequentamong related compared to unrelated recipients.Major/bidirectionally ABO-mismatched recipientsrequired more RBC transfusions than ABO-matched recip-ients, though ABO-mismatching did not affect other NMAHCT outcomes. It was also hypothesized that NMA con-ditioning would be associated with less neutropenia afterday 28 following engraftment. However, while NMA con-ditioning had protective effects on anemia and thrombo-cytopenia after day 28 there was no significant reductionof neutropenia either overall or in the context of ganci-clovir use.48 Elderly patients appear to be more prone tocumulative toxicities of post-HCT drug regimens, butNMA conditioning, optimized HLA matching, and higherdoses of CD34+ cell infusions reduced the risk of cytopeniaafter day 28. Multiple studies have shown that the incidence of infec-
tions early after HCT is reduced after RIC and NMA con-ditioning. There is less bacteremia in the first month pre-sumably due to a lesser degree of neutropenias.49 While theincidence of CMV infection is the same in CMV positiverecipients, NMA-HCT was associated with a lower risk ofhigh-grade CMV infection.50
Older donorsAs the age of HCT recipients has increased, the age of
their sibling donors has increased as well. Concern hasbeen raised that increasing donor age might adverselyaffect the functional fitness of hematopoietic cells andthereby impair the marrow recovery after transplantation.Hematopoietic cells are subject to aging mechanisms suchas accumulated DNA damage, telomere shortening, andepigenetic modification. However, studies on the effect ofdonor age on the function of hematopoietic cells haveyielded controversial results, especially the work on stemcell aging in murine model systems. Dutch investigatorscommented on the variable results seen: “the discrepantconclusions of these studies, however, could be partlycaused by (the different) mouse strains used, becausestrain-dependent increases or decreases in primitivehematopoietic cell frequency and function have beenreported.”51 Another concern is related to the longevity ofhematopoietic stem cells which makes them ideal targetsfor mutagenic changes.52 The theoretical possibility wasraised that recipients of aged stem cells might be at anincreased risk of developing malignant clonal disorders.Published clinical results on the effects of aging on stem
cells also vary. An NMPD study from 2001 reported infe-rior survival among patients given grafts from donorsolder than 45 years.53 A French study initially saw no sig-nificant impact of donor age among MDS and AMLpatients undergoing transplantation.54 In contrast, a lateranalysis by the French group found that donor age ≥60years had a significant adverse impact on overall recipientsurvival.55 A CIBMTR analysis from 2013 reported thatoutcomes were superior in recipients of grafts from HLA-identical sibling donors >50 years old compared to thosewith grafts from HLA-matched unrelated donors <50years of age.56 We analyzed the effects of donor age on thespeed of hematopoietic engraftment and donorchimerism, acute and chronic GVHD, and NRM among1,174 patients undergoing myeloablative and 367 patientsundergoing NMA conditioning before HLA-matched relat-ed or unrelated HCT.57 CD34 cell harvests were reduced inolder (60-82 years) donors (median 5.6 × 106 cells/kg) com-
pared to younger (<60 years) donors (median 7.7 × 106cells/kg). However, sustained engraftment rates amongrecipients with older and younger donors were compara-ble. Sustained grafts were seen in 97% and 98% ofpatients given myeloablative and NMA conditioning,respectively, who had younger donors, and 90% and100%, respectively, for those who had older donors. Alsothe tempo of neutrophil and platelet recoveries and donorchimerism did not show significant differences, except foran average 1.3-day delay in neutrophil recovery amongmyeloablative patients with older donors (P=0.04).Moreover, aged stem cells did not convey an increased riskof donor-derived clonal disorders after HCT since nonewere seen. Both myeloablative and NMA recipients witholder sibling donors had significantly less grade 2–4 acuteGVHD compared to recipients with grafts from younger
Nonmyeloablative allogeneic hematopoietic cell transplantation
haematologica | 2016; 101(5) 527
Figure 3. Overall survival. (A) The probability of OS by donor type after myeloab-lative conditioning regimen, adjusted for age and disease risk index. (B) Theprobability of OS by donor type after reduced intensity conditioning regimen,adjusted for disease risk index and secondary AML. (Originally published inBlood. Ciurea SO, Zhang MJ, Bacigalupo AA, et al. Haploidentical transplantwith posttransplant cyclophosphamide vs. matched unrelated donor transplantfor acute myeloid leukemia. Blood. 2015;126(8):1033-1040. ©The AmericanSociety of Hematology).
A
B

unrelated donors. Rates of grade 3 and 4 acute GVHD,chronic GVHD, and NRM among recipients with olderdonors were not significantly different from those seen inrecipients with younger donors. We concluded from thissingle-center study that grafts from donors ≥60 years ofage did not adversely affect outcomes of HCT comparedto grafts from younger donors <60 years of age.
RelapseRelapse or progression of the underlying malignancy has
remained the principal cause of failure of allogeneic HCT.This has been especially true in patients who for reasonsof age or comorbidities have been conditioned with NMAregimens, where cure of malignancy depends almostentirely on GVT effects. The following sections will dis-cuss outcomes with a minimal-intensity conditioning reg-imen and use the results as a basis for proposing ways toreduce relapse or progression.We maintained the NMA FLU/low-dose TBI platform
for patients with advanced hematologic malignanciesbecause most of our patients either did not need or wouldnot tolerate higher dose regimens, and because the regi-men best defines the limits of GVT effects.15,58 PowerfulGVT effects were seen across all disease stages except forALL in CR2 and beyond, where all patients progressed. Asshown in Figure 2, between 45% and 75% of patientsexperienced sustained remissions depending on the natureand stage of the underlying malignancy. Overall 5-yearrelapse mortality was 34.5%. Seventy percent of relapseor progression occurred in year 1 and much of the remain-der in year 2 after HCT. We hypothesize that early diseaserelapse or progression was due to blunted GVT effectsfrom early post-transplantation immune compromise.Later, as the donor immune system was being built up andimmunosuppressive drugs tapered and then discontinued,the “brakes were taken off” the immune cells, enablingGVT effects. This hypothesis is indirectly supported byformer extensive immune function studies showingrecovery of antibody responses to neoantigens, such asbacteriophage fX174 and keyhole limpet hemocyanin,among others, as well as cellular immunity within 1-3years after HCT.59 Consistent with this hypothesis, relapserates in most diseases were markedly reduced in years 3-5. The options for decreasing the still existing relapse or
progression risk are limited. Increasing the intensity, andthereby the toxicity, of the conditioning regimen may beproblematic for at least two reasons. One is that a majorityof patients did not relapse and, therefore, would beexposed to unnecessary toxicity. The other is that mostpatients were elderly and/or had comorbidities which pre-empted dose escalation. Also, more than one-fifth ofpatients had failed preceding high-dose HCT and anotherone-fifth had planned autologous HCT, and receivinganother high-dose HCT regimen might be too toxic.Given these limitations, we envision two principalapproaches for reducing the risk of relapse or progressionin elderly or medically infirm patients. One approach is based on the hypothesis that delaying
disease relapse or progression until the grafted immunesystem is recovered sufficiently to generate GVT effectswould increase cure rates. Such a delay would be accom-plished with well-tolerated drugs or antibody-drug conju-gates which, even though not curative on their own,would pave the way for curative GVT effects. An example
of such an approach has been the treatment of patientswith Ph1+ ALL in first remission with a tyrosine kinaseinhibitor for one year after HCT.60 The overall 5-year sur-vival rate was 69% and 85% in the subgroup withoutMRD before HCT, which is impressively better than pre-vious results without tyrosine kinase inhibitors.Candidate agents for patients with other malignanciesinclude antibodies to CD20 (NHL) and CD30 (Hodgkinlymphoma), proteosome inhibitors (MM), and the FLT3inhibitors (AML). A second approach would be to reduce the tumor bur-
den before HCT. One way to accomplish this is throughthe use of chimeric antigen receptor (CAR) T-cells inpatients who have B cell lymphoid malignancies express-ing CD19.61,62 Another way is to increase the pre-trans-plant tumor cell kill by low-toxicity, targeted radiationtherapy using a mAb to CD45 coupled to radionuclidesused in addition to the basic FLU/2 Gy TBI regimen. Onepreliminary study summarized early results in 58 patientswith advanced AML or high-risk MDS who were older
Figure 4. Graft-versus-host disease and use of systemic steroids. (A) Cumulativeincidence of use of systemic steroids in arm 1 (n=69), arm 2 (n=71) and arm 3(n=68). (B) Viral infections. Cumulative incidence of cytomegalovirus reactiva-tion in arm 1 (n=69), arm 2 (n=71) and arm 3 (n=68). Originally published inHaematologica (Kornblit B, et al. A randomized phase II trial of tacrolimus,mycophenolate mofetil and sirolimus after non-myeloablative unrelated donortransplantation. Haematologica 2014; 99(10): 1624-1631. ©2014 FerrataStorti Foundation).
A
B
haematologica | 2016; 101(5)528
R. Storb and B.M. Sandmaier

References
1. Yu C, Storb R, Mathey B, et al. DLA-identi-cal bone marrow grafts after low-dose totalbody irradiation: Effects of high-dose corti-costeroids and cyclosporine on engraftment.Blood. 1995;86(11):4376-4381.
2. Storb R, Yu C, Wagner JL, et al. Stablemixed hematopoietic chimerism in DLA-identical littermate dogs given sublethaltotal body irradiation before and pharmaco-logical immunosuppression after marrowtransplantation. Blood. 1997;89(8):3048-3054.
3. Hogan WJ, Little M-T, Zellmer E, et al.Postgrafting immunosuppression withsirolimus and cyclosporine facilitates stablemixed hematopoietic chimerism in dogsgiven sublethal total body irradiation beforemarrow transplantation from DLA-identicallittermates. Biol Blood Marrow Transplant.2003;9(8):489-495.
4. Storb R, Yu C, Barnett T, et al. Stable mixedhematopoietic chimerism in dog leukocyteantigen-identical littermate dogs givenlymph node irradiation before and pharma-cologic immunosuppression after marrowtransplantation. Blood. 1999;94(3):1131-1136.
5. Appelbaum FR, Brown P, Sandmaier B, et al.Antibody-radionuclide conjugates as part ofa myeloblative preparative regimen for mar-row transplantation. Blood. 1989;73(8):2202-2208.
6. Pagel JM, Gooley TA, Rajendran J, et al.Allogeneic hematopoietic cell transplanta-tion after conditioning with 131I-anti-CD45antibody plus fludarabine and low-dosetotal body irradiation for elderly patientswith advanced acute myeloid leukemia orhigh-risk myelodysplastic syndrome. Blood.2009;114(27):5444-5453.
7. Gopal AK, Guthrie KA, Rajendran J, et al.90Y-Ibritumomab tiuxetan, fludarabine, andTBI-based nonmyeloablative allogeneic
transplantation conditioning for patientswith persistent high-risk B-cell lymphoma.Blood. 2011;118(4):1132-1139.
8. Sandmaier BM, Bethge WA, Wilbur DS, etal. Bismuth 213-labeled anti-CD45 radioim-munoconjugate to condition dogs for non-myeloablative allogeneic marrow grafts.Blood. 2002;100(1):318-326.
9. Chen Y, Kornblit B, Hamlin DK, et al.Durable donor engraftment after radioim-munotherapy using �-emitter astatine-211-labeled anti-CD45 antibody for conditioningin allogeneic hematopoietic cell transplanta-tion. Blood. 2012;119(5):1130-1138.
10. Nakamae H, Wilbur DS, Hamlin DK, et al.Biodistribution, myelosuppression, and tox-icities in mice treated with an anti-CD45antibody labeled with the �-emittingradionuclides bismuth-213 or astatine-211.Cancer Res. 2009;69(6):2408-2415.
11. Eto M, Mayumi H, Tomita Y, et al. Specificdestruction of host-reactive mature T cells ofdonor origin prevents graft-versus-host dis-ease in cyclophosphamide-induced tolerantmice. J Immunol. 1991;146(5):1402-1409.
12. Luznik L, Engstrom LW, Iannone R, Fuchs EJ.Posttransplantation cyclophosphamide facil-itates engraftment of major histocompatibil-ity complex-identical allogeneic marrow inmice conditioned with low-dose total bodyirradiation. Biol Blood Marrow Transplant.2002;8(3):131-138.
13. Luznik L, O'Donnell PV, Fuchs EJ. Post-transplantation cyclophosphamide for toler-ance induction in HLA-haploidentical bonemarrow transplantation. Semin Oncol.2012;39(6):683-693.
14. Luznik L, O'Donnell PV, Symons HJ, et al.HLA-haploidentical bone marrow transplan-tation for hematologic malignancies usingnonmyeloablative conditioning and high-dose, post-transplantation cyclophos-phamide. Biol Blood Marrow Transplant.2008;14(28):641-650.
15. Storb R, Gyurkocza B, Storer BE, et al.Graft-versus-host disease and graft-versus-
tumor effects after allogeneic hematopoieticcell transplantation. J Clin Oncol. 2013;31(12):1530-1538.
16. Kornblit B, Maloney DG, Storer BE, et al. Arandomized phase II trial of tacrolimus,mycophenolate mofetil and sirolimus afternonmyeloablative unrelated donor trans-plantation. Haematologica. 2014;99(10):1624-1631.
17. Giralt S, Logan B, Rizzo D, et al. Reduced-intensity conditioning for unrelated donorprogenitor cell transplantation: long-termfollow-up of the first 285 reported to theNational Marrow Donor Program. BiolBlood Marrow Transplant. 2007;13(7):844-852.
18. Pulsipher MA, Chitphakdithai P, Logan BR,et al. Donor, recipient, and transplant char-acteristics as risk factors after unrelateddonor PBSC transplantation: beneficialeffects of higher CD34+ cell dose. Blood.2009;114(13):2606-2616.
19. Michallet M, Le QH, Mohty M, et al.Predictive factors for outcomes after reducedintensity conditioning hematopoietic stemcell transplantation for hematological malig-nancies: a 10-year retrospective analysisfrom the Societe Francaise de Greffe deMoelle et de Therapie Cellulaire. ExpHematol. 2008;36(5):535-544.
20. Ho VT, Kim HT, Aldridge J, et al. Use ofmatched unrelated donors compared withmatched related donors is associated withlower relapse and superior progression-freesurvival after reduced-intensity conditioninghematopoietic stem cell transplantation. BiolBlood Marrow Transplant. 2011;17(8): 1196-1204.
21. Soiffer RJ, Lerademacher J, Ho V, et al.Impact of immune modulation with anti-T-cell antibodies on the outcome of reduced-intensity allogeneic hematopoietic stem celltransplantation for hematologic malignan-cies. Blood. 2011;117(25):6963-6970.
22. Blaise D, Farnault L, Faucher C, et al.Reduced-intensity conditioning with
529haematologica | 2016; 101(5)
than 50 years and treated with the anti-CD45 mAb cou-pled to 131I.6 One-year survival was 41%. Another studyadded 90Y coupled to an anti-CD20 mAb to FLU/2 Gy TBIin 40 patients with persistent, high-risk NHL. The estimat-ed 30-month progression-free survival was 51%.7 Severalproperties of the beta-emitting radionuclides 131I and 90Ylimit their effectiveness including their long half-lives of2.5 and 8 days, their relatively low energy of 0.7 and 2.3MeV, and their long path lengths of 0.8-11.3 mm, respec-tively, which result in off-target effects. Additionally, 131Iemits weak gamma radiation during its decay whichnecessitates placing patients in isolation rooms for severaldays. To get around these limitations, we have focusedour attention on an alpha-emitting radionuclide, 211At,which has a half-life of 7.2 hours, high energy (5.9 MeV),and a path length of only 0.04-0.06 mm. This results in theunique ability of killing mAb-targeted cells while causingminimal damage to surrounding tissues. Moreover, thealpha particles cause multiple strand breaks, hence DNArepair mechanisms are inhibited, which reduces the risk ofsecondary cancer. An additional advantage of 211At is thatit is relatively cheap compared to other alternatives. Ourfirst clinical protocol has been firmly based on 15 years of
experience with alpha-emitting radionuclides in a canineallogeneic HCT model.
Conclusions
Allogeneic HCT after RIC or MMA regimens to treatolder or medically infirm patients with advanced hemato-logical malignancies is feasible and effective. This isenabled in large part by GVT effects, and results in curesof appreciable numbers of malignancies. Increasing dis-ease control and decreasing NRM, the latter mostly asso-ciated with or preceded by GVHD, will need to beaddressed in future trials.
FundingThe authors are grateful for research funding from the National
Institutes of Health, Bethesda, MD, USA, grants CA078902,CA018029, CA015704 and HL122173. Further support camefrom the Laura Landro Salomon Endowment Fund. The contentis solely the responsibility of the authors and does not necessarilyrepresent the official views of the National Institutes of Health norits subsidiary Institutes and Centers.
Nonmyeloablative allogeneic hematopoietic cell transplantation

Fludarabin, oral Busulfan, and thymoglobu-lin allows long-term disease control and lowtransplant-related mortality in patients withhematological malignancies. Exp Hematol.2010;38(12):1241-1250.
23. Luger SM, Ringdén O, Zhang M-J, et al.Similar outcomes using myeloablative vsreduced-intensity allogeneic transplantpreparative regimens for AML or MDS.Bone Marrow Transplant. 2012;47(2):203-211.
24. Belkacemi Y, Labopin M, Hennequin C, etal. Reduced-intensity conditioning regimenusing low-dose total body irradiation beforeallogeneic transplant for hematologic malig-nancies: Experience from the EuropeanGroup for Blood and MarrowTransplantation. Int J Radiat Oncol BiolPhys. 2007;67(2):544-551.
25. Scott BL, Pasquini MC, Logan B, et al.Results of a phase III randomized, multi-cen-ter study of allogeneic stem cell transplanta-tion after high versus reduced intensity con-ditioning in patients with myelodysplasticsyndrome (MDS) or acute myeloid leukemia(AML): Blood and Marrow TransplantClinical Trials Network (BMT CTN) 0901.Blood. 2015;126(23):LBA-8.26. Gragert L, Eapen M, Williams E, etal.HLA match likelihoods for hematopoieticstem-cell grafts in the U.S. registry. N Engl JMed. 2014;371(4):339-348.
27. Verneris MR, Lee SJ, Ahn KW, et al. HLAmismatch Is associated with worse out-comes after unrelated donor reduced-Intensity conditioning hematopoietic celltransplantation: an analysis from the centerfor international blood and marrow trans-plant research. Biol Blood MarrowTransplant. 2015;21(10):1783-1789.
28. Nakamae H, Storer BE, Storb R, et al. Low-dose total body irradiation and fludarabineconditioning for HLA class I-mismatcheddonor stem cell transplantation andimmunologic recovery in patients withhematologic malignancies: a multicentertrial. Biol Blood Marrow Transplant.2010;16(3):384-394.
29. Brunstein CG, Fuchs EJ, Carter SL, et al.Alternative donor transplantation afterreduced intensity conditioning: results ofparallel phase 2 trials using partially HLA-mismatched related bone marrow or unre-lated umbilical cord blood grafts. Blood.2011;118(2):282-288.
30. Passweg JR, Baldomero H, Bader P, et al.Hematopoietic SCT in Europe 2013: recenttrends in the use of alternative donors show-ing more haploidentical donors but fewercord blood transplants. Bone MarrowTransplant. 2015;50(4):476-482.
31. Ciurea SO, Zhang MJ, Bacigalupo AA, et al.Haploidentical transplant with posttrans-plant cyclophosphamide vs matched unre-lated donor transplant for acute myeloidleukemia. Blood. 2015;126(8):1033-1040.
32. Sayer HG, Kröger M, Beyer J, et al. Reducedintensity conditioning for allogeneichematopoietic stem cell transplantation inpatients with acute myeloid leukemia: dis-ease status by marrow blasts is the strongestprognostic factor. Bone Marrow Transplant.2003;31(12):1089-1095.
33. Armand P, Kim HT, Logan BR, et al.Validation and refinement of the DiseaseRisk Index for allogeneic stem cell transplan-tation. Blood. 2014;123(23):3664-3671.
34. McCurdy SR, Kanakry JA, Showel MM, etal. Risk-stratified outcomes of nonmyeloab-lative HLA-haploidentical BMT with high-dose posttransplantation cyclophos-phamide. Blood. 2015;125(19):3024-3031.
35. Jaiswal SR, Chakrabarti A, Chatterjee S, etal. Haploidentical peripheral blood stem celltransplantation with post-transplantationcyclophosphamide in children withadvanced acute leukemia with a fludarabine,busulfan and melphalan based conditioning.Biol Blood Marrow Transplant. 2015pii:S1083-8791(15)00737-5.
36. Ghiso A, Raiola AM, Gualandi F, et al. DLIafter haploidentical BMT with post-trans-plant CY. Bone Marrow Transplant.2015;50(1):56-61.
37. Thakar M, Hari PN, Keever-Taylor CA, et al.Donor natural killer (NK) cell immunothera-py following non-myeloablative HLA-hap-loidentical hematopoietic cell transplanta-tion (HCT) improves relapse and progres-sion free survival (PFS) in patients withhematologic malignancies [abstract]. BiolBlood Marrow Transplant. 2016 (epub).
38. McSweeney PA, Niederwieser D, ShizuruJA, et al. Hematopoietic cell transplantationin older patients with hematologic malig-nancies: replacing high-dose cytotoxic thera-py with graft-versus-tumor effects. Blood.2001;97(11):3390-3400.
39. Childs R, Clave E, Contentin N, et al.Engraftment kinetics after nonmyeloablativeallogeneic peripheral blood stem cell trans-plantation: full donor T-cell chimerism pre-cedes alloimmune responses. Blood.1999;94(9):3234-3241.
40. Baron F, Baker JE, Storb R, et al. Kinetics ofengraftment in patients with hematologicmalignancies given allogeneic hematopoieticcell transplantation after nonmyeloablativeconditioning. Blood. 2004;104(8):2254-2262.
41. Baron F, Petersdorf EW, Gooley T, et al.What is the role for donor natural killer cellsafter nonmyeolablative conditioning? BiolBlood Marrow Transplant. 2009;15(5):580-588.
42. Kornblit B, Maloney DG, Storb R, et al.Fludarabine and 2 Gy TBI is superior to 2 GyTBI as conditioning for HLA-matched relat-ed hematopoietic cell transplantation: aphase III randomized trial. Biol BloodMarrow Transplant. 2013;19(9):1340-1347.
43. Hogan WJ, Maris M, Storer B, et al. Hepaticinjury after nonmyeloablative conditioningfollowed by allogeneic hematopoietic celltransplantation: a study of 193 patients.Blood. 2004;103(1):78-84.
44. Parikh CR, Schrier RW, Storer B, et al.Comparison of ARF after myeloablative andnonmyeloablative hematopoietic cell trans-plantation. American Journal of KidneyDiseases. 2005;45(3):502-509.
45. Weiss AS, Sandmaier BM, Storer B, Storb R,McSweeney PA, Parikh CR. Chronic kidneydisease following nonmyeloablativehematopoietic cell transplantation. Am JTransplant. 2006;6(1):89-94.
46. Fukuda T, Hackman RC, Guthrie KA, et al.Risks and outcomes of idiopathic pneumo-nia syndrome after nonmyeloablative andconventional conditioning regimens for allo-geneic hematopoietic stem cell transplanta-tion. Blood. 2003;102(8):2777-2785.
47. Wang Z, Sorror ML, Leisenring W, et al. Theimpact of donor type and ABO incompati-bility on transfusion requirements after non-myeloablative hematopoietic cell transplan-tation. Br J Haematol. 2010;149(1):101-110.
48. Nakamae H, Storer B, Sandmaier BM, et al.Cytopenias after day 28 in allogeneichematopoietic cell transplantation: impactof recipient/donor factors, transplant condi-tions and myelotoxic drugs. Haematologica.2011;96(12):1838-1846.
49. Junghanss C, Marr KA, Carter RA, et al.Incidence and outcome of bacterial and fun-
gal infections following nonmyeloablativecompared with myeloablative allogeneichematopoietic stem cell transplantation: amatched control study. Biol Blood MarrowTransplant. 2002;8(9):512-520.
50. Nakamae H, Kirby KA, Sandmaier BM, et al.Effect of conditioning regimen intensity onCMV infection in allogeneic hematopoieticcell transplantation. Biol Blood MarrowTransplant. 2009;15(6):694-703.
51. de Haan G, Nijhof W, Van Zant G. Mousestrain-dependent changes in frequency andproliferation of hematopoietic stem cellsduring aging: correlation between lifespanand cycling activity. Blood. 1997;89(5):1543-1550.
52. Rossi DJ, Jamieson CH, Weissman IL. Stemscells and the pathways to aging and cancer(Review). Cell. 2008;132(4):681-696.
53. Kollman C, Howe CWS, Anasetti C, et al.Donor characteristics as risk factors in recip-ients after transplantation of bone marrowfrom unrelated donors: the effect of donorage. Blood. 2001;98(7):2043-2051.
54. Robin M, Porcher R, Ades L, et al. Matchedunrelated or matched sibling donors result incomparable outcomes after non-myeloabla-tive HSCT in patients with AML or MDS.Bone Marrow Transplant. 2013; 48(10):1296-1301.
55. Peffault de Latour R, Brunstein CG, PorcherR, et al. Similar overall survival using sibling,unrelated donor, and cord blood grafts afterreduced-intensity conditioning for olderpatients with acute myelogenous leukemia.Biol Blood Marrow Transplant. 2013;19(9):1355-1360.
56. Alousi AM, Le-Rademacher J, Saliba RM, etal. Who is the better donor for olderhematopoietic transplant recipients: anolder-aged sibling or a young, matched unre-lated volunteer? Blood. 2013;121(13):2567-2573.
57. Rezvani AR, Storer BE, Guthrie KA, et al.Impact of donor age on outcome after allo-geneic hematopoietic cell transplantation.Biol Blood Marrow Transplant. 2015;21(1):105-112.
58. Storb R, Gyurkocza B, Storer BE, et al.Allogeneic hematopoietic cell transplanta-tion following minimal intensity condition-ing: predicting acute graft-versus-host dis-ease and graft-versus-tumor effects. BiolBlood Marrow Transplant. 2013;19(5):792-798.
59. Witherspoon RP, Storb R, Ochs HD, et al.Recovery of antibody production in humanallogeneic marrow graft recipients:Influence of time posttransplantation, thepresence or absence of chronic graft-versus-host disease, and antithymocyte globulintreatment. Blood. 1981;58(2):360-368.
60. Ram R, Storb R, Sandmaier BM, et al. Non-myeloablative conditioning with allogeneichematopoietic cell transplantation for thetreatment of high-risk acute lymphoblasticleukemia. Haematologica. 2011;96(8):1113-1120.
61. Davila ML, Riviere I, Wang X, et al. Efficacyand toxicity management of 19-28z CAR Tcell therapy in B cell acute lymphoblasticleukemia. Science Transl Med. 2014;6(224):224ra25.
62. Turtle CJ, Berger C, Sommermeyer D, et al.Anti-CD19 chimeric antigen receptor-modi-fied T cell therapy for B cell non-Hodgkinlymphoma and chronic lymphocyticleukemia: Fludarabine and cyclophos-phamide lymphodepletion improves in vivoexpansion and persistence of CAR-T cellsand clinical outcomes [abstract]. Blood.2015;126(23):184.
R. Storb and B.M. Sandmaier
530 haematologica | 2016; 101(5)

haematologica | 2016; 101(5) 531
Received: December 10, 2015.
Accepted: January 28, 2016.
©2016 Ferrata Storti Foundation
Check the online version for the most updatedinformation on this article, online supplements,and information on authorship & disclosures:www.haematologica.org/content/101/5/531
Material published in Haematologica is cov-ered by copyright. All rights reserved to theFerrata Storti Foundation. Copies of articlesare allowed for personal or internal use.Permission in writing from the publisher isrequired for any other use.
Correspondence: [email protected]
Role of the tumor microenvironment in matureB-cell lymphoid malignanciesNathan H. Fowler,1 Chan Yoon Cheah,1,2,3 Randy D. Gascoyne,4 John Gribben,5Sattva S. Neelapu,1 Paolo Ghia,6,7 Catherine Bollard,8 Stephen Ansell,9 MichaelCurran,1 Wyndham H. Wilson,10 Susan O’Brien,11 Cliona Grant,12 RichardLittle,13 Thorsten Zenz,14 Loretta J. Nastoupil,1 and Kieron Dunleavy10
1Department of Lymphoma/Myeloma, The University of Texas MD Anderson Cancer Center,Houston, TX, USA; 2Department of Haematology, Pathwest Laboratory Medicine WA and SirCharles Gairdner Hospital, Perth, Western Australia; 3University of Western Australia, Perth;4British Columbia Cancer Research Centre, Vancouver, British Columbia, Canada;5Department of Haemato-Oncology, Barts Cancer Institute, London, UK; 6Università Vita-Salute San Raffaele, Division of Experimental Oncology, IRCCS Istituto Scientifico SanRaffaele, Milan, Italy; 7Department of Onco-Hematology, Ospedale San Raffaele, Milan,Italy; 8Children’s Research Institute, Washington, DC, USA; 9Division of Hematology, MayoClinic, Rochester, MN, USA; 10Lymphoid Malignancies Branch, National Cancer Institute,Bethesda, MD, USA; 11University of California, Irvine, CA, USA; 12St. James’ Hospital, Dublin,Ireland; 13Cancer Therapeutic Evaluation Program, National Cancer Institute, Bethesda,MD, USA; and 14University of Heidelberg, Germany
Ferrata StortiFoundation
EUROPEANHEMATOLOGYASSOCIATION
Haematologica 2016Volume 101(5):531-540
REVIEW ARTICLE
doi:10.3324/haematol.2015.139493
The tumor microenvironment is the cellular and molecular environ-ment in which the tumor exists and with which it continuouslyinteracts. In B-cell lymphomas, this microenvironment is intriguing
in that it plays critical roles in the regulation of tumor cell survival and pro-liferation, fostering immune escape as well as the development of treat-ment resistance. The purpose of this review is to summarize the proceed-ings of the Second Annual Summit on the Immune Microenvironment inHematologic Malignancies that took place on September 11-12, 2014 inDublin, Ireland. We provide a timely overview of the composition andbiological relevance of the cellular and molecular microenvironment inter-face and discuss the role of interactions between the microenvironmentand neoplastic cells in a variety of B-cell lymphomas. In addition, wefocus on various novel therapeutic strategies that target the tumormicroenvironment, including agents that modulate B-cell receptor path-ways and immune-checkpoints, chimeric antigen receptor T cells andimmunomodulatory agents.
ABSTRACT
Introduction
Recent advances in the understanding of the pathogenesis of hematologic malig-nancies have focused attention on the role of the tumor microenvironment. In B-cell lymphomas, the cellular infiltrate intimately associated with the malignantlymphocytes, and the molecules that can be released or trapped within it, may aidtumor cell proliferation and survival as well as escape from immunosurveillance.1
Recognition of the microenvironment’s importance has paved the way for thedevelopment of exciting novel strategies that target the microenvironment and itsinteractions with neoplastic cells. In particular, drugs targeting B-cell receptor (BCR)signaling and programmed death-1 (PD-1) pathways as well as chimeric antigenreceptor (CAR) T-cell therapy represent promising advances in lymphoma treat-ment. The purpose of this review is to summarize the proceedings of the SecondAnnual Summit on the Role of the Immune Microenvironment in B-cellLymphomas that took place in Dublin, Ireland on September 11-12, 2014. Themanuscript reflects the meeting’s structure: the first half is devoted to an overviewof the tumor microenvironment in various lymphoma subtypes, and the remainingis a discussion of novel therapeutic approaches targeting the tumor microenviron-

ment and practical aspects concerning the design and con-duct of studies evaluating these agents.
Overview of the microenvironment in B-cellmalignancies
The tumor microenvironment of B-cell lymphomas ishighly variable with regards to both spatial arrangementand composition of cells, including immune and inflam-matory cells, blood and lymphatic vascular networks andthe extracellular matrix. The cellular composition of themicroenvironment generally mirrors that of the normaltissue at the site of development, the exception being clas-sical Hodgkin lymphoma (see below). Tumor cells retain adegree of dependence on interactions with non-malignantcells and stromal elements of the tumor microenviron-ment for survival and proliferation.2 However, tumor cellsalso use these interactions to generate immunosuppressivemechanisms that promote tumor escape from immunesurveillance and lead to disease progression.2-4 Increasingdata indicate a critical role for the tumor microenviron-ment in mediating treatment resistance.5 The cellular com-position and spatial characteristics of the microenviron-ment demonstrate significant heterogeneity depending ona number of factors, including the lymphoma subtype.Scott and Gascoyne have proposed three major modelsthat divide up the broad range of tumor microenviron-ments described in B-cell lymphomas (Figure 1).2 The first,re-education, is typified by follicular lymphoma (FL), inwhich malignant cells retain dependence on the microen-vironment for survival and proliferation signals; the sec-ond, recruitment, is observed in classical Hodgkin lym-phoma (cHL) in which the infrequent Reed-Sternberg cellsare surrounded by an extensive support milieu of non-malignant cells that is distinct from the composition ofnormal lymphoid tissue; the third, effacement, is seen inBurkitt lymphoma (BL) and to some extent in diffuse largeB-cell lymphoma (DLBCL), whereby genetic aberrations,such as translocation of MYC, within the malignant celllead to autonomous, microenvironment-independentgrowth and survival.2 These tumors rely little on themicroenvironment, which is sparse when compared to themicroenvironment in cHL. Thus, the extent to which dif-ferent histological subtypes of lymphoid malignancy aresusceptible to agents targeting the immune microenviron-ment is likely to vary depending on the degree to whichthe tumor cells are dependent on external stimuli forgrowth or proliferation. In the following section, we pro-vide an overview of the current understanding of thestructure, composition and function of the tumor microen-vironment in B-cell lymphomas and chronic lymphocyticleukemia (CLL).
Aggressive lymphomas
Diffuse large B-cell lymphoma DLBCL is the most common type of non-Hodgkin lym-
phoma and is recognized as a heterogeneous disease withdistinct molecular subtypes that are derived from differentstages of B-cell differentiation.6,7 Alizadeh et al. firstdescribed gene expression profiling to define distinct sub-types of DLBCL: activated B cells and germinal center Bcells.6 Seminal work by the Leukemia/LymphomaMolecular Profiling Project further described two stromal
signatures (termed stromal-1 and -2) in the tumormicroenvironment, present in both activated and germinalcenter subtypes, which were predictive of outcome.8Although key genetic lesions may explain some of thisdisparity, other factors, such as the microenvironment,likely play an important role. The contribution of thetumor microenvironment to the pathogenesis and tumorsurvival of DLBCL is poorly understood; however, severalrecent studies have yielded intriguing findings and shedsome light on the microenvironment’s possible roles. Onerecent study in DLBCL demonstrated that 29% of caseshave mutations or deletions resulting in inactivation of theβ2-microglobulin gene (B2M) and 21% feature inactiva-tions in the CD58 gene (CD58), two molecules that arecritically involved in the immune recognition of tumorcells by circulating T-lymphocytes and natural killer (NK)cells, respectively.9 The immune escape from these impor-tant immune cells (circulating T-lymphocytes and NKcells) implicates the evasion of immune recognition asplaying an important role in the pathogenesis of DLBCL.Thus, in the majority of cases of DLBCL these two genealterations may be co-selected during lymphomagenesisto avoid cytotoxic circulating T-lymphocytes and NK cells.Many studies have looked at the role of PD-1 and PD-
L1, which are expressed in many aggressive B-cell lym-phomas and have also been associated with mechanismsof immune evasion.3,10-12 The MHC class II transactivatorCIITA is commonly fused to PD-L1 and PD-L2, which canresult in a decrease in HLA-DR expression.10 A study bySteidl et al. looked at rearrangements of CIITA in B-celllymphomas;10 combined with PD-L1 copy number gainsand translocations independent of CIITA, this fusionresulted in T-cell exhaustion and immune escape. In addi-tion, translocations and copy-number gains of PD-L1/2appear to be a dominant mechanism of immune escape inprimary mediastinal B-cell lymphoma (PMBL).13-15 Kiyasuet al. studied 1253 DLBCL biopsies and found tumor cell,but not microenvironmental, expression of PD-L1 wasassociated with adverse overall survival, a difference thatwas present even among the subgroup of patients treatedwith R-CHOP or similar regimens.16 Tumor PD-L1 expres-sion was significantly associated with non-germinal centerB-cell phenotype. Other studies have investigated the role of chemokines
and cytokines such as CCL22, CCL17, GAL-1 and TGF-βvis-à-vis how they recruit and/or retain immunosuppres-sive cells such as M2 macrophages, regulatory T cells(Tregs), and exhausted T cells, and in that way contribute tothe pathogenesis of B-cell lymphomas.2,17,18 Riihijarvi et al.found that both CD68 mRNA levels and CD68+ tumor-associated macrophages, detected by immunohistochem-istry, were adverse prognostic factors for overall survivalamong patients treated uniformly with chemotherapy in aprospective clinical trial.19 In contrast, among patientstreated with chemo-immunotherapy, the impact of CD68+tumor-associated macrophages was reversed, such thatpatients with high CD68+ tumor-associated macrophageshad improved overall survival. This interesting observa-tion led the authors to speculate that rituximab may alterthe function of tumor-associated macrophages from hav-ing a pro-survival effect to an anti-tumor one.
Mantle cell lymphomaThe molecular hallmark of mantle cell lymphoma
(MCL) is the t(11;14) translocation, which results in con-
N.H. Fowler et al.
532 haematologica | 2016; 101(5)

stitutive expression of cyclin D1, leading to cell cyclederegulation. However, extrinsic microenvironment-derived signals also play a role in the pathogenesis of thisdisease.20 MCL is biologically characterized by a tendencytoward extranodal dissemination, mediated by attractionand retention through a highly regulated process involvingchemokine gradients and adhesion molecules such asVLA-4, CCR7, CXCR5 and CXCR4.21 Through this mech-anism, MCL cells interact with stromal cells such asfibroblasts and macrophages. Adhesion to stromal ele-ments is an important mechanism of chemoresistance,and is likely a reason for the incurability of patients fol-lowing chemotherapy.22 Another means by which MCLcells are protected from chemotherapy is through inter-leukin (IL)-6 secretion, which may be secreted by the MCLcells themselves or by bone marrow stromal cells.23 IL-6activates the JAK/STAT3 and PI3K/Akt pathways, knownto be key regulators of MCL growth and survival. Relative to other lymphoma subtypes, the precise com-
position of the MCL tumor microenvironment is not wellcharacterized. Macrophages have been described in MCLalthough, in contrast to FL and cHL, systematic evaluationof their prognostic or pathogenic implications is lacking.24Studies in small series have suggested that increased num-bers of macrophages are associated with aggressive clini-cal behavior.25,26 Two studies indicate that MCL cellsinduce microenvironmental changes to evade the hostimmune response. Firstly, intratumoral biopsies showedthat CD4+CD25+Foxp3+ Tregs are present in MCL, wherethey likely contribute to a reduction of anti-tumor cyto-toxicity.18 Secondly, PD-L1 (B7-H1) was shown to beexpressed by MCL cell lines, in which it resulted inimpaired T-cell proliferation after tumor exposure, inhibit-ed specific anti-tumor T-cell responses and impaired T-cell-mediated tumor cell killing.27 The negative PI3K regu-lator PTEN is often inactivated by phosphorylation inMCL.28 This, along with antigenic stimulation via the BCR,resulted in constitutive activation of Syk, Btk and PI3k-Akt, which are critical in MCL disease progression andmaintenance.29 Inhibition of Syk and Btk has been shownto inhibit BCR-mediated adhesion of MCL to bone mar-row stromal cells and to increase apoptosis.30
Hodgkin lymphomaThe tumor microenvironment in cHL has been exten-
sively studied, with four variant morphological patternsdescribed: nodular sclerosing, mixed cellularity, lympho-cyte-rich and lymphocyte-depleted. Neoplastic HodgkinReed-Sternberg (HRS) cells account for <5% of the tumor,with the remaining cells comprising B and T cells,eosinophils, neutrophils, mast cells, fibroblasts andmacrophages.31 These cells are attracted by chemokinessecreted by HRS cells such as CCL17 (TARC) andCCL12.32,33 HRS cells also secrete cytokines such asmacrophage migration inhibition factor, which inducesmacrophage M2 polarization,34 and IL-9, which promotesmast cell differentiation (which in turn results in angiogen-esis and fibrosis).35 Thus, HRS cells both attract and inducethe differentiation of immune cells resulting in a tumormicroenvironment favorable for tumor cell growth andsurvival.36The importance of the tumor microenvironment in cHL
was illustrated in studies by two independent groups whoused gene expression profiling to demonstrate overexpres-sion of genes associated with macrophages in biopsies
taken from patients who experienced treatment failure.37This tied in neatly with the findings of immunohisto-chemical studies, in which increased number of CD68+cells in diagnostic biopsy specimens was prognostic ofinferior progression-free survival and disease-specific sur-vival in patients treated with doxorubicin, bleomycin, vin-blastine and dacarbazine, independently of establishedclinical and laboratory parameters.38 The adverse prognos-tic impact of CD68 expression on overall survival was val-idated in another study from Barts Cancer Institute.39CD68 is not specific for macrophages, as it stains othermyeloid cells, and some fibroblasts.40 Increased numbersof CD163+ cells [whose expression is restricted to M2polarized (immunosuppressive) macrophages] has beensuggested by some studies to be a superior adverse prog-nostic marker.41-43 An interesting recent study showed thatpatients with Hodgkin lymphoma have higher numbers ofcirculating myeloid-derived suppressor cells in theirperipheral blood than have healthy controls, and thatincreased levels of CD34+ myeloid-derived suppressorcells were predictive of inferior progression-free survival.44 With regard to lymphocyte subsets in the tumor
microenvironment, increased numbers of non-follicular Bcells are associated with favorable survival, indicating thatthey likely play an important role in the immunologicalcontrol of cHL.39,45,46 Somewhat counter-intuitively,increased numbers of FOXP3+ Tregs have been associatedwith superior progression-free and overall survival.39,47,48while increased numbers of granzyme B+ cytotoxic T cellshave the opposite effect on survival.47,48 Although thesefindings require validation in larger, prospectively treatedcohorts of patients, they suggest that Tregs have a contrast-ing function in cHL compared with solid tumors, such asdirect suppression of HRS cells.
Indolent lymphomas
Follicular lymphomaIn FL and mucosal-associated lymphoid tissue (MALT)
lymphoma, tumor cells appear to depend heavily on themicroenvironment for survival and proliferation.2 Geneexpression profiling of tumor infiltrating lymphocytes(TIL) in FL revealed two immune response signatureswhich predicted disparate clinical outcomes.49 Interactionsbetween TIL and tumor cells can result in modulation ofthe immune response, which can have prognostic implica-tions.50-54 For example, studies have shown that high num-bers of PD1+ TIL are prognostically favorable, whilepatients with ≤5% PD1+ TIL had a higher risk of histolog-ical transformation to DLBCL.55 In another study fromVancouver, the follicular localization of Tregs was found tobe an adverse prognostic factor for overall survival andtransformation risk.56Tumor-associated macrophages also appear to predict
an unfavorable clinical course.52 Analysis of the geneexpression profiles of CD4+ and CD8+ FL TIL revealedaltered gene expression that resulted in impaired actinpolymerization and immune synapse formation anddecreased cytotoxicity and T-cell motility, leading to T-cellexhaustion and immunosuppression.57-60 This altered geneexpression in TIL has prognostic significance with respectto overall survival and time to transformation.57 In terms ofthe potential therapeutic implications of these findings inT cells, an interesting study demonstrated that FL cells
The microenvironment in B-cell lymphoid malignancies
haematologica | 2016; 101(5) 533

with T-cell immunological synapse dysfunction can berepaired with the immunomodulatory agent lenalido-mide.59
Marginal zone lymphomaExtranodal marginal zone lymphomas (MZL) of MALT
provide a classical illustration of the role of the microenvi-ronment in lymphomagenesis through B-cell antigen stim-ulation. Chronic infections may provide antigenic stimula-tion, which results in different manifestations of MZL atvarious anatomic sites. Examples include gastric MALT andHelicobacter pylori,61 splenic MZL and hepatitis C,62 ocularadnexal MZL and Chlamydophilia psittaci,63 and cutaneousMZL and Borrelia.64 Eradication of the implicated micro-organism leads to lymphoma regression in many cases,supporting antigenic dependence.65 The occurrence of sec-ondary genetic lesions, in particular t(11;18), has been asso-ciated with poor responses to eradication therapy for gas-tric MALT lymphoma, presumably due to the develop-ment of independence from the microenvironment forgrowth and survival.66 Although splenic MZL generally hasan indolent course, up to one-third of patients experiencerapid disease progression. Dense infiltrates of CD40+ cellswithin the bone marrow correlate with inferior prognosis,likely through interactions with CD40L with surroundingcells in the tumor microenvironment (including mast cells,helper T cells, dendritic cells, macrophages and B cells)resulting in immune cell activation through phosphoryla-tion of STAT3 and resultant secretion of TNF/IL-6 – the neteffect of which is the induction of a microenvironmentfavoring tumor growth and survival.67
Chronic lymphocytic leukemiaStudies examining tumor escape in CLL differ as to
whether changes in expression of classical and non-classi-cal human leukocyte antigens by tumor cells can modulatethe interactions of NK- and T-cell subpopulations with tar-get cells.68 In CLL, T-cell dysfunction is mediated by
expression of inhibitory molecules such as CD200,CD270, PD-L1 and B7-H3 on tumor cells, with predomi-nant influences mediated by PD-L1 expression.69,70Expression of these molecules has been linked to a poorprognosis in patients with CLL.69 Interestingly, reducingexpression of these genes in tumor cells can improve T-cellfunction. In addition, treatment of TIL with lenalidomidehas been shown to reverse the signs of T-cell exhaustionand improve T-cell function.69BCL-2 expression71 has been suggested to be in part con-
trolled by miR-15/16 expression, but alternative microen-vironmental interactions may be associated with BCL-2upregulation and increased cell survival in CLL.72 Indeed,BCL-2 can be up-regulated by CD40/CD40L interactions,as shown by the increased expression upon culture withsoluble CD40L. This interaction may potentially occur inthe infiltrated lymphoid tissues and in particular in theproliferation centers where CD4+ T cells can be found inclose proximity to leukemic B cells. Moreover, additionalstudies have shown that co-culture of CLL cells and stro-mal cells results in up-regulation of BCL-2 expression,thereby providing survival and drug-resistance signals toCLL cells.73 Investigations into the types of stromal cellsthat may mediate these interactions show that monocytescontribute to CLL survival and mediate expansion of CLLcells.74,75 Analyses in murine models show that depletingmonocyte levels can decrease CLL burden in the mice.74Similarly, the stimulation of surface receptors, includingToll-like receptors76 and BCR, is able to induce upregula-tion of BCL-2 and other anti-apoptotic molecules suggest-ing that a wide array of signals from the microenviron-ment can indeed be responsible for the regulation of apop-tosis. All these signals translate into activation of down-stream signaling pathways, including the MAPK and theNF-κB pathways, which contribute to the survival ofleukemic cells. ERK is constitutively active in approxi-mately 50% of CLL patients,77 likely due to the stimulationby anergizing antigenic elements, while SYK and NF-κB
N.H. Fowler et al.
534 haematologica | 2016; 101(5)
Table 1. Overview of lymphoma subtypes, examples of impact of tumor microenvironment on outcome and novel agents of potential therapeuticrelevance.Lymphoma subtype Key tumor microenvironment elements, prognostic impact Therapeutic agents
Hodgkin lymphoma Increased macrophage gene expression, CD68+ infiltrate (adverse)37 PD-1 inhibitors153
Increased myeloid derived suppressor cells (adverse)44
Increased Treg (favorable)39, 47, 48
Increased non-follicular B cells (favorable)39
Increased cytotoxic T cells (adverse)47, 48
Diffuse large B-cell lymphoma Increased CD68+ TAM and CD68 mRNA (adverse in patients treated Rituximab19
with chemotherapy, favorable in patients treated with PD-1 inhibitors89
chemo-immunotherapy)19
Increased tumor microenvironment PD-L1 expression16
Follicular lymphoma Immune response signature-1 (favorable)49 Lenalidomide59
Increased TAM53 Lenalidomide and rituximab132
Increased PD1+ TIL (favorable)55 PD-1 inhibitors90
Intra- or peri-follicular Treg (adverse)56
Marginal zone lymphoma Dense infiltrates of CD40+ cells (adverse)67
Mantle cell lymphoma Increased TAM associated with aggressive clinical behavior25, 26 BTK inhibitors108
Tumor cell adhesion to stromal elements (adverse)21
Chronic lymphocytic leukemia Tumor-stromal interactions73 Lenalidomide154
Induction of myeloid derived suppressor cells75 BTK inhibitors106
Promotion of BCR signaling and NFκB activation78 PI3K inhibitors121
PD-1: programmed cell death-1; PD-L1, programmed cell death ligand-1; TAM: tumor associated macrophage; TIL: tumor infiltrating lymphocyte.

are upregulated in virtually all cases of CLL, with manypatients having recurrent mutations within the NF-κBpathway78,79 in addition to induction by the microenviron-ment.
Novel therapies targeting the microenvironment
The following section focuses on several novel classes ofagents that therapeutically exploit the dependence of lym-phoma cells on microenvironmental stimuli as part of theirmechanism of action.
Checkpoint inhibitorsPD-1 limits the response of activated T cells at sites of
infection and prevents autoimmunity.80,81 Binding of PD-1by its ligands PD-L1 and PD-L2 produces inhibitory signalsthat ultimately result in apoptosis of activated T cells, the
so-called “immune checkpoint”.82 However, PD-1 is alsopresent on other immune cells including Treg, B and NKcells. Thus, PD-1 blockade enhances anti-tumor cytotoxi-city through increased NK-cell killing and Treg
suppression.83,84 Tumor cells are able to exploit the path-way in a similar manner by expressing PD-L1 on TIL.85 Invitro experimental models indicate that PD-L1 expressionby tumors results in the impairment of anti-tumorresponses.86 Antibodies targeting the PD-1 axis thus“release the brakes” from effector T cells and promoteanti-tumor cytotoxicity.87 Antibody-dependent cell-medi-ated cytotoxicity (ADCC) of tumor cells expressing PD-1or PD-L1 does not appear to be a mechanism of action forthese agents, as PD-1/PD-L1 surface expression by tumorcells or tumor microenvironment does not seem to be nec-essary for their activity.88 Various agents targeting the PD-1 axis are under development; however, preliminary data
The microenvironment in B-cell lymphoid malignancies
haematologica | 2016; 101(5) 535
Figure 1. Schematic diagram of the typical microenvironment of the three B-cell lymphoma subtypes that represent the extremes of the spectrum of tumormicroenvironment — recruitment, re-education and effacement. These lymphoma subtypes represent the range of tumor cell content, from ~1% in cHL to typicallymore than 90% in BL. The other B-cell lymphomas fall within this range, as shown for the most common B-cell lymphomas (center). Typically, the ratio of malignantcells to microenvironmental cells increases across the range, from cHL to BL, as shown. DLBCL, diffuse large B-cell lymphoma; FOXP3, forkhead box protein P3; HRS,Hodgkin Reed–Sternberg; MALT, mucosa-associated lymphoid tissue; MCL, mantle cell lymphoma; TFH, follicular T helper; TH, T helper; TFR, follicular regulatory T.Reproduced from Scott and Gascoyne2 with permission from Nature Publishing Group.

on three agents are currently available. The investigationalagent pidilizumab is a humanized IgG1 monoclonal anti-body directed against PD-1, which has been explored inphase II studies in DLBCL89 and FL.90 Pidilizumab increasedin CD4+CD25+PD-L1+ activated T helper cells and PD-1ligand-bearing monocytes in a phase II study in DLBCL,89and in a phase II study of pidilizumab and rituximab inpatients with FL a 41-gene signature representing immuneactivation correlated with improved progression-free sur-vival.90 In both studies, pidilizumab was well tolerated andappeared to increase efficacy relative to historic controls.Pembrolizumab (humanized) and nivolumab (fullyhuman), both investigational in hematologic malignancies,are IgG4 antagonistic anti-PD-1 monoclonal antibodieswith outstanding activity in heavily pre-treated Hodgkinlymphoma.91,92 Preliminary results regarding nivolumabshow promise in a variety of subtypes of non-Hodgkinlymphomas93 and phase II studies in multiple histologicaltypes are planned or underway.
Chimeric antigen receptor T-cell therapy Much has been written about the success of investiga-
tional anti-CD19 CAR T-cell therapy in relapsed/refracto-ry acute lymphoblastic leukemia, CLL and DLBCL.94-96This technology uses gene-modified autologous T cellswith antigen specificity for CD19, expressed mainly onthe surface of B cells.97 CD19 represents a near optimaltumor-associated antigen to target, as its restricted expres-sion minimizes off-target toxicity. One of the problemswith CAR T-cell therapy is to overcome the immunosup-pressive tumor microenvironment that includes M2 polar-ized macrophages, Tregs, and myeloid-derived suppressorcells.98 Investigators have approached this problem bymodifying the CAR T-cell construct number in a numberof customized ways, including the incorporation of pro-inflammatory cytokines such as IL-12,99 expression ofdominant negative TGF-β,100 anti-apoptotic Fas-knock-downs101 and the expression of survival signals such as Bcl-xl.102 An alternate approach would be to combine CAR Tcells with agents targeting the PD-1 axis to enhance theanti-tumor cytotoxicity.
B-cell receptor pathway inhibitors B cells depend on signals mediated through the BCR to
govern a variety of cellular processes including prolifera-tion, apoptosis and differentiation.103 Deregulation of theBCR pathway is thought to be central to the pathogenesisof many B-cell lymphomas.104 The BCR signaling cascadeinvolves numerous tyrosine kinases including Btk, Syk andPI3K, and small molecule inhibitors targeting these kinaseshave been developed. Ibrutinib is a selective, small molecule that irreversibly
binds to Btk.105 Ibrutinib has excellent activity in CLL,106,107MCL108 and Waldenström macroglublinemia109 and hasgained regulatory approval for the treatment of relapsed orrefractory patients with these diseases and also for first-linetherapy in patients with del(17p) CLL. Although themechanism of action of ibrutinib involves direct effects onmalignant B cells, including induction of apoptosis and dis-ruption of cell adhesion and migration,110 the effects on thetumor microenvironment are also important. Btk regulatesNK cell function in response to antigen presentation.111However, ibrutinib also inhibits Itk, which is involved inNK cell effector function following FcR-mediated engage-ment.112 Interestingly, while some preclinical studies have
shown that ibrutinib may antagonize ADCC induced byanti-CD20 monoclonal antibodies such as rituximab, in theclinical setting ibrutinib in combination with rituximab ishighly active.113,114 More selective Btk inhibitors that spareItk do not appear to have the same antagonism and mayprove more effective in combinations. Through Itk inhibi-tion, ibrutinib also influences T-cell polarization towardtype 1 T helper cells and effector T cells.115 Preclinical workby Levy et al. at Stanford also suggests that ibrutinibpotently enhances immunological tumor control when co-administered with a TLR9 agonist through stimulation ofantigen-presenting cells in the tumor microenvironment.116The same group also described how ibrutinib enhanced theT-cell anti-tumor activity of PD-L1 inhibitors, a findingwith clear implications for combination studies.117 Btkplays a role in polarizing macrophages to an M1 (inflam-matory) phenotype; as mice deficient in Btk are skewedtowards M2 (immunosuppressive) polarization, whichsuggests a theoretical potential for ibrutinib to induce anunhelpful change in the microenvironment.118 However,we are unaware of data regarding macrophage polarizationin ibrutinib-treated patients. Several PI3K inhibitors with various isoform specificities
are in development. The most advanced, idelalisib, is aselective inhibitor of the p110δ isoform of PI3K. It hasdemonstrated excellent clinical activity in patients withrelapsed/refractory CLL/small lymphocytic lymphomaand FL, indications for which it has gained approval fromboth the Food and Drug Administration (FDA) and theEuropean Medicines Agency (EMA).119-121 PI3Kδ isexpressed by both normal and malignant lymphoid cells,and PI3k inhibition by idelalisib in vitro leads to inductionof apoptosis.122 Like ibrutinib, idelalisib interferes withpro-survival microenvironment-derived signals, chemo-taxis and adhesion.123,124 Its antagonism of ADCC inducedby anti-CD20 monoclonal antibodies is weaker that thatof ibrutinib in vitro.125 Idelalisib does not appear cytotoxicto T-cell subsets;126 however, the investigational dual PI3Kp110γ and p110δ inhibitor duvelisib (IPI-145) reduces theviability of T and NK cells and impairs T-cell production ofpro-inflammatory cytokines.127
Immunomodulatory drugsImmunomodulatory drugs exert pleiotropic effects both
directly on lymphoma cells and on the immune microen-vironment. Lenalidomide (FDA-approved for multiplemyeloma and relapsed MCL) has activity in a range oflymphoma subtypes both as a single agent128-131 and incombination with rituximab, particularly in MCL andFL.132-137 The molecular mechanism of action of lenalido-mide has only recently been described in detail.Immunomodulatory drugs bind to the E3 ubiquitin ligasecereblon (CRBN), which is re-directed by lenalidomide toinduce proteosomal degradation of the transcription fac-tors Ikaros (IKZF1) and Aiolos (IKZF3).138-140 These tran-scription factors provide pro-survival signals for tumorcells and suppress IL-2 production. The binding ofimmunomodulatory drugs to CRBN therefore blocks sur-vival signals to tumor cells and leads to increased IL-2 pro-duction and enhancement of T-cell co-stimulation.138Furthermore, lenalidomide induces type 1 T helper cellpolarization,141 reduces Treg cells, increases antigen presen-tation to effector T-cell populations,142 repairs the immunesynapse between tumor cells and cytotoxic T cells,69restores impaired T-cell motility and interferes with com-
N.H. Fowler et al.
536 haematologica | 2016; 101(5)

munication between endothelial and tumor cells, reducingneoangiogenesis.143 Lenalidomide also induces a change inthe tumor microenvironment from an M2 macrophageimmunosuppressive state to a pro-inflammatory statethrough polarization of macrophages toward an M1 phe-notype.144 Lenalidomide augments the ADCC of anti-CD20 monoclonal antibodies145,146 and lowers the activa-tion threshold of NK cells.147 The multitude of mechanismsby which lenalidomide is able to alter the tumor microen-vironment into a hostile one for lymphoma provides a sat-isfactory explanation for the activity observed in the clinic– an excellent illustration of the potential benefits of tar-geting the lymphoma cell niche.
Future directions
Novel combinations It is unlikely that any one agent or modulator of a single
pathway will prove successful in inhibiting tumor cell sur-vival over the long-term in B-cell lymphoproliferative dis-eases. Effective curative strategies will likely require opti-mal synergistic combinations of effective agents.However, the large number of possible combinations, lim-ited resources and paucity of patients for clinical trialsmake it an imperative to prioritize and develop thosecombinations that are most likely to be curative.Designing logical combinations with strong pre-clinicalrationales is, therefore, a priority of translational researchin hematologic malignancies. Strategies that include thetargeting of various steps of the cancer-immunity cycle148will be imperative. For example, drugs targeting the PD-1axis enhance the host anti-tumor response and may belogically used in combination with many of the aforemen-tioned novel agents.148 Furthermore, “precision immunolo-gy” should consider the immunological milieu of bothhost and tumor. For example, highly immunogenic tumors(such as cHL) may benefit from rational strategies thatinclude immunostimulatory combinations such as PD-1/PD-L1 inhibitors plus T-cell priming treatments.149 Incontrast, immunologically inert lymphomas may be betterapproached with strategies such as CAR T cells in combi-nation with agents such as monoclonal antibodies.150Caution in developing such combination studies is
required and vigilant monitoring for clinical or laboratoryadverse events is essential. Two studies using the combi-nation of lenalidomide, rituximab and idelalisib inrelapsed/refractory FL were recently terminated due to an
unexpected frequency and severity of hepatotoxicity,including two deaths.151,152 These episodes highlight theneed to incorporate correlative studies into all multi-agentinvestigational protocols to survey for unexpected toxici-ties as well as to understand tumor biology and the rea-sons for treatment resistance better.
Monitoring the microenvironment during therapyAlthough researchers typically obtain a snapshot of the
microenvironment at the time of diagnostic biopsy, thedevelopment of processes that enable dynamic assess-ment is important. Although tumors with a circulatingphase, such as CLL, are comparatively easy to assess atserial time-points from blood samples, obtaining biopsiesduring treatment poses major logistic challenges in mostpatients with lymphoma. To address this challenge, novelstrategies that can assess circulating tumor DNA andmutational analyses in the peripheral blood are welcomedand should be incorporated in future studies aimed atdeveloping therapies directed at the microenvironment.
Conclusion
Improved understanding of tumor biology and the roleof the tumor microenvironment has led to advances in thediagnosis, classification, prognostication and novel treat-ment of patients with hematologic malignancies. In partic-ular, translational research leading to drugs that target theinteraction between the tumor microenvironment andmalignant cells has provided many promising newapproaches to cancer therapy. Ongoing dynamic and cor-relative studies of tumor biology and the contribution ofthe tumor microenvironment in the context of novel drugdevelopment should be encouraged to identify optimaltherapies for various lymphomas and improve the curabil-ity of these diseases.
AcknowledgmentsThis manuscript was developed, in part, based on discussions
from the Second Annual Summit on the ImmuneMicroenvironment in Hematologic Malignancies that took placeon September 11-12, 2014, in Dublin, Ireland. The SecondAnnual Summit was sponsored by a grant from AbbVie Inc.,Acerta Pharma, Celgene Corporation, F. Hoffman-La RocheLTD, Infinity Pharmaceuticals, Inc., Pharmacyclics, Inc., and TGTherapeutics, Inc. Project management support for this manu-script was provided by BioConnections, LLC.
The microenvironment in B-cell lymphoid malignancies
haematologica | 2016; 101(5) 537
References
1. Shaffer AL 3rd, Young RM, Staudt LM.Pathogenesis of human B cell lymphomas.Annu Rev Immunol. 2012;30:565-610.
2. Scott DW, Gascoyne RD. The tumourmicroenvironment in B cell lymphomas. NatRev Cancer. 2014;14(8):517-534.
3. Andersen MH. The targeting of immuno-suppressive mechanisms in hematologicalmalignancies. Leukemia. 2014;28(9):1784-1792.
4. Brusa D, Serra S, Coscia M, et al. The PD-1/PD-L1 axis contributes to T-cell dysfunc-tion in chronic lymphocytic leukemia.Haematologica. 2013;98(6):953-963.
5. Meads MB, Gatenby RA, Dalton WS.Environment-mediated drug resistance: amajor contributor to minimal residual dis-ease. Nat Rev Cancer. 2009;9(9):665-674.
6. Alizadeh AA, Eisen MB, Davis RE, et al.Distinct types of diffuse large B-cell lym-phoma identified by gene expression profil-ing. Nature. 2000;403(6769):503-511.
7. Dunleavy K, Roschewski M, Wilson WH.Precision treatment of distinct molecularsubtypes of diffuse large B-cell lymphoma:ascribing treatment based on the molecularphenotype. Clin Cancer Res. 2014;20(20):5182-5193.
8. Lenz G, Wright G, Dave SS, et al. Stromalgene signatures in large-B-cell lymphomas.N Engl J Med. 2008;359(22):2313-2323.
9. Challa-Malladi M, Lieu YK, Califano O, etal. Combined genetic inactivation of beta2-microglobulin and CD58 reveals frequentescape from immune recognition in diffuselarge B cell lymphoma. Cancer Cell.2011;20(6):728-740.
10. Steidl C, Shah SP, Woolcock BW, et al. MHCclass II transactivator CIITA is a recurrentgene fusion partner in lymphoid cancers.Nature. 2011;471(7338):377-381.
11. Andorsky DJ, Yamada RE, Said J, et al.Programmed death ligand 1 is expressed bynon-Hodgkin lymphomas and inhibits theactivity of tumor-associated T cells. ClinCancer Res. 2011;17(13):4232-4244.
12. Chen BJ, Chapuy B, Ouyang J, et al. PD-L1expression is characteristic of a subset of

aggressive B-cell lymphomas and virus-asso-ciated malignancies. Clin Cancer Res.2013;19(13):3462-3473.
13. Twa DD, Chan FC, Ben-Neriah S, et al.Genomic rearrangements involving pro-grammed death ligands are recurrent in pri-mary mediastinal large B-cell lymphoma.Blood. 2014;123(13):2062-2065.
14. Dunleavy K, Steidl C. Emerging biologicalinsights and novel treatment strategies inprimary mediastinal large B-cell lymphoma.Semin Hematol. 2015;52(2):119-125.
15. Twa DD, Mottok A, Chan FC, et al.Recurrent genomic rearrangements in pri-mary testicular lymphoma. J Pathol.2015;236(2):136-141.
16. Kiyasu J, Miyoshi H, Hirata A, et al.Expression of programmed cell death ligand1 is associated with poor overall survival inpatients with diffuse large B-cell lymphoma.Blood. 2015;126(19):2193-2201.
17. Ishida T, Ishii T, Inagaki A, et al. Specificrecruitment of CC chemokine receptor 4-positive regulatory T cells in Hodgkin lym-phoma fosters immune privilege. CancerRes. 2006;66(11):5716-5722.
18. Yang ZZ, Novak AJ, Stenson MJ, et al.Intratumoral CD4+CD25+ regulatory T-cell-mediated suppression of infiltrating CD4+ Tcells in B-cell non-Hodgkin lymphoma.Blood. 2006;107(9):3639-3646.
19. Riihijarvi S, Fiskvik I, Taskinen M, et al.Prognostic influence of macrophages inpatients with diffuse large B-cell lymphoma:a correlative study from a Nordic phase IItrial. Haematologica. 2015;100(2):238-245.
20. Perez-Galan P, Dreyling M, Wiestner A.Mantle cell lymphoma: biology, pathogene-sis, and the molecular basis of treatment inthe genomic era. Blood. 2011;117(1):26-38.
21. Burger JA, Ford RJ. The microenvironmentin mantle cell lymphoma: cellular andmolecular pathways and emerging targetedtherapies. Semin Cancer Biol. 2011;21(5):308-312.
22. Kurtova AV, Tamayo AT, Ford RJ, et al.Mantle cell lymphoma cells express high lev-els of CXCR4, CXCR5, and VLA-4 (CD49d):importance for interactions with the stromalmicroenvironment and specific targeting.Blood. 2009;113(19):4604-4613.
23. Zhang L, Yang J, Qian J, et al. Role of themicroenvironment in mantle cell lym-phoma: IL-6 is an important survival factorfor the tumor cells. Blood. 2012;120(18):3783-3792.
24. Cohen PL, Kurtin PJ, Donovan KA, et al.Bone marrow and peripheral blood involve-ment in mantle cell lymphoma. Br JHaematol. 1998;101(2):302-310.
25. Argatoff LH, Connors JM, Klasa RJ, et al.Mantle cell lymphoma: a clinicopathologicstudy of 80 cases. Blood. 1997;89(6):2067-2078.
26. Pittaluga S, Wlodarska I, Stul MS, et al.Mantle cell lymphoma: a clinicopathologicalstudy of 55 cases. Histopathology.1995;26(1):17-24.
27. Wang L, Qian J, Lu Y, et al. Immune evasionof mantle cell lymphoma: expression of B7-H1 leads to inhibited T-cell response to andkilling of tumor cells. Haematologica.2013;98(9):1458-1466.
28. Dal Col J, Zancai P, Terrin L, et al. Distinctfunctional significance of Akt and mTORconstitutive activation in mantle cell lym-phoma. Blood. 2008;111(10):5142-5151.
29. Buggy JJ, Elias L. Bruton tyrosine kinase(BTK) and its role in B-cell malignancy. IntRev Immunol. 2012;31(2):119-132.
30. Bernard S, Danglade D, Gardano L, et al.
Inhibitors of BCR signalling interrupt thesurvival signal mediated by the micro-envi-ronment in mantle cell lymphoma. Int JCancer. 2015;136(12):2761-2774.
31. Swerdlow SH CE, Harris N, et al. WHOClassification of Tumors of theHematopoeitic and Lymphoid Tissues.Lyon: IARC, 2008.
32. Plattel WJ, van den Berg A, Visser L, et al.Plasma thymus and activation-regulatedchemokine as an early response marker inclassical Hodgkin's lymphoma.Haematologica. 2012;97(3):410-415.
33. Hedvat CV, Jaffe ES, Qin J, et al.Macrophage-derived chemokine expressionin classical Hodgkin's lymphoma: applica-tion of tissue microarrays. Mod Pathol.2001;14(12):1270-1276.
34. Ma Y, Visser L, Roelofsen H, et al.Proteomics analysis of Hodgkin lymphoma:identification of new players involved in thecross-talk between HRS cells and infiltratinglymphocytes. Blood. 2008;111(4):2339-2346.
35. Glimelius I, Edstrom A, Amini RM, et al. IL-9 expression contributes to the cellular com-position in Hodgkin lymphoma. Eur JHaematol. 2006;76(4):278-283.
36. Liu Y, Sattarzadeh A, Diepstra A, et al. Themicroenvironment in classical Hodgkin lym-phoma: an actively shaped and essentialtumor component. Semin Cancer Biol.2014;24:15-22.
37. Sanchez-Aguilera A, Montalban C, de laCueva P, et al. Tumor microenvironmentand mitotic checkpoint are key factors in theoutcome of classic Hodgkin lymphoma.Blood. 2006;108(2):662-668.
38. Steidl C, Lee T, Shah SP, et al. Tumor-associ-ated macrophages and survival in classicHodgkin's lymphoma. N Engl J Med.2010;362(10):875-885.
39. Greaves P, Clear A, Coutinho R, et al.Expression of FOXP3, CD68, and CD20 atdiagnosis in the microenvironment of classi-cal Hodgkin lymphoma is predictive of out-come. J Clin Oncol. 2013;31(2):256-262.
40. Pulford KA, Sipos A, Cordell JL, et al.Distribution of the CD68 macrophage/myeloid associated antigen. Int Immunol.1990;2(10):973-980.
41. Klein JL, Nguyen TT, Bien-Willner GA, et al.CD163 immunohistochemistry is superiorto CD68 in predicting outcome in classicalHodgkin lymphoma. Am J Clin Pathol.2014;141(3):381-387.
42. Zaki MA, Wada N, Ikeda J, et al. Prognosticimplication of types of tumor-associatedmacrophages in Hodgkin lymphoma.Virchows Arch. 2011;459(4):361-366.
43. Tan KL, Scott DW, Hong F, et al. Tumor-associated macrophages predict inferior out-comes in classic Hodgkin lymphoma: a cor-relative study from the E2496 Intergrouptrial. Blood. 2012;120(16):3280-3287.
44. Romano A, Parrinello NL, Vetro C, et al.Circulating myeloid-derived suppressor cellscorrelate with clinical outcome in Hodgkinlymphoma patients treated up-front with arisk-adapted strategy. Br J Haematol.2015;168(5):689-700.
45. Panico L, Tenneriello V, Ronconi F, et al.High CD20+ background cells predict afavorable outcome in classical Hodgkin lym-phoma and antagonize CD68+macrophages. Leuk Lymphoma. 2014:1-7.
46. Tudor CS, Distel LV, Eckhardt J, et al. B cellsin classical Hodgkin lymphoma are impor-tant actors rather than bystanders in thelocal immune reaction. Hum Pathol.2013;44(11):2475-2486.
47. Alvaro T, Lejeune M, Salvado MT, et al.
Outcome in Hodgkin's lymphoma can bepredicted from the presence of accompany-ing cytotoxic and regulatory T cells. ClinCancer Res. 2005;11(4):1467-1473.
48. Kelley TW, Pohlman B, Elson P, et al. Theratio of FOXP3+ regulatory T cells togranzyme B+ cytotoxic T/NK cells predictsprognosis in classical Hodgkin lymphomaand is independent of bcl-2 and MALexpression. Am J Clin Pathol. 2007;128(6):958-965.
49. Dave SS, Wright G, Tan B, et al. Prediction ofsurvival in follicular lymphoma based onmolecular features of tumor-infiltratingimmune cells. N Engl J Med. 2004;351(21):2159-2169.
50. Alvaro T, Lejeune M, Salvado MT, et al.Immunohistochemical patterns of reactivemicroenvironment are associated with clini-cobiologic behavior in follicular lymphomapatients. J Clin Oncol. 2006;24(34):5350-5357.
51. Dave SS, Wright G, Tan B, et al. Prediction ofsurvival in follicular lymphoma based onmolecular features of tumor-infiltratingimmune cells. N Engl J Med. 2004;351(21):2159-2169.
52. de Jong D, Fest T. The microenvironment infollicular lymphoma. Best Pract Res ClinHaematol. 2011;24(2):135-146.
53. Farinha P, Masoudi H, Skinnider BF, et al.Analysis of multiple biomarkers shows thatlymphoma-associated macrophage (LAM)content is an independent predictor of sur-vival in follicular lymphoma (FL). Blood.2005;106(6):2169-2174.
54. Glas AM, Knoops L, Delahaye L, et al. Gene-expression and immunohistochemical studyof specific T-cell subsets and accessory celltypes in the transformation and prognosis offollicular lymphoma. J Clin Oncol.2007;25(4):390-398.
55. Carreras J, Lopez-Guillermo A, Roncador G,et al. High numbers of tumor-infiltratingprogrammed cell death 1-positive regulatorylymphocytes are associated with improvedoverall survival in follicular lymphoma. JClin Oncol. 2009;27(9):1470-1476.
56. Farinha P, Al-Tourah A, Gill K, et al. Thearchitectural pattern of FOXP3-positive Tcells in follicular lymphoma is an independ-ent predictor of survival and histologic trans-formation. Blood. 2010;115(2):289-295.
57. Kiaii S, Clear AJ, Ramsay AG, et al. Follicularlymphoma cells induce changes in T-cellgene expression and function: potentialimpact on survival and risk of transforma-tion. J Clin Oncol. 2013;31(21):2654-2661.
58. Myklebust JH, Irish JM, Brody J, et al. HighPD-1 expression and suppressed cytokinesignaling distinguish T cells infiltrating follic-ular lymphoma tumors from peripheral Tcells. Blood. 2013;121(8):1367-1376.
59. Ramsay AG, Clear AJ, Kelly G, et al.Follicular lymphoma cells induce T-cellimmunologic synapse dysfunction that canbe repaired with lenalidomide: implicationsfor the tumor microenvironment andimmunotherapy. Blood. 2009;114(21):4713-4720.
60. Ramsay AG, Gribben JG. The kiss of deathin FL. Blood. 2011;118(20):5365-5366.
61. Wotherspoon AC, Ortiz-Hidalgo C, FalzonMR, et al. Helicobacter pylori-associatedgastritis and primary B-cell gastric lym-phoma. Lancet. 1991;338(8776):1175-1176.
62. Suarez F, Lortholary O, Hermine O, et al.Infection-associated lymphomas derivedfrom marginal zone B cells: a model of anti-gen-driven lymphoproliferation. Blood.2006;107(8):3034-3044.
N.H. Fowler et al.
538 haematologica | 2016; 101(5)

63. Ferreri AJ, Guidoboni M, Ponzoni M, et al.Evidence for an association betweenChlamydia psittaci and ocular adnexal lym-phomas. J Natl Cancer Inst. 2004;96(8):586-594.
64. Kutting B, Bonsmann G, Metze D, et al.Borrelia burgdorferi-associated primarycutaneous B cell lymphoma: complete clear-ing of skin lesions after antibiotic pulse ther-apy or intralesional injection of interferonalfa-2a. J Am Acad Dermatol. 1997;36(2 Pt2):311-314.
65. Wotherspoon AC, Doglioni C, Diss TC, etal. Regression of primary low-grade B-cellgastric lymphoma of mucosa-associatedlymphoid tissue type after eradication ofHelicobacter pylori. Lancet. 1993;342(8871):575-577.
66. Inagaki H, Nakamura T, Li C, et al. GastricMALT lymphomas are divided into threegroups based on responsiveness toHelicobacter pylori eradication and detec-tion of API2-MALT1 fusion. Am J SurgPathol. 2004;28(12):1560-1567.
67. Franco G, Guarnotta C, Frossi B, et al. Bonemarrow stroma CD40 expression correlateswith inflammatory mast cell infiltration anddisease progression in splenic marginal zonelymphoma. Blood. 2014;123(12):1836-1849.
68. Guillaume N, Marolleau JP. Is immuneescape via human leukocyte antigen expres-sion clinically relevant in chronic lympho-cytic leukemia? Focus on the controversies.Leuk Res. 2013;37(4):473-477.
69. Ramsay AG, Clear AJ, Fatah R, et al.Multiple inhibitory ligands induce impairedT-cell immunologic synapse function inchronic lymphocytic leukemia that can beblocked with lenalidomide: establishing areversible immune evasion mechanism inhuman cancer. Blood. 2012;120(7):1412-1421.
70. Riches JC, Davies JK, McClanahan F, et al. Tcells from CLL patients exhibit features of T-cell exhaustion but retain capacity forcytokine production. Blood. 2013;121(9):1612-1621.
71. von Bergwelt-Baildon M, Maecker B,Schultze J, et al. CD40 activation: potentialfor specific immunotherapy in B-CLL. AnnOncol. 2004;15(6):853-857.
72. Allegra D, Bilan V, Garding A, et al.Defective DROSHA processing contributesto downregulation of MiR-15/-16 in chroniclymphocytic leukemia. Leukemia.2014;28(1):98-107.
73. Kurtova AV, Balakrishnan K, Chen R, et al.Diverse marrow stromal cells protect CLLcells from spontaneous and drug-inducedapoptosis: development of a reliable andreproducible system to assess stromal celladhesion-mediated drug resistance. Blood.2009;114(20):4441-4450.
74. Hanna B, McClanahan F, Zaborsky N, et al.Targeting dysfunctional myeloid cells delaysdisease development and improves immunefucntion in a CLL mouse model. Blood.2014;124(21):3298.
75. Jitschin R, Braun M, Buttner M, et al. CLL-cells induce IDOhi CD14+HLA-DRlomyeloid-derived suppressor cells that inhibitT-cell responses and promote TRegs. Blood.2014;124(5):750-760.
76. Fonte E, Apollonio B, Scarfo L, et al. In vitrosensitivity of CLL cells to fludarabine maybe modulated by the stimulation of Toll-likereceptors. Clin Cancer Res. 2013;19(2):367-379.
77. Muzio M, Apollonio B, Scielzo C, et al.Constitutive activation of distinct BCR-sig-naling pathways in a subset of CLL patients:
a molecular signature of anergy. Blood.2008;112(1):188-195.
78. Herishanu Y, Perez-Galan P, Liu D, et al. Thelymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activa-tion, and tumor proliferation in chronic lym-phocytic leukemia. Blood. 2011;117(2):563-574.
79. Mansouri L, Sutton LA, Ljungstrom V, et al.Functional loss of IkappaBepsilon leads toNF-kappaB deregulation in aggressivechronic lymphocytic leukemia. J Exp Med.2015;212(6):833-843.
80. Freeman GJ, Long AJ, Iwai Y, et al.Engagement of the PD-1 immunoinhibitoryreceptor by a novel B7 family member leadsto negative regulation of lymphocyte activa-tion. J Exp Med. 2000;192(7):1027-1034.
81. Nishimura H, Nose M, Hiai H, et al.Development of lupus-like autoimmune dis-eases by disruption of the PD-1 gene encod-ing an ITIM motif-carrying immunorecep-tor. Immunity. 1999;11(2):141-151.
82. Seo SK, Seo HM, Jeong HY, et al. Co-inhibitory role of T-cell-associated B7-H1and B7-DC in the T-cell immune response.Immunol Lett. 2006;102(2):222-228.
83. Francisco LM, Salinas VH, Brown KE, et al.PD-L1 regulates the development, mainte-nance, and function of induced regulatory Tcells. J Exp Med. 2009;206(13):3015-3029.
84. Terme M, Ullrich E, Aymeric L, et al. IL-18induces PD-1-dependent immunosuppres-sion in cancer. Cancer Res.2011;71(16):5393-5399.
85. Ahmadzadeh M, Johnson LA, Heemskerk B,et al. Tumor antigen-specific CD8 T cellsinfiltrating the tumor express high levels ofPD-1 and are functionally impaired. Blood.2009;114(8):1537-1544.
86. Iwai Y, Ishida M, Tanaka Y, et al.Involvement of PD-L1 on tumor cells in theescape from host immune system andtumor immunotherapy by PD-L1 blockade.Proc Natl Acad Sci USA. 2002;99(19):12293-12297.
87. Brahmer JR, Drake CG, Wollner I, et al.Phase I study of single-agent anti-pro-grammed death-1 (MDX-1106) in refractorysolid tumors: safety, clinical activity, phar-macodynamics, and immunologic corre-lates. J Clin Oncol. 2010;28(19):3167-3175.
88. Weber JS, Kudchadkar RR, Yu B, et al.Safety, efficacy, and biomarkers of nivolum-ab with vaccine in ipilimumab-refractory or-naive melanoma. J Clin Oncol.2013;31(34):4311-4318.
89. Armand P, Nagler A, Weller EA, et al.Disabling immune tolerance by pro-grammed death-1 blockade with pidilizum-ab after autologous hematopoietic stem-celltransplantation for diffuse large B-cell lym-phoma: results of an international phase IItrial. J Clin Oncol. 2013;31(33):4199-4206.
90. Westin JR, Chu F, Zhang M, et al. Safety andactivity of PD1 blockade by pidilizumab incombination with rituximab in patientswith relapsed follicular lymphoma: a singlegroup, open-label, phase 2 trial. LancetOncol. 2014;15(1):69-77.
91. Moskowitz CH, Ribrag V, Michot J-M, et al.PD-1 blockade with the monoclonal anti-body pembrolizumab (MK-3475) in patientswith classical Hodgkin lymphoma afterbrentuximab vedotin failure: preliminaryresults from a phase 1b study (KEYNOTE-013). Blood. 2014;124(21):290.
92. Ansell SM, Lesokhin AM, Borrello I, et al.PD-1 Blockade with nivolumab in relapsedor refractory Hodgkin's lymphoma. N Engl JMed. 2015;372(4):311-319.
93. Lesokhin AM, Ansell SM, Armand P, et al.Preliminary results of a phase I study ofnivolumab (BMS-936558) in patients withrelapsed or refractory lymphoid malignan-cies. Blood (ASH Annual MeetingAbstracts). 2014;124(21):291.
94. Porter DL, Levine BL, Kalos M, et al.Chimeric antigen receptor-modified T cellsin chronic lymphoid leukemia. N Engl JMed. 2011;365(8):725-733.
95. Maude SL, Frey N, Shaw PA, et al. Chimericantigen receptor T cells for sustained remis-sions in leukemia. N Engl J Med.2014;371(16):1507-1517.
96. Kochenderfer JN, Dudley ME, Kassim SH, etal. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignan-cies can be effectively treated with autolo-gous T cells expressing an anti-CD19chimeric antigen receptor. J Clin Oncol.2015;33(6):540-549.
97. Kochenderfer JN, Feldman SA, Zhao Y, et al.Construction and preclinical evaluation ofan anti-CD19 chimeric antigen receptor. JImmunother. 2009;32(7):689-702.
98. Vesely MD, Kershaw MH, Schreiber RD, etal. Natural innate and adaptive immunity tocancer. Annu Rev Immunol. 2011;29:235-271.
99. Pegram HJ, Lee JC, Hayman EG, et al.Tumor-targeted T cells modified to secreteIL-12 eradicate systemic tumors withoutneed for prior conditioning. Blood.2012;119(18):4133-4141.
100.Foster AE, Dotti G, Lu A, et al. Antitumoractivity of EBV-specific T lymphocytestransduced with a dominant negative TGF-beta receptor. J Immunother. 2008;31(5):500-505.
101.Dotti G, Savoldo B, Pule M, et al. Humancytotoxic T lymphocytes with reduced sen-sitivity to Fas-induced apoptosis. Blood.2005;105(12):4677-4684.
102.Eaton D, Gilham DE, O'Neill A, et al.Retroviral transduction of human peripheralblood lymphocytes with Bcl-X(L) promotesin vitro lymphocyte survival in pro-apoptot-ic conditions. Gene Ther. 2002;9(8):527-535.
103.Niiro H, Clark EA. Regulation of B-cell fateby antigen-receptor signals. Nat RevImmunol. 2002;2(12):945-956.
104.Kuppers R. Mechanisms of B-cell lymphomapathogenesis. Nat Rev Cancer. 2005;5(4):251-262.
105.Honigberg LA, Smith AM, Sirisawad M, etal. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is effica-cious in models of autoimmune disease andB-cell malignancy. Proc Natl Acad Sci USA.2010;107(29):13075-13080.
106.Byrd JC, Furman RR, Coutre SE, et al.Targeting BTK with ibrutinib in relapsedchronic lymphocytic leukemia. N Engl JMed. 2013;369(1):32-42.
107.Byrd JC, Brown JR, O'Brien S, et al. Ibrutinibversus ofatumumab in previously treatedchronic lymphoid leukemia. N Engl J Med.2014;371(3):213-223.
108.Wang ML, Rule S, Martin P, et al. TargetingBTK with ibrutinib in relapsed or refractorymantle-cell lymphoma. N Engl J Med.2013;369(6):507-516.
109.Treon SP, Tripsas CK, Meid K, et al. Ibrutinibin previously treated Waldenstrom'smacroglobulinemia. N Engl J Med.2015;372(15):1430-1440.
110.Ponader S, Chen SS, Buggy JJ, et al. TheBruton tyrosine kinase inhibitor PCI-32765thwarts chronic lymphocytic leukemia cellsurvival and tissue homing in vitro and invivo. Blood. 2012;119(5):1182-1189.
The microenvironment in B-cell lymphoid malignancies
haematologica | 2016; 101(5) 539

111.Ni Gabhann J, Spence S, Wynne C, et al.Defects in acute responses to TLR4 in Btk-deficient mice result in impaired dendriticcell-induced IFN-gamma production by nat-ural killer cells. Clin Immunol. 2012;142(3):373-382.
112.Khurana D, Arneson LN, Schoon RA, et al.Differential regulation of human NK cell-mediated cytotoxicity by the tyrosine kinaseItk. J Immunol. 2007;178(6):3575-3582.
113.Kohrt HE, Sagiv-Barfi I, Rafiq S, et al.Ibrutinib antagonizes rituximab-dependentNK cell-mediated cytotoxicity. Blood.2014;123(12):1957-1960.
114.Burger JA, Keating MJ, Wierda WG, et al.Safety and activity of ibrutinib plus ritux-imab for patients with high-risk chroniclymphocytic leukaemia: a single-arm, phase2 study. Lancet Oncol. 2014;15(10):1090-1099.
115.Dubovsky JA, Beckwith KA, Natarajan G, etal. Ibrutinib is an irreversible molecularinhibitor of ITK driving a Th1-selective pres-sure in T lymphocytes. Blood. 2013;122(15):2539-2549.
116.Sagiv-Barfi I, Kohrt HE, Burckhardt L, et al.Ibrutinib enhances the antitumor immuneresponse induced by intratumoral injectionof a TLR9 ligand in mouse lymphoma.Blood. 2015;125(13):2079-2086.
117.Sagiv-Barfi I, Kohrt HE, Czerwinski DK, etal. Therapeutic antitumor immunity bycheckpoint blockade is enhanced by ibruti-nib, an inhibitor of both BTK and ITK. ProcNatl Acad Sci USA. 2015;112(9):E966-972.
118.Ni Gabhann J, Hams E, Smith S, et al. Btkregulates macrophage polarization inresponse to lipopolysaccharide. PLoS One.2014;9(1):e85834.
119.Gopal AK, Kahl BS, de Vos S, et al. PI3Kδinhibition by idelalisib in patients withrelapsed indolent lymphoma. N Engl J Med.2014;370(11):1008-1018.
120.Flinn IW, Kahl BS, Leonard JP, et al. Idelalisib,a selective inhibitor of phosphatidylinositol3-kinase-delta, as therapy for previouslytreated indolent non-Hodgkin lymphoma.Blood. 2014;123(22):3406-3413.
121.Brown JR, Byrd JC, Coutre SE, et al.Idelalisib, an inhibitor of phosphatidylinosi-tol 3-kinase p110delta, for relapsed/refracto-ry chronic lymphocytic leukemia. Blood.2014;123(22):3390-3397.
122.Lannutti BJ, Meadows SA, Herman SE, et al.CAL-101, a p110delta selective phos-phatidylinositol-3-kinase inhibitor for thetreatment of B-cell malignancies, inhibitsPI3K signaling and cellular viability. Blood.2011;117(2):591-594.
123.Hoellenriegel J, Meadows SA, Sivina M, etal. The phosphoinositide 3'-kinase deltainhibitor, CAL-101, inhibits B-cell receptorsignaling and chemokine networks in chron-ic lymphocytic leukemia. Blood.2011;118(13):3603-3612.
124.Maffei R, Bulgarelli J, Fiorcari S, et al.Endothelin-1 promotes survival andchemoresistance in chronic lymphocyticleukemia B cells through ETA receptor. PLoSOne. 2014;9(6):e98818.
125.Roit FD, Engelberts PJ, Taylor RP, et al.Ibrutinib interferes with the cell-mediatedanti-tumor activities of therapeutic CD20antibodies: implications for combinationtherapy. Haematologica. 2015;100(1):77-86.
126.Herman SE, Gordon AL, Wagner AJ, et al.
Phosphatidylinositol 3-kinase-delta inhibitorCAL-101 shows promising preclinical activ-ity in chronic lymphocytic leukemia byantagonizing intrinsic and extrinsic cellularsurvival signals. Blood. 2010;116(12):2078-2088.
127.Dong S, Guinn D, Dubovsky JA, et al. IPI-145 antagonizes intrinsic and extrinsic sur-vival signals in chronic lymphocyticleukemia cells. Blood. 2014;124(24):3583-3586.
128.Witzig TE, Vose JM, Zinzani PL, et al. Aninternational phase II trial of single-agentlenalidomide for relapsed or refractoryaggressive B-cell non-Hodgkin's lymphoma.Ann Oncol. 2011;22(7):1622-1627.
129.Wiernik PH, Lossos IS, Tuscano JM, et al.Lenalidomide monotherapy in relapsed orrefractory aggressive non-Hodgkin's lym-phoma. J Clin Oncol. 2008;26(30):4952-4957.
130.Habermann TM, Lossos IS, Justice G, et al.Lenalidomide oral monotherapy produces ahigh response rate in patients with relapsedor refractory mantle cell lymphoma. Br JHaematol. 2009;145(3):344-349.
131.Zinzani PL, Vose JM, Czuczman MS, et al.Long-term follow-up of lenalidomide inrelapsed/refractory mantle cell lymphoma:subset analysis of the NHL-003 study. AnnOncol. 2013;24(11):2892-2897.
132.Fowler NH, Davis RE, Rawal S, et al. Safetyand activity of lenalidomide and rituximabin untreated indolent lymphoma: an open-label, phase 2 trial. Lancet Oncol.2014;15(12):1311-1318.
133.Wang M, Fowler N, Wagner-Bartak N, et al.Oral lenalidomide with rituximab inrelapsed or refractory diffuse large cell, follic-ular and transformed lymphoma: a phase IIclinical trial. Leukemia. 2013;27(9):1902-1909.
134.Wang M, Fayad L, Wagner-Bartak N, et al.Lenalidomide in combination with ritux-imab for patients with relapsed or refractorymantle-cell lymphoma: a phase 1/2 clinicaltrial. Lancet Oncol. 2012;13(7):716-723.
135.Kimby E, Martinelli G, Ostenstad B, et al.Rituximab plus lenalidomide improves thecomplete remission rate in comparison withrituximab monotherapy in untreated follicu-lar lymphoma patients in need of therapy.Primary endpoint analysis of the random-ized phase-2 trial SAKK 35/10. Blood.2014;124(21):799.
136.Martin P, Jung S-H, Johnson JL, et al.CALGB 50803 (Alliance): a phase II trial oflenalidomide plus rituximab in patientswith previously untreated follicular lym-phoma. ASCO Meeting Abstracts. 2014;32(15_suppl):8521.
137.Ruan J, Martin P, Shah BD, et al. Sustainedremission with the combination biologicdoublet of lenalidomide plus rituximab asinitial treatment for mantle cell lymphoma: amulti-center phase II study report. Blood(ASH Annual Meeting Abstracts). 2014;124(21):625.
138.Gandhi AK, Kang J, Havens CG, et al.Immunomodulatory agents lenalidomideand pomalidomide co-stimulate T cells byinducing degradation of T cell repressorsIkaros and Aiolos via modulation of the E3ubiquitin ligase complex CRL4(CRBN.). Br JHaematol. 2014;164(6):811-821.
139.Lu G, Middleton RE, Sun H, et al. The
myeloma drug lenalidomide promotes thecereblon-dependent destruction of Ikarosproteins. Science. 2014;343(6168):305-309.
140.Kronke J, Udeshi ND, Narla A, et al.Lenalidomide causes selective degradationof IKZF1 and IKZF3 in multiple myelomacells. Science. 2014;343(6168):301-305.
141.Lee BN, Gao H, Cohen EN, et al. Treatmentwith lenalidomide modulates T-cellimmunophenotype and cytokine produc-tion in patients with chronic lymphocyticleukemia. Cancer. 2011;117(17):3999-4008.
142.Henry JY, Labarthe MC, Meyer B, et al.Enhanced cross-priming of naive CD8+ Tcells by dendritic cells treated by theIMiDs® immunomodulatory compoundslenalidomide and pomalidomide.Immunology. 2013;139(3):377-385.
143.Maffei R, Fiorcari S, Bulgarelli J, et al.Endothelium-mediated survival of leukemiccells and angiogenesis-related factors areaffected by lenalidomide treatment inchronic lymphocytic leukemia. ExpHematol. 2014;42(2):126-136 e121.
144.Fiorcari S, Martinelli S, Bulgarelli J, et al.Lenalidomide interferes with tumor-pro-moting properties of nurse-like cells inchronic lymphocytic leukemia.Haematologica. 2015;100(2):253-262.
145.Zhang L, Qian Z, Cai Z, et al. Synergisticantitumor effects of lenalidomide and ritux-imab on mantle cell lymphoma in vitro andin vivo. Am J Hematol. 2009;84(9):553-559.
146.Wu L, Adams M, Carter T, et al. lenalido-mide enhances natural killer cell and mono-cyte-mediated antibody-dependent cellularcytotoxicity of rituximab-treated CD20+tumor cells. Clin Cancer Res. 2008;14(14):4650-4657.
147.Lagrue K, Carisey A, Morgan DJ, et al.Lenalidomide augments actin remodellingand lowers NK cell activation thresholds.Blood. 2015;126(1):50-60.
148.Chen DS, Mellman I. Oncology meetsimmunology: the cancer-immunity cycle.Immunity. 2013;39(1):1-10.
149.Ansell SM, Hurvitz SA, Koenig PA, et al.Phase I study of ipilimumab, an anti-CTLA-4 monoclonal antibody, in patients withrelapsed and refractory B-cell non-Hodgkinlymphoma. Clin Cancer Res. 2009;15(20):6446-6453.
150.Kochenderfer JN, Rosenberg SA. Chimericantigen receptor-modified T cells in CLL. NEngl J Med. 2011;365(20):1937-1938.
151.Cheah CY, Nastoupil LJ, Neelapu SS, et al.Lenalidomide, idelalisib, and rituximab areunacceptably toxic in patients withrelapsed/refractory indolent lymphoma.Blood. 2015;125(21):3357-3359.
152.Smith S, Pitcher BN, Jung SH, et al.Unexpected and serious toxicity observedwith combined idelalisib, lenalidomideand rituximab in relapsed/refractory B celllymphomas: Alliance A051201 andA051202 (Abstract 3091). Blood. 2014;124(21):3091.
153.Ansell SM, Lesokhin AM, Borrello I, et al.PD-1 blockade with nivolumab in relapsedor refractory Hodgkin's lymphoma. N Engl JMed. 2015;372(4):311-319.
154.Chanan-Khan A, Miller KC, Musial L, et al.Clinical efficacy of lenalidomide in patientswith relapsed or refractory chronic lympho-cytic leukemia: results of a phase II study. JClin Oncol. 2006;24(34):5343-5349.
N.H. Fowler et al.
540 haematologica | 2016; 101(5)

haematologica | 2016; 101(5) 541
Received: November 12, 2015.
Accepted: January 20, 2016.
©2016 Ferrata Storti Foundation
Check the online version for the most updatedinformation on this article, online supplements,and information on authorship & disclosures:www.haematologica.org/content/101/5/541
Material published in Haematologica is cov-ered by copyright. All rights reserved to theFerrata Storti Foundation. Copies of articlesare allowed for personal or internal use.Permission in writing from the publisher isrequired for any other use.
Correspondence: [email protected]@tuftsmedicalcenter.org
Chronic myeloid leukemia: reminiscences and dreamsTariq I. Mughal,1 Jerald P. Radich,2 Michael W. Deininger,3 Jane F. Apperley,4Timothy P. Hughes,5 Christine J. Harrison,6 Carlo Gambacorti-Passerini,7Giuseppe Saglio,8 Jorge Cortes,9 and George Q. Daley10
1Tufts University Medical Center, Boston, MA, USA; 2Fredrick Hutchinson Cancer Center,University of Washington, Seattle, WA, USA; 3University of Utah, Huntsman CancerInstitute, Salt Lake City, UT, USA; 4Imperial College London, Hammersmith Hospital, UK;5University of Adelaide, Australia; 6Newcastle University, Newcastle-upon-Tyne, UK;7University of Milano-Bicocca, Monza, Italy; 8University of Turin, Italy; 9MD AndersonCancer Center, Houston, TX, USA; and 10Boston Children’s Hospital, Harvard MedicineSchool, Boston, MA, USA
Ferrata StortiFoundation
EUROPEANHEMATOLOGYASSOCIATION
Haematologica 2016Volume 101(5):541-558
REVIEW ARTICLE
doi:10.3324/haematol.2015.139337
With the deaths of Janet Rowley and John Goldman in December2013, the world lost two pioneers in the field of chronic myeloidleukemia. In 1973, Janet Rowley, unraveled the cytogenetic
anatomy of the Philadelphia chromosome, which subsequently led to theidentification of the BCR-ABL1 fusion gene and its principal pathogeneticrole in the development of chronic myeloid leukemia. This work was alsoof major importance to support the idea that cytogenetic changes weredrivers of leukemogenesis. John Goldman originally made seminal contri-butions to the use of autologous and allogeneic stem cell transplantationfrom the late 1970s onwards. Then, in collaboration with Brian Druker, heled efforts to develop ABL1 tyrosine kinase inhibitors for the treatment ofpatients with chronic myeloid leukemia in the late 1990s. He also led theglobal efforts to develop and harmonize methodology for molecular moni-toring, and was an indefatigable organizer of international conferences.These conferences brought together clinicians and scientists, and accelerat-ed the adoption of new therapies. The abundance of praise, tributes andtestimonies expressed by many serve to illustrate the indelible impressionsthese two passionate and affable scholars made on so many people’s lives.This tribute provides an outline of the remarkable story of chronic myeloidleukemia, and in writing it, it is clear that the historical triumph of biomed-ical science over this leukemia cannot be considered without appreciatingthe work of both Janet Rowley and John Goldman.
ABSTRACT
Introduction: the power of targeted therapy
The biology and treatment of patients with chronic myeloid leukemia (CML), arare heterogeneous clonal hematopoietic stem cell disorder characterized by a con-sistent cytogenetic abnormality (the Philadelphia chromosome) and the presence ofthe BCR-ABL1 fusion gene, must surely be ranked as one of the most successfulcancer medicine stories of the past century. The BCR-ABL1 fusion gene encodes theoncoprotein BCR-ABL1 (also referred to as p210 or BCR-ABL) with a constitutiveactive tyrosine kinase activity that is the primary cause of the chronic phase ofCML.1,2 The discovery in 1996 that this kinase activity could be pharmacologicallyinactivated by a modified 2-phenylaminopyrimidine paved the way for the suc-cessful introduction of imatinib (also known as STI571, glivec, or gleevec) as an ini-tial oral treatment for newly diagnosed CML patients.3 Imatinib, now termed a 1st-generation tyrosine kinase inhibitor (TKI), substantially and durably reduces thenumber of CML cells in the chronic phase at a daily oral dose of 400 mg, and hasimproved the 10-year survival rates from less than 20% to around 83% (Figure 1).4

The greatest advance is in those patients who achieve acomplete cytogenetic response (CCyR) within two yearsof starting imatinib leading to life spans indistinguishablefrom the general population.5 These impressive resultswith imatinib therapy have had profound effects on thenatural history of CML and its prevalence. Current esti-mates suggest that in the USA, where about 5500 newcases are diagnosed annually, the prevalence will increaseto about 120,000 by 2020 and to about 200,000 by 2050.6
However, imatinib is far from perfect, with only approx-imately 60% of patients remaining on the standard dailydose of 400 mg after six years due to either lack of drugtolerance or drug resistance.7 Imatinib is inducing respons-es also in the more advanced phases of CML, but theseresponses are not durable. There are now four newerTKIs, three so-called 2nd-generation inhibitors and one 3rd-generation inhibitor, all of which are more potent thanimatinib in in vitro assays. Of the 2nd-generation drugs, nilo-tinib (also known as AMN107) and dasatinib (also knownas BMS-354825) are licensed in the US and many otherparts of the world for patients with CML in the chronicphase as first-line and subsequent therapies, while bosu-tinib (also known as SKI-606), is currently licensed forCML patients resistant or refractory to first-line drugs andis anticipated to be approved for first-line use in the nearfuture. The 3rd-generation inhibitor ponatinib (also knownas AP24534), is the newest and is licensed for CMLpatients who either have a T315I mutation or who fail torespond to any of the other currently approved TKIs.Current experience suggests both nilotinib and dasatinibachieve deeper and faster molecular responses than ima-tinib, but the precise benefits of such responses remain anenigma. Thus far, there is little evidence of a statisticallysignificant improvement in overall survival (OS), thoughlong-term follow up confirmed a superior rate of freedomfrom progression compared with patients with less deepmolecular responses at the same time points.8
The advent of TKIs in the treatment of CML has openeda new era of precision medicine for diverse malignanciesin which relatively non-specific and often toxic drugs aregradually being replaced by safer and better toleratedagents whose mechanism of action is precisely defined,and for which the treatment algorithm is guided by indi-vidual patient genomic information.9 Indeed, many TKIshave activity against other tyrosine kinases and could,therefore, be useful in treating patients whose malignan-cies harbor these gene mutations. In this review, we dis-cuss the various milestones in the study, diagnosis, moni-toring and treatment of CML, and speculate on the notionof cure and candidates for future therapy.10
Cytogenetics and molecular biology
Claims of priority can almost always be challenged butit is generally agreed that Alfred Velpeau in France be cred-ited with the first detailed description of what must havebeen leukemia in 1827.11 As a result of astute clinical obser-vations, he described a 63-year old florist and lemonadesalesman who presented with gross hepatosplenomegalyand was noted to have “globules of pus” in his blood. Theprecise diagnosis, however, remained elusive. The firstplausible story of what we now know as CML probably
began in 1845 almost simultaneously by John Bennett inEdinburgh and Rudolph Virchow in Berlin.12,13 They bothpublished accurate case reports and probably neither wereaware of the other’s publication until later. Major progressin both the therapy and, indeed, the understanding of thedisease did not occur until 1960. Figure 2 depicts the prin-cipal milestones in the study and treatment of CML.
Janet Rowley defines the cytogenetics of thePhiladelphia chromosomeFollowing the discovery by Joe Tjo and Albert Levan in
1956 that humans have 46 chromosomes, many effortswere directed to the study of chromosomal abnormalitiesin human cancers.14 By 1959, reports pertaining to thepresence of constitutional abnormalities related to partic-ular phenotypes began to appear, the most well knownbeing the association of the gain of chromosome 21 inpatients with Down syndrome.15 The work of PeterNowell and David Hungerford led in 1960 to the discov-ery of the Philadelphia (Ph) chromosome.16 These investi-gators were tinkering with cytological techniques, whichrevealed metaphase spreads in bone marrow by acciden-tally rinsing slides with water. Among a series of bonemarrow samples from patients with leukemia were 2males with CML, in which they observed a “minute”chromosome. From cutting out the chromosomes fromphotographs of metaphases and laying them in rowsaccording to centromere position and size, they deducedthat this abnormal chromosome was a deletion of the Ychromosome. As these 2 patients had received therapy,there was some debate that the chromosomal abnormalityhad resulted from chromosomal damage induced by thetreatment. Following additional work, they speculatedthat the chromosomal abnormality was probably not con-stitutive and may well be causally associated to CML.
At around the same time, Balkie and colleagues madethe same discovery in Edinburgh, Scotland.17 Theyshowed the presence of the same “small” chromosome inbone marrow and blood samples, but not in skin cells.With this observation, they were able to conclude that theabnormal chromosome was an acquired abnormalityassociated with the leukemia, particularly as the bonemarrow and blood samples contained a high level ofmyeloblasts. In addition, a number of their patients wereuntreated, thus refuting the claim that the abnormalitywas therapy induced. They concluded that this small
T.I. Mughal et al.
542 haematologica | 2016; 101(5)
Janet Rowley and John Goldman.

chromosome was derived from the group of small acro-centric chromosomes, known as chromosomes 21 and 22.As Down syndrome is associated with an increased risk ofdeveloping leukemia, although not CML, they made theassumption that the small chromosome must have arisenfrom a chromosome 21 and that the most likely explana-tion was a deletion. The Ph chromosomal abnormalitywas heralded as the first consistent cytogenetic abnormal-ity in a human malignancy and the superscript ‘1’ wasadded, Ph1, on the premise that additional abnormalitieswould be discovered. This did not occur and the super-script had been dropped by 1990. The formal recognitionthat a human cancer might be caused by an acquired chro-mosomal aberration, vindicated the hypothesis postulatedby Theodore Boveri in Germany in 1914 that cancer maybe caused by acquired chromosomal abnormalities.18 Thenext important observations which established that CMLwas a stem cell-derived clonal disease came from PhillipFialkow and colleagues in 1967.19 They applied a genetictechnique developed by Ohno et al. based on X chromo-some mosaicism in females, and by demonstrating poly-morphism in the X-linked glucose-6-phosphatase dehy-drogenase locus, established the clonal nature of CML.20
With the advent of the new chromosomal banding tech-niques in the early 1970s, it became possible to accuratelyidentify the individual chromosome for the first time.Janet Rowley from Chicago used these techniques, whichshe had learnt whilst on a visit to Oxford, UK. Amongsamples from patients with hematologic malignancies col-lected over previous years were those from patients withCML in whom the Ph chromosome was present. Whilstlaboriously comparing the chromosomal preparationsmade in the conventional manner with those preparedusing these novel approaches, she noted on chromosome9 “a duly fluorescein segment resembling the end of chromosome22, but equally other chromosomes”. This remarkable observa-tion of the balanced reciprocal translocation of geneticmaterial between the long arms of chromosomes 9 and 22,t(9;22)(q34;q11) was published with difficulty in Nature inJune 1973 (Figure 3).21 There were some valuable quota-tions in that paper which remain unchanged to this day:“the mechanism for the production of such a specific chromosomaltranslocation (if this is the correct explanation of these findings) isnot clear”; “this would constitute the only specific translocation in
humans that has been identified”; “this abnormality is involved ininitiation rather than a consequence”.
The Molecular Biology StoryJanet Rowley’s seminal work in deciphering the Ph
chromosome provided the framework for the unravelingof the genomic architecture, structure and function of theoncogene driving CML, which would become known asBCR-ABL1. These molecular events began in 1982, whenNora Heisterkamp et al. in Rotterdam, the Netherlands,observed that c-Abl, the human homolog of v-Abl, theoncogene of a murine leukemia virus first described byAbelson in 1970, localized to human chromosome 9.21-24This discovery rekindled interest in a possible role of c-Ablin Ph-positive leukemia, even after attempts to demon-strate transforming capacity for c-Abl had proven unsuc-cessful.
The proof that c-abl was implicated in the Ph transloca-tion was achieved on the basis of somatic cell hybridsgenerated by fusions of murine or hamster cell lines withcells from CML patients and healthy controls. These linescontained the rearranged chromosomes from the Phtranslocation or their normal counterparts. Southern blotanalysis was performed on DNA from the various
Chronic myeloid leukemia: reminiscences and dreams
haematologica | 2016; 101(5) 543
Figure 2. Milestones in the study and treatment of chronic myeloid leukemia.
Figure 1. Survival with chronic myeloid leukemia over time (1993-2013): theGerman CML-Study Group experience. Courtesy of Prof H Kantarjian; adapted,with permission, from Harrison’s Principles of Internal Medicine, 2014.
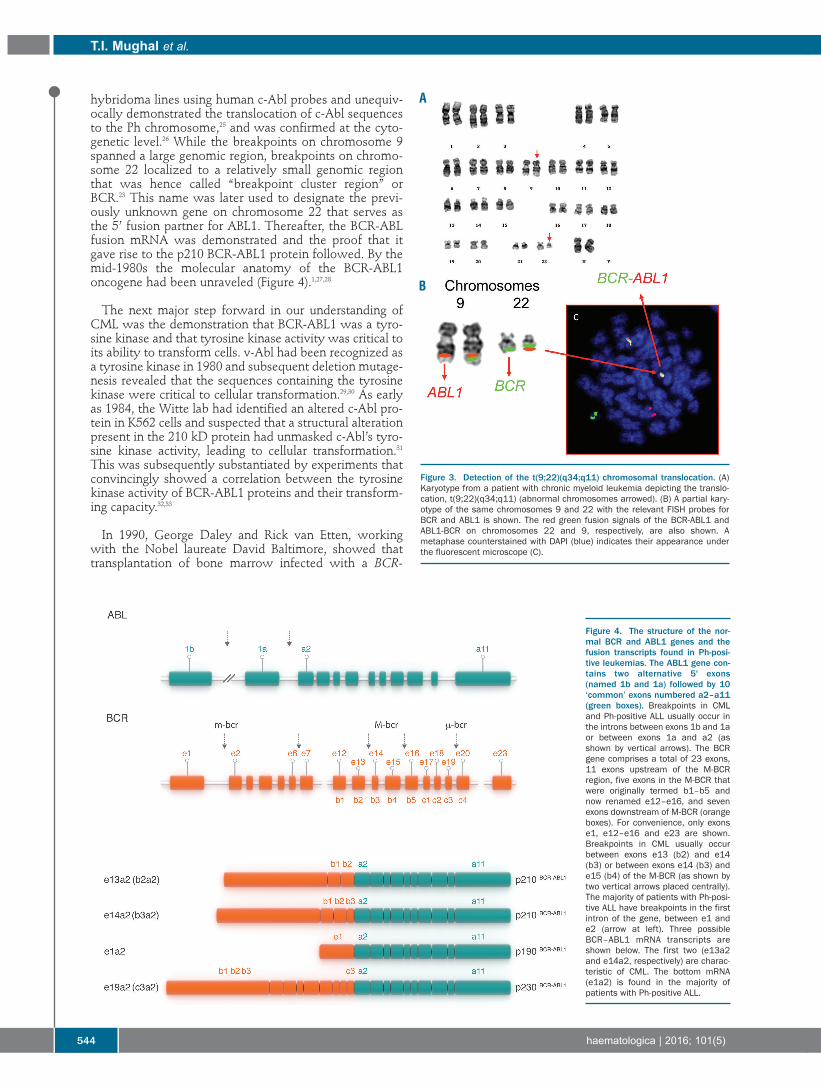
hybridoma lines using human c-Abl probes and unequiv-ocally demonstrated the translocation of c-Abl sequencesto the Ph chromosome,25 and was confirmed at the cyto-genetic level.26 While the breakpoints on chromosome 9spanned a large genomic region, breakpoints on chromo-some 22 localized to a relatively small genomic regionthat was hence called “breakpoint cluster region” orBCR.23 This name was later used to designate the previ-ously unknown gene on chromosome 22 that serves asthe 5’ fusion partner for ABL1. Thereafter, the BCR-ABLfusion mRNA was demonstrated and the proof that itgave rise to the p210 BCR-ABL1 protein followed. By themid-1980s the molecular anatomy of the BCR-ABL1oncogene had been unraveled (Figure 4).1,27,28
The next major step forward in our understanding ofCML was the demonstration that BCR-ABL1 was a tyro-sine kinase and that tyrosine kinase activity was critical toits ability to transform cells. v-Abl had been recognized asa tyrosine kinase in 1980 and subsequent deletion mutage-nesis revealed that the sequences containing the tyrosinekinase were critical to cellular transformation.29,30 As earlyas 1984, the Witte lab had identified an altered c-Abl pro-tein in K562 cells and suspected that a structural alterationpresent in the 210 kD protein had unmasked c-Abl’s tyro-sine kinase activity, leading to cellular transformation.31This was subsequently substantiated by experiments thatconvincingly showed a correlation between the tyrosinekinase activity of BCR-ABL1 proteins and their transform-ing capacity.32,33
In 1990, George Daley and Rick van Etten, workingwith the Nobel laureate David Baltimore, showed thattransplantation of bone marrow infected with a BCR-
T.I. Mughal et al.
544 haematologica | 2016; 101(5)
Figure 3. Detection of the t(9;22)(q34;q11) chromosomal translocation. (A)Karyotype from a patient with chronic myeloid leukemia depicting the translo-cation, t(9;22)(q34;q11) (abnormal chromosomes arrowed). (B) A partial kary-otype of the same chromosomes 9 and 22 with the relevant FISH probes forBCR and ABL1 is shown. The red green fusion signals of the BCR-ABL1 andABL1-BCR on chromosomes 22 and 9, respectively, are also shown. Ametaphase counterstained with DAPI (blue) indicates their appearance underthe fluorescent microscope (C).
Figure 4. The structure of the nor-mal BCR and ABL1 genes and thefusion transcripts found in Ph-posi-tive leukemias. The ABL1 gene con-tains two alternative 5' exons(named 1b and 1a) followed by 10‘common’ exons numbered a2–a11(green boxes). Breakpoints in CMLand Ph-positive ALL usually occur inthe introns between exons 1b and 1aor between exons 1a and a2 (asshown by vertical arrows). The BCRgene comprises a total of 23 exons,11 exons upstream of the M-BCRregion, five exons in the M-BCR thatwere originally termed b1–b5 andnow renamed e12–e16, and sevenexons downstream of M-BCR (orangeboxes). For convenience, only exonse1, e12–e16 and e23 are shown.Breakpoints in CML usually occurbetween exons e13 (b2) and e14(b3) or between exons e14 (b3) ande15 (b4) of the M-BCR (as shown bytwo vertical arrows placed centrally).The majority of patients with Ph-posi-tive ALL have breakpoints in the firstintron of the gene, between e1 ande2 (arrow at left). Three possibleBCR–ABL1 mRNA transcripts areshown below. The first two (e13a2and e14a2, respectively) are charac-teristic of CML. The bottom mRNA(e1a2) is found in the majority ofpatients with Ph-positive ALL.
A
B
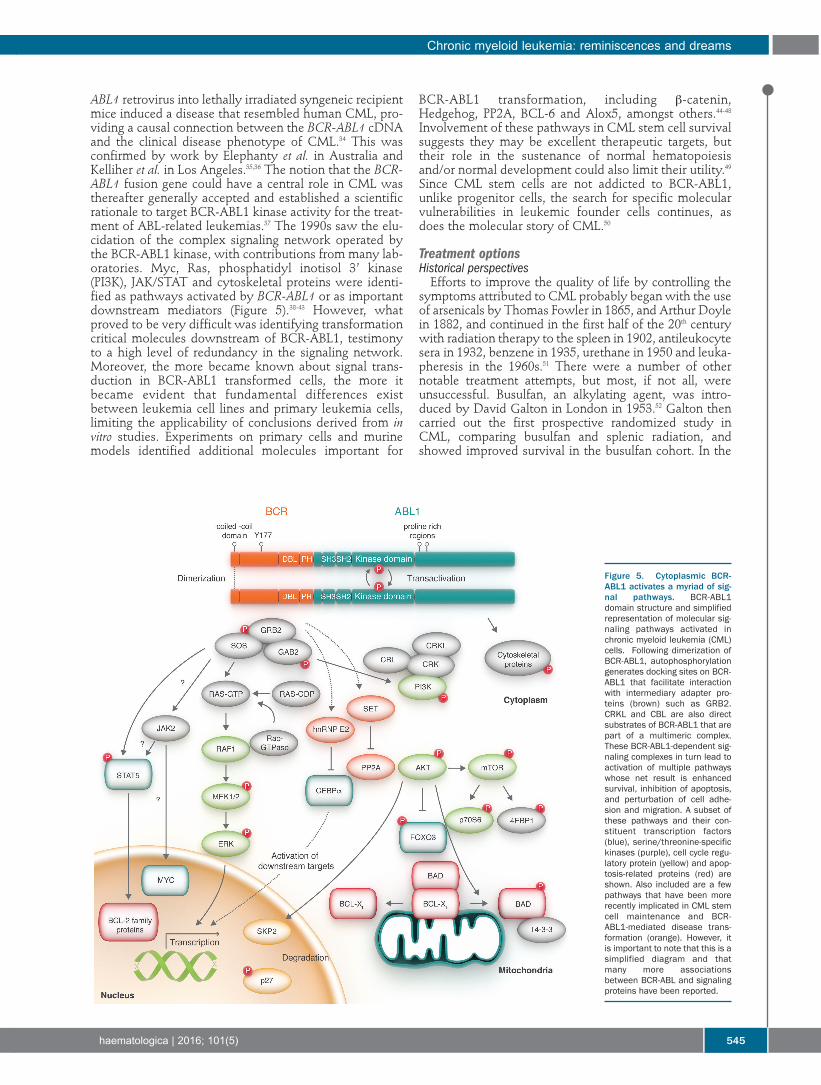
ABL1 retrovirus into lethally irradiated syngeneic recipientmice induced a disease that resembled human CML, pro-viding a causal connection between the BCR-ABL1 cDNAand the clinical disease phenotype of CML.34 This wasconfirmed by work by Elephanty et al. in Australia andKelliher et al. in Los Angeles.35,36 The notion that the BCR-ABL1 fusion gene could have a central role in CML wasthereafter generally accepted and established a scientificrationale to target BCR-ABL1 kinase activity for the treat-ment of ABL-related leukemias.37 The 1990s saw the elu-cidation of the complex signaling network operated bythe BCR-ABL1 kinase, with contributions from many lab-oratories. Myc, Ras, phosphatidyl inotisol 3’ kinase(PI3K), JAK/STAT and cytoskeletal proteins were identi-fied as pathways activated by BCR-ABL1 or as importantdownstream mediators (Figure 5).38-43 However, whatproved to be very difficult was identifying transformationcritical molecules downstream of BCR-ABL1, testimonyto a high level of redundancy in the signaling network.Moreover, the more became known about signal trans-duction in BCR-ABL1 transformed cells, the more itbecame evident that fundamental differences existbetween leukemia cell lines and primary leukemia cells,limiting the applicability of conclusions derived from invitro studies. Experiments on primary cells and murinemodels identified additional molecules important for
BCR-ABL1 transformation, including β-catenin,Hedgehog, PP2A, BCL-6 and Alox5, amongst others.44-48Involvement of these pathways in CML stem cell survivalsuggests they may be excellent therapeutic targets, buttheir role in the sustenance of normal hematopoiesisand/or normal development could also limit their utility.49Since CML stem cells are not addicted to BCR-ABL1,unlike progenitor cells, the search for specific molecularvulnerabilities in leukemic founder cells continues, asdoes the molecular story of CML.50
Treatment optionsHistorical perspectives Efforts to improve the quality of life by controlling the
symptoms attributed to CML probably began with the useof arsenicals by Thomas Fowler in 1865, and Arthur Doylein 1882, and continued in the first half of the 20th centurywith radiation therapy to the spleen in 1902, antileukocytesera in 1932, benzene in 1935, urethane in 1950 and leuka-pheresis in the 1960s.51 There were a number of othernotable treatment attempts, but most, if not all, wereunsuccessful. Busulfan, an alkylating agent, was intro-duced by David Galton in London in 1953.52 Galton thencarried out the first prospective randomized study inCML, comparing busulfan and splenic radiation, andshowed improved survival in the busulfan cohort. In the
Chronic myeloid leukemia: reminiscences and dreams
haematologica | 2016; 101(5) 545
Figure 5. Cytoplasmic BCR-ABL1 activates a myriad of sig-nal pathways. BCR-ABL1domain structure and simplifiedrepresentation of molecular sig-naling pathways activated inchronic myeloid leukemia (CML)cells. Following dimerization ofBCR-ABL1, autophosphorylationgenerates docking sites on BCR-ABL1 that facilitate interactionwith intermediary adapter pro-teins (brown) such as GRB2.CRKL and CBL are also directsubstrates of BCR-ABL1 that arepart of a multimeric complex.These BCR-ABL1-dependent sig-naling complexes in turn lead toactivation of multiple pathwayswhose net result is enhancedsurvival, inhibition of apoptosis,and perturbation of cell adhe-sion and migration. A subset ofthese pathways and their con-stituent transcription factors(blue), serine/threonine-specifickinases (purple), cell cycle regu-latory protein (yellow) and apop-tosis-related proteins (red) areshown. Also included are a fewpathways that have been morerecently implicated in CML stemcell maintenance and BCR-ABL1-mediated disease trans-formation (orange). However, itis important to note that this is asimplified diagram and thatmany more associationsbetween BCR-ABL and signalingproteins have been reported.

mid-1960s, busulfan was replaced by hydroxycarbamide(previously hydroxyurea), a ribonucleotide reductaseinhibitor, following recognition that busulfan is mutagenic,and a randomized study confirming the superiority ofhydroxycarbamide, though neither drug was able toreduce the proportion of Ph positive cells or prolong overallsurvival.53,54 Interferon alpha (IFN-α) was introduced intothe clinics in the mid-1980s and proved popular, despitefrequent side-effects such as flu-like symptoms andfatigue.55 In the early 1990s, several randomized studiescomparing IFN-α or interferon-α n1 (wellferon) withhydroxycarbamide or busulfan were undertaken anddemonstrated an improvement in overall survival by about2-3 years with IFN-α.56-58 In addition, a French study testingthe addition of cytarabine to IFN-2b found this to result inan increased proportion of patients achieving a cytogeneticresponse.59 Thereafter, interferon, either alone or in combi-nation with cytarabine, replaced hydroxycarbamide as thepreferred treatment for CML in the chronic phase.60 Theprecise mode of action of IFN-α remains unclear, but isprobably related to its immunomodulatory properties. IFN-α was replaced by imatinib as the preferred treatment forpatients with CML in the chronic phase in the summer of2001 following a randomized study comparing imatinibwith IFN-α plus cytarabine.61-63 The results were veryimpressive and established the firm position of imatinib,and also constituted the final proof of the importance ofthe BCR-ABL1 oncoprotein to CML. The introduction ofimatinib was rapidly followed by the development of thenext generation tyrosine kinase inhibitors (TKIs).
Prognostic and predictive factorsVarious efforts have been made to establish criteria
definable at diagnosis that may help to predict responseto therapy and survival for individual patients.Historically, the Sokal score was developed in 1984 forpatients treated with busulfan, and the Hasford (alsoknown as the Euro) score in 1998 for patients treated withIFN-α.64,65 Both scoring systems have since been con-firmed to be useful in the TKI era. Stratifying patients intogood-, intermediate-, and poor-risk categories may assistin the decision-making process regarding appropriatetreatment options. In 2011, a simpler and TKI-specificscore, the European Treatment and Outcome Study(EUTOS), was proposed but requires further confirmationbefore it can be widely used.66 More recently, theresponse to TKIs at a given time point, disease risk andstage, BCR-ABL1 genotype, the presence of comorbidi-ties, financial aspects and local monitoring capabilities areall being increasingly used to personalize treatment.67
The Imatinib StoryIt is remarkable how, in 1994, against a background of
considerable skepticism about any possible clinical value ofTKIs, Brian Druker in Portland, Oregan, and collaboratingscientists at Ciba-Geigy (now Novartis) in Basel,Switzerland, developed a compound, imatinib, that couldreverse the clinical and hematologic features of CML.3,68Imatinib, a 2-phenylaminopyrimidine, inhibits the enzy-matic action of the activated BCR-ABL1 tyrosine kinase byoccupying the ATP-binding pocket of the tyrosine kinasecomponent of the BCR-ABL1 oncoprotein, thereby block-ing the capacity of the enzyme to phosphorylate and acti-vate downstream effector molecules that cause theleukemic phenotype. It also binds to an adjacent part of thekinase domain in a manner that holds the ABL-activationloop of the oncoprotein in an inactive configuration (Figure6).69 The International Randomized Study of Interferon andSTI571 (IRIS) demonstrated that imatinib induced ‘cumula-tive best’ CCyR, equivalent to a 2-log reduction in BCR-ABL1 transcripts level, in 82% of all previously untreatedpatients with CML in the chronic phase.60,70 About 2% ofall patients in the chronic phase progress to advanced-phase disease each year, which contrasts with estimatedannual progression rates of more than 15% for patientstreated with hydroxycarbamide and about 10% forpatients receiving IFN-α, either with or without cytara-bine.4,71 The 8-year event-free survival was 83% and theestimated overall survival was 93% (corrected for CML-related deaths only), confirming the notion that imatinib
T.I. Mughal et al.
546 haematologica | 2016; 101(5)
Table 1. Definitions of response.Type of response Definition
CHR Complete hematologic response Normal differential, WBC and platelets within the normal rangeMCyR Major cytogenetic response 0%-35% Ph+ marrow metaphasesCCyR Complete cytogenetic response 0% Ph+ marrow metaphasesMMR Major molecular response BCR-ABL1/ABL1 ratio ≤ 0.1% (international scale)MR4.0 BCR-ABL1/ABL1 ratio ≤ 0.01% (international scale): this is a 4-log reductionMR4.5 BCR-ABL1/ABL1 ratio ≤ 0.003% (international scale): this is a 4.5-log reductionCMR Complete molecular response Undetectable BCR-ABL1 (test of sensitivity ≥ 4.5 logs)
WBC: white blood cells.
Figure 6. Imatinib binds an Inactive ABL1 conformation. Adapted, with permis-sion, from Schindler et al. Science 2000.

substantially prolongs overall survival compared with his-torical patients who received IFN-α or hydroxycar-bamide.72 A substantial proportion of the patients in CCyRalso achieve a 3-log reduction or more in BCR-ABL1 tran-scripts (referred to as MMR, or MR3), and this proportionseems to have continued to increase steadily with time onimatinib. A minority of patients achieve a deeper molecu-lar response with more than 4-log or 4.5-log reduction inBCR-ABL1 transcripts [referred to as MR4.0 and MR,4.5respectively; MR4.5 was previously referred to as a completemolecular response (CMR)] (Table 1).73,74 These results wereconfirmed by independent single centers as well as compa-ny-led registration studies.75 It should also be said that thesuccess of these and other CML treatment studies epito-mize the critical importance of an optimal molecular mon-itoring methodology (see below).
The standard starting daily dose of imatinib is 400 mgfor newly diagnosed patients in the chronic phase, but theoptimal dose is not known and no maximum tolerateddose was established in the initial phase I study.60 Severalsingle-arm studies suggest that higher doses, up to 800 mgdaily, might give better results with a greater proportion ofpatients achieving CCyR and MMR.76,77 Such studies alsosuggest better PFS and transformation-free survival, butwith potentially more side-effects, particularly myelosup-pression. Amongst randomized studies, the TOPS(Tyrosine Kinase Inhibitor Optimization and Selectivity)study showed imatinib 800 mg to induce MR3 more rapid-ly than imatinib 400 mg, but at one year there was no sta-tistically significant difference.78 In contrast, there is per-suasive evidence from the recent randomized German(CML IV) study that optimized high-dose imatinib allowsmost patients to achieve MR4.5, and this may provide animproved therapeutic basis for treatment discontinua-tions.79 A subset analysis from this randomized study alsoshowed a greater benefit for patients over 65 years ofage.80 Another recent randomized intergroup phase IIstudy also demonstrated deeper molecular responses inthe 800 mg daily arm compared with 400 mg daily, withMR4 of 25% and 10%, respectively, with a trend forimproved progression-free and overall survival, but withsubstantially more grade 3 and 4 side-effects.81 There isalso some evidence that imatinib 600 mg daily is toleratedin more than 80% of CML patients and results in superiorcytogenetic and molecular responses at 12 and 24 monthscompared to the conventional 400 mg daily dose.82 It isalso of interest to note that in the German CML IV study,the median daily dose of imatinib was actually 628 mg,lending additional support to the 600 mg dose strategy.
Regardless of the dose of imatinib, the current safetyanalysis of imatinib is quite impressive, with very fewpotentially serious long-term side-effects noted after tenyears or more continuous use.83 Table 1 depicts the relativetoxicities of all currently available TKIs for CML. Whenthe drug is used at the standard starting daily dose of 400mg, most adverse effects occur within the first two yearsof starting therapy, and are generally mild to moderate(grades 1 and 2). Most of these include lethargy, nausea,headache, various skin reactions (including Steven-Johnson syndrome), infraorbital edema, bone pains, andsometimes, generalized fluid retention. In general theseeffects are easily manageable and potentially reversible.Significant cytopenias, in particular neutropenia and/or
thrombocytopenia and sometimes anemia occur less com-monly and usually in the first 6-12 months of therapy.Liver chemistry can also be abnormal, and this may, onrare occasions, progress to liver failure. Rare incidences ofprolongation of the QTc interval on the electrocardiographhave also been reported. It is possible that some patients,such as older patients and those with other co-morbiditiessuch as impaired cardiac function, might be more suscep-tible to toxicity. The longer-term follow-up studies do,however, indicate an adverse effect on the quality of life,particularly in younger female patients, and other uniqueeffects, such as effects on bone growth and mineralizationand gynecomastia.84-86 Finally, although there appears to beno definitive evidence to suggest exposure to imatinibincreases the risk of developing a second malignancy, it isreasonable for specialists to remain cautious and followpatients on long-term treatment carefully.87
The next generation-TKI story Further research into the imatinib story has shown that
only about 60% of CML patients remain on the standarddosages of imatinib after six years, implying that about40% have required an alternative treatment or higherdoses of imatinib.88 In addition, a population-based reportfound that only half of newly diagnosed patients withCML in chronic phase were in CCyR and receiving ima-tinib at two years after starting treatment.89 The main rea-sons for this are secondary (acquired) resistance which inmost cases results from the expansion of subclones withpoint mutations in the BCR-ABL1 kinase domain (KD).90-92A variety of other resistance mechanisms have also beendescribed, including poor adherence, amplification of theBCR-ABL1 fusion gene, relative overexpression of BCR-ABL1 protein, and overexpression of the multidrug resist-ant P-glycoprotein (MDR1).
Point mutations in the kinase domain of BCR-ABL1 thatconfer resistance to imatinib code for amino acid substitu-tions that may preclude entry of imatinib into the ATP-binding pocket or, in general, the inhibitory action of ima-tinib. The precise position of the mutation appears to dic-tate the degree of resistance to the drug. Some mutationsare associated with minor degrees of drug resistance, whilethe T315I (also referred to as the gatekeeper) mutation con-fers a very high level of resistance.93 The precise signifi-cance, and indeed the kinetics, of the over 100 currentlywell-characterized mutations have only partially been char-acterized (Figure 7).94 It is also possible, though not con-firmed in vivo, that resistant mutant clones could enhancethe fitness of sensitive clones by altering their microenvi-ronment by generating paracrine factors, such as IL-3.95
Primary resistance to imatinib appears to be very rare,and when observed may be related to poor drug compli-ance, poor gastrointestinal absorption, p450 cytochromepolymorphisms, interactions with other medications, orabnormal drug efflux and influx at the cellular level due tolow drug influx transporter (OCT1).96 The recognition ofimatinib’s qualified success led efforts to develop the next-generation TKIs and other alternative treatments. Initialefforts focused on two 2nd-generation TKIs: nilotinib anddasatinib (Figure 8).97
Nilotinib was designed as a chemical modification ofimatinib in an effort to increase its selectivity and activity.
Chronic myeloid leukemia: reminiscences and dreams
haematologica | 2016; 101(5) 547

Indeed, nilotinib has little activity against other kinasesinhibited by imatinib, such as KIT and PDGFRA/B.98Nilotinib is taken orally twice daily with food restrictionsdue to its bioavailibity being affected adversely by high fatintake. Like imatinib, it acts as an ATP-competitiveinhibitor by binding to the closed (inactive) conformationof the ABL1 kinase domain, but with a much higher affin-ity. In vitro studies suggest that nilotinib is approximately30- to 50-fold more potent than imatinib. Nilotinib is alsoactive in 32 of the currently 33 imatinib-resistant cell lineswith mutant BCR-ABL1, but like imatinib has no activityagainst the T315I mutation.99
Dasatinib is a thiazole-carboxamide structurally unrelat-ed to imatinib which binds to the ABL1 kinase domainregardless of the conformation of the activation loop (i.e.whether open or closed).100,101 It also inhibits some of theSrc family kinases that are involved in signal transductionin lymphoid cells and results in NK-cell expansion. Pre-clinical studies showed that dasatinib is 300-fold morepotent than imatinib and is active against 18 of 19 testedimatinib-resistant BCR-ABL1 mutants, with the notableexception of the T315I mutant.99
In 2004, both drugs entered studies of patients whowere resistant or intolerant to imatinib at standarddosages. The efficacy, but not the toxicity, of both drugswas fairly similar, with about 45% of the imatinib-resis-tant patients achieving CCyR and a 4-year overall survivalof 78%. The results for the imatinib-intolerant groupwere slightly better for both drugs.99-109 All responses weresimilar in patients with or without mutations, except forthe cohort with T315I mutation, where no responseswere noted with either drug. Though these results areimpressive, it is interesting that only one-third of theresponding patients remained on nilotinib or dasatinib atfive years, which means that two-thirds of patientsrequired a further change of therapy. In an analysis of asub-set receiving dasatinib for imatinib-resistant/intoler-ant disease, it was noted that dasatinib maintaineddurable efficacy irrespective of the presence or absence ofpre-existing KD mutations.110
The most common nilotinib treatment-related toxicitywas myelosuppression (although this was less pro-nounced than that observed with most other TKIs) fol-lowed by headaches, pruritus, and rashes (Table 2).Overall, 22% of the patients in the chronic phase experi-enced thrombocytopenia, with 19% having either grade 3or 4 severity; 16% had neutropenia and a further 16% hadanemia; metabolic effects included hyperglycemia.Following longer follow up, an increased incidence of car-diovascular events, in particular peripheral arterial disease,was noted, although many affected patients had predis-posing risk factors.111 About 19% of all patients experiencearthralgia, and about 14% experience fluid retention, par-ticularly pleural effusions, and rarely pericardial effusionsand other unique effects, such as panniculitis.104,112 In thedasatinib-treated cohort, hematologic toxicity was morecommon, with neutropenia and/or thrombocytopeniaoccurring in one-half of all patients and anemia in 20%.108Non-hematologic toxicity includes diarrhea, headaches,superficial edema, pleural effusions, and occasional peri-cardial effusions. Grades 3/4 side effects were rare; grades3/4 pleural effusions occurred in 6% of patients.
Following these encouraging results, both nilotinib anddasatinib entered clinical trials for first-line therapy ofnewly diagnosed patients in 2006. Nilotinib at twodosages, either 300 mg/day or 400 mg/day, was testedagainst imatinib 400 mg/day in the Evaluating NilotinibEfficacy and Safety in Newly Diagnosed Patients(ENESTnd) randomized study, with the rate of MR3 at 12months as the primary end point.113 Dasatinib was testedat a dose of 100 mg/day in a trial known as Dasatinib ver-sus Imatinib Study in Treatment-Naïve CML Patients(DASISION), with confirmed CCyR at 12 months as theprimary end point.114 Both of these primary end pointswere met: ENESTnd accorded higher rates of MR3 at 12months for both doses of nilotinib compared with ima-tinib (44% and 43% vs. 22%; P<0.001) and DASISIONshowed that dasatinib resulted in more frequent con-firmed CCyR at 12 months compared with imatinib (77vs. 66%; P=0.007). Both drugs were licensed for first-lineuse in patients with CML in the chronic phase in 2010.Table 3 summarizes the latest updates from both trials.
T.I. Mughal et al.
548 haematologica | 2016; 101(5)
Figure 7. Mutations in thekinase domain of ABL1identified in tyrosinekinase inhibitors (TKI)resistant chronic myeloidleukemia cells. The 10most frequent mutations,accounting for approxi-mately 70% of TKI-resis-tant CML patients arehighlighted in red.

Many of the secondary end points were also met in bothtrials, and the overall results suggested that front-line ther-apy with dasatinib or nilotinib (at either dose) achieves ear-lier and higher molecular response rates, in particular fasterand deeper molecular responses (MR3, MR4 and beyond),that in turn appear to decrease the rates of progression tothe advanced phases of CML.115-117 Nilotinib was associatedwith hyperglycemia, hypercholesterolemia, increasedtriglycerides, and an increased incidence of cardiovascular,cerebrovascular and peripheral arterial occlusive dis-ease.118,119 Dasatinib was associated with substantial hema-tologic toxicity, pleural effusions and, infrequently, pericar-dial effusions and pulmonary hypertension (Table 2).120Discontinuation rates for disease progression or treatmentfailure for any cause appears to be similar at around 33%-38% at three years for both drugs, with the caveat that thedefinitions of progression and the duration of follow upprior to censoring in these two large studies were not uni-form. A recent independent North American consortia trialcomparing daily dasatinib 100 mg to daily imatinib 400 mgproduced very similar results to DASISION in terms of effi-cacy and safety.121 Collectively, neither of these two stud-ies, nor the ENESTnd or the companion ENESTcmr stud-ies, demonstrated a survival advantage for a 2nd-generationTKI being used for first-line therapy of a newly diagnosedpatient with CML in chronic phase, despite the superiorearly molecular responses and the subsequent MR4.5
responses.122 In addition, the associated cardiovascular tox-icity in all three trials has been higher than that seen withimatinib.
The third and newest of the 2nd-generation TKIs, bosu-tinib, an oral dual ABL1 and SRC inhibitor, is chemicallydifferent from both dasatinib and nilotinib, and appears tobe able to overcome binding impediments conferred by
several kinase domain mutations to imatinib, nilotinib, ordasatinib (Figure 8).123 Phase II studies of once daily bosu-tinib 500 mg/day in CML patients who were either resist-ant or intolerant to imatinib demonstrated a CCyR of47%, an overall survival at two years of 88% in the ima-tinib-resistant cohort, and a remarkable 98% in the ima-tinib intolerant cohort; three years later, 40% of patientsremained on bosutinib.124-127 The principal side-effectsincluded diarrhea, abnormal liver chemistry and variousskin rashes, all of which were easily manageable withdose reduction and/or concomitant medications (Table 2).Based on these results, the drug was approved in 2012 forthe treatment of adult CML patients with chronic phase oradvanced phase disease who were resistant to prior TKItherapy. The drug was then tested in the phase IIIBosutinib Efficacy and safety in CML (BELA) study, innewly diagnosed patients with CML in the chronicphase.128 Since the primary end point was not met, andwith the CCyR results being similar in both arms of thestudy (70% for bosutinib and 68% for imatinib) the drugwas not approved for first-line use. It is, however, of inter-est that the MR3 rates at 12 months were significantlyimproved at 41% with bosutinib compared to 27% withimatinib, and the drug discontinuation rate was 37% attwo years for bosutinib and 29% for imatinib.Furthermore, the risk of transformation to the advancedphases was significantly lower for bosutnib. These latestmolecular results lend some support for the drug’s futurecandidacy as first-line therapy.
Ponatinib is a 3rd-generation TKI which has an interest-ing chemical structure based on a modification of a purinescaffold and a central triple carbon-carbon bond with asubstructure beyond the bond that is similar to imatinib(Figure 8).129 The drug inhibits ABL1, SRC and a variety ofother kinases, including KIT, PDFGRA, FGFR1 andFLT3.130,131 It was developed initially for patients who wereconsidered to have become resistant to TKIs as a result ofa T315I subclone. This feature is attributed to the com-pound’s unique structure which allows it to bind andinhibit ABL1 with no steric hindrance due to the T315Imutation.132,133 The drug was tested in the Ponatinib Ph-positive acute lymphoblastic leukemia (ALL) and CML
Table 2. Adverse events related to tyrosine kinase inhibitors in patients withchronic myeloid leukemia.
Imatinib Dasatinib Nilotinib Bosutinib Ponatinib
Peripheral edemas ++Pulmonary hypertension +Effusions +++ + ++Diarrhea + +++Rash + + ++ + ++Nausea + +Hyperglycemia ++PAOD +++ ++Arterial thrombosis + ++ +++Venous thrombosis ++Asthenia ++Skin fragility ++Muscle cramps ++
PAOD: peripheral arterial occlusive disease.
Figure 8. Chemical structures of imatinib, nilotinib, dasatinib, bosutinib andponatinib.
Chronic myeloid leukemia: reminiscences and dreams
haematologica | 2016; 101(5) 549

Evaluation (PACE) phase II study in which 449 patientswith CML in the chronic and advanced phases and Ph-positive ALL with resistant to or intolerance from dasa-tinib or nilotinib or with the T315I mutation wereenrolled. The patients received once daily 45 mg pona-tinib, and the results indicated that the drug had consider-able activity in all patients, including those in advancedphase disease, regardless of base-line kinase domain muta-tions and the responses seemed to be durable.134,135 Thestudy results showed that there were 46% CCyR (40%without T315I; 66% with T315I) and 34% MR3 (27%without T315I; 56% with T315I). The most common side-effect was thrombocytopenia, rash, dry skin and abdomi-nal pain, and platelet dysfunction was also noted (Table2).136 Serious thrombotic events were observed in 9%, butconsidered to be treatment-related in 3%. The study drug-discontinuation rate due to toxicity was 12%.
The Evaluation of Ponatinib versus Imatinib in CML(EPIC) phase III randomized study of ponatinib and ima-tinib in newly diagnosed patients began in 2012 and pre-liminary results suggest that the drug accords high rates ofearly molecular response and MR3 compared with ima-tinib.137 The drug was licensed in December 2012 forpatients with CML in the chronic or advanced phasesresistant or intolerant to prior TKI therapy and Ph-chro-mosome positive ALL resistant or intolerant to prior TKItherapy. This approval constituted ponatinib to be theonly licensed TKI with activity against the T315I sub-clone. Unfortunately, in October 2013, concerns aboutexcessive arterial vascular events led to the suspension ofthe drug and the manufacturer elected to discontinue theEPIC study.118,119 In early 2014, despite these serious risks,ponatinib was re-licensed exclusively for the treatment ofadult patients with T315I-positive CML in all phases orT315I-positive Ph-chromosome positive ALL and adultpatients with CML in all phases or Ph-chromosome posi-tive ALL for whom no other TKI therapy was indicated.The precise mechanisms of ponatinib-related arterial vas-cular events, and indeed those associated with nilotinib,which seem to occur at a considerably lower frequency,still remain elusive.
There is continuing interest in developing effectivetreatments for T315I-positive CML, which are of addi-tional interest given the challenges with ponatinib.Omecetaxine (formerly called homoharringtonine) is asemi-synthetic plant alkaloid that enhances apoptosis ofCML cells. It has actually been under investigation in CMLand other myeloid malignancies since the 1970s.138 Theresults of recent studies were encouraging, with modestactivity noted in patients with CML in the chronic andadvanced phases, including some with the T315I sub-clone. The drug was licensed in 2014 for use in patientswith CML (all phases) who were resistant or intolerant totwo or more TKIs. Another candidate drug that hasshown some activity in T315I subclone is HS-438, whichhas been tested in clinical trials.139
The allogeneic stem cell transplantation story“Thy bones are marrowless, thy blood is cold” (The Tragedy
of Macbeth: William Shakespeare, 1606).Though the original concept of bone marrow transplan-
tation was probably first advocated by Thomas Fraser in1894, when he famously recommended that patients eat
bone marrow “sandwiches; flavored by port-wine” (toimprove taste), sporadic attempts at marrow transplanta-tion were taken much earlier.140 The modern era of bonemarrow transplantation [now stem cell transplantation(SCT)] did not begin until a basic understanding of thehistocompatibility system was gained in 1958. Much ofthe pioneering work thereafter was carried out by theNobel laureate E. Donnall Thomas in Seattle, resulting inthe first successful allo-SCT using syngeneic donors in1979.141,142 In 1978, John Goldman in London showed thatmarrow-populating stem cells were present in the periph-eral blood of untreated CML patients.143 This led to the useof an autograft for patients ineligible for an allo-SCT, andthough in some patients Ph-negative hematopoiesis wasrestored, very few patients remained Ph-negative forextended periods.
Subsequent efforts in allo-SCT using sibling and volun-teer unrelated donors were increasingly successful, as aresult of the recognition that the graft-versus-leukemia(GvL) effect plays a major role in eradicating CML afterallo-SCT, and improvements in the conditioningregimens.144,145 This coupled with the availability ofhematopoietic stem cells from a variety of sources,improvements in SCT technology, and a better under-standing and treatment of the alloimmmune-mediatedgraft-versus-host disease (GvHD) led to significantlyimproved results for the majority of patients transplantedin the chronic phase, and indeed made SCT more widelyavailable to higher risk and also older patients.146-148 Thepotential to accord long-term survival and probable curefor patients with CML in the chronic phase was firmlyestablished by the early 1990s, and an allograft was thenconsidered the first-line treatment for all eligible patientsin the chronic phase.149 The use of donor lymphocyte infu-sions to treat early relapse after allograft by exploiting theGvL effect became popular in the mid-1990s, and con-firmed the importance of the donor derived immune sys-tem to overcome residual leukemic cells.150,151
The major factors influencing survival are patient age,disease phase at time of SCT, disease duration, degree ofhistocompatibility between donor and recipient, and gen-der of donor.152 The best results are achieved following afull intensity (conventional) allograft, using an HLA-iden-tical sibling donor or a suitable matched unrelated donor;the 5-year leukemia-free survival is 80% and 60%, respec-tively (Figure 9).149 The results following a reduced intensi-ty regiment are generally inferior.153 There is a roughly20% chance of transplant-related mortality and a 15%chance of relapse. The possible major complicationsinclude GvHD, reactivation of infection withcytomegalovirus or other viruses, idiopathic pneumonitisand sinusoidal obstruction syndrome (previously knownas veno-occlusive disease of the liver). Post-transplantmolecular monitoring studies suggest that most, but notall, patients who are negative for BCR-ABL1 transcripts atfive years following the allograft, remain negative for longperiods, and it is likely that, in the majority of thesepatients, the CML may truly have been eradicated.154,155
Since 1999, the numbers of allografts performed forCML have dramatically decreased, interestingly, somethree years prior to the licensing of imatinib for CML. Thistrend has continued, and the use of allo-SCT is now
T.I. Mughal et al.
550 haematologica | 2016; 101(5)
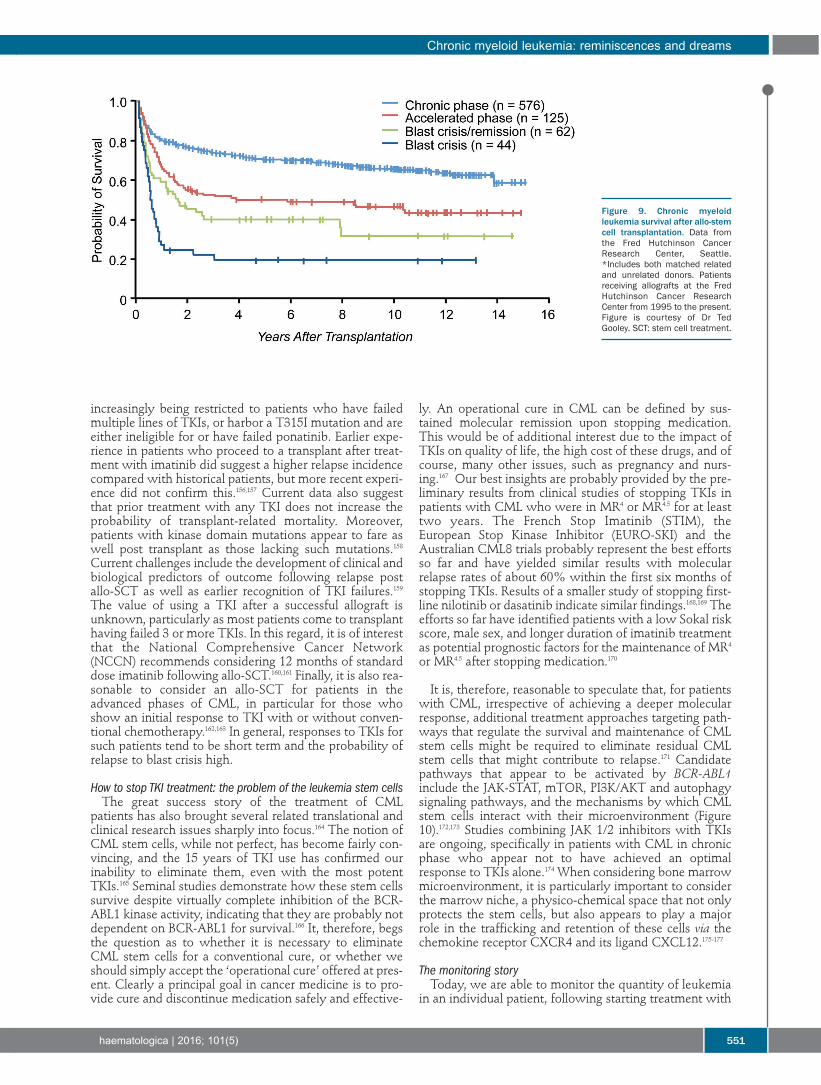
increasingly being restricted to patients who have failedmultiple lines of TKIs, or harbor a T315I mutation and areeither ineligible for or have failed ponatinib. Earlier expe-rience in patients who proceed to a transplant after treat-ment with imatinib did suggest a higher relapse incidencecompared with historical patients, but more recent experi-ence did not confirm this.156,157 Current data also suggestthat prior treatment with any TKI does not increase theprobability of transplant-related mortality. Moreover,patients with kinase domain mutations appear to fare aswell post transplant as those lacking such mutations.158Current challenges include the development of clinical andbiological predictors of outcome following relapse postallo-SCT as well as earlier recognition of TKI failures.159The value of using a TKI after a successful allograft isunknown, particularly as most patients come to transplanthaving failed 3 or more TKIs. In this regard, it is of interestthat the National Comprehensive Cancer Network(NCCN) recommends considering 12 months of standarddose imatinib following allo-SCT.160,161 Finally, it is also rea-sonable to consider an allo-SCT for patients in theadvanced phases of CML, in particular for those whoshow an initial response to TKI with or without conven-tional chemotherapy.162,163 In general, responses to TKIs forsuch patients tend to be short term and the probability ofrelapse to blast crisis high.
How to stop TKI treatment: the problem of the leukemia stem cellsThe great success story of the treatment of CML
patients has also brought several related translational andclinical research issues sharply into focus.164 The notion ofCML stem cells, while not perfect, has become fairly con-vincing, and the 15 years of TKI use has confirmed ourinability to eliminate them, even with the most potentTKIs.165 Seminal studies demonstrate how these stem cellssurvive despite virtually complete inhibition of the BCR-ABL1 kinase activity, indicating that they are probably notdependent on BCR-ABL1 for survival.166 It, therefore, begsthe question as to whether it is necessary to eliminateCML stem cells for a conventional cure, or whether weshould simply accept the ‘operational cure’ offered at pres-ent. Clearly a principal goal in cancer medicine is to pro-vide cure and discontinue medication safely and effective-
ly. An operational cure in CML can be defined by sus-tained molecular remission upon stopping medication.This would be of additional interest due to the impact ofTKIs on quality of life, the high cost of these drugs, and ofcourse, many other issues, such as pregnancy and nurs-ing.167 Our best insights are probably provided by the pre-liminary results from clinical studies of stopping TKIs inpatients with CML who were in MR4 or MR4.5 for at leasttwo years. The French Stop Imatinib (STIM), theEuropean Stop Kinase Inhibitor (EURO-SKI) and theAustralian CML8 trials probably represent the best effortsso far and have yielded similar results with molecularrelapse rates of about 60% within the first six months ofstopping TKIs. Results of a smaller study of stopping first-line nilotinib or dasatinib indicate similar findings.168,169 Theefforts so far have identified patients with a low Sokal riskscore, male sex, and longer duration of imatinib treatmentas potential prognostic factors for the maintenance of MR4
or MR4.5 after stopping medication.170
It is, therefore, reasonable to speculate that, for patientswith CML, irrespective of achieving a deeper molecularresponse, additional treatment approaches targeting path-ways that regulate the survival and maintenance of CMLstem cells might be required to eliminate residual CMLstem cells that might contribute to relapse.171 Candidatepathways that appear to be activated by BCR-ABL1include the JAK-STAT, mTOR, PI3K/AKT and autophagysignaling pathways, and the mechanisms by which CMLstem cells interact with their microenvironment (Figure10).172,173 Studies combining JAK 1/2 inhibitors with TKIsare ongoing, specifically in patients with CML in chronicphase who appear not to have achieved an optimalresponse to TKIs alone.174 When considering bone marrowmicroenvironment, it is particularly important to considerthe marrow niche, a physico-chemical space that not onlyprotects the stem cells, but also appears to play a majorrole in the trafficking and retention of these cells via thechemokine receptor CXCR4 and its ligand CXCL12.175-177
The monitoring storyToday, we are able to monitor the quantity of leukemia
in an individual patient, following starting treatment with
Chronic myeloid leukemia: reminiscences and dreams
haematologica | 2016; 101(5) 551
Figure 9. Chronic myeloidleukemia survival after allo-stemcell transplantation. Data fromthe Fred Hutchinson CancerResearch Center, Seattle.*Includes both matched relatedand unrelated donors. Patientsreceiving allografts at the FredHutchinson Cancer ResearchCenter from 1995 to the present.Figure is courtesy of Dr TedGooley. SCT: stem cell treatment.

TKI, with considerable precision. First by an examinationof the peripheral blood we confirm normalization of theblood count, second by bone marrow metaphase cytoge-netic we confirm CCyR and finally by measuring numbersof BCR-ABL1 transcripts in the blood or marrow by quan-titative reverse transcriptase PCR (RQ-PCR) we confirm amolecular response (MR). The use of fluorescence in situhybridization (FISH) to detect a BCR-ABL1 fusion gene ininterphase cells is more sensitive than metaphase cytoge-netics but much less sensitive than RQ-PCR.
Molecular monitoring was initially developed in 1988 asa qualitative assay to detect early relapse following anallograft and was thereafter replaced by quantitative PCR,which is now generally considered the optimal methodfor monitoring patients with CML during treatment.178-181Unfortunately, there remains widespread inconsistency inRQ-PCR results. This is mainly due to interlaboratory dif-ferences in technology and methodology employed sincemolecular monitoring was popularized in the early ima-tinib era. The RQ-PCR standardization project was startedby John Goldman in 2006 in Bethesda, Maryland, toaddress some of these challenges.182 The results areexpressed as a ratio of BCR-ABL1 copy numbers to copynumbers of a control gene (x 100% on a log scale) or as alog10 reduction from standardized value of 0 for untreatedpatients. In practice, the recommended way of expressionis to use a laboratory-specific conversion factor to convertthe value obtained in a given laboratory to a value of theInternational Scale (IS), where 100% is the value for a spe-cific cohort of untreated patients studied in 2002, based on30 newly diagnosed patients with CML in the chronicphase, tested in three laboratories.183 Patients who achievea transcript number of 0.1% on the IS, which is equivalentto a 3-log reduction from the baseline for untreatedpatients, are said to have achieved a MR3, and those with-out detectable transcripts have achieved a MR4.5, as dis-cussed in the section on treatment (Table 3).184
Despite the many efforts towards harmonization ofmolecular methods, widespread inconsistency remains.185It is likely that some of the intrinsic difficulties related tothe complex time consuming methodology whichrequires specialized skills and knowledge may be over-come by the new automated BCR-ABL1 assay that is con-tained within a single-use microfluidic cartridge, using aspecialized instrument, such as the Cepheid GeneXpert.186 Itis of interest that this specialized equipment was initiallydeveloped for bioterrorism assays following the anthrax
attacks in the USA soon after the September 11 attacks in2001. In this system, RNA extraction and real-time PCR isprepared. This system incorporates conversion to theBCR-ABL1 international reporting scale and has the samesensitivity as usual quantitative methods. This system isespecially attractive for hospitals where only sporadicCML cases are treated.
Further improvements include the introduction of dig-ital PCR; in particular with regards to the assessment ofthe impact of deep molecular response, which is increas-ingly recognized as an effective clinical strategy to allowfor discontinuing TKI therapy safely in some patients,even in the presence of minimal residual disease.187 Thisis a conceptual approach where a sample is partitionedinto thousands of separate reactions. This partitioningcan be performed either by sorting into different reactionwells by pumps and valves (Fluidigm), or by diluting thesample into separate micelles (BioRad). Either methodseems to increase sensitivity by over a log compared toconventional RQ-PCR. This powerful digital tool appearsto be particularly attractive to help improve efforts todiscontinue TKI therapy safely in candidate patients whohave been in MR4, MR4.5 or MR5. It is likely that furtherimprovements will be made by the application of thenext generation DNA sequencing approaches.188Conversely, many efforts are addressing suitablemethodology and technology for wider and, importantly,more affordable use of RQ-PCR.189
Another important test in molecular monitoring of CMLpatients is BCR-ABL1 kinase domain mutation analysis inthose who have acquired TKI resistance and who mightrequire an alternative treatment, or those who progress toadvanced phase disease. This test also helps to determinethe clinical consequences of clonal diversity of BCR-ABL1and the co-existence of subclones. Next generationsequencing (NGS) techniques appear to be superior to thecurrent Sanger sequencing, in particular for the identifica-tion of compound mutations, which might be more fre-quent in acquired resistance to 2nd- and 3rd-generationTKIs.190 Compound mutations are two or more mutationsin the same BCR-ABL1 allele, and not multiple clones withdifferent mutations. It is of interest that while over 100different point mutations have now been described, only12 positions appear to be involved in compound muta-tions, many of which include the T315I mutation. Newtechnologies incorporating computer modeling help usunderstand how the leukemic cells develop clever tactics
T.I. Mughal et al.
552 haematologica | 2016; 101(5)
Table 3. DASISION and ENESTnd: summary of data from different studies.Dasatinib Imatinib Nilotinib Nilotinib Imatinib 100 mg qd 400 mg qd 300 mg bid 400 mg bid 400 mg qdn = 259 n = 260 n = 282 n = 281 n = 283
Cumulative MR³ at 4 years 74%* 46% 77%* 77%* 60%MR4 by 3 years 36%# 22% 50%* 44%* 26%MR4.5 by 3 years 22%# 12% 32%* 28%# 15%Progression to AP/BC (ITT) 8 (3.1%) 13 (5%) 9 (3.2%)‡ 6 (2.1%)§ 19 (6.7%)Overall survival 92.9% 92.1% 95.1% 97.0% 94.0%
Progression-free survival 90% 90.2% 96.9% 98.3% 94.7%ITT: intention to treat;*versus imatinib, P<0.0001; #versus imatinib, P<0.003; ‡versus imatinib, P=0.05; §versus imatinib, P=0.007.

to evade selective pressures of the more potent TKIs, inparticular ponatinib, and result in structural changeswhich limit or exclude TKI binding.132,133
Expert panel definition of response and failure to respond to TKI treatmentThe recommendations of an expert panel of hematolo-
gists convened under the aegis of the EuropeanLeukemiaNet (ELN) up-dated a series of recommendationsin 2013, designed to help the clinician in optimal manage-ment of CML and to benefit from the 15 years of experi-ence with TKI treatment in patients with CML (Table 4).191
The panel had been providing such recommendationssince 2005 when the original consensus report was collat-ed. They recommended as initial treatment with imatinib,nilotinib or dasatinib, with response being assessed byRQ-PCR and/or conventional cytogenetics at three, sixand 12 months. Optimal response was defined as BCR-ABL1 transcript levels of less than 10% at three months,less than 1% at six months, and less than 0.1% from 12months onwards, whereas more than 10% at six monthsand more than 1% from 12 months onward define failureand required alternative treatment. Interestingly, the panelalso considered a partial cytogenetic response (PCyR) at
Chronic myeloid leukemia: reminiscences and dreams
haematologica | 2016; 101(5) 553
Table 4. European LeukemiaNet 2013 Guidelines: response to first-line treatment with imatinib, nilotinib or dasatinib. Optimal Warning Failure
Baseline NA - HIGH RISK, NA- CCA/Ph+ (major route)
3 months - BCR-ABL ≤ 10% and/or - BCR-ABL >10% - No CHR- Ph+ ≤ 35% and/or and/or
- Ph + 36%-95% - Ph + > 95%6 months - BCR-ABL < 1% - BCR-ABL 1-10% and/or - BCR-ABL > 10%
and/or Ph + 1%-35% and/or- Ph+ 0 - Ph + > 35%
12 months - BCR-ABL ≤ 0.1% - BCR-ABL 0.1%-1 % - BCR-ABL > 1% and/or
- Ph + > 0%Then, and at any time - BCR-ABL ≤ 0.1% - BCR-ABL 0.1%-1% - Loss of CHR, loss
of CCYR, confirmed loss of MMR, mutations and CCA/Ph+
NA: not applicable.
Figure 10. Targeting chronicmyeloid leukemia cells at dif-ferent levels.

three months and a CCyR from six months onwards ascomprising optimal response, whereas no cytogeneticresponse at three months, less than PCyR at six months,and less than CCyR from 12 months onward define fail-ure. This reflects the opinions of several other CMLexperts, highlighting the notion of a CCyR, rather than adeep molecular response, being associated with sur-vival.5,192 The panel felt that despite the notion of an earlymolecular response being a clear predictor of outcome andimpending risk, a change of therapy was not mandatorysince current studies do not suggest that a change of ther-apy at three months changes the outcome. In tandem,some experts consider that halving BCR-ABL1 transcriptswithin 76 days together with the achievement of a BCR-ABL1 less than 10% by six months, in addition to a CCyRat one year and beyond, may be reasonable milestones forchange of therapy.193-196
Future prospects and conclusions
The CML success story has unfolded over a relativelyshort period of time and the efforts of Janet Rowley andJohn Goldman have been crucial to our understanding ofthe biology of what is now considered a genetically simplecancer. Their work has provided vital insights that haveresulted in the success of molecularly targeted therapy, notonly for CML patients, but also for other malignancies.197Two decades since Brian Druker’s initial studies with ima-tinib, a personalized treatment algorithm is available for thenewly diagnosed patient with CML. Treatment involves achoice of three first-line orally administered drugs and twoeffective next-line therapies that should be used based onrisk stratification, co-morbidities, the side-effects profileand the BCR-ABL1 genotype. Furthermore, drug access andthe cost of TKI therapy are significant issues on the agendaof world-wide healthcare, given the increased prevalence ofCML across the globe.165,198 Currently, there is little differ-ence in the pricing structure of the licensed first-line drugs,but this should change dramatically now that generic ima-tinib becomes available as the patent for Gleevec (imatinibmesylate) expired in the US in 2015 and in Europe expiresin 2016. Regardless of the initial choice of TKI, the vastmajority of patients achieve a durable CCyR, with a lifes-
pan approaching that of the general population. In mostinstances the medication must be continued indefinitely,and a principal challenge now is to develop strategies tostop TKIs safely and effectively. For the moment, it is prob-ably best to discontinue the TKI therapy only within theframework of a clinical trial.
It seems crucial to improve our understanding of the var-ious resistance mechanisms, in particular the emerging roleof the bone marrow microenvironment and stem cell niche,and to assess the importance of the persistence of BCR-ABL1 by PCR, even in patients who have confirmed MR4.5
and beyond.199 Challenges also remain in the optimal mon-itoring of patients with CML on treatment, in particularwith regard to the interlaboratory discrepancy in results,and indeed, harmonizing results to the international stan-dard. The monitoring technology would also benefit frombeing further simplified, and importantly, by being moreaffordable. Last, but not least, the ELN 2013 recommenda-tions should be up-dated to harmonize expert opinions. Arecent French report expressed some concern in not beingable to validate the current recommendations with regardsto identifying optimal response, though treatment failurewas confirmed for a cohort of 180 patients being treatedwith imatinib.200 Our understanding of the mechanisms andtreatment of patients with advanced phase disease remainslimited. In addition, questions remain with regard to the ini-tiating biological event, at least in some patients with CMLin chronic phase.201 Clearly, the CML story is richly studdedwith insight, innovation and scientific breakthroughs.Arguably, however, there is much work to be done in orderto pay tribute to, and to continue the story that was initiat-ed by Janet Rowley and John Goldman.
AcknowledgmentsThe authors wish to thank Alpine Oncology Foundation, in
particular Dr. Alpa Parmar, for organizing the Janet Rowley andJohn Goldman Special Colloquium during the 19th EHAMeeting and Novartis Oncology Global for their support. TIMwishes to thank Professor Bob Lowenberg for his mentorship andhelp in arranging this event, Professor Christine Chomienne(President of EHA-2014) for her opening address and theInternational CML Foundation. JFA acknowledges the support ofNIHR Biomedical Research Centre funding
T.I. Mughal et al.
554 haematologica | 2016; 101(5)
References
1. Ben-Neriah Y, Daley GQ, Mes-Masson AM,et al. The chronic myelogenous leukemia-specific P210 protein is the product of thebcr/abl hybrid gene. Science. 1986;233(4760):212-214.
2. Goldman JM. Melo JV. Targeting the BCR-ABL tyrosine kinase in chronic myeloidleukemia. N Engl J Med. 2001;344(14):1084-1086.
3. Druker BJ, Tamura S, Buchdunger E, et al.Effects of a selective inhibitor of the Abltyrosine kinase on the growth of Bcr-Ablpositive cells. Nat Med. 1996;2(5):561-566.
4. Druker BJ, Guilhot F, O’Brien SG, et al. Five-year Follow-up of Patients ReceivingImatinib for Chronic Myeloid Leukemia. NEngl J Med. 2006;355(23):2408-2417.
5. Kantarjian H, Cortes JE. Complete cytoge-
netic response, not deep molecular response,is associated with survival in chronicmyeloid leukemia. J Clin Oncol. 2014;32(27):3077.
6. Huang XL, Cortes J, Kantarjian H.Estimations of the increasing prevalence andplateau prevalence of chronic myeloidleukemia in the era of tyrosine kinaseinhibitor therapy. Cancer. 2012;118(12):3123-3127.
7. Mughal TI, Cortes J, Cross NCP, et al.Chronic myeloid leukemia – some topicalissues. Leukemia. 2007;21(7):1347-1352.
8. Falchi L, Kantarjian HM, Wang X, et al.Significance of deeper molecular responsesin patients with chronic myeloid leukemiain early chronic phase treated with tyrosinekinase inhibitors. Am J Hematol. 2013;88(12):1024-1029.
9. Mughal TI, Goldman JM. An essay in cancermedicine: Lessons learned from patients
with chronic myeloid leukaemia. Clin Med.2005;1:2-7.
10. Cheah CY, Burbury K, Apperley JF, et al.Patients with myeloid malignancies bearingPDGFRB fusion genes achieve durable long-term remissions with imatinib. Blood.2014;123(23):3574-3577.
11. Velpeau A. [Sur la resorption du puseat surl’alteration du sang dans les maladiesClinique de persection nenemant.] Premierobservation. Rev Med. 1827;2:216.
12. Bennett JH. Case of hypertrophy of thespleen and liver in which death took placefrom suppuration of the blood. Edinb MedSurg J. 1845;64:413-423.
13. Virchow R. [Weisses Blut]. Froriep’s Notzien1845;36:151-156.
14. Tjio JH, Levan A. The chromosome numberof man. Hereditas. 1956;42:1-6.
15. Lejeune J, Turpin R, Gautier M.[Chromosomic diagnosis of mongolism].

Archives Francaises de Pediatrie. 1959;16:962-963.
16. Nowell PC, Hungerford DA. A minute chro-mosome in human granulocytic leukemia.Science. 1960;132:1497.
17. Baikie AG, Court-Brown WM, Buckton KE,Harnden DG, Jacobs PA, Tough IM. A possi-ble specific chromosome abnormality inhuman chronic myeloid leukaemia. Nature.1960;188:1165-1166.
18. Boveri T. [Frage der Entstehung malignerTumoren.] Jena: Gustav Fischer;1914.
19. Fialkow PJ, Garler SM, Yoshida A. Clonalorigin of chronic myelocytic leukemia inman. Proc Natl Acad Sci USA. 1967;58(4):1468-1471.
20. Ohno S, Makino S. The single X-nature ofsex chromatin in man. Lancet. 1961;1(7168):78-79.
21. Rowley JD. Letter: A new consistent chro-mosomal abnormality in chronic myeloge-nous leukaemia identified by quinacrine flu-orescence and Giemsa staining. Nature.1973;243(5405):290-293.
22. Abelson HT, Rabstein LS. Lymphosarcoma:virus-induced thymic-independent diseasein mice. Cancer Res. 1970;30(8):2213-2222.
23. Groffen J, Stephenson JR, Heisterkamp N,de Klein A, Bartram CR, Grosveld G.Philadelphia chromosomal breakpoints areclustered within a limited region, bcr, onchromosome 22. Cell. 1984;36(1):93-99.
24. Heisterkamp N, Groffen J, Stephenson JR, etal. Chromosomal localization of human cel-lular homologues of two viral oncogenes.Nature. 1982;299(5885):747-749.
25. de Klein A, van Kessel AG, Grosveld G, et al.A cellular oncogene is translocated to thePhiladelphia chromosome in chronic myelo-cytic leukaemia. Nature. 1982;300(5894):765-767.
26. Bartram CR, de Klein A, Hagemeijer A, et al.Translocation of c-abl oncogene correlateswith the presence of a Philadelphia chromo-some in chronic myelocytic leukaemia.Nature. 1983;306(5940):277-280.
27. Stam K, Heisterkamp N, Grosveld G, et al.Evidence of a new chimeric bcr/c-abl mRNAin patients with chronic myelocyticleukemia and the Philadelphia chromosome.N Engl J Med. 1985;313(23):1429-1433.
28. Shtivelman E, Lifshitz B, Gale RP, et al.Alternative splicing of RNAs transcribedfrom the human abl gene and from the bcr-abl fused gene. Cell. 1986;47(2):277-284.
29. Witte ON, Dasgupta A, Baltimore D.Abelson murine leukaemia virus rotein isphosphorylated in vitro to form phosphoty-rosine. Nature. 1980;283(5750):826-831.
30. Prywes R, Foulkes JG, Baltimore D. Theminimum transforming region of v-abl is thesegment encoding protein-tyrosine kinase. JVirol. 1985;54(1):114-122.
31. Konopka JB, Watanabe SM, Witte ON. Analteration of the human c-abl protein inK562 leukemia cells unmasks associatedtyrosine kinase activity. Cell. 1984;37(3):1035-1042.
32. Daley GQ, Baltimore D. Transformation ofan interleukin 3-dependent hematopoeticcell line by chronic myelogenous leukemia-specific P210 bcr/abl protein. Proc Natl AcadScience. 1988;85(23):9312-9316.
33. Lugo TG, Pendergast AM, Muller AJ, et al.Tyrosine kinase activity and transformationpotency of bcr-abl oncogene products.Science. 1990;247(4946):1079-1082.
34. Daley GQ, van Etten, Baltimore D.Induction of chronic myelogenous leukemiain mice by P210 BCR/ABL gene of thePhiladelphia chromosome. Science. 1990;
247(4944):824-830.35. Elephanty AG, Hariharan IK, Cory S. Bcr-
abl, the hallmark of chronic myeloidleukeima in man, induces multiple hemo-poietic neoplasms in mice. EMBO J.1990;9(4):1069-1078.
36. Kelliher MA, McLaughlin J, Witte ON,Rosenberg N. Induction of a Chrnic myeloidleukemia-like Syndrome in Mice with v-abland BCR/ABL. Proc Natl Acad Sci USA.1990;87(17):6649-6653.
37. Anafi M, Gazit A, Gilon C, et al. Selectiveinteractions of transforming and normal ablproteins with ATP, tyrosine-copolymer sub-strates, and tyrphostins. J Biol Chem.1992;267(7):4518-4523.
38. Sawyers CL, Callahan W, Witte ON.Dominant negative MYC blocks transfor-mation by ABL oncogenes. Cell. 1992;70(6):901-910.
39. Pendergast AM, Quilliam LA, Cripe LD, etal. BCR-ABL-induced oncogenesis is mediat-ed by direct interaction with the SH2domain of the GRB-2 adaptor protein. Cell.1993;75(1):175-185.
40. Neshat MS, Raitano AB, Wang HG, et al.The survival function of the Bcr-Abl onco-gene is mediated by Bad- dependent and -independent pathways: roles for phos-phatidylinositol 3- kinase and Raf. Mol CellBiol. 2000;20(4):1179-1186.
41. Sillaber C, Gesbert F, Frank DA, et al. STAT5activation contributes to growth and viabili-ty in Bcr/Abl- transformed cells. Blood.2000;95(6):2118-2125.
42. Salgia R, Sattler M, Pisick E, et al.p210BCR/ABL induces formation of com-plexes containing focal adhesion proteinsand the protooncogene product p120c-Cbl.Exp Hematol. 1996;24(2):310-313.
43. Sattler M, Mohi MG, Pride YB, et al. Criticalrole for Gab2 in transformation byBCR/ABL. Cancer Cell. 2002;1(5):479-492.
44. Zhao C, Blum J, Chen A, et al. Loss of beta-catenin impairs the renewal of normal andCML stem cells in vivo. Cancer Cell.2007;12(6):528-541.
45. Dierks C, Beigi R, Guo GR, et al. Expansionof Bcr-Abl-positive leukemic stem cells isdependent on Hedgehog pathway activa-tion. Cancer Cell. 2008;14(3):238-249.
46. Neviani P, Harb JG, Oaks JJ, et al. PP2A-acti-vating drugs selectively eradicate TKI-resis-tant chronic myeloid leukemic stem cells. JClin Invest. 2013;123(10):4144-4157.
47. Hurtz C, Hatzi K, Cerchietti L, et al. BCL6-mediated repression of p53 is critical forleukemia stem cell survival in chronicmyeloid leukemia. J Exp Med. 2011;208(11):2163-2174.
48. Chen Y, Hu Y, Zhang H, et al. Loss of theAlox5 gene impairs leukemia stem cells andprevents chronic myeloid leukemia. NatGenet. 2009;41(7):783-792.
49. O'Hare T, Zabriskie MS, Eiring AM, et al.Pushing the limits of targeted therapy inchronic myeloid leukaemia. Nat Rev Cancer.2012;12(8):513-526.
50. Corbin AS, Agarwal A, Loriaux M, et al.Human chronic myeloid leukemia stem cellsare insensitive to imatinib despite inhibitionof BCR-ABL activity. J Clin Invest.2011;121(1):396-409.
51. Mughal T, Goldman J. Chronic myeloidleukemia: a historical perspective. In:Mughal T, Goldman J, (eds.) ChronicMyeloproliferative Disorders. Paul Street,London: Informa Healthcare 2008: pp1-16.
52. Dalton DAG. Myelran in chronic myeloidleukaemia. Lancet. 1953;1:208.
53. Kennedy BJ, Yarbo JW. Metabolic and thera-
peutic effects of hydroxyurea in chronicmyelogenous leukemia. Trans Assoc AmPhysicians. 1965;78:391-399.
54. Hehlmann R, Heimpel H, Hasford J, et al.Randomized comparison of busulfan andhydroxyurea in chronic myelogenousleukemia: prolongation of survival byhydroxyurea. The German CML StudyGroup. Blood. 1993;82(2):398-407.
55. Talpaz M, Kantarjian H, McCredie K, et al.Hematologic remission and cytogeneticimprovement induced by recombinanthuman interferon alpha A in chronic myel-ogenous leukemia. N Engl J Med. 1986;314(17):1065-1069.
56. Hehlmann R, Heimpel H, Hasford J, et al; onbehalf of the German CML Study Group.Randomized comparison of interferon-alphawith busulphan and hydroxyurea in chronicmyelogenous leukemia. Blood.1994;84(12):4064-4077.
57. The Italian Cooperative Study Group onChronic Myeloid Leukemia. Interferon alfa-2a as compared with conventionalchemotherapy for the treatment of chronicmyeloid leukemia. N Engl J Med. 1994;330(12):820-825.
58. Allan NC, Richards SM, Shepherd PC. UKMedical Research Council randimised, mul-ticentre trial of interferon-alpha n1 forchronic myeloid leukaemia: improved sur-vival irrespective of cytogenetic response.The UK Medical Research Council’sWorking Parties for Therapeutic Trials inAdult Leukaemia. Lancet. 1995;345(8962):1392-1397.
59. Guilhot F, Chastang C, Michallet M, et al; onbehalf of the French Chronic MyeloidLeukemia Study Group. Interferon alfa-2bcombined with cytarabine versus interferonalone in chronic myelogenous leukemia. NEngl J Med. 1997;337(4):223-329.
60. Silver RT, Woolf SH, Hehlmann R, et al. Anevidence-based analysis of the effect ofbusulfan, hydoxyurea, interferon, and allo-geneic bone marrow transplantation in treat-ing the chronic phase of chronic myeloidleukemia: developed for the AmericanSociety of Hematology. Blood.1999;94(5):1517-1536.
61. Druker BJ, Talpaz M, Resta DJ, et al. Efficacyand safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloidleukemia. N Engl J Med. 2001;344(14):1031-1037.
62. Mughal TI, Goldman JM. Chronic myeloidleukaemia: A therapeutic challenge. AnnOncol. 1995;6(7):637-644.
63. Mughal TI, Goldman JM. Chronic Myeloidleukemia: Current Status and Controversies.Oncology (Williston Park). 2004;18(7):837-847.
64. Sokal JE, Cox EB, Baccarani M, et al.Prognostic discrimination in “good-risk”chronic granulocytic leukemia. Blood.1984;63(4):789-799.
65. Hasford J, Pfirrmann M, Hehlmann R, et al.A new prognostic score for survival ofpatients with chronic myeloid leukaemiatreated with interferon alpha. J Natl CancerInst. 1998;90(11):850-858.
66. Hoffman VS, Baccarani M, Lindoerfer D, etal. The EUTOS prognostic score: review andvalidation in 1288 patients with CML treat-ed frontline with imatinib. Leukemia.2013;27(10):2016-2022.
67. Mughal TI, Barbui T, Abdel-Wahab O, et al.Novel insights into the biology and treat-ment of chronic myeloproliferative neo-plasms. Leuk Lymphoma. 2015;56(7):1938-1948.
Chronic myeloid leukemia: reminiscences and dreams
haematologica | 2016; 101(5) 555

68. Druker BJ, Lydon NB. Lessons learned fromthe development of an abl tyrosine kinaseinhibitor for chronic myelogenous leukemia.J Clin Invest. 2000;105(1):3-7.
69. Deininger MW, Buchdunger E, Druker BJ.The development of imatinib as a therapeu-tic agent for chronic myeloid leukemia.Blood. 2005;105(7):2640-2653.
70. Kantarjian H, Sawyers C, Hochhaus A, et al.Hematologic and cytogenetic responses toimatinib mesylate in chronic myelogenousleukemia. N Engl J Med. 2002;346(9):645-652.
71. Hochhaus A, Druker B, Sawyers C, et al.Favorable long-term follow-up results over 6years for response, survival, and safety withimatinib mesylate therapy in chronic-phasechronic myeloid leukemia after failure ofinterferon-alpha treatment. Blood.2008;111(3):1039-1043.
72. Deininger MW, O’Brien SG, Guilhot F, et al.International Randomized Study ofInterferon vs STI571 (IRIS) 8 years Follow-up: Sustained Survival and Low Risk forProgression or Events in Patients withNewly Diagnosed Chronic MyeloidLeukemia in Chronic Phase Treated withImatinib. ASH Annual Meeting Abstracts2009;114:1126-.
73. Baccarani M, Soverini S. MolecularResponse in CML: Where is the bar? Blood.2014;124(4):469-471.
74. Mughal TI, Vannucchi AM, Soverini S, et al.Preclinical and clinical issues in chronicmyeloproliferative neoplasms.Haematologica. 2014;99(5):797-801.
75. Kantarjian H, O’Brien S, Jabbour E, et al.Improved survival in chronic myeloidleukemia since the introduction of imatinib:a single-institution historical experience.Blood. 2012;119(9):1981-1987.
76. Kantarjian H, Talpaz M, O’Brien S, et al.High-dose imatinib mesylate therapy innewly diagnosed Philadelphia chromosome-positive chronic phase chronic myeloidleukemia. Blood. 2004;103(8):2873-2878.
77. Cortes JE, Kantarjian HM, Goldberg SL, et al.High-dose imatinib in newly diagnosedchronic-phase chronic myeloid leukemia:high rates of rapid cytogenetic and molecu-lar responses. J Clin Oncol. 2009;27(28):4754-4759.
78. Cortes JE, Baccarani M, Guilhot F, et al.Phase III, randomized. Open-label study ofdaily imatinib mesylate 400mg versus800mg in patients with newly diagnosed,previously untreated chronic myeloidleukemia in chronic phase using molecularend points: tyrosine kinase inhibitor opti-mization and selectivity study. J Clin Oncol.2010;28(3):424-430.
79. Hehlmann R, Muller MC, Lauseker M, et al.Deep Molecular Response Is Reached by theMajority of Patients Treated With Imatinib,Predicts Survival, and Is Achieved MoreQuickly by Optimized High-Dose Imatinib:Results From the Randomized CML-StudyIV. J Clin Oncol. 2014;32(5):415-423.
80. Proetel U, Pletsch N, Lauseker M, et al.Older patients with chronic myeloidleukemia (>65 years) profit more from high-er imatinib doses than younger patients: asubanalysis of the randomized CML-StudyIV. Ann Hematol. 2014;93(7):1167-1176.
81. Deininger MW, Kopecky KJ, Radich JP, et al.Imatinib 800mg daily induces deeper molec-ular responses than imatinib 400mg daily:results of SWOG S0325, an intergroup ran-domized PHASE II trial in newly diagnosedchronic phase chronic myeloid leukaemia.Br J Haematol. 2014;164(2):223-232.
82. Preudhomme C, Guilhot J, Nicolini FE, et al.Imatinib plus peginterferon alfa-2a in chron-ic myeloid leukemia. N Engl J Med.2010;363(26):2511-2521.
83. Gambacorti-Passerini C, Antolini L, MahonFX, et al. Multicenter independent assess-ment of outcomes in chronic myeloidleukemia patients treated with imatinib. JNat Cancer Inst. 2011;103(7):553-561.
84. Mughal TI, Schreiber A. Long term toxicityof imatinib when used as a first-line therapyin chronic myeloid leukemia. Biologics.2010;4:315-323.
85. Efficace F, Baccarani M, Breccia M, et al.Chronic fatigue is the most important factorlimiting health-related quality of life ofchronic myeloid leukemia patients treatedwith imatinib. Leukemia. 2013;27(7):1511-1519.
86. Gambacorti-Passerini C, Tornaghi L,Cavagnini F, et al. Gynaecomastia in menwith chronic myeloid leukaemia after ima-tinib. Lancet. 2003;361(9373):1954-1956.
87. Verma D, Kantarjian H, Strom SS, et al.Malignancies occurring during therapy withtyrosine kinase inhibitors (TKIs) for chronicmyeloid leukemia (CML) and other hemato-logic malignancies. Blood. 2011;118(16):4353-4358.
88. de Lavallade H, Apperley JF, Khorashad JS, etal. Imatinib for newly diagnosed patientswith chronic myeloid leukemia: incidence ofsustained responses in an intention-to-treatanalysis. J Clin Oncol. 2008;26(20):3358-3363.
89. Lucas CM, Wang L, Austin GM, et al. A pop-ulation study of imatinib in chronic myeloidleukemia demonstrates lower efficacy thanin clinical trials. Leukemia. 2008;22(10):1963-1966.
90. Apperley JF. Chronic myeloid leukaemia.Lancet. 2015;385(9976):1447-1459.
91. Gorre ME, Mohammed M, Ellwood K, et al.Clinical resistance to STI-571 cancer therapycaused by BCR-ABL gene mutation oramplification. Science. 2001;293(5531):876-680.
92. Apperley JF. Part I: mechanisms of resistanceto imatinib in chronic myeloid leukemia.Lancet Oncol. 2007;8(11):1018-1029.
93. Khorashad JS, de Lavallade H, Apperley JF, etal. Finding of kinase domain mutations inpatients with chronic phase chronic myeloidleukemia responding to imatinib may iden-tify those at risk of disease progression. JClin Oncol. 2008;26(29):4806-4813.
94. Soverini S, Branford S, Nicolini FE, et al.Implications of BCR-ABL1 kinase domain-mediated resistance in chronic myeloidleukemia. Leuk Res. 2014;38(1):10-20.
95. Eiring AM, Deininger MW. Individualizingkinase-targeted cancer therapy: the para-digm of chronic myeloid leukemia. GenomeBiol. 2014;15(9):461.
96. Watkins DB, Hughes TP, White DL. OCT1and imatinib transport in CML: is it clinicallyrelevant? Leukemia. 2015;29(10):1960-1969.
97. Goldman JM. How I treat chronic myeloidleukemia in the imatinib era. Blood.2007;110(8):2828-2837.
98. Weisberg E, Manley PW, Breitenstein W, etal. Characterization of AMN107, a selectiveinhibitor of native and mutant Bcr-Abl.Cancer Cell. 2005;7(2):129-141.
99. Bradeen HA, Eide CA, O’Hare T, et al.Comparison of imatinib mesylate, dasatinib(BM-354825) and nilotinib (AMN107) in anN-ethyl-N-nitrosourea (ENU)-based muta-genesis screen: high efficacy of drug combi-nations. Blood. 2006;108(7):2332-2338.
100.Tokarski JS, Newitt JA, Chang CYJ, et al.
The structure of dasatinib (BMS-354825)bound to activated ABL kinase domain elu-cidates its inhibitory activity against ima-tinib-resitant ABL mutants. Cancer Res.2006;66(11):5790-5797.
101.Schittenhelm MM, Shiraga S, Schroeder A,et al. Dasatinib (BMS-354825), a dualSRC/ABL kinase inhibitor inhibits the kinaseactivity of wild-type, juxtamembrane, andactivation loop mutant KIT isoforms associ-ated with human malignancies. Cancer Res.2006;66(1):473-481.
102.Milojkovic D, Apperley JF, Gerrad G, et al.Responses to second-line tyrosine kinaseinhibitors are durable: an intention-to-treatanalysis in chronic myeloid leukemiapatients. Blood. 2012;119(8):1838-1843.
103.Kantarjian H, Giles F, Wunderle L, et al.Nilotinib in imatinib-resistant CML andPhiladelphia chromosome-positive ALL. NEngl J Med. 2006;354(24):2542-2551.
104.Giles FJ, le Coutre PD, Pinilla-Ibarz J, et al.Nilotinib in imatinib-resistant or imatinib-intolerant patients with chronic myeloidleukemia in chronic phase: 48-month fol-low-up results of a phase II study. Leukemia.2013;27(1):107-112.
105.Talpaz M, Shah NP, Kantarjian H, et al.Dasatinib in imatinib-resistant Philadelphiachromosome-positive leukemias. N Engl JMed. 2006;354(24):2531-2541.
106.Hochhaus A, Baccarani M, Deininger M, etal. Dasatinib induces durable cytogeneticresponses in patients with chronic myeloge-nous leukemia in chronic phase with resist-ance or intolerance to imatinib. Leukemia.2008;22(6):1200-1206.
107.Shah NP, Kantarjian HM, Kim DW, et al.Intermittent target inhibition with dasatinibwith dasatinib 100mg once daily preservesefficacy and improves toerability in ima-tinib-resistant and – intolerant chronic-phasechronic myeloid leukemia. J Clin Oncol.2008;26(19):3204-3212.
108.Shah NP, Guilhot F, Cortres JE, et al. Long-term outcome with dasatinib after imatinibfailure in chronic-phase chronic myeloidleukemia: follow-up of a phase 3 study.Blood. 2014;123(15):2317-2324.
109.Brümmendorf TH, Shah NP, Cortes JE, et al.Long-term efficacy and safety of dasatinib100mg once daily (QD) in patients withimatinib resistant/intolerant chronic-phasechronic myeloid leukemia (CML-CP): 5-yearfolow-up from CA180-034. Onkologie.2011;34:263.
110.Müller MC, Cortes JE, Kim DW, et al.Dasatinib treatment of chronic-phase chron-ic myeloid leukemia: analysis of responsesaccording to preexisting BCR-ABL muta-tions. Blood. 2009;114(24):4944-4953.
111.Le Coutre P, Rea D, Abruzzese E, et al.Severe peripheral arterial disease duringnilotinib therapy. J Natl Cancer Inst.2011;103(17):1347-1348.
112.Assouline S, Laneuville P, Gambacorti-Passerini C. Panniculitis during dasatinibtherapy for imatinib-resistant chronic myel-ogenous leukemia. N Engl J Med. 2006;354(24):2623-2624.
113.Saglio G, Kim DW, Issaragrisil S, et al.Nilotinib versus imatinib for newly diag-nosed chronic mye;loid leukemia. N Engl JMed. 2010;362(24):2251-2259.
114.Kantarjian H, Shah NP, Hochhaus A, et al.Dasatinib versus imatinib in newly diag-nosed chronic-phase chronic myeloidleukemia. N Engl J Med. 2010;362(24):2260-2270.
115.Larson RA, Hochhaus A, Clark RE, et al.Nilotinib vs imatinib in patients with newly
T.I. Mughal et al.
556 haematologica | 2016; 101(5)

diagnosed Philadelphia chromosome-posi-tive chronic myeloid leukemia in chronicphase: ENESTnd 3-year follow-up.Leukemia. 2012;26(10):2197-2203.
116. Jabbour E, Kantarjian HM, Saglio G, et al.Early response with dasatinib or imatinib inchronic myeloid leukemia: 3-year follow-upfrom a randomized phase 3 trial (DASI-SION). Blood. 2014;123(4):494-500.
117.Tefferi A. Nilotinib treatment-associatedaccelerated atherosclerosis: when is the riskjustified? Leukemia. 2013;27(9):1939-1940.
118.Giles FJ, Mauro MJ, Hong F, et al. Rates ofperipheral arterial occlusive disease inpatients with chronic myeloid leukemia inthe chronic phase treated with imatinib,nilotinib, or non-tyrosine kinase therapy: aretrospective cohort analysis. Leukemia.2013;27(6):1310-1315.
119.Montani D, Bergot E, Gunther S, et al.Pulmonary arterial hypertension in patientstreated with dasatinib. Circulation.2012;125(17):2128-2137.
120.Radich JP, Kopecky KJ, Appelbaum FR, et al.A randomized trial of dasatinib 100mg ver-sus imatinib 400mg in newly diagnosedchronic-phase chronic myeloid leukemia.Blood. 2012;120(19):3898-3905.
121.Hughes TP, Saglio G, Kantarjian HM, et al.Early molecular response predicts outcomesin patients with chronic myeloid leukemiain chronic phase treated with frontline nilo-tinib or imatinib. Blood. 2014;123(9):494-500.
122.Hughes TP, Lipton JH, Spector N, et al. Deepmolecular responses achieved in patientswith CML-CP who are switched to nilotinibafter long-term imatinib. Blood.2014;124(5):729-736.
123.Remsing LL, Rix U, Colinge J, et al. Globaltarget profile of the kinase inhibitor bosu-tinib in primary chronic myeloid cells.Leukemia. 2009;23(3):477-480.
124.Cortes JE, Kantarjian HM, BrummendorfTH, et al. Safety and efficacy of bosutinib(SKI-606) in chronic phase Philadelphiachromosome-positive chronic myeloidleukemia patients with resistance or intoler-ance to imatinib. Blood. 2011;118(17):4567-4576.
125.Kantarjian HM, Cortes JE, Kim DW, et al.Bosutinib safety and management of toxici-ty in leukemia patients with resistance orintolerance to imatinib and other tyrosinekinase inhibitors. Blood. 2014;123(9):1309-1318.
126.Gambacorti-Passerini C, Brummendorf TH,Kim DW, et al. Bosutinib efficacy and safetyin chronic phase chronic myeloid leukemiaafter imatinib resistance or intolerance: min-imum 24-month follow-up. Am J Hematol.2014;89(7):732-742.
127.Lipton JH, Cortes JE, Khoury HJ, et al. Long-term bosutinib in patients with chronicphase chronic myeloid leukemia after priorimatiniib failure. J Clin Oncol. 2015;33(15):7076.
128.Brümmendorf TH, Cortes JE, de Souza CA,et al. Bosutinib versus imatinib in newlydiagnsoed chronic-phase chronic myeloidleukaemia: results from the 24-month fol-low-up of the BELA trial. Br J Haemtol.2015;168(1):69-81.
129.O’Hare T, Shakespeare WC, Zhu X, et al.AP24534, a pan-BCR-ABL inhibitor forchronic myeloid leukemia, potentiallyinhibits the T315I mutant and overcomemu-tation-based resistance. Cancer Cell. 2009;16(5):401-412.
130.Goldman JM. Ponatinib for ChronicMyeloid Leukemia. N Engl J Med 2012;
367(22):2148-2149.131.Lierman E, Smits S, Cools J, et al. Ponatinib
is active against imatinib-resistant mutantsof FIP1L1-PDGFRA and KIT, and againstFGFR1-derived fusion kinases. Leukemia.2012;26(7):1693-1695.
132.Zabriskie M, Eide CA, Tantravahisk, et al.BCR-ABL1 compound mutations combiningkey kinase domain positions confer clinicalresistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell. 2014;26(3):428-442.
133.Deininger MW, Hodgson JG, Shah NP, et al.Compound mutations in BCR-ABL1 are notmajor drivers of primary or secondary resist-ance to ponatinib in CP-CML patients.Blood. 2015 Nov 24. [Epub ahead of print]
134.Cortes JE, Kantarjian HM, Shah NP, et al.Ponatinib in refractory Philadelphia chromo-some-positive leukemias. N Engl J Med.2012;367(22):2075-2088.
135.Cortes JE, Kim D-W, Pinella-Ibarz J, et al. Aphase 2 trial of ponatinib in Philadelphiachromosome-positive leukemias. N Engl JMed. 2013;369(19):1783-1796.
136.Neelkantan P, Marin D, Laffan M, et al.Platelet dysfunction associated with pona-tinib, a new pan BCR-ABL inhibitor withefficacy for CML resistant to multiple tyro-sine kinase inhibitor therapy.Haematologica. 2012;97(9):1444.
137.Lipton JH, Chuah C, Guerci-Bresler A, et al.EPIC: A phase III trial of ponatinib (PON)versus imatinib (IM) in patients with newlydiagnosed CR-CML. Blood. 2014;124(21):519.
138.Gandhi V, Plunkett W, Cortes JE, et al.Omacetaxine: A protein translation inhibitorfor treatment of chronic myelogenousleukemia. Clin Cancer Res. 2014;20(7):1735-1740.
139.Yun SM, Jung KH, Kim SJ, et al. HS-438, anew inhibitor of imatinib-resistant BCR-ABL T315I mutation in chronic myeloidleukemia. Cancer Lett. 2014;348(1-2):50-60.
140.Mughal TI, Kavita R. Port-wine flavouredsandwiches and haematopoetic stem celltransplantation. Lancet Oncology. 2009;10(9):926.
141.Appelbaum FR. Hematopoietic-cell trans-plantation at 50. N Engl J Med. 2007;357(15):1472-1475.
142.Mughal T. Chronic leukemias. Classicpapers in Hematologic Malignancies. EdThomas, Nathan, Goldman. Pub: TaylorFrancis, London and New York. 2001;139-44.
143.Goldman JM, Catovsky D, Galton DA.Reversal of blast-cell crisis in C.G.L. bytransfusion of stored autologous buffy-coatcells. Lancet. 1978;1(8061):437-438.
144.Goldman JM, Baughan AS, McCarthy DM,et al. Marrow transplantation for patients inthe chronic phase of chronic granulocyticleukaemia. Lancet. 1982;2(8299):623-625.
145.Hansen JA, Gooley TA, Martin PJ, et al. Bonemarrow transplants from unrelated donorsfor patients with chronic myeloid leukemia.N Engl J Med. 1998;338(14):962-968.
146.Gluckman E, Broxmeyer HA, Auerbach AD,et al. Hematopoietic reconstitution in apatient with Fanconi’s anemia by means ofumbilical-cord blood from an HLA-identicalsibling. N Engl J Med. 1989;321(17):1174-1178.
147.Goldman JM, Apperley JF, Jones L, et al.Bone Marrow Transplantation for patientswith Chronic Myeloid Leukemia. N Engl JMed. 1986;314(4):202-207.
148.Apperley JF, Jones J, Hale G, et al. Bone mar-row transplantation for patients with chron-
ic myeloid leukaemia: T-cell depletion withCampath-1 reduces the incidence of graft-versus-host disease but may increase the riskof leukaemic relapse. Bone MarrowTransplant. 1986;1(1):53-66.
149. Innes AJ, Milojkovic D, Apperley JF.Allogeneic transplantation for CML in theTKI era: striking the right balance. Nat RevClin Oncol. 2015 Nov 17 [Epub ahead ofprint]
150.Kolb HJ, Mittermüller J, Clemm C, et al.Donor leukocyte transfusions for treatmentof recurrent chronic myelogenous leukemiain marrow transplant patients. Blood.1990;76(12):2462-2465.
151.McKinnon S, Papadopoulos EB, CarabasiMH, et al. Adoptive immunotherapy evalu-taing escalating doses of donor leukocytesfor relapse of chronic myeloid leukemiaafter bone marrow transplantation: separa-tion of graft-versus-leukemia responsesfrom graft-versus-host disease. Blood. 1995;86(4):1261-1268.
152.Gratwohl A, Hermans J, Goldman JM, et al.Risk assessment for patients with chronicmyeloid leukemia before allogeneic blood ormarrow transplantation. Lancet. 1998;352(9134):1087-1082.
153.Crawley C, Szydlo R, Lalancette M, et al.Outcomes of reduced-intensity transplanta-tion for chronic myeloid leukemia: an analy-sis of prognostic factors from the ChronicLeukemia Working Party of the EBMT.Blood. 2005;106(9):2969-2976.
154.Mughal TI, Yong A, Szydlo R, et al. Theprobability of long-term leukemia free sur-vival for patients in molecular remission 5years after allogeneic stem cell transplanta-tion for chronic myeloid leukemia in chronicphase. Br J Haematol. 2001;115(3):569-574.
155.Radich JP, Gooley T, Bryant E et al. The sig-nificance of bcr-abl molecular detection inchronic myeloid leukemia patients “late,” 18months or more after transplantation. Blood.2001;98(6):1701-1707.
156.Sauselle S, Lauseker M, Gratwohl A, et al.Allogeneic hematopoietic stem cell trans-plantation (allo SCT) for chronic myeloidleukemia in the imatinib era: evaluation ofits impact within a subgroup of the random-ized German CML Study IV. Blood.2010;115(10):1880-1885.
157.Oyekunle A, Zander A, Binder M, et al.Outcome of allogeneic SCT in patients withchronic myeloid leukemia in the era of tyro-sine kinase inhibitor therapy. Ann Hematol.2013;92(4):487-496.
158. Jabbour E, Cortes JE, Santos FP, et al. Resultsof allogeneic hematopoietic stem cell trans-plantation for chronic myelogenousleukemia patients who failed tyrosine kinaseinhibitors after developing BCR-ABL1kinase domain mutations. Blood.2011;117(13):3641-3647.
159. Jain NA, Ito S, Tian X, et al. Clinical and bio-logical predictors of outcome followingrelapse of CML post-allo-SCT. BoneMarrow Transplant. 2015;50(8):1138-1140.
160.Shimoni A, Volchek Y, Koren-Michowitz M,et al. Phase 1/2 study of nilotinib prophylax-is after allogeneic stem cell transplantation inpatients with advanced chronic myeloidleukemia or Philadelphia chromosome-posi-tive acute lymphoblastic leukemia. Cancer.2015;121(6):863-871.
161.O’Brien S, Abboud CN, Akhtari M, et al.Clinical Practice Guidelines in Oncology.Chronic Myelogenous Leukemia, Version 3.National Comprehensive Cancer Network.[Last accessed June 2 2014].
162.Sawyers CL, Hochhaus A, Feldman E, et al.
Chronic myeloid leukemia: reminiscences and dreams
haematologica | 2016; 101(5) 557

Imatinib induces hematologic and cytoge-netic responses in patients with chronicmyelogenous leukemia in myeloid blast cri-sis: results of a phase II study. Blood.2002;99(10):3530-3539.
163.Apperley JF, Cortes JE, Kim DW, et al.Dasatinib in the treatment of chronicmyeloid leukemia in accelerated phase afterimatinib failure: the START-A trial. J ClinOncol. 2009;27(21):3472-3479.
164.Bjorkholm M, Ohm L, Eloranta S, et al.Success story of targeted therapy in chronicmyeloid leukemia: a population-based studyof patients diagnosed in Sweden from 1973to 2008. J Clin Oncol. 2011;29(18):2514-2520.
165.Gallipoli P, Abraham SA, Holyoake TL.Hurdles Toward a Cure for CML: The CMLStem Cell. Hematol Oncol Clin North Am.2011;25(5):951-966.
166.Copland M, Hamilton A, Elrick LJ, et al.Dasatinib (BMS-354825) targets an earlierprogenitor population than imatinib in pri-mary CML but does not eliminate the quies-cent fraction. Blood. 2006;107(11):4532-4539.
167.CML Experts. The price of drugs for chronicmyeloid leukemia (CML) is a reflection ofthe unsustainable prices of cancer drugs:from the perspective of a large group ofCML experts. Blood. 2013;121(22):4439-4442.
168.Mahon F, Rea D, Guilhot J, et al.Discontinuation of imatinib in patients withchronic myeloid leukemia who have main-tained complete molecular remission for atleast 2 years: the prospective multicentreStop Imatinib (STIM) trial. Lancet Oncol.2010;11(11):1029-1035.
169.Ross DM, Branford S, Seymour JF, et al.Safety and efficacy of imatinib cessation forCML patients with stable undetectable min-imal residual disease: results from theTWISTER study. Blood. 2013;122(4):515-522.
170.Branford S, Yeung DT, Ross DM, et al. Earlymolecular response and female sex stronglypredict stable undetectable BCR-ABL1, thecriteria for imatinib discontinuation inpatients with CML. Blood. 2013;121(19):3818-3824.
171.Ahmed W, van Etten RA. Alternativeapproaches to eradicating the malignantclone in chronic myeloid leukemia: tyrosine-kinase inhibitor combinations and beyond.Hematology Am Soc Hematol EducProgram. 2013;2013:189-200.
172.O’Hare T, Zabriske MS, Eiring AM,Deininger MW. Pushing the limits of target-ed therapy in chronic myeloid leukemia. NatRev Cancer. 2012;12(8):513-526.
173.Bhatia R. Altered microenvironment regula-tion of CML stem cells. Leuk Supplements.2014;S1-S2.
174.Gallipoli P, Cook A, Rhodes S, et al.JAK2/STAT5 inhibition by nilotinib withruxolitinib contributes to the elimination ofchronic phase CML CD34+ cells in vitro andin vivo. Blood 2014;124(9):1492-1501.
175.Zhang B, Ho YW, Maeda T, et al. Altered
microenvironmental regulation of leukemicand normal stem cells in chronic myeloge-nous leukemia. Cancer Cell. 2012;21(4):577-592.
176.Weisberg E, Azab AK, Manley PW, et al.Inhibition of CXCR4 in CML cells disruptstheir interaction with the bone marrowmicroenvironment and sensitizes them tonilotinib. Leukemia. 2012;26(5):985-990.
177.Krause DS, Scadden DT. A hostel for thehostile: the bone marrow niche in hemato-logic neoplasms. Haematologica.2015;100(11):1376-1387.
178.Kawasaki ES, Clark SS, Coyne MY, et al.Diagnosis of chronic myeloid and acutelymphocytic leukemias by detection ofleukemia-specific mRNA sequences ampli-fied in vitro. Proc Natl Acad Sci USA.1988;85(15):5698-5702.
179.Sawyers CL, Timpson L, Kawasaki ES, ClarkSS, Witte ON, Champlin R. Molecularrelapse in chronic myelogenous leukemiapatients after bone marrow transplantationdetected by polymerase chain reaction. ProcNatl Acad Sci. 1990;87(2):563-567.
180.Hughes TP, Morgan GJ, Martiat P, GoldmanJM. Detection of residual leukemia afterbone marrow transplant for chronic myeloidleukemia: role of polymerase chain reactionin predicting relapse. Blood. 1991;77(4):874-878.
181.Cross NC, Feng L, Chase A, Bungey J,Hughes TP, Goldman JM. Competitive poly-merase chain reaction to estimate the num-ber of BCR-ABL transcripts in chronicmyeloid leukemia patients after bone mar-row transplantation. Blood. 1993;82(6):1929-1936.
182.Hughes T, Deininger M, Hochhaus A, et al.Monitoring CML patients responding totreatment with tyrosine kinase inhibitors:review and recommendations for harmoniz-ing current methodology for detecting BCR-ABL transcripts and kinase domain muta-tions and for expressing results. Blood.2006;108(1):28-37.
183.Hughes TP, Kaeda J, Branford S, et al.Frequency of major molecular responses toimatinib or interferon alfa plus cytarabine innewly diagnosed chronic myeloid leukemia.N Engl J Med. 2003;349(15):1423-1432.
184.Cross NCP, White HE, Müller MC, Saglio G,Hochhaus A. Standardized definitions ofmolecular response in chronic myeloidleukemia. Leukemia. 2012;26(10):2172-2175.
185.Cross NCP, White HE, Colomer D, et al.Laboratory recommendations for scoringdeep molecular responses following treat-ment for chronic myeloid leukemia.Leukemia 2015;29(5):999-1003.
186.Winn-Deen ES, Helton B, Van Atta R, et al.Development of an integrated assay fordetection of Bcr-Abl RNA. Clin Chem.2007;53(9):1593-1600.
187.Oehler VG, Wuin J, Ramakrishnan R, et al.Absolute quantitative detection of ABL tyro-sine kinase domain point mutations inchronic myeloid leukemia using a novelnanofluidic platform and mutation-specificPCR. Leukemia. 2009;23(2):396-399.
188.Ross D, Branford S. Seymour JF, et al.Patients with chronic myeloid leukemiawho maintain a complete molecular remis-sion after stopping imatinib treatment haveevidence of persistent leukemia by DNAPCR Leukemia. 2010;24(10):1719-1724.
189.LaBarre P, Hawkins KR, Gerlach J, et al. Asimple, inexpensive device for nucleic acidamplification without electricity-towardinstrument-free molecular diagnostics inlow-resource settings. PLoS One. 2011;6(5):e19738.
190.Soverini S, Hochhaus A, Nicolini FE, et al.BCR-ABL kinase domain mutation analysisin chronic myeloid leukemia patients treatedwith tyrosine kinase inhibitors: recommen-dations from an expert panel on behalf ofEuropean LeukemiaNet. Blood. 2011;118(5):1208-1215.
191.Baccarani M, Deininger MW, Rosti G, et al.European LeukemiaNet Recommendationsfor the management of Chronic MyeloidLeukemia. Blood. 2013;122(6):872-884.
192.San-Miguel JF, Kantarjian HM. Improvedunderstanding of disease biology and treat-ment: Multiple Myeloma and ChronicMyeloid Leukaemias in 2014. Nat Rev ClinOncol. 2015;12(2):71-72.
193.Branford S, Yeung DT, Parker WT, et al.Prognosis for patients with CML and>10%BCR-ABL1 after 3 months of imatinibdepends on the rate of BCR-ABL1 decline.Blood. 2014;124(4):511-518.
194.Branford S, Roberts N, Yeung DT, et al.Any BCR-ABL reduction below 10% at 6months of therapy significantly improvesoutcome for CML patients with a poorresponse at 3 months. Blood. 2013;122(21):(Abstract 254).
195.Kim DD, Hamad N, Lee HG, et al. BCR/ABLlevel at 6 months identifies good risk CMLsubgroup after failing early molecularresponse at 3 months following imatinibtherapy for CML in chronic phase. Am JHematol. 2014;89(6):626-632.
196.Neelakantan P, Gerrad C, Lucas C, et al.Combining BCR-ABL1 transcript levels at 3and 6 months in chronic myeloid leukemia:implications for early intervention strategies.Blood. 2013;121(14):2739-2742.
197.The Lancet Haematology. Chronic myeloidLeukaemia: time to push for a cure? Lancet.2015;2(5):e175.
198.Mathisen MS, Kantarjian HM, Cortes JE,Jabbour E. Practical issues surrounding theexplosion of tyrosine kinase inhibitors forthe management of chronic myeloidleukemia. Blood Rev. 2014;28(5):179-187.
199.Holyoake Tl, Helgason GV. Do we needmore drugs for CML? Immunol Rev.2015;263(1):106-123.
200.Etienne G, Dulucq S, Lascaux A, et al. ELN2013 response status criteria: Relevance forde novo imatinib chronic phase chronicmyelpoid leukemia patients? Am J Hematol.2015;90(1):37-41.
201.Ross TS, Mgbemena VE. Re-evaluating therole of BCR/ABL in chronic myelogenousleukemia. Mol Cell Oncol. 2014;1(3):e963450.
T.I. Mughal et al.
558 haematologica | 2016; 101(5)

haematologica | 2016; 101(5) 559
Received: January 13, 2016.
Accepted: February 26, 2016.
Pre-published: March 4, 2016.
©2016 Ferrata Storti Foundation
Check the online version for the most updatedinformation on this article, online supplements,and information on authorship & disclosures:www.haematologica.org/content/101/6/559
Material published in Haematologica is cov-ered by copyright. All rights reserved to theFerrata Storti Foundation. Copies of articlesare allowed for personal or internal use.Permission in writing from the publisher isrequired for any other use.
Correspondence: [email protected]
Ferrata StortiFoundation
EUROPEANHEMATOLOGYASSOCIATION
Haematologica 2016Volume 101(5):559-565
ARTICLERed Cell Biology & its Disorders
doi:10.3324/haematol.2016.142273
Phosphatidylserine is localized exclusively to the inner leaflet of themembrane lipid bilayer of most cells, including erythrocytes. Thisasymmetric distribution is critical for the survival of erythrocytes in
circulation since externalized phosphatidylserine is a phagocytic signal forsplenic macrophages. Flippases are P-IV ATPase family proteins that active-ly transport phosphatidylserine from the outer to inner leaflet. It has not yetbeen determined which of the 14 members of this family of proteins is theflippase in human erythrocytes. Herein, we report that ATP11C encodes amajor flippase in human erythrocytes, and a genetic mutation identified ina male patient caused congenital hemolytic anemia inherited as an X-linkedrecessive trait. Phosphatidylserine internalization in erythrocytes with themutant ATP11C was decreased 10-fold compared to that of the control,functionally establishing that ATP11C is a major flippase in human erythro-cytes. Contrary to our expectations phosphatidylserine was retained in theinner leaflet of the majority of mature erythrocytes from both controls andthe patient, suggesting that phosphatidylserine cannot be externalized aslong as scramblase is inactive. Phosphatidylserine-exposing cells werefound only in the densest senescent cells (0.1% of total) in which scram-blase was activated by increased Ca2+ concentration: the percentage ofthese phosphatidylserine-exposing cells was increased in the patient’ssenescent cells accounting for his mild anemia. Furthermore, the finding ofsimilar extents of phosphatidylserine exposure by exogenous Ca2+-activat-ed scrambling in both control erythrocytes and the patient’s erythrocytesimplies that suppressed scramblase activity rather than flippase activitycontributes to the maintenance of phosphatidylserine in the inner leaflet ofhuman erythrocytes.
ATP11C is a major flippase in human erythrocytes and its defect causes congenitalhemolytic anemiaNobuto Arashiki,1 Yuichi Takakuwa,1 Narla Mohandas,2 John Hale,2 KenichiYoshida,3 Hiromi Ogura,4 Taiju Utsugisawa,4 Shouichi Ohga,5 Satoru Miyano,6Seishi Ogawa,3 Seiji Kojima,7 and Hitoshi Kanno,4,8
1Department of Biochemistry, School of Medicine, Tokyo Women’s Medical University,Japan; 2Red Cell Physiology Laboratory, New York Blood Center, NY, USA; 3Department ofPathology and Tumor Biology, Graduate School of Medicine, Kyoto University, Japan;4Department of Transfusion Medicine and Cell Processing, School of Medicine, TokyoWomen’s Medical University, Japan; 5Department of Pediatrics, Graduate School ofMedicine, Yamaguchi University, Japan; 6Laboratory of DNA Information Analysis,Human Genome Center, Institute of Medical Science, The University of Tokyo, Japan;7Department of Pediatrics, Graduate School of Medicine, Nagoya University, Japan; and8Division of Genomic Medicine, Department of Advanced Biomedical Engineering andScience, Graduate School of Medicine, Tokyo Women's Medical University, Japan
ABSTRACT
Introduction
In human erythrocytes, phosphatidylserine (PS) is present exclusively in the innerleaflet of the membrane lipid bilayer as a result of ATP-dependent active transport(flipping) of aminophospholipids (such as PS and phosphatidylethanolamine) fromthe outer to inner leaflet. PS interacts with spectrin, a cytoskeletal protein under-neath erythrocyte membranes, to maintain membrane deformability and mechani-cal stability of the erythrocytes1 and protects spectrin from glycation, which

decreases the membrane deformability necessary for tra-versing narrow capillaries and the splenic sinuses.2 Moreimportantly, preventing surface exposure of PS is criticalfor erythrocyte survival: exposure of PS on the outer sur-face of the membrane at the end of the 120-day lifespan oferythrocytes is a phagocytic signal for splenicmacrophages to remove the senescent cells.3-5 Indeed inthese cells, the Ca2+ concentration is elevated to activatelipid scrambling, with consequent surface exposure of PS,which is recognized as an “eat-me signal” bymacrophages.4-7 Besides being exposed on normal senes-cent cells, PS is exposed prematurely by sickle erythro-cytes and thalassemic erythrocytes, resulting in a short-ened life span of the red blood cells and consequenthemolytic anemia in these disorders.7-10 Maintenance, reg-ulation, and disruption of the asymmetric PS distributionare, therefore, important for both erythrocyte survival anddeath. While it has been well established that PS distribu-tion is determined by flippase and scramblase activities,the molecular identities of these activities in human ery-throcytes have not been defined. Furthermore, the relativecontributions of these two activities in maintaining theasymmetric distribution of PS under physiological andpathological states are not well understood.Flippases are members of the P-IV ATPase family of pro-
teins composed of 10 transmembrane domains (Figure
1A).11,12 They contribute to localization of PS in the innerleaflet of erythrocyte membranes through ATP-dependentactive transport of aminophospholipids (such as PS) fromthe outer to inner leaflet.13-15 However, the flippase inhuman erythrocytes has not yet been definitively identi-fied. Among the 14 family members, ATP8A1, ATP8A2,ATP11A, and ATP11C were previously shown to trans-port PS in the plasma membrane.12,16-19 ATP11C has beenimplicated as one of the candidates in murine erythrocytesbased on the finding that its mutation in mice resulted inanemia with stomatocytosis.20 However, there are differ-ences between the characteristics of human and murineerythrocytes, including their life span, cell volume and cellhemoglobin content. Furthermore, recent studies havedocumented significant differences in gene expressionduring human and murine erythropoiesis.21 For example,GLUT1 which is abundantly expressed in human erythro-cytes is not expressed by murine erythrocytes. As such itis important to identify the major flippase in human ery-throcytes.In the present study, we identified a point mutation in
ATP11C through whole-exome sequencing of DNA froma male patient with mild hemolytic anemia without mor-phological abnormalities. Detailed analyses establishedthat this mutation is responsible for hemolysis and relatedclinical features and that ATP11C is a major flippase in
N. Arashiki et al.
560 haematologica | 2016; 101(5)
Figure 1. Schematic representationof ATP11C, erythrocyte morphology,and genotyping for the ATP11Cmutation. (A) Schematic of ATP11Cand the site of a c.1253C>A mis-sense mutation coding forThr418Asn. (B) Phase-contrastmicroscopy images (×1,000; pre-pared from original images withoutany modifications) of Giemsa-stained blood from the healthy con-trol and proband. No morphologicalabnormality was observed. (C) Wavedata from direct sequencing ofgenomic DNA for exon 13, whichincludes the coding region ofThr418. The sequence of the anti-sense strand corresponding to thesense strand represented in (A) isdisplayed. Arrows indicate positionof the mutations.
A B
C

human erythrocyte membranes. By defining the contribu-tion of ATP11C to PS distribution when scramblase isinactive under physiologically low Ca2+ concentrationsand when it is activated under elevated high Ca2+ concen-trations, as in senescent cells, we established that sup-pressed scramblase activity rather than flippase activitycontributes to the maintenance of PS in the inner leaflet ofhuman erythrocytes.
Methods
This study was approved by the Ethics Committee for HumanGenome/Gene Analysis Research of Tokyo Women’s MedicalUniversity (#223D). The healthy volunteer, the proband, and theproband’s mother provided informed consent for blood samplecollection and the blood was used in all of the studies outlined.Suppliers of all reagents and laboratory instruments, and the com-position of buffer solutions are given in the Online SupplementaryInformation.
Whole-exome sequencingWhole-exome sequencing was performed as reported previous-
ly.22 Briefly, genomic DNA was extracted from leukocytes, andcoding sequences were enriched with a SureSelect Human AllExon V4 kit and used for massively parallel sequencing with theHiSeq 2000 platform with 100-bp paired-end reads. Candidategermline variants were detected through our in-house pipeline forwhole exome-sequencing analysis. Single nucleotide variants withan allele frequency >0.25 and insertion-deletions with an allele fre-quency >0.1 were called. Identified variants were verified bySanger sequencing of polymerase chain reaction amplicons (detailsof the methods are given in the Online Supplementary Information).
Measurement of phosphatidylserine flipping activity inerythrocytesTo measure PS flipping activity, 1 μL of 1 mg/mL Fluorescent PS
(NBD-PS) was added to 1 mL suspension of washed erythrocytesat a hematocrit of 5% in phosphate-buffered saline with glucose(PBS-G) and incubated for 0–20 min at 37 °C. Twenty microlitersof the erythrocyte suspension were washed with 1 mL PBS-Gwith 1% bovine serum albumin [BSA; BSA (+)] to remove NBD-PS remaining in the outer leaflet.20 To measure loaded NBD-PSafter the 20-min incubation, incubated erythrocytes were washedwith PBS-G in the absence of BSA [20 min BSA (-)]. NBD-derivedfluorescence associated with variously treated erythrocytes wasmeasured by fluorescence activated cell sorting (FACS). For allsamples, 100,000 cells were analyzed. To determine the basal levelof flippase-independent flipping activity, the cell suspension wastreated with 5 mM N-ethylmaleimide (NEM) in PBS-G for 20 minat 37 °C, which irreversibly and non-specifically inactivates flip-pases.9
Analysis of phosphatidylserine-exposing cellsPS exposed on the erythrocyte cell surface was analyzed by
Ca2+-dependent binding of fluorescently labeled annexin V.1
Washed, unfractionated erythrocytes and senescent erythrocytesfractionated by density centrifugation, as we reported previously,23
were suspended in nine volumes of Tris-buffered saline with glu-cose (TBS-G). To control the intracellular Ca2+ concentration, ery-throcytes were treated for 20 min at 37 °C with 2 μM A23187,5 aCa2+ ionophore, in TBS-G; accurate final concentrations of freeCa2+ were determined by adding 1 mM EGTA and calculating theconcentration of CaCl2 using Calcon free software (e.g., 0.958 mMfor a final 1 μM). After incubation, the erythrocytes were washed
three times with 1 mL TBS-G including 1% BSA to removeA23187 from the erythrocyte membranes. Ten microliters of thepacked erythrocytes were suspended in 1 mL TBS-G including 5mM CaCl2. Thereafter, 1 μL 0.25 mg/mL fluorescein isothio-cyanate (FITC)-conjugated annexin V was added, and the fluores-cence on erythrocytes quantified by FACS as described above. Thecut-off for identification of PS-positive cells was set at a fluores-cent signal value 20-fold higher than that detected in the absenceof FITC-annexin V.
Results
Clinical history and analyses of a male patient withcongenital hemolytic anemia
A male proband was born at full-term after a clinicallynormal pregnancy without any apparent anomalies. Atthe age of 4 years, his mother noted that the boy had pig-mented urine, but medical advice was not sought until hewas 13 years old, when he was diagnosed with unknowncongenital hemolytic anemia (Table 1). Neither mental norgrowth retardation was noted, and he began studyingcomputer sciences at the age of 18. The marriage betweenhis parents was not consanguineous and his parents andsiblings (a sister and a brother) are healthy. Laboratorydata indicated mild hemolytic anemia without any partic-ular morphological abnormality of the erythrocytes (Table1, Figure 1B). The leukocyte count was within the normalrange, and the platelet count was slightly low. Withregards to the lymphocytes, CD2-positive T-lymphocytesaccounted for 84% (normal range, 72-90%) and CD20-positive B-lymphocytes for 12% (normal range, 7-30%).Extensive laboratory analyses investigating erythrocytedeformability, membrane proteins and lipids, erythrocyteenzymes, and hemoglobins failed to elucidate a cause,such as hereditary spherocytosis, erythrocyte enzyme
ATP11C, a major flippase in human erythrocytes
haematologica | 2016; 101(5) 561
Table 1. Clinical laboratory parameters for the proband at age 13 and 19. Age 13 Age 19
White blood cell count (109/�L) 2.3 4.0Red blood cell count (1012/�L) 3.80 3.76Hemoglobin (g/dL) 11.8 12.3Hematocrit (%) 36.4 39.3MCV (fL) 95.8 105MCH (pg) 31.1 32.7MCHC (%) 32.4 31.3RDW-CV (%) 13.2 12.4Platelet count (109/�L) 125 149Reticulocytes (%) 1.4 1.7Haptoglobin (mg/dL) 10 17Total bilirubin (mg/dL) 1.8 1.8Direct bilirubin (mg/dL) 0.8 0.6Serum total protein (g/dL) 7.7 7.2Aspartate aminotransferase (IU/L) 23 16Alanine aminotransferase (IU/L) 13 11Lactate dehydrogenase (IU/L) 183 138γ-Glutamyl transpeptidase (IU/L) 10 9MCV: mean corpuscular volume; MCH; mean corpuscular hemoglobin; MCHC: meancorpuscular hemoglobin concentration; RDW: red cell distribution width.

deficiency, or unstable hemoglobinopathy, of the hemoly-sis (Online Supplementary Figure S1, Online SupplementaryTable S1). To identify the molecular etiology of the hemolytic ane-
mia in the proband, we performed whole-exome sequenc-ing as described previously22 and identified 349 candidatevariants: 46 indels and 303 non-synonymous singlenucleotide variants: none of which was previously knownto be causative genes for hemolytic anemia (OnlineSupplementary Table S2). We focused on ATP11C since itsdefect has been associated with anemia in mice.20 We iden-tified a missense mutation in ATP11C (GenBank AccessionNumber NM_001010986) on the X chromosome,c.1253C>A, corresponding to p.Thr418Asn. The proband ishemizygous and the mother is heterozygous for this muta-tion, determined by direct sequencing (Figure 1C).
Flippase activity in the patient’s erythrocytesFlipping activity was measured by monitoring PS inter-
nalization using flow cytometry. Fluorescent PS (NBD-PS) was loaded exogenously onto membranes of control,the patient’s, and maternal erythrocytes in similaramounts and incubated for up to 20 min at 37 °C [Figure2A; BSA (-)]. After the indicated times, NBD-PS remainingin the outer leaflet was extracted with 1% BSA such thatthe remaining cell-associated fluorescence represented PSthat flipped to the inner leaflet (Figure 2). In control ery-throcytes, all cells were clearly NBD-positive after 5 min,indicating that NBD-PS translocated from the outer toinner leaflet by flippase activity (Figure 2A). In contrast,the patient’s erythrocytes showed very little NBD fluores-cence even after 20 min, indicating dramatically decreasedflippase activity. Quantitative analyses confirmed theimportance of ATP11C for PS internalization: 35% ofloaded PS was transported to the inner leaflet at 20 min innormal erythrocytes, but only ~3% (10-fold decrease) wastransported in the patient’s erythrocytes (Figure 2B). Toconfirm the contribution of ATP11C in normal cells, ery-throcytes were treated with NEM, a non-specific flippaseinhibitor,9 before loading NBD-PS. NEM-treated controlerythrocytes showed very little PS internalization, con-firming the importance of ATP11C for flipping activity(Figure 2B, Online Supplementary Figure S2). The residualflippase activity in the patient’s erythrocytes was alsodiminished by NEM treatment (Figure 2B). The maternal erythrocytes comprised two populations:
55-60% showed exactly the same peak positions as thecontrol for all incubation periods, and the other 40–45%were identical to those of the patient (Figure 2A). The for-mer possessed normal PS transport activity, and the latterlacked flippase activity. The total internalized PS was 55-60% of the control amount, reflecting the proportion oferythrocytes with normal activity (Figure 2B). The pres-ence of two populations suggests random inactivation ofthe X chromosome in the erythroblast populations.
Phosphatidylserine-exposing erythrocytes in the circulationTo understand the mechanisms underlying the
proband’s “mild” hemolytic anemia, the percentages ofPS-exposing erythrocytes in whole blood samples and cellfractions enriched for senescent cells were measured byanalyzing the binding of FITC-conjugated annexin V to PSon the cell surface using flow cytometry (Figure 3).Senescent cells were collected as the densest fraction
(0.1% of total cells) by density gradient centrifugation.23There was no apparent difference in the proportion of thedense cell populations among the different blood samplesstudied (data not shown). The percentage of PS-positivecells among total erythrocytes was slightly higher for thepatient (8.86%) than in the control (6.32%) and increasedfurther in the patient’s senescent erythrocytes: 15.86%versus 9.17% in the control. Maternal erythrocytes had aprofile similar to that of the control cells. PS-positive ery-throcytes were greatly increased only in the densest senes-cent cells, suggesting that PS exposure did not occur untilvery late stages of the erythrocytes’ lifespan.
Phosphatidylserine exposure promoted by Ca2+-activatedscrambling in the patient's erythrocytesIn senescent erythrocytes, PS exposure on the cell sur-
face is promoted by the Ca2+-activated scramblase, whichtranslocates phospholipids, including PS, between theinner and outer leaflets.4-7 To examine whether ATP11Cprevents Ca2+-activated PS externalization, the proportionof PS-exposing erythrocytes was measured under differentCa2+ concentrations controlled by treatment with A23187,a Ca2+ ionophore (Figure 4). With increasing Ca2+ concen-trations up to 50 μM, the proportion of PS-positive cellsincreased. The control and patient’s erythrocytes exhibit-ed similar annexin V binding profiles with no apparent dif-ferences in the percentages of PS-positive cells at all Ca2+concentrations tested.
Discussion
In the present study ATP11C was identified as a majorflippase molecule of human erythrocyte membranesthrough whole-exome sequencing of a male patient withan ATP11C missense mutation on X chromosome,c.1253C>A, corresponding to p.Thr418Asn. This muta-tion is not recorded in SNP databases (dbSNP132 and 135)or our in-house database for Japanese patients with con-genital anemia due to bone marrow failure, red cell apla-sia, or hemolytic anemia. The patient’s erythrocytesshowed 10-fold less flipping activity compared with con-trol cells, clearly demonstrating that ATP11C is a majorflippase in human erythrocytes. Thr418 is near Asp412,the phosphorylation site for forming the 4-aspartyl phos-phate intermediate essential for active transport of PS.24The amino acid sequence between Asp412 and Thr418 isconserved among all P-type ATPases. We, therefore,hypothesized that this mutation could disturb the func-tional activity of ATP11C. It should be noted that theresidual flipping activity (~3%) in the patient’s red cellsmay arise from other flippases such as ATP8A1, ATP8A2,and ATP11A. RNAseq analyses of normal human ery-throblasts generated from CD34-positive cells in an in vitroculture system21 demonstrated that mRNA of the threecandidate flippase genes, ATP8A1, ATP11A, and ATP11C,were indeed expressed at all stages of human terminal ery-throid differentiation (Online Supplementary Figure S3A).Alternatively, the missense mutation may not completelyabolish the enzymatic activity of ATP11C. This residualactivity may contribute to PS internalization during ery-thropoiesis, especially in the patient’s erythrocytes.Investigation of the molecular basis of the congenitalhemolytic anemia in the proband (Table 1) using severaldiagnostic tests for already-known hemolytic anemias
N. Arashiki et al.
562 haematologica | 2016; 101(5)

failed to elucidate the cause of hemolysis as hereditaryspherocytosis, a red cell enzyme deficiency, or unstablehemoglobinopathy (Online Supplementary Figure S1, OnlineSupplementary Table S1). The identification of a mutationin the gene encoding the flippase, ATP11C, enabled us todiscover a new candidate gene responsible for human con-genital hemolytic anemia.Although we predicted that loss of flipping activity could
lead to a definitive increase of PS-exposing (positive) ery-throcytes in the majority of the patient’s circulating cells,this was not found to be the case. PS was retained in theinner leaflet of the vast majority of both control erythro-cytes and those from the patient, suggesting that PS is notexposed on the erythrocyte cell surface as long as scram-blase is inactive, regardless of flippase activity. The propor-tion of PS-exposing cells increased only in the densestsenescent cells (0.1% of total) in which scramblase was acti-vated to transport PS from the inner to outer leaflet byincreased Ca2+ concentration. The proportion increased fur-ther in the patient’s senescent cells with deficiency of flip-pase activity, indicating that ATP11C does play a role inactive transport of externalized PS back to the inner leafletto some extent in senescent erythrocytes. The distinctincrease in PS-exposing cells in a small population of senes-
cent cells from the patient indicated that PS exposureoccurred at a very late stage of the patient’s erythrocytelifespan. It should be emphasized that PS-positive cells arecontinuously removed from the circulation by phagocytosisand those remaining in the circulation reflect the populationthat has not yet been cleared. The increased percentage ofPS-positive senescent cells in the patient’s circulation does,therefore, indicate persistent and mild hemolysis, corre-sponding to the clinical symptoms such as mild jaundice. Interestingly, the maternal erythrocytes comprised two
populations; 55-60% possessed normal PS transport activi-ty, and 40-45% lacked flippase activity, like those of thepatient, suggesting random inactivation of the X chromo-some in the erythroblast populations. The proportion of PS-positive circulating erythrocytes in the mother was similarto that in controls and the woman is not anemic. Thesefindings imply that hemolytic anemia in the proband withATP11Cmutation is inherited as an X-linked recessive trait. A balance between flipping and scrambling activities
maintains the asymmetric distribution of PS. Our findingthat the proportion of PS-positive cells in which lipid scram-bling was promoted by exogenous Ca2+ incorporation up to50 μM was very similar between control erythrocytes andthe patient’s erythrocytes implies that ATP11C cannot com-
ATP11C, a major flippase in human erythrocytes
haematologica | 2016; 101(5) 563
Figure 2. Flipping activity of erythrocytes with the ATP11Cmutation. (A) Primary NBD-derived fluorescence data fromflow cytometry. Left: NBD-PS loaded onto erythrocyte mem-branes for 20 min without BSA treatment [20 min BSA (-)].Right: time-dependent internalization of NBD-PS with BSAtreatment to remove NBD-PS remaining in the outer leaflet.Events are indicated with arbitrary units. (B) Quantitation ofthe proportion of internalized NBD-PS calculated by meanfluorescence obtained from (A). The values were obtainedby dividing internalized NBD-PS by loaded NBD-PS in eachindividual. NEM-treated cells from control (C + NEM), thepatient’s (P + NEM), and maternal erythrocytes (M + NEM)were also analyzed to confirm the contribution of ATP11C toobserved flipping activity. Primary flow data are shown inOnline Supplementary Figure S2.
A
B

pete sufficiently with Ca2+-activated PS scrambling to main-tain PS asymmetry. In normal erythrocytes, the Ca2+ con-centration increases transiently under shear stress-induceddeformation during passage through narrow vessels andgradually increases during red cell senescence.25,26 Underthese conditions, scramblase is activated to scramble PSfrom the inner to outer leaflet, and the concerted effort ofATP11C and other flippases may not be sufficient to pre-vent Ca2+-activated PS externalization in these cells.Together, our findings imply that suppression of scramblaseactivity rather than flippase activity is the major contributorto maintenance of PS in the inner leaflet of normal erythro-cytes and that PS externalization as an “eat-me signal”depends primarily on scramblase activity at the end of theerythrocytes’ lifespan. Asymmetric PS distribution is important in other human
cells. For instance, in platelet membranes PS is distributedin the inner leaflet probably by flippase activity under stat-ic conditions and exposed to the cell surface by Ca2+-acti-vated scrambling via TMEM16F when blood coagulationis initially activated.27,28 Based on our findings concerningerythrocyte senescence, flippase activity cannot fully com-pensate for significantly increased scrambling activity. No morphological change was observed in human ery-
throcytes with ATP11C mutation, while Atp11c mutantmice have anemia with stomatocytosis.20 Other differ-ences between the human and murine systems is thatwhile there is 10-fold less flippase activity in maturehuman erythrocytes with mutant ATP11C, flippase activi-ty is nearly normal in mature erythrocytes from Atp11cmutant mouse. In addition, while mRNA levels of bothATP11A and ATP11C were very similar in human ery-throblasts, only Atp11c mRNA was highly expressed inmice, with no expression of Atp11a (Online SupplementaryFigure S3). These findings suggest that total flippase activ-ity might be significantly decreased or absent in the ery-
N. Arashiki et al.
564 haematologica | 2016; 101(5)
Figure 4. PS cell surface exposure in Ca2+-loaded erythrocytes with the ATP11C mutation. Effect of Ca2+-stimulated scrambling on PS exposure in control andpatient’s erythrocytes. Ca2+ was introduced by a Ca2+ ionophore, A23187, for 20 min at 37 °C. After removal of the Ca2+ ionophore, PS-exposed cells were analyzedby monitoring FITC-annexin V binding to erythrocytes.
Figure 3. PS cell surface exposure in circulating erythrocytes with the ATP11Cmutation. Exposed PS was detected by Ca2+-dependent specific binding of FITC-annexin V. Unfractionated erythrocytes (total) and fractionated erythrocytesobtained from density gradient centrifugation (senescence) for the control,patient, and mother were suspended in isotonic buffer including 5 mM CaCl2
before adding FITC-annexin V. Primary data obtained from flow cytometry analy-ses of FITC-derived fluorescence on erythrocytes are displayed. Events areshown with arbitrary units. The values in each panel indicate the proportion ofPS-positive cells.

throblasts of Atp11c mutant mice with resultant PS expo-sure on the outer membrane with subsequent gradual flip-ping back of the PS to the inner leaflet due to other flippas-es in mature erythrocytes, inducing the stomatocyticshape change.29,30 On the other hand, the flippase activityis presumably maintained in the erythroblasts of humansubjects with ATP11C deficiency, due to compensation byother flippases including ATP11A. As a result, mature ery-throcytes with ATP11C deficiency may maintain thebiconcave disc shape because most PS is located in theinner leaflet from the erythroblast stage to the mature ery-throcyte stage. Anemia in the mutant mouse may resultfrom PS-positive erythroblasts being possibly eliminated(ineffective erythropoiesis) as previously documented inthe case of pyruvate kinase deficient mice.31 Based on theexpression of ATP8A1 in mature murine erythrocytes,32 itis likely that flippase activity in murine erythrocytes is pri-marily driven by ATP8A1 while ATP11C is the primary
flippase in human erythrocytes.In summary, our analyses of a patient with mild
hemolytic anemia identified ATP11C as a major flippasein human erythrocytes and showed that genetic mutationof ATP11C causes congenital mild hemolytic anemiainherited as an X-linked recessive trait. We suggest thatthe contribution of ATP11C to the maintenance of PS inthe inner leaflet is important in senescent cells whenscramblase is active but very subtle under physiological,low Ca2+ concentrations when scramblase is inactive.
AcknowledgmentsWe thank the proband and his mother who made this work
possible. We would also like to thank Editage (www.editage.jp)for English language editing. This work was supported by JSPSKAKENHI grant number 25460375 (YT) and 25461609 (HK),by AMED for the Practical Research Project for Rare/IntractableDiseases grant number 27280301 (for the research group organ-
ATP11C, a major flippase in human erythrocytes
haematologica | 2016; 101(5) 565
References
1. Manno S, Takakuwa Y, Mohandas N.Identification of a functional role for lipidasymmetry in biological membranes: phos-phatidylserine-skeletal protein interactionsmodulate membrane stability. Proc NatlAcad Sci USA. 2002;99(4):1943-1948.
2. Manno S, Mohandas N, Takakuwa Y. ATP-dependent mechanism protects spectrinagainst glycation in human erythrocytes. JBiol Chem. 2010;285(44):33923-33929.
3. Crosby WH. Siderocytes and the spleen.Blood. 1957;12(2):165-170.
4. Lauber K, Blumenthal SG, Waibel M,Wesselborg S. Clearance of apoptotic cells:getting rid of the corpses. Mol Cell.2004;14(3):277-287.
5. Basse F, Stout JG, Sims PJ, Wiedmer T.Isolation of an erythrocyte membrane pro-tein that mediates Ca2+-dependent transbi-layer movement of phospholipid. J BiolChem. 1996;271(29):17205-17210.
6. Bratosin D, Estaquier J, Petit F, et al.Programmed cell death in mature erythro-cytes: a model for investigating death effec-tor pathways operating in the absence ofmitochondria. Cell Death Differ.2001;8(12):1143-1156.
7. Boas FE, Forman L, Beutler E.Phosphatidylserine exposure and red cellviability in red cell aging and in hemolyticanemia. Proc Natl Acad Sci USA. 1998;95(6):3077-3081.
8. Chiu D, Lubin B, Roleofsen B, van DeenenLL. Sickled erythrocytes accelerate clottingin vitro: an effect of abnormal membranelipid asymmetry. Blood. 1981;58(2):398-401.
9. Kuypers FA, Lewis RA, Hua M, et al.Detection of altered membrane phospho-lipid asymmetry in subpopulations ofhuman red blood cells using fluorescentlylabeled annexin V. Blood. 1996;87(3):1179-1187.
10. Kuypers FA, Yuan J, Lewis RA, et al.Membrane phospholipid asymmetry inhuman thalassemia. Blood. 1998;91(8):3044-3051.
11. Paulusma CC, Oude Elferink RPJ. The type4 subfamily of P-type ATPases, putativeaminophospholipid translocases with a role
in human disease. Biochim Biophys Acta.2005;1741(1–2):11-24.
12. Lopez-Marques RL, Theorin L, PalmgrenMG, Pomorski TG. P4-ATPases: lipid flip-pases in cell membranes. Pflugers Arch.2014;466(7):1227-1240.
13. Seigneuret M, Devaux, PF. ATP-dependentasymmetric distribution of spin-labeledphospholipids in the erythrocyte mem-brane: Relation to shape changes. Proc NatlAcad Sci USA. 1984;81(12):3751-3755.
14. Zachowski A, Favre E, Cribier S, Hervé P,Devaux PF. Outside-inside translocation ofaminophospholipids in the human erythro-cyte membrane is mediated by a specificenzyme. Biochemistry. 1986;25(9):2585-2590.
15. Daleke DL, Lyles JV. Identification andpurification of aminophospholipid flippases.Biochim Biophys Acta. 2000;1486(1):108-127.
16. Zhou X, Graham TR. Reconstitution ofphospholipid translocase activity with puri-fied Drs2p, a type-IV P-type ATPase frombudding yeast. Proc Natl Acad Sci USA.2009;106(39):16586-16591.
17. Coleman JA, Kwok MC, Molday RS.Localization, purification, and functionalreconstitution of the P4-ATPase Atp8a2, aphosphatidylserine flippase in photorecep-tor disc membranes. J Biol Chem.2009;284(47):32670-32679.
18. van der Velden LM, Wichers CG, vanBreevoort AE, et al. Heteromeric interactionsrequired for abundance and subcellularlocalization of human CDC50 proteins andclass 1 P4-ATPases. J Biol Chem.2010;285(51):40088-40096.
19. Takatsu H, Tanaka G, Segawa K, et al.Phospholipid flippase activities and sub-strate specificities of human type IV P-typeATPases localized to the plasma membrane.J Biol Chem. 2014;289(48):33543-33556.
20. Yabas M, Coupland LA, Cromer D, et al.Mice deficient in the putative phospholipidflippase ATP11C exhibit altered erythrocyteshape, anemia, and reduced erythrocyte lifespan. J Biol Chem. 2014;289(28):19531-19537.
21. An X, Schulz VP, Li J, et al. Global transcrip-tome analyses of human and murine termi-nal erythroid differentiation. Blood.
2014;123 (22):3466-3477.22. Kunishima S, Okuno Y, Yoshida K, et al.
ACTN1 mutation cause congenitalmacrothrombocytopenia. Am J Hum Genet.2013;92(3):431-438.
23. Arashiki N, Kimata N, Manno S, MohandasN, Takakuwa Y. Membrane peroxidationand methemoglobin formation are both nec-essary for band 3 clustering: mechanisticinsights into human erythrocyte senescence.Biochemistry. 2013;52(34):5760-5769.
24. Vestergaard AL, Coleman JA, Lemmin T, etal. Critical roles of isoleucine-364 and adja-cent residues in a hydrophobic gate controlof phospholipid transport by the mam-malian P4-ATPase ATP8A2. Proc Natl AcadSci USA. 2014;111(14):E1334-E1343.
25. Johnson RM, Tang K. Induction of a Ca(2+)-activated K+ channel in human erythrocytesby mechanical stress. Biochim Biophys Acta.1992;1107(2):314-318.
26. Brain MC, Pihl C, Robertson L, Brown CB.Evidence for a mechanosensitive calciuminflux into red cells. Blood Cells Mol Dis.2004;32(3):349-352.
27. Castoldi E, Collins PW, Williamson PL,Bevers EM. Compound heterozygosity for 2novel TMEM16F mutations in a patientwith Scott syndrome. Blood. 2011;117(16):4399-4400.
28. Lhermusier T, Chap H, Payrastre B. Plateletmembrane phospholipid asymmetry: fromthe characterization of a scramblase activityto the identification of an essential proteinmutated in Scott syndrome. J ThrombHaemost. 2011;9(10):1883-1891.
29. Daleke DL, Huestis WH. Incorporation andtranslocation of aminophospholipids inhuman erythrocytes. Biochemistry. 1985;24(20):5406-5416.
30. Daleke DL, Huestis WH. Erythrocyte mor-phology reflects the transbilayer distributionof incorporated phospholipids. J Cell Biol.1989;108(4):1375-1385
31. Aizawa S, Harada T, Kanbe E, et al.Ineffective erythropoiesis in mutant micewith deficient pyruvate kinase activity. ExpHematol. 2005;33(11):1292-1298.
32. Soupene E, Kuypers FA. Identification of anerythroid ATP-dependent aminophospho-lipid transporter. Br J Haematol. 2006;133(4):436-438.

566 haematologica | 2016; 101(5)
Received: September 14, 2015.
Accepted: December 18, 2015.
Pre-published: December 24, 2015.
©2016 Ferrata Storti Foundation
Check the online version for the most updatedinformation on this article, online supplements,and information on authorship & disclosures:www.haematologica.org/content/101/5/566
Material published in Haematologica is cov-ered by copyright. All rights reserved to theFerrata Storti Foundation. Copies of articlesare allowed for personal or internal use.Permission in writing from the publisher isrequired for any other use.
Correspondence: [email protected]
Sickle cell anemia is a manifestation of a single point mutation inhemoglobin, but inflammation and pain are the insignia of this dis-ease which can start in infancy and continue throughout life. Earlier
studies showed that mast cell activation contributes to neurogenicinflammation and pain in sickle mice. Morphine is the common analgesictreatment but also remains a major challenge due to its side effects andability to activate mast cells. We, therefore, examined cannabinoid recep-tor-specific mechanisms to mitigate mast cell activation, neurogenicinflammation and hyperalgesia, using HbSS-BERK sickle and cannabi-noid receptor-2-deleted sickle mice. We show that cannabinoids mitigatemast cell activation, inflammation and neurogenic inflammation in sicklemice via both cannabinoid receptors 1 and 2. Thus, cannabinoids influ-ence systemic and neural mechanisms, ameliorating the disease pathobi-ology and hyperalgesia in sickle mice. This study provides ‘proof of prin-ciple’ for the potential of cannabinoid/cannabinoid receptor-based thera-peutics to treat several manifestations of sickle cell anemia.
Cannabinoid receptor-specific mechanisms to alleviate pain in sickle cell anemia viainhibition of mast cell activation and neurogenic inflammation Lucile Vincent, Derek Vang, Julia Nguyen, Barbara Benson, Jianxun Lei,and Kalpna Gupta
Vascular Biology Center, Division of Hematology, Oncology and Transplantation,Department of Medicine, University of Minnesota, Minneapolis, MN, USA
ABSTRACT
Ferrata StortiFoundation
EUROPEANHEMATOLOGYASSOCIATION
Haematologica 2016Volume 101(5):566-577
ARTICLE Red Cell Biology & Its Disorders
doi:10.3324/haematol.2015.136523
Introduction
Sickle-cell anemia (SCA) is one of the most common inherited disorders and isassociated with both unpredictable recurrent acute pain and chronic pain1.Morphine, an opioid, has been the drug of choice for the treatment of severe painassociated with SCA.1,2 However, morphine is highly histaminergic, and is knownto activate mast cells.2 We showed earlier that mast cells contribute to neurogenicinflammation and hyperalgesia in sickle mice.3 We also found that cannabinoidsmitigate chronic and hypoxia/reoxygenation (H/R)-evoked acute hyperalgesia insickle mice.4,5 Cannabinoids have anti-inflammatory effects and provide protectionfrom ischemia/reperfusion injury.6-10 Since pain is a manifestation of complex sicklepathobiology including inflammation, vascular dysfunction and ischemia/reperfu-sion injury, we investigated cannabinoid receptor-specific modulation of vascularfunction, inflammation and hyperalgesia.Cannabinoid receptors, CB1R and CB2R, are expressed in both the central nerv-
ous system and non-central nervous system tissues, including inflammatory cells.11-15 CB1R and CB2R activation on mast cells has been shown to inhibit degranulationand inflammation, respectively.16 Activation of CB2R peripherally generates anantinociceptive response in inflammatory and neuropathic pain.17 CB2R is involvedin neuroinflammation and the CB2R agonist, JWH-133, mitigates stress-relatedneuroinflammation-dependent pathologies.18,19 Selective activation of peripheralcannaboid receptors is appealing because it would avoid neuropsychiatric adverseeffects associated with activation of CB1R in the central nervous system.Sickle mice display neurogenic inflammation and hyperalgesia via a mast-cell-
dependent mechanism.3 Cannaboid receptors are important modulators of vascularfunction with an anti-ischemic effect and direct anti-inflammatory effects byinhibiting mast cell degranulation.19 Since vascular dysfunction, ischemia/reperfu-

sion injury and inflammation are hallmark features ofSCA, we hypothesized that targeting specific cannaboidreceptors may have beneficial effects on sickle pathobiol-ogy and pain. We used transgenic HbSS-BERK mice, here-after referred to as sickle mice, which show features ofpain and inflammation similar to patients with SCA,4,5,20and sickle mice with deletion of CB2R, to examine thecontribution of each cannaboid receptor in mast cell acti-vation, neurogenic inflammation, and pain.
Methods
The procedures are described in detail in the OnlineSupplementary Methods.
AnimalsSickle (HbSS-BERK) and control mice (HbAA-BERK): BERK trans-
genic mice are murine α and β globin knockouts that expresshuman sickle hemoglobin (S), demonstrating severe sickle cell dis-ease, or normal (A) hemoglobin.4,5,21
CB2R knockout (CB2R-/-) mice: CB2R-/- mice (Stock # 005786;Jackson Laboratory, Bar Harbor, ME, USA) were backcrossed withBERK mice to obtain sickle and control mice without CB2R(HbSS/CB2R-/-; HbAA/CB2R-/-), and littermates with CB2R(HbSS/CB2R+/+; HbAA/CB2R+/+). Sickle or control mice with CB2R-/-
or CB2R+/+ were identified by polymerase chain reaction withprimers specific for the CB2R (Cnr2) gene (Jackson Laboratory).Sickle (HbSS) and control (HbAA) mice were bred and phenotypedfor sickle and normal human hemoglobin by iso-electric focusing4
and genotyping for the knockout and hemoglobin transgenes(Transnetyx, Cordova, TN, USA). All experiments were per-formed following protocols approved by the University ofMinnesota’s Institutional Animal Care and Use Committee.
TreatmentsThe cannabinoid receptor agonist, CP55,940, (Tocris Bioscience,
Bio-Techne, Minneapolis, MN, USA), was prepared in 2%dimethylsulfoxide (DMSO) and 98% normal saline. Mice weretreated daily with 0.3 mg/Kg CP55,940 or 2% DMSO in salineintraperitoneally in a volume of 25 μL/10 g of body weight. To evaluate the contribution of individual cannaboid receptors,
mice were treated with ACEA (Tocris Bioscience), a CB1R selec-tive agonist (Ki = 1.4 nM), or JWH-133 (National Institute on DrugAbuse-NIDA, USA), a CB2R selective agonist (Ki = 3.4 nM).22 Micereceived 1 mg/Kg ACEA or JWH-133 prepared in 2% DMSO and98% normal saline intraperitoneally in a volume of 25 μL/10 g ofbody weight.
Pain-related behaviorsMice were acclimatized to each test protocol in a quiet room at
constant temperature and tested for thermal- (heat and cold),mechanical-, and deep tissue-hyperalgesia (grip force), andcatalepsy (bar test).4
Hyoxia/reoxygenationMice were exposed to hypoxia with 8% O2 and 92% N2 for 3 h
followed by re-oxygenation in room air for 1 h.5
Neurogenic inflammationPlasma extravasation in response to vehicle (10% ethanol,
7.5% Tween in saline), capsaicin (1.6%), or substance P (100nM) injected intradermally in the dorsal skin was assessed bythe Miles assay using Evans blue dye (Sigma-Aldrich, St. Louis,MO, USA).3
Blood flow measurementBlood flow in the dorsal skin was measured with a laser
Doppler blood perfusion monitor (LaserfloR Model BPM 403,Vasamedics, Inc., St. Paul, MN, USA).23
Mast cell activationAt the endpoint of the study, skin punch biopsies (4 mm) were
incubated for indicated times and the culture medium was ana-lyzed for cytokines (Q-Plex™ Array; Quansys Biosciences, Inc.,Logan, UT, USA) and neuropeptides by enzyme-linkedimmunosorbent assays.3 Degranulating mast cells in skin sectionswere quantified and cultured mast cells from skin were immuno-stained for co-expression of mast cell specific c-kit/CD117 (BDBioscience, San Jose, CA, USA), FcεR1 (eBioscience, San Diego,CA, USA) and tryptase3 (Santa Cruz Biotechnology, Inc., SantaCruz, CA, USA).
Hematopathology of bloodHematocrit, total hemoglobin, complete blood counts and red
cell indices (% sickle red blood cells) were determined as previous-ly described.3
Statistical analysisAll data were analyzed using Prism software (v 5.0a, GraphPad
Prism Inc., San Diego, CA, USA). Repeated measures analysis ofvariance (ANOVA) with the Bonferroni correction was used tocompare the responses between treatments. A summary of thesignificance analysis of ANOVA [F(DFn, DFd) values] is given inOnline Supplementary Table S1. A P-value of <0.05 was consideredstatistically significant. All data are presented as mean ± standarderror of mean (SEM).
Results
Cannabinoids mitigate chronic hyperalgesia in sicklemice
Similar to chronic pain in SCA, HbSS-BERK sickle micedemonstrate tonic hyperalgesia4,5,20 compared to HbAA-BERK control mice and H/R evoked acute hyperalgesiasimulating the pain of a vaso-occlusive crisis.5 Earlier weshowed that a single injection of CP55,940, a non-selectivecannabinoid receptor agonist, at a dose of 0.3 mg/Kgrelieved tonic deep tissue as well as CFA-induced mechan-ical hyperalgesia in these sickle mice.4 Chronic painrequires repeated treatment, which can result in tolerance;we, therefore, examined whether chronic treatment withCP55,940 had a sustained analgesic effect over a period oftime. Daily treatment with CP55,940 significantly reduceddeep tissue, mechanical and thermal hyperalgesia in sicklemice (Figure 1A-F). The effect of CP55,940 was sustainedover a period of 3 weeks. Due to the elaborate number ofvalues for each test and each time point, statistical signifi-cance between vehicle and CP55,940 for each time pointand for CP55,940 as compared to baseline (before treat-ment) are indicated in the figures and legends. Chronictreatment did not lead to catalepsy since the bar test didnot show a significant difference between animals treatedwith CP55,940 or vehicle (Figure 1G).
Cannabinoids mitigate hyperalgesia via cannabinoidreceptorsUsing pharmacological and genetic approaches we ana-
lyzed whether cannabinoids relieved chronic and acute
Mechanisms of cannabinoid analgesia in sickle mice
haematologica | 2016; 101(5) 567
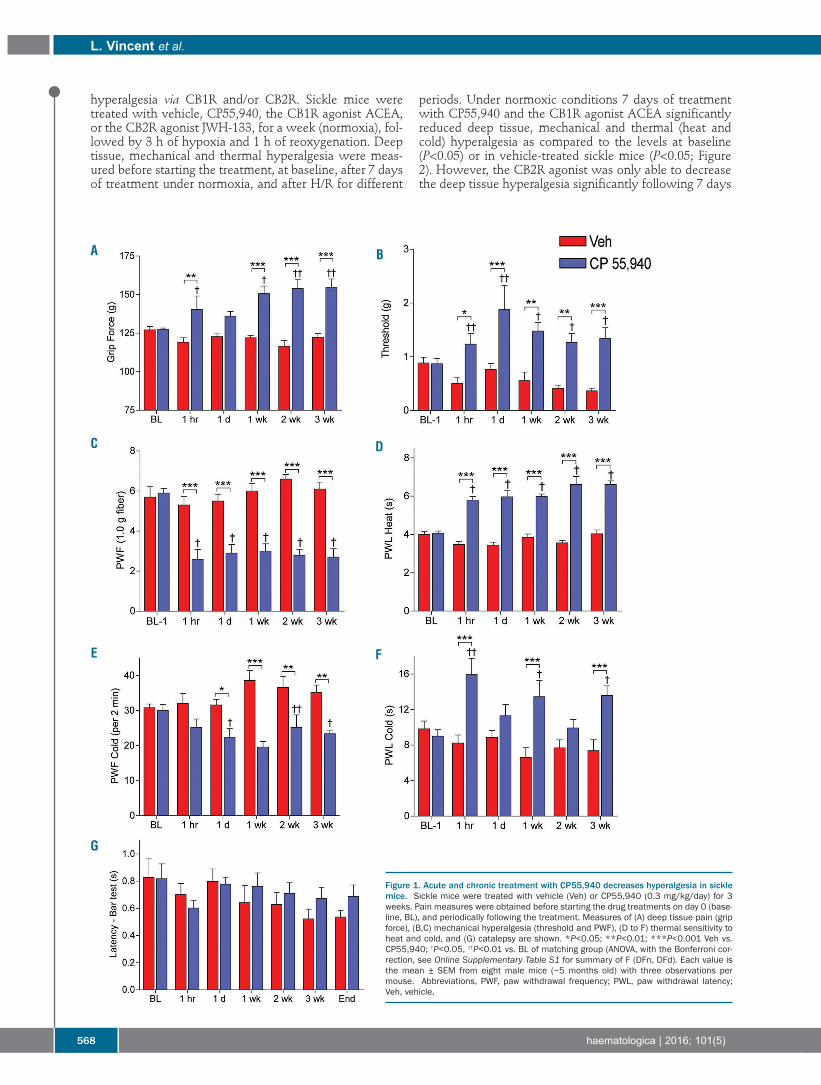
hyperalgesia via CB1R and/or CB2R. Sickle mice weretreated with vehicle, CP55,940, the CB1R agonist ACEA,or the CB2R agonist JWH-133, for a week (normoxia), fol-lowed by 3 h of hypoxia and 1 h of reoxygenation. Deeptissue, mechanical and thermal hyperalgesia were meas-ured before starting the treatment, at baseline, after 7 daysof treatment under normoxia, and after H/R for different
periods. Under normoxic conditions 7 days of treatmentwith CP55,940 and the CB1R agonist ACEA significantlyreduced deep tissue, mechanical and thermal (heat andcold) hyperalgesia as compared to the levels at baseline(P<0.05) or in vehicle-treated sickle mice (P<0.05; Figure2). However, the CB2R agonist was only able to decreasethe deep tissue hyperalgesia significantly following 7 days
L. Vincent et al.
568 haematologica | 2016; 101(5)
Figure 1. Acute and chronic treatment with CP55,940 decreases hyperalgesia in sicklemice. Sickle mice were treated with vehicle (Veh) or CP55,940 (0.3 mg/kg/day) for 3weeks. Pain measures were obtained before starting the drug treatments on day 0 (base-line, BL), and periodically following the treatment. Measures of (A) deep tissue pain (gripforce), (B,C) mechanical hyperalgesia (threshold and PWF), (D to F) thermal sensitivity toheat and cold, and (G) catalepsy are shown. *P<0.05; **P<0.01; ***P<0.001 Veh vs.CP55,940; †P<0.05, ††P<0.01 vs. BL of matching group (ANOVA, with the Bonferroni cor-rection, see Online Supplementary Table S1 for summary of F (DFn, DFd). Each value isthe mean ± SEM from eight male mice (~5 months old) with three observations permouse. Abbreviations, PWF, paw withdrawal frequency; PWL, paw withdrawal latency;Veh, vehicle.
A B
C D
E F
G

of treatment (P<0.05 versus baseline or vehicle; Figure 2A).The CB2R agonist did not show a significant effect onmechanical or thermal (heat and cold) hyperalgesia (Figure2B-D). Thus, under normoxic conditions representative ofchronic pain in SCA, the CB1R agonist as well as the non-selective cannaboid receptor agonist CP55,940 appear tobe uniformly effective in attenuating different pain pheno-types including deep tissue, mechanical and thermalhyperalgesia in sickle mice. On the other hand, the CB2Ragonist only mitigated deep tissue hyperalgesia, suggest-ing that CB1R agonism is critical for treating phenotypical-ly diverse chronic pain in SCA. Earlier we found that H/R-evoked acute deep tissue
hyperalgesia in sickle mice was attenuated by a singleinjection of CP55,940.4 Here we examined whether treat-ment with cananbinoids could prevent HR-evoked hyper-algesia. Pre-treatment of mice with CP55,940, and theCB1R agonist for 7 days decreased tonic hyperalgesia andalso prevented H/R-evoked deep tissue, mechanical andthermal hyperalgesia (Figure 2A-D). However, treatmentwith the CB2R agonist decreased tonic as well as H/R-
evoked deep tissue hyperalgesia (Figure 2A) but did notreduce tonic or H/R-evoked mechanical or thermal (heatand cold) hyperalgesia (Figure 2B-D). Furthermore, to determine the contribution of either
CB1R or CB2R to the analgesia provided by CP55,940, wetreated CB2R-deleted (HbSS CB2R-/-) and intact CB2R(HbSS CB2R+/+) sickle mice with a single dose of CP55,940under normoxia (Figure 3). Control CB2R-/- and sickleCB2R-/- mice did not differ in baseline hyperalgesia ascompared to control CB2R+/+ and sickle CB2R+/+, respec-tively. An increase in grip force was observed in controlCB2R+/+ mice following CP55,940 treatment, but not incontrol CB2R-/- mice (Figure 3A). CP55,940 had no effecton mechanical or cold sensitivity in control CB2R-/- mice orCB2R+/+ mice (Figure 3B,D). Conversely, CP55,940increased heat sensitivity in control CB2R-/- mice but hadno effect on control CB2R+/+ (Figure 3C). CP55,940 treat-ment did not lead to catalepsy since the bar test (Figure 3E)did not show a significant difference from baseline in anygroup. Sickle CB2R-/- and sickle CB2R+/+ mice displayed similar
Mechanisms of cannabinoid analgesia in sickle mice
haematologica | 2016; 101(5) 569
Figure 2. Cannabioids attenuate hypoxia/reoxygenation-evoked hyperalgesia in a receptor-specific manner. Sickle mice (HbSS) were treated with vehicle (Veh),CP55,940, CB1R agonist (ACEA) or CB2R agonist (JWH-133) for 7 days. All mice were then treated with 3 h of hypoxia and 1 h of reoxygenation (H/R). Pain measureswere obtained before starting the drug treatments on day 0 (baseline, BL) and at the conclusion of drug treatments, day 7 (D7) prior to H/R, immediately after H/Rand periodically up to 24 h after H/R. Measures of (A) deep pain, (B) mechanical hyperalgesia and (C-D) thermal sensitivity to heat and cold are shown. ¶P<0.05,¶¶P<0.01 vs. BL of matching group; †P<0.05 vs. Day 7 (D7) of matching group; *P<0.05, **P<0.01 vs. Veh of matched time point. (Two-way ANOVA, with theBonferroni correction, see Online Supplementary Table S1 for the summary of F (DFn, DFd). Each value is the mean ± SEM from five male mice (4-5 months old)with three observations per mouse. Abbreviations, H/R, hypoxia/reoxygenation.
A B
C D

pain behaviors at baseline and a significant decrease inhyperalgesia following CP55,940 treatment (Figure 3A-D).However, significantly greater relief from heat (P<0.001)and deep tissue hyperalgesia (P<0.01) was observed insickle CB2R-/- mice compared to sickle CB2R+/+ mice fol-lowing CP55,940 treatment. Thus, CB1R or CB2R may beused to variable extents to respond to cannabinoid therapyin cases of different pain phenotypes. Together, these datasuggest that under conditions of both chronic and acutepain, activation of CB1R is critical to attenuate hyperalge-sia, and CB2R may partly contribute to cananbinoid anal-gesia, perhaps by modulating inflammatory sickle patho-biology.
Cannabinoids attenuate mast cell activation in sicklecell anemiaWe recently reported that mast cell activation occurs in
SCA and contributes to hyperalgesia and observed a cor-relative increase in dorsal skin blood flow with neurogenicinflammation and mast cell activation.3 We, therefore,analyzed whether cannabinoids influence vascular flowand mast cell activation. Sickle mice treated withCP55,940 daily for 3 weeks showed a significant decreasein dorsal skin blood flow 1 h after CP55,940 injection andthe decrease persisted for the entire duration of treatment(P<0.01 versus vehicle 1 h after treatment and P<0.001 1day, and 1, 2 and 3 weeks; Figure 4A). Treatment with
L. Vincent et al.
570 haematologica | 2016; 101(5)
Figure 3. Cannabinoid analgesia is modulated in CB2R-knockout sickle mice.HbAA-CB2R+/+, HbAA-CB2R-/-, HbSS-CB2R+/+ and HbSS-CB2R-/- mice were treated witha single injection of CP55,940 (0.3 mg/kg, i.p.). Pain measures were obtained beforestarting the drug treatments (at baseline, BL), and periodically after the injection.Measures of (A) deep pain, (B) mechanical hyperalgesia and (C-D) thermal sensitivityto heat and cold, and (E) catalepsy are shown. *P<0.05, **P<0.01, ***P<0.001 vs.HbAA-CB2-R+/+ at matching time point; P<0.05, P<0.01 vs. HbSS-CB2-R+/+ at match-ing time point; ¶P<0.05, ¶¶P<0.01 vs. BL of matching group (ANOVA, with theBonferroni correction, see Online Supplementary Table S1 for the summary of F(DFn, DFd). Each value is the mean ± SEM from five mice (3 males and 2 females,~4.5 months old) with three observations per mouse.
A B
C D
E

CP55,940 significantly decreased activation (degranula-tion) of mast cells in sickle mice compared to sickle micetreated with vehicle (Figure 4B-D). Sickle mice treatedwith CP55,940 showed about 40% less activated mastcells compared to those treated with the vehicle (P<0.01;Figure 4D). Similarly, mast cells isolated from the skin ofsickle mice treated with CP55,940 exhibited lowerimmunoreactivity for c-kit, FcεRI and tryptase (Figure 4E)and released significantly less substance P and tryptase ascompared to mast cells from vehicle-treated mice (P<0.05for both; Figure 4F-G). Earlier we showed that mast cellactivation contributes to inflammation in sickle mice byenhancing the release of several cytokines or chemokines.3We observed that, compared to vehicle treatment,CP55,940 treatment of sickle mice for 3 weeks significant-ly decreased the cytokines released from skin biopsies (IL-1α, IL-6, TNF-α, MCP-1; P<0.01, Figure 2H). Consistentwith decreased mast cell activation, treatment withCP55,940 lowered the levels of granulocte macrophagecolony-stimulating factor (GM-CSF) and regulated on acti-vation, normal T-cell expressed and secreted (RANTES),two chemokines involved in mast cell recruitment andfunction,24,25 by at least 35% (P<0.01). GM-CSF plays acritical role in regulating leukocyte counts, which are oftenelevated in SCA.26 We have previously reported leukocy-tosis in sickle mice and have shown that mast cells play arole in this process.3 Treatment with CP55,940 significant-ly decreased white blood cell counts and sickle red bloodcells, compared to the effect of vehicle, both under nor-moxia and following H/R incitement (Table 1). ThusCP55,940 treatment dampens the inflammatory responseand sickling of red blood cells by decreasing the activationof mast cells.
Hypoxia/reoxygenation-induced mast cell activation is attenuated by cannabinoids in a receptor-specificmannerNext we determined cannaboid receptor-specific inhibi-
tion of mast cell activation in sickle mice under normoxiaand H/R. Sickle mice showed a trend towards increasedmast cell activation following H/R injury as compared tonormoxia (Figure 5A,B). Additionally, treatment with
CP55,940 for 7 days led to a significant reduction in mastcell activation, both under normoxia and following H/R,compared to vehicle under the respective conditions(P<0.05 for each condition; Figure 5B). Although the CB1Ragonist ACEA caused appreciable inhibition of mast cellactivation, the CB2R agonist JWH-133 produced a signifi-cant decrease in degranulating mast cells (P<0.05).Consistent with the inhibitory effect on mast cell activa-tion, administration of CP55,940, compared to treatmentwith only the vehicle, significantly reduced plasmatryptase, β-hexosaminidase and serum amyloid proteinafter H/R injury in sickle mice (P<0.05; Figure 5C). Thelevel of serum substance P was elevated after H/R injurycompared to the level in normoxia in sickle mice (P<0.05;Figure 5D). CP55,940 treatment decreased the levels ofsubstance P both under normoxia and following H/Rinjury (P<0.01; Figure 5D,E). Following H/R, the CB2Ragonist significantly reduced substance P levels as com-pared to the levels in vehicle-treated mice (P<0.05; Figure5E). The CB1R agonist tended to decrease serum sub-stance P but the difference was not statistically significant.Together, these data suggest that H/R-evoked mast cellactivation leading to neuroinflammation is predominantlymediated by CB2R.
Cannabinoids reduce neurogenic inflammationEarlier we found that mast cell activation contributes to
neurogenic inflammation in sickle mice.3 Considering theH/R-induced mast cell activation described above, weexamined the role of cannaboid receptors in relieving neu-rogenic inflammation. Evans blue leakage increased signif-icantly in the skin of sickle mice following H/R incitementcompared to the leakage in normoxia (P<0.05; Figure 6A).CP55,940 decreased Evans blue leakage in sickle miceunder normoxia as well as following H/R (P<0.001; Figure6A). Evoked leakage of Evans blue by intradermal injec-tion of capsaicin or substance P is higher in sickle micethan in control mice under normoxia.3 Treatment withCP55,940 or CB1R and CB2R agonists significantlyreduced Evans blue leakage evoked by capsaicin(CP55,940, P<0.001; CB1R and CB2R P<0.05) and sub-stance P (CP55,940, CB1R and CB2R P<0.001) as com-
Mechanisms of cannabinoid analgesia in sickle mice
haematologica | 2016; 101(5) 571
Table 1. The effect of CP55,940 on hematologic parameters in SCA.NORMOXIA HYPOXIA/REOXYGENATION
HbAA-BERK HbSS-BERK HbAA-BERK HbSS-BERKParameter Veh CP55,940 Veh CP55,940 Veh CP55,940 Veh CP55,940
Peripheral blood
RBC (109/L) 11.4 ± 0.3 11.2 ± 0.1 10.0 ± 0.3 10.2 ± 0.1 11.0 ± 0.3 10.2 ± 0.1 9.1 ± 0.2 9.3 ± 0.3Total Hb (g/dL) 12.7 ± 0.6 12.7 ± 0.5 10.2 ± 0.4* 9.8 ± 0.3* 12.9 ± 0.7 12.5 ± 0.3 9.6 ± 0.3# 9.9 ± 0.6Hematocrit (%) 45.2 ± 1.0 44.4 ± 0.9 41.6 ± 1.0** 41.5 ± 0.8 45.6 ± 1.1 43.5 ± 0.9 40.8 ± 1.1## 40.1 ± 0.9##
WBC (109/L) 7.3 ± 0.4 7.1 ± 0.3 18.7 ± 0.5*** 15.1 ± 0.4**¶ 8.9 ± 0.3 7.9 ± 0.3 22.8 ± 0.8¶### 16.9 ± 0.9##°°Neutrophils (109/L) 1.7 ± 0.2 1.5 ± 0.2 7.8 ± 0.2*** 6.4 ± 0.2**¶ 2.1 ± 0.2 1.9 ± 0.2 9.0 ± 0.2### 5.6 ± 0.2#°�Lymphocytes (109/L) 4.3 ± 0.4 4.3 ± 0.3 6.3 ± 0.4* 5.7 ± 0.3 5.3 ± 0.4 4.9 ± 0.3 7.5 ± 0.5# 6.0 ± 0.6Monocytes (109/L) 0.2 ± 0.1 0.3 ± 0.1 1.1 ± 0.1* 0.5 ± 0.1 0.5 ± 0.1 0.4 ± 0.1 1.4 ± 0.2# 0.6 ± 0.1°
RBC indicesSickle RBC (% total) n/a n/a 28.9 ± 1.8 18.4 ± 1.2¶¶ n/a n/a 37.9 ± 1.8¶¶ 23.8 ± 1.5¶°°°
Complete blood counts were measured in whole blood after 7 days of treatment with vehicle (Veh) or CP55,940. On day 7, mice were separated into two groups: the normoxiagroup (control condition) or the hypoxia/reoxygenation group in wich mice were treated with 3 h of hypoxia and 1 h of reoxygenation (H/R). Blood was collected on day 8, (24h after the end of treatment or after H/R). RBC: red blood cells; Hb: hemoglobin; WBC: white blood cells; n/a: not applicable. *P<0.05, **P<0.01, ***P<0.001 vs. HbAA-BERK VehNormoxia; ¶P<0.05, ¶¶P<0.01 vs. HbSS- BERK Veh Normoxia; #P<0.05, ##P<0.01, ###P<0.001 vs. HbAA-BERK Veh H/R; °P<0.05, °°P<0.01, °°°P<0.001 vs. HbSS-BERK Veh H/R. n =five male mice in each group. Data are mean ± SEM (ANOVA, with the Bonferroni correction).

pared to vehicle (Figure 6B,C). Thus CP55,940 reducesH/R-mediated neurogenic inflammation via both CB1Rand CB2R. Since neurogenic inflammation is orchestratedby peripheral nerves in conjunction with mast cell activa-tion, it is likely that CB2R predominantly mediates thecannabinoid response on mast cells as indicated above,while CB1R mediates the response on peripheral nerves.
Discussion
Pain in SCA may be a result of vascular dysfunction,
inflammation and direct neural injury, involving multipletargets. Moreover, the unique acute pain due to a “crisis”in addition to chronic pain further adds to the complexityand heterogeneity of SCA pain as compared to severe painin other conditions. It is not, therefore, surprising that cur-rent pain management strategie, requiring identification oftherapeutic modalities acting on multiple targets peripher-ally and in the central nervous system are not alwayseffective. Cannabinoid receptors are unique targetsbecause of their peripheral and central activity at a multi-cellular level.Given the psychotropic effects of CB1R, attention is
L. Vincent et al.
572 haematologica | 2016; 101(5)
Figure 4. CP55,940 reduces mast cell activation.Mice were treated with CP55,940 (0.3 mg/kg/day,i.p.) or vehicle for 3 weeks and analyzed asdescribed. (A) Measures of cutaneous blood flow inthe dorsal skin. **P<0.01, ***P<0.001; †P<0.01vs. respective baseline (BL) before starting thetreatments. Each value is the mean ± SEM fromeight male mice (~5 months old) with three obser-vations per mouse. (B,C) Representative images oftoluidine blue stained dorsal skin sections of HbSSmice treated for 3 weeks with vehicle (Veh) orCP55,940 (CP). Each image is representative ofimages from five mice per condition. Scale bar =100 μm. (D) Ratio of degranulating/total mastcells. *P<0.01; †P<0.05, ††P<0.001 vs. HbAA Veh.(E) Representative confocal images of skin mastcells in culture stained for c-kit/CD117 (red), FcεRI(green), and tryptase (blue). Scale bar = 5 μm. n=5.(F,G) Substance P and tryptase in mast cell condi-tioned medium. *P<0.05; **P<0.01; †P<0.05 vs.HbSS Veh. (H) Skin biopsies were incubated in cul-ture medium for 24 h and cytokines released inconditioned medium were analyzed. HbSS BERKVeh and HbSS CP55,940 are represented by redand blue bars, respectively. Values are expressedas a percent of HbSS Veh. *P<0.05, **P<0.01 vs.HbSS Veh (ANOVA, with the Bonferroni correction,see Online Supplementary Table S1 for the sum-mary of F (DFn, DFd). Each value is the mean ±SEM from five male mice (~5 months old).
A B C
E
D
F G
H

being focused on the possibility of targeting CB2R, whichdoes not have psychotropic effects.27,28 CB2R agonistsand/or knockout mice provide compelling evidence thatCB2R activation mitigates neuropathic and inflammatorypain, and is protective against ischemia/reperfusion injuryby decreasing the endothelial expression of adhesion mol-ecules and secretion of chemokines,15,29,30 and by attenuat-
ing leukocyte adhesion to the endothelium, trans-endothelial migration, and interrelated oxidative-nitrosative damage,31,32 all of which are consistent with thepathobiology of SCA. We show here that targeting thecannabinoid receptors is effective in reducing inflamma-tion, mast cell activation and neurogenic inflammation,which orchestrate pain.
Mechanisms of cannabinoid analgesia in sickle mice
haematologica | 2016; 101(5) 573
Figure 5. CP55,940 reduces hypoxia/reoxygenation-evoked mast cell activation. Mice were treated with vehicle (Veh), CP55,940 (CP), CB1R agonist (CB1-R Ag,ACEA) or CB2R agonist (CB2-R Ag, JWH-133) for 1 week followed by normoxia (N) or hypoxia/reoxygenation (H/R) and analyzed as described. (A) Representativeimages of toluidine blue stained dorsal skin sections of HbSS mice. Each image is representative of images from five male mice per condition. Scale bar = 50 μm.(B) Ratio of degranulating/total mast cells. *P<0.05 vs. HbSS Veh H/R. (C) Levels of tryptase, β-hexosaminidase (β-hex) and serum amyloid protein (SAP) after H/R.*P<0.05, **P<0.01 vs. HbSS Veh normo, †P<0.05 vs. HbSS Veh H/R (ANOVA, with the Bonferroni correction, see Online Supplementary Table S1 for the summaryof F (DFn, DFd). (D-E) Levels of substance P in HbSS mice in normoxia or after H/R injury. Substance P expressed as the percentage of HbAA Veh in normoxia (C,E)or HbSS Veh (D). Each value in (B-E) is the mean ± SEM from five male mice, ~5 months old.
A
B
C
D E

Sickle mice exhibit spontaneous musculoskeletal painand cutaneous hyperalgesia to mechanical, heat and coldstimuli.4,5 These symptoms recapitulate the pain pheno-type observed in patients with SCA.1,20 Previously weobserved that an acute dose of CP55,940 attenuated deeptissue hyperalgesia and mechanical hyperalgesia inducedby complete Freud’s adjuvant (CFA) in sickle mice.4,5 Ourpresent observations that sickle mice exhibit sustainedanalgesia over 3 weeks of chronic treatment withCP55,940 suggest that tolerance to cannabinoid analgesiadoes not develop. Cannabinoids have been found to be protective against
ischemia/reperfusion injury.33 CP55,940 prevented sicklinginduced by H/R in sickle mice, suggesting that some of theanalgesic effects of cannabinoids could be due to theireffect on sickle pathobiology. Furthermore, treatmentwith specific CB1R (ACEA) and CB2R (JWH-133) agonistsreduced deep hyperalgesia, but only the CB1R agonistwas able to reduce mechanical and thermal (heat and cold)hyperalgesia following H/R. Complementary to theseobservations, CP55,940 treatment had an antihyperalgesiceffect in HbSS CB2R-/- mice on mechanical and thermal
(heat and cold) hyperalgesia but not on deep tissue hyper-algesia under normoxia. Cannabinoid analgesia is, there-fore, mediated through both CB1R and CB2R, which isspecific to the sickle pain phenotype. Thus, cannaboidreceptors agonists not only have an analgesic effect butalso have a systemic effect on the disease pathophysiolo-gy because pre-treatment with cannabinoids for a weekprevented H/R-induced hyperalgesia. Together with ourearlier studies demonstrating that CP55,940 is effective indecreasing chronic and CFA-induced hyperalgesia,4 thepresent findings highlight the analgesic potential ofcannabinoids to relieve different pain phenotypes undernormoxia (representing chronic pain) and under H/R (rep-resenting the pain of a vaso-occlusive crisis). Importantly,the present data support the use of both CB1R and CB2Ragonists for overall analgesia but, depending on the char-acteristics of the pain, one or the other agonist may poten-tially be more useful. Mast cell activation contributes to sickle pathophysiolo-
gy by mediating inflammation and pain.3 Inflammatorymediators, proteases including tryptase and pro-inflam-matory cytokines are released from mast cells upon activa-
L. Vincent et al.
574 haematologica | 2016; 101(5)
Figure 6. CP55,940 reduces hypoxia/reoxygenationinjury-evoked neurogenic inflammation. (A)Spontaneous Evans blue leakage in HbSS treated withvehicle (Veh, red), or CP55,940 (0.3 mg/kg/day, blue) for1 week, under normoxia or after hypoxia/reoxygenation(H/R). **P<0.0001; *P<0.05. (B) Evans blue leakageevoked by injection of saline, capsaicin, or substance P inthe dorsal skin of HbSS mice treated with vehicle,CP55,940 (0.3 mg/kg), CB1R agonist (CB1-R Ag, ACEA)or CB2R agonist (CB2-R Ag, JWH-133) for 7 days followedby H/R. *P<0.05, **P<0.001 vs. Veh for each treatment;†P<0.05 vs. CP55,940 (ANOVA, with the Bonferroni cor-rection, see Online Supplementary Table S1 for the sum-mary of F (DFn, DFd)). (C) Images showing Evans blueleakage in the dorsal skin of sickle mice after H/R. Eachimage represents reproducible images from five malemice; and each value is the mean ± SEM from five malemice, ~5 months old.
A B
C

tion and contribute to heightened inflammation in SCA.Tryptase, in addition to enhancing inflammation and neu-rogenic inflammation, activates protease activated recep-tor 2 (PAR2) on peripheral nerve endings and promotesnociception.3,34,35 Thus, the sickle microenvironment favorspersistent mast cell activation, consecutively causing noci-ceptor sensitization, which in turn aggravates hyperalge-sia. Indeed our recent studies showed nociceptor sensiti-zation and activation of the p38MAPK pathway in thespinal cords of sickle mice, suggestive of central sensitiza-tion.36 The cannabinoid receptors CB1R and CB2R arefound on mast cells.37,38 Since, mast cells produce endo-cannabinoids, including anandamide, palmitoyl -ethanolamide, and 2-arachidonylglycerol, a potentialautocrine regulatory loop may exist.39 Mast cells are tightlycontrolled by the endocannabinoid system in the skinthereby limiting excessive activation and maturation.Human mucosal-type mast cells use CB1R-mediated sig-naling to limit degranulation and maturation from progen-itor mast cells.37 Mast cell activation was attenuated fol-lowing CP55,940 treatment with a correlative decrease intryptase, substance P and cytokines released from the skinand in cutaneous blood flow. Significantly higher acetyl-choline-induced forearm blood flow has been reported insickle patients as compared to normal subjects, and signif-icantly increased blood flow was observed in females ascompared to male sickle patients.40 Sickle females wereresponsive to blood flow inhibition with the nitric oxidesynthase inhibitor, NG-monomethyl-L-arginine, but sicklemales were not, suggesting that gender-based nitric oxide-dependent and -independent mechanisms are involved.Since, CP55,940 inhibited blood flow in male mice in ourstudy, it may be acting via nitric oxide-independent mech-anisms, but may also inhibit nitric oxide-dependent bloodflow in females, an aspect that requires further examina-tion.Mast cell activation also occurs in response to
ischemia/reperfusion injury.41 Factors associated with mastcell activation were also reduced in H/R-incited sicklemice following CP55,940 treatment. GM-CSF and whiteblood cell counts are elevated in SCA patients26 and insickle mice, and are both further increased by H/R incite-ment.3 Our finding that CP55,940 decreased GM-CSF lev-els, leukocyte counts and also sickle red blood cells hasimportant implications for improving vaso-occlusive crisesand the accompanying pain. A direct effect of CP55,940on reducing sickling of red blood cells is an exciting possi-bility, but the reduction could also be due to an indirecteffect, which warrants further investigation. Importantly,the observed inhibitory effect of both CB1R and CB2Ragonists on neurogenic inflammation and mast cell activa-tion suggests the beneficial effect of cannabinoids on com-plex inflammatory and vascular sickle pathobiology andassociated conditions. Several studies support the analgesic effect of cannabi-
noids in humans.42,43 Sativex, a cannabis-derived oromu-cosal spray, containing equal proportions of THC andcannabidiol has been shown to be effective in treatingsymptoms of multiple sclerosis, including spasticity andneuropathic pain.44,45 Sativex is also being tested in twophase 3 trials for cancer pain and neuropathic pain.46Furthermore, Abrams et al.47 showed that using vaporizedcannabis in conjunction with opioids augments the anal-gesic effects of opioids. Unfortunately, side effects associ-ated with higher doses such as sedation, dizziness, blurred
vision, impaired cognitive functioning and the risk ofaddiction limit the use of cannabinoids for therapy.However, targeting the CB1R and CB2R receptors simul-taneously in the periphery would minimize the sideeffects and concurrently help in managing pain. A recentreport by Khasabova et al.48 described that the activation ofperipheral CB1R and CB2R synergistically reduced tumor-evoked hyperalgesia. A questionnaire-based study evalu-ating the use of marijuana in sickle patients found that52% of patients who indulged in marijuana used it toreduce or prevent acute or chronic pain.49,50 Pain in SCAcould be of mixed type, including nociceptive, neuropath-ic and inflammatory mechanisms with the involvement ofboth peripheral and central nociceptor sensitization.1 TheCB1R agonist was able to improve hyperalgesia signifi-cantly in sickle mice, and the CB2R agonist significantlyattenuated mast cell activation and neurogenic inflamma-tion, which may improve the condition of the systemicdisease, consequently reducing pain. Since, deep hyperal-gesia was mitigated by the CB2R agonist (JWH-133), tar-geting both CB1R and CB2R simultaneously may be ofadvantage in treating the complex, mixed type of pain thattypically occurs in SCA. CP55,940, via the CB2R, wasdemonstrated to stimulate serotonin 2A receptor activityin the pre-frontal cortex of rats, suggesting of an influenceon cognitive and mood disorders.51 The effect of cannabi-noids on neuropsychiatric conditions in SCA does, there-fore, require consideration.Interestingly we did not see an increase in hyperalgesia
with the deletion of CB2R in either control or sickle mice.Earlier studies in CB2R-deleted C57BL/6 mice, comparedto wild-type C57BL/6 animals, did not show an effect onbaseline hyperalgesia in paw withdrawal latency inresponse to heat or mechanical allodynia induced usingvon Frey filaments or in a tail withdrawal assay.28,52 In thisstudy on CB2R-/- mice, an effect on morphine-inducedantinociception was observed only in the early inflamma-tory phase of formalin-induced nociception, whichdiminished later (after 60 min). Similarly, we observed anincrease in paw withdrawal latency in control CB2R-/- fol-lowing CP55,940 treatment, which could be due to anincrease in inflammation in CB2R-/- and may demonstratean anti-inflammatory effect of CP55,940 perhaps viaCB1R. An increase in grip force in control mice occurredfollowing CP55,940 treatment but not in the controlCB2R-/-, suggesting that CB2R is required to alleviatedeep hyperalgesia. Similar to our observations of noeffect of CP55,940 on mechanical hyperalgesia but anincrease in heat-provoked paw withdrawal latency incontrol CB2R-/- mice, in a previous study WIN 55,212-2 (apotent cannaboid receptor agonist) did not influencemechanical hyperalgesia but led to an increase in heat-provoked paw withdrawal latency in CB2R-/- C57BL/6mice in a model of neuropathic pain.28 These data suggesta role of CB2R in the anti-allodynic effect in a neuropathicpain model. In the sickle mice we observed a uniformeffect of CP55,940 on deep tissue, mechanical and ther-mal hyperalgesia. This shows the diverse pathobiology ofsickle pain, perhaps involving inflammation and neuropa-thy, making both CB1R and CB2R agonists necessary toachieve analgesia.CB1R-mediated psychotropic effects and utilization of
smoked cannabis are major deterrants to the use ofcannabis as a medicine.53 However, the recent discovery ofcannaboid receptor-specific agonists and delivery following
Mechanisms of cannabinoid analgesia in sickle mice
haematologica | 2016; 101(5) 575

vaporization provide advantages to the use of cannabinoidsin the medical setting, following well-controlled clinical tri-als.47 Societal stigma against “marijuana” also calls for thedevelopment of cannabis-derived medications in user-friendly drug-delivery systems to dignify their use.Evidence-based knowledge about cannabis-derived med-ications, their dosage and side effects needs to be acquiredin disease-specific, pre-clinical and clinical investigations, asemphasized recently.53,54 It is noteworthy that in the statesof the USA in which cannabis has been legalized for med-ical use, the mean annual opioid overdose mortality ratesbetween 1999 and 2010 were reduced by 24.8% (95% CI,-37.5% to -9.5%; P=0.003).55 Pain in SCA is associated witha poor quality of life and increased morbidity and opioids,with all their side effects, remain the mainstay of therapy.1Our observations in a pre-clinical setting of SCA provide acompelling rationale to examine the potential of cannaboid
receptor-specific agonists and cannabinoids to treat painand ameliorate the associated pathobiology in SCA.
AcknowledgmentsFunding: this work was supported by NIH RO1 grants
HL68802 and 103773 and UO1 HL117664 and Institute forEngineering in Medicine grants to KG. Confocal imaging wasperformed using the Olympus FluoView 1000 IX2 instrument atthe University of Minnesota - University Imaging Centers,http://uic.umn.edu. The authors would like to thank Stefan Kren,Katherine NH Johnson, Sugandha Rajput, Ritu Jha and SusanThompson for breeding, genotyping, phenotyping mice and/ortechnical assistance; Drs. Robert P. Hebbel and David Archer forproviding breeder sickle mice; Drs. David Largaespada andAnindya Bagchi for advice on mouse genetics; Dr Donald A.Simone for a critical review of the manuscript; and Michael J.Franklin and Carol Taubert for editorial assistance.
L. Vincent et al.
576 haematologica | 2016; 101(5)
References
1. Ballas SK, Gupta K, Adams-Graves P. Sicklecell pain: a critical reappraisal. Blood.2012;120(18):3647-3656.
2. Gupta M, Msambichaka L, Ballas SK, GuptaK. Morphine for the treatment of pain insickle cell disease. Sci World J. 2015;2015:540154.
3. Vincent L, Vang D, Nguyen J, et al. Mast cellactivation contributes to sickle cell pathobi-ology and pain in mice. Blood. 2013;122(11):1853-1862.
4. Kohli DR, Li Y, Khasabov SG, et al. Pain-related behaviors and neurochemical alter-ations in mice expressing sickle hemoglobin:modulation by cannabinoids. Blood.2010;116(3):456-465.
5. Cain DM, Vang D, Simone DA, Hebbel RP,Gupta K. Mouse models for studying pain insickle disease: effects of strain, age, andacuteness. Br J Haematol. 2012;156(4):535-544.
6. Klein TW. Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat RevImmunol. 2005;5(5):400-411.
7. Nagarkatti P, Pandey R, Rieder SA, HegdeVL, Nagarkatti M. Cannabinoids as novelanti-inflammatory drugs. Future Med Chem.2009;1(7):1333-1349.
8. Pacher P, Bátkai S, Kunos G. The endo-cannabinoid system as an emerging target ofpharmacotherapy. Pharmacol Rev.2006;58(3):389-462.
9. Hillard CJ. Role of cannabinoids and endo-cannabinoids in cerebral ischemia. CurrPharm Des. 2008;14(23):2347-2361.
10. Zhang M, Adler MW, Abood ME, Ganea D,Jallo J, Tuma RF. CB2 receptor activationattenuates microcirculatory dysfunction dur-ing cerebral ischemic/reperfusion injury.Microvasc Res. 2009;78(1):86-94.
11. Di Marzo V, Stella N, Zimmer A.Endocannabinoid signalling and the deterio-rating brain. Nat Rev Neurosci. 2015;16(1):30-42.
12. Van Sickle MD, Duncan M, Kingsley PJ, etal. Identification and functional characteriza-tion of brainstem cannabinoid CB2 recep-tors. Science. 2005;310(5746):329-332.
13. Munro S, Thomas KL, Abushaar M.
Molecular characterization of a peripheralreceptor for cannabinoids. Nature. 1993;365(6441):61-65.
14. Matsuda LA, Lolait SJ, Brownstein MJ,Young AC, Bonner TI. Structure of acannabinoid receptor and functional expres-sion of the cloned cDNA. Nature.1990;346(6284):561-564.
15. Rajesh M, Pan H, Mukhopadhyay P, et al.Pivotal advance: Cannabinoid-2 receptoragonist HU-308 protects against hepaticischemia/reperfusion injury by attenuatingoxidative stress, inflammatory response, andapoptosis. J Leuko Biol. 2007;82(6):1382-1389.
16. Small-Howard AL, Shimoda LM, Adra CN,Turner H. Anti-inflammatory potential ofCB1-mediated cAMP elevation in mast cells.Biochem J. 2005;388(Pt 2):465-473.
17. Ibrahim MM, Deng H, Zvonok A, et al.Activation of CB2 cannabinoid receptors byAM1241 inhibits experimental neuropathicpain: Pain inhibition by receptors not pres-ent in the CNS. Proc Natl Acad Sci USA.2003;100(18):10529-10533.
18. Zoppi S, Madrigal JL, Caso JR, et al.Regulatory role of the cannabinoid CB2receptor in stress- induced neuroinflamma-tion in mice. Br J Pharmacol.2014;171(11):2814-2826.
19. Pini A, Mannaioni G, Pellegrini-GiampietroD, et al. The role of cannabinoids ininflammatory modulation of allergic respi-ratory disorders, inflammatory pain andischemic stroke. Curr Drug Targ.2012;13(7):984-993.
20. Brandow AM, Stucky CL, Hillery CA,Hoffmann RG, Panepinto JA. Patients withsickle cell disease have increased sensitivityto cold and heat. Am J Hematol.2013;88(1):37-43.
21. Paszty C, Brion CM, Manci E, et al.Transgenic knockout mice with exclusivelyhuman sickle hemoglobin and sickle cell dis-ease. Science. 1997;278(5339):876-878.
22. Pertwee RG. Pharmacology of cannabinoidreceptor ligands. Curr Med Chem.1999;6(8):635-664.
23. McCulloch KM, Ji SA, Raju TNK. Skinblood-flow changes during routine nurseryprocedures. Early Hum Dev. 1995;41(2):147-156.
24. Mattoli S, Ackerman V, Vittori E, Marini M.Mast cell chemotactic activity of RANTES.Biochem Biophys Res Commun. 1995;209(1):316-321.
25. Galli SJ, Borregaard N, Wynn TA.Phenotypic and functional plasticity of cellsof innate immunity: macrophages, mast cellsand neutrophils. Nat Immunol.2011;12(11):1035-1044.
26. Conran N, Saad SO, Costa F, Ikuta T.Leukocyte numbers correlate with plasmalevels of granulocyte–macrophage colony-stimulating factor in sickle cell disease. AnnHematol. 2007;86(4):255-261.
27. Murineddu G, Deligia F, Dore A, Pinna G,Asproni B, Pinna GA. Different classes ofCB2 ligands potentially useful in the treat-ment of pain. Recent Pat CNS Drug Disc.2013;8(1):42-69.
28. Desroches J, Charron S, Bouchard JF,Beaulieu P. Endocannabinoids decrease neu-ropathic pain-related behavior in micethrough the activation of one or bothperipheral CB1 and CB2 receptors.Neuropharmacol. 2014;77:441-452.
29. Murikinati S, Juttler E, Keinert T, et al.Activation of cannabinoid 2 receptors pro-tects against cerebral ischemia by inhibitingneutrophil recruitment. FASEB J.2010;24(3):788-798.
30. Montecucco F, Lenglet S, Braunersreuther V,et al. CB(2) cannabinoid receptor activationis cardioprotective in a mouse model ofischemia/reperfusion. J Mol Cell Cardiol.2009;46(5):612-620.
31. Pacher P, Hasko G. Endocannabinoids andcannabinoid receptors in ischaemia-reperfu-sion injury and preconditioning. Br JPharmacol. 2008;153(2):252-262.
32. Zhang M, Martin BR, Adler MW, et al.Modulation of cannabinoid receptor activa-tion as a neuroprotective strategy for EAEand stroke. J Neuroimmune Pharmacol.2009;4(2):249-259.
33. Tuma RF, Steffens S. Targeting the endo-cannabinod system to limit myocardial andcerebral ischemic and reperfusion injury.Curr Pharm Biotechnol. 2012;13(1):46-58.
34. Galli SJ, Tsai M. IgE and mast cells in allergicdisease. Nat Med. 2012;18(5):693-704.
35. Oliveira SM, Silva CR, Ferreira J. Critical roleof protease-activated receptor 2 activation

by mast cell tryptase in the development ofpostoperative pain. Anesthesiology.2013;118(3):679-690.
36. Cataldo G, Rajput S, Gupta K, Simone DA.Sensitization of nociceptive spinal neuronscontributes to pain in a transgenic model ofsickle cell disease. Pain. 2015;156(4):722-730.
37. Sugawara K, Zakany N, Hundt T, et al.Cannabinoid receptor 1 controls humanmucosal-type mast cell degranulation andmaturation in situ. J Allergy Clin Immunol.2013;132(1):182-193.
38. Stander S, Schmelz M, Metze D, Luger T,Rukwied R. Distribution of cannabinoidreceptor 1 (CB1) and 2 (CB2) on sensorynerve fibers and adnexal structures inhuman skin. J Dermatol Sci. 2005;38(3):177-188.
39. Bisogno T, Maurelli S, Melck D,DePetrocellis L, DiMarzo V. Biosynthesis,uptake, and degradation of anandamide andpalmitoylethanolamide in leukocytes. J BiolChem. 1997;272(6):3315-3323.
40. Gladwin MT, Schechter AN, Ognibene FP, etal. Divergent nitric oxide bioavailability inmen and women with sickle cell disease.Circulation. 2003;107(2):271-278.
41. Reichel CA, Lerchenberger M, Uhl B, et al.Plasmin inhibitors prevent leukocyte accu-mulation and remodeling events in the
postischemic microvasculature. Plos One.2011;6(2):e17229.
42. Williamson EM, Evans FJ. Cannabinoids inclinical practice. Drugs. 2000;60(6):1303-1314.
43. Croxford JL, Miller SD. Immunoregulationof a viral model of multiple sclerosis usingthe synthetic cannabinoid R(+)WIN55,212. JClin Invest. 2003;111(8):1231-1240.
44. Barnes MP. Sativex®: clinical efficacy andtolerability in the treatment of symptoms ofmultiple sclerosis and neuropathic pain. ExpOpin Pharmacother. 2006;7(5):607-615.
45. Collin C, Ehler E, Waberzinek G, et al. Adouble-blind, randomized, placebo-con-trolled, parallel-group study of Sativex, insubjects with symptoms of spasticity due tomultiple sclerosis. Neurol Res. 2010;32(5):451-459.
46. Brower V. New pain drugs in pipeline, butchallenges to usage remain. J Natl CancerInst. 2012;104(7):503-505.
47. Abrams DI, Couey P, Shade SB, Kelly ME,Benowitz NL. Cannabinoid-opioid interac-tion in chronic pain. Clin Pharmacol Ther.2011;90(6):844-851.
48. Khasabova IA, Chandiramani A, Harding-Rose C, Simone DA, Seybold VS. Increasing2-arachidonoyl glycerol signaling in theperiphery attenuates mechanical hyperalge-
sia in a model of bone cancer pain.Pharmacol Res. 2011;64(1):60-67.
49. Howard J, Anie KA, Holdcroft A, Korn S,Davies SC. Cannabis use in sickle cell dis-ease: a questionnaire study. Br J Haematol.2005;131(1):123-128.
50. Elikkottil J, Gupta P, Gupta K. The analgesicpotential of cannabinoids. J Opioid Manag.2009;5(6):341-357.
51. Franklin JM, Carrasco GA. Cannabinoidreceptor agonists upregulate and enhanceserotonin 2A (5-HT(2A)) receptor activityvia ERK1/2 signaling. Synapse. 2013;67(3):145-159.
52. Desroches J, Bouchard JF, Gendron L,Beaulieu P. Involvement of cannabinoidreceptors in peripheral and spinal morphineanalgesia. Neuroscience. 2014;261:23-42.
53. Volkow ND, Baler RD, Compton WM,Weiss SRB. Adverse health effects of mari-juana use. N Engl J Med. 2014;370(23):2219-2227.
54. Wilkinson ST, D'Souza DC. Problems withthe medicalization of marijuana. JAMA.2014;311(23):2377-2378.
55. Bachhuber MA, Saloner B, CunninghamCO, Barry CL. Medical Cannabis laws andopioid analgesic overdose mortality in theUnited States, 1999-2010. JAMA InternMed. 2014;174(10):1668-1673.
Mechanisms of cannabinoid analgesia in sickle mice
haematologica | 2016; 101(5) 577

578 haematologica | 2016; 101(5)
Received: November 7, 2015.
Accepted: February 15, 2016.
Pre-published: February 26, 2016.
©2016 Ferrata Storti Foundation
Check the online version for the most updatedinformation on this article, online supplements,and information on authorship & disclosures:www.haematologica.org/content/101/5/578
Material published in Haematologica is cov-ered by copyright. All rights reserved to theFerrata Storti Foundation. Copies of articlesare allowed for personal or internal use.Permission in writing from the publisher isrequired for any other use.
Correspondence: [email protected]
Ferrata StortiFoundation
EUROPEANHEMATOLOGYASSOCIATION
Haematologica 2016Volume 101(5):578-586
ARTICLE Blood Transfusion
doi:10.3324/haematol.2015.139139
Metabolic pathways that correlate with post-transfusion circulation of stored murine redblood cells Karen de Wolski,1 Xiaoyoun Fu,1,2 Larry J. Dumont,3 John D. Roback,4 HayleyWaterman,1 Katherine Odem-Davis,1 Heather L. Howie,1 and James C. Zimring1,2,5
1Bloodworks NW Research Institute, Seattle, WA, USA; 2University of WashingtonDepartment of Internal Medicine, Division of Hematology, Seattle, WA, USA; 3GeiselSchool of Medicine at Dartmouth, Lebanon; 4Department of Pathology and LaboratoryMedicine, Emory University School of Medicine, Atlanta, GA, USA; and 5University ofWashington Department of Laboratory Medicine and Department of Internal Medicine,Division of Hematology, Seattle, WA, USA
Transfusion of red blood cells is a very common inpatient proce-dure, with more than 1 in 70 people in the USA receiving a redblood cell transfusion annually. However, stored red blood cells
are a non-uniform product, based upon donor-to-donor variation in redblood cell storage biology. While thousands of biological parameterschange in red blood cells over storage, it has remained unclear whichchanges correlate with function of the red blood cells, as opposed tobeing co-incidental changes. In the current report, a murine model of redblood cell storage/transfusion is applied across 13 genetically distinctmouse strains and combined with high resolution metabolomics toidentify metabolic changes that correlated with red blood cell circula-tion post storage. Oxidation in general, and peroxidation of lipids in par-ticular, emerged as changes that correlated with extreme statistical sig-nificance, including generation of dicarboxylic acids and monohydroxyfatty acids. In addition, differences in anti-oxidant pathways known toregulate oxidative stress on lipid membranes were identified. Finally,metabolites were identified that differed at the time the blood was har-vested, and predict how the red blood cells perform after storage, allow-ing the potential to screen donors at time of collection. Together, thesefindings map out a new landscape in understanding metabolic changesduring red blood cell storage as they relate to red blood cell circulation.
ABSTRACT
Introduction
Transfusion of stored red blood cells (RBCs) is amongst the most frequent inpa-tient therapies; for example, in the United States, approximately 1 out of every 70people are transfused each year. However, while the process of RBC collection,storage, and transfusion is well-controlled, there remains substantial variability inthe quality of RBC units, presumably as a result of varying donor characteristics.1 Itis understood that some donors’ RBCs consistently store poorly, and there are cur-rently no methodologies to identify such donors (other than autologous 51-Cr sur-vival studies).1 Thus, measuring and standardizing the quality of stored RBCsremains elusive.The biological changes that occur during RBC storage, collectively called the
“storage lesion” consist of myriad cellular and biochemical alterations.2,3 While thecatalog of changes known to occur with RBC storage continues to grow into thethousands (with the application of omics technologies), it remains unclear whichchanges correlate to clinical performance of transfused RBCs and which are coinci-dental. Both historical and more recent data have demonstrated that RBCs change

during storage and the underlying metabolism of RBCs areboth heritable traits in humans.4-9 Much attention has been paid in recent years to the con-
cern that transfusion of longer stored RBC units may resultin worse medical outcomes. These concerns are largelyfueled by numerous retrospective studies reporting such aneffect.10 Recently, several randomized controlled trials, inparticular in clinical settings, have been completed, and nodifference was observed between groups.11-13 Thereremains considerable debate surrounding this issue;14 how-ever, this is unrelated to the goal of providing the best RBCunits available, and to storing RBCs so as to generate themost efficacious product. The goal of the current study isto elucidate donor biology that affects RBC storage, and theability of the RBC to circulate post transfusion.To identify biochemical components of the storage lesion
that are correlated with RBC performance, in particular theability of stored RBCs to circulate post transfusion, we ana-lyzed 13 commonly available inbred strains of mice.Herein, we report significant variation amongst strains,both with respect to post-transfusion recovery of storedRBCs, and also the basic metabolomics of stored RBCs.Moreover, significant correlations between biochemicalpathways and RBC storage are identified. In particular,lipid metabolism and oxidation (and underlying anti-oxi-dant pathways), emerged as a dominant theme in differ-ences in RBC storage from genetically distinct murinedonors. Together, these findings help to distinguish bio-chemical components of the storage lesion that correlatewith RBC function in a mouse model. These studies pro-vide mechanistic insight into the biology of RBC storage,define the landscape of murine specifics for ongoing basicresearch, and also highlight novel hypotheses to provide arational basis for subsequent human studies.
Methods
MiceThe following strains of mice were purchased from Jackson
Labs (Bar Harbor, ME, USA): KK/HIJ, LG/J, AKR/J, FVB/NJ,C3H/HeJ, DBA/2J, NOD/ShiLtJ, 129X1/SvJ, 129S1/SvImJ, A/J,BTBR/ T+ tf/J, Balb/cByJ, C57Bl/6J). All were female and used forblood donation at 12-15 weeks of age. UbiC-GFP male mice,which are on a C57BL/6 background, were bred to FVB/NJfemales in the Bloodworks NW Research Institute (BWNWRI)Vivarium and offspring were used as RBC recipients at 24-28weeks of age. HOD mice, used as a fresh tracer population fortransfused RBCs, were likewise bred in the BWNWRI Vivarium.The HOD mouse was first described on an FVB background,15 buthas now been backcrossed onto C57BL6J for greater than 20 gen-erations. All mice were maintained on standard rodent chow andwater in a temperature- and light-controlled environment. Allexperiments were performed according to approved InstitutionalAnimal Care and Use Committee (IACUC) procedures.
Collection and storage of bloodWhole blood (600 μl) was collected via cardiac puncture into 84
μl CPDA-1 (12.3%) in a sterile 1.7 mL snapcap microcentrifugetube. Hematocrits were adjusted to approximately 75% (byremoval of supernatant) and samples were stored in Eppendorftubes for seven days at 4ºC. After storage, 50 μl stored RBCs wereresuspended in 510 μl PBS and 5 μl of fresh HOD packed RBCswere added to the suspension as an internal control. The mixtureof RBCs was then directly transfused into FVB/NJ x UbiC-GFP
recipients by intravenous tail vein injection. The remaining sam-ple of stored RBCs was snap frozen in liquid nitrogen for futuremetabolomics analysis. Ratios of donor blood to HOD tracerRBCs was enumerated, both at baseline in the cells to be trans-fused (pre-transfusion mixture) and also in peripheral bloodacquired from recipients 24 h after transfusion (post-transfusionsamples collected into ACD). Pre-transfusion and post-transfusionRBCs were washed three times with PBS, and stained for 30 minwith 0.5 µg anti-Fy3 (clone MIMA29) in 50 µl PBS. Stained cellswere then washed three times with PBS and incubated with 0.2 µgAPC goat-anti-mouse Igs (BD cat. 550826) in 50 µl PBS for 30 min,which stains RBCs bound with MIMA-29, and thus identifiesHOD tracer RBCs. Cells were then washed three times, re-sus-pended in PBS, and analyzed by flow cytometry; 500 HOD+events were counted for each sample. This approach utilizesMIMA-29 to stain HOD tracer RBCs with a color that is differentthan the GFP RBCs, which fluoresce spontaneously. Forward andside scatter were used to exclude fragmented or lysed RBC frag-ments, such that counts reflected intact RBCs. Final RBC survivalwas calculated by the formula:
(Circulating Stored RBC/HOD RBC of post-transfusion sample) /(Stored RBC/HOD pre-transfusion sample)
For experiments studying “fresh RBCS”, the RBCs were collect-ed and processed identically to stored RBCs, with the only differ-ence being they were used after collection and without furtherstorage.
Ultrahigh performance liquid chromatography-tandemmass spectroscopy and gas chromatography-mass spectroscopyDetails of ultrahigh performance liquid chromatography-tan-
dem mass spectroscopy (UPLC-MS/MS) and gas chromatography-mass spectroscopy (GC-MS) are available in the OnlineSupplementary Appendix. Mass Spec was carried out by a commer-cial vendor (Metabolon Inc., NC, USA).
Statistical analysisComparisons of recoveries between strains were performed
separately on each experiment using one-way ANOVA followedby Tukey’s multiple comparisons test, with a single pooled vari-ance. In the case of stored RBCs, the data were first log10-trans-formed to approximate a normal distribution; the data from thefresh RBCs required no data transformation. Analyses were per-formed using Graphpad PRISM 6 software. Linear correlationswere summarized by Pearson’s coefficient with P values by F-testsand q-values to account for multiple testing in the evaluation ofstatistical significance.
Results
Strain dependent variation in post-transfusion recoveries of stored red blood cellsTo test the genetic variation in RBC storage phenotype
amongst mice, a panel of inbred strains was analyzed.These strains were chosen due to commercial availability,well characterized biology and resolved genetic sequence.Consideration was also given to sampling different phylo-genetic arms (Figure 1A). A well-characterized murinemodel of RBC storage was utilized17 with minor modifica-tions (see Methods). RBCs from each of the indicated teststrains were collected, processed, and either transfused as“fresh” RBCs or stored for seven days. The post-transfu-sion circulation of test RBCs, 24 h after transfusion (24-h
Metabolic predictors of RBC storage in mice
haematologica | 2016; 101(5) 579

recovery) was determined by transfusing test RBCs intoGFP-F1 recipients and enumerating GFP negative popula-tions in peripheral blood 24 h post transfusion. Thisapproach allows analysis of RBC recovery without havingto risk altering the RBCs through any labeling procedure.To isolate donor biology as a variable, a single commontransfusion recipient was utilized for all donor strains; inparticular, an F1 cross between UbiC-GFP and FVB mice(GFP-F1). RBCs from UbiC-GFP mice express high levelsof green fluorescent protein (GFP) in RBCs and are on aC57BL/6 background. Thus, the GFP-F1 mice are het-erozygous at all loci between B6 and FVB mice and have aGFP transgene.To control for differences in transfusion volume and
phlebotomy, and also to allow enumeration on a cell bycell basis, a “fresh tracer” control RBC population wasadded to each test RBC population prior to transfusion(Figure 1B, left panel). The tracer population consisted ofHOD RBCs, which express an easily detectable transgeneon RBCs. Essentially no GFP negative events are observedin untransfused recipient mice (Figure 1B, upper rightpanel). The ratio of test RBCs to tracer RBCs 24 h aftertransfusion (Figure 1B, lower right panel) was then correct-ed to the pre-transfusion ratio. Recovery was evaluated foreach strain for both seven days of storage and in freshlyisolated RBCs.Three independent experiments were performed for
stored RBCs from each of the indicated strains, and the
results of each experiment are shown (Figure 1C).Whereas there was relative consistency in a given strainacross experiments, there was substantial strain-to-strainvariability in RBC recoveries after storage; no statisticallysignificant differences in 24-h recovery were observedfrom freshly isolated samples from the 13 different strains(Figure 1D). Extensive ANOVA comparisons betweeneach strain, for both stored and fresh RBCs, were per-formed and multiple differences of statistical significancewere noted only for stored RBCs (Online SupplementaryTable S1).
General metabolomic analysis of strain variation in redblood cell storageFor each experiment, prior to transfusion, a sample of the
RBCs was snap-frozen. Samples were subsequently sub-jected to analysis of metabolites by LC-MS/MS, resulting inthe resolution and relative quantification of 520 compoundsof known identity. Principal component analysis (PCA)showed a clear distinction between fresh and stored RBCsamples within each of the 13 mouse strains, as a functionof the RBC metabolome (Figure 2). In addition, fresh sam-ples from the C57Bl/6J strain demonstrated limited segrega-tion from the general sample groupings and suggest thatthis strain may exhibit a base-line metabolic profile that fur-ther differentiates it from other strains. However, there wasotherwise no clear distinction in general metabolomesbetween strains as a function of storage.
K. de Wolski et al.
580 haematologica | 2016; 101(5)
Figure 1. Twenty-four hour recovery of fresh and stored red blood cells (RBCs) across 13 inbred strains of mice. (A) Phylogenetic distribution of mouse strains usedfor this study (reproduced from Genome Res 2004:14:1806-11 with kind permission of Dr. Petkov and Genome Research16). (B) Representative flow cytometry plotsare shown to indicate the enumeration of fresh tracer and test RBCs in a pre-transfusion sample (left panel). Recipient GFP+ RBCs are shown, including empty gateswhere fresh tracer and test RBCs appear (upper right panel). The final mixture in a recipient mouse 24 h after transfusion is shown (lower right panel). (C) Individualresults of three out of three experiments of stored RBCs is shown. (D) Individual results of three out of three experiments of fresh RBCs is shown (only 2 replicateswere obtained for fresh RBCs for KK/HiJ, LG/J and the C3H/HeJ mouse strains).
A
C
B
D

Glucose metabolism of red blood cell storage acrossinbred mouse strainsAnalysis of RBC storage has traditionally focused on
generation of ATP as a necessary energy source for main-tenance of RBC physiology. In addition, much attentionhas been paid to generation and maintenance of 2,3-DPG,due to its ability to regulate oxygen affinity by hemoglo-bin. Analysis of glycolytic metabolites across strainsdemonstrated decreases in glucose during storage in allcases (Figure 3A). Likewise, in all strains, 2,3-DPGdropped substantially at seven days storage (Figure 3B).The end point of glycolysis (lactate) had a similar increasein stored RBCs from each strain (Figure 3C). ATP was notdetected as a metabolite by this approach, and thus wasnot assessed in the current study.
Metabolites and pathways that correlate with redblood cell storageTo investigate which metabolites (and metabolic path-
ways) may be associated with the ability of RBCs to sur-vive post transfusion, metabolites in stored RBC unitswere chosen based upon the following criteria (correlationgreater than 0.5 or less than -0.5, P<0.05, q value <0.01;see Online Supplementary Table S2 for a full list of com-pounds). Using these criteria, 11 metabolites had a posi-tive correlation with RBC storage (Online SupplementaryTable S2, stored RBCs, positive correlation), 9 of the 11metabolites identified were lipids, although various lipidsubspecies were identified, including free fatty acids(polyunsaturated, and monohydroxy), lysolipids, andglycerol species. Also noted was vitamin E (alpha-toco-pherol), a common cellular anti-oxidant most involvedwith oxidative stress in the lipid compartment. 12-HETEis an arachidonic acid (AA) metabolite that has biologicalproperties. Finally, tryptophan was noted. As examples ofthe typical distributions, both of the fatty acids docos-
apentaenoate (DPA) and docosahexaenoate (DHA)showed a pattern (in general) of decrease over storage instrains that stored poorly and increase in storage of strainsthat stored well (Figure 4A and C) resulting in a positivecorrelation with final levels and 24-h RBC recovery (Figure4B and D).A total of 49 metabolites had a negative correlation with
stored RBCs that fit the above criteria, the majority ofwhich were lipid species (Online Supplementary Table S2,stored RBCs, negative correlation). Of these, 19 wereeither dicarboxylic acids (DCAs) or monohydroxy fattyacids (MHAs), known to be associated with lipid oxida-tion and peroxidation, 10 of which had inverse correla-tions greater than 0.80 with both P values and q values lessthan 0.0001. In all of these cases, levels were low at timeof collection and increased over storage, to a greater extentin strains that stored poorly. Representative box plots ofthe relative amounts of a DCA (dodecanedioate) and aMHA (16-hydroxypalmitate) are shown (Figure 4E and G)along with the correlation plots between these analytesand 24-h RBC recovery (Figure 4F and H). Of note, amongthe MHAs identified are products with known biologicalfunction, including 13-HODE, 9-HODE and the dihy-droxy fatty acid (9,10-DiHOME). In addition, 4-hydroxy-2-nonenal fit these criteria, and is a well-known productand mediator of lipid peroxidation.18 5-HETE, which is aneicosanoid, was also observed to have a similar pattern(Figure 4I and J). Of note, a related species (12-HETE)showed an opposite correlation (Figures 4K and L),although to a less dramatic effect. Two lysolipids and amonoacylglycerol were also observed (OnlineSupplementary Table S2). In addition to lipid species, nega-tive correlates also included metabolites involved in glu-tathione metabolism (4-hydroxy-nonenal-glutathione andmethionine sulfoxide).To test the hypothesis that base-line metabolite levels,
Metabolic predictors of RBC storage in mice
haematologica | 2016; 101(5) 581
Figure 2. Principal component analysis of metabolites measuredin fresh and stored red blood cells (RBCs). PCA for the indicatedstrains (based on color) are shown for both fresh (spheres) andstored (cylinders) RBCs.

present at time of blood collection, would predict qualityof RBC storage over time, correlations were calculatedbetween metabolite levels in freshly collected RBCs andpost-transfusion recovery after storage (OnlineSupplementary Table S2, fresh RBCs). The same cut-off values of significance were used as above (correlation >0.5or <-0.5, P<0.05, q value <0.01). Fourteen metabolites metthese criteria with regards to positive correlation, in a vari-ety of pathways, including amino acid metabolism,pyrimidine metabolism, and the urea cycle (OnlineSupplementary Table S2, fresh RBCs, positive correlation).Aspartate is presented as an example of a positively corre-lating analyte (Figure 4M). Twenty-four metabolites metthe criteria with a negative correlation (OnlineSupplementary Table S2, fresh RBCs, negative correlation).Like the negative correlation of metabolites after storage,a substantial clustering of fatty acid metabolites was
observed, including 9 long chain fatty acids of differentcomposition, polyunsaturated fatty acids (linoleate andderivatives), and monoacylglycerols. As an example,palmitate (16:1) is shown (Figure 4N). In addition, toco-pherol, 4-hydroxy-nonenal-glutathione, and other aminoacid metabolites were observed, which is expanded on inthe discussion of anti-oxidant pathways below. Finally,correlations were analyzed for the fold change of a givenanalyte over storage by calculating the ratio in fresh RBCscompared to seven days of storage (Online SupplementaryTable S2, ratio of stored to fresh). The analysis of foldchange reveals the same general patterns as observed byanalyzing data from seven days of storage, with 17 ratioshaving a positive correlation and 33 ratios having a nega-tive correlation, which fit the same significance criteria asabove. In general, the same classes of compoundsemerged, with a few notable differences, discussed below.
K. de Wolski et al.
582 haematologica | 2016; 101(5)
Figure 3. Products of glycolysis are commonacross strains and do not correlate with 24-hrecovery. (A) Glucose levels were equivalent infresh red blood cells (RBCs) from each strain anddecreased commonly after storage. (B) 2,3-DPGwas equivalent across strains and uniformlydecreased after seven days of storage. (C) Lactate was uniformly low in fresh RBCs andincreased equivalently, over storage, for allstrains analyzed. Open boxes represent levels attime of collection whereas gray boxes indicate lev-els after storage. All levels of metabolites are rel-ative concentrations based upon areas under thepeaks and are averages for all three experimentsshown in Figure 1.
A
B
C

Analysis of common anti-oxidant pathwaysDue to the preponderance of lipid peroxidation prod-
ucts, we analyzed differences in common anti-oxidantpathways. Alpha-tocopherol (vitamin E) is a majorhydrophobic anti-oxidant that mainly exerts its effects in
lipid membranes. Levels of alpha-tocopherol in freshRBCs negatively correlated with recoveries of stored RBCs(Figure 5A and F, top panel). At the same time, levels ofalpha-tocopherol in stored RBCs positively correlatedwith recoveries (Figure 5F, middle panel). In other words,
Metabolic predictors of RBC storage in mice
haematologica | 2016; 101(5) 583
Figure 4. Levels of the indicatedmetabolite are shown in fresh(white) and stored (gray) sam-ples, as are the correlations ofthe metabolite with 24-h redblood cell (RBC) recovery. (A andB) DPA, (C and D) DHA, (E and F)dodecanedioate, (G and H) 16-hydroxypalmitate, (I+J) 5-HETE,(K and L) 12-HETE. Correlationsof levels of aspartate (M) andpalmitate (N) in freshly isolatedRBCs are shown versus the 24-hRBC recovery after seven days ofstorage. Open boxes representlevels at time of collection where-as gray boxes indicate levelsafter storage. Correlation calcula-tions represent Pearson’s coeffi-cient. All levels of metabolites arerelative concentrations basedupon areas under the peaks andare averages for all three experi-ments shown in Figure 1.Correlation plots show combineddata from all three of the indicat-ed experiments in Figure 1.
A
C
E
G
I
K
B
D
F
H
J
L
M N

the less alpha-tocopherol present at the time of collectionand the more alpha-tocopherol that remained after stor-age, the better the RBC recoveries. These data are incon-sistent with a simple model of increased alpha-tocopherolproviding increased resistance to oxidation. However, jux-taposition of starting and ending levels of alpha-toco-pherol showed that strains that stored poorly started withhigher alpha-tocopherol levels than strains that storedwell; however, by the end of storage, strains that storedpoorly had lower levels of alpha-tocopherol than strainsthat stored well (Figure 5A). Strains that stored well hadlittle change in alpha-tocopherol at all, whereas poorlystoring strains rapidly depleted their alpha-tocopherol.This trend becomes clear when the correlation(s) are ana-lyzed with regards to the ratio of alpha-tocopherol fromfresh RBCs to stored RBCs (Figure 5F, bottom panel).These data are equally consistent with poorly storingstrains generating more oxidative stress, having decreasedanti-oxidant regeneration, or both. Alternatively, alpha-tocopherol could represent non-causal association and bea surrogate marker.Although less dramatic than the pattern seen with
alpha-tocopherol, GSH showed a similar trend (Figures 5Band G). Since the most common anti-oxidant pathway ofGSH is for 2 GSH molecules to form GSSG; GSSG levelswere examined (Figure 5C); however, no obvious patternemerged to correlate with changes in GSH levels. In con-trast, a clear trend was seen with the analysis of 4-hydroxy-nonenal-glutatione, a common product of GSHanti-oxidant activity upon a product of lipid peroxidation(Figure 5D). Of note, increases in 4-hydroxy-nonenal-glu-tathione from fresh RBCs to stored RBCs correlated withpoor RBC recoveries (Online Supplementary Table S2 anddata not shown). Because vitamin C serves as an intermedi-ate between GSH and alpha-tocopherol, vitamin C levelswere examined, but no meaningful trend was observed(Figure 5E) and ascorbate levels did not correlate with RBCstorage (data not shown). Finally, although its correlationwas 0.48 (and thus technically below the 0.5 cut off), N-acetylcysteine (which is both an antioxidant and also aGSH precursor) had levels in stored RBCs that correlatedwell with 24-h RBC recovery (Figure 5H).
Discussion
The current report makes the observation that, similarto donor-to-donor variation in storage of human RBCs,genetically distinct stains of mice have a wide range ofRBC storage biology regarding both metabolome andpost-storage circulation. The ability of an RBC to circulatedoes not guarantee its full function (e.g. traversing mico-capillary beds, delivering oxygen to tissues, removingCO2, etc.); however, it seems a fair statement that an RBCthat does not circulate will certainly not function. Thus,24-h recovery is a meaningful, if not all encompassing,metric and is currently used as licensing criteria for RBCstorage systems.The current studies are carried out in a tractable animal
model; however, like all models, it suffers the potentialthat it may not translate into human biology.Nevertheless, it serves to generate new knowledge ofmammalian RBC storage biology which may translateinto humans, and which provides (from amongst thenumerous changes in RBCs during storage) a focused list
of compounds and pathways to test in human RBC stor-age. It is worth noting that lipid peroxidation is a well-known component of storage of human RBCs.19-23 In addi-tion, a number of studies have been reported regardingmetabolomics of human RBCs and these show heritabledifferences amongst donors and different storage condi-tions;6,7,24-28 to the best of our knowledge no human studieshave been reported that have combined metabolomicsand in vivo recovery.It is worth noting that, in the current study, the stored
RBCs were not leukoreduced. The majority of (althoughcertainly not all) stored RBCs in humans are leukoreduced.Given the volume of mouse blood required for leukore-ducction, this approach was not considered feasible in thesetting of broad screening of multiple strains. However,we have previously reported metabolomics analysis of B6and FVB strains, using filter leukoreduced products. In thissetting, the same lipid oxidation pathways are associatedwith poor storage.29 In addition, dicarboxylic acids remainassociated with poor storage in leukoreduced RBCs (datanot shown). Thus, while some of the associated changemay be due to contaminating leukocytes and/or platelets,the major findings of lipid peroxidation persist even withleukoreduction. A second consideration is that a 7-daystorage time was chosen for this study, since this allowedthe widest range of differences between strains to beobserved, while still allowing for the best storing strains tohave recoveries of greater than 75% (in line with FDAstandards for human RBC storage).To control for potential differences in recipient phago-
cytic biology, a single common recipient strain of micewas used for all donors in the current study. This approachalso allowed an experimental design in which donor RBCsdid not have to be manipulated or labeled prior to transfu-sion. However, there is a theoretical risk that one is cross-ing alloimmune barriers between strains, which couldaccount for some differences in survival. Naturally occur-ring RBC alloantibodies (analogous to ABO in humans)have not been described in mice, and 24-h recovery is typ-ically too short a period of time for adaptive humoralresponses to occur; thus, we did not predict any problemswith alloimmunity in the current studies. In support ofthis notion, there were no statistically significant differ-ences in 24-h recovery of freshly isolated RBCs, thus indi-cating that, even if alloantibodies were present, they hadno clear functional outcome. Nevertheless, one must givetheoretical consideration to possible effects of crossingstrain barriers.The biological underpinnings that regulate lipid oxida-
tion across strains are unclear; however, alpha-tocopheroland GSH are candidates for being involved in the underly-ing processes that lead to lipid peroxidation. Alpha-toco-pherol does have the ability to inhibit chemical oxidationof RBCs;30 however, the extent to which such is a normalpathway during RBC storage is unclear and has only beentested in an in vitro setting.31 Of interest, it has recentlybeen reported that vitamin C and N-acetylcysteine miti-gate oxidative stress in in vitro human RBC studies.32 In vivostudies in mice have shown that vitamin C supplementa-tion can improve storage as measured by 24-h recovery.33The generation of products of lipid oxidation not only
gives insight into underlying RBC storage biology, butmay in themselves represent a biologically significantcomponent of transfused RBCs. Among the lipid oxida-tion products that were observed to both increase with
K. de Wolski et al.
584 haematologica | 2016; 101(5)

storage and negatively correlate with RBC recoveries are5-HETE and the bioactive lipids 9,10-DiHOME, and (13-HODE+9-HODE). In addition, 4-hydroxy-2-nonenal hasbiological and signaling properties, in addition to being acommon indicator of lipid peroxidation. Interestingly, incontrast to 5-HETE, 12-HETE had a significant positivecorrelation to RBC storage. Both 5-HETE and 12-HETE arearachidonic acid metabolites generated by separatelipoxygenase enzymes. Bioactive lipids are known to beinvolved in complex biologies including inflammation,coagulation, vascular tone and immunity. Bioactive lipidshave been implicated in the pathogenesis of transfusion-related acute lung injury (TRALI), a potentially lethal
sequela of blood transfusion.34-36 It is also worth consider-ing the source of substrates for lipid oxidation pathways,such as eicosanoid generation. Levels of both medium andlong chain fatty acids were strongly correlated with RBCrecovery, and lysolipids and monoacylglycerols increasedwith storage, suggesting release of the observed free fattyacids from glycerophospholipid breakdown. It is unclear ifrelease of free fatty acids from phospholipids precedeslipid oxidation as a separate step or is the result of lipidperoxidation, but it is clear that both correlate.Increases in long chain fatty acids at the time of RBC col-
lection strongly correlated with the post-storage RBC recov-ery. Of these, 4 particular long chain fatty acids had a nega-
Metabolic predictors of RBC storage in mice
haematologica | 2016; 101(5) 585
Figure 5. Anti-oxidant pathways during red blood cell (RBC) storage. Levels of alpha-tocopherol in freshly collected RBCs and after storage are shown (A). Correlationplots are shown for alpha-tocopherol for levels in fresh, stored, and the ration of fresh/stored (F). Levels of GSH in freshly collected RBCs and after storage are shown (B).Correlation plots are shown for GSH for levels in fresh, stored, and the ration of fresh/stored (G). Levels of GSSG (C), 4-hydroxy-nonenal-glutathione (D), and ascorbate (E)are shown in freshly collected RBCs and after storage. Correlations between the ratio of N-acetylcysteine (fold change) is shown (H). Open boxes represent levels at timeof collection; gray boxes indicate levels after storage. Correlation calculations represent Pearson’s coefficient. All levels of metabolites are relative concentrations basedupon areas under the peaks and are averages for all three experiments shown in Figure 1. All horizontal axes labeled 24-h recovery represent RBCs circulating 24-h posttransfusion, as described in Methods. Correlation plots show combined data from all three of the indicated experiments in Figure 1.
A
B
C
D
E
GF
H

tive correlation both in fresh RBCs and stored RBCs (palmi-tate, palmitoleate,10-heptadecenoate, and cis-vaccenate).There was no significant change in the levels of these 4 longchain fatty acids between time of collection and after stor-age. Although one cannot rule out a simultaneous increasein production and consumption resulting in an unalteredsteady state, a more likely explanation is that increased lipidbreakdown (or processes that are associated with it), as afunction of the normal RBC biology, predispose RBCs topoor storage. In contrast to the above long chain fatty acids,omega-3 fatty acids DPA and DHA had a positive correla-tion with 24-h recovery when measured after storage. EPAcan be converted into DPA, which then can be converted toDHA. EPA was detected in this panel, but had no correla-tion to RBC recoveries. These findings are of potential prac-tical value, as they may serve as criteria to evaluate howRBCs will store through screening at time of donation.In summary, a model emerges from the current studies
in which lipid peroxidation is associated with poor 24-hrecovery. Future experimental studies in mice will berequired to test the functional relevance of the tocopherol-ascorbate-GSH axis and lipid peroxidation. Human stud-ies will also be required to assess the extent to which theobservations generated herein predict human RBC storagebiology. The further resolution of these issues is a muchneeded step in advancing the ability to predict and controlthe quality of stored human RBCs.
AcknowledgmentsWe would like to acknowledge the technical and scientific staff
at Metabolon Inc., who carried out the mass spectroscopy analy-sis of the RBC specimens.
FundingThis work was supported, in part, by a grant from the
National Heart Lung and Blood Institute (HL095479-05).
K. de Wolski et al.
586 haematologica | 2016; 101(5)
References
1. Dumont LJ, AuBuchon JP. Evaluation ofproposed FDA criteria for the evaluation ofradiolabeled red cell recovery trials.Transfusion. 2008;48(6):1053-1060.
2. Hess JR. Red cell changes during storage.Transfus Apher Sci. 2010;43(1):51-59.
3. Hess JR. Red cell storage. J Proteomics.2010;73(3):368-373.
4. Brewer GJ. Genetic and population studiesof quantitative levels of adenosine triphos-phate in human erythrocytes. BiochemGenet. 1967;1(1):25-34.
5. Dern RJ, Wiorkowski JJ. Studies on thepreservation of human blood. IV. Thehereditary component of pre- and poststor-age erythrocyte adenosine triphosphatelevels. J Lab Clin Med. 1969;73(6):1019-1029.
6. van 't Erve TJ, Doskey CM, Wagner BA, etal. Heritability of glutathione and relatedmetabolites in stored red blood cells. FreeRadic Biol Med. 2014;76:107-113.
7. van 't Erve TJ, Wagner BA, Martin SM, et al.The heritability of metabolite concentra-tions in stored human red blood cells.Transfusion. 2014;54(8):2055-2063.
8. van't Erve TJ, Wagner BA, Martin SM, et al.The heritability of hemolysis in storedhuman red blood cells. Transfusion. 2015;55(6):1178-1185.
9. van 't Erve TJ, Wagner BA, Ryckman KK,Raife TJ, Buettner GR. The concentration ofglutathione in human erythrocytes is a her-itable trait. Free Radic Biol Med. 2013;65:742-749.
10. Zimring JC. Established and theoretical fac-tors to consider in assessing the red cellstorage lesion. Blood. 2015;125(14):2185-2190.
11. Fergusson DA, Hebert P, Hogan DL, et al.Effect of fresh red blood cell transfusions onclinical outcomes in premature, very low-birth-weight infants: the ARIPI randomizedtrial. JAMA. 2012;308(14):1443-1451.
12. Lacroix J, Hebert PC, Fergusson DA, et al.Age of transfused blood in critically illadults. N Engl J Med. 2015;372(15):1410-1418.
13. Steiner ME, Ness PM, Assmann SF, et al.Effects of red-cell storage duration onpatients undergoing cardiac surgery. N EnglJ Med. 2015;372(15):1419-1429.
14. Qu L, Triulzi DJ. Clinical effects of redblood cell storage. Cancer Control. 2015;22(1):26-37.
15. Desmarets M, Cadwell CM, Peterson KR,Neades R, Zimring JC. Minor histocompat-ibility antigens on transfused leukoreducedunits of red blood cells induce bone mar-row transplant rejection in a mouse model.Blood. 2009;114(11):2315-2322.
16. Petkov PM, Ding Y, Cassell MA, et al. Anefficient SNP system for mouse genomescanning and elucidating strain relation-ships. Genome Res. 2004 Sep;14(9):1806-1811.
17. Gilson CR, Kraus TS, Hod EA, et al. A novelmouse model of red blood cell storage andposttransfusion in vivo survival.Transfusion. 2009;49(8):1546-1553.
18. Uchida K. 4-Hydroxy-2-nonenal: a productand mediator of oxidative stress. Prog LipidRes. 2003;42(4):318-343.
19. Arun P, Padmakumaran Nair KG,Manojkumar V, Deepadevi KV, LakshmiLR, Kurup PA. Decreased hemolysis andlipid peroxidation in blood during storagein the presence of nicotinic acid. Vox Sang.1999;76(4):220-225.
20. Karon BS, van Buskirk CM, Jaben EA,Hoyer JD, Thomas DD. Temporal sequenceof major biochemical events during bloodbank storage of packed red blood cells.Blood Transfus. 2012;10(4):453-461.
21. Knight JA, Searles DA, Blaylock RC. Theeffect of metal chelators on lipid peroxida-tion in irradiated erythrocytes. Ann ClinLab Sci. 1992;22(6):417-422.
22. Knight JA, Voorhees RP, Martin L. Theeffect of metal chelators on lipid peroxida-tion in stored erythrocytes. Ann Clin LabSci. 1992;22(4):207-213.
23. Knight JA, Voorhees RP, Martin L, AnstallH. Lipid peroxidation in stored red cells.Transfusion. 1992;32(4):354-357.
24. D'Alessandro A, Blasi B, D'Amici GM,Marrocco C, Zolla L. Red blood cell sub-populations in freshly drawn blood: appli-cation of proteomics and metabolomics toa decades-long biological issue. BloodTransfus. 2013;11(1):75-87.
25. D'Alessandro A, Hansen KC, Silliman CC,Moore EE, Kelher M, Banerjee A.Metabolomics of AS-5 RBC supernatantsfollowing routine storage. Vox Sang.2015;108(2):131-140.
26. D'Alessandro A, Nemkov T, Hansen KC,
Szczepiorkowski ZM, Dumont LJ. Redblood cell storage in additive solution-7preserves energy and redox metabolism: ametabolomics approach. Transfusion.2015;55(12):2955-2966.
27. D'Alessandro A, Nemkov T, Kelher M, etal. Routine storage of red blood cell (RBC)units in additive solution-3: a comprehen-sive investigation of the RBC metabolome.Transfusion. 2015;55(6):1155-1168.
28. Roback JD, Josephson CD, Waller EK, et al.Metabolomics of ADSOL (AS-1) red bloodcell storage. Transfus Med Rev.2014;28(2):41-55.
29. Zimring JC, Smith N, Stowell SR, et al.Strain-specific red blood cell storage,metabolism, and eicosanoid generation in amouse model. Transfusion. 2014;54(1):137-148.
30. Claro LM, Leonart MS, Comar SR, doNascimento AJ. Effect of vitamins C and Eon oxidative processes in human erythro-cytes. Cell Biochem Funct. 2006;24(6):531-535.
31. Leonart MS, Weffort-Santos AM, MunozEM, Higuti IH, Fortes VA, Nascimento AJ.Effect of vitamin E on red blood cell preser-vation. Braz J Med Biol Res. 1989;22(1):85-86.
32. Pallotta V, Gevi F, D'Alessandro A, Zolla L.Storing red blood cells with vitamin C andN-acetylcysteine prevents oxidative stress-related lesions: a metabolomics overview.Blood Transfus. 2014;12(3):376-387.
33. Stowell SR, Smith NH, Zimring JC, et al.Addition of ascorbic acid solution to storedmurine red blood cells increases posttrans-fusion recovery and decreases microparti-cles and alloimmunization. Transfusion.2013;53(10):2248-2257.
34. Silliman CC, Moore EE, Kelher MR, KhanSY, Gellar L, Elzi DJ. Identification of lipidsthat accumulate during the routine storageof prestorage leukoreduced red blood cellsand cause acute lung injury. Transfusion.2011;51(12):2549-2554.
35. Silliman CC, Bjornsen AJ, Wyman TH, etal. Plasma and lipids from stored plateletscause acute lung injury in an animal model.Transfusion. 2003;43(5):633-640.
36. Silliman CC, Voelkel NF, Allard JD, et al.Plasma and lipids from stored packed redblood cells cause acute lung injury in an ani-mal model. J Clin Invest. 1998;101(7):1458-1467.

haematologica | 2016; 101(5) 587
Received: September 15, 2015.
Accepted: January 14, 2016.
Pre-published: January 22, 2016.
©2016 Ferrata Storti Foundation
Check the online version for the most updatedinformation on this article, online supplements,and information on authorship & disclosures:www.haematologica.org/content/101/5/587
Material published in Haematologica is cov-ered by copyright. All rights reserved to theFerrata Storti Foundation. Copies of articlesare allowed for personal or internal use.Permission in writing from the publisher isrequired for any other use.
Correspondence: [email protected]
Impaired killing of Candida albicans by granulocytes mobilized for transfusion purposes: a role for granule components Roel P. Gazendam,1 Annemarie van de Geer,1 John L. van Hamme,1 Anton T.J.Tool,1 Dieke J. van Rees,1 Cathelijn E.M. Aarts,1 Maartje van den Biggelaar,1Floris van Alphen,1 Paul Verkuijlen,1 Alexander B. Meijer,1 Hans Janssen,2 DirkRoos,1 Timo K. van den Berg1 and Taco W. Kuijpers1,31Sanquin Research, and Landsteiner Laboratory, Academic Medical Center, University ofAmsterdam; 2The Netherlands Netherlands Cancer Institute, Division of Cell Biology,Amsterdam; 3Emma Children’s Hospital, Academic Medical Center, University ofAmsterdam, Amsterdam, The Netherlands
Ferrata StortiFoundation
EUROPEANHEMATOLOGYASSOCIATION
Haematologica 2016Volume 101(5):587-596
ARTICLEBlood Transfusion
doi:10.3324/haematol.2015.136630
Granulocyte transfusions are used to treat neutropenic patientswith life-threatening bacterial or fungal infections that do notrespond to anti-microbial drugs. Donor neutrophils that have
been mobilized with granulocyte-colony stimulating factor (G-CSF) anddexamethasone are functional in terms of antibacterial activity, but lessis known about their fungal killing capacity. We investigated the neu-trophil-mediated cytotoxic response against C. albicans and A. fumigatusin detail. Whereas G-CSF/dexamethasone-mobilized neutrophilsappeared less mature as compared to neutrophils from untreated con-trols, these cells exhibited normal ROS production by the NADPH oxi-dase system and an unaltered granule mobilization capacity upon stim-ulation. G-CSF/dexamethasone-mobilized neutrophils efficiently inhib-ited A. fumigatus germination and killed Aspergillus and Candida hyphae,but the killing of C. albicans yeasts was distinctly impaired. Followingnormal Candida phagocytosis, analysis by mass spectrometry of purifiedphagosomes after fusion with granules demonstrated that major con-stituents of the antimicrobial granule components, including major basicprotein (MBP), were reduced. Purified MBP showed candidacidal activ-ity, and neutrophil-like Crisp-Cas9 NB4-KO-MBP differentiated intophagocytes were impaired in Candida killing. Together, these findingsindicate that G-CSF/dexamethasone-mobilized neutrophils for transfu-sion purposes have a selectively impaired capacity to kill Candida yeasts,as a consequence of an altered neutrophil granular content.
ABSTRACT
Introduction
The intensified use of chemotherapy and immunosuppressive treatment modal-ities and related neutropenia results in increased morbidity and mortality due tobacterial and fungal infections.1,2 Invasive fungal infections in particular are charac-terized by mortality rates of up to 90%, and this is in a large part due to the grow-ing resistance to antifungals.1-3 Granulocyte transfusions are administered to critical-ly ill patients with neutropenia or neutrophil dysfunction and infections that do notrespond to antimicrobial therapy.4,5 Granulocyte-colony stimulating factor (G-CSF)and dexamethasone treatment of donors increases the yield of granulocytes fortransfusion (GTX), but it also recruits a distinct pool of neutrophils from the bonemarrow with an altered gene expression profile.6 We previously found that certaingenes known to be involved in the antifungal immune response were downregulat-ed in G-CSF/dexamethasone-mobilized neutrophils.6 However, it is not knownwhether this altered gene expression profile also impacts the cytotoxic response

against the clinically relevant fungal pathogens, Aspergillusfumigatus and Candida albicans. In general, human neutrophil killing mechanisms
include reactive oxygen species (ROS) production by theNADPH oxidase system and non-oxidative cytotoxicmechanisms.7,8 G-CSF has been shown in vitro to enhanceneutrophil chemotaxis, phagocytosis and NADPH oxidaseactivation,9,10 whereas dexamethasone exerts immunosup-pressive effects on human and murine neutrophilfunction.11,12 We and others have shown that neutrophilsfrom G-CSF/dexamethasone-treated donors display pro-longed survival rates, intact NADPH oxidase activationand a normal antimicrobial response against gram-positiveand gram-negative bacteria.13-16 Nevertheless, G-CSF-mobilized donor neutrophils have been reported to con-tain reduced levels of lactoferrin for example, derived fromthe specific granules, as compared to neutrophils fromuntreated controls.17 During granulopoiesis granular pro-teins are synthesized, and when released by the matureneutrophil these proteins employ cytotoxic activity orlimit the availibility of nutrients for the pathogen.7,18 Thesegranule-dependent cytotoxic mechanisms are pivotal inthe host defense against fungal pathogens. It has, forinstance, been shown that the human neutrophil inhibi-tion of A. fumigatus germination depends on specific gran-ule-derived lactoferrin, which mediates the sequestrationof iron.19 Granular extracts from human neutrophils, con-taining in particular cathepsin G and major basic protein(MBP), but also azurocidin and defensins, demonstratedcandidacidal activity.20,21 Previously, we found that genesinvolved in the antifungal response, including the genethat encodes for CARD9, were downregulated in the G-CSF/dexamethasone-mobilized neutrophils.6 HumanCARD9 deficiency is characterized by invasive fungalinfection and impaired neutrophil candidacidal activity.22In the present study we have investigated the killing of
fungi by G-CSF/dexamethasone-mobilized neutrophils indetail. Our results demonstrate that G-CSF/dexametha-sone-mobilized neutrophils have immature characteris-tics, produce normal amounts of ROS, efficiently inhibit A.fumigatus germination and kill their hyphae. However, thekilling of C. albicans was substantially impaired in G-CSF/dexamethasone-mobilized neutrophils relative totheir normal counterparts. Analyses of the phagosomesafter fusion with granules revealed reduced levels ofantimicrobial proteases, including MBP, in G-CSF/dexam-ethasone-mobilized neutrophils. Interestingly, MBP isrequired for the killing of Candida and contributes to theobserved killing defect in G-CSF/dexamethasone-mobi-lized neutrophils.
Methods
Cell isolation and study approval Heparinized venous blood was collected from healthy granulo-
cyte donors, with or without G-CSF/dexamethasone treatment.Donors received G-CSF (600 μg subcutaneously) and dexametha-sone (8 mg orally), 16 to 20 hours before blood donation. Thestudy was approved by the Sanquin Research Ethical MedicalCommittee (Amsterdam, The Netherlands) and in accordancewith the Declaration of Helsinki (version Seoul 2008). The granulocytes were isolated by centrifugation of heparin
blood over isotonic Percoll with a specific density of 1.076 g/mland after lysis of the erythrocytes with isotonic
NH4Cl-KHCO3-EDTA solution resuspended in Hepes-bufferedsaline solution (Hepes-buffer).22
Killing of microorganisms The microbicidal activity of granulocytes was assessed for
Candida albicans (strain SC5314) and a clinical isolate of Aspergillusfumigatus. The microorganisms were grown under aerobic condi-tions at 30°C for 7 days on potato dextrose agar (Aspergillus)(Neogen, Lansing, Michigan, USA) or overnight in Luria-Bertanibroth (LB) (Candida). Hereafter, the Aspergillus yeasts were collected by centrifuga-
tion, washed twice in PBS and resuspended in RPMI 1640 medium(Life Technologies, Bleiswijk, The Netherlands). Opsonizationwas performed with 10% v/v human pooled serum for 15 min-utes, at 37°C. For the neutrophil-mediated inhibition of germina-tion, the same number of Aspergillus yeast cells were incubatedwith an increasing number of neutrophils (0.25, 0.5, 1.0 or 1.5 *105
cells/ml, E:T 1:2000, 1:1000, 1:500 or 1:350, respectively) in a 96-well plate overnight at 37°C in RPMI 1640 medium containing L-glutamine and 10% (v/v) FCS (Life Technolgies). Subsequently,the neutrophils were lysed in water/NaOH, pH 11.0 and incubat-ed with MTT (3-[4,5-dimethylthiazol-2-yl]-2,5- diphenyltetrazoli-um bromide; thiazolyl blue) (Sigma). After the addition of acidicisopropanol (0.04 M HCl) the optical density was measured in theplate reader at 570 nm (Tecan, Männedorf, Switzerland) and the A.fumigatus hyphae viability was calculated as compared to the incu-bation without neutrophils (i.e. 100%). To assess the A. fumigatus and the C. albicans hyphae killing, neutrophils (0-1x105
cells) were cultured for one hour (Aspergillus) or 2 hours (Candida)on a preformed monolayer at 37 °C. Hereafter, the cells were lysedin water/NaOH, pH 11.0 and incubated with MTT. Theabsorbance of the acidic isopropanol-diluted samples was meas-ured on the plate reader (Tecan) and the viability calculated as apercentage of the viability after incubation without neutrophils.To determine the neutrophil killing of Candida, the yeasts were
collected by centrifugation, washed twice in PBS and resuspended
R.P. Gazendam et al.
588 haematologica | 2016; 101(5)
Table 1. Distinct composition of the G-CSF/dexamethasone-mobilizedphagosomes after fusion with granules.Protein Function
Major Basic Protein Homolog (MBPH) C-type lectin, cytotoxinResistin (RETN) Pro-inflammatoryPoly(rC)-binding protein 1 (PCBP1) RNA bindingSerine/arginine-rich RNA bindingsplicing factor 4 (SRSF4) Adenylyl cyclase-associated Receptor resistin, protein 1 (CAP1) filament dynamicsLipocalin-2 (LCN2) Ferric siderophore, metalloproteaseMajor Basic Protein (MBP) C-type lectin, cytotoxinEosinophil peroxidase (EPX) Peroxidase activityPeptidoglycan recognition Peptidoglycan receptorprotein 1 (PGLYRP1) Vesicle-associated membrane Vesicular fusionprotein 8 (VAMP8) Grancalcin (GCA) Pro-inflammatory
Neutrophils from healthy controls and G-CSF/dexamethasone-treated donors were stim-ulated with C. albicans for 45 minutes; subsequently, the phagosomes were isolated andanalyzed by Mass Spectrometry. The proteins that were significantly decreased in theneutrophil phagosomes from the G-CSF/dexamethasone-treated donors as compared tountreated controls are shown. N=5, FDR = 0.05 and S = 0.6

in Hepes-medium. After opsonization with 10% (v/v) pooledserum for 15 min, at 37°C, the Candidawas added at a ratio of 4 :1 neutrophil (5x106 cells/ml). At the desired time points, 100-μlsamples were diluted in 2.5 ml of water/NaOH, pH 11.0. At theend of the incubation period, the number of viable microorgan-isms in each sample was determined by the pourplate method inLB agar. The colony-forming units (CFU) were determined afterovernight incubation at 37°C, and the percentage of killing wascalculated as described.22
The recombinant proteins for the candidacidal experimentswere major basic protein (MBP) (kind gift from prof. G.J. Gleich,Utah, USA, recombinant protein produced in our lab) and majorbasic protein homologue (recombinant protein produced in ourlab, detailed methodology in the Online Supplementary Appendix).
Immunostaining and FACS analysis The expression of surface-bound receptors on granulocytes
was assayed in total leukocyte samples by flow cytometry(FACS), with the commercially available antibodies againsthuman-CD11b (clone 44A, ATCC, Rockville, MD, USA), CD32(clone AT10, AbD Serotec, Oxford, UK), CD16 (clone 3G8, BDPharmingen, Breda, the Netherlands), EMR3 (clone 3D7, AbD,Puchheim, Germany), CXCR4 (Clone 44717, R&D systems,Oxford, UK) and gp91phox (clone 7D5, MBL, Woburn, MA, USA).As a secondary antibody, Alexa488 rabbit anti-mouse-IgG(Molecular Probes, Bleiswijk, the Netherlands) was used.Samples were analyzed on an LSRII flow cytometer equippedwith FACSDiva software (BD Biosciences). Cells were gatedbased on their forward and side scatter, and 10,000 gated eventswere collected per sample.
Degranulation assaysNeutrophils (2×106/ml) were incubated in Hepes buffer at 37ºC
in a shaking water-bath before adding the (priming) agents PAF (1 μM, 5 minutes, Sigma, Steinheim, Germany) or cytochalasin B (5 μg/ml, 5 minutes, Sigma) were added. Subsequently, the cellswere stimulated with fMLP (1 μM, Sigma, 15 minutes). After stim-ulation, the cells were put on ice, washed with Hepes buffer once,and subsequently stained with antibodies against neutrophil gran-ule markers: CD63-PE (IgG1, 435); CD66b-FITC (IgG1, CLB-B13.9). Data are expressed as mean fluorescence intensities (MFI).The cells were analyzed on an LSRII flow cytometer equippedwith FACSDiva software (BD Bioscience). The release of elastaseand lactoferrin was evaluated with ELISA kits (HyCult Biotech)according to the manufacturer’s instructions. The proteolyticactivity was determined by incubating neutrophils (2.5×106/ml inHepes buffer) with DQ-Green BSA (10 μg/ml, Molecular Probes).Upon stimulation with cytochalasin B (5 μg/ml, Sigma)/ fMLP (1 μM) the fluorescence was monitored at 30-second intervals for1 hour by infinitiPRO2000 plate reader (Excitation 485 nm;Emission 535 nm) (Tecan).
StatisticsStatistical analysis was performed with GraphPad Prism version
6.00 for Windows (GraphPad Software, San Diego, CA, USA). MSdata were analyzed with Proteome Discoverer Software (ThermoScientific, version 1.4), Scaffold (Proteome Software, version 4.0)and MaxQuant (FDR set at 0.05 and S0.6, version 1.4.1.2). Datawere evaluated by paired, two-tailed student’s t-test, two-wayANOVA with post hoc Bonferroni test and by the Mann-Whitneytest. The results are presented as the mean ± SEM, as indicated.Data were considered significant when P<0.05.
Supplemental methodsDetailed methodology of the Online Supplementary Figures is
described in the Online Supplementary Appendix.
Mobilized neutrophils and impaired Candida killing
haematologica | 2016; 101(5) 589
Figure 1. Maturation and NADPH oxidase activity in G-CSF/dexamethasone-mobi-lized neutrophils. (A) Neutrophils from untreated controls or G-CSF/dexamethasone-treated controls were stained for the expression of maturation markers EMR3,CXCR4, CD16, CD32, CD11b and the NADPH oxidase component gp91phox by flowcytometry, left panel. Morphological characteristics were assessed on a cytospin,right panel. The arrows indicate a multi-lobular control neutrophil or a band-shapedG-CSF/dexamethasone-mobilized neutrophil. (B) To measure the production of hydro-gen peroxide, control and G-CSF/dexamethasone-mobilized neutrophils were stimu-lated with various stimuli: zymosan, serum-treated zymosan, phorbol-12-myristate-13-acetate (PMA), or platelet-activating factor (PAF) followed by formyl-Met-Leu-Phe(fMLP), in the presence of Amplex Red and horseradish peroxidase. Results aremeans ± SEM, N=5. *P< 0.05 compared to untreated controls.
A
B

Results
G-CSF/dexamethasone treatment recruits immatureneutrophils with normal NADPH oxidase activity andgranule mobilization capacityPreviously, we found that the G-CSF/dexamethasone-
mobilized neutrophils demonstrated an altered geneexpression profile, and this could either be due to therecruitment of a relatively immature population of neu-trophils or direct gene-regulatory effects of G-CSF/dexam-ethasone. A single administration of subcutaneous G-CSFis combined with an oral dose of dexamethasone to obtainan optimal number of neutrophil mobilization for transfu-sion.23 We isolated neutrophils from healthy donors treated with
G-CSF and dexamethasone, which resulted in a ~10-foldincrease in circulating neutrophils (Online SupplementaryFigure S1). The chemokine receptor CXCR4 involved inneutrophil retention in the bone marrow was reduced onthe surface of G-CSF/dexamethasone-mobilized neu-trophils as compared to control neutrophils (Figure 1A, leftpanel).24 The G-CSF/dexamethasone-mobilized neutrophilsdemonstrated band-shaped nuclei as compared to the mul-tilobular nuclei observed in neutrophils from healthy con-trols (Figure 1A, right panel). The G-CSF/dexamethasone-mobilized neutrophils also showed low surface expressionof the late neutrophil maturation markers EMR3 and CD16,but normal levels of the early myeloid maturation markersCD11b and CD32, when compared to expression levels oncirculating neutrophils from untreated controls (Figure 1A,left panel). Given the fact that the proteins involved in theantimicrobial functions of neutrophils, including the
NADPH oxidase and the different intracellular granules, aregradually formed during granulopoiesis,18 it was of interestto assess these in G-CSF/dexamethasone-mobilized neu-trophils. Surface expression of gp91phox, i.e. the catalyticplasma membrane component of the NADPH oxidaseenzyme complex, was normal when detected with themAb 7D5 (Figure 1A). The functional NADPH oxidaseactivity upon cell activation was also comparable betweencontrol and G-CSF/dexamethasone-mobilized neutrophils(Figure 1B).14Furthermore, the mobilization of azurophilic granules
was measured by the membrane expression of CD63 andthe release of elastase and MPO upon stimulation withcytochalasin-B/fMLP (Figure 2A-C). The mobilization ofspecific granules was evaluated by the membrane expres-sion of CD66b and the release of lactoferrin upon stimula-tion with PAF/fMLP (Figure 2D,E). The overall serine pro-tease activity in the extracellular medium was determined(i.e. DQ BSA fluorescence upon proteolytic cleavage)(Figure 2F). All were found to be intact in G-CSF/dexam-ethasone-mobilized neutrophils as compared to normalneutrophils. Finally, immuno-EM analysis demonstratedthe normal appearance and frequency of myeloperoxidase(MPO)-positive azurophilic granules in the G-CSF/dexam-ethasone-mobilized neutrophils (Online SupplementaryFigure S2). Therefore, it appears that although the G-CSF/dexamethasone-mobilized neutrophils show signs ofimmaturity with respect to their nuclear morphology andthe expression of certain surface markers, both theNADPH oxidase activity and the presence and mobiliza-tion of azurophilic and specific granule markers appearedto be unaltered.
R.P. Gazendam et al.
590 haematologica | 2016; 101(5)
Figure 2. The mobilization and proteolytic activity of azurophilic and specific granules.Neutrophils from untreated controls and G-CSF/dexamethasone-treated donors werestimulated with cytochalasin-B/fMLP or PAF/fMLP, and the plasma membrane expressionof CD63 (A) as well as the extracellular concentration of elastase, MPO (B,C) (azurophilicgranule markers) and CD66b as well as lactoferrin (specific granule markers) (D,E) weremeasured by flow cytometry or ELISA. (F) Proteolytic activity was measured in the extra-cellular medium of untreated neutrophils and G-CSF/dexamethasone-mobilized neu-trophils upon stimulation with cytochalasin-B/fMLP, PAF/fMLP or in TX-100 cell lysate byDQ-Green BSA assay. Results are means ± SEM, N=5.
A B C D E
F
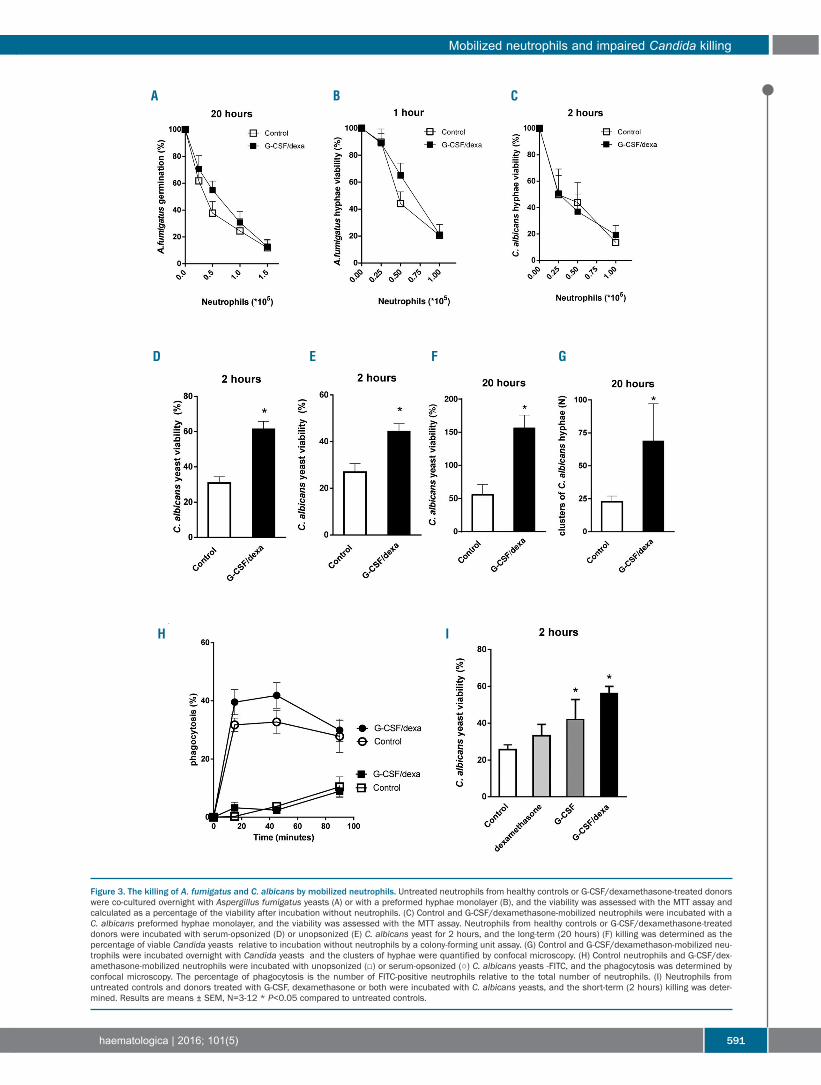
Mobilized neutrophils and impaired Candida killing
haematologica | 2016; 101(5) 591
Figure 3. The killing of A. fumigatus and C. albicans by mobilized neutrophils. Untreated neutrophils from healthy controls or G-CSF/dexamethasone-treated donorswere co-cultured overnight with Aspergillus fumigatus yeasts (A) or with a preformed hyphae monolayer (B), and the viability was assessed with the MTT assay andcalculated as a percentage of the viability after incubation without neutrophils. (C) Control and G-CSF/dexamethasone-mobilized neutrophils were incubated with aC. albicans preformed hyphae monolayer, and the viability was assessed with the MTT assay. Neutrophils from healthy controls or G-CSF/dexamethasone-treateddonors were incubated with serum-opsonized (D) or unopsonized (E) C. albicans yeast for 2 hours, and the long-term (20 hours) (F) killing was determined as thepercentage of viable Candida yeasts relative to incubation without neutrophils by a colony-forming unit assay. (G) Control and G-CSF/dexamethason-mobilized neu-trophils were incubated overnight with Candida yeasts and the clusters of hyphae were quantified by confocal microscopy. (H) Control neutrophils and G-CSF/dex-amethasone-mobilized neutrophils were incubated with unopsonized (□) or serum-opsonized (○) C. albicans yeasts -FITC, and the phagocytosis was determined byconfocal microscopy. The percentage of phagocytosis is the number of FITC-positive neutrophils relative to the total number of neutrophils. (I) Neutrophils fromuntreated controls and donors treated with G-CSF, dexamethasone or both were incubated with C. albicans yeasts, and the short-term (2 hours) killing was deter-mined. Results are means ± SEM, N=3-12 * P<0.05 compared to untreated controls.
D E F G
H I
A B C

Antifungal activity by G-CSF/dexamethasone-mobilizedneutrophilsNext, we determined directly the cytotoxic capacity
against A. fumigatus and C. albicans. Invasive infectionsstart with the germination of yeasts into hyphae thatenables them to invade tissues and spread via the blood-stream, which forms the basis for their pathogenicity.25Therefore we assessed both the intracellular killing ofyeasts by neutrophils, which functions to prevent germi-nation, as well as the extracellular destruction of pre-formed hyphae.The neutrophils from G-CSF/dexamethasone-treated
donors normally inhibited the A. fumigatus germinationafter overnight incubation with the yeasts as compared tountreated controls (Figure 3A). The G-CSF/dexametha-sone-mobilized neutrophils also efficiently degraded amonolayer of preformed A. fumigatus hyphae (Figure 3B).A preformed monolayer of C. albicans hyphae was also aseffectively degraded by the G-CSF/dexamethasone-mobi-lized neutrophils as by control neutrophils (Figure 3C).However, we observed that G-CSF/dexamethasone-mobi-lized neutrophils showed a clear and distinctive defect inboth the short-term (2 hours) and long-term (20 hours)killing of the C. albicans yeasts as compared to the neu-trophils from untreated controls (Figure 3D-F). In addition,the G-CSF/dexamethasone-mobilized neutrophils wereless able to inhibit the C. albicans yeast germination in anovernight assay (Figure 3G). This defect in yeast killingcould not be explained by changes in the phagocyticcapacity, since the phagocytosis and killing of both unop-sonized and serum-opsonized C. albicans yeasts was com-pletely normal (Figure 3H).We assessed whether the in vivo treatment with G-CSF
or dexamethasone seperately could be held responsible for
the Candida killing defect of the G-CSF/dexamethasone-mobilized neutrophils. Neutrophils were isolated fromhealthy donors treated with G-CSF or dexamethasoneseparately, each of which resulted in a ~7- or ~2-foldincrease in circulating neutrophils, respectively (OnlineSupplementary Figure S1). When compared to the neu-trophils from untreated controls, we observed that thedexamethasone-mobilized neutrophils were not impairedin the killing of C. albicans yeasts or any of the other fungalkilling tests performed (Figure 3I), whereas the neutrophilsfrom G-CSF-treated donors showed a significant C. albi-cans killing defect (Figure 3I), although not exactly to thesame extent as in the case of donor-derived neutrophilsmobilized with both G-CSF and dexamethasone (Figure3I). Taken together, a selective C. albicans yeast killing defect
was observed for G-CSF/dexamethasone-mobilized neu-trophils, whereas these neutrophils showed a normalcytotoxic response against the Aspergillus yeasts andhyphae, as well as against preformed Candida hyphae.
Candida-induced phagosome formation To obtain further insight into the Candida yeast killing
defect of G-CSF/dexamethasone-mobilized neutrophilsupon normal phagocytosis, we decided to explore the con-tents of the Candida phagosome in more detail. Under nor-mal conditions of phagocytosis the granules fuse with thephagosome containing internalized pathogens, therebycreating a cytotoxic environment for the degradation ofmicrobes.26,27 To determine the cytotoxic composition ofthe phagosome after fusion with granules, we magnetical-ly labeled Candida yeast, and - after synchronized phago-cytosis and lysis of the neutrophils - we isolated thephagosomes and measured their composition by mass
R.P. Gazendam et al.
592 haematologica | 2016; 101(5)
Figure 4. Distinct composition of the G-CSF/dexamethasone-mobilized neutrophil phagosomes after fusion with granules. (A) Neutrophils from healthy controls andG-CSF/dexamethasone-treated donors were stimulated with C. albicans for 45 minutes, the phagosomes were isolated and analyzed by mass spectrometry. The pro-teins that were significantly decreased in the phagosomes from the G-CSF/dexamethasone-treated donors as compared to untreated controls are shown in the heatmap. The red boxes show upregulated and green downregulated proteins in G-CSF/dexamethasone-mobilized phagosomes compared to controls. (B) The differen-tially expressed proteins between the control and G-CSF/dexamethasone-mobilized phagosomes are depicted in a volcano plot. N=5, FDR = 0.05 and S = 0.6
A
B

spectrometry, according to a previously reportedmethod.28First, confocal analyses confirmed that the isolated
phagosomes after granule fusion were highly positive forCandida (green), MPO (red) and elastase (yellow) (OnlineSupplementary Figure S3A). Secondly, kinetic analysesshowed that the number of elastase peptides in Candidaphagosomes similarly increased with time, which con-firmed the normal phagocytosis by G-CSF/dexametha-sone-mobilized neutrophils and indicates phagosomalmaturation (Figure 3H, Online Supplementary Figure S3B,C). Comparison of the G-CSF/dexamethasone-mobilized
and control neutrophil Candida-phagosomes after fusionwith granules for some of the known componentsshowed a similar expression of e.g. the membrane-expressed integrin CR3 (CD11b/CD18, αMβ2) (OnlineSupplementary Figure S3C), which is critically involved inthe recognition, uptake and killing of C. albicans.8 This is
clearly consistent with the comparable phagocytosis ofCandida yeasts by G-CSF/dexamethasone-mobilized andcontrol neutrophils (see above). In addition, cytochromeb558 of the NAPDH oxidase system was identified (OnlineSupplementary Figure S3C), which reinforces the normalROS production upon uptake of Candida yeasts. Finally,the phagosomes after fusion with granules also containeda variety of components that were derived from the vari-ous granules in neutrophils, i.e. MPO (azurophilic), elas-tase (azurophilic), lactoferrin (specific) and MMP9 (gelati-nase) (Online Supplementary Figure S3C), and thereappeared to be no differences in the fusion of these gran-ules with the phagosome upon comparison of G-CSF/dex-amethasone-mobilized and control neutrophil phago-somes.
We subsequently evaluated whether there were differ-ences in the phagosomal composition between
Mobilized neutrophils and impaired Candida killing
haematologica | 2016; 101(5) 593
Figure 5. MBP candidacidal activity and impairedCandida killing by neutrophil-like NB4 MBP-KOcells. (A) Overnight incubation of Candida with puri-fied MBP or with buffer only, and assessment of ger-mination by microscopy. (B) Recombinant MBP (50ng/ml), MBPH (50 ng/mL) or buffer only were incu-bated for 2 hours with Candida and the viability wasdetermined by the colony-forming unit assay. (C)Neutrophil-like NB4-WT, NB4-scrambled or NB4MBP-KO cells were incubated with C. albicansyeasts, and the percentage of viable Candidayeasts was calculated relative to incubation withoutcells by a colony-forming unit assay. (D) Neutrophil-like NB4-WT or NB4 MBP-KO cells were incubatedwith C. albicans hyphae, and the viability was calcu-lated relative to incubation without cells by the MTTassay. (E) Neutrophil-like NB4-WT or NB4 MBP-KOcells were incubated with A. fumigatus conidia, andthe germination was determined as the percentageof viable A. fumigatus hyphae relative to incubationwithout cells by a MTT assay. Results are means ±SEM, N=2-3, *P<0.05.
A
B
D
C
E

G-CSF/dexamethasone-treated donors and untreated con-trols that could potentially explain the observed killingdefect in G-CSF/dexamethasone-mobilized neutrophils.In total 11 neutrophil-derived proteins were identified tobe significantly decreased in the G-CSF/dexamethasone-mobilized phagosomes after fusion with granules (Figure4). The neutrophil-derived proteins that we observed to bedecreased in the G-CSF/dexamethasone-mobilized phago-somes have been described to be involved in variousaspects of cellular innate immunity, including cytotoxicactivity, vesicular fusion, pro-inflammatory activation andactin-filament rearrangement (Table 1).20,29-32 In the G-CSF/dexamethasone-mobilized phagosomes afterfusion with granules, 79 proteins were signficantly upreg-ulated, including 65 Candida-derived proteins and 14 pro-teins of human origin. Several of these host proteins areknown to be involved in vesicular trafficking and as a neg-ative regulator of phagosomal formation, e.g. Rap1A andRab27A (Online Supplementary Table S1).33,34We focused on the most pronounced differences
between the G-CSF/dexamethasone-mobilized and con-trol phagosomes after fusion with granules. The proteinsmajor basic protein (MBP, PRG2) and major basic proteinhomolog (MBPH, PRG3) were virtually absent in the G-CSF/dexamethasone-mobilized phagosomes after fusionwith granules (Figure 4). MS analyses of whole cell neu-trophil lysates demonstrated that MBP, MBPH and EPXwere also significantly reduced in neutrophils from G-CSF/dexamethasone-treated donors as compared tohealthy controls (Online Supplementary Figure S4).Interestingly, MBP has a C-type lectin domain, and uponcleavage of the propeptide, becomes cytotoxic.35,36 Upontesting the candidacidal effect of MBP and MBPH in theabsence of neutrophils, we found that incubation for 2hours or overnight of purified MBP or MBPH withCandida yeast resulted in strongly decreased yeast viabili-ty and germination (Figure 5A,B). The addition of MBP orMBPH did not affect the viability of A. fumigatus (OnlineSupplementary Figure S5). We used the Crispr-Cas9 tech-nique to generate MBP knock-outs in NB4 cells (NB4-MBP-KO), which become neutrophil-like uponstimulation with ATRA (Online Supplementary Figure S6).37Both the Crispr technique and the knock-out of the pro-tein MBP in particular did not interfere with importantcytotoxic responses, including the ROS production by theNADPH oxidase system and Candida phagocytosis (OnlineSupplementary Figure S6). The neutrophil-like NB4-MBP-KO cells demonstrated a complete Candida killing defectwhen compared to neutrophil-like NB4-WT or NB4 cellsthat were transfected with a scrambled construct against anon-mammalian protein (Figure 5C). The neutrophil-likeMBP knock-out cells normally killed Candida hyphae andinhibited the Aspergillus conidia germination, as also didthe wild-type neutrophil-like NB4 cells (Figure 5D,E).These experiments further indicate that the killing ofCandida depends on the presence of MBP and MBPH inthe phagosome to contribute to the cytotoxic activity.
Discussion
In the present study we determined the cytotoxic activ-ity against Candida albicans and Aspergillus fumigatus byneutrophils mobilized with G-CSF and dexamethasonefor transfusion purposes. G-CSF/dexamethasone-mobi-
lized neutrophils efficiently inhibited A. fumigatus germi-nation and killed both the Aspergillus and Candida hyphae.However, the early and late killing of C. albicans yeastswere impaired by G-CSF/dexamethasone-mobilized neu-trophils relative to normal neutrophils. Analyses of thephagosomes after fusion with granules revealed reducedlevels of antimicrobial proteases, including MBP, in G-CSF/dexamethasone-mobilized neutrophils.Interestingly, MBP was required for the killing of Candidaand contributes to the observed killing defect in G-CSF/dexamethasone-mobilized neutrophils. G-CSF has been shown in vitro to enhance neutrophil
functions in terms of chemotaxis, phagocytosis andNADPH oxidase activation,9,10 whereas dexamethasonehas immunosuppressive effects.11,12 The incubation of neu-trophils with dexamethasone prevents A. fumigatushyphae killing and the addition of G-CSF restores thedefect.12 We found that the neutrophils from the G-CSF/dexamethasone-treated donors normally killed amonolayer of Aspergillus hyphae. An explanation for thisdiscrepancy in results could be that Roilodes et al. addedthe dexamethasone in vitro, whereas the donors in ourstudy were treated with a single dose of dexamethasoneand/or G-CSF overnight in vivo. It has been described thatneutrophils from donors treated for 5 consecutive dayswith G-CSF demonstrated normal MPO levels butdecreased lactoferrin levels.17 The neutrophil-mediatedinhibition of Aspergillus yeasts germination depends oniron-sequestration by lactoferrin.19 After one day of donorpretreatment we found normal levels of both MPO andlactoferrin in the G-CSF/dexamethasone-mobilized neu-trophil phagosomes. In line with this observation the G-CSF/dexamethasone-mobilized neutrophils were com-pletely able to inhibit the germination of A. fumigatus. Theneutrophil killing of Aspergillus hyphae depends on ROSproduction by the NADPH oxidase system.38 Both theROS production and A. fumigatus hyphae killing was nor-mal by the G-CSF/dexamethasone-mobilized neutrophils.The G-CSF/dexamethasone-mobilized neutrophilsshowed an effective cytotoxic response in the inhibitionof A. fumigatus germination and the killing of the hyphae.The G-CSF/dexamethasone-mobilized neutrophils
were able to phagocytose C.albicans, but showed a cleardefect in the intracellular killing. Analyses of the Candida-phagosomes revealed that several proteins were reducedin the G-CSF/dexamethasone-mobilized cells, whereasthe aforementioned granule markers lactoferrin, MPO andelastase were found in comparable levels to controls. Themost significant differences were MBP and MBP homolog(MBPH), present in controls and virtually absent in G-CSF/dexamethasone-mobilized phagosomes after fusionwith granules and in whole G-CSF/dexamethasone-mobi-lized neutrophils. Since G-CSF/dexamethasone treatmentrecruits an immature pool of neutrophils, some granulecomponents, including MBP, may not have been fully syn-thesized. MBP is mostly known as a marker foreosinophils. Borregaard et al. have already reported thatMBPH is also present in various granules of neutrophils,39while we have now confirmed by Immuno-EM analysisthat neutrophil granules do contain MBP (OnlineSupplementary Figure S7). Both MBP and MBPH have beendemonstrated to desintegrate membranes and exertantimicrobial activity.35,36 Gabay et al. investigated theantimicrobial properties of purified granule-derived pro-teins and found that MBP is one of the most potent candi-
R.P. Gazendam et al.
594 haematologica | 2016; 101(5)

dacidal proteins, e.g. it is 70-fold more toxic thandefensins.20 Purified human MBP also displayed strong in vitro inhibition of Candida germination under our condi-tions, which confirmed its fungicidal activity. Moreover, ina knock-out cell model to support the role of MBP, theneutrophil-like NB4-MBP-KO cells were found to be com-pletely impaired in Candida killing without any effect onphagocytosis and ROS production. The results in this neu-trophil-like cell model confirmed that MBP is involved inCandida killing.In addition to MBPH, the analyses of the phagosomes
identified several other proteins that were significantlydecreased in the G-CSF/dexamethasone-mobilized phago-somes after fusion with granules. Eosinophil peroxidase(EP) is not strictly eosinophil specific39 and found to be dif-ferentially expressed between G-CSF/dexamethasone-mobilized and control neutrophils, as well as in theirphagosomes after granule fusion (Figure 4). Peroxidaseactivity is important, as neutrophils from MPO-deficientpatients fail to kill Candida.21 Although no difference in themajor azurophil granule protein MPO was detected, wecannot exclude a contribution of EPO to the observedCandida killing defect in the G-CSF/dexamethasone-mobi-lized cells. The hormone resistin and its receptor adenylylcyclase-associated protein 1 (CAP1) were also decreased inthe G-CSF/dexamethasone-mobilized phagosomes afterfusion with granules. Resistin is produced by granulocytesupon activation and has pro-inflammatory effects.29However, Candida killing improved only slightly whenresistin was added, and was observed in both control andG-CSF/dexamethasone-mobilized neutrophils (OnlineSupplementary Figure S8). The G-CSF/dexamethasone-mobilized phagosomes after fusion with granules alsoshowed reduced levels of the calcium-binding proteingrancalcin and lipocalin-2. Although little is known abouttheir exact role in humans, neutrophils from the respectiveknock-out mice showed normal candidacidal respons-es.40,41The number of defensin-1 peptides were slightly
decreased in the G-CSF/dexamethasone-mobilized phago-somes after fusion with granules (Online SupplementaryFigure S3C). Defensins are derived from azurophilic gran-ules and have been described to be cytotoxic for Candidaalbicans.20 Although MBP proteins (PRG2) contributes to a
very large extent, it may be the combined reduction ofgranule-derived antimicrobial proteins in the G-CSF/dex-amethasone-mobilized neutrophil phagosomes thataggravates the Candida killing defect. It would be a rele-vant topic of future investigations to determine whetherG-CSF or dexamethasone administration results indecreased expression of these granule-derived antimicro-bial proteins.Furthermore, the killing of Candida hyphae by the
G-CSF/dexamethasone-mobilized neutrophils was nor-mal. We have investigated the neutrophil-mediated killingmechanisms of Candida yeasts and hyphae. It appearedthat both the NADPH oxidase system and the phagoso-mal maturation are required for the neutrophil-mediatedkilling of Candida yeasts, whereas these toxic mechanismsare redundant in the killing of Candida hyphae (data notshown).8 This may explain why G-CSF/dexamethasone-mobilized neutrophils show a selective killing defect forCandida yeasts but not hyphae.In conclusion, we have investigated the killing of
A. fumigatus and C. albicans by G-CSF/dexamethasone-mobilized neutrophils in detail. Our results demonstratethat G-CSF/dexamethasone-mobilized neutrophils pro-duce normal amounts of ROS, efficiently inhibit A. fumigatus germination and kill their hyphae. However,the killing of C. albicans yeasts was substantially impairedin G-CSF/dexamethasone-mobilized neutrophils relativeto their normal counterpart. Analyses of the phagosomesafter fusion with granules revealed reduced levels ofantimicrobial proteins and in particular MBP in G-CSF/dexamethasone-mobilized neutrophil phago-somes, which contribute to the observed Candida killingdefect. On some occasions, the Candida yeast form alsoplays a critical role in fungal dissemination, e.g. Candidaglabrata yeasts do not form hyphae but cause severe infec-tions.42 In critically ill neutropenic patients with a Candidasepsis, the indications for G-CSF/dexamethasone neu-trophil transfusions may not alter, because these neu-trophils are still capable to help kill the invasive Candidahyphae when antifungals seem ineffective.
FundingRPG was supported by the Landsteiner Foundation for Blood
Transfusion Research (LSBR 1706) awarded to TWK.
Mobilized neutrophils and impaired Candida killing
haematologica | 2016; 101(5) 595
References
1. Denning DW, Bromley MJ. InfectiousDisease. How to bolster the antifungalpipeline. Science. 2015;347(6229):1414-1416.
2. Gudlaugsson O, Gillespie S, Lee K, et al.Attributable mortality of nosocomial can-didemia, revisited. Clin Infect Dis.2003;37(9):1172-1177.
3. Johnston DL, Lewis V, Yanofsky R, et al.Invasive fungal infections in paediatricacute myeloid leukaemia. Mycoses.2013;56(4):482-487.
4. Drewniak A, Kuijpers TW. Granulocytetransfusion therapy: randomization afterall? Haematologica. 2009;94(12):1644-1648.
5. Marfin AA, Price TH. GranulocyteTransfusion Therapy. J Intensive Care Med.
2013;30(2):79-88.6. Drewniak A, van Raam BJ, Geissler J, et al.
Changes in gene expression of granulocytesduring in vivo granulocyte colony-stimulat-ing factor/dexamethasone mobilization fortransfusion purposes. Blood. 2009;113(23):5979-5998.
7. Brown GD. Innate antifungal immunity:the key role of phagocytes. Annu RevImmunol. 2011;29:1-21.
8. Gazendam RP, van Hamme JL, Tool AT, etal. Two independent killing mechanisms ofCandida albicans by human neutrophils:evidence from innate immunity defects.Blood. 2014;124(4):590-597.
9. Kitagawa S, Yuo A, Souza LM, Saito M,Miura Y, Takaku F. Recombinant humangranulocyte colony-stimulating factorenhances superoxide release in humangranulocytes stimulated by the chemotacticpeptide. Biochem Biophys Res Commun.
1987;144(3):1143-1146.10. Roilides E, Walsh TJ, Pizzo PA, Rubin M.
Granulocyte colony-stimulating factorenhances the phagocytic and bactericidalactivity of normal and defective humanneutrophils. J Infect Dis. 1991;163(3):579-583.
11. Nohmi T, Abe S, Tansho S, Yamaguchi H.Suppression of anti-Candida activity ofmurine and human neutrophils by gluco-corticoids. Microbiol Immunol. 1994;38(12):977-982.
12. Roilides E, Uhlig K, Venzon D, Pizzo PA,Walsh TJ. Prevention of corticosteroid-induced suppression of human polymor-phonuclear leukocyte-induced damage ofAspergillus fumigatus hyphae by granulo-cyte colony-stimulating factor and gammainterferon. Infect Immun. 1993;61(11):4870-4877.
13. Dale DC, Liles WC, Llewellyn C, Rodger E,

Price TH. Neutrophil transfusions: kineticsand functions of neutrophils mobilizedwith granulocyte-colony-stimulating factorand dexamethasone. Transfusion. 1998;38(8):713-721.
14. Drewniak A, Boelens JJ, Vrielink H, et al.Granulocyte concentrates: prolonged func-tional capacity during storage in the pres-ence of phenotypic changes.Haematologica. 2008;93(7):1058-1067.
15. Mochizuki K, Kikuta A, Ohto H, et al.Extended storage of granulocyte concen-trates mobilized by G-CSF with/withoutdexamethasone and collected by bag sepa-ration method. Transfus Med. 2007;17(4):296-303.
16. van Raam BJ, Drewniak A, Groenewold V,van den Berg TK, Kuijpers TW.Granulocyte colony-stimulating factordelays neutrophil apoptosis by inhibitionof calpains upstream of caspase-3. Blood.2008;112(5):2046-2054.
17. Leavey PJ, Sellins KS, Thurman G, et al. Invivo treatment with granulocyte colony-stimulating factor results in divergenteffects on neutrophil functions measured invitro. Blood. 1998;92(11):4366-4374.
18. Borregaard N, Sehested M, Nielsen BS,Sengelov H, Kjeldsen L. Biosynthesis ofgranule proteins in normal human bonemarrow cells. Gelatinase is a marker of ter-minal neutrophil differentiation. Blood.1995;85(3):812-817.
19. Zarember KA, Sugui JA, Chang YC, Kwon-Chung KJ, Gallin JI. Human polymor-phonuclear leukocytes inhibit Aspergillusfumigatus conidial growth by lactoferrin-mediated iron depletion. J Immunol.2007;178(10):6367-6373.
20. Gabay JE, Scott RW, Campanelli D, et al.Antibiotic proteins of human polymor-phonuclear leukocytes. Proc Natl Acad SciUSA. 1989;86(14):5610-5614.
21. Lehrer RI. Functional aspects of a secondmechanism of candidacidal activity byhuman neutrophils. J Clin Invest. 1972;51(10):2566-2572.
22. Drewniak A, Gazendam RP, Tool AT, et al.Invasive fungal infection and impaired neu-
trophil killing in human CARD9 deficiency.Blood. 2013;121(13):2385-2392.
23. Liles WC, Rodger E, Dale DC. Combinedadministration of G-CSF and dexametha-sone for the mobilization of granulocytes innormal donors: optimization of dosing.Transfusion. 2000;40(6):642-644.
24. Devi S, Wang Y, Chew WK, et al.Neutrophil mobilization via plerixafor-mediated CXCR4 inhibition arises fromlung demargination and blockade of neu-trophil homing to the bone marrow. J ExpMed. 2013;210(11):2321-2336.
25. Kumamoto CA, Vinces MD. Contributionsof hyphae and hypha-co-regulated genes toCandida albicans virulence. Cell Microbiol.2005;7(11):1546-1554.
26. Kinchen JM, Ravichandran KS. Phagosomematuration: going through the acid test.Nat Rev Mol Cell Biol. 2008;9(10):781-795.
27. Zhao XW, Gazendam RP, Drewniak A, etal. Defects in neutrophil granule mobiliza-tion and bactericidal activity in familialhemophagocytic lymphohistiocytosis type5 (FHL-5) syndrome caused bySTXBP2/Munc18-2 mutations. Blood.2013;122(1):109-111.
28. Lonnbro P, Nordenfelt P, Tapper H.Isolation of bacteria-containing phago-somes by magnetic selection. BMC CellBiol. 2008;9:35.
29. Jiang S, Park DW, Tadie JM, et al. Humanresistin promotes neutrophil proinflamma-tory activation and neutrophil extracellulartrap formation and increases severity ofacute lung injury. J Immunol. 2014;192(10):4795-4803.
30. Jones LC, Moussa L, Fulcher ML, et al.VAMP8 is a vesicle SNARE that regulatesmucin secretion in airway goblet cells. JPhysiol. 2012;590(Pt 3):545-562.
31. van EM, van Roomen CP, Renkema GH, etal. Characterization of human phagocyte-derived chitotriosidase, a component ofinnate immunity. Int Immunol. 2005;17(11):1505-1512.
32. Xu P, Roes J, Segal AW, Radulovic M. Therole of grancalcin in adhesion of neutrophils.Cell Immunol. 2006;240(2):116-121.
33. Pizon V, Desjardins M, Bucci C, Parton RG,Zerial M. Association of Rap1a and Rap1bproteins with late endocytic/phagocyticcompartments and Rap2a with the Golgicomplex. J Cell Sci. 1994;107( Pt 6):1661-1670.
34. Yokoyama K, Kaji H, He J, et al. Rab27anegatively regulates phagocytosis by pro-longation of the actin-coating stage aroundphagosomes. J Biol Chem. 2011;286(7):5375-5382.
35. Lehrer RI, Szklarek D, Barton A, Ganz T,Hamann KJ, Gleich GJ. Antibacterial prop-erties of eosinophil major basic protein andeosinophil cationic protein. J Immunol.1989;142(12):4428-4434.
36. Plager DA, Loegering DA, Weiler DA, et al. Anovel and highly divergent homolog ofhuman eosinophil granule major basic pro-tein. J Biol Chem. 1999;274(20):14464-14473.
37. Sanjana NE, Shalem O, Zhang F. Improvedvectors and genome-wide libraries forCRISPR screening. Nat Methods. 2014;11(8):783-784.
38. Rex JH, Bennett JE, Gallin JI, Malech HL,Melnick DA. Normal and deficient neu-trophils can cooperate to damageAspergillus fumigatus hyphae. J Infect Dis.1990;162(2):523-528.
39. Rorvig S, Ostergaard O, Heegaard NH,Borregaard N. Proteome profiling of humanneutrophil granule subsets, secretory vesi-cles, and cell membrane: correlation withtranscriptome profiling of neutrophil pre-cursors. J Leukoc Biol. 2013;94(4):711-721.
40. Ferreira MC, Whibley N, Mamo AJ,Siebenlist U, Chan YR, Gaffen SL.Interleukin-17-induced protein lipocalin 2 isdispensable for immunity to oral candidia-sis. Infect Immun. 2014;82(3):1030-1035.
41. Roes J, Choi BK, Power D, Xu P, Segal AW.Granulocyte function in grancalcin-defi-cient mice. Mol Cell Biol. 2003;23(3):826-830.
42. Zaoutis TE, Greves HM, Lautenbach E,Bilker WB, Coffin SE. Risk factors for dis-seminated candidiasis in children with can-didemia. Pediatr Infect Dis J. 2004; 23(7):635-641.
R.P. Gazendam et al.
596 haematologica | 2016; 101(5)

haematologica | 2016; 101(5) 597
Received: October 26, 2015.
Accepted: February 12, 2016.
Pre-published: February 17, 2016.
©2016 Ferrata Storti Foundation
Check the online version for the most updatedinformation on this article, online supplements,and information on authorship & disclosures:www.haematologica.org/content/101/5/597
Material published in Haematologica is cov-ered by copyright. All rights reserved to theFerrata Storti Foundation. Copies of articlesare allowed for personal or internal use.Permission in writing from the publisher isrequired for any other use.
Correspondence: [email protected]
Long-term serial xenotransplantation of juvenilemyelomonocytic leukemia recapitulates humandisease in Rag2–/–γc–/– mice Christopher Felix Krombholz,1,2 Konrad Aumann,3 Matthias Kollek,1,2 DanielaBertele,1 Silvia Fluhr,1,4 Mirjam Kunze,5 Charlotte M. Niemeyer,1,6Christian Flotho,1,6* and Miriam Erlacher,1,6*
1Department of Pediatrics and Adolescent Medicine, Division of Pediatric Hematologyand Oncology, University Medical Center, Freiburg; 2Faculty of Biology, University ofFreiburg; 3Department of Pathology, University Medical Center, Freiburg; 4HermannStaudinger Graduate School, University of Freiburg; 5Department of Obstetrics andGynecology, University Medical Center, Freiburg; and 6The German Cancer Consortium,Heidelberg, Germany *These authors contributed equally to this work
Ferrata StortiFoundation
EUROPEANHEMATOLOGYASSOCIATION
Haematologica 2016Volume 101(5):597-606
ARTICLEMyeloproliferative Disorders
doi:10.3324/haematol.2015.138545
Juvenile myelomonocytic leukemia is a clonal malignant disease affect-ing young children. Current cure rates, even with allogeneichematopoietic stem cell transplantation, are no better than 50%-60%.
Pre-clinical research on juvenile myelomonocytic leukemia is urgentlyneeded for the identification of novel therapies but is hampered by theunavailability of culture systems. Here we report a xenotransplantationmodel that allows long-term in vivo propagation of primary juvenilemyelomonocytic leukemia cells. Persistent engraftment of leukemiccells was achieved by intrahepatic injection of 1x106 cells into newbornRag2–/–γc–/– mice or intravenous injection of 5x106 cells into 5-week oldmice. Key characteristics of juvenile myelomonocytic leukemia werereproduced, including cachexia and clonal expansion of myelomonocyt-ic progenitor cells that infiltrated bone marrow, spleen, liver and,notably, lung. Xenografted leukemia cells led to reduced survival ofrecipient mice. The stem cell character of juvenile myelomonocyticleukemia was confirmed by successful serial transplantation that result-ed in leukemia cell propagation for more than one year. Independenceof exogenous cytokines, low donor cell number and slowly progressingleukemia are advantages of the model, which will serve as an importanttool to research the pathophysiology of juvenile myelomonocyticleukemia and test novel pharmaceutical strategies such as DNA methyl-transferase inhibition.
ABSTRACT
Introduction
Juvenile myelomonocytic leukemia (JMML) is a malignant myeloproliferative dis-order of infancy and early childhood with an aggressive clinical course. Clinicalsymptoms are caused by hematopoietic insufficiency and excessive proliferation ofleukemic monocytes and granulocytes, leading to hepatosplenomegaly, lym-phadenopathy, skin rash and respiratory failure.1-3 JMML is caused by hyperactiva-tion of the RAS signaling pathway due to acquired activating mutations in theKRAS, NRAS or PTPN11 genes,4-7 or due to acquired loss of heterozygosity of theconstitutionally deficient NF1 gene in patients with neurofibromatosis type 1 or ofthe CBL gene in the Noonan-like “CBL syndrome”.8-13 JMML is rapidly fatal unlessallogeneic hematopoietic stem cell transplantation (HSCT) is performed, but eventhis approach is burdened with a significant risk of recurrence.14,15
A serious obstacle to research into JMML is the lack of suitable experimentalmodels, impeding the development and pre-clinical evaluation of novel therapeutic

approaches. Primary JMML leukemia cells cannot bemaintained in culture as they differentiate and becomeapoptotic.16 An immortalized cell line derived from JMMLcells has not yet been successfully established.17 The gen-eration of induced pluripotent stem cell lines originatingfrom JMML cells was reported, but conceptually such sys-tems are limited by their artificial nature and the risk offurther transformation during reprogramming.18 Several ofthe “canonical” JMML mutations that deregulate the RASsignaling pathway were studied in genetically engineeredmouse models, successfully inducing myeloproliferativedisorders in the experimental animals.19-28 Those were,however, still murine leukemias, and critical disease char-acteristics of JMML such as recurrent monosomy 7 or ele-vated fetal hemoglobin are not simulated in transgenicsystems.Xenotransplantation into murine hosts offers the unique
possibility of basic and translational research into livingprimary JMML cells, while at the same time propagatingand multiplying this precious clinical material. However,earlier attempts at JMML xenotransplantation were com-promised by difficult leukemia cell engraftment (presum-ably owing to residual natural killer cell activity of the hoststrains) or rapid demise of engrafted animals within a fewweeks, and not all reports documented the xenologousengraftment of long-term leukemia-initiating cells via suc-cessful serial transplantation.29-31 In addition, the experi-ments depended on high input cell numbers (up to 5x107
cells), a considerable practical obstacle concerning the lim-ited availability of primary clinical JMML material, and oncostly repeated application of human granulocyte-macrophage colony-stimulating factor (GM-CSF).Here we report the suitability of the Rag2–/–γc–/– mouse
strain for the reproduction of primary human JMML inrecipient animals. The system is characterized by goodphenotypic imitation of typical disease features, longduration of xenologous engraftment, quantitative expan-sion of leukemic cell material outside the human organ-ism, and the possibility of retransplantation to furtherexpand cell numbers and extend the duration of experi-ments without additional input of cryopreserved material.Not least, the data support the stem cell character of long-term leukemia-initiating cells in JMML.
Methods
Primary cellsHuman cells were collected after obtaining informed consent
from parents or legal guardians and approval from institutionalreview committees. Samples from JMML patients were collectedin the context of the European Working Group of MDS inChildhood (EWOG-MDS). Clinical information is provided inOnline Supplementary Table S1. Single cell suspensions obtainedfrom mashed spleens were subjected to density gradient centrifu-gation (Ficoll) to separate and cryopreserve mononuclear cells(MNC). Where indicated, MNC were depleted from CD3+ T cells(MACS immunobeads, Miltenyi; <0.15% remaining T cells). Cordblood was obtained from healthy newborns and CD34+ cells wereenriched by the MACS technique (Miltenyi; purity >90%).
XenotransplantationAll experiments were approved by local authorities and fol-
lowed the German “Tierversuchsgesetz”. Rag2–/–γc–/– BALB/c mice32
were maintained in a specific pathogen-free environment.
Newborn mice were irradiated with 2.5 Gy within their first fourdays of life. Eight hours after irradiation, JMML MNC werethawed and 1x106 viable cells were injected intrahepatically (30μl). Alternatively, 5-week old mice were irradiated with 3 Gy andtransplanted intravenously with 5x106 viable MNC. Single cell sus-pensions were obtained from bone marrow (BM), spleen andblood. Liver, kidney and lung were digested with collagenase Dand DNase (Roche) followed by density gradient centrifugation.For serial transplantation, 1-4x106 BM cells from engrafted mice(containing 60%-70% human cells) were injected into recipients.
Flow cytometryCell suspensions were subjected to red blood cell lysis and
stained with antibodies listed in Online Supplementary Table S2.Cytometric Bead Array kits (human inflammatory and Th1/Th2cytokines; BD) were used according to the manufacturer’s instruc-tions. A FACSCalibur (BD) was used; analyses were performedusing FlowJo (FlowJo) and Cyflogic (Cyflo). The gating strategy isshown in Online Supplementary Figure S1.
ImmunohistochemistryOrgans were fixed in 4% buffered formalin, and sternums were
decalcified. After paraffin-embedding, sections were deparaf-finized in xylene and graded alcohols. H&E and chloracetateesterase staining followed standard protocols.Immunohistochemical staining was performed after specific anti-gen retrieval in “low pH target retrieval solution” (Dako) for 30min. Primary and secondary antibodies are listed in OnlineSupplementary Table S2. The EnVision FLEX System or the AP-K5005 system were used for visualization (Dako). Sections werecounterstained with hematoxylin (Dako) and mounted.
Genetic analysisHuman-specific PCR for PTPN11 was performed on
hematopoietic cells isolated from murine organs (forward primerATCCGACGTGGAAGATGAGA, reverse primer TCAGAG-GTAGGATCTGCACAGT). Human HL60 cells and hematopoiet-ic cells from non-transplanted mice were used as positive and neg-ative controls. PCR products were sequenced bidirectionally(BigDye Terminator kit, Life Technologies; ABI 3730xl or 3130xlcapillary sequencers).
PyrosequencingHuman-specific PTPN11 PCR products were generated as
above using a biotinylated reverse primer and pyrosequenced on aPyromark Q24 (Qiagen) using sequencing primer ACATCAA-GATTCAGAACACT. The wild-type/mutant allelic ratio ofPTPN11 point mutations was calculated using PyroMark Q24 soft-ware v.2.0 (Qiagen).
Statistical analysisCharts show mean values and standard errors of the mean
(SEM). Mann-Whitney test, Kaplan-Meier analysis and Mantel-Cox log rank test were used (Statview 4.1 software). P<0.05 wasconsidered statistically significant.
Results
Xenotransplantation of human JMML cells intoRag2–/–γc–/– mice results in leukemic engraftmentWe chose Rag2 and interleukin-2 receptor gamma chain
double-deficient mice (Rag2–/–γc–/–) as recipients for theJMML xenografts. The genetic defect of this strain leads tonear-complete abolishment of residual T cell, B cell, and
C.F. Krombholz et al.
598 haematologica | 2016; 101(5)

natural killer cell activity,32 a prerequisite for successfulJMML xenotransplantation.31 The mice were transplantedwith MNC isolated from splenectomy preparations of 5children with JMML. Flow cytometry showed that thepre-transplantation MNC samples consisted of a medianof 21% CD34+ stem/progenitor cells (range 1%-65%),44% CD33+ myeloid cells (range 18%-89%), 13% CD3+T cells (range 3%-23%) and 27% CD19+ B cells (range9%-31%). The cellular viability upon thawing and the cellcomposition of xenotransplanted material from individualpatients is shown in Online Supplementary Figure S2. Basedon our own previous experience with a xenotransplanta-tion system for healthy human hematopoiesis using thesame host strain,33 we started by using intrahepatic injec-tion of graft cells into newborn mice (1x106 viable JMMLMNC per mouse) as route of transplantation. To see if theprocedure could be simplified and make the model lessdependent on the timely birth of pups, we also transplant-ed 5-week old mice via conventional intravenous injection;these mice received 5x106 JMML MNC to compensate forthe higher body weight at that age. The xenografted cellswere monitored in all mice by biweekly collection ofblood and flow cytometry of human CD45+ cells. Whereasmost animals were sacrificed at elected time points (rang-ing from 10 to 20 weeks after transplantation) for pheno-
type analysis and harvest of JMML cells, a subset of micewas euthanized only when in poor condition so as to learnabout the natural disease course.We defined the level of human engraftment in a given
murine organ as the proportion of human CD45+ cellswithin the total population of murine and human CD45+cells. Following an accepted convention in xenotransplan-tation models,30,34 the occurrence of 0.5% or more humanCD45+ cells in the murine BM was scored as successfulengraftment. Using these definitions, JMML MNC from 4children (Patients #1, #2, #3 and #5) engrafted into recipi-ent mice with an overall leukemic engraftment rate of58/82 mice (Figure 1), not counting 16 animals with non-leukemic T-cell engraftment (see below). Transplantationfrom one patient was unsuccessful (Patient #4, n=9 recipi-ent mice) (Figure 1). In total, 64% (58/91) mice engrafted.Levels of human engraftment were variable and there wasno correlation between percentage of human CD45+ cellsand time from transplantation or condition of the mice.However, we cannot exclude the possibility that leukemicengraftment in mice might have reached higher levels insome mice if sacrificed later.To confirm the presence of JMML cells and rule out the
possibility that co-transplanted healthy hematopoieticstem cells were engrafted into recipient mice, human
JMML xenograft in Rag2–/–γc–/– mice
haematologica | 2016; 101(5) 599
Figure 1. Sustained engraftment of xenotransplanted juvenile myelomonocytic leukemia (JMML) cells in Rag2–/–γc–/– mice. Spleen mononuclear cells (MNC) fromJMML Patients #1 to #5 were transplanted into sublethally irradiated mice (1x106 cells per mouse via intrahepatic injection or 5x106 cells per mouse via intravenousinjection). Hematopoietic cells were obtained from indicated organs at 7-37 weeks post transplant. Human cell engraftment as assessed by flow cytometry of CD45+
cells is shown for animals transplanted from Patient #1 (n=31 mice), Patient #2 (n=37 mice), Patient #3 (n=10 mice) and Patient #5 (n=4 mice). The level of humanengraftment was defined as proportion of human CD45+ cells within the total population of murine and human CD45+ cells. Cells from Patient #4 consistently failedto engraft (n=9 mice). The dotted line represents the definition of successful engraftment (≥0.5% human CD45+ cells).
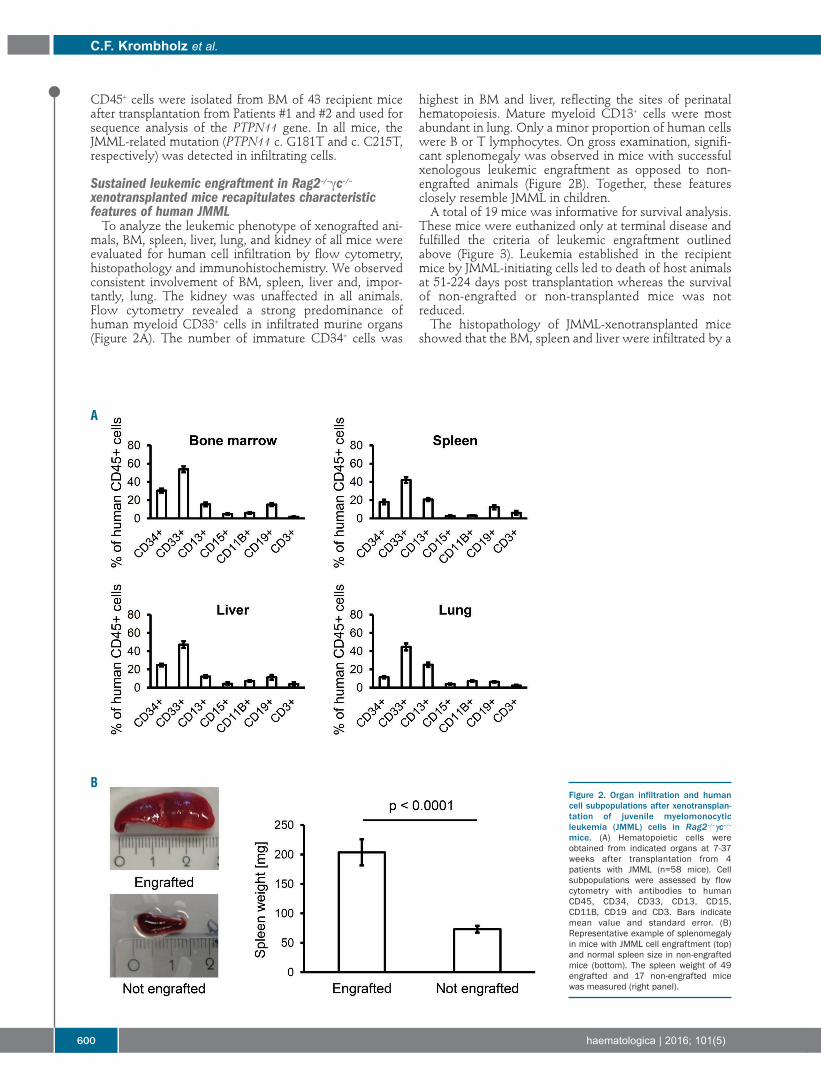
CD45+ cells were isolated from BM of 43 recipient miceafter transplantation from Patients #1 and #2 and used forsequence analysis of the PTPN11 gene. In all mice, theJMML-related mutation (PTPN11 c. G181T and c. C215T,respectively) was detected in infiltrating cells.
Sustained leukemic engraftment in Rag2–/–γc–/–xenotransplanted mice recapitulates characteristic features of human JMML To analyze the leukemic phenotype of xenografted ani-
mals, BM, spleen, liver, lung, and kidney of all mice wereevaluated for human cell infiltration by flow cytometry,histopathology and immunohistochemistry. We observedconsistent involvement of BM, spleen, liver and, impor-tantly, lung. The kidney was unaffected in all animals.Flow cytometry revealed a strong predominance ofhuman myeloid CD33+ cells in infiltrated murine organs(Figure 2A). The number of immature CD34+ cells was
highest in BM and liver, reflecting the sites of perinatalhematopoiesis. Mature myeloid CD13+ cells were mostabundant in lung. Only a minor proportion of human cellswere B or T lymphocytes. On gross examination, signifi-cant splenomegaly was observed in mice with successfulxenologous leukemic engraftment as opposed to non-engrafted animals (Figure 2B). Together, these featuresclosely resemble JMML in children. A total of 19 mice was informative for survival analysis.
These mice were euthanized only at terminal disease andfulfilled the criteria of leukemic engraftment outlinedabove (Figure 3). Leukemia established in the recipientmice by JMML-initiating cells led to death of host animalsat 51-224 days post transplantation whereas the survivalof non-engrafted or non-transplanted mice was notreduced.The histopathology of JMML-xenotransplanted mice
showed that the BM, spleen and liver were infiltrated by a
C.F. Krombholz et al.
600 haematologica | 2016; 101(5)
Figure 2. Organ infiltration and humancell subpopulations after xenotransplan-tation of juvenile myelomonocyticleukemia (JMML) cells in Rag2–/–γc–/–
mice. (A) Hematopoietic cells wereobtained from indicated organs at 7-37weeks after transplantation from 4patients with JMML (n=58 mice). Cellsubpopulations were assessed by flowcytometry with antibodies to humanCD45, CD34, CD33, CD13, CD15,CD11B, CD19 and CD3. Bars indicatemean value and standard error. (B)Representative example of splenomegalyin mice with JMML cell engraftment (top)and normal spleen size in non-engraftedmice (bottom). The spleen weight of 49engrafted and 17 non-engrafted micewas measured (right panel).
A
B
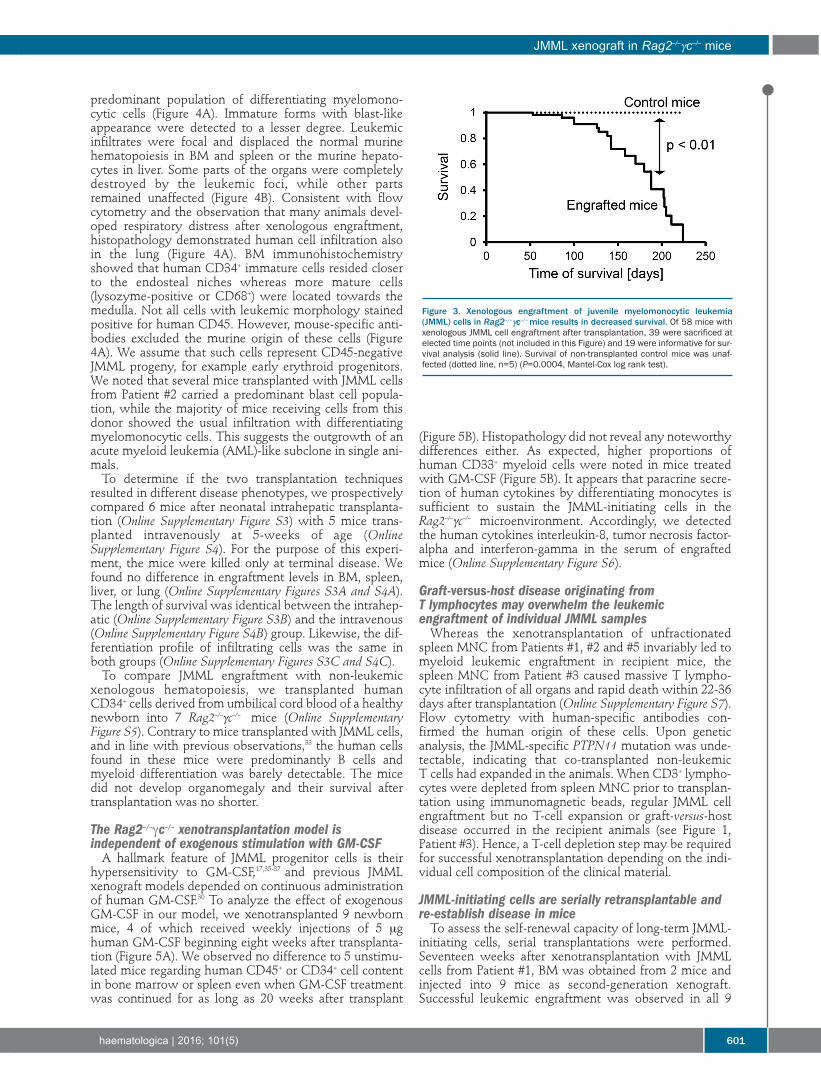
predominant population of differentiating myelomono-cytic cells (Figure 4A). Immature forms with blast-likeappearance were detected to a lesser degree. Leukemicinfiltrates were focal and displaced the normal murinehematopoiesis in BM and spleen or the murine hepato-cytes in liver. Some parts of the organs were completelydestroyed by the leukemic foci, while other partsremained unaffected (Figure 4B). Consistent with flowcytometry and the observation that many animals devel-oped respiratory distress after xenologous engraftment,histopathology demonstrated human cell infiltration alsoin the lung (Figure 4A). BM immunohistochemistryshowed that human CD34+ immature cells resided closerto the endosteal niches whereas more mature cells(lysozyme-positive or CD68+) were located towards themedulla. Not all cells with leukemic morphology stainedpositive for human CD45. However, mouse-specific anti-bodies excluded the murine origin of these cells (Figure4A). We assume that such cells represent CD45-negativeJMML progeny, for example early erythroid progenitors.We noted that several mice transplanted with JMML cellsfrom Patient #2 carried a predominant blast cell popula-tion, while the majority of mice receiving cells from thisdonor showed the usual infiltration with differentiatingmyelomonocytic cells. This suggests the outgrowth of anacute myeloid leukemia (AML)-like subclone in single ani-mals.To determine if the two transplantation techniques
resulted in different disease phenotypes, we prospectivelycompared 6 mice after neonatal intrahepatic transplanta-tion (Online Supplementary Figure S3) with 5 mice trans-planted intravenously at 5-weeks of age (OnlineSupplementary Figure S4). For the purpose of this experi-ment, the mice were killed only at terminal disease. Wefound no difference in engraftment levels in BM, spleen,liver, or lung (Online Supplementary Figures S3A and S4A).The length of survival was identical between the intrahep-atic (Online Supplementary Figure S3B) and the intravenous(Online Supplementary Figure S4B) group. Likewise, the dif-ferentiation profile of infiltrating cells was the same inboth groups (Online Supplementary Figures S3C and S4C).To compare JMML engraftment with non-leukemic
xenologous hematopoiesis, we transplanted humanCD34+ cells derived from umbilical cord blood of a healthynewborn into 7 Rag2–/–γc–/– mice (Online SupplementaryFigure S5). Contrary to mice transplanted with JMML cells,and in line with previous observations,33 the human cellsfound in these mice were predominantly B cells andmyeloid differentiation was barely detectable. The micedid not develop organomegaly and their survival aftertransplantation was no shorter.
The Rag2–/–γc–/– xenotransplantation model is independent of exogenous stimulation with GM-CSFA hallmark feature of JMML progenitor cells is their
hypersensitivity to GM-CSF,17,35-37 and previous JMMLxenograft models depended on continuous administrationof human GM-CSF.30 To analyze the effect of exogenousGM-CSF in our model, we xenotransplanted 9 newbornmice, 4 of which received weekly injections of 5 μghuman GM-CSF beginning eight weeks after transplanta-tion (Figure 5A). We observed no difference to 5 unstimu-lated mice regarding human CD45+ or CD34+ cell contentin bone marrow or spleen even when GM-CSF treatmentwas continued for as long as 20 weeks after transplant
(Figure 5B). Histopathology did not reveal any noteworthydifferences either. As expected, higher proportions ofhuman CD33+ myeloid cells were noted in mice treatedwith GM-CSF (Figure 5B). It appears that paracrine secre-tion of human cytokines by differentiating monocytes issufficient to sustain the JMML-initiating cells in theRag2–/–γc–/– microenvironment. Accordingly, we detectedthe human cytokines interleukin-8, tumor necrosis factor-alpha and interferon-gamma in the serum of engraftedmice (Online Supplementary Figure S6).
Graft-versus-host disease originating from T lymphocytes may overwhelm the leukemic engraftment of individual JMML samplesWhereas the xenotransplantation of unfractionated
spleen MNC from Patients #1, #2 and #5 invariably led tomyeloid leukemic engraftment in recipient mice, thespleen MNC from Patient #3 caused massive T lympho-cyte infiltration of all organs and rapid death within 22-36days after transplantation (Online Supplementary Figure S7).Flow cytometry with human-specific antibodies con-firmed the human origin of these cells. Upon geneticanalysis, the JMML-specific PTPN11 mutation was unde-tectable, indicating that co-transplanted non-leukemic T cells had expanded in the animals. When CD3+ lympho-cytes were depleted from spleen MNC prior to transplan-tation using immunomagnetic beads, regular JMML cellengraftment but no T-cell expansion or graft-versus-hostdisease occurred in the recipient animals (see Figure 1,Patient #3). Hence, a T-cell depletion step may be requiredfor successful xenotransplantation depending on the indi-vidual cell composition of the clinical material.
JMML-initiating cells are serially retransplantable andre-establish disease in miceTo assess the self-renewal capacity of long-term JMML-
initiating cells, serial transplantations were performed.Seventeen weeks after xenotransplantation with JMMLcells from Patient #1, BM was obtained from 2 mice andinjected into 9 mice as second-generation xenograft.Successful leukemic engraftment was observed in all 9
JMML xenograft in Rag2–/–γc–/– mice
haematologica | 2016; 101(5) 601
Figure 3. Xenologous engraftment of juvenile myelomonocytic leukemia(JMML) cells in Rag2–/–γc–/–mice results in decreased survival. Of 58 mice withxenologous JMML cell engraftment after transplantation, 39 were sacrificed atelected time points (not included in this Figure) and 19 were informative for sur-vival analysis (solid line). Survival of non-transplanted control mice was unaf-fected (dotted line, n=5) (P=0.0004, Mantel-Cox log rank test).

mice (Figure 6A). The survival of recipient mice was simi-lar to that of primary recipients (Figure 6B). The secondaryrecipients developed splenomegaly and showed predomi-nant myeloid infiltration (Figure 6C). The overall level oforgan infiltration with human cells and the lineage distri-
bution of human cell progeny were comparable to first-generation xenograft mice. At ten weeks after xenotransplantation with JMML
MNCs from Patient #2, 8 secondary recipients were trans-planted with BM harvested from 4 mice. Four of the sec-
C.F. Krombholz et al.
602 haematologica | 2016; 101(5)
Figure 4. Histopathology demonstrates human myelomonocytic infiltration in murine tissues. (A) Histology and immunohistochemistry of bone marrow, spleen, liverand lung at time of terminal illness revealed leukemic infiltration with differentiating myelomonocytic cells that replaced regular murine tissue. Immunostaining withantibodies to murine CD45 and human CD45 confirmed the human origin of the leukemic cells (top panel, first row). Immature CD34-positive cells were located inthe peritrabecular region and had a blast-like appearance. By contrast, lysozyme- and CD68-positive, more differentiated myelomonocytic cells were found in thecenter of the medullary cavity (top panel, second and third row). The murine spleen was infiltrated by myelomonocytic cells positive for human CD45 and lysozymebut negative for murine CD45 (middle panel). Human myelomonocytic cells were also found in murine liver and lung (bottom panel). (B) Focal displacement of murinehematopoiesis by human CD45-positive myelomonocytic cells. Dotted frames indicate areas with higher magnification shown on the right.
A
B

ondary recipients showed successful engraftment. BMcells from these mice were then used for tertiary trans-plantation and led to engraftment in 8 of 8 mice (OnlineSupplementary Figure S8). Again, the level of leukemicorgan infiltration and length of survival were comparableto first-generation xenograft mice. Mutation analysisdemonstrated that the leukemia-specific PTPN11 muta-tion was invariably present when human cells wereretrieved from serially engrafted mice, regardless of infil-trated organ or graft generation (data not shown). This ruledout the possibility that the leukemic clone might havebeen lost after repeated xenotransplantation and that long-lived non-leukemic progenitors might have prevailed. Inaddition, quantitative pyrosequencing was employed tocompare the mutant allele fraction between source mate-rial (spleen MNC) and purified human CD45+ cells
retrieved from BM cells of serially xenografted mice(Figure 7). We observed that close to 100% of human cellswere of leukemic origin in primary, secondary, or tertiaryrecipients.
Discussion
The need for a pre-clinical model of JMML that can beused for basic research, biomarker identification and drugtesting prompted us to establish a xenotransplantationsystem for this leukemia in immunodeficient mice. Otherinvestigators have previously xenografted JMML cells butreported various difficulties.29-31 Lapidot et al., using SCIDmice as host strain, observed a rapid decline in well-beingand cachexia as soon as 2-4 weeks after xenotransplanta-
JMML xenograft in Rag2–/–γc–/– mice
haematologica | 2016; 101(5) 603
Figure 5. Application of exogenous gran-ulocyte-macrophage colony-stimulatingfactor (GM-CSF) increases myeloid dif-ferentiation without affecting overallengraftment. (A) Schematic diagram ofthe experimental set up. Nine mice weretransplanted intrahepatically with 1x106
juvenile myelomonocytic leukemia(JMML) mononuclear cells of Patient #2.Five weeks later, human cell engraftmentwas confirmed by flow cytometry ofCD45+ peripheral blood cells. Mice weredivided into two experimental groupsmatched for level of engraftment inperipheral blood. Four mice receivedweekly injections of 5 μg recombinanthuman GM-CSF, while saline was admin-istered in 5 mice. Applications werestarted eight weeks after transplanta-tion. The animals were analyzed 12weeks later. (B) The human CD45+ cellengraftment, the proportion of CD34+
progenitor cells and the proportion ofCD33+ myeloid cells were determined inbone marrow (left) and spleen (right).Levels of human CD45+ and CD34+ cellswere comparable between untreated(open symbols) and treated (filled sym-bols) animals. The proportion of CD33+
cells was significantly higher in bonemarrow and spleen of treated animals(P=0.01, Mann-Whitney test).
A
B

tion of JMML cells.30 Their experiments involved continu-ous treatment of host mice with human GM-CSF. In theabsence of exogenous human GM-CSF the leukemic cellsdid not engraft at all. Nakamura et al. reported highly vari-able levels of myeloid engraftment in NOD/SCID/γc–/–mice (average 18.7% CD33+ of human CD45+ cells in thebone marrow, range 9.4%-37.7%), although the numberof cells transplanted was fairly high (107 cells per mouse).31In addition, those previous reports on JMML xenotrans-plantation lacked a detailed analysis of the natural diseasecourse of recipient mice as all animals were sacrificed nolater than 12 weeks after transplantation.29-31 However, wefelt that a system with a more chronic disease coursewould be desirable if JMML were to be modeled in exper-imental animals for pre-clinical research.In an attempt to overcome the obstacles discussed
above we chose Rag2–/–γc–/– mice as hosts and intrahepaticinjection into newborn recipients as mode of transplanta-tion. We favored this technique because of an earlierdescription by Traggiai and the documented suitability fortransplantation of healthy cord blood-derived CD34+
cells.33,38 Adopting an intrahepatic strategy seemed espe-cially appropriate for JMML since it is a disease of earlychildhood, frequently affects the liver and most probablyoriginates from fetal hematopoietic cells, which appear tobe supported better by neonatal than adult tissues.39,40 Inaddition, intravenous injection would be technically chal-lenging in newborn mice. When we later compared the
phenotype of mice transplanted intrahepatically with thatof animals xenografted at older age via the intravenousroute we found no significant differences in level of organinfiltration, length of survival after transplantation, orother aspects of the ensuing leukemia. Although we didnot perform systematic titrations of input cell numbers,we believe that the intrahepatic technique might be thebetter choice if clinical samples with limited cell numberwere to be transplanted. Juvenile myelomonocytic leukemia cells from 4 patients
readily engrafted in the mice whereas transplantationfrom one child was unsuccessful. We can only speculatewhether this failure relates to low amounts of JMML-ini-tiating cells in the spleen MNC preparations (OnlineSupplementary Table S1) or to poor material quality (i.e.latency between splenectomy and cryopreservation).After successful engraftment, the xenotransplanted micedisplayed symptoms similar to those observed in childrenwith JMML, including hepatosplenomegaly, cachexia andpulmonary infiltration with respiratory distress. Detailedanalysis of murine hematopoietic organs revealed focalinfiltration by human myelomonocytic CD33+ cells. CD13expression indicated the presence of cells at more maturestages of differentiation and was stronger in spleen andlung than BM and liver, consistent with the physiologicalroute of myeloid differentiation. Immature CD34+
leukemic cells were located in the endosteal regions ofbone indicating that they shared hematopoietic niches
C.F. Krombholz et al.
604 haematologica | 2016; 101(5)
Figure 6. Analysis of secondary recip-ient mice after serial xenotransplan-tation. (A) Seventeen weeks afterxenotransplantation with juvenilemyelomonocytic leukemia (JMML)cells from Patient #1, bone marrow(BM) cells from 2 mice were re-trans-planted into 9 second-generationmice (1x106 cells per animal). The sec-ondary recipient animals were sacri-ficed for analysis when terminally sick(165-328 days after transplantation).The level of human engraftment wasdefined as proportion of humanCD45+ cells within the total populationof murine and human CD45+ cells.Open triangles indicate first-genera-tion (donor) mice; closed diamonds,second-generation recipients. Thedotted line represents the definitionof successful engraftment (≥0.5%human CD45+ cells). (B) Secondaryrecipient mice (solid line) had signifi-cantly reduced survival compared tountransplanted control mice (dottedline, n=5) (P<0.01, Mantel-Cox logrank test). (C) Hematopoietic cellswere obtained from indicated organsand cell subpopulations wereassessed by flow cytometry with anti-bodies to human CD45, CD34, CD33,CD13, CD15, CD11B, CD19 and CD3.Bars indicate mean value and stan-dard error.
A
C
B

with their normal counterparts, similar to a recent obser-vation in AML.41 The lineage composition of engraftedJMML cell progeny clearly differed from that of xenotrans-planted cord blood-derived healthy CD34+ cells, where Bcells predominated and only minor myeloid populationswere observed. Importantly, xenotransplanted miceshowed a chronic disease course with a median survival ofmore than 20 weeks. This makes it possible to evaluate
pharmaceuticals with delayed activity, in particular theDNA-hypomethylating agent azacitidine which hasrecently gained clinical interest for use in JMML.42,43 In con-trast to previous JMML xenotransplantation models,Rag2–/–γc–/– mice efficiently sustained engrafting JMML cellsin the absence of exogenous human GM-CSF. Weeklyapplication of human GM-CSF enhanced myeloid differ-entiation but did not influence the total level of engraft-ment or time to leukemia. Serial transplantation of JMML cells confirmed the pres-
ence of long-term JMML-initiating cells with self-renewalcapacity. Phenotype and disease kinetics were similar inprimary, secondary and tertiary recipients. Importantly,typical morphology with differentiating myelomonocyticcells was preserved over time and no disease accelerationwas observed. Serial transplantability is not only impor-tant to the scientific concept of leukemia-initiating cells,but is also a valuable tool for the expansion of primaryJMML cells from a practical perspective. Using theRag2–/–γc–/– JMML system and serial retransplantation wehave maintained JMML cells in vivo for 1.5 years in total.In the process, unmanipulated clinical JMML material wasexpanded rather than consumed. This has not so far beenfeasible by in vitro culture.In summary, we present a novel xenotransplantation
model of JMML that closely mimics human disease. Weare confident that the model will be useful to further char-acterize the JMML-initiating cell, amplify scarce and valu-able clinical material, and complement the recently evolv-ing early-phase clinical trials for novel pharmaceuticalstrategies such as epigenetic therapy.
AcknowledgmentsWe are grateful to A. Meier, N. Fischer and B. Stopp for tech-
nical assistance and to N. Krause, B. Müller and K. Thumm foranimal care. We thank H. Pahl and H. Eibel for insightful dis-cussions.
FundingGerman Research Foundation (CRC 992-C05 to CF and
SPP1463 FL345/4-2 to CF) and Freiburg Institute for AdvancedStudies (fellowship to ME).
JMML xenograft in Rag2–/–γc–/– mice
haematologica | 2016; 101(5) 605
Figure 7 (right). Leukemic allele frequency was maintained during serialtransplantations. Pyrosequencing was performed on human CD45+ cellsobtained from murine bone marrow to determine the mutant allele frequencyof PTPN11 c.C215T (Patient #1, upper panel) and PTPN11 c.G181T (Patient #2,lower panel) in serially transplanted animals. Since the leukemic mutationswere heterozygous, a 50% allele frequency corresponds to 100% leukemiccells.
References
1. Niemeyer CM, Arico M, Basso G, et al.Chronic myelomonocytic leukemia in child-hood: a retrospective analysis of 110 cases.European Working Group onMyelodysplastic Syndromes in Childhood(EWOG-MDS). Blood. 1997;89(10):3534-3543.
2. Locatelli F, Niemeyer CM. How I treat juve-nile myelomonocytic leukemia. Blood.2015;125(7):1083-1090.
3. Chang TY, Dvorak CC, Loh ML. Bedside tobench in juvenile myelomonocyticleukemia: insights into leukemogenesis froma rare pediatric leukemia. Blood. 2014;124(16):2487-2497.
4. Flotho C, Valcamonica S, Mach-Pascual S, etal. RAS mutations and clonality analysis inchildren with juvenile myelomonocyticleukemia (JMML). Leukemia. 1999;13(1):32-37.
5. Tartaglia M, Niemeyer CM, Fragale A, et al.
Somatic mutations in PTPN11 in juvenilemyelomonocytic leukemia, myelodysplasticsyndromes and acute myeloid leukemia. NatGenet. 2003;34(2):148-150.
6. Kratz CP, Niemeyer CM, Castleberry RP, etal. The mutational spectrum of PTPN11 injuvenile myelomonocytic leukemia andNoonan syndrome/myeloproliferative dis-ease. Blood. 2005;106(6):2183-2185.
7. Miyauchi J, Asada M, Sasaki M, et al.Mutations of the N-ras gene in juvenilechronic myelogenous leukemia. Blood.1994;83(8):2248-2254.
8. Bader JL, Miller RW. Neurofibromatosis andchildhood leukemia. J Pediatr. 1978;92(6):925-929.
9. Stiller CA, Chessells JM, Fitchett M.Neurofibromatosis and childhoodleukaemia/lymphoma: a population-basedUKCCSG study. Br J Cancer. 1994;70(5):969-972.
10. Side LE, Emanuel PD, Taylor B, et al.Mutations of the NF1 gene in children withjuvenile myelomonocytic leukemia without
clinical evidence of neurofibromatosis, type1. Blood. 1998;92(1):267-272.
11. Steinemann D, Arning L, Praulich I, et al.Mitotic recombination and compound-het-erozygous mutations are predominant NF1-inactivating mechanisms in children withjuvenile myelomonocytic leukemia and neu-rofibromatosis type 1. Haematologica.2010;95(2):320-323.
12. Loh ML, Sakai DS, Flotho C, et al. Mutationsin CBL occur frequently in juvenilemyelomonocytic leukemia. Blood.2009;114(9): 1859-1863.
13. Niemeyer CM, Kang MW, Shin DH, et al.Germline CBL mutations cause develop-mental abnormalities and predispose tojuvenile myelomonocytic leukemia. NatGenet. 2010;42(9):794-800.
14. Locatelli F, Nollke P, Zecca M, et al.Hematopoietic stem cell transplantation(HSCT) in children with juvenilemyelomonocytic leukemia (JMML): resultsof the EWOG-MDS/EBMT trial. Blood.2005;105(1):410-419.

15. Bergstraesser E, Hasle H, Rogge T, et al.Non-hematopoietic stem cell transplanta-tion treatment of juvenile myelomonocyticleukemia: a retrospective analysis and defi-nition of response criteria. Pediatr BloodCancer. 2007;49(5):629-633.
16. Sakashita K, Kato I, Daifu T, et al. In vitroexpansion of CD34(+)CD38(-) cells understimulation with hematopoietic growth fac-tors on AGM-S3 cells in juvenilemyelomonocytic leukemia. Leukemia.2015;29(3):606-614.
17. Gualtieri RJ, Castleberry RP, Gibbons J, et al.Cell culture studies and oncogene expres-sion in juvenile chronic myelogenousleukemia. Exp Hematol. 1988;16(7):613-619.
18. Gandre-Babbe S, Paluru P, Aribeana C, et al.Patient-derived induced pluripotent stemcells recapitulate hematopoietic abnormali-ties of juvenile myelomonocytic leukemia.Blood. 2013;121(24):4925-4929.
19. Jacks T, Shih TS, Schmitt EM, et al. Tumourpredisposition in mice heterozygous for atargeted mutation in Nf1. Nat Genet.1994;7(3):353-361.
20. Hawley RG, Fong AZ, Ngan BY, Hawley TS.Hematopoietic transforming potential ofactivated ras in chimeric mice. Oncogene.1995;11(6):1113-1123.
21. MacKenzie KL, Dolnikov A, Millington M,Shounan Y, Symonds G. Mutant N-rasinduces myeloproliferative disorders andapoptosis in bone marrow repopulatedmice. Blood. 1999;93(6):2043-2056.
22. Araki T, Mohi MG, Ismat FA, et al. Mousemodel of Noonan syndrome reveals celltype- and gene dosage-dependent effects ofPtpn11 mutation. Nat Med. 2004;10(8):849-857.
23. Tuveson DA, Shaw AT, Willis NA, et al.Endogenous oncogenic K-ras(G12D) stimu-lates proliferation and widespread neoplas-tic and developmental defects. Cancer Cell.2004;5(4):375-387.
24. Kim A, Morgan K, Hasz DE, et al. Beta com-mon receptor inactivation attenuates myelo-proliferative disease in Nf1 mutant mice.Blood. 2007;109(4):1687-1691.
25. Cutts BA, Sjogren AK, Andersson KM, et al.
Nf1 deficiency cooperates with oncogenicK-RAS to induce acute myeloid leukemia inmice. Blood. 2009;114(17):3629-3632.
26. Naramura M, Nandwani N, Gu H, Band V,Band H. Rapidly fatal myeloproliferative dis-orders in mice with deletion of Casitas B-celllymphoma (Cbl) and Cbl-b in hematopoieticstem cells. Proc Natl Acad Sci USA.2010;107(37):16274-16279.
27. Wang J, Liu Y, Li Z, et al. Endogenous onco-genic Nras mutation promotes aberrantGM-CSF signaling in granulocytic/monocyt-ic precursors in a murine model of chronicmyelomonocytic leukemia. Blood. 2010;116(26):5991-6002.
28. Chang YI, You X, Kong G, et al. Loss ofDnmt3a and endogenous Kras cooperate toregulate hematopoietic stem and progenitorcell functions in leukemogenesis. Leukemia.2015;29(9):1847-1856.
29. Iversen PO, Lewis ID, Turczynowicz S, et al.Inhibition of granulocyte-macrophagecolony-stimulating factor prevents dissemi-nation and induces remission of juvenilemyelomonocytic leukemia in engraftedimmunodeficient mice. Blood. 1997;90(12):4910-4917.
30. Lapidot T, Grunberger T, Vormoor J, et al.Identification of human juvenile chronicmyelogenous leukemia stem cells capable ofinitiating the disease in primary and second-ary SCID mice. Blood. 1996;88(7):2655-2664.
31. Nakamura Y, Ito M, Yamamoto T, et al.Engraftment of NOD/SCID/gammac(null)mice with multilineage neoplastic cells frompatients with juvenile myelomonocyticleukaemia. Br J Haematol. 2005;130(1):51-57.
32. Goldman JP, Blundell MP, Lopes L, et al.Enhanced human cell engraftment in micedeficient in RAG2 and the commoncytokine receptor gamma chain. Br JHaematol. 1998;103(2):335-342.
33. Labi V, Bertele D, Woess C, et al.Haematopoietic stem cell survival and trans-plantation efficacy is limited by the BH3-only proteins Bim and Bmf. EMBO MolMed. 2013;5(1):122-136.
34. Notta F, Mullighan CG, Wang JC, et al.Evolution of human BCR-ABL1 lym-phoblastic leukaemia-initiating cells. Nature.2011;469(7330):362-367.
35. Gualtieri RJ, Emanuel PD, Zuckerman KS, etal. Granulocyte-macrophage colony-stimu-lating factor is an endogenous regulator ofcell proliferation in juvenile chronic myel-ogenous leukemia. Blood. 1989;74(7):2360-2367.
36. Emanuel PD, Bates LJ, Castleberry RP,Gualtieri RJ, Zuckerman KS. Selective hyper-sensitivity to granulocyte-macrophagecolony-stimulating factor by juvenile chron-ic myeloid leukemia hematopoietic progeni-tors. Blood. 1991;77(5):925-929.
37. Freedman MH, Cohen A, Grunberger T, etal. Central role of tumour necrosis factor,GM-CSF, and interleukin 1 in the pathogen-esis of juvenile chronic myelogenousleukaemia. Br J Haematol. 1992;80(1):40-48.
38. Traggiai E, Chicha L, Mazzucchelli L, et al.Development of a human adaptive immunesystem in cord blood cell-transplanted mice.Science. 2004;304(5667):104-107.
39. Wulf-Goldenberg A, Keil M, Fichtner I,Eckert K. Intrahepatic transplantation ofCD34+ cord blood stem cells into newbornand adult NOD/SCID mice induce differen-tial organ engraftment. Tissue Cell.2012;44(2):80-86.
40. Arora N, Wenzel PL, McKinney-Freeman SL,et al. Effect of developmental stage of HSCand recipient on transplant outcomes. DevCell. 2014;29(5):621-628.
41. Boyd AL, Campbell CJ, Hopkins CI, et al.Niche displacement of human leukemicstem cells uniquely allows their competitivereplacement with healthy HSPCs. J ExpMed. 2014;211(10):1925-1935.
42. Furlan I, Batz C, Flotho C, et al. Intriguingresponse to azacitidine in a patient with juve-nile myelomonocytic leukemia and mono-somy 7. Blood. 2009;113(12):2867-2868.
43. Cseh A, Niemeyer CM, Yoshimi A, et al.Bridging to transplant with azacitidine injuvenile myelomonocytic leukemia: a retro-spective analysis of the EWOG-MDS studygroup. Blood. 2015;125(14):2311-2313.
C.F. Krombholz et al.
606 haematologica | 2016; 101(5)

haematologica | 2016; 101(5) 607
Received: August 12, 2015.
Accepted: January 22, 2016.
Pre-published: January 27, 2016.
©2016 Ferrata Storti Foundation
Check the online version for the most updatedinformation on this article, online supplements,and information on authorship & disclosures:www.haematologica.org/content/101/5/607
Material published in Haematologica is cov-ered by copyright. All rights reserved to theFerrata Storti Foundation. Copies of articlesare allowed for personal or internal use.Permission in writing from the publisher isrequired for any other use.
Correspondence: [email protected]
Association of acute myeloid leukemia’smost immature phenotype with risk groupsand outcomesJonathan M. Gerber,1 Joshua F. Zeidner,2 Sarah Morse,3 Amanda L. Blackford,3Brandy Perkins, Breann Yanagisawa,3 Hao Zhang,3 Laura Morsberger,3 JudithKarp,3 Yi Ning,3 Christopher D. Gocke,3 Gary L. Rosner,3 B. Douglas Smith,3 andRichard J. Jones3
1Levine Cancer Institute, Charlotte, NC; 2Lineberger Comprehensive Cancer Center,University of North Carolina, Chapel Hill, NC; and 3The Sidney Kimmel ComprehensiveCancer Center at Johns Hopkins, Johns Hopkins University, Baltimore, MD, USA
*JMG and JFZ contrbuted equally to this work.
Ferrata StortiFoundation
EUROPEANHEMATOLOGYASSOCIATION
Haematologica 2016Volume 101(5):607-616
ARTICLEAcute Myeloid Leukemia
doi:10.3324/haematol.2015.135194
The precise phenotype and biology of acute myeloid leukemia stemcells remain controversial, in part because the “gold standard”immunodeficient mouse engraftment assay fails in a significant frac-
tion of patients and identifies multiple cell-types in others. We sought toanalyze the clinical utility of a novel assay for putative leukemia stem cellsin a large prospective cohort. The leukemic clone’s most primitivehematopoietic cellular phenotype was prospectively identified in 109newly-diagnosed acute myeloid leukemia patients, and analyzed againstclinical risk groups and outcomes. Most (80/109) patients harboredCD34+CD38– leukemia cells. The CD34+CD38– leukemia cells in 47 of the80 patients displayed intermediate aldehyde dehydrogenase expression,while normal CD34+CD38– hematopoietic stem cells expressed high levelsof aldehyde dehydrogenase. In the other 33/80 patients, the CD34+CD38–
leukemia cells exhibited high aldehyde dehydrogenase activity, and most(28/33, 85%) harbored poor-risk cytogenetics or FMS-like tyrosine kinase 3internal tandem translocations. No CD34+ leukemia cells could be detectedin 28/109 patients, including 14/21 patients with nucleophosmin-1 muta-tions and 6/7 acute promyelocytic leukemia patients. The patients withCD34+CD38– leukemia cells with high aldehyde dehydrogenase activitymanifested a significantly lower complete remission rate, as well as poorerevent-free and overall survivals. The leukemic clone’s most immature phe-notype was heterogeneous with respect to CD34, CD38, and ALDHexpression, but correlated with acute myeloid leukemia risk groups andoutcomes. The strong clinical correlations suggest that the most immaturephenotype detectable in the leukemia might serve as a biomarker for “clin-ically-relevant” leukemia stem cells. ClinicalTrials.gov: NCT01349972.
ABSTRACT
Introduction
More than two decades ago, Lapidot et al. reported that acute myeloid leukemia(AML) cells capable of engrafting immunodeficient mice expressed a CD34+CD38– nor-mal hematopoietic stem cell (HSC) phenotype.1 These so-called leukemia stem cells(LSCs) gave rise to partially differentiated progeny that constituted the bulk of theleukemia, but possessed only limited proliferative potential.2 More recently, leukemiccells of varying surface phenotypes, even within the same patient, have been shown tobe capable of engrafting immunodeficient mice, the generally accepted “gold standard”for LSC activity.3,4 However, this traditional approach for LSC identification has provento be somewhat elusive. Not only is the assay cumbersome and non-quantitative,5 but

in a significant fraction of AML patients no leukemia cell sub-set will engraft5-10 even using the newer mouse models.11 Thisinability to confirm the identity of LSCs in many patients isat least in part the reason that the clinical relevance of LSCsremains uncertain.12 Regardless of their tumorigenic potential in immunodefi-
cient mice, leukemic cells that persist after therapy [i.e. min-imal residual disease (MRD)] are arguably the most clinicallyimportant.13,14 We recently showed that MRD during com-plete remission (CR) was enriched for CD34+CD38– leukemiccells, and their presence after therapy was highly associatedwith subsequent clinical relapse.13 Others found thatCD34+CD38– leukemia cell frequency correlated with prog-nosis.7,15 Thus, accumulating evidence now suggests that ini-tial clinical responses likely reflect the behavior of the bulkleukemia, while long-term survival/cure requires the eradica-tion of LSCs.7,13-15 We also showed that the leukemicCD34+CD38– cells from most patients, particularly thosewith core-binding factor (CBF) AMLs, could be separatedfrom normal HSCs by their expression of aldehyde dehydro-genase 1 (ALDH). Normal HSCs exhibited high ALDHexpression (CD34+CD38–ALDHhigh), while the putative LSCsexpressed intermediate levels (CD34+CD38–ALDHint).13 Thesefindings have recently been confirmed.16,17Clinical outcomes in AML are highly diverse with some
patients curable with standard therapies, others initiallyrefractory to all known therapies, and the majority eventuallyrelapsing and succumbing to the disease after initially achiev-ing CRs. While patient factors such as age and performancestatus may influence the heterogeneous outcomes, theunderlying biology - currently best reflected by cytogeneticand molecular markers - is the major determinant. AML’shighly diverse biology suggests that the LSCs are also hetero-geneous. Accordingly, our previous report identified twopatients, both primary refractory to induction, whose puta-tive LSCs demonstrated high ALDH expression indistin-guishable from normal HSCs.13 We could not detect anyCD34+ leukemia cells in two other patients.13 Other groupshave also described heterogeneous CD34 and ALDH expres-sion in AMLs.8,16-20Since no leukemia subset from many patients will engraft
immunodeficient mice,5-11 and no leukemic CD34+CD38–cells can be identified in some patients,4,13,15,16,21 other meansfor LSC identification are needed to allow for their study clin-ically.14 Based on our smaller study of mostly CBF AMLpatients,13 we hypothesized that the most primitivehematopoietic cell phenotype that could be found inleukemia cells might have important clinical relevance. Thus,we prospectively assessed the leukemia’s most immaturephenotype in a multi-institutional randomized clinical trialcomparing two induction therapies in patients lacking favor-able-risk cytogenetics: standard cytarabine-based “7+3" ther-apy22 and a novel regimen called FLAM (flavopiridol, cytara-bine, mitoxantrone).23,24 To fully assess heterogeneity of theleukemic clone’s most immature phenotype, we also studiedpatients who initially agreed to the trial but were ultimatelyineligible because they were found to have favorable-riskcytogenetics. Here we find that the most primitivehematopoietic cellular phenotype present in leukemia cells isnot only heterogeneous for CD34, CD38, and ALDH expres-sion, but also that this phenotypic heterogeneity correlateswith both AML risk groups and outcomes. Moreover, therobust clinical correlations suggest that the most immaturephenotype detectable in the leukemia might serve as a bio-marker for “clinically-relevant” LSCs.
Methods
PatientsPatients aged 18-70 with newly-diagnosed AML, excluding CBF
AMLs and APL, were eligible for this multicenter clinical study (clin-icaltrials.gov NCT01349972).24 Patients were randomized 2:1 toFLAM or the standard “7+3” regimens, respectively.24 Patients whoachieved complete or partial responses to the first cycle were eligibleto receive a second cycle of FLAM or high-dose cytarabine (HiDAC),and/or could undergo allogeneic bone marrow transplantation(alloBMT) as per physician discretion. Johns Hopkins patients whowere study ineligible because their cytogenetics proved favorablewere also included in this analysis. Informed consent for participa-tion in NCT01349972, as well as for the bone marrow donations bythe patients not treated on trial, was obtained in accordance with theDeclaration of Helsinki as approved by the Johns HopkinsInstitutional Review Board.
Isolation of cellsSpecimens were collected between April 2011 and April 2013.
Marrow mononuclear (MMNC) and CD34+ cell subsets were iden-tified and isolated as previously described.13,25 At least 500,000 cellsfrom each AML specimen were then stained with Aldefluor(Aldagen, Durham, NC, USA) to assess ALDH activity according tothe manufacturer’s instructions utilizing diethylaminobenzaldehyde(DEAB) controls. Next, cells were labeled with monoclonal phyco-erythrin-conjugated anti-CD34 and allophycocyanin (APC)-conjugated anti-CD38 (BD Biosciences, San Jose, CA, USA)and analyzed with a MoFlo cell sorter (Beckman Coulter, Brea, CA,USA). Gating for CD34 and CD38 populations was based on clearlydistinguishable populations, or in the absence of such, the negativeantibody control.25 A representative example of gating is shown inOnline Supplementary Figure S1.
Fluorescence in situ hybridization (FISH) and molecularanalysesFor patients with cytogenetic abnormalities detectable by FISH,
250-1000 cell aliquots were sorted directly onto slides and fixed with3:1 methanol-glacial acetic acid (Sigma-Aldrich, St. Louis, MO,USA). FISH was performed and analyzed by the Johns HopkinsKimmel Cancer Center Cytogenetics Core, using probes specific forthe patients’ known cytogenetic abnormalities per manufacturer’sguidelines (Abbot Molecular, Des Plaines, IL, USA) as we previouslydescribed.13 Real-time polymerase chain reaction for FLT3 internaltandem duplication (ITD) (qPCR) and NPM1 mutations (reversetranscriptase-qPCR) was performed by Johns Hopkins MolecularHematopathology Laboratory.
Data analysisThe AML’s most immature phenotype was scored in a blinded
fashion by RJJ, BP, and SM as we previously described.13 Any differ-ences in scoring were to be decided by a simple majority, but therewas complete concordance on all observations. The samples werethen de-identified by the Johns Hopkins Kimmel Cancer CenterSpecimen Accessioning Core for statistical analysis. Clinical out-comes were determined by the NCT01349972 clinical study team24
blinded to the AML phenotypic data. Event-free survival (EFS) wasdefined as the date of treatment to the occurrence of persistentAML, relapse, or death. Poor-risk cytogenetics [> 3 clonal abnormal-ities, -5, 5q-, -7, -7q, t(3;3), inv 3, non-t(9;11) 11q23 excluding t(6;9),t(9;22)] and molecular abnormalities (FLT3-ITD mutation) were clas-sified according to the European LeukemiaNet reporting system.26
Statistical analysisP-values for differences in categorical data were determined by
J.M. Gerber et al.
608 haematologica | 2016; 101(5)

Fisher’s exact tests or T-tests, and for differences in outcome, strati-fied by treatment arm (FLAM vs. 7+3), by Mantel-Haenszel tests.Overall survival (OS) and EFS were estimated using the Kaplan-Meier method. Differences in OS and EFS according to the leukemicclone’s most primitive hematopoietic cellular phenotypes were ana-lyzed with hazard ratios (HR) from Cox proportional hazards mod-els that adjust for treatment arm, and tested for significance usinglikelihood ratio tests. Analyses were completed using R version3.1.1.27
Results
Patient characteristicsThe leukemia clone’s most primitive hematopoietic cellu-
lar phenotype was assessed in all patients entered inNCT0134997224 with adequate bone marrow specimens foranalyses. Of the 147 patients entered in the clinical trial, bonemarrow samples from 98 patients were analyzed. The mainreason for patients not being analyzed was the absence of aresearch sample because not enough cells could be obtainedwith the diagnostic marrow (43 patients). The specimenarrived in the laboratory but was not adequate for analysis in4 patients, and consent for the laboratory study was with-drawn in 2 patients. Over the same time frame, sevenpatients with CBF AML and 14 with APL were newly diag-nosed and treated at Johns Hopkins. Bone marrow samplesfrom 4 of the CBF patients and 7 of the APL patients wereavailable for analysis. The clinical characteristics of the 98patients on trial and the 11 favorable-risk patients not eligiblefor the trial are shown in Table 1.
The leukemia’s most immature phenotype was heterogeneousWe defined the most immature phenotype present in the
AML based on CD34, CD38, and ALDH expression, as wepreviously described.25,28,29 As CD34+CD38–ALDHhigh
HSCs16,28,30 differentiate into more committed progenitors,both CD34 and ALDH expression decrease while CD38expression increases.29,31-33 Thus, CD34+CD38-ALDHint,CD34+CD38+, and CD34- cells were considered increasinglymore differentiated phenotypes. The leukemic versus normalorigin of the hematopoietic phenotypes was determined bycytogenetic (FISH) or molecular (FLT3-ITD or NPM1) mark-ers when present. CD34+ cells comprised a median of 12% (range 0.07 - 81%)
of total MMNCs from the 98 patients in NCT01349972. In
22/98 of the patients, the AML phenotype was clinicallydetermined to be CD34- by standard flow cytometry crite-ria:7,16,34 i.e., CD34+ cells represented < 1% of the MMNCs (Table 1, Online Supplementary FigureS2A). In all 22 patients with < 1% CD34+ cells in theMMNCs, the small fraction (mean + SEM - 0.52+0.08) ofCD34+ cells was completely CD38-ALDHhigh (Table 2, Figure1A), and displayed low forward (FSC) and side (SSC) scatteron flow cytometry (data not shown). Only a small percentage(2.2+1.6%) of the CD34+CD38-ALDHhigh cells contained theleukemia-specific marker present in the five CD34–leukemias with cytogenetics detectable by FISH (Table 2).Likewise, when an AML with < 1% CD34+ cells was FLT3-ITD or NPM1-mutated (14/22 patients), the CD34+ cells didnot harbor the mutation (Figure 1B). CD34+ cells comprised a mean of 25.3+3.1% of MMNCs
in the 76 patients from NCT01349972 who harbored CD34+leukemia cells; the CD34+CD38– cells comprised 44.8+3.4%of the CD34+ cells in these patients. In 43 of these 76 patients,the majority (65.1+3.4%) of CD34+CD38–cells were ALDHint
(Table 2, Online Supplementary Figure S2B), while the ALDHhigh
population represented 1.7+0.5% of the CD34+CD38– cells(Figure 2A, Table 2). In the 11/43 cases with leukemia-specificcytogenetics scorable by FISH, we confirmed that theCD34+CD38–ALDHint cells were predominantly leukemic(Table 2). In contrast, the small number ofCD34+CD38–ALDHhigh cells predominantly lacked the FISHmarker that characterized the leukemia (Table 2). Likewise,when AMLs with prominent CD34+CD38–ALDHint popula-tions exhibited FLT3-ITD mutations (3 patients) or wereNPM1-mutated (4 cases), the CD34+CD38–ALDHint cellsexhibited the mutation while the CD34+CD38–ALDHhigh cellsdid not (Figure 2B). The CD34+CD38–ALDHhigh HSCs exhib-ited much lower FSC (data not shown) and SSC than theCD34+CD38–ALDHint AML cells (Figure 2A). These data areconsistent with the CD34+CD38–ALDHhigh cells representingnormal HSCs as we previously demonstrated.13 The 4 CBFpatients also displayed prominent leukemic CD34+CD38–ALDHint populations harboring, and small CD34+CD38–ALDHhigh fractions lacking, the FISH abnormality (Tables 1, 2).
Only ALDHhigh CD34+CD38– cells were present in 26 of the76 patients in NCT01349972 with > 1% CD34+ cells (Tables1, 2 and Figure 3A). In the 14 patients with leukemia-specificmutations scorable by FISH, the CD34+CD38–ALDHhigh pop-ulation contained mostly (78+6.7%) leukemic cells (Table 2).Similarly, this population was mostly leukemic in those
Relevance of the AML most primitive phenotype
haematologica | 2016; 101(5) 609
Figure 1. Assessment of CD34+ cellsfrom an NPM1 and FLT3-ITD mutat-ed AML patient with < 1% CD34+
cells. (A) Representative flow cyto-metric staining pattern of ALDHactivity by CD34 is displayed onMMNCs from patient. All the CD34+
cells are CD34+CD38–ALDHhigh. TheCD34+ALDHhigh cells are shown inrectangle. (B) FLT3-ITD status of iso-lated cell fractions. The CD34– blastsharbored the FLT3-ITD mutation,while the CD34+ cells exclusively dis-played the 330bp wild-type gene.
A B
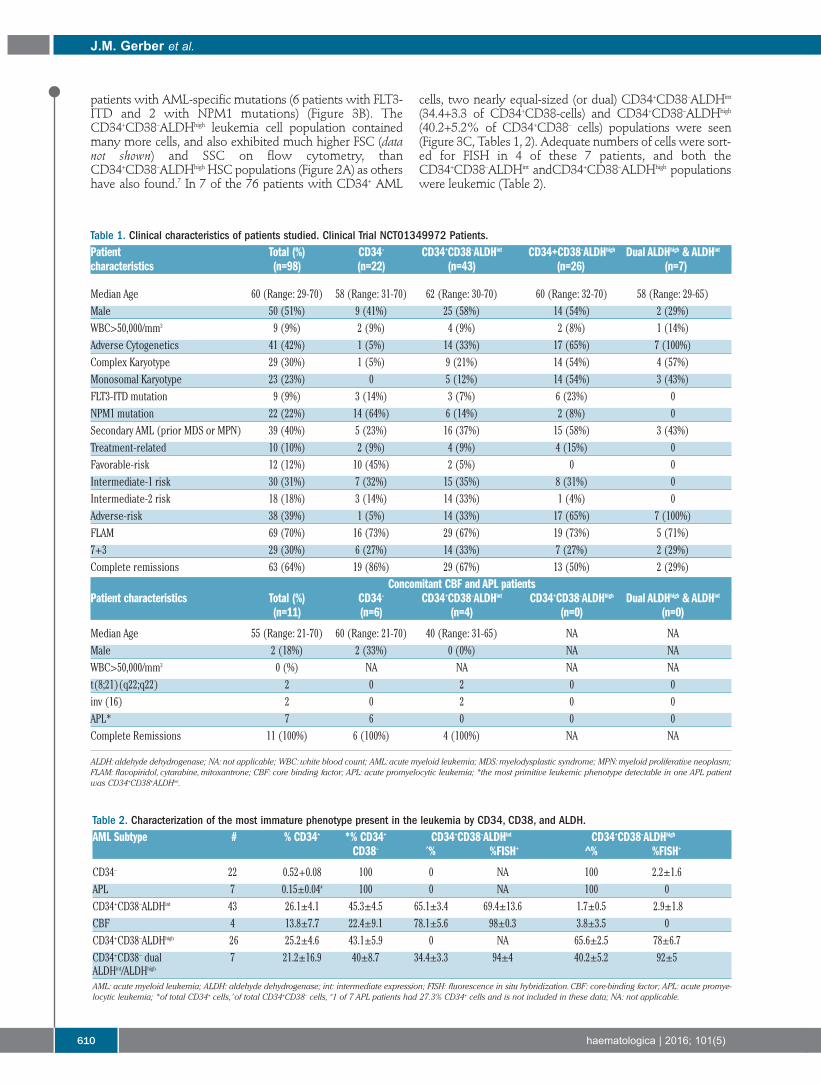
patients with AML-specific mutations (6 patients with FLT3-ITD and 2 with NPM1 mutations) (Figure 3B). TheCD34+CD38–ALDHhigh leukemia cell population containedmany more cells, and also exhibited much higher FSC (datanot shown) and SSC on flow cytometry, thanCD34+CD38–ALDHhigh HSC populations (Figure 2A) as othershave also found.7 In 7 of the 76 patients with CD34+ AML
cells, two nearly equal-sized (or dual) CD34+CD38–ALDHint
(34.4+3.3 of CD34+CD38-cells) and CD34+CD38–ALDHhigh
(40.2+5.2% of CD34+CD38– cells) populations were seen(Figure 3C, Tables 1, 2). Adequate numbers of cells were sort-ed for FISH in 4 of these 7 patients, and both theCD34+CD38–ALDHint andCD34+CD38–ALDHhigh populationswere leukemic (Table 2).
J.M. Gerber et al.
610 haematologica | 2016; 101(5)
Table 1. Clinical characteristics of patients studied. Clinical Trial NCT01349972 Patients.Patient Total (%) CD34– CD34+CD38–ALDHint CD34+CD38–ALDHhigh Dual ALDHhigh & ALDHint
characteristics (n=98) (n=22) (n=43) (n=26) (n=7)
Median Age 60 (Range: 29-70) 58 (Range: 31-70) 62 (Range: 30-70) 60 (Range: 32-70) 58 (Range: 29-65)Male 50 (51%) 9 (41%) 25 (58%) 14 (54%) 2 (29%)WBC>50,000/mm3 9 (9%) 2 (9%) 4 (9%) 2 (8%) 1 (14%)Adverse Cytogenetics 41 (42%) 1 (5%) 14 (33%) 17 (65%) 7 (100%)Complex Karyotype 29 (30%) 1 (5%) 9 (21%) 14 (54%) 4 (57%)Monosomal Karyotype 23 (23%) 0 5 (12%) 14 (54%) 3 (43%)FLT3-ITD mutation 9 (9%) 3 (14%) 3 (7%) 6 (23%) 0NPM1 mutation 22 (22%) 14 (64%) 6 (14%) 2 (8%) 0Secondary AML (prior MDS or MPN) 39 (40%) 5 (23%) 16 (37%) 15 (58%) 3 (43%)Treatment-related 10 (10%) 2 (9%) 4 (9%) 4 (15%) 0Favorable-risk 12 (12%) 10 (45%) 2 (5%) 0 0Intermediate-1 risk 30 (31%) 7 (32%) 15 (35%) 8 (31%) 0Intermediate-2 risk 18 (18%) 3 (14%) 14 (33%) 1 (4%) 0Adverse-risk 38 (39%) 1 (5%) 14 (33%) 17 (65%) 7 (100%)FLAM 69 (70%) 16 (73%) 29 (67%) 19 (73%) 5 (71%)7+3 29 (30%) 6 (27%) 14 (33%) 7 (27%) 2 (29%)Complete remissions 63 (64%) 19 (86%) 29 (67%) 13 (50%) 2 (29%)
Concomitant CBF and APL patientsPatient characteristics Total (%) CD34– CD34+CD38–ALDHint CD34+CD38–ALDHhigh Dual ALDHhigh & ALDHint
(n=11) (n=6) (n=4) (n=0) (n=0)
Median Age 55 (Range: 21-70) 60 (Range: 21-70) 40 (Range: 31-65) NA NAMale 2 (18%) 2 (33%) 0 (0%) NA NAWBC>50,000/mm3 0 (%) NA NA NA NAt(8;21)(q22;q22) 2 0 2 0 0 inv (16) 2 0 2 0 0 APL* 7 6 0 0 0 Complete Remissions 11 (100%) 6 (100%) 4 (100%) NA NA
ALDH: aldehyde dehydrogenase; NA: not applicable; WBC: white blood count; AML: acute myeloid leukemia; MDS: myelodysplastic syndrome; MPN: myeloid proliferative neoplasm;FLAM: flavopiridol, cytarabine, mitoxantrone; CBF: core binding factor; APL: acute promyelocytic leukemia; *the most primitive leukemic phenotype detectable in one APL patientwas CD34+CD38+ALDHint.
Table 2. Characterization of the most immature phenotype present in the leukemia by CD34, CD38, and ALDH.AML Subtype # % CD34+ *% CD34+ CD34+CD38–ALDHint CD34+CD38–ALDHhigh
CD38– ^% %FISH+ ^% %FISH+
CD34– 22 0.52+0.08 100 0 NA 100 2.2±1.6APL 7 0.15±0.04# 100 0 NA 100 0CD34+CD38–ALDHint 43 26.1±4.1 45.3±4.5 65.1±3.4 69.4±13.6 1.7±0.5 2.9±1.8CBF 4 13.8±7.7 22.4±9.1 78.1±5.6 98±0.3 3.8±3.5 0CD34+CD38–ALDHhigh 26 25.2±4.6 43.1±5.9 0 NA 65.6±2.5 78±6.7CD34+CD38– dual 7 21.2±16.9 40±8.7 34.4±3.3 94±4 40.2±5.2 92±5ALDHint/ALDHhigh
AML: acute myeloid leukemia; ALDH: aldehyde dehydrogenase; int: intermediate expression; FISH: fluorescence in situ hybridization. CBF: core-binding factor; APL: acute promye-locytic leukemia; *of total CD34+ cells, ^of total CD34+CD38– cells, #1 of 7 APL patients had 27.3% CD34+ cells and is not included in these data; NA: not applicable.

Absence of detectable CD34+ AML cells is associated withNPM1 mutations or APLOf the 22 patients in NCT01349972 with < 1% CD34+ cells
in the MMNCs, 14 harbored NPM1 mutations compared to8 of the 76 patients with CD34+ AML cells (Table 1; OnlineSupplementary Figure S2A, S2B, P<0.001). Of the 12 patientswith NPM1 mutations as the sole abnormality, no CD34+leukemia cells could be detected in 11 (Online SupplementaryFigure S2A) and one harbored CD34+ CD38–ALDHint leukemiacells (Online Supplementary Figure S2B) (P<0.002). The onlytwo patients in the series with t(9;11) were among the other8 non-NPM1-mutated patients in this CD34– group(P<0.001), as were 4 patients with normal cytogenetics(Online Supplementary Figure S2A). Only one CD34– patientharbored poor-risk cytogenetics, and 3 of the CD34– NPM1-mutated patients also manifested FLT3-ITD mutations(Online Supplementary Figure S2A). Of the 8 CD34+NPM1-mutated patients, 6 had a predominant population ofCD34+CD38–ALDHint (5 had additional detectable mutations)and 2 (both with complex cytogenetics) hadCD34+CD38–ALDHhigh leukemia cells (Online SupplementaryFigure S2B).Of the 7 APL patient specimens available for study, 6 also
had < 1% (0.15+0.04) CD34+ cells (Tables 1, 2). These 6patients showed exactly the same pattern as the other AMLswith <1% CD34+: i.e., the CD34+ cells were exclusivelyCD38–ALDHhigh and lacked the t(15;17) by FISH (Table 2).CD34+ cells comprised 27.3% of the MMNCs in the otherAPL patient (Table 2); very few (0.9%) of the CD34+ cellsfrom this patient were CD38–, and they all lacked t(15;17) byFISH. In contrast, the CD34+CD38+ cells did harbor thetranslocation.
CD34+CD38–ALDHhigh leukemia cells are associated withpoor-risk AMLOf the 26 patients in NCT01349972 displaying a predom-
inant CD34+CD38–ALDHhigh leukemic population, 17 had
poor-risk cytogenetics and an additional 4 patients had FLT3-ITD mutations (Table 1). All 7 of the patients with dualCD34+CD38–ALDHint and CD34+CD38– ALDHhigh leukemiapopulations also harbored poor-risk cytogenetics: 4 had high-ly complex cytogenetic changes, two del 7q, and one del 5q(Table 1). Thus, 28/33 (85%) patients withCD34+CD38–ALDHhigh AML cells harbored poor-risk geneticmarkers, while only 4 of the 22 (18%) patients with <1%CD34+ cells and 16 out of 43 (37%) patients with predomi-nant CD34+CD38–ALDHint populations harbored poor-riskcytogenetics or FLT3-ITD mutations (P<0.0001). The patientswith CD34+CD38–ALDHhigh AML cells were also more likelyto have AML arising out of myelodysplastic syndrome ormyeloproliferative disease (18/33, 55%) than theCD34+CD38–ALDHint and CD34– groups (21/65, 32%,P=0.04).
The leukemias’ most primitive hematopoietic cell phenotype correlates with outcomesNot surprisingly, given the strong association with poor-
risk genetics, patients harboring CD34+CD38–ALDHhigh
leukemic populations displayed relative drug resistance.There was a significantly lower CR rate for patients harbor-ing CD34+CD38–ALDHhigh leukemic populations when com-pared to patients with CD34+CD38–ALDHint or no CD34+cells AML cells (Table 3, P=0.007). The CR rates for thepatients with CD34+CD38–ALDHhigh leukemic populationswere similar with FLAM (11/24, 46%) and 7+3 (4/9, 44%).However, there was a trend for more CRs on the FLAM arm(36/45, 80%) than on the 7+3 arm (12/20, 60%) in the other65 patients (P=0.1).We next studied if the most immature phenotype present
in the leukemia also showed a correlation with survival. OSwas significantly different according to most immatureleukemia phenotype present in the leukemia (P=0.02, Table3, Figure 4A), with patients harboring CD34+CD38–ALDHhigh
AML cells demonstrating the worst OS. There was also a sig-
Relevance of the AML most primitive phenotype
haematologica | 2016; 101(5) 611
Figure 2. Prominent ALDHint population of CD34+CD38– cells from a patient with FLT3-ITD AML. (A) Representative flow cytometric staining pattern of ALDH activityby side scatter (SSC) is displayed for CD34+CD38– cells isolated from patient. (B) FLT3-ITD status of isolated cell fractions. The CD34+CD38–ALDHint population (oval)harbored the FLT3-ITD mutation, while the CD34+CD38–ALDHhigh cells (square) exclusively displayed the 330bp wild-type gene.
A B

nificant difference in EFS according to the most primitiveleukemia phenotype (P<0.001, Table 3, Figure 4B). The EFSprobability at 1-year was 61% (95% CI, 41-90%), 45% (95%CI 29-69%), and 19% (95% CI 8-47%) for patients withoutdetectable CD34+ leukemia cells and those withCD34+CD38–ALDHint and CD34+CD38–ALDHhigh leukemiacells, respectively (P<0.001). As others have found a strong correlation between just
leukemic CD34+CD38– cell numbers (without using ALDHexpression) at diagnosis and outcome,7,15 we analyzed theprognostic impact of total CD34+CD38–numbers. There wasa trend for total CD34+CD38– cell numbers at diagnosis tocorrelate with outcome. For patients with detectable CD34+AML cells in trial NCT01349972, CD34+CD38– cells repre-sented 5.6+1.5% of the MMNCs for those who entered a CRcompared to 11+3% in those who did not (P=0.08, t-test). Ofthose patients with < 5% CD34+CD38– cells, 23% remainedevent-free compared to 9.5% with > 5% CD34+CD38– cells(P=0.2, Fisher's exact test). The mean frequency ofCD34+CD38– cells was the same in both the ALDHint andALDHhigh groups at 7.6+1.8% and 7.5+2.5%, respectively.The type of postremission therapy was not specifically
mandated on this trial, and many of the patients went ontoalloBMT (Table 3). AlloBMT was very effective in all patientsin NCT01349972, regardless of their most primitiveleukemia phenotype. Of the patients with ALDHhigh leukemiacells, 8/15 who achieved a CR underwent alloBMT in CR1and 5 remain alive and disease-free (Table 3). Similarly, 16 ofthe CD34+CD38–ALDHint and 6 of the CD34– patients under-went alloBMT in CR1, and 7 and 5 patients remain alive anddisease-free, respectively (Table 3, P=0.2). In contrast, theoutcomes of the patients who did not receive alloBMT inCR1, with most receiving cytarabine-based consolidationtherapy, differed significantly by the most primitive pheno-type present in leukemia cells. Seven patients withCD34+CD38–ALDHhigh leukemia cells did not undergoalloBMT in CR1, and all relapsed including 3 with normalcytogenetics and wild-type FLT3/NPM1 (Table 3). In con-trast, 4/13 (3/9 with normal cytogenetics and wild-typeFLT3/NPM1) patients with CD34+CD38–ALDHint leukemiacells and 7/13 (1 of 2 with normal cytogenetics and wild-typeFLT3/NPM1) CD34– patients who did not undergo alloBMTremain alive and disease-free in CR1 (Table 3, P=0.06).
Discussion
The failure of CRs to reliably translate into cures in AML35,36can be explained by the LSC paradigm. However, the trueclinical relevance of LSCs remains the focus of considerabledebate.3-20,37 Several groups have shown that CD34+CD38–leukemia cell numbers present at diagnosis have strong prog-nostic significance, providing support for a clinical relevancefor LSCs.7,15 Patients with increased numbers of CD34+CD38–at diagnosis in clinical trial NCT01349972 showed a trendtoward worse outcomes. Our inability to show a strongerclinical correlation between CD34+CD38– leukemia cell num-bers at diagnosis and outcome may relate to the exclusion offavorable-risk cytogenetic-risk groups from the study. Wealso did not use the same methodology as others whoshowed a stronger correlation; we analyzed only totalCD34+CD38– numbers, while others further refined theCD34+CD38– subset to include the expression of leukemicstem cell associated markers7 or CD123.15 We did find thatthe most immature hematopoietic cellular phenotype pres-ent in leukemia cells was heterogeneous, ranging fromCD34– to that of primitive HSCs (i.e., CD34+CD38–ALDHhigh),but was relatively consistent across AML risk groups.Perhaps most importantly, the strong association betweenthe leukemic clone’s most immature phenotype and out-come in this prospective patient cohort supports further test-ing of this clinical biomarker in future studies.The vast majority of AML patients (80/109) in our series
harbored CD34+CD38– leukemia cells, as initially reportedby Lapidot et al.1 Moreover, we confirmed our prior data13that the majority of non poor-risk AMLs, including all of theCBF patients, harbored CD34+CD38– leukemia cells thatcould be separated from normal HSCs by their lowerALDH activity. However, 33 out of 98 (34%) of patientsfrom NCT01349972 harbored CD34+CD38–ALDHhigh
leukemia cells. This group of patients was more likely toharbor poor-risk cytogenetics or FLT3-ITD mutations, andhad a statistically lower chance of achieving CRs than theother AML patients. Importantly, the presence ofCD34+CD38–ALDHhigh leukemia cells was associated with asignificantly lower EFS and OS, even when no unfavorablegenetic or cytogenetic abnormalities could be identified.Even though patients with CD34+CD38–ALDHhigh LSCs did
J.M. Gerber et al.
612 haematologica | 2016; 101(5)
Table 3. Clinical outcomes of patients in NCT01349972 by leukemia’s most primitive hematopoietic cellular phenotype.CD34+CD38–ALDHhigh CD34+CD38–ALDHint CD34– P*(including dual) (n=43) (n=22)
(n=33)
Complete remission 15/33 (45%) 29/43 (67%) 19/22 (86%) 0.007Median OS (months) 9.4 (95% CI: 6-36) 18.7 (95% CI: 13-36) Not reached 0.02
HR – 1.3 (95% CI: 0.7-1.4) HR – 1 HR - 0.4 (95% CI: 0.2–1)Median EFS (months) 2.2 (95% CI: 2-6) 11.3 (95% CI, 4-36) 13 (95% CI: 4-36) 0.001
HR = 2.2 (95% CI: 1.2-3.9) HR - 1 HR - 0.6 (95% CI: 0.3-1.3)AlloBMT in CR1 8/15 (53%) 16/29 (55%) 6/20 (30%)Continuously EF 5/8 (63%) 7/16 (44%) 5/6 (83%) 0.2Median EFS (months) Not reached Not reached 23.1 (95% CI: 23 – 35)No BMT in CR1 7/15 (47%) 13/29 (45%) 13/20 (65%)Continuously EF 0/7 4/13 (31%) 7/13 (54%) 0.06Median EFS (months) 4.6 (95% CI: 3 – 32) 11.9 (95% CI: 5 – 32) Not reached
ALDH: aldehyde dehydrogenase; int: intermediate expression; EF: event (relapse or death)-free; EFS: event-free survival; OS: overall survival; alloBMT: allogeneic bone marrow trans-plantation; CR1: first complete remission; HR: hazard ratio; CI: confidence interval; *P values for Fisher’s exact tests.

poorly overall, 5/8 of those patients who got to alloBMTremain alive and disease-free. Several groups have alsodescribed ALDHhigh LSCs in a fraction of AML patients whoappeared to have a worse prognosis.8,17-20 No CD34+ leukemia cells could be detected in 22/98
patients from NCT01349972. As others have alsodescribed,7,16,34 the CD34+ cells in these patients represented<1% of the cells at diagnosis, were exclusivelyCD38–ALDHhigh, and lacked the leukemic mutation. Thus, theCD34+ cells in such patients likely represented residual nor-mal HSCs. NPM1 mutations were detected in 14 (64%) ofthe 22 AML patients who lacked detectable CD34+ cells, and11/12 AML patients with NPM1 mutation as a sole abnor-mality were in this group. No CD34+ cells were detected in6/7 APL patients, as others have also found.38 The one APLpatient in this series with CD34+ AML cells only had thet(15;17) detected in the CD34+CD38+ cells. Other groupshave reported that CD34 expression is a bad prognostic fac-tor for both NPM1-mutated AMLs39 and APLs;40-42 the smallnumbers of these patients in our cohort may have hindereddemonstrating similar statistical significance. The phenotype of the LSCs in NPM1-mutated AML has
been somewhat controversial. Two other groups also foundthat most NPM1-mutated AMLs were CD34–, with theCD34+ cells lacking leukemia mutations.4,16 However, Martelliet al. found that the NPM1 mutation was present inCD34+CD38– cells, and these cells generated AML in immun-odeficient mice.43 Interestingly, CD34+ cells represented>1.5% of the MMNCs in all the NPM1-mutated AMLs trans-planted into mice in that report.43We also found the mutationin the CD34+CD38– cells from all 8 NPM1-mutated AMLpatients with >1% CD34+ MMNCs. Of note, 7 of thesepatients had cytogenetic or FLT3-ITD mutations in additionto NPM1. Thus, it appears that the most immature leukemiccell in NPM1-mutated AMLs can be either CD34+ or CD34–;it is possible that the differences can be explained by the factthat Martelli et al. did not perform immunodeficient mousetransplants with any of the 18 patients in their series harbor-ing < 1% CD34+ cells.43 Despite our inability to detect AML cells by PCR in the
small population of CD34+ cells present in the diagnosticmarrows of the CD34– AMLs, a very small number (2.2%) ofCD34+CD38–ALDHhigh cells had the FISH marker that charac-
terized the AML (Table 2). Zeijlemaker et al. recently suggest-ed that although the vast majority of AML patients with <1% CD34+ cells in their diagnostic marrow lacked CD34+AML cells, a small number did harbor neoplastic CD34+cells.21 It is similarly possible that CD34+CD38–ALDHhigh AMLcells may be present at very low levels in the patients whoseleukemias’ most immature phenotype appeared to beCD34+CD38–ALDHint; however, based on the low FSC/SSCof these CD34+CD38–ALDHhigh cells , we believe that thesesmall leukemic populations by FISH represent flow sortingcontamination. We also previously found that theCD34+CD38–ALDHhigh cells present in AMLs harboring largeCD34+CD38–ALDHint populations only produced normalhematopoiesis when transplanted into immunocompro-mised mice.13 Importantly, the phenotype of the most primi-tive hematopoietic cells found to harbor predominatelyleukemia-specific mutations correlated with AML riskgroups and outcomes.Our data raise the possibility that the most immature phe-
notype present in leukemia may be a function of the stage ofhematopoietic differentiation at which the leukemogenicmutation develops. Those AMLs harboring leukemia cellssharing a phenotype with primitive normal HSCs(CD34+CD38–ALDHhigh) had the worst prognosis, while CBFand intermediate-risk AMLs’ most primitive phenotype wasthat of more differentiated hematopoietic progenitors(CD34+CD38–ALDHint). The most immature hematopoieticphenotype found in the most favorable-risk AMLs, APLs andthose with NPM1-mutations as sole abnormalities, expressedeven more differentiated phenotypes: CD34+CD38+ andCD34–CD38+. These findings suggest that the leukemia clones’ most
primitive hematopoietic cellular phenotype might serve as abiomarker for risk-stratifying patients at diagnosis. About 30-40% of AML patients lack any cytogenetic or usual geneticprognostic factors,44 and even when present such prognosticfactors may not be available for days or weeks. The mostimmature phenotype present in leukemia cells can be readilydetermined in essentially all patients by flow cytometrywithin hours of diagnosis. Rapid risk-stratification may beparticularly useful for patients harboringCD34+CD38–ALDHhigh leukemia cells, which appear to iden-tify high-risk patients often refractory to induction
Relevance of the AML most primitive phenotype
haematologica | 2016; 101(5) 613
Figure 3. Prominent ALDHhigh populations of CD34+CD38– cells. (A) Representative flow cytometric staining pattern of ALDH activity by side scatter (SSC) is displayedfor CD34+CD38– cells isolated from patient. The CD34+CD38-ALDHhigh cells represented essentially all of the total CD34+CD38- cells. (B) FLT3-ITD status of isolatedcell fractions. The CD34+CD38-ALDHhigh population harbored the FLT3-ITD mutation. (C) Representative flow cytometric staining pattern of ALDH activity by side scatter(SSC) is displayed for CD34+CD38– cells isolated from a patient with dual CD34+CD38–ALDHint and CD34+CD38–ALDHhigh populations.
A B C
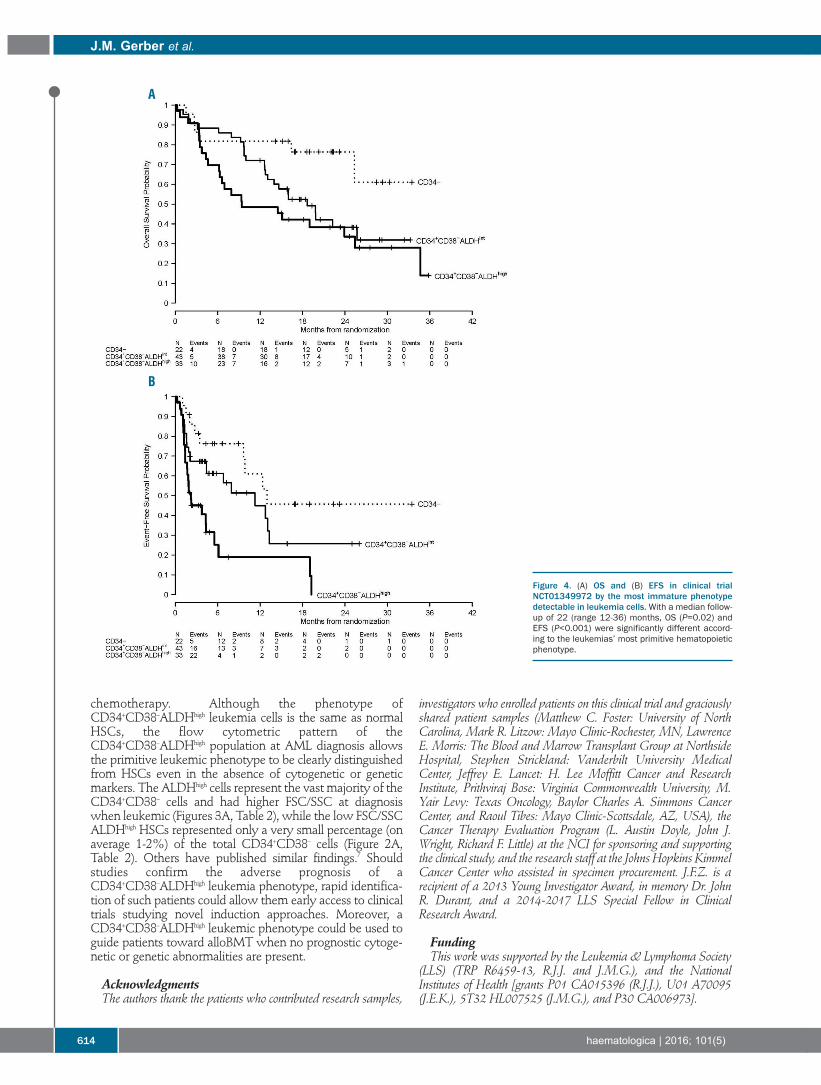
chemotherapy. Although the phenotype ofCD34+CD38–ALDHhigh leukemia cells is the same as normalHSCs, the flow cytometric pattern of theCD34+CD38–ALDHhigh population at AML diagnosis allowsthe primitive leukemic phenotype to be clearly distinguishedfrom HSCs even in the absence of cytogenetic or geneticmarkers. The ALDHhigh cells represent the vast majority of theCD34+CD38– cells and had higher FSC/SSC at diagnosiswhen leukemic (Figures 3A, Table 2), while the low FSC/SSCALDHhigh HSCs represented only a very small percentage (onaverage 1-2%) of the total CD34+CD38– cells (Figure 2A,Table 2). Others have published similar findings.7 Shouldstudies confirm the adverse prognosis of aCD34+CD38–ALDHhigh leukemia phenotype, rapid identifica-tion of such patients could allow them early access to clinicaltrials studying novel induction approaches. Moreover, aCD34+CD38–ALDHhigh leukemic phenotype could be used toguide patients toward alloBMT when no prognostic cytoge-netic or genetic abnormalities are present.
AcknowledgmentsThe authors thank the patients who contributed research samples,
investigators who enrolled patients on this clinical trial and graciouslyshared patient samples (Matthew C. Foster: University of NorthCarolina, Mark R. Litzow: Mayo Clinic-Rochester, MN, LawrenceE. Morris: The Blood and Marrow Transplant Group at NorthsideHospital, Stephen Strickland: Vanderbilt University MedicalCenter, Jeffrey E. Lancet: H. Lee Moffitt Cancer and ResearchInstitute, Prithviraj Bose: Virginia Commonwealth University, M.Yair Levy: Texas Oncology, Baylor Charles A. Simmons CancerCenter, and Raoul Tibes: Mayo Clinic-Scottsdale, AZ, USA), theCancer Therapy Evaluation Program (L. Austin Doyle, John J.Wright, Richard F. Little) at the NCI for sponsoring and supportingthe clinical study, and the research staff at the Johns Hopkins KimmelCancer Center who assisted in specimen procurement. J.F.Z. is arecipient of a 2013 Young Investigator Award, in memory Dr. JohnR. Durant, and a 2014-2017 LLS Special Fellow in ClinicalResearch Award.
FundingThis work was supported by the Leukemia & Lymphoma Society
(LLS) (TRP R6459-13, R.J.J. and J.M.G.), and the NationalInstitutes of Health [grants P01 CA015396 (R.J.J.), U01 A70095(J.E.K.), 5T32 HL007525 (J.M.G.), and P30 CA006973].
J.M. Gerber et al.
614 haematologica | 2016; 101(5)
Figure 4. (A) OS and (B) EFS in clinical trialNCT01349972 by the most immature phenotypedetectable in leukemia cells. With a median follow-up of 22 (range 12-36) months, OS (P=0.02) andEFS (P<0.001) were significantly different accord-ing to the leukemias’ most primitive hematopoieticphenotype.
A
B

Relevance of the AML most primitive phenotype
haematologica | 2016; 101(5) 615
References1. Lapidot T, Sirard C, Vormoor J, et al. A cell
initiating human acute myeloid leukaemiaafter transplantation into SCID mice. Nature.1994;367(6464):645-648.
2. Bonnet D, Dick JE. Human acute myeloidleukemia is organized as a hierarchy thatoriginates from a primitive hematopoieticcell. Nat Med. 1997;3(7):730-737.
3. Sarry JE, Murphy K, Perry R, et al. Humanacute myelogenous leukemia stem cells arerare and heterogeneous when assayed inNOD/SCID/IL2Rgammac-deficient mice. JClin Invest. 2011;121(1):384-395.
4. Taussig DC, Vargaftig J, Miraki-Moud F, et al.Leukemia-initiating cells from some acutemyeloid leukemia patients with mutatednucleophosmin reside in the CD34(-) frac-tion. Blood. 2010;115(10):1976-1984.
5. Pearce DJ, Taussig D, Zibara K, et al. AMLengraftment in the NOD/SCID assay reflectsthe outcome of AML: implications for ourunderstanding of the heterogeneity of AML.Blood. 2006;107(3):1166-1173.
6. Cheung AM, Chow HC, Kwong YL, Liang R,Leung AY. FLT3/internal tandem duplicationsubclones in acute myeloid leukemia differ intheir engraftment potential in NOD/SCIDmice. Leuk Res. 2010;34(1):119-122.
7. Terwijn M, Zeijlemaker W, Kelder A, et al.Leukemic stem cell frequency: a strong bio-marker for clinical outcome in acute myeloidleukemia. PLoS One. 2014;9(9):e107587.
8. Cheung AM, Wan TS, Leung JC, et al.Aldehyde dehydrogenase activity inleukemic blasts defines a subgroup of acutemyeloid leukemia with adverse prognosisand superior NOD/SCID engrafting poten-tial. Leukemia. 2007;21(7):1423-1430.
9. Monaco G, Konopleva M, Munsell M, et al.Engraftment of acute myeloid leukemia inNOD/SCID mice is independent of CXCR4and predicts poor patient survival. Stem Cells.2004;22(2):188-201.
10. Ailles LE, Gerhard B, Kawagoe H, Hogge DE.Growth characteristics of acute myelogenousleukemia progenitors that initiate malignanthematopoiesis in nonobese diabetic/severecombined immunodeficient mice. Blood.1999;94(5):1761-1772.
11. Feuring-Buske M, Gerhard B, Cashman J,Humphries RK, Eaves CJ, Hogge DE.Improved engraftment of human acutemyeloid leukemia progenitor cells in beta 2-microglobulin-deficient NOD/SCID miceand in NOD/SCID mice transgenic forhuman growth factors. Leukemia.2003;17(4):760-763.
12. Rombouts WJ, Martens AC, Ploemacher RE.Identification of variables determining theengraftment potential of human acutemyeloid leukemia in the immunodeficientNOD/SCID human chimera model.Leukemia. 2000;14(5):889-897.
13. Gerber JM, Smith BD, Ngwang B, et al. Aclinically relevant population of leukemicCD34+CD38 - cells in acute myeloidleukemia. Blood. 2012;119(15):3571-3577.
14. Ghiaur G, Gerber J, Jones RJ. Concise review:Cancer stem cells and minimal residual dis-ease. Stem Cells. 2012;30(1):89-93.
15. Vergez F, Green AS, Tamburini J, et al. High
levels of CD34+CD38low/-CD123+ blastsare predictive of an adverse outcome in acutemyeloid leukemia: a Groupe Ouest-Est desLeucemies Aigues et Maladies du Sang(GOELAMS) study. Haematologica.2011;96(12):1792-1798.
16. Schuurhuis GJ, Meel MH, Wouters F, et al.Normal hematopoietic stem cells within theAML bone marrow have a distinct and higherALDH activity level than co-existingleukemic stem cells. PLoS One. 2013;8(11):e78897.
17. Hoang VT, Buss EC, Wang W, et al. The rar-ity of ALDH(+) cells is the key to separationof normal versus leukemia stem cells byALDH activity in AML patients. Int J Cancer.2015;137(3):525-536.
18. Pearce DJ, Taussig D, Simpson C, et al.Characterization of cells with a high alde-hyde dehydrogenase activity from cordblood and acute myeloid leukemia samples.Stem Cells. 2005;23(6):752-760.
19. Ran D, Schubert M, Pietsch L, et al.Aldehyde dehydrogenase activity among pri-mary leukemia cells is associated with stemcell features and correlates with adverse clin-ical outcomes. Exp Hematol.2009;37(12):1423-1434.
20. Ran D, Schubert M, Taubert I, et al.Heterogeneity of leukemia stem cell candi-dates at diagnosis of acute myeloid leukemiaand their clinical significance. Exp Hematol.2012;40(2):155-165.
21. Zeijlemaker W, Kelder A, Wouters R, et al.Absence of leukaemic CD34 cells in acutemyeloid leukaemia is of high prognosticvalue: a longstanding controversy deci-phered. Br J Haematol. 2015;171(2):227-238.
22. Estey EH. How to manage high-risk acutemyeloid leukemia. Leukemia.2012;26(5):861-869.
23. Karp JE, Blackford A, Smith BD, et al. Clinicalactivity of sequential flavopiridol, cytosinearabinoside, and mitoxantrone for adultswith newly diagnosed, poor-risk acute myel-ogenous leukemia. Leuk Res. 2010;34(7):877-882.
24. Zeidner JF, Foster MC, Blackford AL et al.Randomized multicenter phase 2 study offlavopiridol (alvocidib), cytarabine, andmitoxantrone (FLAM) versuscytarabine/daunorubicin (7+3) in newly diag-nosed acute myeloid leukemia.Haematologica. 2015;100(9):1172-9.
25. Gerber JM, Qin L, Kowalski J et al.Characterization of chronic myeloidleukemia stem cells. Am J Hematol.2011;8631-37.
26. Dohner H, Estey EH, Amadori S et al.Diagnosis and management of acute myeloidleukemia in adults: recommendations froman international expert panel, on behalf of theEuropean LeukemiaNet. Blood.2010;115(3):453-474.
27. R: R Core Team. R: A language and environ-ment for statistical computing. R Foundationfor Statistical Computing, Viena, Austria.2014.
28. Ghiaur G, Yegnasubramanian S, Perkins B,Gucwa JL, Gerber JM, Jones RJ. Regulation ofhuman hematopoietic stem cell self-renewalby the microenvironment's control of retinoicacid signaling. Proc Natl Acad Sci USA.
2013;110(40):16121-16126.29. Gerber JM, Gucwa JL, Esopi D et al.
Genome-wide comparison of the transcrip-tomes of highly enriched normal and chronicmyeloid leukemia stem and progenitor cellpopulations. Oncotarget. 2013;4(5):715-728.
30. Storms RW, Trujillo AP, Springer JB et al.Isolation of primitive human hematopoieticprogenitors on the basis of aldehyde dehy-drogenase activity. Proc Natl Acad Sci U S A.1999;96(16):9118-9123.
31. Krause DS, Fackler MJ, Civin CI, May WS.CD34: structure, biology, and clinical utility.Blood. 1996;87(1):1-13.
32. Jones RJ, Barber JP, Vala MS, et al.Assessment of aldehyde dehydrogenase inviable cells. Blood. 1995;85(10):2742-2746.
33. Jones RJ, Collector MI, Barber JP, et al.Characterization of mouse lymphohe-matopoietic stem cells lacking colony-form-ing activity. Blood. 1996;88(2):487-491.
34. van der Pol MA, Feller N, Roseboom M, et al.Assessment of the normal or leukemic natureof CD34+ cells in acute myeloid leukemiawith low percentages of CD34 cells.Haematologica. 2003;88(9):983-993.
35. Appelbaum FR, Gundacker H, Head DR, etal. Age and acute myeloid leukemia. Blood.2006;107(9):3481-3485.
36. Cheson BD, Bennett JM, Kopecky KJ, et al.Revised recommendations of theInternational Working Group for Diagnosis,Standardization of Response Criteria,Treatment Outcomes, and ReportingStandards for Therapeutic Trials in AcuteMyeloid Leukemia. J Clin Oncol.2003;21(24):4642-4649.
37. Becker MW, Jordan CT. Leukemia stem cellsin 2010: current understanding and futuredirections. Blood Rev. 2011;25(2):75-81.
38. Turhan AG, Lemoine FM, Debert C, et al.Highly purified primitive hematopoietic stemcells are PML-RARA negative and generatenonclonal progenitors in acute promyelocyticleukemia. Blood. 1995; 85(8):2154-2161.
39. Dang H, Chen Y, Kamel-Reid S, Brandwein J,Chang H. CD34 expression predicts anadverse outcome in patients with NPM1-pos-itive acute myeloid leukemia. Hum Pathol.2013;44(10):2038-2046.
40. Lee JJ, Cho D, Chung IJ, et al. CD34 expres-sion is associated with poor clinical outcomein patients with acute promyelocyticleukemia. Am J Hematol. 2003;73(3):149-153.
41. Breccia M, De Propris MS, Stefanizzi C, et al.Negative prognostic value of CD34 antigenalso if expressed on a small population ofacute promyelocitic leukemia cells. AnnHematol. 2014;93(11):1819-1823.
42. Ahmad EI, Akl HK, Hashem ME, ElgoharyTA. The biological characteristics of adultCD34+ acute promyelocytic leukemia. MedOncol. 2012;29(2):1119-1126.
43. Martelli MP, Pettirossi V, Thiede C, et al.CD34+ cells from AML with mutated NPM1harbor cytoplasmic mutated nucleophosminand generate leukemia in immunocompro-mised mice. Blood. 2010;116(19):3907-3922.
44. Walker AR ,Marcucci G. Management ofpatients with cytogenetically normal acutemyeloid leukemia who have neither favor-able nor unfavorable markers. J Natl ComprCanc Netw. 2014;12(4):527-534.

616 haematologica | 2016; 101(5)
Received: October 6, 2015.
Accepted: February 3, 2016.
Pre-published: February 8, 2016.
©2016 Ferrata Storti Foundation
Check the online version for the most updatedinformation on this article, online supplements,and information on authorship & disclosures:www.haematologica.org/content/101/5/616
Material published in Haematologica is cov-ered by copyright. All rights reserved to theFerrata Storti Foundation. Copies of articlesare allowed for personal or internal use.Permission in writing from the publisher isrequired for any other use.
Correspondence: [email protected]
Adoptive transfer of chimeric antigen receptor-transduced T cells is apromising strategy for cancer immunotherapy. The CD38 molecule,with its high expression on multiple myeloma cells, appears a suit-
able target for antibody therapy. Prompted by this, we used three differentCD38 antibody sequences to generate second-generation retroviral CD38-chimeric antigen receptor constructs with which we transduced T cells fromhealthy donors and multiple myeloma patients. We then evaluated the pre-clinical efficacy and safety of the transduced T cells. Irrespective of thedonor and antibody sequence, CD38-chimeric antigen receptor-transducedT cells proliferated, produced inflammatory cytokines and effectively lysedmalignant cell lines and primary malignant cells from patients with acutemyeloid leukemia and multi-drug resistant multiple myeloma in a cell-dose,and CD38-dependent manner, despite becoming CD38-negative during cul-ture. CD38-chimeric antigen receptor-transduced T cells also displayed sig-nificant anti-tumor effects in a xenotransplant model, in which multiplemyeloma tumors were grown in a human bone marrow-like microenviron-ment. CD38-chimeric antigen receptor-transduced T cells also appeared tolyse the CD38+ fractions of CD34+ hematopoietic progenitor cells, mono-cytes, natural killer cells, and to a lesser extent T and B cells but did notinhibit the outgrowth of progenitor cells into various myeloid lineages and,furthermore, were effectively controllable with a caspase-9-based suicidegene. These results signify the potential importance of CD38-chimeric anti-gen receptor-transduced T cells as therapeutic tools for CD38+ malignanciesand warrant further efforts to diminish the undesired effects of thisimmunotherapy using appropriate strategies.
Pre-clinical evaluation of CD38 chimeric antigen receptor engineered T cellsfor the treatment of multiple myelomaEsther Drent,1,2 Richard W.J. Groen,1,3 Willy A. Noort,1,3 Maria Themeli,1 JeroenJ. Lammerts van Bueren,6 Paul W.H.I. Parren,6,7,8 Jürgen Kuball,4 ZsoltSebestyen,5 Huipin Yuan,9 Joost de Bruijn,9,10 Niels W.C.J. van de Donk,1Anton C.M. Martens,1,3,5 Henk M. Lokhorst,1,4 and Tuna Mutis1,2
1Department of Hematology, VU University Medical Center, Amsterdam, theNetherlands; 2Departments of Clinical Chemistry and Hematology, Utrecht, theNetherlands; 3Department of Cell Biology, University Medical Center, Utrecht, theNetherlands; 4Department of Hematology, University Medical Center, Utrecht, theNetherlands; 5Department of Immunology, University Medical Center Utrecht, theNetherlands; 6Genmab, Utrecht, the Netherlands; 7Department of Cancer andInflammation Research, Institute of Molecular Medicine, University of SouthernDenmark, Odense, Denmark; 8Department of Immunohematology and BloodTransfusion, Leiden University Medical Center, the Netherlands; 9Xpand BiotechnologyBV, Bilthoven, the Netherlands; 10The School of Engineering and Materials Science,Queen Mary University of London, UK
ABSTRACT
Ferrata StortiFoundation
EUROPEANHEMATOLOGYASSOCIATION
Haematologica 2016Volume 101(5):616-625
ARTICLE Plasma Cell Disorders
doi:10.3324/haematol.2015.137620
Introduction
Multiple myeloma (MM), a malignant disorder of antibody-producing clonalplasma cells, is the second most common hematologic neoplasia worldwide.1
Despite four decades of drug innovation, MM remains incurable with chemother-apy. Furthermore, the prognosis of MM patients who become refractory to recent-ly developed novel agents is very poor.2 On the other hand, clinical and experimen-tal data collected over the past decades suggest that MM could be successfullytreated through (cellular) immunotherapy.3,4 The curative potential of cellular

immunotherapy in MM is illustrated by the induction oflong-term sustained remissions after allogeneic stem celltransplantation or donor lymphocyte infusions in a subsetof patients.5,6 A highly appealing and more specificimmunotherapy strategy for cancer is the adoptive trans-fer of cytotoxic T cells that are genetically engineered toexpress chimeric antigen receptors (CAR).7,8 A CAR is anartificial hybrid receptor, in which the antigen-recognizingdomain of a tumor-reactive monoclonal antibody is fusedwith T-cell signaling domains. Upon retroviral or lentiviraltransduction of cytotoxic T cells, CAR expressed on thecell surface redirect the cytotoxic T cells toward the origi-nal target of the antibody in a non-HLA-restrictedmanner,7,8 making it possible to apply the therapy regard-less of the patient’s HLA type. Currently the most success-ful CAR-approaches are based on targeting the CD19 mol-ecule, which is broadly expressed in several B-cell malig-nancies but not on the malignant plasma cells frompatients with MM. Among a few potential CAR candi-dates for MM,9 the CD38 molecule, with its high and uni-form expression on malignant plasma cells, has long beensuggested a suitable target for antibody therapy of MM.The utility of CD38 as a suitable target has been support-ed by the results of recently initiated clinical trials inwhich MM patients were safely and effectively treatedwith the CD38-specific human monoclonal antibodydaratumumab.10Encouraged by these clinical results, we started to
explore the feasibility of development of a CART-cell ther-apy based on targeting the CD38 molecule. Using variableheavy and light chain sequences of three different humanCD38 antibodies, we generated three different CD38-CAR. We transduced T cells from healthy individuals andMM patients with the CD38-CAR and evaluated them foressential functions such as antigen-specific proliferationand cytokine production, for in vitro and in vivo anti-tumorefficacy and for potential undesired effects such as target-ing normal CD38+ cell fractions in the peripheral bloodand bone marrow. We also evaluated the feasibility ofcontrolling CD38-CART cells by introducing a caspase-9-based suicide gene.
Methods
Bone marrow mononuclear cells from patients with multiple myeloma or acute myeloid leukemia
Bone marrow mononuclear cells containing 5-20% malignantplasma cells or ~50% acute myeloid leukemia (AML) blasts wereisolated from bone marrow aspirates of MM/AML patientsthrough Ficoll-Paque density centrifugation and cryopreserved inliquid nitrogen until use. All bone marrow and blood samplingfrom the patients was performed after informed consent andapproved by the institutional medical ethical committee.
Peripheral blood mononuclear cells from healthy individuals Peripheral blood mononuclear cells were isolated from the buffy
coats of healthy blood-bank donors by Ficoll-Paque density cen-trifugation after informed consent and approval by the institution-al medical ethical committee.
Retroviral constructsThe sequences of three different human CD38 antibodies,
which are distinct from, but display similar affinities to the recent-ly documented daratumumab10 (Online Supplementary Table S1)were kindly provided by Genmab. Cloning methods are describedin the Online Supplementary Methods.
Retroviral chimeric antigen receptor transductioninto T cellsTransduction methods are described in the Online Supplementary
Methods.
Flow cytometry-based cell lysis assaysTo detect the lysis of various cell subsets by CART cells in
mononuclear cells from whole bone marrow or peripheral blood,serial dilutions of CART cells were incubated with CFSE-labeledbone marrow mononuclear cells or peripheral blood mononuclearcells for 24 h. The cells were then harvested, stained for differentCD markers and topro3 or LIVE/DEAD® Fixable Near-IR (LifeTechnologies L10119) and were quantitatively analyzed throughvolume-equalized measurements using a FACS Canto flowcytometer. For each cell subset identified with a CD marker,CFSE+, viable+/Topro3- cells were counted as surviving target cells.Percentage cell lysis in a treated sample was calculated as followsand only if the analyzed target cell population contained >500viable cells in the untreated samples. % lysis cells = 1 − (absolutenumber of surviving cells in treated wells / absolute number of sur-viving cells in untreated wells) × 100%.
Bioluminescence imaging-based cell lysis assays To determine the lysis of Luc-GFP-transduced human malignant
cell lines by CD38-CART cells, serial dilutions of mock or CD38-CART cells were co-incubated with the malignant cell lines. Theluciferase signal produced by surviving malignant cells was deter-mined after 16-24 h with a SpectraMax luminometer (MolecularDevices) within 15 min after the addition of 125 μg/mL beetleluciferin (Promega).11 The percent lysis was then calculated as inthe flow-based cytotoxicity assay above.
Experimental animalsRAG2-/-γc-/- mice used in this study were originally obtained
from the Amsterdam Medical Center (AMC, Amsterdam, theNetherlands). The mice were bred and maintained in filter topcages under specified pathogen-free conditions at the CentralAnimal Facility (GDL, Utrecht University, Utrecht, theNetherlands) and received sterile water and radiation-sterilizedfood pellets ad libitum.
In vivo efficacy of CD38-chimeric antigen receptor-transduced T cells against multiple myeloma tumorsgrowing in a humanized microenvironmentTo create a human bone marrow-like environment in mice,
hybrid scaffolds were coated in vitro with human mesenchymalstromal cells. After a week of in vitro culture, humanized scaffoldswere seeded with CD38+ UM9 cells and implanted subcutaneous-ly into the mice, as described previously,11,12 and in the OnlineSupplementary Methods.
Results
Generation of CD38-chimeric antigen receptor-transduced T cells
We used the variable heavy and light chain sequences ofthree different CD38 antibodies with CD38 binding affini-ties comparable to that of daratumumab (Online
CD38-CART cells for multiple myeloma treatment
haematologica | 2016; 101(5) 617
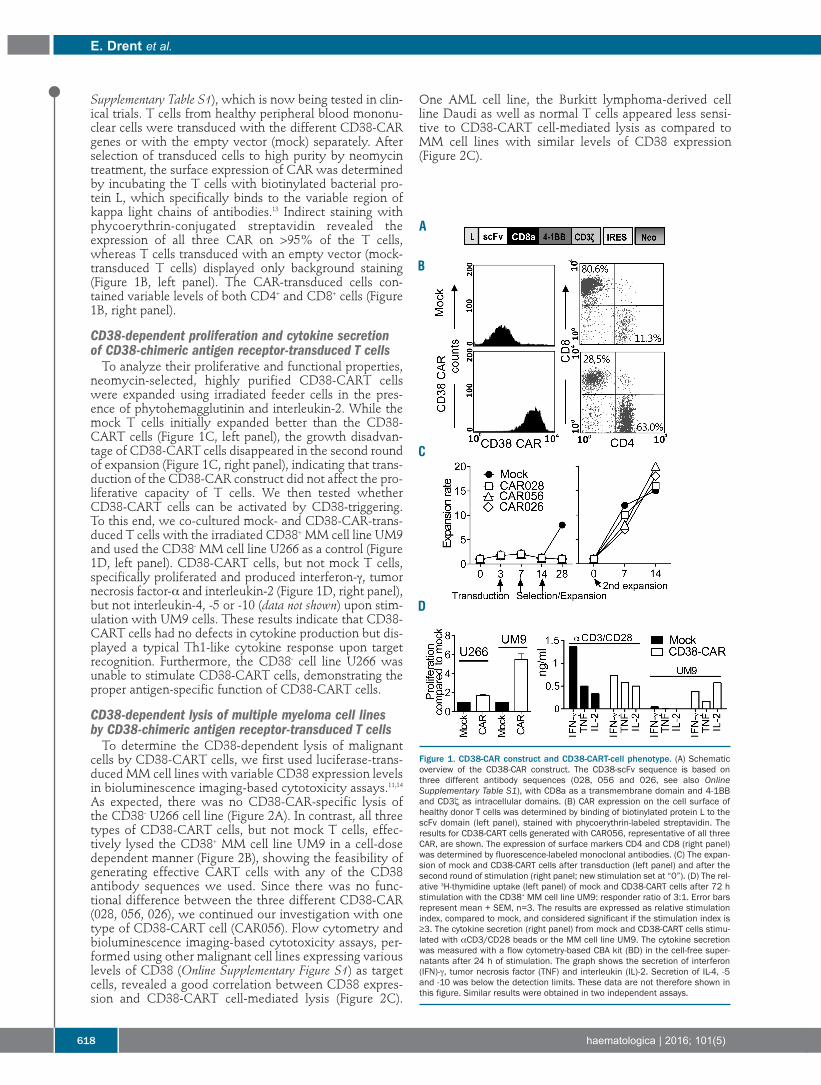
Supplementary Table S1), which is now being tested in clin-ical trials. T cells from healthy peripheral blood mononu-clear cells were transduced with the different CD38-CARgenes or with the empty vector (mock) separately. Afterselection of transduced cells to high purity by neomycintreatment, the surface expression of CAR was determinedby incubating the T cells with biotinylated bacterial pro-tein L, which specifically binds to the variable region ofkappa light chains of antibodies.13 Indirect staining withphycoerythrin-conjugated streptavidin revealed theexpression of all three CAR on >95% of the T cells,whereas T cells transduced with an empty vector (mock-transduced T cells) displayed only background staining(Figure 1B, left panel). The CAR-transduced cells con-tained variable levels of both CD4+ and CD8+ cells (Figure1B, right panel).
CD38-dependent proliferation and cytokine secretionof CD38-chimeric antigen receptor-transduced T cellsTo analyze their proliferative and functional properties,
neomycin-selected, highly purified CD38-CART cellswere expanded using irradiated feeder cells in the pres-ence of phytohemagglutinin and interleukin-2. While themock T cells initially expanded better than the CD38-CART cells (Figure 1C, left panel), the growth disadvan-tage of CD38-CART cells disappeared in the second roundof expansion (Figure 1C, right panel), indicating that trans-duction of the CD38-CAR construct did not affect the pro-liferative capacity of T cells. We then tested whetherCD38-CART cells can be activated by CD38-triggering.To this end, we co-cultured mock- and CD38-CAR-trans-duced T cells with the irradiated CD38+ MM cell line UM9and used the CD38- MM cell line U266 as a control (Figure1D, left panel). CD38-CART cells, but not mock T cells,specifically proliferated and produced interferon-γ, tumornecrosis factor-α and interleukin-2 (Figure 1D, right panel),but not interleukin-4, -5 or -10 (data not shown) upon stim-ulation with UM9 cells. These results indicate that CD38-CART cells had no defects in cytokine production but dis-played a typical Th1-like cytokine response upon targetrecognition. Furthermore, the CD38- cell line U266 wasunable to stimulate CD38-CART cells, demonstrating theproper antigen-specific function of CD38-CART cells.
CD38-dependent lysis of multiple myeloma cell lines by CD38-chimeric antigen receptor-transduced T cells To determine the CD38-dependent lysis of malignant
cells by CD38-CART cells, we first used luciferase-trans-duced MM cell lines with variable CD38 expression levelsin bioluminescence imaging-based cytotoxicity assays.11,14As expected, there was no CD38-CAR-specific lysis ofthe CD38- U266 cell line (Figure 2A). In contrast, all threetypes of CD38-CART cells, but not mock T cells, effec-tively lysed the CD38+ MM cell line UM9 in a cell-dosedependent manner (Figure 2B), showing the feasibility ofgenerating effective CART cells with any of the CD38antibody sequences we used. Since there was no func-tional difference between the three different CD38-CAR(028, 056, 026), we continued our investigation with onetype of CD38-CART cell (CAR056). Flow cytometry andbioluminescence imaging-based cytotoxicity assays, per-formed using other malignant cell lines expressing variouslevels of CD38 (Online Supplementary Figure S1) as targetcells, revealed a good correlation between CD38 expres-sion and CD38-CART cell-mediated lysis (Figure 2C).
One AML cell line, the Burkitt lymphoma-derived cellline Daudi as well as normal T cells appeared less sensi-tive to CD38-CART cell-mediated lysis as compared toMM cell lines with similar levels of CD38 expression(Figure 2C).
E. Drent et al.
618 haematologica | 2016; 101(5)
Figure 1. CD38-CAR construct and CD38-CART-cell phenotype. (A) Schematicoverview of the CD38-CAR construct. The CD38-scFv sequence is based onthree different antibody sequences (028, 056 and 026, see also OnlineSupplementary Table S1), with CD8a as a transmembrane domain and 4-1BBand CD3ζ as intracellular domains. (B) CAR expression on the cell surface ofhealthy donor T cells was determined by binding of biotinylated protein L to thescFv domain (left panel), stained with phycoerythrin-labeled streptavidin. Theresults for CD38-CART cells generated with CAR056, representative of all threeCAR, are shown. The expression of surface markers CD4 and CD8 (right panel)was determined by fluorescence-labeled monoclonal antibodies. (C) The expan-sion of mock and CD38-CART cells after transduction (left panel) and after thesecond round of stimulation (right panel; new stimulation set at “0”). (D) The rel-ative 3H-thymidine uptake (left panel) of mock and CD38-CART cells after 72 hstimulation with the CD38+ MM cell line UM9: responder ratio of 3:1. Error barsrepresent mean + SEM, n=3. The results are expressed as relative stimulationindex, compared to mock, and considered significant if the stimulation index is≥3. The cytokine secretion (right panel) from mock and CD38-CART cells stimu-lated with αCD3/CD28 beads or the MM cell line UM9. The cytokine secretionwas measured with a flow cytometry-based CBA kit (BD) in the cell-free super-natants after 24 h of stimulation. The graph shows the secretion of interferon(IFN)-γ, tumor necrosis factor (TNF) and interleukin (IL)-2. Secretion of IL-4, -5and -10 was below the detection limits. These data are not therefore shown inthis figure. Similar results were obtained in two independent assays.
A
B
C
D
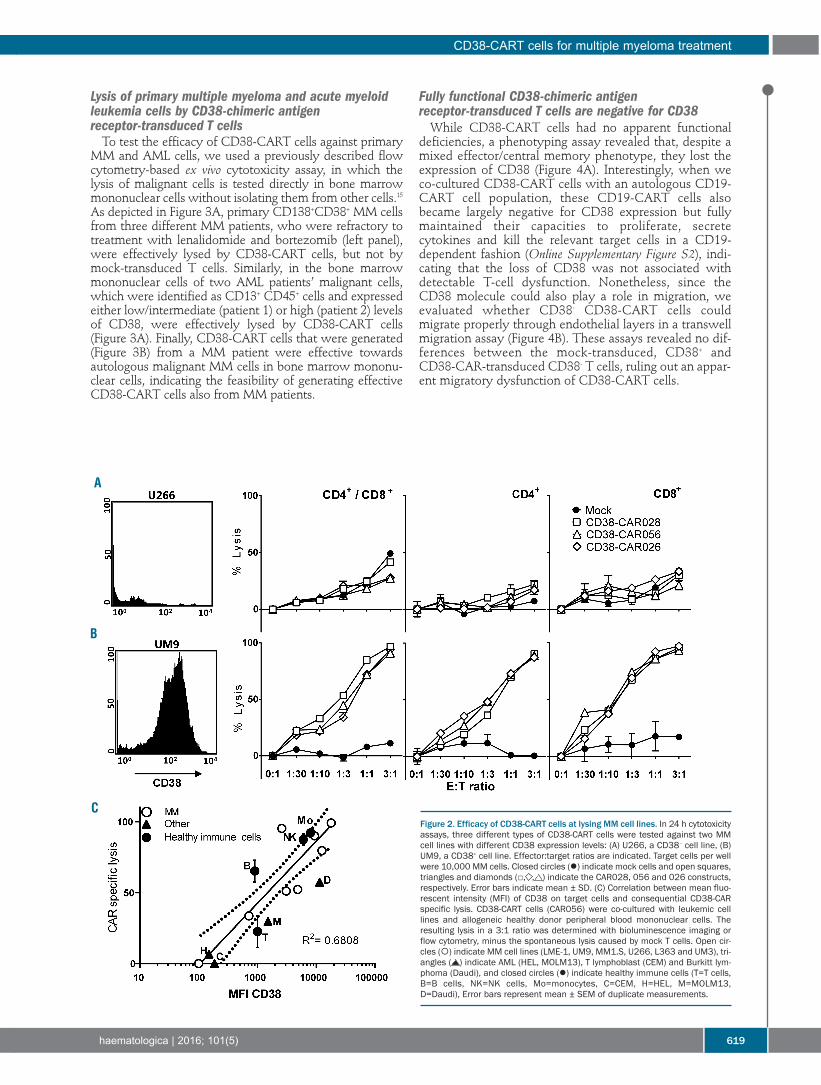
Lysis of primary multiple myeloma and acute myeloidleukemia cells by CD38-chimeric antigenreceptor-transduced T cells To test the efficacy of CD38-CART cells against primary
MM and AML cells, we used a previously described flowcytometry-based ex vivo cytotoxicity assay, in which thelysis of malignant cells is tested directly in bone marrowmononuclear cells without isolating them from other cells.15As depicted in Figure 3A, primary CD138+CD38+ MM cellsfrom three different MM patients, who were refractory totreatment with lenalidomide and bortezomib (left panel),were effectively lysed by CD38-CART cells, but not bymock-transduced T cells. Similarly, in the bone marrowmononuclear cells of two AML patients’ malignant cells,which were identified as CD13+ CD45+ cells and expressedeither low/intermediate (patient 1) or high (patient 2) levelsof CD38, were effectively lysed by CD38-CART cells(Figure 3A). Finally, CD38-CART cells that were generated(Figure 3B) from a MM patient were effective towardsautologous malignant MM cells in bone marrow mononu-clear cells, indicating the feasibility of generating effectiveCD38-CART cells also from MM patients.
Fully functional CD38-chimeric antigenreceptor-transduced T cells are negative for CD38While CD38-CART cells had no apparent functional
deficiencies, a phenotyping assay revealed that, despite amixed effector/central memory phenotype, they lost theexpression of CD38 (Figure 4A). Interestingly, when weco-cultured CD38-CART cells with an autologous CD19-CART cell population, these CD19-CART cells alsobecame largely negative for CD38 expression but fullymaintained their capacities to proliferate, secretecytokines and kill the relevant target cells in a CD19-dependent fashion (Online Supplementary Figure S2), indi-cating that the loss of CD38 was not associated withdetectable T-cell dysfunction. Nonetheless, since theCD38 molecule could also play a role in migration, weevaluated whether CD38- CD38-CART cells couldmigrate properly through endothelial layers in a transwellmigration assay (Figure 4B). These assays revealed no dif-ferences between the mock-transduced, CD38+ andCD38-CAR-transduced CD38- T cells, ruling out an appar-ent migratory dysfunction of CD38-CART cells.
CD38-CART cells for multiple myeloma treatment
haematologica | 2016; 101(5) 619
Figure 2. Efficacy of CD38-CART cells at lysing MM cell lines. In 24 h cytotoxicityassays, three different types of CD38-CART cells were tested against two MMcell lines with different CD38 expression levels: (A) U266, a CD38– cell line, (B)UM9, a CD38+ cell line. Effector:target ratios are indicated. Target cells per wellwere 10,000 MM cells. Closed circles () indicate mock cells and open squares,triangles and diamonds (□, , ) indicate the CAR028, 056 and 026 constructs,respectively. Error bars indicate mean ± SD. (C) Correlation between mean fluo-rescent intensity (MFI) of CD38 on target cells and consequential CD38-CARspecific lysis. CD38-CART cells (CAR056) were co-cultured with leukemic celllines and allogeneic healthy donor peripheral blood mononuclear cells. Theresulting lysis in a 3:1 ratio was determined with bioluminescence imaging orflow cytometry, minus the spontaneous lysis caused by mock T cells. Open cir-cles () indicate MM cell lines (LME-1, UM9, MM1.S, U266, L363 and UM3), tri-angles ( ) indicate AML (HEL, MOLM13), T lymphoblast (CEM) and Burkitt lym-phoma (Daudi), and closed circles () indicate healthy immune cells (T=T cells,B=B cells, NK=NK cells, Mo=monocytes, C=CEM, H=HEL, M=MOLM13,D=Daudi), Error bars represent mean ± SEM of duplicate measurements.
A
B
C

In vivo efficacy of CD38-chimeric antigen receptor-transduced T cells against multiple myeloma tumorsgrowing in a humanized microenvironment To substantiate the in vitro results, we questioned
whether the CD38- CART cells could mediate anti-MMeffects in vivo after systemic injection in our recently devel-oped model in Rag2-/-γc-/- mice, in which a humanizedbone marrow-like niche for MM cells is generated by sub-cutaneous implantation of ceramic scaffolds coated withhuman bone marrow stromal cells11,12 (Figure 4). Thus, weimplanted such scaffolds seeded with luciferase-trans-duced UM9 MM cells in the back of the mice (6 scaffoldsper mouse). Upon detection of the luciferase signal by bio-luminescence imaging, we treated the mice with intra-venous injections of CD38-CART cells using a previouslyestablished treatment scheme.16 Mock-transduced T cellswere used as controls. As illustrated in Figure 4B, in thecontrol group treated with mock T cells, tumors showed
fast progression. Although not curative, treatment of thetumor-bearing mice with CD38-CART cells induced a sig-nificant anti-tumor effect (Figure 4B,C) underscoring thepotential of CD38-CART cells to properly infiltrate andlyse MM tumors growing in their natural, protectiveniche. Post mortem analyses revealed that the remainingCD138+ tumors were still positive for CD38 (Figure 4D),thus ruling out tumor escape due to “antigen loss” vari-ants.
Impact of CD38-chimeric antigen receptor-transduced T cells on CD38+ normal hematopoietic cells andhematopoietic progenitor cellsBesides the high levels expressed in MM cells, the CD38
molecule is expressed at intermediate levels on a subset ofhematopoietic progenitor cells17 and on a fraction of nor-mal hematopoietic cells including activated T cells, naturalkiller cells, B cells and monocytes. We, therefore, evaluat-
E. Drent et al.
620 haematologica | 2016; 101(5)
Figure 3. Efficacy of CD38-CART cells generated from healthy individuals atlysing primary MM cells. (A) Bone marrow-derived mononuclear cells fromthree MM patients, all three refractory to lenalidomide and bortezomib, andbone marrow mononuclear cells from two AML patients were co-incubatedwith no, mock- or CD38-CART cells generated from healthy peripheral bloodmononuclear cells for 16 h. Closed circles () indicate mock and opensquares (□) indicate CAR056T cells (representative of all CAR). The graphsdepict the resulting lysis of CD138+/CD38+ cells (MM) orCD13+/CD7+/CD45dim/CD38+ cells (AML1, moderate CD38 expression) andCD33+/CD133+/CD45dim/CD38+ cells (AML2, high CD38 expression) in threeeffector:target cell ratios. The percent lysis in these flow cytometry assayswas calculated as described in the Methods section. (B) Efficacy of CD38-CART cells generated from a MM patient: CAR expression on the cell surfaceof the patient’s T cells was determined by flow cytometry with protein L stain-ing (see also Figure 1). (C) Bone marrow-derived mononuclear cells from theMM patient were co-incubated with autologous mock- or CD38-CART cells for16 h. The graph depicts resulting lysis of CD138+/CD38+ cells at two ratios,determined in flow cytometry-based assays.
A
B C

ed the possible negative impact of CART cells on these cellsubsets by co-incubating unsorted bone marrow mononu-clear cells with CD38-CART cells. CD38-CART cellsappeared to eliminate the CD38+ fractions of mature T, B,natural killer and monocyte cell subsets (Figure 5A) andthe CD38+ fraction of CD34+ cells (Figure 5B) in a 4 hassay. The lysis of CD34+CD38+ cells did not, however,have any influence on the development of colony-formingunits of monocytes or of granulocytes in a 14-dayhematopoietic precursor cell colony-forming assay18,19
(Figure 5C,D).
Specific elimination of CD38-chimeric antigenreceptor-transduced T cells using a suicide gene (iCasp9)Although CD38-CART cells did not lyse the CD38- frac-
tions of mature hematopoietic cells and did not inhibit theoutgrowth of these cell populations, a cautious approachtoward the clinical application of this construct is stillrequired. As a first step towards safer application ofCD38-CART cells, we tested the possibility of controllingthem with a suicide gene based on the inducible caspase-9 (iCasp9) gene that is activated with a small dimerizermolecule AP20187 (B/B).20 Thus, we inserted an iCasp9
CD38-CART cells for multiple myeloma treatment
haematologica | 2016; 101(5) 621
Figure 4. Tumor growth in mock- and CD38-CART-cell-treated mice. (A) Analysis of CD38-CART cells after 2 weeks of in vitro culture, with fluorescence-labeled mono -clonal antibodies for CD45RA and CD62L and CD38. (B) Leukocyte transmigration assay, in which mock and CART cells were cultured in a transwell system in theinserts with human umbilical vein endothelial cells, which were activated with tumor necrosis factor (TNF)-α. Spontaneous TNFα-induced transmigration was com-pared to active migration induced by 10% human serum in the lower compartment. % migrated cells = [Relative Fluorescence Units (RFU) of cells in lower compart-ment / RFU of total cells in both compartments] * 100%. (C) Analysis of tumor load in mice by quantification of bioluminescent imaging measurements. Each groupcontained six mice, each harboring six scaffolds. Results are mean tumor load (cpm/cm2) of six mice per group. Closed circles () indicate mock cells and opensquares (□) indicate CAR056 cells. The error bars represent mean + SEM, n=6. The differences between groups were analyzed after week 6 using an unpairedStudent T test, P<0.0001 (D) Bioluminescent imaging of mice on the right side; mice were implanted with fully humanized bone marrow stromal cell scaffolds eachcoated with 1×106 UM9-GFP-Luc tumor cells. At 7, 9 and 13 days after implantation, mice were injected intravenously with 20×106 mock or CD38-CART cells. (E)Representative immunohistochemistry figure: remaining tumors were stained with CD38 and CD138 antibody, T = tumor, sc = scaffold.
A B C
D
E

vector containing a green fluorescent protein (GFP) mark-er gene into the CD38-CART cells by retroviral transduc-tion. Around 50% of the CD38-CART cells were trans-duced, as detected by GFP expression (Figure 6A, upperpanel). When tested without sorting the iCasp9-trans-duced (GFP+) cells, all iCasp9-transduced, GFP+, but noneof the iCasp9-non-transduced, GFP-CD38-CART cellswere eliminated upon incubation with the dimerizerAP20187 (Figure 6A, lower panel). As expected, thedimerizer treatment also resulted in a proportionaldecrease in the lysis of the MM cell line UM9. (Figure
6B). There was still some remaining lysis due to the sur-viving iCasp9-negative CD38-CART cells, indicating thattriggering the suicide gene did not induce bystanderdamage to the cells in the close vicinity. When testedafter sorting for GFP+ cells (Figure 6C,D), almost all GFP+
cells died after treatment with the dimerizer (Figure 6C)and there was no CD38-specific lysis left (Figure 6D),confirming the results obtained in previous studies,20,21and suggesting the possibility of controlling CD38-CARTcells using the iCasp9 suicide gene without undesiredconsequences.
E. Drent et al.
622 haematologica | 2016; 101(5)
Figure 5. The impact of CD38-CART cells on non-malignant hematopoietic cells in bone marrow and outgrowth of hematopoietic cell lineages. (A) Bone marrowmononuclear cells from three MM patients were co-incubated with none, mock- or CD38-CART cells for 16 h. The graphs depict the resulting lysis of the total or theCD38+ fractions of CD3+ (T cells), CD56+ (mainly natural killer cells), CD14+ (monocytes) and CD19+ (B cells) subsets at three effector:target rations, determined withflow cytometry and calculated as described in the Methods section. Results are from three individual experiments combined. Closed circles () indicate mock cellsand open squares (□) indicate CAR056 cells. Error bars represent mean ± SEM, n=3. (B) CD34+ fraction of bone marrow mononuclear cells from healthy donors wasco-incubated with none, mock- or CD38-CART cells for 4 h at different target:effector cell ratios before being transferred into the semisolid hematopoietic progenitorcell culture medium. After incubation, cells were analyzed by flow cytometry for surviving CD34+ cells with CD38 expression. The graphs depict the resulting lysis ofthe total or the CD38+ fraction of CD34+ cells. Closed circles () indicate mock cells and open squares (□) indicate CAR056 cells. (C) After 14 days of culture inplastic dishes, colony-forming unit-monocytes (CFU-M), and CFU-granulocytes (CFU-G) were visible. (D) The numbers of CFU-M and CFU-G colonies were determinedmicroscopically. Results of a representative experiment are shown as mean ± SD.
A C
D
B
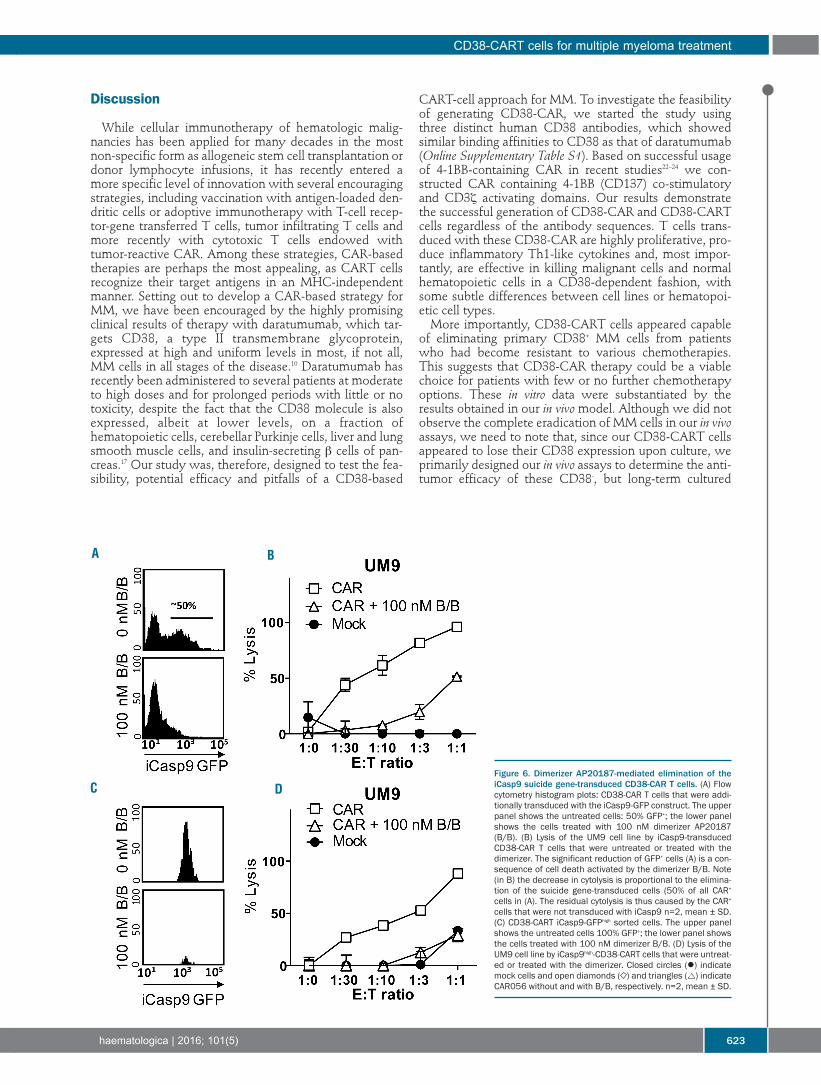
Discussion
While cellular immunotherapy of hematologic malig-nancies has been applied for many decades in the mostnon-specific form as allogeneic stem cell transplantation ordonor lymphocyte infusions, it has recently entered amore specific level of innovation with several encouragingstrategies, including vaccination with antigen-loaded den-dritic cells or adoptive immunotherapy with T-cell recep-tor-gene transferred T cells, tumor infiltrating T cells andmore recently with cytotoxic T cells endowed withtumor-reactive CAR. Among these strategies, CAR-basedtherapies are perhaps the most appealing, as CART cellsrecognize their target antigens in an MHC-independentmanner. Setting out to develop a CAR-based strategy forMM, we have been encouraged by the highly promisingclinical results of therapy with daratumumab, which tar-gets CD38, a type II transmembrane glycoprotein,expressed at high and uniform levels in most, if not all,MM cells in all stages of the disease.10 Daratumumab hasrecently been administered to several patients at moderateto high doses and for prolonged periods with little or notoxicity, despite the fact that the CD38 molecule is alsoexpressed, albeit at lower levels, on a fraction ofhematopoietic cells, cerebellar Purkinje cells, liver and lungsmooth muscle cells, and insulin-secreting β cells of pan-creas.17 Our study was, therefore, designed to test the fea-sibility, potential efficacy and pitfalls of a CD38-based
CART-cell approach for MM. To investigate the feasibilityof generating CD38-CAR, we started the study usingthree distinct human CD38 antibodies, which showedsimilar binding affinities to CD38 as that of daratumumab(Online Supplementary Table S1). Based on successful usageof 4-1BB-containing CAR in recent studies22–24 we con-structed CAR containing 4-1BB (CD137) co-stimulatoryand CD3ζ activating domains. Our results demonstratethe successful generation of CD38-CAR and CD38-CARTcells regardless of the antibody sequences. T cells trans-duced with these CD38-CAR are highly proliferative, pro-duce inflammatory Th1-like cytokines and, most impor-tantly, are effective in killing malignant cells and normalhematopoietic cells in a CD38-dependent fashion, withsome subtle differences between cell lines or hematopoi-etic cell types. More importantly, CD38-CART cells appeared capable
of eliminating primary CD38+ MM cells from patientswho had become resistant to various chemotherapies.This suggests that CD38-CAR therapy could be a viablechoice for patients with few or no further chemotherapyoptions. These in vitro data were substantiated by theresults obtained in our in vivomodel. Although we did notobserve the complete eradication of MM cells in our in vivoassays, we need to note that, since our CD38-CART cellsappeared to lose their CD38 expression upon culture, weprimarily designed our in vivo assays to determine the anti-tumor efficacy of these CD38-, but long-term cultured
CD38-CART cells for multiple myeloma treatment
haematologica | 2016; 101(5) 623
Figure 6. Dimerizer AP20187-mediated elimination of theiCasp9 suicide gene-transduced CD38-CAR T cells. (A) Flowcytometry histogram plots: CD38-CAR T cells that were addi-tionally transduced with the iCasp9-GFP construct. The upperpanel shows the untreated cells: 50% GFP+; the lower panelshows the cells treated with 100 nM dimerizer AP20187(B/B). (B) Lysis of the UM9 cell line by iCasp9-transducedCD38-CAR T cells that were untreated or treated with thedimerizer. The significant reduction of GFP+ cells (A) is a con-sequence of cell death activated by the dimerizer B/B. Note(in B) the decrease in cytolysis is proportional to the elimina-tion of the suicide gene-transduced cells (50% of all CAR+
cells in (A). The residual cytolysis is thus caused by the CAR+
cells that were not transduced with iCasp9 n=2, mean ± SD.(C) CD38-CART iCasp9-GFPhigh sorted cells. The upper panelshows the untreated cells 100% GFP+; the lower panel showsthe cells treated with 100 nM dimerizer B/B. (D) Lysis of theUM9 cell line by iCasp9high-CD38-CART cells that were untreat-ed or treated with the dimerizer. Closed circles () indicatemock cells and open diamonds ( ) and triangles ( ) indicateCAR056 without and with B/B, respectively. n=2, mean ± SD.
A B
C D

CD38-CART cells. This may have negatively influencedthe anti-tumor efficacy, since it is known that long-termcultured T cells rapidly lose their in vivo persistence capac-ities.25,26 In addition, and perhaps even more importantly,in our model, unlike all previously reported CAR studies,the human MM tumors were grown to larger masses in afully humanized bone marrow microenvironment. TheMM microenvironment is known to provide essential sig-nals for survival, growth and, more importantly, immuneresistance of MM cells.11,12,27,28 Since our model includessome of the microenvironment-related aspects, our resultssuggest that the efficacy of CART-cell treatment could beimproved if the therapy were to be combined withimmune checkpoint inhibitors and/or with survivinand/or MCL-1 inhibitors which are effective modifiers ofcell adhesion-mediated immune resistance induced by thetumor microenvironment.11Unlike a number of earlier reports, which mainly
focused on the anti-tumor efficacy of CD38-CART cells,29–31 we devoted a considerable part of our investigation toidentifying the potential drawbacks and risks of CD38-CART-cell therapy. Although CD38-CART cells eliminat-ed the CD38+ fractions of immune cell subsets as well asthe CD38+ fraction of hematopoietic progenitor cells, weobserved no inhibition of the outgrowth of hematopoieticlineages from CD34+CD38- progenitor cells. Furthermore,CD38-CART cells did not induce complete depletion ofmature hematopoietic cells in the periphery. The CD38-fractions of important immune cells, such as B and T cells,were also unaffected. These results suggest that the thera-py will spare sufficient numbers of T and B cells for theseto maintain their functions. However, since CD38 is awell-known T-cell activation molecule, and has also beenimplicated in chemotaxis,32 T-cell development,33 dendriticcell trafficking and humoral immune responses,34 it wouldbe relevant to determine whether an intact immuneresponse would be possible in the absence of CD38. Apartial solution to this issue came from the analyses ofCD38-CART cells: remarkably, we discovered that theCD38-CART cells, regardless of which single chain variantfragment was used, became completely devoid of CD38expression on their surface in various independently gen-erated batches of cells. The loss of CD38 was thus unlike-ly to be caused by a genetic defect, but was most probablydue to the “self lysis” of the CD38+ fractions, which wasalso described in another CD38-CAR study.29 Our CD38-CD38-CART cells, however, had no growth disadvantage,had a highly activated status, displayed CD38-dependentproliferation, cytokine production, and cytotoxic activitiesand showed no other detectable functional aberrancies.This was also the case for CD19-CART cells whichbecame CD38- after co-culture with CD38-CART cells(Online Supplementary Figure S2). Furthermore CD38-CART cells did not show any defects in transmigrationassays and they also mediated significant anti-MM effects
in vivo, thus indicating their capacity to migrate properlyand infiltrate into the MM niches and to kill them. Thus,it seems likely that: (i) not all activated T cells have to beCD38+, and (ii) CD38 expression is not essential for T cellsto fulfill their functions. This latter conclusion is also sup-ported by the fact that there is still no evidence, even fromCD38 knockout mice,32 that CD38-deficient effector Tcells are functionally defective. On the other hand, the relatively broad expression of
the target antigen of CD38-CART cells increases the riskof the so-called “cytokine release syndrome” due to mas-sive activation of CART cells, as has been observed in pre-vious trials with ERBB2- and CD19-CART cells.35–37Although the interleukin-6 receptor antagonist tocilizum-ab appears to reduce cytokine release syndrome38 it wouldstill be desirable to minimize the occurrence of suchsevere side effects. Furthermore, since we cannot rule outtoxicities occurring due to the possible attack of non-hematopoietic CD38+ cells, development of an optimalCD38-CART-cell therapy would require the improvementof the target-specificity as well as the in vivo control ofCD38-CART cells, and probably also in the case of otherCART-cell approaches targeting the kappa light chain,39CD138,40 Lewis Y antigen,41 BCMA,42 CS1,43,44 andCD44v6.45 One future option to improve the target-speci-ficity could be optimization of the target cell affinity ofCART cells. In addition, suicide genes may enable the invivo control of adoptively transferred CART cells. Indeed,in our first attempt to improve the safety profile of CD38-CART cells we observed that the iCasp9 gene20,46 can effec-tively control CART cells. These results, which are inagreement with those of other studies,20,45,47 provide posi-tive prospects for future clinical trials. The safety profile ofCART cells could also be improved by the generation ofinducible CAR constructs or using the recently developeddual CAR technologies. Considering all the data together, we conclude that
CD38-CART cells are powerful immunotherapeutic toolsand can be beneficial, especially for MM patients whohave no other chemotherapy options. These results war-rant further studies aimed at diminishing the undesiredeffects of CD38-CART cells against normal CD38+ cellsthrough optimizing the formers’ CD38 affinity andimproving in vivo controllability.
Acknowledgments We thank Dr. C. June for providing the sequence for the 4-1BB-
CD3ζ transgene, Dr. D. Spencer for the inducible caspase-9 plas-mid (15567), Dr. M. Sadelain for providing viral supernatant forCD19-CAR, Drs G.J. Ossenkoppele, A.A. van de Loosdrecht andS. Zweegman for critically reading the manuscript and suggestions,R. de Jong-Korlaar, M. Emmelot and L. Lubbers for technicalassistance with in vivo experiments. The RAG2-/-γc-/- mice used inthis study were originally obtained from the Amsterdam MedicalCenter, Amsterdam, the Netherlands.
E. Drent et al.
624 haematologica | 2016; 101(5)
References
1. Kyle RA, Rajkumar SV. Multiple myeloma.N Engl J Med. 2004;351(18):1860–1873.
2. Kumar SK, Lee JH, Lahuerta JJ, et al. Risk ofprogression and survival in multiple myelo-
ma relapsing after therapy with IMiDs andbortezomib: a multicenter InternationalMyeloma Working Group study. Leukemia.2012;26(1):149-157.
3. Kröger N, Damon L, Zander AR, et al.Secondary acute leukemia following mitox-antrone-based high-dose chemotherapy forprimary breast cancer patients. Bone
Marrow Transplant. 2003;32(12):1153-1157. 4. Bensinger WI, Buckner CD, Anasetti C, et al.
Allogeneic marrow transplantation for mul-tiple myeloma: an analysis of risk factors onoutcome. Blood. 1996;88(7):2787-2793.
5. Kröger N, Miyamura K, Bishop MR.Minimal residual disease following allogene-ic hematopoietic stem cell transplantation.

Biol Blood Marrow Transplant. 2011;17(1Suppl):S94-100.
6. Lokhorst HM, Schattenberg A, CornelissenJJ, et al. Donor lymphocyte infusions forrelapsed multiple myeloma after allogeneicstem-cell transplantation: predictive factorsfor response and long-term outcome. J ClinOncol. 2000;18(16):3031-3037.
7. Brentjens RJ, Davila ML, Riviere I, et al.CD19-targeted T cells rapidly induce molec-ular remissions in adults with chemothera-py-refractory acute lymphoblastic leukemia.Sci Transl Med. 2013;5(177):177ra38.
8. Grupp SA, Kalos M, Barrett D, et al.Chimeric antigen receptor–modified T cellsfor acute lymphoid leukemia. N Engl J Med.2013;368(16):1509-1518.
9. Garfall AL, Fraietta JA, Maus M V.Immunotherapy with chimeric antigenreceptors for multiple myeloma. DiscovMed. 2014;17(91):37-46.
10. Lokhorst HM, Plesner T, Laubach JP, et al.Targeting CD38 with daratumumabmonotherapy in multiple myeloma. N Engl JMed. 2015;373(13):1207-1219.
11. De Haart SJ, van de Donk NWCJ, MinnemaMC, et al. Accessory cells of the microenvi-ronment protect multiple myeloma from T-cell cytotoxicity through cell adhesion-medi-ated immune resistance. Clin Cancer Res.2013;19(20):5591-5601.
12. Groen RWJ, Noort WA, Raymakers RA, etal. Reconstructing the human hematopoieticniche in immunodeficient mice: opportuni-ties for studying primary multiple myeloma.Blood. 2012;120(3):e9-e16.
13. Zheng Z, Chinnasamy N, Morgan RA.Protein L: a novel reagent for the detectionof chimeric antigen receptor (CAR) expres-sion by flow cytometry. J Transl Med.2012;10(1):29.
14. McMillin DW, Delmore J, Weisberg E, et al.Tumor cell-specific bioluminescence plat-form to identify stroma-induced changes toanticancer drug activity. Nat Med.2010;16(4):483-489.
15. Van der Veer MS, de Weers M, van Kessel B,et al. Towards effective immunotherapy ofmyeloma enhanced elimination of myelomacells by combination of lenalidomide withthe human CD38 monoclonal antibodydaratumumab. Haematologica. 2011;96(2):284-290.
16. Spaapen RM, Groen RWJ, van denOudenalder K, et al. Eradication ofmedullary multiple myeloma by CD4+cytotoxic human T lymphocytes directed ata single minor histocompatibility antigen.Clin Cancer Res. 2010;16(22):5481-5488.
17. Malavasi F, Deaglio S, Funaro A, et al.Evolution and function of the ADP ribosylcyclase/CD38 gene family in physiologyand pathology. Physiol Rev. 2008;88(3):841-886.
18. Mutis T, Schrama E, van Luxemburg-HeijsSA, et al. HLA class II restricted T-cell reac-tivity to a developmentally regulated anti-gen shared by leukemic cells and CD34+early progenitor cells. Blood. 1997;90(3):1083-1090.
19. Miller CL, Lai B. Human and mousehematopoietic colony-forming cell assays.
Methods Mol Biol. 2005;290:71-89. 20. Straathof KC, Pulè MA, Yotnda P, et al. An
inducible caspase 9 safety switch for T-celltherapy. Blood. 2005;105(11):4247-4254.
21. Tey S-K, Dotti G, Rooney, Cliona M et al.Inducible caspase 9 suicide gene to improvethe safety of allodepleted T cells after hap-loidentical stem cell transplantation. BiolBlood Marrow Transplant. 2007;13(8):913-924.
22. Kalos M, Levine BL, Porter DL, et al. T cellswith chimeric antigen receptors have potentantitumor effects and can establish memoryin patients with advanced leukemia. SciTransl Med. 2011;3(95):95ra73.
23. Frigault MJ, Lee J, Basil MC, et al.Identification of chimeric antigen receptorsthat mediate constitutive or inducible prolif-eration of T cells. Cancer Immunol Res.2015;3(4):356-367.
24. Imai C, Mihara K, Andreansky M, et al.Chimeric receptors with 4-1BB signalingcapacity provoke potent cytotoxicity againstacute lymphoblastic leukemia. Leukemia.2004;18(4): 676-684.
25. Spaulding C, Guo W, Effros RB. Resistanceto apoptosis in human CD8+ T cells thatreach replicative senescence after multiplerounds of antigen-specific proliferation. ExpGerontol. 1999;34(5):633-644.
26. Akbar AN, Henson SM. Are senescence andexhaustion intertwined or unrelatedprocesses that compromise immunity? NatRev Immunol. 2011;11(4):289-295.
27. Mitsiades CS, Mitsiades NS, Richardson PG,Munshi NC, Anderson KC. Multiple myelo-ma: A prototypic disease model for the char-acterization and therapeutic targeting ofinteractions between tumor cells and theirlocal microenvironment. J Cell Biochem.2007;101(4):950-968.
28. Meads MB, Hazlehurst LA, Dalton WS. Thebone marrow microenvironment as a tumorsanctuary and contributor to drug resistance.Clin Cancer Res. 2008;14(9): 2519-2526.
29. Mihara K, Yanagihara K, Takigahira M, etal. Activated T-cell-mediated immunother-apy with a chimeric receptor against CD38in B-cell non-Hodgkin lymphoma. JImmnotherapy. 2009;32(7):737-743.
30. Bhattacharyya J, Mihara K, Kitanaka A, et al.T-cell immunotherapy with a chimericreceptor against CD38 is effective in eradi-cating chemotherapy-resistant B-cell lym-phoma cells overexpressing survivin inducedby BMI-1. Blood Cancer J. 2012;2(6):e75.
31. Mihara K, Bhattacharyya J, Kitanaka A, et al.T-cell immunotherapy with a chimericreceptor against CD38 is effective in elimi-nating myeloma cells. Leukemia. 2012;32(7):737-743.
32. Partida-Sánchez S, Cockayne DA, Monard S,et al. Cyclic ADP-ribose production by CD38regulates intracellular calcium release, extra-cellular calcium influx and chemotaxis in neu-trophils and is required for bacterial clearancein vivo. Nat Med. 2001;7(11):1209-1216.
33. Bean AG, Godfrey DI, Ferlin WG, et al.CD38 expression on mouse T cells: CD38defines functionally distinct subsets of alphabeta TCR+CD4-CD8- thymocytes. IntImmunol. 1995;7(2):213-221.
34. Partida-Sánchez S, Goodrich S, Kusser K, etal. Regulation of dendritic cell trafficking bythe ADP-ribosyl cyclase CD38: impact onthe development of humoral immunity.Immunity. 2004;20(3):279-291.
35. Morgan R a, Yang JC, Kitano M, et al. Casereport of a serious adverse event followingthe administration of T cells transducedwith a chimeric antigen receptor recognizingERBB2. Mol Ther. 2010;18(4):843-851.
36. Kochenderfer JN, Dudley ME, Feldman SA,et al. B-cell depletion and remissions ofmalignancy along with cytokine-associatedtoxicity in a clinical trial of anti-CD19chimeric-antigen-receptor-transduced Tcells. Blood. 2012;119(12):2709-2720.
37. Casucci M, Hawkins RE, Dotti G, BondanzaA. Overcoming the toxicity hurdles ofgenetically targeted T cells. Cancer ImmunolImmunother. 2015;64(1):123-130.
38. Maude SL, Barrett D, Teachey DT, GruppSA. Managing cytokine release syndromeassociated with novel T cell-engaging thera-pies. Cancer J. 2014;20(2):119-122.
39. Vera J, Savoldo B, Vigouroux S, et al. Tlymphocytes redirected against the kappalight chain of human immunoglobulin effi-ciently kill mature B lymphocyte-derivedmalignant cells. Blood. 2006;108(12):3890-3897.
40. Jiang H, Zhang W, Shang P, et al.Transfection of chimeric anti-CD138 geneenhances natural killer cell activation andkilling of multiple myeloma cells. MolOncol. 2014;8(2):297-310.
41. Peinert S, Prince HM, Guru PM, et al. Gene-modified T cells as immunotherapy for mul-tiple myeloma and acute myeloid leukemiaexpressing the Lewis Y antigen. Gene Ther.2010;17(5):678-686.
42. Carpenter RO, Evbuomwan MO, PittalugaS, et al. B-cell maturation antigen is a prom-ising target for adoptive T-cell therapy ofmultiple myeloma. Clin Cancer Res.2013;19(8):2048-2060.
43. Chu J, Deng Y, Benson DM, et al. CS1-spe-cific chimeric antigen receptor (CAR)-engi-neered natural killer cells enhance in vitroand in vivo antitumor activity againsthuman multiple myeloma. Leukemia.2014;28(4):917-927.
44. Chu J, He S, Deng Y, et al. Genetic modifica-tion of T cells redirected toward CS1enhances eradication of myeloma cells. ClinCancer Res. 2014;20(15):3989-4000.
45. Casucci M, Nicolis di Robilant B, Falcone L,et al. CD44v6-targeted T cells mediatepotent antitumor effects against acutemyeloid leukemia and multiple myeloma.Blood. 2013;122(20):3461-3472.
46. Hoyos V, Savoldo B, Quintarelli C, et al.Engineering CD19-specific T lymphocyteswith interleukin-15 and a suicide gene toenhance their anti-lymphoma/leukemiaeffects and safety. Leukemia. 2010;24(6):1160-1170.
47. Budde LE, Berger C, Lin Y, et al. Combininga CD20 chimeric antigen receptor and aninducible caspase 9 suicide switch toimprove the efficacy and safety of T celladoptive immunotherapy for lymphoma.PLoS One. 2013;8(12):e82742.
CD38-CART cells for multiple myeloma treatment
haematologica | 2016; 101(5) 625

626 haematologica | 2016; 101(5)
Received: August 18, 2015.
Accepted: December 23, 2015.
Pre-published: December 31, 2015.
©2016 Ferrata Storti Foundation
Check the online version for the most updatedinformation on this article, online supplements,and information on authorship & disclosures:www.haematologica.org/content/101/5/626
Material published in Haematologica is cov-ered by copyright. All rights reserved to theFerrata Storti Foundation. Copies of articlesare allowed for personal or internal use.Permission in writing from the publisher isrequired for any other use.
Correspondence: [email protected]
Natural killer cells are key cells of the innate immune system.Natural killer cell receptor repertoires are diversified by a sto-chastic expression of killer-cell-immunoglobulin-like receptors
and lectin-like receptors such as NKG2 receptors. All individuals harbora subset of natural killer cells expressing NKG2A, the inhibitory check-point receptor for HLA-E. Most neoplastic and normal hematopoieticcells express HLA-E, the inhibitory ligand of NKG2A. A novel anti-human NKG2A antibody induced tumor cell death, suggesting that theantibody could be useful in the treatment of cancers expressing HLA-E.We found that immunodeficient mice, co-infused with human primaryleukemia or Epstein-Barr virus cell lines and NKG2A+ natural killer cells,pre-treated with anti-human NKG2A, were rescued from disease pro-gression. Human NKG2A+ natural killer cells reconstituted in immuno -deficient mice after transplantation of human CD34+ cells. These naturalkiller cells are able to kill engrafted human primary leukemia or Epstein-Barr virus cell lines by lysis after intraperitoneal administration of anti-human NKG2A. Thus, this anti-NKG2A may exploit the anti-leukemicaction of the wave of NKG2A+ natural killer cells recovering afterhematopoietic stem cell transplants or adoptive therapy with naturalkiller cell infusions from matched or mismatched family donors afterchemotherapy for acute leukemia, without the need to search for a nat-ural killer cell alloreactive donor.
Effects of anti-NKG2A antibody administrationon leukemia and normal hematopoietic cellsLoredana Ruggeri,1 Elena Urbani,1 Pascale André,2 Antonella Mancusi,1Antonella Tosti,1 Fabiana Topini,1 Mathieu Bléry,2 Lucia Animobono,1François Romagné,3 Nicolai Wagtmann,2 and Andrea Velardi1
1Division of Hematology and Clinical Immunology and Bone Marrow TransplantationProgram, Department of Medicine, University of Perugia, Italy; 2Innate Pharma,Marseille, France; and 3Division of Immunology, University of Marseille, France
ABSTRACT
Ferrata StortiFoundation
EUROPEANHEMATOLOGYASSOCIATION
Haematologica 2016Volume 101(5):626-633
ARTICLE Cell Therapy & Immunotherapy
doi:10.3324/haematol.2015.135301Introduction
Natural killer (NK) cells play a critical role in host defense against infections andtumors by secreting cytokines and killing infected or transformed cells. Activationof NK-cell effector functions is regulated by activating and inhibitory receptors thatrecognize ligands on potential target cells. NK cell-mediated killing is efficientwhen target cells abundantly express stress- or transformation-induced ligands foractivating NK receptors, and few or no major histocompatibility complex (MHC)-class I molecules, which are ligands for inhibitory receptors on NK cells. In humans,a family of killer cell immunoglobulin-like receptors (KIR) bind distinct subgroupsof human leukocyte antigen (HLA) class I allotypes. KIR are clonally expressed onNK cells, creating a repertoire of NK cells with specificities for different HLA classI molecules. Due to extensive genetic polymorphisms, there are significant varia-tions in the repertoire of KIR+ NK cells among individuals in the population.Another inhibitory receptor, with broad specificity, the CD94-NKG2A complex,recognizes HLA-E, a non-classical MHC class I molecule. CD94-NKG2A and itsHLA-E ligand exhibit very limited polymorphism. CD94-NKG2A is expressed pri-marily on NK cells that do not express an inhibitory KIR for a self-HLA class I, soit fills gaps in the KIR repertoire. However, some NK cells co-express CD94-NKG2A and one or more inhibitory KIR with different MHC class I specificities.1-3
The NKG2A receptor is also expressed on T cells.

Individuals harbor NK cells in their repertoire that mayexpress, as the only inhibitory receptor, a single KIR thatis inhibited by one self-MHC class I KIR ligand. Targetcells that lack this KIR ligand do not block NK cell activa-tion, and are killed. The clinical relevance of such missingself-recognition was demonstrated in adult patients withacute myeloid leukemia (AML) and in children with acutelymphoblastic leukemias (ALL).4-9 Haploidentical stem celltransplantation from KIR ligand mismatched donors (NKalloreactive donors) was associated with a reduced risk ofrelapse and increased survival rates.4-8 Unfortunately, NKalloreactive donors cannot be identified for about 50% ofpatients who express each of the main three groups of KIRligands (HLA-C group 1 and 2 and Bw4 specificity) whichblock all the NK cells in the donor repertoire. To extendthe benefits of NK cell alloreactivity to these patientsanother strategy had to be found. A human anti-KIR mon-oclonal antibody (lirilumab) was generated to bind to allKIR2D inhibitory receptors specific for groups 1 and 2HLA-C alleles. In vitro and murine model studies showedthat lirilumab efficiently promoted NK cell alloreactivityand killing of otherwise resistant HLA-C group 1+ or group2+ targets, such as normal and tumor cells.10-13 Phase I clin-ical trials demonstrated that the anti-inhibitory KIR mAbis safe.14 Phase II clinical trials with lirilumab are ongoing.Another approach has been to generate and explore the
role of an anti-human NKG2A antibody. Every individualpossesses NKG2A+ NK cells which are always blocked byHLA-E. Since HLA-E is expressed by most normal andneoplastic hematopoietic cells, these are protected fromkilling by CD94-NKG2A+ NK cells.1-3Stem cell transplantation remains the only curative
treatment option for many patients with acute leukemia.Interestingly, in the immediate post-transplant period,most reconstituting NK cells are NKG2A+.15 Nguyen andGodal have already demonstrated in vitro that anti-NKG2Aantibody treatment is able to reconstitute NKG2A+ NK celllysis against acute leukemia cells.16,17 Administering ananti-NKG2A monoclonal antibody could strengthen manyof the benefits of NK cell alloreactivity and potentiate theanti-leukemic action of NK cells recovering afterhematopoietic transplants or of NK cell infusions frommatched or mismatched family donors without the needto search for an NK alloreactive donor. We have generated a novel, humanized anti-NKG2A
therapeutic monoclonal antibody that is being developedfor treatment of solid tumors such as ovarian cancer andhematologic malignancies. In this study, we investigatedthe potential clinical role of this new therapeutic mono-clonal antibody in vitro and in humanized mouse models.
Methods
Therapeutic anti-NKG2A monoclonal antibodyThe murine anti-human NKG2A monoclonal antibody clone
Z270 was generated and characterized as previously described.18
Details of the generation and characterization of humanized Z270will be reported elsewhere. In brief, the murine Z270 monoclonalantibody was humanized by grafting the Kabat complementaritydetermining regions onto a human acceptor framework, andexpressed in Chinese hamster ovary cells. Recombinant human-ized clones were screened to identify those that retained bindingto CD94-NKG2A with similar affinity as the original murine mon-oclonal antibody. Clones were then counter-screened on CD94-
NKG2C and CD94-NKG2E, to ensure specificity for CD94-NKG2A. The selected humanized clone, designated humZ270, orIPH2201, was expressed as an IgG4 with a single point mutationin the Fc heavy chain to prevent formation of half-antibodies.
Cell isolationAll neoplastic cells were obtained from patients’ bone marrow
aspirates or peripheral blood. All the normal lympho-hematopoi-etic cell types were obtained from healthy donors. Patients anddonors provided prior written informed consent to the use of theirbiological material in accordance with the Declaration of Helsinki. Neoplastic cells (if >95% of all cells) were obtained from periph-
eral blood or marrow samples after Ficoll-Hypaque gradient sepa-ration.Human T and B cells and monocytes were purified from periph-
eral blood mononuclear cells on a Ficoll-Hypaque gradient andenriched by human T and B isolation kits or anti-CD14+
microbeads, respectively, and immunomagnetic selection(Miltenyi Biotec, Bergisch Gladbach, Germany). Dendritic cellswere obtained as described elsewhere.19
Human NK cells were purified from peripheral blood mononu-clear cells on a Ficoll-Hypaque gradient, then enriched by a humanNK isolation kit and immunomagnetic selection (Miltenyi Biotec).Single KIR+/NKG2A- NK cells were cloned and used as controls forNK cell alloreactivity assay as previously described.7 NKG2A+/KIR-
NK cells were depleted of KIR2DL1/2/3+ and KIR3DL1+ cells usinganti-KIR2DL2/L3/S2 (clone CH-L, IgG2b) (BD Biosciences SanJosé, CA, USA), anti-KIR2DL1 (clone #143211, IgG1) (R&DSystems Inc., Minneapolis, MN, USA) and KIR3DL1 (MiltenyiBiotech) phycoerythrin (PE)-conjugated monoclonal antibodiesand negative selection by anti-PE immunomagnetic microbeads(Miltenyi Biotech). NKG2A+/KIR- NK cells were stimulated by 1%phytohemagglutinin (Biochrom, Berlin, Germany) and 250 IU/mLinterleukin-2 (Novartis Farma S.p.A., Origgio, Italy), and expandedfor up to 7 days. At the end of culture, before their use, the finalpurity of the NKG2A+ NK cells was >95%.CD34+ stem cells were obtained from healthy donors’ peripher-
al blood after mobilization with granulocyte colony-stimulatingfactor, leukapheresis and positive selection by immunomagneticmicrobeads conjugated with anti-human CD34+ monoclonal anti-body (Miltenyi Biotec).
Epstein-Barr virus cell linesHLA-E+ Epstein-Barr virus (EBV)-transformed B-cell lines, which
were resistant to NKG2A+ NK cell lysis, were a kind gift from theEuropean Collection for Biomedical Research (ECBR). Anti-humanHLA-E-PE (IgG1, clone 3D12, eBioscience, San Diego, CA, USA)was used to estimate HLA-E expression on EBV cell lines and allthe other normal and neoplastic human hematopoietic cells byflow cytometry.
In vitro cytotoxicity assaysNKG2A+/KIR- NK cells were pre-treated with humanized anti-
human NKG2A antibody or with an isotype control antibody (10μg/1x106 cells/mL). Single KIR+/NKG2A- and KIR-/NKG2A+ NKcells were screened for alloreactivity by standard 51Cr release cyto-toxicity assays at an increasing effector-to-target (E:T) ratio (from1:1 to 20:1) against KIR ligand mismatched HLA-E+ B and T cells,monocytes, dendritic cells, EBV cell lines, chronic lymphaticleukemia (CLL) cells, T-cell ALL, AML and multiple myeloma(MM) cells.
Mouse modelsColonies of non-obese diabetic - severe combined immunodefi-
ciency (NOD-SCID) mice and NOD-scidIL2rgtm (NSG) mice
Antileukemic effect of anti-human NKG2A antibody
haematologica | 2016; 101(5) 627

were bred at the University of Perugia Animal House. Breederswere obtained from Jackson Laboratory (Bar Harbor, Maine,USA).All experiments were performed in accordance with the
National Ethic Approval Document for animal experimentation. Female 10-week old mice were irradiated with 3.5 Gy. The next
day NOD-SCID mice received an intravenous co-infusion of pri-mary AML cells (12x106) or EBV-transformed B-cell line (12x106)and NKG2A+ non-alloreactive, interleukin-2-activated NK cells(1x106) that had been pre-treated with anti-human NKG2A mon-oclonal antibody (10 μg/1x106 cells/mL) at the E:T ratio of 1:12.Isotype control antibody-pretreated NK cells were infused in con-trol mice at the E:T ratio of 1:12.Mice that succumbed to leukemia or EBV lymphoproliferative
disease were assessed for AML or EBV organ infiltration by flowcytometry analysis with a specific panel of anti–human mono-clonal antibodies which previously characterized the neoplasticcells (see below). In a model of engrafted disease, we infused the same mouse
strain with AML or EBV cell lines. When bone marrow engraft-ment was around 20-30%, mice were given escalating doses ofinterleukin-2-activated NKG2A+ NK cells that had been pre-treat-ed with anti-human NKG2A monoclonal antibody (10 μg/1x106
cells/mL) (from 1 to 10x106 per mouse, intravenously). Mice thatdied of leukemia or lymphoma were assessed for AML or EBVorgan infiltration by flow cytometry analysis using a specific panelof anti-human monoclonal antibodies (see below).In other mouse models, the day after irradiation, female 10-
week old NSG mice were given 10x106 human CD34+ hematopoi-etic stem cells intravenously. At day 20 mice were infused intra-venously with 5x106 HLA-E+ EBV cells or AML cells. When CD34+
stem cells had differentiated into CD56+/CD3-/NKG2A+ NK cells,mice received an intraperitoneal administration of 200, 250 or 300µg anti-human NKG2A monoclonal antibody. Control mice wereleft untreated or treated with the same doses of isotype controlantibody.From day 40 onwards mice were evaluated for EBV or AML
engraftment with a combination of anti-human CD45 monoclonalantibody and monoclonal antibody specific for AML or the EBVcell line (anti-CD20, anti-CD19, anti-IgM, anti-kappa, anti-lamb-da, anti-CD23, anti-CD3, anti-CD33, anti-CD34, anti-CD56, anti-CD117, anti-CD8, CD4, CD34 monoclonal antibodies,eBioscience). Mice that succumbed to EBV lymphoproliferative disease or
leukemia were assessed for EBV or AML organ infiltration by flowcytometry analysis with a combination of anti-human monoclonalantibodies (see above). Mice that survived were sacrificed after100 days, and tumor organ infiltration analyzed with the sameantibody combination.
Statistical analysesThe Student t test was used to compare variables and was
applied by Graphpad Prism 5. The Kaplan-Meier method wasused to evaluate murine survival. All P values are two-sided andconsidered statistically significant at P values <0.05.
Results
In vitro treatment with anti-human NKG2A antibodytriggers NKG2A+ natural killer cell lysis of HLA-E+
hematopoietic lineage targets
In order to assess the susceptibility of normal and neo-plastic hematopoietic lineage targets to alloreactive NK
cell lysis, we generated single inhibitory KIR+NKG2A- NKcell clones and evaluated their ability to kill KIR ligand-lacking targets such as B and T cells, monocytes, dendriticcells, EBV cell lines, CLL, T-ALL, AML and MM cells.These normal and neoplastic hematopoietic lineage cellsexpressed HLA-E and were resistant to NKG2A+ NK cells.Figure 1 shows that most acute leukemias express HLA-E.All HLA-E+ lympho-hematopoietic cell types were targetsof alloreactive NK cell killing when they did not expressthe appropriate inhibitory KIR ligand for the singleinhibitory KIR receptor expressed by alloreactive NK cellclones (Figure 2A). We pre-treated NKG2A+ NK cells withanti-human NKG2A antibody and assessed their ability tokill otherwise resistant HLA-E+ hematopoietic lineagecells. Treatment with anti-NKG2A monoclonal antibodyconverted NKG2A+KIR- NK cells into cells that were func-tionally “alloreactive” against HLA-E+ lympho-hematopoi-etic cells i.e. killed B and T cells, monocytes, dendriticcells, EBV cell lines, CLL, T-ALL, AML and MM cells. Themost effective lysis was obtained with an E:T ratio of 15:1(Figure 2B). Each cytotoxicity assay was repeated with three targets
for each category of cells and the mean ± standard devia-tion is shown.
In vivo treatment with the anti-human NKG2A antibodyeradicates HLA-E+ leukemia and lymphomaIn order to evaluate the in vivo efficacy of anti-NKG2A
monoclonal antibody at triggering NKG2A+ NK cells to killneoplastic cells, we developed xenogenic murine modelsof human neoplastic disease. NOD-SCID mice thatreceived the HLA-E+ EBV-cell lines or AML cells died ofhigh-grade lymphoma or AML. In these mice, co-infusionof human NKG2A+ non-alloreactive NK cells did not pre-vent engraftment of EBV or AML cells and mice died ofthe diseases. In contrast, infusion of NKG2A+ NK cells that had been
pre-treated with anti-NKG2A monoclonal antibody, pre-vented engraftment of human EBV cell lines and AML cellsand mice survived without symptoms or signs of tumorlocalization (Figure 3A). In fact, mice were sacrificed 100days after cell infusion and cytofluorimetric analysis con-firmed the absence of neoplastic infiltration. We pooledresults from eight experiments with four mice per groupfor each experiment.NKG2A+ NK cell elimination of engrafted human AML
or human EBV cell lines was evaluated in escalating doseexperiments. At least 3x106 NKG2A+ NK cells per mouse,pre-treated with anti-NKG2A, were necessary to rescue80% mice (Figure 3B). Repeating intraperitoneal doses ofantibody did not improve the results because of autolo-gous NK cell killing (fratricide effect).In order to assess the ability of endogenously generated
NKG2A+ NK cells to cure leukemia or lymphoma, wetransplanted NSG mice with 10x106 human CD34+
hematopoietic cells. The transplanted CD34+ hematopoi-etic stem cells differentiated into various hematopoieticlineage cells, including NKG2A+ NK cells.20 Twenty daysafter the CD34+ cell infusion, mice received HLA-E+ EBVcell lines or AML tumor cells. On day 30 after the CD34+
cell infusion, when the numbers of NKG2A+ NK cellsreached a plateau value in the bone marrow and spleen,we treated three groups of mice with 200, 250 or 300 µgof the anti-NKG2A monoclonal antibody. Control micetreated with isotype control monoclonal antibody, and
L. Ruggeri et al.
628 haematologica | 2016; 101(5)
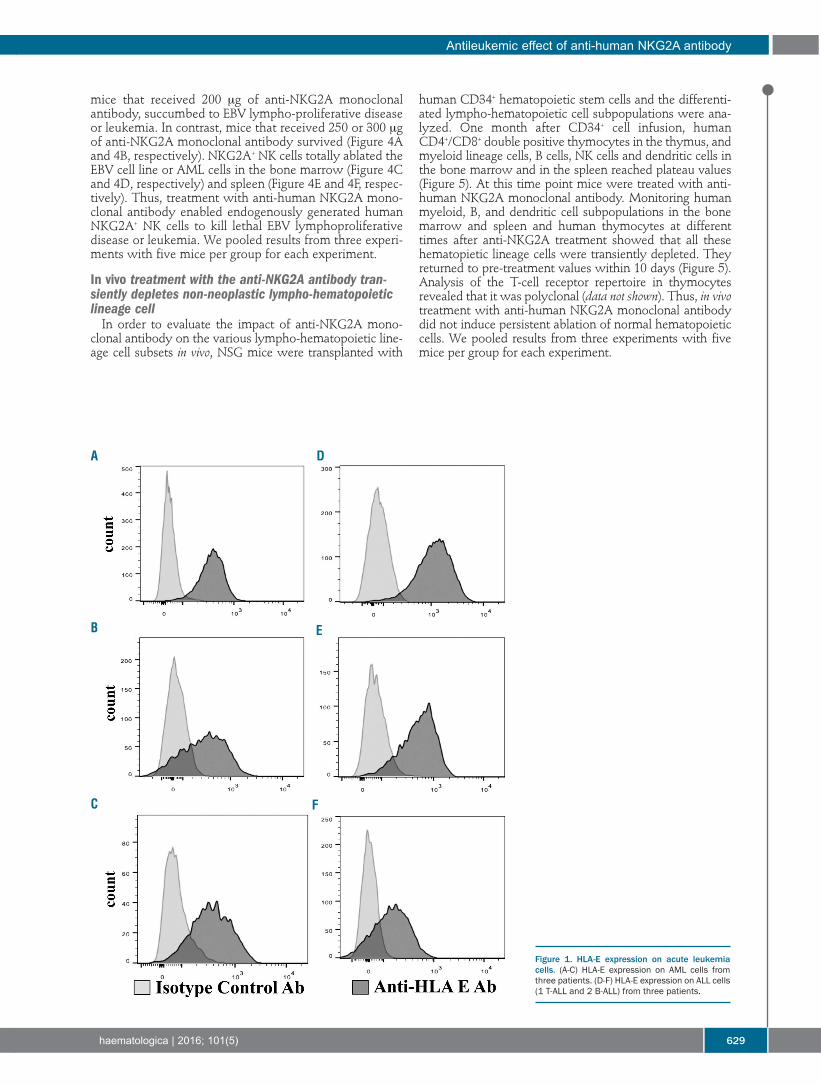
mice that received 200 μg of anti-NKG2A monoclonalantibody, succumbed to EBV lympho-proliferative diseaseor leukemia. In contrast, mice that received 250 or 300 μgof anti-NKG2A monoclonal antibody survived (Figure 4Aand 4B, respectively). NKG2A+ NK cells totally ablated theEBV cell line or AML cells in the bone marrow (Figure 4Cand 4D, respectively) and spleen (Figure 4E and 4F, respec-tively). Thus, treatment with anti-human NKG2A mono-clonal antibody enabled endogenously generated humanNKG2A+ NK cells to kill lethal EBV lymphoproliferativedisease or leukemia. We pooled results from three experi-ments with five mice per group for each experiment.
In vivo treatment with the anti-NKG2A antibody tran-siently depletes non-neoplastic lympho-hematopoieticlineage cellIn order to evaluate the impact of anti-NKG2A mono-
clonal antibody on the various lympho-hematopoietic line-age cell subsets in vivo, NSG mice were transplanted with
human CD34+ hematopoietic stem cells and the differenti-ated lympho-hematopoietic cell subpopulations were ana-lyzed. One month after CD34+ cell infusion, humanCD4+/CD8+ double positive thymocytes in the thymus, andmyeloid lineage cells, B cells, NK cells and dendritic cells inthe bone marrow and in the spleen reached plateau values(Figure 5). At this time point mice were treated with anti-human NKG2A monoclonal antibody. Monitoring humanmyeloid, B, and dendritic cell subpopulations in the bonemarrow and spleen and human thymocytes at differenttimes after anti-NKG2A treatment showed that all thesehematopietic lineage cells were transiently depleted. Theyreturned to pre-treatment values within 10 days (Figure 5).Analysis of the T-cell receptor repertoire in thymocytesrevealed that it was polyclonal (data not shown). Thus, in vivotreatment with anti-human NKG2A monoclonal antibodydid not induce persistent ablation of normal hematopoieticcells. We pooled results from three experiments with fivemice per group for each experiment.
Antileukemic effect of anti-human NKG2A antibody
haematologica | 2016; 101(5) 629
Figure 1. HLA-E expression on acute leukemiacells. (A-C) HLA-E expression on AML cells fromthree patients. (D-F) HLA-E expression on ALL cells(1 T-ALL and 2 B-ALL) from three patients.
A D
E
FC
B
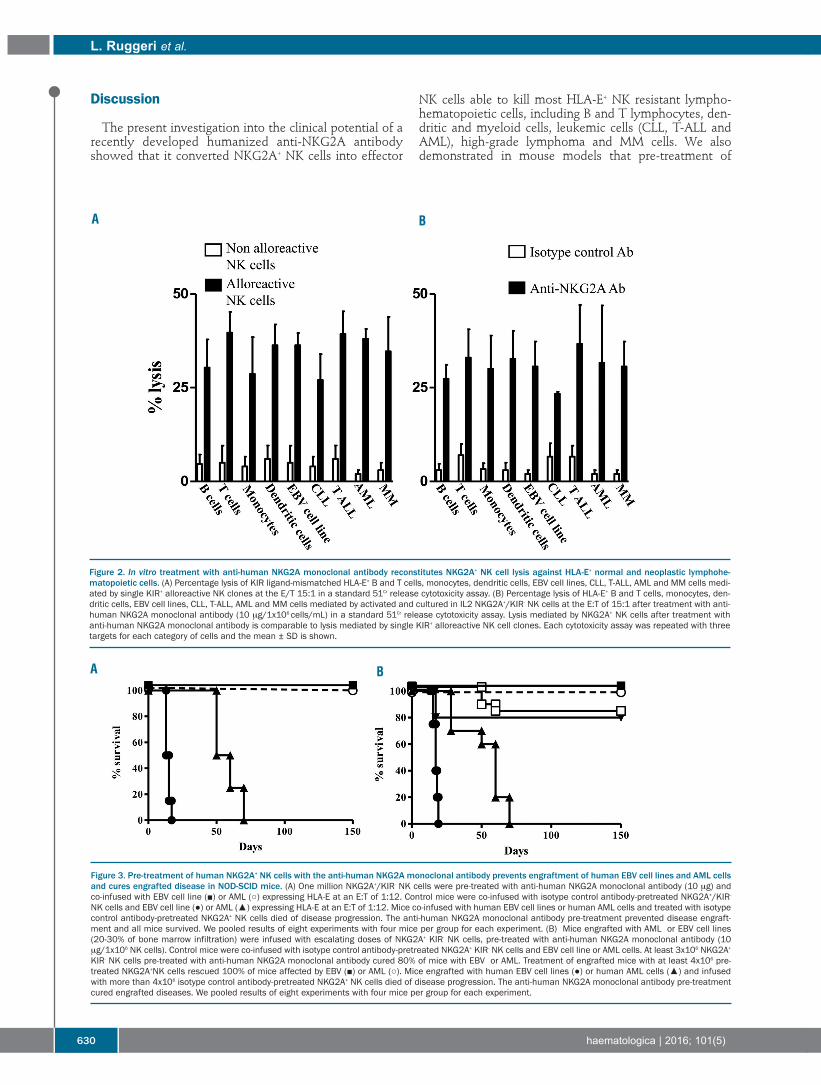
Discussion
The present investigation into the clinical potential of arecently developed humanized anti-NKG2A antibodyshowed that it converted NKG2A+ NK cells into effector
NK cells able to kill most HLA-E+ NK resistant lympho-hematopoietic cells, including B and T lymphocytes, den-dritic and myeloid cells, leukemic cells (CLL, T-ALL andAML), high-grade lymphoma and MM cells. We alsodemonstrated in mouse models that pre-treatment of
L. Ruggeri et al.
630 haematologica | 2016; 101(5)
A B
Figure 3. Pre-treatment of human NKG2A+ NK cells with the anti-human NKG2A monoclonal antibody prevents engraftment of human EBV cell lines and AML cellsand cures engrafted disease in NOD-SCID mice. (A) One million NKG2A+/KIR- NK cells were pre-treated with anti-human NKG2A monoclonal antibody (10 μg) andco-infused with EBV cell line (■) or AML (○) expressing HLA-E at an E:T of 1:12. Control mice were co-infused with isotype control antibody-pretreated NKG2A+/KIR-
NK cells and EBV cell line (●) or AML (▲) expressing HLA-E at an E:T of 1:12. Mice co-infused with human EBV cell lines or human AML cells and treated with isotypecontrol antibody-pretreated NKG2A+ NK cells died of disease progression. The anti-human NKG2A monoclonal antibody pre-treatment prevented disease engraft-ment and all mice survived. We pooled results of eight experiments with four mice per group for each experiment. (B) Mice engrafted with AML or EBV cell lines(20-30% of bone marrow infiltration) were infused with escalating doses of NKG2A+ KIR- NK cells, pre-treated with anti-human NKG2A monoclonal antibody (10μg/1x106 NK cells). Control mice were co-infused with isotype control antibody-pretreated NKG2A+ KIR- NK cells and EBV cell line or AML cells. At least 3x106 NKG2A+
KIR- NK cells pre-treated with anti-human NKG2A monoclonal antibody cured 80% of mice with EBV or AML. Treatment of engrafted mice with at least 4x106 pre-treated NKG2A+NK cells rescued 100% of mice affected by EBV (■) or AML (○). Mice engrafted with human EBV cell lines (●) or human AML cells (▲) and infusedwith more than 4x106 isotype control antibody-pretreated NKG2A+ NK cells died of disease progression. The anti-human NKG2A monoclonal antibody pre-treatmentcured engrafted diseases. We pooled results of eight experiments with four mice per group for each experiment.
A B
Figure 2. In vitro treatment with anti-human NKG2A monoclonal antibody reconstitutes NKG2A+ NK cell lysis against HLA-E+ normal and neoplastic lymphohe-matopoietic cells. (A) Percentage lysis of KIR ligand-mismatched HLA-E+ B and T cells, monocytes, dendritic cells, EBV cell lines, CLL, T-ALL, AML and MM cells medi-ated by single KIR+ alloreactive NK clones at the E/T 15:1 in a standard 51Cr release cytotoxicity assay. (B) Percentage lysis of HLA-E+ B and T cells, monocytes, den-dritic cells, EBV cell lines, CLL, T-ALL, AML and MM cells mediated by activated and cultured in IL2 NKG2A+/KIR- NK cells at the E:T of 15:1 after treatment with anti-human NKG2A monoclonal antibody (10 μg/1x106 cells/mL) in a standard 51Cr release cytotoxicity assay. Lysis mediated by NKG2A+ NK cells after treatment withanti-human NKG2A monoclonal antibody is comparable to lysis mediated by single KIR+ alloreactive NK cell clones. Each cytotoxicity assay was repeated with threetargets for each category of cells and the mean ± SD is shown.

NKG2A+ NK cells with anti-human NKG2A monoclonalantibody prevented engraftment of otherwise lethal EBVcell lines or AML cells.Interestingly, the repertoire of each individual expresses
a certain percentage of NKG2A+ NK cells and, afterhematopoietic stem cell transplantation, a large popula-tion of reconstituting NK cells express the CD94-NKG2Ainhibitory receptor.15 Consequently, the use of humanizedanti-NKG2A antibody could enlarge the NK cell popula-tion that exerts an anti-tumor effect to the benefit ofpatients with hematologic malignancies. Potential side effects such as autoreactivity against
hematopoietic stem cells and subsequent cytopenia coulddevelop, particularly after transplantation. To test thishypothesis, we transplanted mice with human CD34+stem cells and then leukemic cells, which engraftedbecause the stem cells could not develop into mature Tcells or alloreactive single KIR+NKG2A- NK cells.20 Humanhematopoietic stem cells could, however, develop intoNKG2A+ NK cells.20 Anti-NKG2A antibody treatmentreconstituted NKG2A+ NK cell-mediated lysis of HLA-E+
engrafted leukemic cells, rescuing mice from death. The
side effects appear slight as cytopenia of normalhematopoietic cells was transient and mice recoveredquickly. The slight, transient cytopenia in the committedmyeloid line may be due to either alloreactive NK cell frat-ricide or to CD34+ cell conservation. In fact, recurrent dos-ing does not seem to reduce the number of CD34+ cells asengraftment was always successful (data not shown). Onemight hypothesize that they are not a target of alloreactiveNK cells.Interestingly these in vitro and in vivo results are in accor-
dance with previous findings that lirilumab bound to allKIR2D inhibitory receptors for groups 1 and 2 HLA-C alle-les and blocked NK cell inhibitory recognition of self-HLA-C. It activated NK cell killing in vivo, eradicating tumors inmice.10-13 In fact, clinical trials of this fully human anti-KIRantibody as a single agent are ongoing in patients withacute leukemia.14We might hypothesize about using the humanized anti-
NKG2A antibody as an alternative to chemotherapy.Some studies demonstrated safety and a promising clinicalrole of haploidentical alloreactive NK cell infusions incombination with chemotherapy for the treatment of eld-
Antileukemic effect of anti-human NKG2A antibody
haematologica | 2016; 101(5) 631
Figure 4. In vivo treatment with the anti-human NKG2A monoclonal antibody rescues NSG mice engrafted with human CD34+ hematopoietic stem cells and HLA-E+ human AML cells or an EBV cell line. After 3.5 Gy total body irradiation, mice were infused with 10x106 human CD34+ hematopoietic stem cells. After 20 daysthey were infused with an EBV cell line or AML cells. When NKG2A+ NK cells differentiated from CD34+ cells, mice were treated with anti-human NKG2A monoclonalantibody. Mice that received 250 μg (○) or 300 μg (●) of anti-human NKG2A monoclonal antibody survived, control mice (isotype control antibody) (■) or mice thatreceived 200 μg of the antibody (▲) succumbed to EBV lympho-proliferative disease (A) or AML (B). NKG2A+ NK cells ablated the EBV cell line in bone marrow* (C)and spleen (E) and AML cells in bone marrow* (D) and spleen (F). The normal human CD45+ hematopoietic population, which developed from CD34+ cells, was tran-siently depleted after administration of human anti-NKG2A antibody. We pooled results of three experiments with five mice per group for each experiment. * Bonemarrow cell numbers are from two femurs per mouse.
A C
B D F
E

erly or pediatric patients with high-risk acuteleukemias.22,23 We speculate that humanized anti-NKG2Amay be useful in similar settings, in order to reconstitutelysis by NKG2A+ NK cells obtained from non-alloreactivehaploidentical or identical donors. The role of the NKG2A receptor in autoimmune dis-
eases is controversial. Activated NK cells with NKG2Adown-regulation may play a role in the pathogenesis ofpsoriasis.24 However, since reconstituted NK cell lysis bymeans of the anti-NKG2A antibody is also directedagainst activated autologous T and B cells which mediateautoimmune diseases, the antibody might also be envis-aged as therapy against human autoimmune diseases. In amurine model of rheumatoid arthritis, an anti-murineNKG2A (Fab) antibody selectively increased lysis of autol-ogous TH17 and TFH cells, which are the mediators ofrheumatoid arthritis. The antibody blockade of theinhibitory interaction between the NKG2A receptor andits Qa-1 ligand enhanced the NK cell-dependent elimina-
tion of pathogenic T cells, resulting in blockade of diseaseonset or progression.25In vitro and in vivo findings suggest that the humanized
anti-NKG2A antibody described here constitutes aunique, relatively safe, therapeutic approach to malignanthematologic and autoimmune diseases. Phase I/II clinicaltrials with anti-human NKG2A antibody are ongoing inpatients with tumor types known to express HLA-E,including CLL (ClinicalTrials.gov :NCT02557516), head andneck cancer (ClinicalTrials.gov: NCT02331875) and ovariancancer (ClinicalTrials.gov NCT02459301)26 in order to vali-date the present observations and provide hope for those50% of patients with hematologic and solid malignancieswho cannot find alloreactive NK cell donors.
Acknowledgments The authors thank Dr Geraldine Anne Boyd for editorial assis-
tance. LR is a Leukemia and Lymphoma Society Scholar inClinical Research.
L. Ruggeri et al.
632 haematologica | 2016; 101(5)
Figure 5. Treatment with the anti-human NKG2A monoclonal antibody transiently depleted HLA-E+ autologous myeloid, B, T, NK and DC subpopulations in NSGmice engrafted with human CD34+ hematopoietic stem cells. After 3.5 Gy total body irradiation, mice were infused with human CD34+ hematopoietic stem cells.One month after, when NKG2A+ NK cells differentiated from CD34+ cells reached a plateau value, mice were treated with 300 μg of anti-human NKG2A monoclonalantibody. Transient depletion of human myeloid, B, dendritic, and NK cell subpopulations in the bone marrow* (A) and spleen (B) and double negative (DN), singleCD8+ or CD4+, CD4+/CD8+ double positive (DP) thymocytes (C) was followed by recovery of all cell subsets within 10 days. We pooled results of three experimentswith five mice per group for each experiment. *Bone marrow cell numbers are from two femurs per mouse.
A B C
References
1. Vivier E, Raulet DH, Moretta A, et al.Innate or adaptive immunity? The exampleof natural killer cells. Science. 2011;331(6013):44-49.
2. Caligiuri MA. Human natural killer cells.Blood. 2008;112(3):461-469.
3. Moretta L, Moretta A. Unravelling naturalkiller cell function: triggering and inhibitoryhuman NK receptors. EMBO J. 2004;23(2):255-259.
4. Ruggeri L, Capanni M, Casucci M, et al.Role of natural killer cell alloreactivity inHLA-mismatched hematopoietic stem celltransplantation. Blood. 1999;94(1):333-339.
5. Ruggeri L, Capanni M, Urbani E, et al.Effectiveness of donor natural killer cellalloreactivity in mismatched hematopoietictransplants. Science. 2002;295(5562):2097-2100.
6. Karre K. A perfect mismatch. [commen-tary] Science. 2002;295(5562):2029-2031.
7. Ruggeri L, Mancusi A, Capanni M, et al.Donor natural killer cell allorecognition ofmissing self in haploidentical hematopoiet-ic transplantation for acute myeloidleukemia: challenging its predictive value.Blood. 2007;110(1):433-440.
8. Velardi A, Ruggeri L, Mancusi A, Aversa F,Christiansen FT. Natural killer cellallorecognition of missing self in allogeneichematopoietic transplantation: a tool for
immunotherapy of leukemia. Curr OpinImmunol. 2009;21(5):525-530.
9. Pende D, Marcenaro S, Falco M, et al. Anti-leukemia activity of alloreactive NK cells inKIR ligand-mismatched haploidenticalHSCT for pediatric patients: evaluation ofthe functional role of activating KIR andredefinition of inhibitory KIR specificity.Blood. 2009;113(13):3119-3129.
10. Romagne F, Andre P, Spee P, et al. Pre-clin-ical characterization of 1-7F9, a novelhuman anti-KIR therapeutic antibody thataugments NK-mediated killing of tumorcells. Blood. 2009;114(13):2667-2677.
11. Sola C, André P, Lemmers C, et al. Geneticand antibody-mediated reprogramming ofnatural killer cell missing-self recognition in

vivo. Proc Natl Acad Sci USA. 2009;106(31):12879-12884.
12. Benson DM, Jr Bakan CE, Zhang S, et al.IPH2101, a novel anti-inhibitory KIR anti-body, and lenalidomide combine toenhance the natural killer cell versus multi-ple myeloma effect. Blood. 2011;118(24):6387-6391.
13. Kohrt HE, Thielens A, Marabelle A, et al.Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells asmonotherapy and in combination withanti-CD20 antibodies. Blood. 2014;123(5):678-686.
14. Vey N, Bourhis JH, Boissel N, et al. A phase1 trial of the anti-inhibitory KIR mAbIPH2101 for AML in complete remission.Blood. 2012;120(22):4317-4323.
15. Stern M, De Angelis C, Urbani E, et al.Natural killer cell KIR repertoire reconstitu-tion after haploidentical stem cell trans-plantation. Bone Marrow Transplant. 2010;45(11):1607-1610.
16. Godal R, Bachanova V, Gleason M, et al.Natural killer cell killing of acute myeloge-nous leukemia and acute lymphoblasticleukemia blasts by killer cell immunoglob-ulin-like receptor-negative natural killercells after NKG2A and LIR-1 blockade. Biol
Blood Marrow Transplant. 2010;16(5):612-621.
17. Nguyen S, Dhedin N, Vernant JP, et al. NK-cell reconstitution after haploidenticalhematopoietic stem-cell transplantations:immaturity of NK cells and inhibitoryeffect of NKG2A override GvL effect.Blood. 2005;105(10):4135-4142.
18. Moretta A, Vitale M, Sivori S, et al. Humannatural killer cell receptors for HLA-class Imolecules. Evidence that the Kp43 (CD94)molecule functions as receptor for HLA-Balleles. J Exp Med. 1994;180(2):545-555.
19. Mancusi A, Ruggeri L, Urbani E, et al.Haploidentical hematopoietic transplanta-tion from KIR ligand-mismatched donorswith activating KIRs reduces non relapsemortality. Blood. 2015;125(20):3173-82.
20. André MC, Erbacher A, Gille C, et al. Long-term human CD34+ stem cell-engraftednonobese diabetic/SCID/IL-2R gamma(null)mice show impaired CD8+ T cell mainte-nance and a functional arrest of immatureNK cells. J Immunol. 2010;185(5):2710-2720.
21. Miller JS, Soignier Y, Panoskaltsis-MortariA, et al. Successful adoptive transfer and invivo expansion of human haploidenticalNK cells in patients with cancer. Blood.2005;105(8):3051-3057.
22. Rubnitz JE, Inaba H, Ribeiro RC, et al.NKAML: a pilot study to determine thesafety and feasibility of haploidentical nat-ural killer cell transplantation in childhoodacute myeloid leukemia. J Clin Oncol.2010;28(6):955-959.
23. Curti A, Ruggeri L, D'Addio A, et al.Successful transfer of alloreactive hap-loidentical KIR ligand-mismatched naturalkiller cells after infusion in elderly high riskacute myeloid leukemia patients. Blood.2011;118(12):3273-3279.
24. Son SW, Kim EO, Ryu ES, et al.Upregulation of Fas and downregulation ofCD94⁄NKG2A inhibitory receptors on cir-culating natural killer cells in patients withnew-onset psoriasis. Br J Dermatol.2009;161(2):281-288.
25. Leavenworth JW, Wang X, Wenander CS,Spee P, Cantor H. Mobilization of naturalkiller cells inhibits development of colla-gen-induced arthritis. Proc Natl Acad SciUSA. 2011;108(35):14584-14589.
26. Seymour L, Tinker A, Hirte H, WagtmannN, Dodion P. Phase I and dose ranging,phase II studies with IPH2201, a human-ized monoclonal antibody targeting HLA-Ereceptor CD94/NKG2A. Ann Oncol.2015;26 (Suppl. 2):ii3-ii5.
Antileukemic effect of anti-human NKG2A antibody
haematologica | 2016; 101(5) 633

634 haematologica | 2016; 101(5)
Received: October 27, 2015.
Accepted: February 5, 2016.
Pre-published: February 11, 2016.
©2016 Ferrata Storti Foundation
Check the online version for the most updatedinformation on this article, online supplements,and information on authorship & disclosures:www.haematologica.org/content/101/634
Material published in Haematologica is cov-ered by copyright. All rights reserved to theFerrata Storti Foundation. Copies of articlesare allowed for personal or internal use.Permission in writing from the publisher isrequired for any other use.
Correspondence: [email protected]
Ferrata StortiFoundation
EUROPEANHEMATOLOGYASSOCIATION
Haematologica 2016Volume 101(5):634-643
ARTICLE Stem Cell Transplantation
doi:10.3324/haematol.2015.138594
Reduced intensity haplo plus single cord transplant compared to double cord transplant:improved engraftment and graft-versus-host disease-free, relapse-free survivalKoen van Besien,1 Parameswaran Hari,2 Mei-Jie Zhang,2 Hong-Tao Liu,3 WendyStock,3 Lucy Godley,3 Olatoyosi Odenike,3 Richard Larson,3 Michael Bishop,3Amittha Wickrema,3 Usama Gergis,1 Sebastian Mayer,1 Tsiporah Shore,1Stephanie Tsai,1 Joanna Rhodes,1 Melissa M. Cushing,4 Sandra Korman,2 andAndrew Artz1
1Department of Hematology/Oncology and Meyer Cancer Center – Stem Cell TransplantProgram, Weill Cornell Medical College, New York, NY; 2Center for International BoneMarrow Transplant Research, Medical College of Wisconsin, Milwaukee, WI; 3Section ofHematology/Oncology-Hematopoietic Cellular Therapy Program, University of Chicago, Il;and 4Department of Pathology – Cellular Therapy Laboratory, Weill Cornell MedicalCollege, New York, NY, USA
Umbilical cord blood stem cell transplants are commonly used inadults lacking HLA-identical donors. Delays in hematopoieticrecovery contribute to mortality and morbidity. To hasten recov-
ery, we used co-infusion of progenitor cells from a partially matched relat-ed donor and from an umbilical cord blood graft (haplo-cord transplant).Here we compared the outcomes of haplo-cord and double-cord trans-plants. A total of 97 adults underwent reduced intensity conditioning fol-lowed by haplo-cord transplant and 193 patients received reduced inten-sity conditioning followed by double umbilical cord blood transplanta-tion. Patients in the haplo-cord group were more often from minoritygroups and had more advanced malignancy. Haplo-cord recipientsreceived fludarabine-melphalan-anti-thymocyte globulin. Double umbili-cal cord blood recipients received fludarabine-cyclophosphamide andlow-dose total body irradiation. In a multivariate analysis, haplo-cord hadfaster neutrophil (HR=1.42, P=0.007) and platelet (HR=2.54, P<0.0001)recovery, lower risk of grade II-IV acute graft-versus-host disease(HR=0.26, P<0.0001) and chronic graft-versus-host disease (HR=0.06,P<0.0001). Haplo-cord was associated with decreased risk of relapse (HR0.48, P=0.001). Graft-versus-host disease-free, relapse-free survival wassuperior with haplo-cord (HR 0.63, P=0.002) but not overall survival(HR=0.97, P=0.85). Haplo-cord transplantation using fludarabine-melpha-lan-thymoglobulin conditioning hastens hematopoietic recovery with alower risk of relapse relative to double umbilical cord blood transplanta-tion using the commonly used fludarabine-cyclophosphamide-low-dosetotal body irradiation conditioning. Graft-versus-host disease-free andrelapse-free survival is significantly improved. Haplo-cord is a readilyavailable graft source that improves outcomes and access to transplant forthose lacking HLA-matched donors. Trials registered at clinicaltrials.govidentifiers 00943800 and 01810588.
ABSTRACT
Introduction
Allogeneic transplantation with HLA-identical donors is an effective and poten-tially curative therapy for hematologic malignancies. Limited availability of HLA-identical donors, particularly in patients from under-represented minority groups,

has generated interest in transplantation using mis-matched unrelated umbilical cord blood (UCB) stem cells.The promise of cord blood transplantation resides in itsability to provide a source of stem cells that can engraftacross HLA barriers with low rates of graft-versus-host dis-ease (GvHD) and exert potent graft-versus-leukemia (GvL)effects, possibly mediated by contaminating maternalcells.1-5 But cord blood transplantation is hampered by thelow progenitor cell doses in the grafts, and hence oftenvery delayed recovery of neutrophils and platelets, partic-ularly in adult recipients.6,7 This in turn leads to prolongedhospitalization, expense, morbidity and early mortality.Though smaller studies have shown encouraging results,8,9a recent study found that the outcomes of cord bloodtransplantation in older adults were inferior to those of 8/8matched unrelated donor transplant recipients, mostlybecause of increased early treatment-related mortality.7Several recent studies have been conducted to improve
hematopoietic recovery after umbilical cord blood trans-plantation in adults in order to reduce early morbidity andmortality, and possibly health care utilization. DoubleUCB transplantation is perhaps the most commonly usedof these procedures. But in a recently reported random-ized study in pediatric patients, it was not associated withimproved outcomes relative to single cord transplant.10We and others have investigated an alternative strategy:
the use of third-party CD34 selected adult haplo-identicalstem cells to supplement a single UCB stem cell graft.11-15In an initial report using a reduced intensity conditioningapproach, we showed encouraging rates of engraftmentand of long-term outcome.16 We also showed how the ini-tial engraftment of the haplo-identical stem cells was, inthe large majority of cases, ultimately superseded by theoutgrowth of UCB cells. Since then, more than 150 addi-tional such transplants have been performed at two insti-tutions in the US, where they have become the preferredform of alternative donor transplantation. Here we con-ducted a formal comparison with patients undergoingreduced intensity conditioning and double UCB transplan-tation. The comparison group consisted of adult doubleUCB blood transplant recipients who had received themost widely used reduced intensity conditioning regimen.Trials were registered at clinicaltrials.gov identifiers 00943800and 01810588.
Methods
Patients and controlsIn 2007, a prospective study was initiated at the University of
Chicago for haplo-cord transplantation following reduced intensi-ty conditioning (clinicaltrials.gov identifier 00943800). As of 2012,this was followed by a joint prospective study of reduced intensityconditioning conducted by Weill Cornell Medical College andUniversity of Chicago (clinicaltrials.gov identifier 01810588). The pri-mary objective of the latter study was to define the optimal celldose of the umbilical cord blood graft for haplo-cord transplanta-tion, and, if possible, to match for inherited paternal antigens andnon-inherited maternal antigens. Eligibility criteria for both stud-ies were similar.Patients with hematologic malignancies in need of an allogeneic
stem cell transplant (SCT) who lacked an HLA-identical related orunrelated donor were eligible. Additional eligibility criteria includ-ed Eastern Cooperative Oncology Group (ECOG) performancestatus less than or equal to 2, bilirubin less than or equal to 2
mg/dL, creatinine less than 1.5 times the upper limit of normal,preserved heart and lung function, and no evidence of chronicactive hepatitis or cirrhosis. HIV negativity was required, andpregnant females were excluded from the study. The studies wereapproved by the Institutional Review Board of both institutions,and all patients and donors provided written informed consent.The studies were conducted in accordance with the Declaration ofHelsinki and were registered on clinicaltrials.gov.Cases (n=97) include patients consecutively enrolled on these
two studies and receiving reduced intensity conditioning betweenJanuary 2007 and mid-2013. One pediatric patient was excluded,as were 2 patients undergoing transplant for myelofibrosis and thesingle patient with myeloma.The control group consisted of adult patients with leukemia,
lymphoma or myelodysplastic syndrome reported to the Centerfor International Blood and Marrow Transplant Research(CIBMTR) who received a double UCB graft following reducedintensity conditioning using fludarabine, cyclophosphamide, andlow-dose total body irradiation between 2007 and 2011 at UStransplant centers. This is the most widely utilized reduced inten-sity conditioning regimen for cord blood transplantation in adultswith an acceptable treatment-related mortality and is used as theconditioning regimen in several national clinical trials.6,7,17 Patientswith Karnofsky Performance Score (KPS) less than 60, with incom-plete background or follow-up information, who received anti-thymocyte globulin (ATG) or who did not receive a calcineurininhibitor after transplant, were excluded. A total of 193 CIBMTRpatients fulfilled these criteria and were included as a controlgroup. Seven of the 193 control patients had donors that werepoorly matched (HLA 3/6). Their exclusion did not affect theresults of the analysis.
Donors and stem cell processing Cord bloodCord-blood units for haplo-cord were selected based on HLA-
typing and cell count. Grafts were matched for at least 4 of 6 HLAloci by the standard cord criteria (i.e. low resolution for HLA-Aand HLA- B, and high resolution for HLA-DR)18 and contained aminimum cell count of 1x107 nucleated cells per kilogram (kg) ofthe recipient’s body weight before freezing. In contrast with com-mon practice, we prioritized matching over cell dose. As of mid-2012, for graft selection we utilized high-resolution HLA typingfor HLA A, B, C and DR.19
Haploidentical donor The haploidentical donor was a relative. Donors underwent
stem cell mobilization using filgrastim for four consecutive days.Apheresis was started on day 5 and continued daily until at least5x106 CD 34+ cells / recipient kg were collected. After collection,and prior to cryopreservation, haplo-identical grafts were T-celldepleted initially using the Isolex 300i CD34 selection device. Asof early April 2010, the Isolex 300i CD34 selection device was nolonger available, and instead, the Miltenyi CliniMACS device wasused under an Investigational New Device (IND) from the UnitedStates Food and Drug Agency. In the initial protocol (clinicaltrials.govidentifiers 00943800) the cell dose of the haplo-cord donor wasbased on CD3 cell dose (<1x106 CD3 per kgrec).
16 In that study, itwas noted that the administration of very high doses of haploCD34 cells correlated with failure of umbilical cord blood engraft-ment. Subsequently, the cell dose of the haplo graft has been basedon CD34 dose with a target dose of 3-5 x106 CD34 per kgrec.
Donor directed antibodiesAs of the tenth patient enrolled on the initial protocol, UCB and
haplo-identical donor selection was also based on avoidance of
Haplocord vs. double cord transplant
haematologica | 2016; 101(5) 635

K. van Besien et al.
636
Table 1. Pre-transplant characteristics of patients included in the UC/WCMC and CIBMTR study cohorts.Variable UC/WCMC CIBMTR P Haplo+Cord Double UCB Total n 97 193 Age, in years, n (%) 0.1520-29 8 (8) 11 (6) 30-39 15 (15) 13 (7) 40-49 18 (19) 31 (16) 50-59 24 (25) 63 (33) 60-69 29 (30) 70 (36) 70+ 3 (3) 5 (3) Median (range) 54 (20-73) 57 (20-72) 0.03Sex, n (%) 0.30Male 60 (62) 107 (55) Female 37 (38) 86 (45) Weight in kg, n, median (range) n=61 n=185 0.48 80 (49, 136) 79 (46, 146) Sorror Comorbidity Index, n (%) 0.290 32 (33) 52 (27) 1-2 29 (30) 52 (27) 3+ 39 (37) 88 (45) Missing 0 1 (1) N, median (range) 1 (0, 8) 2 (0, 10) 0.13Race, n (%) <0.0001White 59 (61) 152 (79) Black 23 (24) 19 (10) Others 6 (6) 19 (10) Unknown/declined 9 (9) 3 (2) Ethnicity, n (%) 0.05*Hispanic 9 (9) 20 (10) Non-Hispanic 64 (66) 162 (84) Unknown/declined 24 (25) 11 (6) KPS, n (%) 0.04**90-100% 77 (79) 119 (62) 60- 80% 20 (21) 57 (30) Missing 0 17 (9) Disease, n (%) 0.95AML 54 (56) 108 (56) ALL 12 (12) 21 (11) CLL 1 (1) 8 (4) CML 4 (4) 3 (2) Other acute leukemia 1 (1) 3 (2) Other leukemia 2 (2) 0 Myelodysplastic disorders 11 (11) 18 (9) Non-Hodgkin lymphoma 8 (8) 21 (11) Hodgkin lymphoma 4 (4) 11 (6) Disease risk, n (%) <0.0001Low 34 (35) 92 (48) Moderate 21 (22) 68 (35) High 42 (43) 33 (17) Conditioning regimen, n (%) <0.0001TBI + fludarabine + Cy 0 193 (100) Fludarabine + melphalan + ATG 97 (100) 0 GvHD prophylaxis, n (%) 0.17CSA alone 0 4 (2) CSA + MMF 97 (100) 185 (96) CSA + MTX 0 4 (2) HLA-match for CB units,a n (%) <0.00016/6 10 (10) 7 (4) 5/6 64 (66) 59 (31) 4/6 23 (24) 119 (62) ≤ 3/6 0 7 (4) Missing 0 1b (1) TNC cell dose at infusion (x107/kg), n, median (range) -Unit 1 n=97 n=167 1.7 (0.5, 9.0) 2.1 (0.6,5.2) Unit 2 - n=159 2.0 (0.3, 5.1) Sum of units - n=159 4.1 (1.1, 9.2) Year of transplant, n (%) <0.00012007-2009 17 (18) 88 (46) 2010-2013 80 (82) 105 (54)
aFor double UCB blood transplants, degree of HLA-match is defined as the value of the lower HLA-matched unit. HLA-matched data were available for one of two CB units. UC:University of Chicago; WCMC: Weill Cornell Medical College; CIBMTR: Center for International Bone Marrow Transplant Research; KPS: Karnofsky Performance Score. *Calculationexcluding category unknown/declined. **Calculation excluding Category Misssing.
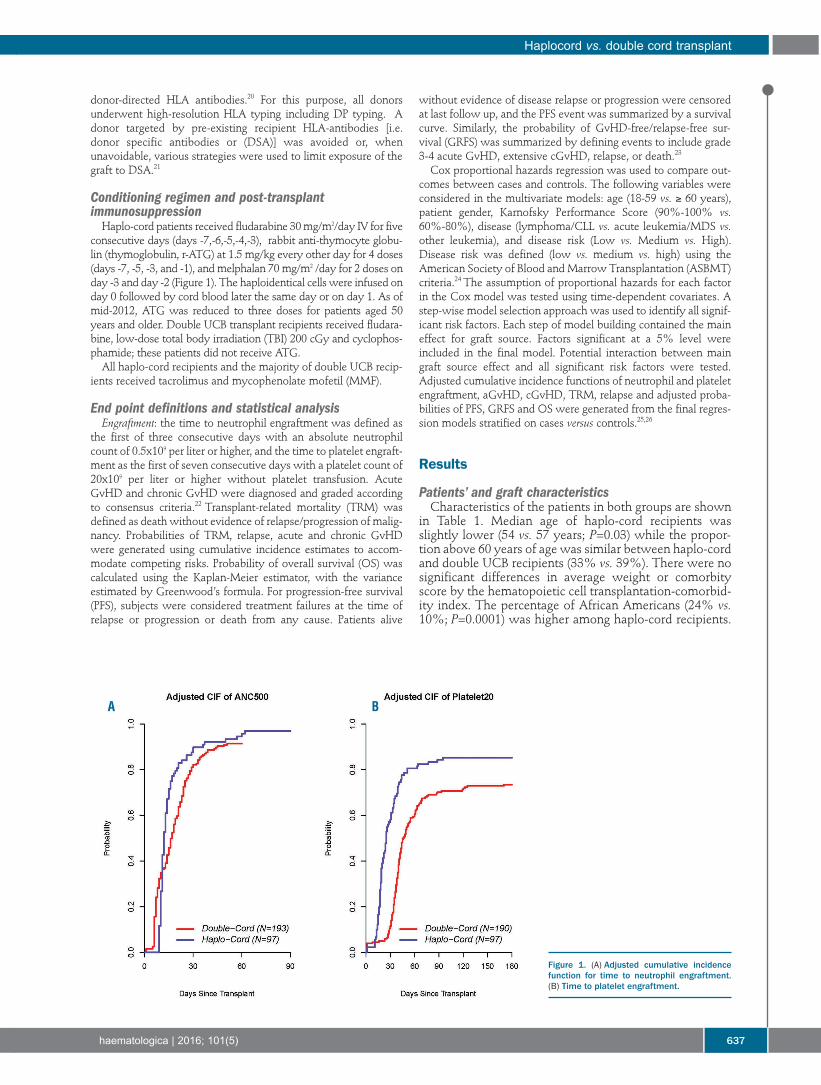
donor-directed HLA antibodies.20 For this purpose, all donorsunderwent high-resolution HLA typing including DP typing. Adonor targeted by pre-existing recipient HLA-antibodies [i.e.donor specific antibodies or (DSA)] was avoided or, whenunavoidable, various strategies were used to limit exposure of thegraft to DSA.21
Conditioning regimen and post-transplant immunosuppressionHaplo-cord patients received fludarabine 30 mg/m2/day IV for five
consecutive days (days -7,-6,-5,-4,-3), rabbit anti-thymocyte globu-lin (thymoglobulin, r-ATG) at 1.5 mg/kg every other day for 4 doses(days -7, -5, -3, and -1), and melphalan 70 mg/m2 /day for 2 doses onday -3 and day -2 (Figure 1). The haploidentical cells were infused onday 0 followed by cord blood later the same day or on day 1. As ofmid-2012, ATG was reduced to three doses for patients aged 50years and older. Double UCB transplant recipients received fludara-bine, low-dose total body irradiation (TBI) 200 cGy and cyclophos-phamide; these patients did not receive ATG. All haplo-cord recipients and the majority of double UCB recip-
ients received tacrolimus and mycophenolate mofetil (MMF).
End point definitions and statistical analysisEngraftment: the time to neutrophil engraftment was defined as
the first of three consecutive days with an absolute neutrophilcount of 0.5x109 per liter or higher, and the time to platelet engraft-ment as the first of seven consecutive days with a platelet count of20x109 per liter or higher without platelet transfusion. AcuteGvHD and chronic GvHD were diagnosed and graded accordingto consensus criteria.22 Transplant-related mortality (TRM) wasdefined as death without evidence of relapse/progression of malig-nancy. Probabilities of TRM, relapse, acute and chronic GvHDwere generated using cumulative incidence estimates to accom-modate competing risks. Probability of overall survival (OS) wascalculated using the Kaplan-Meier estimator, with the varianceestimated by Greenwood’s formula. For progression-free survival(PFS), subjects were considered treatment failures at the time ofrelapse or progression or death from any cause. Patients alive
without evidence of disease relapse or progression were censoredat last follow up, and the PFS event was summarized by a survivalcurve. Similarly, the probability of GvHD-free/relapse-free sur-vival (GRFS) was summarized by defining events to include grade3-4 acute GvHD, extensive cGvHD, relapse, or death.23
Cox proportional hazards regression was used to compare out-comes between cases and controls. The following variables wereconsidered in the multivariate models: age (18-59 vs. ≥ 60 years),patient gender, Karnofsky Performance Score (90%-100% vs.60%-80%), disease (lymphoma/CLL vs. acute leukemia/MDS vs.other leukemia), and disease risk (Low vs. Medium vs. High).Disease risk was defined (low vs. medium vs. high) using theAmerican Society of Blood and Marrow Transplantation (ASBMT)criteria.24 The assumption of proportional hazards for each factorin the Cox model was tested using time-dependent covariates. Astep-wise model selection approach was used to identify all signif-icant risk factors. Each step of model building contained the maineffect for graft source. Factors significant at a 5% level wereincluded in the final model. Potential interaction between maingraft source effect and all significant risk factors were tested.Adjusted cumulative incidence functions of neutrophil and plateletengraftment, aGvHD, cGvHD, TRM, relapse and adjusted proba-bilities of PFS, GRFS and OS were generated from the final regres-sion models stratified on cases versus controls.25,26
Results
Patients’ and graft characteristicsCharacteristics of the patients in both groups are shown
in Table 1. Median age of haplo-cord recipients was slightly lower (54 vs. 57 years; P=0.03) while the propor-tion above 60 years of age was similar between haplo-cordand double UCB recipients (33% vs. 39%). There were nosignificant differences in average weight or comorbityscore by the hematopoietic cell transplantation-comorbid-ity index. The percentage of African Americans (24% vs.10%; P=0.0001) was higher among haplo-cord recipients.
Haplocord vs. double cord transplant
haematologica | 2016; 101(5) 637
Figure 1. (A) Adjusted cumulative incidencefunction for time to neutrophil engraftment.(B) Time to platelet engraftment.
A B
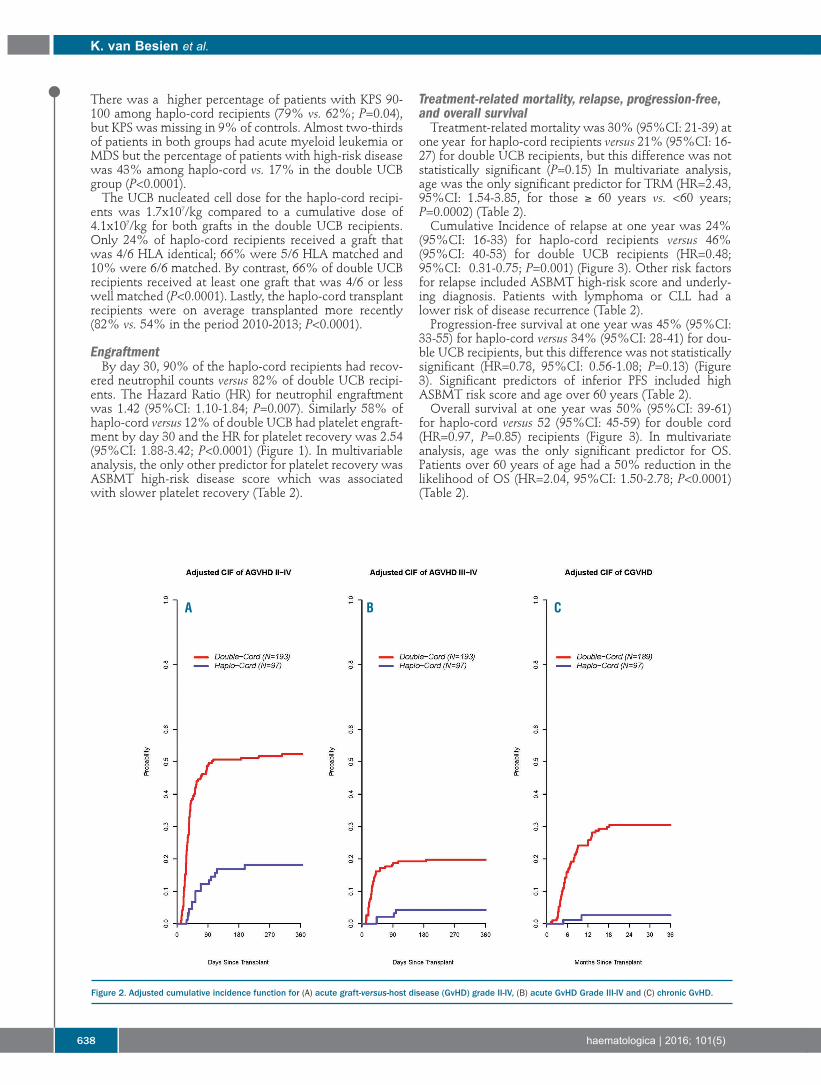
There was a higher percentage of patients with KPS 90-100 among haplo-cord recipients (79% vs. 62%; P=0.04),but KPS was missing in 9% of controls. Almost two-thirdsof patients in both groups had acute myeloid leukemia orMDS but the percentage of patients with high-risk diseasewas 43% among haplo-cord vs. 17% in the double UCBgroup (P<0.0001).The UCB nucleated cell dose for the haplo-cord recipi-
ents was 1.7x107/kg compared to a cumulative dose of4.1x107/kg for both grafts in the double UCB recipients.Only 24% of haplo-cord recipients received a graft thatwas 4/6 HLA identical; 66% were 5/6 HLA matched and10% were 6/6 matched. By contrast, 66% of double UCBrecipients received at least one graft that was 4/6 or lesswell matched (P<0.0001). Lastly, the haplo-cord transplantrecipients were on average transplanted more recently(82% vs. 54% in the period 2010-2013; P<0.0001).
Engraftment By day 30, 90% of the haplo-cord recipients had recov-
ered neutrophil counts versus 82% of double UCB recipi-ents. The Hazard Ratio (HR) for neutrophil engraftmentwas 1.42 (95%CI: 1.10-1.84; P=0.007). Similarly 58% ofhaplo-cord versus 12% of double UCB had platelet engraft-ment by day 30 and the HR for platelet recovery was 2.54(95%CI: 1.88-3.42; P<0.0001) (Figure 1). In multivariableanalysis, the only other predictor for platelet recovery wasASBMT high-risk disease score which was associatedwith slower platelet recovery (Table 2).
Treatment-related mortality, relapse, progression-free,and overall survivalTreatment-related mortality was 30% (95%CI: 21-39) at
one year for haplo-cord recipients versus 21% (95%CI: 16-27) for double UCB recipients, but this difference was notstatistically significant (P=0.15) In multivariate analysis,age was the only significant predictor for TRM (HR=2.43,95%CI: 1.54-3.85, for those ≥ 60 years vs. <60 years;P=0.0002) (Table 2).Cumulative Incidence of relapse at one year was 24%
(95%CI: 16-33) for haplo-cord recipients versus 46%(95%CI: 40-53) for double UCB recipients (HR=0.48;95%CI: 0.31-0.75; P=0.001) (Figure 3). Other risk factorsfor relapse included ASBMT high-risk score and underly-ing diagnosis. Patients with lymphoma or CLL had alower risk of disease recurrence (Table 2).Progression-free survival at one year was 45% (95%CI:
33-55) for haplo-cord versus 34% (95%CI: 28-41) for dou-ble UCB recipients, but this difference was not statisticallysignificant (HR=0.78, 95%CI: 0.56-1.08; P=0.13) (Figure3). Significant predictors of inferior PFS included highASBMT risk score and age over 60 years (Table 2).Overall survival at one year was 50% (95%CI: 39-61)
for haplo-cord versus 52 (95%CI: 45-59) for double cord(HR=0.97, P=0.85) recipients (Figure 3). In multivariateanalysis, age was the only significant predictor for OS.Patients over 60 years of age had a 50% reduction in thelikelihood of OS (HR=2.04, 95%CI: 1.50-2.78; P<0.0001)(Table 2).
K. van Besien et al.
638 haematologica | 2016; 101(5)
Figure 2. Adjusted cumulative incidence function for (A) acute graft-versus-host disease (GvHD) grade II-IV, (B) acute GvHD Grade III-IV and (C) chronic GvHD.
A B C
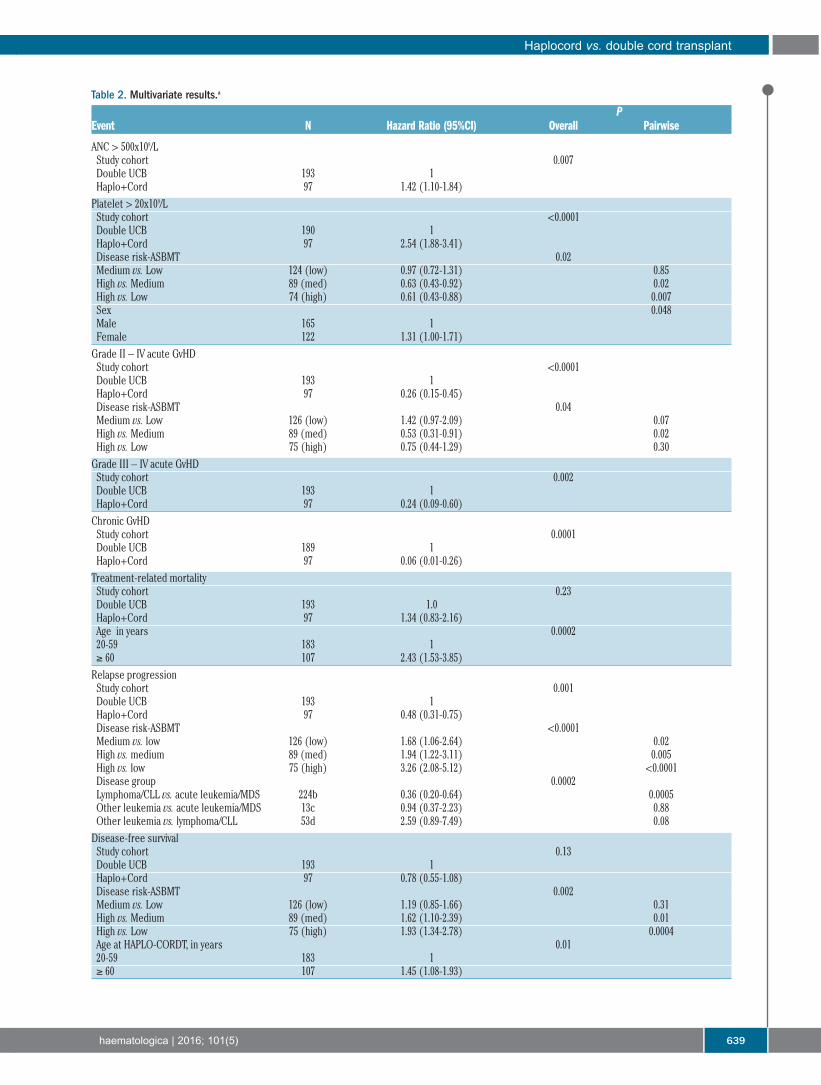
Haplocord vs. double cord transplant
haematologica | 2016; 101(5) 639
Table 2. Multivariate results.a
PEvent N Hazard Ratio (95%CI) Overall Pairwise
ANC > 500x109/L Study cohort 0.007Double UCB 193 1Haplo+Cord 97 1.42 (1.10-1.84)Platelet > 20x109/L Study cohort <0.0001Double UCB 190 1Haplo+Cord 97 2.54 (1.88-3.41)Disease risk-ASBMT 0.02Medium vs. Low 124 (low) 0.97 (0.72-1.31) 0.85High vs. Medium 89 (med) 0.63 (0.43-0.92) 0.02High vs. Low 74 (high) 0.61 (0.43-0.88) 0.007Sex 0.048Male 165 1Female 122 1.31 (1.00-1.71)Grade II – IV acute GvHD Study cohort <0.0001Double UCB 193 1Haplo+Cord 97 0.26 (0.15-0.45)Disease risk-ASBMT 0.04Medium vs. Low 126 (low) 1.42 (0.97-2.09) 0.07High vs. Medium 89 (med) 0.53 (0.31-0.91) 0.02High vs. Low 75 (high) 0.75 (0.44-1.29) 0.30Grade III – IV acute GvHD Study cohort 0.002Double UCB 193 1Haplo+Cord 97 0.24 (0.09-0.60)Chronic GvHD Study cohort 0.0001Double UCB 189 1Haplo+Cord 97 0.06 (0.01-0.26)Treatment-related mortalityStudy cohort 0.23Double UCB 193 1.0Haplo+Cord 97 1.34 (0.83-2.16)Age in years 0.000220-59 183 1≥ 60 107 2.43 (1.53-3.85)Relapse progressionStudy cohort 0.001Double UCB 193 1Haplo+Cord 97 0.48 (0.31-0.75)Disease risk-ASBMT <0.0001Medium vs. low 126 (low) 1.68 (1.06-2.64) 0.02High vs. medium 89 (med) 1.94 (1.22-3.11) 0.005High vs. low 75 (high) 3.26 (2.08-5.12) <0.0001Disease group 0.0002Lymphoma/CLL vs. acute leukemia/MDS 224b 0.36 (0.20-0.64) 0.0005Other leukemia vs. acute leukemia/MDS 13c 0.94 (0.37-2.23) 0.88Other leukemia vs. lymphoma/CLL 53d 2.59 (0.89-7.49) 0.08Disease-free survival Study cohort 0.13Double UCB 193 1Haplo+Cord 97 0.78 (0.55-1.08)Disease risk-ASBMT 0.002Medium vs. Low 126 (low) 1.19 (0.85-1.66) 0.31High vs. Medium 89 (med) 1.62 (1.10-2.39) 0.01High vs. Low 75 (high) 1.93 (1.34-2.78) 0.0004Age at HAPLO-CORDT, in years 0.0120-59 183 1≥ 60 107 1.45 (1.08-1.93)

Graft-versus-host disease and relapse-free survival(GRFS) The cumulative incidence of acute GvHD grade 2-4 by
day 120 was 17% (95%CI: 10%-25%) in the haplo-cordpatients versus 51% (95%CI: 44%-57%) in the doubleUCB group (P<0.0000). Grade 3-4 acute GvHD at day 120was similarly reduced in haplo-cord recipients versus con-trols 4% versus 19% (P<0.0001). Finally, chronic GvHDwas much reduced in haplo-cord versus controls with acumulative incidence at one year of 3% versus 30%(P<0.0000) (Figure 4). Combining these important clinical end points, at one
year 38% of haplo-cord recipients were alive without dis-ease progression or serious GvHD versus 21% of doubleUCB recipients. There was a 37% improvement in hazardrate for GRFS (HR=0.63, 95%CI: 0.47-0.85; P=0.002)(Figure 4). A higher KPS (≥90%) was also associated witha superior GRFS (Table 2).All calculations related to TRM, relapse, PFS, survival,
GvHD and GRFS were repeated after excluding from thecontrol group those patients with under 4/6 HLAmatched grafts or with missing graft HLA information.This had no impact on any of the outcomes (OnlineSupplementary Table S1).
Discussion
Here we conducted a comparison of 97 adults whounderwent haplo-cord transplant with a control group ofpatients reported to the CIBMTR undergoing reducedintensity conditioning and double UCB transplantation.The control group was restricted to patients receiving flu-darabine-cyclophosphamide low-dose TBI conditioning.Originally developed at the University of Minnesota, itappears safer than many other conditioning regimens andhas been widely adopted.7,17 In a recent CIBMTR study, itwas the regimen utilized in over two-thirds of US adultsundergoing non-myeloablative conditioning and UCBtransplant, and therefore a logical choice for our controlgroup. The tolerability of this regimen results in part fromits minimal myelosuppression,27 and typically a minimumUCB cell dose of more than 3x107 nucleated cells is consid-ered a requisite.6,18 For our study patients, we used a regi-men that includes thymoglobulin, and that in addition ismuch more myelosuppressive and may occasionally leadto irreversible myelosuppression.28 Despite this, we
demonstrated more rapid neutrophil recovery and evenmore notably accelerated platelet recovery after haplo-cord transplantation. This should have considerableimpact on duration of hospitalization, transfusion needs,and the expense of alternative donor transplantation ingeneral.29,30 We were also able to achieve this result despiteaccepting lower doses of umbilical cord blood cells, a prac-tice that in other studies of cord blood transplantation hasbeen associated with increased failure rates.18,31,32 We were unable to show a significant improvement in
TRM despite the more rapid engraftment. This is some-what paradoxical, but the benefits of rapid neutrophil andplatelet recovery were possibly offset by the more inten-sive conditioning regimen used for haplo-cord and poten-tially by infections related to thymoglobulin-mediated T-cell depletion. The rate of disease recurrence after haplo-cord trans-
plantation was significantly decreased. Whether thereduction in relapse relates to the difference in condition-ing, rather than to a graft-related effect, cannot be ascer-tained from our data,32,33 but it occurred despite the use ofthymoglobulin in the haplo-cord patients. ATG may benecessary to assure a smooth transition over time betweenthe haplo-graft and umbilical cord blood predominance. Inits absence, severe rejection and prolonged second nadirshave been reported.15,34 The use of ATG has been contro-versial because of concerns over higher rates of diseaserecurrence and increased rates of infections, toxicity andpost-transplant lymphoproliferative disease.35 Increasingevidence, supported by our findings, suggests that manyside-effects are dose related and that with appropriatedosing and monitoring, rabbit ATG is safe and not associ-ated with increased rates of disease recurrence.36 Despitethe reduction in disease recurrence, progression-free andOS were not significantly improved. We also observed a very significant decrease in the inci-
dence of acute and chronic GvHD with haplo-cord trans-plantation. In part, this can be attributed to our use ofATG. The control group did not receive ATG and allpatients received double UCB blood transplantationwhich was recently shown to be associated with moreacute GvHD.10 There may be additional reasons for thedecrease in acute and chronic GvHD. For example, sincethe size of the cord blood unit no longer determines therate of engraftment, we were able to choose smaller, bet-ter matched UCB units; better matching has been shownto be a major determinant of decreased GvHD.19 Lastly,
K. van Besien et al.
640 haematologica | 2016; 101(5)
Overall survivalStudy cohort 0.85Double UCB 193 1.0Haplo+Cord 97 0.97 (0.68-1.36)Age at HAPLO-CORDT, in years <0.000118-59 183 1≥ 60 107 2.04 (1.50-2.78)GvHD/relapse-free survival GRFSStudy cohort 0.002Double UCB 187 1.0Haplo+Cord 97 0.63 (0.46-0.85)Karnofsky Score 0.0260-80% vs. 90-100% 0.65 (0.48-0.89) 0.005
aRisk factors evaluated: age (18-59 years vs. ≥60 years), sex, Karnofsky Performance Score (KPS) (90%-100% vs. 60%-80% vs. missing), disease [(lymphoma/chronic lymphocyticleukemia (CLL) vs. acute leukemia/myelodysplastic syndromes (MDS) vs. other leukemia)], disease risk (low vs. medium vs. high). bAcute leukemia/MDS, n=224. cOther leukemia,n=13. dLymphoma/CLL, n=53.
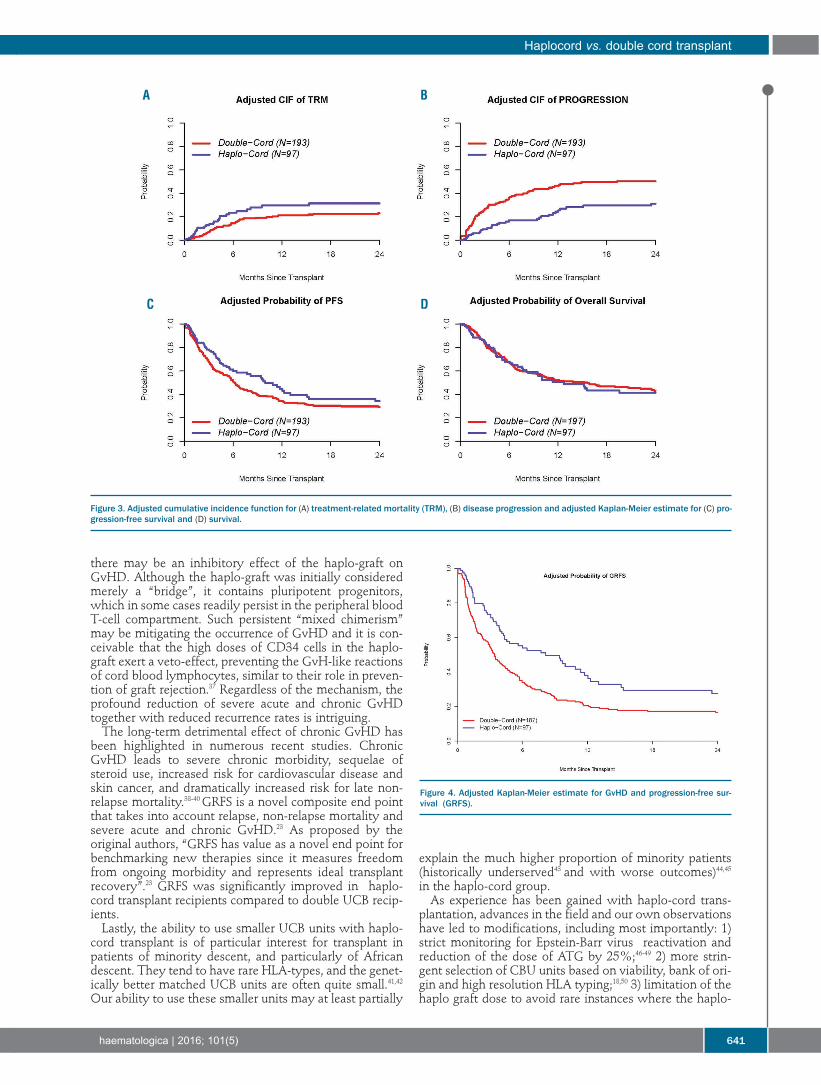
there may be an inhibitory effect of the haplo-graft onGvHD. Although the haplo-graft was initially consideredmerely a “bridge”, it contains pluripotent progenitors,which in some cases readily persist in the peripheral bloodT-cell compartment. Such persistent “mixed chimerism”may be mitigating the occurrence of GvHD and it is con-ceivable that the high doses of CD34 cells in the haplo-graft exert a veto-effect, preventing the GvH-like reactionsof cord blood lymphocytes, similar to their role in preven-tion of graft rejection.37 Regardless of the mechanism, theprofound reduction of severe acute and chronic GvHDtogether with reduced recurrence rates is intriguing. The long-term detrimental effect of chronic GvHD has
been highlighted in numerous recent studies. ChronicGvHD leads to severe chronic morbidity, sequelae ofsteroid use, increased risk for cardiovascular disease andskin cancer, and dramatically increased risk for late non-relapse mortality.38-40 GRFS is a novel composite end pointthat takes into account relapse, non-relapse mortality andsevere acute and chronic GvHD.23 As proposed by theoriginal authors, “GRFS has value as a novel end point forbenchmarking new therapies since it measures freedomfrom ongoing morbidity and represents ideal transplantrecovery”.23 GRFS was significantly improved in haplo-cord transplant recipients compared to double UCB recip-ients. Lastly, the ability to use smaller UCB units with haplo-
cord transplant is of particular interest for transplant inpatients of minority descent, and particularly of Africandescent. They tend to have rare HLA-types, and the genet-ically better matched UCB units are often quite small.41,42Our ability to use these smaller units may at least partially
explain the much higher proportion of minority patients(historically underserved43 and with worse outcomes)44,45in the haplo-cord group. As experience has been gained with haplo-cord trans-
plantation, advances in the field and our own observationshave led to modifications, including most importantly: 1)strict monitoring for Epstein-Barr virus reactivation andreduction of the dose of ATG by 25%;46-49 2) more strin-gent selection of CBU units based on viability, bank of ori-gin and high resolution HLA typing;18,50 3) limitation of thehaplo graft dose to avoid rare instances where the haplo-
Haplocord vs. double cord transplant
haematologica | 2016; 101(5) 641
Figure 3. Adjusted cumulative incidence function for (A) treatment-related mortality (TRM), (B) disease progression and adjusted Kaplan-Meier estimate for (C) pro-gression-free survival and (D) survival.
Figure 4. Adjusted Kaplan-Meier estimate for GvHD and progression-free sur-vival (GRFS).
A B
C D

graft outcompetes the UCB unit;16 and 4) avoidance ofdonors targeted by HLA antibodies, since these were asso-ciated with graft failure.20 The most noteworthy limitations of this analysis relate
to the non-randomized comparison and potential bias oftwo different data sources (i.e. center-specific data relativeto registry data). Adjustment for standard transplantcovariates reduces but does not negate the lack of otherpatient covariates not captured and may influence theresults. GvHD outcomes may have been captured differ-ently for the haplo-cord centers (either better or worse)relative to the registry. We believe differences in relapseand PFS are probably accurate, as we would not expect amajor difference in relapse detection. Ideally, we wouldhave compared our outcomes to patients receiving a simi-lar conditioning regimen, but this turned out to be impos-sible. The fludarabine-melphalan-ATG regimen has onlybeen studied in limited numbers (and with different dos-ing regimens) in double UCB studies.51,52 In the CIBMTRdata-set, fludarabine-alkylator combinations were used infewer than 10% of older adults with AML receivingreduced intensity conditioning and double UCB trans-
plant. Lastly, there is a remote possibility that theobserved advantages in rate of engraftment are simply aresult of better HLA-matching, which was achievedbecause of our CBU unit selection strategy. This is highlyunlikely given the well described predominance of thehaplo-graft early after transplant.53Several competing technologies are under development
involving in vitro expansion of UCB cells or other progeni-tors or methods to enhance homing.54 Additional trials willbe required to determine if any of these procedures will ulti-mately be superior. CD34 selected haplo-identical cells havethe advantage of available technology and rapidity. Haplo-identical transplantation with non-selected cells providesanother readily available, affordable and technically lessburdensome alternative. In parallel phase II studies it result-ed in earlier engraftment than double cord transplant, buthad higher rates of disease recurrence.6 Further studies willbe needed to compare outcomes and of these competingtechnologies, and to further advance the field.
FundingSupported by an unrestricted grant from Miltenyi Biotec.
K. van Besien et al.
642 haematologica | 2016; 101(5)
References1. Gluckman E, Ruggeri A, Volt F, et al.
Milestones in umbilical cord blood trans-plantation. Br J Haematol. 2011;154(4):441-447.
2. Sanz J, Sanz MA, Saavedra S, et al. Cordblood transplantation from unrelated donorsin adults with high-risk acute myeloidleukemia. Biol Blood Marrow Transplant.2010;16(1):86-94.
3. Brunstein CG, Gutman JA, Weisdorf DJ, etal. Allogeneic hematopoietic cell transplan-tation for hematological malignancy: rela-tive risks and benefits of double umbilicalcord blood. Blood. 2010;116(22):4693-4699.
4. Laughlin MJ, Eapen M, Rubinstein P, et al.Outcomes after transplantation of cordblood or bone marrow from unrelateddonors in adults with leukemia. N Engl JMed. 2004;351(22):2265-2275.
5. Barker JN, Scaradavou A, Stevens CE.Combined effect of total nucleated cell doseand HLA match on transplantation outcomein 1061 cord blood recipients with hemato-logic malignancies. Blood. 2010;115(9):1843-1849.
6. Brunstein CG, Fuchs EJ, Carter SL, et al.Alternative donor transplantation afterreduced intensity conditioning: results ofparallel phase 2 trials using partially HLA-mismatched related bone marrow or unre-lated double umbilical cord blood grafts.Blood. 2011;118(2):282-288.
7. Weisdorf D, Eapen M, Ruggeri A, et al.Alternative donor transplantation for olderpatients with acute myeloid leukemia in firstcomplete remission: a center for internation-al blood and marrow transplant research-eurocord analysis. Biol Blood MarrowTransplant. 2014;20(6):816-822.
8. Peffault de Latour R, Brunstein CG, PorcherR, et al. Similar overall survival using sibling,unrelated donor, and cord blood grafts afterreduced-intensity conditioning for olderpatients with acute myelogenous leukemia.Biol Blood Marrow Transplant. 2013;19(9):
1355-1360.9. Eapen M, Rocha V, Sanz G, et al. Effect of
graft source on unrelated donor haemopoi-etic stem-cell transplantation in adults withacute leukaemia: a retrospective analysis.Lancet Oncol. 2010;11(7):653-660.
10. Wagner JE Jr, Eapen M, Kurtzberg J. One-unit versus two-unit cord-blood transplanta-tion. N Engl J Med. 2015;372(3):288-293.
11. Fernandez MN, Regidor C, Cabrera R, et al.Unrelated umbilical cord blood transplantsin adults: Early recovery of neutrophils bysupportive co-transplantation of a low num-ber of highly purified peripheral bloodCD34+ cells from an HLA-haploidenticaldonor. Exp Hematol. 2003;31(6):535-544.
12. Gormley NJ, Wilder J, Khuu H, et al. Co-Infusion of Allogeneic Cord Blood withHaploidentical CD34+ Cells ImprovedTransplant Outcome for Patients withSevere Aplastic Anemia Undergoing CordBlood Transplantation. ASH AnnualMeeting Abstracts. 2011;118:654.
13. Kwon M, Bautista G, Balsalobre P, et al.Haplo-cord transplantation using CD34+cells from a third-party donor to speedengraftment in high-risk patients withhematologic disorders. Biol Blood MarrowTransplant. 2014;20:(12):2015-2022.
14. Liu H, van Besien K. Alternative donor trans-plantation-"mixing and matching": the roleof combined cord blood and haplo-identicaldonor transplantation (haplo-cord SCT) as atreatment strategy for patients lacking stan-dard donors. Curr Hematol Malig Rep.2015;10(1):1-7.
15. Lindemans CA, van Besien K. Topping it up:methods to improve cord blood transplanta-tion outcomes by increasing the number ofCD34+ cells. Cytotherapy. 2015;17(6):723-729.
16. Liu H, Rich ES, Godley L, et al. Reduced-intensity conditioning with combined hap-loidentical and cord blood transplantationresults in rapid engraftment, low GVHD,and durable remissions. Blood. 2011;118(24):6438-6445.
17. Brunstein CG, Eapen M, Ahn KW, et al.Reduced-intensity conditioning transplanta-tion in acute leukemia: the effect of sourceof unrelated donor stem cells on outcomes.Blood. 2012;119(23):5591-5598.
18. Barker JN, Byam C, Scaradavou A. How Itreat: the selection and acquisition of unre-lated cord blood grafts. Blood. 2011;117(8):2332-2339.
19. Eapen M, Klein JP, Ruggeri A, et al. Impact ofallele-level HLA matching on outcomes aftermyeloablative single unit umbilical cordblood transplantation for hematologicmalignancy. Blood. 2014;123(1):133-140.
20. Yoshihara S, Taniguchi K, Ogawa H, Saji H.The role of HLA antibodies in allogeneicSCT: is the type-and-screen strategy neces-sary not only for blood type but also forHLA? Bone Marrow Transplant.2012;47(12):1499-1506.
21. Gergis U, Mayer S, Gordon B, et al. A strat-egy to reduce donor-specific HLA Absbefore allogeneic transplantation. BoneMarrow Transplant. 2014;49(5):722-724.
22. Filipovich AH, Weisdorf D, Pavletic S, et al.National Institutes of Health consensusdevelopment project on criteria for clinicaltrials in chronic graft-versus-host disease: I.Diagnosis and staging working group report.Biol Blood Marrow Transplant.2005;11(12):945-956.
23. Holtan SG, DeFor TE, Lazaryan A, et al.Composite end point of graft-versus-hostdisease-free, relapse-free survival after allo-geneic hematopoietic cell transplantation.Blood. 2015;125(8):1333-1338.
24. American Society of Blood and MarrowTransplantation. ASBMT RFI 2015 -DiseaseClassifications Corresponding to CIBMTRClassifications. Available from:http://c.ymcdn.com/sites/www.asbmt.org/re s o u r c e / r e sm g r / R F I / R F I _ 2 0 1 5 _ -_CIBMTR_Disease_Cl.pdf. Last accessed 22March 2016.
25. Zhang X, Zhang MJ. SAS macros for estima-tion of direct adjusted cumulative incidencecurves under proportional subdistribution

hazards models. Comput MethodsPrograms Biomed. 2011;101(1):87-93.
26. Zhang X, Loberiza FR, Klein JP, Zhang MJ. ASAS macro for estimation of direct adjustedsurvival curves based on a stratified Coxregression model. Comput MethodsPrograms Biomed. 2007;88(2):95-101.
27. Brunstein CG, Barker JN, Weisdorf DJ, et al.Umbilical cord blood transplantation afternonmyeloablative conditioning: impact ontransplantation outcomes in 110 adults withhematologic disease. Blood. 2007;110(8):3064-3070.
28. van Besien K, Smith S, Anastasi J, et al.Irreversible myelosuppression after fludara-bine-melphalan conditioning: observationsin patients with graft rejection. Blood.2004;103(11):4373-4374.
29. Majhail NS, Mothukuri JM, Brunstein CG,Weisdorf DJ. Costs of hematopoietic celltransplantation: comparison of umbilicalcord blood and matched related donor trans-plantation and the impact of posttransplantcomplications. Biol Blood MarrowTransplant. 2009;15(5):564-573.
30. Ramirez P, Brunstein CG, Miller B, Defor T,Weisdorf D. Delayed platelet recovery afterallogeneic transplantation: a predictor ofincreased treatment-related mortality andpoorer survival. Bone Marrow Transplant.2011;46(7):981-986.
31. Laughlin MJ, Barker J, Bambach B, et al.Hematopoietic engraftment and survival inadult recipients of umbilical-cord bloodfrom unrelated donors. N Engl J Med.2001;344(24):1815-1822.
32. Rio B, Chevret S, Vigouroux S, et al.Decreased nonrelapse mortality after unre-lated cord blood transplantation for acutemyeloid leukemia using reduced-intensityconditioning: a prospective phase II multi-center trial. Biol Blood Marrow Transplant.2015;21(3):445-453.
33. de Lima M, Anagnostopoulos A, Munsell M,et al. Non-Ablative versus Reduced IntensityConditioning Regimens in the Treatment ofAcute Myeloid Leukemia and High-RiskMyelodysplastic Syndrome. Dose isRelevant for Long-Term Disease Controlafter Allogeneic Hematopoietic Stem CellTransplantation. Blood. 2004;104(3):865-872.
34. Lindemans CA, te Boome LCJ, Admiraal R,et al. Sufficient Immune-suppression withThymoglobulin is essential for a successfulHaplo-myeloid Bridge in Haplo-Cord Blood
Transplantation. Biol Blood MarrowTransplant. 2015;21(19):1839-1845.
35. Soiffer RJ, Lerademacher J, Ho V, et al.Impact of immune modulation with anti-T-cell antibodies on the outcome of reduced-intensity allogeneic hematopoietic stem celltransplantation for hematologic malignan-cies. Blood. 2011;117(25):6963-6970.
36. Segovia J, van Besien K. Antithymocyteglobulin for graft-versus-host disease pro-phylaxis: mistakenly maligned. LeukLymphoma. 2015;56(4):841-842.
37. Ophir E, Reisner Y. The use of donor-derived veto cells in hematopoietic stem celltransplantation. Front Immunol. 2012;3:93.
38. Storb R, Gyurkocza B, Storer BE, et al. Graft-versus-host disease and graft-versus-tumoreffects after allogeneic hematopoietic celltransplantation. J Clin Oncol.2013;31(12):1530-1538.
39. Bhatia S, Francisco L, Carter A, et al. Latemortality after allogeneic hematopoietic celltransplantation and functional status oflong-term survivors: report from the BoneMarrow Transplant Survivor Study. Blood.2007;110(10):3784-3792.
40. Boyiadzis M, Arora M, Klein J, et al. Impactof chronic graft-versus-host disease on laterelapse and survival on 7489 patients aftermyeloablative allogeneic hematopoietic celltransplantation for leukemia. Clin CancerRes. 2015;21(9):2020-2028.
41. Page KM, Mendizabal A, Betz-Stablein B, etal. Optimizing donor selection for publiccord blood banking: influence of maternal,infant, and collection characteristics on cordblood unit quality. Transfusion. 2014;54(2):340-352.
42. Cairo MS, Wagner EL, Fraser J, et al.Characterization of banked umbilical cordblood hematopoietic progenitor cells andlymphocyte subsets and correlation withethnicity, birth weight, sex, and type ofdelivery: a Cord Blood Transplantation(COBLT) Study report. Transfusion.2005;45(6):856-866.
43. Joshua TV, Rizzo JD, Zhang MJ, et al. Accessto hematopoietic stem cell transplantation:effect of race and sex. Cancer.2010;116(14):3469-3476.
44. Baker KS, Davies SM, Majhail NS, et al. Raceand socioeconomic status influence out-comes of unrelated donor hematopoietic celltransplantation. Biol Blood MarrowTransplant. 2009;15(12):1543-1554.
45. Schwake CJ, Eapen M, Lee SJ, et al.
Differences in characteristics of UShematopoietic stem cell transplantation cen-ters by proportion of racial or ethnic minori-ties. Biol Blood Marrow Transplant.2005;11(12):988-998.
46. Blaes AH, Cao Q, Wagner JE, et al.Monitoring and preemptive rituximab ther-apy for Epstein-Barr virus reactivation afterantithymocyte globulin containing nonmye-loablative conditioning for umbilical cordblood transplantation. Biol Blood MarrowTransplant. 2010;16(2):287-291.
47. Heslop HE. How I treat EBV lymphoprolif-eration. Blood. 2009;114(19):4002-4008.
48. Baron F, Labopin M, Blaise D, et al. Impact ofin vivo T-cell depletion on outcome of AMLpatients in first CR given peripheral bloodstem cells and reduced-intensity condition-ing allo-SCT from a HLA-identical siblingdonor: a report from the Acute LeukemiaWorking Party of the European Group forBlood and Marrow Transplantation. BoneMarrow Transplant. 2014;49(3):389-396.
49. Salem G, Ruppert AS, Elder P, et al. Lowerdose of antithymocyte globulin does notincrease graft-versus-host disease in patientsundergoing reduced-intensity conditioningallogeneic hematopoietic stem cell trans-plant. Leuk Lymphoma. 2015;56(4):1058-1065.
50. Eapen M, Klein JP, Sanz GF, et al. Effect ofdonor-recipient HLA matching at HLA A, B,C, and DRB1 on outcomes after umbilical-cord blood transplantation for leukaemiaand myelodysplastic syndrome: a retrospec-tive analysis. Lancet Oncol. 2011;12(13):1214-1221.
51. Cutler C, Stevenson K, Kim HT, et al.Double umbilical cord blood transplantationwith reduced intensity conditioning andsirolimus-based GVHD prophylaxis. BoneMarrow Transplant. 2011;46(5):659-667.
52. Ballen KK, Spitzer TR, Yeap BY, et al.Double unrelated reduced-intensity umbili-cal cord blood transplantation in adults. BiolBlood Marrow Transplant. 2007;13(1):82-89.
53. van Besien K, Liu H, Jain N, Stock W, Artz A.Umbilical Cord Blood TransplantationSupported by Third-Party Donor Cells:Rationale, Results, and Applications. BiolBlood Marrow Transplant. 2013;19(5):682-691.
54. Lund TC, Boitano AE, Delaney CS, ShpallEJ, Wagner JE. Advances in umbilical cordblood manipulation-from niche to bedside.Nat Rev Clin Oncol. 2015;12(3):163-174.
Haplocord vs. double cord transplant
haematologica | 2016; 101(5) 643

644 haematologica | 2016; 101(5)
Received: November 14, 2015.
Accepted: February 3, 2016
Pre-published: Febraury 8, 2016.
©2016 Ferrata Storti Foundation
Check the online version for the most updatedinformation on this article, online supplements,and information on authorship & disclosures:www.haematologica.org/content/101/5/644
Material published in Haematologica is cov-ered by copyright. All rights reserved to theFerrata Storti Foundation. Copies of articlesare allowed for personal or internal use.Permission in writing from the publisher isrequired for any other use.
Correspondence: [email protected]
Allogeneic bone marrow transplantation is an essential therapy foracquired aplastic anemia and prognosis has recently improved.However, engraftment failure and graft-versus-host disease are
potential fatal complications. Various risk factors for poor prognosishave been identified, such as patient age and human-leukocyte antigendisparity, but the relationship between donor age and prognosis is stillunknown. Therefore, we performed a cohort study to compare theprognosis of unrelated bone marrow transplantation from younger andolder donors using the registry database in Japan. We evaluated 427patients (age 16-72 years) with aplastic anemia who underwent bonemarrow transplantation from younger (≤39 years, n=281) or older (≥40years, n=146) unrelated donors. Overall survival of the older donorgroup was significantly inferior to that of the younger donor group(adjusted hazard ratio 1.64; 95% confidence interval 1.15-2.35; P<0.01).The incidence of fatal infection was significantly higher in the olderdonor group (13.7% vs. 7.5%; P=0.03). Primary engraftment failure andacute graft-versus-host disease were significantly more frequent in theolder donor group (9.7% vs. 5.0%; adjusted hazard ratio 1.30; P=0.01,and 27.1% vs. 19.7%; adjusted hazard ratio 1.56; P=0.03, respectively).Acute graft-versus-host disease was related to a worse prognosis in thewhole cohort. This study showed the inferiority of older donors inaplastic anemia; thus, donor age should be considered when multipledonors are available. A large-scale prospective study is warranted toestablish a better donor selection algorithm for bone marrow transplan-tation in aplastic anemia.
Allogeneic unrelated bone marrow transplantation from older donors results in worse prognosis in recipients with aplasticanemiaYasuyuki Arai,1 Tadakazu Kondo,1 Hirohito Yamazaki,2 Katsuto Takenaka,3Junichi Sugita,4 Takeshi Kobayashi,5 Yukiyasu Ozawa,6 Naoyuki Uchida,7 KojiIwato,8 Naoki Kobayashi,9 Yoshiyuki Takahashi,10 Ken Ishiyama,11 TakahiroFukuda,12 Tatsuo Ichinohe,13 Yoshiko Atsuta,14,15 Takehiko Mori,16 and TakanoriTeshima4 on behalf of the Japan Society for Hematopoietic CellTransplantation
1Department of Hematology and Oncology, Graduate School of Medicine, KyotoUniversity; 2Division of Transfusion Medicine, Kanazawa University Hospital;3Department of Medicine and Biosystemic Science, Kyushu University Graduate Schoolof Medicinal Sciences, Fukuoka; 4Department of Hematology, Hokkaido UniversityGraduate School of Medicine, Sapporo; 5Hematology Division, Tokyo MetropolitanCancer and Infectious Diseases Center Komagome Hospital, Tokyo; 6Department ofHematology, Japanese Red Cross Nagoya First Hospital; 7Department of Hematology,Toranomon Hospital, Tokyo; 8Department of Hematology, Hiroshima Red Cross Hospital& Atomic-Bomb Survivors Hospital; 9Department of Hematology, Sapporo HokuyuHospital; 10Department of Pediatrics, Nagoya University Graduate School of Medicine;11Department of Hematology, Kanazawa University Hospital; 12Hematopoietic Stem CellTransplantation Division, National Cancer Center Hospital, Tokyo; 13Department ofHematology and Oncology, Hiroshima University Hospital; 14Japanese Data Center forHematopoietic Cell Transplantation, Nagoya; 15Department of Healthcare Administration,Nagoya University Graduate School of Medicine; and 16Division of Hematology,Department of Internal Medicine, Keio University School of Medicine, Tokyo, Japan
ABSTRACT
Ferrata StortiFoundation
EUROPEANHEMATOLOGYASSOCIATION
Haematologica 2016Volume 101(5):644-652
ARTICLE Stem Cell Transplantation
doi:10.3324/haematol.2015.139469

Introduction
Allogeneic hematopoietic cell transplantation is aneffective and, therefore, indispensable therapy foracquired aplastic anemia (AA) in adults.1 Patients withAA are eligible for transplant if they are under 40 yearsof age or when they are refractory to immunosuppres-sive therapy;1,2 bone marrow transplantation (BMT) froma human leukocyte antigen (HLA)-matched sibling donoror an unrelated donor is selected according to the donoravailability.2 The prognosis of BMT for AA has recentlyimproved and 5-year overall survival (OS) is as high as72% for younger patients (≤40 years old) and 53% forolder patients (>40 years).3 However, severe complications, such as engraftment
failure, infection, and graft-versus-host disease (GvHD),are problems that need to be addressed in order toimprove the overall prognosis of AA, especially for unre-lated BMT.2,3 Various risk factors are reportedly associat-ed with these complications and poor prognosis, such as older patient age, longer periods from diagnosis totransplantation, HLA-mismatched donors, and femaledonors.2-4 In addition to these, biological speculationfrom previous published studies regarding hematopoieticstem cell repopulation and donor-derived T-cell functionhave suggested that transplantation from older donorsmay result in a higher incidence of engraftment failureand acute GvHD (aGvHD), and, as a result, increasetransplant-related death and lead to inferior OS.According to murine studies, hematopoietic stem cellsfrom older donors do not re-populate as efficiently,5,6 andgrafts from older donors have a higher ratio of memoryT cells to naïve T cells;7 an increase in peripheral bloodmemory T cells has been shown to be related to theoccurrence of aGvHD in humans.8,9 The influence of donor age in unrelated hematopoietic
cell transplantation has long been discussed in variousstudies, and some have shown a relationship betweenolder donor and worse prognosis.10-15 Most of thesecohorts, however, were mainly composed of hematolog-ic malignancies, and AA cases were not included,11-15 or, ifthey were, they made up only a small proportion of thecohort.10 AA should be analyzed independently frommalignant diseases, especially with regard to engraft-ment and GvHD, because the incidence of graft failure ismore often documented in AA, and GvHD more directlyimpacts OS.2 Moreover, engraftment and GvHD areclosely related to pre- or post-transplant tumor load inhematologic malignancies, which is irrelevant to AApatients.16-18 As far as we know, however, no studies haveinvestigated donor age as a candidate risk factor for poorprognosis in transplantation for AA.Therefore, we performed a cohort study to compare
the prognosis of patients with AA who underwent BMTfrom younger donors versus older donors using theJapanese transplant registry database, in particular onengraftment and GvHD. We focused on BMT from unre-lated donors in order to avoid the correlation betweenpatient and donor age; thus, BMT from related siblingdonors were excluded because siblings tend to be bornonly a few years apart.10 Our study should provideimportant insights into donor selection algorithms forBMT in patients with AA.
Methods
Inclusion criteria and clinical procedures in BMTData for adult patients (age >16 years) with AA who under-
went a first allogeneic BMT from unrelated donors betweenJanuary 1 1993 and December 31 2013 were obtained from theTransplant Registry Unified Management Program (TRUMP) inJapan.19 The eligibility criteria for transplantation was in accor-dance with international guidelines and recommendations;1,2
BMT is the first-line treatment for young patients with severeAA with a sibling donor, and the second-line treatment follow-ing immunosuppressive treatment in older patients or in thoseto be grafted from an unrelated donor.2
The unrelated donor selection was based primarily on HLAdisparity, and candidates were nominated among 8/8 or, if notavailable, 7/8 (or lower) HLA-A, B, C, and DR allele matchedvolunteers (age 20-55 years) registered in the Japanese MarrowDonor Program. Data on 10 alleles including HLA-DQ were notavailable. The donor was finally determined after considerationof various factors, such as ABO blood type, sex, and bodyweight; donor age usually has little significance on donor selec-tion in Japan.20
Selection of conditioning regimens and GvHD prophylaxis isat the discretion of attending physicians in each institute, con-sidering disease status, number of transfusions and amountstransfused, patients’ age and performance status, the risk ofinfections, etc.; donor age is not usually considered. Donor-derived serum and/or erythrocytes were depleted from grafts incases of mismatched ABO blood types, and grafts were trans-planted without ex vivo T-cell depletion. Our protocol compliedwith the Declaration of Helsinki and was approved by theTRUMP Data Management Committee and by the EthicsCommittee of Kyoto University where the study was per-formed. Patient information was anonymized, so consent wasnot required.
Data collection and definition of each covariateFrom the registry database, we extracted data on basic pre-
transplant characteristics and post-transplant clinical courses.Donors were categorized into two groups with respect to age(younger vs. older than the 75th percentile; the closest valuewhich is the multiple of 5 was adopted as the cut-off point).Donor age was considered a continuous variable and its influ-ence was analyzed. Conditioning regimens were summarizedaccording to the definitions of myeloablative conditioning(MAC) and reduced-intensity conditioning (RIC), which wereconsistent with those established in the RIC regimen work-shop.21 Data on the use of anti-thymocyte or anti-T-cell globulin(ATG) before and after BMT were also collected. Periodsbetween diagnosis and BMT were calculated from the day ofinitial diagnosis of AA.With respect to the post-transplant clinical course, engraftment
of neutrophils and platelets was defined as the first of three con-secutive days during which neutrophil and platelet counts wereat least 500/μL and 5.0x104/μL without transfusion support,respectively. Diagnosis and classification of GvHD were definedaccording to traditional criteria.22,23 A protective environment,prophylactic administration of antibiotics, and intravenousimmunoglobulin replacement were adopted as standard preven-tion strategies for infection in accordance with the guideline fromthe Japanese Society for Hematopoietic Cell Transplantation.Analytical methods are shown in the Online Supplementary
Appendix.
Prognosis and donor age in unrelated BMT for AA
haematologica | 2016; 101(5) 645
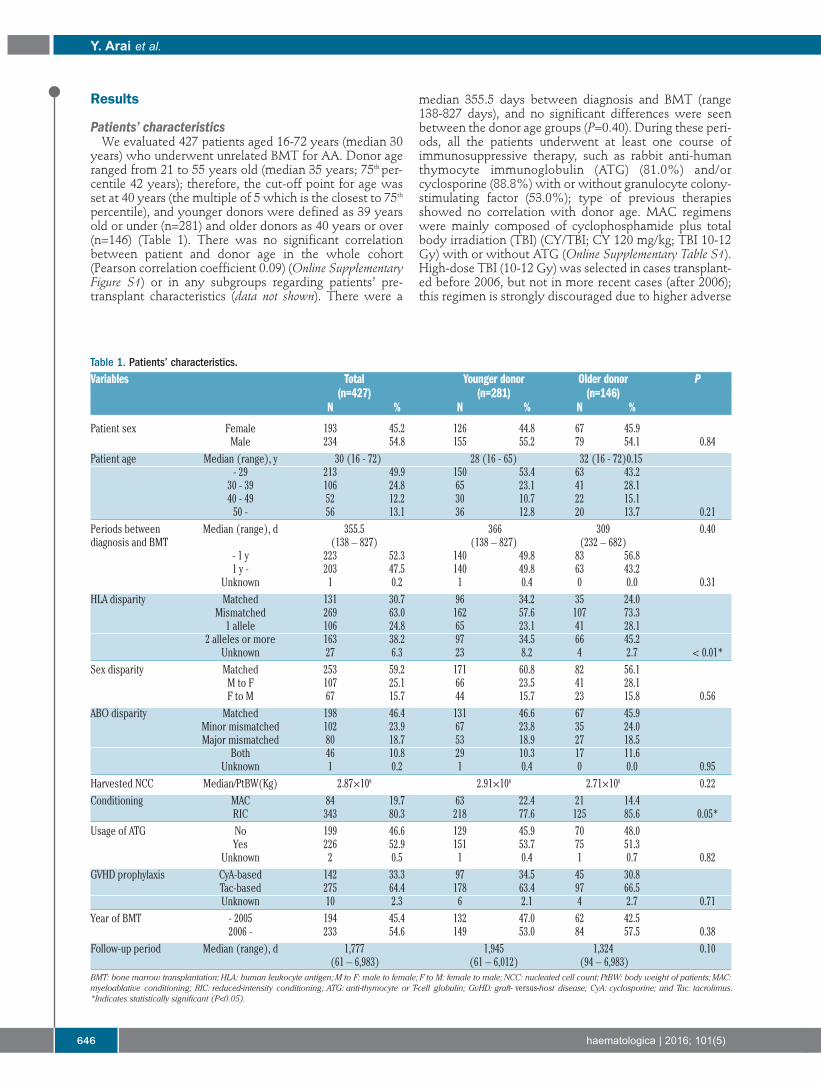
Results
Patients’ characteristicsWe evaluated 427 patients aged 16-72 years (median 30
years) who underwent unrelated BMT for AA. Donor ageranged from 21 to 55 years old (median 35 years; 75th per-centile 42 years); therefore, the cut-off point for age wasset at 40 years (the multiple of 5 which is the closest to 75th
percentile), and younger donors were defined as 39 yearsold or under (n=281) and older donors as 40 years or over(n=146) (Table 1). There was no significant correlationbetween patient and donor age in the whole cohort(Pearson correlation coefficient 0.09) (Online SupplementaryFigure S1) or in any subgroups regarding patients’ pre-transplant characteristics (data not shown). There were a
median 355.5 days between diagnosis and BMT (range138-827 days), and no significant differences were seenbetween the donor age groups (P=0.40). During these peri-ods, all the patients underwent at least one course ofimmunosuppressive therapy, such as rabbit anti-humanthymocyte immunoglobulin (ATG) (81.0%) and/orcyclosporine (88.8%) with or without granulocyte colony-stimulating factor (53.0%); type of previous therapiesshowed no correlation with donor age. MAC regimenswere mainly composed of cyclophosphamide plus totalbody irradiation (TBI) (CY/TBI; CY 120 mg/kg; TBI 10-12Gy) with or without ATG (Online Supplementary Table S1).High-dose TBI (10-12 Gy) was selected in cases transplant-ed before 2006, but not in more recent cases (after 2006);this regimen is strongly discouraged due to higher adverse
Y. Arai et al.
646 haematologica | 2016; 101(5)
Table 1. Patients’ characteristics.Variables Total Younger donor Older donor P
(n=427) (n=281) (n=146)N % N % N %
Patient sex Female 193 45.2 126 44.8 67 45.9Male 234 54.8 155 55.2 79 54.1 0.84
Patient age Median (range), y 30 (16 - 72) 28 (16 - 65) 32 (16 - 72)0.15- 29 213 49.9 150 53.4 63 43.230 - 39 106 24.8 65 23.1 41 28.1 40 - 49 52 12.2 30 10.7 22 15.1 50 - 56 13.1 36 12.8 20 13.7 0.21
Periods between Median (range), d 355.5 366 309 0.40diagnosis and BMT (138 – 827) (138 – 827) (232 – 682)
- 1 y 223 52.3 140 49.8 83 56.8 1 y - 203 47.5 140 49.8 63 43.2
Unknown 1 0.2 1 0.4 0 0.0 0.31HLA disparity Matched 131 30.7 96 34.2 35 24.0
Mismatched 269 63.0 162 57.6 107 73.3 1 allele 106 24.8 65 23.1 41 28.1
2 alleles or more 163 38.2 97 34.5 66 45.2Unknown 27 6.3 23 8.2 4 2.7 < 0.01*
Sex disparity Matched 253 59.2 171 60.8 82 56.1 M to F 107 25.1 66 23.5 41 28.1 F to M 67 15.7 44 15.7 23 15.8 0.56
ABO disparity Matched 198 46.4 131 46.6 67 45.9 Minor mismatched 102 23.9 67 23.8 35 24.0 Major mismatched 80 18.7 53 18.9 27 18.5
Both 46 10.8 29 10.3 17 11.6 Unknown 1 0.2 1 0.4 0 0.0 0.95
Harvested NCC Median/PtBW(Kg) 2.87×108 2.91×108 2.71×108 0.22Conditioning MAC 84 19.7 63 22.4 21 14.4
RIC 343 80.3 218 77.6 125 85.6 0.05*Usage of ATG No 199 46.6 129 45.9 70 48.0
Yes 226 52.9 151 53.7 75 51.3 Unknown 2 0.5 1 0.4 1 0.7 0.82
GVHD prophylaxis CyA-based 142 33.3 97 34.5 45 30.8 Tac-based 275 64.4 178 63.4 97 66.5 Unknown 10 2.3 6 2.1 4 2.7 0.71
Year of BMT - 2005 194 45.4 132 47.0 62 42.5 2006 - 233 54.6 149 53.0 84 57.5 0.38
Follow-up period Median (range), d 1,777 1,945 1,324 0.10(61 – 6,983) (61 – 6,012) (94 – 6,983)
BMT: bone marrow transplantation; HLA: human leukocyte antigen; M to F: male to female; F to M: female to male; NCC: nucleated cell count; PtBW: body weight of patients; MAC:myeloablative conditioning; RIC: reduced-intensity conditioning; ATG: anti-thymocyte or T-cell globulin; GvHD: graft- versus-host disease; CyA: cyclosporine; and Tac: tacrolimus.*Indicates statistically significant (P<0.05).

events.24 On the other hand, RIC consisted of CY (200mg/kg), TBI (2-4 Gy), and/or fludarabine (100-120 mg/m2)with or without ATG (Thymoglobulin), 2.5-10 mg/kg, orrabbit anti-human T-cell immunoglobulin (ATG-F), 10-25mg/kg. GvHD prophylaxis was composed ofcyclosporine- and tacrolimus-based regimens, and bothwere usually coupled with short-term methotrexate(98.6% and 94.2%, respectively). There was no significantdifference between distribution of donor age according toyear of BMT (before vs. after 2006).
Overall survival was significantly worse following BMTfrom older donorsOverall survival of the older donor group was inferior to
that of the younger donor group (65.9% vs. 77.7% at 1year, 54.3% vs. 71.7% at 5 years after BMT) (Figure 1).This difference was significant in the univariate analysis[hazard ratio (HR)] of overall mortality in the older donorgroup compared to the younger donor group, 1.65; 95%confidence interval (CI) 1.19-2.29; P<0.01] (Table 2).Among other variables, older age of patients (≥30 years)(Online Supplementary Figure S2), ABO blood type majormismatch, GvHD prophylaxis with cyclosporine, andBMT before 2006 were associated with a worse survival(P<0.1). In the multivariate analysis, including these fac-tors and the other known confounders (HLA disparity and
conditioning regimens), the older donor group showedsignificantly higher overall mortality (HR 1.64; 95%CI:1.15-2.25; P<0.01) (Table 2).This inferiority of OS in the older donor group (i.e. supe-
riority in the younger donor group) was observed in eachsubgroup according to patient characteristics, with adjust-ed HRs being more than 1 in almost all subgroups (Figure2). This tendency was also confirmed when we confinedthe analysis to only more recent cases (BMT after 2006)transplanted within one year after diagnosis using RICregimen including ATG (n=128; adjusted HR 2.03; 95%CI:0.94-4.39; P=0.07). Moreover, we compared OS betweeneach donor group using Kaplan-Meier curves stratified bysubgroups of patient age, HLA disparity, and conditioningregimens (Online Supplementary Figure S3), because patientage is a known strong prognostic factor,2 and HLA andconditioning regimens were statistically related to donorage in this cohort (Table 1). Differences in survival accord-ing to donor age were also apparent in each subgroup.When treating donor age as a continuous variable (sup-
posing that the increase of one year in donor age has thesame impact on OS), it is significantly related to poorer OSin multivariate analyses adjusted by confounding factors(HR 1.03; 95%CI: 1.01-1.05 per one year increase in age,P<0.01; HR 1.36; 95%CI: 1.08-1.70 per 10 years increasein age) (Online Supplementary Table S2), supporting our
Prognosis and donor age in unrelated BMT for AA
haematologica | 2016; 101(5) 647
Table 2. Overall mortality according to each variable before BMT.Variables Univariate analysis Multivariate analysis
HR 95%CI P HR 95%CI P
Donor age Younger 1.00 reference 1.00 referenceOlder 1.65 1.19 - 2.29 < 0.01* 1.64 1.15 – 2.35 < 0.01*
Patient sex Female 1.00 referenceMale 1.07 0.77 - 1.49 0.68
Patient age - 29 y 1.00 reference 1.00 reference30 y - 1.59 1.14 – 2.21 < 0.01* 1.97 1.34 – 2.90 < 0.01*
Periods between diagnosis and BMT - 1 y 1.00 reference1 y - 1.00 0.72 - 1.38 0.98
HLA disparity Matched 1.00 reference 1.00 referenceMismatched 1.22 0.84 - 1.77 0.29 1.29 0.87 – 1.90 0.21
Sex disparity Matched 1.00 referenceM to F 1.35 0.93 - 1.97 0.12F to M 1.32 0.84 - 2.05 0.23
ABO disparity Matched 1.00 reference 1.00 referenceMinor mismatched 1.25 0.83 - 1.87 0.29 1.31 0.85 – 2.02 0.22Major mismatched 1.46 0.96 - 2.23 0.08 1.53 0.97 – 2.43 0.07
Both 1.06 0.59 - 1.90 0.85 1.31 0.71 – 2.42 0.39Harvested NCC Lower 1.00 reference
Higher 1.14 0.82 - 1.58 0.43Conditioning MAC 1.00 reference 1.00 reference
RIC 0.86 0.58 - 1.26 0.45 0.80 0.51 – 1.26 0.34Usage of ATG No 1.00 reference
Yes 0.85 0.61 - 1.18 0.32GVHD prophylaxis CyA-based 1.00 reference 1.00 reference
Tac-based 0.69 0.49 - 0.97 0.04* 0.73 0.50 – 1.08 0.11Year of BMT - 2005 1.00 reference 1.00 reference
2006 - 0.62 0.44 - 0.87 < 0.01* 0.62 0.42 – 0.93 0.02*HR: hazard ratio; CI: confidence interval. BMT: bone marrow transplantation; HLA: human leukocyte antigen; M to F: male to female; F to M: female to male; NCC: nucleated cellcount; MAC: myeloablative conditioning; RIC: reduced-intensity conditioning; ATG: anti-thymocyte or T-cell globulin; GvHD: graft- versus-host disease; CyA: cyclosporine; and Tac:tacrolimus.*Indicates statistically significant (P<0.05).

findings obtained by analyses treating donor age as thebinary variable, and indicating that donor age is the inde-pendent risk factor.The causes of mortality were summarized and com-
pared between the two donor groups (Table 3). The majorcauses included infection and organ failure in both groups,and the incidence of fatal infections, especially bacterialinfections, was significantly higher in the older donorgroup (13.7% vs. 7.5%; P=0.03). The reasons for mortalitybeyond one year after BMT were also summarized,because OS decreased during this period, especially in theolder donor group. GvHD, infections, and organ failureswere more often documented in patients transplantedfrom older donors, though no significant differences weredetected because of the relatively smaller number ofpatients (Online Supplementary Table S3).
Poorer engraftment and higher incidence of aGvHDwere associated with older donorsIn order to address the causes underlying the differ-
ences in OS and mortality between the younger and theolder donor groups, we compared clinical coursesbetween donor age groups, with a particular focus onengraftment and GvHD because they are critical param-eters that may determine the prognosis of patients withAA after BMT.2 As for engraftment, the older donor group showed a sig-
nificantly lower proportion of neutrophil and plateletengraftment following BMT (Table 4 and Figure 3A).Primary engraftment failure was more frequentlyobserved in the older donor group than in the youngerdonor group (9.7% vs. 5.0%, HR 1.15; P<0.01). Neutrophil
engraftment or engraftment failure was still significantlyhigher in the older donor group after multivariate analysesadjusted for confounding factors such as patient age, HLAdisparity, ABO disparity, harvested NCC, conditioningregimens, and GvHD prophylaxis (adjusted P=0.01)(Table 4). When treating donor age as the continuous vari-able, adjusted HR of engraftment failure per 1-yearincrease in age is 1.01 (95%CI: 1.003-1.03; P<0.01) andthis is 1.16 per 10-year increase (95%CI: 1.03-1.32).With regard to GvHD, grade II-IV aGvHD was signifi-
cantly more frequent in the older donor group (27.1% vs.19.7%; adjusted HR 1.56; P=0.03) (Table 4 and Figure 3B),while there was no significant difference in grade III-IVaGvHD between groups (8.3% vs. 6.9%; adjusted HR1.32; P=0.45); in addition, the incidence of cGvHD wasalmost the same in both groups (24.6% vs. 27.8%; adjust-ed HR 0.91; P=0.66) (Table 4 and Figure 3B). The incidenceof grade II-IV aGvHD in the older donor groups comparedwith the younger donor groups was analyzed in varioussubgroups; the older donor group showed a tendency tohave higher incidence of aGvHD in many subgroups, withhigher HRs in older patients and HLA-mismatched trans-plantation (adjusted HR 2.07 and 1.61, respectively).Among patients diagnosed with grade II aGvHD, 33.5%of them were refractory to the primary corticosteroidadministration, requiring the stronger immunosuppressivetherapies for longer periods, while 66.6% in grade III-IVaGvHD patients underwent secondary therapies.When treating donor age as the continuous variable,
adjusted HR of grade II-IV aGvHD per 1-year increase inage is 1.03 (95%CI: 1.01-1.05; P=0.01) and per 10-yearincrease is 1.34 (95%CI: 1.05-1.72).
Y. Arai et al.
648 haematologica | 2016; 101(5)
Figure 1. Prognosis after BMT in each donor age group. Overall survival (OS) iscalculated with the Kaplan-Meier method in each donor age group, and com-pared with Cox proportional hazards model after being adjusted for confound-ing factors (see Table 2).
Table 3. Comparisons of the causes of mortality in each age group ofdonors.
Younger donor Older donor P(n=281) (n=146)
N % N %
Infection 21 7.5 20 13.7 0.03*Bacteria 10 11Virus 3 2Fungus 8 4Organ failure 23 8.2 17 11.6 0.24Lung 9 4CNS 4 1Liver 2 5Heart 4 4Kidney 3 3GvHD 7 2.5 5 3.4 0.56Acute 5 1chronic 2 4Graft failure 4 1.4 4 2.7 0.34TMA/VOD 2 0.7 2 1.4 0.60Hemorrhage 11 3.9 7 4.8 0.67Secondary malignancy 6 2.1 1 0.7 0.26Others/unknown 9 3.2 7 4.8Total 83 29.5 63 43.2
Any fatal infections and organ failures following GvHD or other post-transplant compli-cations are all categorized into “infection” and “organ failure” as the causes of NRM. TMA: thrombotic microangiopathy; VOD: veno-occlusive disease; CNS: central nervoussystem; GvHD: graft- versus-host disease. *Indicates statistically significant (P<0.05).

Impact of aGvHD on overall survival and its relationship to mortalityIt has been thought that complications with aGvHD
may directly result in poor OS in patients with AAbecause the graft-versus-host effect does not have the samemerit as the graft-versus-leukemia effects observed intransplant for leukemia.17 To confirm this hypothesis inour cohort, we determined OS regarding aGvHD as atime-dependent covariate;25 as a result, aGvHD (grade II-IV) showed a tendency towards a worse prognosis in thewhole cohort (adjusted HR 1.42; 95%CI: 0.95-2.11;P=0.08) and in both donor age groups. Landmark analysis(on day 30 or day 60 after BMT) also showed a trendtowards a worse survival in patients with aGvHD (data notshown). Poor response to immunosuppressive therapieseven in grade II aGvHD can support these data, and thehigher incidence of aGvHD in the older donor group maypartially account for the worse prognosis in this group dueto cases of infection and organ failure (Table 3 and OnlineSupplementary Table S3).
Discussion
This cohort study regarding donor age and prognosis ofunrelated BMT for AA revealed three major findings: 1)OS in transplantation from older donors (>40 years old)was significantly worse than that from younger donors; 2)neutrophil and platelet engraftment was suppressed andengraftment failure was more often observed followingtransplant from older donors; and 3) the older donor grouphad a higher incidence of aGvHD.First, we clearly showed an inferior prognosis in the
older donor group compared to the younger donor group.This result was confirmed by multivariate and varioussubgroup analyses, in order to exclude the influence ofconfounding factors, such as patient age, HLA disparity,conditionings, etc. Our data indicate that older donor agecan be considered an independent risk factor for poorprognosis after unrelated BMT for AA irrespective ofwhether it is treated as the binary covariate or the contin-uous covariate. It should be emphasized that donor age
Prognosis and donor age in unrelated BMT for AA
haematologica | 2016; 101(5) 649
Figure 2. Subgroup analyses of overall survival(OS) with respect to patients’ pre-transplant char-acteristics. OS is compared in each subgroup withrespect to pre-transplant patients’ characteristics.Hazard ratios (HRs) of overall mortality in the olderdonor group are shown in comparison with theyounger donor group (i.e. HR >1 indicates better OSin the younger donor group). Black dots: HRs. Blackbars: 95%CI ranges. CyA: cyclosporine; Tac:tacrolimus.

was not correlated with patient age (which is the strongestprognostic factor2) either in our whole cohort (OnlineSupplementary Figure S1) or in any subgroup of patients’characteristics, such as sex, HLA disparity, conditioningregimens, GvHD prophylaxis, and year of BMT. As far aswe know, so far there have been no reports of a relation-ship between donor age and prognosis in AA patients.This difference in prognosis can be explained in part by
the significantly higher incidence of fatal infection (espe-cially bacterial infection) in the older donor group (Table3), which may have been due to insufficiency or dysfunc-tion of immune cells derived from older donor grafts.Actually, this speculation is supported by previous studiesin mice indicating that recovery of the absolute number oflymphocytes in the early post-transplant period wasdelayed in recipients transplanted from older donors evenafter bone marrow engraftment, suggesting the delayedrecovery of cytotoxic T cells and immunoglobulin-secret-ing B cells (leading to hypogammaglobulinemia).26,27Moreover, suppression of neutrophil function was shownin neutrophils from aged donors due to the decrease insecondary messenger generation, such as diacylglyceroland inositol-triphosphate, and the defect in superoxidegeneration which is essential for bacterial killing.28
Unfortunately, there were no data on lymphocyte charac-teristics and neutrophil function in our dataset, but ourepidemiological data and biological studies in mice sug-gest that controlling severe infection, especially bacterialinfection, might be a key issue in improving prognosis fol-lowing transplantation from older donors.Next, we observed a relationship between older donor
grafts and a higher incidence of primary graft failure inboth analyses, whether treating donor age as the binary oras the continuous variables. Engraftment of donor grafts isan essential factor in transplantation in AA.2 The inferiori-ty in engraftment with older donors in combination withpoor recovery of CD4+ naïve T cells and B cells mentionedabove may increase opportunistic infections and accountfor the worse OS. In addition, higher transplant-relatedmortality following salvage secondary transplant afterengraftment failure generally results in an even worseprognosis.Poor engraftment with older donors has also been
shown in murine transplant models,5,6 and this kind of“aging” in grafts from older donors may be related to age-associated modifications in DNA methylation patterns29and/or shorter length of telomeres in hematopoietic stemor progenitor cells from older donors.30
Y. Arai et al.
650 haematologica | 2016; 101(5)
Figure 3. Cumulative incidence of engraft-ment and graft-versus-host disease (GvHD)in each donor age group. Incidence of (A)neutrophil and platelet engraftment and (B)acute GvHD (aGvHD) (grade II-IV) and chronicGvHD (cGvHD) (all grades) are calculatedwith Gray’s method considering death or sal-vage transplantation (after graft failure) ascompeting risks. Fine-Gray proportional haz-ard models are used to compare these inci-dences; P values are adjusted according toconfounding factors, such as patient age,HLA disparity, ABO disparity, harvested NCC,conditionings, and GvHD prophylaxis.
A
B

Finally, we showed a higher incidence of aGvHD inolder donors in both analyses, whether treating donor ageas the binary or the continuous variable. This may beexplained by the higher ratio of memory T-cell to naïve T-cell subsets in older people;7 recent clinical studies haveshown that peripheral blood CD8+ effector memory Tcells are closely associated with aGvHD in humans,8,9 incontrast to previous findings in a murine model.31 Differentgene expression profiles regarding GvHD, such as trans-forming growth factor-β in CD4+ and CD8+ T cells, werealso shown in older donors.32 In our cohort, all the graftswere injected without ex vivo T-cell depletion; therefore, itmay be speculated that massive amounts of antigen-expe-rienced memory T cells (including those which can recog-nize and attack the recipient-specific major and/or minorhistocompatibility antigens) were injected, especially incases with older donors, which initiated an allo-reactionleading to aGvHD. At the same time, a hyper-acute phaseof aGvHD targeting the bone marrow niche may induceengraftment failure in BMT from an older donor.33 Thesespeculations suggest that appropriate use of ATG could behelpful in overcoming this disadvantage in choosing olderdonor-derived bone marrow grafts.The impact of aGvHD on OS is another important point
that needs to be discussed. In transplantation for hemato-logic malignancies, aGvHD, if not severe and beyond con-trol, can be an indicator for better survival because GvHDmay guarantee graft-versus-tumor effects that can sup-press post-transplant relapse.17 In AA, however, we con-firmed that GvHD, regardless of the severity, does nothave any beneficial effects on patients, and worsens prog-nosis; grade II-IV aGvHD was related to inferior OS inboth donor age groups, and grade III-IV aGvHD increasedmortality to an even greater extent (HR 3.19; P<0.01). Oneof the explanations for this inferior survival is the refrac-toriness of aGvHD in our cohort; more than 30% ofpatients were refractory to the initial steroid therapy evenin grade II aGvHD, and more than 60% of those withgrade III-IV required secondary immunosuppressive ther-apies. Therefore, the higher incidence of aGvHD follow-ing BMT from older donors may also explain the worseprognosis in this group.Graft-versus-host disease was selected as the main cause
of mortality in only a small number of patients, and therewas no difference between donor age groups (Table 3). It
is suspected that most of the patients who experiencedlong-term episodes of GvHD acquired fatal infection ororgan dysfunction after continuous immunosuppressivestatus due to the nature of the GvHD itself or its treat-ment.34,35 Among these patients, the main cause of mortal-ity was recorded as infection or organ failure in our data-base. In summary, we found the inferiority of older donors in
unrelated BMT for AA compared to younger donors(treated as the binary covariate; >40 years vs. >39 years orolder, or the continuous covariate), mainly because of thehigher incidence of engraftment failure and aGvHD in theformer group; these complications can induce fatal infec-tions. This analysis suggests that donor age should receivea special focus as criterion when multiple unrelated donorsare available for AA, and there should be a concertedeffort to recruit younger voluntary candidate BMT donors.Our study, however, was retrospective in design and wasconducted in only one country. In addition, due to the longperiod of patient recruitment, protocols were not neces-sarily compatible with the current guidelines in somepatients; the widely recommended protocol is to trans-plant as soon as possible after diagnosis with a condition-ing regimen including cyclophosphamide, ATG, and low-dose TBI.24 We confirm that our main results can be repro-duced in the subgroup analyses of patients who weretreated according to the current guidelines. Moreover, it isdifficult to carry forward a discussion regarding the choicebetween a younger unrelated donor and an older matchedsibling donor in a retrospective study; therefore, large-scale international prospective studies are needed to vali-date these results and to revise the donor selection algo-rithm for the future.
AcknowledgmentsThe authors would like to thank all the physicians and data
managers at the centers who contributed valuable data on trans-plantation to the Japan Society for Hematopoietic CellTransplantation (JSHCT), Japan Marrow Donor Program(JMDP), and TRUMP.
FundingThis study was supported by research funding from the
Ministry of Education, Science, Sports, and Culture in Japan toT. Kondo.
Prognosis and donor age in unrelated BMT for AA
haematologica | 2016; 101(5) 651
Table 4. Comparisons of clinical courses after BMT in each group of donors.Cumulative incidence (%) HR 95%CI P
Variables Younger Olderdonor donor
EngraftmentNeutrophil 95.0 90.3 0.77 0.63 – 0.94 0.01*Platelet 77.4 63.1 0.76 0.58 – 0.99 0.04*aGvHDGrade II – IV 19.7 27.1 1.56 1.04 – 2.37 0.03*Grade III – IV 6.9 8.3 1.32 0.64 – 2.74 0.45cGvHDAll 27.8 24.6 0.91 0.59 – 1.39 0.66Extensive 14.0 12.4 1.01 0.56 – 1.85 0.96
Hazard ratios (HR) and P values are adjusted with potential confounding factors, such as patient age, HLA disparity, ABO disparity, harvested NCC, conditionings, and graft- versus-host disease (GvHD) prophylaxis. aGVHD: acute GvHD; cGvHD: chronic GvHD. CI: confidence interval.*Indicates statistically significant (P<0.05).

References1. Marsh JC, Ball SE, Cavenagh J, et al.
Guidelines for the diagnosis and manage-ment of aplastic anaemia. Br J Haematol.2009;147(1):43-70.
2. Marotta S, Pagliuca S, Risitano AM.Hematopoietic stem cell transplantation foraplastic anemia and paroxysmal nocturnalhemoglobinuria: current evidence and rec-ommendations. Expert Rev Hematol.2014;7(6):775-789.
3. Gupta V, Eapen M, Brazauskas R, et al.Impact of age on outcomes after bone mar-row transplantation for acquired aplasticanemia using HLA-matched sibling donors.Haematologica. 2010;95(12):2119-2125.
4. Stern M, Passweg JR, Locasciulli A, et al.Influence of donor/recipient sex matchingon outcome of allogeneic hematopoieticstem cell transplantation for aplastic ane-mia. Transplantation. 2006;82(2):218-226.
5. Liang Y, Van Zant G, Szilvassy SJ. Effects ofaging on the homing and engraftment ofmurine hematopoietic stem and progenitorcells. Blood. 2005;106(4):1479-1487.
6. Kamminga LM, van Os R, Ausema A, et al.Impaired hematopoietic stem cell function-ing after serial transplantation and duringnormal aging. Stem Cells. 2005;23(1):82-92.
7. Miller RA. The aging immune system:primer and prospectus. Science.1996;273(5271):70-74.
8. Khandelwal P, Lane A, Chaturvedi V, et al.Peripheral Blood CD38 Bright CD8+Effector Memory T Cells Predict AcuteGraft-versus-Host Disease. Biol BloodMarrow Transplant. 2015;21(7):1215-1222.
9. Loschi M, Porcher R, Peffault de Latour R,et al. High number of memory t cells isassociated with higher risk of acute graft-versus-host disease after allogeneic stemcell transplantation. Biol Blood MarrowTransplant. 2015;21(3):569-574.
10. Kollman C, Howe CW, Anasetti C, et al.Donor characteristics as risk factors inrecipients after transplantation of bonemarrow from unrelated donors: the effectof donor age. Blood. 2001;98(7):2043-2051.
11. Carreras E, Jimenez M, Gomez-Garcia V,et al. Donor age and degree of HLAmatching have a major impact on the out-come of unrelated donor haematopoieticcell transplantation for chronic myeloidleukaemia. Bone Marrow Transplant.2006;37(1):33-40.
12. Mehta J, Gordon LI, Tallman MS, et al.Does younger donor age affect the out-come of reduced-intensity allogeneichematopoietic stem cell transplantation forhematologic malignancies beneficially?Bone Marrow Transplant. 2006;38(2):95-100.
13. Fabre C, Koscielny S, Mohty M, et al.Younger donor's age and upfront tandemare two independent prognostic factors forsurvival in multiple myeloma patientstreated by tandem autologous-allogeneicstem cell transplantation: a retrospectivestudy from the Societe Francaise de Greffede Moelle et de Therapie Cellulaire (SFGM-TC). Haematologica. 2012;97(4):482-490.
14. Finke J, Schmoor C, Bethge WA, et al.Prognostic factors affecting outcome afterallogeneic transplantation for hematologi-cal malignancies from unrelated donors:results from a randomized trial. Biol BloodMarrow Transplant. 2012;18(11):1716-1726.
15. Servais S, Porcher R, Xhaard A, et al. Pre-transplant prognostic factors of long-termsurvival after allogeneic peripheral bloodstem cell transplantation with matchedrelated/unrelated donors. Haematologica.2014;99(3):519-526.
16. Arai Y, Aoki K, Takeda J, et al. Clinical sig-nificance of high-dose cytarabine added tocyclophosphamide/total-body irradiationin bone marrow or peripheral blood stemcell transplantation for myeloid malignan-cy. J Hematol Oncol. 2015;8(102).
17. Arai Y, Takeda J, Aoki K, et al. Efficiency ofhigh-dose cytarabine added to CY/TBI incord blood transplantation for myeloidmalignancy. Blood. 2015;126(3):415-422.
18. Olsson R, Remberger M, Schaffer M, et al.Graft failure in the modern era of allogeneichematopoietic SCT. Bone MarrowTransplant. 2013;48(4):537-543.
19. Atsuta Y, Suzuki R, Yoshimi A, et al.Unification of hematopoietic stem celltransplantation registries in Japan andestablishment of the TRUMP System. Int JHematol. 2007;86(3):269-274.
20. Kanda J, Fuji S, Kato S, et al. Decision analy-sis for donor selection in stem cell trans-plantation-HLA-8/8 allele-matched unrelat-ed donor vs HLA-1 AG mismatched relateddonor. Blood Cancer J. 2014;4:e263.
21. Giralt S, Ballen K, Rizzo D, et al. Reduced-intensity conditioning regimen workshop:defining the dose spectrum. Report of aworkshop convened by the center for inter-national blood and marrow transplantresearch. Biol Blood Marrow Transplant.2009;15(3):367-369.
22. Glucksberg H, Storb R, Fefer A, et al.Clinical manifestations of graft-versus-hostdisease in human recipients of marrowfrom HL-A-matched sibling donors.Transplantation. 1974;18(4):295-304.
23. Filipovich AH, Weisdorf D, Pavletic S, et al.National Institutes of Health consensusdevelopment project on criteria for clinicaltrials in chronic graft-versus-host disease: I.Diagnosis and staging working group
report. Biol Blood Marrow Transplant.2005;11(12):945-956.
24. Socie G. Allogeneic BM transplantation forthe treatment of aplastic anemia: currentresults and expanding donor possibilities.Hematology Am Soc Hematol EducProgram. 2013;2013:82-86.
25. Iacobelli S, Committee ES. Suggestions onthe use of statistical methodologies in stud-ies of the European Group for Blood andMarrow Transplantation. Bone MarrowTransplant. 2013;48 Suppl 1:S1-37.
26. Hirayama M, Azuma E, Jiang Q, et al. Thereconstitution of CD45RBhiCD4+ naive Tcells is inversely correlated with donor agein murine allogeneic haematopoietic stemcell transplantation. Br J Haematol.2000;111(2):700-707.
27. Azuma E, Hirayama M, Yamamoto H,Komada Y. The role of donor age in naiveT-cell recovery following allogeneichematopoietic stem cell transplantation:the younger the better. Leuk Lymphoma.2002;43(4):735-739.
28. Lipschitz DA, Udupa KB, Indelicato SR,Das M. Effect of age on second messengergeneration in neutrophils. Blood.1991;78(5):1347-1354.
29. Poglio S, Cahu X, Uzan B, et al. Rapid child-hood T-ALL growth in xenograft modelscorrelates with mature phenotype and NF-kappaB pathway activation but not withpoor prognosis. Leukemia. 2015;29(4):977-980.
30. Gadalla SM, Wang T, Haagenson M, et al.Association between donor leukocytetelomere length and survival after unrelatedallogeneic hematopoietic cell transplanta-tion for severe aplastic anemia. JAMA.2015;313(6):594-602.
31. Moncrieffe H, Coles M, Stockinger B. Theinfluence of CD4 T-cell subsets on controlof CD4 T-cell-mediated graft-versus-hostdisease. Immunology. 2008;125(4):459-468.
32. Baron C, Somogyi R, Greller LD, et al.Prediction of graft-versus-host disease inhumans by donor gene-expression profil-ing. PLoS Med. 2007;4(1):e23.
33. Shono Y, Ueha S, Wang Y, et al. Bone mar-row graft-versus-host disease: early destruc-tion of hematopoietic niche after MHC-mis-matched hematopoietic stem cell transplan-tation. Blood. 2010;115(26):5401-5411.
34. Arai Y, Yamashita K, Mizugishi K, et al.Risk factors for hypogammaglobulinemiaafter allo-SCT. Bone Marrow Transplant.2014;49(6):859-861.
35. Arai Y, Yamashita K, Mizugishi K, et al.Serum neutrophil extracellular trap levelspredict thrombotic microangiopathy afterallogeneic stem cell transplantation. BiolBlood Marrow Transplant. 2013; 19(12):1683-1689.
Y. Arai et al.
652 haematologica | 2016; 101(5)