Download - Achalasia in father and son

Achalasia in Father and Son D O N A L D M A C K L E R , M D , and R I C H A R D S C H N E I D E R , M D
Achalasia has not been previously reported in father and son. A man, age 38, and his father, age 66, presented two years apart with symptoms o f dysphagia and aspiration. The diagnosis o f achalasia was made on the basis o f x-ray and endoscopic findings and on esophageal motility studies performed on the father. Both responded well to pneumatic dilatation and have been asymptomatic for 24 months and 6 months, respectively. These cases again suggest a genetic basis for achalasia.
Although the etiology of achalasia has never been fully defined, genetic factors may play a role, as suggested by a few reports of familial occurrence, especially among siblings (1). This inheritance may be through an autosomal recessive gene, as sup- ported by a clustering of cases in families with in- breeding (2-4) and in studies with inbred wire fox t e r r i e r s (5). In 1972, K i l p a t r i c k and Mil les r e p o r t e d a m o t h e r and a d a u g h t e r w i th acha l a s i a , the first k n o w n d o c u m e n t a t i o n o f a c h a l a s i a in a p a r e n t and chi ld (6). H e r e w e d e s c r i b e a s e c o n d e x a m p l e o f fa- mil ial a c h a l a s i a wi th ve r t i c a l t r a n s m i s s i o n , this t ime f rom fa the r to son. W e r e v i e w e v i d e n c e suppor t ing an a u t o s o m a l r e c e s s i v e m o d e o f i nhe r i t ance for th is
d i sease .
CASE REPORTS
Case 1. This 38-year-old Caucasian male had experi- enced progressive dysphagia for 12 months, with regurgi- tation of undigested food and a 20-pound weight loss. He also noted nocturnal reflux of a frothy mucus associated with choking. The patient has two s ibl ing--a healthy old- er brother and a younger sister With systemic lupus ery- thematosis; both siblings denied dysphagia, aspiration, and regurgitation. These three children were not the prod- uct of a consanguineous marriage.
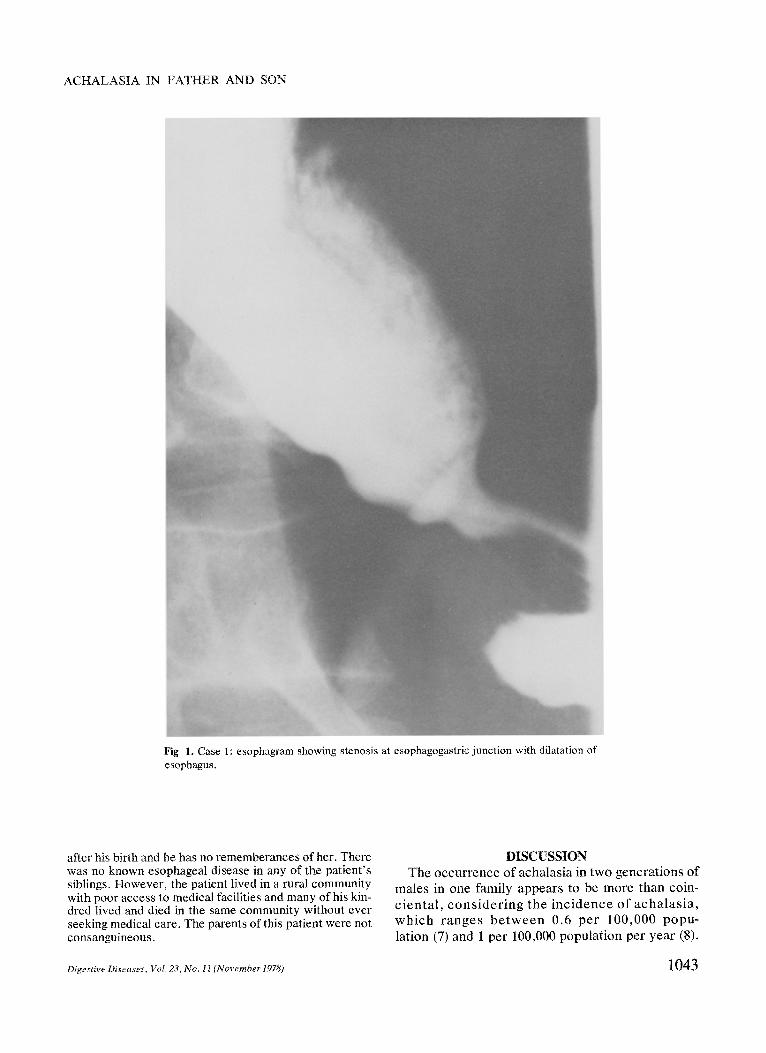
Barium swallow showed that the distal esophagus tapered to a 2- or 3-mm lumen and that the esophagus con- tained retained food (Figure 1). Esophagoscopy revealed normal mucosa and a narrowed esophagagastric junction
From the Department of Gastroenterology, Department of Medicine, St. Thomas Hospital, and The Department of Gastro- enterology, Vanderbilt University Medical School.
Address for reprint requests: Dr. Richard Schneider, Box 380, St. Thomas Hospital, Nashville, Tennessee 37202.
that opened easily to allow passage of the scope. A #50 Fr. Maloney dilator also passed through this narrowed segment easily. The patient was dilated with a Browne- McHardy bag dilator to a pressure of 10 lb/in. 2 for 20 sec two successive times during one session. He has been asymptomatic for 24 months.
Case 2. The father of the first patient was admitted 18 months after his son. This 66-year-old man complained of dysphagia for at least 3 years prior to admission. He was edentulous and had subsisted on a pureed and liquid diet for many years prior to the onset of his dysphagia. At night he would often be awakened by coughing and would regurgitate his food at these times. Ten day prior to ad- mission he aspirated a large amount of his liquid dinner and was seen at another hospital with aspiration pneu- monia.
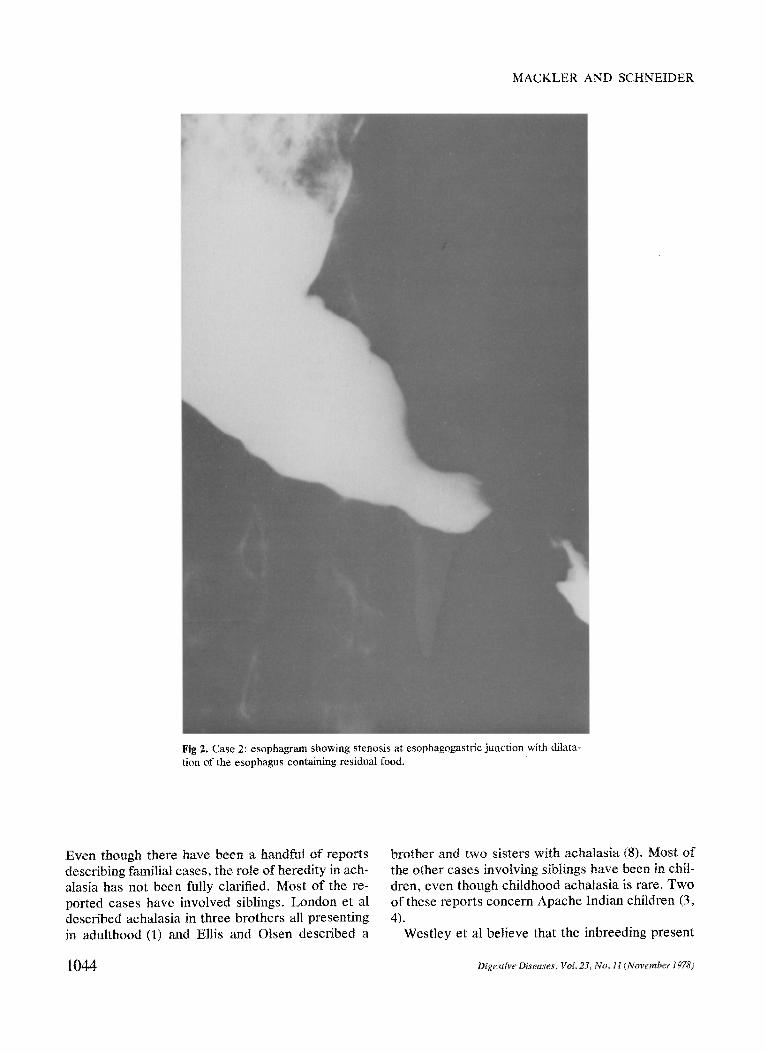
On admission, chest x-ray showed a massively dilated esophagus with an air fluid level at the carina. An upper- gastrointestinal series with barium swallow revealed a markedly dilated and tortuous esophagus with narrowing at the esophagogastric junction (Figure 2).
At esophagoscopy the patient 's esophagus was saccu- lar and partially filled with foul-smelling semisolid food and secretions, preventing adequate visualization of the lower esophageal lumen. After passing a #50 Fr. Ma- loney dilator through the esophagogastric junction with- out difficulty and advancing the retained material into the stomach, the endoscope entered the stomach easily. Esophageal motility studies were performed with a Hon- eywe l l a p p a r a t u s wi th i n t r a lumina l t r a n s d u c e r s . The lower esophageal sphincter produced a pressure of 40 mm Hg and relaxed only partially with swallowing. He propa- gated no peristalsis at all with swallows. He underwent a pneumatic dilatation with a Browne-McHardy bag which was inflated twice to 10 lb/in, z for approximately 30 sec. On follow-up 6 months later he had total relief of his dys- phagia, although he was still endentulous and preferred to imbibe his liquid and pureed diet.
His father died of pneumonia. His mother died shortly
1042 Digestive Diseases, Vol. 23, No. 11 (November 1978)
0002-9211/78/1100-1042505.00/1 �9 Digestive Disease Systems, Inc.
ACHALASIA IN FATHER AND SON
Fig 1. Case 1: esophagram showing stenosis at esophagogastric junction with dilatation of esophagus.
after his birth and he has no rememberances of her. There was no known esophageal disease in any of the patient's siblings. However, the patient lived in a rural community with poor access to medical facilities and many of his kin- dred lived and died in the same community without ever seeking medical care. The parents of this patient were not consanguineous.
DISCUSSION The occurrence of achalasia in two generations of
males in one family appears to be more than coin- c ien ta l , c o n s i d e r i n g the i nc idence o f acha la s i a , w h i c h r anges b e t w e e n 0.6 per 100,000 popu- lation (7) and 1 per 100,000 population per year (8).
Digestive Diseases, Vol, 23, No. l l (November 1978) 1043
MACKLER AND SCHNEIDER
Fig 2. Case 2: esophagram showing stenosis at esophagogastfic junction with dilata- tion of the esophagus containing residual food.
Even though there have been a handful of reports describing familial cases, the role of heredity in ach- alasia has not been fully clarified. Most of the re- ported cases have involved siblings. London et al described achalasia in three brothers all presenting in adulthood (l) and Ellis and Olsen described a
brother and two sisters with achalasia (8). Most of the other cases involving siblings have been in chil- dren, even though childhood achalasia is rare. Two of these reports concern Apache Indian children (3, 4).
Westley et al believe that the inbreeding present
1044 Digestive Diseases, Vol. 23, No. l l (November 1978)

ACHALASIA IN FATHER AND SON
in Apache Indians allowed the expression of a rare gene in the six cases of achalasia they reported in one Apache kindred. Inbreeding among wire fox terriers also results in a high frequency of achalasia, confirmed by radiographic studies, as reported by Osborne et al (5). They analyzed pedigrees accord- ing to Wright's coefficient of inbreeding and found that their dogs with achalasia had approximately 10% more homozygous loci than expected in a ran- dom-mating population. Both groups conclude from existing data that the inheritance of achalasia is probably the result of expression of a pair of mutant genes which are recessively inherited, and that these genes are fully penetrant in the homozygous state.
Assuming an autosomal recessive mode of inher- itance, the son in our study would have inherited one allele from his father, who has two alleles for achalasia, and one allele from his mother, who is a heterozygous carrier and therefore does not have achalasia. Assuming an incidence of achalasia of 1 in 100,000 population, the frequency of this mutant gene would be close of 1/300. The chance of a known homozygote marrying a carrier spouse is 1/ 150 (the carrier frequency). Random mating would thus result in a child with achalasia in 1/300 births-- well within statistical probability. The lack of a ge- netic marker makes further studies on the inher- itance of achalasia difficult, especially since we are unable to detect carriers.
Prior to this report, the only other example of vertically transmitted achalasia was in a mother and daughter (6). Whereas this previous example could have been attributed to either autosomal or to X- linked dominant inheritance, X-linked inheritance is ruled out by this report of father-to-son transmis- sion. In our patients and in the other reported fami- lies, no environmental or infectious agent could be implicated. None of these patients have lived in or traveled to an endemic area for Chagas disease. Further supporting a genetic factor is the fact that other diseases with autosomal recessive inheritance have been repor ted in associa t ion with ach- alasia (9). Although the familial aggregation of ach- alasia may be ascribed to multifactorial causation, McKusick feels there are enough case reports of achalasia occurring in multiple sibling members to justify placing its inheritance under mendelian ge- netic control and to make multifactorial inheritance less likely (10).
It may be valuable to screen the siblings and chil- dren of patients with achalasia by barium swallow
and manometry studies. Many persons tolerate ach- alasia for long periods of time and present initially in an advanced state with a saccular sigmoid esoph- agus which is hard to treat adequately. Both siblings of patient 1 refused barium studies and manometry to detect the presence of early achalasia, and there- fore in asymptomatic family members early screen- ing may not always be successful or possible. In ad- dition, although the presence of a widened, saccu- lar, aperis tal t ic esophagus would be highly suggestive of achalasia, it would nevertheless be difficult to justify pneumatic dilatation or surgical intervention in the individual without dysphagia or aspiration. Continued follow-up and surveillance would then be indicated so that the patient could be treated at the earliest opportunity once symptoms arise and before the development of aspiration and carcinoma. The usefulness of this screening needs to be determined.
ACKNOWLEDGMENTS
We are indebted to Dr. Eric Engel, Director" of the Ge- netics Center, Vanderbilt University Medical School, for aiding with the genetic calculations and reviewing the manuscript. We also thank Dr. Herman Kaplan for refer- ring these patients.
REFERENCES
1. London FA, Raab DE, Fuller J, Olsen AM: Achalasia in three siblings; a rare occurrence. Mayo Clin Pror 52:97- 100, 1977
2. Dayalan N, Chettur L, Ramakrishnan MS: Achalasia of the cardia in sibs. Arch Dis Child 47:115-118, 1972
3. Westley CR, Herbst JJ, Goldman S, Wiser WC: Infantile achalasia: Inherited as an autosomal recessive disorder. J Pediatr 87:243-246, 1975
4. Singh H, Gupta HL, Sethi RS, Khetartal SK: Cardiac ach- alasia in childhood. Postgrad Med J 45:327-335, 1969
5. Osborne CA, Clifford DH, Jessen C: Hereditary esophageal achalasia in dogs. J Am Vet Med Assoc 151:572-581, 1967
6. Kilpatriek ZA, Milles SS: Achalasia in mother and daughter. Gastroenterology 62:1042-1046, 1972
7. Earlam RJ, Ellis FH Jr, Nobrega FT: Achalasia of the esoph- agus in a small urban community. Mayo Clin Proc 44:478- 483, 1969
8. Ellis FH Jr, Olsen AM: Achalasia in the Esophagus. Phila- delphia, WB Saunders, 1969 pp 66-68
9. Rozycki DL, Ruben FJ, Rapin I, Spiro AJ: Autosomal re- cessive deafness: Associated with short stature, vitiligo, muscle wasting and achalasia. Arch Otolaryngol 93:194-197, 1971
10. McKusick VA: Mendelian Inheritance in Man. Catalogs of autosomal dominant, autosomal necessive and X-linked phenotypes. Baltimore, John Hopkins University Press, 1975, p 332
Digestive Diseases, Vol. 23, No. 11 (November 1978) 1045







