
Multiple sequence alignments and motif discovery
Tutorial 5

• Multiple sequence alignment– ClustalW– Muscle
• Motif discovery– MEME– Jaspar
Multiple sequence alignments and motif discovery

• More than two sequences– DNA– Protein
• Evolutionary relation– Homology Phylogenetic tree– Detect motif
Multiple Sequence Alignment
GTCGTAGTCG-GC-TCGACGTC-TAG-CGAGCGT-GATGC-GAAG-AG-GCG-AG-CGCCGTCG-CG-TCGTA-AC
A
D B
CGTCGTAGTCGGCTCGACGTCTAGCGAGCGTGATGCGAAGAGGCGAGCGCCGTCGCGTCGTAAC

• Dynamic Programming– Optimal alignment– Exponential in #Sequences
• Progressive– Efficient– Heuristic
Multiple Sequence Alignment
GTCGTAGTCG-GC-TCGACGTC-TAG-CGAGCGT-GATGC-GAAG-AG-GCG-AG-CGCCGTCG-CG-TCGTA-AC
A
D B
CGTCGTAGTCGGCTCGACGTCTAGCGAGCGTGATGCGAAGAGGCGAGCGCCGTCGCGTCGTAAC

ClustalW
“CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice”, J D Thompson et al

ClustalW
• Progressive– At each step align two existing alignments or
sequences– Gaps present in older alignments remain fixed
-TGTTAAC-TGT-AAC-TGT--ACATGT---CATGT-GGC

ClustalW - Inputhttp://www.ebi.ac.uk/Tools/clustalw2/index.html
Input sequences
Gap scoring
Scoring matrix
Email address
Output format

ClustalW - Output
Match strength in decreasing order: * : .

ClustalW - Output

ClustalW - Output

ClustalW - Output

ClustalW - Output
Pairwise alignment scores
Building alignment
Final score
Building tree

ClustalW - Output

ClustalW Output
Sequence names Sequence positions
Match strength in decreasing order: * : .

ClustalW - Output

ClustalW - Output
Branch length

ClustalW - Output

ClustalW - Output
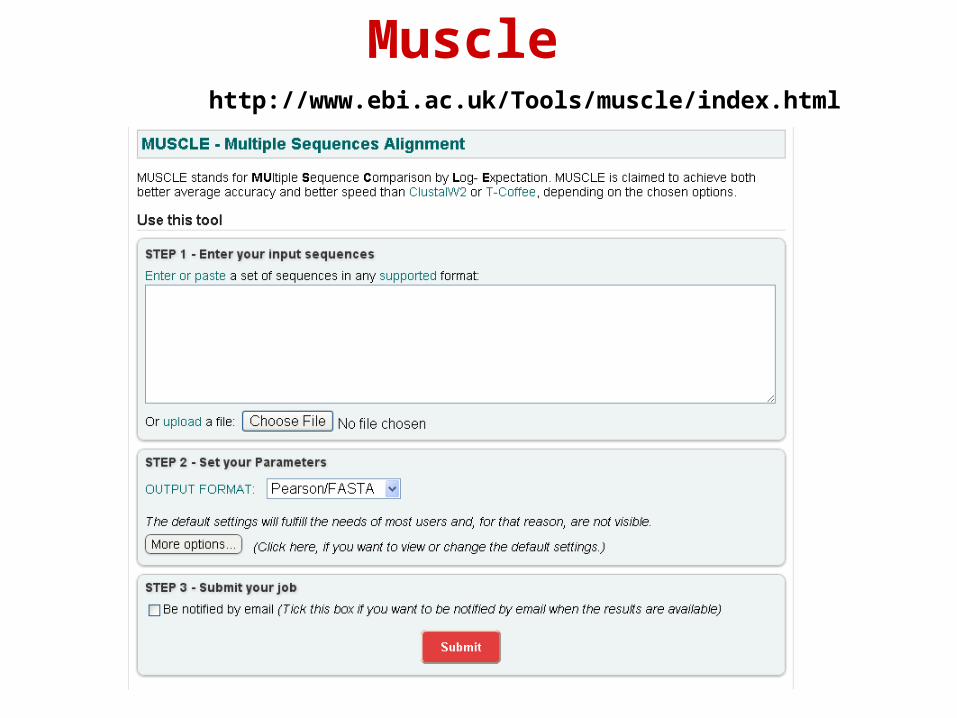
http://www.ebi.ac.uk/Tools/muscle/index.html
Muscle

Muscle - output
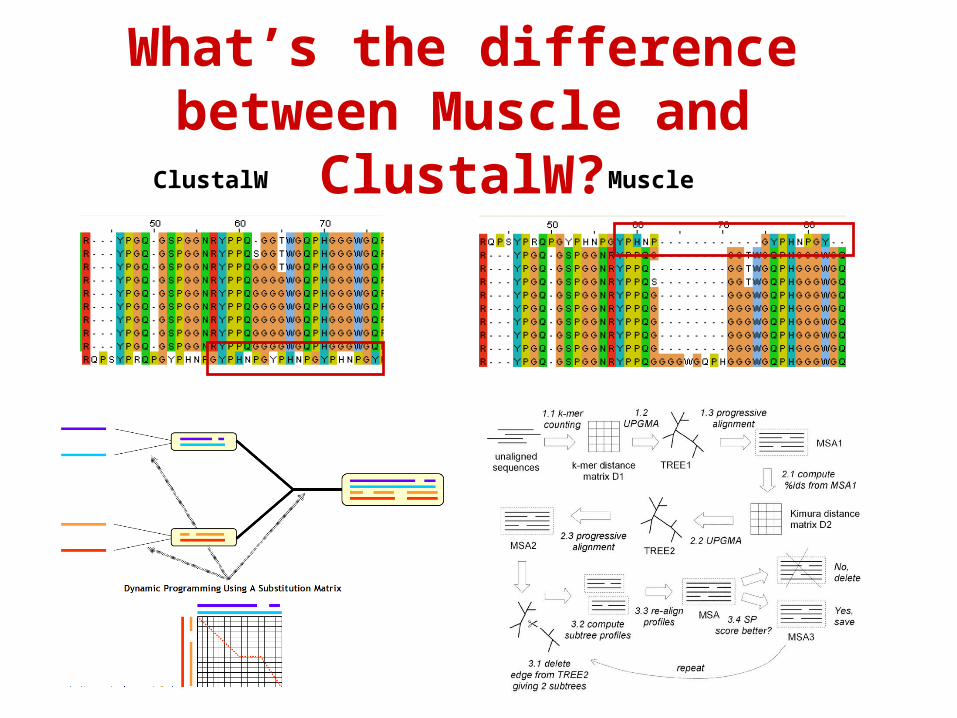
What’s the difference between Muscle and ClustalW?
ClustalW Muscle

http://www.megasoftware.net/index.html

Can we find motifs using multiple sequence alignment?
1 2 3 4 5 6 7 8 9 10
A 0 0 0 0 0 0.5 1/6 1/3 0 0
D 0 0.5 1/3 0 0 1/6 5/6 1/6 0 1/6
E 0 0 2/3 1 0 0 0 0 1 5/6
G 0 1/6 0 0 1 1/3 0 0 0 0
H 0 1/6 0 0 0 0 0 0 0 0
N 0 1/6 0 0 0 0 0 0 0 0
Y 1 0 0 0 0 0 0.5 0.5 0 0
1 3 5 7 9..YDEEGGDAEE....YDEEGGDAEE....YGEEGADYED....YDEEGADYEE....YNDEGDDYEE....YHDEGAADEE.. * :** *:
MotifA widespread pattern with a biological significance

Can we find motifs using multiple sequence alignment?
YES! NO

MEME – Multiple EM* for Motif finding
• http://meme.sdsc.edu/• Motif discovery from unaligned sequences
– Genomic or protein sequences• Flexible model of motif presence (Motif can be absent in
some sequences or appear several times in one sequence)
*Expectation-maximization

MEME - InputEmail addres
s
Input file (fasta file)
How many times in each
sequence?
How many motifs?
How many sites?
Range of motif
lengths

MEME - Output
Motif score

MEME - Output
Motif length
Number of times
Motif score

MEME - Output
Low uncertainty
=
High information content

MEME - Output
Multilevel Consensus

Sequence names
Position in sequence
Strength of match
Motif within sequence
MEME - Output

Overall strength of motif matches
Motif location in the input sequence
MEME - OutputSequence names

MAST
• Searches for motifs (one or more) in sequence databases:– Like BLAST but motifs for input– Similar to iterations of PSI-BLAST
• Profile defines strength of match– Multiple motif matches per sequence– Combined E value for all motifs
• MEME uses MAST to summarize results: – Each MEME result is accompanied by the MAST result for
searching the discovered motifs on the given sequences.
http://meme.sdsc.edu/meme4_4_0/cgi-bin/mast.cgi

MEME - InputEmail
address
Input file (motifs)
Database

JASPAR
• Profiles – Transcription factor binding sites– Multicellular eukaryotes– Derived from published collections of experiments
• Open data accesss

JASPAR• profiles
– Modeled as matrices.– can be converted into PSSM for scanning genomic
sequences.
1 2 3 4 5 6 7 8 9 10
A 0 0 0 0 0 0.5 1/6 1/3 0 0
D 0 0.5 1/3 0 0 1/6 5/6 1/6 0 1/6
E 0 0 2/3 1 0 0 0 0 1 5/6
G 0 1/6 0 0 1 1/3 0 0 0 0
H 0 1/6 0 0 0 0 0 0 0 0
N 0 1/6 0 0 0 0 0 0 0 0
Y 1 0 0 0 0 0 0.5 0.5 0 0

Search profile
http://jaspar.genereg.net/

scoreorganism logoName of gene/protein







