co-expression of antibody fab heavy and light chain genes from separate evolved compatible replicons...
TRANSCRIPT

Journal of Immunological Methods 317 (2006) 56–63www.elsevier.com/locate/jim
Research paper
Co-expression of antibody fab heavy and light chain genes fromseparate evolved compatible replicons in E. coli
Brandon Leonard a,b, Vikram Sharma a, Vaughn Smider a,⁎
a IntegriGen, Inc., 42 Digital Dr. Suite 6, Novato, CA 94949, USAb Program in Biomedical Engineering, University of California, San Diego, San Diego, CA, USA
Received 19 April 2006; received in revised form 17 August 2006; accepted 7 September 2006Available online 4 October 2006
Abstract
Antibody molecules bind to antigen with six complementary determining region (CDR) loops, three of which are located oneach variable heavy (VH) and light (VL) chains. Discovery and optimization of antibodies that bind antigen using in vitrotechniques require diversification of one or more of these CDRs. Since antibodies are dimeric, simultaneous diversification ofheavy and light chains on separate genetic elements would allow “chain shuffling” to occur simply and efficiently. Efficientexpression of antibody VH and VL requires that the two separate replicons be compatible with one another, but also have similarproperties, such as copy number in E. coli. Standard plasmids that are compatible with one another in E. coli exist at widelyvariable copy numbers. Recently we described the isolation of ColE1 mutants that have similar copy numbers but differentincompatibility characteristics. Thus, new compatibility groups in the ColE1 family were established. Herein we describe the E.coli expression of VH and VL genes to form a functional Fab. The ability to express antibody heavy and light chains from separatebut compatible high copy plasmids should allow new opportunities in antibody engineering, such as rapid chain shuffling andgeneration of more complex antibody libraries.© 2006 Elsevier B.V. All rights reserved.
Keywords: Antibody engineering; Fab expression; Compatible plasmids; Origin of replication; ColE1
1. Introduction
There are now 18 therapeutic recombinant antibodieson the U.S. market (Reichert et al., 2005). Discovery ofrecombinant antibody therapeutics can involve tradi-tional hybridoma technology (Kohler and Milstein,1975), followed by PCR cloning of the heavy and lightchain genes (Larrick et al., 1989), or can be done purelyin vitro using a variety of display techniques (Hoogen-
⁎ Corresponding author. Present address: Department of MolecularBiology, The Scripps Research Institute, SR202, 10550 N. TorreyPines Rd., La Jolla, CA 92037, USA.
E-mail address: [email protected] (V. Smider).
0022-1759/$ - see front matter © 2006 Elsevier B.V. All rights reserved.doi:10.1016/j.jim.2006.09.006
boom, 2005). With regards to the latter, phage display(Smith and Petrenko, 1997), E. coli surface display(Daugherty et al., 1999), yeast display (Boder andWittrup, 1997), ribosome display (Hanes and Pluckthun,1997), and mRNA display (Wilson et al., 2001) have allbeen used as vehicles for expressing large antibodylibraries and selecting antigen binders to various targets.Additionally, these techniques are also used to “affinitymature” antibody leads, in a process analogous to so-matic hypermutation, which occurs naturally in thevertebrate immune response.
E. coli is a workhorse organism for the discovery,expression, and purification of antibody fragments. In-deed, both phage display and E. coli surface display

57B. Leonard et al. / Journal of Immunological Methods 317 (2006) 56–63
utilize this bacterium. Furthermore biochemical charac-terization and structural biology are often done usingFab fragments produced in E. coli (Ulrich et al., 1995).The backbones of the plasmid and phagemid vectorsused in these systems are based on those of some of theoriginal recombinant DNA work (Bolivar et al., 1977).In general they use standard promoters like lac or tac todrive antibody expression, leader sequences like omp orpho to target the antibody into the non-reducing peri-plasm, and origins of replication like ColE1, which isfound in several standard vectors like pBluescript, thepUC series, or the lower copy pBR322.
Antibody molecules are dimeric proteins composed ofa heavy chain and light chain, both of which undergosomatic diversity generating processes (Smider and Chu,1997). The diversity of antibodies arises both from the V(D)J recombination of each V-region gene, as well as thecombinatorial association of different heavy and lightchains (Lewis, 1994). InE. coli, Fab fragments consistingof the variable and first constant domain of each chain areoften produced in the periplasm. Standard expressionsystems in E. coli use a single vector, often with onepromoter driving a bicistronic mRNA (Ulrich et al.,1995). These vectors require independent cloning of theheavy and light chain genes, making chain shuffling orhigh throughput analysis of different heavy and lightchain combinations difficult. The dimeric nature ofantibodies suggests that their discovery and expressionsystems may be optimized by VH and VL co-expressionfrom separate vectors, such that any heavy chain could bepaired with any light chain in the cell, without indepen-dent cloning events (Collet et al., 1992). Recent analysisindicates this type of system would allow achievement ofa far deeper complexity of antibody libraries with fewercloning steps and less technical difficulty than standardlibraries based on a bicistronic system (Ostermeier andBenkovic, 2000). This possibility could only be realizedthrough the use of compatible plasmids.
Plasmid compatibility in E. coli is determined by theorigin of replication (Novick, 1987). Most standardplasmid vectors use the ColE1 origin of replication.Plasmids compatible with some ColE1 origins, such aspMB1 and p15A, however, exist at a much lower copynumber in cells. Thus, although attempts to co-expressheavy and light chains from different plasmids havebeen made (Collet et al., 1992), these methods have notbeen widely adopted due to the disparity in copy number(and lack of equal expression control) between thedivergent compatible plasmids. This difficulty ismagnified by the fact that overexpression of a singleantibody heavy or light chain is often toxic to the cell,and optimization of Fab production often necessitates
balancing heavy and light chain synthesis (Humphreyset al., 2002).
We recently described the directed evolution of theColE1 replication origin (Kim et al., 2005). Among themutants discovered, several displayed compatibilitywith wild-type ColE1, and this property could be tran-sferred by cloning the mutant origins into new plasmids.At least two new incompatibility groups of ColE1 wereestablished. Since compatible ColE1 plasmids would bevaluable to the co-expression of any dimeric protein,including antibodies, we evaluated the possibility ofproducing functional antibody Fab by co-expressingheavy and light chain genes from separate vectors in E.coli using the new mutant ColE1 origins. Herein wereport that plasmid levels of cells co-cultured with twocompatible ColE1 plasmids are at equivalent and highcopy numbers, that heavy and light chains can beexpressed separately at levels similar to expression froma single plasmid, and that the antibody is functional inbinding antigen. This compatible plasmid system couldoffer significant benefits in antibody discovery, chainshuffling, and high throughput protein production.
2. Materials and methods
2.1. Strains and plasmids
E. coli XL1-Blue (Stratagene, La Jolla, CA) was usedfor cloning and Top 10F (Invitrogen, Carlsbad, CA) wasused for antibody expression. The pVHVL-wt-Ampvectorcontains the RA7 heavy and light chain genes fused to theCH1 and Cλ2 constant regions, respectively. Fab expres-sion is driven by the lac promoter which produces abicistronic mRNA. Vectors pVHVL-wt-Amp, pBS-3.3-Kan (Kim et al., 2005), and pBS-wt-Amp (pBluescript IISK-, Stratagene) were used in vector construction.
2.2. Construction of pVL-wt-Amp
The pVHVL-wt-Amp vector is shown on the left inFig. 1. The heavy chain gene was removed from pVHVL-wt-Amp using the restriction enzyme Nco I, whichcleaves in the leader sequences of both VH and VL. TheDNA (1 μg) was resolved on a 1% agarose gel and the∼5 kb band was isolated by gel purification usingQIAEXII Agarose Gel Extraction (Qiagen, Chatsworth,CA). The vector (∼100 ng) was then recircularized at15 °C overnight using 40 units of T4 ligase (NewEnglandBiolabs) in 10 μl. Resulting recombinant plasmids wereanalyzed by Nco I restriction digest, and correct sequenceconfirmed by sequencing from the pCT-F primer(CCATGATTACGCCAAGCTT TGGAGCC).

Fig. 1. Plasmids used in the present study. Left, is a schematic of a bicistronic Fab expression vector, pVHVL-wt-Amp, where both the VH and VL aredriven by one promoter on a single plasmid. Middle, shows pVH-3.3-Kan, a new VHCH1 vector residing on the high-copy ColE1 compatible origin3.3. Right, shows a light chain expression vector residing on a ColE1 origin, which is compatible with the 3.3 mutant. This vector expresses lambdalight chains, however a similar vector that expresses kappa constant region has also been constructed (not shown). The two compatible vectors can beselected on amp/kan plates, and can be co-cultured stably without antibiotic. Abbreviations are AmpR, β-lacatmase; KanR, aminoglycosidephosphotransferase; L, leader peptide; LacP, lac promoter. Restriction sites are indicated.
58 B. Leonard et al. / Journal of Immunological Methods 317 (2006) 56–63
2.3. Construction of pVH-3.3-Kan
A Hind III restriction digest was done on pVHVL-wt-Amp to obtain the VH insert. The DNA (1 μg) wasresolved on a 1% agarose gel and the 780 bp VH-CH1band was isolated by gel purification using QIAEXIIAgarose Gel Extraction. The pBS-3.3-Kan vector waslinearized with Hind III, gel purified as described above,and then dephosphorylated with Antarctic Phosphataseaccording to the manufacturer's instructions (NewEngland Biolabs, Beverly, MA). The insert and the vec-tor were then ligated. Recombinant colonies were anal-yzed by PCR using the −50 primer (TTGTGAGCGGATAACAATTTC) and the CH1.1 reverse primer(CCTTGACCAGGCAGCCCAGG). Clones with thecorrect orientation of insert were identified by amplifi-cation of a 675 bp band. The correct sequence wasconfirmed by sequencing with the T7 primer.
2.4. Cotransformations
Transformations were with chemically competent E.coli, using 0.01 μg of each plasmid in both single anddouble transformations. Efficiency was similar with oneor two plasmids. Transformants were plated on LBagar plates containing either 100 μg/ml carbenicillin,25 μg/ml kanamycin, or both antibiotics.
2.5. DNA analysis
Doubly resistant colonies were cultured overnight,and induced as described below. DNAwas minipreppedfrom these cultures (Qiagen, Chatsworth, CA), digested
by Not I restriction, and resolved by 0.8% agarose gelelectrophoresis.
2.6. Antibody preparation (per l)
A 5 ml overnight culture of Top10F E. coli tran-sformed with each DNA was added to 1 l of TerrificBroth (TB) containing 0.4% glucose, supplementedwith the appropriate antibiotics and incubated for 6 to12 h at 30 °C (O.D.600=0.5). The cultures were centri-fuged for 10 min at 3000 g, the pellet resuspended in 1 lof fresh TB (without glucose) containing 0.1 mM IPTG(Pierce, Rockford, IL), and incubated overnight at25 °C. Cultures were harvested by centrifuging for30 min at 3500 g, and resuspending the pellet in 20 ml of20 mM Tris pH 7.4, 1 mM EDTA, and 40% sucrose, andincubated at 4 °C with gentle agitation for 2 h. Thesamples were centrifuged for 30 min at 10,000 g and thesupernatants were saved as the periplasmic extract. Theextracts were used directly, or were dialyzed (10,000MWCO) against 20 mM Tris, 500 mM NaCl for furtherpurification by IMAC. Dialysis buffers were changed3 times over the course of 2 days.
2.7. Slot blot
Prewet Whatman paper was placed under a prewetPure Nitrocellulose Transfer and Immobilization Mem-brane (Schleicher and Schuell Inc., Keene, NH) in a slotblot apparatus (Schleicher and Schuell Inc., Keene,NH). Between 20 μl to 50 μl of each sample were usedin each slot, and adsorbed to the membrane by vacuumforce. Phosphate buffered saline containing 0.05%

59B. Leonard et al. / Journal of Immunological Methods 317 (2006) 56–63
tween (PBST) and 2% nonfat milk (Biorad Laboratories,Hercules, CA) was used to block the membrane for15 min at room temperature. A 1:1000 dilution of eitheranti-lambda antibodies (Sigma-Aldrich Co., St. Louis,Mo) or anti-Fab antibodies (US Biological, Swampscott,Massachusetts) conjugated to horseradish peroxidase(HRP) were used to detect light chain or Fab, respec-tively. The membrane was washed 5 times with PBSTand then developed with the metal enhanced DABsubstrate kit (Pierce, Rockford, Ill).
2.8. IMAC nickel purification
The dialyzed periplasmic extract was applied to 1 mlof packed Probond resin (Invitrogen, Carlsbad). Theresin was washed with five column volumes of 20 mMTris pH 7.4/500 mM NaCl, then with five column vol-umes of 20 mM Tris buffer. Protein was eluted in stepfractions containing one column volume of 10 mM,25 mM, 50 mM, 100 mM, 200 mM, 300 mM, 400 mMor 500 mM imidazole in 20 mM Tris buffer.
All fractions were slot blotted as described aboveusing both the anti-lambda and anti-Fab antibodies fordetection to determine which fraction contained thehighest concentration of antibody. The fractions with thehighest concentrations were used in later experiments.The antibody in these fractions were N80% pure bySDS-PAGE and coomassie staining.
2.9. Western blot
Proteins were resolved on a 15% SDS-polyacryl-amide resolving gel (5% stacking gel) in a minigelapparatus (Hoeffer Scientific Instruments, San Fran-cisco, CA) at 100 V for 1.5 hr. Rainbow Marker (Amer-sham, Piscataway, NJ) was used as a molecular weightstandard. Protein was transferred to a nitrocellulosemembrane in a Trans-blot apparatus (Bio-Rad) accord-ing to the manufacturer's instructions, and developed asdescribed above for the slot blot.
2.10. ELISA
High binding assay plates (Costar, Corning, NY) werecoated with 50 μl of TNF (Biosource, Camarillo, CA),BSA (New England Biolads Inc., Ipswich, MA) and β-Interferon (Sigma-Aldrich Co., St. Louis, Mo) at 1 μg/mlin PBS and incubated at 4 °C overnight. Plates wereblocked with 200 μl PBST containing 2% nonfat milk(Bio-Rad Laboratories, Hercules, CA) for 1 h at 37 °C.Antibody (0.5 μg in 50 μl) was added to the wells andincubated for 1 h at 37 °C. The plates were washed four
times with 150 μl PBST. Antibody was detected with50 μl of a 1 to 1000 dilution of anti-6xHis-HRP (US-Biological, Swampscott, Massachusetts) and incubatedfor 45 min at 37 °C. The plate was washed 5 times withPBST and developed with 50 μl of Quanta Blue (Pierce,Rockford, IL) per well according to the manufacturersinstructions. Fluorescent signal was detected at excitation334 nM/ emission 420 nM.
3. Results
To test whether new compatible ColE1-derivedplasmids would allow functional expression of antibodygenes, we constructed vectors that separately expressedthe heavy and light chain genes of the anti-TNFα FabRA7. The RA7 antibody is of human origin and utilizes aheavy chain V-region derived from VH 3–30 germline,and light chain V-region derived from Vλ2. It specifi-cally binds a non linear epitope in human TNFα with aKD of 30 nM. A vector (pVHVL-wt-Amp) expressingboth VH and VL in Fab format from a single mRNA on awild-type ColE1 backbone was used to construct othervectors and was also used as a control (Fig. 1, left). Avector which expresses the heavy chain of RA7 (pVH-3.3-Kan) was constructed on a plasmid containing the3.3 origin of replication mutant (Kim et al., 2005), and akanamycin resistance selectable marker (Fig. 1, middle).A second plasmid (pVL-wt-Amp) containing the lightchain gene on a wild-type ColE1 origin and ampicillinresistance selectable marker was also constructed (Fig. 1,right). The two vectors both utilize the lac promoter, thusexpression of VH and VL is dependent on the concen-tration of the inducer IPTG. These plasmids, and theirvector alone counterparts (e.g. no antibody gene present)were transformed into E. coli singly, or co-transformedwith a plasmid containing the appropriate antibodychain, origin, and antibiotic selection partner. Thus, ineach co-transformation an AmpR wild-type ColE1vector was always paired with a KanR 3.3 mutant vector.We have already demonstrated that co-transformation oftwo plasmids containing the same origin (either wild-type ColE1 or 3.3) does not lead to stable colonyformation (Kim et al., 2005). Co-transformants wereplated on amp, kan, or amp/kan containing plates. Onlycells that were co-transformed produced doubly resistantcolonies.
3.1. Plasmid content of Fab expressing cells
In order to analyze the DNA plasmid content of Fabproducing cells, colonies were picked and grown in asmall scale culture (5 ml) under inducing conditions (see

60 B. Leonard et al. / Journal of Immunological Methods 317 (2006) 56–63
Materials and Methods). Plasmid minipreps were per-formed on 1.5 ml of each culture to analyze the DNAcontent of the cells (Fig. 2). Plasmids from singly ordoubly transformed cells were analyzed by agaroseelectrophoresis, ethidium bromide staining, and UVvisualization. To clearly determine the presence of bothplasmids, they were digested with Not I, which cleaveseach plasmid only once. The linear species of pVHVL-wt-Amp was seen to migrate at 5.9 kb, as expected. Thesingly transformed pVH-3.3-Kan migrated at a singleband at 4.4 kb, and the digested pVL-wt-Amp migratedas a single band at 5.2 kb. Plasmid derived from cellsdoubly transformed with pVH-3.3-Kan and pVL-wt-Amp produced two bands when digested with Not I, atthe exact positions of the individually transformedminipreps (Fig. 2). The relative intensity of stainingbetween the two plasmids was similar, indicating thatboth plasmids are maintained at similar copy numbers.Similar results were found when the vector alone con-structs were analyzed, indicating that antibody geneexpression had no detectible effect on copy number or
Fig. 2. Plasmid content of cells containing compatible replicons. DNAwas miniprepped from cultures induced to express Fab. Plasmid wasundigested or cleaved with Not I (a single-cutter for all plasmids) asindicated. When co-cultured plasmid was analyzed (lanes 7–8 and 13–14) equivalent amounts of each plasmid were present regardless ofwhether the plasmids expressed antibody (lanes 7–8), or were devoidof Fab genes (lanes 13–14). Similar DNA levels were seen for thesingle vectors alone.
plasmid maintenance. Although miniprep DNA analysisindicated that the plasmid content of the co-culturedplasmids was similar, it was formally possible that theplasmids segregated unequally during cell division, suchthat by the time the minipreps were made, 50% of thecells contained one plasmid and 50% the other. Weaddressed this issue by plating aliquots of the cells onamp, kan, or amp/kan plates after overnight growth andinduction. We found that N95% of colonies derivedfrom plating co-cultured cells retained amp/kan resis-tance after growth and induction. Thus, we concludethat the antibody expressing compatible plasmids arestably maintained within single cells during growth andantibody gene expression.
3.2. Fab expression
Periplasmic extracts were analyzed for the presenceof heavy or light chains. We employed an anti-λantibody to specifically detect light chains, or an anti-Fab antibody which recognizes both heavy and lightchains. The pVHVL-wt-Amp plasmid clearly producedlight chain (Fig. 3, lane 1) and heavy chain (Fig. 3,lane 8). However two distinct bands were seen bywestern blot when detected with anti-λ, and three dis-tinct bands when detected by anti-Fab. Expression of VL
alone from pVL-wt-Amp was significantly less thanpVHVL-wt-Amp (Fig. 3, lanes 2 and 9). Similarly, pVH-3.3-Kan alone produced very little VH when expressedalone. However when pVH-3.3-Kan was co-culturedwith pVL-wt-Amp, both VL and VH levels were verynear that produced by the pVHVL-wt-Amp cells. Asexpected, none of the vector alone cultures produceddetectible Fab, indicating that the secondary antibodiesused in the western blot do not cross react withendogenous E. coli proteins.
The VH and VL expression resulted in two bands(∼31 and 26 kDa) reactive with anti-λ antibodies, andthree bands (∼31, 26, and 24 kDa) reactive with anti-Fab antibodies. Cross reactivity of the antibodies withendogenous E. coli proteins was ruled out by the use ofseveral negative controls in Fig. 3 (e.g. vector alone inlanes 5–7 and 12–14). We reasoned that the ∼26 kDaVL band was a degradation product of full-length VL,removing the C-terminal region including the 6xHis tag.This was confirmed in the experiment shown in Fig. 4.Extract from cells containing pVH-3.3-Kan and pVL-wt-Amp was purified over a metal affinity column andanalyzed using anti-λ and anti-6xHis (Fig. 4). Neitherextract nor purified Fab contained the ∼26 kDa bandwhen analyzed by anti-6xHis (lanes 3 and 4), whereasanalysis by anti-λ showed that extract contained both

Fig. 3. Efficient expression of antibody VH and VL genes in E. coli. Cells that expressed Fab from a single plasmid (lanes 1 and 8), or that expressedVL alone (lanes 2 and 9), VH alone (lanes 3 and 10), or Fab from two plasmids (lanes 4 and 11), or the relevant vectors without VL or VH genes, wereanalyzed for the presence of lambda constant region (left) or with an anti-Fab antibody (right) in a western blot. Fab from two vectors was expressedto similar levels as a single vector (compare lanes 4 and 11 with lanes 1 and 8). Expression in the periplasm was greatly diminished for VL alone (lane2) or VH alone (lane 10). The lower VL band (∼26 kDa in lanes 1 and 4) is a degradation product of the 30 kDa VL band, removing the C-terminuscontaining the 6xHis tag (Fig. 4, and data not shown). These bands are also seen in the anti-Fab gel, migrating above the ∼24 kDa heavy chain.
Fig. 4. Identification of a degradation product of Fab VL. Periplasmicextract (lanes 1 and 3) or IMAC purified Fab (lanes 2 and 4) wasanalyzed by western blot using anti-lambda (lanes 1 and 2) or anti−6xHis (lanes 3 and 4).
61B. Leonard et al. / Journal of Immunological Methods 317 (2006) 56–63
bands, but IMAC purified antibody only contained thefull length band. Taken together these results indicatethat the ∼26 kDa band contains degraded antibodywithout the 6xHis tag. When antibody is purified usingIMAC, this band is removed due to it lacking the metalbinding histidine tail.
3.3. Separately expressed VH and VL form functional Fab
Since we could clearly express VH and VL fromseparate compatible replicons on a small scale, weprepared Fab from 1 L cultures and analyzed its func-tionality. Yield of Fab was 0.55 mg/L for the singleconstruct, and 0.45 mg/L for the cells containing twoplasmids. Antibody molecules contain disulfide bondsthat covalently link the VH to the VL of the Fab throughthe constant regions. To determine whether these bondsformed, we performed western blots in the absence orpresence of the reducing agent dithiothreitol (DTT) andcompared antibody derived from a single vector or fromthe co-cultured compatible vectors. Antibody derivedfrom the single vector (Fig. 5A, lane 1) or separatecompatible vectors (Fig. 5A, lane 2) was seen to migrateas ∼55 kDa and ∼30 kDa bands in the absence of DTT.The presence of DTT resulted in disappearance of the∼55 kDa band in both cases (Fig. 5A, lanes 3 and 4),indicating the presence of disulfide bonds in the un-reduced sample.
We then tested the antigen binding ability of theantibody by ELISA. The RA7 antibody is specific forTNFα. In ELISAwe tested binding to TNFα, as well asthe negative control antigens bovine serum albumin(BSA) and β-interferon (β-IFN). Fab derived from thesingle vector showed specific signal on TNFα but notBSA or β-IFN (Fig. 5B, left). This pattern was also seenwith the antibody derived from separate compatibleplasmids (Fig. 5B, middle). As a further negative con-trol, we replaced the λ light chain gene of the RA7
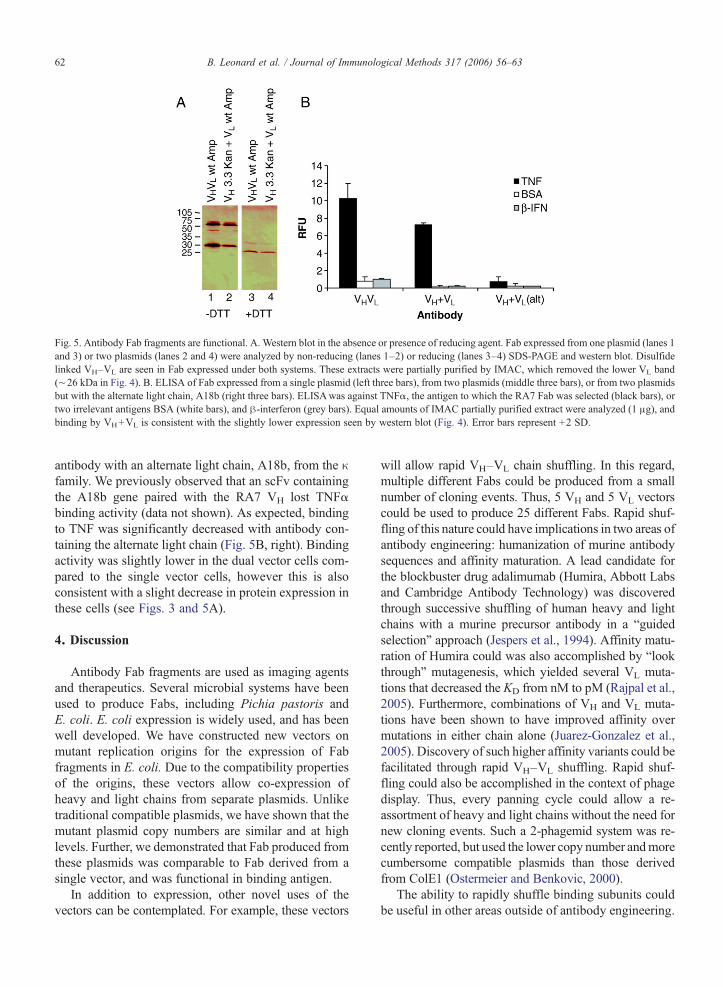
Fig. 5. Antibody Fab fragments are functional. A. Western blot in the absence or presence of reducing agent. Fab expressed from one plasmid (lanes 1and 3) or two plasmids (lanes 2 and 4) were analyzed by non-reducing (lanes 1–2) or reducing (lanes 3–4) SDS-PAGE and western blot. Disulfidelinked VH–VL are seen in Fab expressed under both systems. These extracts were partially purified by IMAC, which removed the lower VL band(∼26 kDa in Fig. 4). B. ELISA of Fab expressed from a single plasmid (left three bars), from two plasmids (middle three bars), or from two plasmidsbut with the alternate light chain, A18b (right three bars). ELISAwas against TNFα, the antigen to which the RA7 Fab was selected (black bars), ortwo irrelevant antigens BSA (white bars), and β-interferon (grey bars). Equal amounts of IMAC partially purified extract were analyzed (1 μg), andbinding by VH+VL is consistent with the slightly lower expression seen by western blot (Fig. 4). Error bars represent +2 SD.
62 B. Leonard et al. / Journal of Immunological Methods 317 (2006) 56–63
antibody with an alternate light chain, A18b, from the κfamily. We previously observed that an scFv containingthe A18b gene paired with the RA7 VH lost TNFαbinding activity (data not shown). As expected, bindingto TNF was significantly decreased with antibody con-taining the alternate light chain (Fig. 5B, right). Bindingactivity was slightly lower in the dual vector cells com-pared to the single vector cells, however this is alsoconsistent with a slight decrease in protein expression inthese cells (see Figs. 3 and 5A).
4. Discussion
Antibody Fab fragments are used as imaging agentsand therapeutics. Several microbial systems have beenused to produce Fabs, including Pichia pastoris andE. coli. E. coli expression is widely used, and has beenwell developed. We have constructed new vectors onmutant replication origins for the expression of Fabfragments in E. coli. Due to the compatibility propertiesof the origins, these vectors allow co-expression ofheavy and light chains from separate plasmids. Unliketraditional compatible plasmids, we have shown that themutant plasmid copy numbers are similar and at highlevels. Further, we demonstrated that Fab produced fromthese plasmids was comparable to Fab derived from asingle vector, and was functional in binding antigen.
In addition to expression, other novel uses of thevectors can be contemplated. For example, these vectors
will allow rapid VH–VL chain shuffling. In this regard,multiple different Fabs could be produced from a smallnumber of cloning events. Thus, 5 VH and 5 VL vectorscould be used to produce 25 different Fabs. Rapid shuf-fling of this nature could have implications in two areas ofantibody engineering: humanization of murine antibodysequences and affinity maturation. A lead candidate forthe blockbuster drug adalimumab (Humira, Abbott Labsand Cambridge Antibody Technology) was discoveredthrough successive shuffling of human heavy and lightchains with a murine precursor antibody in a “guidedselection” approach (Jespers et al., 1994). Affinity matu-ration of Humira could was also accomplished by “lookthrough” mutagenesis, which yielded several VL muta-tions that decreased the KD from nM to pM (Rajpal et al.,2005). Furthermore, combinations of VH and VL muta-tions have been shown to have improved affinity overmutations in either chain alone (Juarez-Gonzalez et al.,2005). Discovery of such higher affinity variants could befacilitated through rapid VH–VL shuffling. Rapid shuf-fling could also be accomplished in the context of phagedisplay. Thus, every panning cycle could allow a re-assortment of heavy and light chains without the need fornew cloning events. Such a 2-phagemid system was re-cently reported, but used the lower copy number andmorecumbersome compatible plasmids than those derivedfrom ColE1 (Ostermeier and Benkovic, 2000).
The ability to rapidly shuffle binding subunits couldbe useful in other areas outside of antibody engineering.

63B. Leonard et al. / Journal of Immunological Methods 317 (2006) 56–63
For example many proteins are dimers, and the ability torapidly express subunits from different vectors couldfacilitate rapid mutagenesis studies or directed evolu-tion. Systems that could benefit from compatible vectorsinclude polyketide synthetase subunits in antibiotic pro-duction (Cane et al., 1998), or tRNA/synthetase pairs inunnatural amino acid incorporation into target proteins(Xie and Schultz, 2005). Additionally, high throughputstructural studies of dimeric proteins could be facilitatedby rapid subunit cloning and purification using a twoplasmid system, which could be part of the significantoptimization required to clone, express, purify, andcrystallize proteins in large scale structural genomicsprojects (Studier, 2005).
Acknowledgements
The authors thank Bill Heriot and Martin Baker fortechnical support, and Richard Lerner and Kirk Trislerfor useful discussions.
References
Boder, E.T., Wittrup, K.D., 1997. Yeast surface display for screeningcombinatorial polypeptide libraries. Nat. Biotechnol. 15, 553.
Bolivar, F., Rodriguez, R.L., Greene, P.J., Betlach,M.C., Heyneker, H.L.,Boyer, H.W., 1977. Construction and characterization of new cloningvehicles. II. A multipurpose cloning system. Gene 2, 95.
Cane, D.E., Walsh, C.T., Khosla, C., 1998. Harnessing the biosyntheticcode: combinations, permutations, and mutations. Science 282, 63.
Collet, T.A., Roben, P., O'Kennedy, R., Barbas III, C.F., Burton, D.R.,Lerner, R.A., 1992. A binary plasmid system for shuffling combina-torial antibody libraries. Proc. Natl. Acad. Sci. U. S. A. 89, 10026.
Daugherty, P.S., Olsen, M.J., Iverson, B.L., Georgiou, G., 1999.Development of an optimized expression system for the screeningof antibody libraries displayed on the Escherichia coli surface.Protein Eng. 12, 613.
Hanes, J., Pluckthun, A., 1997. In vitro selection and evolution offunctional proteins by using ribosome display. Proc. Natl. Acad.Sci. U. S. A. 94, 4937.
Hoogenboom, H.R., 2005. Selecting and screening recombinantantibody libraries. Nat. Biotechnol. 23, 1105.
Humphreys, D.P., Carrington, B., Bowering, L.C., Ganesh, R., Sehdev,M., Smith, B.J., King, L.M., Reeks, D.G., Lawson, A., Popplewell,A.G., 2002. A plasmid system for optimization of Fab' productionin Escherichia coli: importance of balance of heavy chain and lightchain synthesis. Protein Expr. Purif. 26, 309.
Jespers, L.S., Roberts, A., Mahler, S.M.,Winter, G., Hoogenboom,H.R.,1994. Guiding the selection of human antibodies from phage displayrepertoires to a single epitope of an antigen. Biotechnology (NY) 12,899.
Juarez-Gonzalez, V.R., Riano-Umbarila, L., Quintero-Hernandez, V.,Olamendi-Portugal, T., Ortiz-Leon, M., Ortiz, E., Possani, L.D.,Becerril, B., 2005. Directed evolution, phage display and combina-tion of evolved mutants: a strategy to recover the neutralizationproperties of the scFv version of BCF2 a neutralizing monoclonalantibody specific to scorpion toxin Cn2. J. Mol. Biol. 346, 1287.
Kim, D., Rhee, Y., Rhodes, D., Sharma, V., Sorenson, O., Greener, A.,Smider, V., 2005. Directed evolution and identification of controlregions of ColE1 plasmid replication origins using only nucleotidedeletions. J. Mol. Biol. 351, 763.
Kohler, G., Milstein, C., 1975. Continuous cultures of fused cellssecreting antibody of predefined specificity. Nature 256, 495.
Larrick, J.W., Danielsson, L., Brenner, C.A., Abrahamson,M., Fry, K.E.,Borrebaeck, C.A., 1989. Rapid cloning of rearranged immunoglob-ulin genes from human hybridoma cells using mixed primers and thepolymerase chain reaction. Biochem. Biophys. Res. Commun. 160,1250.
Lewis, S., 1994. The mechanism of V(D)J joining: lessons frommolecular, immunological, and comparative analyses. Adv.Immunol. 56, 27.
Novick, R.P., 1987. Plasmid incompatibility. Microbiol. Rev. 51, 381.Ostermeier, M., Benkovic, S.J., 2000. A two-phagemid system for the
creation of non-phage displayed antibody libraries approachingone trillion members. J. Immunol. Methods 237, 175.
Rajpal, A., Beyaz, N., Haber, L., Cappuccilli, G., Yee, H., Bhatt, R.R.,Takeuchi, T., Lerner, R.A., Crea, R., 2005. A general method forgreatly improving the affinity of antibodies by using combinatoriallibraries. Proc. Natl. Acad. Sci. U. S. A. 102, 8466.
Reichert, J.M., Rosensweig, C.J., Faden, L.B., Dewitz, M.C., 2005.Monoclonal antibody successes in the clinic. Nat. Biotechnol. 23,1073.
Smider, V., Chu, G., 1997. The end-joining reaction in V(D)Jrecombination. Semin. Immunol. 9, 189.
Smith, G.P., Petrenko, V.A., 1997. Phage display. Chem. Rev. 97, 391.Studier, F.W., 2005. Protein production by auto-induction in high
density shaking cultures. Protein Expr. Purif. 41, 207.Ulrich, H.D., Patten, P.A., Yang, P.L., Romesberg, F.E., Schultz, P.G.,
1995. Expression studies of catalytic antibodies. Proc. Natl. Acad.Sci. U. S. A. 92, 11907.
Wilson, D.S., Keefe, A.D., Szostak, J.W., 2001. The use of mRNAdisplay to select high-affinity protein-binding peptides. Proc. Natl.Acad. Sci. U. S. A. 98, 3750.
Xie, J., Schultz, P.G., 2005. Adding amino acids to the geneticrepertoire. Curr. Opin. Chem. Biol. 9, 548.