guillain-barré syndrome -dr sajith sebastian
TRANSCRIPT

GUILLAIN-BARRÉ
SYNDROME
Dr Sajith Sebastian Resident DNB

Guillain-Barré syndrome is an acute inflammatory demyelinating polyneuropathy characterized by progressive muscle weakness and areflexia
It has an annual incidence of 0.6 to 2.4 cases per 100,000 population and occurs at all ages and in both sexes
With the marked decline in the incidence of polio, Guillain-Barré syndrome is now the most common cause of acute flaccid paralysis in healthy people

Worldwide, the annual incidence is about 0.6–4 occurrences per 100,000 people.
Men are one and a half times more likely to be affected than women.
The incidence increases with age; there are approximately 1 cases per 100,000 people aged below 30 years and about 4 cases per 100,000 in those older than 75 years.
The incidence of GBS during pregnancy is 1.7 cases per 100,000 of the population.
Congenital and neonatal Guillain–Barré syndrome have also been reported

PATHOGENESIS Peripheral nerve demyelization in GBS is
believed to be immunologically mediated
Humoral factors and cell-mediated immune phenomena have been implicated in the damage of myelin and/or the myelin-producing Schwann cells


Guillain-Barré syndrome has been reported to follow vaccinations epidural anesthesia thrombolytic agents
It has been associated with some systemic processes, such as Hodgkin's disease SLE Sarcoidosis, and infection with Campylobacter, Lyme disease,
EBV, CMV, HSV, mycoplasma, and recently acquired HIV infection

Campylobacter infectionCampylobacter infection is the most commonly identified precipitant of Guillain-Barré syndrome
A case-control study involving 103 patients with the disease found that 26% of affected individuals had evidence of recent C. jejuni infection compared with 2% of household and 1% of age-matched controls
Seventy percent of those infected with C. jejuni reported a diarrheal illness within 12 weeks before the onset of the neurologic illness
Campylobacter jejuni infection and Guillain-Barre syndromeN Engl J Med 1995 Nov 23;333(21):1374-9

PATHOPHYSIOLOGY
Guillain-Barré is the result of a cell-mediated immune attack on peripheral nerve myelin proteins.
These changes may be caused by cross-reacting antibodies to GM1 ganglioside (present in high concentrations in peripheral nerve myelin) formed in response to similar epitopes expressed by the infecting Campylobacter strain
However, mechanisms other than molecular mimicry may be associated with the production of antibodies to GM1 ganglioside

CELLULAR & HUMORAL IMMUNE MECHANISMS

The main lesions are acute inflammatory demyelinating neuropathy and, particularly in patients with Campylobacter-associated disease
With the autoimmune attack there is an influx of macrophages and other immune-mediated agents that attack myelin, cause inflammation and destruction, and leave the axon unable to support nerve conduction.

The Guillain-Barré syndrome variant known as Miller Fisher syndrome, in which the cranial nerves are affected, is also associated with Campylobacter infection
In these patients cross-reacting antibodies to GQ1b ganglioside, which is present in cranial nerve myelin, have been found

CLINICAL FEATURES
Two-thirds of patients develop the neurologic symptoms 2-4 weeks after what appears to be a benign respiratory or gastrointestinal infection
The initial symptoms are fine paresthesias in the toes and fingertips, followed by lower extremity weakness that may ascend over hours to days to involve the arms, cranial nerves, and in severe cases the muscles of respiration

Early in the course, patients frequently complain of aching or sciatica-like lower back or leg pain
At some point during their illness, up to 25 percent of patients require mechanical ventilation
More than 90% of patients reach the nadir of their function within two to four weeks, with return of function occurring slowly over weeks to months

PHYSICAL EXAMINATION Symmetric limb weakness with diminished or absent
reflexes
Minimal loss of sensation despite paresthesias
Signs of autonomic dysfunction are present in 50 percent of patients, including Cardiac dysrhythmias (asystole, bradycardia, sinus
tachycardia, and atrial/ventricular tachyarrhythmias) Orthostatic hypotension Transient or persistent hypertension Paralytic ileus Bladder dysfunction Abnormal sweating

DIAGNOSTIC STUDIES Nerve conduction studies: Findings depend on subtype of GBS. – The majority show demyelinating pattern while –
some patients may show evidence of axonal loss with little or no demyelination.
Mostly they demonstrate a variety of abnormalities indicating evolving multifocal demyelination Slowed nerve conduction velocities Partial motor conduction block Abnormal temporal dispersion Prolonged distal latencies
A normal study after several days of symptoms, makes the diagnosis of Guillain-Barré syndrome unlikely

Inflammatory markers :ESR is usually raised and CRP is sometimes
Evidence of SIADH or renal dysfunction
Antiganglioside antibodies 1. Anti-GM1 • It is positive in 25% of pts and
is a with worse outcome 2. Anti-GD1a • AMAN subtype of GBS 3. Anti-GQ1b • Miller- Fisher syndrome

Respiratory function tests: – These may show reduced vital capacity, maximal
inspiratory and expiratory pressures.
Arterial blood gases may indicate progressive respiratory failure.
Infection screen : Campylobacter jejuni, Cytomegalovirus, Epstein-Barr virus, Herpes simplex virus, Mycoplasma pneumoniae. HIV antibodies
Radiological: An MRI of the spine may show selective anterior spinal nerve root enhancement with gadolinium and will exclude cervical nerve impingement




SUBTYPES
Acute inflammatory demyelinating polyradiculo -neuropathy = AIDP
Acute motor axonal neuropathy=AMAN •
Acute motor and sensory axonal neuropathy= AMSAN
Miller fisher syndrome =MFS Acute Panautonomic Neuropathy


AIDP Most common form
Accounts for around 85– 90% of cases.
The clinical features are of symmetrical ascending motor weakness with hypo- or areflexia.
Severe cases may develop secondary axonal damage

2/3s have identifiable preceding event
50% begin with paresthesias followed by weakness in legs; 10% begin with arm weakness; rarely begins in face
Ophthalmoplegia: partial 15%, total 5%
Autonomic dysfunction in 65%, arrhythmias, hypotension, urinary
retention in 10-15%,

Progresses for days to 4 weeks
15% with severe disability
Mortality 3-5%
CSF: protein may be normal early, elevated in 90% by clinical nadir, cells< 10 in 95%, >50 suggests HIV
EDX: prolonged F & distal motor latencies, conduction block 30-40% in routine studies

ACUTE MOTOR AXONAL NEUROPATHY (AMAN) More common in Japan ,China, amongst young
people and in the summer months. It has an association with precedent infection
with Campylobacter jejuni. Clinical features are similar to AIDP but tendon
reflexes may be preserved. Electrophysiological testing may distinguish
from other variants as selective motor nerve and axonal involvement is demonstrated.
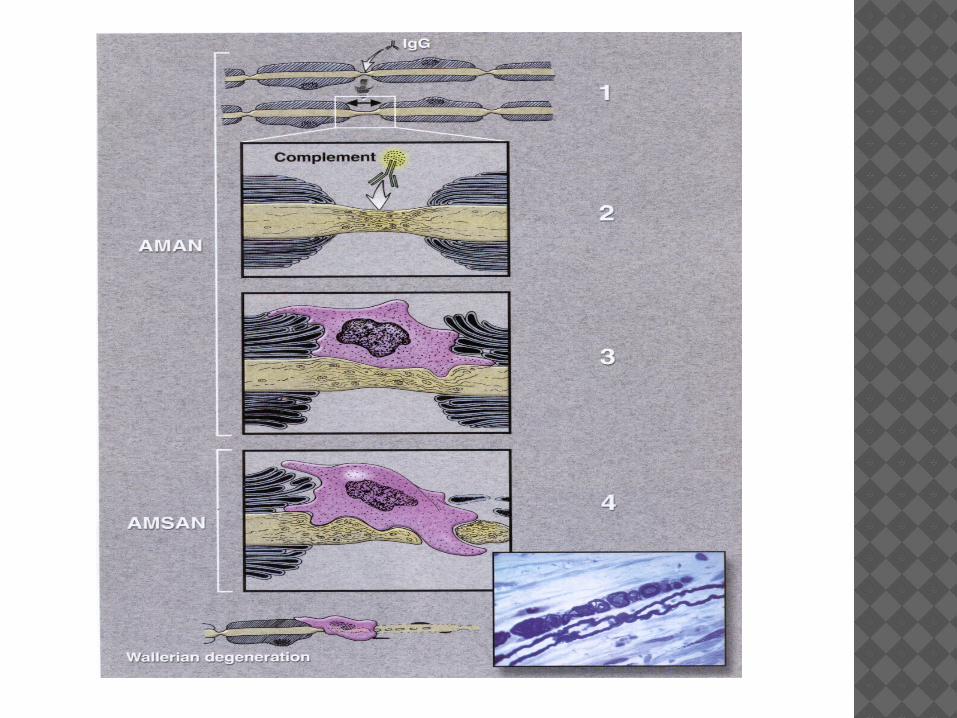
In AMAN the pathological process involves binding of antibodies to ganglioside antigens on the axon cell membrane, macrophage invasion, inflammation and axonal damage.

Pathology: axonal plasmalemma at nodes of Ranvier
sometimes limited to physiologic dysfunction c nodal lengthening. May go on to extension through axonal basal lamina. Most axons recover s Wallerian degeneration
Prognosis similair to AIDP Mortality <5%
EDX: reduced CMAPs c normal F & distal motor latencies and sensory studies. Fibrillations in 2-3 weeks

ACUTE MOTOR AND SENSORY AXONAL NEUROPATHY (AMSAN) variant of GBS in which both motor and sensory
fibres are involved and which can be demonstrated on electrophysiological studies.
It is more severe and associated with prolonged or even partial recovery.
Commonly preceded by diarrhea esp. c. jejuni
Abrupt onset of weakness c rapid progression to quadriplegia & respiratory insufficiency
Clinical features are similar to AMAN but also involve sensory symptoms.

The underlying pathological process is similar to that for AMAN (i.e. antibody mediated axonal damage).
Longer recovery, more residual & mortality 10-15%
EDX: no response in some motor nerves, decreased amplitude of the CMAPs, fibrillations on needle study, absent SNAPs


MILLER FISHER SYNDROME (MFS) Ataxia Areflexia Ophthalmoplegia.
Benign variant and usually not requiring Immuno-therapy
25% of patients may develop limb weakness. Electrophysiological studies show primarily sensory
conduction failure. Antiganglioside antibodies to GQ1b are found in 90% of
patients and are associated with ophthalmoplegia . There have been limited pathological studies in MFS but
demyelination of nerve roots has been demonstrated.

Studies show preferential location of anti-GQ1b to cerebellar molecular layer & Cranial Nerves 3,4 & 6
May act at N-M junction depleting acetylcholine from nerve terminals

ACUTE PANAUTONOMIC NEUROPATHY
Manifests over 1-2 weeks but may be of subacute onset
Frequent preceding infection DTRs lost in 1/3, distal sensory changes
1/4 Albumino cytologic dissociation EDX: NCVs usually normal Recovery is gradual and incomplete


TREATMENT The main modalities of therapy for
Guillain-Barré syndrome includePlasmapheresis andAdministration of intravenous immune
globulin
Supportive care and monitoring.


IV IG-within 2 or possibly 4 weeks of the onset of neuropathic symptoms;
Corticosteroids are not recommended for the management of GBS.
sequential treatment with PE followed by IV IG or immunoabsorption followed IV IG is not recommended for patients with GBS
PE and IV IG are treatment options for children with severe GBS


Supportive Care • Airway • respiratory • Cardiovascular • Gastrointestinal • Neurological • Psychological Rehabilitation

CAUSES OF DEATH
ARDS, sepsis, pulmonary emboli, and unexplained cardiac arrest.
Other Factors associated with a poorer outcome includeOlder ageSevere, rapidly progressive diseaseProlonged mechanical ventilation (>1
month)Persistent, severely abnormal findings on
electromyography

PROGNOSIS
The majority of patients with GBS either recover completely or are left with only minor deficits eg, distal numbness or foot-drop
However, 5 to 10 percent of patients will suffer permanent disabling weakness, imbalance, or sensory loss
3 to 8 percent of patients die despite intensive care
There may be a recurrence in 2–5% of cases. The mortality from GBS ranges from 2–12%.

Thank You