gut microbiota protects against gastrointestinal...
TRANSCRIPT

Microenvironment and Immunology
Gut Microbiota Protects against GastrointestinalTumorigenesis Caused by Epithelial Injury
Yu Zhan1, Po-Ju Chen1, WilliamD. Sadler1, FuyuanWang1, Sara Poe3, Gabriel N�u~nez2, Kathryn A. Eaton4, andGrace Y. Chen1
AbstractInflammation is a critical player in the development of both colitis-associated and sporadic colon cancers.
Several studies suggest that the microbiota contribute to inflammation and tumorigenesis; however, studies tounderstand the role of the microbiota in colon tumor development in germ-free (GF) mice are limited. Wetherefore studied the effects of the microbiota on the development of inflammation and tumors in GF andconventionally raised specific pathogen-free (SPF) mice treated with azoxymethane (AOM) and dextran sulfatesodium (DSS).We discovered that GFmice developed significantlymore and larger tumors comparedwith that inSPF mice after AOM and DSS treatment despite the lack of early acute inflammation in response to chemicallyinduced injury by DSS. Although the extent of intestinal epithelial damage and apoptosis was not significantlydifferent in GF and SPF mice, there was a delay in intestinal epithelial repair to DSS-induced injury in GF miceresulting in a late onset of proinflammatory and protumorigenic responses and increased epithelial proliferationand microadenoma formation. Recolonization of GF mice with commensal bacteria or administration oflipopolysaccharide reduced tumorigenesis. Thus, although commensal bacteria are capable of driving chronicinflammation and tumorigenesis, the gut microbiota also have important roles in limiting chemically inducedinjury and proliferative responses that lead to tumor development. Cancer Res; 73(24); 7199–210. �2013 AACR.
IntroductionColorectal cancer is the third most common cancer in the
United States. One of the major risk factors for the develop-ment of colorectal cancer is the presence of chronic inflam-mation, which occurs in patients with inflammatory boweldisease (1). Even in cases of sporadic colon cancer, inflamma-tory mediators have clearly been associated with tumor pro-motion within the tumor microenvironment (2, 3). Recently,there has been significant interest in the role of the gutmicrobiota in the development of intestinal inflammation andcancer. Epithelial barrier defects associated with adenomaformation in mice harboring the ApcMin/þ mutation inCDX2-expressing colon cells result in bacterial translocationinto tumors and enhancement of inflammation-mediatedtumor growth, suggesting that the gut microbiota promoteinflammation important for tumor progression (4). Severalstudies also suggest that disruption of the normal microbiota
that results in dysbiosis is associated with colitis and carci-nogenesis (5–7). Thus, the current dogma is that the gutmicrobiota contributes to colitis and tumorigenesis, which isconsistent with observations that inflammation and tumordevelopment in several mouse models is abrogated in germ-free conditions or with antibiotic depletion of intestinalmicrobes (8–11). Notably, both interleukin (IL)-2–deficientand IL-10–deficient mice, which under conventional condi-tions develop spontaneous colitis, have significantly reducedor absent intestinal inflammation in germ-free conditions (12,13), and furthermore, deficiency in MyD88, an adaptor proteindownstream of Toll-like receptor (TLR) signaling that isinvolved in bacterial sensing, ameliorated both inflammationand tumor development in IL-10–deficient mice (8, 9). In theApcMin/þ mouse model of spontaneous colon tumorigenesis,deletion of theMyD88 gene results in fewer intestinal tumors aswell (14). Altogether, these studies suggest a detrimental effectby the gut microbiota in promoting intestinal inflammationand tumorigenesis. However, a beneficial role for commensalbacteria in suppressing carcinogenesis has also been demon-strated. For example, Lactobacillus and Bifidobacterium havebeen shown to have anticarcinogenic effects through suchactivities as enyzmatic detoxification of carcinogens, produc-tion of short-chain fatty acids that promote intestinal homeo-stasis, and regulation of epithelial proliferation and apoptosis(15). Similarly, TLR signaling, presumably through commensalbacteria, has been implicated in increased resistance to chem-ically induced colitis and promotion of intestinal epithelialrepair (16, 17). In addition, mice deficient in bacterial sensors,such as members of the Nod-like receptor (NLR) family have
Authors' Affiliations: 1Division of Hematology andOncology, Departmentof Internal Medicine, 2Department of Pathology, Comprehensive CancerCenter, 3Unit for Laboratory Animal Medicine, and 4Department of Micro-biology and Immunology, University of Michigan Medical School, Univer-sity of Michigan, Ann Arbor, Michigan
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
Corresponding Author: Grace Y. Chen, University of Michigan CancerCenter, 1500 EastMedical Center Drive, Ann Arbor, MI 48109. Phone: 734-936-5310; Fax: 734-647-9654; E-mail: [email protected]
doi: 10.1158/0008-5472.CAN-13-0827
�2013 American Association for Cancer Research.
CancerResearch
www.aacrjournals.org 7199
on June 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 28, 2013; DOI: 10.1158/0008-5472.CAN-13-0827

significantly more inflammation-induced tumors than wild-type mice (5, 18–23).
To determine the role of the gut microbiota in colontumorigenesis, we tested germ-free (GF) mice in the azoxy-methane (AOM)/dextran sulfate sodium (DSS)mousemodel ofinflammation-associated tumorigenesis. In this model, GF orconventional, specific pathogen-free (SPF) mice were given asingle intraperitoneal injection of the carcinogen, AOM, fol-lowed by multiple rounds of DSS, which injures the intestinalepithelial and induces colitis (24, 25). In contrast with othermouse models, we found that the presence of gut bacteria wascritical for suppressing tumorigenesis as GF mice developedmore tumors than SPF mice. The absence of commensalbacteria in GF mice was associated with poor inflammatoryresponses to resolve intestinal injury, resulting initially in ahypoproliferative epithelium and delayed regeneration of theepithelium. Epithelial proliferation did eventually occur in GFmice after DSS-induced injury, but was associated with sig-nificantly elevated proinflammatory and protumorigenic med-iators as well as abnormal epithelial restitution with micro-adenoma formation. The sterile inflammation that occurs inGFmice likely ismediated byMyD88-TRIF asGFmice deficientin both genes have fewer tumors. Our data suggest a criticalrole for the gut microbiota in promoting timely epithelialrepair in response to intestinal injury to prevent dysregulatedinflammation and epithelial proliferation. These findings aresignificant in that they demonstrate that commensal bacteriado not act solely as drivers of damaging inflammation andtumorigenesis, but highlight instead their beneficial role inmaintaining intestinal health and homeostasis to preventtumorigenesis.
Materials and MethodsMice
SPF C57BL/6J mice were originally purchased from JacksonLaboratory and bred in-house. GF C56BL/6J mice were alsooriginally obtained from Jackson Laboratory, rederived intoGFconditions, and bred and maintained GF in the University ofMichigan GF Mouse facility. GF MyD88-TRIF doubly deficientmice were obtained as a kind gift from Kathy McCoy. GF micewere housed in bubble isolators and are free of all bacteria,fungi, viruses, and parasites. Sterility was verified by regularinterval aerobic and anaerobic cultures as well as Gram stainsof feces and bedding. Both SPF and GFmice were fed the sameautoclaved chowdiet. Adult (6- to 12-week-old)micewere usedfor all experiments. All animal studies were approved by theUniversity Committee on Use and Care of Animals.
Tumor inductionMice were injected with 10 mg/kg AOM (Sigma-Aldrich) i.p.
on day 0 followed 5 days later by a 5-day course of 1% or 1.5%DSS depending on the lot of DSS in the drinking water. DSSwater was sterilized by 0.2 mm filtration. Mice were thenallowed to recover for 16 days with untreated drinking water.The 5 days of DSS followed by 16 days of untreated drinkingwater was repeated at least two times. Mice were sacrificed 3weeks after the last cycle of DSS for tumor counting. Tumors in
the colon were counted with the assistance of a magnifier andmeasured by calipers.
Assessment of inflammationColons were harvested from mice, flushed free of feces, and
jelly-rolled for formalin fixation and paraffin embedding. Five-micron sections were used for hematoxylin and eosin staining.Histologic assessment was performed in a blinded fashionusing a previously described scoring system, but modified asfollows (19). Sections were scored on a 3 to 4 point scale forthree parameters—inflammation/cellular infiltration, epithe-lial lesions, and epithelial regeneration—that were summedtogether. For inflammation, severity and distribution wereseparately assessed and combined into one score; assessmentof the epithelium was evaluated by averaging the severity ofcrypt loss or ulceration over 15 fields; epithelial hyperplasiawas scored on the basis of severity and distribution.
Apoptosis and proliferationColon sections from formalin-fixed, paraffin-embedded
were assessed for apoptotic cells by terminal deoxynucleotidyltransferase–mediated dUTP nick end labeling (TUNEL) assayusing the ApoAlert DNA Fragmentation Assay kit (ClontechLaboratories, Inc.). For tumors, number of apoptotic cells werecounted and averaged over 3 to 5 high power fields (HPF), andfor colon tissue sections of mice treated with DSS, the numberof apoptotic surface epithelial cells per crypt was counted overapproximately 150 crypts. Epithelial proliferation was assessedby Ki67 staining and proliferation index was assessed bycounting the number of Ki67þ cells per crypt in approximately50 well-aligned crypts.
Cytokine expressionColonic tissue was homogenized and total RNA isolated
using the Nucleospin RNA kit (Machery-Nagel). cDNA syn-thesis was performed using iScript (Bio-Rad) and cDNA wasused for quantitative PCR using the SYBR Green Master Mix(Applied Biosystems) on the ABI 7900HT (Applied Biosys-tems). Ct values were normalized to the housekeeping geneb-actin. Primer sequences are available in SupplementaryMethods.
Treatment of mice with lipopolysaccharideGFmice were administered sterilely filtered lipopolysaccha-
ride (LPS, E. coli O26:B6, Sigma) at 1 mg/mL in the drinkingwater beginning at 1 week before the administration of DSS(day-4 AOM/DSS protocol) and continued throughout theduration of the experiment. This concentration was selectedon the basis of the results of the study conducted by Rakoff-Nahoum and colleagues, which demonstrated decreased mor-tality of commensal-depleted SPFmicewith this concentrationof LPS (16).
Statistical analysisData are represented as means � SEM. Comparison of
tumor counts, cytokine expression, proliferation, and apopto-sis between SPF and GF mice were performed using anunpaired Student t test. The presence or absence of adenomas
Zhan et al.
Cancer Res; 73(24) December 15, 2013 Cancer Research7200
on June 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 28, 2013; DOI: 10.1158/0008-5472.CAN-13-0827

in SPF andGFmicewas assessed by a Fisher exact test. P values<0.05 were considered statistically significant.
ResultsThe gut microbiota is important for epithelial injury-associated colon tumor suppressionTodirectly interrogate the role of the gutmicrobiota in colon
tumorigenesis, we used a well-established inflammation-asso-ciated colon cancer model (24), which mimics human colitis-associated colon cancer, but also has features resemblingsporadic colon cancer, namely the prevalence of mutationsaffecting the Wnt signaling pathway and the progression ofadenomatous polyps to carcinomas (26, 27). In this model,mice are injected with a single intraperitoneal injection of theexperimental carcinogen AOM followed by repeated rounds ofwater containing DSS, which causes epithelial injury, increased
intestinal permeability, resulting in bacterial translocation intothe mucosa, and commensal-driven inflammatory responses.GF C57BL/6 mice are particularly susceptible to DSS-inducedinjury and we observed 100% mortality with 5 days of 2.5% or2% DSS together with AOM and was associated with completeloss of crypts in a significant proportion of the distal colonobserved microscopically in moribund GF mice (Supplemen-tary Fig. S1). However, with lower concentrations of DSS, 100%survival of GF mice can be achieved. After treatment withAOM/DSS, GF mice developed significantly more adenoma-tous tumors that were larger in size than that in conventionallyhoused SPF mice (Fig. 1A–C). As described previously withtumors associated with the AOM/DSS model, tumors in GFmice were premalignant adenomatous polys associated withnuclear b-catenin localization similar to that observed in SPFmice (Fig. 1D and Supplementary Fig. S2; ref. 26). Altogether,these results strongly suggest that the gut microbiota can
SPF GF
GF
SPF
0
2
4
6
8
10
12
WT SPF WT GF
Num
ber
of tu
mors
*
WT SPF
WT GF
Fra
ction o
f to
tal
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
<1 mm 1–2 mm >2 mm
*
BA
C
DTumor size
Figure 1. GF mice develop moretumors compared with SPF mice.A, representative photographs ofthe distal rectum and anus of SPFand GF mice after treatment withAOM and 4 cycles of 1.5% DSS.B, number of tumors in age- andsex-matched B6 GF (n ¼ 14) andSPFmice (n¼ 20). �,P < 0.05. Data,means � SEM. C, graph of tumorsize in GF and SPF mice afterAOM/DSS treatment. Data, means� SEM. D, representativemicrographs of adenomatoustumors in SPF and GF mice afterAOM/DSS treatment.Magnification, �200.
Intestinal Bacteria Suppress Tumorigenesis
www.aacrjournals.org Cancer Res; 73(24) December 15, 2013 7201
on June 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 28, 2013; DOI: 10.1158/0008-5472.CAN-13-0827

protect the host against the development of colon tumorssecondary to chemically induced epithelial injury and chal-lenges to genomic integrity by chemical carcinogenesis.
Increased tumorigenesis in GF mice is not associatedwith alterations in levels of epithelial apoptosis
Increased host susceptibility to inflammation-inducedtumorigenesis has been associated with increased epithelialdestruction that promotes excessive proinflammatory, pro-tumorigeneic responses (19, 22, 23). Alternatively, enhancedcellular survival may also lead to increased tumor develop-ment (18). To investigate the first possibility, we assessedlevels of DSS-induced apoptosis along the surface epitheliumof the colon during the first round of DSS (day 8), whichprecedes the development of mucosal erosion and ulcera-tion (19) and upon completion of DSS (day 10). At both ofthese time points, we observed similar numbers of apoptoticcells within the surface epithelium, suggesting no differencesin early DSS-induced damage in SPF and GF mice (Fig. 2A).Consistently, GF mice did not have significant losses inweight compared with SPF mice during the initial roundsof DSS (Supplementary Fig. S3). Similar to the early lesions,evaluation of tumors on day 98 after 4 rounds of DSS in SPFand GF mice also demonstrated no significant differences inlevels of apoptotic cells within tumors (Fig. 2B), suggestingthat the gut microbiota does not suppress tumor develop-
ment by affecting epithelial apoptosis either before or aftertumorigenesis.
GF mice exhibit impaired early inflammatory responsesto intestinal injury followed by delayed inflammationand production of proinflammatory, protumorigenicmediators
The development of tumors typically correlates with theextent of inflammation during the acute inflammatoryresponse after the first round of DSS (19, 21, 23). We, therefore,examined the colons of AOM/DSS-treated SPF and GF miceimmediately after the first round of DSS at the peak ofinflammatory responses and 1–2 weeks following when theepithelium has typically undergone restitution in mice in thismodel (19, 23). Inflammation was scored histologically basedon the extent of inflammatory cell infiltration, mucosalerosion, and extent of regenerating gland formation, or hyper-plasia (see Materials and Methods). During the acute inflam-matory phase (days 12–13), SPF mice had significantly higherhistologic scores (Fig. 3A). Consistently, SPF mice exhibitedincreased recruitment of inflammatory cells compared withthat in GF mice, particularly Gr1þ and CD11bþ cells, repre-sentative of both neutrophils and macrophages, within thecolon lamina propria (Supplementary Fig. S4), consistent withprevious reports (17). The increased histologic score andinflammatory cell infiltration in SPF mice was accompanied
0
5
10
15
20
25
30
35
40
45
GFSPF
Num
ber
of apopto
tic c
ells
/HP
F
A
BP = 0.54
SPF GF
Day 8
Day 10
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
Day 8 Day 10
Num
ber
of apopto
tic c
ells
/cry
pt
SPF
GF
Figure 2. Chemically induced earlyepithelial apoptosis and tumorapoptosis are not affected by thegut microbiota. A, representativemicrographs (�200) of colonsections from SPF and GF mice ondays 8 and 10 of AOM/1.5% DSStreatment (3 and 5 days after thestart of the first round of DSS,respectively) after TUNEL staining(left). Graph of number of apoptoticcells/crypt with approximately150 crypts counted (right). B,representative micrographs (�200;right) of tumor sections at the endof AOM/DSS treatment stained byTUNEL assay (top) and withpropidium iodide as a counterstain(bottom). Graph of average numberof TUNELþ cells per high powerfield (5 fields counted) in GF andSPF tumors (n ¼ 4–5 mice/each;left). Data, means � SEM.
Zhan et al.
Cancer Res; 73(24) December 15, 2013 Cancer Research7202
on June 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 28, 2013; DOI: 10.1158/0008-5472.CAN-13-0827

by an elevation in the production of inflammatory cytokinesand chemokines within the colon that are important forimmune cell recruitment and wound repair, such as CXCL1,MIP-2, IL-6, IL-22, and Reg3g as assessed by real-time PCR(Fig. 4). In contrast, upregulation of these cytokines andchemokines are significantly impaired during the acute inflam-matory phase in GF mice on day 12 (Fig. 4).In the AOM/DSSmodel, resolution of intestinal damage and
inflammation typically occurs 1 to 2 weeks after the first roundof DSS (23), just before the second round of DSS as reflected inthe decreasing histologic scores in SPF mice (Fig. 3A) and
evidence of regenerating epitheliumwith hyperplasia on day 13(Fig. 3B). GFmice, on the other hand, continue to demonstrateevidence of persistent intestinal damage on day 13 (Fig. 3B)with loss of crypts and absence of hyperproliferative epithe-lium. By the second week on day 18 or day 26 just before thesecond round of DSS, SPF mice have nearly restituted theirepithelium back to baseline; however, the colons of GF micecontinue to have persistent mucosal damage and delayedformation of regenerating glands, resulting in higher histologicscores compared with SPF at these later time points (Fig. 3Aand B). Associated with the higher histologic scores for GF
SPF
Day 0
Day 26
Day 13
Day 18
GFB
1
2
3
4
5
6
7
8
9
10Hyperplasia
Epithelium
Inflammation
0
Day 0 Day 12 Day 13 Day 18 Day 26
His
tolo
gic
score
Day 8 Day 10SPF SPFSPFSPFSPFSPFGF GFGFGFGFGFGFSPF
*
**
*
*
A
Figure 3. GF mice exhibit delayedcolonic inflammation. A, histologicscores encompassing extent ofinflammation, epithelial damage,andepithelial hyperplasia inGFandSPF mice (n ¼ 5 mice/group/timepoint) at various time pointsduring AOM/DSS treatment. B,representative micrographs (�200)of colons harvested from age- andsex-matched GF and SPF mice atvarious time points during AOM/DSS treatment.Day13, 3daysafterthe completion of the first round ofDSS; Day 26, start of the secondround of DSS. Arrows, mucosalerosion with inflammatory cellinfiltration; double arrows,submucosal edema; dashedarrows, regenerating epithelium/hyperplasia. �, P < 0.05; Data,means � SEM.
Intestinal Bacteria Suppress Tumorigenesis
www.aacrjournals.org Cancer Res; 73(24) December 15, 2013 7203
on June 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 28, 2013; DOI: 10.1158/0008-5472.CAN-13-0827

mice, there is also a delayed, but significantly higher upregula-tion in proinflammatory mediators as well as factors involvedin epithelial remodeling and growth such as the matrix metal-loproteinase (MMP)-12, c-myc, and the EGF family memberepiregulin compared with that in SPF mice on day 17 (Fig. 4).
Delayed hyperproliferation in GFmice is associated withearly microadenoma formation
We next examined levels of epithelial proliferation in thecolons of SPF and GFmice early (day 12) and late (day 26) afterthe first round of DSS by Ki67 staining. During the acuteinflammatory phase immediately after completion of the firstround of DSS (day 12), when upregulation of inflammatory
cytokines and recruitment of immune cells occurred in SPFmice, there was an increased number of Ki67þ epithelial cellsassociated with epithelial regeneration and subsequent near-complete resolution of inflammation by day 26 (Fig. 5A). Incontrast, the colons of GF mice were in a hypoproliferativestate with no evidence of any epithelial regeneration imme-diately after completion of thefirst roundofDSS ondays 12 and13 (Fig. 5A), consistent with previous reports (17). However, onday 26, more than 2 weeks after completion of the first cycle ofDSS and just before the start of second round of DSS, when thecolons of SPF mice have essentially normalized morphologi-cally, we observed instead significantly elevated levels ofepithelial proliferation in GF mouse colons as demonstrated
0
5,000
10,000
15,000
20,000
Day 0 Day 12 Day 17
MIP-2
0
10
20
30
40
50
60
70
Day 0 Day 12 Day 17
ccl3
0
100
200
300
400
Day 0 Day 12 Day 17
Epiregulin
0
100
200
300
400
500
Day 0 Day 12 Day 17
IL-22
0
1,000
2,000
3,000
4,000
5,000
6,000
7,000
Day 0 Day 12 Day 17
*
**
0
200
400
600
800
1,000
Day 0 Day 12 Day 17
IL-6
0
50
100
150
200
250
300
350
Day 0 Day 12 Day 17
MMP12
0
5
10
15
20
Day 0 Day 12 Day 17
TNF-a
0
1
2
3
4
5
Day 0 Day 12 Day 17
c-myc
0
50
100
150
200
250
300
Day 0 Day 12 Day 17Rel
ativ
e m
RN
A e
xpre
ssio
n KC
SPF
GF
*
0
500
1,000
1,500
2,000
Day 0 Day 12 Day 17
Reg3g
IL-Ib
**
*
*
*
*
*
*
*
* **
*
*
*
Figure 4. GF mice have impairedinitial inflammatory responsesfollowed by delayed increase inproinflammatory, protumorigenicfactors. Relative mRNA expressionof various factors associated withintestinal repair and inflammationon day 12 (2 days after thecompletion of the first round ofDSS) and day 17 (1 week after thecompletion of the first round ofDSS) of AOM/1.5% DSS treatmentin age- and sex-matched GF andSPFmice asmeasuredby real-timePCR. Expression values werenormalized with respect tothe housekeeping gene b-actin(n¼ 5–6 mice/group). Data, means� SEM. �, P < 0.05; ��, P ¼ 0.09.
Zhan et al.
Cancer Res; 73(24) December 15, 2013 Cancer Research7204
on June 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 28, 2013; DOI: 10.1158/0008-5472.CAN-13-0827

by increased Ki67 staining within the epithelium (Fig. 5A).More importantly, the delayed hyperplasia in GF mice was notassociated with normalization of the epithelium; rather, weobserved formation of microadenomas within the mucosa ofGFmice by day 26 in the distal rectum that were not present inSPFmice (Fig. 5B), specificallywith nomicroadenomas presentin the SPF mice group and microadenomas present in 100% ofthe GF mice group (P < 0.05; Fisher exact test, n ¼ 5 mice/group). However, in established tumors, there were no differ-ences in proliferative activity between SPF and GF tumors(Supplementary Fig. S5). Together, these results suggest thatthe gut microbiota is important for promoting normal inflam-mation necessary for repair of damaged epithelium to preventaberrant and delayed inflammatory and epithelial growthresponses that lead to tumorigenesis.
GFmicedeficient inTLRreceptor signalinghave reducedtumorigenesisDespite the absence of bacterial-driven inflammatory
responses in GF mice, inflammation and the upregulation ofproinflammatory mediators still occur albeit late. In GF mice,this upregulation is clearly commensal-independent, andtherefore must arise from endogenous signals that may be
produced during tissue injury. TLRs, although primarily rec-ognized as bacterial sensors, are also capable of recognizingendogenous ligands that are released during cell death andinjury to mediate sterile inflammation (28–30). Moreover,MyD88 signaling is associated with induction of tumor-pro-moting factors and promotes spontaneous intestinal tumor-igenesis in ApcMin/þmice (14). We therefore hypothesized thatin GF mice, pathologic activation of TLR signaling duringsterile inflammation by persistent tissue injury results inincreased tumorigenesis. To test this hypothesis, we treatedB6 GF mice deficient in both MyD88 and TRIF (MyD88-TRIFDKO), adaptor proteins downstream of all TLRs, with AOM/DSS. Downregulation of all TLR signaling in GF MyD88-TRIFDKO was associated with reduced number and size of tumorscompared with that in GF wild-type (WT) mice (Fig. 6A–C)although was not sufficient to limit tumor development to thesame extent as that in SPFWTmice (Fig. 6A and B), suggestingthat other pathways are also involved in tumor suppression.
The gut microbiota and its products limit AOM/DSS-induced tumorigenesis in GF mice
Wenext determinedwhether recolonization ofGFmicewithcommensal bacteria by co-housing with SPF mice was
0
5
10
15
20
25
30
Day 0 Day 12 Day 26
Pro
lifera
tion index
SPF
GF
GFSPF
Day 0
Day 26
Day 12
A
B
*
*
Figure 5. Delayed proliferativeresponses in GF mice areassociated with earlymicroadenoma formation. A,representativemicrographs ofKi67immunoreactivity in colon sections(�200) from age- and sex-matchedGF and SPF mice at time pointsearly (day 12) and late (day 26) afterthe completion of the first round of1.5% DSS (N ¼ 4–5 mice/group/time point; left). Graph of numberof Ki67þ cells per crypt(approximately 30–50 cryptscounted) in GF and SPF mice atvarious time points after the firstround of DSS during AOM/DSStumor induction protocol (right).Student t test was used todetermine significance. �, P < 0.05;data, means � SEM. B,representative micrographs of day26 (just before the start of thesecond roundofDSS)Ki67-stainedGF colon sections at �100 (left)and �200 (right) magnification.Arrows, microadenomas.
Intestinal Bacteria Suppress Tumorigenesis
www.aacrjournals.org Cancer Res; 73(24) December 15, 2013 7205
on June 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 28, 2013; DOI: 10.1158/0008-5472.CAN-13-0827

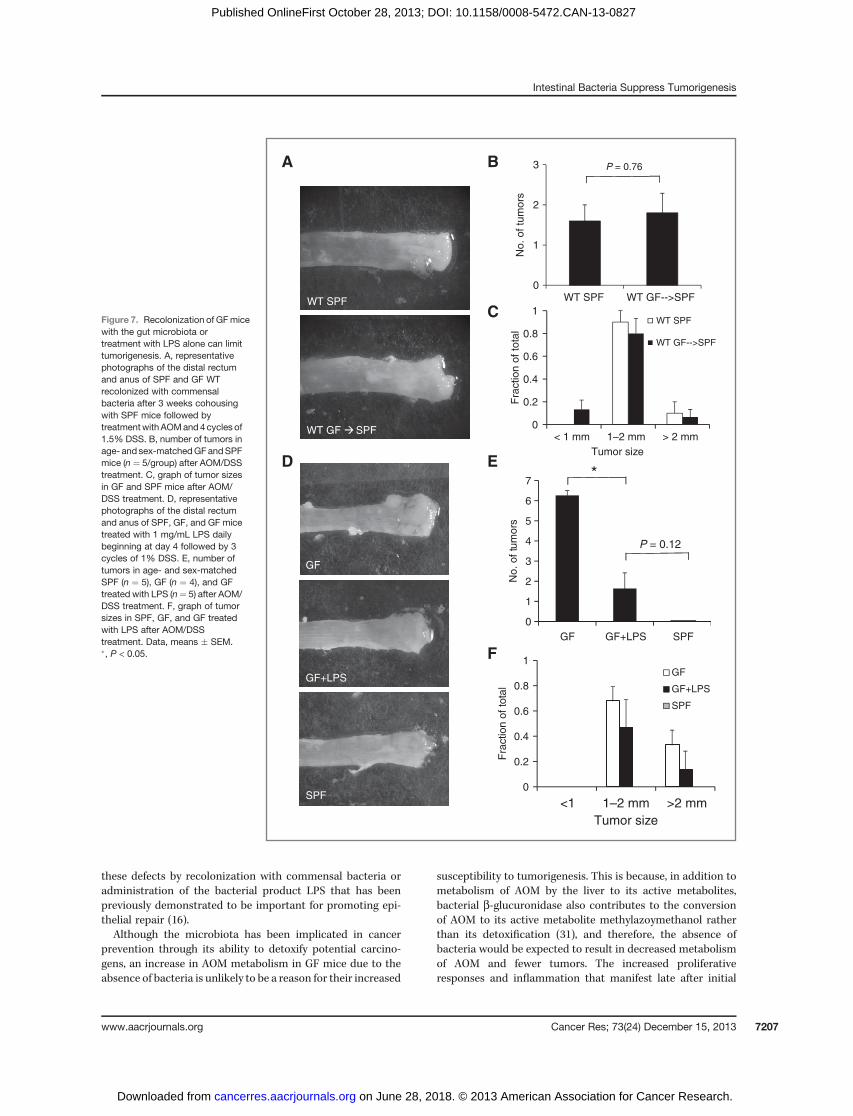
sufficient to protect mice fromDSS-induced injury and tumor-igenesis. After cohousing GF mice with SPF mice for 3 weeksfollowed by AOM/DSS treatment, 100% survival of conventio-nalized GFmice was achieved with 2%DSS that was previouslyassociated with 100% mortality in GF mice (SupplementaryFigs. S1 and S6A), and weight changes in conventionalized GFmice with AOM/DSS treatment more closely followed that ofSPF mice (Supplementary Fig. S6B). Importantly, the numberof tumors that developed in recolonized GF mice after AOM/DSS was no longer significantly different from that in SPFmice(Fig. 7A). Furthermore, tumors were similar in size betweenrecolonized GF and SPFmice (Fig. 7B). The similarity in tumordevelopment between conventionalized GFmice and SPFmicewas likely due to similar recovery times from DSS-inducedinjury as observed by insignificant differences in histologicscores after the first cycle of DSS (Supplementary Fig. S7A andS7B) and in the kinetics of proinflammatory/proliferativemarker induction as measured by real-time PCR between SPFand conventionalized GF mice (Supplementary Fig. S7C).These results suggest that colonization of GF mice by micro-biota is sufficient to limit DSS-induced injury and promotenormal inflammatory responses to restore epithelial restitu-tion and protect against tumorigenesis.
It has previously been demonstrated that LPS producedby commensal bacteria increases resistance to DSS-induced
injury by promoting inflammation and epithelial repair (16).We therefore wanted to determine whether administering LPSto GF mice would also protect against AOM/DSS-inducedtumorigenesis. Indeed, continuous administration of LPS inthe drinking water of GF significantly reduced the number oftumors in GF mice although the size of tumors that ultimatelydeveloped was not significantly different (Fig. 7E and F).
DiscussionIn this study, we used GF mice to determine the importance
of the gutmicrobiota in suppressing colon tumorigenesis usingthe AOM/DSSmodel.We demonstrated that inGFmice devoidof any microbiota, there is delayed upregulation of inflamma-tory responses associated with poor healing and restitution ofDSS-induced epithelial damage. Despite the initial hypoproli-ferative state observed in GF mice, there is eventually, even inthe absence of bacteria, a delayed induction of proinflamma-tory mediators and growth factors that leads to dysregulatedepithelial proliferation and microadenoma formation withoutcomplete epithelial restitution. This delayed upregulation ofproinflammatory and proliferative factors during sterileinflammation in GF mice likely occurs in part through MyD88and/or TRIF as GF MyD88-TRIF DKO mice developed fewertumors than GF WT mice. GF mice can also be rescued from
GF WT
SPF WT
GF MyD88-TRIF DKO
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
<1 mm 1–2 mm
Tumor size
>2 mm
Fra
ction o
f to
tal
GF WT
GF MyD88-TRIF DKO
* *
0
1
2
3
4
5
6
GF WT GF MyD88-
TRIF DKO
SPF WT
No. of tu
mors
*
**
A B
C
Figure 6. GF MyD88-TRIF DKOmice develop fewer tumors thanGF mice. A, representativephotographs of the distal rectumand anus of C57BL/6 SPF WT(n ¼ 9), GF WT (n ¼ 11), and GFMyD88-TRIFDKOmice (n¼9) aftertreatmentwithAOMand3cyclesof1% DSS. B, number of tumors inage- and sex-matched SPF WT,GF WT, and GF MyD88-TRIF DKOmice with AOM/DSS treatment. C,graph of tumor size in SPF WT, GFWT, and GF MyD88-TRIF DKOmice. Data, means � SEM. � and��, P < 0.05.
Zhan et al.
Cancer Res; 73(24) December 15, 2013 Cancer Research7206
on June 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 28, 2013; DOI: 10.1158/0008-5472.CAN-13-0827
these defects by recolonization with commensal bacteria oradministration of the bacterial product LPS that has beenpreviously demonstrated to be important for promoting epi-thelial repair (16).Although the microbiota has been implicated in cancer
prevention through its ability to detoxify potential carcino-gens, an increase in AOM metabolism in GF mice due to theabsence of bacteria is unlikely to be a reason for their increased
susceptibility to tumorigenesis. This is because, in addition tometabolism of AOM by the liver to its active metabolites,bacterial b-glucuronidase also contributes to the conversionof AOM to its active metabolite methylazoymethanol ratherthan its detoxification (31), and therefore, the absence ofbacteria would be expected to result in decreased metabolismof AOM and fewer tumors. The increased proliferativeresponses and inflammation that manifest late after initial
WT SPF
WT GF �SPF
0
1
2
3
WT GF-->SPFWT SPF
No. of tu
mors
Fra
ction o
f to
tal
A B
C
P = 0.76
0
0.2
0.4
0.6
0.8
1
< 1 mm 1–2 mm > 2 mm
WT SPF
WT GF-->SPF
Tumor size
0
0.2
0.4
0.6
0.8
1
1–2 mm<1 >2 mm
Fra
ction o
f to
tal
GF
GF+LPS
SPF
Tumor size
No. of tu
mors
GF
GF+LPS
SPF
D E
F
0
1
2
3
4
5
6
7
SPFGF+LPSGF
*
P = 0.12
Figure 7. Recolonization of GFmicewith the gut microbiota ortreatment with LPS alone can limittumorigenesis. A, representativephotographs of the distal rectumand anus of SPF and GF WTrecolonized with commensalbacteria after 3 weeks cohousingwith SPF mice followed bytreatmentwithAOMand4cycles of1.5% DSS. B, number of tumors inage- and sex-matchedGFandSPFmice (n ¼ 5/group) after AOM/DSStreatment. C, graph of tumor sizesin GF and SPF mice after AOM/DSS treatment. D, representativephotographs of the distal rectumand anus of SPF, GF, and GF micetreated with 1 mg/mL LPS dailybeginning at day 4 followed by 3cycles of 1% DSS. E, number oftumors in age- and sex-matchedSPF (n ¼ 5), GF (n ¼ 4), and GFtreated with LPS (n¼ 5) after AOM/DSS treatment. F, graph of tumorsizes in SPF, GF, and GF treatedwith LPS after AOM/DSStreatment. Data, means � SEM.�, P < 0.05.
Intestinal Bacteria Suppress Tumorigenesis
www.aacrjournals.org Cancer Res; 73(24) December 15, 2013 7207
on June 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 28, 2013; DOI: 10.1158/0008-5472.CAN-13-0827

DSS administration in GF mice are also unlikely to be due todifferences in levels of DSS-induced intestinal epithelial dam-age as epithelial apoptosis and resultant epithelial damageearly after the initial DSS treatment were not statisticallydifferent between SPF and GF mice (Figs. 2 and 3). Rather,the persistence of intestinal epithelial damage associated withimpaired activation of inflammatory, wound repair pathwayslikely results in inappropriate proliferative responses later onthat are further fueled by repeated DSS-induced damage andinflammation from additional cycles of DSS.
In our tumor studies with our colony of C56BL/6J GF mice,we reduced the concentration of DSS to enable GF mice tosurvive multiple rounds of DSS. At these lower concentrationsof DSS, our analysis of colons at multiple time points reveal anearly defect in inflammatory, wound-healing responses in GFmice that may have not been evident in other studies withother colonies of GF mice where higher concentrations of DSSresulted in significant damage and inflammation (32–34).Withour colony, concentrations above 2% resulted in 100% mor-tality, but examination of their colons histologically showedsignificant mucosal damage and submucosal edema (Supple-mentary Fig. S1). It would be interesting to determine whetherspecific bacterial populations previously demonstrated to haveprotective effects against colitis or bacterial products areeffective in increasing survival in our colony of GF mice withhigher concentrations of DSS, and whether the mechanisminvolves decreasing inflammation and damage or promotingtimely epithelial repair.
Our studies demonstrate an essential function for commen-sal bacteria in the prevention of colon tumorigenesis byfacilitating epithelial repair. These results are in contrast toearlier reports of decreased inflammation-associated tumor-igenesis in other mouse models such as the Il-10�/�/AOM orApcMin/þ mouse model in which under GF conditions, inflam-mation and tumorigenesis are abrogated in the absence ofbacteria (9, 35). The difference in outcome between these twomodels may be due to epithelial injury as a prominent featureof the AOM/DSS model, resulting in dependence on woundrepair pathways for limiting tumor development. Thus, withthe AOM/DSSmodel, in the context of chronic epithelial injury,intestinal bacteria are critical for triggering "normal" inflam-matory responses necessary for timely repair of injury andinhibition of tumorigenesis. Consistently, after the first cycle ofDSS, GF mice exhibited decreased levels of recruitment ofinflammatory cells (both Gr1þ and CD11bþ), representingboth neutrophils and macrophages, which have been demon-strated to be associated with effective wound repair and arepoorly recruited in GF andMyD88�/� mice after DSS-inducedintestinal injury (17, 36). Furthermore, GF mice that arerecolonized with commensal bacteria demonstrate upregula-tion of factors involved in epithelial repair and restitution ofthe epithelium similarly to SPFmice (Fig. 7 and SupplementaryFig. S7). LPS, a major component of intestinal bacteria that isrecognized by TLR4 and signals through the downstreamadaptorMyD88, has been previously demonstrated to promotethe induction of cytoprotective factors, such as CXCL1, TNF-a,and IL-6, during physiologic inflammatory responses to DSS-induced injury (16), and was also capable of reducing tumor
development in AOM/DSS-treated GF mice (Fig. 7D–F). It isalso important to note, however, that the LPS used in this studywas not highly purified, and may contain contaminatingbacterial components that signal through other pattern rec-ognition receptors (37). Furthermore, although the differencein tumors numbers in GF mice treated with LPS followed byAOM and 1% DSS was not statistically significantly differentfrom that in SPF mice with the number of experimental miceused, tumors still developed, whereas SPF mice developednone (Fig. 7E). It is therefore possible that other bacterialactivities will also contribute to epithelial repair and tumorsuppression. For example, other bacterial sensingmechanismssuch as through the NLRs are also important for promotingwound repair and curtailing aberrant inflammatory responsesduring colitis-associated tumorigenesis (5, 6, 19, 20, 22, 23, 38,39). Alternatively, the gut microbiota may also help promoteintestinal epithelial homeostasis through the production ofmetabolic byproducts such as short-chain fatty acids, ratherthan through its direct immuostimulatory activities. This isconsistent with studies demonstrating that short-chain fattyacids ameliorate DSS-induced colitis when administered to GFmice (33).
Despite the absence of bacterial-driven inflammatoryresponses in GF mice, sterile inflammation can still occur.However, this results in pathologic proliferation and earlymicroadenoma formation rather than epithelial restitution.This phenomenon is associated with upregulation of inflam-matory mediators, such as CXCL1, MMP12, IL-6, that althoughare important initially for wound repair, are also implicated intumor promotion (40–43). Similarly, IL-22, which was poorlyinduced in germ-free mice and is important for repair, issignificantly upregulated at later time points, which has beenassociated with tumor promotion (44). In addition, the aber-rant, late inflammatory response is associated with upregula-tion of factors such as c-myc and epiregulin, which are involvedin proliferation and tumorigenesis. In GF mice, this upregula-tionmust arise from endogenous signals thatmay be producedduring tissue injury in the absence of bacteria, resulting insterile inflammation. Our results suggest that these sterileinflammatory responses that may predispose to tumor devel-opment are mediated through MyD88 and TRIF as GFMyD88-TRIF DKO mice developed fewer tumors than GF WT mice.MyD88 and TRIF are adaptor proteins that are downstream ofthe TLRs, which in addition to recognizing bacteria, alsorespond to molecules released during cell death, as can occurwithDSS-induced injury (28–30).Moreover,MyD88 signaling isassociated with induction of tumor-promoting factors (14).Thus, our data suggests that in GF mice, persistent tissuedamage results in inappropriate, pathologic activation of theMyD88 and/or TRIF signaling pathway that promote sterileinflammation, epithelial proliferation, and tumorigenesis.Although MyD88 is downstream of TLRs, the IL-1R/IL-18Rpathways also utilizeMyD88 as an adaptor protein (45–47), andtherefore these non-TLR pathways may also be involved inpromoting inflammation and tumorigenesis in GF mice. Inaddition, since GF MyD88-TRIF DKO mice still developmore tumors compared with SPF WT mice, it is also likelythat other pathways that remain to be identified contribute to
Zhan et al.
Cancer Res; 73(24) December 15, 2013 Cancer Research7208
on June 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 28, 2013; DOI: 10.1158/0008-5472.CAN-13-0827

tumorigenesis. It is also interesting to note that SPFMyD88 KOmice have been previously reported to have more tumors thanSPF WTmice with a higher concentration of DSS than used inthe current study (48), and may be explained in part by thepresence of commensal bacteria driving inflammation andtumorigenesis in SPF MyD88 KO mice.Our findings highlight the importance of commensal-
driven inflammatory responses to properly initiate intestinalrepair responses in the presence of chemically inducedinjury that is critical for preventing late tumorigenesis. Whatwill be important to determine is whether specific bacterialpopulations or delivery of bacterial products aside from LPSare also capable of limiting tumorigenesis by promotingwound repair and the context by which these occur. Ourgerm-free model system will enable us to address thesequestions and also allow us to develop strategies thatharness the beneficial activities of the gut microbiota toprevent the development of dysregulated inflammation andcolon cancer.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: G. Nunez, G.Y. ChenDevelopment of methodology: P.-J. Chen, F. Wang, G.Y. ChenAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): Y. Zhan, F. Wang, S. Poe, K.A. Eaton, G.Y. ChenAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): P.-J. Chen, F. Wang, G. Nunez, K.A. Eaton, G.Y. ChenWriting, review, and/or revision of the manuscript: G. Nunez, K.A. Eaton,G.Y. ChenAdministrative, technical, or material support (i.e., reporting or orga-nizing data, constructing databases): P.-J. Chen, W.D. Sadler, F. Wang, S. PoeStudy supervision: S. Poe, G.Y. Chen
AcknowledgmentsThe authors thank theUniversity ofMichiganMicroscopy and ImagingAnalysis
and DNA Sequencing MicroArray Core Facilities, the Comprehensive CancerCenter Research Histology and Immunoperoxidase Laboratory, and the Unit forLaboratory AnimalMedicinePathologyCore for Animal Research for their services.
Grant SupportThis work was supported by the American Cancer Society Research Scholar
Grant, NIH K08 CA13318, R01 CA166879, and the American Recovery andReinvestment Act Supplement P30 CA4659-22S3 (G.Y. Chen).
The costs of publication of this article were defrayed in part by the payment ofpage charges. This article must therefore be hereby marked advertisementin accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
ReceivedMarch 22, 2013; revised September 23, 2013; acceptedOctober 9, 2013;published OnlineFirst October 28, 2013.
References1. Ekbom A, Helmick C, Zack M, Adami HO. Ulcerative colitis and
colorectal cancer. A population-based study. N Engl J Med 1990;323:1228–33.
2. Terzic J, Grivennikov S, Karin E, Karin M. Inflammation and coloncancer. Gastroenterology 2010;138:2101–14.
3. Schetter AJ, Nguyen GH, Bowman ED, Mathe EA, Yuen ST, HawkesJE, et al. Association of inflammation-related and microRNA geneexpression with cancer-specific mortality of colon adenocarcinoma.Clin Cancer Res 2009;15:5878–87.
4. Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, JauchD, et al. Adenoma-linked barrier defects and microbial productsdrive IL-23/IL-17-mediated tumour growth. Nature 2012;491:254–8.
5. Couturier-Maillard A, Secher T, Rehman A, Normand S, De ArcangelisA, Haesler R, et al. NOD2-mediated dysbiosis predisposes mice totransmissible colitis and colorectal cancer. J Clin Invest 2013;123:700–11.
6. Elinav E, Strowig T, KauAL, Henao-Mejia J, Thaiss CA, Booth CJ, et al.NLRP6 inflammasome regulates colonic microbial ecology and risk forcolitis. Cell 2011;145:745–57.
7. Sobhani I, Tap J,Roudot-Thoraval F, Roperch JP, Letulle S, LangellaP,et al. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoSONE 2011;6:e16393.
8. Rakoff-Nahoum S, Hao L, Medzhitov R. Role of toll-like receptorsin spontaneous commensal-dependent colitis. Immunity 2006;25:319–29.
9. Uronis JM,MuhlbauerM,HerfarthHH,RubinasTC, JonesGS, JobinC.Modulation of the intestinal microbiota alters colitis-associated colo-rectal cancer susceptibility. PLoS ONE 2009;4:e6026.
10. Kado S, Uchida K, Funabashi H, Iwata S, Nagata Y, Ando M, et al.Intestinal microflora are necessary for development of spontaneousadenocarcinoma of the large intestine in T-cell receptor beta chain andp53 double-knockout mice. Cancer Res 2001;61:2395–8.
11. Balish E,Warner T. Enterococcus faecalis induces inflammatory boweldisease in interleukin-10 knockout mice. Am J Pathol 2002;160:2253–7.
12. Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, BalishE, et al. Resident enteric bacteria are necessary for development of
spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun 1998;66:5224–31.
13. SchultzM, TonkonogySL,SellonRK, VeltkampC,Godfrey VL,KwonJ,et al. IL-2-deficient mice raised under germfree conditions developdelayed mild focal intestinal inflammation. Am J Physiol 1999;276:G1461–72.
14. Rakoff-Nahoum S, Medzhitov R. Regulation of spontaneous intestinaltumorigenesis through the adaptor proteinMyD88. Science 2007;317:124–7.
15. Arthur JC, Jobin C. The struggle within: microbial influences oncolorectal cancer. Inflamm Bowel Dis 2011;17:396–409.
16. Rakoff-NahoumS,Paglino J, Eslami-Varzaneh F, EdbergS,MedzhitovR. Recognition of commensal microflora by toll-like receptors isrequired for intestinal homeostasis. Cell 2004;118:229–41.
17. Pull SL, Doherty JM, Mills JC, Gordon JI, Stappenbeck TS. Activatedmacrophages are an adaptive element of the colonic epithelial pro-genitor niche necessary for regenerative responses to injury. Proc NatlAcad Sci U S A 2005;102:99–104.
18. HuB, Elinav E,Huber S, BoothCJ, Strowig T, Jin C, et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 andNLRC4. Proc Natl Acad Sci U S A 2010;107:21635–40.
19. Chen GY, Shaw MH, Redondo G, Nunez G. The innate immunereceptor Nod1 protects the intestine from inflammation-inducedtumorigenesis. Cancer Res 2008;68:10060–7.
20. Zaki MH, Vogel P, Body-Malapel M, LamkanfiM, Kanneganti TD. IL-18 production downstream of the Nlrp3 inflammasome confersprotection against colorectal tumor formation. J Immunol 2010;185:4912–20.
21. Zaki MH, Vogel P, Malireddi RK, Body-Malapel M, Anand PK, Bertin J,et al. The NOD-like receptor NLRP12 attenuates colon inflammationand tumorigenesis. Cancer Cell 2011;20:649–60.
22. Allen IC, TeKippe EM, Woodford RM, Uronis JM, Holl EK, Rogers AB,et al. The NLRP3 inflammasome functions as a negative regulator oftumorigenesis during colitis-associated cancer. J Exp Med 2010;207:1045–56.
23. Chen GY, Liu M, Wang F, Bertin J, Nunez G. A functional role for Nlrp6in intestinal inflammation and tumorigenesis. J Immunol 2011;186:7187–94.
Intestinal Bacteria Suppress Tumorigenesis
www.aacrjournals.org Cancer Res; 73(24) December 15, 2013 7209
on June 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 28, 2013; DOI: 10.1158/0008-5472.CAN-13-0827

24. Tanaka T, Kohno H, Suzuki R, Yamada Y, Sugie S, Mori H. A novelinflammation-related mouse colon carcinogenesis model induced byazoxymethane and dextran sodium sulfate. Cancer Sci 2003;94:965–73.
25. Kitajima S, Takuma S, Morimoto M. Changes in colonic mucosalpermeability in mouse colitis induced with dextran sulfate sodium.Exp Anim 1999;48:137–43.
26. Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, et al.IKKbeta links inflammation and tumorigenesis in a mouse model ofcolitis-associated cancer. Cell 2004;118:285–96.
27. Suzuki R, Kohno H, Sugie S, Tanaka T. Sequential observations on theoccurrence of preneoplastic and neoplastic lesions in mouse colontreated with azoxymethane and dextran sodium sulfate. Cancer Sci2004;95:721–7.
28. Erridge C. Endogenous ligands of TLR2 and TLR4: agonists or assis-tants? J Leukoc Biol 2010;87:989–99.
29. Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA,et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor4, promoting lethal, endotoxin-induced shock. Nat Med 2007;13:1042–9.
30. Chen GY, Nunez G. Sterile inflammation: sensing and reacting todamage. Nat Rev Immunol 2010;10:826–37.
31. Suaeyun R, Kinouchi T, Arimochi H, Vinitketkumnuen U, Ohnishi Y.Inhibitory effects of lemon grass (Cymbopogon citratus Stapf) onformation of azoxymethane-induced DNA adducts and aberrant cryptfoci in the rat colon. Carcinogenesis 1997;18:949–55.
32. Hudcovic T, Stepankova R, Kozakova H, Hrncir T, Tlaskalova-Hogen-ovaH. Effects ofmonocolonizationwith Escherichia coli strainsO6K13and Nissle 1917 on the development of experimentally induced acuteand chronic intestinal inflammation in germ-free immunocompetentand immunodeficient mice. Folia Microbiol (Praha) 2007;52:618–26.
33. Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, et al.Regulation of inflammatory responsesbygutmicrobiota andchemoat-tractant receptor GPR43. Nature 2009;461:1282–6.
34. Pils MC, Bleich A, Prinz I, Fasnacht N, Bollati-Fogolin M, Schippers A,et al. Commensal gut flora reduces susceptibility to experimentallyinduced colitis via T-cell-derived interleukin-10. Inflamm Bowel Dis2011;17:2038–46.
35. Li Y, Kundu P, SeowSW, deMatos CT, Aronsson L, Chin KC, et al. Gutmicrobiota accelerate tumor growth via c-jun and STAT3 phosphor-ylation in APCMin/þ mice. Carcinogenesis 2012;33:1231–8.
36. Malvin NP, Seno H, Stappenbeck TS. Colonic epithelial response toinjury requires Myd88 signaling in myeloid cells. Mucosal Immunol2012;5:194–206.
37. Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L,et al. An essential role for NOD1 in host recognition of bacterialpeptidoglycan containing diaminopimelic acid. Nat Immunol 2003;4:702–7.
38. Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD.The NLRP3 inflammasome protects against loss of epithelial integrityand mortality during experimental colitis. Immunity 2010;32:379–91.
39. Normand S, Delanoye-Crespin A, Bressenot A, Huot L, Grandjean T,Peyrin-Biroulet L, et al. Nod-like receptor pyrin domain-containingprotein 6 (NLRP6) controls epithelial self-renewal and colorectal car-cinogenesis upon injury. Proc Natl Acad Sci U S A 2011;108:9601–6.
40. Allavena P, Germano G, Marchesi F, Mantovani A. Chemokines incancer related inflammation. Exp Cell Res 2011;317:664–73.
41. PopivanovaBK,KitamuraK,WuY,KondoT, KagayaT, KanekoS, et al.Blocking TNF-alpha in mice reduces colorectal carcinogenesis asso-ciated with chronic colitis. J Clin Invest 2008;118:560–70.
42. Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S,et al. IL-6 and Stat3 are required for survival of intestinal epithelial cellsand development of colitis-associated cancer. Cancer Cell 2009;15:103–13.
43. Wang D, Wang H, Brown J, Daikoku T, Ning W, Shi Q, et al. CXCL1induced by prostaglandin E2 promotes angiogenesis in colorectalcancer. J Exp Med 2006;203:941–51.
44. Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, et al. IL-22BP is regulated by the inflammasome andmodulates tumorigenesisin the intestine. Nature 2012;491:259–63.
45. Muzio M, Ni J, Feng P, Dixit VM. IRAK (Pelle) family member IRAK-2and MyD88 as proximal mediators of IL-1 signaling. Science1997;278:1612–5.
46. Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z. MyD88: an adapterthat recruits IRAK to the IL-1 receptor complex. Immunity 1997;7:837–47.
47. Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M,et al. Targeted disruption of theMyD88 gene results in loss of IL-1- andIL-18-mediated function. Immunity 1998;9:143–50.
48. Salcedo R,Worschech A, CardoneM, Jones Y, Gyulai Z, Dai RM, et al.MyD88-mediated signaling prevents development of adenocarcino-mas of the colon: role of interleukin 18. J Exp Med 2010;207:1625–36.
Zhan et al.
Cancer Res; 73(24) December 15, 2013 Cancer Research7210
on June 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 28, 2013; DOI: 10.1158/0008-5472.CAN-13-0827

2013;73:7199-7210. Published OnlineFirst October 28, 2013.Cancer Res Yu Zhan, Po-Ju Chen, William D. Sadler, et al. Caused by Epithelial InjuryGut Microbiota Protects against Gastrointestinal Tumorigenesis
Updated version
10.1158/0008-5472.CAN-13-0827doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2013/10/28/0008-5472.CAN-13-0827.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/73/24/7199.full#ref-list-1
This article cites 48 articles, 15 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/73/24/7199.full#related-urls
This article has been cited by 5 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/73/24/7199To request permission to re-use all or part of this article, use this link
on June 28, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst October 28, 2013; DOI: 10.1158/0008-5472.CAN-13-0827